Новые соединения, содержащие их фармацевтические композиции и способы их применения
Номер патента: 13371
Опубликовано: 30.04.2010
Авторы: Тхупари Джаган, Медгалчи Сьюзен М., Таунсенд Крэйг А., Кухаджа Фрэнсис П., Макфэдден Джилл М.






Формула / Реферат
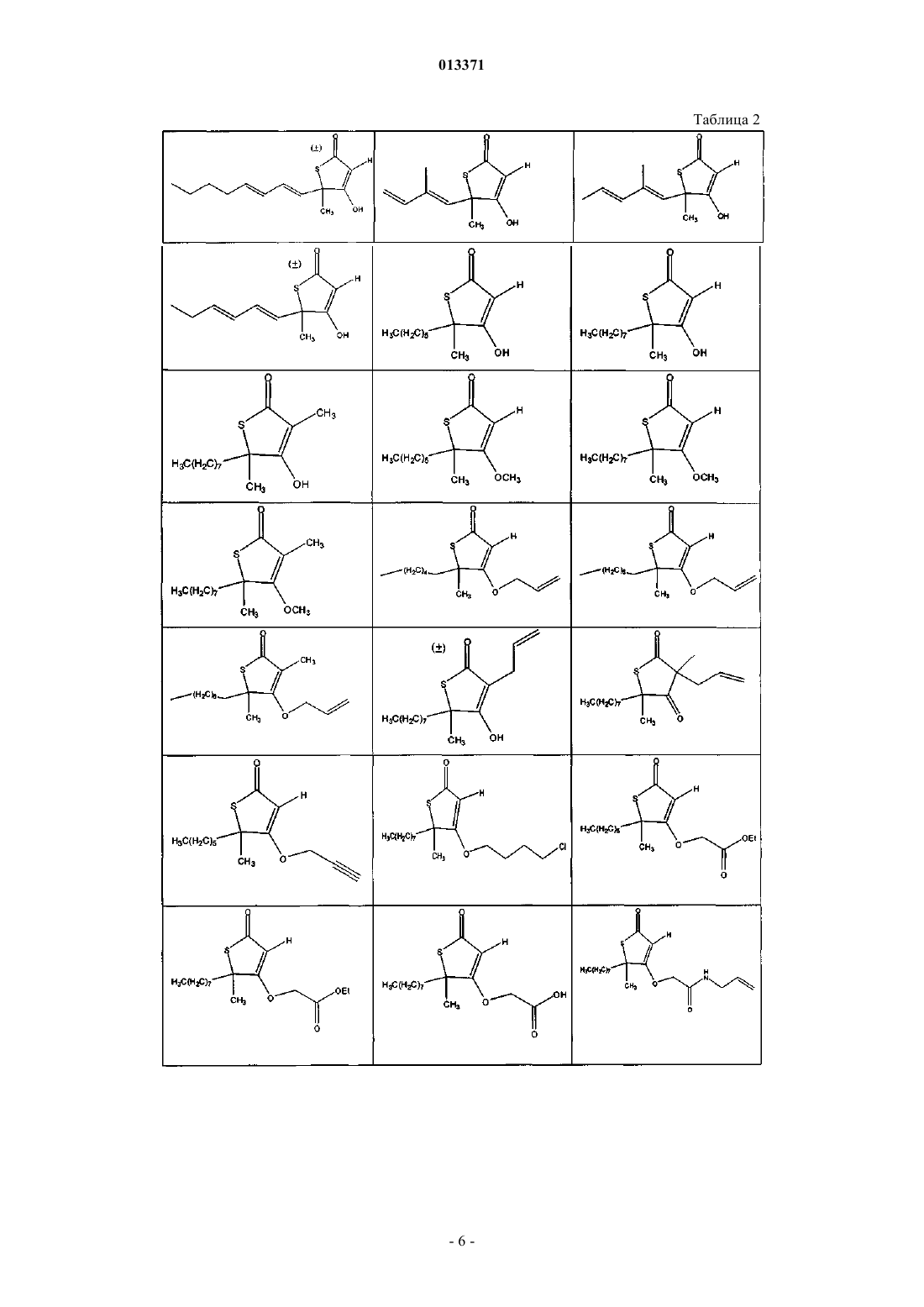
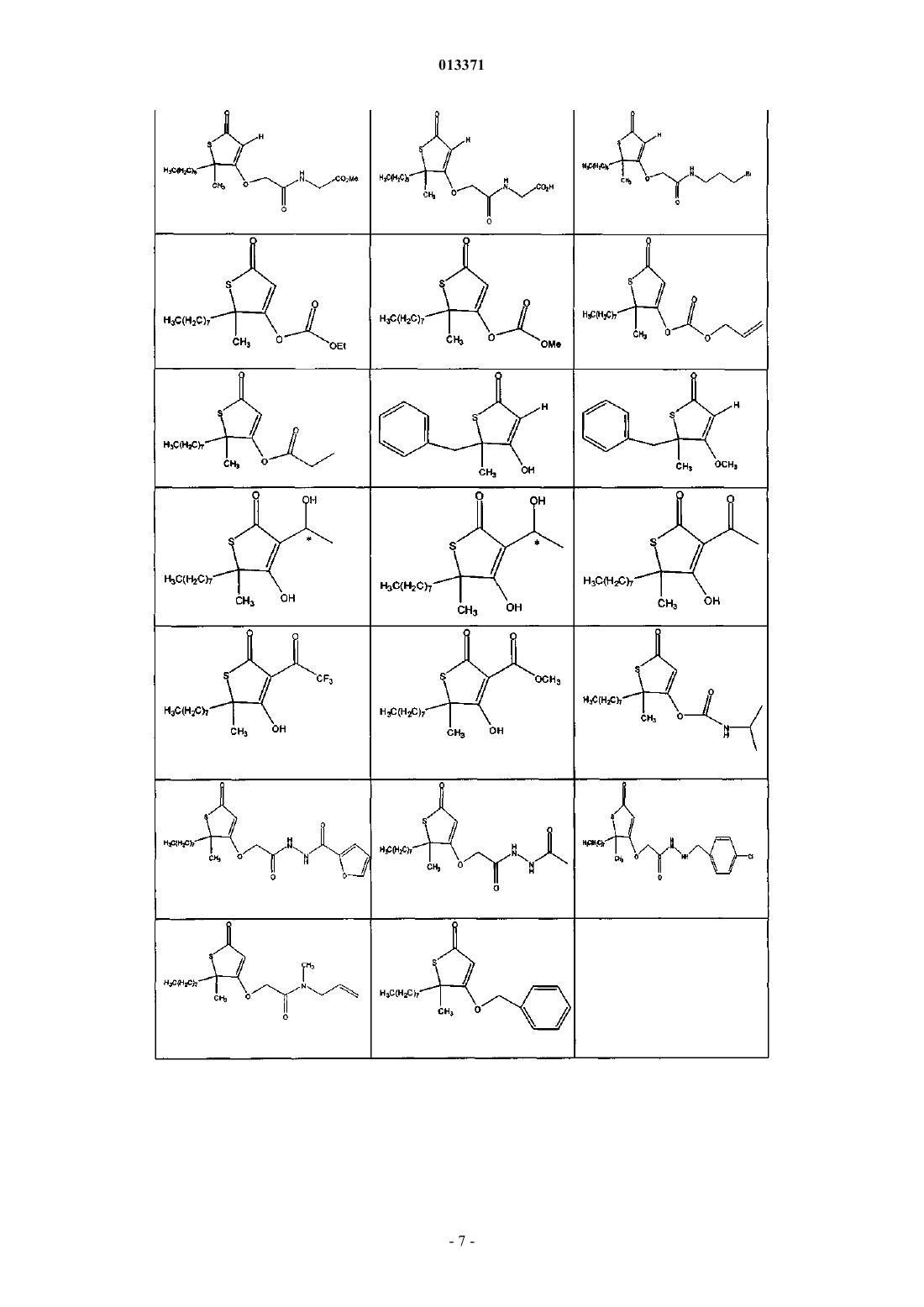
1. Соединение, выбранное из группы, включающей



2. Соединение формулы

3. Фармацевтическая композиция, содержащая фармацевтический разбавитель и соединение по п.2.
4. Способ лечения рака, включающий введение пациенту, нуждающемуся в этом, композиции по п.3.
5. Соединение формулы

6. Фармацевтическая композиция, содержащая фармацевтический разбавитель и соединение по п.5.
7. Способ лечения рака, включающий введение пациенту, нуждающемуся в этом, композиции по п.6.
8. Соединение формулы

9. Фармацевтическая композиция, содержащая фармацевтический разбавитель и соединение по п.8.
10. Способ лечения рака, включающий введение пациенту, нуждающемуся в этом, композиции по п.9.
11. Соединение формулы I

где R1 представляет собой Н;
R2 представляет собой -OCH2C(O)NHR5, где R5 представляет собой C1-C10-арил, содержащий атом галогена;
R3 представляет собой -CH3;
R4 представляет собой -н-C6-C8-алкил.
12. Фармацевтическая композиция, содержащая фармацевтический разбавитель и соединение по п.11.
13. Способ лечения рака, включающий введение пациенту, нуждающемуся в этом, композиции по п.12.
Текст
013371 Предшествующий уровень техники Синтаза жирных кислот. Жирные кислоты имеют три основные роли в физиологии клеток. Во-первых, они являются строительными блоками биологических мембран. Во-вторых, производные жирных кислот служат в качестве гормонов и внутриклеточных мессенджеров. В-третьих, и что представляет особую важность для настоящего изобретения, жирные кислоты являются топливными молекулами, которые могут храниться в жировой ткани в виде триацилглицеридов, которые известны также как нейтральные жиры. Существует четыре основных фермента, вовлеченных в путь синтеза жирных кислот: синтаза жирных кислот (FAS), алкинил-KoA-карбоксилаза (ACC), малат-дегидрогеназа и цитрат-лиаза. Главный фермент FAS катализирует NADPH-зависимую конденсацию предшественников малонил-KoA и алкинил-KoA с продуцированием жирных кислот. NADPH представляет собой восстановитель, который обычно служит в качестве важного донора электронов в двух точках реакционного цикла FAS. Три других фермента (т.е. ACC, малат-дегидрогеназа и цитрат-лиаза) продуцируют необходимые предшественники. В синтез жирных кислот вовлечены также другие ферменты, например ферменты, продуцирующиеFAS имеет определенный Комиссией по ферментам (Е.С.)2.3.1.85 и известна также как синтетаза жирных кислот, лигаза жирных кислот, а также имеет ее систематическое название ацилKoA:малонил-KoA С-ацилтрансфераза (декарбоксилирование, оксоацил- и еноил-восстановление и гидролиз сложного тиоэфира). Существует семь различных ферментов, или каталитических доменов, вовлеченных в катализируемый FAS синтез жирных кислот: алкинилтрансацилаза, малонилтрансацилаза,бета-кетоацилсинтетаза (конденсирующий фермент), бета-кетоацилредуктаза, бета-гидроксиацилдегидраза, еноилредуктаза и тиоэстераза (Wakil, S. J., Biochemistry, 28: 4523-4530, 1989). Все семь из указанных ферментов вместе образуют FAS. Хотя катализируемый FAS синтез жирных кислот подобен у низших организмов, например, таких как бактерии, и высших организмов, например, таких как микобактерии, дрожжи и люди, однако имеются некоторые существенные различия. У бактерий семь ферментативных реакций осуществляются семью отдельными полипептидами, которые являются несвязанными. Они относятся к классу FAS II типа. И наоборот, ферментативные реакции у микобактерии, дрожжей и людей осуществляются многофункциональными полипептидами. Так, например, дрожжи имеют комплекс, состоящий из двух отдельных полипептидов, тогда как у микобактерии и людей все семь реакций осуществляются одним полипептидом. Они относятся к классу FAS I типа. Ингибиторы FAS. Было показано, что различные соединения ингибируют синтазу жирных кислот (FAS). ИнгибиторыFAS могут быть идентифицированы по способности соединения ингибировать ферментативную активность очищенной FAS. Активность FAS может быть определена измерением включения меченного радиоактивным изотопом предшественника (т.е. алкинил-KoA или малонил-KoA) в жирные кислоты или спектрофотометрическим измерением окисления NADPH (Dils, et al., Methods Enzymol., 35: 74-83). В табл. 1 перечислены некоторые ингибиторы FAS. Таблица 1 Типичные ингибиторы ферментов, участвующих в пути синтеза жирных кислот Из четырех ферментов, участвующих в пути синтеза жирных кислот, FAS является предпочтительной мишенью для ингибирования вследствие того, что она действует только в пути синтеза жирных кислот, тогда как три других фермента вовлечены в другие клеточные функции. Поэтому ингибирование одного из трех других ферментов, вероятно, оказывает влияние на нормальные клетки. Из семи ферментативных стадий, осуществляемых FAS, стадия, катализируемая конденсирующим ферментом (т.е. бетакетоацил-синтетазой) и еноилредуктазой, является наиболее простым кандидатом для ингибиторов, которые замедляют или останавливают синтез жирных кислот. Конденсирующий фермент комплекса FAS полностью охарактеризован с точки зрения структуры и функции. Активный центр конденсирующего фермента содержит критический цистеинтиол, который является мишенью антилипидемических реаген-2 013371 тов, например, таких как ингибитор церуленин. Предпочтительные ингибиторы конденсирующего фермента включают широкий диапазон химических соединений, включающих алкилирующие реагенты, окислители и реагенты, способные подвергаться дисульфидному обмену. Связующий карман фермента предпочитает Е, Е диены с длинной цепью. В принципе эффективным ингибитором конденсирующего фермента может быть реагент, содержащий диен в боковой цепи и группу, которая проявляет химическую активность к анионам тиолата. Примером такого соединения является церуленин [(2S,3R)-2,3-эпокси-4-оксо-7,10-додекадиеноиламид] Церуленин ковалентно связывается с критической цистеинтиольной группой в активном центре конденсирующего фермента синтазы жирных кислот, инактивируя указанную ключевую ферментативную стадию (Funabashi, et al., J. Biochem., 105: 751-755, 1989). Хотя отмечалось, что церуленин обладает и другими активностями, однако они проявляются или у микроорганизмов, которые не могут быть релевантными моделями человеческих клеток (например, ингибирование синтеза холестерина в грибах,Omura (1976), Bacteriol. Rev., 40: 681-697, или ослабленный синтез РНК (RNA) у вирусов, Perez, et al.(1991), FEBS, 280: 129-133); происходят в основном при повышенных концентрациях лекарственного средства (ингибирование вирусной HIV протеазы при 5 мг/мл, Moelling, et al. (1990), FEBS, 261: 373-377) или могут быть прямым результатом ингибирования эндогенного синтеза жирных кислот (ингибирование процессинга антигена в В-лимфоцитах и макрофагах, Falo et al. (1987), J. Immunol., 139: 3918-3923). В некоторых данных предполагается, что церуленин не ингибирует специфически миристоилирование белков (Simon, et al., J. Biol. Chem., 267: 3922-3931, 1992). Еще несколько ингибиторов FAS раскрыты в заявке на патент США 08/096908 и ее CIP, поданной 24 января 1994 г., описания которых включены в данный патент посредством ссылки. Включены ингибиторы синтазы жирных кислот, цитрат-лиазы, KoA-карбоксилазы и малат-дегидрогеназы.Antibiotics 39:1211-1218, 1986) описывают триаксин С (иногда называемый WS-1228A), природный ингибитор ацил-KoA-синтетазы, который является продуктом Streptomyces sp. SK-1894. Химическая структура триаксина С: 1-гидрокси-3-(Е,Е,Е-2',4',7'-ундекатриенилидин)триазен. Триаксин С при концентрации 8,7 мкМ вызывает 50% ингибирование ацил-KoA-синтетазы печени крыс; родственное соединение триаксин А ингибирует ацил-KoA-синтетазу посредством механизма, который является конкурентным жирным кислотам с длинной цепью. Ингибирование ацил-KoA-синтетазы является токсичным для клеток животных. Tomoda et al. (Tomoda et al., J. Biol. Chem. 266: 4212-4219, 1991) утверждают, что триаксин С при концентрации 1,0 мкМ вызывает ингибирование роста клеток Раджи (Raji) и, кроме того,как было показано, ингибирует рост клеток Веро (Vero) и Хела (Hela). Tomoda et al. также утверждают,что ацил-KoA-синтетаза оказывает существенное влияние на клетки животных и что ингибирование фермента оказывает летальное действие. В патенте США 5981575 (описание которого включено в данное изобретение посредством ссылки) показано семейство соединений (гамма-замещенные-альфа-метилен-бета-карбокси-гамма-бутиролактоны), ингибирующих синтез жирных кислот и рост опухолевых клеток и вызывающих снижение веса. Соединения, раскрытые в патенте '575, имеют несколько преимуществ по сравнению с природным продуктом церуленином при терапевтических применениях: (1) не содержат высокоактивную эпоксидную группу церуленина; (2) являются стабильными и растворимыми в водном растворе; (3) могут быть получены двухстадийной реакцией синтеза и поэтому легко получаются в больших количествах и (4) легко метятся тритием для получения высокой специфической активности в биохимических и фармакологических анализах. Синтез данного семейства соединений, которые являются ингибиторами синтазы жирных кислот, их применение в качестве средств для лечения опухолевых клеток, экспрессирующих FAS, и их применение в качестве средств для снижения веса описано в патенте США '575. В патенте '575 раскрыто также применение ингибиторов синтазы жирных кислот для системного снижения массы адипоцитов(числа адипоцитов или их размера) и в качестве средства для снижения веса. Основными центрами синтеза жирных кислот у мышей и людей являются печень (см. Roncari, Can.(Goldrick et al., 1974, Clin. Sci. Mol. Med., 46-469-79). Ингибиторы синтеза жирных кислот в качестве антимикробных средств. Церуленин был первоначально выделен в качестве потенциального противогрибкового антибиотика из культурального бульона Cephalosporium caerulens. Структурно церуленин охарактеризован как амид(2R,3S)-эпокси-4-оксо-7,10-транс, трансдодекановой кислоты. Было показано, что механизм его действия включает ингибирование за счет необратимого связывания бета-кетоацил-АСР-синтазы, являющейся конденсирующим ферментом, необходимым для биосинтеза жирных кислот. Церуленин характеризуется, прежде всего, как противогрибковое средство, обладающее активностью против Candida иSaccharomyces sp. Кроме того, была показана его активность in vitro против бактерий, актиномицетов и-3 013371 микобактерий и отсутствие таковой против Mycobacterium tuberculosis. Активность ингибиторов синтеза жирных кислот, и церуленина в частности, не оценивалась против простейших, таких как Toxoplasmagondii, или других инфекционных эукариотических патогенных организмов, таких как PneumocystisSchistosoma. Инфекционные заболевания, которые являются особенно восприимчивыми к лечению, представляют собой заболевания, вызывающие повреждения на наружных доступных поверхностях инфицированного животного. Наружные доступные поверхности включают все поверхности, которые могут быть достигнуты неинвазивными средствами (без разреза или прокола кожи), включающие саму кожную поверхность, слизистые оболочки, такие, которые покрывают назальную, ротовую, желудочно-кишечную или мочеполовую поверхности, и легочные поверхности, такие как альвеолярные мешочки. Восприимчивые к лечению заболевания включают: (1) кожные микозы или дерматомикозы, в особенности вызванныеTrichomonas vaginalis, и (6) легочные заболевания, в особенности вызванные Mycobacteriura tuberculosis,Aspergillus или Pneumocystis carinii. Вызывающие инфекцию микроорганизмы, которые являются восприимчивыми к обработке ингибиторами синтеза жирных кислот, включают Mycobacterium tuberculosis,в особенности полирезистентные к лекарственным средствам штаммы и простейшие, такие какToxoplasma. Для ингибирования роста микробных клеток может быть использовано любое соединение, которое ингибирует синтез жирных кислот. Однако вводимые больному соединения не должны быть в равной степени токсичными для больного и целевых микробных клеток. Соответственно, выгодно выбрать ингибиторы, которые только или предпочтительно воздействуют на целевые микробные клетки. Эукариотические микробные клетки, которые зависят от их собственных эндогенно синтезируемых жирных кислот, будут экспрессировать FAS I типа. Это доказывается как тем фактом, что ингибиторыFAS являются ингибиторами роста, так и тем фактом, что экзогенно добавленные жирные кислоты могут защищать от ингибиторов FAS нормальные клетки больного, но не указанные микробные клетки. Поэтому для лечения инфекций могут быть использованы средства, предотвращающие синтез жирных кислот клетками. В эукариотах жирные кислоты синтезируются с участием FAS I типа и с использованием в качестве субстратов алкинил-KoA, малонил-KoA и NADPH. Таким образом, другие ферменты, которые могут использовать субстраты на указанном пути, могут также оказывать влияние на скорость синтеза жирных кислот и поэтому являются значимыми для микробов, которые зависят от эндогенно синтезируемых жирных кислот. Ингибирование экспрессии или активности любого из указанных ферментов будет оказывать влияние на рост микробных клеток, зависимых от эндогенно синтезируемых жирных кислот. Продукт FAS I типа в различных организмах является разным. Так, например, в грибах S.cerevisiae продукты предпочтительно представляют собой пальмитат и стеарат, стереофицированные в кофермент А. В Mycobacterium smegmatis продукты представляют собой KoA эфиры насыщенных жирных кислот,углеродная цепь которых содержит от 16 до 24 атомов углерода. Указанные липиды часто дополнительно перерабатываются для пополнения клеток, нуждающихся в различных липидных компонентах. Можно ожидать, что ингибирование ключевых стадий при последующей переработке или утилизации жирных кислот будет ингибировать клеточную функцию в любом случае: зависит ли клетка от эндогенных жирных кислот или утилизируется жирная кислота, поступающая с внешней поверхности клеток,и поэтому ингибиторы указанных последующих "даунстрим" стадий не могут быть достаточно избирательными для микробных клеток, зависимых от эндогенных жирных кислот. Однако было обнаружено,что введение ингибитора синтазы жирных кислот I типа в такие микробы делает их более восприимчивыми к ингибированию ингибиторами последующего процессинга и/или утилизации жирных кислот. Введение ингибитора синтеза жирных кислот в комбинации с одним или более ингибиторами последующих стадий биосинтеза липидов и/или утилизации будет оказывать, вследствие синергизма, избирательное действие на микробные клетки, зависимые от эндогенно синтезируемых жирных кислот. Предпочтительные комбинации включают ингибитор FAS и алкинил-KoA-карбоксилазы или FAS и ингибитор MAS. Когда установлено, что млекопитающее инфицировано клетками организма, которые экспрессируют FAS I типа или если FAS найдена в биологической жидкости больного, млекопитающее или больной могут быть подвергнуты лечению введением ингибитора синтеза жирных кислот (патент США 5614551). Ингибирование нейропептида-Y для подавления аппетита и стимуляции снижения веса раскрыто в международной патентной заявкеPCT/US01/05316, описание которой включено в данный патент посредством ссылки. Однако в указанной заявке не описывается или не раскрывается какое-либо из соеди-4 013371 нений, раскрытых в настоящем изобретении. Стимулирование карнитинпальмитоилтрансферазы-1 (СРТ-1) для стимуляции снижения веса раскрыто в заявке на патент США 60/354480, которая включена в данное описание посредством ссылки. В указанной заявке не описывается или не раскрывается какое-либо из соединений, раскрытых в данной заявке. Применение ингибиторов FAS для ингибирования роста раковых клеток раскрыто в патенте США 5759837, описание которого включено в данное изобретение посредством ссылки. В указанном патенте не описывается или не раскрывается какое-либо из соединений, раскрытых в данном описании. Сущность изобретения Обнаружены новые соединения - производные тиофен-2-она, имеющие множество терапевтически ценных свойств, включающих, например, ингибирование FAS, ингибирование NPY, стимуляцию СРТ-1,способность вызывать снижение веса и проявляющих противораковые свойства. Другой объект данного изобретения относится к фармацевтическим композициям, содержащим фармацевтический разбавитель и производное тиофен-2-она. Еще один объект изобретения относится к способам лечения рака у животных и людей введением фармацевтической композиции, содержащей фармацевтический разбавитель и производное тиофен-2-она. Краткое описание чертежей Фиг. 1 показывает схему синтеза для получения тиолактамицина. Фиг. 2 показывает схему синтеза для получения некоторых соединений в соответствии с изобретением. Фиг. 3 показывает схему синтеза для получения некоторых соединений в соответствии с изобретением. Фиг. 4 показывает схему синтеза для получения некоторых соединений в соответствии с изобретением. Фиг. 5 показывает схему синтеза для получения некоторых соединений в соответствии с изобретением. Фиг. 6 показывает схемы синтеза для получения некоторых соединений в соответствии с изобретением. Фиг. 7 показывает схему синтеза для получения соединения в соответствии с изобретением. Фиг. 8 показывает схему синтеза для получения некоторых соединений в соответствии с изобретением. Фиг. 9 показывает две схемы синтеза для получения некоторых соединений в соответствии с изобретением. Фиг. 10 показывает схему синтеза для получения некоторых соединений в соответствии с изобретением. Фиг. 11 показывает результаты тестирования in vivo некоторых соединений в соответствии с изобретением на их способность снижать вес. Фиг. 12 показывает результаты тестирования in vivo соединения в соответствии с изобретением на противораковую активность. Подробное описание изобретения Соединения изобретения могут быть получены традиционными способами. Синтез ряда соединений описан в примерах. Соединения могут быть применены для лечения рака. Один вариант изобретения представлен соединениями, выбранными из следующей группы.-7 013371 Другой вариант изобретения относится к соединению формулы Следующий вариант изобретения относится к фармацевтической композиции, содержащей фармацевтический разбавитель и соединение формулы Следующий вариант настоящего изобретения относится к способу лечения рака, включающему введение пациенту, нуждающемуся в этом, вышеуказанной фармацевтической композиции. Изобретение также относится к фармацевтической композиции, содержащей фармацевтический разбавитель и соединение формулы а также способу лечения рака, включающему введение пациенту, нуждающемуся в этом, такой фармацевтической композиции. Следующий вариант изобретения относится к фармацевтической композиции, содержащей фармацевтический разбавитель и соединение формулы а также к способу лечения рака путем введения пациенту, нуждающемуся в этом, указанной фармацевтической композиции. Другой вариант изобретения относится к соединению формулы IR2 представляет собой ОСН 2 С(O)NHR5, где R5 представляет собой C1-C10-арил, содержащий атом галогена;R4 представляет собой -н-C6-C8-алкил. Следующий вариант настоящего изобретения относится к фармацевтической композиции, содержащей фармацевтический разбавитель и соединение формулы I. Кроме того, изобретение касается способа лечения рака, включающего введение пациенту, нуждающемуся в этом, фармацевтической композиции, содержащей фармацевтический разбавитель и соединение формулы I.-8 013371 Композиции настоящего изобретения могут быть представлены для введения людям и другим животным в виде единичных дозированных форм, таких как таблетки, капсулы, пилюли, порошки, гранулы,стерильные парентеральные растворы или суспензии, пероральные растворы или суспензии, эмульсии типа масло-в-воде и типа вода-в-масле, содержащие подходящие количества соединения, суппозитории и суспензии или растворы в текучей среде. Используемые в данном описании термины фармацевтический разбавитель и фармацевтический носитель имеют одно и то же значение. Для перорального введения могут быть получены либо твердые, либо жидкие единичные дозированные формы. Для приготовления твердых композиций, таких как таблетки, соединение может быть смешано с традиционными ингредиентами, такими как тальк, стеарат магния, дикальцийфосфат, алюмосиликат магния, сульфат кальция, крахмал, лактоза, акация, метилцеллюлоза, и функционально подобными материалами, используемыми в качестве разбавителей или носителей. Капсулы получают смешиванием соединения с инертным фармацевтическим разбавителем и заполнением смеси в твердые желатиновые капсулы соответствующего размера. Мягкие желатиновые капсулы получают механическим включением суспензии соединения с приемлемым растительным маслом, вазелиновым маслом или другим инертным маслом в желатиновую капсулу. Могут быть получены жидкие единичные дозированные формы для перорального введения, такие как сиропы, эликсиры и суспензии. Эти формы могут быть растворены в водном наполнителе вместе с сахаром, ароматическими корригентами и консервантами с образованием сиропа. Суспензии с водным наполнителем могут быть получены с помощью суспендирующего средства, такого как акация, трагакант, метилцеллюлоза и подобных соединений. Жидкие единичные дозированные формы для парентерального введения могут быть получены с использованием соединения и стерильного наполнителя. При получении растворов соединение может быть растворено в воде для инъекций и затем стерилизовано с помощью фильтра перед заполнением раствора в подходящий сосуд или ампулу, которые герметически запаивают. В наполнителе могут быть растворены адъюванты, такие как местный анестетик, консервант и буферные вещества. После заполнения в сосуд композиция может быть заморожена, и под вакуумом удалена вода. Затем лиофилизованный порошок может быть отвешен в сосуд и восстановлен перед использованием. Рассматриваемые для соединений изобретения клинические терапевтические показания включают(2) рак, возникающий во многих тканях, клетки которых избыточно экспрессируют синтазу жирных кислот; и (3) ожирение, обусловленное потреблением продуктов с избыточным количеством калорий. Определение доз и продолжительность лечения будут зависеть от множества факторов, включающих (1) возраст больного, массу тела и функционирование органов (например, функционирование печени и почек); (2) природу заболевания и степень распространения подвергаемого лечению болезненного процесса, а также любой имеющийся значимый сопутствующий патологический процесс и осуществляемую сопутствующую лекарственную терапию и (3) параметры, зависящие от лекарственного средства, такие как путь введения, частота и продолжительность дозирования, необходимые для осуществления лечения,и терапевтический индекс лекарственного средства. Обычно дозы выбирают таким образом, чтобы достичь уровней в сыворотке от 1 до 100 нг/мл с целью получения эффективных концентраций в целевом участке, равных примерно от 1 до 10 мкг/мл. Примеры Изобретение проиллюстрировано, но при этом не ограничено, следующими примерами. Ряд соединений в соответствии с изобретением синтезировали, как описано ниже. Биологическую активность некоторых соединений профилировали следующим образом. Каждое соединение тестировали на (1) ингибирование очищенной человеческой FAS; (2) ингибирование активности синтеза жирных кислот в целых клетках и (3) цитотоксичность против культивируемых клеток карциномы человеческой молочной железы MCF-7, которые, как известно, обладают высокими уровнями FAS и активностью синтеза жирных кислот, с использованием кристаллического фиолетового и анализов ХТТ. Затем выбранные соединения с низкими уровнями цитотоксичности тестировали на снижение веса у мышей Balb/C. Кроме того, типичное соединение из группы, которая показывала значительное снижение веса и низкие уровни цитотоксичности, тестировали на его действие на окисление жирных кислот и активность в отношении карнитин-пальмитоилтрансферазы-1 (СРТ-1), а также гипоталамическую экспрессию NPY нозернанализом мышей Balb/C. Некоторые соединения тестировали также на активность против грамположительных и/или грамотрицательных бактерий. К раствору (S)-тиомолочной кислоты 1 (4,0 г, 37,7 ммоль) в пентане (24 мл) добавляли триметилалкинилальдегид (4,5 мл, 41,5 ммоль) и трифторуксусную кислоту (TFA) (48 мкл). Раствор кипятили с обратным холодильником в течение 20 ч с использованием ловушки Дина-Старка. После охлаждения удаляли растворитель с получением смеси цис:транс-изомеров (2,5:1) соединений 1 и 2 (6,4 г, 99%). В результате перекристаллизации (смесь пентан/Et2O (8:1), -78 С) получали чистое соединение 1.(с, 1 Н). Рацемическое соединение 1 получали также из -тиомолочной кислоты. Общая методика A.(3,3 мл, 1,4 М в н-гексане) и образовавшийся раствор перемешивали в течение 30 мин при 0 С и затем охлаждали до -78 С. Затем с помощью канюли при -78 С добавляли по каплям соединение 1 (800 мг,4,6 ммоль) в ТГФ и полученный раствор перемешивали при -78 С в течение 30 мин. При -78 С добавляли через канюлю транс-2-метил-2-бутеналь (0,4 мл, 4,6 ммоль) в ТГФ (1,4 мл). После перемешивания при-78 С в течение 1,5 ч добавляли 1 н. HCl (25 мл) и раствор экстрагировали Et2O (330 мл). Объединенные органические слои сушили (MgSO4), фильтровали и выпаривали. В результате флэш-хроматографии(смесь 10% EtOAc/гексан, rf=0,1) получали соединение 3 (955 мг, 81%) в виде смеси диастереоизомеров 1,6:1. 1 Н ЯМР (300 МГц, CDCl3) основной диастереоизомер:0,99 (с, 9 Н), 1,40 (с, 3 Н), 1,63 (д, J=6,7 Гц,3 Н), 1,69 (м, 3 Н), 4,36 (с, 1 Н), 5,25 (с, 1 Н), 5,60-5,65 (м, 1 Н); второстепенный диастереоизомер: 0,98 (с,9 Н), 1,59 (с, 3 Н), 1,63 (д, J=6,7 Гц, 3 Н), 1,72 (м, 3 Н), 4,25 (с, 1 Н), 5,07 (с, 1 Н), 5,60-5,64 (м, 1 Н); 13 С ЯМР (75 МГц, CDCl3) основной диастереоизомер:12,5, 13,2, 24,3, 24,8, 60,7, 81,8, 87,9, 126,3,133,8, 178,3. ИК (ATR) 3466, 1743 см-1. Вычислено для C13H22O3S: С, 60,4; Н, 8,58. Найдено С, 60,4; Н, 8,60. Изсоединения 1 (800 мг, 4,59 ммоль) и 2-трансоктеналя (0,58 мл, 5,1 ммоль), следуя общей методике А, после флэш-хроматографии (смесь 10% EtOAc/гексан) получали соединение 4 (1,1 г, 81%) в виде смеси диастереоизомеров 1,2:1. 1 Н ЯМР (300 МГц, CDCl3) основной диастереоизомер:0,85 (т, J=7,2 Гц, 3 Н), 0,97 (ш.с, 9 Н), 1,181,35 (с, 6 Н), 1,56 (с, 3 Н), 2,00-2,08 (м, 2 Н), 2,38 (д, J=5 Гц, 1 Н), 4,15-4,19 (м, 1 Н), 5,13 (с, 1 Н), 5,45-5,59- 10013371 Изсоединения 1 (800 мг, 4,59 ммоль) и 2-трансгексеналя (0,58 мл, 5,1 ммоль), следуя общей методике А, после флэш-хроматографии (смесь 10% EtOAc/гексан) получали соединение 5 (813 мг, 65%) в виде смеси диастереоизомеров 2,4:1. 1 Н ЯМР (300 МГц, CDCl3)0,87 (т, J=7,3 Гц, 3 Н), 0,99 (с, 9 Н), 1,38-1,45 (м, 2 Н), 1,41 (с, 3 Н), 2,02 Изсоединения 1 (800 мг, 4,59 ммоль) и 2-метил-2-пентеналя (0,58 мл, 5,0 ммоль), следуя общей методике А, после флэш-хроматографии (смесь 10% EtOAc/гексан) получали соединение 6 (884 мг, 71%) в виде смеси диастереоизомеров 1,8:1. 1 Н ЯМР (300 МГц, CDCl3)0,93-0,99 (м, 12 Н), 1,40 (с, 3 Н), 1,68 (с, 3 Н), 2,01-2,06 (м, 2 Н), 4,33 (д,J=6,9 Гц, 1 Н), 5,24 (с, 1 Н), 5,48-5,54 (м, 1 Н); 13 С ЯМР (75 МГц, CDCl3)12,6, 13,8, 20,9, 21,1, 24,8, 35,4, 60,6, 81,8, 87,9, 132,6, 133,9, 178,3; ИК (NaCl) 2961, 1767 см-1. Вычислено для C14H24O3S: С, 61,7; Н, 8,88. Найдено С, 61,6; Н, 8,90. Общая методика В. К раствору соединения 3 (3,23 г, 12,5 ммоль) в Cl(CH2)2Cl (115 мл), охлажденному до 0 С, добавляли NEt3 (4,2 мл, 30 ммоль) и 2,4-динитробензилсульфенилхлорид (6,6 г, 28,2 ммоль). Раствор нагревали до комнатной температуры в течение 30 мин или до тех пор, пока ТСХ (смесь 10% EtOAc/гексан, rf=0,55 основной, rf=0,48 второстепенный) не покажет завершение образования диастереоизомерных сложных эфиров сульфеновой кислоты. Затем смесь кипятили с обратным холодильником при 90 С в течение 4 ч или до тех пор, пока ТСХ не покажет завершение конверсии сложного эфира сульфеновой кислоты. После охлаждения до 0 С добавляли пентан (50 мл) и образовавшуюся смесь фильтровали через рыхлый слой целита и выпаривали. В результате флэш-хроматографии (смесь 2% EtOAc/гексан, rf=0,4) получали чистое соединение 7 (2,3 г, 75%). Изсоединения 4 (306 мг, 1,00 ммоль), следуя общей методике В, после флэш-хроматографии Изсоединения 5 (690 мг, 2,53 ммоль), следуя общей методике В, после флэш-хроматографии Изсоединения 6 (500 мг, 2,51 ммоль), следуя общей методике В, после флэш-хроматографииHRMS (EI) m/z вычислено для C14H22O2S (M+) 254,1341, найдено 254,1309. Общая методика С. Сложный этиловый эфир 2-(R)-2,4-диметил-2-тиопропионилгекса-3,5-диеновой кислоты (12) Непосредственно к раствору соединения 7 (250 мг, 1,0 ммоль) в EtOH (3,9 мл) добавляли карбонат цезия (332 мг, 1,0 ммоль). Через 20 мин смесь вливали в смесь NH4Cl (насыщенный)/1 н. HCl (15 мл, 3:1) и экстрагировали Et2O (320 мл). Объединенные органические слои сушили (MgSO4), фильтровали и выпаривали с получением неочищенного соединения 11. К соединению 11 добавляли CH2Cl2 (7,5 мл) и раствор охлаждали до 0 С. Добавляли NEt3 (0,14 мл, 1,0 ммоль) и пропионилхлорид (0,09 мл, 1,0 ммоль) и раствор перемешивали при 0 С. Через 40 мин добавляли NH4Cl (насыщенный) (20 мл) и образовавшуюся смесь экстрагировали CH2Cl2 (315 мл). Объединенные органические слои сушили (MgSO4),фильтровали и выпаривали. После флэш-хроматографии (смесь 5% EtOAc/гексан, rf=0,4) получали чистое соединение 12 (261 мг, 72%).HRMS (EI) m/z вычислено для C13H20O3S (M+) 256,1133, найдено 256,1127. Сложный этиловый эфир -2-тиоалкинил-2-метилдека-3,5-диеновой кислоты (13) Из соединения 8 (200 мг, 0,71 ммоль) и алкинилхлорида (55 мкл, 0,78 ммоль), следуя общей методике С, после флэш-хроматографии (смесь 5% EtOAc/гексан) получали соединение 13 (119 г, 59%). 1 Н ЯМР (300 МГц, CDCl3)0,84-0,89 (м, 3 Н), 1,23 (т, J=7,1 Гц, 3 Н), 1,28-1,38 (м, 4 Н), 1,71 (с, 3 Н),2,01-2,08 (м, 2 Н), 2,23 (с, 3 Н), 4,18 (кв., J=7,1 Гц, 2 Н), 5,66-5,76 (м, 2 Н), 5,89-6,03 (м, 1 Н), 6,20 (дд, J=10,3,15,3 Гц, 1 Н); 13 С ЯМР (75 МГц, CDCl3)13,8, 13,9, 22,2, 22,8, 29,9, 31,2, 32,3 56,1, 61,9, 128,4, 129,2, 132,2, 137,1,171,6, 194,6; ИК (NaCl) 2930, 1737, 1694 см-1;HRMS (ES) m/z вычислено для C15H24O3SNa+ (M+Na+) 307,1338, найдено 307,1339. Сложный этиловый эфир -2-тиоалкинил-2-метилокта-3,5-диеновой кислоты (14) Из соединения 9 (353 мг, 1,39 ммоль) и алкинилхлорида (98 мл, 1,39 ммоль), следуя общей методике С, после флэш-хроматографии (смесь 5% EtOAc/гексан) получали соединение 14 (142 г, 40%). 1 Н ЯМР (300 МГц, CDCl3)0,83 (т, J=7,3 Гц, 3 Н), 1,24 (т, J=7,1 Гц, 3 Н), 1,72 (с, 3 Н), 2,03-2,17 (м,- 12013371 2 Н), 2,25 (с, 3 Н), 4,17 (кв., J=7,1 Гц, 2 Н), 5,72-5,81 (м, 2 Н), 5,95-6,04 (дд, J=10, 15 Гц, 1 Н) 6,18-6,27 (дд,J=10, 15 Гц, 1 Н); 13 С ЯМР (75 МГц, CDCl3)13,2, 13,9, 22,8, 25,6, 30,2, 56,1, 61,9, 128,2, 128,4, 132,1, 138,5, 171,6,194,8; ИК (NaCl) 2929, 1736, 1693 см-1;HRMS (ES) m/z вычислено для C13H20O3SNa+ (M+Na+) 279,1025, найдено 279,1032. Сложный этиловый эфир -2-тиоалкинил-2,4-диметилгепта-3,5-диеновой кислоты (15) Из соединения 10 (369 мг, 1,46 ммоль) и алкинилхлорида (103 мкл, 1,46 ммоль), следуя общей методике С, после флэш-хроматографии (смесь 5% EtOAc/гексан) получали соединение 15 (271 г, 77%). 1 Н ЯМР (300 МГц, CDCl3)1,26 (т, J=7,1 Гц, 3 Н), 1,74 (д, J=6,6 Гц, 3 Н), 1,81 (с, 3 Н), 1,85 (с, 3 Н),2,25 (с, 3 Н), 4,17 (кв., J=7,1 Гц, 2 Н), 5,56 (с, 1 Н), 5,65-5,73 (д.кв., J=6,6, 16 Гц, 1 Н), 5,99 (д, J=16 Гц, 1H); 13 С ЯМР (75 МГц, CDCl3)13,8, 14,1, 18,2, 26,2, 30,5, 55,6, 62,0, 125,2, 128,3, 135,7, 138,5, 172,2,194,8; ИК (NaCl) 2926, 1737, 1694 см-1;HRMS (EI) m/z вычислено для C13H20O3S (M+) 256,1133, найдено 256,1118. Сложный этиловый эфир -2-тиоалкинил-2,4-диметилгекса-3,5-диеновой кислоты (16) Из -соединения 7 (380 мг, 1,56 ммоль) и алкинилхлорида (110 мкл, 1,56 ммоль), следуя общей методике С, после флэш-хроматографии (смесь 5% EtOAc/гексан) получали соединение 16 (230 г, 61%). 1 Н ЯМР (300 МГц, CDCl3)1,25 (т, J=7,1 Гц, 3 Н), 1,84 (с, 3 Н), 1,87 (с, 3 Н), 2,24 (с, 3 Н), 4,21 (кв.,J=7,1 Гц, 2H), 5,03 (д, J=10,6 Гц, 1 Н), 5,21 (д, J=17,3 Гц, 1 Н), 5,74 (с, 1 Н), 6,26-6,35 (дд, J=10,6, 17,3 Гц,1 Н); 13 С ЯМР (75 МГц, CDCl3)12,9, 13,9, 25,9, 30,1, 55,8, 62,0, 113,3, 131,3, 138,3, 141,3, 182,3, 194,6; ИК (NaCl) 2982, 1735, 1692 см-1. Общая методика D. 5-(R)-4-Гидрокси-3,5-диметил-5-(2-метилбута-1,3-диенил)-5-Н-тиофен-2-он (17) (тиолактамицин) К соединению 12 (315 мг, 1,23 ммоль) в ТГФ (18,5 мл) при -78 С добавляли LiHMDS (3,1 мл,3,1 ммоль, 1,0 М в ТГФ) и раствору давали возможность медленно нагреваться до -5 С. Затем раствор вливали в 1 н. HCl (25 мл) и экстрагировали EtOAc (315 мл). Объединенные органические слои сушили(MgSO4), фильтровали и выпаривали. Полученную неочищенную смесь переносили в NaHCO3 (насыщенный, 15 мл) и экстрагировали Et2O (310 мл). Затем водный слой подкисляли до рН 3 (рН бумаги) 1 н.HCl и экстрагировали Et2O (310 мл) и EtOAc (210 мл). Объединенные органические слои сушили(MgSO4), фильтровали и выпаривали с получением чистого соединения 17 (182 мг, 70%, 96%, энант. избыток). После перекристаллизации из смеси гексан/ацетон (3:1) получали оптически обогащенное соединение 17. Из соединения 13 (62 мг, 0,22 ммоль), следуя общей методике D, получали соединение 18 (21 мг,41%). 1 Н ЯМР (300 МГц, CDCl3) кетотаутомер:0,88 (т, J=6,9 Гц, 3 Н), 1,19-1,41 (м, 4 Н), 1,75 (с, 3 Н), 2,03- 13013371 2,19 (м, 2 Н), 3,22 (д, J=21 Гц, 1 Н), 3,51 (д, J=21 Гц, 1H), 5,67 (д, J=15 Гц, 1H), 5,80 (дт, J=7,17 Гц, 1H), 6,02 Из соединения 14 (364 мг, 0,46 ммоль), следуя общей методике D, получали соединение 19 (180 мг,60%). 1 Н (300 МГц, CDCl3 существует в виде смеси кето:енольный таутомер 2,3:1) кетотаутомер:1,00 (т,J=7,4 Гц, 3 Н), 1,76 (с, 3 Н), 2,09-2,16 (м, 2 Н), 3,21 (д, J=21 Гц, 1 Н), 3,52 (д, J=21 Гц, 1H), 5,70 (д, J=15 Гц,1 Н), 5,86 (дт, J=15, 6 Гц, 1 Н), 6,02 (дд, J=10, 15 Гц, 1 Н), 6,38 (дд, J=15,10 Гц, 1H); 1 Н ЯМР (300 МГц, MeOD) енольный таутомер:1,09 (т, J=7,4 Гц, 3 Н), 1,87 (с, 3 Н), 2,14-2,29 (м,2 Н), 5,78 (д, J=15 Гц, 1 Н), 5,87 (дт, J=15, 6,57 Гц, 1 Н), 6,09-6,18 (м, 1 Н), 6,38 (дд, J=10,2, 15 Гц, 1 Н); 13 С ЯМР (75 МГц, MeOD) енольный таутомер:14,1, 25,2, 26,9, 61,0, 101 (m), 129,7, 131,7, 132,7,138,9, 188,9, 197,1; ИК (NaCl) 2965, 1592 см-1; Из соединения 15 (226 мг, 0,9 ммоль), следуя общей методике D, получали соединение 20 (95 мг,49%). 1 Из соединения 16 (181 мг, 0,75 ммоль), следуя общей методике D, получали соединение 21 (66 мг,45%). 1 Н ЯМР (300 МГц, CDCl3) кетотаутомер:1,78 (с, 3 Н), 1,86 (с, 3 Н), 3,43 (д, J=5,6 Гц, 2H), 5,12 (д,J=10,6 Гц, 1H), 5,27 (д, J=17,3 Гц, 1 Н), 5,83 (с, 1 Н), 6,27-6,37 (дд, J=10,6, 17,3 Гц, 1 Н); 1 Н ЯМР (300 МГц, MeOD) енольный таутомер:1,79 (с, 3 Н), 1,84 (с, 3 Н), 5,04 (д, J=10,7 Гц, 1 Н),5,25 (д, J=17,3 Гц, 1 Н), 5,66 (с, 1 Н), 6,36 (дд, J=10,7, 17,3 Гц, 1 Н); 13 С ЯМР (75 МГц, MeOD)12,6, 30,4, 59,0, 102 (m), 116,9, 131,4, 140,6, 142,3, 189,9, 197,3; Из соединения 31 (1,4 мг, 5,0 ммоль), следуя общей методике D, получали соединение 22 (500 мг,- 14013371 45%). К смеси LiHMDS (6,2 мл, 6,20 ммоль, 1 М в ТГФ) в ТГФ (9,7 мл) при -78 С добавляли по каплям с помощью канюли -соединение 1 (1,00 г, 5,75 ммоль) в ТГФ (9,60 мл) и образовавшийся раствор перемешивали в течение 30 мин при -78 С. Затем при -78 С добавляли через канюлю октилтрифлат (1,63 г,6,20 ммоль) в ТГФ (4 мл). После перемешивания при -78 С в течение 2 ч добавляли 1 н. HCl (10 мл) и раствор экстрагировали Et2O (315 мл). Объединенные органические слои сушили (MgSO4), фильтровали и выпаривали. После флэш-хроматографии (смесь 2% EtOAc/гексан) получали чистое соединение 23 в виде смеси отдельных диастереоизомеров 2:1-6:1 (1,33 г, 81%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=6,5 Гц, 3 Н), 0,99 (с, 9 Н), 1,24-1,26 (м, 12 Н), 1,54 (с, 3 Н), 1,721,84 (м, 2 Н), 5,13 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)13,9, 22,6, 24,9, 25,1, 25,9, 29,2, 29,3, 29,5, 31,8, 35,2, 41,2, 55,3, 86,7,177,7; ИК (NaCl) 3443, 2929, 1829, 1769 см-1. Вычислено для C16H30O2S: С, 67,0; Н, 10,6. Найдено С, 66,3; Н, 10,5. Из -соединения 1 (500 мг, 2,87 мл) и гексилтрифлата (738 мг, 2,87 ммоль), следуя общей методике Е, получали соединение 24 (557 мг, 75%) в виде смеси отдельных диастереоизомеров 2:1-6:1. 1 Н ЯМР (300 МГц, CDCl3)0,87 (т, J=6,5 Гц, 3 Н), 0,99 (0, 9H), 1,24-1,29 (м, 8 Н), 1,54 (с, 3 Н), 1,721,80 (м, 2 Н), 5,13 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)13,9, 22,5, 24,9, 24,9, 25,1, 25,9, 29,1, 31,6, 41,2, 55,3, 86,7, 177,8; ИК (NaCl). Вычислено для C14H26O2S: С, 65,1; Н, 10,1. Найдено С, 64,5; Н, 10,1.HRMS (EI) m/z вычислено для C14H26O2S+ (M+) 258,1654, найдено 286,1653. Общая методика F. Сложный этиловый эфир -2-алкинилсульфанил-2-метилдекановой кислоты (26) К соединению 23 (650 мг, 2,27 ммоль) в EtOH (14,1 мл) добавляли NaOEt (2,1M) (2,16 мл,4,54 ммоль) (свежеполученный с использованием металлического Na (200 мг, 8,3 ммоль) в EtOH (4,0 мл) и раствору давали возможность перемешиваться при комнатной температуре. Через 2 ч раствор вливали в NH4Cl (насыщенный)/1 н. HCl (25 мл, 3:1) и полученную смесь экстрагировали Et2O (320 мл). Затем объединенные органические слои промывали Н 2 О (325 мл), сушили (MgSO4), фильтровали и выпаривали с получением неочищенного соединения 25. К растворенному в CH2Cl2 (26 мл) соединению 25 при 0 С добавляли NEt3 (0,5 мл, 3,49 ммоль) и алкинилхлорид (0,3 мл, 3,49 ммоль). Через 40 мин при 0 С добавляли NH4Cl (насыщенный) (30 мл) и раствор экстрагировали CH2Cl2. Объединенные органические слои сушили (MgSO4), фильтровали и выпаривали. После флэш-хроматографии (смесь 5% EtOAc/гексан) получали чистое соединение 26 (542 мг, 79%). 1H ЯМР (300 МГц, CDCl3)0,87 (т, J=6,9 Гц, 3 Н), 1,22-1,27 (м, 15 Н), 1,61 (с, 3 Н), 1,75-1,84 (м, 2 Н),2,26 (с, 3 Н), 4,18 (кв., J=7,1 Гц, 2H); 13 С ЯМР (75 МГц, CDCl3)13,9, 14,1, 22,6, 23,4, 24,4, 29,1, 29,2, 29,6, 30,3, 31,8, 38,3, 55,8, 61,5,173,1, 195,8; ИК (NaCl) 3430, 1868, 1693, 1644 см-1; Вычислено для C15H28O3S: С, 62,5; Н, 9,78. Найдено С, 62,6; Н, 9,83. Сложный этиловый эфир -2-алкинилсульфанил-2- метилоктановой кислоты (28) Из соединения 24 (940 мг, 3,63 ммоль) и алкинилхлорида (0,3 мл, 3,63 ммоль), следуя общей методике F, после флэш-хроматографии (смесь 5% EtOAc/гексан) получали соединение 28 (727 мг, 77%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=6,9 Гц, 3 Н), 1,22-1,27 (м, 11 Н), 1,61 (с, 3 Н), 1,75-1,79 (м, 2 Н),2,25 (с, 3 Н), 4,17 (кв., J=7 Гц, 2H); 13 С ЯМР (75 МГц, CDCl3)13,9, 14,1, 22,4, 23,4, 24,4, 29,3, 30,3, 31,5, 38,4, 55,7, 61,5, 173,0, 194,7; ИК (NaCl) 3449, 1736, 1694 см-1; Вычислено для C13H24O3S: С, 59,9; Н, 9,29. Найдено С, 60,6; Н, 9,44. Сложный этиловый эфир -2-метил-2-пропионилсульфанилдекановой кислоты (30) Из соединения 23 (613 мг, 2,14 ммоль) и пропионилхлорида (0,19 мл, 2,14 ммоль), следуя общей методике F, после флэш-хроматографии (смесь 5% EtOAc/гексан) получали соединение 30 (484 мг, 75%). 1 Н ЯМР (300 МГц, CDCl3)0,84 (т, J=6,9 Гц, 3 Н), 1,10 (т, J=7,5 Гц, 3 Н), 1,19-1,24 (м, 15 Н), 1,58 (с,3 Н), 1,72-1,77 (м, 2 Н), 2,48 (кв., J=7,5 Гц, 2 Н), 4,17 (кв., J=7 Гц, 2 Н); 13 С ЯМР (75 МГц, CDCl3)9,45, 14,1, 14,1, 22,6, 23,5, 24,5, 29,1, 29,3, 29,7, 31,8, 36,9, 38,5, 55,5,61,4, 173,2, 199,2. Вычислено для C16H30O3S: С, 63,5; Н, 10,0. Найдено С, 63,7; Н, 10,0. Сложный этиловый эфир -2-алкинилсульфанил-2-метил-3- фенилдекановой кислоты (31) Из 5-бензил-2-трет-бутил-5-метил-[1,3]-оксатиолан-4-она 1 (1,2 г, 4,7 ммоль), следуя общей методике F, после флэш-хроматографии (смесь 5% EtOAc/гексан) получали соединение 31 (954 мг, 76%). 1 Н ЯМР (300 МГц, CDCl3)1,19 (т, J=7 Гц, 3 Н), 1,55 (с, 3 Н), 2,26 (с, 3 Н), 3,13 (кв., J=13 Гц, 2 Н),4,13 (кв., J=7 Гц, 2 Н), 7,1 (м, 2 Н), 7,2 (м, 3 Н); 13 С ЯМР (75 МГц, CDCl3)14,0, 23,1, 30,3, 43,6, 56,3, 61,7, 127,2, 128,1, 130,7, 135,4, 172,8, 194,8. Общая методика G. К соединению 26 (500 мг, 1,7 ммоль) в толуоле (27 мл) при -78 С добавляли LiHMDS (4,3 мл,4,3 ммоль, 1,0 М в ТГФ) и раствору давали возможность медленно нагреваться до -5 С. Раствор затем вливали в 1 н. HCl (40 мл) и экстрагировали EtOAc (325 мл). Объединенные органические слои сушили Из соединения 28 (715 мг, 2,75 ммоль), следуя общей методике G, после флэш-хроматографии Из соединения 30 (469 мг, 1,55 ммоль) и NaHMDS (3,87 мл, 3,87 ммоль, 1,0 М в ТГФ), следуя общей методике G, получали соединение 34 (397 мг, 70%). 1 Н ЯМР (300 МГц, CDCl3) енольный таутомер:0,86 (т, J=6,8 Гц, 3 Н), 1,23 (с, 11 Н), 1,30-1,45 (м,1 Н), 1,59 (с, 3 Н), 1,74 (с, 3 Н), 1,84-1,88 (м, 2 Н); 13 С ЯМР (75 МГц, CDCl3)7,48, 14,0, 22,6, 25,2, 25,9, 29,2, 29,4, 29,6, 31,8, 38,5, 58,2, 110,5, 180,9,198,0; ИК (NaCl) 2927, 1601 см-1. Общая методика Н.NaH (14 мг, 0,35 ммоль, 60% в минеральном масле) и раствору давали возможность нагреваться и перемешиваться при 0 С в течение 30 мин. Затем непосредственно добавляли диметилсульфат (50 мкл,0,55 ммоль) и смеси давали возможность нагреваться и перемешиваться при комнатной температуре в течение 2,5 ч. Добавляли смесь NH4Cl (насыщенный)/1 н. HCl (3:1, 10 мл) и раствор экстрагировали Et2O(310 мл). Объединенные органические слои промывали Н 2 О (315 мл), сушили (MgSO4), фильтровали и выпаривали. В результате флэш-хроматографии (смесь 15% EtOAc/гексан) получали чистое соединение 35 (59 мг, 80%). 1 Н ЯМР (300 МГц, CDCl3)0,85 (т, J=7 Гц, 3 Н), 1,07-1,18 (м, 1 Н), 1,23 (с, 10 Н), 1,43-1,49 (м, 1 Н),1,61 (с, 3 Н), 1,74-1,81 (м, 2 Н), 3,81 (с, 3 Н), 5,29 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)14,0, 22,6, 25,1, 26,4, 29,1, 29,3, 29,5, 31,8, 38,8, 59,3, 59,4, 101,3, 187,3,193,8. Вычислено для C14H24O2S: С, 65,6; Н, 9,50. Найдено С, 65,8; Н, 9,50. Из соединения 33 (40,3 мг, 0,19 ммоль) и диметилсульфата (35 мкл, 0,37 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 15% EtOAc/гексан) получали соединение 36 (25 мг, 58%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=6,7 Гц, 3 Н), 1,08-1,13 (м, 1 Н), 1,24 (с, 6 Н), 1,35-1,39 (м, 1 Н),1,61 (с, 3 Н), 1,75-1,82 (м, 2 Н), 3,81 (с, 3 Н), 5,30 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)14,0, 22,5, 25,1, 26,4, 29,2, 31,5, 38,9, 59,4, 101,3, 187,3, 193,8. Из соединения 34 (40 мг, 0,16 ммоль), KH (27 мг, 0,20 ммоль, 30% в минеральном масле) и диметилсульфата (30 мкл, 0,31 ммоль), следуя общей методике Н, получали соединение 37 (30 мг, 71%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=7 Гц, 3 Н), 1,06-1,09 (м, 1 Н), 1,24 (с, 10 Н), 1,41-1,48 (м, 1 Н),1,55 (с, 3 Н), 1,71-1,79 (м, 2 Н), 1,98 (с, 3 Н), 4,09 (с, 3 Н); 13 С ЯМР (75 МГц, CDCl3)9,59, 14,1 22,6, 25,2, 26,5, 29,2, 29,4, 29,6, 31,8, 38,9, 58,7, 59,8, 111,3,180,2, 195,7; Из соединения 22 (50 мг, 0,23 ммоль) и диметилсульфата (44 мкл, 0,45 ммоль), следуя общей методике Н, получали соединение 38 (38 мг, 74%). 1 Н ЯМР (300 МГц, CDCl3)1,65 (с, 3 Н), 3,1 (кв., J=7 Гц, 2 Н), 3,84 (с, 3 Н), 5,19 (с, 1 Н), 7,21 (м, 5 Н); 13 С ЯМР (75 МГц, CDCl3)26,0, 45,0 59,3, 59,9, 101,9, 127,2, 128,0, 130,4, 135,9, 186,5, 192,9. Сложный этиловый эфир -5-метил-5-октил-2-оксотиофен-4-илоксиуксусной кислоты (39) Из соединения 32 (39 мг, 0,16 ммоль) и этилбромацетата (36 мкл, 0,32 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 15% EtOAc/гексан) получали соединение 39 (39 мг, 73%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=6 Гц, 3 Н), 1,24 (с, 11 Н), 1,29 (т, J=7 Гц, 3 Н), 1,47-1,48 (м, 1 Н),1,68 (с, 3 Н), 1,85-1,88 (м, 2 Н), 4,25 (кв., J=7 Гц, 2 Н), 4,54 (с, 2 Н), 5,20 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)14,1, 14,1, 22,6, 25,1, 26,4, 29,2, 29,3, 29,5, 31,8, 38,8, 59,7, 61,9, 67,9,102,3, 166,2, 185,3, 193,4; ИК (NaCl) 2928, 1762, 1682, 1612 см-1. Вычислено для C17H28O4S: С, 62,2; Н, 8,59. Найдено С, 62,2; Н, 8,67. Сложный этиловый эфир -5-метил-5-гексил-2-оксотиофен-4-илоксиуксусной кислоты (40) Из соединения 33 (20 мг, 0,09 ммоль) и этилбромацетата (20 мкл, 0,2 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 15% EtOAc/гексан) получали соединение 40 (18 мг, 67%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=6,8 Гц, 3 Н), 1,24-1,27 (м, 7 Н), 1,32 (т, J=7 Гц, 3 Н), 1,47-1,48 Из соединения 32 (47 мг, 0,18 ммоль) и 3-йод-1-хлорбутана (40 мкл, 0,36 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 20% EtOAc/гексан) получали соединение 41 (46 мг, 85%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=7 Гц, 3H), 1,07-1,27 (м, 1 Н), 1,24 (с, 10 Н), 1,48-1,51 (м, 1 Н),1,62 (с, 3 Н), 1,75-1,82 (м, 2 Н), 1,89-1,98 (м, 4 Н), 3,59 (т, J=5,9 Гц, 2 Н), 3,95-3,98 (м, 2 Н), 5,28 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)14,1, 22,6, 25,1, 26,0, 26,5, 29,0, 29,2, 29,3, 29,5, 29,7, 31,8, 44,1, 59,6,71,7, 101,6, 186,1, 193,8. Вычислено для C17H29ClO2S: С, 61,3; Н, 8,78. Найдено С, 61,9; Н, 9,01. Из соединения 33 (36 мг, 0,17 ммоль) и 3-йод-1-хлорбутана (40 мкл, 0,34 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 20% EtOAc/гексан) получали соединение 42 (32 мг, 75%). 1 Н ЯМР (400 МГц, CDCl3)0,86 (т, J=5,1 Гц, 3 Н), 1,09-1,14 (м, 1 Н), 1,25 (с, 6 Н), 1,44-1,53 (м, 1 Н),1,63 (с, 3 Н), 1,77-1,85 (м, 2 Н), 1,90-2,00 (м, 4 Н), 3,59 (т, J=4,5 Гц, 2H), 3,95-3,99 (м, 2 Н), 5,28 (с, 1 Н); Из соединения 32 (31 мг, 0,12 ммоль) и аллилбромида (21 мкл, 0,25 ммоль), следуя общей методике Н, получали смесь соединений 43 и 44, 3:1 (26 мг, 74%), которая могла быть разделена и очищена с использованием флэш-хроматографии (смесь 15% EtOAc/гексан). Из соединения 33 (270 мг, 1,3 ммоль) и аллилбромида (0,2 мл, 2,52 ммоль), следуя общей методике Н, получали смесь соединения 45 и соединения 46 2,3:1 (205 мг, 58%), которая могла быть разделена и очищена с использованием флэш-хроматографии (смесь 15% EtOAc/гексан). Из соединения 34 (70 мг, 0,27 ммоль) и аллилбромида (47 мкл, 0,55 ммоль), следуя общей методике Н, получали смесь соединений 47 и 48, 2,3:1 (данные С-алкилирования не показаны) (67 мг, 82%), которая могла быть разделена и очищена с использованием флэш-хроматографии (смесь 20% EtOAc/гексан). Из соединения 33 (45 мг, 0,21 ммоль) и пропаргилбромида (37 мкл, 0,21 ммоль), следуя общей методике Н, получали соединение 49 (21 мг, 40%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=7 Гц, 3 Н), 1,11-1,20 (м, 1 Н), 1,24 (с, 6 Н), 1,41-1,49 (м, 1 Н),1,63 (с, 3 Н), 1,76-1,86 (м, 2 Н), 2,59 (т, J=2,5 Гц, 1H), 4,62 (д, J=3,7 Гц, 1 Н), 4,63 (д, J=3,7 Гц, 1 Н), 5,43 (с,1 Н). Сложный трет-бутиловый эфир -5-метил-5-октил-2- оксотиофен-4-илоксиуксусной кислоты (50) Из соединения 32 (60 мг, 0,25 ммоль) и трет-бутилбромацетата (73 мкл, 0,49 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 15% EtOAc/гексан) получали соединение 50 (62 мг,70%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=7 Гц, 3 Н), 1,24 (с, 12 Н), 1,49 (с, 9 Н), 1,68 (с, 3 Н), 1,83-1,86 (м,2 Н), 4,43 (с, 2 Н), 5,19 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)14,0, 22,6, 25,2, 26,3, 28,1, 29,2, 29,3 29,5, 31,8, 38,9, 59,7, 68,5, 83,4,102,1, 165,2, 185,5, 193,4. Вычислено для C19H32O4S: С, 64,0; Н, 9,05. Найдено С, 64,1; Н, 9,08. Сложный трет-бутиловый эфир -5-метил-5-гексил-2-оксотиофен-4-илоксиуксусной кислоты (51) Из соединения 33 (169 мг, 0,79 ммоль) и трет-бутилбромацетата(0,23 мл, 1,58 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 15% EtOAc/гексан) получали соединение 51 (206 мг,80%). 1 Из соединения 22 (150 мг, 0,68 ммоль) и трет-бутилбромацетата (0,20 мл, 1,36 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 20% EtOAc/гексан) получали соединение 52 (159 мг,74%). 1 Н ЯМР (300 МГц, CDCl3)1,49 (с, 9 Н), 1,69 (с, 3 Н), 3,17 (с, 2 Н), 4,44 (кв., J=8 Гц, 2 Н), 5,13 (с, 1 Н),7,24 (м, 5 Н); 13 С ЯМР (75 МГц, CDCl3)25,8, 28,1, 45,0, 60,1, 68,4, 83,6, 102,6, 127,2, 128,1, 130,5, 135,9, 165,3,184,9, 192,8. Общая методика I. К соединению 50 (65 мг, 0,18 ммоль), растворенному в CH2Cl2 (1,4 мл), добавляли трифторуксусную кислоту (TFA) (0,7 мл) и раствор перемешивали при комнатной температуре в течение 4 ч. Выпаривали растворители и неочищенную смесь подвергали хроматографии (смесь 20% EtOAc/2% СН 3 СО 2 Н/гексан) с получением чистого соединения 53 (48 мг, 89%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=6,9 Гц, 3 Н), 1,24 (с, 11 Н), 1,47-1,48 (м, 1 Н),1,68 (с, 3 Н), 1,84- 20013371 1,88 (м, 2 Н), 4,62 (с, 2 Н), 5,31 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)14,1, 22,6, 25,1, 26,1, 29,2, 29,3 29,5, 31,8, 38,9, 60,1, 67,7, 102,4, 169,8,185,8, 195,4; ИК (NaCl) 3442, 1645 см-1. Вычислено для C15H24O4S: С, 59,9; Н, 8,05. Найдено С, 60,0; Н, 8,09. Из соединения 51 (177 мг, 0,54 ммоль) и трифторуксусной кислоты (TFA) (2,61 мл), следуя общей методике I, после флэш-хроматографии (смесь 20% EtOAc/2% СН 3 СО 2 Н/гексан) получали соединение 54 Из соединения 52 (117 мг, 0,35 ммоль) и трифторуксусной кислоты (TFA) (1,4 мл), следуя общей методике I, после флэш-хроматографии (смесь 30% EtOAc/2% СН 3 СО 2 Н/гексан) получали соединение 55 К охлажденному раствору (0 С) соединения 53 (64 мг, 0,21 ммоль) в CH2Cl2 (1,1 мл) добавляли гидрохлорид 1-[3-диметиламино)пропил]-3-этилкарбодиимида (EDC) (49 мг, 0,25 ммоль), DMAP (3 мг,0,02 ммоль) и аллиламин (18 мкл, 0,25 ммоль) и смеси давали возможность нагреваться до комнатной температуры и перемешиваться в течение 12 ч. Раствор вливали в раствор 1 н. HCl (насыщенный) (1:3) и экстрагировали Et2O (310 мл). Объединенные органические слои сушили (MgSO4), фильтровали и выпаривали с получением неочищенного соединения 56. После флэш-хроматографии (смесь 50%EtOAc/гексан) получали чистое соединение 56 (50 мг, 66%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=7 Гц, 3 Н), 1,12-1,22 (м, 1 Н), 1,24 (с, 10 Н), 1,41-1,51 (м, 1 Н),1,68 (с, 3 Н), 1,82-1,87 (м, 2 Н), 3,98 (каж.т, J=6 Гц, 2 Н), 4,50 (с, 2 Н), 5,20 (д, J=10 Гц, 1H), 5,22 (д,J=17,3 Гц, 1H), 5,35 (с, 1 Н), 5,80-5,90 (ддд, J=6, 10, 17 Гц, 1 Н), 6,19 (ш.с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)14,0, 22,6, 25,3, 26,5, 29,2, 29,4, 29,5, 31,8, 39,1, 41,6, 59,3, 70,3, 103,4,117,2, 133,2, 165,3, 183,9, 192,8. Вычислено для C18H29O3S: С, 63,7; Н, 8,61. Найдено С, 63,4; Н, 8,67. Общая методика J. К раствору соединения 54 (42,4 мг, 0,15 ммоль) в CH3CN (0,86 мл) добавляли трис-(2-оксо-3-оксазолинил)фосфиноксид 3 (91 мг, 0,20 ммоль), гидрохлорид метилглицината (19,7 мг,0,16 ммоль) и NEt3 (43 мкл, 0,31 ммоль) и раствору давали возможность перемешиваться при комнатной температуре в течение 20 мин, смесь вливали в раствор смеси NH4Cl (насыщенный)/1 н. HCl (10 мл) и экстрагировали Et2O (310 мл). Объединенные органические слои сушили (MgSO4), фильтровали, выпаривали и подвергали хроматографии (смесь 40-50% EtOAc/гексан) с получением чистого соединения 57 К соединению 57 (22 мг, 0,06 ммоль), растворенному в смеси ТГФ/Н 2 О (0,5 мл, 3:1) и охлажденному до 0 С, добавляли LiOH (3 мг, 0,07 ммоль) и полученному раствору давали возможность перемешиваться в течение 45 мин. Затем смесь вливали в раствор HCl (10 мл, 1 н.) и экстрагировали Et2O (310 мл). Объединенные органические слои сушили (MgSO4), фильтровали и выпаривали с получением неочищенного соединения 58. После флэш-хроматографии (смесь 50% EtOAc/2% СН 3 СО 2 Н/гексан) получали чистое соединение 58 (19 мг, 86%). 1 Н ЯМР (300 МГц, CDCl3)0,85 (т, J=6,7 Гц, 3 Н), 1,25 (с, 7 Н), 1,48-1,52 (м, 2 Н), 1,68 (с, 3 Н), 2,082,10 (м, 2 Н), 4,05 (с, 2 Н), 4,56 (с, 2 Н), 5,41 (с, 1 Н). Из соединения 54 (61 мг, 0,22 ммоль) и гидробромида 1-аминопропанола (50 мг, 0,23 ммоль), следуя общей методике J, после флэш-хроматографии (смесь 50% EtOAc/гексан) получали соединение 59 Из соединения 55 (72 мг, 0,26 ммоль) и аллиламина (21 мкл, 0,28 ммоль), следуя общей методике J,после флэш-хроматографии (градиент 10-50% EtOAc/гексан) получали соединение 60 (39 мг, 47%). 1 Н ЯМР (300 МГц, CDCl3)1,73 (с, 3 Н), 3,17 (с, 2 Н), 3,93 (м, 2 Н), 4,41 (с, 2 Н), 5,22 (м, 2 Н), 5,24 (с,1 Н), 5,80 (м, 1 Н), 5,83 (с, 1 Н), 7,24 (м, 5 Н). 13 С ЯМР (75 МГц, CDCl3)26,0, 41,6, 45,4, 59,7, 70,3, 103,9 117,1, 127,5, 128,3, 130,2, 133,3, 135,6,165,3, 183,4, 192,0. Общая методика K.LiHMDS (0,58 мл, 0,58 ммоль, 1 M в ТГФ) и раствору давали возможность перемешиваться при -78 С в течение 30 мин. Затем добавляли этилхлорформиат (60 мкл, 0,62 ммоль) и смесь переносили в баню с льдом и давали возможность медленно нагреваться до комнатной температуры. Через 1 ч смесь вливали при комнатной температуре в раствор смеси HCl (1 н.)/NH4Cl (насыщенный) (10 мл) и экстрагировалиEt2O (310 мл). Объединенные органические слои сушили (MgSO4), фильтровали, выпаривали и хроматографировали (смесь 20% EtOAc/гексан) с получением чистого соединения 61 (111 мг, 91%). 1 Н ЯМР (300 МГц, CDCl3)0,85 (т, J=6,9 Гц, 3 Н), 1,12-1,17 (м, 11 Н), 1,38 (т, J=7 Гц, 3 Н), 1,42-1,50 Из соединения 32 (73 мг, 0,30 ммоль) и метилхлорформиата (37 мкл, 0,48 ммоль), следуя общей методике K, после флэш-хроматографии (смесь 20% EtOAc/гексан) получали соединение 62 (63 мг, 70%). 1 Из соединения 32 (51,5 мг, 0,21 ммоль) и аллилхлорформиата (33 мкл, 0,32 ммоль), следуя общей методике K, после флэш-хроматографии (смесь 15% EtOAc/гексан) получали соединение 63 (46,3 мг,67%). 1 Н ЯМР (300 МГц, CDCl3)0,85 (т, J=7,3 Гц, 3 Н), 1,16-1,23 (ш.с, 10 Н), 1,41-1,51 (м, 2 Н), 1,67 (с,3 Н), 1,81-1,87 (м, 2 Н), 4,74 (каж.дт, J=6,13 Гц, 2H), 5,37 (каж.д.кв., J=10,3, 1,02 Гц, 1 Н), 5,44 (каж.д.кв.,J=15,9, 1,02 Гц, 1 Н), 5, 90-6,0 (м, 1 Н), 6,39 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)14,0, 22,6, 25,2, 25,8, 29,1, 29,2, 29,4, 31,8, 38,4, 60,1, 70,2, 112,9, 120,6,130,23, 150,0, 175,5, 193,7. ИК (NaCl) 2927, 1782, 1691, 1606 см-1. Вычислено для C17H26O4S: С, 62,5; Н, 8,03. Найдено С, 62,6; Н, 8,07. Из соединения 32 (40 мг, 0,17 ммоль) и пропионилхлорида (20 мкл, 0,22 ммоль), следуя общей методике K, после флэш-хроматографии (смесь 15% EtOAc/гексан) получали соединение 64 (23,1 мг, 47%). 1 Из соединения 22 (50 мг, 0,23 ммоль) и этилхлорформиата (35 мкл, 0,36 ммоль), следуя общей методике K, получали соединение 65 (67 мг, 88%). 1 Н ЯМР (300 МГц, CDCl3)1,31 (т, J=7 Гц, 3 Н), 1,69 (с, 3 Н), 3,15 (с, 2 Н), 4,36 (кв., J=7 Гц, 2H), 6,33- 23013371 К соединению 32 (247 мг, 1,02 ммоль), растворенному в гексане, добавляли триэтиламин (0,23 мл,1,68 ммоль) и триметилсилилхлорид (0,21 мл, 1,64 ммоль) и раствору давали возможность перемешиваться при комнатной температуре в течение 4 ч, смесь фильтровали через рыхлый слой целита и выпаривали с получением 5-метил-5-октил-4-триметилсиланилокси-5 Н-тиофен-2-она. К раствору TiCl4(0,7 мл, 0,7 ммоль) в CH2Cl2 (1,95 мл) при -78 С добавляли ацетальдегид (54 мкл, 0,97 ммоль) и полученному раствору давали возможность перемешиваться при -78 С в течение 5 мин. Затем с помощью канюли в раствор смеси TiCl4/ацетальдегид добавляли 5-метил-5-октил-4-триметилсиланилокси-5 Н-тиофен-2 он, растворенный в CH2Cl2 (0,4 мл), с получением при этом смеси ярко-оранжевого цвета. Образовавшейся смеси давали возможность нагреваться и перемешиваться в течение 20 мин при 0 С, смесь вливали в NH4Cl (насыщенный) (15 мл) и экстрагировали CH2Cl2 (315 мл). Органические слои объединяли,сушили (MgSO4), фильтровали и выпаривали. После флэш-хроматографии (смесь 10% EtOAc/гексан) получали чистые соединение 66 (34 мг) и соединение 67 (24 мг) (50%). К соединению 32 (94 мг, 0,38 ммоль) в CH2Cl2 (1,9 мл) при 0 С добавляли NEt3 (58 мкл,0,42 ммоль), диметиламинопиридин (DMAP) (19 мг, 0,15 ммоль) и уксусный ангидрид (43 мкл,0,47 ммоль). Раствор перемешивали при 0 в течение 15 мин, затем давали возможность нагреваться и перемешиваться при комнатной температуре в течение 2-14 ч или до тех пор, пока ТСХ не покажет завершение реакции, смесь вливали в смесь NH4Cl (насыщенный)/HCl (1 н.) (3:1, 8 мл) и экстрагировалиCH2Cl2 (310 мл). Органические слои объединяли, сушили (MgSO4), фильтровали и выпаривали с получением неочищенного соединения 68. После флэш-хроматографии (смесь 30% EtOAc/2% АсОН/гексан)(rf=0,44) получали чистое соединение 68 (83 мг, 78%). 1 Н ЯМР (300 МГц, CDCl3)0,84 (м, 3 Н), 1,22 (ш.с, 10 Н), 1,48 (м, 2 Н), 1,65 (с, 3 Н), 1,77-1,92 (м, 2 Н),2,55 (с, 3 Н); 13 С ЯМР (75 МГц, CDCl3)13,9, 22,6, 23,8, 25,1, 26,3, 29,1, 29,2, 29,5, 31,7, 39,4, 59,7, 109,7, 190,5,195,5, 204,9. Из соединения 32 (90 мг, 0,37 ммоль), ангидрида трифторуксусной кислоты (114 мкл, 0,81 ммоль),диметиламинопиридина (DMAP) (18 мг, 0,15 ммоль) и NEt3 (108 мкл, 0,77 ммоль), следуя общей методике L, после флэш-хроматографии (смесь 40% гексан/10% ТГФ/2% AcOH/EtOAc) получали соединение 69HRMS (EI) m/z вычислено для C15H21F3O3S+ (M+) 338,1158, найдено 338,1171. Сложный метиловый эфир 4-гидрокси-5-метил-5-октил-2-оксо-2,5-дигидротиофен-3-карбоновой кислоты (70)- 24013371 Из соединения 32 (91 мг, 0,37 ммоль), метилхлорформиата (63 мкл, 0,81 ммоль), диметиламинопиридина (DMAP) (23 мг, 0,18 ммоль) и NEt3 (108 мкл, 0,77 ммоль), следуя общей методике L, после флэшхроматографии (30% EtOAc/2% АсОН/гексан - 10% ТТФ/2% AcOH/EtOAc) получали соединение 70(66 мг, 59%, 79% в расчете на извлеченные исходные продукты). 1 Н ЯМР (300 МГц, MeOD)0,86 (т, J=6,9 Гц, 3 Н), 1,20 (ш.с, 12 Н), 1,35 (с, 3 Н), 1,55 (м, 1 Н), 1,711,75 (м, 1 Н), 3,59 (с, 3 Н); 13 С ЯМР (75 МГц, MeOD)13,3, 21,8, 24,4, 27,0, 28,5, 28,6, 29,0, 30,2, 31,0, 50,4, 58,3, 124,6, 168,1,187,7, 196,7.HRMS (EI) m/z вычислено для C15H24O4S+ (M+) 300,1389, найдено 300,1375. 2-Метил-2-октил-5-оксо-2,5-дигидротиофен-3-иловый эфир изопропилкарбаминовой кислоты (71) К соединению 32 (46 мг, 0,19 ммоль), растворенному в гексане, добавляли триэтиламин (43 мкл,0,31 ммоль) и триметилсилилхлорид (36 мкл, 0,29 ммоль) и раствору давали возможность перемешиваться при комнатной температуре в течение 4 ч, смесь фильтровали через рыхлый слой целита и выпаривали с получением 5-метил-5-октил-4-триметилсиланилокси-5 Н-тиофен-2-она, который повторно растворяли в CH2Cl2 (0,4 мл). К полученной смеси добавляли изопропилизоцианат (19,2 мл, 0,19 ммоль) и раствору давали возможность перемешиваться при комнатной температуре в течение 2 ч. Добавляли NH4Cl (насыщенный) (5 мл) и смесь экстрагировали CH2Cl2 (310 мл). Органические слои объединяли, сушили(MgSO4), фильтровали и выпаривали с получением неочищенного соединения 71. После флэшхроматографии (смесь 20% EtOAc/2% АсОН/гексан) получали чистое соединение 71 (35 мг, 60%). 1 Н ЯМР (300 МГц, CDCl3)0,85 (т, J=7,0 Гц, 3 Н), 1,14-1,24 (м, 17 Н), 1,45 (м, 1 Н), 1,63 (с, 3 Н), 1,761,79 (м, 2 Н), 3,81-3,88 (м, 1 Н), 5,16 (д, J=7 Гц, 1 Н), 6,33 (с, 1 Н); 13 С ЯМР (75 МГц, CDCl3)13,9, 20,4, 22,5, 22,8, 25,1, 25,9, 29,1, 29,3, 29,5, 31,8, 38,7, 44,0, 60,2,111,6, 149,7, 176,2, 194,5. Общая методика M.(6,0 мг, 0,05 ммоль) и гидразид 2-фуранкарбоновой кислоты (54 мг, 0,43 ммоль). Образовавшуюся смесь перемешивали при 0 С в течение 30 мин, затем ей давали возможность нагреваться до комнатной температуры и перемешиваться в течение 12 ч. Раствор вливали в NH4Cl (10 мл, насыщенный) и экстрагировали CH2Cl2 (310 мл). Объединенные органические слои сушили (Na2SO4), фильтровали и выпаривали с получением неочищенного соединения 72. После флэш-хроматографии (смесь 10% EtOAc/гексан) получали чистое соединение 72 (91 мг, 68%). 1 Из соединения 53 (100 мг, 0,33 ммоль) и уксусного гидразида (26,8 мг, 0,36 ммоль), следуя общей методике М, после флэш-хроматографии (смесь 2% AcOH/EtOAc) получали соединение 73 (70,4 мг,60%). 1 Н ЯМР (400 МГц, CDCl3)0,85 (т, J=7,2 Гц, 3 Н), 1,23 (м, 11 Н), 1,48-1,52 (м, 1 Н), 1,67 (с, 3 Н), 1,841,86 (м, 2 Н), 2,07 (с, 3 Н), 4,64 (с, 2 Н), 5,42 (с, 1 н); Из соединения 53 (83 мг, 0,28 ммоль) и N-метил-N-аллиламина (29 мкл, 0,30 ммоль), следуя общей методике М, после флэш-хроматографии (смесь 40% EtOAc/гексан) получали соединение 75 (51 мг,52%). 1 Н ЯМР (300 МГц, CDCl3)0,83 (т, J=6,9 Гц, 3 Н), 1,22 (м, 11 Н), 1,43-1,47 (м, 1 Н), 1,67 (с, 3 Н), 1,821,86 (м, 2 Н), ротамер 1: 2,91 (с, 3 Н), ротамер 2: 2,95 (с, 3 Н), ротамер 1: 3,84 (д, J=4,8 Гц, 2H), ротамер 2: 3,98 (д, J=6 Гц, 2 Н), ротамер 1: 4,62 (с, 2 Н), ротамер 2: 4,65 (с, 2 Н), 5,12-5,28 (м, 2 Н), ротамер 1: 5,18 (с,1 Н), ротамер 2: 5,25 (с, 1 Н), 5,65-5,81 (м, 1 Н); 13 С ЯМР (100 МГц, CDCl3)14,0, 22,5, 25,1, 26,2, 29,1, 29,3, 29,4, 31,7, 33,4 (ротамер 2: 33,9), 38,8,50,2 (ротамер 2: 51,0), 59,7, 69,0 (ротамер 2: 69,3), 102,3, 117,4 (ротамер 2: 118,2), 131,6 (ротамер 2: 131,8), 164,5 (ротамер 2: 164,9), 185,5 (ротамер 2: 185,6), 193,4. Из соединения 32 (50 мг, 0,21 ммоль) и бензилбромида (37 мл, 0,31 ммоль), следуя общей методике Н, после флэш-хроматографии (смесь 15% EtOAc/гексан) получали соединение 76 (49 мг, 75%). 1 Н ЯМР (300 МГц, CDCl3)0,86 (т, J=6,9 Гц, 3 Н), 1,24 (м, 11 Н), 1,41-1,48 (м, 1 Н), 1,66 (с, 3 Н), 1,791,86 (м, 2 Н), 4,98 (с, 2 Н), 5,39 (с, 1 Н), 7,31-7,42 (м, 5 Н); 13 С ЯМР (100 МГц, CDCl3)14,1, 22,6, 25,0, 26,4, 29,1, 29,3, 29,4, 31,8, 38,8, 59,7, 74,0, 102,2, 127,6,128,8, 128,8, 134,3, 185,8, 194,1; ИК (NaCl) 2928, 1681, 1610 см-1. Ссылки 1. Strijtveen, В.; Kellogg, R.M. Tetrahedron, 1987, 43, 5039-5054. 2. Sasaki, H.; Oishi, H.; Hayashi, Т.; Matsuura, I.; Ando K.; Sawada, M. J. Antibiotics, 1982. 3. Kunieda, Т.; Nagamatsu, Т.; Higuchi, Т.; Hirobe, M. Tetrahedron Lett. 1988, 29, 2203-2206. Биологические и биохимические методы Очистка FAS от клеток карциномы человеческой молочной железы ZR-75-1. Человеческую FAS очищали от культивированных клеток карциномы человеческой молочной железы ZR-75-1, полученных из Коллекции американских типовых культур. В методике, применяемой Linnet al., 1981 и Kuhajda et al., 1994, используется гипотонический лизис, последовательные осаждения полиэтиленгликолем (ПЭГ) и анионообменная хроматография. Клетки ZR-75-1 выращивали при 37 С в культуральной среде RPMI, содержащей 10% фетальной телячьей сыворотки, пенициллин и стрептомицин, в атмосфере, содержащей 5% СО 2.- 26013371 Десять колб Т 150 со слитыми клетками лизировали с использованием 1,5 мл лизирующего буфераIgepal Са-630) и образованную смесь гомогенизировали на льду, применяя 20 усилий. Лизат центрифугировали в роторе JA-20 (Beckman) со скоростью 20000 об/мин в течение 30 мин при 4 С и супернатант контактировали с 42 мл лизирующего буфера. К супернатанту медленно добавляли раствор 50% ПЭГ(PEG) 8000 в лизирующем буфере до получения конечной концентрации 7,5%. После встряхивания в течение 60 мин при 4 С раствор центрифугировали в роторе JA-20 (Бекман) со скоростью 15000 об/мин в течение 30 мин при 4 С. Затем к супернатанту добавляли твердый ПЭГ 8000 до получения конечной концентрации 15%. После повторения встряхивания и центрифугирования, как указывалось выше, осадок после центрифугирования ресуспендировали на протяжении ночи при 4 С в 10 мл буфера А (20 мМK2HPO4, рН 7,4). После отфильтровывания 0,45 мкМ белковый раствор помещали на анионообменную колонку Mono Q 5/5 (Pharmacia). Колонку промывали в течение 15 мин буфером А со скоростью 1 мл/мин и связанное вещество элюировали с 60 мл линейным градиентом в течение 60 мин до получения 1 М KCl. FAS (MM (MW) 270 kD) обычно вымывается при концентрации KCl 0,25 М в трех 0,5 мл фракциях, как было установлено с использованием 4-15% SDS-PAGE и красителя Кумасси G-250 (BioRad). Концентрация белка в FAS определялась с использованием реагента для анализа белка + реагент Кумасси (Pierce) в соответствии с инструкциями производителя и использованием в качестве стандартаBSA (альбумина бычьей сыворотки). В результате осуществления данной методики получали, по существу, чистые препараты FAS (95%), как было установлено с помощью окрашенных красителем Кумасси гелей. Измерение ферментативной активности FAS и определение IC50 соединений. Активность FAS измеряли путем непрерывного спектрофотометрического контроля, зависимого от малонил-KoA окисления NADPH в 96-луночных планшетах при OD340 (Dils et al. и Arslanian et al., 1975). Каждая лунка содержала 2 мкг очищенной FAS, 100 мМ K2HPO4, pH 6,5, 1 мМ дитиотреита (Sigma) и 187,5 мкМ -NADPH (Sigma). Приготавливали исходные растворы ингибиторов в ДМСО с концентрациями 2, 1 и 0,5 мг/мл, которые обеспечивали конечные концентрации 20, 10 и 5 мкг/мл, когда в лунку добавляли 1 мкл исходного раствора. В каждом эксперименте использовали церуленин (Sigma) в качестве положительного контроля, а также ДМСО контроль, ингибиторы и бланки (фермент FAS отсутствовал) (слепой опыт), причем все из них использовали в двух повторах. Анализ осуществляли на масс-спектрометре и спектрофотометре фирмы Molecular Devices. Планшет, содержащий FAS, буферы, ингибиторы и контроли, помещали в спектрофотометр, нагретый до 37 С. С использованием протокола кинетики в лунки (в двух повторах) помещали бланки, содержащие 100 мкл 100 мМ K2HPO4, pH 6,5, и осуществляли считывание планшета при OD340 в течение 5 мин с 10-секундными интервалами для измерения независимого от малонил-KoA окисления NADPH. Планшет удаляли из спектрофотометра и в каждую лунку, кроме бланков, добавляли малонил-KoA (конечная концентрация в лунке 67,4 мкМ) и алкинил-KoA (конечная концентрация в лунке 61,8 мкМ). Для измерения зависимого от малонил-KoA окисления NADPH планшет опять считывали, как указывалось выше, с использованием протокола кинетики. Разница между OD340 для зависимого от малонил-KoA и независимого от малонил-KoA окисления NADPH представляла собой специфическую активность FAS. Вследствие чистоты препарата FAS, независимое от малонил-KoA окисление NADPH являлось незначительным.IC50 соединений изобретения против FAS определялась построением графика зависимости OD340 от каждой тестируемой концентрации ингибитора, проведением линейной регрессии и вычислением линии наибольшего соответствия, значений r2 и 95% доверительных интервалов. Концентрация соединения, дающая 50% ингибирование FAS, представляла собой IC50. Строили графики зависимости OD340 от времени, используя компьютерное обеспечение SOFT макс. PRO (Molecular Devices), для каждой концентрации соединения. Компьютеризацию линейной регрессии, линии наибольшего соответствия, r2 и 95% доверительных интервалов осуществляли с использованием версии 3.0 Prism (компьютерное обеспечение Graph Pad). Анализ роста клеток с использованием кристаллического фиолетового. Анализ с использованием кристаллического фиолетового определяет рост клеток, но не цитотоксичность. В данном анализе применялось окрашивание кристаллическим фиолетовым фиксированных в 96-луночных планшетах клеток с последующей солюбилизацией и измерением OD340 на спектрофотометре. OD340 соответствует росту клеток в единицу времени измерения. Клетки обрабатывали представляющими интерес соединениями или контрольным наполнителем и вычисляли IC50 для каждого соединения. Для измерения цитотоксичности отдельных соединений против клеток карциномы, 5104 клеток карциномы человеческой молочной железы MCF-7, полученных из Коллекции американских типовых культур, высевали в ячейки 24-ячеечных пластин в DMEM среду, содержащую 10% фетальную телячью сыворотку, пенициллин и стрептомицин. После инкубирования в течение ночи при 37 С в атмосфере,содержащей 5% СО 2, в ячейки (в трех повторах) объемом 1 мкл добавляли подлежащие тестированию соединения, растворенные в ДМСО, имеющие следующие концентрации: 50, 40, 30 20 и 10 мкг/мл. В- 27013371 случае необходимости тестировали дополнительные концентрации. В ячейки (в трех повторах) добавляли 1 мкл контрольного наполнителя ДМСО. В качестве положительных контролей использовали С 75 с концентрациями 10 и 5 мкг/мл (в трех повторах). Через 72 ч инкубирования клетки окрашивали 0,5 мл красителя кристаллического фиолетового(0,5% в 25% метаноле) в каждой ячейке. Через 10 мин ячейки промывали, сушили на воздухе и затем солюбилизировали с использованием 0,5 мл 10% додецилсульфата натрия при встряхивании в течение 2 ч. После переноса 100 мкл из каждой ячейки в 96-луночные планшеты осуществляли считывание планшета при OD340 на масс-спектрометре и спектрофотометре фирмы Molecular Devices. Вычисляли средние значения OD340 с использованием компьютерного обеспечения SOFT макс. Pro (Molecular Devices) и определяли значения IC50 методом линейной регрессии с использованием компьютерной версии 3.02 Prism(компьютерное обеспечение Graph Pad, Сан-Диего). Анализ цитотоксичности ХТТ. Анализ цитотоксичности ХТТ является нерадиоактивной альтернативой анализу цитотоксичности с высвобождением [51Cr]. ХТТ является тетразолиевой солью, которая восстанавливается до формазанового красителя под воздействием только метаболически активных, жизнеспособных клеток. Восстановление ХТТ определяли спектрофотометрически в виде OD490-OD650. Для измерения цитотоксичности отдельных соединений против клеток карциномы, 9103 клеток карциномы человеческой молочной железы MCF-7, полученных из Коллекции американский типовых культур, высевали в лунки 96-луночных планшетов в DMEM среду, содержащую 19% фетальную телячью сыворотку, инсулин, пенициллин и стрептомицин. После инкубирования при 37 С в атмосфере, содержащей 5% CO2, в лунки (в трех повторах) объемом 1 мкл добавляли подлежащие тестированию соединения, растворенные в ДМСО, имеющие следующие концентрации: 80, 40, 20, 10, 5, 2,5, 1,25 и 0,625 мкг/мл. В случае необходимости тестировали дополнительные концентрации. В лунки (в трех повторах) добавляли 1 мкл ДМСО, который представлял собой контрольный наполнитель. В качестве положительных контролей использовали С 75 с концентрациями 40, 20, 10, 15, 12,5, 10 и 5 мкг/мл в трех повторах. Через 72 ч инкубирования клетки инкубировали в течение 4 ч с ХТТ реагентом в соответствии с инструкциями производителя (комплект II (ХТТ) для пролиферации клеток, Roche). Планшеты считывали при OD490 и OD650 на масс-спектрометре и спектрофотометре фирмы Molecular Devices. Три лунки,содержащие ХТТ реагент без клеток, служили в качестве планшетного бланка. Данные представляли в виде OD490-OD650. Средние значения и стандартную ошибку среднего вычисляли с использованием компьютерного обеспечения SOFT макс. Pro (Molecular Devices).IC50 соединений определялась как концентрация лекарственного средства, приводящая к уменьшению OD490 - OD6so на 50% по сравнению с контролями. OD490-OD650 вычисляли с использованием компьютерного обеспечения SOFT макс. Pro (Molecular Devices) для каждой концентрации соединения. IC50 рассчитывали линейной регрессией, построением графика зависимости активности FAS, выраженной в виде процента относительно значения для контроля, от концентрации лекарственного средства. Линейную регрессию, линию наибольшего соответствия, значения r2 и 95% доверительные интервалы определяли с использованием версии 3.0 Prism (компьютерное обеспечение Graph Pad). Измерение включения [14 С] ацетата в общие липиды и определение IC50 соединений. В данном анализе определялось включение [14 С] ацетата в общие липиды, которое является мерой активности пути синтеза жирных кислот in vitro. Он использовался для измерения ингибирования синтеза жирных кислот in vitro. Культивированные, как указывалось выше, клетки карциномы человеческой молочной железыMCF-7 высевали в ячейки 24-ячеечных пластин при концентрации 5104 клеток в ячейке. После инкубирования в течение ночи подлежащие тестированию соединения, солюбилизированные в ДМСО, добавляли в ячейки при концентрациях 5, 10 и 20 мкг/мл, причем в трех повторах, при этом в случае необходимости тестировали более низкие концентрации. В лунки (в трех повторах) добавляли ДМСО, который представлял собой контрольный наполнитель. В качестве положительных контролей использовали С 75 с концентрациями 5 и 10 мкг/мл в трех повторах. Через 4 ч инкубирования в каждую ячейку добавляли 0,25 мкКи [14 С] ацетата (объем 10 мкл). Через 2 ч дополнительного инкубирования из ячеек удаляли отсасыванием среду и в каждую ячейку добавляли 800 мкл смеси хлороформ:метанол (2:1) и 700 мкл 4 мМ MgCl2. Содержимое каждой ячейки переносили в пробирки Эппендорфа объемом 1,5 мл и на полной скорости вращали в течение 2 мин в высокоскоростной микроцентрифуге Эппендорфа 5415D. После удаления водного (верхнего) слоя в каждую пробирку дополнительно добавляли 700 мкл смеси хлороформ:метанол (2:1) и 500 мкл 4 мМ MgCl2 и затем центрифугировали, как указывалось выше, в течение 1 мин. Водный слой удаляли пипеткой Пастера и отбрасывали. В каждую пробирку дополнительно добавляли 400 мкл смеси хлороформ:метанол(2:1) и 200 мкл 4 мМ MgCl2, затем центрифугировали и отбрасывали водный слой. Нижнюю (органическую) фазу переносили в сцинтилляционную пробирку и сушили при 40 С под N2 газом. После сушки добавляли 3 мл сцинтиллятора (АРВNBC 5104) и осуществляли количественное определение 14 С в пробирках. С использованием сцинтилляционного счетчика Бекмана рассчитывали средние значенияIC50 соединений определялась как концентрация лекарственного средства, приводящая к 50% уменьшению включения [14 С] ацетата в липиды по сравнению с контролями. Она определялась построением графика зависимости среднего значения cpm от каждой тестируемой концентрации ингибитора,осуществлением линейной регрессии и вычислением линии наибольшего соответствия, значений r2 и 95% доверительных интервалов. Средние значения cpm вычисляли с помощью сцинтилляционного счетчика Бекмана (модель LS 6500) для каждой концентрации соединения. Компьютеризацию линейной регрессии, линии наибольшего соответствия, r2 и 95% доверительных интервалов осуществляли с использованием версии 3.0 Prism (компьютерное обеспечение Graph Pad). Анализ карнитинпальмитоилтрансферазы-1 (СРТ-1). СРТ-1 катализирует АТФ-зависимый перенос длинноцепочечных жирных кислот из ацил-KoA в ацилкарнитин, который ингибируется малонил-KoA. Поскольку для активности СРТ-1 требуется митохондриальная мембрана, ферментативная активность измерялась в способных к пропусканию клетках или митохондриях. В данном анализе способные к пропусканию клетки использовались для измерения переноса [метил-14 С] L-карнитина в органически растворимое производное ацилкарнитина. Клетки MCF-7 высевали на 24-ячеечные пластины в DMEM среду с 10% фетальной телячьей сывороткой при концентрации 106 клеток, взятые в трех повторах: для контролей, лекарственных средств и малонил-KoA. За 2 ч до завершения анализа добавляли лекарственные средства при указанных концентрациях, полученных из исходных растворов в ДМСО с концентрацией 10 мг/мл, контрольный наполнитель, состоящий из ДМСО и не содержащий лекарственного средства. Поскольку малонил-KoA не может поступать в интактные клетки, его добавляли к клеткам только в используемом для анализа буфере, которые не были предварительно инкубированы с лекарственными средствами. После инкубирования в течение ночи при 37 С среду удаляли и заменяли 700 мкл используемого в анализе буфера, состоящего из 50 мМ имидазола, 70 мМ KCl, 80 мМ сахарозы, 1 мМ EGTA, 2 мМ MgCl2, 1 мМ DTT, 1 мМ KCN,1 мМ АТФ, 0,1% бычьего сывороточного альбумина, не содержащего жирных кислот, 70 мкМ пальмитоил-KoA, 0,25 мкКи [метил-14 С] L-карнитина, 40 мкг дигитонина с лекарственным средством,контрольного наполнителя ДМСО или 20 мкМ малонил-KoA. Концентрации лекарственных средств и ДМСО в используемом для анализа буфере являются такими же, как те, которые использовались при 2-часовом предварительном инкубировании. После инкубирования в течение 6 мин при 37 С реакцию прекращали добавлением 500 мкл охлажденной льдом 4 М перхлорной кислоты. Затем клетки собирали и центрифугировали при 13000g в течение 5 мин. Осадок после центрифугирования опять промывали 500 мкл охлажденной льдом перхлорной кислоты и центрифугировали. Образованный осадок ресуспендировали в 800 мкл dH2O и экстрагировали 150 мкл бутанола. Бутанольную фазу, представляющую собой производное ацилкарнитина, определяли количественно с помощью жидкостной сцинтилляции. Проверка новых ингибиторов FAS на их способность к снижению веса. Для проверки новых ингибиторов на их способность к снижению исходного веса использовали мышей Balb/C (Jackson Labs). Животных поселяли в отапливаемые помещения с 12-часовым циклом чередования дня и ночи и мышам давали корм и воду без ограничения. Для каждого тестируемого соединения с контрольным наполнителем в трех повторах в эксперименте использовали трех мышей. При проведении экспериментов трех мышей для каждого тестируемого соединения помещали отдельно в клетку. Соединения разбавляли в ДМСО до концентрации 10 мг/мл и вводили внутрибрюшинно дозу 60 мг/кг примерно в 100 мкл ДМСО или только наполнитель. Мышей наблюдали и ежедневно взвешивали; вычисляли средние массы и стандартные ошибки с использованием программного обеспечения Excel(Microsoft). Эксперимент продолжали до тех пор, пока обработанные животные достигали своего веса до обработки. Выбранные соединения тестировали на животных, помещенных в метаболические клетки. Дозирование животным было идентично дозированию животным в экспериментах по проверке ингибиторов, в которых трех животных помещали в одну метаболическую клетку. Ежедневно измеряли вес животных,потребление ими пищи и воды и продуцирование мочи и фекалий. Результаты тестирования соединений 21 и 44 показаны на фиг. 11. Антимикробные свойства. Для оценки антимикробной активности соединений использовали анализ микроразбавления бульона. Соединения тестировали при двукратных последовательных разбавлениях и концентрации, ингибирующие видимый рост (OD600 при 10% контроля), определяли как MIC. Тестируемые микроорганизмы включали Staphylococcus aureus (ATCC29213), Enterococcus faecalis (ATCC29212), Pseudomonasaeruginosa (ATCC27853) и Escherichia coli (ATCC25922). Анализ осуществляли в двух питательных средах: бульон Мюллера-Хинтона и триптиказо-соевый бульон. Чашку или пластину с кровяным агаром (Т-соя (Tsoy)/5% крови овцы) засевали замороженной биомассой, сохраняемой в Т-соевом бульоне, содержащем 10% глицерина, и инкубировали всю ночь при 37 С. Колонии суспендировали в стерильном бульоне с тем, чтобы помутнение соответствовало помутнению 0,5 стандарта Mc Farland. Инокулят разбавляли стерильным бульоном (Мюллер-Хинтон или- 29013371 триптиказо-соевый) при соотношении 1:10 и 195 мкл распределяли по лункам 96-луночных планшетов. Подлежащие тестированию соединения, растворенные в ДМСО, добавляли в лунки (в двух повторах) в объеме 5 мкл при следующих концентрациях: 25, 12,5, 6,25, 3,125, 1,56 и 0,78 мкл/мл. В случае необходимости тестировали дополнительные концентрации. В лунки (в двух повторах) добавляли 5 мкл ДМСО,который представлял собой контрольный наполнитель. В каждый опыт включали последовательные разбавления положительных контрольных соединений: ванкомицина (Е. faecalis и S. aureus) и тобрамицина(Е. coli и P. aeruginosa). Через 24 ч инкубирования при 37 С осуществляли считывание пластин при OD600 на массспектрометре и спектрофотометре фирмы Molecular Devices. Вычисляли средние значения OD600 с использованием компьютерного обеспечения SOFT макс. Pro (Molecular Devices) и определяли значенияMIC методом линейной регрессии с использованием компьютерной версии 3.02 Prism (компьютерное обеспечение Graph Pad, Сан-Диего). MIC определялась как концентрация соединения, необходимая для получения показания OD600, эквивалентного показанию для 10% контрольного наполнителя. Тестирование противоопухолевой активности in vivo. Результаты данного эксперимента показаны на фиг. 12. Для исследования противоопухолевых действий соединения 36 in vivo использовали расположенные сбоку у мышей-самок (Harlan) подкожные ксенотрансплантаты линии человеческих клеток рака ободочной кишки НСТ-116. Все эксперименты,проводимые на животных, выполнялись в соответствии с руководством по уходу за животными, помещенными в ветеринарную лечебницу. 107 Клеток НСТ-116 (0,1 мл клеточной массы) ксенотрансплантировали из культуры в DMEM, дополненной 10% FBS, 10 бестимусным мышам (не имеющим вилочковой железы). Обработка начиналась в то время, когда через 4 дня после инокуляции развивались поддающиеся измерению опухоли. Соединение 36 (10 мг/кг) разбавляли в 20 мкл ДМСО и вводили внутрибрюшинно (i.p.). Пять животных получали JMM-II-265 внутрибрюшинно в дни, указанные стрелками на фиг. 11,и пять получали контрольный ДМСО. Опухоли измеряли в указанные дни. Отклонения в результатах представляли собой стандартную ошибку среднего. Результаты биологического тестирования (табл. 3). Таблица 3
МПК / Метки
МПК: C07D 333/32, A61K 31/38
Метки: соединения, композиции, содержащие, применения, способы, фармацевтические, новые
Код ссылки
<a href="https://eas.patents.su/30-13371-novye-soedineniya-soderzhashhie-ih-farmacevticheskie-kompozicii-i-sposoby-ih-primeneniya.html" rel="bookmark" title="База патентов Евразийского Союза">Новые соединения, содержащие их фармацевтические композиции и способы их применения</a>
Новые соединения, фармацевтические композиции, содержащие их, и способы их использования

Номер патента: 10484
Опубликовано: 30.10.2008
Авторы: Макфэдден Джилл М., Кухаджа Фрэнсис П., Таунсенд Крэйг А., Медгалчи Сьюзен М., Тхупари Джаган Н.
МПК: A61K 31/34, C07D 307/56
Метки: композиции, использования, фармацевтические, их, соединения, содержащие, способы, новые
Формула / Реферат:
1. Соединения формулы I в которой R1 означает C1-С20алкил или группу =СН2; R2 означает C1-С20алкил; X1 означает NHR4, где R4 представляет собой C1-С20алкил, С3алкенил и необязательно содержит карбоксильную группу, спиртовую группу, или когда R1 означает группу =СН2, и R2 означает С8алкил, X1 означает группу -NHCH2COOMe или группу -NHCH2COOtrBu. 2. Соединения по п.1, в которых R1 представляет C1-С10алкил или группу =СН2. 3. Соединения по п.2, в...
Новые соединения имидазолина, способ их получения и содержащие их фармацевтические композиции

Номер патента: 4536
Опубликовано: 24.06.2004
Авторы: Гобер Алан, Ньюмен-Танкреди Адриан, Миллан Марк, Лякост Жан-Мишель, Корди Алекс
МПК: C07D 233/06, A61P 25/24, A61K 31/4164...
Метки: композиции, фармацевтические, новые, имидазолина, соединения, способ, получения, содержащие
Формула / Реферат:
1. Соединения формулы (I) где A представляет кольцо бензола, незамещенное или замещенное 1-4 одинаковыми или различными группами, выбранными из неразветвленного или разветвленного (C1-C6)алкила, неразветвленного или разветвленного (C1-C6)алкокси, гидрокси, полигалоген (C1-C6)алкила, у которого алкильная часть не разветвлена или разветвлена, циано, нитро, амино, алкиламино, диалкиламино, тиоалкила, сульфонилалкила, сульфинилалкила, карбокси,...
Новые соединения циклогептена и содержащие их фармацевтические композиции

Номер патента: 4859
Опубликовано: 26.08.2004
Авторы: Хикман Джон, Доре Жильбер, Касара Патрик, Ле-Дигаре Тьерри, Гильбо Никола, Пьерре Ален, Такер Гордон
МПК: A61P 35/00, C07D 233/64, A61K 31/4174...
Метки: фармацевтические, циклогептена, соединения, содержащие, новые, композиции
Формула / Реферат:
1. Соединения формулы (I) где X представляет собой связь или группу, выбираемую из алкилена, CO, S(O)m *-S(O)n-, A1-, *-CO-A1-, -A1-S(O)n-A'1- и -A1-CO-A'1- (где A1 и A'1 одинаковы или различны, представляют собой алкиленовую группу и n представляет собой 0, 1 или 2), символ * указывает точку присоединения этих групп к циклогептену, Y представляет собой арильную, гетероарильную, циклоалкильную или гетероциклоалкильную группу, каждая из этих...
Новые соединения 2,3-метаноаминокислоты, способ их получения и содержащие их фармацевтические композиции

Номер патента: 2645
Опубликовано: 29.08.2002
Авторы: Де Нантей Гийом, Рюпэн Ален, Верберен Тони, Глоанек Филипп
МПК: A61K 38/05, C07D 209/52, A61P 7/02...
Метки: фармацевтические, соединения, новые, получения, содержащие, 2,3-метаноаминокислоты, композиции, способ
Формула / Реферат:
1. Соединение формулы (I) в которой n равно 2 или 3, R1 представляет собой (С3-С8)циклоалкильную группу, необязательно замещенную фенильную группу или линейную либо разветвленную (C1-C6)алкильную группу, необязательно замещенную одной или более одинаковыми или различными группами, выбранными из галогена, (С3-C8)циклоалкила и необязательно замещенного фенила, R2 представляет собой аминогруппу, амидиногруппу, необязательно замещенную одной или...
Новые гидразидные соединения, способ их получения и содержащие их фармацевтические композиции

Номер патента: 2442
Опубликовано: 25.04.2002
Авторы: Делла Зюана Одиль, Бутэн Жан, Монхе Вега Антонио, Кеньяр Даниель-Энри, Дюо Жак, Альдана Мораса Игнанио
МПК: C07C 271/20, A61K 31/165, A61P 3/04...
Метки: композиции, способ, гидразидные, соединения, фармацевтические, получения, содержащие, новые
Формула / Реферат:
1. Соединения формулы (I) R-NH-A-CO-NH-NH-(W)n-Z (I) где n равен 0 или 1, W обозначает группу -СО- или группу S(O)r, где r равен 0, 1 или 2, Z обозначает группу, выбранную из необязательно замещенного арила, необязательно замещенного арилалкила, необязательно замещенного гетероарила и необязательно замещенного гетероарилалкила, R обозначает группу, выбранную из: Z1-T-CO- Z1-O-T-CO- Z1-T-O-CO- Z1-T-S(O)q- где Z1 обозначает необязательно...
Предыдущий патент: Средство для профилактики стоматологических заболеваний
Следующий патент: Термоотверждаемая связующая композиция, способ связывания волокон и стекловолокнистый материал
Случайный патент: Беспотенциальный разъем, встроенный в планку крепления оружейных аксессуаров