Иммуноконъюгат и его применение для лечения рака
Номер патента: 24844
Опубликовано: 31.10.2016
Авторы: Чзан Цянь, Пань Чинь, Дервин Дэниел Д., Гангвар Санжив




























Формула / Реферат
1. Иммуноконъюгат или его фармацевтически приемлемая соль, в котором соединение формулы (II)

где X представляет собой Cl, Br или тозилат;
ААа и каждый AAb независимо выбираются из группы, состоящей из аланина, β-аланина, γ-аминомасляной кислоты, аргинина, аспарагина, аспарагиновой кислоты, γ-карбоксиглутаминовой кислоты, цитруллина, цистеина, глутаминовой кислоты, глутамина, глицина, гистидина, изолейцина, лейцина, лизина, метионина, норлейцина, норвалина, орнитина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина;
m равно 0, 1, 2, 3, 4 или 5;
Y представляет собой спейсерный фрагмент, причем Y выбран из группы, состоящей из

и их комбинаций,
где индекс q равен 0 или 1, независимо в каждом случае, а индекс r равен от 1 до 24, независимо в каждом случае; и
Z представляет собой реакционноспособную функциональную группу, способную к конъюгации с антителом, где -Z представляет собой -NH2, -ОН, -ONH2, -СО2Н, -SH, циклооктин, -N3,

сконъюгировано через группу Z к антителу или к антигенсвязывающему фрагменту такого антитела.
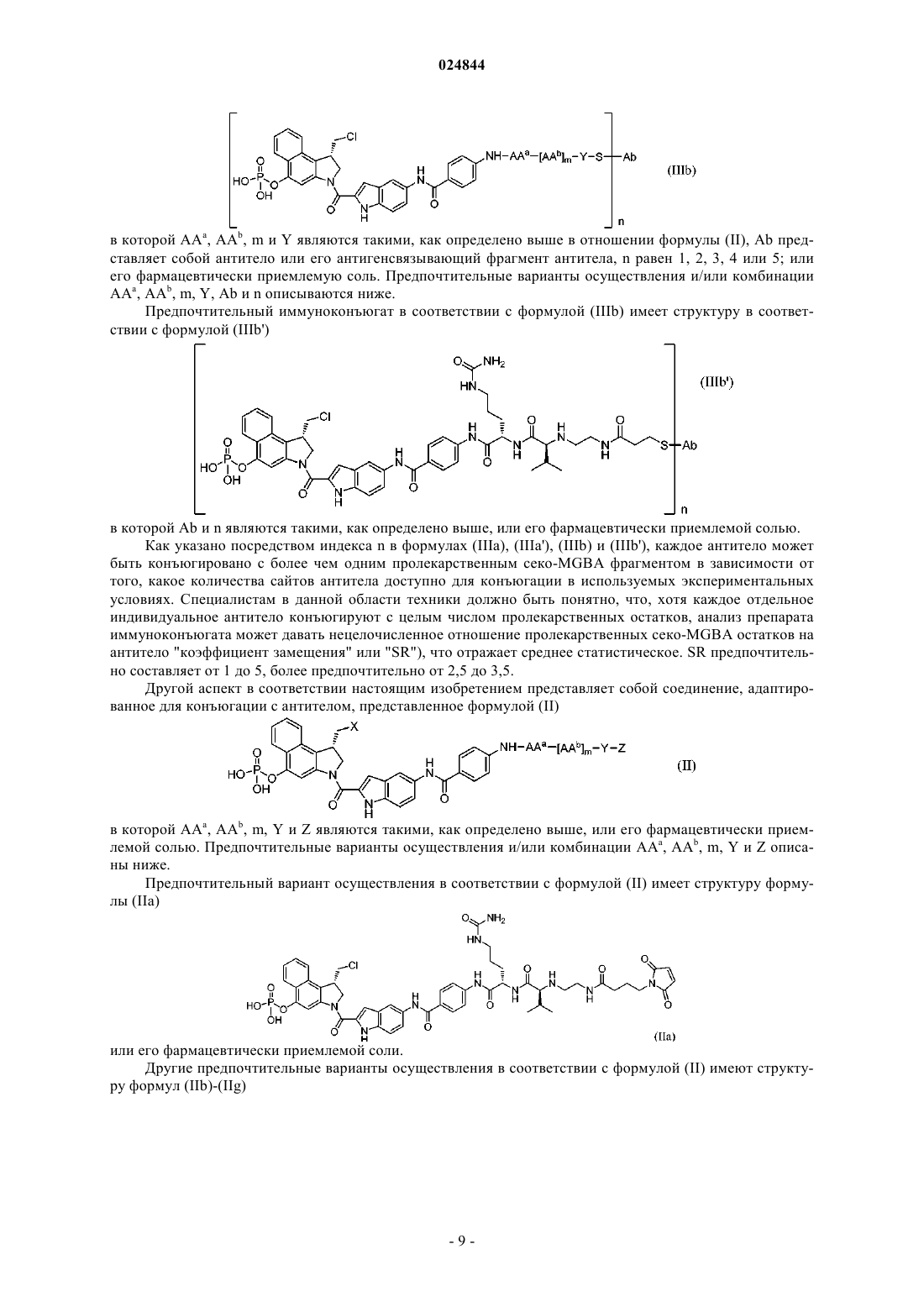
2. Иммуноконъюгат по п.1 или его фармацевтически приемлемая соль, где иммуноконъюгат имеет структуру, соответствующую формуле (IIIa)

где ААа и каждый AAb независимо выбираются из группы, состоящей из аланина, β-аланина, γ-аминомасляной кислоты, аргинина, аспарагина, аспарагиновой кислоты, γ-карбоксиглутаминовой кислоты, цитруллина, цистеина, глутаминовой кислоты, глутамина, глицина, гистидина, изолейцина, лейцина, лизина, метионина, норлейцина, норвалина, орнитина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина;
m равно 0, 1, 2, 3, 4 или 5;
Y представляет собой спейсерный фрагмент, причем Y выбран из группы, состоящей из

и их комбинаций,
где индекс q равен 0 или 1, независимо в каждом случае, а индекс r равен от 1 до 24, независимо в каждом случае;
n равно 1, 2, 3, 4 или 5 и
Ab представляет антитело или его антигенсвязывающий фрагмент.
3. Иммуноконъюгат по п.1 или его фармацевтически приемлемая соль, где иммуноконъюгат имеет структуру, соответствующую формуле (IIIa')

где n равно 1, 2, 3, 4 или 5 и
Ab представляет антитело или его антигенсвязывающий фрагмент.
4. Иммуноконъюгат по п.3, в котором антитело представляет собой человеческое моноклональное антитело, которое распознает человеческий антиген, выбранный из группы, состоящей из CD70, мезотелина, ПСМА, CD19, глипикана-3, В7Н4, RG-1, CD22 и PTK7.
5. Соединение или его фармацевтически приемлемая соль, где соединение имеет структуру в соответствии с формулой (II)

где X представляет собой Cl, Br или тозилат;
ААа и каждый AAb независимо выбираются из группы, состоящей из аланина, β-аланина, γ-аминомасляной кислоты, аргинина, аспарагина, аспарагиновой кислоты, γ-карбоксиглутаминовой кислоты, цитруллина, цистеина, глутаминовой кислоты, глутамина, глицина, гистидина, изолейцина, лейцина, лизина, метионина, норлейцина, норвалина, орнитина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина;
m равно 0, 1, 2, 3, 4 или 5;
Y представляет собой спейсерный фрагмент, причем Y выбран из группы, состоящей из

и их комбинаций,
где индекс q равен 0 или 1, независимо в каждом случае, а индекс r равен от 1 до 24, независимо в каждом случае; и
Z представляет собой реакционноспособную функциональную группу, способную к конъюгации с антителом, где -Z представляет собой -NH2, -ОН, -ONH2, -СО2Н, -SH, циклооктин, -N3

6. Соединение по п.5, в котором ААа выбирается из группы, состоящей из цитруллина, лизина, аспарагиновой кислоты, глутаминовой кислоты, серина, изолейцина, лейцина и треонина и m равно 0, 1 или 2.
7. Соединение по п.5, где -ААа-[AAb]m- представляет собой дипептид, выбранный из группы, состоящей из Val-Cit, Phe-Cit, Phe-Lys, Val-Lys, Val-Glu, Val-Asp, Val-Ser и Val-Gly, каждый дипептид, читаемый в направлении от N- к -C.
8. Соединение по п.5, имеющее структуру в соответствии с формулой (IIa)

или его фармацевтически приемлемая соль.
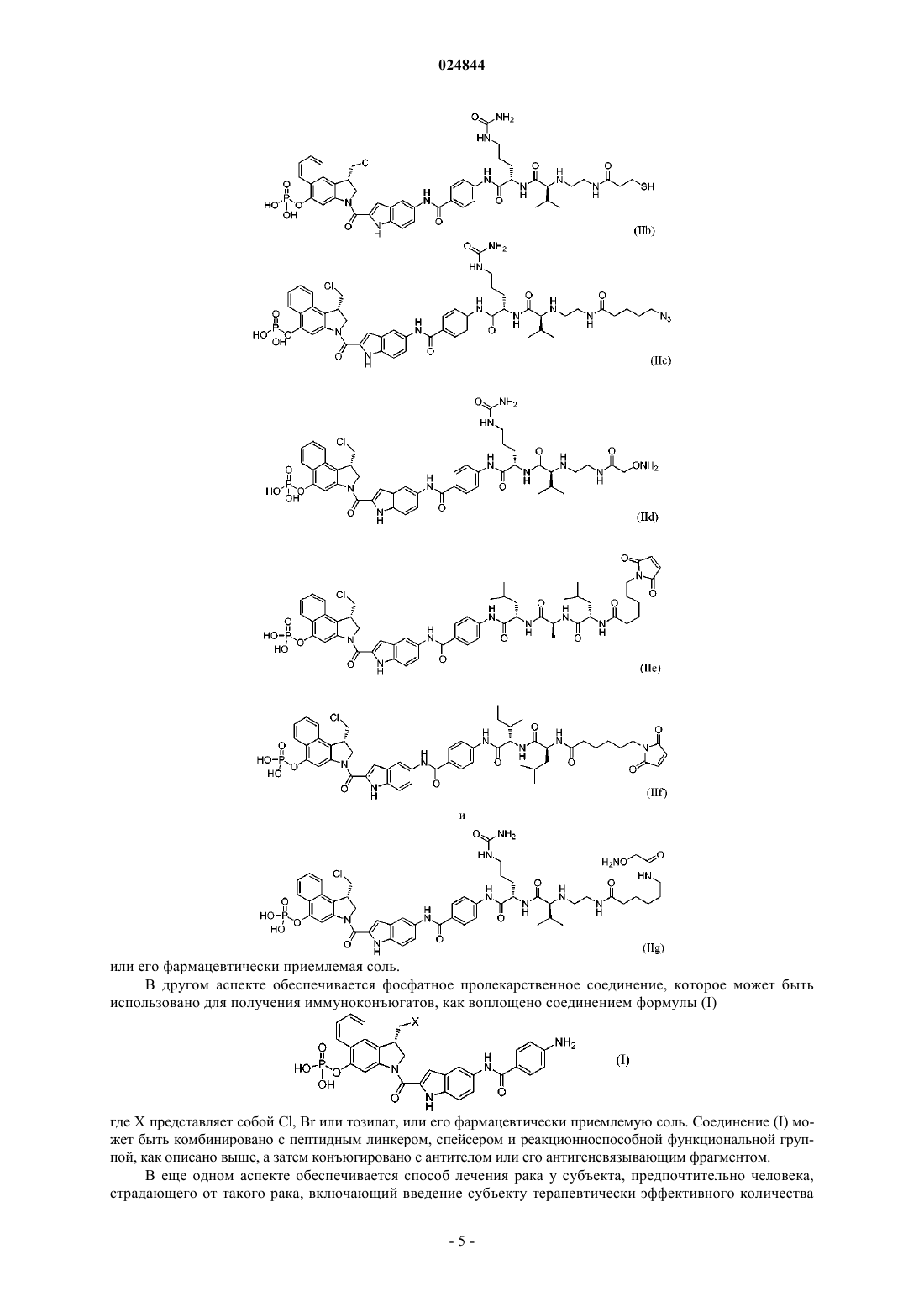
9. Соединение по п.5, имеющее структуру, выбранную из группы, состоящей из формул (IIb)-(IIg)


или его фармацевтически приемлемая соль.
10. Соединение в соответствии с формулой (I)

где X представляет собой Cl, Br или тозилат,
или его фармацевтически приемлемая соль.
11. Способ лечения рака у субъекта, страдающего от такого рака, включающий введение субъекту терапевтически эффективного количества иммуноконъюгата, имеющего структуру по п.3.
12. Способ по п.11, в котором рак представляет собой рак почки, рак поджелудочной железы, рак яичников, лимфому, рак толстой кишки, мезотелиому, рак желудка, рак легкого, рак предстательной железы, аденокарциному, рак печени или рак молочной железы.
13. Способ по п.11, в котором рак характеризуется раковыми клетками, которые экспрессируют человеческие CD70, мезотелин, ПСМА, CD19, глипикан-3, В7Н4, RG-1, CD22 или PTK7.
Текст
ИММУНОКОНЪЮГАТ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА Предложен иммуноконъюгат или его фармацевтически приемлемая соль, в котором соединение формулы (II), где X представляет собой Cl, Br или тозилат, Y представляет собой спейсерный фрагмент, Z представляет собой реакционноспособную функциональную группу, способную к конъюгации с антителом, сконъюгирировано с антителом или с антигенсвязывающим фрагментом антитела. Также предложен способ лечения рака у субъекта с помощью такого иммуноконъюгата. Область техники Настоящее изобретение относится к терапевтическим иммуноконъюгатам и способам применения таких иммуноконъюгатов для лечения рака. Уровень техники Натуральные продукты СС-1065 и дуокармицины являются мощными цитотоксическими агентами,которые связываются с малой бороздкой ДНК. Они характеризуются конденсированным циклопропановым кольцом (пунктирная рамка в структурах ниже), которое дестабилизируется посредством конформационных изменений, вызываемых при связывании в малой бороздке, и реагирует с адениновым основанием ДНК в реакции алкилирования с раскрытием кольца. Повреждение ДНК может быть непоправимым, что приводит к гибели клеток. Активность этих натуральных продуктов стимулировало исследования, направленные на разработку аналогов, полезных в качестве противоопухолевых препаратов. См., например, Cacciari et al. 2000 иSuckling 2004 (полные ссылки на документы, приведенные в настоящем изобретении в виде первого автора или изобретателя и года, перечислены в конце этого описания. Такие перечисленные документы включены в настоящее описание посредством ссылки). Аналоги имеют либо конденсированный циклопропановый фармакофор, либо эквивалент с открытым кольцом (секо), как показано в структурах ниже,где Ar представляет собой ароматическое кольцо, которое обычно представляет собой фенил или пиррол,и X представляет собой уходящую группу, такую как хлор или бром. секо-Форма может превращаться в циклопропановую форму посредством элиминирования НХ, и этот процесс может происходить либо in В дальнейшем в настоящем изобретении термин "агент, связывающийся с малой бороздкой" или"MGBA" будет использоваться для ссылки на тип соединения СС-1065/дуокармицин, имеющих конденсированное циклопропановое кольцо или его секо-форму, хотя известны и другие типы соединений, связывающиеся с малой бороздкой ДНК. Иммуноконъюгаты представляют область текущего большого интереса в противоопухолевой терапии. В иммуноконъюгате фрагмент лекарственного препарата конъюгирован (ковалентно связан) с антителом, антиген которого уникально экспрессируется или сверхэкспрессируется раковой клеткой ("опухоль-ассоциированный антиген"). В связывании с его антигеном антитело функционирует как нацеливающий агент для доставки фрагмента лекарственного препарата к раковой клетки с высокой специфичностью. Антиген может представлять собой белок на поверхности раковой клетки. После связывания антитела с антигеном комплекс антиген-иммуноконъюгат интернализуется и в конечном счете попадает внутрь везикулярного тела, такого как лизосомы, где ковалентный линкер между фрагментом лекарственного препарата и антитела расщепляется, высвобождая молекулу лекарственного препарата, которая оказывает свой цитотоксический эффект. С другой стороны, ассоциированный с опухолью антиген может быть антигеном, который секретируется опухолевыми клетками в близлежащее экстрацеллюлярное пространство. Предпочтительно, чтобы ковалентный линкер был сконструирован таким образом, чтобы расщепление осуществлялось посредством фактора, предпочтительно находящимся в лизосоме, но не в плазме. Одним из таких факторов является низкое значение лизосомального рН, так что ковалентный линкер может быть чувствительной к кислоте группой, такой как гидразон. Другим таким фактором является обычно высокая внутриклеточная концентрация глутатиона, которая обеспечивает расщепление ковалентных дисульфидных линкеров с помощью механизма дисульфидного обмена. Еще один из таких факторов представляет собой присутствие лизосомальных ферментов, таких как катепсин В, который может расщеплять пептидные линкеры, сконструированные, чтобы быть предпочтительными субстратами(Dubowchik et al. 2002). Их активность делает MGBAs привлекательными кандидатами в качестве остатков лекарственного препарата в иммуноконъюгате. Иллюстративные раскрытия, касающиеся MGBAs и их использования в иммуноконъюгатах, включают Boyd et al. 2008 and 2010; Chen et al. 2010; Gangwar et al. 2008; Ng et al. 2002, 2006a, 2006b, 2009a, 2009b, and 2010; и Sufi et al. 2010. Существует возможность случайного расщепления ковалентного линкер, в то время как иммуноконъюгат все еще находится в системном кровотоке и еще не доставлен к целевой раковой клетке, в результате чего происходит преждевременное высвобождение лекарственного фрагмента, что создает риск системной токсичности. Такой риск представляет особый интерес, если остаток лекарственного препарата является сильнодействующим цитотоксином, таким как MGBA. Однако, если иммуноконъюгат использует секо-MGBA, риск может быть уменьшен путем дериватизации фенольной гидроксильной группы для блокирования превращения в циклопропановую форму. Таким образом, в случае случайного расщепления то, что высвобождается, является неактивным производным секо-MGBA. Посредством выбора производного, которое расщепляется внутри лизосомы, функции производной пролекарственной группы и обеспечивает фактор защищнности: два расщепления, линкера и пролекарственной группы,должно произойти перед тем, как высвободится активный цитотоксин. Одна из таких пролекарственных групп представляет собой карбамат, который может быть расщеплен лизосомальными и/или цитозольными карбоксиэстеразами, как показано ниже (Ra и Rb представляют родовые радикальные группы.) Иллюстративные раскрытия, относящиеся к секо-MGBA иммуноконъюгатам, являющимися пролекарствами с карбаматной группой см. в Aristoff et al. 1991; Boger et al. 1999; Другой пролекарственной группой, которую можно использовать с секо-MGBAs, является фосфат,и в этом случае расщепляющим ферментом является фосфатаза, находящаяся внутри лизосомы и/или в цитозоле. Иллюстративные раскрытия, относящиеся к фосфатным пролекарственным группам в секоMGBAs, включают Boyd et al. 2010, Chen et al. 2009, Glazier 2003, King et al. 2011, Kutyavin et al. 1997, Ngal. 2010). Оно имеет карбаматную пролекарственную группу; валин-цитруллин (Val-Cit, перечисленные в направлении от N- к С-концу, т.е. от амино (NH2) конца к карбокси (СО 2 Н) концу) дипептидный линкер,сконструированный для расщепления катепсином В (Dubowchik et al. 2002); и малеимидную группу для иммуноконъюгации посредством присоединения посредством реакции по Михаэлю к сульфгидрильным группам антитела. Иммуноконъюгат соединения (А) и антитела анти-CD70 проходят клинические испытания. Сущность изобретения Мы заметили, что в некоторых случаях карбамат-пролекарственный секо-MGBA иммуноконъюгат,такой как полученный из соединения (А), не является столь мощным противоопухолевым средством, как хотелось бы. Мы предполагаем, что снижение эффетивности связано с недостаточной активностью карбоксиэстеразы внутри некоторых типов раковых клеток и как следствие неэффективного декарбамоилирования. Таким образом, желательно разработать пролекарственные секо-MGBA иммуноконъюгаты,которые не зависят от карбоксиэстеразы для их активации. В одном аспекте данное изобретение относится к иммуноконъюгату, содержащему антитело (или его антигенсвязывающий фрагмент) и фосфатно-пролекарственному секо-MGBA соединения. Удаление фосфатной пролекарственной группы является не зависимым от активности карбоксиэстеразы в клеткемишени, что позволяет избежать недостатки, указанные выше. Напротив, соединения в соответствии с настоящим изобретением зависят от фосфатазы, которая, как показали наши эксперименты, присутствует на достаточном уровне в целом ряде опухолевых клеток человека. Один вариант осуществления представляет собой иммуноконъюгат, в котором соединение формулы (II) где X представляет собой Cl, Br или тозилат; ААа и каждый AAb независимо выбираются из группы, состоящей из аланина, -аланина, аминомасляной кислоты, аргинина, аспарагина, аспарагиновой кислоты, -карбоксиглутаминовой кислоты, цитруллина, цистеина, глутаминовой кислоты, глутамина, глицина, гистидина, изолейцина, лейцина,лизина, метионина, норлейцина, норвалина, орнитина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина;Y представляет собой спейсерный фрагмент, причем Y выбран из группы, состоящей изZ представляет собой реакционноспособную функциональную группу, способную к конъюгации с антителом, где -Z представляет собой -NH2, -ОН, -ONH2, -СО 2 Н, -SH, циклооктин, -N3, сконъюгировано через группу Z к антителу или к антигенсвязывающему фрагменту такого антитела. В одном варианте осуществления изобретения иммуноконъюгат имеет структуру, соответствующую формуле (IIIa) В другом варианте осуществления изобретения иммуноконъюгат имеет структуру, соответствующую формуле (IIIa')Ab представляет антитело или его антигенсвязывающий фрагмент. Предпочтительно, чтобы антитело представляло бы собой человеческое моноклональное антитело,которое распознает человеческий антиген, выбранный из группы, состоящей из CD70, мезотелина,ПСМА, CD19, глипикана-3, В 7 Н 4, RG-1, CD22 и PTK7. В другом аспекте обеспечивается фосфатное пролекарственное секо-MGBA соединение, адаптированное для конъюгации к антителу или его антигенсвязывающему фрагменту. Такое соединение содержит остаток фосфатного пролекарственного секо-MGBA, ферментативно расщепляемый пептидный линкер, спейсерный фрагмент и реактивную функциональную группу для присоединения к антителу или его антигенсвязывающему фрагменту. Один вариант осуществления представляет собой соединение формулы (II) или его фармацевтически приемлемую соль где X представляет собой Cl, Br или тозилат; ААа и каждая AAb независимо выбираются из группы, состоящей из аланина, -аланина, аминомасляной кислоты, аргинина, аспарагина, аспарагиновой кислоты, -карбоксиглутаминовой кислоты, цитруллина, цистеина, глутаминовой кислоты, глутамина, глицина, гистидина, изолейцина, лейцина,лизина, метионина, норлейцина, норвалина, орнитина, фенилаланина, пролина, серина, треонина, триптофана, тирозина и валина;Y представляет собой спейсерный фрагмент, причем Y выбран из группы, состоящей изZ представляет собой реакционноспособную функциональную группу, способную к конъюгации с антителом, где Z представляет собой -NH2, -ОН, -ONH2, -СО 2 Н, -SH, циклооктин, -N3, В одном варианте осуществления ААа выбирается из группы, состоящей из цитруллина, лизина, аспарагиновой кислоты, глутаминовой кислоты, серина, изолейцина, лейцина и треонина и m равно 0, 1 или 2. В одном варианте осуществления изобретения -AAa-[AAb]m- представляет собой дипептид, выбранный из группы, состоящей из Val-Cit, Phe-Cit, Phe-Lys, Val-Lys, Val-Glu, Val-Asp, Val-Ser и Val-Gly, каждый дипептид, читаемый в направлении от N- к -C. В одном варианте осуществления изобретения соединение имеет структуру в соответствии с формулой (IIa) или его фармацевтически приемлемая соль. В одном варианте осуществления изобретения соединение имеет структуру, выбранную из группы,состоящей из формул (IIb)-(IIg) или его фармацевтически приемлемая соль. В другом аспекте обеспечивается фосфатное пролекарственное соединение, которое может быть использовано для получения иммуноконъюгатов, как воплощено соединением формулы (I) где X представляет собой Cl, Br или тозилат, или его фармацевтически приемлемую соль. Соединение (I) может быть комбинировано с пептидным линкером, спейсером и реакционноспособной функциональной группой, как описано выше, а затем конъюгировано с антителом или его антигенсвязывающим фрагментом. В еще одном аспекте обеспечивается способ лечения рака у субъекта, предпочтительно человека,страдающего от такого рака, включающий введение субъекту терапевтически эффективного количества иммуноконъюгата в соответствии с настоящим изобретением. В одном варианте осуществления изобретения иммуноконъюгат имеет структуру, соответствующую формуле (IIIa'). Рак может быть раком, чьи клетки экспрессируют антиген, выбранные из группы, состоящей из CD70, мезотелина, ПСМА, CD19,глипикана-3, В 7 Н 4, RG-1, CD22 и PTK7. Примеры раковых заболеваний, которые можно лечить, включают рак почки, рак поджелудочной железы, рак яичников, лимфому, рак толстой кишки, мезотелиому,рак желудка, рак легкого, рак предстательной железы, аденокарциному, рак печени и рак молочной железы. Краткое описание чертежей На фиг. 1 А и 1 В в комбинации показывают синтез соединений в соответствии с настоящим изобретением, имеющих малеимидные реакционноспособные функциональные группы. Фиг. 2 показывает синтез другого соединения в соответствии с настоящим изобретением. На фиг. 3A-3D в комбинации показывают синтез еще одного соединения в соответствии с настоящим изобретением. Фиг. 4A-4R сравнивают ингибиторные свойства фосфатных пролекарственных иммуноконъюгатов на клеточную пролиферацию в соответствии с настоящим изобретением по сравнению с ингибиторными свойствами карбаматных пролекарственных иммуноконъюгатов, используя анализ пролиферации по включению 3 Н тимидина. Линии исследованных клеток включали следующие раковые клеточные линии человека: 786-0 (рак почки), простатический специфический мембранный антиген (ПСМА), HPAC (рак поджелудочной железы), 0VCAR3 (рак яичников), Ramos (лимфома Беркитта), Н 226 (мезотелиома), N87(рак желудка), Н 292 (мукоэпидермоидная легочная карцинома), H1650 (аденокарцинома), Hep 3b (гепатоцеллюлярная карцинома), Hep G2 (гепатоцеллюлярная карцинома), НСС-1954 (рак молочной железы),MDA-MB-468 (рак молочной железы), НСТ 116 (рак толстой кишки) и Н 520 (рак легких). Кроме того,была использована трансфицированная клеточная линия СНО (яичника китайского хомячка), экспрессирующая мезотелин. Фиг. 5 изображает хроматографические профили, показывающие дефосфорилирование фосфатного пролекарственного соединения в соответствии с настоящим изобретением ферментами микросом печени человека. Фиг. 6 представляет собой график зависимости количества дефосфорилированного продукта от концентрации микросом в течение 1 ч. Фиг. 7 показывает синтез еще одного соединения в соответствии с настоящим изобретением, имеющего азидную реакционноспособную функциональную группу для конъюгации с антителом. Фиг. 8 показывает синтез еще одного соединения в соответствии с настоящим изобретением, имеющего гидроксиламиновую реакционноспособную функциональную группу, для конъюгации с антителом. Фиг. 9 А и 9 В в комбинации показывают синтез соединения в соответствии с настоящим изобретением, имеющего трипептид Leu-Ala-Leu. Фиг. 10 А и 10 В в комбинации показывают синтез соединения в соответствии с настоящим изобретением, имеющего дипептид Leu-Ile. Фиг. 11 А и 11 В в комбинации показывают синтез другого соединения в соответствии с настоящим изобретением. Фиг. 12A-12I показывают результаты различных исследований с ксенотрансплантатом. Подробное описание изобретения Определения."Антитело" означает целые антитела и любой антигенсвязывающий фрагмент (т.е. "антигенсвязывающая часть") или их отдельные цепи. Целое антитело представляет собой гликопротеин, содержащий по меньшей мере две тяжелые (Н) цепи и две легкие (L) цепи, соединенные между собой дисульфидными связями. Каждая тяжелая цепь содержит вариабельную область тяжелой цепи (Vh) и константную область тяжелой цепи, содержащую три домена CH1, CH2 и CH3. Каждая легкая цепь содержит вариабельную область легкой цепи (VL или VK) и константная область легкой цепи содержит один единственный домен CL. VH и VL области могут быть дополнительно подразделены на гипервариабельные области, называемые области, определяющие комплементарность (CDR), перемежаясь с более консервативными каркасными областями (FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных от амино- к карбоксиконцу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3 и FR4. Вариабельные области содержат связывающий домен, который взаимодействует с антигеном. Константные области могут опосредовать связывание антитела с тканями или факторами хозяина, включая различные клетки иммунной системы (например, эффекторные клетки) и первый компонент (C1q) классической системы комплемента. Про антитело говорят "специфически связывающееся" с антигеном X, если указанное антитело связывается с антигеном X с KD 510-8 М или менее, более предпочтительно от 110-8 М или менее, более предпочтительно 610-9 М или менее, более предпочтительно от 310-9 М или менее, еще более предпочтительно от 210-9 М или менее. Антитело может быть химерное, гуманизированное или предпочтительно человеческое. Константные области тяжелой цепи могут быть сконструированы так,-6 024844 чтобы влиять на тип или степень гликозилирования, увеличивать время полураспада антитела, для повышения или снижения взаимодействия с эффекторными клетками или системой комплемента или для модуляции других свойств. Конструирование может осуществляться посредством замены, вставки или делеции одной или более аминокислот или замещением домена доменом из другого типа иммуноглобулина или комбинацией вышеперечисленного."Фрагмент антитела" и "антигенсвязывающий фрагмент" антитела (или просто "часть антитела") означает один или несколько фрагментов антитела, которые сохраняют способность специфически связываться с антигеном. Было показано, что антигенсвязывающая функция антитела может быть осуществлена фрагментами антитела полной длины, например (i) фрагментом Fab, моновалентным фрагментом,состоящим из VL, VH, CL и CH1 доменов; (ii), F(ab')2-фрагментом, бивалентным фрагментом, содержащим два Fab-фрагмента, связанных дисульфидным мостиком в шарнирной области; (iii) Fab'-фрагментом, который является, по существу, Fab с частью шарнирной области (см., например, Abbas et al., Cellular andMolecular Immunology, 6th Ed., Saunders Elsevier 2007); (iv) Fd фрагментом, состоящим из VH и CH1 доменов; (v) Fv фрагментом, состоящим из VL и VH доменов одного плеча антитела, (vi) фрагментом dAb(Ward et al., (1989) Nature 341:544-546), который состоит из домена VH; (vii) изолированной области, определяющей комплементарность (CDR), а также (viii) нанотела тяжелой цепи вариабельной области, содержащей один вариабельный домен и два константных домена. Кроме того, хотя два домена фрагментаFV, VL и VH, кодируются отдельными генами, они могут быть соединены с использованием рекомбинантных способов с помощью синтетического линкера, который позволяет им быть составленными в виде одноцепочечного белка, в котором VL и VH области соединяются с образованием моновалентных молекул (известных как одноцепочечные Fv или scFv), см., например, Bird et al. (1988) Science 242:423426; и Huston et al. (1988) Proc. Natl. Acad. Sci. USA 85:5879-5883). Такие одноцепочечные антитела также включены в термин "антигенсвязывающий фрагмент" или "антигенсвязывающий фрагмент" антитела."Изолированное антитело" означает антитело, которое, по существу, не содержит другие антитела,имеющие различные антигенные специфичности (например, изолированное антитело, которое специфически связывает антиген X, является, по существу, свободным от антител, которые специфически связывают антигены, отличные от антигена X). Изолированное антитело, которое специфически связывает антиген X, может, однако, иметь перекрестную реактивность к другим антигенам, таким как антиген молекулы X из других видов. В некоторых вариантах осуществления изолированное антитело специфически связывается с человеческим антигеном X и не вступает в перекрестную реакцию с другим (не человеческим) антигеном X. Более того, изолированное антитело может быть, по существу, свободным от другого клеточного материала и/или химических веществ."Моноклональное антитело" или "композиция моноклонального антитела" означает препарат молекул антител, композицию, состоящую из единственного типа молекул, которая демонстрирует единственную специфичность связывания и сродство к конкретному эпитопу."Человеческое антитело" означает антитело, имеющее вариабельные области, в которых как каркасная, так и CDR-области (и константная область, если она присутствует), происходят из последовательностей иммуноглобулина зародышевой линии человека. Человеческие антитела могут включать последующие модификации, в том числе природные или синтетические модификации. Человеческие антитела могут включать в себя аминокислотные остатки, не кодируемые последовательностями иммуноглобулина зародышевой линии человека (например, мутации, введенные случайным образом или с помощью сайт-специфического мутагенеза in vitro или посредством соматической мутации in vivo). Тем не менее, "человеческое антитело" не включает в себя антитела, в которых последовательности CDR, полученные из зародышевой линии других видов млекопитающих, таких как мыши, были привиты на человеческие каркасные последовательности. Выражение "человеческое моноклональное антитело" означает антитело, демонстрирующее единственную специфичность связывания, которое имеет вариабельные области, в которых как каркасная, так и CDR-области происходят из последовательностей иммуноглобулина зародышевой линии человека. В одном варианте осуществления человеческие моноклональные антитела продуцируются гибридомой,которая включает В-клетки, полученные из трансгенного нечеловеческого животного, например трансгенной мыши, геном которой содержит трансген человеческой тяжелой цепи и трансген легкой цепи,вставленные в иммортализованную клетку. Если для конкретных стереоизомеров специально не указано (например, утолщенной или пунктирной связью в соответствующем стереоцентре в структурной формуле, посредством изображения двойной связи как имеющей Е- или Z-конфигурацию в структурной формуле, или с помощью номенклатуры, обозначающей стереохимию), все стереоизомеры включаются в объем настоящего изобретения, как чистые соединения, а также их смеси. Если не указано иное, индивидуальные энантиомеры, диастереомеры,геометрические изомеры, а также их комбинации и смеси все входят в объем настоящего изобретения. Специалистам в данной области техники будет понятно, что соединения могут иметь таутомерные формы (например, кето- и енольной формы), резонансные формы и цвиттерионные формы, эквивалентные тем, которые изображены в структурных формулах, использованных в настоящем изобретении, и что структурные формулы охватывают такие таутомерные, резонансные или цвиттерионные формы. Термин "фармацевтически приемлемая соль" означает соль соединения, пригодную для фармацевтического препарата. Когда соединение имеет одну или несколько основных групп, соль может быть солью присоединения кислоты, такой как сульфат, гидробромид, тартрат, мезилат, малеат, цитрат, фосфат,ацетат, памоат (эмбонат), гидроиодид, нитрат, гидрохлорид, лактат, метилсульфат, фумарат, бензоат,сукцинат, лактобионат, суберат, тозилат и тому подобное. Когда соединение имеет одну или больше кислотных групп, соль может быть солью, такой как соль кальция, калия, соль магния, меглуминовая соль,аммонийная соль, цинковая соль, соль пиперазина, трометаминовая соль, литиевая соль, соль холина,соль диэтиламина, соль 4-фенилциклогексиламина, бензатиновая соль, натриевая соль, соль тетраметиламмония и тому подобное. Полиморфные кристаллические формы и сольваты также включены в объем в соответствии настоящим изобретением. Что касается фосфатных групп, конкретные примеры фармацевтически приемлемых солей включают натриевые, калиевые и аммониевые соли. Композиции. В одном аспекте композиция в соответствии настоящим изобретением является иммуноконъюгатом, в котором соединение формулы (I) где X представляет собой нуклеофильно замещаемую уходящую группу или его фармацевтически приемлемую соль, конъюгирована с помощью пептидного линкера с антителом или его антигенсвязывающим фрагментом. Конъюгация предпочтительно проводится через его 4-NH2 группу. Пептидиный линкер предпочтительно представляет собой дипептид, выбранный из Val-Cit, Phe-Cit, Phe-Lys, ValLys, Glu-Val, Val-Asp, Val-Ser и Val-Gly, причем каждый дипептид читается в направлении от N- к Сконцу. В одном вариант осуществления иммуноконъюгат имеет структуру, представленную формулой в которой ААа, AAb, m, и Y являются такими, как определено выше в отношении формулы (II), Ab представляет собой антитело или антигенсвязывающий фрагмент антитела, n = 1, 2, 3, 4 или 5; или его фармацевтически приемлемую соль. Предпочтительные варианты осуществления и/или комбинации ААа,AAb, m, Y, Ab и n описаны ниже. Предпочтительный иммуноконъюгат согласно формуле (IIIa) имеет структуру формулы (IIIa') в которой Ab и n являются такими, как определено выше, или его фармацевтически приемлемой солью. Таким образом, этот вариант осуществления имеет дипептидный линкер Val-Cit с пониманием специалистами в данной области того, что в структурной формуле (IIIa') дипептид изображен в ориентации от С- кN-концу при чтении в направлении слева направо. В другом варианте осуществления иммуноконъюгат имеет структуру, представленную формулой в которой ААа, AAb, m и Y являются такими, как определено выше в отношении формулы (II), Ab представляет собой антитело или его антигенсвязывающий фрагмент антитела, n равен 1, 2, 3, 4 или 5; или его фармацевтически приемлемую соль. Предпочтительные варианты осуществления и/или комбинации ААа, AAb, m, Y, Ab и n описываются ниже. Предпочтительный иммуноконъюгат в соответствии с формулой (IIIb) имеет структуру в соответствии с формулой (IIIb') в которой Ab и n являются такими, как определено выше, или его фармацевтически приемлемой солью. Как указано посредством индекса n в формулах (IIIa), (IIIa'), (IIIb) и (IIIb'), каждое антитело может быть конъюгировано с более чем одним пролекарственным секо-MGBA фрагментом в зависимости от того, какое количества сайтов антитела доступно для конъюгации в используемых экспериментальных условиях. Специалистам в данной области техники должно быть понятно, что, хотя каждое отдельное индивидуальное антитело конъюгируют с целым числом пролекарственных остатков, анализ препарата иммуноконъюгата может давать нецелочисленное отношение пролекарственных секо-MGBA остатков на антитело "коэффициент замещения" или "SR"), что отражает среднее статистическое. SR предпочтительно составляет от 1 до 5, более предпочтительно от 2,5 до 3,5. Другой аспект в соответствии настоящим изобретением представляет собой соединение, адаптированное для конъюгации с антителом, представленное формулой (II) в которой ААа, AAb, m, Y и Z являются такими, как определено выше, или его фармацевтически приемлемой солью. Предпочтительные варианты осуществления и/или комбинации ААа, AAb, m, Y и Z описаны ниже. Предпочтительный вариант осуществления в соответствии с формулой (II) имеет структуру формулы (IIa) или его фармацевтически приемлемой соли. Другие предпочтительные варианты осуществления в соответствии с формулой (II) имеют структуру формул (IIb)-(IIg) или их фармацевтически приемлемой соли. В формулах (I) и (II) X представляет собой нуклеофильнозамещаемую группу, предпочтительно Cl,Br или тозилат, более предпочтительно Cl. В формулах (II), (IIa) и (IIIb), ААа и каждая AAb независимо выбирается из группы, состоящей из аланина, -аланина, -аминомасляной кислоты, аргинина, аспарагина, аспарагиновой кислоты, карбоксиглутаминовой кислоты, цитруллина (Cit), цистеина, глутаминовой кислоты, глутамина, глицина,гистидина, изолейцина, лейцина, лизина, метионина, норлейцина, норвалина, орнитина, фенилаланина,пролина, серина, треонина, триптофана, тирозина и валина. Предпочтительно ААа и AAb выбираются из группы, состоящей из аргинина, аспарагиновой кислоты, цитруллина, глутаминовой кислоты, глутамина,глицина, гистидина, лизина, фенилаланина, серина, треонина, триптофана, тирозина и валина. Фрагмент -AAa-[AAb]m- в формулах (II), (IIIa) и (IIIb) представляет собой пептидный линкер, где последовательные аминокислоты соединены пептидной связью. Предпочтительно, чтобы одна из них была бы расщепляемой посредством внутриклеточного или экстрацеллюлярного фермента, связанного с клеткой-мишенью. Фермент может быть, например, катепсином В, катепсином Е, легумаином или CD10. Пептидный линкер предпочтительно состоит из от одной до шести, более предпочтительно от одной до трех и наиболее предпочтительно от одной до двух аминокислот. ААа предпочтительно представляет собой Cit,Lys, Glu, Ser, Ile, Leu, Thr или более предпочтительно Cit или Lys, особенно в комбинации с m, равным 0,1 или 2. Суффикс m в формулах (II), (IIIa) и (IIIb) равен 0, 1, 2, 3, 4 или 5. Предпочтительно m равен 0, 1 или 2, более предпочтительно 1. Таким образом, -AAa-[AAb]m- представляет собой линкер из одной аминокислоты, дипептидный линкер, трипептидный линкер и так далее, в зависимости от значения m, причем с ААа на С-конце и AAb на N-конце. Где m равно 1, так что ААа и AAb в комбинации образуют дипептид,предпочтительные дипептиды представляют собой Val-Cit, Phe-Cit, Phe-Lys, Val-Lys, Glu-Val, Val-Asp,Val-Ser и Val-Gly, причем каждый дипептид представлен в обычном направлении от N-конца к С-концу. Дипептидный линкер Val-Cit является особенно предпочтительным. Там, где пептидный линкер состоит из одной аминокислоты (то есть m = 0), то он предпочтительно представляет собой Cit, Glu, Lys и Ser,как описано в публикации Chen et al. 2010, раскрытие которой включено в настоящее изобретение посредством ссылки. Предпочтительно, чтобы вышеупомянутый дипептидные и одинопептидные линкеры расщеплялись бы катепсином В. Другим ферментом, который может быть использован для расщепления пептидных линкеров, является легумаин, лизосомные цистеиновые протеазы, которые предпочтительно расщепляют при Ala-Ala-Asn. Пептиды Leu-Ala-Leu и Leu-Ile предназначены для расщепления посредством CD10 и катепсином Е соответственно. Спейсер Y в формулах (II), (IIIa) и (IIIb) обеспечивает пространственное разделение между пролекарственным секо-MGBA и антителом (или его антигенсвязывающим фрагментом), чтобы исключить стерические препятствия связывания антигена посредством последнего или стерического препятствия расщеплению пептидного линкера последним. Кроме того, спейсер Y может быть использован, чтобы придать повышенную растворимость или уменьшение агрегационных свойств конъюгатов. Спейсер Y может содержать один или более модульных сегментов, которые могут быть собраны в любом количестве комбинаций. Примерами подходящих сегментов спейсера Y являются и их комбинаций, где индекс q равен 0 или 1, независимо в каждом случае, а индекс r равен от 1 до 24,предпочтительно от 2 до 4, независимо в каждом случае. Эти сегменты могут быть объединены, как показано ниже Спейсер Y предпочтительно обеспечивает линейное расстояние от 5 до 20 атомов, более предпочтительно от 5 до 15 атомов, между аминокислотой ААа или AAb , к которой она присоединена, и реакционноспособной функциональной группой Z. В другом предпочтительном вариант осуществления спейсер Y присоединен к ААа или AAb в зависимости от обстоятельств посредством отличной от карбоксильной ацильной группы, непосредственно присоединяемой к -аминогруппе ААа или AAb. Предпочтительно, чтобы спейсер Y соединялся с аминогруппой ААа или AAb в зависимости от обстоятельств через метиленовую группу в спейсере Y. В формуле (II) Z представляет собой реакционноспособную функциональную группу, способную к конъюгации с антителом. Предпочтительно Z представляет собой -NH2, -ОН, -ONH2 (гидроксиламин),-СО 2 Н, -SH, циклооктин, азид (-N3) Группа -ОН может быть этерифицирована карбоксильной группой на антителе, например боковой цепью аспарагиновой или глутаминовой кислоты. Группа -СО 2 Н может быть этерифицирована -ОН группой или амидирована аминогруппой (например, на боковой цепи лизина) на антителе. Малеимидная группа может быть конъюгирована группой -SH на антителе (например, от цистеина или из химической модификации антител, которая ввела сульфгидрильную функцию) в реакции присое- 11024844N-гидроксисукцинимидная группа функциональнирует как активированная карбоксильная группа и может быть удобно амидирована посредствим реакции с аминогруппой (например, от лизина). Группа -SH особенно полезна для конъюгации, когда антитело модифицировано, для того чтобы ввести в него малеимидную группу в реакции присоединения по Михаэлю, которая является "зеркальным отражением" по отношению к описанной выше. Антитела могут быть модифицированы, чтобы иметь малеимидную группу N-сукцинимидил-4-(малеимидометил)циклогексанкарбоксилатом (SMCC) или его сульфированным вариантом сульфо-SMCC, причем оба реагента производятся фирмой SigmaAldrich. Азид и циклооктин являются комплементарными (взаимодействующими) функциональными группами, которые могут быть конъюгированы с помощью так называемой "клик-химии", в которой азид присоединяется через напряженную связь алкинациклооктина, образуя 1,2,3-триазоловое кольцо. Азид может быть Z-группой в соединении формулы (II) и циклооктин может быть расположен на антителе или на его антигенсвязывающем фрагменте или наоборот. Циклооктиновая группа может быть обеспечена с помощью реагента DIBO (доступного от Invitrogen/Molecular Probes, Eugene, Oregon). Способы введения неприродных аминокислот в антитела могут быть использованы с неприродными аминокислотами, обеспечивая функциональные группы для конъюгации с реакционноспособной функциональной группой. Например, неприродная аминокислота n-ацетилфенилаланин может быть инкорпорирована в антитело, как предлагается в Tian et al, WO 2008/030612 A2 (2008). Кетогруппа в nацетилфенилаланине может оказаться в сайте конъюгации посредством образования оксимов взаимодействием с гидроксиламинными реакционноспособными функциональными группами. Другие неприродные аминокислоты также могут быть инкорпорированы с использованием технологии ReCODE отAmbrx, Inc (La Jolla, California). В приведенных выше иммуноконъюгатах антитело предпочтительно является антителом против антигена, ассоциированного с опухолью, что позволяет иммуноконъюгату избирательно или предпочтительно нацеливать на раковые клетки. Примеры таких антигенов включают мезотелин, простатический специфический мембранный антиген (ПСМА), CD19, CD22, CD30, CD70, В 7 Н 4 (также известный как О 8 Е), протеинтирозинкиназу 7 (PTK7), глипикан-3, RG1, CTLA 4 и также CD44. Антитело может быть от животного (например, мыши), химерное, гуманизированное или предпочтительно человеческое. Антитело предпочтительно является моноклональным, особенно человеческим моноклональным антителом. Получение человеческих моноклональных антител против некоторых из вышеупомянутых антигенов раскрыто в Korman et al., US 2009/0074660 A1 (B7H4); Rao-Naik et al.,8097703 B2 (CD19); King et al., US 2010/0143368 Al (CD22); Keler et al., US 7387776 B2 (2008) (CD30);(CD44); раскрытие которых включено в настоящее описание посредством ссылки. Предпочтительно,чтобы антитело представляло бы собой человеческое моноклональное антитело против CD70, мезотелин,CD19, глипикан-3, В 7 Н 4, RG-1, CD22 или PTK7. Другой аспект в соответствии настоящим изобретением представляет собой пролекарственное соединение формулы (I) или его фармацевтически приемлемая соль, где X представляет собой нуклеофильнозамещаемую уходящую группу, которая предпочтительно представляет собой Cl, Br или тозилат и более предпочтительноCl. Соединение формулы (I) может быть конъюгировано с антителом или его антигенсвязывающим фрагментом, что дает терапевтически полезный иммуноконъюгат посредством соответствующего линкера, спейсера и реакционноспособной функциональной группы, как описано выше. Предпочтительный вариант осуществления формулы (I), в которой X представляет собой Cl, соответствует структуре формулы (Ia) Биологическая активность Проводили серию экспериментов пролиферации клеток, в которых сравнивали способность иммуноконъюгатов, полученных из соединения (IIa), и известное соединение (А). В каждом случае использованная методика представляла собой анализ включения Н-тимидина, процедурные детали которой обеспечиваются в приведенных ниже примерах. Поскольку иммуноконъюгаты идентичны, за исключением пролекарственной группы - фосфат против карбамата - и то же самое лекарственное вещество формулы (IV) высвобождается после расщепления пептидного линкера и удаления пролекарственной группы, эксперименты обеспечивают сравнение эффекта от пролекарственной группы на потенцию иммуноконъюгата. Эти параллельные результаты показывают, что,неожиданно, иммуноконъюгаты соединения (IIa) значительно более эффективны, чем у соединения (А),хотя одно и то же конечное активное соединение (IV) участвует в каждом конкретном случае. Фиг. 4 А показывает ингибирующую активность двух иммуноконъюгатов в отношении пролиферации 786-0 клеток, которые представляют собой клетки рака почки человека, экспрессирующие CD70. В каждом случае конъюгированное антитело представляло собой 2 Н 5, моноклональное антитело человека анти-CD70. Соединение (IIa) иммуноконъюгат не только имело более низкую EC50 (0,1000 нМ по сравнению с 0,5725 нМ), но также имело более высокий общий процент ингибирования, о чем свидетельствует глубина ее кривой ингибирования (полная информация о последовательности антитела 2 Н 5 раскрыта в Coccia et al. 2010 и Terret et al. 2012b; раскрытие которой включено в настоящем изобретении посредством ссылки. Последовательности VH CDR1, CDR2 и CDR3 и VK CDR1, CDR2 и CDR3 для антитела 2 Н 5 приведены в SEQ ID NO:1, SEQ ID NO:2, SEQ ID NO:3, SEQ ID NO:4, SEQ ID NO:5 и SEQ IDNO:6 соответственно). Фиг. 4 В представляет аналогичное исследование, но с использованием клеток HPAC, которые являются мезотелин-экспрессирующими клетками рака поджелудочной железы человека. В каждом случае конъюгированными антителами были 6 А 4, антимезотелин человеческими моноклональными антителами. В этом эксперименте были получены два различных конъюгата соединения (IIa) с коэффициентом замещения 1,25 и 3. (Концентрации цитотоксина на оси X корректируется с учетом коэффициент замещения.) Фиг. 4 В показывает, что иммуноконъюгат соединения (А), по существу, неактивное, в то время как иммуноконъюгаты соединения (IIa) имели приблизительно наномолярные EC50. Следует также отметить, что выше коэффициент замещения иммуноконъюгата соединения (IIa), тем он показывает более высокий общий процент ингибирования (более глубокая кривая). (Полная информация о последовательности антитела 6 А 4 раскрыта в Terrett et al. 2011b, раскрытие которого включено посредством ссылки. Последовательности VH CDR1, CDR2 и CDR3 и VK CDR1, CDR2 и CDR3 для антитела 6 А приведены вSEQ ID NO:7, SEQ ID NO:8, SEQ ID NO:9, SEQ ID NO:10, SEQ ID NO: 11 и SEQ ID NO:12 соответственно). Фиг. 4 С представляет другое аналогичное исследование на этот раз с использованием СНОмезоклеток, которые представляют собой клетки яичника китайского хомячка, трансфицированные для экспрессии мезотелина. Конъюгированным антителом представляло собой опять же 6 А 4. Иммуноконъюгат соединения (А) был, по существу, неактивным, в то время как иммуноконъюгат соединения (IIa) имел EC50 около 2 нМ. Фиг. 4D представляет еще одно аналогичное исследование, на этот раз с использованием клетокOVCAR3, которые представляют собой клетки рака яичников человека, экспрессирующие мезотелин. Конъюгированным антителом представляло собой опять же 6 А 4. EC50 для иммуноконъюгата соединения(IIa) было приблизительно в 1000 раз ниже, чем для иммуноконъюгата соединения (А). Фиг. 4 Е представляет еще одно аналогичное исследование, на этот раз с использованием клетокRamos, которые представляют собой клетки лимфомы Беркитта человека, экспрессирующие CD19. Конъюгированным антителом представляло собой 21D4, анти-CD19 моноклональное антитело человека. Вновь отмечается различия между иммуноконъюгатами соединений (А) и (IIa), причем первый является,по существу, неактивным, тогда как второй имел субнаномолярную EC50. (Полная информация о последовательности антитела 21D4 раскрыта в King et al. 2010a и Rao-Naik et al. 2012; описания которых включены в настоящее изобретение посредством ссылки. Последовательности VH CDR1, CDR2 и CDR3 иID NO:16, SEQ ID NO:17 и SEQ ID NO:18 соответственно) Фиг. 4F представляет еще одно аналогичное исследование, на этот раз с клетками NCI-H226 (Н 226),которые представляют собой клетки, экспрессирующие мезотелин, полученные из мезотелиомы легких человека. Конъюгированным антителом представляло собой опять же 6 А 4. Кроме того, в качестве изотипического контроля соединение (IIa) было конъюгировано с 2 А 10, которое является человеческим моноклональным антителом против простатического специфического мембранного антигена (ПСМА). Как и ожидалось, иммуноконъюгат ПСМА обладал очень незначительной активностью или не обладал вовсе,так как клетки Н 226 не экспрессируют ПСМА. Иммуноконъюгат соединения (А) так же обладал низкой или нулевой активностью. В противоположность этому каждый из двух иммуноконъюгатов соединения(IIa) обладали субнаномолярными ЕС 50. Как и в предыдущих примерах, иммуноконъюгат с более высокой степенью замещения демонстрировал более высокий общий процент ингибирования пролиферации.(Полная информация о последовательности антитела 2 А 10 раскрыта в Huang et al. 2009 и Cardarelli et al. 2011; описания которых включены в настоящее изобретение посредством ссылки. ПоследовательностиID NO:20, SEQ ID NO:21, SEQ ID NO:22, SEQ ID NO:23 и SEQ ID NO:24 соответственно). Фиг. 4G показывает активность иммуноконъюгата антимезотелин антитела 6 А 4 и соединения (IIa) против клеток карциномы желудка человека N87, которые экспрессируют мезотелин. Наблюдались субнаномолярные EC50. Фиг. 4 Н показывает активность иммуноконъюгатов антитела 6 А 4 и соединения (IIa) при различных коэффициентах замещения (SR) против клеток Н 292, которые представляют собой клетки мукоэпидермоидной легочной карциномы человека, экспрессирующие мезотелин. Иммуноконъюгат анти-ПСМА антитела 2 А 10 и соединения (IIa) использовали в качестве контроля. Значения EC50 показаны в таблице ниже. Они показывают, что существует значительное увеличение активности, когда SR увеличивается до приблизительно двух или выше. Фиг. 4I показывает исследование, аналогичное исследованию на фиг. 4 Н, но против клеток H1650,которые представляют собой клетки аденокарциномы человека, экспрессирующие мезотелин. Величины ЕС 50 представлены в табл. II. Опять же, скачок активности был отмечен при SR выше приблизительно двух. Фиг. 4J и 4K представляют исследования иммуноконъюгатов в соответствии с настоящим изобретением в отношении двух клеточных линий гепатоцеллюлярной карциномы человека Hep 3 В и Hep G2,обе из которых экспрессируют глипикан-3. Сравнивали эффективность человеческого моноклонального антитела анти-глипикан-3 4 А 6 одного и в конъюгированном виде с соединением (IIa) вместе с контрольным иммуноконъюгатом анти-CD70 антитела 2 Н 5 и соединения (IIa). (Ни Hep 3 В, ни Hep G2 клетки не экспрессируют CD70). Результаты показывают, что иммуноконъюгат 4 А 6 был очень эффективен, приблизительно в 1000 раз более активен, чем контрольный иммуноконъюгат CD70. Одно антиглипикан-3 антитело 4 А 6 было неактивным. (Информация о последовательности для антитела 4 А 6 раскрыта в Terrettet al. 2010b, раскрытие которого включено посредством ссылки. Последовательности VH CDR1, CDR2 иNO:27, SEQ ID NO:28, SEQ ID NO:29 и SEQ ID NO:30 соответственно) Фиг. 4L, 4M и 4N представляют исследования активности иммуноконъюгата анти-В 7 Н 4 человеческого моноклонального антитела 2 А 7 и соединения (IIa) на клеточных линиях MDA-MB-468, НСС-1954 и OVCAR3. Первые две линии представляют собой клетки рака молочной железы человека, а третья является линией раковых клеток яичника человека, каждая из которых экспрессирует В 7 Н 4, но не CD70. Иммуноконъюгат анти-ПСМА антитела 2 А 10 и соединения IIa использовали в качестве контроля. Как можно видеть из чертежей, существует некоторая специфическая активность против клеточных линий рака молочной железы, но незначительная активность или ее отсутствие против клеток OVCAR3 (полная информация о последовательности антитела 2 А 7 раскрыта в Korman et al. 2009 and Terrett et al. 2011a; описания которых включены в настоящее изобретение посредством ссылки. Последовательности VHNO:32, SEQ ID NO:33, SEQ ID NO:34, SEQ ID NO:35 и SEQ ID NO:36 соответственно). Фиг. 4O представляет исследования активности иммуноконъюгатов анти-ПСМА человеческого моноклонального антитела 2 А 10 и анти-RG-1 человеческого моноклонального антитела 19G9, каждое конъюгированное с соединением (IIa), против раковых клеток простаты человека LNCap, которые экспрессируют как ПСМА, так и RG-1. Сравнительные данные в качестве контроля обеспечивались в виде иммуноконъюгата антимезотелинового антитела 6 А 4 и соединения (IIa). Можно видеть, что конъюгаты как ПСМА, так и RG-1 были эффективными, каждый с субнаномолярными EC50 (полная информация о последовательности антитела 19G9 раскрыта в King et al. 2011, раскрытие которой включено в настоящее изобретение посредством ссылки. Последовательности VH CDR1, CDR2 и CDR3 и VL CDR1, CDR2 иID NO:41 и SEQ ID NO:42 соответственно). Фиг. 4 Р представляет исследования активности иммуноконъюгатов анти-CD19 человеческого моноклонального антитела 21D4 и анти-CD22 человеческого моноклонального антитела 12 С 5, каждое конъюгированное с соединением (IIa), против клеток Ramos, которые экспрессируют как CD19, так иCD22. Сравнительные данные обеспечивались в виде иммуноконъюгата анти-6 А 4 мезотелин антитела и анти-CD70 антитела 2 Н 5 с соединением (IIa). Иммуноконъюгаты как анти-CD19, так и анти-CD22 показали прекрасную эффективность, каждый с субнаномолярными EC50 (полная информация о последовательности антитела 12 С 5 раскрыта в King et al. 2010b, раскрытие которого включено посредством ссылки. Последовательности VH CDR1, CDR2 и CDR3 и VK CDR1, CDR2 и CDR3 для антител 12 С 5 приведены в SEQ ID NO:43, SEQ ID NO:44, SEQ ID NO:45, SEQ ID NO:46, SEQ ID NO:47 и SEQ ID NO:48 соответственно). Фиг. 4Q показывает активность иммуноконъюгата анти-PTK7 человеческого моноклонального антитела 4D5 с соединением (IIa) против клеток НСТ 116, которые представляют собой клетки карциномы толстой кишки человека. Иммуноконъюгат имел ЕС 50 равную 1,87 нМ, в то время как контрольный иммуноконъюгат на основе анти-ПСМА антитела 2 А 10 был гораздо менее эффективным, с EC50 равным 951 нМ (полная информация о последовательности антитела 4D5, раскрыта в Terrett et al. 2010 а и Terrettet al. 2012a; описания которых включены в настоящее изобретение посредством ссылки. Последовательности VH CDR1, CDR2 и CDR3 и VK CDR1, CDR2 и CDR3 для антитела 4D5 приведены в SEQ ID NO:49,SEQ ID NO:50, SEQ ID NO:51, SEQ ID NO:52, SEQ ID NO:53 и SEQ ID NO:54 соответственно). Фиг. 4R показывает активность иммуноконъюгата вышеупомянутого анти-PTK7 антитела 4D5 против клеток Н 520, которые представляют собой плоскоклеточную карциному легких человека. Иммуноконъюгат PTK7 имел EC50 равную 0,224 нМ, в то время как контрольный иммуноконъюгат ПСМА снова имел гораздо меньшую эффективность, с EC50 равную 61,6 нМ. Приведенные выше результаты показывают, что неожиданно природа пролекарственной группы фосфат по сравнению карбамата - играет существенную роль в эффективности иммуноконъюгатов, которые по всем остальным параметрам идентичны. Не вдаваясь в теорию, наша гипотеза состоит в том, что разные клетки человека (в том числе раковые клетки) содержат различные уровни карбоксиэстеразы и что многие, если не большинство, содержат недостаточные уровни карбоксиэстеразы для расщепления карбаматной пролекарственной группы эффективным образом. Однако пролекарственные иммуноконъюгаты с фосфатной группой не страдают от такой проблемы, по-видимому, потому что в большинстве,если не во всех клетках, уровень активности фосфатазы является достаточно высоким для эффективного расщепления фосфатной группы. Применение. Иммуноконъюгаты в соответствии с настоящим изобретением можно применять для лечения заболеваний, таких как, но не ограничиваясь этим, гиперпролиферативные заболевания, в том числе рак головы и шеи, который включают опухоли головы, шеи, полости носа, околоносовых пазух, носоглотки,ротовой полости, ротоглотки, гортани, глотки, слюнных желез и параганглиомы; рак печени и желчных протоков, в частности гепатоцеллюлярной карциномы; рака кишечника, особенно рака толстой кишки; рак яичников, мелкоклеточный рак и немелкоклеточный рак легких (МРЛ и НМРЛ); рак молочной железы, саркомы, такие как фибросаркомы, злокачественная фиброзная гистиоцитома, эмбриональная рабдомиосаркома, лейомиосаркома, нейрофибросаркома, остеосаркома, синовиальная саркома, липосаркома и альвеолярная саркома мягких тканей; лейкозы, такие как острый промиелоцитарный лейкоз (ХМЛ), острый миелобластный лейкоз (ОМЛ), острый лимфобластный лейкоз (ALL), и хронический миелолейкоз(ХМЛ), опухоли центральной нервной системы, в частности рака мозга, множественная миелома (ММ),лимфомы, такие как лимфома Ходжкина, лимфоплазмацитоидная лимфома, фолликулярная лимфома,лимфома лимфоидной ткани слизистых оболочек, лимфома клеток мантийной зоны, В-крупноклеточная лимфома, лимфома Беркитта, и Т-клеточная анапластическая крупноклеточная лимфома. В клиническом отношении применение способов и использование описанных в настоящем изобретении композиций приводит к уменьшению размеров или количества злокачественных опухолей и/или снижению сопутствующих симптомов (где это применимо). В патологическом отношении применение способа и применение композиций, описанных в настоящем изобретении, будет производить патологически соответствующий ответ, такой как ингибирование пролиферации раковых клеток, уменьшение размеров рака или опухоли, предотвращение дальнейшего метастазирования и ингибирование опухолевого ангиогенеза. Способ лечения таких заболеваний, включает введение терапевтически эффективного количества предлагаемой в изобретении комбинации пациенту. Способ может быть повторен, если необходимо. В частности,рак может быть раком, клетки которого экспрессируют CD70, мезотелин, CD19, глипикан-3, В 7 Н 4, RG-1,CD22 или PTK7. В предпочтительном варианте осуществления рак, который лечат, является раком почки, раком поджелудочной железы, раком яичников, лимфомой, раком толстой кишки, мезотелиомой,раком желудка, раком легкого, раком предстательной железы, аденокарциномой, раком печени или раком молочной железы. Иммуноконъюгаты в соответствии с настоящим изобретением могут вводиться в комбинации с другими терапевтическими агентами, в том числе антителами, алкилирующими агентами, ингибиторами ангиогенеза, антиметаболитами, ДНК-расщепляющими агентами, ДНК-сшивающими агентами, интеркаляторами ДНК, агентами, связывающимися в малой бороздке ДНК, ендиинами, ингибиторами белка теплового шока 90, ингибиторами гистондеацетилазы, иммуномодуляторами, стабилизаторами микротрубочек, аналогами нуклеозидов (пуриновых или пиримидиновых), ингибиторами ядерного экспорта, ингибиторами протеасомы, ингибиторами топоизомеразы (I или II), ингибиторами тирозинкиназы и ингибиторами сериновой/треониновой киназы. Специфические терапевтические агенты включают адалимумаб,ансамитоцин Р 3, ауристатин, бендамустин, бевацизумаб, бикалутамид, блеомицин, бортезомиб, бусульфан, каллистатин А, камптотецин, капецитабин, карбоплатин, кармустин, цетуксимаб, цисплатин, кладрибин, цитарабин, криптофицины, дакарбазин, дазатиниб, даунорубицин, доцетаксел, доксорубицин,дуокармицин, динемицин А, эпотилоны, этопозид, флоксуридин, флударабин, 5-фторурацил, гефитиниб,гемцитабин, ипилимумаб, гидроксимочевина, иматиниб, инфликсимаб, интерфероны, интерлейкины, лапахон, леналидомид, иринотекан, майтансин, мехлоретамин, мелфалан, 6-меркаптопурин, метотрексат,митомицин С, нилотиниб, оксалиплатин, паклитаксел, прокарбазин, субероиланилид гидроксамовой кислоты (SAHA), 6-тиогуанидин, тиотепа, тенипозид, топотекан, трастузумаб, трихостатин, винбластин,винкристин и виндезин. Иммуноконъюгаты в соответствии с настоящим изобретением могут быть приготовлены и введены с использованием способов, обычных для биологических препаратов. Композиции могут содержать вспомогательные вещества, такие как приводятся в Gennaro, ed., Remington: The Science and Practice ofPharmacy, 20th Ed. (Lippincott WilliamsWilkins 2003), раскрытие которого включено в настоящее изобретении посредством ссылки. Типичные вспомогательные вещества включают, без ограничения, буферные агенты (например, фосфаты, ацетаты, трис(гидроксиметил)аминометан (Трис, солюбилизаторы и эмульгаторы (например, полисорбат), консерванты (например, тимеросал, бензиловый спирт), соли(например, NaCl, KCl) хелатирующие агенты (например, ЭДТА), углеводороды (например, сахароза,глюкоза, мальтоза, трегалоза), носители (например, альбумин), аминокислоты и их соответствующие соли: гидрохлориды, цитраты, сорбит, декстран и тому подобное. Иммуноконъюгаты могут быть представлены в виде лиофилизированных порошков с или без вспомогательных веществ, которые затем могут быть растворены в носителе, таком как стерильная вода для инъекций, раствор хлорида натрия для инъекций, или раствор глюкозы для инъекций, с добавлением наполнителей или без. С другой стороны, иммуноконъюгат может быть предоставлен в виде концентрированного раствора, необязательно включающей вспомогательные вещества, который затем разбавляют до нужной концентрации перед введением. Альтернативные формы включают дисперсии, микроэмульсии и липосомы. Предпочтительно фармацевтическая композиция пригодна для внутривенного (IV), внутримышечного, подкожного, парентерального, спинального или эпидермального введения (например, путем инъекции или инфузии). Термин "парентеральное введение" означает способы введения, отличные от энтерального и местного введения, обычно путем инъекции, и включают, без ограничения, внутривенную,внутримышечную, внутриартериальную, интратекальную, внутрикапсулярную, внутриглазничную,внутрисердечную, внутрикожную, внутрибрюшинную, транстрахеальную, подкожную, внутрикожную,внутрисуставную, субкапсулярную, субарахноидальную, интраспинальную, эпидуральную и интрастернальную инъекцию и инфузию. Предпочтительные способы введения включают IV инфузию, IV болюсную, подкожную, внутримышечную."Терапевтически эффективное количество" предпочтительно приводит к уменьшению выраженности симптомов заболевания, увеличению частоты и продолжительности бессимптомных периодов болезни или предотвращению патологии или инвалидности из-за болезни. Например, для лечения опухоли у субъектов "терапевтически эффективное количество" предпочтительно ингибирует рост опухоли по меньшей мере приблизительно на 20%, более предпочтительно по меньшей мере приблизительно на 40%, еще более предпочтительно по меньшей мере приблизительно на 60% и еще более предпочтительно по меньшей мере приблизительно на 80% по сравнению с нелеченными субъектами. Терапевтически эффективное количество терапевтического соединения может уменьшить размер опухоли или иным образом улучшать симптомы у субъекта, который обычно представляет собой человека, но может быть другим млекопитающим. Режимы дозирования могут корректироваться, чтобы обеспечить терапевтический ответ. Например,может быть введена одна доза, могут быть введены несколько раздельных доз в течение долгого времени или доза может быть пропорционально уменьшена или увеличена, как указано в конкретной ситуации. Особенно предпочтительным является приготовление парентеральных композиций в единичной дозированной форме для облегчения введения и равномерности дозирования. "Лекарственная форма" относится к физически дискретным единицам, пригодным в качестве единичных доз для субъектов, подлежащего лечению, и каждая единица содержит заданное количество активного соединения, рассчитанное на получение желаемого терапевтического эффекта, в ассоциации с требуемым фармацевтическим носителем. Устройства, такие как предварительно заполненные шприцы, двухкамерные шприцы и автоинжекторы могут быть использованы. Доза будет варьироваться в зависимости от заболевания, которое лечат, характеристик пациента,таких как возраст, пол и генотип, и стадии лечения. Как правило, доза может составлять от приблизительно 0,5 мг/кг до приблизительно 300 мг/кг. Примеры Применение в соответствии с настоящим изобретением может стать более понятным посредством следующих примеров, которые приведены в качестве иллюстрации, но не как ограничения. Пример 1 - соединение (IIa). В этом примере описан синтез соединения 12 в соответствии с настоящим изобретением (также упоминаемое в настоящем изобретении как соединение (IIa. Фиг. 1 А и 1 В в совокупности показывают схему его синтеза. Соединение 2. Трифторуксусную кислоту (TFA Fisher, 5 мл) добавляли к раствору соединения 1 (1 г, 2,36 ммоль) в безводном дихлорметане (ДХМ Sigma-Aldrich, 5 мл). Реакционную смесь перемешивали при комнатной температуре (RT, приблизительно 25 С) в течение 30 мин. Анализ с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) показал, что реакция завершена. Реакционную смесь концентрировали и сушили в высоком вакууме в течение ночи с получением 1,15 г соединения 2 в виде зеленоватого твердого вещества (TFA соль). Соединение 4. ди-трет-Бутилдикарбонат Вос)2 О, 10,24 г, 46,97 ммоль) в безводном метаноле(Acros, 100 мл) и палладий (Aldrich, 10% на активированном угле, 400 мг) добавляли к суспензии этилового эфира 5-нитроиндол-2-карбоксилата (соединение 3, Acros, 10 г, 42,7 ммоль). Реакционную смесь гидрировали в атмосфере Н 2 (30 фунтов на кв.дюйм) в течение 8 ч. Реакционную смесь фильтровали и фильтрат концентрировали и сушили в высоком вакууме с получением соединения 4 (10,5 г, 81%) в виде желтого твердого вещества. 1(с, 9 Н), 1,33 (т, 3 Н). Соединение 5. Раствор моногидрата LiOH (Sigma-Aldrich) в воде (150 мл, 0,57 М) добавляли к раствору соединения 4 (10,5 г, 34,5 ммоль) в ацетонитриле (150 мл). Реакционную смесь нагревали при 40 С в течение ночи, после чего гидролиз был завершен согласно анализу ВЭЖХ. Реакционную смесь разбавляли водой (300 мл) и концентрировали в вакууме для удаления ацетонитрила. Полученный раствор экстрагировали EtOAc (2150 мл) и органические слои отбрасывали. Водный слой подкисляли до рН 4 добавлением 10% раствора KHSO4 (Aldrich). Полученную смесь снова экстрагировали этилацетатом (3150 мл). Органические слои от трех экстракций позже объединяли, сушили над безводным MgSO4 и концентрировали в вакууме с получением соединения 5 в виде твердого вещества коричневого цвета (9,7 г,- 17024844H-ЯМР (ДМСО)12,84 (с, 1H), 11,65 (с, 1H), 9,20 (с, 1H), 7,73 (с, 1H), 7,23 (м, 2 Н), 6,92 (с, 1 Н),1,42 (с, 9 Н); МС (ESI) m/z 299 (М+Na)+. Соединение 6. Получали раствор соединения 5 (779 мг, 2,83 ммоль) и N,N,N',N'-тетраметил-О-(7 азабензотриазол-1-ил)урония гексафторфосфата (HATU Oakwood, 1,07 г, 2,83 ммоль) в безводном N,Nдиметилформамиде (ДМФА Sigma-Aldrich, 10 мл). N,N-Диизопропилэтиламин (DIPEA) добавляли для доведения рН реакционного раствора до значения выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. Неочищенное соединение 2 (1,15 г, оценивается как 2,36 ммоль) с последующим добавлением дополнительно DIPEA, чтобы довести рН реакционной смеси до значения выше 8. Реакционную смесь перемешивали при комнатной температуре в течение ночи. ВЭЖХ-анализ показал, что реакция была почти завершена. Реакционную смесь разбавляли этилацетатом и промывали водой и затем солевым раствором. Органические фазы концентрировали до суспензии с 2 г силикагеля. Суспензию очищали на 40 г колонке CombiFlash с 0-50% градиентом EtOAc в гексане с получением соединения 6 в виде желтого твердого вещества (1,05 г, 76%). 1(м, 1H), 3,90 (м, 1H), 1,48 (с, 9 Н). Соединение 7. Реакционная смесь, состоящую из соединения 6 (780 мг, 1,34 ммоль) и Pd/C (SigmaAldrich, 10%, 150 мг), в безводном ДХМ (Sigma-Aldrich, 10 мл) и безводном метаноле (5 мл) перемешивали в атмосфере под водородом из баллона в течение ночи. ВЭЖХ анализ показал, что реакция завершена. Реакционную смесь фильтровали через CELITE и фильтрат концентрировали, получая соединение 7 в виде светло-желтого твердого вещества (562 мг, 86%), которое использовали на следующей стадии без дополнительной очистки. Соединение 9. Раствор соединения 7 (260 мг, 0,53 ммоль), ди-трет-бутилдиэтилфосфорамидита(Sigma-Aldrich, 0,58 мл, 2,1 ммоль) и тетразола (Sigma-Aldrich, 0,45 М в ацетонитриле 14 мл) в тетрагидрофуране (ТГФ Sigma-Aldrich, безводный, 25 мл) перемешивали при комнатной температуре в течение 2 ч. ВЭЖХ анализ показал, что исходное вещество 7 полностью превратилось в соединение 8. Без выделения соединения 8 прибавили раствор м-хлорпербензойной кислоты (mCPBA, Sigma-Aldrich, 365 мг, 2,12 ммоль) в ДХМ (безводный, Sigma-Aldrich, 10 мл). Полученную реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. ВЭЖХ анализ показал, что реакция завершена. Реакционную смесь концентрировали с 0,5 г силикагеля с образованием суспензии. Суспензию подвергали очистке на колонке 4 г CombiFlash с 10-50% градиентом EtOAc в гексане. Фракции, содержащие продукт, объединяли и сушили в высоком вакууме, получая соединение 9 в виде белого твердого вещества (212 мг, 59%). Соединение 10. TFA (Fisher, 3 мл) добавляли к раствору соединения 9 (212 мг, 0,031 ммоль) в безводном ДХМ (Sigma-Aldrich, 3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. ВЭЖХ анализ показал, что реакция завершена. Реакционную смесь концентрировали и сушили в высоком вакууме в течение ночи, получая соединение 10 в виде зеленоватого твердого вещества (271 мг). 1 Н-ЯМР в виде соли ТФУ (ДМСО-d6):11,81 (с, 1H), 8,42 (с, 1H), 8,12 (д, 1H), 7,94 (д, 1H), 7,55 (м,1H), 7,42 (м, ЗН), 7,18 (с, 1H), 7,02 (д, 1H), 4,78 (т, 1H), 4,68 (д, 1H), 4,32 (с, 1H), 4,02 (м, 1H), 3,90 (м, 1H). Соединение 12. Значение рН раствора соединения 11 (179 мг, 0,25 ммоль предположительно моноТФУ-соли, полученной в соответствии с Sufi et al. 2010, раскрытие которой включено в настоящее изобретение посредством ссылки), и HATU (Oakwood, 84,4 мг, 0,222 ммоль) в безводном ДМФА (SigmaAldrich, 3 мл) доводили до значения выше 8 добавлением DIPEA. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. ВЭЖХ-анализ показал, что большая часть соединения 11 была активирована. Соединение 10 (130 мг, 0,222 ммоль предположительно моно-ТФУ-соль) добавляли к реакционной смеси. Значение рН доводили до значения выше 8 добавлением DIPEA. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. ВЭЖХ-анализ показал, что реакция завершена. Реакционную смесь вводили в препаративную ВЭЖХ и очищали с 10-100% градиентом ацетонитрила в воде (с 0,1% TFA) в качестве элюента. Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 12 в виде белого твердого вещества (138 мг, 55%). Масс-спектр: [М+Н] 1055. Специалистам в данной области техники будет понятно, что синтез, описанный выше и на фиг. 1 А и 1 В, включает конвергентную сборку предшественников фрагментов а и а' и что альтернативные стратегии синтеза могут быть использованы, например, посредством сборки фрагментов предшественниковb, b' и b" или фрагментов с, с' и с", как показано ниже. Последние две стратегии проиллюстрированы вSufi et al. 2010, раскрытие которого включено посредством ссылки, и могут быть адаптированы для синтеза соединений в соответствии с настоящим изобретением с соответствующими изменениями. Пример 2 - соединение (Ia). Этот пример относится к синтезу соединения 14, также упоминаемого в настоящем изобретении как соединение (Ia). Схема синтеза приведена на фиг. 2. Соединение 13. DIPEA (Aldrich, 17 мкл, 01 ммоль) добавляли к раствору соединения 10 а (SigmaAldrich, 15,7 мг, 0,051 ммоль) и HATU (Oakwood, 19,4 мг, 0,051 ммоль) в безводном ДМФА (Acros, 1 мл). Значение рН реакционной смеси было выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. ВЭЖХ анализ показал, исходное вещество было полностью активировано. Соединение 10 (30 мг, 0,051 ммоль предположительно, моно-ТФУ-соль) добавляли к этому раствору. Добавляли дополнительно DIPEA, чтобы довести рН реакционной смеси до значения выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. ВЭЖХ анализ показал, что реакция завершена. Сырой продукт очищали препаративной ВЭЖХ с 10-100% градиентом ацетонитрила в воде(0,1% ТФУ по объему) в качестве элюента с получением соединения 13 (25 мг, 71%) в виде не совсем белого твердого вещества. Соединение 14. ТФУ (VWR, 1 мл) добавляли к раствору соединения 13 (25 мг, 0,036 ммоль) в безводном ДХМ (Acros, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. ВЭЖХ анализ показал, что реакция завершена. Реакционную смесь концентрировали и очищали препаративной ВЭЖХ с 10-65% градиентом ацетонитрила в воде (0,1% ТФУ по объему) в качестве элюента, с получением соединения 14 (21 мг, 82% предположительно, моно-ТФУ-соль) в виде не совсем белого твердого вещества. 1(ДМСО-d6):5,909 (с, 1 Н), MS: [M + Н] 591. Пример 3 - соединение (IIb). Фиг. 3 A-3D показывают в комбинации синтез соединения 28, также упоминаемое в настоящем изобретении как соединение (IIb). Специалистам в данной области техники будет понятно, что общая стратегия, используемая в настоящем изобретении соответствует образцу b/b'/b", описанному выше в примере 1. Фиг. 3 А показывает синтез первого промежуточного соединения 18. Соединение 17. Раствор трет-бутилового эфира (2-оксоэтил)карбамата (соединение 16, Aldrich, 5 г,31,4 ммоль) в метаноле (безводный, Acros, 15 мл) добавляли к раствору (S)-метил 2-амино-3 метилбутаноата (соединение 15, Bachem, соль HCl, 5,26 г, 31,4 ммоль), ацетат натрия (Aldrich, 10,3 г,82,06 ммоль) в метаноле (безводный, Acros, 65 мл). Затем добавляли NaBH3CN (Aldrich, 3,95 г, 62,8 ммоль). Реакционную смесь перемешивали при температуре 22,5 С в течение 16 ч и концентрировали до суспензии. Суспензию растворяли в воде (100 мл) и полученный раствор экстрагировали этилацетатом(3100 мл). Органические фазы объединяли, промывали солевым раствором (1100 мл), концентрировали и сушили в вакууме с получением соединения 17 (масло, 9,4 г неочищенного). [М + Н] 275. Соединение 18. Раствор гидрата LiOH (Aldrich, 2,88 г, 68,6 ммоль) в воде (40 мл) добавляли к раствору сырого соединения 17 (9,4 г) в метаноле (40 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи и разбавляли водой (40 мл). Полученный раствор экстрагировали ДХМ (40 мл). Водный слой отделяли и подкисляли раствором KHSO4 (Aldrich, 20% в воде) до рН 5. Суспензию фильтровали и твердое вещество сушили в высоком вакууме, получая соединение 18 (4,85 г, 60,2% из расчета на соединение 15) в виде белого твердого вещества. Масс-спектр: [М + Н] 261. 1H-ЯМР (ДМСО-d6)6,82 (т, 1H), 3,06 (м, 2 Н), 2,83 (д, 1H), 2,62 (м, 2 Н), 2,31 (м, 1H), 1,88 (м, 1H),1,38 (с, 9 Н), 0,86 (д, 6 Н). Фиг. 3 В показывает синтез второго промежуточного соединения 22. Соединение 21. трет-Бутил-4-аминобензойную кислоту (соединение 19, Fluka, 8,75 г, 45,3 ммоль),N-(3-диметиламинопропил)-N'-этилкарбодиимид ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли EtoAc (600 мл) и полученный раствор промывали раствором Na2CO3 (5%, 200 мл), насыщенным раствором NaHCO3 (1200 мл) и солевым раствором (1200 мл). Объединенные органические фазы сушили над безводным MgSO4 и концентрировали, получая неочищенное соединение 21 в виде желтого твердого вещества. 1(синглет, 2 Н), 4,10-4,29 (м, 4 Н), 2,90-3,05 (м, 2 Н), 1,32-1,70 (м, 13 Н), МС: [М + Н] 573. Соединение 22. Пиперидин (Aldrich, 14 мл) добавляли к раствору сырого соединения 21 в безводном ДМФА (Acros, 140 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. ВЭЖХ анализ показал, что реакция завершена. Реакционную смесь концентрировали и остаток растворяли в EtOAc (600 мл). Органическую фазу промывали насыщенным раствором соли (1200 мл), сушили над безводным MgSO4 и концентрировали с силикагелем до суспензии. Суспензию очищали флэшхроматографией с 0-20% градиентом метанола в ДХМ в качестве элюента с получением соединения 22 в виде белого твердого вещества (9,5 г, выход 72% из расчета на соединение 20). 1H-ЯМР (ДМСО-d6)7,82 (д, 2 Н), 7,72 (д, 2 Н), 5,97 (т, 1H), 5,38 (с, 2 Н), 3,43 (м, 1H), 2,98 (т, 2 Н),1,30-1,62 (м, 13 Н), МС: [М + Н] 351. Фиг. 3 С показывает синтез третьего промежуточного соединения 25. Соединение 23. DIPEA (Aldrich) добавляли к раствору соединения 18 (1,5 г, 5,77 ммоль), бензотриазол-1-илокси-трис(диметиламино)фосфоний гексафторфосфата (ВОР, Bachem, 2,87 г, 6,5 ммоль) и соединения 22 (1,84 г, 5,26 ммоль) в ДМФА (Acros, безводный, 15 мл) в количестве, достаточном для доведения рН раствора до значения выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. ВЭЖХ анализ показал, что реакция завершена. ДХМ (100 мл) и насыщенный растворNaHCO3 (100 мл) добавляли к реакционной смеси. Водный слой экстрагировали ДХМ (225 мл). Органические фазы объединяли и концентрировали с силикагелем (8 г) до суспензии. Суспензию очищали на 80 г колонке CombiFlash с 0-10% градиентом метанола в ДХМ в качестве элюента. Содержащие продукт фракции объединяли, концентрировали и сушили в высоком вакууме с получением соединения 23 в виде белого твердого вещества (2,1 г, 68%). Масс-спектр: [М + Н] 593. Соединение 24. HCl в диоксане (Aldrich, N 4, 7 мл) добавляли к раствору соединения 23 (1,8 г, 3,03 ммоль) в ацетонитриле (квалификации для ВЭЖХ, Aldrich, 20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. ВЭЖХ анализ показал, что реакция завершена. Продукт собирали фильтрацией и сушили в высоком вакууме в течение ночи, получая соединение 24 в виде почти белого твердого вещества (1,45 г, 94%). Масс-спектр: [М + Н] 437. Соединение 25. DIPEA добавляли к раствору соединения 24 (200 мг, 0,39 ммоль, предположительно, двойная соль HCl) и (Вос)2 О (94,2 мг, 0,43 ммоль) в ДМФА (безводный, 2 мл) в количестве, достаточном для приведения рН реакционной смеси к значению выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. ВЭЖХ анализ показал, что реакция завершена. Добавляли диэтиловый эфир (20 мл) и смесь перемешивали при комнатной температуре в течение 15 мин. Продукт выделяли в виде полутвердого вещества. Растворитель декантировали и полученный продукт сушили в высоком вакууме в течение ночи, получая соединение 25 в виде белого твердого вещества (178 мг, 85%,HCl соль). Масс-спектр: [М + Н] 537. 1(м, 6 Н). Фиг. 3D показывает завершение синтеза соединения 28, также упоминаемого в настоящем изобретении как соединение (IIb). Соединение 26. DIPEA (Aldrich, 0,142 мл, 0,816 ммоль) добавляли к раствору соединения 25 (394 мг, 0,734 ммоль) и HATU (Oakwood, 186 мг, 0,489 ммоль) в ДМФА (Acros, безводный, 3 мл). Значение рН реакционной смеси было выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. Прибавляли соединение 10 (пример 1, 192 мг, 0,407 ммоль). Прибавляли дополнительноDIPEA для поддержания рН реакционной смеси выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь вводили в препаративную ВЭЖХ и очищали элюированием 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт,объединяли и лиофилизировали с получением соединения 26 (205 мг, 51%) в виде белого твердого вещества. Масс-спектр: [М + Н] 990. Соединение 27. ТФУ (VWR, 5 мл) добавляли к раствору соединения 26 (205 мг, 0,207 ммоль) в ДХМ (Acros, безводный, 5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали и лиофильно сушили с получением соединения 27 в виде его ТФУ-соли (210 мг, 100%). Масс-спектр: [М + Н] 890. Соединение 28. DIPEA (Aldrich, 0,147 мл, 0,842 ммоль) добавляли к раствору 3 меркаптопропионовой кислоты (Aldrich, 59,6 мг, 0,562 ммоль), соединения 27 (100 мг, 0,112 ммоль) и ВОР (Aldrich, 124 мг, 0,281 ммоль) в ДМФА (Acros, безводный, 2 мл). Значение рН реакционной смеси было выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь вводили в препаративную ВЭЖХ и очищали элюированием 10-65% градиентом ацетонитрила в воде (с 0,01% ТФУ) с получением соединения 28 (45 мг, 45,5%). Масс-спектр: [М + Н] 978. Пример 4 - получение иммуноконъюгатов. Ниже приведена в качестве иллюстрации общая процедура для получения иммуноконъюгатов в соответствии настоящим изобретением, основанная на введении свободных тиольных групп на антитело реакцией с лизиновой s-аминогруппы с 2-иминотиоланом, с последующей реакцией с малеимидсодержащим пролекарственным фрагментом, таким как соединение (IIa). Первоначально заменяли буфер в растворе антитела на 0,1 М фосфатный буфера (рН 8,0), содержащий 50 мМ NaCl и 2 мМ диэтилентриаминпентауксусную кислоту (DTPA) и концентрировали до 5-10 мг/мл. Тиолирование достигалось за счет добавления 2-иминотиолана к антителу. Количество 2-иминотиолана, которое должно быть добавлено, может быть определено в предварительном эксперименте, и оно изменяется от антитело к антителу. В предварительном эксперименте осуществляли титрование добавлением возрастающих количеств 2 иминотиолана к антителу и последующей инкубацией с антителами в течение 1 ч при комнатной температуре 25 С), антитело обессоливали в 50 мМ HEPES буфере, рН 6,0 с помощью колонки SEPHADEXG-25 и число введенных тиоловых групп быстро определяли реакцией с дитиодипиридином (DTDP). Реакция тиольных групп с DTDP приводило к освобождению тиопиридина, что можно контролировать спектроскопически при 324 нм. Как правило, использовали образцы с концентрацией белка 0,5-1,0 мг/мл. Оптическая плотность при 280 нм могла быть использована для точного определения концентрации белка в образцах, а затем аликвоту каждой пробы (0,9 мл) инкубировали с 0,1 мл DTDP (5 мМ исходный раствор в этаноле) в течение 10 мин при комнатной температуре. Чистый образец одного буфера плюсDTDP также инкубировали параллельно. Через 10 мин измеряли поглощение при 324 нм и количественно определяли число тиоловых групп с использованием коэффициента экстинкции для тиопиридина равного 19800 М-1. Как правило, желательным является уровень тиолирования около трех тиоловых групп на антитело. Например, для некоторых антител это может быть достигнуто посредством добавления 15-кратного молярного избытка 2-иминотиолана с последующей инкубацией при комнатной температуре в течение 1 ч. Антитело затем инкубировали с 2-иминотиоланом в желаемом молярном соотношении и затем обессоливали в буфере для конъюгации (50 мМ, рН 6,0 HEPES буфер, содержащий 5 мМ глицина и 2 мМ DTPA). Тиолированное вещество выдерживали на льду, пока количество введенных тиолов не определят количественно, как описано выше. После проверки количества введенных тиолов, фрагмент лекарственное средство-линкер добавляли при 3-кратном молярном избытке, считая на тиол. Реакции конъюгации дают возможность протекать в буфере для конъюгации, также содержащем конечную концентрацию, равную 5 % диметилсульфоксида(ДМСО), или подобного альтернативного растворителя. Как правило, исходный раствор лекарственное средство-линкер растворяется в 100% ДМСО. Исходный раствор добавляли непосредственно к тиолированному антителу, которое имеет достаточно ДМСО для доведения конечной концентрации до 10% или предварительно разбавленного в буфере для конъюгации, содержащим конечную концентрацию в 10% ДМСО, с последующим добавлением к равному объему тиолированного антитела. Реакционную смесь конъюгации инкубировали при комнатной температуре в течение 2 ч при перемешивании. После инкубации реакционную смесь конъюгации центрифугировали и фильтровали через фильтр 0,2 мкм. Очистка конъюгата могла быть достигнута посредством хроматографии с использованием ряда способов. В одном способе конъюгат очищали с использованием гель-фильтрационной хроматографии на SEPHACRYL S200, предварительно уравновешенным 50 мМ HEPES буфером, рН 7,2, содержащей 5 мМ глицина и 50 мМ NaCl. Хроматографию осуществляли с линейной скоростью потока 28 см/ч. Фракции, содержащие конъюгат собирали, объединяли и концентрировали. В альтернативном способе очистка могла быть достигнута путем ионообменной хроматографии. Условия варьировались от антитело к антителу и должны быть оптимизированы в каждом случае. Например, реакционную смесь конъюгат антитело-лекарственная субстанция наносли на колонку с SP- SEPHAROSE, предварительно уравновешенную в 50 мМ HEPES, рН 5,5, содержащим 5 мМ глицина. Конъюгат антитела элюировали с использованием градиента 0-1 М NaCl в уравновешивающем буфере при рН 5,5. Соответствующие фракции, содержащие иммуноконъюгат, объединяли и диализовали против буфера для композиции (50 мМ, рН 7,2 HEPES буфер, содержащий 5 мМ глицина и 100 мМ NaCl). В соответствии с описанной выше процедурой иммуноконъюгаты соединения (IIa) получали с антиПСМА человеческим моноклональным антителом (2 А 10, Huang et al. 2009 и Cardarelli et al. 2011); антимезотелин человеческим моноклональным антителом (6 А 4, Terrett et al. 2009a), анти-CD70 человеческим моноклональным антителом (2 Н 5, Terrett et al. 2009a), и анти-CD19 человеческим моноклональным антителом (21D4, Rao-Naik et al. 2009). Иммуноконъюгаты сравнения с соединением (А) были получены подобным образом. Специалистам в данной области техники очевидно, что вышеописанные условия и методология являются иллюстративными и не ограничивающими, и что другие подходы для конъюгации известны в данной области техники и могут быть использованы в настоящем изобретении. Пример 5 - исследование инкорпорации 3 Н-тимидина. Этот пример в целом описывает процедуру, используемую для исследования антипролиферативной активности иммуноконъюгатов в соответствии настоящим изобретением. Линии опухолевых клеток человека были получены из Американской коллекции типовых культур (АТСС), P.O. Box 1549, Manassas,VA 20108, USA, и культивировались в соответствии с инструкциями АТСС. Клетки СНО-мезо получали посредством трансфекции клеток СНО ДНК, содержащей ген человеческого мезотелина, и подвергнутый селекции для (получения) стабильных клонов, экспрессирующих человеческий мезотелин. Клетки высевали при 1,0104 клеток/лунку в 96-луночных планшетах для 3 часового анализа Н тимидина в указанном порядке. Серийные разведения (1:3) иммуноконъюгатов добавляли к лункам. Планшеты инкубировали в течение 72 ч. Планшеты загружали 1,0 мкКи 3 Н-тимидина на лунку в течение последних 24 ч общего периода инкубации собирали и считывали на сцинтилляционном счетчике Top Count Scintillation Counter(Packard Instruments, Meriden, CT). Величины EC50 - концентрация, при которой агент ингибирует или уменьшает пролиферацию клеток на 50% от максимального ингибирования - определяли с помощью программного обеспечения PRISM, версия 4.0 (GraphPad Software, La Jolla, CA, США). Антипролиферативные результаты показаны на фиг. 4A-4F, рассмотренных выше. Пример 6 - дефосфорилирование с помощью клеточных лизатов. Этот пример демонстрирует, что фосфатные пролекарственные соединения секо-MGBA в соответствии с настоящим изобретением могут дефосфорилироваться посредством лизатов человеческих опухолевых клеток. Клетки 786-0 в виде замороженных гранул с предполагаемым количеством 107 клеток на пробирку для вортекса ресуспендировали и гомогенизировали в 500 мкл буфера для лизиса (25 мМ ацетат натрия,1 мМ ЭДТА, 0,25 М сахарозы, 0,1% Triton Х-100, рН 5,5). Гомогенизация включает перемешивание на вортексе в течение 30 с, после чего следует одна минута охлаждении на льду. Для каждой пробирки выполняли по три цикла вортекса и охлаждения. Образцы клеток затем дополнительно смешивали, пропуская через иглу 19 три раза. После гомогенизации ДНК клеточного лизата гидролизовали, используя ДНКазу BENZONASE. Образцы сначала вносили в 1 мМ MgSO4. После перемешивания 2 мкл ДНКазы BENZONASE (неразбавленной) добавляли в каждую ампулу лизата (4 мкл/мл, об./об., конечная концентрация). Затем образцы хранили при комнатной температуре в течение 15 мин, а затем охлаждали на льду. На этой стадии об успешной активности ДНКазы свидетельствовало появление хлопьевидного осадка. Затем образцы центрифугировали при максимальной скорости в микроцентрифуге в течение 5 мин для удаления клеточного дебриса. Супернатант замораживали при 70 С для последующего использования. Концентрацию белка в лизате определяли с помощью Pierce BCA Protein Determination methodology (Thermo Scientific). Было найдено, что образцы в этом исследовании содержали 2,85 мг/мл белка. Получали 2000 мкМ исходный раствор соединения формулы (Ia). Лизат разбавляли до 2,1 мг/мл в буфере для лизиса. Для реакции добавляли 5 мкл исходного раствора соединения (Ia) к 95 мкл лизата. Конечные концентрации составляли 100 мкМ соединения (Ia) в лизате, содержащем 2 мг/мл белка. Буфер, содержащий 25 мМ ацетата натрия, 1 мМ ЭДТА, 0,25 М сахарозы, 0,1% Triton100 и 1 мМ MgSO4 обеспечивал буферизацию при рН 5,5. Использовали отрицательные контроли с буфером для лизиса вместо клеточного лизата с ДНКазойBENZONASE и также добавляли MgSO4. Исследуемые и контрольные образцы помещали в нагревательный блок, установленный на уровне 37 С. Через определенные промежутки времени (5 мин, 1, 2, 4 и 8 ч) отбирали 50 мкл аликвоты и реакции в каждой аликвоте останавливали добавлением 150 мкл (3 Х) холодного этанола. Образцы хранили на льду в течение 1 ч и центрифугировали для удаления белка. Супернатант сохраняли для UPLC (сверхпроизводителъная жидкостная хроматография) анализа, при следующих условиях: Колонка: Waters HSS Т 3 2,150 мм С 18 UPLC. Подвижная фаза: А буфер: вода/0,1% TFA; В буфер: ацетонитрил/0,1% TFA. Система ВЭЖХ: Waters UPLC. Объем инъекции: 4 мкл. Элюирование: от 25 до 40% В, через 1,8 мин. Расход: 1 мл/мин. Обнаружение: A340. За реакцией следили с помощью мониторинга за продукцией соединений (IVa) и (IVb), которые являются секо- и циклопропановой формами дефосфорилированного соединения (Ia) соответственно. Было установлено, что дефосфорилирование происходит достаточно быстро, заканчиваясь полностью в течение 8 ч. Поскольку рН реакционной смеси составляла 5,5, близкая к найденной в лизосомальных органеллах, результаты показывают вовлечение кислой фосфатазы в реакцию дефосфорилирования. Аналогичный эксперимент осуществляли с использованием лизатов клеток Н 226. Было обнаружено, что дефосфорилирование полностью заканчивалось в течение 3 ч. Пример 7 - дефосфорилирование с помощью микросом печени. Этот пример демонстрирует дефосфорилирование соединения (Ia) с помощью ферментов микросом печени человека и мыши. Микросомы печени человека, полученные из пулированных печеночных источников, получали изXenotech (Part Number H-0630). Они поставлялись с номинальной концентрацией белка 20 мг/мл в 20% растворе сахарозы. Содержание белка проверяли с помощью ВСА (бицинхониновая кислота) анализа с использованием реагента от Thermo-Fisher. Микросомы печени человека разбавляли до 8,42 мг/мл в реакционном буфере (100 мМ Трис-HCl, 1 мМ MgCl2, 0,9% NaCl, pH 7,4). Количественно разбавление было спроектировано для обеспечения точной доставки 0,8 мг в 95 мкл конечного 100 мкл реакционного объема. Исходный раствор затем серийно разбавляли 1:1 по объему, получая десять сток-растворов для доставки от 8 до 0,0156 мг/мл микросом в сток-растворе. Получали исходный раствор 2000 мкМ соединения формулы (Ia). Для реакций по 95 мкл каждого сток-раствора микросом уравновешивали при 37 С в нагревательном блоке. Аликвоту в 5 мкл стокраствора соединения (Ia) затем добавляли в каждую реакционный флакон в шахматном порядке в точности через 30-секундные интервалы. Конечная концентрация соединения (Ia) была 100 мкМ. Образцы выдерживали для протекания реакции при температуре в течение 1 ч, после чего реакцию останавливали добавлением 100 мкл холодного этанола. Остановку осуществляли снова точно через 30-секундные интервалы, так что в каждом флаконе реакция протекала в течение ровно 1 ч. Образцы готовили в двух повторах. Белок осаждали на льду в течение 30 мин. Супернатант собирали после центрифугирования. Хроматографический анализ UPLC проводили с использованием тех же условий, как описано в предыдущем примере, снова контролируя получение соединений (IVa) и (IVb). Хроматограммы, показывающие превращение соединения (Ia) в соединения (IVa) и (IVb) как функцию концентрации микросом, показаны на фиг. 5. Фиг. 6 представляет собой график зависимости количества соединений (IVa) и (IVb), получаемых из соединения (Ia) через 60 мин как функцию концентрации микросом. Скорость образования была, по существу, линейной для концентраций микросом от 0,00156 до 0,0125 мг/мл. Используя данные в этом диапазоне, рассчитали скорость образования соединений (IVa) и (IVb), равную 0,0054 мкмоль/мин/мг. Используя такую же методику, но с микросомами печени мыши (Xenotech, Part No. М-3000), получали скорость равную 0,0018 мкмоль/мин/мг. Пример 8 - соединение (IIc). Этот пример и фиг. 7 описывают синтез соединения 30, также упоминаемое в настоящем изобретении как соединение (IIc). Соединение 30. DIPEA (Aldrich, 7,85 мкл, 0,045 ммоль) добавляли к раствору соединения 27 (20 мг,0,022 ммоль), 5-азидопентаноевой кислоты 29 (Bachem, 6,43 мг, 0,045 ммоль) и ВОР (Aldrich, 19,87 мг,0,045 ммоль) в ДМФА (Acros, безводный, 0,5 мл). Значение рН реакционной смеси оставалась выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь наносили на колонку препаративной ВЭЖХ и очищали элюированием 10-65% градиентом ацетонитрила в воде (с 0,1% TFA), получая соединение 30 (5 мг, 21,9%). Масс-спектр: [М + Н] 1015. Соединение 30 (IIc) имеет азидную реакционноспособную функциональную группу, что делает его пригодным для конъюгации использующей "клик-химию". Пример 9 - соединения (IId). Этот пример и фиг. 8 описывают синтез соединения 34, также упоминаемое в настоящем изобретении как соединение (IId). Соединение 32. Дициклогексилкарбодиимид (DCC Novabiochem, 277 мг, 1,098 ммоль) добавляли к раствору 1-гидроксипирролидин-2,5-диона (Acros, 126 мг, 1,098 ммоль), (Вос-аминокси)уксусной кислоты 31 (TCI, 200 мг, 1,046 ммоль) в 1,4-диоксане (Aldrich, безводный, 2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Реакционную смесь фильтровали через CELITE и фильтрат концентрировали. Остаток растворяли в EtOAc (30 мл), промывали насыщенным растворомNaHCO3 (20 мл) и водой (20 мл). Органические фазы концентрировали и сушили в высоком вакууме с получением соединения 32 (275 мг, 91,2%). 1 Н-ЯМР (400 МГц, CDCl3):7,66 (с, 1H), 4,78 (с, 2 Н), 2,87 (с, 4 Н), 1,49 (с, 9 Н). Соединение 33. DIPEA (Aldrich, 36,5 мкл, 0,21 ммоль) добавляли к раствору соединения 32 (20 мг,0,07 ммоль) и соединения 27 (31 мг, 0,035 ммоль) в ДМФА (Aldrich, безводный, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь очищали с помощью препаративной ВЭЖХ с 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 33 (24 мг, 64,5%). Масс-спектр: [М + Н] 1063. Соединение 34. ТФУ (Acros, 0,5 мл) добавляли к раствору соединения 33 (24 мг, 0,023 ммоль) в ДХМ (Aldrich, безводный, 0,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. Реакционную смесь очищали с помощью препаративной ВЭЖХ с 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 34 (10 мг, 45,7%). Масс-спектр: [М + Н] 963. Соединение 34 (IId) имеет гидроксиламиновые реакционноспособные функциональные группы, что делает его пригодным для конъюгации посредством образования оксима с антителом, модифицированным, чтобы содержать кетогруппу, например, посредством включения неприродной аминокислоты пацетилфенилаланина. Пример 10 - соединение (IIe). Этот пример и фиг. 9 А-9 В в комбинации описывают синтез соединения 48, также называемое в настоящем изобретении соединением (IIe). Соединение 37. Хлорид меди(II) (Aldrich, 1,252 г, 9,31 ммоль) добавляли к раствору трет-бутил-4 аминобензойной кислоты 36 (Fluka, 1,5 г, 7,76 ммоль), Fmoc-защищенного лейцина 35 (Bachem, 2 г, 5,66 ммоль), EDC (Fluka, 1,786 г, 9,31 ммоль) и трет-бутанола (Chem-Impex, 1,426 г, 9,31 ммоль) в ДМФА(Aldrich, безводный, 15 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь обрабатывали этилацетатом (30 мл), водой (30 мл) и солевым раствором (30 мл). Органические фазы концентрировали и сушили в высоком вакууме с получением соединения 37 (3,7 г). Масс-спектр: [М + Н] 529. Соединение 38. Пиперидин (Aldrich, 3 мл) добавляли к раствору неочищенного соединения 37 (3,7 г) в ДМФА (Aldrich, безводный, 15 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 ч и затем очищали с помощью препаративной ВЭЖХ, элюируя 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 38 (1,7 г, 70,8% за две стадии). Масс-спектр: [М + Н] 307. Соединение 41. DIPEA (Aldrich, 0,97 мл, 5,5 ммоль) добавляли к раствору трет-бутилового эфира аланина гидрохлорида 39 (Bachem, 0,4 г, 2,22 ммоль) и соединения 40 (Bachem, 1 г, 2,22 ммоль) в ДМФА(Aldrich, безводный, 20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и смесь обрабатывали этилацетатом (100 мл), водой (50 мл) и солевым раствором (50 мл). Органические фазы концентрировали и сушили в высоком вакууме с получением соединения 41 (1,3 г). Масс-спектр:[М + Н] 481. Соединение 42. ТФУ (Acros, 10 мл) добавляли к раствору неочищенного соединения 41 (1,3 г) в ДХМ (Acros, безводный, 10 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 ч, концентрировали и очищали препаративной ВЭЖХ, элюируя 10-100% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 42 (0,68 г, 72,2% за две стадии). Масс-спектр: [М + Н] 425. Соединение 43. DIPEA (Aldrich, 0,246 мл, 1,413 ммоль) добавляли к раствору соединения 38 (289 мг, 0,842 ммоль), соединения 42 (200 мг, 0,471 ммоль) и ВОР (Bachem, 313 мг, 0,707 ммоль) в ДМФА(Aldrich, безводный, 3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и обрабатывали этилацетатом (30 мл), водой (20 мл) и солевым раствором (20 мл). Органические фазы концентрировали и очищали на колонке с 4 г CombiFlash, элюируя 0-60% градиентом EtOAc в гексане. Органические фазы объединяли, концентрировали и сушили в высоком вакууме с получением соединения 43 (126 мг, 37,5 %). Масс-спектр: [М + Н] 713. Соединение 44. ТФУ (Acros, 3 мл) добавляли к раствору соединения 43 (126 мг) в ДХМ (Acros, безводный, 3 мл). Реакционную смесь выдерживали при комнатной температуре в течение 0,5 ч, концентрировали и очищали препаративной ВЭЖХ, элюируя 10-100% градиентом ацетонитрила в воде (с 0,1%TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 44 (84 мг, 72%). Масс-спектр: [М + Н] 657. Соединение 45. DIPEA (Aldrich, 21 мкл, 0,122 ммоль) добавляли к раствору соединения 44 (40 мг,0,061 ммоль), HATU (Oakwood, 18,53 мг, 0,049 ммоль) в ДМФА (Aldrich, безводный, 1 мл). Значение рН реакционной смеси было выше 8. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин. К этой реакционной смеси добавляли соединение 10 (34,5 мг, 0,073 ммоль) с последующим добавлением DIPEA (Aldrich, 21 мкл, 0,122 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и очищали препаративной ВЭЖХ, элюируя 10-100% градиентом ацето- 24024844 нитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 45 (20 мг, 30%). Масс-спектр: [М + Н] 1110. Соединение 46. Пиперидин (Aldrich, 0,2 мл, 2,02 ммоль) добавляли к раствору соединения 45 (20 мг) в ДМФА (Acros, безводный, 0,5 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 ч. Реакционную смесь очищали препаративной ВЭЖХ, элюируя 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 46 (8 мг, 50%). Масс-спектр: [М + Н] 888. Соединение 48. DIPEA (Aldrich, 10 мкл, 0,006 ммоль) добавляли к раствору соединения 46 (8 мг,0,009 ммоль) и N-сукцинимидил-6-малеимидогексаноата 47 (TCI, 5,55 мг, 0,018 ммоль) в ДМФА (Acros,безводный, 1 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 ч. Реакционную смесь очищали препаративной ВЭЖХ, элюируя 10-100% градиентом ацетонитрила в воде (с 0,1%TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 48 (2 мг, 20%). Масс-спектр: [М + Н] 1081. Трипептид Leu-Ala-Leu соединения 48 (IIe) является субстратным мотивом для фермента CD10, так что конъюгаты, полученные с этим соединением, должны быть чувствительными к расщеплению посредством CD10. Пример 11 - соединение (IIf). Этот пример и фиг. 10 А-10 В в комбинации описывают синтез соединения 56, также называемого в настоящем изобретении как соединение (IIf). Соединение 50. Хлорид меди (II) (Aldrich, 0,913 г, 6,79 ммоль) добавляли к раствору трет-бутил-4 аминобензойной кислоты 36 (Fluka, 1,312 г, 6,79 ммоль), Fmoc-защищенного изолейцина 49 (Bachem, 2 г,5,66 ммоль), EDC (Fluka, 1,302 г, 6,79 ммоль) и трет-бутанола (Chem-Impex, 1,040 г, 6,79 ммоль) в ДМФА(Aldrich, безводный, 20 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь обрабатывали этилацетатом (30 мл), водой (30 мл) и солевым раствором (30 мл). Объединенные органические фазы концентрировали и очищали на колонке с 40 г CombiFlash,элюируя 0-20% градиентом этилацетата в гексане. Содержащие продукт фракции объединяли, концентрировали и сушили в высоком вакууме с получением соединения 50 (0,85 г, 28,4%). Масс-спектр: [М + Н] 529. Соединение 51. Пиперидин (Aldrich, 0,5 мл) добавляли к раствору сырого соединения 50 (3,7 г) в ДМФА (Aldrich, безводный, 5 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 ч. Реакционную смесь обрабатывали этилацетатом (20 мл), водой (15 мл) и солевым раствором (15 мл). Органические фазы объединяли, концентрировали и очищали на колонке с 12 г CombiFlash, элюируя 0-75% градиента EtOAc в гексане. Содержащие продукт фракции объединяли, концентрировали и сушили в высоком вакууме с получением соединения 51 (0,38 г, 76,9%). Масс-спектр: [М + Н] 307. Соединение 52. DIPEA (Aldrich, 0,228 мл, 1,305 ммоль) добавляли к раствору соединения 51 (0,2 г,0,653 ммоль) и соединения 40 (Bachem, 294 мг, 0,653 ммоль) в ДМФА (Aldrich, безводный, 3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь обрабатывали этилацетатом (50 мл), водой (20 мл) и солевым раствором (20 мл). Объединенные органические фазы концентрировали и очищали на колонке с 4 г CombiFlash, элюируя 0-40% градиентом EtOAc в гексане. Фракции, содержащие продукт, концентрировали с получением остатка, который сушили в высоком вакууме, получая соединение 52 (178 мг, 42,5%). Масс-спектр: [М + Н] 642. Соединение 53. ТФУ (Acros, 3 мл) добавляли к раствору соединения 52 (178 мг, 0,277 ммоль) в ДХМ (Acros, безводный, 3 мл). Реакционную смесь выдерживали при комнатной температуре в течение 0,5 ч. Реакционную смесь концентрировали и сушили посредством лиофилизации с получением соединения 53 (145 мг, 89,5%). Масс-спектр: [М + Н] 586. Дипептид Leu-Ile соединения 56 (IIf) является субстратным мотивом для фермента катепсина Е, поэтому иммуноконъюгаты, полученные с этим соединением, должны быть чувствительными к расщеплению катепсином Е. Пример 12 - соединение (IIg). Этот пример и фиг. 11 А-11 В в комбинации описывают синтез соединения 67, также называемого в настоящем изобретении как соединение (IIg). Соединение 57. Хлорид меди(II) (Aldrich, 0,913 г, 6,79 ммоль) добавляли к раствору метилового эфира 4-аминобензойной кислоты 56 (Aldrich, 0,913 г, 6,05 ммоль), Fmoc-защищенного цитруллина 20(Bachem, 2 г, 5,04 ммоль), EDC (Fluka, 1,160 г, 6,05 ммоль) и трет-бутанола (Chem-Impex, 0,926 г, 6,05 ммоль) в ДМФА (Aldrich, безводный, 20 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь обрабатывали этилацетатом (100 мл), водой (30 мл) и солевым раствором (30 мл). Органические фазы концентрировали и очищали на колонке с 40 г CombiFlash,элюируя градиентом 0-20% метанола в ДХМ. Содержащие продукт фракции объединяли, концентрировали и сушили в высоком вакууме с получением соединения 57 (1,825 г, 68,2%). Масс-спектр: [М + Н] 531. Соединение 58. Пиперидин (Aldrich, 0,2 мл) добавляли к раствору соединения 57 (1,25 г, 3,437 ммоль) в ДМФА (Aldrich, безводный, 3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь обрабатывали этилацетатом (40 мл), водой (15 мл) и солевым раствором (15 мл). Органические фазы объединяли, концентрировали и очищали на колонке с 12 г CombiFlash, элюируя 0-30% градиентом метанола в ДХМ. Содержащие продукт фракции объединяли, концентрировали и сушили в высоком вакууме с получением соединения 58 (0,778 г, 73,1%). Масс-спектр:[М + Н] 309. Соединение 59. DIPEA (Aldrich, 1,0 мл, 5,71 ммоль) добавляли к раствору соединения 58 (778 мг,2,52 ммоль), соединения 18 (655 мг, 2,52 ммоль) и ВОР (Bachem, 1,12 г, 2,52 ммоль) в ДМФА (Aldrich безводный, 10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь обрабатывали этилацетатом (50 мл), водой (20 мл) и солевым раствором (20 мл). Объединенные органические фазы концентрировали и очищали на колонке с 40 г CombiFlash, элюируя 0-15% градиентом метанола в ДХМ. Органические фазы объединяли, концентрировали и сушили в высоком вакууме с получением соединения 59 (1,06 г, 76%). Масс-спектр: [М + Н] 551. Соединение 60. ТФУ (Acros, 5 мл) добавляли к раствору соединения 59 (1,06 г, 1,92 ммоль) в ДХМ(Acros, безводный, 5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. Реакционную смесь концентрировали и сушили посредством лиофилизации с получением соединения 60 в виде его ТФУ-соли (1,09 г). Масс-спектр: [М + Н] 451. Соединение 62. DIPEA (Aldrich, 1,02 мл, 5,838 ммоль) добавляли к раствору соединения 60 (500 мг,1,109 ммоль), 6-трет-бутоксикарбонил)амино)гексановой кислоты 61 (Fluka, 450 мг, 1,946 ммоль) и ВОР (Bachem, 0,720 г, 1,629 ммоль) в ДМФА (Aldrich, безводный, 10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь обрабатывали этилацетатом (60 мл),водой (30 мл) и солевым раствором (30 мл). Объединенные органические фазы концентрировали и очищали на колонке с 12 г CombiFlash, элюируя градиентом 0-20% метанола в ДХМ. Органические фазы объединяли, концентрировали и сушили в высоком вакууме с получением соединения 62 (427 мг, 58%). Масс-спектр: [М + Н] 664. Соединение 63. Раствор LiOH (Aldrich, 150 мг в 10 мл воды) добавляли к раствору соединения 62(0,427 г, 0,643 ммоль) в ацетоне (Baker, 10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и нейтрализовали уксусной кислотой (Fisher, ледяная, 0,3 мл). Реакционную смесь очищали препаративной ВЭЖХ, элюируя 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 63 (325 мг, 77,8%). Масс-спектр: [М + Н] 650. Соединение 64. DIPEA (Aldrich, 27 мкл, 0,153 ммоль) добавляли к раствору соединения 63 (59,5 мг,0,092 ммоль), HATU (Oakwood, 23,21 мг, 0,061 ммоль) в ДМФА (Aldrich, безводный, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. К полученному раствору добавляли соединение 10 (36 мг, 0,076 ммоль), с последующим дополнительным DIPEA (Aldrich, 27 мкл, 0,153 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и очищали препаративной ВЭЖХ, элюируя 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 64 (38 мг, 55,7%). Масс-спектр: [М + Н] 1103. Соединение 65. ТФУ (Acros, 1 мл) добавляли к раствору соединения 64 (38 мг, 0,034 ммоль) в ДХМ(Acros, безводный, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 ч. Реакционную смесь концентрировали и сушили посредством лиофилизации с получением соединения 65 в виде его ТФУ соли (43 мг). Масс-спектр: [М + Н] 1003. Соединение 66. DIPEA (Aldrich, 29 мкл, 0,159 ммоль) добавляли к раствору соединения 32 (15,4 мг,0,053 ммоль) и соединения 65 (43 мг, 0,043 ммоль) в ДМФА (Aldrich, безводный, 1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь очищали препаративной ВЭЖХ, элюируя 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 66 (22 мг, 43,5%). Масс-спектр: [М + Н] 1176. Соединение 67. Раствор соединения 66 (22 мг, 0,019 ммоль) в 4 н. HCl в растворе 1,4-диоксана (Aldrich, 10 мл) перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали и очищали препаративной ВЭЖХ, элюируя 10-65% градиентом ацетонитрила в воде (с 0,1% TFA). Фракции, содержащие продукт, объединяли и лиофилизировали с получением соединения 67 (12 мг,58,7%). Масс-спектр: [М + Н] 1076. Пример 13 - результаты in vivo. Фиг. 12A-12I демонстрируют эффективность in vivo иммуноконъюгатов в соответствии с настоящим изобретением в ксенотрансплантатных исследованиях на мышах, в отношении различных типов рака. Исследования клеток OVCAR3. Пять миллионов клеток рака яичников OVCAR3, ресуспендированных в 0,1 мл фосфатно-солевом буфере (PBS), плюс 0,1 мл матригеля имплантировали подкожно в паховую область SCID (тяжлая комбинированная иммунная недостаточность) мышей. Измерения опухоли,- 26024844 начинали через 23 дня, и мышей рандомизировали на группы по 6 мышей в каждой со средним размером опухоли равном 90 мм 3, оцениваемом как LWH/2 опухолей. Через 24 дня после имплантации опухоли мышам вводили однократно внутрибрюшинно тестируемое соединение. Фиг. 12 А показывает, что в отношении клеток OVCAR3, иммуноконъюгаты антимезотелин антитела 6 А 4 с соединением (IIa) подавляли рост опухоли, особенно при более высокой дозе цитотоксина, равной 0,314 мкмоль/кг. Сравнительные данные для соответствующего иммуноконъюгата анти-CD19 антитела 21D4 также представлены в качестве изотипического контроля. Исследования клеток N87. Два с половиной миллиона клеток рака желудка N87, ресуспендированных в 0,1 мл PBS плюс 0,1 мл матригель, имплантировали подкожно в паховую область SCID мышей. Измерения опухоли начинали через 12 дней и мышей рандомизировали на группы по 7 мышей в каждой со средним размером опухоли равном 110 мм 3, оцениваемом как LWH/2 опухолей. На 14 день после имплантации опухоли мышам вводили однократно внутрибрюшинно тестируемые соединения. Фиг. 12 В показывает, что в отношении клеток N87 иммуноконъюгат антимезотелин антитела 6 А 4 с соединением(IIa) в значительной степени ингибирует рост опухоли. Сравнительные данные для одного буфера композиции или иммуноконъюгата анти-CD19 антитела 21D4 и соединения (IIa) представлены, последние в качестве изотипического контроля. Исследования клеток Н 226. Пять миллионов клеток мезотелиомы Н 226, ресуспендированных в 0,1 мл PBS, содержащим 0,1 мл матригеля, имплантировали подкожно в паховую область SCID мышей. Измерения опухоли начинали через 14 дней и мышей рандомизировали на группы по 9 мышей в каждой со средним размером опухоли равном 110 мм 3, оцениваемом как LWH/2 опухолей. На 15 день после имплантации опухоли мышам вводили однократно внутрибрюшинно тестируемые соединения. Фиг. 12 С показывает дозозависимый эффект иммуноконъюгата антимезотелин антитела 6 А 4 и соединения (IIa) на рост опухоли. SR в каждом случае был 3,6. Исследования клеток Н 1650. Два с половиной миллиона клеток опухоли легких H1650 (аденокарцинома), ресуспендированных в 0,1 мл PBS плюс 0,1 мл матригеля, имплантировали подкожно в паховую область мышей SCID. Измерения опухоли начинали через 7 дней и мышей рандомизировали на группы по 6 мышей в каждой со средними размерами опухоли 110 мм 3, оцениваемом как LWH/2 опухолей. На 9, 14 и 21 день после имплантации опухоли мышам вводили однократно внутрибрюшинно тестируемые соединения. Фиг. 12D показывает ингибирование опухоли иммуноконъюгатом антимезотелин антитела 6 А 4 и соединения (IIa). Сравнительные данные контроля в виде носителя и иммуноконъюгата анти-CD19 антитела 21D4 и соединения (IIa) представлены, последние в качестве изотипического контроля. Исследования клеток Hep 3 В. Четыре миллиона опухолевых клеток печени Hep 3 В, ресуспендированных в 0,1 мл PBS, плюс 0,1 мл матригеля, имплантировали подкожно в паховую область SCID мышей. Измерения опухоли начинали через 10 дней и мышей рандомизировали на группы по 7 мышей в каждой со средним размеров опухоли равном 90 мм 3, оцениваемом как LWH/2 опухолей. На 11 день после имплантации опухоли мышам вводили однократно внутрибрюшинно тестируемые соединения. Фиг. 12 Е показывает дозозависимый ингибирующий эффект на рост опухоли иммуноконъюгата антиглипикан 3 антитела 4 А 6 и соединения (IIa). Самый сильный эффект наблюдался при дозе 0,1 мкмоль/кг. Сравнительные данные для одного носителя, для одного антитела 4 А 6 или для иммуноконъюгата антиCD19 антитела 21D4 и соединения (IIa) также показаны, последний в качестве изотипического контроля. Фиг. 12F представляет собой график из того же исследования, но демонстрирующий способность иммуноконъюгата антитела 4 А 6-соединение (IIa) облегчать ассоциированную с опухолью кахексию. Также представлены данные для иммуноконъюгата анти-CD19-соединение (IIa), показывающие, что конъюгат анти-CD19, что хотя он снижает рост опухоли (на фиг. 12 Е), но не облегчает кахексию (на фиг. 12F). Исследования клеток LNCaP. Два с половиной миллиона клеток опухоли простаты LNCaP, ресуспендированных в 0,1 мл PBS плюс 0,1 мл матригель, имплантировали подкожно в паховую область SCID мышей. Измерения опухоли начинали через 30 дней и мышей рандомизировали на группы по 7 мышей в каждой со средними размерами опухоли равном 150 мм 3, оцениваемом как LWH/2 опухолей. На 31 день после имплантации опухоли мышам вводили однократно внутрибрюшинно тестируемые соединения. Фиг. 12G показывает ингибирование роста опухоли иммуноконъюгатом анти-ПСМА антители 2 А 10 и соединения (IIa) по сравнению с буфером композиции и иммуноконъюгатом анти-CD19 антитела 21D4 и соединения (IIa) в качестве изотипического контроля. В каждом случае концентрация цитотоксина составляла 0,1 мкмоль/кг. Фиг. 12 Н показывает другой набор результатов из того же исследовании с иммуноконъюгатом анти-RG-1 антитела 19G9 и соединения (IIa) с изотипическим контролем иммуноконъюгата анти-CD19. Исследования клеток Raji. Десять миллионов клеток человеческой лимфомы Беркитта Raji, ресуспендированных в 0,1 мл PBS, плюс 0,1 мл матригель, имплантировали подкожно в паховую областьSCID мышей. Измерения опухоли начинали через 6 дней и мышей рандомизировали на группы по 8 мышей в каждой со средним размером опухоли, равном 90 мм 3, оцениваемом как LWH/2 опухолей. Через 7 дней после имплантации опухоли мышам вводили однократно внутрибрюшинно тестируемые соединения. Фиг. 12I показывает ингибирующее действие на рост опухоли иммуноконъюгатов антимезотелин антитела 6 А 4, анти-CD19 антитела 21D4, анти-CD22 антитела 12 С 5 и анти-CD70 антитела 1F4, каждое с соединением (IIa) (полная информации о последовательности антитела 1F4 раскрыта в Coccia et al. 2010,раскрытие которой включено в настоящее изобретение посредством ссылки. Последовательности VHNO:56, SEQ ID NO:57, SEQ ID NO:58, SEQ ID NO:59 и SEQ ID NO:60 соответственно). Приведенное выше подробное описание в соответствии настоящим изобретением включает пассажи, которые являются главным образом или исключительно связанными с конкретными частями или аспектами изобретения. Следует понимать, что это сделано для ясности и удобства, и что конкретный признак может относиться более чем к одному пассажу, в котором он раскрыт, и что настоящее раскрытие охватывает все соответствующие комбинации информации, найденной в различных пассажах. Кроме того, хотя различные чертежи и описания относятся к конкретным вариантам осуществления изобретения, следует понимать, что если специфический признак раскрыт в контексте конкретного чертежа или варианта осуществления, такой признак также может быть использован в соответствующей степени в контексте другого чертежа или варианта осуществления в комбинации с другим признаком или в изобретении в целом. Кроме того, хотя настоящее изобретение особенно описывается в отношении некоторых предпочтительных вариантов осуществления, настоящее изобретение не ограничивается такими предпочтительными вариантам осуществления. Напротив, объем изобретения определяется посредством прилагаемой формулой изобретения. Ссылки Полный перечень ссылок для следующих ссылок, процитированных в сокращенном виде посредством первого автора (или изобретателя) и даты ранее в этом описании, приведены ниже. Каждая из этих ссылок включена в настоящее изобретение в качестве ссылки во всех отношениях.Zhao et al., US 7655660 B2 (2010). Перечень последовательностей Списки последовательностей, включенные в настоящее изобретение посредством ссылки во всей своей полноте под названием "SEQT11770WOPCT.txt", содержат от SEQ ID NO:1 до SEQ ID NO:60,которые включают в себя последовательности нуклеиновых кислот и/или аминокислот, описанные в настоящем изобретении. Список последовательностей был представлен в текстовом формате ASCII черезEFS-Web, и таким образом, представляет собой как бумажную, так и пригодною для ввода в компьютер формы. Перечень последовательностей была впервые создан с использованием PatentIn 3.5 27 апреля 2012 г. и составляет 10 кБ. В следующей таблице приведены описания последовательностей, раскрытых в этой заявке.
МПК / Метки
МПК: A61P 35/00, A61K 47/48, C07D 403/06
Метки: рака, применение, иммуноконъюгат, лечения
Код ссылки
<a href="https://eas.patents.su/30-24844-immunokonyugat-i-ego-primenenie-dlya-lecheniya-raka.html" rel="bookmark" title="База патентов Евразийского Союза">Иммуноконъюгат и его применение для лечения рака</a>
Способ определения стволовых клеток рака молочной железы и применение антагонистов cxcr1 для лечения рака

Номер патента: 23466
Опубликовано: 30.06.2016
Авторы: Джинестьер Кристоф, Уича Макс С.
МПК: G01N 33/574
Метки: молочной, антагонистов, применение, определения, способ, стволовых, рака, клеток, cxcr1, железы, лечения
Формула / Реферат:
1. Способ определения наличия стволовых клеток злокачественной солидной опухоли у пациента с раком молочной железы, где способ включает определение экспрессии белка CXCR1 в образце ткани опухоли, взятом у пациента с раком молочной железы, где наличие белка CXCR1 в образце ткани указывает на наличие стволовых клеток злокачественной солидной опухоли в молочной железе.2. Способ по п.1, где указанная стадия определения включает приведение указанного...
N-α-(бензилоксикарбонил)-l-γ-глутамил-3-[[2-[[бис[бис(2-хлорэтил)амино]фосфонил]окси]этил]сульфонил]-l-аланил-2(r)-фенилглицин или его соль, фармацевтическая композиция, содержащая это соединение, применение этого соединения для лечения рака и способ лечения рака с помощью этого соединения

Номер патента: 16052
Опубликовано: 30.01.2012
Авторы: Боуланджер Уилльям А., Жичкин Павел Э., Шоу Стивен Р., Меклер Харолд, Кьэрсгорд Ханс Й., Гевель Ронан И., Иди Деннис Л., Колиер Стивен Дж., Истам Стивен А., Эрнандес-Абад Педро Э., Херр Джейсон Р., Поломски Роберт Э.
МПК: A61P 35/00
Метки: фармацевтическая, композиция, содержащая, это, соединение, помощью, n-α-(бензилоксикарбонил)-l-γ-глутамил-3-[[2-[[бис[бис(2-хлорэтил)амино]фосфонил]окси]этил]сульфонил]-l-аланил-2(r)-фенилглицин, рака, способ, применение, этого, лечения, соединения, соль
Формула / Реферат:
1. Соединение формулыили его соль.2. Фармацевтическая композиция, включающая соединение по п.1 или его соль.3. Применение соединения по п.1 или его соли для приготовления лекарственного средства, предназначенного для лечения рака.4. Способ лечения рака у млекопитающего, включающий введение млекопитающему соединения по п.1 или его...
Соединения 4-пиридинона и их применение для лечения рака

Номер патента: 17736
Опубликовано: 28.02.2013
Авторы: Борзиллери Роберт М., Цай Чжэнь-Вэй
МПК: A61P 35/00, C07D 401/12, A61K 31/4427...
Метки: рака, лечения, применение, 4-пиридинона, соединения
Формула / Реферат:
1. Соединение, имеющее формулу (I)или его соль,где G представляет собой Н, -СНХ-ОР(=О)(ОН)2 или -CHX-OC(=O)Z;X представляет собой Н или C1-С2-алкил, возможно замещенный одним или более заместителем, выбранным из ОН, галогена, циано и/или -NR1R2;Z представляет собой C1-С6-алкил или 5-6-членный гетероцикло, содержащий один атом азота в качестве гетероатома, возможно замещенный одним или более заместителем, выбранным из С1-С4-алкила, ОН, галогена,...
Фосфаплатины и их применение для лечения рака

Номер патента: 21526
Опубликовано: 30.07.2015
Автор: Боуз Ратиндра Н.
МПК: A61K 31/282, A61K 33/24, A61P 35/00...
Метки: применение, рака, фосфаплатины, лечения
Формула / Реферат:
1. Комплекс фосфаплатины, выбранный из группы, состоящей из:(i) энантиомерно чистого (1R,2R)-пиро-DACH-2, имеющего формулу (I),или его фармацевтически приемлемой соли или сольвата;(ii) энантиомерно чистого (1S,2S)-пиро-DACH-2, имеющего формулу (II),или его фармацевтически приемлемой соли или сольвата;(iii) цис-пиро-DACH-2, имеющего формулу (III),или его фармацевтически приемлемой соли или сольвата;(iv) энантиомерно чистого (1R,2R)-пиро-DACH-4,...
Производные дифенилоксиндол-2-она и их применение для лечения рака

Номер патента: 13209
Опубликовано: 30.04.2010
Авторы: Престегор Мортен, Батчер Стивен Питер, Крог-Енсен Кристиан, Коултер Томас Стивен, Уддин Мохаммед, Ренье Серж, Педерсен Ханс Христиан, Монталбетти Кристиан, Фелдинг Якоб, Линде Вигго
МПК: A61K 31/407, A61K 31/404, A61K 31/4188...
Метки: дифенилоксиндол-2-она, рака, производные, лечения, применение
Формула / Реферат:
1. Применение соединения общей формулы (I)где R1, R2, R3и R4 независимо выбраны из водорода, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила, гидрокси, необязательно замещенного С1-6-алкокси, необязательно замещенного С2-6-алкенилокси, карбокси, необязательно замещенного C1-6-алкоксикарбонила, необязательно замещенного C1-6-алкилкарбонила, необязательно замещенного С1-6-алкилкарбонилокси, формила, амино, моно- и...
Предыдущий патент: Соединения, связывающие фарнезоидный x-рецептор (nr1h4) и модулирующие его активность
Следующий патент: Замещенные хинолины и их применение в качестве лекарственных средств
Случайный патент: Терморегулирующий материал, устройство и способ его изготовления