Способ получения 1,1,1,2-тетрафторэтана
Номер патента: 715
Опубликовано: 28.02.2000
Авторы: Маккоун Джефри Уоррен, Джэскот Стенли Майкл, Чиу Юон, Смит Эддисон Майлз, Фриденсон Джей Филип, Керри Густаво, Клеммер Пол Ген, Танг Хсуех Санг, Визо Немезик Рогелио





Формула / Реферат
1. Способ получения 1,1,1,2-тетрафторэтана, включающий:
а) осуществление первой стадии реакции, заключающейся в выпаривании первой возвращенной в цикл композиции, состоящей из фтористого водорода и 1,1,1-трифтор-2-хлорэтана при мольном отношении фтористого водорода к 1,1,1-трифтор-2-хлорэтану, равном, по меньшей мере, примерно 1:1, и реакции этой композиции при подходящих условиях в присутствии катализатора фторирования с образованием первого продукта реакции, содержащего 1,1,1,2-тетрафторэтан; и
б) осуществление второй стадии реакции, заключающейся в выпаривании второй композиции, состоящей из фтористого водорода, трихлорэтилена и первого продукта реакции (а), таким образом, что мольное отношение фтористого водорода к трихлорэтилену составляет, по меньшей мере, примерно 3:1, причем вторая стадия реакции проводится в присутствии катализатора фторирования и при температуре, которая выше чем на первой стадии реакции, с получением второго продукта реакции, включающего смесь 1,1,1,2-тетрафторэтана, 1,1,1-трифтор-2-хлорэтана, фтористого водорода, трихлорэтилена и хлористого водорода.
2. Способ по п.1, дополнительно включающий последующую стадию выделения 1,1,1,2-тетрафторэтана.
3. Способ по п.1, дополнительно включающий последующие стадии:
в) выделения хлористого водорода путем первой отгонки из второго продукта реакции со стадии (б);
г) выделения продукта, содержащего 1,1,1,2-тетрафторэтан, путем второй отгонки из смеси, полученной на стадии (в), и получения возвращаемой в цикл смеси 1,1,1-трифтор-2-хлорэтана, трихлорэтилена и фтористого водорода из второго отгона и добавление возвращаемой в цикл смеси в качестве питательной загрузки на стадии (а); и
д) выделения практически чистого 1,1,1,2-тетрафторэтана из продукта со стадии (г).
4. Способ по п.1, в котором вторую стадию (б) реакции проводят при температуре, которая, по меньшей мере, на 5шС выше чем на первой стадии (а) реакции.
5. Способ по п.3, дополнительно включающий добавление части возвращаемой в цикл смеси 1,1,1-трифтор-2-хлорэтана, трихлорэтилена и фтористого водорода из второго отгона в качестве питательной смеси на вторую стадию (б) реакции.
6. Способ по п.1, дополнительно включающий дополнительную стадию удаления НС1 из первого продукта реакции до стадии (б).
7. Способ по п.1, в котором катализатор фторирования, используемый на стадиях (а) и (б) выбран из группы, включающей окислы хрома, алюминия, кобальта, марганца, никеля и железа, гидроокиси, галогениды, оксигалогениды и неорганические соли этих металлов, Сr2О3/Аl2О3, Сr2O3/АlF3, Сr2О3/уголь, СоСl2/Cr2О3/Аl2O3, NiCl2/Cr2О3/Al2O3, CoCl2/AlF3 и NiСl2/AlF3.
8. Способ по п.1, в котором катализатор фторирования, используемый на стадиях (а) и (б) представляет собой Сr2О3.
9. Способ по п.1, в котором катализатор фторирования, используемый на стадиях (а) и (б) представляет собой Сr2О3, активность которого поддерживают пропусканием потока кислорода.
10. Способ по п.1, в котором первую стадию (а) реакции проводят при температуре в интервале от примерно 300шС до примерно 375шС; вторую стадию (б) реакции проводят при температуре в интервале от примерно 305шС до примерно 380шС; вторую стадию (б) реакции проводят при температуре, которая выше на примерно 5-30шС чем температура на первой стадии (а) реакции; катализатор фторирования, используемый на стадиях (а) и (б) представляет собой Cr2О3, активность которого поддерживают пропусканием потока кислорода; время контакта первой и второй композиций с катализатором фторирования на стадиях (а) и (б) находится в интервале от примерно 5 с до примерно 60 с; стадии (а) и (б) проводят при давлении примерно 50-150 ф/дюйм2 (344,7-1034,2 кПа); мольное отношение фтористого водорода к 1,1,1-трифтор-2-хлорэтану на стадии (а) находится в интервале от примерно 3:1 до примерно 60:1 и мольное отношение фтористого водорода к трихлорэтилену на второй стадии (б) реакции находится в интервале от примерно 5:1 до примерно 80:1.
Текст
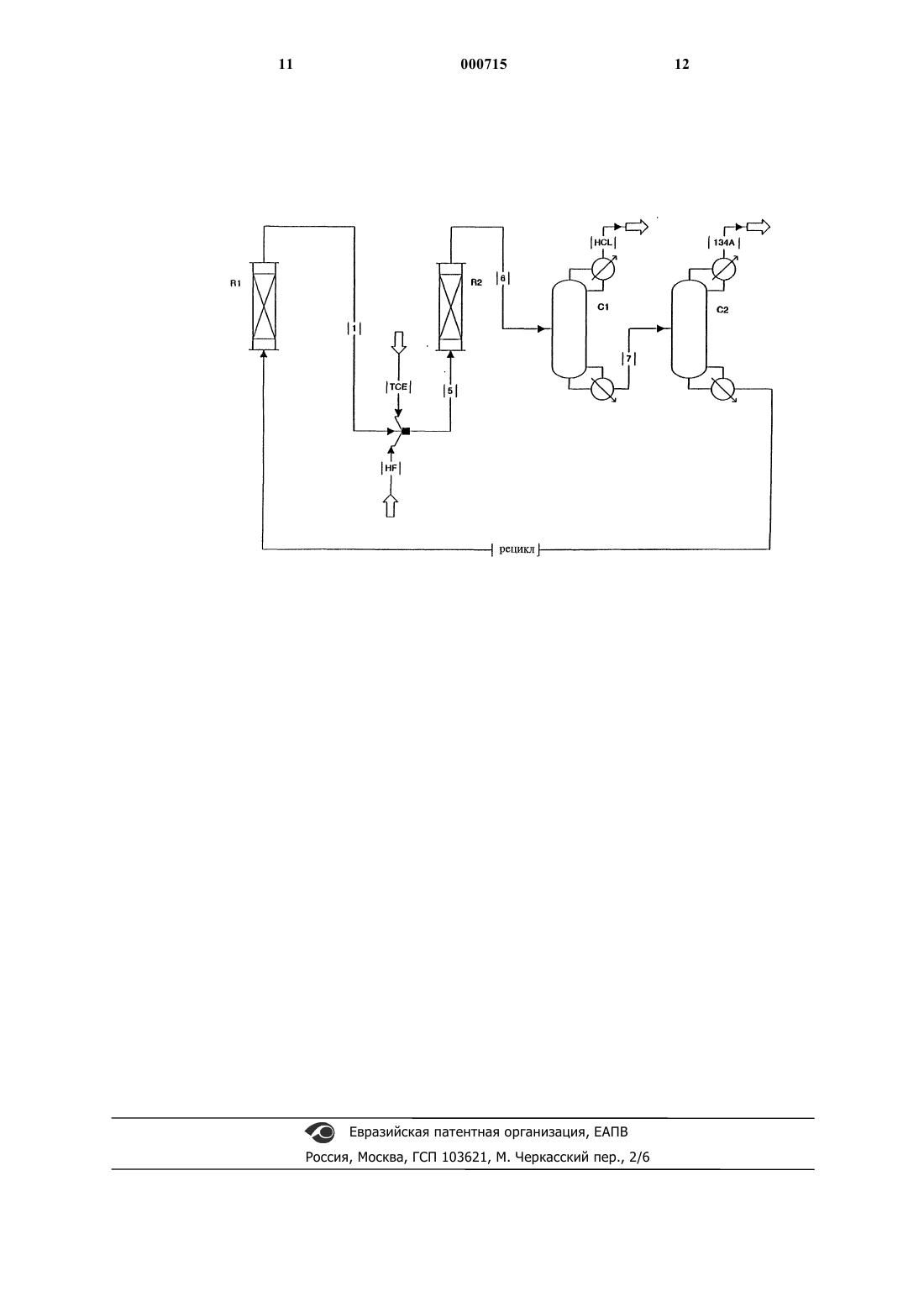
1 Область изобретения Данное изобретение относится к способу получения 1,1,1,2-тетрафторэтана (HFC-134a). В особенности оно относится к способу полученияHFC-134a парофазным каталитическим фторированием 1,1,1-трифтор-2-хлорэтана (HCFC133a) фтористым водородом в первом реакторе и перемещением полученного продукта во второй реактор вместе с трихлорэтиленом (ТСЕ) и фтористым водородом (HF). Вторая реакция проводится в присутствии катализатора фторирования при более высокой температуре, чем первая реакция. Описание предшествующего уровня техники Известно, что HFC-134a является очень полезным соединением для замены вредных для окружающей среды хлорфторуглеводородных охлаждающих агентов. Оно также пригодно в качестве вспенивающего агента и газавытеснителя в аэрозольной упаковке. Известны разнообразные способы получения HFC-134a. В патентах США 5243105 и 5395996 описан двухстадийный способ получения HFC-134a,заключающийся в реакции трихлорэтилена с фтористым водородом с образованием HCFC133a. Затем HCFC-133a реагирует с фтористым водородом на второй стадии с образованиемHFC-134a. В этих источниках указано, что взаимодействие трихлорэтилена с фтористым водородом для получения HCFC-133a нужно проводить при более низкой температуре, чем реакцию HCFC-133a с фтористым водородом. Последовательность стадий и различие в температурных условиях противоположны тем, которые используются согласно данному изобретению. В патентах США 5243107 и 5382722 описано взаимодействие HCFC-133a и HF в первой реакционной зоне и последующее пропускание продукта реакции во вторую реакционную зону вместе с трихлорэтиленом. Эта вторая реакционная зона находится при более низкой температуре, чем первая зона. И в этом случае это противоположно температурным условиям согласно данному изобретению. Согласно патентам США 5334786 и 5395988 получаютHFC-134a взаимодействием трихлорэтилена с фтористым водородом с образованием HCFC133a с последующим фторированием HCFC133a. Этот способ требует разбавления трихлорэтилена и фтористого водорода азотом или аргоном, которые инертны по отношению к реагентам, а также требует наличия трех реакторов. В патенте США 4158675 раскрыто получение 1,1,1,2-тетрафторэтана HFC-134a парофазным каталитическим фторированием HCFC-133a фтористым водородом в первом реакторе. При указанных условиях получается как примесь нежелательный 1,1-дифтор-2-хлорэтилен, который реагирует с фтористым водородом. Ранее стояла проблема достижения сравнительно высоких выходов HFC-134a без одно 000715 2 временного получения в больших количествах побочных продуктов, которые нужно обрабатывать и от которых нужно избавляться. Данное изобретение предусматривает способ, в котором промежуточные продукты возвращаются в цикл на стадии синтеза, тем самым увеличивается эффективность процесса. Более высокая температура в реакторе на второй стадии обеспечивает ряд преимуществ. Они включают более высокий выход HCFC-133a и более высокую степень конверсии ТСЕ. Следовательно, можно использовать меньший реактор и меньшее количество катализатора. Соответственно, снижаются затраты и капиталовложения. Становится возможным достижение высокой степени конверсии трихлорэтилена, приближающейся к 100%. Более высокие степени конверсии ТСЕ могут устранить возможность разделения фаз в потоке рецикла. Они также приводят к уменьшению количеств ТСЕ, подаваемых в первый реактор, что способствует уменьшению образования НС 1 в первом реакторе и тем самым увеличению равновесных количеств HFC-134a, образующегося в первом реакторе. Количество побочных хлорфторуглеводородов значительно снижается или они вообще не получаются. Способ также приводит к получению полезныхHFC-125 и HFC-143-a, являющихся побочными продуктами, вместо HFC-123/124 и HCFC141b/142b, соответственно. HFC-125 и HFC143-a, которые также являются полезными охлаждающими агентами, не приводят к разрушению озона, в то время как HFC-123/124 и HCFC141b/142b разрушают озон и снимаются с производства. Экономится энергия, так как для отделения технического продукта HFC-134a отHCFC-133 а и фтористого водорода, которые возвращаются в первый реактор, не требуется охлаждения. Кроме того, поскольку реакция трихлорэтилена и фтористого водорода является экзотермической, выделяющееся при этом тепло используется для поддержания более высокой температуры во втором реакторе. СУЩНОСТЬ ИЗОБРЕТЕНИЯ Изобретение предусматривает способ получения 1,1,1,2-тетрафторэтана, который включает: а) осуществление первой стадии реакции,состоящей в выпаривании первой возвращенной в цикл композиции, содержащей фтористый водород и 1,1,1-трифтор-2-хлорэтан при мольном отношении фтористого водорода к 1,1,1 трифтор-2-хлорэтану, равном, по меньшей мере,примерно 1:1, и взаимодействие этой композиции в подходящих условиях в присутствии катализатора фторирования для получения первого продукта реакции, представляющего собой 1,1,1,2-тетрафторэтан; и б) осуществление второй стадии реакции,состоящей в выпаривании второй композиции,содержащей фтористый водород, трихлорэтилен и первый продукт реакции со стадии а), таким 3 образом, что мольное отношение фтористого водорода к трихлорэтилену равняется, по меньшей мере, примерно 3:1, причем вторая стадия реакции проводится в присутствии катализатора фторирования и при температуре, которая выше, чем на первой стадии реакции, с получением второго продукта реакции, включающего 1,1,1,2-тетрафторэтан,1,1,1-трифтор-2-хлорэтан, фтористый водород, трихлорэтилен и хлористый водород. Согласно предпочтительному варианту выделяют 1,1,1,2-тетрафторэтан. Это может быть сделано при осуществлении стадий: в) выделения хлористого водорода путем первой отгонки из второго продукта реакции со стадии (б); г) выделения продукта, представляющего собой 1,1,1,2-тетрафторэтан, путем второй отгонки из смеси, полученной на стадии (в), и получения возвращаемой в цикл смеси 1,1,1 трифтор-2-хлорэтана, трихлорэтилена и фтористого водорода из второго отгона и добавление этой смеси на стадии (а); и д) выделения практически чистого 1,1,1,2 тетрафторэтана из продукта со стадии (г). Другой вариант изобретения предусматривает возможное добавление части возвращаемой в цикл смеси 1,1,1-трифтор-2-хлорэтана, трихлорэтилена и фтористого водорода из второго отгона в качестве питающей смеси на второй стадии реакции (б). Еще один вариант предусматривает выделение НСl из первого продукта реакции в первом реакторе. На чертеже представлено схематическое изображение способа по изобретению. ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНОГО ВАРИАНТА Первая стадия (а) при получении HFC-134a представляет собой выпаривание и взаимодействие первой возвращенной в цикл композиции,состоящей из фтористого водорода и HCFC133a, в первом реакторе. Этот реактор обозначен как R1 на чертеже. В то время как здесь можно применять свежие фтористый водород иHCFC-133a, в способе заявляется композиция,содержащая возвращенный в цикл продукт со стадии (б). Мольное отношение фтористого водорода к HCFC-133a устанавливается равным,по меньшей мере, примерно 1:1, предпочтительно от примерно 1:1 до примерно 100:1, более предпочтительно от примерно 2:1 до примерно 80:1 и наиболее предпочтительно от примерно 3:1 до примерно 60:1. Можно использовать мольные отношения более, чем примерно 100:1, однако это менее экономично. Испарившуюся композицию нагревают в первом реакторе предпочтительно до температуры от примерно 250 до примерно 425 С, более предпочтительно от примерно 280 до примерно 400 С и наиболее предпочтительно от примерно 300 до примерно 375 С. Температуру в реакторе измеряют на выходе. Давление в реакторе не являет 000715 4 ся критическим. Рабочее давление составляет предпочтительно от примерно 0 до примерно 200 ф/дюйм 2 и еще предпочтительнее от примерно 50 до примерно 150 ф/дюйм 2. Первый реактор является предпочтительно адиабатическим, заполненным катализатором фторирования. Пары органического соединения контактируют с катализатором фторирования в течение примерно 1-100 с, более предпочтительно в течение примерно 3-70 с и наиболее предпочтительно в течение примерно 5-60 с. Для целей данного изобретения время контакта представляет собой время, которое требуется для прохождения газообразных реагентов через слой катализатора при условии, что этот слой является на 100% пористым. Можно использовать любой известный катализатор фторирования. Такие катализаторы включают окислы хрома, алюминия, кобальта, марганца, никеля и железа, гидроокиси, галоидные соединения, оксигалогениды и неорганические соли этих металлов,Cr2 О 3/Аl2 О 3,Сr2 О 3/АlF3,Сr2 О 3/уголь,СоСl2/Сr2 О 3/Аl2 О 3, NiCl2/Сr2 О 3/Al2O3, CoCl2/АlF3 и NiСl2/АlF3. Катализаторы окись хрома/окись алюминия описаны в патенте США 5155082. Окись хрома может быть кристаллической или аморфной. Предпочтительной является аморфная окись хрома. Окись хрома (Cr2 О 3) является коммерчески доступной и продается в виде гаммы продуктов с частицами различного размера. Катализатор содержится в количестве,необходимом для осуществления реакции. Согласно предпочтительному варианту, через окись хрома для поддержания активности катализатора пропускают небольшое количество газообразного кислорода иди воздуха. Количество подаваемого в реактор воздуха или кислорода составляет предпочтительно от примерно 0,01 до примерно 30 мол.% кислорода в расчете на общее количество органических веществ,подаваемых в реактор. Более предпочтительно подавать от примерно 0,05 до примерно 20 мол.% и наиболее предпочтительно от примерно 0,1 до примерно 10 мол.%. Полученная реакционная смесь включает HFC-134a, HCFC-133a,фтористый водород, НСl и небольшие количества других побочных продуктов. Стадия (б) проводится предпочтительно одновременно со стадией (а) и включает выпаривание второй композиции, содержащей фтористый водород, ТСЕ и первый продукт реакции, полученный на стадии (а), который перетекает из первого реактора R1 по линии 1 во второй реактор R2, как показано на чертеже. HF,ТСЕ и первый продукт реакции, полученный на стадии (а), соединяются и перетекают по линии 5 в реактор R2, как показано на чертеже. Согласно данному способу композиция, реагирующая на стадии (б), содержит свежую порцию ТСЕ и HF и может содержать возвращаемый в цикл продукт со стадии (г), описанной ниже. Эту вторую композицию нагревают до 5 температуры от примерно 255 до примерно 430 С, более предпочтительно от примерно 285 до примерно 405 С и наиболее предпочтительно от примерно 305 до примерно 380 С. Температуру в реакторе также измеряют на выходе. Важным признаком изобретения является проведение второй стадии (б) реакции при температуре, которая выше, чем температура на первой стадии (а). Согласно предпочтительному варианту, разница температур на стадии (а) и стадии(б) колеблется от примерно 5 до примерно 130 С или более предпочтительно от примерно 5 до примерно 60 С и наиболее предпочтительно от примерно 5 до примерно 30 С. Давление в реакторе не является критическим. Рабочее давление может составлять, предпочтительно, от примерно 0 до примерно 200 ф/дюйм 2 и более предпочтительно от примерно 50 до примерно 150 ф/дюйм 2. Вторая стадия также осуществляется в присутствии катализатора фторирования,который может быть любым из перечисленных для первой стадии (а). На второй стадии (б) реакции мольное отношение HF к ТСЕ может меняться от примерно 3:1 до примерно 100:1 или,предпочтительнее, от примерно 4:1 до примерно 90:1 и, более предпочтительно, от примерно 5:1 до примерно 80:1. Можно использовать мольные отношения более 100, но это менее экономично. В то время как первый продукт реакции подается во второй реактор на стадию (б),1,1,1,2-тетрафторэтан в основном получается на первой стадии. Он затем проходит через второй реактор. Установлено, что во второй реакции могут принимать участие побочные продукты,образующиеся во время первой реакции и не являющиеся HFC-134a. Продукт второй реакции в основном содержит HCFC-133a и вместе с непрореагировавшим HF возвращается в цикл в первый реактор, где образуется HFC-134a. Согласно предпочтительному варианту на обеих стадиях (а) и (б) продукт проходит вниз через слой катализатора. Катализатор предпочтительно предварительно обрабатывать и активировать, а также регенерировать после продолжительного использования в реакторе. Предварительную обработку можно осуществлять путем нагревания катализатора до примерно 250430 С в токе HF, разбавленного газообразным азотом, для достижения высокой активности катализатора. Предпочтительно для поддержания каталитической активности в каждый реактор во время работы непрерывно подавать кислород. Кислород подают со скоростью, достаточной для обеспечения мольного отношения кислорода к органическим веществам от примерно 0 до примерно 0,1 или, предпочтительно,от примерно 0,005 до примерно 0,05. Если катализатор дезактивирован, его можно регенерировать нагреванием до примерно 250-430 С в токе азота, содержащего низкую концентрацию кислорода с последующим охлаждением. Каждую стадию (а) и (б) можно проводить в любом под 000715 6 ходящем реакционном сосуде, но он должен быть изготовлен из материалов, стойких к коррозионному действию фтористого водорода,таких как Hastalloy, Inconel и Monel. На следующей стадии (в) выделяется хлористый водород путем первой отгонки из продукта второй реакции на стадии (б). Продукт второй реакции протекает по линии 6 и подвергается такой отгонке в колонне Сl, как показано на чертеже, с получением отгона и кубового остатка. Цель отгонки заключается в отделении хлористого водорода от баланса продукта второй реакции. Это осуществляют с использованием обычной дистилляционной колонны методом, хорошо известным специалистам. Отгонку предпочтительно осуществлять при давлении от примерно 5 ф/дюйм 2 до примерно 500 ф/дюйм 2,предпочтительно от примерно 10 до примерно 400 ф/дюйм 2 и наиболее предпочтительно от примерно 50 до примерно 300 ф/дюйм 2. Давление в дистилляционной колонне определяет рабочую температуру отгонки. Отогнанная часть включает практически весь хлористый водород,а кубовый остаток содержит баланс продукта второй реакции. Кубовый остаток затем подвергают второй перегонке, его передают по линии 7 в колонну С 2, как показано на чертеже. Стадия (г) предусматривает выделение продукта стадии (в), содержащего HFC-134a также в обычной дистилляционной колонне методом, хорошо известным специалисту, таким,как описано выше, с получением дистиллята и кубового остатка. Дистиллят состоит практически из HFC-134a и других полезных фторуглеводородных побочных продуктов реакции, таких как HFC-125 и HFC-143a. HCFC-133a, фтористый водород и ТСЕ кубовый остаток возвращаются в цикл на стадию (а), как показывает линия рецикла на чертеже. Стадия (д) предусматривает выделение композиции, содержащей практически чистый HFC-134a, из продукта,полученного на стадии (г) и других полезных фторуглеводородных побочных продуктов, таких как HFC-125 и HFC-143a. Это осуществляется путем обычной отгонки или другими известными методами. Следующие ниже примеры иллюстрируют стандартный способ и методики предсказания физических свойств и служат для иллюстрации изобретения. Примеры 1-3. Эти примеры показывают влияние температуры на производительность и величину конверсии. В трех различных опытах смесь ТСЕ иHCFC-133a подают в изотермический реактор с насадкой при 260, 320 и 360 С соответственно. Насадкой служит катализатор окись хрома. Мольное отношение HCFC-133a к ТСЕ составляет примерно 3,3. HF подают отдельно. Мольное отношение HF к ТСЕ равно примерно 13. Подают также воздух в количестве 1,4 мол.% О 2 в расчете на органические вещества. Давление в реакторе равно 200 ф/дюйм 2. Скорости подачи органических веществ и HF регулируют таким образом, чтобы получить время контакта, равное 20, 10 и 5 с соответственно. Результаты приведены ниже. Таблица 1 При- Темп., Время мер С контакта, с 1 2 3 Пример 3 был рассчитан с использованием кинетической модели реакции, полученной на основе экспериментальных данных. Эти данные свидетельствуют, что чем выше температура реакции во втором реакторе, тем выше конверсия ТСЕ и тем выше выход HCFC-133a. Примеры 4, 5 и сравнительный пример 6. Эти примеры демонстрируют влияние более высокой температуры во втором реакторе на величину степени конверсии ТСЕ, выход 133 а и получение полезных побочных продуктов с использованием интегрированной системы, показанной на чертеже. Примеры 4 и 5 иллюстрируют процесс, осуществляемый в адиабатических реакторах, содержащих катализатор окись хрома. ТСЕ и HF подают во второй реактор (R2),как показано на чертеже. НСl выводится из колонны для НСl и тяжелая фракция, состоящая из 134a/HF/TCE/133a и других побочных продуктов, подается в колонну с сырым 134 а, откуда 134 а/125/143 а/124 отбираются, a 133a/HF/TCE возвращаются в цикл в первый реактор, как показано на чертеже. Условия реакции и параметры приведены в табл. 2, как и степень конверсии ТСЕ, выход 133 а и количества полезных побочных продуктов. Таблица 2 Пример 4 5 Сравн. 6 Темп. на выходе в 329 333 350 Данные для сравнительного примера 6 получены в сравнительных условиях с использованием компьютерной стимуляции, осуществляемой на основе многочисленных экспериментальных данных. Как указано в данной заявке и как видно из вышеприведенных примеров, более высокая степень конверсии ТСЕ и более высокий выход получаются в том случае, когда температура во втором реакторе (R2) выше, чем в первом реакторе (R1). Концентрация ТСЕ в рецикле составляет примерно 0 (около 19% в расчете на общее количество органических веществ) (в сравнительном примере 6%). Из табл.2 видно, что образуются полезные HFC-побочные продукты. Пример 7. Этот пример демонстрирует экономию энергии при отделении НС 1 перед выделением сырого HFC-134a из HCFC-133a и HF, которые возвращаются в первый реактор. Оборудование для конденсации в колонне для рецикла используют для отделения сырогоHFC-134a от рецикла HCFC-133a и HF, возвращаемого в реактор, рассчитывают с использованием модели дистилляционного процесса, разработанной на основе теоретических положений и лабораторных измерений равновесного состояния пар-жидкость. Расчет показывает, что когда полученный в процессе реакции НСl удаляется вместе с HFC-134a, в колонне для рецикла температура конденсации очень низка, поэтому для получения отгона в этой колонне требуется охлаждение. Однако, когда в начале удаляется НСl в соответствии с данным изобретением, можно избежать необходимости охлаждения для получения отгона в колонне, тем самым экономятся капитальные затраты на систему охлаждения, а также снижается стоимость энергии, необходимой для работы. Когда HFC-134a и НСl отделяют вместе от HCFC-133a и HF при обычном рабочем давлении 150 ф/дюйм 2 (1034,2 кПа), требуется дополнительный расход энергии примерно 350 л.с. на тонну HFC-134a. В колонне для рецикла не требуется охлаждения, когда вначале удаляется НСl. Экономия энергии еще 9 больше, если реакторы работают при более низком давлении. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения 1,1,1,2-тетрафторэтана, включающий: а) осуществление первой стадии реакции,заключающейся в выпаривании первой возвращенной в цикл композиции, состоящей из фтористого водорода и 1,1,1-трифтор-2-хлорэтана при мольном отношении фтористого водорода к 1,1,1-трифтор-2-хлорэтану, равном, по меньшей мере, примерно 1:1, и реакции этой композиции при подходящих условиях в присутствии катализатора фторирования с образованием первого продукта реакции,содержащего 1,1,1,2 тетрафторэтан; и б) осуществление второй стадии реакции,заключающейся в выпаривании второй композиции, состоящей из фтористого водорода, трихлорэтилена и первого продукта реакции (а),таким образом, что мольное отношение фтористого водорода к трихлорэтилену составляет, по меньшей мере, примерно 3:1, причем вторая стадия реакции проводится в присутствии катализатора фторирования и при температуре, которая выше чем на первой стадии реакции, с получением второго продукта реакции, включающего смесь 1,1,1,2-тетрафторэтана, 1,1,1 трифтор-2-хлорэтана, фтористого водорода,трихлорэтилена и хлористого водорода. 2. Способ по п.1, дополнительно включающий последующую стадию выделения 1,1,1,2-тетрафторэтана. 3. Способ по п.1, дополнительно включающий последующие стадии: в) выделения хлористого водорода путем первой отгонки из второго продукта реакции со стадии (б); г) выделения продукта, содержащего 1,1,1,2-тетрафторэтан, путем второй отгонки из смеси, полученной на стадии (в), и получения возвращаемой в цикл смеси 1,1,1-трифтор-2 хлорэтана, трихлорэтилена и фтористого водорода из второго отгона и добавление возвращаемой в цикл смеси в качестве питательной загрузки на стадии (а); и д) выделения практически чистого 1,1,1,2 тетрафторэтана из продукта со стадии (г). 4. Способ по п.1, в котором вторую стадию(б) реакции проводят при температуре, которая, 000715 10 по меньшей мере, на 5 С выше чем на первой стадии (а) реакции. 5. Способ по п.3, дополнительно включающий добавление части возвращаемой в цикл смеси 1,1,1-трифтор-2-хлорэтана, трихлорэтилена и фтористого водорода из второго отгона в качестве питательной смеси на вторую стадию(б) реакции. 6. Способ по п.1, дополнительно включающий дополнительную стадию удаления НС 1 из первого продукта реакции до стадии (б). 7. Способ по п.1, в котором катализатор фторирования, используемый на стадиях (а) и(б), выбран из группы, включающей окислы хрома, алюминия, кобальта, марганца, никеля и железа, гидроокиси, галогениды, оксигалогениды и неорганические соли этих металлов,Сr2 О 3/Аl2 О 3,Сr2O3/АlF3,Сr2 О 3/уголь,СоСl2/Cr2 О 3/Аl2O3, NiCl2/Cr2 О 3/Al2O3, CoCl2/AlF3 и NiСl2/AlF3. 8. Способ по п.1, в котором катализатор фторирования, используемый на стадиях (а) и(б), представляет собой Сr2 О 3. 9. Способ по п.1, в котором катализатор фторирования, используемый на стадиях (а) и(б), представляет собой Сr2 О 3, активность которого поддерживают пропусканием потока кислорода. 10. Способ по п.1, в котором первую стадию (а) реакции проводят при температуре в интервале от примерно 300 С до примерно 375 С; вторую стадию (б) реакции проводят при температуре в интервале от примерно 305 С до примерно 380 С; вторую стадию (б) реакции проводят при температуре, которая выше на примерно 5-30 С чем температура на первой стадии (а) реакции; катализатор фторирования,используемый на стадиях (а) и (б), представляет собой Cr2 О 3, активность которого поддерживают пропусканием потока кислорода; время контакта первой и второй композиций с катализатором фторирования на стадиях (а) и (б) находится в интервале от примерно 5 с до примерно 60 с; стадии (а) и (б) проводят при давлении примерно 50-150 ф/дюйм 2 (344,7-1034,2 кПа); мольное отношение фтористого водорода к 1,1,1-трифтор-2-хлорэтану на стадии (а) находится в интервале от примерно 3:1 до примерно 60:1 и мольное отношение фтористого водорода к трихлорэтилену на второй стадии (б) реакции находится в интервале от примерно 5:1 до примерно 80:1.
МПК / Метки
МПК: C07C 19/08
Метки: способ, 1,1,1,2-тетрафторэтана, получения
Код ссылки
<a href="https://eas.patents.su/7-715-sposob-polucheniya-1112-tetraftoretana.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения 1,1,1,2-тетрафторэтана</a>
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Дельтиль Мишель, Бонне Алан, Мазюри Алан
МПК: C07H 17/08
Метки: 5-0-дезозаминил-6-0-метилэритронолида, получения, продуктов, производные, способ, применение, биологически, активных
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Мейллян Пьер, Бьенейм Юг, Ансель Жан-Эрик
МПК: A61K 31/355, B01J 31/24, C07C 39/19...
Метки: фенолов, использованием, замещенных, cпособ, витамина, способ, получения
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Чандер Мадхави С., Свинделл Чарльз С., Систи Николас Дж.
МПК: C07D 305/14
Метки: промежуточное, способ, промежуточного, соединения, соединение, получения, паклитаксела
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Трициклические соединения, способ их получения, способы получения оптически активных или рацемических производных колхицина и тиохолкицина с использованием трициклических соединений и промежуточныепродукты синтеза

Номер патента: 93
Опубликовано: 25.06.1998
Авторы: Тороманофф Эдмон, Пронин Дидье, Диолез Кристиан, Мазюри Алан, Миддендорп Мишель, Брион Франсис, Шаппер Бернадетт, Мари Кристиан
МПК: C07C 43/21, C07D 317/44
Метки: оптически, рацемических, использованием, тиохолкицина, колхицина, трициклических, получения, способ, активных, соединения, соединений, трициклические, способы, производных, промежуточныепродукты, синтеза
Формула / Реферат:
1. Трициклические соединения общей формулы I в которой либо а) оба R1 и R2 представляют собой алкильную группу, a R3 представляет собой атом водорода или группу A-SO2-, либо б) оба R2 и R3 представляют собой атом водорода или алкил, a R1 представляет собой группу A-SO2-, либо в) все три: R1, R2 и R3 представляют собой атом водорода или все три представляют собой алкил, либо г) R1 представляет собой группу А-SO2- или атом водорода, a...
Способ получения бензо[b]тиофенов, промежуточные соединения и способ их получения.

Номер патента: 682
Опубликовано: 28.02.2000
Авторы: Хоард Дэвид В., Льюк Уэйн Д.
МПК: C07D 333/62
Метки: способ, бензо[b]тиофенов, получения, соединения, промежуточные
Формула / Реферат:
1. Соединение формулы где R1 представляет водород, C1-C4алкокси, C1-C4-алкоксигруппу, замещенную от 1 до 3 фенильной или замещенной фенильной группами, галоген или амино, R2 представляет водород, C1-C4алкокси, С1-С4-алкоксигруппу, замещенную от 1 до 3 фенильной или замещенной фенильной группами, галоген или амино. 2. Соединение по п.1, в котором R1 представляет водород, C1-C4 алкокси или С1-С4-алкоксигруппу, замещенную от 1 до 3...
Предыдущий патент: Водная силикатная композиция.
Следующий патент: Устройство для очистки волокнистого материала
Случайный патент: Фунгицидные смеси, содержащие замещенные анилиды 1-метилпиразол-4-ил-карбоновой кислоты