Гетероциклические соединения, лекарственные средства, содержащие указанные соединения, их применение и способы их получения
Номер патента: 25158
Опубликовано: 30.11.2016
Авторы: Хеккель Армин, Фраттини Сара, Хампрехт Дитер, Клей Йёрг














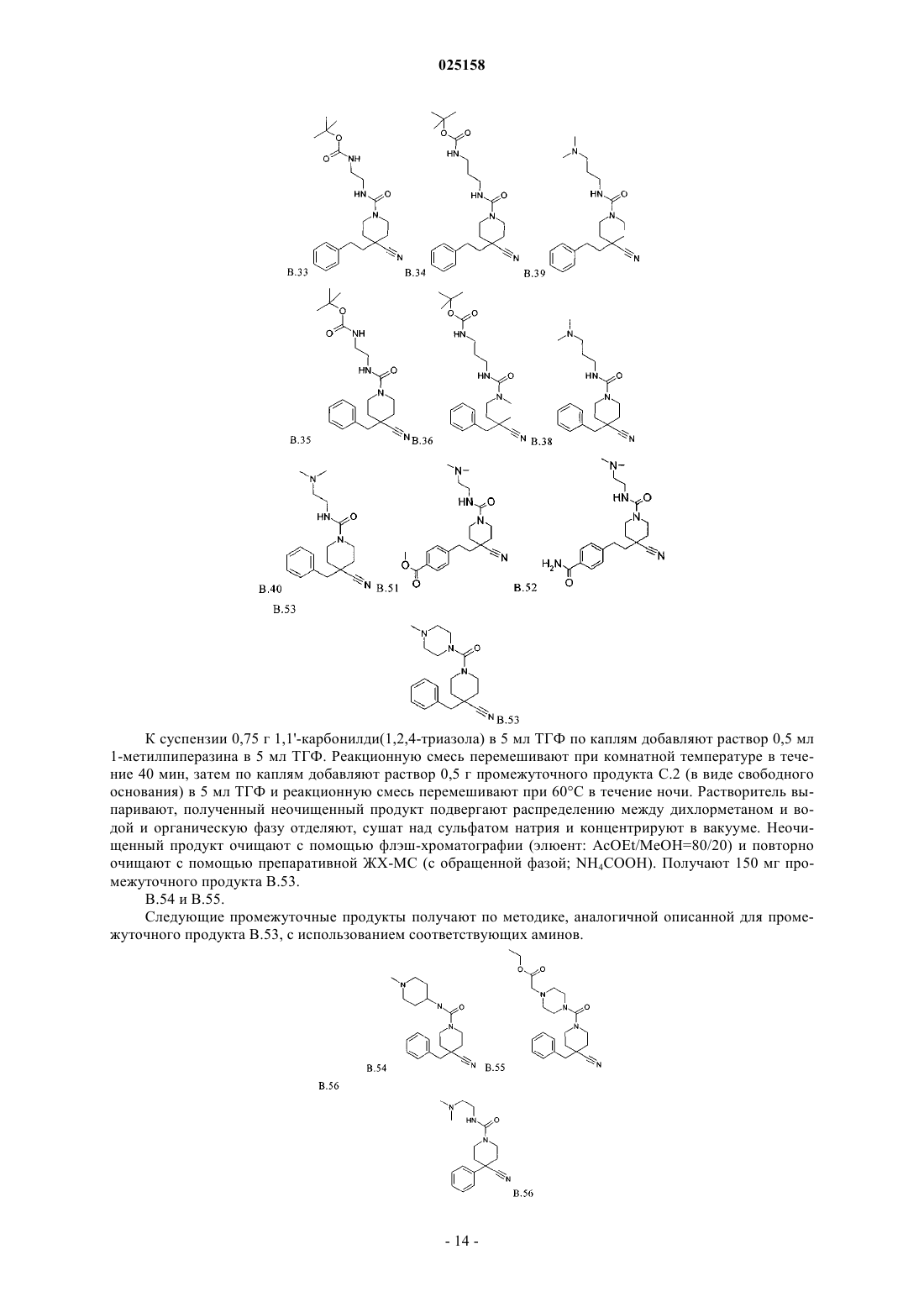










Формула / Реферат
1. Соединение формулы (IA)

в которой
А обозначает связь, -СН2- или -СН2СН2-,
R1 выбран из группы, включающей водород, C1-C6-алкил, C1-C4-алкил-SO2-, C1-С4-алкил-NH-СО-, H2M-СО-, H2N-С1-С4-алкил-, H2N-C1-C4-алкил-СО-, H2N-C1-C4-алкил-NH-CO-, фенил-СО-, фенил-СН2-СО-, фенил-СН2-, C1-C6-алкил-СО-, C1-С6-алкил-О-C1-C4-алкил-СО-, (CH3)2N-C1-C4-алкил-, (CH3)2N-C1-C4-алкил-NH-СО-, (CH3)3N+-C1-C4-алкил-NH-CO-, (СН3)3N+-С1-С4-алкил-N(С1-С4-алкил)-СО-, (СН3)3N+-С2-С4-алкил-, (СН3)3N+-С1-С4-алкил-СО-, H2N-C(NH)-NH-C1-C6-NH-СО-, C1-C6-алкил-О-СО-, C1-C6-алкил-О-СО-C1-C4-алкил-, C1-C6-алкил-О-СО-С1-С4-алкил-СО-, C1-C6-алкил-О-СО-C1-C4-алкил-NH-СО-, C1-C6-алкил-О-СО-NH-C1-C4-алкил-, C1-C6-алкил-О-СО-NH-С1-С4-алкил-СО-, C1-C6-алкил-O-CO-NH-С1-С4-алкил-NH-СО-, НОСО-C1-C4-алкил-, НОСО-C1-C4-алкил-СО-, НОСО-С1-С4-алкил-NH-СО-, H2N-CNH- и H2NC(NH)NH-C1-C6-алкил-СО-, или выбран из группы, включающей формулы (c1)-(с5):


R2 независимо друг от друга выбраны из группы, включающей водород, галоген, CN, C1-C4-алкил и C1-алкил-О-,
R6 независимо друг от друга выбраны из группы, включающей водород, галоген, CN, C1-C4-алкил и C1-алкил-О-,
R3 выбраны из группы, включающей водород, галоген, CN и C1-C4-алкил,
R4 независимо друг от друга выбраны из группы, включающей водород, галоген, CN, C1-C4-алкил, C1-C4-алкил-ОСО-, -COOR4.1 и -CONR4.2R4.3, где
R4.1 обозначает водород или C1-C4-алкил,
R4.2 обозначает водород или C1-C4-алкил,
R4.3 обозначает водород или C1-C4-алкил или
R3 и R4 вместе обозначают -O-C1-С3-алкил-О-,
R7 обозначает водород,
и его фармакологически приемлемые соли присоединения с кислотой.
2. Соединение по п.1, отличающееся тем, что R2, R3, R4, R6, R7 обозначают водород, и его фармакологически приемлемые соли присоединения с кислотой.
3. Соединение по любому из пп.1, 2, отличающееся тем, что А обозначает -СН2СН2- и Е, D обозначают -СН2-, и его фармакологически приемлемые соли присоединения с кислотой.
4. Соединение по любому из пп.1-3, отличающееся тем, что R1 обозначает водород или выбран из группы, включающей C1-C6-алкил, C1-C4-алкил-SO2-, C1-C4-алкил-NH-СО-, H2N-CO-, H2N-С1-С4-алкил-, H2N-С1-С4-алкил-СО-, H2N-С1-С4-алкил-NH-СО-, фенил-СО-, фенил-СН2-СО-, фенил-СН2-, С1-С6-алкил-СО-, С1-С6-алкил-О-С1-С4-алкил-СО-, (СН3)2N-С1-С4-алкил-, (CH3)2N-C1-С4-алкил-NH-СО-, (СН3)3N+-С1-С4-алкил-NH-СО-, (СН3)3N+-С2-С4-алкил-, (СН3)3N+-С1-С4-алкил-СО-, H2N-C(NH)-NH-C1-C6-NH-CO-, C1-C6-алкил-О-СО-, C1-C6-алкил-О-СО-C1-C4-алкил-, C1-C6-алкил-О-СО-C1-C4-алкил-СО-, C1-C6-алкил-О-СО-C1-C4-алкил-NH-СО-, C1-C6-алкил-О-СО-NH-C1-C4-алкил-, C1-C6-алкил-О-СО-NH-C1-C4-алкил-СО-, C1-C6-алкил-О-СО-NH-C1-C4-алкил-NH-СО-, НОСО-C1-C4-алкил-, НОСО-C1-C4-алкил-СО-, НОСО-C1-C4-алкил-NH-СО- и H2N-CNH-, и его фармакологически приемлемые соли присоединения с кислотой.
5. Соединение по любому из пп.1-3, отличающееся тем, что R1 выбран из группы приведенных ниже формул (c1)-(с5):

и его фармакологически приемлемые соли присоединения с кислотой.
6. Соединение по любому из пп.1-5, отличающееся тем, что соединение выбрано из группы, включающей соединения (1)-(4) и (6)-(12):



и его фармакологически приемлемые соли присоединения с кислотой.
7. Соединение по п.6, где соединение представляет собой соединение (1)

и его фармакологически приемлемые соли присоединения с кислотой.
8. Соединение по п.6, где соединение представляет собой соединение (2)

и его фармакологически приемлемые соли присоединения с кислотой.
9. Соединение по п.6, где соединение представляет собой соединение (3)

и его фармакологически приемлемые соли присоединения с кислотой.
10. Соединение по п.6, где соединение представляет собой соединение (4)

и его фармакологически приемлемые соли присоединения с кислотой.
11. Соединение по п.6, где соединение представляет собой соединение (6)

и его фармакологически приемлемые соли присоединения с кислотой.
12. Соединение по п.6, где соединение представляет собой соединение (7)

и его фармакологически приемлемые соли присоединения с кислотой.
13. Соединение по п.6, где соединение представляет собой соединение (8)

и его фармакологически приемлемые соли присоединения с кислотой.
14. Соединение по п.6, где соединение представляет собой соединение (9)

и его фармакологически приемлемые соли присоединения с кислотой.
15. Соединение по п.6, где соединение представляет собой соединение (10)

и его фармакологически приемлемые соли присоединения с кислотой.
16. Соединение по п.6, где соединение представляет собой соединение (11)

и его фармакологически приемлемые соли присоединения с кислотой.
17. Соединение по п.6, где соединение представляет собой соединение (12)

и его фармакологически приемлемые соли присоединения с кислотой.
18. Применение соединения по любому из пп.1-17 или его фармацевтически приемлемой соли для лечения заболевания, выбранного из группы, включающей респираторные заболевания или нарушения и аллергические заболевания дыхательных путей.
19. Применение соединения по любому из пп.1-17 или его фармацевтически приемлемой соли для лечения заболевания, выбранного из группы, включающей хронический бронхит, острый бронхит, бронхит, вызванный бактериальной или вирусной инфекцией, или грибами, или гельминтами, аллергический бронхит, токсический бронхит, хронический обструктивный бронхит (COPD), астму (наследственную или аллергическую), астму у детей, бронхоэктаз, аллергический альвеолит, аллергический или неаллергический ринит, хронический синусит, кистозный фиброз или муковисцидоз, дефицит альфа-1-антитрипсина, кашель, эмфизему легких, интерстициальные заболевания легких, альвеолит, гиперреактивные дыхательные пути, полипы в носу, отек легких, пневмонит различной этиологии.
20. Фармацевтическая композиция, содержащая по меньшей мере одно соединение по любому из пп.1-17 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
21. Комбинации лекарственных средств, которые кроме одного или большего количества соединений по любому из пп.1-17 содержат в качестве дополнительных активных веществ одно или большее количество соединений, выбранных из следующих категорий: другие ингибиторы ENaC, бета-миметики, антихолинергетики, кортикостероиды, ингибиторы PDE4, антагонисты LTD4, ингибиторы EGFR, агонисты допамина, H1-антигистамины, антагонисты PAF, ингибиторы киназы MAP, ингибиторы MPR4, ингибиторы iNOS, ингибиторы SYK, корректоры трансмембранного регулятора муковисцидоза (CFTR) и усиливающие факторы CFTR или их двойные или тройные их комбинации.
Текст
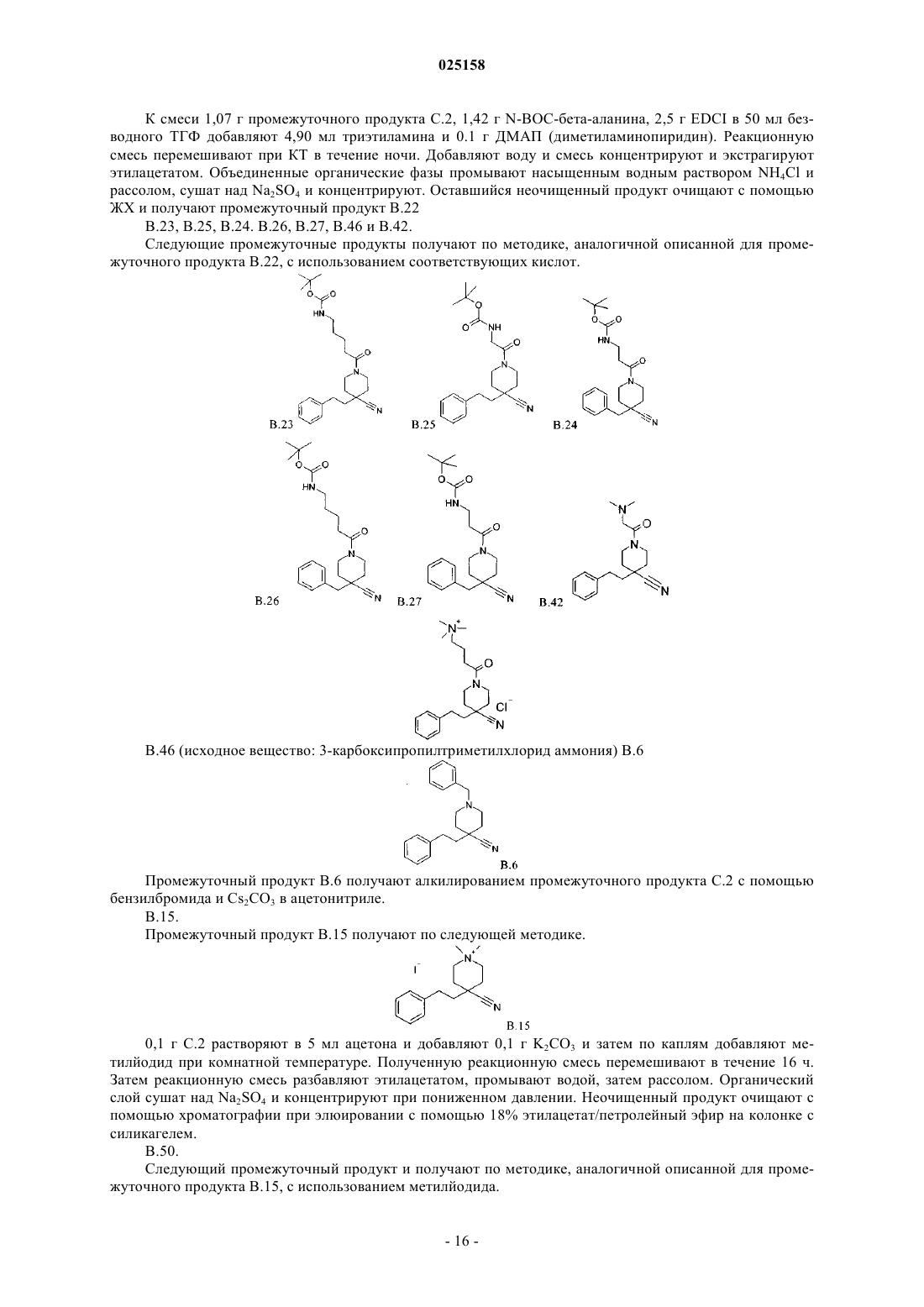
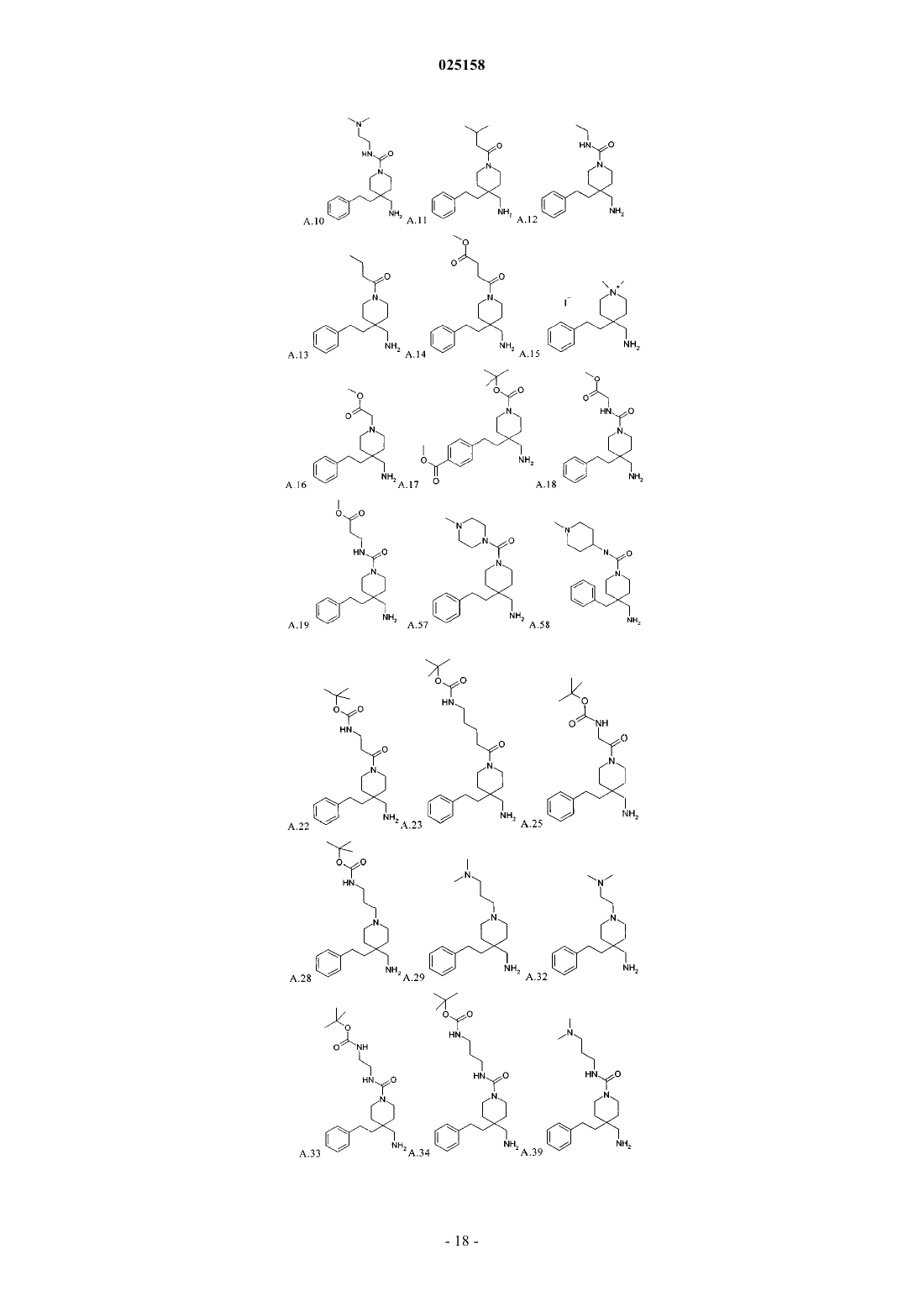
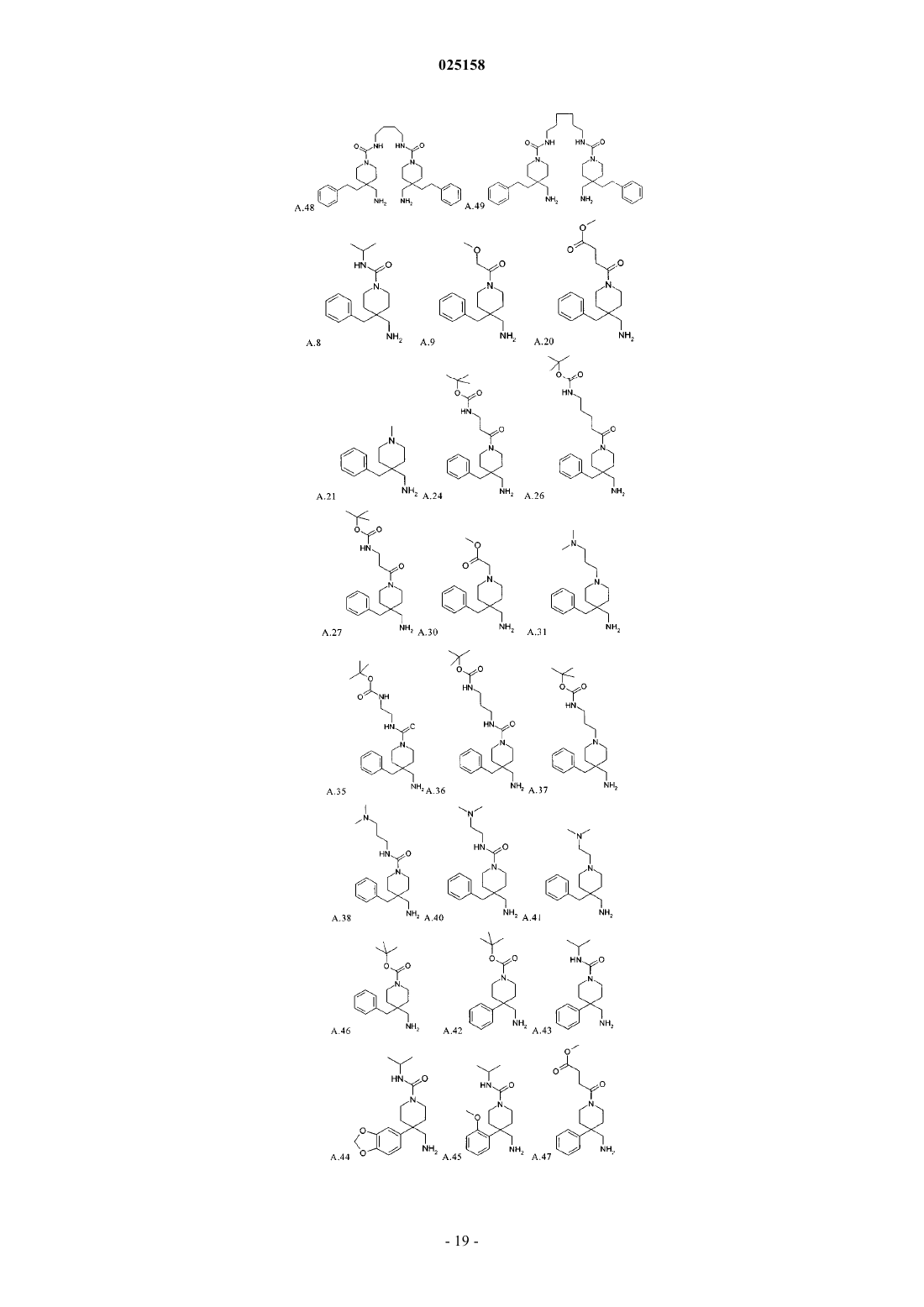
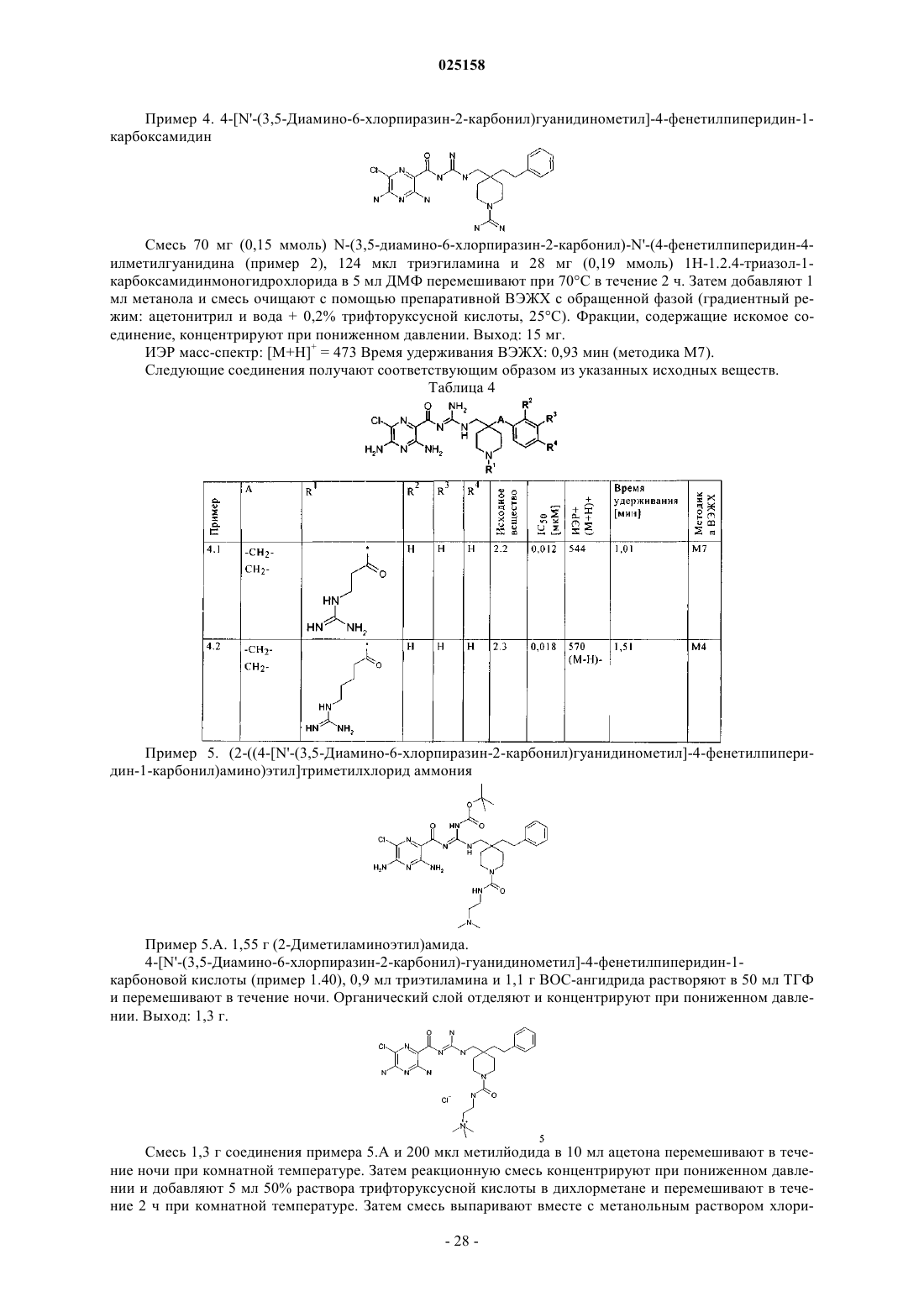
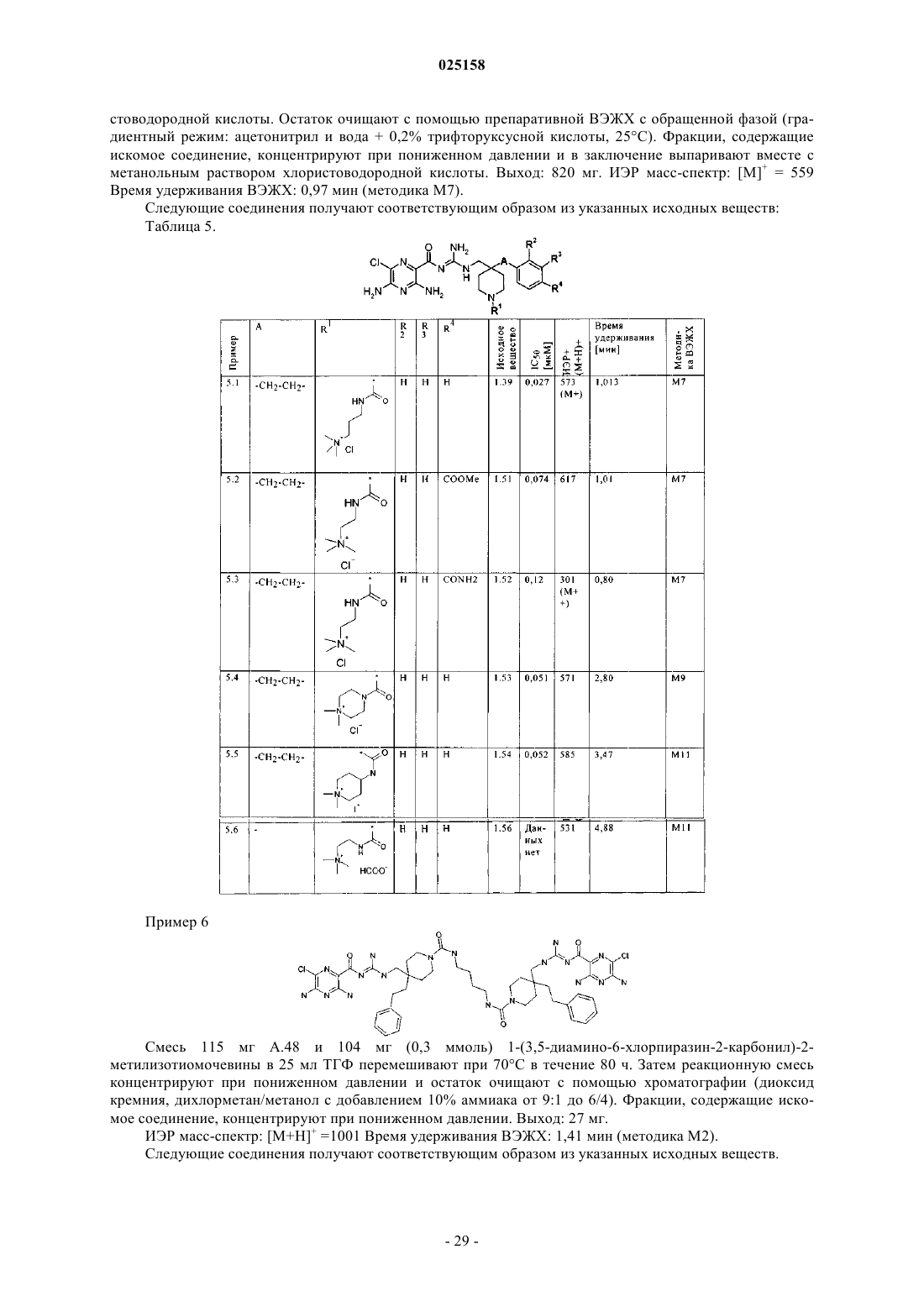
В патенте описаны соединения общей формулы (I) и их таутомеры и соли, предпочтительно их фармацевтически приемлемые соли с неорганическими или органическими кислотами и основаниями, которые обладают ценными фармакологическими характеристиками, в особенности ингибирующим воздействием на эпителиальные натриевые каналы, их применение для лечения заболеваний, в особенности заболеваний легких и дыхательных путей.(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) 1. Область техники, к которой относится изобретение Изобретение относится к соединениям общей формулы (IA) и их фармацевтически приемлемым солям с неорганическими или органическими кислотами, которые обладают ценными фармакологическими характеристиками, в особенности ингибирующим воздействием на эпителиальные натриевые каналы, к их применению для лечения заболеваний, в особенности заболеваний легких и дыхательных путей. 2. Уровень техники Соединения амилоридного типа в предшествующем уровне техники известны, как активные вещества, использующиеся, например, для лечения заболеваний легких и дыхательных путей (J.Med.Chem. 49(2006) 4098-4115). В WO 08135557 раскрыты соединения аналогичной структуры, обладающие ингибирующей активностью по отношению к ENaC (эпителиальные натриевые каналы). Задачей настоящего изобретения является получение новых соединений, которые можно использовать терапевтически для лечения патофизиологических процессов, которые поддаются лечению путем блокады эпителиальных натриевых каналов, в особенности для лечения легких и дыхательных путей. 3. Подробное описание изобретения Согласно изобретению неожиданно было установлено, что указанная выше задача решается с помощью соединений формулы (I) и (IC), предлагаемых в настоящем изобретении. Поэтому настоящее изобретение относится к соединению формулы (IA)H2NC(NH)NH-C1-C6-алкил-CO-, или выбран из группы, включающей формулы (с 1)-(с 5):R2 независимо друг от друга выбраны из группы, включающей водород, галоген, CN, С 1-С 4-алкил и С 1-алкил-О-;R6 независимо друг от друга выбраны из группы, включающей водород, галоген, CN, С 1-С 4-алкил иR4 независимо друг от друга выбраны из группы, включающей водород галоген, CN, C1-C4-алкил,C1-C4-алкил-ОСО-, -COOR4.1 и -CONR4.2R4.3,где R4.1 обозначает водород или C1-C4-алкил;R4.2 обозначает водород или C1-C4-алкил;R4.3 обозначает водород или C1-C4-алкил илиR3 и R4 вместе обозначают -О-C1-C3-алкил-О-; и его фармакологически приемлемые соли присоединения с кислотой. Особенно предпочтительными являются соединения формулы (IA), в которой R2, R3, R4, R6 и R7 обозначают водород. Также особенно предпочтительными являются соединения формулы (IA), в которой А обозначает-СН 2 СН 2-, и Е, D обозначают -СН 2-. Другим вариантом осуществления настоящего изобретения является применение соединения формулы (IA) или его фармацевтически приемлемой соли для лечения респираторных заболеваний или нарушений и аллергических заболеваний дыхательных путей. Предпочтительно соединения формулы (IA) или их фармацевтически приемлемую соль применяют для лечения заболевания, выбранного из группы, включающей хронический бронхит, острый бронхит,бронхит, вызванный бактериальной или вирусной инфекцией, или грибами, или гельминтами, аллергический бронхит, токсический бронхит, хронический обструктивный бронхит (COPD), астму (наследственную или аллергическую), астму у детей, бронхоэктаз, аллергический альвеолит, аллергический или неаллергический ринит, хронический синусит, кистозный фиброз или муковисцидоз, дефицит альфа-1 антитрипсина, кашель, эмфизему легких, интерстициальные заболевания легких, альвеолит, гиперреактивные дыхательные пути, полипы в носу, отек легких, пневмонит различной этиологии, например, вызванный радиацией или вызванный аспирацией или инфекционный пневмонит, предпочтительно хронический бронхит, острый бронхит, бронхит, хронический обструктивный бронхит (COPD), астму (наследственную или аллергическую), муковисцидоз и астму у детей, предпочтительно хронический бронхит,COPD и муковисцидоз. Фармацевтическая композиция содержит по меньшей мере одно соединение, предлагаемое в настоящем изобретении, или их фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Другим вариантом осуществления настоящего изобретения являются комбинации лекарственных средств, которые кроме одного или большего количества соединений, предлагаемых в настоящем изобретении, содержат в качестве дополнительных активных веществ одно или большее количество соединений, выбранных из следующих категорий другие ингибиторы ENaC, бета-миметики, антихолинергетики, кортикостероиды, ингибиторы PDE4, антагонисты LTD4, ингибиторы EGFR, агонисты допамина, Н 1 антигистамины, антагонисты PAF, ингибиторы киназы MAP, ингибиторы MPR4, ингибиторы iNOS, ингибиторы SYK и трансмембранный регулятор муковисцидоза (CFTR) и усиливающие факторы CFTR,предпочтительно VX-770 и VX-809 или их двойные или тройные комбинации. 4. Использующиеся термины и определения Терминам, специально не определенным в настоящем изобретении, следует придавать значения,которые им должен придавать специалист в данной области техники в соответствии с раскрытием и контекстом. Однако, если не указано иное, то при использовании в описании приведенные ниже термины обладают указанными значениями и используются указанные ниже обозначения. В группах, радикалах или фрагментах, определенных ниже, количество атомов углерода часто указывается перед группой, например, C1-C6-алкил обозначает алкильную группу или радикал, содержащий от 1 до 6 атомов углерода. Обычно в отдельных группах, таких как НО, H2N, OS, O2S, NC (цианогруппа), НООС, F3C и т.п.,специалист в данной области техники может определить положение (положения) присоединения радикала к молекуле по свободным валентностям самой группы. Для объединенных групп, содержащих две или большее количество подгрупп, последняя или первая названная группа с дефисом на конце является положением присоединения радикала, например, заместитель "арил-C1-C3-алкил-" показывает, что арильная группа связана с С 1-С 3-алкильной группой, а последняя связана с ядром или группой, к которой присоединен заместитель. Если соединение, предлагаемое в настоящем изобретении описано с помощью химического названия и в виде формулы, то случае любых различий определяющей является формула. Знак звездочки можно использовать в субформулах для обозначения связи, которая соединена с ядром молекулы, как это определено. Например, термин "3-карбоксипропильная группа" означает следующий заместитель: где карбоксигруппа присоединена к третьему атому углерода пропильной группы. Термины "1 метилпропил-", "2,2-диметилпропил-" или "циклопропилметил-" означают следующие группы: Знак звездочки можно использовать в субформулах для обозначения связи, которая соединена с ядром молекулы, как это определено. Многие их приведенных ниже терминов можно использовать в определении формулы или группы многократно и в каждом случае они обладают одним из значений, приведенных выше, независимо друг от друга. Термин "замещенный" при использовании в настоящем изобретении означает, что любой один или большее количество атомов водорода у указанного атома заменены на указанную выбранную группу при условии, что не превышается нормальная валентность указанного атома и что замещение приводит к стабильному соединению. Термин "необязательно замещенный" в объеме настоящего изобретения означает указанную выше группу, необязательно замещенную низшей молекулярной группой. Примерами низших молекулярных групп, считающихся химическим значимыми, являются группы, содержащие 1-200 атомов. Предпочтительно, чтобы такие группы не оказывали неблагоприятного влияния на фармакологическую эффективность соединений. Например, группы могут включать: Линейные или разветвленные углеродные цепи, в которые необязательно включены гетероатомы,необязательно замещенные кольцами, гетероатомами или другими обычными функциональными группами. Ароматические или неароматические кольцевые системы, состоящие из атомов углерода и необязательно гетероатомов, которые, в свою очередь, могут быть замещены функциональными группами. Целый ряд ароматических или неароматических кольцевых систем, состоящих из атомов углерода и необязательно гетероатомов, которые могут быть связаны одной или большим количеством углеродных цепей, в которые необязательно включены гетероатомы, необязательно замещенные кольцами, гетероатомами или другими обычными функциональными группами. Выражение "лечение" или "лечить" означает терапевтическое лечение пациентов, у которых уже развилось одно или большее количество указанных патологических состояний в явной, острой или хронической форме, включающее симптоматическое лечение для снятия симптомов патологического состояния или этиотропное лечение для обращения или частичного обращения патологического состояния или для задержки прогрессирования патологического состояния, если это возможно, в зависимости от патологического состояния и его тяжести. Таким образом, выражение "лечение заболевания" при использовании в настоящем изобретении означает лечение и ведение пациента, у которого развилось заболевание, патологическое состояние или нарушение. Задачей лечения является борьба с заболеванием,патологическим состоянием или нарушением. Лечение включает введение активных соединений для устранения заболевания или борьбы с заболеванием, патологическим состоянием или нарушением, а также облегчение симптомов или осложнений, связанных с заболеванием, патологическим состоянием или нарушением. Выражение "фармацевтически приемлемое" используется в настоящем изобретении для указания таких соединений, материалов, композиций и/или дозированных форм, которые в соответствии с основными положениями медицины являются подходящими для использования при соприкосновении с тканями людей и животных без проявления чрезмерной токсичности, раздражающего воздействия, аллергической реакции или других затруднений или осложнений при разумном соотношении польза/риск. При использовании в настоящем изобретении "фармацевтически приемлемые соли" означает производные раскрытых соединений, в которых исходное соединение изменено путем образования его солей с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, но не ограничиваются только ими, соли неорганических или органических кислот с основными остатками, такими как аминогруппы; соли щелочных металлов или органических соединений с кислотными остатками, такими как карбоксигруппы и т.п. Например, такие соли включают соли, образованные из аммиака, L-аргинина,бетаина, бенетамина, бензатина, гидроксида кальция, холина, диметиламиноэтанола, диэтаноламинаDL-миндальной кислоты, метансульфоновой кислоты, галактаровой кислоты, нафталин-1,5 дисульфоновой кислоты, нафталин-2-сульфоновой кислоты, 1-гидрокси-2-нафтойной кислоты, никотиновой кислоты, азотной кислоты, октановой кислоты, олеиновой кислоты, оротовой кислоты, щавелевой кислоты, пальмитиновой кислоты, памоевой кислоты (эмбоновой кислоты), фосфорной кислоты, пропионовой кислоты, (-)-L-пироглутаминовой кислоты, салициловой кислоты, 4-аминосалициловой кислоты, себациновой кислоты, стеариновой кислоты, янтарной кислоты, серной кислоты, дубильной кислоты,(+)-L-винной кислоты, тиоциановой кислоты, п-толуолсульфоновой кислоты и ундециленовой кислоты. Другие фармацевтически приемлемые соли можно получить с катионами металлов, таких как алюминий,кальций/литий, магний, калий, натрий, цинк и т. п. (см. также публикацию Pharmaceutical salts, Berge,S.M. et al., J Pharm. Sci., (1977), 66, 1-19). Если содержится не основной остаток, такой как аминогруппа,например, четвертичное аммониевое соединение, то указанные выше кислоты могут образовать фармацевтически приемлемые противоионы. Другими примерами являются гидрокарбонат, карбонат и карбонат. Как должен понимать специалист в данной области техники, соли, содержащие многовалентные ионы, могут существовать в разных стезиометрических соотношениях в зависимости от того, обладает ли многовалентный ион в единичным или кратным зарядом. Например, зарядовое состояние многоосновной кислоты зависит от степени ее депротонирования. Фармацевтически приемлемые соли, предлагаемые в настоящем изобретении, можно синтезировать из исходного соединения, которое содержит основной или кислотный фрагмент, по обычным химическим методикам. Обычно такие соли можно получить по реакции этих соединений в форме свободной кислоты или основания со стехиометрическим количеством соответствующего основания или кислоты в воде или органическом растворителе или в их смеси; обычно являются предпочтительными неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил или их смесь. Соли кислот, не указанных выше, которые, например, применимы для очистки или выделения соединений, предлагаемых в настоящем изобретении, (например, трифторацетаты), также входят в объем настоящего изобретения. Термин "галоген" при использовании в настоящем изобретении означает галогенидный заместитель, выбранный из группы, включающей фтор, хлор, бром или йод. Термин "C1-Cn-алкил", где n является целым числом, равным от 2 до n, по отдельности или в комбинации с другим радикалом означает ациклический насыщенный, разветвленный или линейный углеводородный радикал, содержащий от 1 до n атомов С. Например, термин C1-C5-алкил включает радикалы Н 3 С-, Н 3 С-СН 2-, Н 3 С-СН 2-СН 2-, Н 3 С-СН(СН 3)-, Н 3 С-СН 2-СН 2-СН 2-, Н 3 С-СН 2-СН(СН 3)-, Н 3 С-СН(СН 3)СН 2-, Н 3 С-С(СН 3)2-, Н 3 С-СН 2-СН 2-СН 2-СН 2-, Н 3 С-СН 2-СН 2-СН(СН 3)-, Н(С-СН 2-СН(СН 3)-СН 2-, H3CСН(СН 3)-СН 2-СН 2-, Н 3 С-СН 2-С(СН 3)2-, H3C-C(CH3)2-CH2-, Н 3 С-СН(СН 3)-СН(СН 3)- и Н 3 С-СН 2 СН(СН 2 СН 3)-. Термин "C3-Cn-циклоалкил", где n является целым числом, равным от 4 до n, по отдельности или в комбинации с другим радикалом означает циклический насыщенный неразветвленный углеводородный радикал, содержащий от 3 до n атомов С. Например, термин С 3-С 7-циклоалкил включает циклопропил,циклобутил, циклопентил, циклогексил и циклогептил. Во всех случаях расхождение между структурой и ее названием определяющей является структура. 5. Предпочтительные варианты осуществления Символ А обозначает связь или -СН 2 СН 2-. Символ D обозначает -СН 2-. Символ Е обозначает -СН 2-.R1 выбран из группы приведенных ниже формул (c1)-(с 5): Особенно предпочтительно, если R обозначает водород или выбран из группы, включающей C1-C6 алкил, C1-C4-алкил-SO2-, C1-C4-алкил-NH-СО-, H2N-CO-, H2N-C1-C4-алкил-, H2N-C1-C4-алкил-CO-, H2NС 1-С 4-алкил-NH-СО-, фенил-СО-, фенил-СН 2-СО-, фенил-СН 2-, C1-C6-алкил-СО-, C1-C6-алкил -О-C1-С 4 алкил-СО-, (СН 3)2N-С 1-С 4-алкил-, (СН 3)2N-C1-C4-алкил-NH-СО-, (CH3)3N+-C1-C4-алкил-NH-СО-, (СН 3)3X- обозначает любой анион, образующий фармацевтически приемлемую соль, предпочтительно выбранный из группы, включающей CF3-COO-, Cl-, I-, Br-, НСОО- и СН 3-СОО-, наиболее предпочтительноR4.1 обозначает водород или C1-C4-алкил, предпочтительно водород или C1-C2-алкил, особенно предпочтительно водород или метил,R4.2 обозначает водород или C1-C4-алкил, предпочтительно водород или C1-C2-алкил, особенно предпочтительно водород или метил,R4.3 обозначает водород или C1-C4-алкил, предпочтительно водород или C1-C2-алкил, особенно предпочтительно водород или метил, илиR3 и R4 вместе обозначают -O-C1-С 3-алкил-О-; предпочтительно -О-C1-C2-алкил-О-. Заместитель R6 выбран из группы, включаюЕдей водород, галоген, CN или C1-C4-алкил, предпочтительно водород. Любое из определений R1-R6, приведенных выше, можно объединить друг с другом и получить вариант осуществления настоящего изобретения. 6. Получение Приведенные ниже методики являются подходящими для получения соединений общей формулы(IA). Соединения, предлагаемые в настоящем изобретении, можно получить по методикам синтеза, которые известны специалисту в данной области техники и описаны в литературе по органическому синтезу. Общие методики для стадий введения или удаления защитных групп описаны, например, в публикации:and Sons, inc. Соединения предпочтительно получают по методикам, аналогичным более полно описанным ниже в настоящем изобретении, в частности, как это описано в экспериментальном разделе. Соединения общей формулы (I) можно получить по реакции S-метилизотиомочевин формулы (II) с первичными аминами формулы (III) в растворителе, таком как ТГФ (тетрагидрофуран), ацетонитрил или ДМФ (диметилформамид), или в смеси растворителей, предпочтительно в присутствии основания, в особенности если первичный амин (III) используют в виде соли присоединения с кислотой, предпочтительно при температуре от 18 до 90 С. Соединения общей формулы (I) можно превратить в соединения общей формулы (Ia) по реакции сBOC2O в присутствии основания, предпочтительно триэтиламина, в растворителе, таком как, например,ТГФ. Соединения общих формул (I) и (Ia) можно модифицировать по методикам синтеза, которые известны специалисту в данной области техники и описаны в литературе по органическому синтезу, предпочтительно с помощью стадий защиты функциональных групп или удаления защитных групп или гидрирование. Кроме того, группу R1 в соединениях общей формулы (Ia) можно модифицировать при условиях, несовместимых в ацилгуанидиновой группой, содержащейся в соединениях общей формулы I,предпочтительно алкилированием третичных аминогрупп с получением четвертичных аммониевых соединений. Соединения общей формулы (Ia) можно превратить в соединения общей формулы (I) путем удале-5 025158 ния фрагмента ВОС при стандартных условиях удаления защитной группы в кислой среде. Предпочтительной реализацией схемы 1 является схема 1.1. Схема 1.1 Соединения общей формулы (II) можно получить по реакции S-метилизотиомочевины (которую можно получить in situ из ее соли путем добавления основания) с 1-(трет-бутилкарбамоил)проп-1-ен-2 илкарбоксилатом общей формулы (IV) в растворителе, таком как ДХМ (дихлорметан), ТГФ или смесь этих растворителей, предпочтительно при температуре от -10 до 25 С. Соединения общей формулы (IV) можно полечить из соответствующей карбоновой кислоты общей формулы (V) и соли 2-трет-бутил-5-метилизоксазолия общей формулы (VI), которую можно использовать в виде изолированной соли (например, в виде гексафторфосфата; X = PF6) или получить in situ из трет-бутанола, 5-метилизоксазола и трифторметансульфоновой кислоты. Последнюю реакцию предпочтительно проводят в растворителе, таком как ДМФ, или в смеси растворителей путем добавления триэтиламин или другого основания, предпочтительно при охлаждении до 0-10 С. Соединения общей формулы (III) можно получить из соединений общей формулы (XV) восстановлением цианогруппы, предпочтительно путем гидрирования с использованием никеля Ренея в качестве катализатора под давлением водорода в присутствии избытка аммиака в растворителе, таком как например, метанол. Группу Rc в соединениях общей формулы (XV) можно модифицировать по методикам синтеза, которые известны специалисту в данной области техники и описаны в литературе по органическому синтезу, предпочтительно с помощью стадий защиты функциональных групп или удаления защитных групп, этерификации, амидирования или гидрирования. В зависимости от природы Rc этот фрагмент можно удалить по методикам синтеза, которые известны специалисту в данной области техники и описаны в литературе по органическому синтезу, предпочтительно путем удаления защитной группы с получением соединений общей формулы (XVI). Соединения общей формулы (XVI) можно превратить в соединения общей формулы (XV) по методикам синтеза, которые известны специалисту в данной области техники и описаны в литературе по органическому синтезу, предпочтительно с помощью ацилирования, алкилирования или восстановительного аминирования. Соединения общей формулы (XV), в которой D обозначает -СН 2- или -СН 2-СН 2-, можно получить по реакции алкилирующих реагентов общей формулы (VI) с 4-цианопиперидинами общей формулы (VII) в растворителе, таком как ТГФ, где соединение общей формулы (VII) депротонируют с помощью основания, предпочтительно ДАЛ (диизопропиламид лития), n-BuLi или NaH, предпочтительно при температуре от -80 до 0 С, и в которой LG обозначает отщепляющуюся группу, предпочтительно Cl, Br, I, мезилат или тозилат, и в которой G обозначает ацильный фрагмент, предпочтительно группу ВОС. Соединения общей формулы (XV), в которой А обозначает связь, можно получить двойным алкилированием фенилацетонитрилов общей формулы (IX) бис-хлорэтиламинами общей формулы (VIII) с добавлением основания, предпочтительно NaH, в растворителе, предпочтительно ДМФ, в которой Rc обозначает ацильный фрагмент, предпочтительно группу ВОС. Схема 3 Соединения общей формулы (X), в которой L обозначает цепь, содержащую по меньшей мере 2 атома углерода, можно получить по реакции (XI) с кислотой, предпочтительно ТФК (трифторуксусная кислота) или HCl, в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФ или вода, предпочтительно при температуре от 10 С и 50 С. Соединения общей формулы (XII), в которой L обозначает цепь, содержащую по меньшей мере 2 атома углерода, можно получить с помощью последовательности реакций, начиная с (XIII), вводя в него ВОС-ангидрид в качестве защитной группы, кватернизируя алкилгалогенидом, предпочтительно алкилйодидом, в растворителе, таком как ацетон, ТГФ, диоксан или дихлорметан, предпочтительно при температуре от 10 С и 50 С с последующим удалением защитной группы с помощью кислот. Соединения общей формулы (XIV), в которой L обозначает цепь, содержащую по меньшей мере 2 атома углерода, можно получить по реакции соединений общей формулы (X) с 1H-1.2.4-триазол-1 карбоксамидином или S-метилизотиомочевиной в ДМФ предпочтительно при температуре от 50 С и 90 С. 7. Примеры 7.1. Синтез промежуточных продуктов. Промежуточный продукт А.61. 3,5-Диамино-6-хлорпиразин-2-карбоновая кислота(6 моль/л в воде; 240 мл; 1,44 моля) кипятят с обратным холодильником в течение 3 ч. Смеси дают охладиться до КТ (комнатная температура) и затем нейтрализуют путем добавления хлористоводородной кислоты (6 моль/л в воде; примерно 240 мл). Добавляют воду (200 мл). Образовавшийся осадок отфильтровывают с отсасыванием, промывают водой и сушат при 60 С. Выход: 99,6 г (107% от теоретического значения) C5H5ClN4O2 ИЭР (ионизация электрораспылением) масс-спектр: m/z = 189 [М+Н]+; m/z= 187[М-Н]-. Промежуточный продукт А.62. 3,5-Диамино-6-бромпиразин-2-карбоновую кислоту получают из метил-3,5-диамино-6-бромпиразин-2-карбоксилата (который получают из метил-3,5-диамино-6-хлорпиразин-2-карбоксилата, как описа-9 025158 но в публикации J.Med.Chem. 10 (1967) 66-75) по методике, аналогичной описанной для синтеза промежуточного продукта А.61. Промежуточный продукт В.61. 1-(Трет-бутилкарбамоил)проп-1-ен-2-ил 3,5-диамино-6-хлорпиразин-2-карбоксилат Стадия 1. Смесь трет-бутанола (21,0 мл; 226 ммоль) и 5-метилизоксазола (18,0 мл; 221 ммоль) охлаждают в бане со льдом. При постоянном охлаждении по каплям добавляют трифторметансульфоновую кислоту(20,0 мл; 221 ммоль), Полученную смесь перемешивают в течение 1 ч без дополнительного охлаждения. Стадия 2. К раствору или суспензии 3,5-диамино-6-хлорпиразин-2-карбоновой кислоты (промежуточный продукт А.61; 14,0 г; 74,2 ммоль) и триэтиламина (31,0 мл; 222 ммоль) в ДМФ (100 мл) добавляют смесь,полученную на стадии 1. Полученную смесь перемешивают в течение 4 ч при КТ. При перемешивании добавляют воду со льдом. Образовавшийся осадок отфильтровывают с отсасыванием, промывают водой и сушат при 65 С и получают искомое соединение. Выход: 18,2 г (75% от теоретического значения) C13H18ClN5O3 ИЭР масс-спектр: m/z = 328 [М+Н]+; Стадия 1. Смесь 2-метил-2-бутанола (5,75 мл; 51 ммоль) и 5-метилизоксазола (4,42 мл; 51 ммоль) охлаждают в бане со льдом. При постоянном охлаждении по каплям добавляют трифторметансульфоновую кислоту(4,84 мл; 54 ммоль). Полученную смесь перемешивают в течение ночи без дополнительного охлаждения. Стадия 2. К раствору или суспензии 3,5-диамино-6-бромпиразин-2-карбоновой кислоты (промежуточный продукт А.62; 5,00 г; 21,5 ммоль) и триэтиламина (7,48 мл; 54 ммоль) в ДМФ (50 мл) при охлаждении в бане со льдом по каплям добавляют смесь, полученную на стадии 1. Полученную смесь перемешивают в течение 4 ч при КТ, затем выливают в воду со льдом. Образовавшийся осадок отфильтровывают с отсасыванием, промывают водой и сушат при 50 С и получают искомое соединечие. Выход: 7,53 г (91% от теоретического значения) C14H20BrN5O3 ИЭР масс-спектр: m/z = 386 [М+Н]+; m/z = 384 [М-Н]Промежуточный продукт С.61. 3,5-Диамино-6-хлор-N-[(метилсульфанил)метанимидоил]пиразин-2 карбоксамид К NaOH (1 моль/л в веде; 9,2 мл; 9,2 ммоль) добавляют S-метилизотиосульфат мочевины (1,78 г; 6,1 ммоль. Смесь перемешивают до полного растворения. Добавляют смесь ТБМЭ (трет-бутилметиловый эфир)/ТГФ (1:1; 30 мл) и затем 1-(трет-бутилкарбамоил)проп-1-ен-2-ил 3,5-диамино-6-хлорпиразин-2 карбоксилат (промежуточный продукт В.61; 2,00 г; 6,10 ммоль) и смесь перемешивают при КТ в течение ночи, затем добавляют воду (6 мл). Образовавшийся осадок отфильтровывают с отсасыванием, последовательно промывают водой, метанолом и затем диэтиловым эфиром и затем сушат при 50 С и получают искомое соединение. Выход: 1,33 г (84% от теоретического значения) C7H9ClN6OS ИЭР масс-спектр: m/z = 261 [М+Н]+; ммоль. Смесь перемешивают до полного растворения. ТБМЭ/ТГФ (1:1; 100 мл) и затем добавляют 1-(2 мгтил-2-бутилкарбамоил)проп-1-ен-2-ил 3,5-диамино-6-бромпиразин-2-карбоксилат (промежуточный продукт В.62; 7,52 г; 19,5 ммоль) и смесь перемешивают при КТ в течение ночи, затем добавляют воду(100 мл). Образовавшийся осадок отфильтровывают с отсасыванием, промывают смесью ТГФ/вода (1:2) и затем сушат при 50 С и получают искомое соединение. Выход: 5,44 г (92% от теоретического значения) C7H9BrN6OS ИЭР масс-спектр: m/z = 305 [М+Н]+. К раствору 21,10 г диизопропиламина в 300 мл безводного ТГФ по каплям при -78 С добавляют 83,7 мл 2,5 М раствор н-бутиллития в ТГФ. Полученный раствор перемешивают при этой температуре в течение 30 мин. Затем по каплям добавляют раствор 40,00 г 1-N-BOC-4-цианопиперидина в 300 мл ТГФ. После перемешивания в течение 1 ч по каплям добавляют 51,99 мл (2-бромэтил)-бензола. После добавления реакционной смеси дают нагреваться до комнатной температуры и перемешивают в течение ночи. Для остановки реакции добавляют 100 мл воды. ТГФ удаляют и получают взвесь, которую подвергают распределению между этилацетатом и водой. После разделения органический слой промывают насыщенным водным раствором NH4Cl и рассолом, сушат над Na2SO4 и концентрируют при пониженном давлении. Очистка с помощью колоночной хроматографии дает 57,23 г промежуточного продукта D.2. ТСХ (ЭА/ПЭ 1/8) Rf: 0,4. Промежуточный продукт D.8 получают по методике, аналогичной описанной для промежуточного продукта D.2, с использованием бензилбромида в качестве алкилирующего реагента. ТСХ (этилацетат Промежуточный продукт С.17 и получают по методике, аналогичной описанной для промежуточного продукта D.2, с использованием 4-(2-бромэтил)бензойной кислоты в качестве алкилирующего реагента. ТСХ (МеОН/дихлорметан (ДХМ) 5/95) Rf: 0,4. С.18 Промежуточный продукт С.18 и получают путем обработки С.17 аммиаком и TBTU в дихлорметане. В.17 К раствору 4,00 г промежуточного продукта С.17 в 40 мл сухого ДМФ по каплям добавляют 5,40 гK2CO3 и затем 2,40 мл метилйодида. Реакционную смесь перемешивают при комнатной температуре в течение 12 ч. Затем добавляют воду и смесь экстрагируют диэтиловым эфиром. Органические фазы объединяют, сушат над Na2SO4 и выпаривают. Полученный неочищенный продукт очищают с помощью ЖХ К раствору 2,00 г N-BOC-N,N-бис(2-хлорэтил)амина и 1,10 г (2-метоксифенил)-ацетонитрида в 1 5 мл ТГФ и 5 мл ДМФ порциями при КТ добавляют 0,78 г NaH и смесь перемешивают при 55 С в течение 16 ч. Реакцию останавливают путем добавления холодной воды и экстрагируют этилацетатом. Органический слой промывают рассолом и водой, сушат над Na2SO4 фильтруют и выпаривают при пониженном давлении. Неочищенное твердое вещество растирают со смесью CHCl3 и эфира, фильтруют и сушат и получают 1,0 г промежуточного продукта D.45. ТСХ (ЭА:ПЭ 3/7) Rf: 0,6.D.43 и D.44 получают по методике, аналогичной описанной для промежуточного продукта D.45, с использованием соответствующих бензилцианидов. Смесь 2,50 г пиперидина D.2 и 40 мл 25% ТФК в дихлорметане перемешивают в течение 1,5 ч при комнатной температуре. Растворитель выпаривают, добавляют метанольный раствор хлористоводородной кислоты и растворитель повторно выпаривают и получают 2,53 г промежуточного продукта С.2. ИЭР-МС m/z: 215. Соответствующую соль С.2 с ТФК получают путем очистки неочищенного продукта с помощью препаративной ВЭЖХ с обращенной фазой с использованием ТФК в качестве модификатора. С.8, С.43, С.44 и C45. Следующие промежуточные продукты получают по методике, аналогичной описанной для промежуточного продукта С.2, с использованием модифицированной обработки: нейтрализации реакционной смеси насыщенным раствором NaHCO3 с последующей обработкой водой. Смесь 2,30 г пиперидингидрохлорида С.2, 1,08 мл 2-изоцианатопролана, 1,99 мл триэтиламина и 50 мл ТГФ перемешивают при 50 С в течение 2 ч. Реакционную смесь концентрируют при пониженном давлении и добавляют воду и в заключение экстрагируют дихлорметаном. Объединенные органические фазы сушат над MgSO4 и выпаривают и получают 1,65 г промежуточного продукта В.2. ИЭР-МС m/z: 300, 344. В.4, В.8, В.12, В.18, В.19, В.43, В.44, В.45, В.48 и В.49. Следующие промежуточные продукты получают по методике, аналогичной описанной для промежуточного продукта В.2, с использованием соответствующих изоцианатов. ДХМ можно использовать в качестве альтернативного растворителя. 0,22 мл Триэтиламина медленно добавляют к смеси 0,34 г пиперидинтрифторацетата С.2, 0,32 г трифосгена и 5 мл дихлорметана. Смесь перемешивают при комнатной температуре в течение 4 ч и добавляют 0,17 мл N,N-диметилэтилендиамина и реакционную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь концентрируют при пониженном давлении, добавляют смесь ДМФ, метанола и ТФК и полученную смесь фильтруют и очищают с помощью препаративной ВЭЖХ с обращенной фазой. Фракции, содержащие продукт, объединяют и выпаривают. Полученный остаток переносят в дихлорметан и добавляют 4 н. раствор NaOH. Органическую фазу отделяют с помощью картриджа для разделения фаз. Выпаривание дает 0,22 г промежуточного продукта В. 10. ИЭР-MCm/z: 329. В.33 В.34 В.39 В.35 В.36 В.38 В.40, В.51, В.52, В.53, В.54, В.55 и В.56. Следующие промежуточные продукты получают по методике, аналогичной описанной для промежуточного продукта В.10, с использованием соответствующих аминов. К суспензии 0,75 г 1,1'-карбонилди(1,2,4-триазола) в 5 мл ТГФ по каплям добавляют раствор 0,5 мл 1-метилпиперазина в 5 мл ТГФ. Реакционную смесь перемешивают при комнатной температуре в течение 40 мин, затем по каплям добавляют раствор 0,5 г промежуточного продукта С.2 (в виде свободного основания) в 5 мл ТГФ и реакционную смесь перемешивают при 60 С в течение ночи. Растворитель выпаривают, полученный неочищенный продукт подвергают распределению между дихлорметаном и водой и органическую фазу отделяют, сушат над сульфатом натрия и концентрируют в вакууме. Неочищенный продукт очищают с помощью флэш-хроматографии (элюент: AcOEt/MeOH=80/20) и повторно очищают с помощью препаративной ЖХ-МС (с обращенной фазой; NH4COOH). Получают 150 мг промежуточного продукта В.53. В.54 и В.55. Следующие промежуточные продукты получают по методике, аналогичной описанной для промежуточного продукта В.53, с использованием соответствующих аминов. В.56 получают по методике, аналогичной использованной для получения В.53, с использованием в качестве исходных веществ имеющихся в продаже 4-циано-4-фенилпиперидина и N,N-диметилэтилендиамина. В.3 Промежуточный продукт В.3 получают с помощью восстановительного аминирования промежуточного продукта С.2 с помощью формальдегида и NaCNBH3 в ТГФ. В.7 и В.21. Следующие промежуточные продукты получают по методике, аналогичной описанной для промежуточного продукта В.3, с использованием соответствующих карбонильных соединений. Промежуточный продукт В.5 получают с помощью ацилирования промежуточного продукта С.2 бензоилхлоридом и гриэтиламином в дихлорметане.B.1, B.11, В.13, В.14, В.9 В.20 и В.47. Следующие промежуточные продукты получают по методике, аналогичной описанной для промежуточного продукта В.5, с использованием соответствующих хлорангидридов кислот. К смеси 1,07 г промежуточного продукта С.2, 1,42 г N-BOC-бета-аланина, 2,5 г EDCI в 50 мл безводного ТГФ добавляют 4,90 мл триэтиламина и 0.1 г ДМАП (диметиламинопиридин). Реакционную смесь перемешивают при КТ в течение ночи. Добавляют воду и смесь концентрируют и экстрагируют этилацетатом. Объединенные органические фазы промывают насыщенным водным раствором NH4Cl и рассолом, сушат над Na2SO4 и концентрируют. Оставшийся неочищенный продукт очищают с помощью ЖХ и получают промежуточный продукт В.22 В.23, В.25, В.24. В.26, В.27, В.46 и В.42. Следующие промежуточные продукты получают по методике, аналогичной описанной для промежуточного продукта В.22, с использованием соответствующих кислот. Промежуточный продукт В.6 получают алкилированием промежуточного продукта С.2 с помощью бензилбромида и Cs2CO3 в ацетонитриле. В.15. Промежуточный продукт В.15 получают по следующей методике. 0,1 г С.2 растворяют в 5 мл ацетона и добавляют 0,1 г K2CO3 и затем по каплям добавляют метилйодид при комнатной температуре. Полученную реакционную смесь перемешивают в течение 16 ч. Затем реакционную смесь разбавляют этилацетатом, промывают водой, затем рассолом. Органический слой сушат над Na2SO4 и концентрируют при пониженном давлении. Неочищенный продукт очищают с помощью хроматографии при элюировании с помощью 18% этилацетат/петролейный эфир на колонке с силикагелем. В.50. Следующий промежуточный продукт и получают по методике, аналогичной описанной для промежуточного продукта В.15, с использованием метилйодида. В.16, В.28, В.29, В.30, В.31, В.32 и В.37. Следующие промежуточные продукты получают по методике, аналогичной описанной для промежуточного продукта В.6, с использованием соответствующих аминов и соответствующих алкилгалогенидов в качестве алкилирующих реагентов, K2CO3 и ацетона. Суспензию 1,83 г нитрила В.2, 0,40 г никеля Ренея и 40 мл метанольного раствора аммиака гидрируют при комнатной температуре и давлении Н 2, равном 3 бар, в течение 23 ч. В случае неполного превращения дополнительно добавляют катализатор и растворитель и гидрирование продолжают в течение 54 при 50 С. Катализатор удаляют фильтрованием и фильтрат выпаривают и получают 1,95 г промежуточного продукта А.2. ИЭР-МС m/z: 304.A1, А.3, А.4, А.5, А.6, А.7, А.10, А.11, А.12, А.13, А.14, А.15, А.16, А.17, А.18, А.19, А.22, А.23,А.25, А.28, А.29, А.32, А.33, А.34, А.39, А.48, А.51, А.52, А.53, А.54, А.55, А.56 и А.57. Следующие промежуточные продукты получают из соответствующих нитрилы по методике, аналогичной описанной для промежуточного продукта А.2. К раствору 50 мг промежуточного продукта В.55 в 3 мл ТГФ по каплям добавляют 0,1 мл комплекса борана с тетрагидрофураном. Реакционную смесь перемешивают при комнатной температуре в течение 30 мин, затем при 40 С в течение 3 ч. Повторно добавляют 0,1 мл комплекса борана с тетрагидрофураном и реакционную смесь перемешивают при 50 С в течение ночи. Реакционную смесь подвергают распределению между дихлорметаном и водой, органическую фазу промывают насыщенным водным раствором NaHCO3, сушат с помощью картриджа для разделения фаз и концентрируют в вакууме и получают 42 мг промежуточного продукта А.59. А.60 К раствору 1 г промежуточного продукта В.56 в 20 мл ТГФ при перемешивании при -78 С по каплям добавляют 1,2 мл 2 М раствора алюмогидрида лития в ТГФ. Смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Растворитель концентрируют, реакционную смесь подвергают распределению между дихлорметаном и водой и органическую фазу сушат с помощью картриджа для разделения фаз и концентрируют в вакууме и получают 500 мг неочищенного продукта. 200 мг Этого неочищенного продукта очищают с помощью препаративной ЖХ-МС (с обращенной фазой; NH4COOH). Получают 60 мг чистого промежуточного продукта А.60. 7.2. Примеры синтеза. Пример 1. Трет-бутиловый эфир 4-[N'-(3,5-диамино-6-хлорпиразин-2-карбонид)-гуанидинометил]4-фенетилпиперидин-1-карбоновой кислоты Смесь 80 мг (0,3 ммоль) трет-бутилового эфира 4-аминометил-4-фенетилпиперидин-1 карбоциклической кислоты (А.55) и 104 мг (0,3 ммоль) 1-(3,5-диамино-6-хлорпиразин-2-карбонил)-2 метилизотиомочевины (промежуточный продукт С.61) в 2 мл ацетонитрила перемешивают при 70 С в течение 48 ч. Затем реакционную смесь концентрируют при пониженном давлении и остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой (градиентный режим: ацетонитрил и вода + 0,2% трифторуксусной кислоты, 25 С). Фракции, содержащие искомое соединение, концентрируют при пониженном давлении. Выход: 116 мг. ИЭР масс-спектр: [М+Н]+ = 531 Время удерживания ВЭЖХ 2,51 мин (методика M1). Следующие соединения получают соответствующим образом из указанных исходных веществ. Таблица 1 К раствору 140 мг (0,36 ммоль) промежуточного продукта А.57 в 2,5 мл ДМФ добавляют 0,13 млN,N-диизопропилэтиламина. Реакционную смесь перемешивают при комнатной температуре в течение 10 мин затем добавляют 85 мг (0,33 ммоль) 1-(3,5-диамино-6-хлорпиразин-2-карбонил)-2-метилизотиомочевины. Реакционную смесь нагревают при 70 С в течение 3 ч. Растворитель удаляют и полученный неочищенный продукт очищают с помощью флэш-хроматографии (первый элюент: AcOEt/MeOH=90/10 для удаления примесей; второй элюент дихлорметан/MeOH/NH3 от 90/10/0,1 до 50/50/0,1) и получают искомый продукт. Выход: 55 мг. IC50[мкМ] = 0,014 ИЭР масс-спектр: [М+Н]+ =557. Время удерживания ВЭЖХ: 5,60 мин (методика М 9). Следующие соединения получают соответствующим образом из указанных исходных веществ: Таблица 1.1 Смесь 50 мг (0,3 ммоль) трет-бутилового эфира 4-[N'-(3,5-диамино-6-хлорпиразин-2-карбонил)гуанидинометил]-4-фенетилпиперидин-1-карбоновой кислоты (пример 1) в 1 мл дихлорметана перемешивают с 250 мкл трифторуксусной кислоты при комнатной температуре в течение 2 ч. Затем реакционную смесь концентрируют при пониженном давлении. Выход: 40 мг. ИЭР масс-спектр: [М+Н]+ = 431 Время удерживания ВЭЖХ: 1,78 мин (методика M1). Следующие соединения получают соответствующим образом из указанных исходных веществ. Таблица 2 Смесь 145 мг (0,23 ммоль) метилового эфира 4-[N'-(3,5-диамино-6-хлорпиразин-2-карбонил)гуанидинометил]-4-фенетилпиперидин-1-ил -уксусной кислоты (пример 1 16) в 5 мл метанола и 235 мкл 4 н. NaOH перемешивают при 50 С в течение 1 ч. Затем раствор подкисляют с помощью 470 мкл 4 н. HCl и концентрируют при пониженном давлении. Остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой (градиентный режим: ацетонитрил и вода + 0,2% трифторуксусной кислоты, 25 С). Фракции, содержащие искомое соединение, концентрируют при пониженном давлении. Выход: 35 мг. ИЭР масс-спектр: [М+Н]+ = 489. Время удерживания ВЭЖХ: 1,33 мин (методика М 2). Следующие соединения получают соответствующим образом из указанных исходных веществ: Таблица 3 Смесь 70 мг (0,15 ммоль) N-(3,5-диамино-6-хлорпиразин-2-карбонил)-N'-(4-фенетилпиперидин-4 илметилгуанидина (пример 2), 124 мкл триэгиламина и 28 мг (0,19 ммоль) 1 Н-1.2.4-триазол-1 карбоксамидинмоногидрохлорида в 5 мл ДМФ перемешивают при 70 С в течение 2 ч. Затем добавляют 1 мл метанола и смесь очищают с помощью препаративной ВЭЖХ с обращенной фазой (градиентный режим: ацетонитрил и вода + 0,2% трифторуксусной кислоты, 25 С). Фракции, содержащие искомое соединение, концентрируют при пониженном давлении. Выход: 15 мг. ИЭР масс-спектр: [М+Н]+ = 473 Время удерживания ВЭЖХ: 0,93 мин (методика М 7). Следующие соединения получают соответствующим образом из указанных исходных веществ. Таблица 4 Пример 5.A. 1,55 г (2-Диметиламиноэтил)амида. 4-[N'-(3,5-Диамино-6-хлорпиразин-2-карбонил)-гуанидинометил]-4-фенетилпиперидин-1 карбоновой кислоты (пример 1.40), 0,9 мл триэтиламина и 1,1 г ВОС-ангидрида растворяют в 50 мл ТГФ и перемешивают в течение ночи. Органический слой отделяют и концентрируют при пониженном давлении. Выход: 1,3 г. Смесь 1,3 г соединения примера 5.A и 200 мкл метилйодида в 10 мл ацетона перемешивают в течение ночи при комнатной температуре. Затем реакционную смесь концентрируют при пониженном давлении и добавляют 5 мл 50% раствора трифторуксусной кислоты в дихлорметане и перемешивают в течение 2 ч при комнатной температуре. Затем смесь выпаривают вместе с метанольным раствором хлори- 28025158 стоводородной кислоты. Остаток очищают с помощью препаративной ВЭЖХ с обращенной фазой (градиентный режим: ацетонитрил и вода + 0,2% трифторуксусной кислоты, 25 С). Фракции, содержащие искомое соединение, концентрируют при пониженном давлении и в заключение выпаривают вместе с метанольным раствором хлористоводородной кислоты. Выход: 820 мг. ИЭР масс-спектр: [М]+ = 559 Время удерживания ВЭЖХ: 0,97 мин (методика М 7). Следующие соединения получают соответствующим образом из указанных исходных веществ: Таблица 5. Смесь 115 мг А.48 и 104 мг (0,3 ммоль) 1-(3,5-диамино-6-хлорпиразин-2-карбонил)-2 метилизотиомочевины в 25 мл ТГФ перемешивают при 70 С в течение 80 ч. Затем реакционную смесь концентрируют при пониженном давлении и остаток очищают с помощью хроматографии (диоксид кремния, дихлорметан/метанол с добавлением 10% аммиака от 9:1 до 6/4). Фракции, содержащие искомое соединение, концентрируют при пониженном давлении. Выход: 27 мг. ИЭР масс-спектр: [М+H]+ =1001 Время удерживания ВЭЖХ: 1,41 мин (методика М 2). Следующие соединения получают соответствующим образом из указанных исходных веществ.
МПК / Метки
МПК: A61P 11/00, C07D 401/14, A61K 31/497, C07D 401/12
Метки: способы, применение, гетероциклические, соединения, содержащие, лекарственные, средства, получения, указанные
Код ссылки
<a href="https://eas.patents.su/30-25158-geterociklicheskie-soedineniya-lekarstvennye-sredstva-soderzhashhie-ukazannye-soedineniya-ih-primenenie-i-sposoby-ih-polucheniya.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероциклические соединения, лекарственные средства, содержащие указанные соединения, их применение и способы их получения</a>
Дейтерированные производные катехоламина и лекарственные средства, содержащие указанные соединения

Номер патента: 17983
Опубликовано: 30.04.2013
Авторы: Шнайдер Франк, Алкен Рудольф-Гизберт
МПК: C07B 59/00, C07C 229/26
Метки: производные, катехоламина, дейтерированные, лекарственные, указанные, содержащие, средства, соединения
Формула / Реферат:
1. Дейтерированные производные катехоламина общей формулы Iгде R1 представляет собой H;R2 представляет собой D;R3 представляет собой H, метил или этил;R4 представляет собой H;R5 представляет собой Н или D;R6 представляет собой Н или D, где оба остатка R6 не являются одновременно D и где оба остатка R6 не являются одновременно H,их стереоизомеры, энантиомеры или диастереомеры в оптически чистой форме, а также их физиологически приемлемые соли.2....
Бициклические гетероциклы, содержащие эти соединения лекарственные средства, их применение и способ их получения

Номер патента: 4981
Опубликовано: 28.10.2004
Авторы: Химмельсбах Франк, Юнг Биргит, Лангкопф Эльке, Баум Анке, Золка Флавио, Метц Томас
МПК: A61P 35/00, C07D 239/94, A61K 31/517...
Метки: содержащие, средства, эти, лекарственные, способ, применение, соединения, гетероциклы, получения, бициклические
Формула / Реферат:
1. Бициклические гетероциклы общей формулы I в которой Ra обозначает атом водорода или C1-C4алкильную группу, Rb обозначает фенильную, бензильную или 1-фенилэтильную группу, в которых фенильное ядро замещено соответственно остатками R1-R3, где R1 и R2 могут иметь идентичные либо разные значения и каждый представляет собой соответственно атом водорода, фтора, хлора, брома или иода, C1-C4алкильную, гидрокси-, C1-C4алкокси-,...
Производные хиназолина, содержащие эти соединения лекарственные средства, их применение и способ их получения

Номер патента: 6317
Опубликовано: 27.10.2005
Авторы: Золка Флавио, Лангкопф Эльке, Баум Эльке, Юнг Биргит, Химмельсбах Франк, Блех Штефан
МПК: A61P 35/00, A61K 31/505, C07D 239/94...
Метки: эти, получения, соединения, лекарственные, применение, производные, средства, содержащие, способ, хиназолина
Формула / Реферат:
1. Производные хиназолина общей формулы в которой Ra обозначает бензильную, 1-фенилэтильную или 3-хлор-4-фторфенильную группу, Rb обозначает диметиламино-, N-метил-N-этиламино-, диэтиламино-, N-метил-N-изопропиламино-, N-метил-N-циклопропиламино-, N-метил-N-(2-метоксиэтил)амино-, N-этил-N-(2-метоксиэтил)амино-, бис(2-метоксиэтил)амино-, морфолино-, N-метил-N-(тетрагидрофуран-3-ил)амино-, N-метил-N-(тетрагидрофуран-2-илметил)амино-,...
Бициклические гетероциклы, содержащие эти соединения лекарственные средства, их применение и способ их получения

Номер патента: 5679
Опубликовано: 28.04.2005
Авторы: Лангкопф Эльке, Юнг Биргит, Блех Штефан, Золка Флавио, Химмельсбах Франк
МПК: A61K 31/517, A61P 35/00, C07D 239/94...
Метки: содержащие, средства, получения, бициклические, гетероциклы, эти, способ, применение, лекарственные, соединения
Формула / Реферат:
1. Бициклические гетероциклы общей формулы в которой Ra обозначает бензильную или 1-фенилэтильную группу либо замещенную остатками R1 и R2 фенильную группу, при этом R1 представляет собой атом водорода, фтора, хлора или брома, метильную, трифторметильную, циано- или этинильную группу и R2 представляет собой атом водорода или фтора, и один из заместителей Rb или Rc обозначает R3-(CH2)m-O-группу, а другой из заместителей Rb или Rc обозначает...
Замещённые глюкопиранозилом бензольные производные, содержащие эти соединения лекарственные средства, их применение и способ их получения

Номер патента: 11158
Опубликовано: 27.02.2009
Авторы: Эккхардт Маттиас, Томас Лео, Барсумян Эдуард Леон, Химмельсбах Франк, Айккельманн Петер
МПК: C07D 309/10, A61K 31/351, A61P 3/00...
Метки: применение, глюкопиранозилом, эти, замещённые, способ, средства, получения, соединения, бензольные, лекарственные, содержащие, производные
Формула / Реферат:
1. Замещенные глюкопиранозилом бензольные производные общей формулы I в которой R1 выбран среди значений группы A; а когда R3 выбран среди значений группы B, дополнительно может также представлять собой водород, фтор, хлор, бром, йод, C1-C4алкил, замещенную 1-3 атомами фтора метильную группу, замещенную 1-5 атомами фтора этильную группу, C1-C4алкоксигруппу, замещенную 1-3 атомами фтора метоксигруппу, замещенную 1-5 атомами фтора этоксигруппу,...
Предыдущий патент: Способ переработки отходящего газа синтеза фишера-тропша
Следующий патент: Складной механизм
Случайный патент: Автомобильная шина