Способ получения азациклоалкилалканоилпсевдотетрапептидов
Номер патента: 2724
Опубликовано: 29.08.2002
Авторы: Ванасс Бенуа Ж., Леон Патрик, Лиу Роберт К., Стэммлер Роберт, Д'нетто Джеффри А., Доби Кристоф, Лявинь Мишель, Менсел Джеймс Дж.



























Формула / Реферат
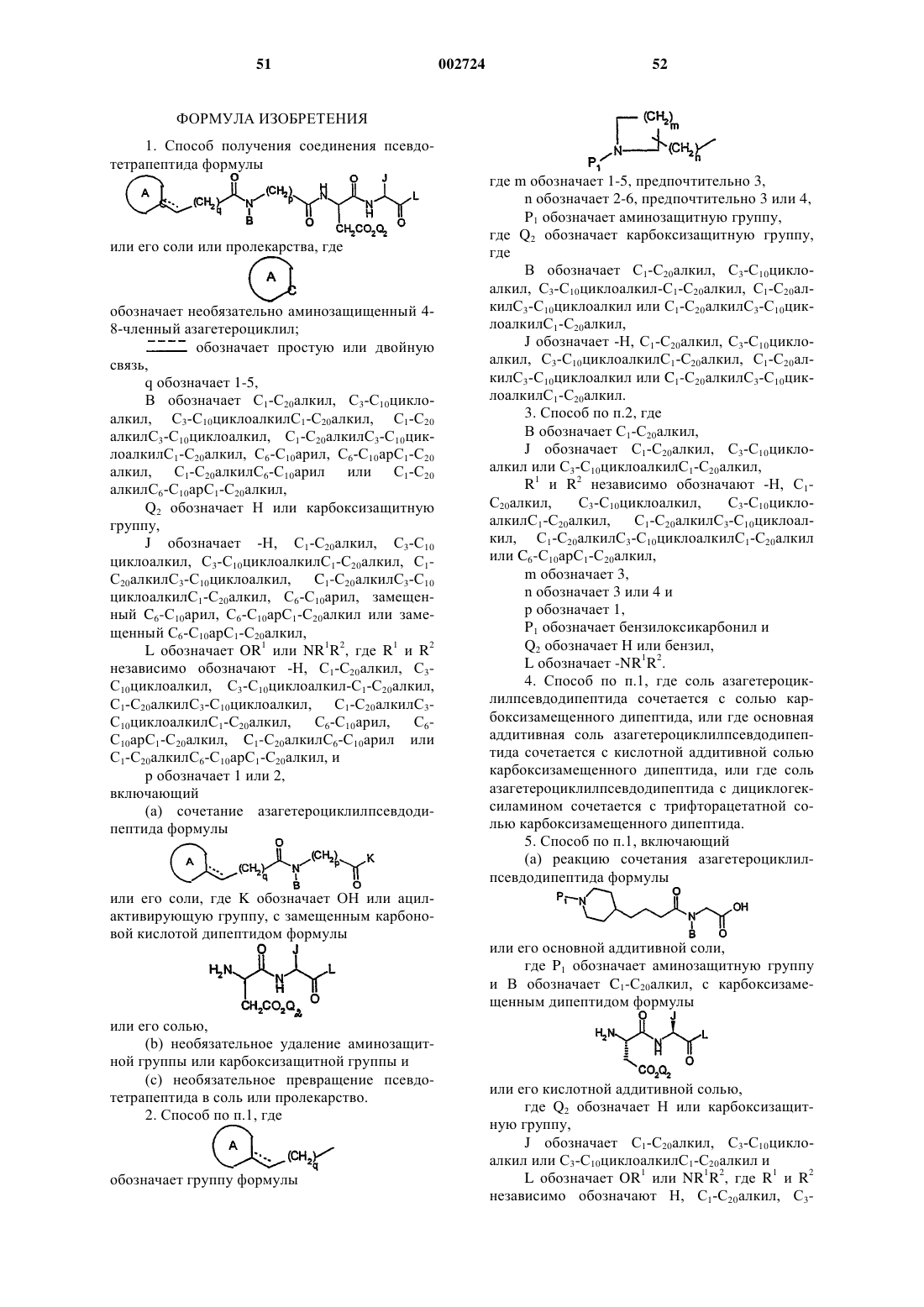
1. Способ получения соединения псевдотетрапептида формулы

или его соли или пролекарства, где

обозначает необязательно аминозащищенный 4-8-членный азагетероциклил;
![]() обозначает простую или двойную связь,
обозначает простую или двойную связь,
q обозначает 1-5,
В обозначает С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкилС1-С20алкил, С1-С20алкилС3-С10циклоалкил, С1-С20алкилС3-С10циклоалкилС1-С20алкил, С6-С10арил, С6-С10арС1-С20алкил, С1-С20алкилС6-С10арил или С1-С20алкилС6-С10арС1-С20алкил,
Q2 обозначает Н или карбоксизащитную группу,
J обозначает -Н, С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкилС1-С20алкил, С1-С20алкилС3-С10циклоалкил, C1-С20алкилС3-С10циклоалкилС1-С20алкил, С6-С10арил, замещенный C6-С10арил, С6-С10арС1-С20алкил или замещенный С6-С10арС1-С20алкил,
L обозначает OR1 или NR1R2, где R1 и R2 независимо обозначают -Н, С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкил-С1-С20алкил, С1-С20алкилС3-С10циклоалкил, С1-С20алкилС3-С10циклоалкилС1-С20алкил, С6-С10арил, С6-С10арС1-С20алкил, C1-С20алкилС6-С10арил или С1-С20алкилС6-С10арС1-С20алкил, и
р обозначает 1 или 2,
включающий
(а) сочетание азагетероциклилпсевдодипептида формулы

или его соли, где K обозначает ОН или ацил-активирующую группу, с замещенным карбоновой кислотой дипептидом формулы

или его солью,
(b) необязательное удаление аминозащитной группы или карбоксизащитной группы и
(c) необязательное превращение псевдотетрапептида в соль или пролекарство.
2. Способ по п.1, где

обозначает группу формулы

где m обозначает 1-5, предпочтительно 3,
n обозначает 2-6, предпочтительно 3 или 4,
P1 обозначает аминозащитную группу,
где Q2 обозначает карбоксизащитную группу, где
В обозначает С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкил-С1-С20алкил, С1-С20алкилС3-С10циклоалкил или С1-С20алкилС3-С10циклоалкилС1-С20алкил,
J обозначает -Н, С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкилС1-С20алкил, С1-С20алкилС3-С10циклоалкил или C1-С20алкилС3-С10циклоалкилС1-С20алкил.
3. Способ по п.2, где
В обозначает С1-С20алкил,
J обозначает С1-С20алкил, С3-С10циклоалкил или С3-С10циклоалкилС1-С20алкил,
R1 и R2 независимо обозначают -Н, С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкилС1-С20алкил, С1-С20алкилС3-С10циклоалкил, С1-С20алкилС3-С10циклоалкилС1-С20алкил или C6-С10арС1-С20алкил,
m обозначает 3,
n обозначает 3 или 4 и
р обозначает 1,
P1 обозначает бензилоксикарбонил и
Q2 обозначает Н или бензил,
L обозначает -NR1R2.
4. Способ по п.1, где соль азагетероциклилпсевдодипептида сочетается с солью карбоксизамещенного дипептида, или где основная аддитивная соль азагетероциклилпсевдодипептида сочетается с кислотной аддитивной солью карбоксизамещенного дипептида, или где соль азагетероциклилпсевдодипептида с дициклогексиламином сочетается с трифторацетатной солью карбоксизамещенного дипептида.
5. Способ по п.1, включающий
(а) реакцию сочетания азагетероциклилпсевдодипептида формулы

или его основной аддитивной соли,
где P1 обозначает аминозащитную группу и В обозначает C1-С20алкил, с карбоксизамещенным дипептидом формулы

или его кислотной аддитивной солью,
где Q2 обозначает Н или карбоксизащитную группу,
J обозначает С1-С20алкил, С3-С10циклоалкил или С3-С10циклоалкилС1-С20алкил и
L обозначает OR1 или NR1R2, где R1 и R2 независимо обозначают Н, С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкилС1-С20алкил, С1-С20алкилС3-С10циклоалкил, С1-С20алкилС3-С10циклоалкилС1-С20алкил или арС1-С20алкил,
с образованием псевдотетрапептида формулы

(b) необязательное удаление аминозащитной группы или карбоксизащитной группы, и
(c) необязательное превращение псевдотетрапептида в его соль или пролекарство.
6. Способ по п.5, где
P1 обозначает бензилоксикарбонил,
Q2 обозначает бензил,
В обозначает этил,
J обозначает циклогексилметил и
L обозначает NH2.
7. Способ по п.5, где
P1 обозначает бензилоксикарбонил,
Q2 обозначает Н,
В обозначает этил,
J обозначает циклогексилметил и
L обозначает NH2.
8. Способ получения замещенного циклогексилметилом пептида формулы

где Q2 обозначает Н или карбоксизащитную группу и
Q3 обозначает Н или аминозащитную группу, включающий
(а) восстановление замещенного фенилметилом пептида формулы

(b) необязательное удаление аминозащитной группы или карбоксизащитной группы.
9. Способ по п.8, где восстановление осуществляют путем каталитической гидрогенизации, предпочтительно используя платиновый катализатор, такой как окись платины или платина на алюминии.
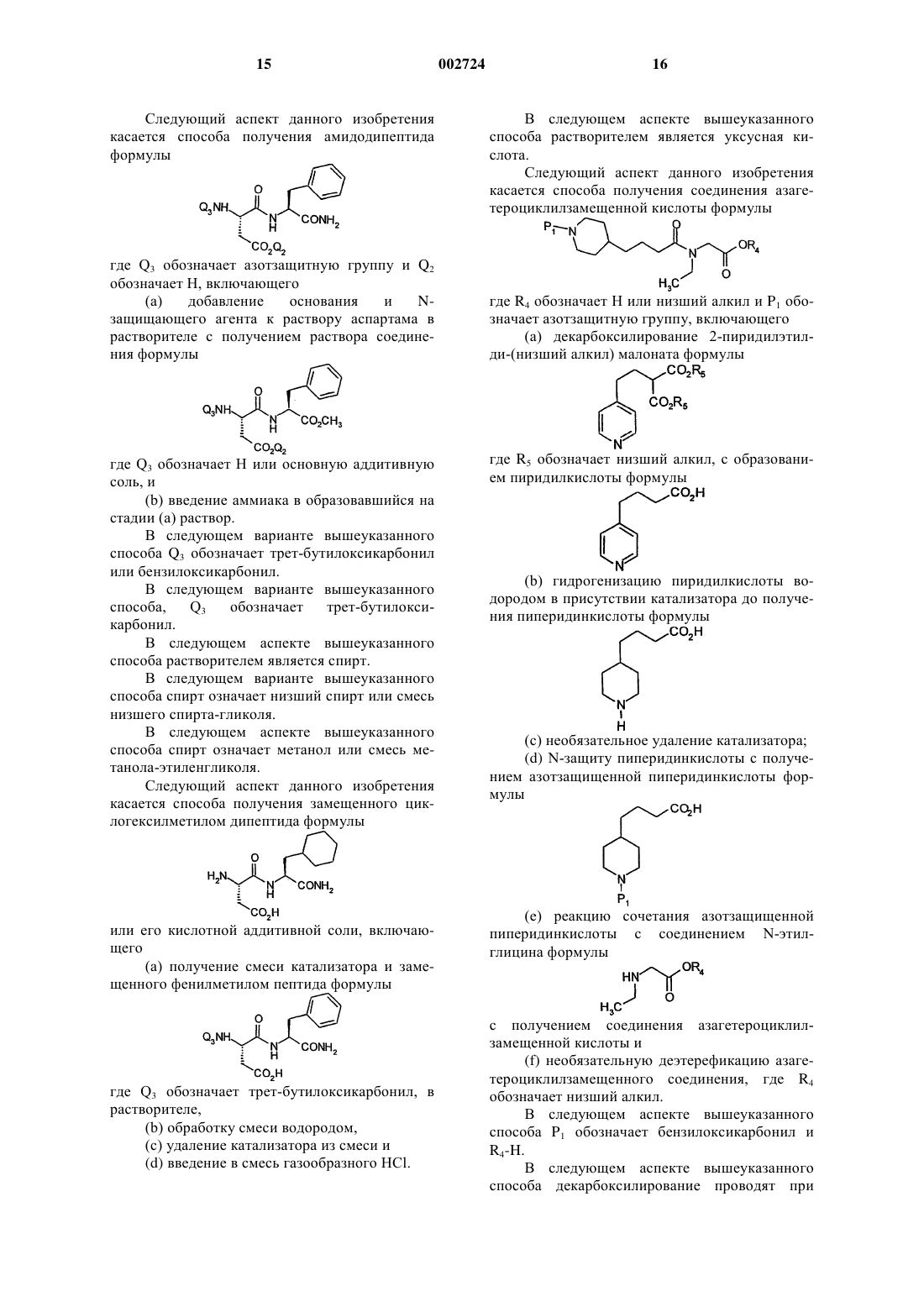
10. Способ получения амидодипептида формулы

где Q2 обозначает Н или основную аддитивную соль, или карбоксизащитную группу,
Q3 обозначает Н или аминозащитную группу,
включающий амидирование пептидного сложного эфира формулы

где R3 обозначает C1-С10алкил.
11. Способ по п.10, где амидирование осуществляют, используя раствор аммиака в спирте, таком как С1-С4cпирт, или используя аммиак в смеси растворителей С1-С4cпирт-С1-С3гликоль, которая включает метанол и этиленгликоль.
12. Способ получения соединения защищенного аспартама формулы

где Q2 обозначает Н или карбоксизащитную группу,
Q3 обозначает аминозащитную группу и
R3 обозначает C1-С10алкил,
включающий введение аминозащитной группы в соединение аспартама формулы

13. Способ по п.12, где
Q3 обозначает бензилоксикарбонил или трет-бутилоксикарбонил,
Q2 обозначает Н.
14. Способ получения амидодипептида формулы

Q3 обозначает аминозащитную группу и
Q2 обозначает Н,
включающий
(а) добавление основания и аминозащищающего агента к раствору аспартама в растворителе с получением раствора соединения формулы

где Q3 обозначает Н или основную аддитивную соль, и
(b) введение аммиака в образовавшийся на стадии (а) раствор.
15. Способ по п.14, где Q3 обозначает трет-бутилоксикарбонил или бензилоксикарбонил, растворителем является C1-С3cпирт или смесь С1-С3cпирт-С1-С3гликоль.
16. Способ по п.15, где растворителем является метанол или смесь метанол-этиленгликоль.
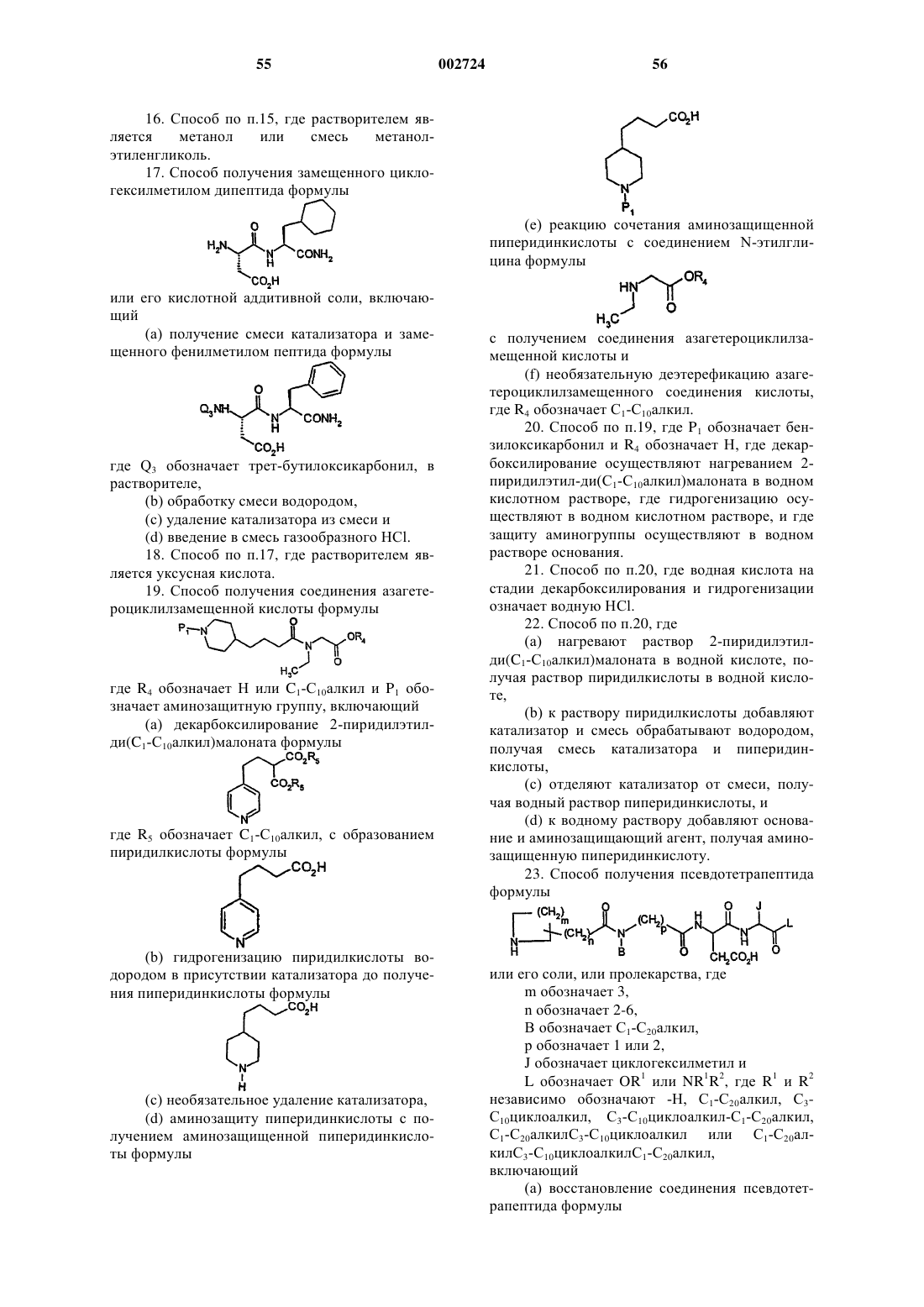
17. Способ получения замещенного циклогексилметилом дипептида формулы

или его кислотной аддитивной соли, включающий
(а) получение смеси катализатора и замещенного фенилметилом пептида формулы

где Q3 обозначает трет-бутилоксикарбонил, в растворителе,
(b) обработку смеси водородом,
(c) удаление катализатора из смеси и
(d) введение в смесь газообразного НСl.
18. Способ по п.17, где растворителем является уксусная кислота.
19. Способ получения соединения азагетероциклилзамещенной кислоты формулы

где R4 обозначает Н или C1-С10алкил и P1 обозначает аминозащитную группу, включающий
(а) декарбоксилирование 2-пиридилэтил-ди(C1-С10алкил)малоната формулы

где R5 обозначает C1-С10алкил, с образованием пиридилкислоты формулы

(b) гидрогенизацию пиридилкислоты водородом в присутствии катализатора до получения пиперидинкислоты формулы

(c) необязательное удаление катализатора,
(d) аминозащиту пиперидинкислоты с получением аминозащищенной пиперидинкислоты формулы

(е) реакцию сочетания аминозащищенной пиперидинкислоты с соединением N-этилглицина формулы

с получением соединения азагетероциклилзамещенной кислоты и
(f) необязательную деэтерефикацию азагетероциклилзамещенного соединения кислоты, где R4 обозначает С1-С10алкил.
20. Способ по п.19, где P1 обозначает бензилоксикарбонил и R4 обозначает Н, где декарбоксилирование осуществляют нагреванием 2-пиридилэтил-ди(С1-С10алкил)малоната в водном кислотном растворе, где гидрогенизацию осуществляют в водном кислотном растворе, и где защиту аминогруппы осуществляют в водном растворе основания.
21. Способ по п.20, где водная кислота на стадии декарбоксилирования и гидрогенизации означает водную НСl.
22. Способ по п.20, где
(a) нагревают раствор 2-пиридилэтил-ди(C1-С10алкил)малоната в водной кислоте, получая раствор пиридилкислоты в водной кислоте,
(b) к раствору пиридилкислоты добавляют катализатор и смесь обрабатывают водородом, получая смесь катализатора и пиперидинкислоты,
(c) отделяют катализатор от смеси, получая водный раствор пиперидинкислоты, и
(d) к водному раствору добавляют основание и аминозащищающий агент, получая аминозащищенную пиперидинкислоту.
23. Способ получения псевдотетрапептида формулы

или его соли, или пролекарства, где
m обозначает 3,
n обозначает 2-6,
В обозначает С1-С20алкил,
р обозначает 1 или 2,
J обозначает циклогексилметил и
L обозначает OR1 или NR1R2, где R1 и R2 независимо обозначают -Н, С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкил-С1-С20алкил, С1-С20алкилС3-С10циклоалкил или С1-С20алкилС3-С10циклоалкилС1-С20алкил,
включающий
(а) восстановление соединения псевдотетрапептида формулы

где

обозначает пиридил или

где m обозначает 3 и P1 обозначает Н или аминозащитную группу,
Q2 обозначает Н или карбоксизащитную группу,
J обозначает фенилметил,
(b) необязательное удаление аминозащитной группы или карбоксизащитной группы, и
(c) необязательное превращение псевдотетрапептида в соль или пролекарство.
24. Способ по п.23, где восстановление осуществляют путем каталитической гидрогенизации.
25. Способ по п.24, где

обозначает

где m обозначает 3,
P1 обозначает аминозащитную группу, предпочтительно неустойчивую к гидрогенизации,
n обозначает 3,
р обозначает 1 и
Q2 обозначает карбоксизащитную группу, предпочтительно неустойчивую к гидрогенизации.
26. Способ по п.25, где действие каталитического восстановления состоит в одновременном восстановлении и удалении P1 и Q2.
27. Способ по п.24, где

обозначает пиридил и
Q2 обозначает карбоксизащитную группу, предпочтительно неустойчивую к гидрогенизации.
28. Способ по п.27, где действие каталитического восстановления состоит в одновременном восстановлении и удалении Q2.
29. Соединение псевдодипептида формулы

или его соль,
где P1 обозначает аминозащитную группу,
В обозначает С1-С20алкил, С3-С10циклоалкил, С3-С10циклоалкил-С1-С20алкил, С1-С20алкилС3-С10циклоалкил, С1-С20алкилС3-С10циклоалкилС1-С20алкил, С6-С10арил, С6-С10арС1-С20алкил, C1-С20алкилС6-С10арил или С1-С20алкилС6-С10арС1-С20алкил, и
R4 обозначает Н или C1-С10алкил.
30. Соединение псевдодипептида по п.29,
где P1 обозначает аминозащитную группу, такую как бензилоксикарбонил,
В обозначает С1-С20алкил, такой как этил, и
R4 обозначает Н.
Текст
1 Область изобретения Данное изобретение касается способа конвергентного получения азациклоалканоилпсевдотетрапептидов, включающего сочетание дипептида с псевдодипептидом. Это изобретение касается также промежуточных соединений и способов получения промежуточных соединений, используемых при получении псевдотетрапептида. Предпосылки изобретения Азациклоалкилалканоилпсевдотетрапептиды, как, к примеру, N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамид, обладают противотромбозной активностью, включая ингибирование агрегации тромбоцитов и образования тромбов у млекопитающих, и применимы для профилактики и лечения тромбоза, связанного с патологическими состояниями, такими как инфаркт миокарда, удар, периферическое поражение артерии и диссеминированное внутрисосудистое свертывание. См. публикацию РСТ патентной заявкиWO 95/10295. До сих пор эти псевдотетрапептиды получали последовательным синтезом из С-концевой аминокислоты, используя стандартные методики синтеза пептидов в твердой фазе или в растворе. Однако последовательное сочетание аминокислот мало подходит для получения лекарственного средства в больших объемах, поскольку это вынуждает к организации производства с линейной (последовательной) технологией. Таким образом, существует необходимость альтернативного подхода к синтезу псевдотетрапептидов. Такой способ должен существенно повышать универсальность и эффективность промышленного получения. Конвергентный подход может обеспечить получение специальных модификаций псевдотетрапептида за счет проведения специфического химического превращения на одном из реакционноспособных участков, а не по всему псевдотетрапептиду и позволит одновременно получить ряд аналогов псевдотетрапептида. Краткое описание изобретения Настоящее изобретение касается способа получения псевдотетрапептида формулы I или его соли или пролекарства, где обозначает необязательно N-защищенный азагетероциклил; обозначает простую или двойную связь;J обозначает -Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил или замещенный аралкил; или его соли, где К обозначает ОН или ацилактивирующую группу,с карбоксизамещенным дипептидом формулы или его солью,(b) необязательное удаление аминозащитной группы или карбоксизащитной группы с получением псевдотетрапептида и(c) необязательное превращение псевдотетрапептида в соль или пролекарство. Подробное описание изобретения Определение терминов. Подразумевается, что если не оговорено особо, то нижеперечисленные термины, как они используются выше и далее во всем описании настоящего изобретения, имеют следующие значения."Азагетероциклил" означает карбоциклическую систему с 4-8-членным насыщенным,ненасыщенным или ароматическим кольцом,где один из атомов углерода, но не углерод фрагмента заменен атомом азота. Когда атом азота включен в циклическую систему посредством двух простых связей, он необязательно замещен азотзащитной группой P1. Характерные примеры азагетероциклилгрупп включают пиперидинил,N-трет-бутоксикарбонилпиперидинил, N-бензилоксикарбонилпиперидинил, пирролидинил,N-тpeт-бутоксикарбонилпирролидинил,Nбензилоксикарбонилпирролидинил, пирролил,пиридинил и тому подобные. Предпочтительными азагетероциклилгруппами являются пи 3 ридил, N-трет-бутоксикарбонилпиперидин-4-ил и N-бензилоксикарбонилпиперидин-4-ил."Aлкил" означает насыщенную алифатическую углеводородную группу, которая может быть линейной или разветвленной, приблизительно, с 1-20 атомами углерода в цепи. Под"разветвленной" понимается, что низшая алкильная группа, такая как метил, этил или пропил, соединена с линейной алкильной цепью. Предпочтительными линейными или разветвленными алкильными группами являются "низшие алкильные" группы, представляющие собой линейные или разветвленные алкильные группы с 1-10 атомами углерода. Более предпочтительно, алкильные группы содержат от 1 до 6 атомов углерода."Циклоалкил" означает насыщенную карбоциклическую группу с одним или более кольцами и содержит порядка 3-10 атомов углерода. Предпочтительные циклоалкильные группы включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и декагидронафтил."Циклоалкилалкил" означает алкильную группу, замещенную циклоалкильной группой. Предпочтительные циклоалкилалкильные группы включают циклопентилметил, циклогексилметил,циклогексилэтил,декагидронафт-1 илметил и декагидронафт-2-илметил."Алкилциклоалкил" означает циклоалкильную группу, замещенную алкильной группой. Характерные примеры алкилциклоалкильных групп включают 1-, 2-, 3- или 4-метил- или этилциклогексил."Алкилциклоалкилалкил" означает алкильную группу, замещенную алкилциклоалкильной группой. Характерные примеры алкилциклоалкилалкильных групп включают 1-, 2-, 3 или 4-метил- или этил- циклогексилметил или 1-, 2-, 3- или 4-метил- или этилциклогексилэтил."Арил" означает фенильную или нафтилгруппу."Замещенный арил" означает фенил- или нафтилгруппу, замещенную одним или более заместителями арильных групп, и заместители могут быть одинаковыми или различными, где"заместитель арильных групп" включает алкил,алкенил, алкинил, арил, аралкил, гидрокси, алкокси, аралкокси, гидроксиалкил, ацил, формил,карбокси, алкеноил, ароил, галоген, нитро, тригалоидметил, циано, алкоксикарбoнил, арилоксикарбонил, аралкоксикарбонил, ациламино,ароиламино, карбамоил, алкилкарбамоил, диалкилкарбамоил, арилкарбамоил, аралкилкарбамоил, алкилсульфонил, алкилсульфинил, арилсульфонил, арилсульфинил, аралкилсульфонил,аралкилсульфинил или -N-RaRb, где Ra и Rb независимо обозначают водород, алкил, арил или аралкил. Предпочтительными заместителями арильных групп являются алкил, гидрокси, алкокси, галоген и тригалоидметил."Аралкил" означает алкильную группу, замещенную арильным радикалом. Предпочтительные аралкильные группы включают бензил,нафт-1-илметил, нафт-2-илметил и фенетил."Замещенный аралкил" означает аралкильную группу, замещенную по арилу одним или более заместителями арильных групп."Спирт" означает вышеуказанную алкильную группу с 1-10 атомами углерода, замещенную одной или более гидроксильными группами. Термин "низший спирт" означает спирт с 14 атомами углерода, замещенными одной гидроксильной группой. Характерные примеры спиртов включают метанол, этанол, 2-пропанол,1-бутанол и тому подобные. "Гликоль" означает алкил, замещенный двумя или более гидроксильными группами. Характерные примеры гликолей включают этиленгликоль, пропиленгликоль и тому подобные. Термин "простой эфир" означает соединение формулы R-O-R', где R и R' обозначают низший алкил. R и R' могут быть соединены через один или большее число атомов метиленовых групп или через дополнительный атом кислорода. Характерные примеры простых эфиров включают диэтиловый эфир, метилтретбутиловый эфир, тетрагидрофуран, диоксан и тому подобные."Полярный апротонный" означает растворители, не содержащие гидроксильных групп,но имеющие сравнительно большой дипольный момент. Характерные примеры полярных апротонных растворителей включают ацетонитрил,N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), 1,1-диметоксиэтан (ДМЭ), гексаметилфосфорный триамид (ГМФА) и тому подобные."Алкоксид" означает основание формулы М-ОН, где М обозначает щелочной металл, который выбирают из натрия, кальция, лития и калия."Карбонат" означает основание формулы М 2 СО 3, где М выбирают из магния, натрия,кальция, лития и калия."Бикарбонат" означает основание формулы МНСО 3, где М выбирают из натрия, кальция,лития и калия."Природная аминокислота" означает соединение карбоновой кислоты с аминогруппой в-положении по отношению к карбоксильной группе, т.е. соединение формулы H2N-CHRCO2H, где R обозначает -Н, алкил, циклоалкил,циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил,замещенный аралкил, или-CH2CO2Q, где Q определен в этом описании. Предпочтительными аминокислотами являются такие, где R - циклогексилметил. Предпочтительным аминокислотам соответствует L-стереохимия при углероде."Пептид" или "полипептид" означает полимер, в котором мономерами являются остатки природных или неприродных аминокислот, соединенные вместе посредством амидных связей."Пептидная основная цепь" означает последовательность амидных связей, через которые соединены аминокислотные остатки."Аминокислотный остаток" означает звенья отдельной аминокислоты, включенные в пептиды по изобретению."Псевдопептид" означает пептид, в котором в пептидную основную цепь включен один или более мономеров неприродной аминокислоты. Характерные примеры неаминокислотных мономеров включают 1-[(фенилметокси)карбонил]-4-пиперидинбутановую кислоту и 4-пиридинмасляную кислоту."Неприродная аминокислота" означает соединение карбоновой кислоты с аминогруппой,находящейся в положении, отличающемся ототносительно карбоксильной группы. Предпочтительные неприродные аминокислоты по изобретению включают соединения формулы NH2(СН 2)2 СО 2 Н и обозначает азагетероциклил. Характерные примеры предпочтительных неприродных аминокислот включают 4-пиперидинбутановую кислоту, 3-(4-пиперидинилметилен)пропионовую кислоту и 4-пиридинмасляную кислоту."N-защитная группа" или "азотзащитная группа" или "аминозащитная группа" означают легко удаляемую группу, которая известна в данной области как группа, защищающая аминогруппу от нежелательной реакции в ходе синтеза, и избирательно устранима. Применение Nзащитных групп для защиты групп от нежелательных реакций в ходе синтеза является хорошо известным приемом в данной области и подобные защитные группы общеизвестны, сравни, к примеру, с Т.Н. Greene and P.G.M. Wuts,Protective Groups in Organic Synthesis, 2nd edition,John WileySons, New York (1991), упоминаемым здесь в качестве ссылки. Предпочтительными N-защитными группами являются ацил,включающие формил, ацетил, хлорацетил, трихлорацетил, о-нитрофенилацетил, о-нитрофеноксиацетил, трифторацетил, ацетоацетил, 4 хлорбутирил, изобутирил, о-нитроциннамоил,пиколиноил, ацилизотиоцианат, аминокапроил,бензоил и тому подобные, и ацилокси, включающие метоксикарбонил, 9-флуоренилметоксикарбонил, 2,2,2-трифторэтоксикарбонил,2-триметилсилилэтоксикарбонил,винилоксикарбонил, аллилоксикарбонил, трет-бутилоксикарбонил (ВОС), 1,1-диметилпропинилокси 002724"Кислотонеустойчивая N-защитная группа" и "кислотонеустойчивая азотзащитная группа" означают вышеуказанную N-защитную группу, которая легко удаляется при обработке кислотой, хотя остается относительно устойчивой к другим реагентам. Предпочтительной кислотонеустойчивойN-защитную группу, которая легко удаляется гидрированием, хотя остается относительно устойчивой к другим реагентам. Предпочтительной неустойчивой к гидрированию Nзащитной группой является бензилоксикарбонил (CBZ)."Неустойчивая к металлам азотзащитная группа" означает вышеуказанную азотзащитную группу, которая легко удаляется металлами. Предпочтительной неустойчивой к металлам азотзащитной группой является аллил, который удаляется обработкой Pd(0)."N-Защищающий агент" означает реагент,используемый для введения N-защитной группы в молекулярную частицу. Такие защитные группы обычно вводят путем замещения уходящей от N-защищающего агента группы на требующий защиты нуклеофильный атом азота. Характерные примеры N-защищающих агентов включают ацил- и арилгалогениды, включающие ацетилхлорид и бензоилхлорид и тому подобные; ацил- и арилангидриды, включающие уксусный ангидрид, трифторуксусный ангидрид и бензойный ангидрид и тому подобные; формиаты, включающие бензилхлорформиат; и карбонаты, такие как ди-трет-бутилдикарбонат и бензил-N сукцинимидилкарбонат."Карбоксизащитная группа" и "кислотная защитная группа" означают легко отщепляемую группу, которая известна из данной области, как защищающая в ходе синтеза карбоксильную(-CO2H) группу от нежелательной реакции, и избирательно устранима. Применение карбоксизащитных групп является хорошо известным приемом в данной области и подобные защитные группы общеизвестны, сравни, например, с Т.Н. Greene and P.G.M. Wuts, Protective GroupsSons, New York (1991), упоминаемым здесь в качестве ссылки. Примеры карбоксизащитных групп включают сложные эфиры, такие как метоксиметил, метилтиометил, тетрагидропиранил, бензилоксиметил, замещенный и незамещенный фенацил, 2,2,2-трихлорэтил, третбутил, циннамил, замещенный и незамещенный бензил, триметилсилил, аллил и тому подобные, 7 и амиды и гидразиды, включающие N,Nдиметил,7-нитроиндолил,гидразид,Nфенилгидразид и тому подобные. Особенно предпочтительными карбоксизащитными группами являются трет-бутил и бензил."Неустойчивая к гидрированию карбоксизащитная группа" и "неустойчивая к гидрированию кислотная защитная группа" означают вышеуказанную кислотную защитную группу, которая легко удаляется гидрированием, хотя остается относительно устойчивой к другим реагентам. Предпочтительной неустойчивой к гидрированию кислотной защитной группой является бензил."Кислотонеустойчивая карбоксизащитная группа" и "кислотонеустойчивая кислотная защитная группа" означают вышеуказанную кислотную защитную группу, которая легко удаляется при обработке кислотой, хотя остается относительно устойчивой к другим реагентам. Предпочтительной кислотонеустойчивой кислотной защитной группой является трет-бутил."Неустойчивая к металлам карбоксизащитная группа" и "неустойчивая к металлам кислотная защитная группа" означают вышеуказанную кислотную защитную группу, которая легко удаляется при обработке металлами. Предпочтительными неустойчивыми к металлам кислотными защитными группами являются фенацил и аллил, которые удаляются обработкой Zn и Pd(0)."Ацил-активирующая группа" означает группу, которая, при замещении на нее гидроксильной группы карбоновой кислоты, делает карбонильную группу более восприимчивой к нуклеофильному воздействию, тем самым способствуя замещению гидроксильной группы нуклеофилами. Характерные примеры ацилактивирующих групп включают галоген (т.е. ацилгалогениды); сложные эфиры карбоновых кислот с гидроксибензотриазолом, N-гидроксисукцинимидом, пентафторфенолом, п-нитрофенолом и тому подобным; симметричные ангидриды, асимметричные ангидриды, полученные,к примеру, по реакции карбоновой кислоты с изопропилхлорформиатом, этилхлорформиатом,изобутилхлорформиатом и тому подобным; Nкарбоксиангидриды; продукты реакции карбоновой кислоты с карбодиимидами, такими как дициклогексил-, диизопропил- и N,N-диметилпропилэтилкарбодиимид; и производные, полученные по реакции карбоновой кислоты с (бензотриазол-1-илокси)трис-(диметиламино)фосфороний-гексафторфосфатом, (бензотриазол-1 илокси)трис-(пирролидино)фосфороний-гексафторфосфатом,2-(1 Н-бензотриазол-1-ил)1,1,3,3-тетраметилуроний-тетрафторборатом, 2(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний-гексафторфосфатом и дифенилфосфоразидатом. Термин "соль", используемый в сочетании с псевдотетрапептидом, псевдодипептидом и 8 дипептидом включает кислотные и основные аддитивные соли. Когда псевдотетрапептид, псевдодипептид и дипептид замещены основной группой, то,необязательно, образуются кислотные аддитивные соли, которые могут быть более удобной для применения формой. Кислоты, которые могут быть использованы для получения кислотных аддитивных солей, включают, предпочтительно, те кислоты, которые дают, при смешении со свободным основанием, фармацевтически приемлемые соли, то есть соли, анионы которых являются нетоксичными для пациента при фармацевтических дозах солей, с тем, чтобы положительные воздействия, присущие свободному основанию, не ослаблялись побочными действиями, связанными с анионами. Хотя фармацевтически приемлемые соли указанных основных соединений предпочтительны, все кислотные аддитивные соли пригодны в качестве источников для получения свободной основной формы, даже если конкретная соль, сама по себе, требуется только как промежуточный продукт, как, к примеру, когда соль получают только в целях очистки и идентификации, или когда ее используют в качестве промежуточного соединения при получении фармацевтически приемлемой соли способом ионного обмена. Фармацевтически приемлемыми солями, входящими в объем изобретения, являются соли, производные следующих кислот: неорганических кислот, таких как соляная кислота, серная кислота,фосфорная кислота и сульфаминовая кислота; и органических кислот, таких как уксусная кислота, лимонная кислота, молочная кислота, винная кислота, малоновая кислота, метансульфокислота, этансульфокислота, бензолсульфокислота, птолуолсульфокислота, циклогексилсульфаминовая кислота, хинная кислота и тому подобные. В число соответствующих им кислотных аддитивных солей входят галоидводородные соли, к примеру, гидрохлорид и гидробромид, сульфат,фосфат, нитрат, сульфамат, ацетат, трифторацетат, цитрат, лактат, тартрат, малонат, оксалат,салицилат, пропионат, сукцинат, фумарат, малеат, метилен-бисгидроксинафтоаты, гентизаты, мезилаты, изотионаты и ди-п-толуоилтартраты, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, циклогексилсульфамат и хинат соответственно. Согласно еще одной отличительной особенности изобретения, кислотные аддитивные соли псевдотетрапептида, псевдодипептида или дипептида получают по реакции свободного основания с соответствующей кислотой с применением или адаптацией известных способов. К примеру, кислотные аддитивные соли пептидов по данному изобретению могут быть получены либо растворением свободного основания в водном или водно-спиртовом растворе или в смесях других пригодных водных растворителей, содержащих соответствующую кислоту, и 9 выделением соли путем упаривания раствора,либо по реакции свободного основания и кислоты в органическом растворителе, в этом случае соли разделяют непосредственно или они могут быть получены путем концентрации раствора. Псевдотетрапептид, псевдодипептид или дипептид могут быть регенерированы из солей с применением или адаптацией известных способов. К примеру, псевдотетрапептид, псевдодипептид или дипептид могут быть регенерированы из их кислотных аддитивных солей при обработке основанием, к примеру, водным раствором бикарбоната натрия или водным раствором аммиака. Когда псевдотетрапептид, псевдодипептид или дипептид замещены кислотной группой,могут быть получены основные аддитивные соли, которые могут быть более удобной для применения формой. Основания, которые могут быть использованы для получения основных аддитивных солей, включают предпочтительно те основания, которые дают в сочетании со свободной кислотой фармацевтически приемлемые соли, то есть соли, катионы которых являются нетоксичными для организма млекопитающих при фармацевтических дозах солей, с тем, чтобы положительные воздействия, присущие свободной кислоте, не ослаблялись побочными действиями, связанными с катионами. Фармацевтически приемлемые соли, включающие, к примеру, соли щелочных и щелочно-земельных металлов или соли аминов, и входящие в объем изобретения, включают соли, полученные из следующих оснований: гидрида натрия, гидроокиси натрия, гидроокиси калия, гидроокиси кальция, гидроокиси алюминия, гидроокиси лития, гидроокиси магния, гидроокиси цинка,аммиака, триметиламмония, триэтиламмония,этилендиамина, N-метилглюкамина, лизина,аргинина,орнитина,холина,N,N'дибензилэтилендиамина, хлорпрокаина, диэтаноламина, прокаина, N-бензилфенетиламина,диэтиламина, дициклогексиламина, пиперазина,трис-(гидроксиметил)аминометана, гидроокиси тетраметиламмония и тому подобных. Соли псевдотетрапептида, псевдодипептида или дипептида с металлами могут быть получены при контактировании гидрида, гидроокиси, карбоната или схожих с ними реакционноспособных соединений выбранного металла, в водном или органическом растворителе, с псевдотетрапептидом, псевдодипептидом или дипептидом в форме свободной кислоты. Используемым водным растворителем может быть вода или смесь воды с органическим растворителем предпочтительно спиртом, таким как метанол или этанол, кетоном, таким как ацетон, алифатическим простым эфиром, таким как тетрагидрофуран, или сложным эфиром, таким как этилацетат. Такие реакции обычно проводят при температуре окружающей среды, но при жела 002724 10 нии их можно проводить при нагревании или охлаждении. Соли аминов могут быть получены контактированием амина в водном или органическом растворителе с псевдотетрапептидом,псевдодипептидом или дипептидом в форме свободной кислоты. Подходящие водные растворители включают воду или смеси воды со спиртами, такими как метанол или этанол, простыми эфирами, такими как тетрагидрофуран,нитрилами, такими как ацетонитрил, или кетонами, такими как ацетон. Подобным образом могут быть получены соли аминокислот. Псевдотетрапептид, псевдодипептид или дипептид могут быть регенерированы из их основных аддитивных солей с применением или адаптацией известных способов, например, при обработке основной аддитивной соли кислотой,например, соляной кислотой. Соли псевдотетрапептида, псевдодипептида или дипептида полезны также в целях очистки псевдотетрапептида, псевдодипептида или дипептида, к примеру, на основании использования различий в растворимости между солями и исходными пептидами, побочными продуктами и/или исходными веществами, по методикам, хорошо известным специалистам в соответствующей области. Термин "фармацевтически приемлемый сложный эфир" означает сложные эфиры, гидролизующиеся in vivo, и включает сложные эфиры, которые легко разрушаются в теле человека, давая исходный азациклоалкилалканоилпсевдотетрапептид или его соль. Подходящие сложноэфирные группы включают, к примеру,сложные эфиры, полученные из фармацевтически приемлемых алифатических карбоновых кислот, особенно, алкановых, алкеновых, циклоалкановых и алкан-дионовых кислот, в которых каждый алкил- или алкенил-радикал предпочтительно имеет не более 6 атомов углерода. Конкретные примеры сложных эфиров включают формиаты, ацетаты, пропионаты, бутираты,акрилаты и этилсукцинаты. Термин "фармацевтически приемлемое пролекарство" означает пептид, который на основании тщательной медицинской оценки считается пригодным для фармацевтического применения и не вызывает у пациента токсической,аллергической реакции, раздражения и тому подобного, и является эффективным для предполагаемого применения, термин также включает фармацевтически приемлемый сложный эфир, равно как и цвитерионную форму, где она возможна, пептидов по изобретению. Термин"пролекарство" означает пептид, который, быстро преобразовываясь in vivo, дает исходный пептид, например, за счет гидролиза в крови. Фармацевтически приемлемые пролекарства в соответствии с изобретением описаны в Higuchi and V. Stella, Pro-drugs as Novel DeliveryDrug Design, American Pharmaceutical Association and Pergamon Press, 1987, приведенных здесь в качестве ссылок. Псевдотетрапептид, псевдодипептид или дипептид могут в дополнение к хиральным центрам в основной цепи пептида содержать асимметрические центры. Эти асимметрические центры могут независимо быть либо R, либо S конфигурации. Для специалистов в соответствующей области также очевидно, что некоторые пептиды формулы I могут обладать геометрической изомерией. Геометрические изомеры включают цис- и транс-формы пептидов по изобретению, имеющих алкенильные группы. Настоящее изобретение включает получение отдельных геометрических изомеров и стереоизомеров и их смесей. Такие изомеры могут быть выделены из их смесей с применением или адаптацией известных способов, к примеру, хроматографическими способами и способами перекристаллизации, или их получают раздельно из соответствующих изомеров их промежуточных соединений, к примеру, с применением или адаптацией описанных здесь способов. Предпочтительные варианты воплощения Предпочтительные псевдотетрапептиды,получаемые с применением способа по данному изобретению, имеют формулу II где P1 обозначает азотзащитную группу; В обозначает алкил, циклоалкил, циклоалкилалкил,алкилциклоалкил, алкилциклоалкилалкил; Q2 обозначает Н или карбоксизащитную группу P2;R2 независимо обозначают -Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; m обозначает 3; n обозначает 3 или 4. Более предпочтительными псевдотетрапептидами, получаемыми с применением способа по данному изобретению, являются те псевдотетрапептиды формулы II, в которых В обозначает алкил; J обозначает алкил, циклоалкил или циклоалкилалкил; L обозначает OR1 илиm обозначает 3; n обозначает 3 или 4, р обозначает 1. Еще более предпочтительными псевдотетрапептидами, получаемыми с применением способа по данному изобретению, являются псевдотетрапептиды формулы III где P1 обозначает азотзащитную группу; В обозначает алкил; Q2 обозначает Н или карбоксизащитную группу Р 2; J обозначает алкил, циклоалкил или циклоалкилалкил; L обозначает OR1 или NR1R2, где R1 и R2 независимо обозначают-Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил. Наиболее предпочтительными соединениями, получаемыми с применением способа по данному изобретению, являются те соединения формулы III, в которых В обозначает алкил; J обозначает циклоалкилалкил и L обозначаетNR1R2, где R1 и R2 независимо обозначают Н или алкил. Другой аспект данного изобретения касается способа, где Q2 обозначает карбоксизащитную группу. Следующий аспект данного изобретения касается способа, где P1 обозначает бензилоксикарбонил и Q2 обозначает бензил. Следующий аспект данного изобретения касается способа, где P1 обозначает бензилоксикарбонил и Q2 обозначает Н. Следующий аспект данного изобретения касается способа, где P1 обозначает бензилоксикарбонил, Q2 обозначает бензил и L обозначаетNR1R2. Следующий аспект данного изобретения касается способа, по которому соль азагетероциклилпсевдодипептида подвергается реакции сочетания с солью карбоксизамещенного дипептида. Следующий аспект данного изобретения касается способа, по которому основная аддитивная соль азагетероциклилпсевдодипептида подвергается реакции сочетания с кислотной аддитивной солью карбоксизамещенного дипептида. Следующий аспект данного изобретения касается способа, по которому соль азагетероциклилпсевдодипептида с дициклогексиламином подвергается реакции сочетания с трифторацетатной солью карбоксизамещенного дипептида. Следующий аспект данного изобретения касается способа, включающего(а) реакцию сочетания азагетероциклилпсевдодипептида формулы или его основной аддитивной соли, где P1 обозначает азотзащитную группу и В обозначает алкил, с карбоксизамещенным дипептидом формулы или его кислотной аддитивной солью, где Q2 обозначает H или карбоксизащитную группу; J обозначает алкил, циклоалкил или циклоалкилалкил и L обозначает OR1 или NR1R2, где R1 и(b) необязательное удаление аминозащитной группы или карбоксизащитной группы и(c) необязательное превращение псевдотетрапептида в соль или пролекарство. В следующем варианте вышеуказанного способа, P1 обозначает бензилоксикарбонил иQ2 обозначает бензил. В следующем варианте вышеуказанного способа, P1 обозначает бензилоксикарбонил иQ2 обозначает Н. В следующем варианте вышеуказанного способа, P1 обозначает бензилоксикарбонил; Q2 обозначает бензил; В обозначает этил; J обозначает циклогексилметил и L обозначает NH2. В следующем варианте вышеуказанного способа, P1 обозначает бензилоксикарбонил; Q2 обозначает Н; В обозначает этил; J обозначает циклогексилметил и L обозначает NH2. Следующий аспект данного изобретения касается способа получения замещенного циклогексилметилом дипептида формулы где Q2 обозначает Н или карбоксизащитную группу и Q2 обозначает Н или азотзащитную группу, включающий(а) восстановление замещенного фенилметилом пептида формулы(b) необязательное удаление азотзащитной группы или карбоксизащитной группы. В следующем аспекте вышеуказанного способа восстановление осуществляют путем каталитической гидрогенизации. 14 В следующем аспекте вышеуказанного способа каталитическую гидрогенизацию осуществляют, используя платиновый катализатор. В следующем аспекте вышеуказанного способа платиновым катализатором является окись платины или платина на алюминии. Следующий аспект данного изобретения касается способа получения амидодипептида формулы где Q2 обозначает Н или основную аддитивную соль, или карбоксизащитную группу, и Q3 обозначает Н или азотзащитную группу, включающего амидирование пептидного сложного эфира формулы где R3 обозначает низший алкил. В следующем аспекте вышеуказанного способа амидирование осуществляют, используя раствор аммиака в спирте. В следующем аспекте вышеуказанного способа спирт означает низший спирт. В следующем аспекте вышеуказанного способа амидирование осуществляют, используя раствор аммиака в смеси растворителей низшего спирта-гликоля. В следующем аспекте вышеуказанного способа смесь растворителей из низшего спирта-гликоля включает метанол и этиленгликоль. Следующий аспект данного изобретения касается способа получения соединения защищенного аспартама формулы где Q2 обозначает Н или карбоксизащитную группу; Q3 обозначает азотзащитную группу и В следующем варианте вышеуказанного способа, Q3 обозначает бензилоксикарбонил или трет-бутилоксикарбонил. В следующем варианте вышеуказанного способа, Q2 обозначает Н. 15 Следующий аспект данного изобретения касается способа получения амидодипептида формулы где Q3 обозначает азотзащитную группу и Q2 обозначает Н, включающегоNзащищающего агента к раствору аспартама в растворителе с получением раствора соединения формулы где Q3 обозначает Н или основную аддитивную соль, и(b) введение аммиака в образовавшийся на стадии (а) раствор. В следующем варианте вышеуказанного способа Q3 обозначает трет-бутилоксикарбонил или бензилоксикарбонил. В следующем варианте вышеуказанного способа,Q3 обозначает трет-бутилоксикарбонил. В следующем аспекте вышеуказанного способа растворителем является спирт. В следующем варианте вышеуказанного способа спирт означает низший спирт или смесь низшего спирта-гликоля. В следующем аспекте вышеуказанного способа спирт означает метанол или смесь метанола-этиленгликоля. Следующий аспект данного изобретения касается способа получения замещенного циклогексилметилом дипептида формулы или его кислотной аддитивной соли, включающего(а) получение смеси катализатора и замещенного фенилметилом пептида формулы где Q3 обозначает трет-бутилоксикарбонил, в растворителе,(b) обработку смеси водородом,(c) удаление катализатора из смеси и 16 В следующем аспекте вышеуказанного способа растворителем является уксусная кислота. Следующий аспект данного изобретения касается способа получения соединения азагетероциклилзамещенной кислоты формулы где R4 обозначает Н или низший алкил и P1 обозначает азотзащитную группу, включающего(b) гидрогенизацию пиридилкислоты водородом в присутствии катализатора до получения пиперидинкислоты формулы(d) N-защиту пиперидинкислоты с получением азотзащищенной пиперидинкислоты формулы(е) реакцию сочетания азотзащищенной пиперидинкислоты с соединением N-этилглицина формулы с получением соединения азагетероциклилзамещенной кислоты и(f) необязательную деэтерефикацию азагетероциклилзамещенного соединения, где R4 обозначает низший алкил. В следующем аспекте вышеуказанного способа P1 обозначает бензилоксикарбонил иR4-H. В следующем аспекте вышеуказанного способа декарбоксилирование проводят при 17 нагревании 2-пиридилэтил-ди-(низший алкил) малоната в водном кислотном растворе. В следующем аспекте вышеуказанного способа водная кислота означает водную НСl. В следующем аспекте вышеуказанного способа гидрогенизацию осуществляют в водном кислотном растворе. В следующем аспекте вышеуказанного способа водная кислота означает водную НСl. В следующем аспекте вышеуказанного способа N-защиту осуществляют в водном растворе основания. В следующем аспекте вышеуказанного способа P1 обозначает бензилоксикарбонил. В следующем аспекте вышеуказанного способа(а) нагревают раствор 2-пиридилэтил-ди(низший алкил)малоната в водной кислоте, получая раствор пиридилкислоты в водной кислоте;(b) к раствору пиридилкислоты добавляют катализатор и смесь обрабатывают водородом,получая смесь катализатора и пиперидинкислоты;(c) отделяют катализатор от смеси, получая водный раствор пиперидинкислоты, и(d) к водному раствору добавляют основание и N-защищающий агент, получая Nзащищенную пиперидинкислоту. В следующем аспекте вышеуказанного способа водная кислота означает водную НСl. В следующем аспекте вышеуказанного способа R4 обозначает Н. В следующем аспекте вышеуказанного способа P1 обозначает бензилоксикарбонил. Следующий аспект данного изобретения касается способа получения псевдотетрапептида формулы или его соли или пролекарства, где m обозначает 3; n обозначает 2-6; В обозначает алкил; р обозначает 1 или 2; J обозначает циклогексилметил и L обозначает OR1 или NR1R2, где R1 и где m обозначает 3 и P1 обозначает Н или азотзащитную группу; Q2 обозначает Н или карбоксизащитную группу и J обозначает фенилметил;(b) необязательное удаление азотзащитной группы или карбоксизащитной группы и(c) необязательное превращение псевдотетрапептида в соль или пролекарство. В следующем аспекте вышеуказанного способа восстановление осуществляют каталитической гидрогенизацией. В следующем аспекте вышеуказанного способа где m обозначает 3 и P1 обозначает азотзащитную группу; n обозначает 3; р обозначает 1 и Q2 обозначает карбоксизащитную группу. В следующем аспекте вышеуказанного способа P1 обозначает неустойчивую к гидрированию азотзащитную группу и Q2 обозначает неустойчивую к гидрированию карбоксизащитную группу. В следующем аспекте вышеуказанного способа действие каталитического восстановления состоит в одновременном восстановлении и удалении P1 и Q2. В следующем аспекте вышеуказанного способа обозначает пиридил и Q2 обозначает карбоксизащитную группу. В следующем аспекте вышеуказанного способа Q2 обозначает неустойчивую к гидрированию карбоксизащитную группу. В следующем аспекте вышеуказанного способа действие каталитической гидрогенизации состоит в одновременном восстановлении и удалении Q2. Следующий аспект данного изобретения касается псевдотетрапептида формулы где или его соли или пролекарства, где обозначает пиридил или обозначает пиридил илиR1 и R2 независимо обозначают -Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил,алкилциклоалкилалкил. Следующий аспект данного изобретения касается вышеуказанного псевдотетрапептида,где где m обозначает 3; P1 обозначает азотзащитную группу; n обозначает 3; р обозначает 1 и Q2 обозначает неустойчивую к гидрированию карбоксизащитную группу. Следующий аспект данного изобретения касается псевдодипептида формулы или его соли, где P1 обозначает Н или азотзащитную группу; В обозначает алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоaлкилалкил, арил, аралкил, алкиларил или алкиларалкил и R4 обозначает Н или низший алкил. Следующий аспект данного изобретения касается вышеуказанного псевдодипептида, гдеP1 обозначает азотзащитную группу; В обозначает алкил и R1 обозначает Н. Следующий аспект данного изобретения касается вышеуказанного псевдодипептида, гдеP1 обозначает бензилоксикарбонил и В обозначает этил. Данное изобретение иллюстрируется последующими схемами и наглядными примерами, приведенными в целях иллюстрации, но не в порядке ограничения объема изобретения. Подразумевается, что нижеперечисленные сокращения, как они использованы дальше,имеют следующие значения: ВОС (третбутилоксикарбонил), CBZ или Z (бензилоксикарбонил), Gly (глицин), Asp (аспарагиновая кислота), Obzl (бензилокси), TFA (трифторуксусная кислота), Cha (-циклогексилаланин),EtOAc (этилацетат), DMF (диметилформамид),DCC (дициклогексилкарбодиимид), НОВТ (гидроксибензотриазол), TBTU (2-1 Н-бензотриазол 1-ил)-1,1,3,3-тетраметилуронийтетрафторборат),DI (деионизованная вода), PNP (п-нитрофенол), 002724(дициклогексилмочевина), NММ (N-метилморфолин) и МТВЕ (метил-трет-бутиловый эфир). Как показано на схеме 1, настоящее изобретение включает реакцию сочетания азагетероциклилпсевдодипептида формулы IV с карбоксизамещенным дипептидом формулы V с образованием псевдотетрапептида формулы I. Имеется в виду, что в схеме I: В, J, К, L, т, m, q иQ2 имеют вышеуказанные значения. В ходе получения азагетероциклилпсевдодипептидов формулы I или их промежуточных соединений может быть желательно или необходимо предотвратить перекрестные реакции между химически активными заместителями,присутствующими на природных или псевдоаминокислотах или пептидах. Заместители могут быть защищены по известным методикам стандартными защитными группами, которые впоследствии могут быть удалены или сохранены, по мере необходимости, что приводит к желаемым продуктам или промежуточным соединениям (см., к примеру. Green, "ProtectiveGroups in Organic Synthesis", Wiley, New York 1981). Также может быть необходимо или желательно осуществление избирательной защиты или снятия защиты, что позволяет осуществить превращение или удаление существующих заместителей, а также последующую реакцию,приводящую к заданному конечному продукту. Схема 1 Вышеуказанную реакцию сочетания азагетероциклилпсевдодипептида формулы IV с карбоксизамещенным дипептидом формулы V проводят в присутствии органического основания,такого как N-метилморфолин, диизопропилэтиламин или триэтиламин. Подходящие для реакции сочетания растворители включают дихлорметан, толуол, N,N-диметилформамид, этилацетат, ацетонитрил, диметилацетамид, N-метилпирролидон и воду и их смеси. Время реакции сочетания ограничивается интервалом приблизительно от 30 мин до 24 ч, в зависимости от вступающих в реакцию сочетания дипептида и псевдодипептида, активирующего агента, растворителя и температуры. Сочетание осуществляется при температуре приблизительно от-10 до 50 С предпочтительно при температуре окружающей среды. Карбоксильную группу соединения IV предпочтительно активируют с помощью соответствующего активирующего агента. Характерные примеры активирующих агентов включают изопропилхлорформиат в(НОВТ), бис(2-оксо-3-оксазолидинил)-фосфонийхлорид (ВОР-С 1) в присутствии триэтиламина, 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийтетрафторборат (TBTU) в присутствии диизопропилэтиламина, N-гидроксисукцинимид в присутствии N,N'-дициклогексилкарбодиимида (DCC) и тому подобные. Эту активацию предпочтительно осуществляют при температуре приблизительно от 0 до 10 С за период времени приблизительно от 5 мин до 5 ч. В случаях, когда соединение с ацилактивированной группой стабильно, оно может быть получено и выделено заранее для использования в реакции сочетания. Ацилактивирующие группы, пригодные для получения поддающихся выделению псевдодипептидов формулы IV, где К обозначает ацилактивирующую группу,включают сложные эфиры с гидроксибензотриазолом, N-гидроксисукцинимид, пентафторфенол, п-нитрофенол, симметричные ангидриды, ацилгалогениды и тому подобные. В вышеуказанной реакции сочетания, где К обозначает ОН, азагетероциклилпсевдодипептид формулы IV может быть использован в форме свободной карбоновой кислоты или в форме основной аддитивной соли карбоновой кислоты. Предпочтительные основные аддитивные соли включают соли с аминами, такими как дициклогексиламин и триэтиламин, и соли с металлами, такими как натрий или калий. Особенно предпочтительна основная аддитивная соль с дициклогексиламином. Когда используют основную аддитивную соль, кислоту выделяют в свободном виде in situ перед реакцией сочетания путем взаимодействия с подходящей кислотой. Аналогично, дипептид V может быть использован в виде свободного основания, кислотной аддитивной соли или основной аддитивной соли. Предпочтительны кислотные аддитивные соли. Пригодные кислотные аддитивные соли включают гидрохлорид, гидробромид,трифторацетат, ацетат и тозилат. Особенно предпочтительные кислотные аддитивные соли включают гидрохлорид и трифторацетат. Применение кислотных и основных аддитивных солей особенно удобно, поскольку это дает возможность очистки блок-образующих азагетероциклилпсевдодипептидов и карбоксизамещенных дипептидов IV и V с помощью кислотно-щелочных способов экстракции, а также путем перекристаллизации соли, исключая тем самым необходимость очистки, к примеру, путем хроматографии, и позволяет использовать промежуточные соединения в виде водных растворов, что делает ненужным выделение отдельных промежуточных соединений. В особенно предпочтительном варианте реакции сочетания Q2 обозначает Н. Неожидан 002724 22 ное открытие того, что сочетание протекает при полном отсутствии защиты карбоксильной группы в боковой цепи, дает способу некоторые преимущества. Общее число стадий уменьшается, поскольку исключаются реакции введения и снятия защиты, и, кроме того, непрореагировавшая кислота позволяет проводить выделение и очистку дипептида V и продукта реакции сочетания I способом кислотно-щелочной экстракции или путем перекристаллизации основной аддитивной соли, как указано выше. Псевдотетрапептид формулы I, в котором цикл А является ненасыщенным, может быть превращен в насыщенное производное путем селективного восстановления двойных связей цикла, к примеру, каталитическим гидрированием с применением окиси платины. Любые азотзащитные группы P1 и карбоксизащитные группы Р 2, присутствующие в псевдотетрапептиде I, также могут быть удалены, как описано ниже. Соединения формулы II предпочтительно получают, как показано на схеме 2. На схеме 2,m, n, В, К, J и L имеют вышеуказанные значения; P1 обозначает кислотонеустойчивую Nзащитную группу,такую как третбутилоксикарбонил или неустойчивую к гидрированию N-защитную группу, такую как бензилоксикарбонил; и Q2 обозначает Н, кислотонеустойчивую карбоксизащитную группу, такую как трет-бутил, неустойчивую к основаниям карбоксизащитную группу, такую как метил или этил, неустойчивую к гидрированию карбоксизащитную группу, такую как бензил или карбоксизащитную группу, легко расщепляемую металлами, такими как Zn или Pd(0), например,фенацил. Схема 2 Согласно вышеуказанной схеме 2, псевдодипептид формулы VI вступает в реакцию сочетания с дипептидом V, как описано в приведенной выше схеме 1. Продукт сочетания II очищают перекристаллизацией из органического растворителя или смеси органических растворителей, предпочтительно этилацетата-гептана,или путем осаждения основной аддитивной соли. Предпочтительной основной аддитивной солью является соль с дициклогексиламином. Образование соли обычно осуществляют в ин 23 тервале температур приблизительно от температуры окружающей среды до 75 С. Подходящие для получения соли растворители включают спирты, сложные эфиры и полярные апротонные растворители, в смеси с водой или в отсутствии воды. Предпочтительной системой растворителей для получения соли является ацетонитрил-вода. После сочетания VI и V с получением II,азотзащитную группу P1 и, если присутствует,карбоксизащитную группу Р 2 удаляют последовательно или за один прием, получая тетрапептид или псевдопептид VII. Кислотонеустойчивые защитные группы предпочтительно удаляют с помощью трифторуксусной кислоты. Неустойчивые к гидрогенизации защитные группы предпочтительно удаляют гидрогенизацией над палладием на углероде. Неустойчивые к металлам защитные группы удаляют обработкой Zn или Pd(0). Растворителями при реакции гидрирования предпочтительно являются низшие спирты, такие как метанол, этанол или пропанол, или спирто-водные смеси, такие как этанол-вода или пропанол-вода. Псевдотетраептид VII очищают перекристаллизацией из низших спиртов или низшего спирта и воды,предпочтительно из метанола или этанола, или смесей метанола с водой или этанола с водой. Получение псевдотетрапептидов формулыIII по способу настоящего изобретения представлено схемой 3. На схеме 3 P1 обозначает неустойчивую к гидрированию N-защитную группу; Q2 обозначает Н или неустойчивую к гидрированию карбоксизащитную группу, В обозначает алкил; J обозначает алкил, циклоалкил или циклоалкилалкил и L обозначает OR1 или NR1R2, где R1 и R2 независимо обозначают Согласно вышеуказанной схеме 3, псевдодипептид формулы VIII активируют и подвергают реакции сочетания с дипептидом IX, как указано выше на схеме 1. Предпочтительные активирующие агенты включают 2-(1 Нбензотриазол-1-ил)-1,1,3,3-тетраметилуронийтетрафторборат (TBTU) в присутствии диизопропилэтиламина и N-гидроксисукцинимид в присутствии N,N'-дициклогексилкарбодиимида(DCC). Активацию предпочтительно выполняют при температуре приблизительно от 0 до 10 С за период времени приблизительно от 5 мин до 5 ч. В реакции сочетания, указанной в приведенной выше схеме 3, псевдодипептид VIII предпочтительно используют в виде основной аддитивной соли. Предпочтительные основные аддитивные соли включают соли с аминами,такими как дициклогексиламин и триэтиламин,и соли с металлами, такими как натрий или калий. Особенно предпочтительна основная аддитивная соль с дициклогексиламином. Дипептид IX предпочтительно используют в виде кислотной аддитивной соли. Подходящие кислотные аддитивные соли включают гидрохлорид, гидробромид, трифторацетат, ацетат и тозилат. Особенно предпочтительными кислотными аддитивными солями являются гидрохлорид и трифторацетат. Продукт сочетания III очищают перекристаллизацией из органического растворителя или смеси органических растворителей, предпочтительно этилацетата-гептана, или путем осаждения основной аддитивной соли. Предпочтительной основной аддитивной солью является соль с дициклогексиламином. Образование соли обычно проводят при температурном интервале приблизительно от температуры окружающей среды до 75 С. Подходящие для получения соли растворители включают спирты,простые эфиры, сложные эфиры и полярные апротонные растворители в смеси с водой или в отсутствии воды. Предпочтительной системой растворителей для получения соли является смесь ацетонитрила-воды. В особенно предпочтительном варианте реакции сочетания по схеме 3 Q2 обозначает Н. После сочетания псевдодипептида VIII и дипептида IX с получением продукта III, неустойчивые к гидрированию защитные группы P1 и Q2 удаляют за один прием гидрированием над палладием на углероде. Растворителями для реакции гидрирования предпочтительно являются спирты, такие как метанол или этанол, или спиртово-водные смеси, такие как этанол-вода. Тетрапептид или псевдопептид Х очищают перекристаллизацией из низшего спирта или смеси низшего спирта-воды, предпочтительно из метанола или этанола, или смеси метанола-воды или этанола-воды. Получение типичного псевдодипептидаVIII, где P1 обозначает бензилоксикарбонил и В обозначает этил, кратко описывается схемой 4. Очевидно, что синтетический подход, описанный в схеме 4, легко распространить на псевдодипептиды формулы VIII, где P1 и В соответственно отличаются от бензилоксикарбонила и этила. Согласно вышеуказанной схеме 4, диэтиловый эфир 2-(4-пиридил)этилмалоновой кислоты (XI, полученный, к примеру, конденсацией диэтилмалоната с 4-винилпиридином) декарбоксилируют, нагревая в водной кислоте и получая 4-(4-пиридил)бутановую кислоту XII. Затем пиридиновый цикл восстанавливают каталитической гидрогенизацией, получая 4(пиперидин-4-ил)бутановую кислотуXIII. Предпочтительными являются платиновые катализаторы, такие как платина на углероде,окись платины (IV) и платина на алюминии. Платина на алюминии особенно предпочтительна, так как катализатор легко может быть регенерирован. Гидрогенизацию осуществляют в спирте, водном спирте или воде при давлении водорода приблизительно от атмосферного давления до 5 бар и при температуре в интервале приблизительно от комнатной до 70 С. При повышенном давлении водорода могут быть использованы более низкие температуры. Затем защищают N атом пиперидила, к примеру, по реакции XIII с водным основанием и бензилхлорформиатом или бензил-N-сукцинимидилкарбонатом, получая XIV. Реакция сочетанияN-этилглицина и последующее омыление сложного эфира водным алкоголятом дают VIII. Сочетание обычно выполняют, активируя карбоксильную группу соединения XIV путем превращения в хлорангидрид кислоты и последующим добавлением N-этилглицина или этилового эфира N-этилглицина в присутствии основания. Могут также использоваться кислотные аддитивные соли, предпочтительно трифторацетат,N-этилглицина или этилового эфира Nэтилглицина. Подходящие основания включают амины, такие как диизопропилэтиламин или водный алкоголят. По наиболее предпочтительному способу получения VIII, диэтиловый эфир 2-(4 пиридил)этилмалоновой кислоты поглощают водной кислотой, предпочтительно НСl, и декарбоксилируют, нагревая до температуры кипения с обратным холодильником. Затем к полученному раствору 4-(4-пиридил)бутановой кислоты XIII добавляют вышеуказанный катализатор и смесь подвергают каталитической гидрогенизации, получая водный раствор 4-(4 002724 26 пиперидил)бутановой кислоты XIV. После чего катализатор отфильтровывают, кислотный раствор XIV делают щелочным с помощью водного алкоголята и вводят вышеуказанную бензилоксикарбонилзащитную группу. Затем 1[(фенилметокси)карбонил]-4-пиперидинбутановую кислоту экстрагируют органическим растворителем и конденсируют с вышеуказаннымN-этилглицином, получая VIII. Вышеупомянутый способ особенно полезен, поскольку позволяет избежать выделения промежуточных продуктов и включает только одну стадию очистки. Различные промежуточные продукты переносят из одной реакционной стадии в другую в виде водных или органических растворов, тем самым, снижая стоимость и повышая эффективность производства. Получение дипептида формулы IX, где Q2 обозначает Н, J обозначает циклогексилметил иL обозначает NH2, кратко описывается схемой 5. Основные особенности способа, описанного схемой 5, включают не описанную прежде защиту аспартама кислотонеустойчивой или неустойчивой к гидрированию защитной группой Q3 с последующим мягким амидированием аммиаком, протекающим с сохранением стереохимии. Неожиданное ускорение реакции амидирования происходит, когда реакцию проводят в смеси растворителей из метанола-этиленгликоля. Схема 5 В соответствии со схемой 5 аспартам защищается мобильной кислотной N-защитной группой, предпочтительно бутилоксикарбонилом, или неустойчивой к гидрированию защитной группой, предпочтительно бензилоксикарбонилом. Трет-бутилоксикарбонильную защитную группу вводят по реакции аспартама с дитрет-бутилдикарбонатом в присутствии амина,предпочтительно триэтиламина, алкоголята,предпочтительно гидроокиси лития, или карбоната. Защиту обычно осуществляют приблизительно при интервале температур от температуры окружающей среды до 50 С в растворителе из низшего спирта, таком как метанол, или в смеси растворителей из низшего спиртагликоля, такой как метанол-этиленгликоль. 27 Затем защищенный аспартам амидируют,обрабатывая раствор защищенного аспартама газообразным аммиаком в спирте, что дает XVI. Подходящие для амидирования спирты включают низшие спирты, гликоли или их смеси. Предпочтительными спиртами являются метанол, этиленгликоль и смеси метанолаэтиленгликоля. Амидирование осуществляют приблизительно при интервале температур от температуры окружающей среды до 65 С за время приблизительно от 6 ч до 2 дней. Защищенный аспартам может быть выделен и очищен перед амидированием или предпочтительно амидирован путем пропускания газообразного аммиака через полученную на стадии защиты реакционную смесь. Бензольный цикл соединения XVI затем восстанавливают каталитической гидрогенизацией, получая XVII. Предпочтительны платиновые катализаторы, такие как окись платины(IV) и платина на алюминии. Платина на алюминии является особенно предпочтительной в качестве катализатора, поскольку катализатор легко может быть регенерирован. Гидрогенизацию проводят в низшем спирте, предпочтительно бутаноле, или C1-10-насыщенной органической кислоте, предпочтительно уксусной кислоте. Гидрогенизацию осуществляют при давлении водорода приблизительно от 2 до 5 бар и в интервале температур приблизительно от 40 до 80 С. Можно использовать более низкие температуры, если повысить давление водорода. Когда Q3 обозначает неустойчивую к гидрированию защитную группу, снятие защиты с целью получения IX выполняют одновременно с вышеуказанным восстановлением бензольного кольца. Когда Q3 обозначает кислотонеустойчивую защитную группу, такую как третбутилоксикарбонил, снятие защиты предпочтительно производят, обрабатывая раствор соединения XVII газообразным НСl. В особенно предпочтительном варианте кислотонеустойчивую защитную группу удаляют введением газообразного НСl в полученную на стадии гидрирования реакционную смесь. Для специалистов в данной области совершенно очевидно, что настоящее изобретение полностью пригодно для получения объектов,составляющих предмет изобретения, и достижения целей и преимуществ, указанных выше, а также не перечисленных здесь, но вытекающих из них. Указанные здесь соединения, композиции и способы приведены в качестве иллюстрации предпочтительных вариантов воплощения,и предназначены служить характерными примерами, а не ограничивающими объем настоящего изобретения. Пример 1. Получение N-[N-этил-N-[1 оксо-4-(4-пиперидинил)бутил]глицил]-(L)-аспартил-3-циклогексил-(L)-аланинамида. 28 Способ А. Стадия 1. Фенилметиловый эфир N-[Nэтил-N-[1-оксо-4-[1-[(фенилметокси)карбонил]4-пиперидинил]бутил]глицил-(L)аспартил-3 циклогексил-(L)-аланинамида. К перемешиваемому с помощью магнитной мешалки при 0-5 С раствору 25,9 г (66,4 ммоль) N-этил-N-[1-оксо-4-[1-[(фенилметокси) карбонил]-4-пиперидинил]бутил]глицина в 70 мл диметилформамида добавляют 20,9 г (65 ммоль) 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийтетрафторбората, а последующим добавлением по каплям 17,6 г (130 ммоль) диизопропилэтиламина. Полученный гомогенный раствор выливают в перемешиваемую смесь 31,8 г (65 ммоль) трифторацетата (L)-аспартил-3-циклогексил-(L)-аланинамидфенилметилового эфира в 30 мл диметилформамида при 0-5 С. Добавляют диизопропилэтиламин до получения нейтрального - слабо щелочного рН. Полученную смесь убирают из охлаждающей бани и перемешивают при 23 С в течение ночи. Смесь разбавляют водой и экстрагируют 4 порциями этилацетата. Объединенные этилацетатные экстракты промывают 0,5 н. водной уксусной кислотой (3 х), раствором соли, насыщенным водным бикарбонатом натрия (3 х) и раствором соли (2 х), сушат над сульфатом магния,фильтруют и концентрируют в вакууме, получая масло, затвердевающее при стоянии в светлокоричневое твердое вещество (50,97 г, 90,9% чистоты). Порцию 36 г твердого вещества перекристаллизовывают из этилацетата/гептана, получая указанное в заглавии соединение [24 г,химическая чистота 95% (чистый для анализа)].N-[N-Этил-N-[1-оксо-4-(4 пиперидинил)бутил]глицил]-(L)аспартил-3 циклогексил-(L)-аланинамид. Фенилметиловый эфир N-[N-этил-N-[1 оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицил-(L)аспартил-3-циклогексил-(L)-аланинамида, полученный как на стадии 1, растворяют в метаноле и добавляют 10% палладий/углерод. Смесь встряхивают в атмосфере водорода при 50 фунт/кв.дюйм в течение порядка 18 ч. Смесь фильтруют через слой целита и фильтрат упаривают в вакууме,получая N-[N-этил-N-[1-оксо-4-(4-пиперидинил) бутил]глицил-(L)аспартил-3-циклогексил(L)-аланинамид. бутил]глицил]-(L)аспартил-3-циклогексил(L)-аланинамид. В стеклянный трубчатый реактор на 2300 л при температуре порядка 30 С к суспензии дициклогексиламинаN-этил-N-[1-оксо-4-[1[(фенилметокси)карбонил]-4-пиперидинил] бутил]глицина, (112,5 кг) в толуоле (550 кг) и воде (570 кг) добавляют водную серную кислоту(210 кг). После декантации толуольный раствор промывают водным раствором серной кислоты(210 кг) и водой (390 кг). Смесь декантируют и органическую фазу промывают водой (390 кг) и сушат перегонкой до содержания остаточной воды ниже 0,5%. К толуольному раствору добавляют ацетонитрил (220 кг) и Nгидроксисукцинимид (27 кг). Полученную суспензию охлаждают приблизительно до 5 С в атмосфере азота и добавляют за 1 ч раствор дициклогексилкарбодиимида (45 кг) в толуоле (35 кг), дополнительно перемешивают образовавшуюся смесь в течение 5-6 ч. К смеси добавляют(L)аспартил-3-циклогексил-(L)-аланинамид гидрохлорид (75 кг) и триэтиламин (80 кг) и перемешивание продолжают около 2 ч. Реакционную смесь разбавляют водой (370 кг). Полученную суспензию фильтруют, плотный осадок на фильтре промывают водой (30 кг) и объединенные фильтраты переносят в реактор из нержавеющей стали на 2300 литров. К раствору добавляют этилацетат (347 кг). После декантации органической фазы водную фазу подкисливают соляной кислотой (515 кг) и экстрагируют этилацетатом (347 кг). Органическую фазу дважды промывают 20% водным хлористым аммонием (109 кг) и концентрируют в вакууме. Добавляют ацетонитрил (1560 кг) и воду (45 кг),раствор нагревают приблизительно до 75 С и добавляют за 1 ч дициклогексиламин (35 кг). В раствор вносят затравку, N-[N-этил-N-[1-оксо-4[1-[(фенилметокси)карбонил]-4-пиперидинил] бутил]глицил]-(L)аспартил-3-циклогексил(L)-аланинамид и выдерживают его в течение одного часа при 75 С. Затем суспензию охлаждают до 20 С в течение 6 ч и фильтруют. Плотный осадок на фильтре трижды промывают ацетонитрилом (100 кг) и сушат при пониженном давлении при температуре порядка 40 С, что дает указанное в заглавии соединение (125 кг) в виде соли с дициклогексиламином. Стадия 2. N-[N-Этил-N-[1-оксо-4-(4-пиперидинил)бутил]глицил]-(L)аспартил-3 циклогексил-(L)-аланинамид. После продувки гидрогенизатора азотом суспензию N-[N-этил-N-[1-оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицил(L)аспартил-3-циклогексил-(L)-аланинамиддициклогексиламина (124 кг) и влажного палладия на углероде (2 кг, влажность 50%, 5%) в смеси абсолютного этанола (440 кг) и очищенной воды (42 кг) гидрируют приблизительно при 25 С при давлении водорода 2 бар в течение 30 1 ч. После продувки азотом катализатор фильтруют и промывают смесью абсолютного этанола(65 кг) и очищенной воды (5 кг). Фильтрат нагревают приблизительно до 60 С, добавляют ацетон (425 кг) и вносят в смесь в качестве затравки N-[N-этил-N-[1-оксо-4-(4-пиперидинил) бутил]глицил]-(L)аспартил-3-циклогексил(L)-аланинамид (1,1 кг). Суспензию охлаждают до 20 С и фильтруют на встряхиваемом фильтре-осушителе. Плотный осадок на фильтре промывают абсолютным этанолом (100 кг) и сушат в вакууме при температуре порядка 40C, что дает указанное в заглавии соединение (61 кг) в виде белого кристаллического вещества. Способ С. Стадия 1. Фенилметиловый эфир N-этилN-[1-оксо-4-(4-пиридинил)бутил]глицил-(L)-аспартил-(L)-фенилаланинамида. В колбе на 1 л в атмосфере N2 готовят раствор 15,1 г (0,06 моль) N-этил-N-[1-оксо-4-(4 пиридинил)бутил]глицина в 200 мл дихлорметана и охлаждают до 1 С. Получают раствор 6,95 мл (0,055 моль) трет-бутилхлорформиата в 25 мл дихлорметана, охлаждают до 1 С и добавляют по каплям за 45 мин, поддерживая температуру смеси при 1 С. Смесь перемешивают при 1 С в течение 3 ч. В отдельной колбе на 800 мл растворяют 20 г (0,0498 моль) моно(гидрохлорида) фенилметилового эфира (L)-аспартил-(L)-фенилаланинамида в 200 мл дихлорметана и смесь обрабатывают, добавляя по каплям 15,8 мл (0,1 моль) триэтиламина, получают раствор, который добавляют при 1 С за 1 ч к полученному ранее ангидриду. Смесь перемешивают при -2 С в течение 1 ч, затем промывают 250 мл воды, 250 мл насыщенного водного бикарбоната натрия, 250 мл 0,14 М водной соляной кислоты, затем 250 мл воды. Органическую фазу контролируют с помощью ЖХВР на наличие непрореагировавшего исходного вещества и повторяют цикл промывки до тех пор, пока не перестанет наблюдаться дальнейшее уменьшение соответствующих ЖХВР-пиков. Органический слой сушат над сульфатом магния, фильтруют, затем концентрируют в вакууме, оставшееся масло помещают в вакуум для максимального удаления растворителя, получают 23,5 г [выход 79,3% химическая чистота 96,8% (чистый для анализа)] фенилметилового эфира Nэтил-N-[1-оксо-4-(4-пиридинил)бутил]глицил(L)аспартил-(L)-фенилаланинамида. Стадия 2. N-Этил-N-[1-оксо-4-(4-пиперидинил)бутил]глицил-(L)аспартил-3-циклогексил-(L)-аланинамид. К раствору 76 мг фенилметилового эфира(0,126 ммоль) в 1 мл смеси 9:1 изопропанола/воды добавляют 6 мг Pt2O-H2O (79-84% Pt),затем 1,6 мкл 1 н. водной соляной кислоты. Смесь перемешивают и подвергают воздейст 31 вию водорода при 4 бар в течение 5 ч 40 мин при 23 С, после чего в течение 1 ч 45 мин при 60 С. Смесь фильтруют и фильтрат анализируют ЖХВР, чтобы убедиться в 100% расходовании исходного вещества, получают N-этил-N-[1 оксо-4-(4-пиперидинил)бутил]глицил-(L)-аспартил-3-циклогексил-(L)-аланинамид со следами циклогексилметилового эфира N-этил-N[1-оксо-4-(4-пиридинил)бутил]глицил-(L)-аспартил-(L)-фенилаланинамида. Способ D. Стадия 1. Фенилметиловый эфир N-этилN-[1-оксо-4-[1-[(фенилметокси)карбонил]-4 пиперидинил]бутил]глицил]-(L)аспартил-(L)фенилаланинамида. К раствору 11,1 г (28,4 ммоль) N-этил-N[1-оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицина в 44 мл этилацетата добавляют 6,13 г (28,7 ммоль) твердого дициклогексилкарбодиимида, поддерживая температуру между 25 и 33C, за это время образуется осадок (дициклогексилмочевина). В отдельной колбе суспензию 10,97 г (27 ммоль) моно(гидрохлорида)фенилметилового эфира (L)аспартил-(L)-фенилаланинамида в 88 мл этилацетата обрабатывают 4,2 мл (29,7 ммоль) триэтиламина. Смесь перемешивают в течение 15 мин, обеспечивая одновременное растворение дипептида и осаждение триэтиламингидрохлорида. Суспензию добавляют к суспензии активированного N-этил-N-[1-оксо-4-[1[(фенилметокси)карбонил]-4-пиперидинил] бутил]глицина, полученной ранее, при температуре между 27 и 29 С. Смесь перемешивают в течение 1 ч, за это время суспензия становится густой. Добавляют еще 100 мл этилацетата и смесь перемешивают дополнительно в течение 1 ч. Добавляют порцию воды в 60 мл, получая смесь из двух легко смешивающихся жидких фаз и суспендированной твердой дициклогексилмочевины,которую отфильтровывают. Фильтрат промывают 60 мл насыщенного водного бикарбоната натрия, 60 мл 1 н. водной соляной кислоты и 60 мл насыщенного водного хлористого натрия, затем органическую фазу сушат над сульфатом натрия. Смесь фильтруют и концентрируют в вакууме, остаток нагревают до 50 С при 1 мм Нg, получая 17,6 г фенилметилового эфира N-этил-N-[1-оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицил-(L)аспартил-(L)-фенилаланинамида. Образец в 400 мг загружают для предварительной промывки (2 н. водной соляной кислотой, водой,затем 1:1 водой/этанолом) на Dowex 50WX2 (10 г) и собирают, элюируя 50 мл смеси 70/30 этанола/воды, потом для предварительной промывки (2 н. гидроокисью натрия, водой, затем 1:1 этанолом/водой) на Amberlyst A26 (10 г), элюируя 50 мл смеси 70:30 этанола/воды. Содержащий продукт элюат концентрируют в вакууме,осуществляя впоследствии дальнейшее удале 002724 32 ние растворителя в вакууме при 50 С (5 мм Hg),получают 350 мг фенилметилового эфира Nэтил-N-[1-оксо-4-[1-[(фенилметокси)карбонил]4-пиперидинил]бутил]глицил]-(L)аспартил(L)-фенилаланинамида. Стадия 2. N-Этил-N-[1-оксо-4-(4-пиперидинил)бутил]глицил-(L)аспартил-3-циклогексил-(L)-аланинамид. В сухую 2 мл сапфировую ампулу для ЯМР вносят 25 мг (0,03 ммоль) фенилметилового эфира N-этил-N-[1-оксо-4-[1-[(фенилметокси) карбонил]-4-пиперидинил]бутил]глицил-(L)-аспартил-(L)-фенилаланинамида, 12 мг Pt2OH2O (79-84% Pt) и 1 мл смеси уксусной кислоты/2 н. водной соляной кислоты 85/12, в отношении объемов. Смесь перемешивают при давлении водорода 4 бар в течение 5 ч при температуре окружающей среды. Затем смесь фильтруют, используя фильтр Millipore Milex. ЖХВР-анализ фильтрата показывает 100% Nэтил-N-[1-оксо-4-(4-пиперидинил)бутил]глицил-(L)аспартил-3-циклогексил-(L)-аланинамида. Способ Е. В колбу на 100 мл, снабженную магнитной мешалкой, помещают 1,12 г (1,51 ммоль) фенилметилового эфира N-этил-N-[1-оксо-4-[1[(фенилметокси)карбонил]-4-пиперидинил] бутил]глицил-(L)аспартил-(L)-фенилаланинамида, 107 мг 5% Pd/C и 10 мл смеси 9:12 пропанола/воды. Колбу продувают аргоном при 40 С, затем смесь нагревают до 50 С, энергично встряхивая, и колбу продувают несколько раз водородом. Смесь встряхивают при 50 С в атмосфере водорода при давлении 1 бар в течение 5 ч, затем охлаждают до температуры окружающей среды, фильтруют и фильтрат концентрируют в вакууме, получая 713 мг (выход 90%)N-этил-N-[1-oксo-4-(4-пиперидинил)бутил] глицил-(L)аспартил-(L)-фенилаланинамида. Порцию в 50 мг помещают вместе с 50 мгRh/Al2O3 (Engelhard) и 1 мл уксусной кислоты в стеклянную ампулу и смесь встряхивают в течение 17 ч при 80 С в атмосфере водорода при давлении 4 бар. Анализ ЖХВР по площади пика показывает 100% N-этил-N-[1-оксо-4-(4-пиперидинил)бутил]глицил-(L)аспартил-3-циклогексил-(L)-аланинамида. Способ F.N-Этил-N-[1-оксо-4-(4-пиперидинил) бутил]глицил-(L)аспартил-3-циклогексил(L)-аланинамид получают по методике вышеуказанного способа А, за тем исключением, что заменяют N-этил-N-[1-оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинилметилен]пропил] глицин на N-этил-N-[1-оксо-4-[1-[(фенилметоксикарбонил)-4-пиперидинилбутил]глицин. Пример 2. Получение N-[N-этил-N-[1 оксо-4-пиперидинил]бутил]глицил-(L)-аспартил-3-циклогексил-(L)-аланина. 33 Стадия 1. Бисфенилметиловый эфир N-[Nэтил-N-[1-оксо-4-[1-[(фенилметокси)карбонил]4-пиперидинил]бутил]глицил-(L)аспартил-3 циклогексил-(L)-аланина. К перемешиваемой суспензии N-этил-N-[1 оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицина (0,858 г, 1,5 ммоль) в 4 г диметилформамида при 5C добавляют одной порцией 2-(1 Н-бензотриазол-1-ил)-1,1,3,3 тетраметилуронийтетрафторборат (0,482 г, 1,5 ммоль) и смесь перемешивают в течение 3 мин при 5 С. Добавляют диизопропилэтиламин(0,482 г, 1,5 ммоль) и полученную гетерогенную смесь добавляют с помощью пипетки Пастера к раствору 0,755 г (1,5 ммоль) моно(гидрохлорида) бисфенилметилового эфира (L)-аспартил-3-циклогексил-(L)-аланина в 4 г диметилформамида. Дополнительно 1 г диметилформамида используют для споласкивания и еще одну порцию, 0,16 г (0,5 ммоль) диизопропилэтиламина используют для того, чтобы сделать смесь слабо щелочной. Охлаждающую баню убирают и суспензию перемешивают при температуре окружающей среды в течение 20 ч. Полученную оранжевую, гетерогенную смесь распределяют между метил-трет-бутиловым эфиром и Н 2 О. Водную фазу экстрагируют метил-трет-бутиловым эфиром. Объединенные органические фазы последовательно промывают 1 н. водным НСl, водой, насыщенным воднымNаНСО 3 и раствором соли, сушат над MgSO4 и концентрируют в вакууме, получая бисфенилметиловый эфир N-[N-этил-N-[1-oкco-4-[1[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицил]-(L)аспартил-3-циклогексил-(L)аланина (1,21 г) в виде смолы янтарного цвета.Pd/C (Degussa тип Е 101 NO/W, 50% воды по весу) в колбу вибратора Парра на 500 мл. Смесь встряхивают в течение ночи при 40-50 фунт/кв.дюйм водорода и затем фильтруют. Фильтрат концентрируют, получая N-[N-этилN-[1-оксо-4-пиперидинил]бутил]глицил-(L)-аспартил-3-циклогексил-(L)-аланин (6,47 г) в виде стекловидного твердого вещества. 4-пиперидинил]бутил]глицил]-(L)аспартил 4-циклогексил-2-(L)-аминобутановой кислоты. К перемешиваемому с помощью магнитной мешалки при 0-5 С раствору 26,38 г (44,4 ммоль) N-этил-N-[1-оксо-4-[1-[(фенилметокси) карбонил]-4-пиперидинил]бутил]глицина в 70 мл диметилформамида добавляют 15,7 г (48,8 ммоль) 2-(1 Н-бензотриазол-1-ил)-1,1,3,63 тетраметилуронийтетрафторбората, и затем добавляют по каплям 12,9 г (99,6 ммоль) диизопропилэтиламина. Полученный раствор выливают в суспензию 19 г (48,4 ммоль) моно(трифторацетата) фенилметилового эфира(L)аспартил-4-циклогексил-2-(L)-аминобутановой кислоты в 20 мл диметилформамида при 0-5 С. Полученную смесь убирают из охлаждающей смеси и оставляют перемешиваться при 23 С в течение ночи, что дает гомогенный раствор. Смесь разбавляют водой, что приводит к выделению оранжевого масла. Добавляют метил-трет-бутиловый эфир и воду, удаляют органический слой, а водный слой дважды экстрагируют метил-трет-бутиловым эфиром. Объединенные органические слои дважды промывают 1 н. водной соляной кислотой, дважды 1 н. водной гидроокисью натрия и дважды раствором соли, сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая масло,которое оставляют в глубоком вакууме на два дня. Масло (33,4 г) чистят на силикагеле (80% этилацетат-гептан), получая бисфенилметиловый эфир N-[N-этил-N-[1-оксо-4-[1[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицил](L)аспартил-4-циклогексил-2-(L)-аминобутановой кислоты (27,8 г, 73%). Стадия 2. N-[N-Этил-N-[1-оксо-4-(4-пиперидинил)бутил]глицил]-(L)аспартил-4 циклогексил-2-(L)-аминобутановая кислота. Раствор 1,2 г (1,4 ммоль) бисфенилметилового эфира N-[N-этил-N-[1-окco-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил] глицил]-(L)аспартил-4-циклогексил-2-(L)аминобутановой кислоты, полученного как на стадии 1, в 12 мл диоксана выливают в колбу вибратора Парра на 500 мл, содержащую 83 мг 10% Pd/C в 5 мл воды и 3 мл диоксана. Реакционную смесь встряхивают в течение ночи при температуре окружающей среды (около 23 С) при 45 фунт/кв.дюйм водорода. Смесь фильтруют через слой целита в стеклянной воронке для спекшегося материала и плотный осадок на фильтре промывают смесью диоксана и воды. Раствор лиофилизуют и полученный пушистый белый твердый продукт вновь растворяют в минимальном количестве воды и снова лиофилизуют, получая указанное в заглавии соединение в виде пушистого белого твердого веществаMS (FAB) m/z 539, 561 (M+Na)+. Пример 4. Получение трифторацетата этилового эфира N-этилглицина.(465 ммоль) этанола и 5,17 г (42,3 ммоль) 4 диметиламинопиридина в 600 мл дихлорметана,при 3 С, добавляют 47 г (46,2 ммоль) триэтиламина и впоследствии, порциями, 89,1 г (46,5 ммоль) 3-N,N-димeтиламинопропилэтилкарбодиимид гидрохлорида. Перемешиваемой смеси дают нагреться до температуры окружающей среды и перемешивают в течение ночи. Реакционную смесь промывают водой, насыщенным водным бикарбонатом натрия и раствором соли,сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая этиловый эфирN-этилглицина. Перемешиваемый раствор 30,8 г (113 ммоль) этилового эфира N-[(1,1-диметилэтокси) карбонил]-N-этилглицина в 50 мл дихлорметана при 3 С обрабатывают 200 мл 1:1 (в отношении объемов) трифторуксусной кислотой в дихлорметане. Реакционную смесь перемешивают 2 ч, пока она нагреется до температуры окружающей среды. Реакционную смесь концентрируют в вакууме и оставшееся чрезвычайно подвижное (маловязкое) масло перегоняют в глубоком вакууме, получая заданное соединение (55,2 г), которое используют без дополнительной очистки. Пример 5. Получение N-этилглицина. К смеси этиламина (25 кг) и изопропанола(100 кг) в стекляном трубчатом реакторе на 250 л, при 5 С, в атмосфере азота добавляют раствор глиоксиловой кислоты (25 кг) в изопропаноле (25 кг), за 2-3 ч, и раствор гидрируют в присутствии палладия на углероде (влажность 50%, 5%, 2,5 кг) в течение 3 ч при 50 мбар водорода. Затем реакционную смесь фильтруют и фильтрат концентрируют. К суспензии добавляют изопропанол (80 кг) и твердый продукт фильтруют, дважды промывают изопропанолом(7 кг) и сушат в вакууме приблизительно при 40 С, получая N-этилглицин (21 кг) в виде белого кристаллического вещества. Пример 6. Получение N-этил-N-[1-оксо-4[1-[(фенилметокси)карбонил]-4-пиперидинил] бутил]глицина. Способ А. Стадия 1. 1-[(Фенилметокси)карбонил]-4 пиперидинбутановая кислота. К 100 г (622 ммоль) диэтилмалоната при 25 С добавляют 88 г (272 ммоль) 21% (в отношении веса) этанольного этилата натрия в течение 20 мин, поддерживая при этом температуру 23-25 С. Смесь перемешивают в течение 10 мин и добавляют 26,18 г (248 ммоль) 4 винилпиридина. Смесь нагревают до 85 С и 36 перемешивают 3 ч, а затем перемешивают 15 ч при комнатной температуре. Реакционную смесь концентрируют при 44 С, удаляя этанол,и полученное желтое масло перегоняют в глубоком вакууме для удаления оставшегося растворителя. Масло распределяют между метилтрет-бутиловым эфиром и 2,2 н. водным НСl. pН водной фазы доводят до 6 с помощью 2 н. водной гидроокиси натрия. Слои разделяют и органическую фазу дважды промывают раствором соли. Затем органическую фазу экстрагируют 2,2 н. водным НСl (3 х). Объединенные водные экстракты нагревают с обратным холодильником при температуре кипения в течение ночи. Полученный раствор (225 мл, предположительно 248 ммоль) выливают на 2,5 г гидрата окиси платины в колбу вибратора Парра на 500 мл и встряхивают в течение ночи при 47-55 фунт/кв.дюйм водорода. Смесь фильтруют и фильтрат охлаждают до 10 С и обрабатывают 417 мл водной гидроокиси натрия, поддерживая реакционную температуру, равной или ниже 10 С и доводя рН до 14 (требуется около 40 мин). К перемешиваемой смеси добавляют 100 мл тетрагидрофурана с последующим добавлением в течение 30 мин 68 г (273 ммоль) бензилN-сукцинимидилкарбоната в 150 мл тетрагидрофурана, поддерживая при этом температуру 8 С. Смесь нагревают до температуры окружающей среды и перемешивают в течение ночи. Смесь экстрагируют метил-трет-бутиловым эфиром(3 х),этилацетатом,метил-третбутиловым эфиром, этилацетатом и метил-третбутиловым эфиром. Водную фазу охлаждают до 5 С, подкисливают до рН 1,9, медленно добавляя 27 мл концентрированной соляной кислоты,и экстрагируют метил-трет-бутиловым эфиром(3 х). Объединенные органические слои от экстракции промывают раствором соли, сушат сульфатом магния, фильтруют и концентрируют в вакууме, получая 1-[(фенилметокси)карбонил]-4-пиперидинбутановую кислоту (42 г, 55% выход из винилпиридина).MS (FAB) m/z 306. Стадия 2. Этиловый эфир N-этил-N-[1 оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицина. К перемешиваемому при 3 С раствору 40,7 г (133,3 ммоль) 1-[(фенилметокси)карбонил]-4 пиперидинбутановой кислоты в 667 мл дихлорметана добавляют 0,5 мл диметилформамида и по каплям 66,8 мл 2 М оксалилхлорида в дихлорметане. Во время добавления реакционную смесь выдерживают при 3-4 С, затем перемешивают при этой температуре в течение ночи. Реакционную смесь концентрируют в вакууме. Полученное масло цвета меда и 133,3 ммоль трифторацетата этилового эфира N-этилглицина растворяют в 250 мл дихлорметана при 3 С. К перемешиваемой смеси добавляют 52,84 г (408 ммоль) диизопропилэтиламина. Смеси дают нагреться до температуры окружающей среды, 37 перемешивая в течение 2 ч, к этому времени ЖХВР показывает завершение реакции. Реакционную смесь концентрируют в вакууме и полученное масло растворяют в метил-третбутиловом эфире. Метил-трет-бутилэфирный раствор промывают водой (2 х) и объединенные водные слои экстрагируют метил-третбутиловым эфиром. Объединенные органические растворы промывают 1 н. водной гидроокисью натрия (3 х), раствором соли (2 х), сушат над сульфатом магния и концентрируют в вакууме,получая этиловый эфир N-этил-N-[1-оксо-4-[1[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицина (46,69 г), который используют без дополнительной очистки.MS (FAB) m/z 419 (М+Н)+. Стадия 3. N-Этил-N-[1-оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил] глицин. К раствору 46,7 г (112 ммоль) этилового эфира N-этил-N-[1-оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицина в тетрагидрофуране при температуре окружающей среды добавляют 200 мл 1 н. водной гидроокиси натрия и гетерогенную смесь энергично перемешивают в течение 1 ч. Реакционную смесь концентрируют в вакууме и оставшуюся водную смесь разбавляют водой и экстрагируют метил-трет-бутиловым эфиром (3 х). Водный слой подкисливают до рН 3 с помощью бисульфата калия, что приводит к образованию масла. Смесь экстрагируют, метил-трет-бутиловым эфиром (3 х). Объединенные органические слои промывают раствором соли (2 х), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученное масло перегоняют в глубоком вакууме, что дает N-этил-N-[1-оксо-4-[1[(фенилметокси)карбонил]-4-пиперидинил] бутил]глицин (35,5 г).MS (FAB) 391 (М+Н)+. Способ В. К раствору, при 0 С, 2,1 г (6,88 ммоль) 1[(фенилметокси)карбонил]-4-пиперидинбутановой кислоты в 36 мл дихлорметана добавляют 3,6 мл 2 М оксалилхлорида в дихлорметане и 0,25 мл диметилформамида. Реакционную смесь перемешивают при 0 С в течение 2,5 ч. Реакционную смесь концентрируют в вакууме и остаток дважды азеотропно перегоняют с толуолом. Водный раствор 2,06 г (19,9 ммоль) Nэтилглицина в 18,7 мл воды охлаждают до 5 С и добавляют порциями 3,78 С (35 ммоль) твердого карбоната натрия и затем раствор 6,88 ммоль(допускается неочищенный продукт) хлорангидрида 1-[(фенилметокси)карбонил]-4-пиперидинбутановой кислоты в 8 мл тетрагидрофурана. Смесь нагревают до температуры окружающей среды и перемешивают в течение ночи. Реакционную смесь разбавляют водой и экстрагируют этилацетатом (3 х). Водную фазу подкисливают до рН 2 водным бисульфатом калия и экстрагируют метил-трет-бутиловым эфиром. 38 Органический слой концентрируют в вакууме,получая 2,21 г масла (MS отвечает заданному продукту). Масло очищают, используя препаративную обращенно-фазовую ЖХВР (С-18,2 Х 250 см, размер частицы 15 мк, размер пор 300 ангстрем), с применением водного ацетонитрила с добавкой 0,1%, в отношении объемов,трифторуксусной кислоты, при градиенте 3645% ацетонитрила. Продукт-содержащие фракции объединяют и раствор замораживают и лиофилизуют. Образовавшееся масло поглощают этилацетатом, раствор концентрируют в вакууме и полученное масло выдерживают в глубоком вакууме, получая N-этил-N-[1-оксо-4-[1[(фенилметокси)карбонил]-4-пиперидинил] бутил]глицин (1,01 г, выход 36%).MS (FAB) m/z 391 (М+Н)+. Способ С. Стадия 1. Гидрохлорид диэтилового эфира 2-(4-пиридил)этилмалоновой кислоты. В атмосфере азота смесь 20% этилата натрия в этаноле (263,2 кг) и диэтилмалоната (703 кг) в стеклянном трубчатом реакторе на 1600 л перегоняют при интервале температур от окружающей среды до 108 С. Добавляют 4 винилпиридин (80,5 кг) за 3 ч и реакционную смесь перемешивают 4 ч. Затем реакционную смесь охлаждают до температуры окружающей среды и добавляют воду (358 кг) за период времени порядка 1 ч. pН доводят до 4,5 с помощью 33% водной хлористо-водородной кислоты и водный слой экстрагируют метил-третбутиловым эфиром (216 кг). Затем добавляют воду (357,5 кг) и двухфазную смесь подкисляют до рН 1 с помощью 16,5% водной хлористоводородной кислоты (29,6 кг). Слои разделяют и водный раствор гидрохлорида диэтилового эфира 2-(4-пиридил)этилмалоновой кислоты используют как он есть. Стадия 2. Гидрохдорид 4-(4-пиридил) бутановой кислоты. Полученный на стадии 1 водный раствор гидрохлорида диэтилового эфира 2-(4 пиридил)этилмалоновой кислоты перегоняют при атмосферном давлении и температуре 105 С для удаления метил-трет-бутилового эфира и этанола и добавляют 33% водную хлористо-водородную кислоту (67 кг) за период 50 мин. Реакционную смесь перемешивают около 6 ч при температуре около 105 С и затем концентрируют, получая примерно 530 кг дистиллята. Смеси дают охладиться до 60 С и добавляют уксусную кислоту (591 кг). Перегонку продолжают при пониженном давлении (10 мм Нg),чтобы уровень воды в реакционной смеси достиг 6,6%. К суспензии добавляют ацетон (374 кг) и суспензию охлаждают до 15 С за 3 ч. Суспензию перемешивают при 15 С в течение 1 ч и осадок фильтруют, дважды промывают ацетоном (94 кг) и сушат в вакууме в течение 24 ч при 40 С, получая гидрохлорид 4-(4-пиридил) бутановой кислоты (126 кг). Затем твердый про 39 дукт растворяют в воде (300 кг), получая 30% в отношении веса водный раствор, который используют как он есть в стадии 3. Стадия 3. Гидрохлорид 4-(4-пиперидил) бутановой кислоты. К 30% водному раствору гидрохлорида 4(4-пиридил)бутановой кислоты, полученному на стадии 2 (208,9 кг), и воды (145 кг) добавляют платину на углероде (влажность 50%, 5%, 2,52 кг). Смесь гидрируют приблизительно при 70 С при атмосферном давлении в течение 15 ч и затем оставляют охлаждаться до температуры окружающей среды. Катализатор фильтруют и промывают водой (20 кг). Полученный водный 16% раствор гидрохлорида 4-(4-пиперидил) бутановой кислоты (388,5 кг) используют как он есть в стадии 4. Стадия 4. 1-[(Фенилметокси)карбонил]-4 пиперидинбутановая кислота. Либо твердый гидрохлорид 4-(4-пиперидил)масляной кислоты (124 кг) растворяют в воде (600 кг), либо 16% водный раствор гидрохлорида 4-(4-пиперидил)масляной кислоты, полученный на стадии 3 (777 кг), разбавляют водной гидроокисью натрия (289 кг) в стеклянном трубчатом реакторе на 1600 л, охлажденном до 5 С. Добавляют бензилхлорформиат (112 кг) за 2-3 ч и раствор нагревают приблизительно до 25 С. Реакционную смесь экстрагируют метилтрет-бутиловым эфиром (476 кг). Органический слой промывают водой (240 кг) и сушат азеотропной перегонкой при атмосферном давлении в атмосфере азота. Полученный раствор 1[(фенилметокси)карбонил]-4-пиперидинбутановой кислоты в толуоле используют как есть на стадии 5. Вес продукта в растворе 179 кг. Количество: около 35%, в отношении веса, в толуоле. Стадия 5. Дициклогексиламин N-этил-N[1-оксо-4-[1-[(фенилметокси)карбонил]-4-пиперидинил]бутил]глицина. К раствору 1-[(фенилметокси)карбонил]-4 пиперидинбутановой кислоты в толуоле, полученному на стадии 4 (253 кг), в атмосфере азота добавляют тионилхлорид (42 кг). Реакционную смесь разбавляют метил-трет-бутиловым эфиром (85 кг) и перемешивают около 24 ч приблизительно при 15 С в стеклянном трубчатом реакторе на 630 л. Этот раствор добавляют приблизительно за 4-5 ч к двухфазной смеси воды(177 кг), 30% водной гидроокиси натрия (227 кг), N-этилглицина (36 кг) и метил-третбутилового эфира (65 кг) в стеклянном трубчатом реакторе на 1600 л и смесь перемешивают около 30 мин. Затем двухфазную смесь разбавляют водой (100 кг) и подкисляют до рН 6,5 хлористо-водородной кислотой. Водную фазу промывают метил-трет-бутиловым эфиром (65 кг) и добавляют к водной фазе этилацетат (157 кг). Двухфазную смесь подкисляют до рН 4 с помощью хлористо-водородной кислоты (84 кг). Слои разделяют и добавляют к органической фазе этилацетат (385 кг). Органическую фазу 40 нагревают приблизительно до 60 С и добавляют раствор дициклогексиламина (63 кг) в этилацетате (230 кг). В смесь вводят затравку и охлаждают приблизительно до 10 С. Твердый продукт фильтруют, дважды промывают этилацетатом(150 кг) и сушат в вакууме приблизительно при 40 С, получая указанное в заглавии соединение(139 кг) в виде белого кристаллического вещества. Пример 7. Получение N-этил-N-[1-оксо-4(4-пиридинил)бутил]глицина. Стадия 1. Моно(гидрохлорид)фенилметилового эфира N-этилглицина. К 250 мл 2 М раствора этиламина в тетрагидрофуране за 0,5 ч добавляют 47,2 г (0,2 моль) бензилбромацетата в 50 мл тетрагидрофурана,поддерживая температуру реакционной смеси при 22-26 С. Затем реакционную смесь охлаждают до 2 С, при этой температуре гидробромид этиламина кристаллизуется и его собирают фильтрацией. Фильтрат концентрируют в вакууме при 30 С, получая 39,4 г желтого остатка. Остаток поглощают при взбалтывании 350 мл 2 пропанола, что дает белый твердый продукт,который собирают фильтрацией. К фильтрату добавляют при сильном взбалтывании 59 мл 3,6 н. хлористо-водородной кислоты в 2 пропаноле, смесь перемешивают и образовавшийся белый твердый продукт собирают фильтрацией. Остаток сушат, получая 32,5 г твердого продукта. Этот продукт растирают со 100 мл тетрагидрофурана, затем 100 мл 2-пропанола и сушат, получая 30 г (0,13 моль, выход 65%, химическая чистота 87%) моно(гидрохлорида) фенилметилового эфира N-этилглицина. Стадия 2. Фенилметиловый эфир N-этилN-[1-оксо-4-(4-пиридинил)бутил]глицина. Раствор 32,5 г (0,16 моль) 4-пиридинмасляной кислоты и 17,53 мл (0,174 моль) Nметилморфолина в 300 мл дихлорметана в атмосфере азота охлаждают до 0 С и добавляют в течение 0,5 ч предварительно охлажденный(0 С) раствор 18,75 мл (0,152 моль) пивалоилхлорида в 100 мл дихлорметана, выдерживая реакционную смесь при 0 С. В отдельной колбе на 500 мл растворяют 33,5 г (0,145 моль) моно(гидробромида) фенилметиловогo эфира Nэтилглицина в 400 мл дихлорметана и добавляют 35 мл (0,48 моль) N-метилморфолина. Эту смесь добавляют к раствору активированной пиридинмасляной кислоты при -0,5 С в течение 2 ч. Спустя 1 ч смесь разбавляют водой, взбалтывают и разделяют слои. Органическую фазу промывают насыщенным водным бикарбонатом натрия, 0,1 М водной хлористо-водородной кислотой и водой, сушат над сульфатом магния,фильтруют и концентрируют в вакууме, получая 46,2 г (0,135 моль, выход 93%) фенилметилового эфира N-этил-N-[1-оксо-4-(4-пиридинил) бутил]глицина в виде желтого масла. Стадия 3. N-Этил-N-[1-оксо-4-(4-пиридинил)бутил]глицин. 41 Раствор 0,46 г (0,135 моль) фенилметилового эфира N-этил-N-[1-оксо-4-(4-пиридинил) бутил]глицина в 300 мл метанола добавляют к 4 г 10% Pd/C (50% по весу воды) в автоклаве на 1 л. Реакционный сосуд трижды вакуумируют,заполняя вакуум азотом, затем еще трижды вакуумируют, заполняя вакуум водородом при атмосферном давлении. Сосуд взвешивают и нагревают до 25 С при встряхивании в течение 2 ч. Смесь фильтруют в азоте, получая светложелтый фильтрат, который концентрируют в вакууме при 40 С до начала появления твердого белого вещества. Добавляют порцию 2 пропанола в 150 мл, что приводит к быстрой кристаллизации продукта. Смесь перемешивают 0,5 ч, затем продукт собирают фильтрацией. Твердый продукт промывают порциями 2 пропанола в 240 мл, затем сушат, получая 19 г продукта. Маточник концентрируют и остаток поглощают 2-пропанолом, что приводит к образованию белого осадка. Смесь перемешивают 0,5 ч, затем фильтруют и твердый продукт промывают 2-пропанолом, затем сушат, получая 5 г продукта,N-этил-N-[1-оксо-4-(4-пиридинил) бутил]глицина, с общим выходом 24 г (0,095 моль; выход 70%; химическая чистота 99 А%). Пример 8. Получение трифторацетата фенилметилового эфира (L)аспартил-3-циклогексил-(L)-аланинамида. Стадия 1. Фенилметиловый эфир N-[(1,1 диметилэтокси)карбонил]-(L)аспартил-3 циклогексил-(L)-аланинамида. К механически перемешиваемому при 18 С раствору 37,5 г (116 ммоль) ВОС-бензил-(L)-аспарагиновой кислоты в 270 мл этилацетата добавляют раствор 17,75 г (116 ммоль) гидроксибензотриазолгидрата в 25 мл диметилформамида. Затем добавляют за 30 мин раствор 24,5 г (119 ммоль) дициклогексилкарбодиимида в 50 мл этилацетата, используя водяную баню для поддержания реакционной температуры около или ниже 25 С. Реакционную смесь перемешивают в течение 1 ч, затем добавляют сироп из 33 г (116 ммоль) 3 циклогексил-(L)-аланинамид-эфир-моно(трифторацетата) в 25 мл диметилформамида при 23 С с последующим добавлением по каплям чистого N-метилморфолина в течение 2 мин. Во время добавления N-метилморфолина реакционной смеси дают нагреться до 30 С. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь фильтруют для удаления дициклогексилмочевины и фильтрат разбавляют этилацетатом,дважды промывают водой, один раз раствором соли, дважды насыщенным водным бикарбонатом натрия и один раз 1:1 смесью 0,5 н. лимонной кислоты/раствора соли. Органическую фазу сушат сульфатом магния, фильтруют и разбавляют гептаном. 42 Смесь оставляют стоять в течение ночи и образующиеся кристаллы собирают фильтрованием, дважды промывают гептаном и сушат в вакууме, получая первую порцию продукта в 37,5 г и вторую порцию продукта в 9,5 г. Обе порции показывают приемлемую чистоту по ЖХВР и в общей сложности дают 47 г фенилметилового эфира N-[(1,1-диметилэтокси)карбонил]-(L)аспартил-3-циклогексил-(L)-аланинамида (выход 85%).(L)аспартил-3-циклогексил-(L)-аланинамида. К механически перемешиваемому раствору 45,4 г (95,5 ммоль) N-[(1,1-диметилэтокси) карбонил]-(L)аспартил-3-циклогексил-(L)аланинамида в 350 мл дихлорметана при 20 С добавляют 67 г (590 ммоль) трифторуксусной кислоты за 15 мин. Реакционную смесь перемешивают 2 ч. Реакционную смесь концентрируют в вакууме, получая оранжево-желтое масло, которое азеотропно перегоняют с дихлорметаном,а затем подвергают глубокому вакууму для сведения к минимуму содержания остаточных растворителей. Концентрат, масло, обрабатывают смесью 1:1 метил-трет-бутилового эфира/гептана, получая белый твердый продукт. Смесь перемешивают в метил-трет-бутиловом эфире в течение 2 ч, затем оставляют стоять на 2 дня. Кристаллы собирают фильтрованием и сушат в вакууме, получая моно(трифторацетат) фенилметилового эфира (L)-(-аспартил-3 циклогексил-(L)-аланинамида (43 г, выход 92%).MS (FAB) m/z 376 (М+Н)+. Пример 9. Получение трифторацетата бисфенилметилового эфира (L)аспартил-4 циклогексил-2-(L)-аминобутановой кислоты. Стадия 1. Фенилметиловый эфир N-[(1,1 диметилэтокси)карбонил]-4-циклогексил-2-(L)аминобутановой кислоты. Раствор 30 г (107,5 ммоль) N-[(1,1 диметилэтокси)карбонил]-(L)-гомофенилаланина и 2 мл уксусной кислоты в 100 мл метанола выливают на 3 г 5% родий/алюминий в колбу вибратора Парра на 500 мл. Реакционную смесь встряхивают в течение ночи при 46-47 фунт/кв.дюйм водорода, затем фильтруют под слоем азота через Целит 545 в стеклянной воронке для спекшегося материала. Слой целита промывают метанолом. Фильтрат концентрируют в вакууме при 30 С. Полученное масло поглощают метил-трет-бутиловым эфиром,дважды промывают водой и один раз раствором соли, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток (предположительно 107,5 ммоль) растворяют в 250 мл дихлорметана и раствор обрабатывают 12 г 43 время образуется белый твердый продукт. После добавления бензилхлорформиата добавляют 1,3 г (10,75 ммоль) 4-диметиламинопиридина одной порцией. Смесь оставляют перемешиваться в течение 2 ч при 0 С. Затем смесь концентрируют в вакууме, остаток поглощают этилацетатом и раствор дважды промывают водой, дважды 1 н. водной хлористо-водородной кислотой, дважды 1 н. водной гидроокисью натрия и один раз раствором соли, сушат над сульфатом магния, фильтруют и концентрируют, получая фенилметиловый эфир (33,8 г) N[(1,1-диметилэтокси)карбонил]-4-циклогексил 2-(L)-аминобутановой кислоты в виде масла.MS (FAB) m/z 748 (M+Na)+. Стадия 2. Моно(трифторацетат)фенилметилового эфира 4-циклогексил-2-(L)-аминобутановой кислоты. К раствору 33 г (28 ммоль) фенилметилового эфира N-[(1,1-диметилэтокси)карбонил]-4 циклогексил-2-(L)-аминобутановой кислоты в 136 мл дихлорметана за 30 мин добавляют 136 мл трифторуксусной кислоты. Реакционную температуру поддерживают ниже 23C. Смесь перемешивают при температуре окружающей среды 3 ч, затем концентрируют в вакууме. Остаток вновь растворяют в дихлорметане и повторно концентрируют. Полученное маловязкое масло поглощают 50 мл метил-трет-бутилового эфира и 350 мл гептана, в раствор вносят затравку, затем его охлаждают до -10 С на 24 ч. Первую порцию продукта собирают фильтрованием, промывают гептаном и сушат в вакууме(20,1 г). Маточник концентрируют в вакууме,затем вновь растворяют в 12 мл метил-третбутилового эфира и 84 мл гептана. Из этого при введении затравки и охлаждении до -10 С получают вторую порцию продукта в 5,2 г. Обе порции показывают приемлемую чистоту по ЖХВР и в общей сложности дают моно(трифторацетат) фенилметилового эфира 4-циклогексил-2-(L)аминобутановой кислоты (25,7 г, выход 75%).MS (FAB) 276 (М+Н)+. Стадия 3. Бисфенилметиловый эфир N[(1,1-диметилэтокси)карбонил]-(L)аспартил 4-циклогексил-2-(L)-аминобутановой кислоты. К механически перемешиваемому при 18 С раствору 20,4 г (63 ммоль) ВОСбензил(L)-аспарагиновой кислоты в 154 мл этилацетата добавляют раствор 9,65 г (63 ммоль) гидроксибензотриазолгидрата в 12 мл диметилформамида. Добавляют за 15 мин раствор 13,3 г(64,5 ммоль) дициклогексилкарбодиимида в 25 мл этилацетата, поддерживая температуру реакционной смеси около или ниже 25 С. Реакционную смесь перемешивают в течение 1 ч, затем добавляют сироп из 24,5 г (63 ммоль) трифторацетата 4-циклогексил-2-(L)-аминобутановой кислоты в 15 мл диметилформамида при 18 С с последующим добавлением по каплям Nметилморфолина в течение 5 мин. Во время до 002724 44 бавления N-метилморфолина реакционной смеси дают нагреться до 25 С. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь фильтруют для удаления дициклогексилмочевины и фильтрат дважды промывают водой,дважды насыщенным водным бикарбонатом натрия, дважды 0,5 н. водной лимонной кислотой и один раз раствором соли, сушат сульфатом магния, фильтруют и концентрируют в вакууме. Полученный смолообразный твердый продукт растворяют в метил-трет-бутиловом эфире при 50 С, раствор фильтруют горячим для удаления небольшого количества мелких частиц белого нерастворимого вещества, затем разбавляют гептаном. Раствор охлаждают до температуры окружающей среды и помещают в морозильную камеру при -10 С на ночь. Полученный твердый продукт собирают фильтрованием, промывают гептаном и сушат в вакууме,получая бисфенилметиловый эфир N-[(1,1 диметилэтокси)карбонил]-(L)-(-аспартил-4 циклогексил-2-(L)-аминобутановой кислотыMS (FAB) m/z 581. Стадия 4. Моно(трифторацетат) бисфенилметилового эфира (L)аспартил-4-циклогексил-2-(L)-аминобутановой кислоты. К механически перемешиваемому раствору 28,8 г (49,5 ммоль) бисфенилметилового эфира N-[(1,1-диметилэтокси)карбонил]-(L)-аспартил-4-циклогексил-2-(L)-аминобутановой кислоты в 300 мл дихлорметана при 20 С добавляют 222 г (1,94 ммоль) трифторуксусной кислоты за 30 мин и реакционную смесь перемешивают 3 ч. Реакционную смесь концентрируют в вакууме, получая масло, которое повторно растворяют в дихлорметане и концентрируют снова. Остаток обрабатывают смесью 1:1 метил-трет-бутилового эфира/гептана, получая белый твердый продукт. Твердый продукт собирают фильтрованием и сушат в вакууме,получая трифторацетат бисфенилметилового эфира(L)аспартил-4-циклогексил-2-(L)аминобутановой кислоты (27,4 г, 46 ммоль, выход 92,9%). Т.пл 143-144 С. Пример 10. Получение гидрохлорида бисфенилметилового эфира(L)аспартил-3 циклогексил-(L)-аланина. Стадия 1. Фенилметиловый эфир N-[(1,1 диметилэтокси)карбонил]-3-циклогексил-(L)аланина. Перемешиваемый мутный раствор 25,6 г(94,5 ммоль) N-[(1,1-диметилэтокси)карбонил]3-циклогексил-(L)-аланина, 11,2 г (104 ммоль) бензилового спирта и 1,15 г (9,45 ммоль) 4 диметиламинопиридина в смеси 210 мл диметилформамида и 150 мл дихлорметана охлаждают до 5 С и добавляют за 5 мин 19,2 г 3-N,Nдиметиламинопропилэтилкарбодиимидгидрохлорида. Затем смесь нагревают до температуры 45 окружающей среды и перемешивают в течение ночи. Реакционную смесь фильтруют для удаления мелкого порошка и фильтрат концентрируют в вакууме. Остаток поглощают водой и экстрагируют этилацетатом (3 х). Объединенные органические растворы промывают 0,5 н. водной лимонной кислотой и дважды водой, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученное масло подвергают глубокому вакууму для сведения к минимуму содержания летучих продуктов, получают фенилметиловый эфирMS (ионное распыление) m/z 362 (М+Н)+. Стадия 2. Моно(трифторацетат) фенилметилового эфира 3-циклогексил-(L)-аланина. К перемешиваемому раствору 35,3 г (94,5 ммоль) фенилметилового эфира N-[(1,1 диметилэтокси)карбонил]-3-циклогексил-(L)аланина в 100 мл дихлорметана при 17 С добавляют 100 мл трифторуксусной кислоты за 15 мин, за это время температура реакционной смеси поднимается до 21 С. Реакционную смесь перемешивают в течение ночи, затем концентрируют в вакууме. Остаток азеотропно перегоняют с дихлорметаном, толуолом и метил-третбутиловым эфиром. Полученное масло подвергают глубокому вакууму, получая трифторацетат фенилметилового эфира 3-циклогексил-(L)аланина (32,6 г).MS (ионное распыление) m/z 262 (M+H)+. Стадия 3. Бисфенилметиловый эфир N[(1,1-диметилэтокси)карбонил]-(L)аспартил 3-циклогексил-(L)-аланина. К механически перемешиваемому раствору 27,6 г (85,5 ммоль) ВОСбензил-(L)аспарагиновой кислоты в 110 мл этилацетата добавляют раствор 13,1 г (85,5 ммоль) гидроксибензотриазолгидрата в 21,4 мл диметилформамида. Добавляют за 15 мин раствор 18,15 г(88 ммоль) дициклогексилкарбодиимида в 80 мл этилацетата, поддерживая температуру реакционной смеси около или ниже 25 С. Реакционную смесь перемешивают в течение 1 ч, затем добавляют раствор 32 г (85,5 ммоль) трифторацетата фенилметилового эфира 3-циклогексил(L)-аланина в 120 мл этилацетата при 20 С с последующим добавлением по каплям 14,6 г Nметилморфолина в течение 5 мин (конечное значение рН 4-5). Во время добавления Nметилморфолина реакционной смеси дают нагреться до 25 С. Реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь фильтруют для удаления дициклогексилмочевины и фильтрат дважды промывают водой, один раз 0,5 н. водной лимонной кислотой и дважды насыщенным водным бикарбонатом натрия, сушат сульфатом магния, фильтруют и концентрируют в вакууме. Полученное масло растворяют в метил-трет-бутиловом эфире и раствор фильтруют 46 для удаления небольшого количества мелких частиц белого нерастворимого вещества. Фильтрат концентрируют в вакууме и полученное масло выдерживают в глубоком вакууме,что дает бисфенилметиловый эфир N-[(1,1 диметилэтокси)карбонил]-(L)аспартил-3 циклогексил-(L)-аланина (32,9 г).MS (ионное распыление) m/z 566 (М+Н),584 (М+Na)+. Стадия 4. Моно(гидрохлорид) бисфенилметилового эфира (L)аспартил-3-циклогексил-(L)-аланина. Хлористо-водородную кислоту барботируют через перемешиваемый при 0 С с помощью магнитной мешалки раствор 17 г (30 ммоль) бисфенилметилового эфира N-[(1,1 диметилэтокси)карбонил]-(L)аспартил-3 циклогексил-(L)-аланина в 60 г этилацетата приблизительно в течение 5 мин. За это время температура раствора поднимается от 5 до 20 С,затем начинает опять понижаться. Ледяную баню удаляют и раствор перемешивают при температуре окружающей среды в течение 90 мин. Избыток НСl удаляют, продувая через раствор азот, после чего смесь затвердевает. Добавляют этилацетат (100 г), чтобы облегчить перемешивание (тиксотропное) и через смесь барботируют азот в течение еще 3 мин. Твердый продукт выделяют фильтрованием на воронке Бюхнера. Плотный осадок на фильтре промывают этилацетатом и сушат в течение ночи в вакууме (20 мбар),продувая(L)аспартил-3-циклогексил-(L)-аланина (10,7 г) в виде твердого белого вещества.(17,5 г), впрыскивая шприцем по 0,2 мл/мин. После чего добавляют аспартам (5 г) за 9 порций при 12-минутных интервалах. Реакционную температуру поддерживают приблизительно при 38 С в течение 30 мин. Реакционную смесь дегазируют в вакууме до тех пор, пока перестанет наблюдаться выделение газа (около 20 мин). Затем добавляют в течение 45 мин аммиак (19 г), погружая трубку под поверхность реакционного слоя и реакционную смесь нагревают в течение ночи приблизительно до 60 С. Реакционную смесь разбавляют водой (280 г) и температуру реакции доводят до 45 С. Добавляют уксусную кислоту (55 г), доводя рН до 5,9. Смесь охлаждают до 40 С, к этому моменту начинает образовываться твердый продукт. Смесь охлаждают до 20 С в течение 2 ч и до 47 бавляют уксусную кислоту (250 г), доводя рН приблизительно до 5, и смесь перемешивают 0,5 ч. Твердый продукт фильтруют, промывают водой (3 х) и сушат в вакууме при 50 С, получаяN-[(1,1-диметилэтокси)карбонил]-(L)аспартил-3-фенил-(L)-аланинамид (53 г) в виде белого кристаллического вещества. Способ В. Смесь аспартама (25 кг) и ди-третбутилдикарбоната (22 кг) в метаноле (250 кг) в стеклянном трубчатом реакторе на 400 л нагревают приблизительно до 30 С и добавляют гидроокись лития (3,6 кг) за 30 мин. Суспензию перемешивают в течение приблизительно 4 ч и добавляют газообразный аммиак (39 кг) в атмосфере азота. Суспензию перемешивают 2 дня и затем концентрируют в вакууме для устранения аммиака и метанола. Добавляют воду (170 кг) и уксусную кислоту (8 кг), получая суспензию,осадок фильтруют и плотный осадок на фильтре дважды промывают водой (25 кг) и сушат в вакууме приблизительно при 40 С, что дает N[(1,1-диметилэтокси)карбонил]-(L)аспартил 3-фенил-(L)-аланинамид (29 кг) в виде белого кристаллического вещества. Пример 12. Получение моно(гидрохлорида) (L)аспартил-3-циклогексил-(L)-аланинамида. Смесь N-[(1,1-диметилэтокси)карбонил](L)аспартил-3-фенил-(L)-аланинамида (129 кг) и окиси платины (IV) (5 кг) или платины на алюминии (влажность 50%, 55, 12 кг) в уксусной кислоте (700 кг), в стеклянном трубчатом реакторе на 1600 л, гидрируют приблизительно при 60 С при 4 бар водорода в течение 3-5 ч. Катализатор фильтруют и промывают уксусной кислотой (20 кг). В стеклянный трубчатый реактор на 400 л в атмосфере азота к 215 кг раствора уксусной кислоты добавляют газообразную хлористо-водородную кислоту (7,8 кг) за период 1-2 ч. Суспензию перемешивают в течение 1 ч и фильтруют. Плотный осадок на фильтре дважды промывают уксусной кислотой (20 кг) и дважды ацетоном (20 кг) и сушат в вакууме приблизительно при 40 С, получая моно(гидрохлорид)(31 кг) в виде белого кристаллического вещества. Пример 13. Получение моно(гидрохлорида) фенилметилового эфира (L)аспартил-(L)фенилаланинамида. Стадия 1. Моно(натриевая) соль метилового эфира N-[(1-,1-диметилэтокси)карбонил]-(L)-аспартил-(L)-фенилаланина. В колбу на 6 л загружают 294,4 г (0,68 моль) аспартама, 2000 мл метанола, 2000 мл воды и 192,96 г (0,88 моль) ди-трет-бутилдикарбоната и смесь охлаждают до 0 С. Добавляют водную гидроокись натрия (10 н., 68,6 мл) за период 15 мин, поддерживая температуру между 0 и 4 С. Затем охлаждение прекращают и 48 смеси дают нагреться до температуры окружающей среды и перемешивают 2 дня. Прозрачный раствор концентрируют в вакууме, получая масло, которое поглощают 500 мл этилацетата и вновь концентрируют, получая 297 г масла. Масло растворяют в этилацетате при 48 С, получая прозрачный раствор. Раствору дают охладиться до комнатной температуры в течение ночи. Кристаллический продукт собирают фильтрованием, промывают 400 мл этилацетата и сушат в вакуумном сушильном шкафу, получая 165 г (0,42 моль, выход 62%) моно(натриевой) соли N-[(1,1-диметилэтокси) карбонил]-(L)аспартил-(L)-фенилаланина. При уменьшении объема маточников получают дополнительно 95,4 г (0,24 моль) твердого продукта (общий выход 97%). Стадия 2. Моно(натриевая) соль N-[(1,1 диметилэтокси)карбонил]-(L)аспартил-(L)фенилаланинамида. В 4 л колбе конденсируют аммиак (255 г,15 моль). В отдельной колбе готовят раствор 260 г (0,659 моль) моно(натриевой) соли метилового эфира N-[(1,1-диметилэтокси)карбонил]-(L)аспартил-(L)-фенилаланина в 1300 мл метанола и добавляют его к аммиаку за 0,5 ч при температуре между -32 и -5 С. Смесь нагревают до температуры окружающей среды в течение 3 ч, затем перемешивают дополнительно 2 ч. Метанол удаляют в вакууме при 40 С, получая 501 г пасты. Пасту поглощают 1 л этилацетата, из которого кристаллизуется продукт. Смесь разбавляют еще 2 л этилацетата и фильтруют. Твердый продукт сушат в вакууме в течение ночи при температуре окружающей среды,затем в вакууме при 50 С в течение 1,5 ч, получая 243,5 г (0,645 моль, выход 98%) моно(натриевой) соли N-[(1,1-диметилэтокси) карбонил]-(L)аспартил-(L)-фенилаланинамида. Стадия 3. Фенилметиловый эфир N-1,1 диметилэтокси)карбонил]-(L)аспартил-(L)фенилаланинамида. В круглодонную колбу на 1 л в атмосфере азота добавляют 241 г (0,635 моль) моно(натриевой) соли N-[(1,1-диметилэтокси)карбонил]-(L)аспартил-(L)-фенилаланинамида и 2500 мл диметилформамида и смесь перемешивают до получения раствора. Добавляют чистый бензилбромид, 75,4 г (0,635 моль) в течение 5 мин при 23-26 С. Смесь перемешивают при температуре окружающей среды в течение 21,5 ч, затем медленно разбавляют 2500 мл воды, что приводит к подъему температуры до 32 С. Смесь перемешивают 1 ч,при этом температура охлаждается до температуры окружающей среды, к этому моменту кристаллизуется твердый продукт. Твердый продукт собирают фильтрованием, промывают водой, тремя порциями по 1 л, и сушат при 50 С в течение 2 дней, получая 232 г (0,495 ммоль, вы 49 ход 78%) фенилметилового эфира N-[(1,1 диметилэтокси)карбонил]-(L)-к-аспартил-(L)фенилаланинамида. Стадия 4. Моно(гидрохдорид) фенилметидового эфира (L)аспартил-(L)-фенилаланинамида. Раствор фенилметилового эфира N-[(1,1 диметилэтокси)карбонил]-(L)аспартил-(L)фенилаланинамида в 4 н. хлористом водороде/этилацетате перемешивают 1 ч при температуре окружающей среды. Полученный твердый продукт собирают фильтрованием и растворяют в 3 л воды. Раствор промывают порциями дихлорметана по 3500 мл. Водный раствор частично упаривают на роторном испарителе при 55 С. Раствору дают охладиться. Кристаллизуется твердый продукт, который собирают фильтрованием в виде двух порций (113,6 г и 34,1 г), получая в общей сложности 147,7 г(0,364 моль, выход 79%) моно(гидрохлорида) фенилметилового эфира (L)аспартил-(L)фенилаланинамида. Пример 14. Получение N-этил-N-[1-оксо-4[1-(фенилметоксикарбонил)-4-пиперидинилен] бутил]глицина. Стадия 1. 1-Фенилметоксикарбонил-4 пиперидон. Смесь 40 кг N-бензилоксикарбонилсукцинимида и 26 кг (175 моль) 4-пиперидонгидрохлоридгидрата в 38,8 кг воды и 88 кг тетрагидрофурана перемешивают приблизительно при 15 С до полного растворения ( 15 мин). К взбалтываемой смеси (экзотермической) добавляют N-метилморфолин (22,8 кг), выдерживая температуру около или ниже 20 С. Реакционную смесь взбалтывают приблизительно при 20 С в течение 2,5 ч, к этому времени ЖХВР указывает на завершение реакции. Смесь разбавляют 115,2 кг метил-трет-бутилового эфира и 38,8 кг воды и взбалтывают приблизительно при 20 С в течение 5 мин. Взбалтывание прекращают, слоям дают разделиться и водный(нижний) слой удаляют и отбрасывают. Органический слой дважды промывают. 129,6 кг воды(взбалтывают 5 мин, разделяют фазы, удаляют/отбрасывают водную (нижнюю) фазу). Органический слой промывают 5,2 кг NaCl в 46,8 кг воды (взбалтывают 5 мин, разделяют фазы,удаляют/отбрасывают водный (нижний) слой). Органический слой обрабатывают 11,5 кгMgSO4, взбалтывая в течение 1 ч, затем смесь фильтруют. Реактор промывают 8 кг метилтрет-бутилового эфира (фильтруют, объединяют основной фильтрат; содержание воды в объединенном фильтрате 0,52%). Объем фильтрата снижают до половины, перегоняя при пониженном давлении при 30 С. Вакуум заполняют азотом и остаток охлаждают до 20 С (содержание остаточной воды в продукте: 0,43%). Остаток разбавляют 57,6 кг метил-трет-бутилового эфира, затем объем смеси снижают снова до поло 002724 50 вины, перегоняя в вакууме при 30 С. Вакуум заполняют азотом и смесь охлаждают до 20 С(содержание остаточной воды в продукте 0,25%). Этот прием повторяют еще 5 раз. Конечный остаток в колбе разбавляют 28,8 кг метил-трет-бутилового эфира и перемешивают 5 мин, затем исследуют на содержание воды и содержание 1-фенилметоксикарбонил-4-пиперидона (вода: 0,05%, испытание на влажность 1 фенилметоксикарбонил-4-пиперидона: влажность 22,66%, 35,36 кг, 155 моль, выход 88,6%). Стадия 2. 4-(1-Фенилметоксикарбонил-4 пиперидинилен)бутановая кислота. К суспензии 82 г 3-карбоксипропилтрифенилфосфонийбромида в 407 мл 1,2 диэтоксиэтана при 14 С добавляют за 25 мин 220 г 20 вес.% трет-бутилата калия в тетрагидрофуране, поддерживая температуру реакционной смеси при 24-28 С. Смесь перемешивают в течение 1 ч, охлаждают до 10 С и добавляют в течение 30 мин, поддерживая охлаждение, раствор 52,5 г 1-фенилметоксикарбонил-4-пиперидона в 246 мл метил-трет-бутилового эфира. По завершению добавления смесь перемешивают при 12 С в течение 10 мин, затем нагревают до 20 С и перемешивают еще 30 мин. Реакционную смесь обрабатывают 410 мл 1 н. водной НСl в течение 10 мин, разбавляют 328 мл метилтрет-бутилового эфира и затем фазы разделяют. Органическую фазу промывают 205 мл воды,затем 210 мл 1 н. водного NaOH. NaOH-слой,содержащий продукт, собирают отдельно, трижды промывают порциями по 189 г этилацетата,подкисливают до рН 3,48 концентрированной НСl, затем экстрагируют 189 мл этилацетата. Отделяют этилацетный слой, промывают 211 мл воды, затем сушат в течение 30 мин над 10 г МgSO4, фильтруют и концентрируют в вакууме. Масляный остаток (50,7 г) кристаллизуют из толуола/гептана, получая в общей сложности 29,46 г (выход 51%) 4-(1-фенилметоксикарбонил-4-пиперидинилен)бутановой кислоты. Масс-спектр: Мрассчит.303, М+1 набл.304. 1N-этил-N-[1-окco-4-[1-(фенилметоксикарбонил)-4-пиперидинилен]бутил]глицин получают, используя способ примера 6, за исключением того,что заменяют 4-(1-фенилметоксикарбонил-4-пиперидинил)бутановую кислоту на 4-(1-фенилметоксикарбонил-4 пиперидинилен)бутановую кислоту. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения псевдотетрапептида формулы или его соли или пролекарства, где обозначает необязательно аминозащищенный 48-членный азагетероциклил; обозначает простую или двойную связь,q обозначает 1-5,В обозначает С 1-С 20 алкил, С 3-С 10 циклоалкил, С 3-С 10 циклоалкилС 1-С 20 алкил, С 1-С 20 алкилС 3-С 10 циклоалкил, С 1-С 20 алкилС 3-С 10 циклоалкилС 1-С 20 алкил, С 6-С 10 арил, С 6-С 10 арС 1-С 20 алкил, С 1-С 20 алкилС 6-С 10 арил или С 1-С 20 алкилС 6-С 10 арС 1-С 20 алкил,Q2 обозначает Н или карбоксизащитную группу,J обозначает -Н, С 1-С 20 алкил, С 3-С 10 циклоалкил, С 3-С 10 циклоалкилС 1-С 20 алкил, С 1 С 20 алкилС 3-С 10 циклоалкил, C1-С 20 алкилС 3-С 10 циклоалкилС 1-С 20 алкил, С 6-С 10 арил, замещенный C6-С 10 арил, С 6-С 10 арС 1-С 20 алкил или замещенный С 6-С 10 арС 1-С 20 алкил,L обозначает OR1 или NR1R2, где R1 и R2 независимо обозначают -Н, С 1-С 20 алкил, С 3 С 10 циклоалкил, С 3-С 10 циклоалкил-С 1-С 20 алкил,С 1-С 20 алкилС 3-С 10 циклоалкил, С 1-С 20 алкилС 3 С 10 циклоалкилС 1-С 20 алкил,С 6-С 10 арил,С 6 С 10 арС 1-С 20 алкил, C1-С 20 алкилС 6-С 10 арил или С 1-С 20 алкилС 6-С 10 арС 1-С 20 алкил, и р обозначает 1 или 2,включающий или его соли, где K обозначает ОН или ацилактивирующую группу, с замещенным карбоновой кислотой дипептидом формулы или его солью,(b) необязательное удаление аминозащитной группы или карбоксизащитной группы и(c) необязательное превращение псевдотетрапептида в соль или пролекарство. 2. Способ по п.1, гдеQ2 обозначает Н или бензил,L обозначает -NR1R2. 4. Способ по п.1, где соль азагетероциклилпсевдодипептида сочетается с солью карбоксизамещенного дипептида, или где основная аддитивная соль азагетероциклилпсевдодипептида сочетается с кислотной аддитивной солью карбоксизамещенного дипептида, или где соль азагетероциклилпсевдодипептида с дициклогексиламином сочетается с трифторацетатной солью карбоксизамещенного дипептида. 5. Способ по п.1, включающий(а) реакцию сочетания азагетероциклилпсевдодипептида формулы или его основной аддитивной соли,где P1 обозначает аминозащитную группу и В обозначает C1-С 20 алкил, с карбоксизамещенным дипептидом формулы или его кислотной аддитивной солью,где Q2 обозначает Н или карбоксизащитную группу,J обозначает С 1-С 20 алкил, С 3-С 10 циклоалкил или С 3-С 10 циклоалкилС 1-С 20 алкил и С 10 циклоалкил, С 3-С 10 циклоалкилС 1-С 20 алкил,С 1-С 20 алкилС 3-С 10 циклоалкил, С 1-С 20 алкилС 3 С 10 циклоалкилС 1-С 20 алкил или арС 1-С 20 алкил,с образованием псевдотетрапептида формулы(b) необязательное удаление аминозащитной группы или карбоксизащитной группы, и(c) необязательное превращение псевдотетрапептида в его соль или пролекарство. 6. Способ по п.5, гдеL обозначает NH2. 8. Способ получения замещенного циклогексилметилом пептида формулы где Q2 обозначает Н или карбоксизащитную группу и(а) восстановление замещенного фенилметилом пептида формулы(b) необязательное удаление аминозащитной группы или карбоксизащитной группы. 9. Способ по п.8, где восстановление осуществляют путем каталитической гидрогенизации, предпочтительно используя платиновый катализатор, такой как окись платины или платина на алюминии. 10. Способ получения амидодипептида формулы где Q2 обозначает Н или основную аддитивную соль, или карбоксизащитную группу,Q3 обозначает Н или аминозащитную группу,включающий амидирование пептидного сложного эфира формулы где R3 обозначает C1-С 10 алкил. 11. Способ по п.10, где амидирование осуществляют, используя раствор аммиака в спирте, таком как С 1-С 4cпирт, или используя аммиак в смеси растворителей С 1-С 4cпирт-С 1 С 3 гликоль, которая включает метанол и этиленгликоль. 12. Способ получения соединения защищенного аспартама формулы где Q2 обозначает Н или карбоксизащитную группу,Q3 обозначает аминозащитную группу иR3 обозначает C1-С 10 алкил,включающий введение аминозащитной группы в соединение аспартама формулыQ3 обозначает бензилоксикарбонил или трет-бутилоксикарбонил,Q2 обозначает Н. 14. Способ получения амидодипептида формулыQ3 обозначает аминозащитную группу и(а) добавление основания и аминозащищающего агента к раствору аспартама в растворителе с получением раствора соединения формулы где Q3 обозначает Н или основную аддитивную соль, и(b) введение аммиака в образовавшийся на стадии (а) раствор. 15. Способ по п.14, где Q3 обозначает третбутилоксикарбонил или бензилоксикарбонил,растворителем является C1-С 3cпирт или смесь С 1-С 3cпирт-С 1-С 3 гликоль. 16. Способ по п.15, где растворителем является метанол или смесь метанолэтиленгликоль. 17. Способ получения замещенного циклогексилметилом дипептида формулы(е) реакцию сочетания аминозащищенной пиперидинкислоты с соединением N-этилглицина формулы или его кислотной аддитивной соли, включающий(а) получение смеси катализатора и замещенного фенилметилом пептида формулы где Q3 обозначает трет-бутилоксикарбонил, в растворителе,(b) обработку смеси водородом,(c) удаление катализатора из смеси и(d) введение в смесь газообразного НСl. 18. Способ по п.17, где растворителем является уксусная кислота. 19. Способ получения соединения азагетероциклилзамещенной кислоты формулы(b) гидрогенизацию пиридилкислоты водородом в присутствии катализатора до получения пиперидинкислоты формулы(c) необязательное удаление катализатора,(d) аминозащиту пиперидинкислоты с получением аминозащищенной пиперидинкислоты формулы с получением соединения азагетероциклилзамещенной кислоты и(f) необязательную деэтерефикацию азагетероциклилзамещенного соединения кислоты,где R4 обозначает С 1-С 10 алкил. 20. Способ по п.19, где P1 обозначает бензилоксикарбонил и R4 обозначает Н, где декарбоксилирование осуществляют нагреванием 2 пиридилэтил-ди(С 1-С 10 алкил)малоната в водном кислотном растворе, где гидрогенизацию осуществляют в водном кислотном растворе, и где защиту аминогруппы осуществляют в водном растворе основания. 21. Способ по п.20, где водная кислота на стадии декарбоксилирования и гидрогенизации означает водную НСl. 22. Способ по п.20, где(a) нагревают раствор 2-пиридилэтилди(C1-С 10 алкил)малоната в водной кислоте, получая раствор пиридилкислоты в водной кислоте,(b) к раствору пиридилкислоты добавляют катализатор и смесь обрабатывают водородом,получая смесь катализатора и пиперидинкислоты,(c) отделяют катализатор от смеси, получая водный раствор пиперидинкислоты, и(d) к водному раствору добавляют основание и аминозащищающий агент, получая аминозащищенную пиперидинкислоту. 23. Способ получения псевдотетрапептида формулы(а) восстановление соединения псевдотетрапептида формулыQ2 обозначает карбоксизащитную группу,предпочтительно неустойчивую к гидрогенизации. 26. Способ по п.25, где действие каталитического восстановления состоит в одновременном восстановлении и удалении P1 и Q2. 27. Способ по п.24, где где m обозначает 3 и P1 обозначает Н или аминозащитную группу,Q2 обозначает Н или карбоксизащитную группу,J обозначает фенилметил,(b) необязательное удаление аминозащитной группы или карбоксизащитной группы, и(c) необязательное превращение псевдотетрапептида в соль или пролекарство. 24. Способ по п.23, где восстановление осуществляют путем каталитической гидрогенизации. 25. Способ по п.24, гдеQ2 обозначает карбоксизащитную группу,предпочтительно неустойчивую к гидрогенизации. 28. Способ по п.27, где действие каталитического восстановления состоит в одновременном восстановлении и удалении Q2. 29. Соединение псевдодипептида формулыR4 обозначает Н или C1-С 10 алкил. 30. Соединение псевдодипептида по п.29,где P1 обозначает аминозащитную группу, такую как бензилоксикарбонил,В обозначает С 1-С 20 алкил, такой как этил,и
МПК / Метки
МПК: C07K 1/02, C07C 227/18
Метки: способ, азациклоалкилалканоилпсевдотетрапептидов, получения
Код ссылки
<a href="https://eas.patents.su/30-2724-sposob-polucheniya-azacikloalkilalkanoilpsevdotetrapeptidov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения азациклоалкилалканоилпсевдотетрапептидов</a>
Способ получения и очистки n-алкилированного производного аспартама

Номер патента: 1948
Опубликовано: 22.10.2001
Автор: Пракаш Индра
МПК: A23L 1/236, C07K 5/075
Метки: аспартама, производного, очистки, n-алкилированного, получения, способ
Формула / Реферат:
1. Способ очистки 1-сложного метилового эфира N-[N-(3,3-диметилбутил)-L-a-аспартил]-L-фенилаланина формулы включающий стадии: (I) получения раствора 1-сложного метилового N-[N-(3,3-диметилбутил)-L-a-acпapтил]-L-фенилаланина в органическом растворителе; и (II) получения раствора вода/органический растворитель из раствора органического растворителя для осаждения 1-сложного метилового эфира N-[N-(3,3-диметилбутил)-L-a-аспартил]-L-фенилаланина...
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Мейллян Пьер, Бьенейм Юг, Ансель Жан-Эрик
МПК: B01J 31/24, A61K 31/355, C07C 39/19...
Метки: способ, замещенных, фенолов, использованием, получения, витамина, cпособ
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Свинделл Чарльз С., Чандер Мадхави С., Систи Николас Дж.
МПК: C07D 305/14
Метки: соединения, паклитаксела, промежуточного, получения, промежуточное, способ, соединение
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Способ получения защищенного 4-аминометилпирролидин-3-она, промежуточные соединения и способ получения 3-аминометил-4-алкоксииминопирролидина

Номер патента: 2499
Опубликовано: 27.06.2002
Авторы: Моон Кванг Юл, Ли Тае Хи, Чанг Джей Хиок, Ким Вон Суп
МПК: C07D 207/24
Метки: 4-аминометилпирролидин-3-она, промежуточные, получения, 3-аминометил-4-алкоксииминопирролидина, защищенного, способ, соединения
Формула / Реферат:
1. Способ получения соединения формулы (1) в которой Р1 и Р2 обозначают защитные группы; включающий а) восстановление соединения формулы (5) где Р1 такой, как определено для формулы (1); в присутствии катализатора никель Ренея в растворителе в атмосфере водорода в присутствии одной или более добавок с получением соединения формулы (6) где Р1 такой, как определено для формулы (1); b) защиту аминогруппы в присутствии одного или более оснований...
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Дельтиль Мишель, Бонне Алан, Мазюри Алан
МПК: C07H 17/08
Метки: способ, активных, биологически, производные, применение, 5-0-дезозаминил-6-0-метилэритронолида, получения, продуктов
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Предыдущий патент: Способы и промежуточные продукты, полезные для получения антифолатов
Следующий патент: Способ сушки пиломатериалов и устройство для его осуществления