Бифенилкарбоксамиды, снижающие уровень липидов



















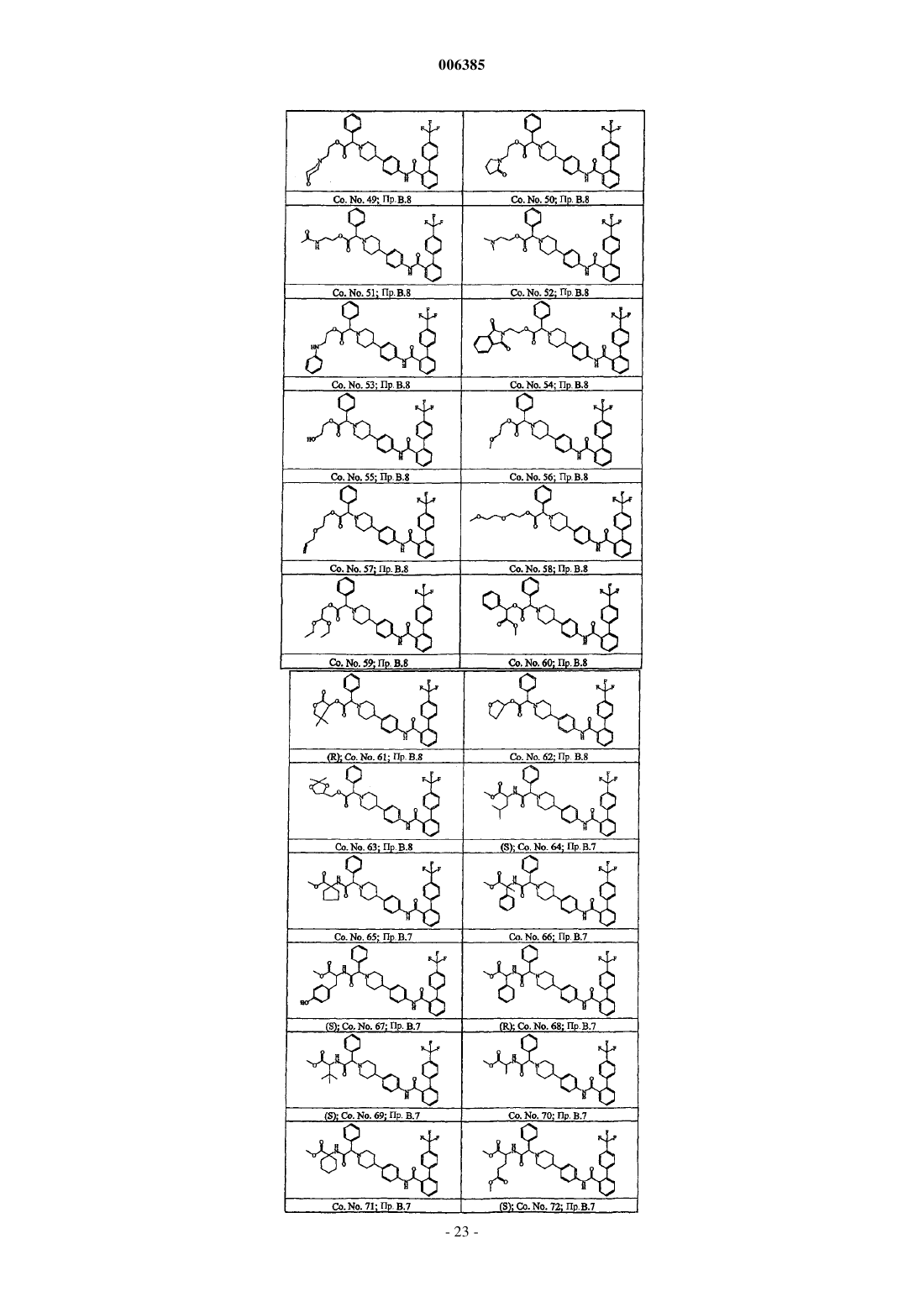
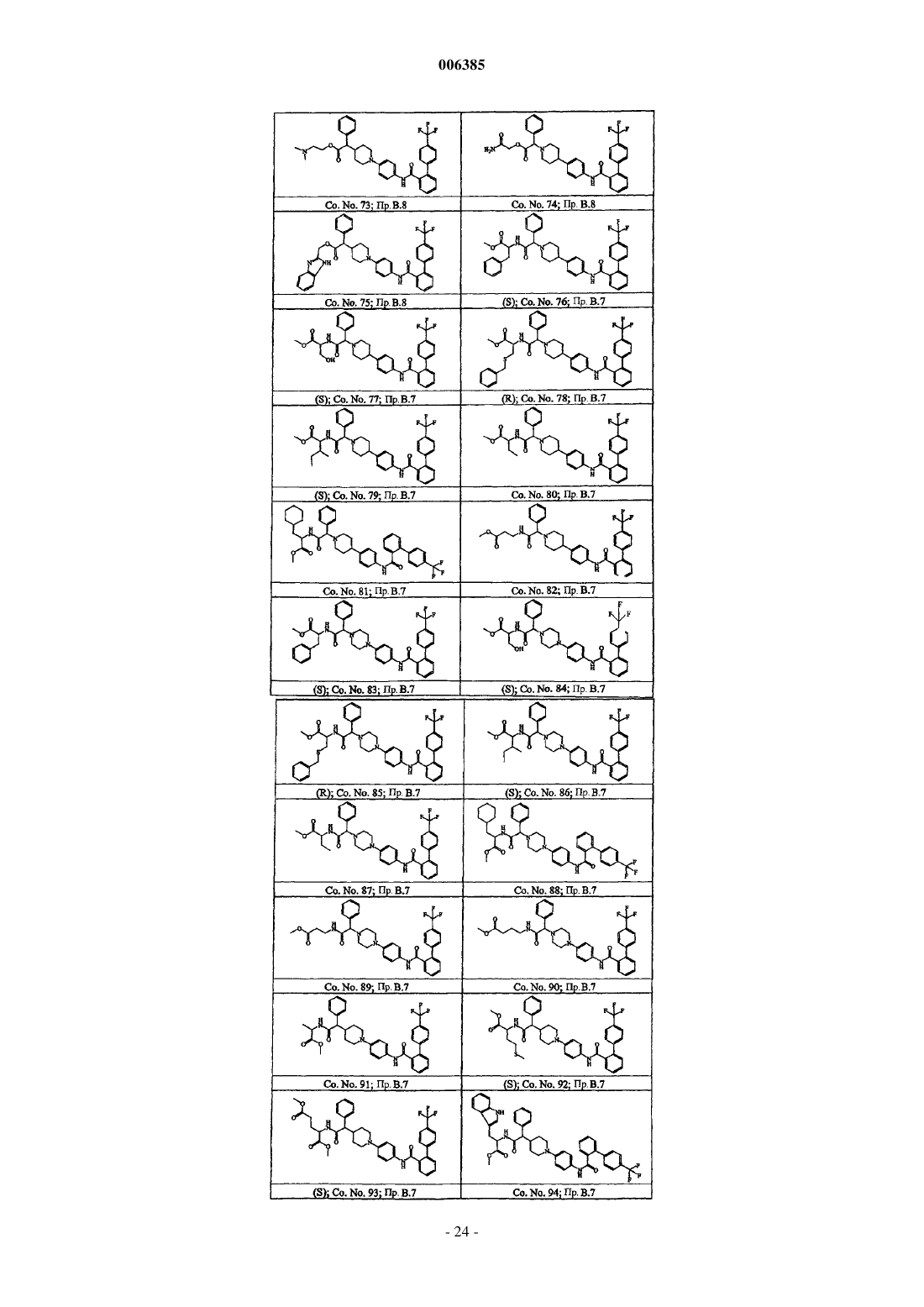
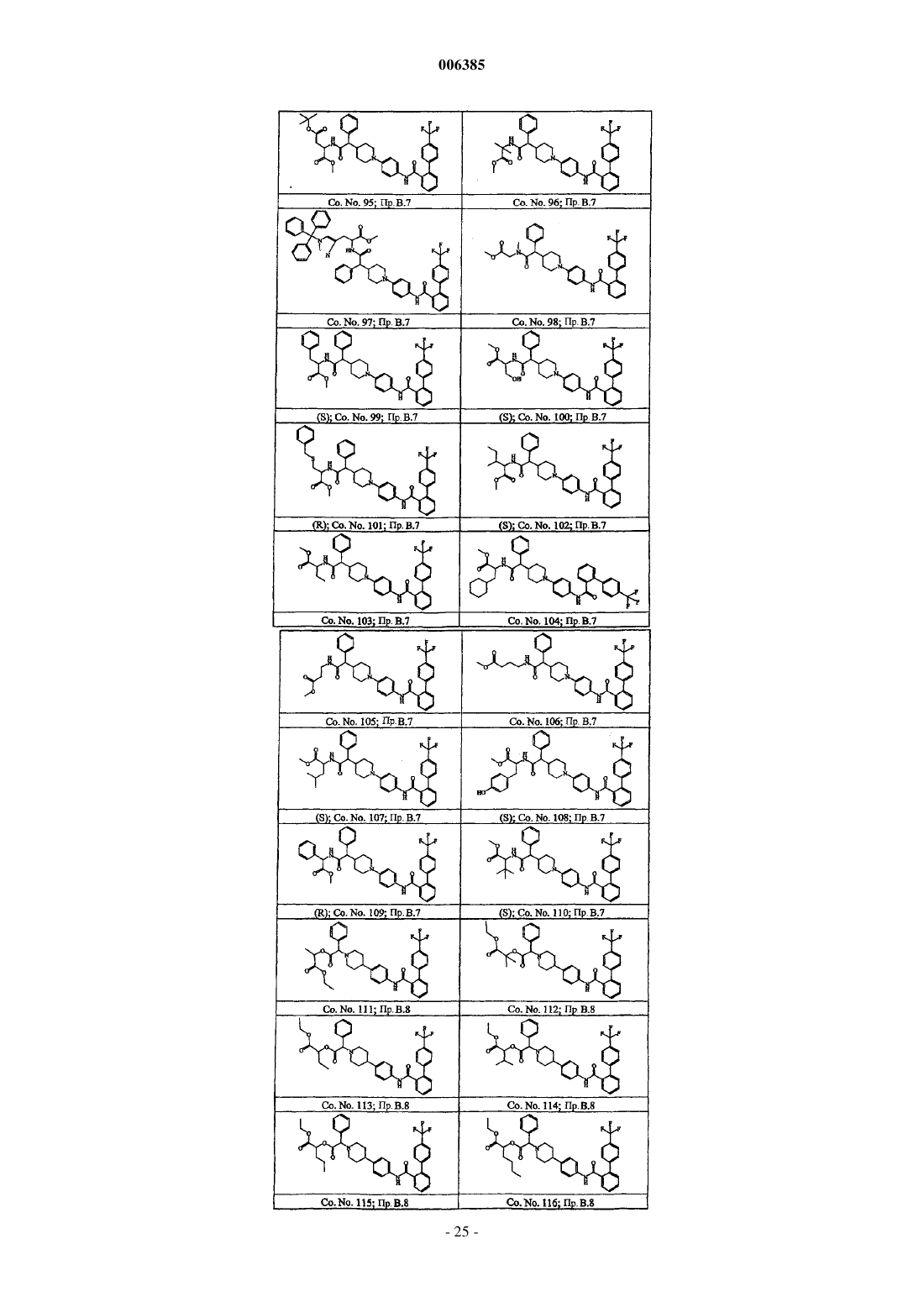
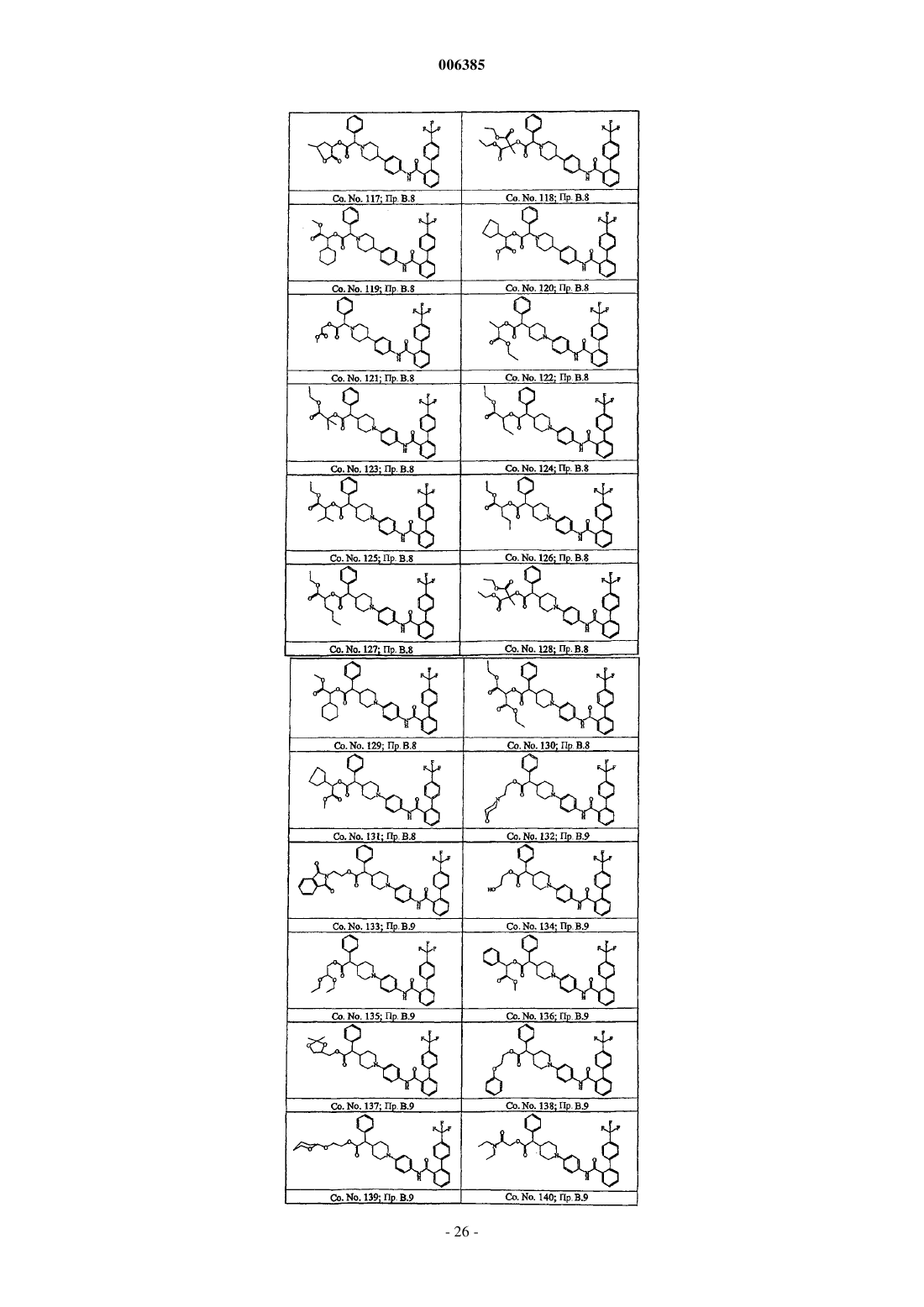

Формула / Реферат
1. Соединения формулы (I)

их N-оксиды, фармацевтически приемлемые кислотно-аддитивные соли и стереохимически изомерные формы,
где p1, p2 и p3, каждый независимо, равен целому числу от 1 до 3;
p4 равен целому числу 0 или 1;
каждый из R1 независимо выбран из водорода, C1-4алкила, C1-4алкилоксигруппы, галогена, гидрокси-, меркапто-, циано-, нитро-, C1-4алкилтиогруппы или полигалогенC1-6алкила, амино-, C1-4алкиламино- и ди(C1-4алкил)аминогруппы;
каждый из R2 независимо выбран из водорода, C1-4алкила, C1-4алкилоксигруппы, галогена или трифторметила;
R3 представляет водород или C1-4алкил;
каждый из R4 независимо выбран из водорода, C1-4алкила, C1-4алкилоксигруппы, галогена или трифторметила;
Z представляет бивалентный радикал формулы

где каждый из X1 и X2 независимо выбран из CH, N или sp2-гибридизованного атома углерода;
Z1 представляет CH2CH2;
Z2 представляет CH2CH2;
A представляет метиленовую группу, замещенную фенилом;
B представляет радикал формулы


где i равно целому числу от 1 до 4;
j равно целому числу от 1 до 4;
k равно целому числу 1 или 2;
Y представляет O или NR9, где R9 представляет водород, C1-6алкил или C1-4алкиламинокарбонил;
R7 представляет водород, C1-6алкил, C2-6алкенил, C2-6алкинил, фенил или фенил, замещенный C1-4алкилом, галогеном, гидроксигруппой или трифторметилом;
R8 представляет C1-6алкил, C2-6алкенил, C2-6алкинил, фенил или фенил, замещенный C1-4алкилом, галогеном, гидроксигруппой или трифторметилом;
каждый из R10 и R11 независимо представляет водород или C1-6алкил;
необязательно R7 и R9, взятые вместе, могут образовывать бивалентный радикал формул -(CH2)3-, -(CH2)4-, -(CH2)5- или -(CH2)6-;
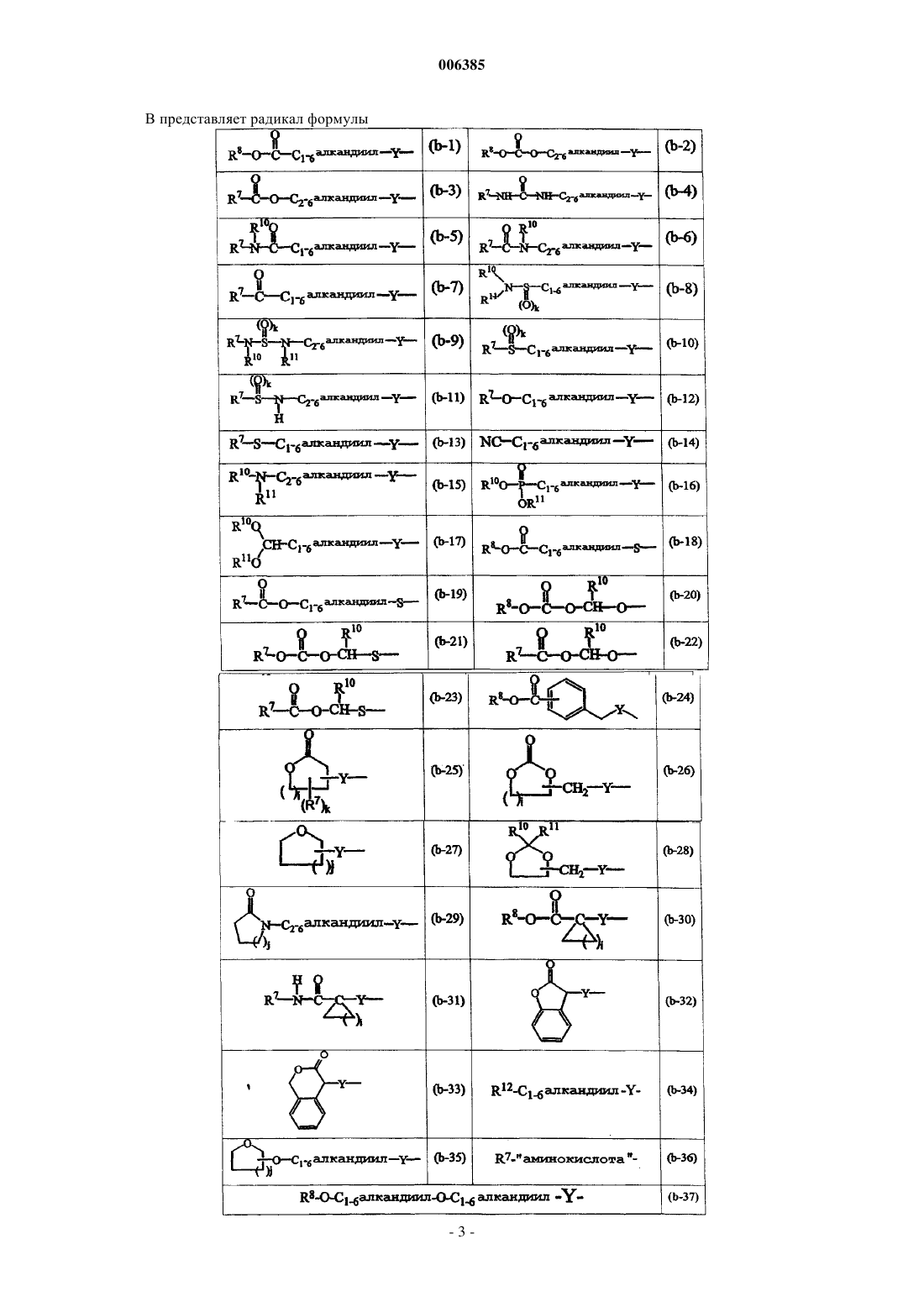
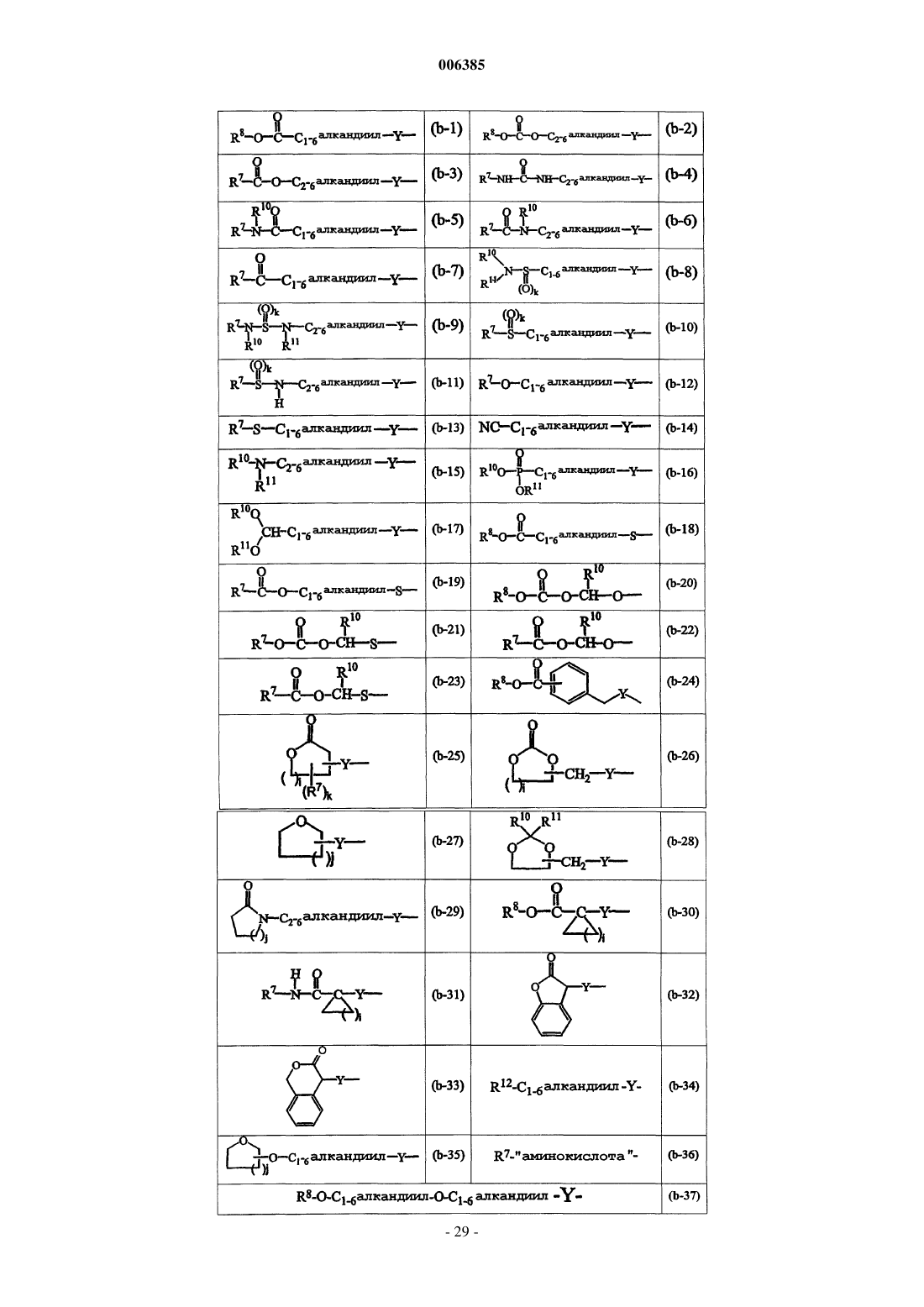
R12 представляет радикал формул

и необязательно в радикале (b-1) C1-6алкандиильный остаток может быть дополнительно замещен фенилом, фенилC1-4алкилом, гидроксифенилC1-4алкилом, C1-4алкилоксикарбонилом, C1-4алкилоксиC1-4алкилом, C1-4алкилтиоC1-4алкилом, фенилC1-4алкилтиоC1-4алкилом, гидроксиC1-4алкилом, тиоC1-4алкилом, C3-6циклоалкилом, C3-6циклоалкилC1-4алкилом или радикалом формул

2. Соединение по п.1, где R1 представляет водород, трет-бутил или трифторметил; R2, R3 и R4 представляют водород.
3. Соединение по любому из пп.1 или 2, где бивалентный радикал A представляет метиленовую группу, замещенную фенилом.
4. Соединение по любому из пп.1-3, где Z представляет бивалентный радикал формулы (a-5), где Z1 и Z2 представляют CH2CH2, а X1 представляет N и X2 представляет CH.
5. Соединение по любому из пп.1-3, где Z представляет бивалентный радикал формулы (a-5), где Z1 и Z2 представляют CH2CH2, а X1 представляет CH и X2 представляет N.
6. Соединение по любому из пп.1-3, где Z представляет бивалентный радикал формулы (a-5), где Z1 и Z2 представляют CH2CH2, а X1 и X2 представляют N.
7. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически активное количество соединения по любому из пп.1-6.
8. Способ получения фармацевтической композиции по п.7, где терапевтически активное количество соединения по любому из пп.1-6 однородно смешивают с фармацевтически приемлемым носителем.
9. Применение соединения по любому из пп.1-6 в качестве лекарственного средства для лечения гиперлипидемии, ожирения и диабета типа II.
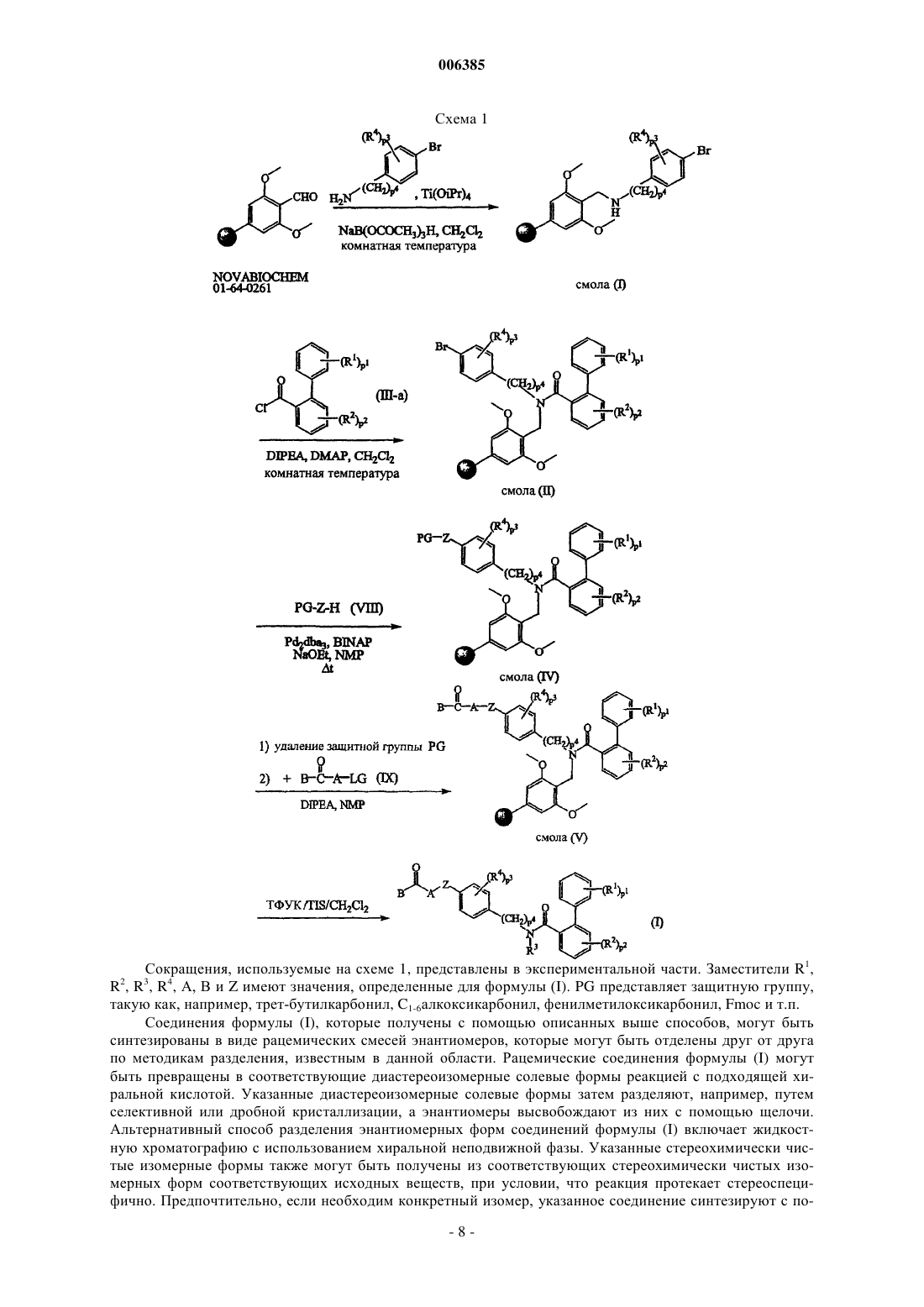
10. Способ получения соединения формулы (I) по п.1, где промежуточное соединение формулы (II), где B, A, Z, R4, p3 и p4 имеют значения, определенные в п.1,

подвергают реакции с бифенилкарбоновой кислотой или галогенангидридом формулы (III), где R1, R2, p1 и p2 имеют значения, определенные для формулы (I), и Q1 выбран из гидроксигруппы и галогена, по меньшей мере, в одном инертном в условиях реакции растворителе и необязательно в присутствии подходящего основания

11. Способ получения соединения формулы (I) по п.1, где промежуточное соединение, имеющее формулу (IV)

где R1, R2, R3, R4, A, Z, p1, p2, p3 и p4 имеют значения, определенные в п.1, а Q2 выбран из галогена и гидроксигруппы, подвергают реакции с промежуточным соединением (V) формулы B-H по меньшей мере в одном инертном в условиях реакции растворителе и необязательно в присутствии по меньшей мере одного подходящего агента сочетания и/или подходящего основания.
12. Способ получения соединения формулы (I) по п.1, где промежуточное соединение формулы (VI), где R1, R2, R3, R4, p1, p2, p3 и p4 имеют значения, определенные в п.1, а Q3 выбран из галогена, B(OH)2, алкилборонатов и их циклических аналогов

подвергают реакции с реагентом формулы (VII), где B, A и Z имеют значения, определенные в п.1, по меньшей мере в одном инертном в условиях реакции растворителе и необязательно в присутствии по меньшей мере одного агента сочетания на основе переходного металла и/или по меньшей мере одного подходящего лиганда

13. Способ получения соединения формулы (I) по п.1, где соединения формулы (I) превращают друг в друга по известным в данной области методикам превращения; или, если это необходимо, соединения формулы (I) превращают в кислотно-аддитивную соль, или, наоборот, кислотно-аддитивную соль соединения формулы (I) превращают в форму свободного основания с помощью щелочи; и, если это желательно, получают их стереохимически изомерные формы.
Текст
006385 Настоящее изобретение относится к новым бифенилкарбоксамидным соединениям, обладающим ингибирующей активностью в отношении аполипопротеина В и сопутствующей снижающей уровень липидов активностью. Изобретение также относится к способам получения таких соединений, фармацевтическим композициям, содержащим указанные соединения, а также к применению указанных соединений в качестве лекарственного средства для лечения гиперлипидемии, ожирения и диабета II типа. Ожирение является причиной серьезных проблем со здоровьем, подобных развитию у взрослых диабета и заболеваний сердца. Кроме того, снижение массы тела становится навязчивой идеей среди все возрастающего числа людей. В настоящее время хорошо осознана причинная связь между гиперхолестеринемией, особенно гиперхолестеринемией, которая связана с повышенными концентрациями в плазме липопротеинов низкой плотности (здесь и далее обозначаемых как LDL) и липопротеинов очень низкой плотности (здесь и далее называемых как VLDL), и преждевременным атеросклерозом и/или сердечно-сосудистым заболеванием. Однако для лечения гиперлипидемии в настоящее время доступно ограниченное число лекарств. Лекарства, используемые преимущественно для устранения гиперлипидемии, включают смолы, усиливающие экскрецию желчных кислот, такие как холестирамин и холестипол, производные фибриновых кислот, такие как безафибрат, клофибрат, фенофибрат, ципрофибрат и гемфиброзил, никотиновую кислоту и ингибиторы синтеза холестерина, такие как ингибиторы HMG Со-фермент-А редуктазы. Неудобство введения (гранулированная форма, которая должна быть диспергирована в воде или апельсиновом соке) и основные побочные эффекты смол (дискомфорт в желудочно-кишечном тракте и запор), усиливающих экскрецию желчных кислот, являются их главными недостатками. Производные фибриновых кислот индуцируют умеренное понижение (на 5-25%) холестерина LDL (за исключением больных с гипертриглицеридемией, у которых изначально низкие уровни имеют тенденцию к повышению), и, хотя обычно они хорошо переносятся, они имеют ряд побочных эффектов, включая возможное появление отторжения, чесотки, усталости, головной боли, бессонницы, мучительной обратимой миопатии и напряжения в группах больших мышц, импотенции и ослабления почечной функции. Никотиновая кислота является сильным агентом, понижающим уровень липидов, который приводит к 15-40%-ному снижению холестерина LDL (и даже к 45-60%-ному снижению в комбинации со смолой, усиливающей экскрецию желчных кислот), но с широким распространением беспокойств, вызванных побочными эффектами, связанными с сосудорасширяющим действием лекарства, таких как головная боль, приливы крови к лицу,сильное сердцебиение, тахикардия и случающиеся время от времени обмороки, а также других побочных эффектов, таких как дискомфорт в желудочно-кишечном тракте, гиперукемия и пониженная переносимость глюкозы. Среди семейства ингибиторов HMG Со-фермент-А редуктазы ловастатин и симвастатин являются неактивными пролекарствами, содержащими лактоновое кольцо, которое гидролизуется в печени с образованием соответствующего активного производного гидроксикислоты. Индуцируя снижение холестерина LDL на 35-45%, они обычно хорошо переносятся, вызывая незначительные побочные эффекты. Однако все еще остается необходимость в новых понижающих уровень липидов агентах с улучшенной эффективностью и/или действующих по другим механизмам, чем названные выше агенты. Липопротеины плазмы представляют собой растворимые в воде комплексы с высокой молекулярной массой, образованные из липидов (холестерин, триглицерид, фосфолипиды) и аполипопротеинов. В зависимости от их плотности (которую измеряют путем ультрацентрифугирования), определено пять основных классов липопротеинов, отличающихся пропорцией липидов и типом аполипопротеинов, которые все имеют свой источник в печени и/или кишечнике. Они включают LDL, VLDL, липопротеины промежуточной плотности (обозначаемые далее как IDL), липопротеины высокой плотности (обозначаемые далее как HDL) и хиломикроны. Определено десять основных аполипопротеинов плазмы человека.VLDL, который выделяется печенью и содержит аполипопротеин В (называемый как Аро-В), подвергается разложению до LDL, который переносит 60-70% всего сывороточного холестерина. Аро-В также является основным протеиновым компонентом LDL. Повышенный уровень холестерина LDL в сыворотке из-за избыточного синтеза или пониженного метаболизма причинно связан с атеросклерозом. Напротив, липопротеины высокой плотности (обозначаемые как HDL), которые содержат аполипопротеин А 1,обладают защитным действием и обратно пропорционально соотносятся с риском коронарного заболевания сердца. Соотношение HDL/LDL, таким образом, является удобным способом оценки атерогенного потенциала профиля содержания липидов в плазме индивидуума. Две изоформы аполипопротеинов (Аро) В, Аро В-48 и Аро В-100, являются важными протеинами при метаболизме липопротеинов человека. Аро В-48, называемый так, потому что, как оказывается, он имеет приблизительно 48% от размера Аро В-100 при анализе на додецилсульфат натрия в полиакриламидных гелях, синтезируется в кишечнике человека. Аро В-48 необходим для сборки хиломикронов и,следовательно, играет обязательную роль при кишечном поглощении пищевых жиров. Аро В-100, который продуцируется в печени человека, необходим для синтеза и секреции VLDL. LDL, которые содержат приблизительно 2/3 холестерина в плазме человека, являются продуктами метаболизма VLDL. Аро В 100 фактически представляет собой единственный протеиновый компонент LDL. Повышенные концентрации Аро В-100 и холестерина LDL в плазме признаны факторами риска для развития атеросклеротического заболевания венечной артерии.-1 006385 Большое число генетических и приобретенных заболеваний может развиваться в результате гиперлипидемии. Они могут быть классифицированы на первичное и вторичное гиперлипидемические состояния. Наиболее обычными причинами вторичных гиперлипидемии являются сахарный диабет, алкогольная и лекарственная зависимость, гипотиреоз, хроническая почечная недостаточность, нефротический синдром, холестаз и булимия. Первичные гиперлипидемии также классифицируют на обычную гиперхолестеринемию, семейную комбинированную гиперлипидемию, семейную гиперхолестеринемию, остаточную гиперлипидемию, синдром хиломиронемии и семейную гипертриглицеридемию. Протеин переноса микросомальных триглицеридов (далее обозначаемый МТР), как известно, катализирует перенос триглицерида и сложных эфиров холестерина предпочтительно к фосфолипидам, таким как фосфатидилхолин. D. Sharp с соавторами в публикации Nature (1993), 365:65, показали, что дефект, вызываемый абеталипопротеинемией, находится в гене МТР. Это указывает на то, что МТР необходим для синтеза Аро-В-содержащих липопротеинов, таких как VLDL, предшественников LDL. Из этого следует, что ингибиторы МТР могли бы ингибировать синтез VLDL и LDL, понижая в результате уровни VLDL, LDL, холестерина и триглицерида у человека. Ингибиторы МТР описаны в канадской патентной заявке 2091102 и в WO 96/26205. Ингибиторы МТР, принадлежащие к классу полиарилкарбоксамидов, также представлены в патенте США 5760246, а также в WO-96/40640 и WO-98/27979. Одной из целей настоящего изобретения является разработка улучшенного лечения пациентов,страдающих ожирением или атеросклерозом, в особенности атеросклерозом сосудов сердца, и, более обобщенно, страдающих заболеваниями, которые связаны с атеросклерозом, такими как ишемическая болезнь сердца, заболевания периферических сосудов и заболевания сосудов мозга. Другая цель настоящего изобретения заключается в том, чтобы вызвать регрессию атеросклероза и ингибировать его клинические последствия, в особенности заболеваемость и смертность. Настоящее изобретение основано на неожиданном открытии, что класс новых бифенилкарбоксамидных соединений действует как селективные ингибиторы МТР, то есть обладает способностью селективно блокировать МТР на уровне кишечной стенки у млекопитающих и, следовательно, является потенциальным кандидатом в качестве лекарственного средства, а именно средства для лечения гиперлипидемии. Кроме того, настоящее изобретение предлагает несколько способов получения таких бифенилкарбоксамидных соединений, а также фармацевтических композиций, содержащих такие соединения. Кроме того, изобретение предлагает определенное количество новых соединений, которые могут быть полезны в качестве промежуточных соединений для получения терапевтически активных бифенилкарбоксамидных соединений, а также способы получения таких промежуточных соединений. И, наконец,изобретение предлагает способ лечения состояния, выбранного из атеросклероза, панкреатита, ожирения,гиперхолестеринемии, гипертриглицеридемии, гиперлипидемии, диабета и диабета II типа, который включает введение терапевтически активного бисфенилкарбоксамидного соединения млекопитающему. Настоящее изобретение раскрывает соединения формулы (I) их N-оксиды, фармацевтически приемлемые кислотно-аддитивные соли и стереохимически изомерные формы,где р 1, р 2 и р 3, каждый независимо равен целому числу от 1 до 3; р 4 равен целому числу 0 или 1; каждый из R1 независимо выбран из водорода, С 1-4 алкила, С 1-4 алкилоксигруппы, галогена, гидрокси-, меркапто-, циано-, нитро-, С 1-4 алкилтиогруппы или полигалогенС 1-6 алкила, амино-, С 1-4 алкиламинои ди(С 1-4 алкил)аминогруппы; каждый из R2 независимо выбран из водорода, С 1-4 алкила, С 1-4 алкилоксигруппы, галогена или трифторметила;R3 представляет водород или С 1-4 алкил; каждый из R4 независимо выбран из водорода, С 1-4 алкила, С 1-4 алкилоксигруппы, галогена или трифторметила;Z представляет бивалентный радикал формулы где каждый из X1 и X2 независимо выбран из СН, N или sp2-гибридизованного атома углерода;-3 006385 где i равно целому числу от 1 до 4;j равно целому числу от 1 до 4;k равно целому числу 1 или 2;R8 представляет C1-6 алкил, С 2-6 алкенил, С 2-6 алкинил, фенил или фенил, замещенный С 1-4 алкилом,галогеном, гидроксигруппой или трифторметилом; каждый из R10 и R11 независимо представляет водород или C1-6 алкил; необязательно R7 и R9, взятые вместе, могут образовывать бивалентный радикал формул -(СН 2)3-,-(СН 2)4-, -(СН 2)5- или -(СН 2)6-; и необязательно в радикале (b-1) С 1-6 алкандиильный остаток может быть дополнительно замещен фенилом, фенилС 1-4 алкилом, гидроксифенилС 1-4 алкилом, С 1-4 алкилоксикарбонилом, С 1-4 алкилоксиС 1-4 алкилом, С 1-4 алкилтиоС 1-4 алкилом, фенилС 1-4 алкилтиоС 1-4 алкилом, гидроксиС 1-4 алкилом, тиоС 1-4 алкилом,С 3-6 циклоалкилом, С 3-6 циклоалкилС 1-4 алкилом или радикалом формул Если не оговорено особо, в приведенных выше определениях и далее галоген обозначает атомы фтора, хлора, брома и йода; С 1-4 алкил обозначает насыщенные углеводородные радикалы с линейной и разветвленной цепью,содержащие от 1 до 4 атомов углерода, такие как, например, метил, этил, пропил, н-бутил, 1-метилэтил,2-метилпропил, 1,1-диметилэтил и т.п.;C1-6 алкил, как подразумевается, включает С 1-4 алкил (определенный выше) и его более высокие гомологи, содержащие 5 или 6 атомов углерода, такие как 2-метилбутил, н-пентил, диметилпропил, нгексил, 2-метилпентил, 3-метилпентил и т.п.; С 2-6 алкенил означает ненасыщенные углеводородные радикалы с линейной и разветвленной цепью,содержащие от 2 до 6 атомов углерода, такие как этенил, пропенил, бутенил, пентенил или гексенил; С 2-6 алкинил означает ненасыщенные углеводородные радикалы с линейной и разветвленной цепью,содержащие от 2 до 6 атомов углерода, такие как этинил, пропинил, бутинил, пентинил или гексинил; С 3-6 циклоалкил объединяет циклопропил, циклобутил, циклопентил и циклогексил; полигалогенС 1-6 алкил означает полигалогензамещенный C1-6 алкил, в частности C1-6 алкил (определенный выше), замещенный 2-13 атомами галогена, например дифторметил, трифторметил, трифторэтил,октафторпентил и т.п.; арил означает моно- и полиароматические группы, такие как фенил, необязательно замещенный одним-тремя заместителями, каждый из которых независимо выбран из нитро-, азидо-, цианогруппы, галогена, гидроксигруппы, C1-6 алкила, С 3-6 циклоалкила, С 1-4 алкилоксигруппы, полигалогенС 1-6 алкила, аминогруппы, моно- или ди(C1-6 алкил)аминогруппы; гетероарил определяют как моно- и полигетероароматические группы, такие как группы, включающие один или более гетероатомов, выбранных из атома азота, кислорода, серы и фосфора, в частности пиридинил, пиразинил, пиримидинил, пиридазинил, триазинил, триазолил, имидазолил, пиразолил,тиазолил, изотиазолил, оксазолил, пирролил, фуранил, тиенил и т.п., включая все их возможные изомерные формы, и необязательно замещенные одним или более заместителями, каждый из которых независимо выбран из нитро-, азидо-, цианогруппы, галогена, гидроксигруппы, C1-6 алкила, С 3-6 циклоалкила, С 14 алкилоксигруппы, полигалогенС 1-6 алкила, аминогруппы, моно- или ди(C1-6 алкил)аминогруппы; С 1-4 алкиламиногруппа означает первичные аминорадикалы, содержащие от 1 до 6 атомов углерода,такие как, например, метиламино, этиламино, пропиламино, изопропиламино, бутиламино, изобутиламино и т.п.; ди(C1-6 алкил)аминогруппа означает вторичные аминорадикалы, содержащие от 1 до 6 атомов углерода, такие как, например, диметиламино, диэтиламино, дипропиламино, диизопропиламино, N-метилN'-этиламино, N-этил-N'-пропиламино и т.п.;-4 006385 С 1-4 алкилтиогруппа означает С 1-4 алкильную группу, присоединенную к атому серы, такую как метилтио, этилтио, пропилтио, изопропилтио, бутилтио и т.п.; используемое в данном описании определение аминокислота используется в общем значении для обозначения встречающихся в природе аминокислот общей формулы R-CH(COOH)-NH2 (то есть глицин,аланин, валин, лейцин, изолейцин, метионин, пролин, фенилаланин, триптофан, серин, треонин, цистеин,тирозин, аспарагин, глютамин, аспарагиновая кислота, эфиры аспарагиновой кислоты, глутаминовая кислота, эфиры глутаминовой кислоты, лизин, аргинин и гистидин), а также не встречающиеся в природе аминокислоты, включая аналоги аминокислот. Таким образом, в данном описании отсылка к аминокислоте означает, например, встречающиеся в природе протеогенные (L)-аминокислоты, а также (D)аминокислоты, химически модифицированные аминокислоты, такие как аналоги аминокислоты, встречающиеся в природе непротеогенные аминокислоты, такие как норлейцин, лантионин или подобные, и химически синтезированные соединения, имеющие свойства, известные в данной области и характеризующиеся аминогруппой, которая может быть введена в часть клетки метаболическим путем. Указанные аминокислоты соединены через их гидроксильную группу с радикалом R7 и через азот с остальной частью молекулы (например, R7-OOC-CRH-NH-). Примерами бивалентного радикала Z формулы (а-5) являются Фармацевтически приемлемые кислотно-аддитивные соли, приведенные выше, как подразумевается, означают терапевтически активные нетоксичные кислотно-аддитивные солевые формы, которые способны образовывать соединения формулы (I). Фармацевтически приемлемые кислотно-аддитивные соли обычно могут быть получены обработкой основной формы подходящей кислотой. Подходящими кислотами являются, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористо-водородная кислота или бромисто-водородная кислота, серная, азотная, фосфорная и подобные им кислоты; или органические кислоты, такие как, например, уксусная кислота, пропановая кислота,гидроксиуксусная кислота, молочная кислота, пировиноградная кислота, щавелевая (то есть этандиовая),малоновая, янтарная (то есть бутандиовая кислота), малеиновая, фумаровая, молочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты. И, наоборот, солевые формы могут быть превращены в форму свободного основания обработкой подходящим основанием. Определение аддитивная соль, используемое в данном описании, также включает сольваты, которые соединения формулы (I), а также их соли способны образовывать. Такие сольваты представляют собой, например, гидраты, алкоголяты и подобные N-оксидные формы соединений формулы (I), которые могут быть получены известными в данной области способами, включают соединения формулы (I), в которых азот окислен до N-оксида. Определение стереохимически изомерные формы, используемое в данном описании, охватывает все возможные изомерные формы, в которых соединения формулы (I) могут существовать. Если не оговорено или не указано особо, химическое название соединений означает смесь всех возможных стереохимически изомерных форм, причем указанные смеси содержат все диастереомеры и энантиомеры базовой молекулярной структуры. Более конкретно, стереогенные центры могут иметь R- или S-конфигурацию; заместители в бивалентных циклических (частично) насыщенных радикалах могут иметь или цис-, или транс-конфигурацию. Если не указано особо, химическое название соединений означает смесь всех возможных стереоизомерных форм, причем указанные смеси содержат все диастереоизомеры и энантиомеры базовой молекулярной структуры. То же самое применимо к описанным в данном описании промежуточным соединениям, используемым для получения конечных продуктов формулы (I). Определения цис- и транс- используются в соответствии с номенклатурой Chemical Abstracts и относятся к положению заместителей в кольцевом фрагменте. Абсолютная стереохимическая конфигурация соединений формулы (I) и промежуточных соединений, используемых при их получении, может быть легко определена квалифицированным специалистом в данной области с помощью хорошо известных способов, таких как, например, дифракция рентгеновских лучей. Более того, некоторые соединения формулы (I) и некоторые промежуточные соединения, используемые при их получении, могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любые полиморфные формы, обладающие свойствами, полезными при лечении состояний,приведенных выше. Группа представляющих интерес соединений включает соединения формулы (I), к которым применимо одно или несколько следующих ограничений:h) Z представляет бивалентный радикал формулы (а-1), где каждый из X1 и X2 представляет азот;i) Z представляет бивалентный радикал формулы (а-2), где X1 представляет азот, a m и m' равны целому числу 1;j) Z представляет бивалентный радикал формулы (а-2), где Х 1 представляет азот, m равно целому числу 2 и m' равно целому числу 1;k) Z представляет бивалентный радикал формулы (а-3), где Х 1 представляет азот, а m и m' равны целому числу 1;l) Z представляет бивалентный радикал формулы (а-3), где Х 1 представляет азот, m равно целому числу 2 и m' равно целому числу 1;m) Z представляет бивалентный радикал формулы (а-4), где m равно целому числу 2 и m' равно целому числу 1;n) Z представляет бивалентный радикал формулы (а-5), где Z1 и Z2 представляют CH2CH2; о) каждый из R5 и R6 независимо представляет водород или метил; р) бивалентный радикал А представляет C1-6 алкандиил, замещенный одной арильной группой, в частности А представляет метиленовую группу, замещенную фенилом;q) В представляет радикал формулы (b-1). Конкретную группу соединений составляют соединения формулы (I), в которой бивалентный радикал А представляет метиленовую группу, замещенную фенилом. Другую конкретную группу соединений составляют соединения формулы (I), в которой Z представляет бивалентный радикал формулы (а-5), где Z1 и Z2 представляют CH2CH2, а Х 1 представляет N иX2 представляет СН. Еще одну конкретную группу соединений составляют соединения формулы (I), в которой Z представляет бивалентный радикал формулы (а-5), где Z1 и Z2 представляют СН 2 СН 2, а X1 представляет СН иX2 представляет N. Еще одну конкретную группу соединений составляют соединения формулы (I), в которой Z представляет бивалентный радикал формулы (а-5), где Z1 и Z2 представляют CH2CH2 и X1 и X2 представляют N. Первый способ получения бифенилкарбоксамидного соединения в соответствии с настоящим изобретением представляет собой способ получения, в котором промежуточный фениленамин, имеющий формулу где В, А, Z и R4 имеют значения, определенные для формулы (I), подвергают реакции с бифенилкарбоновой кислотой или галогенангидридом формулы (III) где заместители R1 и R2 имеют значения, определенные для формулы (I), a Q1 выбран из гидроксигруппы и галогена по меньшей мере в одном инертном в условиях реакции растворителе и необязательно в присутствии подходящего основания, причем указанный способ также необязательно включает превращение соединения формулы (I) в его аддитивную соль и/или получение его стереохимически изомерных форм. В случае, когда Q1 представляет гидроксигруппу, может быть удобно активировать бифенилкарбоновую кислоту формулы (III) добавлением эффективного количества активатора реакции. Неограничивающими примерами таких активаторов реакции являются карбонилдиимидазол, диимиды, такие как N,N'-дициклогексилкарбодиимид или 1-(3-диметиламинопропил)-3-этилкарбодиимид и их функциональные производные. Для такого типа методики ацилирования предпочтительно использовать полярный апротонный растворитель, такой как, например, метиленхлорид. Подходящими основаниями для проведения первого способа являются третичные амины, такие как триэтиламин, триизопропиламин и подобные. Подходящие температуры для проведения первого способа изобретения обычно находятся в интервале приблизительно от 20 до 140 С в зависимости от конкретного используемого растворителя и наиболее часто являются температурами кипения указанного растворителя.-6 006385 Второй способ получения бифенилкарбоксамидного соединения изобретения составляет способ, в котором промежуточное соединение, имеющее формулу (IV) где R1, R2, R3, R4, А и Z имеют значения, определенные для формулы (I), и Q2 выбран из галогена и гидроксигруппы, подвергают реакции с промежуточным соединением (V) формулы В-Н по меньшей мере в одном инертном в условиях реакции растворителе и необязательно в присутствии по меньшей мере одного агента сочетания и/или подходящего основания, причем указанный способ дополнительно необязательно включает превращение соединения формулы (I) в его аддитивную соль и/или получение его стереохимически изомерных форм. В случае, когда Q2 представляет гидроксигруппу, может быть удобно активировать карбоновую кислоту формулы (IV) добавлением эффективного количества активатора реакции. Неограничивающими примерами таких активаторов реакции являются карбонилдиимидазол,диимиды,такие какN,N'-дициклогексилкарбодиимид или 1-(3-диметиламинопропил)-3 этилкарбодиимид и их функциональные производные. В случае использования хирально чистого реагента формулы (V) быстрая и не сопровождаемая энантиомеризацией реакция промежуточного соединения формулы (IV) с указанным промежуточным соединением (V) может быть проведена также в присутствии эффективного количества соединения, такого как гидроксибензотриазол, гексафторфосфат бензотриазолилокситрис(диметиламино)фосфония, гексафторфосфат тетрапирролидинофосфония, гексафторфосфат бромтрипирролидинофосфония или их функциональных производных, как описано у D. Hudson, J. Org. Chem., (1988),53:617. Третий способ получения бифенилкарбоксамидного соединения в соответствии с настоящим изобретением представляет собой способ, в котором промежуточное соединение, имеющее формулу (VI) где R1, R2, R3 и R4 имеют значения, определенные для формулы I, a Q3 выбран из галогена, В(ОН)2, алкилборонатов и их циклических аналогов, подвергают реакции с реагентом, имеющим формулу (VII) где В, А и Z имеют значения, определенные для формулы (I) по меньшей мере в одном инертном в условиях реакции растворителе и необязательно в присутствии по меньшей мере одного агента сочетания на основе переходного металла и/или по меньшей мере одного подходящего лиганда, причем указанный способ также необязательно включает превращение соединения формулы (I) в его аддитивную соль и/или получение его стереохимически изомерных форм. Данный тип реакции, известный в данной области как реакция Бухвальда, ссылки по применению агента сочетания на основе металла и/или подходящих лигандов, например соединений палладия, таких как тетра(трифенилфосфин)палладий, трис(дибензилиденацетон)дипалладий, 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP) и т.п., могут быть найдены, например, в публикациях Tetrahedron Letters, (1996), 37(40), 7181-7184, и J. Am. Chem. Soc., (1996),118:7216. Если Q3 представляет В(ОН)2, алкилборат или его циклический аналог, то в соответствии с публикацией Tetrahedron Letters, (1998), 39:2933-6 в качестве агента сочетания должен быть использован ацетат меди. Соединения формулы (I) могут быть обычно получены с использованием технологии твердофазного синтеза, как представлено ниже на схеме 1. В целом, твердофазный синтез включает взаимодействие при синтезе промежуточного соединения с полимерной подложкой. Нанесенное на полимерную подложку соединение затем проходит через ряд стадий синтеза. После каждой стадии примеси удаляют фильтрованием смолы и ее промывкой несколько раз различными растворителями. На каждой стадии смола может быть разделена для проведения реакции с различными промежуточными соединениями на нескольких стадиях, что обеспечивает в результате синтез большого числа соединений. После последней стадии смолу обрабатывают реагентом или подвергают реакции для отщепления смолы от образца. Более детальное объяснение методик, применяемых в твердофазной химии, приведено, например, в публикациях The Combinatorial Index (В. Bunin, Academic Press) и Novablochem's 1999 CataloguePeptide Сокращения, используемые на схеме 1, представлены в экспериментальной части. Заместители R1,R , R , R4, А, В и Z имеют значения, определенные для формулы (I). PG представляет защитную группу,такую как, например, трет-бутилкарбонил, C1-6 алкоксикарбонил, фенилметилоксикарбонил, Fmoc и т.п. Соединения формулы (I), которые получены с помощью описанных выше способов, могут быть синтезированы в виде рацемических смесей энантиомеров, которые могут быть отделены друг от друга по методикам разделения, известным в данной области. Рацемические соединения формулы (I) могут быть превращены в соответствующие диастереоизомерные солевые формы реакцией с подходящей хиральной кислотой. Указанные диастереоизомерные солевые формы затем разделяют, например, путем селективной или дробной кристаллизации, а энантиомеры высвобождают из них с помощью щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные стереохимически чистые изомерные формы также могут быть получены из соответствующих стереохимически чистых изомерных форм соответствующих исходных веществ, при условии, что реакция протекает стереоспецифично. Предпочтительно, если необходим конкретный изомер, указанное соединение синтезируют с по 2-8 006385 мощью стереоспецифических способов получения. В данных способах предпочтительно используют энантиомерно чистые исходные вещества. Бифенилкарбоксамидные соединения формулы (I), их N-оксидные формы, фармацевтически приемлемые соли и их стереоизомерные формы обладают полезной ингибирующей аполипопротеин В активностью и сопутствующей понижающей уровень липидов активностью. Таким образом, рассматриваемые соединения полезны в качестве лекарственного средства, особенно в способах лечения пациентов, страдающих гиперлипидемией, ожирением, атеросклерозом или диабетом II типа. В частности, рассматриваемые соединения могут быть использованы при производстве лекарственного средства для лечения заболеваний, вызванных избытком липопротеинов очень низкой плотности (VLDL) или липопротеинов низкой плотности (LDL), и в особенности заболеваний, вызванных холестерином, связанным с указанными VLDL и LDL. Убедительно доказана причинная связь между гиперхолестеринемией, особенно гиперхолестеринемией,которая связана с повышенными концентрациями в плазме липопротеинов низкой плотности (LDL) и липопротеинов очень низкой плотности (VLDL), и преждевременным атеросклерозом и/или сердечно-сосудистым заболеванием. VLDL выделяется печенью и содержит аполипопротеин В (Аро-В); эти частицы подвергаются расщеплению при циркуляции до LDL, который переносит приблизительно от 60 до 70% всего сывороточного холестерина. Аро-В также является главным протеиновым компонентом LDL. Повышенные уровни LDLхолестерина в сыворотке вследствие избыточного синтеза или пониженного метаболизма причинно связаны с атеросклерозом. Напротив, липопротеины высокой плотности (HDL), которые содержат аполипопротеин А 1,обладают защитным действием и обратно пропорционально соотносятся с риском коронарного заболевания сердца. Соотношение HDL/LDL, таким образом, является удобным способом оценки атерогенного потенциала профиля содержания липидов в плазме индивидуума. Основной механизм действия соединений формулы (I), как оказывается, включает ингибирование активности МТР (протеин переноса микросомальных триглицеридов) в гепатоцитах и эпителиальных клетках кишечника, что приводит к пониженному продуцированию VLDL и хиломикронов, соответственно. Это является новой и передовой методикой лечения гиперлипидемии и, как ожидается, понижает уровни LDL-холестерина и триглицеридов через снижение печеночного продуцирования VLDL и кишечного продуцирования хиломикронов. Большое число генетических и приобретенных заболеваний может развиваться в результате гиперлипидемии. Они могут быть классифицированы как первичное и вторичное гиперлипидемические состояния. Наиболее обычными причинами вторичных гиперлипидемии являются сахарный диабет, алкогольная и лекарственная зависимость, гипотиреоз, хроническая почечная недостаточность, нефротический синдром, холестаз и булимия. Первичные гиперлипидемии также классифицируют как обычную гиперхолестеринемию, семейную комбинированную гиперлипидемию, семейную гиперхолестеринемию, остаточную гиперлипидемию, синдром хиломиронемии и семейную гипертриглицеридемию. Рассматриваемые соединения также могут быть использованы для предупреждения или лечения больных, страдающих ожирением или атеросклерозом, особенно атеросклерозом сосудов сердца, и, в более общем случае, заболеваниями, которые связаны с атеросклерозом, такими как ишемическая болезнь сердца, периферические сосудистые заболевания, заболевания сосудов мозга. Рассматриваемые соединения могут вызвать регрессию атеросклероза и ингибировать клинические последствия атеросклероза, в особенности заболеваемость и смертность. Из практической применимости соединений формулы (I) следует, что настоящее изобретение также предлагает способ лечения теплокровных млекопитающих, включая людей (в целом называемых пациентами), страдающих заболеваниями, вызванными избытком липопротеинов очень низкой плотности(VLDL) или липопротеинов низкой плотности (LDL), и в особенности заболеваниями, вызванными холестерином, связанным с указанными VLDL и LDL. Таким образом, предлагается способ лечения для оказания помощи пациентам, страдающим состояниями, такими как, например, гиперлипидемия, ожирение,атеросклероз или диабет II типа. Аро-48, синтезируемый кишечником, необходим для агрегатов хиломикронов и, следовательно,выполняет обязательную функцию при кишечной абсорбции пищевых жиров. Настоящее изобретение предлагает бифенилкарбоксамидные соединения, которые действуют как селективные ингибиторы МТР на уровне кишечной стенки. Кроме того, настоящее изобретение предлагает фармацевтические композиции, содержащие по меньшей мере один фармацевтически приемлемый носитель и терапевтически эффективное количество бифенилкарбоксамидного соединения, имеющего формулу (I). Для получения фармацевтических композиций настоящего изобретения эффективное количество конкретного соединения в форме основания или аддитивной соли в качестве активного ингредиента смешивают в виде однородной смеси по меньшей мере с одним фармацевтически приемлемым носителем, причем носитель может иметь большое число форм в зависимости от формы препарата, необходимого для применения. Такие фармацевтические композиции предпочтительно представляют собой единичные дозированные лекарственные формы, подходящие, предпочтительно, для перорального введения, ректального введения, подкожного введения или парентеральных инъекций.-9 006385 Например, при получении композиций в виде пероральных дозированных форм могут быть использованы любые обычные жидкие фармацевтические носители, такие как, например, вода, гликоли, масла,спирты и т.п., в случае жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые фармацевтические носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, диспергирующие агенты и т.п., в случае порошков, пилюль, капсул и таблеток. Вследствие простоты введения таблетки и капсулы являются наиболее предпочтительной пероральной дозированной лекарственной формой, причем в этом случае очевидно использование твердых фармацевтических носителей. В случае композиций для парентеральных инъекций фармацевтический носитель обычно содержит преимущественно воду, хотя могут быть включены другие ингредиенты с целью улучшения растворимости активного ингредиента. Инъецируемые растворы могут быть получены, например, с использованием фармацевтического носителя, содержащего солевой раствор, раствор глюкозы или смесь обоих. Инъецируемые суспензии также могут быть получены путем использования подходящих жидких носителей, суспендирующих агентов и т.п. В композициях, подходящих для подкожного введения, фармацевтический носитель может необязательно включать агент, усиливающий проникновение, и/или подходящий смачивающий агент, необязательно смешанный в небольших пропорциях с подходящими добавками, которые не оказывают сильного вредного действия на кожу. Подходящие добавки могут быть выбраны с целью облегчения применения активного ингредиента на коже и/или содействия получению желаемых композиций. Такие композиции для местного применения могут быть использованы различными путями, например в виде трансдермальной повязки, пластыря или мази. Аддитивные соли соединений формулы (I), благодаря их повышенной растворимости в воде по сравнению с соответствующей основной формой, очевидно, больше подходят для получения водных композиций. Особенно предпочтительно готовить фармацевтические композиции изобретения в виде единичных дозированных лекарственных форм для простоты введения и равномерности дозирования. Определение единичная дозированная лекарственная форма, используемое в данном случае, относится к физически дискретным единицам, походящим для единичного дозирования, причем каждый элемент содержит заранее определенное количество активного ингредиента, рассчитанное для получения желаемого терапевтического эффекта, в комбинации с требуемым фармацевтическим носителем. Примерами таких единичных дозированных лекарственных форм являются таблетки (в том числе таблетки с насечками или с покрытием), капсулы, пилюли, упаковки порошков, облатки, инъецируемые растворы или суспензии, полные чайные ложки, полные столовые ложки и т.п. и их кратные количества. В случае перорального введения фармацевтические композиции настоящего изобретения могут иметь форму твердых дозированных форм, например таблеток (как проглатываемых, так и разжевываемых форм), капсул или гелевых капсул, полученных обычными способами с фармацевтически приемлемыми эксципиентами и носителями, такими как связующие агенты (например, предварительно желатинизированный кукурузный крахмал, поливинилпирролидон, гидроксипропилметилцеллюлоза и т.п.),наполнители (например, лактоза, микрокристаллическая целлюлоза, фосфат кальция и подобные), смазывающие вещества (например, стеарат магния, тальк, диоксид кремния и т.п.), диспергирующие агенты(например, картофельный крахмал, натриевая соль гликолата крахмала и т.п.), смачивающие агенты (например, лаурилсульфат натрия) и т.п. На такие таблетки также может быть нанесено покрытие известными в данной области способами. Жидкие препараты для перорального введения могут иметь форму, например, растворов, сиропов или суспензий, или они могут быть получены в виде сухого продукта для смешивания перед применением с водой и/или другим подходящим жидким носителем. Такие жидкие препараты могут быть получены обычными средствами, необязательно с другими фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, метилцеллюлоза, гидроксипропилметилцеллюлоза или гидрированные пищевые жиры), эмульгирующие агенты (например, лецитин или аравийская камедь), неводные носители (например, миндальное масло, масляные эфиры или этиловый спирт), подслащивающие вещества, корригенты, агенты, маскирующие вкус и запах, и консерванты (например, метилили пропил-п-гидроксибензоаты или сорбиновая кислота). Фармацевтически приемлемые подслащивающие вещества, используемые в фармацевтических композициях изобретения, предпочтительно включают по меньшей мере одно сильное подслащивающее средство, такое как аспартам, ацесульфам-калий, цикламат натрия, алитам, дигидрохальконовое подслащивающее вещество, монеллин, стевиозид сахаралозы (4,1',6'-трихлор-4,1',6'-тридеоксигалактосахароза) или предпочтительно сахарин, натриевую или кальциевую соль сахарина, и необязательно по меньшей мере одно объемное подслащивающее вещество, такое как сорбит, маннит, фруктоза, сахароза,мальтоза, изомальт, глюкоза, сироп гидрированной глюкозы, ксилит, карамель или мед. Сильные подслащивающие вещества обычно используют в низких концентрациях. Например, в случае натриевой соли сахарина указанная концентрация может находиться в интервале приблизительно от 0,04 до 0,1%(масса/объем) из расчета на конечную рецептуру. Объемное подслащивающее вещество можно эффективно использовать в более высоких концентрациях, находящихся в интервале приблизительно от 10 до 35%, предпочтительно приблизительно от 10 до 15% (масса/объем).- 10006385 Фармацевтически приемлемые корригенты, которые могут маскировать горький вкус ингредиентов в рецептурах с низкой дозировкой, предпочтительно представляют собой такие фруктовые вкусовые вещества, как вишневое, малиновое, черносмородиновое или клубничное вкусовое вещество. Хорошие результаты может давать сочетание двух корригентов. В рецептурах с высокой дозировкой могут потребоваться более сильные фармацевтически приемлемые корригенты, такие как Caramel Chocalade, MintCool, Fantasy и т.п. Каждый корригент может присутствовать в конечной композиции в концентрации,находящейся в интервале приблизительно от 0,05 до 1% (масса/объем). Успешно используются комбинации указанных сильных корригентов. Предпочтительно используют корригент, который в рецептуре не подвергается какому-либо изменению или не теряет вкус и/или цвет. Бифенилкарбоксамидные соединения настоящего изобретения могут быть приготовлены для парентерального введения путем инъекции, обычно внутривенной, внутримышечной или подкожной инъекции, например, путем введения ударной дозы или путем непрерывного внутривенного вливания. Рецептуры для инъекций могут быть представлены в виде единичной дозированной лекарственной формы, например в ампулах или контейнерах с несколькими дозами, включая добавленный консервант. Они могут иметь такие формы,как суспензии, растворы или эмульсии в масляном или водном растворителе, и могут содержать рецептурные агенты, такие как изотонирующие, суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может присутствовать в порошковой форме для смешения перед применением с подходящим носителем, например стерильной апирогенной водой. Бифенилкарбоксамидные соединения настоящего изобретения также могут быть приготовлены в виде ректальных композиций, таких как свечи или удерживаемые клизмы, например, содержащие обычную свечную основу, такую как кокосовое масло и/или другие глицериды. Бифенилкарбоксамидные соединения настоящего изобретения могут быть использованы в сочетании с другими фармацевтическими агентами, в частности фармацевтические композиции настоящего изобретения могут также содержать по меньшей мере один дополнительный снижающий уровень липидов агент, обеспечивая, таким образом, так называемую комбинированную понижающую уровень липидов терапию. Указанный дополнительный снижающий уровень липидов агент может представлять собой, например, известное лекарственное средство, обычно используемое для ослабления гиперлипидемии, такое как смола, усиливающая экскрецию желчных кислот, производное фибриновой кислоты или никотиновая кислота, которые приведены выше при описании известного уровня техники. Подходящими дополнительными, понижающими уровень липидов агентами также являются другие ингибиторы биосинтеза холестерина и ингибиторы абсорбции холестерина, в частности ингибиторы HMG-СоА редуктазы и ингибиторы HMG-CoA синтазы, ингибиторы экспрессии гена HMG-CoA редуктазы, ингибиторы СЕТР, ингибиторы АСАТ, ингибиторы скваленсинтетазы и т.п. В комбинированной терапии в соответствии с данным аспектом настоящего изобретения в качестве второго соединения может быть использован любой ингибитор HMG-CoA редуктазы. Определение ингибитор HMG-CoA редуктазы, используемое в данном описании, если не оговорено особо, относится к соединению, которое ингибирует биотрансформацию гидроксиметилглутарил-кофермента А до мевалоновой кислоты, которая катализируется ферментом HMG-CoA редуктазы. Такое ингибирование может быть легко определено квалифицированным специалистом в данной области в соответствии со стандартными способами, например Methods of Enzymoloqy (1981), 71:455-509. Примеры соединений описаны,например, в патенте США 4231938 (включая ловастатин), патенте США 4444784 (включая симвастатин), патенте США 4739073 (включая флувастатин), в патенте США 4346227 (включая правастатин), в ЕР-А-491226 (включая ривастатин) и в патенте США 4647576 (включая аторвастатин). В комбинированной терапии в соответствии с данным аспектом настоящего изобретения в качестве второго соединения может быть использован любой ингибитор HMG-CoA синтазы. Определение ингибитор HMG-CoA синтазы, используемое в данном описании, если не оговорено особо, относится к соединению, которое ингибирует биосинтез гидроксиметилглутарил-кофермента А из ацетил-кофермента А и ацетоксиацетил-кофермента А, который катализируется ферментом HMG-CoA синтазы. Такое ингибирование может быть легко определено квалифицированным специалистом в данной области в соответствии со стандартными методами оценки, например Methods of Enzymology (1985), 110:19-26. Примеры соединений описаны, например, в патенте США 5120729, который относится к производным беталактама, патенте США 5064856, который относится к производным спиролактона, и в патенте США 4847271, который относится к оксетановым соединениям. В комбинированной терапии в соответствии с данным аспектом настоящего изобретения в качестве второго соединения может быть использован любой ингибитор экспрессии гена HMG-CoA редуктазы. Такие агенты могут представлять собой ингибиторы транскрипции HMG-CoA редуктазы, которые блокируют транскрипцию ДНК, или ингибиторы трансляции, которые предупреждают трансляцию мРНК,кодирующую HMG-CoA редуктазу в белке. Такие ингибиторы могут или действовать на транскрипцию или трансляцию напрямую, или могут быть биотрансформированы в соединения, обладающие приведенными выше качествами, с помощью одного или нескольких ферментов в каскаде биосинтеза холестерина,или могут приводить к накоплению метаболитов, имеющих приведенные выше качества. Такое регулирование может быть легко определено квалифицированными специалистами в данной области с помо- 11006385 щью стандартных способов оценки, например Methods of Enzymoloqy (1985), 110:9-19. Примеры соединений описаны, например, в патенте США 5041432 и E.I. Mercer, в Prog. Lip. Res., (1993), 32:357-416. В комбинированной терапии в соответствии с данным аспектом настоящего изобретения в качестве второго соединения может быть использован любой ингибитор СЕТР. Определение ингибитор СЕТР,используемое в описании, если не оговорено особо, относится к соединению, которое ингибирует протеин переноса эфира холестерина (СЕРТ), опосредующий транспорт различных эфиров холестерина и триглицеридов от HDL к LDL и VLDL. Примеры соединений описаны, например, в патенте США 5512548, в J. Antibiot. (1996), 49(8):815-816, и Bioorg. Med. Chem. Lett. (1996), 6:1951-1954. В комбинированной терапии в соответствии с данным аспектом настоящего изобретения в качестве второго соединения может быть использован любой ингибитор АСАТ. Определение ингибитор АСАТ,используемое в описании, если не оговорено особо, относится к соединению, которое ингибирует внутриклеточную этерификацию пищевого холестерина ферментом ацил-СоА:холестеринацилтрансферазы. Такое ингибирование может быть легко определено квалифицированным специалистом в данной области с помощью стандартных способов оценки, например способом Heider et al., Journal of Lipid Research (1983), 24:1127. Примеры соединений описаны, например, в патенте США 5510379, в WO 96/26948 и WO 96/10559. В комбинированной терапии в соответствии с данным аспектом настоящего изобретения в качестве второго соединения может быть использован любой ингибитор скваленсинтетазы. Определение ингибитор скваленсинтетазы, используемое в описании, если не оговорено особо, относится к соединению,которое ингибирует конденсацию двух молекул фарнезилпирофосфата с образованием сквалена, катализируемую ферментом скваленсинтетазы. Такое ингибирование может быть легко определено квалифицированным специалистом в данной области с помощью стандартных способов оценки, например Methodsof Enzymology (1985) 110:359-373. Примеры соединений описаны, например, в публикациях ЕР-0567026,ЕР-0645378 и ЕР-0645377. Квалифицированный специалист в области лечения гиперлипидемии легко определит терапевтически эффективное количество бифенилкарбоксамидного соединения настоящего изобретения по результатам представленных ниже опытов. В общем случае, предполагается, что терапевтически эффективная доза будет составлять приблизительно от 0,001 до 5 мг/кг массы тела, более предпочтительно приблизительно от 0,01 до 0,5 мг/кг массы тела пациента, которого подвергают лечению. Может быть полезно вводить терапевтически эффективную дозу в форме двух или более субдоз через соответствующие интервалы в течение дня. Указанные субдозы могут быть приготовлены в виде единичных дозированных форм, например, каждая из которых содержит приблизительно от 0,1 до 350 мг, более предпочтительно приблизительно от 1 до 200 мг активного ингредиента в единичной дозированной лекарственной форме. Точная дозировка и частота введения зависят от конкретного используемого бифенилкарбоксамидного соединения формулы (I), конкретного состояния, нуждающегося в лечении, серьезности состояния,подвергающегося лечению, возраста, массы и общего физического состояния конкретного пациента, а также от других медикаментов (включая названные выше дополнительные понижающие уровень липидов агенты), которые может принимать пациент, что хорошо известно квалифицированным специалистам в данной области. Кроме того, указанное эффективное ежедневное количество может быть понижено или повышено в зависимости от реакции пациента и/или в зависимости от назначения врача, прописывающего бифенилкарбоксамидные соединения настоящего изобретения. Эффективное ежедневное количество, находящееся в указанном выше интервале, приведено только в качестве руководства. Экспериментальная часть В методиках, описанных далее, используются следующие сокращения: ACN - ацетонитрил,ТГФ - тетрагидрофуран, ДХМ дихлорметан, DIPE - диизопропиловый эфир, ДМФА - N,N-диметилформамид, Ру-ВОР - комплекс (Т-4)-гексафторфосфат(1-)(1-гидрокси-1 Н-бензотриазолато-O)три-1 пирролидинилфосфор(1+) и DIPEA - диизопропилэтиламин. Для некоторых соединений формулы (I) абсолютная стереохимическая конфигурация экспериментально не определена. В этих случаях стереохимически изомерную форму, которая выделяется первой, обозначают А, а второй - В, без дополнительной ссылки на действительную стереохимическую конфигурацию. А. Синтез промежуточных соединений. Пример A.1.a) Смесь 4-[4-(фенилметил)-1-пиперазинил]бензоламина (0,3 моль) и триэтиламина (0,36 моль) в ДХМ (1500 мл) перемешивают при комнатной температуре 15 мин. В течение 30 мин по каплям добавляют 4'-(трифторметил)[1,1'-бифенил]-2-карбонилхлорид (0,36 моль). Смесь перемешивают при комнатной температуре 3 ч и затем дважды промывают водой, затем промывают насыщенным раствором NaCl. Органический слой отделяют, сушат, фильтруют и растворитель выпаривают. Остаток перемешивают вb) Смесь промежуточного соединения (1) (0,19 моль) в метаноле (600 мл) и ТГФ (600 мл) гидрируют в течение ночи с палладием на угле (10%, 3 г) в качестве катализатора. После поглощения водорода (1 экв.) катализатор отфильтровывают и фильтрат упаривают. Остаток растирают с DIPE. Осадок отфильтровы- 12006385 вают и растворяют в воде. Смесь подщелачивают с помощью Na2CO3 и затем экстрагируют ДХМ. Органический слой отделяют, сушат, фильтруют и растворитель выпаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая N-[4-(1-пиперазинил)фенил]-4'-(трифторметил)[1,1'-бифенил]-2-карбоксамид (промежуточное соединение 2).c) Перемешивают смесь промежуточного соединения (2) (0,007 моль) и Na2CO3 (0,007 моль) в ДМФА (50 мл). По каплям добавляют метил 2-бром-2-фенилацетат (0,007 моль). Смесь перемешивают 4 ч. Растворитель выпаривают. Остаток растворяют в ДХМ. Органический слой отделяют, промывают, сушат, фильтруют и растворитель выпаривают. Остаток растирают с 2-пропанолом. Осадок отфильтровывают и сушат, получая 3,34 г метил -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперазинацетата (промежуточное соединение 3).d) Смесь промежуточного соединения 1,3) (0,19 моль) в НСl (36%, 100 мл) перемешивают и кипятят с обратным холодильником 5 ч, затем перемешивают в течение ночи при комнатной температуре. Осадок отфильтровывают и растирают с 2-пропанолом, отфильтровывают и сушат, получая 5 г моногидрохлорида -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперазинуксусной кислоты (промежуточное соединение 4). Пример А.2.a) Метил 2-бром-2-фенилацетат (0,1 моль) добавляют по каплям к смеси 4-(1-пиперазинил)бензонитрила (0,1 моль) и Na2 СО 3 (90,15 моль) в ДМФА (250 мл), перемешивают при комнатной температуре. Реакционную смесь перемешивают в течение ночи. Растворитель выпаривают. Остаток растворяют в ДХМ, промывают, сушат, фильтруют и растворитель выпаривают. Остаток растирают с DIPE, отфильтровывают и сушат, получая 26,5 г метилового эфира 4-(4-цианофенил)фенил-1-пиперазинуксусной кислоты (промежуточное соединение 5).b) Смесь промежуточного соединения (5) (0,079 моль) в смеси метанола, насыщенного NН 3 (600 мл),гидрируют при 14 С в течение ночи с никелем Ренея (1 г) в качестве катализатора. После поглощения водорода (2 экв.) катализатор отфильтровывают и фильтрат упаривают. Остаток растворяют в 2-пропаноле. Смесь подкисляют смесью HCl/2-пропанол и затем перемешивают в течение ночи. Осадок отфильтровывают и сушат, получая 26,7 г гидрохлорида (1:3) 2-пропанолята (1:1) метилового эфира 4-[4-(аминометил)фенил]фенил-1-пиперазинуксусной кислоты (промежуточное соединение 6).c) Перемешивают смесь промежуточного соединения (6) (0,024 моль) в ТГФ (250 мл) и триэтиламина (50 мл). По каплям добавляют 4'-(трифторметил)[1,1'-бифенил]-2-карбонилхлорида (0,026 моль). Смесь перемешивают в течение ночи. Растворитель выпаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, 99/1). Чистые фракции собирают и растворитель выпаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая 7,7 г метилового эфираd) Смесь промежуточного соединения (7) (0,012 моль) в НСl (36%, 100 мл) перемешивают и кипятят с обратным холодильником в течение ночи, затем охлаждают, декантируют и остаток растворяют в метаноле. Растворитель выпаривают. Остаток растирают с DIPE, отфильтровывают и сушат, получая 6,2 г гидрохлорида -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]метил]фенил]-1 пиперазинуксусной кислоты (1:1) (промежуточное соединение 8). Пример А.3. а) Перемешивают смесь 4'-(трифторметил)[1,1'-бифенил]-2-карбоновой кислоты (0,09 моль) в ДХМ(500 мл) и ДМФА (5 мл). Добавляют по каплям этандиолдихлорид (0,09 моль). Смесь перемешивают 1 ч с получением смеси (А). На ледяной бане перемешивают смесь 4-[1-(фенилметил)-4-пиперидинил]бензоламина (0,046 моль) в ДХМ (500 мл) и триэтиламина (20 мл). По каплям добавляют смесь (А). Полученную смесь перемешивают и кипятят с обратным холодильником в течение ночи, затем охлаждают и промывают водой. Органический слой отделяют, сушат, фильтруют и растворитель выпаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент: СН 2 Сl2/СН 3 ОН, 98/2). Чистые фракции собирают и растворитель выпаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат,получая 5,6 г N-[4-[1-(фенилметил)-4-пиперидинил]фенил]-4'-(трифторметил)[1,1'-бифенил]-2-карбоксамида(промежуточное соединение 9, т.пл. 134 С).b) Смесь промежуточного соединения (9) (0,025 моль) в метаноле (250 мл) гидрируют при 50 С в течение ночи с палладием на угле (10%, 2 г) в качестве катализатора. После поглощения водорода (1 экв.) катализатор отфильтровывают и фильтрат упаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая 7,7 г N-[4-(4-пиперидинил)фенил]-4'-(трифторметил)[1,1'-бифенил]-2-карбоксамида (промежуточное соединение 10).c) Смесь промежуточного соединения (10) (0,007 моль) и Na2CO3 (0,007 моль) в ДМФА (50 мл) перемешивают при комнатной температуре. Добавляют по каплям метил 2-бром-2-фенилацетат (0,007 моль). Смесь перемешивают 3 ч. Растворитель выпаривают. Остаток растирают с гексаном, отфильтровывают и сушат, получая 3,37 г метил -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперидинацетата (промежуточное соединение 11, т.пл. 138 С).d) Смесь промежуточного соединения (11) (0,012 моль) в НСl (36%, 100 мл) перемешивают и кипятят с обратным холодильником 6 ч, затем перемешивают в течение ночи при комнатной температуре. Осадок отфильтровывают и растирают с 2-пропанолом. Осадок отфильтровывают и сушат, получая 6,2 г моногидрохлорида -фенил-4-[4-4'-(трифторметил) [1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1 пиперидинуксусной кислоты (промежуточное соединение 12). Пример А.4. а) Перемешивают смесь 4-[4-(фенилметил)-1-пиперазинил]бензоламина (0,12 моль) в ТГФ (300 мл) и триэтиламина (50 мл). Добавляют по каплям [1,1'-бифенил]-2-карбонилхлорид (0,12 моль). Смесь перемешивают в течение ночи. Растворитель выпаривают. Остаток растворяют в ДХМ. Органический слой отделяют, промывают, сушат, фильтруют и растворитель выпаривают. Остаток растирают со смесьюb) Смесь промежуточного соединения (13) (0,1 моль) в метаноле (500 мл) гидрируют 2 ч с палладием на угле (10%, 10 г) в качестве катализатора. После поглощения водорода (1 экв.) катализатор отфильтровывают и фильтрат упаривают. Остаток растирают с 2-пропанолом. Осадок отфильтровывают и сушат, получая 29 г N-[4-(1-пиперазинил)фенил]-[1,1'-бифенил]-2-карбоксамида (промежуточное соединение 14, т.пл. 176 С). Пример А.5.(0,06 моль) и К 2 СО 3 (0,15 моль) в ДМФА (200 мл) перемешивают при 50 С 4 ч, охлаждают, выливают в воду и экстрагируют ДХМ. Органический слой отделяют, промывают водой, сушат, фильтруют и растворитель выпаривают. Остаток кристаллизуют из DIPE. Осадок отфильтровывают и сушат, получая 10,7 гb) Смесь промежуточного соединения (15) (0,036 моль) в НВr (48%, 100 мл) перемешивают и кипятят с обратным холодильником 3 ч, охлаждают, выливают в воду и экстрагируют дважды ДХМ. Органический слой отделяют, промывают водой, сушат, фильтруют и растворитель выпаривают. Остаток растирают с 2-пропанолом. Осадок отфильтровывают и сушат, получая 9,5 г -1-(4-нитрофенил)фенил-4 пиперидинуксусной кислоты (промежуточное соединение 16, т.пл. 216 С).c) К смеси промежуточного соединения (16) (0,0029 моль) в ДХМ (10 мл) добавляют тионилхлорид(0,01 моль). Смесь перемешивают в течение ночи и затем растворитель выпаривают. Остаток растворяют в ДХМ (10 мл). Добавляют метанол (10 мл). Смесь оставляют стоять в течение 4 ч, затем выливают в раствор NаНСО 3 и экстрагируют ДХМ. Органический слой отделяют, сушат, фильтруют и растворитель выпаривают. Остаток растирают со смесью гексан/DIPE. Осадок отфильтровывают и сушат, получая 0,9 гd) Смесь промежуточного соединения (17) (0,0022 моль) в метаноле (100 мл) гидрируют при 50 С с палладием на угле (10%, 0,1 г) в качестве катализатора в присутствии раствора тиофена (4%, 0,1 мл). После поглощения водорода (3 экв.) катализатор отфильтровывают и фильтрат упаривают. Остаток растирают с гексаном. Осадок отфильтровывают и сушат, получая 0,7 г -метил 1-(4-аминофенил)фенил 4-пиперидинацетата (промежуточное соединение 18, т.пл. 125 С).e) Смесь 2-(4-трет-бутилфенил)бензойной кислоты (0,02 моль) и тионилхлорида (0,04 моль) и ДМФА (5 капель) в ДХМ перемешивают и кипятят с обратным холодильником в течение 1 ч. Растворитель выпаривают, добавляют ДХМ (2 х 50 мл) и растворитель снова выпаривают. Остаток растворяют в ДХМ (50 мл) и добавляют к раствору промежуточного соединения (18) (0,02 моль) и DIPEA (0,04 моль) в ДХМ (50 мл). Реакционную смесь перемешивают при комнатной температуре 4 ч. Остаток очищают колоночной хроматографией на силикагеле (элюент: СН 2 Сl2/СН 3 ОН, 99,5/0,5). Чистые фракции собирают и растворитель выпаривают. Остаток растворяют в 2-пропаноле и DIPE и превращают в соль хлористо-водородной кислоты (1:1) с помощью смеси HCl/2-пропанол. Остаток отфильтровывают и сушат, получая 8,7 г гидрохлорида (1:1) 2-пропанолята (1:1) метилового эфира 1-[4-4'-(1,1-диметилэтил)[1,1'-бифенил]2-ил]карбонил]амино]фенил]фенил-4-пиперидинуксусной кислоты (промежуточное соединение 19).a) К раствору 2-бифенилкарбоновой кислоты (0,077 моль) в ДХМ (250 мл) добавляют ДМФА (0,5 мл). Добавляют тионилхлорид (0,154 моль). Смесь перемешивают и кипятят с обратным холодильником 1 ч. Растворитель выпаривают и затем дважды совместно выпаривают с ДХМ (100 мл). Остаток растворяют в ДХМ (100 мл), получая раствор (А). Промежуточное соединение (18) (0,077 моль) и DIPEA (0,154 моль) перемешивают в ДХМ (400 мл). Добавляют раствор (А). Смесь перемешивают при комнатной температуре 3 ч и затем промывают водой. Органический слой отделяют, сушат, фильтруют и выпаривают рас- 14006385 творитель, получая 44 г метилового эфира 1-[4-[([1,1'-бифенил]-2-илкарбонил)амино]фенил]фенил-4 пиперидинуксусной кислоты (промежуточное соединение 21).b) Смесь промежуточного соединения (21) (0,013 моль) в НСl (36%, 200 мл) и диоксана (150 мл) перемешивают и кипятят с обратным холодильником в течение ночи. Реакционную смесь охлаждают и добавляют воду (300 мл). Смесь перемешивают в течение 1 ч и фильтруют. Остаток растворяют в ДХМ и МеОН и растворитель выпаривают. Остаток растирают с DIPE и 2-пропанолом, получая 3,5 г гидрохлорида (1:1) 1-[4-[([1,1'-бифенил]-2-илкарбонил)амино]фенил]фенил-4-пиперидинуксусной кислоты(промежуточное соединение 22). Пример А.7. а) К прозрачному раствору 4'-(трифторметил)[1,1'-бифенил]-2-карбоновой кислоты (0,025 моль) в ДМФА (1 мл) и ДХМ (100 мл) добавляют тионилхлорид (3,6 моль). Смесь перемешивают и кипятят с обратным холодильником 1 ч. Растворитель выпаривают. К остатку добавляют ДХМ (50 мл), затем выпаривают. Остаток растворяют в ДХМ (50 мл) и раствор по каплям добавляют к раствору промежуточного соединения (18) (0,025 моль) в ДХМ (150 мл) и DIPEA (0,049 моль). Реакционную смесь перемешивают 2 ч при комнатной температуре. Добавляют воду и смесь экстрагируют дважды. Отделившийся органический слой сушат, фильтруют и растворитель выпаривают. Остаток перемешивают в DIPE, отфильтровывают, сушат и кристаллизуют из 2-пропанола (150 мл; затем добавляют 300 мл). Смесь перемешивают в течение 2 ч при комнатной температуре. Осадок отфильтровывают, промывают 2-пропанолом и сушат, получая 8,58 г метил -фенил-1-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинацетата (промежуточное соединение 23).b) Смесь промежуточного соединения (23) (0,0014 моль) в концентрированной НСl (25 мл) и диоксане (20 мл) перемешивают и кипятят с обратным холодильником в течение 4 ч, охлаждают и выливают в воду. Смесь экстрагируют ДХМ. Органический слой отделяют, сушат, фильтруют и растворитель выпаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая 0,48 г моногидрохлорида -фенил-1-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинуксуной кислоты (промежуточное соединение 24, 196 С). Пример А.8.a) Промежуточное соединение (23) разделяют на его энантиомеры и очищают с помощью хиральной колоночной хроматографии на Chiralcel OD (1000 , 20 мкм, Daicel; элюент: гексан/этанол/метанол (80/13/7. Первую фракцию очищают высокоэффективной жидкостной хроматографией на RP BDS (HyperprepC18 (100 , 8 мкм; элюент: [(0,5% NH4OAC в H2O)/CH3CN 90/10)]/СН 3 ОН/СН 3 СN (0 мин) 30/70/0, (24 мин) 0/100/0, (24,01 мин) 0/0/100, (32 мин) 30/70/0) и кристаллизуют из 2-пропанола, получая (А)фенил-1[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинацетат (промежуточное соединение 25). Вторую фракцию очищают высокоэффективной жидкостной хроматографией на RP BDS (Hyperprepb) С использованием методики примера А.7b) промежуточное соединение (25) превращают в моногидрохлорид (А)фенил-1-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинуксусной кислоты (промежуточное соединение 27, т.пл. 140 С, []D20=+17,06 (с=9,67 мг/5 мл в СН 3 ОН). с) С использованием методики примера А.7b) промежуточное соединение (26) превращают в моногидрохлорид (В)фенил-1-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинуксусной кислоты (промежуточное соединение 28, т.пл. 135 С, [а]D20=-27,10 (с=11,07 мг/5 мл в СН 3 ОН). Пример А.9.a) Смесь промежуточного соединения (10) (0,019 моль) и Na2 СО 3 (0,019 моль) в ДМФА (125 мл) перемешивают при комнатной температуре. По каплям добавляют метил 2-бром-2-фенилацетат (0,01907 моль). Смесь перемешивают 3 ч. Растворитель выпаривают. Остаток обрабатывают водой и ДХМ. Отделившийся органический слой сушат, фильтруют и растворитель выпаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент: СН 2 Сl2/СН 3 ОН 100/0; 95,5/0,5) и разделяют на энантиомеры с помощью высокоэффективной колоночной хроматографии на Chiralpak AD (элюент: гексан/этанол,70/30). Необходимые фракции собирают и растворитель выпаривают, получая (1 А)-метиловый эфир фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперидинуксусной кислоты (промежуточное соединение 29, т.пл. 158 С, []D20=-28,86 (с=124,95 мг/5 мл в СН 3 ОН после кристаллизации из 2-пропанола и (1 В)-метиловый эфир -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперидинуксусной кислоты (промежуточное соединение 30, т.пл. 160 С, [a]D20=+27,69 (с=24,38 мг/5 мл в СН 3 ОН после кристаллизации из 2-пропанола.b) С использованием методики примера А.7b) промежуточное соединение (29) превращают в (1 А)-фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперидинуксусную кислоту (промежуточное соединение 31, т.пл. 180 С, []D20=-35,90 (с=25,21 мг/5 мл в СН 3 ОН.- 15006385 с) С использованием методики примера А.7b) промежуточное соединение (30) превращают в (1 В)-фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперидинуксусную кислоту (промежуточное соединение 32, т.пл. 135 С, []D20=+35,90 (с=23,94 мг/5 мл в СН 3 ОН. Пример А.10.a) Перемешивают смесь промежуточного соединения (2) (0,019 моль) и Na2CO3 (0,019 моль) в ДМФА (125 мл). По каплям добавляют метил 2-бром-2-фенилацетат (0,019 моль). Смесь перемешивают при комнатной температуре 3 ч. Растворитель выпаривают. Остаток перемешивают в ДХМ и промывают водой. Отделившийся органический слой сушат, фильтруют и растворитель выпаривают. Остаток перемешивают в DIPE. Осадок отфильтровывают, сушат и разделяют с помощью высокоэффективной жидкостной хроматографии Chiralcel OD (элюент: гексан/EtOH, 70/30) на энантиомеры. Необходимые фракции собирают и растворитель выпаривают, получая (1 А)-метиловый эфир -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперазинуксусной кислоты (промежуточное соединение 33) после кристаллизации из 2-пропанола и (1 В)-метиловый эфир -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперазинуксусной кислоты (промежуточное соединение 34) после кристаллизации из 2-пропанола.b) С использованием методики примера А.7b) промежуточное соединение (33) превращают в (1 А)-фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперазинуксусную кислоту (промежуточное соединение 35, т.пл. 230 С, []D20=+60,15 (с=24,52 мг/5 мл в СН 3 ОН.c) С использованием методики примера А.7b) промежуточное соединение (34) превращают в (1 В)-фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперазинуксусную кислоту (промежуточное соединение 36, т.пл. 232 С, []D20=66,37 (с=26,52 мг/5 мл в СН 3 ОН. Пример А.11.a) Добавляют по каплям при перемешивании метил-2-бром-2-фенилацетат (0,1 моль) к смеси промежуточного соединения (14) (0,07 моль) и Na2CO3 (13 г) в ДМФА (300 мл). Смесь перемешивают в течение ночи. Растворитель выпаривают. Остаток кристаллизуют из метанола. Осадок отфильтровывают и сушат, получая 30,2 г метил 4-[4-[([1,1'-бифенил]-2-илкарбонил)амино]фенил]фенил-1-пиперазинацетата (промежуточное соединение 37, т.пл. 125 С).b) Смесь промежуточного соединения (37) (0,053 моль) в НСl (36%, 300 мл) перемешивают и кипятят с обратным холодильником 2 дня, охлаждают, фильтруют и сушат. Остаток растирают с 2-пропаноном. Осадок отфильтровывают и сушат, получая 21,5 г гидрохлорида (1:2) 2-пропанолята (1:1) -фенил-4-[4[([1,1'-бифенил]-2-илкарбонил)амино]фенил]-1-пиперазинуксусной кислоты (промежуточное соединение 38). Пример А.12.a) 2-Бифенилкарбоновую кислоту (0,25 мол) растворяют в ДХМ (500 мл) и ДМФА (0,5 мл). По каплям добавляют тионилхлорид (0,51 моль). Смесь перемешивают и кипятят с обратным холодильником в токе азота. Растворитель выпаривают. Дважды добавляют ДХМ (500 мл). Дважды выпаривают растворитель. Остаток растворяют в ДХМ (200 мл) и затем по каплям при 0 С добавляют к смеси 4-[1-(фенилметил)-4-пиперидинил]бензоламина (0,25 моль) и N-(1-метилэтил)-2-пропанамина (0,75 моль) в ДХМ(800 мл). Температуру смеси доводят до комнатной температуры и затем перемешивают при комнатной температуре в течение ночи в токе азота. Смесь промывают 3 раза водой (800 мл). Органический слой отделяют, сушат, фильтруют и растворитель выпаривают, получая 125 г N-[4-[1-(фенилметил)-4 пиперидинил]фенил]-[1,1'-бифенил]-2-карбоксамида (промежуточное соединение 39).b) Смесь промежуточного соединения (39) (0,145 моль) в метаноле (500 мл) гидрируют при 50 С в течение 2 дней с палладием на угле (10%, 3 г) в качестве катализатора. После поглощения водорода (1 экв.) катализатор отфильтровывают и фильтрат упаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая 49 г N-[4-(4-пиперидинил)фенил]-[1,1'-бифенил]-2-карбоксамида (промежуточное соединение 40).c) Перемешивают смесь промежуточного соединения (40) (0,0084 моль) и Na2CO3 (0,01 моль) в ДМФА (150 мл). По каплям добавляют метил 2-бром-2-фенилацетат (0,01 моль). Смесь перемешивают в течение ночи. Растворитель выпаривают. Остаток растворяют в ДХМ. Органический слой отделяют,промывают, сушат, фильтруют и растворитель выпаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент: ДХМ 100%). Чистые фракции собирают и растворитель выпаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая 2,79 г метилового эфира 4-[4-[([1,1'-бифенил]-2-илкарбонил)амино]фенил]фенил-1-пиперидинуксусной кислоты (промежуточное соединение 41).d) Промежуточное соединение (41) (0,003 моль) в НСl (36%, 25 моль) и диоксане (10 мл) перемешивают и кипятят с обратным холодильником в течение ночи. Растворитель выпаривают. Остаток растирают с DIPE и 2-пропанолом, затем кристаллизуют из ACN. Осадок отфильтровывают, сушат и очищают высокоэффективной жидкостной хроматографией на RP BDS C18 (элюент: (0,5% NH4OAc в H2O/CH3CN 90/10)/МеОН/СН 3 СN 75/25/0; 0/50/50; 0/0/100). Чистые фракции собирают и растворитель выпаривают. Остаток растирают с DIPE, получая 0,5 г 4-[4-[([1,1'-бифенил]-2-илкарбонил)амино]фенил]фенил-1 пиперидинуксусной кислоты (промежуточное соединение 42).- 16006385 Аналогично гидролизом соединения (18) в соответствии с описанным выше способом получают фенил[1-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил)амино]фенил]-4-пиперидинил]ацетил]амино] уксусную кислоту (промежуточное соединение 45). Аналогично гидролизом соединения (12) в соответствии с описанным выше способом получают фенил[4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил)амино]фенил]-1-пиперазинил]ацетил]амино] уксусную кислоту (промежуточное соединение 46). Пример А.13. Смесь метил 2-гидроксиацетата (0,2 моль) в ТГФ (100 мл) перемешивают на ледяной бане. По каплям добавляют 2-хлор-2-фенилацетилхлорид (0,132 моль). Смесь перемешивают в течение ночи, выливают в воду и затем перемешивают 20 мин. Смесь экстрагируют DIPE. Органический слой отделяют,сушат, фильтруют и растворитель выпаривают, получая 30 г 2-метокси-2-оксоэтиловый эфир -хлорбензолуксусной кислоты (промежуточное соединение 43). Пример А.14. Смесь -бромфенилуксусной кислоты (0,1 моль) и тионилхлорида (0,1 моль) в ДХМ (100 мл) и ДМФА(5 капель) перемешивают и кипятят с обратным холодильником 1 ч. Растворитель выпаривают. 3 раза добавляют ДХМ (100 мл). Растворитель выпаривают. Остаток растворяют в ДХМ (100 мл). Смесь охлаждают на льду. Добавляют гидрохлорид метилглицината (0,1 моль). Смесь перемешивают при комнатной температуре 2 ч. Добавляют насыщенный раствор NaHCO3. Смесь перемешивают при комнатной температуре в течение 2 дней. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая 17,8 г метилового эфира [(бромфенилацетил)амино]уксусной кислоты (промежуточное соединение 44). Пример А.15.b) По каплям при перемешивании добавляют 4'-(трифторметил)[1,1'-бифенил]-2-карбонилхлорид(0,12 моль) к смеси промежуточного соединения (47) (0,095 моль) в ДХМ (300 мл) и триэтиламина (50 мл). Смесь перемешивают в течение ночи, выливают в воду и затем перемешивают 30 мин. Органический слой отделяют, промывают, сушат, фильтруют и растворитель выпаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая N-[4-[1,2,3,6-тетрагидро-1-(фенилметил)-4-пиридинил]фенил]-4'-(трифторметил)[1,1'-бифенил]-2-карбоксамид (промежуточное соединение 48). с) Добавляют по каплям при перемешивании 1-хлорэтилхлорформиат (0,078 моль) к смеси промежуточного соединения (48) (0,039 моль) в 1,2-дихлорэтане (500 мл). Смесь перемешивают 30 мин и затем перемешивают и кипятят с обратным холодильником в течение ночи. Растворитель выпаривают. Добавляют метанол(500 мл). Смесь перемешивают и кипятят с обратным холодильником в течение ночи. Растворитель выпаривают. Остаток растирают с DIPE. Осадок отфильтровывают и сушат, получая 20,8 г N-[4-(1,2,3,6-тетрагидро-4-пиридинил)фенил]-4'-(трифторметил)[1,1'-бифенил]-2-карбоксамида (промежуточное соединение 49). Пример А.16. а) Гидробромид (1:1) -фенил-(1-фенилметил)-4-пиперидинуксусной кислоты (1:1) (0,13 моль) перемешивают в ДХМ (300 мл). Добавляют тионилхлорид (0,15 моль). Реакционную смесь перемешивают и кипятят с обратным холодильником 1 ч. Добавляют еще тионилхлорид (0,15 моль), реакционную смесь перемешивают и кипятят с обратным холодильником до полного растворения (приблизительно 3 ч). Растворитель выпаривают. Остаток растворяют в ДХМ (200 мл). Добавляют метанол (50 мл). Смесь перемешивают 1 ч, нейтрализуют раствором NaHCO3. Отделившийся органический слой сушат, фильтруют и растворитель выпаривают. Остаток растворяют в СН 3 ОН/DIPE и превращают в соль хлористо-водородной кислоты (1:1) с помощью смеси HCl/2-пропанол. Образовавшийся осадок отфильтровывают, сушат,снова превращают в свободное основание с помощью раствора NaHCO3 и разделяют на энантиомеры наChiralcel OJ (элюент: метанол). Требуемые фракции собирают и растворитель выпаривают. Каждую из фракций обрабатывают смесью СН 3 ОН/DIPE и превращают в соль хлористо-водородной кислоты с помощью смеси HCl/2-пропанол, получая 16,9 г гидрохлорида (1:1) метилового эфира (А)фенил-1-(фенилметил)-4-пиперидинуксусной кислоты (промежуточное соединение 50, т.пл. 189,9-190,0 С) и 12,2 г гидрохлорида (1:1) метилового эфира (В)фенил-1-(фенилметил)-4-пиперидинуксусной кислоты (промежуточное соединение 51, т.пл. 192,3-193 С).b) Смесь промежуточного соединения (50) (0,047 моль) в метаноле (250 мл) гидрируют при комнатной температуре с палладием на угле (10%, 2 г) в качестве катализатора (2 г). После поглощения водорода (1 экв.) катализатор отфильтровывают и фильтрат упаривают. Остаток растирают с 2-пропанолом, отфильтровывают и сушат, получая 11,5 г (90%) продукта. Фильтрат упаривают, получая 1,1 г гидрохлорида (1:1) метилового эфира (А)фенил-4-пиперидинуксусной кислоты (промежуточное соединение 52).c) Смесь промежуточного соединения (52) (0,046 моль), 1-фтор-4-нитробензола (0,05 моль) и гидрокарбоната натрия (0,15 моль) в N,N-диметилформамиде (100 мл) перемешивают 4 ч при 50 С, затем охлаждают и добавляют воду. Полученную смесь экстрагируют CH2Cl2. Объединенные органические слои- 17006385 промывают водой, сушат, фильтруют и выпаривают растворитель. Остаток растирают со смесьюd) Смесь промежуточного соединения (53) (0,025 моль) в хлористо-водородной кислоте (конц.) (100 мл) перемешивают и кипятят с обратным холодильником 16 ч, затем охлаждают и полученный осадок отфильтровывают, промывают водой (3 раза) и сушат, получая 8,77 г (91%) гидрохлорида (1:1) (А)-1-(4 нитрофенил)фенил-4-пиперидинуксусной кислоты (промежуточное соединение 54, т.пл. 196,4-196,5 С). е) Перемешивают смесь промежуточного соединения (54) (0,013 моль), гидрохлорида (1:1) метилового эфира глицина (98%) (0,026 моль), 1-гидроксибензотриазола (0,015 моль) и 2,6-диметилпиридина(0,052 моль) в CH2Cl2 (100 мл). Добавляют N,N'-метантетраилбис-2-пропанамин (0,026 моль). Реакционную смесь перемешивают 48 ч при комнатной температуре. Смесь промывают Н 2 О. Отделившийся органический слой сушат, фильтруют и растворитель выпаривают. Остаток растирают с 2-пропанолом. Осадок отфильтровывают и сушат, получая 4,37 г (80%) метилового эфира (В)-N-1-(4-нитрофенил)-4 пиперидинил]фенилацетил]глицина (промежуточное соединение 55, т.пл. 165,4-165,5 С).f) Смесь промежуточного соединения (55) (0,01 моль) в метаноле (250 мл) гидрируют при 14 С с палладием на угле (10%, 2 г) в качестве катализатора в присутствии тиофена (4%) (2 мл). После поглощения водорода (3 экв.) катализатор отфильтровывают и фильтрат упаривают, получая 3,81 г (количественный выход) метилового эфира (В)-N-1-(4-аминофенил)-4-пиперидинил]фенилацетил]глицина (промежуточное соединение 56); []D20=+31,37 (с=10,20 мг/5 мл в ДМФА). В. Синтез конечных соединений. Пример В.1. Смесь промежуточного соединения (2) (0,0117 моль) и Nа 2 СО 3 (0,0141 моль) в ДМФА (100 мл) перемешивают при комнатной температуре. По каплям добавляют раствор промежуточного соединения(43) (0,0141 моль) в ДМФА (80 мл). Смесь перемешивают при комнатной температуре 20 ч. Растворитель выпаривают. Остаток перемешивают в ДХМ (150 мл). Органический слой промывают водой, промывают насыщенным раствором NaHCO3 и промывают водой. Объединенный органический слой сушат, фильтруют и растворитель выпаривают. Остаток очищают колоночной хроматографией (элюент:CH2Cl2/CH3OH, 99/1). Нужные фракции собирают и растворитель выпаривают. Остаток перемешивают вDIPE (150 мл). Смесь нагревают до кипения с обратным холодильником и добавляют 2-пропанол (30 мл) и ДХМ (5 мл). Смесь перемешивают и кипятят с обратным холодильником до полного растворения, затем доводят температуру до комнатной. Смесь перемешивают при комнатной температуре 60 ч. Осадок отфильтровывают и сушат в вакууме при 50 С, получая 32 г 2-метокси-2-оксоэтил -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперазинацетата (соединение 14, т.пл. 106 С). Соединение (12, т.пл. 132 С) получают аналогично при взаимодействии промежуточного соединения (2) с промежуточным соединением (44) с использованием описанного выше способа. Соединение (13) получают аналогично при взаимодействии промежуточного соединения (14) с промежуточным соединением (44) с использованием описанного выше способа. Соединение (15) получают аналогично при взаимодействии промежуточного соединения (10) с промежуточным соединением (44) с использованием описанного выше способа. Соединение (16) получают аналогично при взаимодействии промежуточного соединения (14) с промежуточным соединением (43) с использованием описанного выше способа. Пример В.2. Смесь промежуточного соединения (20) (0,0041 моль), гидрохлорида метилглицината (0,008 моль) и триэтиламина (0,025 моль) в ДХМ (50 мл) перемешивают и охлаждают при -25 С. Добавляют Ру-ВОР(0,005 моль) и реакционную смесь перемешивают 4 ч при температуре -25 С. Смеси дают нагреться до комнатной температуры. Остаток очищают колоночной хроматографией на силикагеле (элюент: СН 2 Сl2/СН 3 ОН, 100/0; 99/1). Фракции продукта собирают и растворитель выпаривают. Остаток растирают с DIPE, отфильтровывают, промывают смесью 2-пропанол/Н 2 О (50/50, 3 раза) и сушат, получая 2,1 г гидрата (1:1) 2-пропанолята (1:1) метилового эфира 1-[4-4'-(1,1-диметилэтил)[1,1'-бифенил]-2-ил] карбонил]амино]фенил]-4-пиперидинил]фенилацетил]амино]уксусной кислоты (соединение 22). Пример В.3. Смесь соединения (22) (0,00072 моль) в концентрированной НСl (20 мл) и диоксане (30 мл) перемешивают и кипятят с обратным холодильником. Растворитель выпаривают, добавляют 2-пропанол и снова выпаривают растворитель. Методику повторяют, реакционную смесь перемешивают и кипятят с обратным холодильником 2 ч, охлаждают и экстрагируют дважды ДХМ. Отделившиеся органические слои сушат, растворитель выпаривают и остаток растирают с этилацетатом, отфильтровывают и сушат,получая 0,22 г гидрата (1:1) гидрохлорида (1:1) 1-[4-4'-(1,1-диметилэтил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинил]фенилацетил]амино]уксусной кислоты (соединение 25). Соединение (23) получают аналогично из соединения (15) с использованием описанного выше способа.- 18006385 Пример В.4. Смесь промежуточного соединения (20) (0,0034 моль), этиламина (0,07 моль; 2 М в ТГФ), N,N-диметил-4-пиридинамина (0,001 моль) и триэтиламина (0,0034 моль) в ДХМ (100 мл) перемешивают при комнатной температуре. Добавляют гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида(EDAP) (0,007 моль) и смесь перемешивают при комнатной температуре 6 ч, затем промывают водой. Отделившийся органический слой сушат, фильтруют и растворитель выпаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, градиент 100/0; 99/1; 98/2), получая 4'(1,1-диметилэтил)-N-[4-[4-[2-(этиламино)-2-оксо-1-фенилэтил]-1-пиперидинил]фенил]-[1,1'-бифенил]-2 карбоксамид и 4-фтор-N-[4-(1-пиперидинил)фенил]бензамид [соединение 24). Пример В.5. Смесь промежуточного соединения (4) (0,00016 моль), Ру-ВОР (0,00032 моль) и триэтиламина (0,1 моль) в ДХМ (5 мл) перемешивают 30 мин. Добавляют этаноламин (0,0005 моль) и реакционную смесь перемешивают в течение ночи при 40 С. Реакционную смесь охлаждают. Добавляют воду и смесь перемешивают 15 мин, затем фильтруют через Extrelut и требуемое соединение выделяют с помощью ВЭЖХ,получая соединение (1). Пример В.6. Смесь промежуточного соединения (4) (0,000079 моль), 1,1'-карбонилбис-1 Н-имидазола (0,00024 моль) и триэтиламина (0,00032 моль) в ДХМ (5 мл) перемешивают 30 мин. Полученный раствор добавляют к гидрохлориду метилового эфира (S)-лейцина (0,00016 моль). Реакционную смесь встряхивают в течение 20 ч при комнатной температуре, затем в течение 5 ч при 40 С. Добавляют дополнительное количество гидрохлорида метилового эфира (S)-лейцина (0,00008 моль) в ДХМ (1 мл) и реакционную смесь встряхивают 5 ч при 40 С. Реакционной смеси дают нагреться до комнатной температуры, фильтруют и фильтрат собирают, затем промывают водой (2 мл). Полученную смесь фильтруют через Extrelut и фильтры Extrelut промывают ДХМ (2 х 3 мл). Фильтраты очищают ВЭЖХ (элюент: СН 2 Сl2/СН 3 ОН 90/100). Фракции продукта собирают и растворитель выпаривают, получая 0,041 г (2S)-метилового эфира 4-метил-2-фенил-[4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперазинил] ацетил]амино]пентановой кислоты (соединение 26). Пример В.7. Смесь промежуточного соединения (12) (0,000084 моль), Ру-ВОР (0,00034 моль) и триэтиламина(0,00034 моль) в ДХМ (5 мл) перемешивают 30 мин при комнатной температуре. Полученный прозрачный раствор добавляют к гидрохлориду метилового эфира (S)-лейцина (0,00017 моль). Реакционную смесь встряхивают в течение 24 ч при 40 С. Реакционную смесь фильтруют и остаток на фильтре промывают 1 раз 4 мл ДХМ. К фильтрату добавляют воду (2 мл) и смесь перемешивают 30 мин. Смесь фильтруют черезExtrelut и фильтры Extrelut промывают 2 раза ДХМ (3 мл). Фильтрат упаривают и остаток очищают ВЭЖХ (колонка Waters, с Xterra MS C18; элюент: [(0,5% NH4OAc в Н 2O/СН 3 СN 90/10)]/СН 3 ОН/СН 3 СN(0 мин) 75/25/0, (10 мин) 0/50/50, (16 мин) 0/0/100, (18,10-20,00 мин) 75/25/0), получая (2S)-метиловый эфир 4-метил-2-фенил-[4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперидинил]ацетил]амино]пентановой кислоты (соединение 64). Пример В.8. Добавляют этил 2-бромпропионат (0,0001 моль) к смеси промежуточного соединения (12) (0,000084 моль) и три-н-бутилтиомочевины (ТБТМ) (0,000168 моль) в DIPEA (4 мл). Реакционную смесь встряхивают в течение 2 ч при 70 С. Реакционную смесь охлаждают до комнатной температуры, фильтруют и фильтрат упаривают. Остаток растворяют в ДХМ (4 мл), затем промывают водой (2 мл). Смесь фильтруют черезExtrelut и остаток на фильтре промывают ДХМ (2 х 3 мл). Фильтрат упаривают. Остаток очищают с помощью ВЭЖХ (колонка Waters, с Xterra MS C18; элюент: [(0,5% NH4OAC в Н 2O)/CH3CN 90/10)]/СН 3 СN(0 мин) 85/15, (10 мин) 10/90, (16 мин) 0/100, (18,10-20,00 мин) 85/15), получая 2-этокси-1-метил-2-оксоэтиловый эфир -фенил-4-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-1-пиперидинуксусной кислоты (соединение 111). Пример В.9. Добавляют 2-бромэтанол (1,2 экв., 0,00010 моль) к смеси промежуточного соединения (24)(0,000084 моль) в ДМФА (5 мл) и СsСО 3 (0,00018 моль) и реакционную смесь перемешивают в течение 3 ч при 70 С. Растворитель выпаривают. Остаток распределяют между водой и ДХМ. Экстрагирующий растворитель выпаривают. Остаток очищают ВЭЖХ (колонка Waters, с Xterra MS C18; элюент: [(0,5%NH4OAC в H2O)/CH3CN 90/10) ]/CH3CN (0 мин) 85/15, (10 мин) 10/90, (16 мин) 0/100, (18,10-20,00 мин) 85/15). Фракции продукта собирают и органический растворитель выпаривают. Водные концентраты экстрагируют и растворитель экстракта выпаривают, получая 2-гидроксиэтиловый эфир -фенил-1-[4-4'-(трифторметил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинуксусной кислоты (соединение 134). Пример В.10. Смесь промежуточного соединения (40) (0,00014 моль), промежуточного соединения (44) (0,00016 моль) и Nа 2 СО 3 (0,0016 моль) в ДМФА (5 мл) перемешивают в течение ночи при 60 С. Растворитель выпаривают. Остаток растворяют в ДХМ (10 мл), промывают водой (2 мл), фильтруют через Extrelut и фильт- 19006385 рат упаривают. Остаток очищают ВЭЖХ. Фракции продукта собирают и растворитель выпаривают, получая 0,052 г метилового эфира 4-[4-[([1,1'-бифенил]-2-илкарбонил)амино]фенил]-1-пиперидинил]фенилацетил]амино]уксусной кислоты (соединение 17). Соединение (142) получают аналогично при взаимодействии промежуточного соединения (44) с промежуточным соединением (49) с использованием описанного выше способа. Пример В.11. Смесь промежуточного соединения (24) (0,000084 моль), Ру-ВОР (0,00017 моль) и триэтиламина(0,5 мл) в ДХМ (5 мл) перемешивают 60 мин при комнатной температуре. Добавляют метилгидроксиацетат (0,0001 моль) и реакционную смесь перемешивают 3 ч при 40 С, затем в течение 2 дней при комнатной температуре. Добавляют воду (2 мл). Смесь перемешивают 15 мин, затем фильтруют через Extrelut и органический фильтрат упаривают. Остаток очищают ВЭЖХ. Фракции продукта собирают и растворитель выпаривают, получая 2-метокси-2-оксоэтиловый эфир -фенил-1-[4-4'-(трифторметил)[1,1'бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинуксусной кислоты (соединение 20). Пример В.12. Смесь соединения (97) (0,00081 моль), ДХМ (15 мл) и трифторуксусной кислоты (5 мл) перемешивают при комнатной температуре 2 ч. Растворитель выпаривают. Добавляют ДХМ и выпаривают (3 раза). Остаток перемешивают в DIPE. Осадок отфильтровывают, сушат и очищают высокоэффективной жидкостной хроматографией на Hyperprep C18 BDS (элюент: (0,5% NH4OAc в Н 2 О/СН 3 СN 90/10)/СН 3 ОН/СН 3 СN 45/35/20; 8/47/45; 0/0/100). Нужные фракции собирают и растворитель выпаривают. Остаток перемешивают в DIPE. Осадок отфильтровывают, промывают DIPE и сушат, получая соединение (143). Пример В.13. Перемешивают 4'-(1,1-диметилэтил)[1,1'-бифенил]-2-карбоновую кислоту (0,011 моль) в ДХМ (100 мл). Добавляют тионилхлорид (0,022 моль). Добавляют ДМФА (3 капли) и реакционную смесь перемешивают и кипятят с обратным холодильником 1 ч. Реакционную смесь охлаждают и растворитель выпаривают. Добавляют ДХМ (100 мл) и растворитель снова выпаривают. Остаток растворяют в ДХМ (50 мл) и полученный раствор добавляют к раствору промежуточного соединения (56) (0,0097 моль) в ДХМ (50 мл). Смесь перемешивают. Добавляют гидрокарбонат натрия (насыщенный водный раствор) (50 мл) и смесь перемешивают 2 ч при комнатной температуре. Слои разделяют. Органический слой сушат, фильтруют и растворитель выпаривают. Остаток очищают колоночной хроматографией на силикагеле (элюент:CH2Cl2/CH3OH 98/2). Фракции продукта собирают и растворитель выпаривают. Остаток кристаллизуют из смеси 2-пронанол/DIРЕ, отфильтровывают и сушат (вакуум, 50 С), получая 5,27 г продукта. Полученную фракцию сушат (вакуум, 80 С в течение 2 дней), получая 4,95 г метиловый эфир (В)-N-1-[4-4'(1,1-диметилэтил)[1,1'-бифенил]-2-ил]карбонил]амино]фенил]-4-пиперидинил]фенилацетил]глицина (соединение 145, т.пл. 119,9-120 С). В табл. F-1 перечислены соединения, которые получены в соответствии с приведенными выше примерами. В таблице используются следующие сокращения: С 3 Н 8 О относится к 2-пропаноляту. Таблица F-1C.1. Количественная оценка выделения Аро-В. Клетки HepG2 культивируют в 24-луночных планшетах в среде MEM Rega 3, содержащей 10% сыворотки плода крупного рогатого скота. При 70%-ном слиянии среду заменяют и добавляют испытуемое соединение или носитель (ДМСО, конечная концентрация 0,4%). После выдерживания в течение 24 ч среду переносят в пробирки Эппендорфа и осветляют центрифугированием. Антитело овцы против любого Аро-В добавляют к надосадочной жидкости и смесь выдерживают при 8 С в течение 24 ч. Затем добавляют антитело кролика против овцы и иммунному комплексу дают возможность образовать осадок в течение 24 ч при 8 С. Иммунопреципитат пеллетируют центрифугированием в течение 25 мин при 1320 g и промывают дважды буфером, содержащим 40 мМ Mops, 40 мМ NaH2PO4, 100 мМ NaF, 0,2 мМ DTT, 5 мМ EDTA, 5 мМPMSF. Радиоактивность в пеллетах количественно определяют с помощью сцинтилляционного счетчика. Полученные значения IC50 обобщены в табл. C.1. Таблица С.1 Значения pIC50 (=-log IC50)- 27006385 С.2. Анализ МТР. Активность МТР измеряют с использованием метода анализа, аналогично методу, описанному J.R.Wetterrau, D.B. Zilversmitt, в Chemistry and Physics of Lipids, 38, 205-222 (1985). Для получения донорных и акцепторных везикул соответствующие липиды в хлороформе помещают в стеклянную пробирку для испытаний и сушат в токе N2. К высушенным липидам добавляют буфер, содержащий 15 MM Tris-HCl,pH 7,5, 1 мМ EDTA, 40 мМ NaCl, 0,02% NaN3 (буфер для анализа). Смесь недолго интенсивно перемешивают и затем дают липидам гидратироваться в течение 20 мин на льду. Затем получают везикулы периодической обработкой ультразвуком (Branson 2200) при комнатной температуре в течение максимально 15 мин. Во все препараты везикул включен бутилированный гидрокситолуол в концентрации 0,1%. Смесь для оценки липидного переноса содержит донорные везикулы (40 нмоль фосфатидилхолина, 7,5 мол.% кардиолипина и 0,25 мол.% глицерин-три[1-14C]олеата), акцептор везикул (240 нмоль фосфатидилхолина) и 5 мг BSA в суммарном объеме 675 мкл в пробирке для центрифугирования на 1,5 мл. Добавляют испытуемое соединение, растворенное в ДМСО (конечная концентрация 0,13%). Через 5 мин предварительного выдерживания при 37 С запускают реакцию добавлением МТР в 100 мкл диализного буфера. Реакцию останавливают добавлением 400 мкл целлюлозы DEAE-52, предварительно уравновешенной в 15 мМ Tris-HCl, pH 7,5, 1 мМ EDTA, 0,02% NaN3 (1:1, об./об.). Смесь перемешивают 4 мин и центрифугируют 2 мин при максимальной скорости центрифуги Эппендорфа (4 С) с образованием пеллет связанных DEAE-52 донорных везикул. Аликвоту надосадочной жидкости, содержащую акцепторные липосомы, подсчитывают и импульсы [14 С] используют для расчета процента триглицеридного переноса от донорных к акцепторным везикулам. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединения формулы (I) их N-оксиды, фармацевтически приемлемые кислотно-аддитивные соли и стереохимически изомерные формы,где р 1, р 2 и р 3, каждый независимо равен целому числу от 1 до 3; р 4 равен целому числу 0 или 1; каждый из R1 независимо выбран из водорода, С 1-4 алкила, С 1-4 алкилоксигруппы, галогена, гидрокси-, меркапто-, циано-, нитро-, С 1-4 алкилтиогруппы или полигалогенС 1-6 алкила, амино-, С 1-4 алкиламинои ди(С 1-4 алкил)аминогруппы; каждый из R2 независимо выбран из водорода, С 1-4 алкила, С 1-4 алкилоксигруппы, галогена или трифторметила;R3 представляет водород или С 1-4 алкил; каждый из R4 независимо выбран из водорода, С 1-4 алкила, С 1-4 алкилоксигруппы, галогена или трифторметила;Z представляет бивалентный радикал формулы где каждый из X1 и X2 независимо выбран из СН, N или sp2-гибридизованного атома углерода;- 29006385 где i равно целому числу от 1 до 4;j равно целому числу от 1 до 4;k равно целому числу 1 или 2;R8 представляет C1-6 алкил, С 2-6 алкенил, С 2-6 алкинил, фенил или фенил, замещенный С 1-4 алкилом,галогеном, гидроксигруппой или трифторметилом; каждый из R10 и R11 независимо представляет водород или C1-6 алкил; необязательно R7 и R9, взятые вместе, могут образовывать бивалентный радикал формул -(СН 2)3-,-(СН 2)4-, -(СН 2)5- или -(СН 2)6-; и необязательно в радикале (b-1) C1-6 алкандиильный остаток может быть дополнительно замещен фенилом, фенилС 1-4 алкилом, гидроксифенилС 1-4 алкилом, С 1-4 алкилоксикарбонилом, С 1-4 алкилоксиС 1-4 алкилом, С 1-4 алкилтиоС 1-4 алкилом, фенилС 1-4 алкилтиоС 1-4 алкилом, гидроксиС 1-4 алкилом, тиоС 1-4 алкилом,С 3-6 циклоалкилом, С 3-6 циклоалкилС 1-4 алкилом или радикалом формул 2. Соединение по п.1, где R1 представляет водород, трет-бутил или трифторметил; R2, R3 и R4 представляют водород. 3. Соединение по любому из пп.1 или 2, где бивалентный радикал А представляет метиленовую группу, замещенную фенилом. 4. Соединение по любому из пп.1-3, где Z представляет бивалентный радикал формулы (а-5), где Z1 2 и Z представляют СН 2 СН 2, а X1 представляет N и X2 представляет СН. 5. Соединение по любому из пп.1-3, где Z представляет бивалентный радикал формулы (а-5), где Z1 2 и Z представляют СН 2 СН 2, а X1 представляет СН и X2 представляет N. 6. Соединение по любому из пп.1-3, где Z представляет бивалентный радикал формулы (а-5), где Z1 2 и Z представляют СН 2 СН 2, а X1 и X2 представляют N. 7. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически активное количество соединения по любому из пп.1-6. 8. Способ получения фармацевтической композиции по п.7, где терапевтически активное количество соединения по любому из пп.1-6 однородно смешивают с фармацевтически приемлемым носителем. 9. Применение соединения по любому из пп.1-6 в качестве лекарственного средства для лечения гиперлипидемии, ожирения и диабета типа II. 10. Способ получения соединения формулы (I) по п.1, где промежуточное соединение формулы (II),где В, A, Z, R4, р 3 и р 4 имеют значения, определенные в п.1, подвергают реакции с бифенилкарбоновой кислотой или галогенангидридом формулы (III), где R1,R , р и р 2 имеют значения, определенные для формулы (I), и Q1 выбран из гидроксигруппы и галогена по меньшей мере в одном инертном в условиях реакции растворителе и необязательно в присутствии подходящего основания 2 11. Способ получения соединения формулы (I) по п.1, где промежуточное соединение, имеющее формулу (IV)
МПК / Метки
МПК: C07D 295/15, A61K 31/445, A61P 3/10
Метки: снижающие, уровень, бифенилкарбоксамиды, липидов
Код ссылки
<a href="https://eas.patents.su/30-6385-bifenilkarboksamidy-snizhayushhie-uroven-lipidov.html" rel="bookmark" title="База патентов Евразийского Союза">Бифенилкарбоксамиды, снижающие уровень липидов</a>
Бифенилкарбоксамиды, полезные в качестве снижающих содержание липидов агентов

Номер патента: 6101
Опубликовано: 25.08.2005
Авторы: Мерпул Ливен, Бакс Лео Якобус Йозеф
МПК: C07C 233/80, A61P 3/06, A61K 31/40...
Метки: снижающих, полезные, бифенилкарбоксамиды, липидов, качестве, агентов, содержание
Формула / Реферат:
1. Соединение формулы (I) N-оксиды, фармацевтически приемлемые кислотно-аддитивные соли и их стереохимически изомерные формы, где p1, p2 и p3 означают целые числа, каждое из которых независимо равно 1-3; каждый из R1 независимо выбирают из группы, включающей водород, C1-4-алкил, C1-4-алкилокси, галоген, гидрокси, меркапто, циано, нитро, C1-4-алкилтио или полигалоген-C1-6-алкил, амино, C1-4-алкиламино и ди(C1-4-алкил)амино; каждый из R2...
Выделенное антитело и его фрагмент, способы и набор для оценки уровня перекисного окисления липидов в биологическом образце и способ диагностики заболеваний, обусловленных перекисным окислением липидов

Номер патента: 2520
Опубликовано: 27.06.2002
Авторы: Партхасаратхи Сампатх, Александер Уэйн Р., Медфорд Рассел М.
МПК: A61P 43/00, G01N 33/543, C07K 16/00...
Метки: оценки, набор, окислением, перекисным, обусловленных, липидов, заболеваний, выделенное, биологическом, окисления, способы, образце, фрагмент, диагностики, перекисного, способ, антитело, уровня
Формула / Реферат:
1. Выделенное антитело, которое взаимодействует с антигеном, образовавшимся в результате реакции гидроперекиси липида с первичным амином, и при этом в какой-либо значительной степени не связывается с альдегидмодифицированным белком. 2. Антитело по п.1, отличающееся тем, что конъюгировано с дополнительным агентом. 3. Антитело по п.2, отличающееся тем, что дополнительный агент представляет собой детектируемую метку. 4. Антитело по п.3,...
Полиарилкарбоксамиды, применимые в качестве агентов, снижающих содержание липидов

Номер патента: 5855
Опубликовано: 30.06.2005
Авторы: Бакс Лео Якобус Йозеф, Ван Дер Векен Луи Йозеф Элизабет, Мерпул Ливен, Вьейевуа Марсель, Рувенс Петер Вальтер Мария
МПК: C07D 211/34, A61K 31/495, A61P 3/06...
Метки: липидов, полиарилкарбоксамиды, снижающих, агентов, качестве, применимые, содержание
Формула / Реферат:
1. Соединения полиарилкарбоксамида формулы (I) их N-оксиды, фармацевтически приемлемые соли присоединения (аддитивные) и стереохимически изомерные формы, где Z1 выбран из (CH2)n, где n имеет значения от 1 до 3, CH2CH2O и OCH2CH2; Z2 означает (CH2)m, где m равно 1 или 2, X1 означает O, CH2, CO, NH, CH2O, OCH2, CH2S, SCH2 или прямую связь; X2 и X3, каждый независимо, выбраны из CH, N и sp2 углеродного атома; R1 означает водород или C1-4алкил; Ar1...
Неводные суспензии, снижающие сопротивление течению углеводородов в трубопроводах

Номер патента: 1538
Опубликовано: 23.04.2001
Авторы: Джонстон Рэй Л., Ли Янг Н.
МПК: F17D 1/17, C08K 5/20, C08L 91/06...
Метки: неводные, сопротивление, углеводородов, течению, суспензии, снижающие, трубопроводах
Формула / Реферат:
1. Способ получения стабильной при нагревании неводной суспензии твердого растворимого в углеводородах снижающего трение полиальфаолефина, полученного из олефинов, содержащих от 2 до 30 атомов углерода, и способного снижать сопротивление течению в углеводороде, протекающем по трубопроводу, включающий: (а) тонкий размол указанного твердого полиальфаолефина в присутствии разделяющего агента для получения свободно текущего полиальфаолефина,...
Применение иммуномодулирующей композиции на основе липидов

Номер патента: 2364
Опубликовано: 25.04.2002
Авторы: Шиницкий Мейр, Шенфельд Авнер
МПК: A61P 25/34, A61K 31/66
Метки: иммуномодулирующей, композиции, липидов, основе, применение
Формула / Реферат:
1. Применение липидного препарата, полученного из натурального источника, обогащенного, по меньшей мере, примерно на 10% фосфатидной кислотой, для получения фармацевтической композиции для активации иммунной системы у людей со стрессом. 2. Применение по п.1, при котором указанный стресс является результатом лечения индивидуума по поводу прекращения курения. 3. Применение по п.1, при котором указанный липидный препарат содержит, по меньшей мере,...
Предыдущий патент: Композиция фентанила для интраназального введения
Следующий патент: Фармацевтические композиции эстрогенных агентов
Случайный патент: Способ культивирования орехового дерева с кроной, имеющей форму вертикального шпинделя на опоре