Соединения, связывающие фарнезоидный x-рецептор (nr1h4) и модулирующие его активность







Формула / Реферат
1. Соединение, соответствующее следующей формуле (1), его энантиомер, диастереомер или фармацевтически приемлемая соль

где R выбран из группы, состоящей из COOR6, CONR7R8, тетразолила, SO2NR7R8, SO2-C1-6 алкила, при этом R6 независимо выбран из группы, состоящей из Н или С1-6 алкила, a R7 и R8 независимо друг от друга выбраны из группы, состоящей из Н, C1-6 алкилен-R9, SO2-C1-6 алкила, где R9 выбран из группы, состоящей из СООН и SO3H;
А выбран из группы, состоящей из фенила, пиридила, пиразолила, индазолила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из О-С1-6 алкила, С1-6 алкила;
Q выбран из группы, состоящей из фенила, который возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из галогена;
Y выбран из N или СН;
Z выбран из

где Х = СН;
R1 выбран из группы, состоящей из С3-6 циклоалкила;
R2 и R3 представляют собой галоген.
2. Соединение по п.1, отличающееся тем, что R-A выбран из


3. Соединение по п.1 или 2, отличающееся тем, что Q представляет собой

4. Соединение по любому из пп.1-3, отличающееся тем, что Z представляет собой

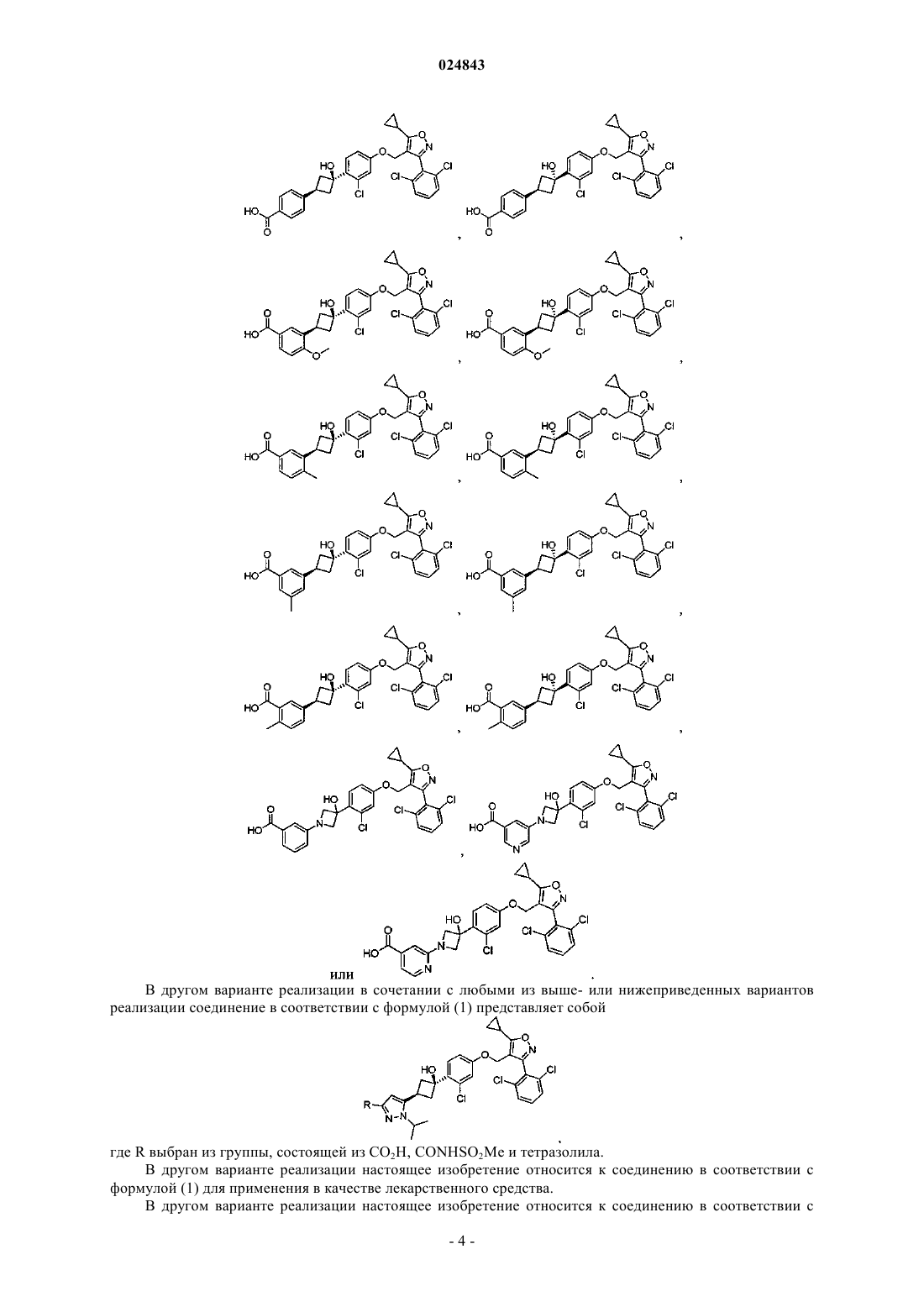
5. Соединение по любому из пп.1-4, выбранное из



6. Фармацевтическая композиция, содержащая соединение по п.1 или его фармацевтически приемлемую соль и по меньшей мере один наполнитель.
7. Соединение формулы (2)

или его фармацевтически приемлемая соль.
8. Фармацевтическая композиция, содержащая соединение по п.7 или его фармацевтически приемлемую соль и по меньшей мере один наполнитель.
9. Соединение формулы (3)

или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, содержащая соединение по п.9 или его фармацевтически приемлемую соль и по меньшей мере один наполнитель.
11. Применение соединения по любому из пп.1-5, 7 и 9 для профилактики и/или лечения заболеваний, опосредованных FXR.
12. Применение по п.11, отличающееся тем, что указанное заболевание выбрано из хронических внутрипеченочных состояний или внепеченочных холестатических состояний;
фиброза печени;
обструктивных или хронических воспалительных заболеваний печени;
цирроза печени;
стеатоза печени и ассоциированных синдромов, холестатических или фиброзных эффектов, ассоциированных с алкогольным циррозом или с вирусными формами гепатита;
печеночной недостаточности или ишемии печени после обширной резекции печени;
ассоциированного с химиотерапией стеатогепатита (CASH);
острой печеночной недостаточности и/или
воспалительных заболеваний кишечника.
13. Применение по п.11, отличающееся тем, что указанное заболевание выбрано из расстройств липидного и липопротеинового обмена;
диабета II типа и клинических осложнений диабета I типа и II типа, включая диабетическую нефропатию, диабетическую нейропатию, диабетическую ретинопатию и другие наблюдаемые эффекты клинического проявления длительного диабета;
состояний и заболеваний, возникающих в результате хронической жировой и фиброзной дегенерации органов вследствие усиленного накопления липидов и, в частности, триглицеридов, и последующей активации профибротических путей, таких как неалкогольная жировая болезнь печени (NAFLD) или неалкогольный стеатогепатит (NASH);
ожирения или метаболического синдрома (сочетания состояний дислипидемии, диабета или аномально высокого индекса массы тела) и/или
острого инфаркта миокарда, острого инсульта или тромбоза, которые возникают в качестве конечного результата хронического обструктивного атеросклероза.
14. Применение по п.11, отличающееся тем, что указанное заболевание выбрано из незлокачественных гиперпролиферативных заболеваний и злокачественных гиперпролиферативных заболеваний, в частности гепатоцеллюлярной карциномы, аденомы и полипоза толстой кишки, аденокарциномы толстой кишки, рака молочной железы, аденокарциномы поджелудочной железы, пищевода Барретта или других форм опухолевых заболеваний желудочно-кишечного тракта и печени.
Текст
(FXR) и действуют как агонисты FXR. Изобретение также относится к применению указанных соединений для получения лекарственного средства для лечения заболеваний и/или состояний посредством связывания указанного ядерного рецептора указанными соединениями и к способу синтеза указанных соединений (1). Z выбран из (а), (b), (с) или (d) Настоящее изобретение относится к соединениям, которые связываются с рецептором NR1H4 (фарнезоидный Х-рецептор, FXR) и действуют как агонисты или модуляторы FXR. Изобретение также относится к применению таких соединений для лечения и/или профилактики заболеваний и/или состояний посредством связывания указанного ядерного рецептора указанными соединениями. Многоклеточные организмы зависят от отработанных механизмов передачи информации между клетками и частями организма. Передаваемая информация может быть крайне сложной и может вызывать изменение генетических программ, участвующих в клеточной дифференцировке, пролиферации или репродукции. Сигналы или гормоны часто представляют собой низкомолекулярные соединения, такие как пептиды, жирные кислоты или производные холестерина. Многие из этих сигналов оказывают свое действие путем изменения в конечном счете транскрипции конкретных генов. Одной хорошо изученной группой белков, которые опосредуют ответ клетки на множество сигналов, является семейство факторов транскрипции, известных как ядерные рецепторы,часто именуемые далее "ЯР". Представители данной группы включают рецепторы стероидных гормонов,витамина D, экдизона, цис- и транс-ретиноевой кислоты, тиреоидного гормона, желчных кислот, производных холестерина, жирных кислот (и других пролифераторов пероксисом), а также так называемые"сиротские" рецепторы, белки, которые структурно похожи на других представителей этой группы, но для которых неизвестны лиганды. Сиротские рецепторы могут свидетельствовать о неизвестных сигнальных путях в клетке или могут представлять собой ядерные рецепторы, функционирующие без активации лигандом. Активация транскрипции некоторыми из этих сиротских рецепторов может происходить в отсутствие экзогенного лиганда и/или посредством путей передачи сигнала, берущих начало от поверхности клетки (D.J. Mangelsdorf et al., Cell. 1995, 83, 835; R.M. Evans, Mol. Endocrinol. 2005, 19,1429). В общем случае, в молекулах ЯР были определены три функциональных домена. Аминоконцевой домен, как полагают, обладает некоторой регуляторной функцией. За ним следует ДНК-связывающий домен, именуемый далее "ДСД", который обычно содержит два цинковых пальца и распознает специфический элемент гормонального ответа, именуемый далее "ЭГО", внутри промоторов отвечающих на действие гормона генов. Конкретные аминокислотные остатки в "ДСД", как было показано, обеспечивают специфичность связывания с последовательностью ДНК (М. Schena and K.R. Yamamoto, Science 1988,241, 965). Лигандсвязывающий домен, именуемый далее "ЛСД", расположен в С-концевых участках молекул известных ЯР. В отсутствие гормона ЛСД, вероятно, препятствует взаимодействию ДСД с соответствующим ЭГО. Связывание гормона, по-видимому, приводит к конформационному изменению в молекуле ЯР и, таким образом, устраняет влияние ЛСД (А.М. Brzozowski et al., Nature 1997, 389, 753). ЯР без ЛСД конститутивно активирует транскрипцию, но на низком уровне. Коактиваторы или транскрипционные активаторы, как полагают, являются связующим звеном между последовательность-специфичными транскрипционными факторами - основным аппаратом транскрипции - и, кроме того, влияют на структуру хроматина клетки-мишени. Несколько белков, таких какSRC-1, ACTR и Grip1, взаимодействуют с ЯР способом, при котором взаимодействие усиливается связыванием лиганда (D.M. Heery et al., Nature 1997, 387, 733; Т. Heinzel et al., Nature 1997, 387, 43; K.W. Nettles and G.L. Greene, Annu. Rev. Physiol. 2005, 67, 309). Модуляторы ядерных рецепторов, такие как стероидные гормоны, влияют на рост и функционирование конкретных клеток за счет связывания с внутриклеточными рецепторами и образования комплексов лигандов с ядерными рецепторами. Комплексы гормонов с ядерными рецепторами далее взаимодействуют с ЭГО в контрольной области конкретных генов и изменяют экспрессию конкретных генов (A.Aranda and A. Pascual, Physiol. Rev. 2001, 81, 1269). Фарнезоидный Х-рецептор альфа (также часто именуемый далее NR1H4, если речь идет о рецепторе человека) представляет собой прототипный ядерный рецептор типа 2, который активирует гены при связывании с промоторным участком целевых генов, образуя гетеродимер с ретиноидным Х-рецептором(В.М. Forman et al., Cell 1995, 81, 687). Соответствующими физиологическими лигандами для NR1H4 являются желчные кислоты (D.J. Parks et al., Science 1999, 284, 1365; M. Makishima et al., Science 1999,284, 1362). Наиболее сильным лигандом является хенодезоксихолевая кислота (ХДХК), которая регулирует экспрессию нескольких генов, участвующих в гомеостазе желчных кислот. Фарнезол и производные, называемые вместе фарнезоидами, как первоначально описано, активируют крысиный ортолог при высокой концентрации, но они не активируют рецептор человека или мыши. FXR экспрессируется в печени, во всех отделах желудочно-кишечного тракта, включая пищевод, желудок, двенадцатиперстную кишку, тонкий кишечник, толстую кишку, в яичниках, надпочечниках и почках. Кроме контроля внутриклеточной экспрессии генов, FXR, по-видимому, также участвует в паракринной и эндокринной передаче сигналов посредством активации экспрессии цитокина фактора роста фибробластов 15 (грызуны) или 19 (обезьяны, люди, J.A. Holt et al., Genes Dev. 2003, 17, 1581; Т. Inagaki et al., Cell. Metab. 2005, 2, 217). Низкомолекулярные соединения, действующие как модуляторы FXR, были предложены в следующих публикациях: WO 2000/037077, WO 2003/015771, WO 2004/048349, WO 2007/076260, WO 2007/092751, WO 2007/140174, WO 2007/140183, WO 2008/051942, WO 2008/157270, WO 2009/005998,-1 024843WO 2009/012125, WO 2008/025539 и WO 2008/025540. Недавно были опубликованы обзоры по другим низкомолекулярным модуляторам FXR (M.L. Crawley, Expert Opin Ther. Pat. 2010, 20, 1047; D. Merk et al.,Future Med. Chem. 2012, 4, 1015). В WO 2011/020615 авторами настоящего изобретения были предложены хиральные соединения циклопропилидена, имеющие следующую общую формулу: где переменные определены так же, как и в настоящей заявке. Задача, лежащая в основе настоящего изобретения, заключается в создании агонистов FXR с улучшенными физико-химическими свойствами, в целом, и сниженной гидрофобностью, улучшенной растворимостью в воде и лучшей мембранной проницаемостью, в частности, по сравнению с соединениями,заявленными в WO 2011/020615. Указанная задача решена с помощью соединения в соответствии со следующей формулой (1), его энантиомера, диастереомера, таутомера, сольвата, пролекарства или фармацевтически приемлемой соли где R выбран из группы, состоящей из COOR6, CONR7R8, тетразолила, SO2NR7R8, C1-6 алкила, SO2-C1-6 алкила и Н, при этом R6 независимо выбран из группы, состоящей из Н или С 1-6 алкила, a R7 и R8 независимо друг от друга выбраны из группы, состоящей из Н, С 1-6 алкила, галоген-C1-6 алкила, С 1-6 алкиленаR9, SO2-C1-6 алкила, где R9 выбран из группы, состоящей из СООН, ОН и SO3H; А выбран из группы, состоящей из фенила, пиридила, пиримидила, пиразолила, индолила, тиенила,бензотиенила, индазолила, бензизоксазолила, бензофуранила, бензотриазолила, фуранила, бензотиазолила, тиазолила, оксадиазолила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из ОН, O-С 1-6 алкила, О-галоген-С 1-6 алкила, С 1-6 алкила, галоген-С 1-6 алкила, С 3-6 циклоалкила и галогена;Q выбран из группы, состоящий из фенила, пиридила, тиазолила, тиофенила, пиримидила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из C1-6 алкила, галоген-С 1-6 алкила, галогена и CF3;R1 выбран из группы, состоящей из водорода, C1-3 алкила, С 3-6 циклоалкила, С 4-5 алкилциклоалкила,где С 1-3 алкил возможно замещен 1-3 заместителями, независимо выбранными из галогена, гидрокси или С 1-6 алкокси;R2 и R3 независимо выбраны из группы, состоящей из водорода, C1-3 алкила, C1-3 галогеналкила, С 1-3 алкокси, С 1-3 галогеналкокси и галогена. В другом варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации R-A в соединении в соответствии с формулой (1) выбран из В другом варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации Q в соединении в соответствии с формулой (1) представляет собой В другом варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации Z в соединении в соответствии с формулой (1) представляет собой В другом варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации соединение в соответствии с формулой (1) выбрано из В другом варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации соединение в соответствии с формулой (1) представляет собой где R выбран из группы, состоящей из СО 2 Н, CONHSO2Me и тетразолила. В другом варианте реализации настоящее изобретение относится к соединению в соответствии с формулой (1) для применения в качестве лекарственного средства. В другом варианте реализации настоящее изобретение относится к соединению в соответствии с формулой (1) для применения для профилактики и/или лечения заболеваний, опосредованных FXR. В другом варианте реализации настоящее изобретение относится к применению соединения в соответствии с формулой (1) для получения лекарственного средства для профилактики и/или лечения заболеваний, опосредованных FXR. В другом варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации заболевание выбрано из хронических внутрипеченочных состояний или некоторых форм внепеченочных холестатических состояний; фиброза печени; обструктивных или хронических воспалительных заболеваний печени; цирроза печени; стеатоза печени и ассоциированных синдромов, холестатических или фиброзных эффектов, ассоциированных с алкогольным циррозом или с вирусными формами гепатита; печеночной недостаточности или ишемии печени после обширной резекции печени; ассоциированного с химиотерапией стеатогепатита (CASH); острой печеночной недостаточности и/или воспалительных заболеваний кишечника. В другом варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации заболевание выбрано из расстройств липидного и липопротеинового обмена; диабета II типа и клинических осложнений диабета I типа и II типа, включая диабетическую нефропатию, диабетическую нейропатию, диабетическую ретинопатию и другие наблюдаемые эффекты клинического проявления длительного диабета; состояний и заболеваний, возникающих в результате хронической жировой и фиброзной дегенерации органов вследствие усиленного накопления липидов и в особенности триглицеридов и последующей активации профибротических путей, таких как неалкогольная жировая болезнь печени (NAFLD) или неалкогольный стеатогепатит (NASH); ожирения или метаболического синдрома(сочетания состояний дислипидемии, диабета или аномально высокого индекса массы тела); и/или острого инфаркта миокарда, острого инсульта или тромбоза, которые возникают в качестве конечного результата хронического обструктивного атеросклероза. В другом варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации заболевание выбрано из незлокачественных гиперпролиферативных заболеваний и злокачественных гиперпролиферативных заболеваний, в частности гепатоцеллюлярной карциномы, аденомы и полипоза толстой кишки, аденокарциномы толстой кишки, рака молочной железы, аденокарциномы поджелудочной железы, пищевода Барретта или других форм опухолевых заболеваний желудочнокишечного тракта и печени. Улучшенные физико-химические свойства были достигнуты за счет введения полярной гидроксильной группы в 1,3-циклобутилиденовую или 1,3-азетидинилиденовую группы, заменяющие прежнее 1,2-циклопропилиденовое кольцо Неожиданно было обнаружено, что полученные соединения сохраняли свою активность по отношению к рецептору FXR, но демонстрировали улучшенные физико-химические свойства, такие как более высокая растворимость в воде и/или мембранная проницаемость. Соединения согласно настоящему изобретению имеют общую химическую структуру в соответствии с формулой (1) по п.1 формулы изобретения. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации настоящее изобретение относится к энантиомеру, диастереомеру или фармацевтически приемлемой соли соединения в соответствии с формулой (1). В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации R в формуле (1) выбран из группы, состоящей из COOR6, CONR7R8, SO2NR7R8 иSO2-C1-6 алкила. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации R6 в формуле (1) представляет собой Н. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации R7 и R8 в формуле (1) независимо друг от друга выбраны из группы, состоящей из Н и SO2-C1-6 алкила. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации R7 в формуле (1) представляет собой Н. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации R8 в формуле (1) представляет собой SO2-C1-6 алкил. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации А выбран из группы, состоящей из фенила, пиридила, пиримидила, пиразолила,индазолила и оксадиазолила. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации А замещен одной или двумя группами, независимо выбранными из C1-6 алкила,более предпочтительно C1-3 алкила. В другом предпочтительном варианте реализации в сочетании с лю-5 024843 быми из выше- или нижеприведенных вариантов реализации А является незамещенным. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации Q представляет собой фенил. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации Q замещен одной или двумя группами, независимо выбранными из галогена, более предпочтительно одной группой, выбранной из галогена, в частности Cl. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации Z представляет собой В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации X = СН. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации R1 представляет собой С 3-6 циклоалкил, в частности циклопропил. В предпочтительном варианте реализации в сочетании с любыми из выше- или нижеприведенных вариантов реализации R2 и R3 независимо выбраны из галогена, в частности Cl. Соединения согласно настоящему изобретению могут находиться в форме пролекарств. Термин"пролекарство" обозначает производное, которое превращается в соединение в соответствии с настоящим изобретением в результате реакции с ферментом, желудочным соком и т.п. при физиологических условиях в живом организме, например в результате реакций окисления, восстановления, гидролиза или т.п., каждая из которых осуществляется ферментативным путем. Примерами пролекарств являются соединения, у которых аминогруппа в соединении согласно настоящему изобретению ацилирована, алкилирована или фосфорилирована с образованием, например, эйкозаноиламино, аланиламино, пивалоилоксиметиламино, или у которых гидроксильная группа ацилирована, алкилирована, фосфорилирована или превращена в борат, например ацетилокси, пальмитоилокси, пивалоилокси, сукцинилокси, фумарилокси,аланилокси, или у которых карбоксильная группа этерифицирована или амидирована. Эти соединения могут быть получены из соединений согласно настоящему изобретению в соответствии с хорошо известными способами. Другие примеры пролекарств представляют собой соединения, в которых карбоксилатная группа соединения согласно настоящему изобретению превращается, например, в алкиловый, ариловый, холиновый сложный эфир, аминоэфир, ацилоксиметиловый сложный эфир, линоленоиловый сложный эфир. Метаболиты соединений согласно настоящему изобретению также находятся в рамках настоящего изобретения. Там, где возможна таутомерия соединений согласно настоящему изобретению или их пролекарств,такая как, например, кетоенольная таутомерия, все индивидуальные формы, такие как, например, кето- и енольная форма, находятся в рамках настоящего изобретения, так же как и их смеси в любом соотношении. То же справедливо для стереоизомеров, таких как, например, энантиомеры, цис/транс-изомеры,конформеры и т.п. При необходимости изомеры могут быть разделены посредством способов, хорошо известных в данной области техники, например, с помощью жидкостной хроматографии. Также возможно разделение энантиомеров путем применения, например, хиральных неподвижных фаз. Кроме того, энантиомеры могут быть выделены путем превращения их в диастереомеры, т.е. сочетания с энантиомерно чистым вспомогательным соединением, последующего разделения полученных диастереомеров и отщепления вспомогательного остатка. Альтернативно, любой энантиомер соединения согласно настоящему изобретению может быть получен посредством стереоселективного синтеза с применением оптически чистых исходных веществ. Другой путь получения чистых энантиомеров из рацемических смесей заключается в применении энантиоселективной кристаллизации с хиральными противоионами. Соединения согласно настоящему изобретению могут находиться в форме фармацевтически приемлемой соли или сольвата. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические основания или кислоты и органические основания или кислоты. В случае, если соединения согласно настоящему изобретению содержат одну или более кислотных или основных групп, изобретение также включает их соответствующие фармацевтически или токсикологически приемлемые соли, в частности их соли, пригодные к применению в фармацевтике. Таким образом, соединения согласно настоящему изобретению, содержащие кислотные группы, могут находиться в виде соединений с этими группами и могут быть применены в соответствии с изобретением, например, в виде солей щелочных металлов, солей щелочно-земельных металлов или аммониевых солей. Более конкретные примеры таких солей включают натриевые соли, калиевые соли, кальциевые соли, соли магния или соли с аммиаком или органическими аминами, такими как, например, этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения согласно настоящему изобретению, содержащие одну или более основных групп, т.е. групп, которые могут быть протонированы, могут находиться и могут быть применены в соответствии с изобретением в форме их солей присоединения с неорганическими или органическими кислотами. Примеры подходящих кислот включают хлороводород, бромоводород, фосфорную кислоту, серную кислоту, азотную кислоту,метансульфоновую кислоту, п-толуолсульфоновую кислоту, нафталиндисульфоновые кислоты, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, пивалевую кислоту, диэтилуксусную кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту,яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты,известные специалисту в данной области техники. Если соединения согласно настоящему изобретению одновременно содержат кислотные и основные группы в молекуле, изобретение также включает, наряду с вышеупомянутыми формами солей, внутренние соли или бетаины (цвиттер-ионы). Соответствующие соли могут быть получены посредством традиционных способов, которые известны специалисту в данной области техники, например, путем приведения соединений согласно изобретению в контакт с органической или неорганической кислотой или основанием в растворителе или диспергирующем агенте или путем анионного обмена или катионного обмена с другими солями. Настоящее изобретение также включает все соли соединений согласно настоящему изобретению, которые вследствие низкой физиологической совместимости непосредственно не подходят для применения в фармацевтических препаратах, но которые могут быть применены, например, в качестве промежуточных продуктов для химических реакций или для получения фармацевтически приемлемых солей. Кроме того, соединения согласно настоящему изобретению могут находиться в форме сольватов,таких как те, которые содержат в качестве сольвата воду, или фармацевтически приемлемые сольваты,такие как спирты, в частности этанол. К тому же, согласно настоящему изобретению предложены фармацевтические композиции, содержащие по меньшей мере одно соединение согласно настоящему изобретению, или его пролекарство, или его фармацевтически приемлемую соль или сольват в качестве активного ингредиента вместе с фармацевтически приемлемым носителем. Термин "фармацевтическая композиция" обозначает один или более активных ингредиентов и один или более инертных ингредиентов, составляющих носитель, а также любой продукт, который получают,прямо или косвенно, в результате сочетания, комплексообразования или агрегации любых двух или более ингредиентов, или в результате диссоциации одного или более ингредиентов, или в результате других типов реакций или взаимодействий одного или более ингредиентов. Соответственно, фармацевтические композиции согласно настоящему изобретению включают любую композицию, полученную путем смешивания по меньшей мере одного соединения согласно настоящему изобретению и фармацевтически приемлемого носителя. Фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать одно или более других соединений в качестве активных ингредиентов, таких как пролекарство или другие модуляторы ядерных рецепторов. Композиции подходят для перорального, ректального, местного, парентерального (включая подкожное, внутримышечное и внутривенное), офтальмологического (глазного), легочного (назальная или буккальная ингаляция) или назального введения, хотя наиболее подходящий путь введения в любом данном случае будет зависеть от природы и тяжести состояний, подлежащих лечению, и от природы активного ингредиента. Они могут быть для удобства представлены в виде лекарственной формы для однократного введения и получены посредством любого из способов, хорошо известных в области фармацевтики. Соединения согласно настоящему изобретению могут быть получены посредством комбинации способов, описанных в схемах I-III. Как показано на схеме I, 4-членный циклический кетон, замещенный заместителем А в положении 3, может вступать в реакцию с металлированным ароматическим или гетероароматическим кольцом M-Q-O-CH2Z (M = металл, например Li) в апротонных растворителях и предпочтительно при низких температурах с получением гидроксилзамещенного 4-членного кольца, содержащего заместители А и Q. В случае когда Y представляет собой СН, возможно образование двух изомеров (А и Q у противолежащих атомов кольца в цис- или транс-положении относительно друг друга). В оптимальных условиях может быть достигнуто образование преимущественно одного из двух изомеров. Указанные два изомера могут быть разделены посредством подходящих способов, известных в данной области техники, таких как, например, хроматография на силикагеле или препаративная обращеннофазовая высокоэффективная жидкостная хроматография (ОФ-ВЭЖХ). На схеме II кратко представлены способы, используемые для получения 4-членных циклических кетонов, необходимых для синтеза соединений согласно настоящему изобретению. В соответствии с вариантом а), промежуточное соединение, содержащее винильную группу, например, полученное путем винилирования соответствующего галогенсодержащего исходного вещества R-A-X (X = галоген), может вступать в реакцию с образующимся in situ ,-дихлоркетеном с образованием 2,2 дихлорциклобутанона. После дегалогенирования, например, в присутствии Zn в уксусной кислоте при кипячении с обратным холодильником получают целевые 3-замещенные циклобутаноны. Альтернативно, винилсодержащие промежуточные соединения могут вступать в реакцию с образующимся in situ незамещенным кетеном с получением в одну стадию целевых промежуточных продуктов на основе циклобутанона. В соответствии с вариантом b) в качестве исходного вещества применяют 3 метиленциклобутанкарбонитрил. Замещенные гетероциклы могут быть построены из цианогруппы за несколько стадий посредством способов, известных специалистам в данной области техники. Целевые циклобутаноны могут быть получены путем окислительного расщепления экзоциклической двойной связи с применением условий и реагентов, известных специалистам в данной области техники, например, с помощью OsO4, озона или RhCl3/NaIO4 в качестве окислителей. Вариант с) представляет способы, применяемые для получения замещенных азетидинонов. Катализируемое Cu или Pd C-N кросс-сочетание 3 гидроксиазетидина с галогенароматическими или галогенгетероароматическими кольцами приводит к получению соответствующих N-замещенных 3-гидроксиазетидинов, которые могут быть превращены в целевые азетидиноны путем окисления. Схема II Схема III иллюстрирует некоторые возможности для модификации заместителей при группе А после образования 4-членных гидроксисодержащих колец. Например, уходящая группа X (например, бромид) может быть замещена цианогруппой, карбоксиэфирной группой, метилсульфонилом или простой тиоэфирной группой посредством катализируемых переходными металлами реакций кросс-сочетания. Полученные производные могут быть далее превращены в другие производные посредством способов,известных специалистам в данной области техники. Например, циано- и сложноэфирная группы могут быть подвергнуты гидролизу в присутствии оснований с получением карбоновой кислоты, которая, в свою очередь, может быть превращена в ацилсульфонамиды. Простой бензиловый тиоэфир можно хлорировать с получением хлорсульфонильного промежуточного соединения, которое вступает в реакцию с аммиаком с образованием соответствующих сульфонамидов. Схема III Таким образом, настоящее изобретение относится к соединениям в соответствии с общей формулой(1), которые связываются с FXR и действуют как агонисты или модуляторы FXR. Изобретение также относится к применению указанных соединений для лечения и/или профилактики заболеваний и/или состояний посредством связывания указанного ядерного рецептора указанными соединениями. Кроме того, настоящее изобретение относится к применению указанных соединений для получения лекарственного средства для лечения и/или профилактики заболеваний и/или состояний посредством связывания указанного ядерного рецептора указанными соединениями. Конкретно, настоящее изобретение относится к применению соединений в соответствии с формулой (1) для получения лекарственного средства для профилактики и/или лечения хронических внутрипеченочных состояний или некоторых форм внепеченочных холестатических состояний; фиброза печени; острых внутрипеченочных холестатических состояний; обструктивных или хронических воспалительных заболеваний, возникающих вследствие неправильного состава желчи; состояний желудочно-кишечного тракта с пониженным усвоением входящих в рацион жира и жирорастворимых витаминов; воспалительных заболеваний кишечника; расстройств липидного и липопротеинового обмена; диабета II типа и клинических осложнений диабета I типа и II типа; состояний и заболеваний, возникающих в результате хронической жировой и фиброзной дегенерации органов вследствие усиленного накопления липидов и в особенности триглицеридов, и последующей активации профибротических путей; ожирения и метаболического синдрома(сочетания состояний дислипидемии, диабета или аномально высокого индекса массы тела); острого инфаркта миокарда, острого инсульта, тромбоза, которые возникают в качестве конечного результата хронического обструктивного атеросклероза; хронических инфекций, вызываемых внутриклеточными бактериями или простейшими паразитами; незлокачественных гиперпролиферативных заболеваний; злокачественных гиперпролиферативных заболеваний, в частности аденокарциномы толстой кишки и гепатоцеллюлярной карциномы; стеатоза печени и ассоциированных синдромов; печеночной недостаточности или нарушения функции печени в результате хронических заболеваний печени или хирургической резекции печени; инфекции гепатита В; инфекции гепатита С и/или холестатических и фиброзных эффектов, ассоциированных с алкогольным циррозом или с вирусными формами гепатита. Лекарственные средства, упомянутые в настоящей заявке, могут быть получены традиционными способами, включая объединение соединения в соответствии с настоящим изобретением и фармацевтически приемлемого носителя. Как предполагают, FXR представляет собой ядерный рецептор желчных кислот. В связи с этим, он модулирует как синтетическую выработку желчных кислот в печени, так и их рециркуляцию в кишечнике (путем регулирования функции белков, связывающих желчные кислоты). Но, помимо участия в физиологии желчных кислот, FXR, по-видимому, вовлечен в регуляцию многих разнообразных физиологических процессов, которые имеют отношение к этиологии и лечению различных заболеваний, таких как холестериновые желчные камни, метаболические расстройства, такие как диабет II типа, дислипидемии или ожирение, хронические воспалительные заболевания, такие как воспалительные заболевания кишечника или хронические внутрипеченочные формы холестаза, и многие другие заболевания (Т. Claudel etFXR регулирует сложный набор подконтрольных генов в печени и желудочно-кишечном тракте. Продукты генов влияют на разнообразные физиологические процессы. В ходе функционального анализа FXR первой проанализированной регуляторной сетью стала регуляция синтеза желчных кислот. В то время как печеночные Х-рецепторы (LXR) индуцируют ключевой фермент превращения холестерина в желчные кислоты, Сур 7 А 1 (цитохром Р 450 7 А 1), посредством индукции регуляторного ядерного рецептора LRH-1, FXR подавляет индукцию Сур 7 А 1 путем активации синтеза мРНК, кодирующей малый гетеродимерный партнер (SHP), дополнительный ядерный рецептор,который доминантно репрессирует LRH-1. Поскольку FXR связывает конечные продукты данного пути,первичные желчные кислоты, такие как холевая кислота (ХК) или хенодезоксихолевая кислота (ХДХК),этот процесс можно рассматривать в качестве примера ингибирования по принципу обратной связи на уровне экспрессии генов (В. Goodwin et al., Mol. Cell 2000, 6, 517; Т.Т. Lu et al., Mol. Cell 2000, 6, 507). Параллельно подавлению синтеза желчных кислот посредством SHP, FXR индуцирует ряд так называемых АВС-транспортеров (АТФ-связывающих кассетных транспортеров), которые отвечают за экспорт токсичных желчных кислот из цитозоля гепатоцита в канальцы, маленькие ответвления желчного протока, где образуется желчь. Данная гепатопротекторная функция FXR впервые стала очевидной при анализе мышей с выключенным геном FXR (С. J. Sinai et al., Cell 2000, 102, 731), когда была показана пониженная или повышенная экспрессия нескольких ABC-транспортеров в печени. В результате дальнейшего детального анализа было обнаружено, что главный экскреторный насос солей желчных кислот BSEP или АВСВ 11 (М. Ananthanarayanan et al., J. Biol. Chem. 2001, 276, 28857; J. R. Plass et al., Hepatology 2002, 35,589), а также ключевой фермент, который опосредует перенос липидов от липопротеинов к фосфолипидам, PLTP (N.L. Urizar et al., J. Biol. Chem. 2000, 275, 39313), и два ключевых канальцевых мембранных транспортера для фосфолипидов, MRP-2 (АВСС 4) (Н.R. Kast et al., J. Biol. Chem. 2002, 277, 2908) и MDR3 (ABCB4) (L. Huang et al., J. Biol. Chem. 2003, 278, 51085), представляют собой прямые мишени для лиганд-управляемой активации транскрипции посредством FXR (кратко изложено в: М. Miyata, J. Pharmacol. Exp. Ther. 2005, 312, 759; G. Rizzo et al., Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5,289). Тот факт, что FXR, по-видимому, является главным рецептором метаболитов и регулятором синтеза, экспорта и рециркуляции желчных кислот, предполагает применение лигандов FXR для индукции выделения желчи и изменения состава желчных кислот в сторону более гидрофильного состава. С разработкой первого синтетического лиганда FXR GW4064 (P.R. Maloney et al., J. Med. Chem. 2000, 43, 2971;T.M. Willson et al., Med. Res. Rev. 2001, 21, 513) в качестве модельного соединения и полусинтетического искусственного лиганда на основе желчной кислоты 6 этил-ХДХК можно анализировать эффекты сверхстимуляции FXR мощными агонистами. Было показано, что оба лиганда индуцируют выделение желчи у животных с перевязанными желчными протоками. Более того, кроме желчегонных эффектов также могут быть продемонстрированы гепатопротекторные эффекты (R. Pellicciari et al., J. Med. Chem. 2002, 45, 3569; Y. Liu et al., J. Clin. Invest. 2003, 112, 1678). Этот гепатопротекторный эффект был далее сужен до противофиброзного эффекта, являющегося результатом репрессии тканевых ингибиторов матриксных металлопротеиназ, TTMP-1 и 2, индукции матриксной металлопротеиназы 2 (ММР-2), расщепляющей отложения коллагена, в звездчатых клетках печени и последующего снижения уровня мРНК коллагена и мРНК трансформирующего ростового фактора бета (TGF-), оба из которых являются профиброзными факторами, под действием агонистов FXR (S. Fiorucci et al., Gastroenterology 2004, 127,1497; S. Fiorucci et al., J. Pharmacol. Exp. Ther. 2005, 314, 584). Кроме того, противохолестатическая активность была продемонстрирована в моделях животных с перевязанными желчными протоками, а также в моделях животных с холестазом, индуцированном эстрогенами (S. Fiorucci et al., J. Pharmacol. Exp.Ther. 2005, 313,604). Генетические исследования показывают, что в наследственных формах холестаза (прогрессирующий семейный внутрипеченочный холестаз = PFIC, тип I-IV) либо снижена ядерная локализация самогоFXR вследствие мутации в гене FIC1 (в случае PFIC I типа, также называемой болезнью Байлера) (F.Chen et al., Gastroenterology 2004, 126, 756; L. Alvarez et al., Hum. Mol. Genet. 2004, 13, 2451), либо снижены уровни экспрессии целевого гена FXR, кодирующего экспортный насос фосфолипидов MDR-3 (в случае PFIC III типа). В совокупности, существует растущая доказательная база относительно того, что соединения, связывающиеся с FXR, будут демонстрировать важное клиническое применение в схеме лечения хронических холестатических состояний, таких как первичный билиарный цирроз (ПБЦ) или первичный склерозирующий холангит (ПСХ) (обзоры в G. Rizzo et al., Curr. Drug Targets Immune Endocr.Targets 2006, 10, 409). Большое влияние, которое активация FXR оказывает на метаболизм и экскрецию желчных кислот,имеет значение не только для холестатических синдромов, но еще большее значение - непосредственнно для терапии, направленной против образования желчных камней. Холестериновые желчные камни образуются из-за низкой растворимости холестерина, который активно выкачивается из клеток печени в просвет канальцев. Именно относительное процентное содержание трех главных компонентов - желчных кислот, фосфолипидов и свободного холестерина, определяет образование смешанных мицелл и, следовательно, условное насыщение раствора свободного холестерина в желчи. Полиморфизмы FXR применяют для картирования локусов количественных признаков в качестве одного фактора, вносящего вклад в развитие желчекаменной болезни (Н. Wittenburg, Gastroenterology 2003, 125, 868). Путем применения синтетического модельного соединения GW4064 для FXR можно показать, что активация FXR приводит к улучшению индекса насыщения холестерином (ИНХ) и непосредственно к прекращению образования желчных камней у мышей линии C75L, подверженных образованию желчных камней, в то время как медикаментозное лечение мышей с выключенным геном FXR не оказывает эффекта на образование желчных камней (A. Moschetta et al., Nature Medicine 2004, 10, 1352). Приведенные результаты позволяют признать FXR как хорошую мишень для разработки низкомолекулярных агонистов, которые могут быть применены для предотвращения образования холестериновых желчных камней или предотвращения повторного образования желчных камней после их хирургического удаления или ударно-волновой литотрипсии (рассмотрено в: S. A. Doggrell, Curr. Opin. Investig.Drugs 2006, 7, 344). Таким образом, в одном варианте реализации изобретения соединение в соответствии с формулой (1) и фармацевтические композиции, содержащие указанное соединение, применяют для профилактики и/или лечения обструктивных или хронических воспалительных расстройств, возникающих вследствие неправильного состава желчи, таких как холелитиаз, также известный как холестериновые желчные камни. Помимо сильного гепатопротекторного и желчегонного, а также противофиброзного эффектов, демонстрируемых FXR при стимулируемой низкомолекулярными соединениями активации в печени, FXR,по-видимому, играет роль в защите кишечника от неопластической трансформации и от развития полипов и их перехода в аденокарциному в пищеварительном тракте (S. Modica et al., Cancer Res. 2008, 68,9589 и R.R. Maran et al., J. Pharmacol. Exp. Ther. 2009, 328, 469). Аналогично ситуации с кишечником,отсутствие FXR приводит к высокому возрастанию риска образования гепатоцеллюлярной карциномы(ГЦК), наиболее распространенной формы рака печени (I. Kim et al., Carcinogenesis 2007, 28, 940 и F.Yang et al., Cancer Res. 2007, 67, 863). В то время как функциональный FXR препятствует образованию аденокарциномы толстой кишки и гепатоцеллюлярной карциномы, активация FXR индуцирует регенерацию печени после гепатэктомии (W. Huang et al., Science 2006, 312, 233). Сочетание гепатопротекторного, антинеопластического эффектов и регенерирующего действия в отношении печени, ассоциированных с активацией FXR, может быть терапевтически использовано для применения агонистов FXR для лечения тяжелых заболеваний печени. В одном варианте реализации соединения в соответствии с изобретением и фармацевтические композиции, содержащие указанные соединения, применяют для лечения заболеваний печени, таких как ГЦК, стимулирования возобновления роста печени и уменьшения интенсивности побочных эффектов, ассоциированных с обширной резекцией печени, циррозом печени вне зависимости от этиологии, и предотвращения или лечения ишемии печени в курсе трансплантации печени или обширного оперативного вмешательства на печени. Со времени открытия первого синтетического агониста FXR и его введения грызунам стало очевидным, что FXR является ключевым регулятором уровня триглицеридов сыворотки (P. Maloney et al., J.Med. Chem. 2000, 43, 2971; T. Willson et al., Med. Res. Rev. 2001, 21, 513). В течение прошедших шести лет были опубликованы накапливающиеся доказательства того, что активация FXR за счет синтетических агонистов приводит к значительному снижению уровня триглицеридов сыворотки, главным образом в форме сниженного уровня ЛПОНП, но также к сниженному уровню общего холестерина сыворотки (Н.R. Kast et al., Mol. Endocrinol. 2001, 15, 1720; N.L. Urizar et al., Science 2002, 296, 1703; G. Lambert etChem. 2004, 279, 2790; S. Bilz et al., Am. J. Physiol. Endocrinol. Metab. 2006, 290, E716). Но снижение уровня триглицеридов в сыворотке не является единственным эффектом. Лечение мышей линий ob/ob или db/db синтетическим агонистом FXR GW4064 приводило к заметному и совместному снижению уровня триглицеридов, общего холестерина, свободных жирных кислот, кетоновых тел, таких как 3-ОН-бутират, в сыворотке. Более того, активация FXR вступает во взаимодействие с внутриклеточным сигнальным путем инсулина в гепатоцитах, что приводит к снижению выработки глюкозы в результате глюконеогенеза в печени, но сопутствует повышению уровня гликогена в печени. Лечение FXR оказывало положительное влияние на чувствительность к инсулину, а также толерантность к глюкозе (K.R. Stayrook et al., Endocrinology 2005, 146, 984; Y. Zhang et al., PNAS 2006, 103, 1006; B. Cariou et al., J. Biol. Chem. 2006, 281, 11039; K. Ma et al., J. Clin. Invest. 2006, 116, 1102; D. Duran-Sandoval etal., Biochimie 2005, 87, 93). Недавно также наблюдали эффект снижения массы тела у мышей, получавших избыточную диету с высоким содержанием липидов (С. Lihong et al., American Diabetes Association(ADA) 66th annual scientific sessions, June 2006, Abstract Number 856-P). Этот эффект потери массы тела может являться результатом индукции FXR FGF-19, фактора роста фибробластов, что, как известно, ведет к снижению массы тела и атлетическому фенотипу (J. Holt et al., Genes Dev. 2003, 17, 1581; E. Tomlinson et al., Endocrinology 2002, 143, 1741). В недавних заявках на патент было продемонстрирован эффект агониста FXR на снижение массы тела (WO 2004/087076; WO 2003/080803). В совокупности данные фармакологические эффекты агонистов FXR могут быть применены в различных терапевтических подходах, как полагают, FXR-связывающие соединения являются хорошими кандидатами для лечения диабета II типа благодаря их действию по повышению чувствительности к инсулину, повышению уровня гликогена и снижению уровня липидов. В одном варианте реализации соединения в соответствии с изобретением и фармацевтические композиции, содержащие указанные соединения, применяют для профилактики и/или лечения диабета II типа, который можно преодолеть за счет FXR-опосредованной положительной регуляции системной чувствительности к инсулину и внутриклеточного сигнального пути инсулина в печени, увеличения усвоения и метаболизма глюкозы периферическими тканями, увеличения запасов гликогена в печени, снижения выработки глюкозы в сыворотку в результате глюконеогенеза в печени. В еще одном варианте реализации указанные соединения и фармацевтические композиции применяют для профилактики и/или лечения хронического внутрипеченочного заболевания, такого как ПБЦ,ПСХ, прогрессирующего семейного холестаза (PFIC), алкогольного цирроза и ассоциированного холестаза и некоторых форм внепеченочных холестатических состояний или фиброза печени. Изобретение также относится к соединению формулы (1) или к фармацевтической композиции, содержащей указанное соединение, для профилактики и/или лечения состояний желудочно-кишечного тракта с пониженным усвоением входящих в рацион жира и жирорастворимых витаминов, которые можно преодолеть за счет повышения уровней желчных кислот и фосфолипидов в кишечнике. В другом варианте реализации указанное соединение или фармацевтическую композицию применяют для предотвращения и/или лечения заболевания, выбранного из группы, состоящей из расстройств липидного и липопротеинового обмена, таких как гиперхолестеринемия, гипертриглицеридемия и атеросклероз как клинически проявляемых состояний, которые могут быть улучшены за счет благоприятного действия FXR по снижению общего холестерина в плазме, снижению триглицеридов в сыворотке, повышению интенсивности превращения холестерина печени в желчные кислоты и повышению клиренса и интенсивности метаболического превращения ЛПОНП и других липопротеинов в печени. В еще одном варианте реализации указанное соединение и фармацевтическую композицию применяют для профилактики и/или лечения заболеваний, при которых сочетание гиполипидемического, анти- 11024843 холестатического и противофиброзного эффектов лекарственных средств, нацеленных на FXR, может быть использовано для лечения стеатоза печени и ассоциированных синдромов, таких как NASH, или для лечения холестатических и фиброзных эффектов, ассоциированных с алкогольным циррозом или с вирусными формами гепатита. Во взаимосвязи с гиполипидемическими эффектами было также показано, что потеря функционального FXR приводит к увеличению заболеваемости атеросклерозом у мышей с выключенным геном АроЕ (Е.A. Hanniman et al., J. Lipid Res. 2005, 46, 2595). Следовательно, агонисты FXR могут иметь клиническое применение в качестве антиатеросклеротических и кардиопротекторных лекарственных средств. Отрицательная регуляция эндотелина-1 в гладкомышечных клетках сосудов также может вносить вклад в указанные полезные терапевтические эффекты (F. He et al., Circ. Res. 2006, 98, 192). Изобретение также относится к соединению в соответствии с формулой (1) или фармацевтической композиции, содержащей указанное соединение, для профилактического и посттравматического лечения сердечно-сосудистых заболеваний, таких как острый инфаркт миокарда, острый инсульт или тромбоз,которые возникают как конечный результат хронического обструктивного атеросклероза. Помимо регулирования образования полипов кишечника и толстой кишки, FXR, по-видимому, экспрессируется в опухолевой ткани и клеточных линиях молочной железы, но не в здоровой ткани молочной железы и, вероятно, взаимодействует с эстрогеновым рецептором (ЭР) в ЭР-положительных клетках рака молочной железы (K.Е. Swales et al., Cancer Res. 2006, 66, 10120 и F. Journe et al., Breast Cancer Res.Treat. 2009, 115, 523). Этот факт позволил бы рассматривать FXR также в качестве потенциальной мишени для лечения пролиферативных заболеваний, в частности метастазирующих форм рака, которые экспрессируют формуFXR, чувствительную к низкомолекулярным соединениям. В еще одном варианте реализации указанные соединения и фармацевтические композиции применяют для профилактики и/или лечения злокачественных гиперпролиферативных заболеваний, таких как различные формы рака, конкретно, некоторые формы рака молочной железы, печени или толстой кишки,при которых воздействие лиганда FXR будет иметь благоприятный эффект. Наконец, FXR по-видимому, также вовлечен в регулирование антибактериальной защиты в кишечнике (Т. Inagaki et al., PNAS. 2006, 103, 3920), хотя точный механизм не предложен. Из этих опубликованных данных, однако, можно сделать вывод, что лечение с применением агонистов FXR может оказывать благоприятное воздействие при лечении воспалительных заболеваний кишечника (ВЗК), в частности, тех форм, при которых затронута верхняя (подвздошная) часть кишечника (например, болезни Крона подвздошной кишки), поскольку данный участок, по-видимому, является местом действия регулирования FXR роста бактерий. При ВЗК тем или иным образом нарушена десенсибилизация адаптивного иммунного ответа в иммунной системе кишечника. Избыточный рост бактерий может в таком случае стать причинным фактором развития хронической воспалительной реакции. Следовательно, подаление роста бактерий за счет FXR-опосредованных механизмов может представлять собой ключевой механизм для предотвращения острых эпизодов воспаления. Таким образом, изобретение также относится к соединению в соответствии с формулой (1) или фармацевтической композиции, содержащей указанное соединение, для предотвращения и/или лечения заболевания, связанного с воспалительными заболеваниями кишечника, такими как болезнь Крона или язвенный колит. FXR-опосредованное восстановление барьерной функции кишечника и снижение некомменсальной бактериальной нагрузки, как полагают, полезны для уменьшения воздействия бактериальных антигенов на иммунную систему кишечника и, следовательно, способны снизить воспалительные реакции. Изобретение также относится к соединению или фармацевтической композиции для профилактики и/или лечения ожирения и ассоциированных расстройств, таких как метаболический синдром (сочетание состояний дислипидемии, диабета и аномально высокого индекса массы тела), которые могут быть преодолены за счет FXR-опосредованного снижения уровней триглицеридов в сыворотке, глюкозы в крови и повышения чувствительности к инсулину и FXR-опосредованного снижения массы тела. В другом варианте реализации соединения или фармацевтическая композиция согласно настоящему изобретению подходят для предотвращения и/или лечения клинических осложнений диабета I типа иII типа. Примеры таких осложнений включают диабетическую нефропатию, диабетическую ретинопатию, диабетические нейропатии или окклюзионную болезнь периферических артерий (ОБПА). Другие клинические осложнения диабета также включены в рамки настоящего изобретения. Кроме того, состояния и заболевания, возникающие в результате хронической жировой и фиброзной дегенерации органов вследствие усиленного накопления липидов и в особенности триглицеридов и последующей активации профибротических путей, также можно предотвращать и/или лечить путем применения соединений или фармацевтической композиции согласно настоящему изобретению. Такие состояния и заболевания включают NASH и хронические холестатические состояния в печени, гломерулосклероз и диабетическую нефропатию в почках, макулярную дегенерацию и диабетическую ретинопатию в глазах и нейродегенеративные заболевания, такие как болезнь Альцгеймера в головном мозге или диабетические нейропатии в периферической нервной системе. Для практического применения соединения согласно настоящему изобретению могут быть скомбинированы в качестве активного ингредиента в однородную смесь с фармацевтическим носителем в соответствии со стандартными фармацевтическими методиками составления смесей. Носитель может принимать широкое разнообразие форм в зависимости от формы препарата, требуемой для введения, например, пероральной или парентеральной (включая внутривенную). При получении композиций для пероральной лекарственной формы могут быть использованы любые из традиционных фармацевтических сред, такие как, например, вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и т.п. в случае пероральных жидких препаратов, таких как, например, суспензии, эликсиры и растворы; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие вещества, связующие вещества, разрыхлители и т.п. в случае пероральных твердых препаратов, таких как, например, порошки, твердые и мягкие капсулы и таблетки, причем твердые пероральные препараты являются предпочтительнее жидких препаратов. Благодаря легкости их введения таблетки и капсулы являются более предпочтительной пероральной лекарственной формой для однократного введения, в случае которой, очевидно, применяют твердые фармацевтические носители. При необходимости, таблетки могут быть покрыты оболочкой с помощью стандартных способов с применением водных или неводных сред. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Процентное содержание активного соединения в этих композициях может, несомненно, варьироваться и может для удобства составлять от примерно 2 до примерно 60% от массы единицы лекарственной формы. Количество активного соединения в таких терапевтически подходящих композициях таково, что эффективная доза будет достигнута. Активные соединения могут также быть введены интраназально в виде, например, жидких капель или спрея. Таблетки, пилюли, капсулы и т.п. могут также содержать связующее вещество, такое как трагакантовая камедь, аравийская камедь, кукурузный крахмал или желатин; вспомогательные вещества, такие как фосфат дикальция; разрыхлитель, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, такое как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. В случае, когда лекарственная форма для однократного введения представляет собой капсулу, она может содержать, в дополнение к веществам вышеуказанных типов, жидкий носитель, такой как жирное масло. Другие различные вещества могут присутствовать в виде оболочек или для модификации физической формы единицы лекарственной формы. Например, таблетки могут быть покрыты шеллаком, сахаром или и тем, и другим. Сироп или эликсир может содержать, в дополнение к активному ингредиенту,сахарозу в качестве подсластителя, метил- и пропилпарабены в качестве консервантов, краситель и ароматизатор, такой как вишневый или апельсиновый ароматизатор. Поскольку соединения согласно настоящему изобретению в основном представляют собой карбоновые кислоты или подобные их анионные изостеры, и поскольку хорошо известно, что солевые формы ионных лекарственных соединений могут существенно влиять на биодоступность лекарственных соединений, соединения согласно настоящему изобретению могут также быть использованы в виде солей с различными противокатионами с получением перорально доступного состава. Такие фармацевтически приемлемые катионы могут представлять собой, среди прочих, моно- или бивалентные ионы, такие как ионы аммония, щелочных металлов - натрия или калия, или щелочно-земельных металлов - магния или кальция, некоторых фармацевтически приемлемых аминов, таких как трис(гидроксиметил)аминометан,этилендиамин, диэтиламин, пиперазин или другие, или некоторых катионных аминокислот, таких как лизин или аргинин. Соединения согласно настоящему изобретению также можно вводить парентерально. Растворы или суспензии этих активных соединений могут быть получены в воде, подходящим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также могут быть получены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. При обычных условиях хранения и применения эти препараты содержат консервант для предотвращения роста микроорганизмов. Фармацевтические формы, подходящие для инъекционного применения, включают стерильные водные растворы или дисперсии и стерильные порошки для экстемпорального приготовления стерильных растворов или дисперсий для инъекций. Во всех случаях форма должна быть стерильной и должна быть жидкой до такой степени, чтобы ее можно легко вводить с помощью шприца. Она должна быть стабильной в условиях производства и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), подходящие их смеси и растительные масла. Любой подходящий путь введения может быть использован для обеспечения млекопитающего, в частности, человека, эффективной дозой соединения согласно настоящему изобретению. Например, могут быть использованы пероральный, ректальный, местный, парентеральный, глазной, легочный, назальный и подобные им пути введения. Лекарственные формы включают таблетки, пастилки, дисперсии,суспензии, растворы, капсулы, кремы, мази, аэрозоли и т.п. Предпочтительно соединения согласно настоящему изобретению вводят перорально. Эффективная доза применяемого активного ингредиента может варьироваться в зависимости от конкретного применяемого соединения, способа введения, состояния, подлежащего лечению, и тяжести состояния, подлежащего лечению. Такая доза может быть легко определена специалистом в данной области техники. При лечении или предотвращении FXR-опосредованных состояний, для которых показаны соединения согласно настоящему изобретению, удовлетворительные результаты получают, как правило, когда соединения согласно настоящему изобретению вводят в суточной дозе от примерно 0,1 до примерно 100 мг на 1 кг массы тела животного, предпочтительно в виде однократной суточной дозы или в виде отдельных доз от двух до шести раз в сутки или в форме с замедленным высвобождением. Для большинства крупных млекопитающих общая суточная доза составляет от примерно 1,0 до примерно 1000 мг,предпочтительно от примерно 1 до примерно 50 мг. В случае взрослого человека массой 70 кг общая суточная доза будет, как правило, составлять от примерно 7 до примерно 350 мг. Этот режим дозирования можно корректировать для обеспечения оптимального терапевтического ответа. Соединения согласно настоящему изобретению могут быть получены в соответствии со способами,описанными в последующих схемах и примерах, с применением соответствующих веществ и далее проиллюстрированы с помощью следующих конкретных примеров. Более того, путем применения способов,описанных в настоящей заявке, в сочетании с обычными знаниями в данной области техники легко могут быть получены дополнительные соединения согласно настоящему изобретению, заявленные в настоящем описании. Соединения, проиллюстрированные в примерах, однако не следует расценивать в качестве образующих единственный род, который считается предметом настоящего изобретения. Примеры также иллюстрируют детали получения соединений согласно настоящему изобретению. Специалистам в данной области техники без труда будет понятно, что известные изменения условий и процессов следующих препаративных методик могут быть применены для получения этих соединений. Соединения согласно настоящему изобретению в общем случае выделяют в виде их фармацевтически приемлемых солей, таких как соли, описанные выше. Основания со свободной аминогруппой, соответствующие выделенным солям, могут быть получены путем нейтрализации подходящим основанием, таким как водный гидрокарбонат натрия, карбонат натрия, гидроксид натрия и гидроксид калия, и экстракции полученного из солевой формы основания со свободной аминогруппой органическим растворителем с последующим упариванием. Основание со свободной аминогруппой, выделенное таким образом, можно далее превращать в другую фармацевтически приемлемую соль путем растворения в органическом растворителе с последующим добавлением соответствующей кислоты и последующим упариванием, осаждением или кристаллизацией. Свободные карбоновые кислоты, соответствующие выделенным солям, могут быть получены путем нейтрализации подходящей кислотой, такой как водная хлороводородная кислота, гидросульфат натрия, дигидрофосфат натрия, и экстракции, полученной из солевой формы кислоты со свободной карбоксильной группой органическим растворителем с последующим упариванием. Карбоновую кислоту, выделенную таким образом, можно далее превращать в другую фармацевтически приемлемую соль путем растворения в органическом растворителе с последующим добавлением соответствующего основания и последующим упариванием, осаждением или кристаллизацией. Получение соединений согласно настоящему изобретению проиллюстрировано ниже. Если на схемах не указано иное, переменные имеют значения, описанные выше. Представленные ниже примеры предназначены для иллюстрации конкретных вариантов реализации изобретения. Подходящие исходные вещества, структурные элементы и реагенты, применяемые для описанных ниже синтезов, доступны на коммерческой основе, например в Sigma-Aldrich или Acros Organics, или могут быть получены обычным способом с помощью методик, описанных в литературе, например в "March's Advanced OrganicCH2Cl2 (ДХМ) (200 мл) добавляли по каплям SOCl2 (40 мл, 336 ммоль). Полученную смесь перемешивали при комнатной температуре (кт) в течение 2 ч и растворители удаляли при пониженном давлении. Остаток растворяли в N,N-диметилформамиде (ДМФА) (200 мл) и к этому раствору добавляли 4-бром-3 хлорфенол (9,7 г, 47 ммоль), К 2 СО 3 (40 г, 290 ммоль) и NaI (12 г, 80 ммоль). Смесь перемешивали при 60 С в течение ночи, затем охлаждали до кт, разбавляли водой (1000 мл) и экстрагировали этилацетатом(ЭА) (500 мл 3). Объединенные органические фазы промывали солевым раствором (500 мл 3), сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали посредством флэш-хроматографии на силикагеле (КХ) с получением указанного в заголовке соединения 1 а (19 г, 88%) в виде белого твердого вещества. Стадия 1: метиловый эфир 3-(2,2-дихлор-3-оксоциклобутил)бензойной кислоты (1b). В трехгорлой круглодонной колбе, снабженной холодильником, верхнеприводной мешалкой и капельной воронкой с компенсатором давления, растворяли в атмосфере азота метил-3-винилбензоат (5 г,31 ммоль) в сухом Et2O (150 мл). В эту колбу добавляли цинковую пыль (6 г, 3 экв.) и реакционную смесь обрабатывали ультразвуком в течение 30 мин. По прошествии этого времени добавляли по каплям раствор трихлорацетилхлорида (8,7 мл, 2,5 экв.) в сухом Et2O (50 мл), продолжая обработку ультразвуком в течение еще 30 мин. В течение процесса реакционную смесь нагревали до 35 С. Обработку ультразвуком продолжали в течение 2,5 ч при кипячении с обратным холоильником, завершение реакции определяли посредством 1 Н ЯМР-спектроскопии. Реакционную смесь оставляли охлаждаться до кт и реакцию останавливали водой (50 мл). Добавление воды осуществляли по каплям с некоторым количеством перерывов на несколько минут из-за протекания замедленной экзотермической реакции. После перемешивания в воде в течение 20 мин реакционную смесь фильтровали через слой целита и промывали Et2O. Органический слой промывали порциями воды (2250 мл), насыщенным раствором бикарбоната натрия(2250 мл) и солевым раствором (1250 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта 1b в виде темно-желтого вязкого масла (8,7 г неочищенного продукта). Стадия 2: метиловый эфир 3-(3-оксоциклобутил)бензойной кислоты (1 с). Неочищенное соединение 1b (8,7 г) растворяли в ледяной уксусной кислоте (55 мл) в круглодонной колбе в атмосфере азота. В эту колбу добавляли цинковую пыль (4,6 г, 2,2 экв.) и реакционную смесь перемешивали и нагревали до 120 С в течение 3 ч. После охлаждения до кт смесь фильтровали через слой целита, остаток на фильтре промывали порциями ЭА. Объединенный раствор концентрировали при пониженном давлении перед растворением в ЭА (500 мл), промывали солевым раствором (150 мл 2) и затем сушили над сульфатом натрия, фильтровали и концентрировали снова. Неочищенную смесь перемешивали в течение 5 мин в хлороформе (250 мл) и фильтровали через воронку с фильтром Шотта. Фильтрат концентрировали с получением неочищенного продукта в виде бледно-желтого масла. Неочищенный продукт очищали посредством КХ с (ПЭ/ЭА = 9:1, ПЭ = петролейный эфир) с получением целевого продукта 1 с (2,5 г, 38% на 2 стадии) в виде бледно-желтого масла. Стадия 3: метиловый эфир 3-(3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)-3-гидроксициклобутил)бензойной кислоты (1). К перемешиваемому раствору соединения 1 а (1,67 г, 3,5 ммоль) в сухом ТГФ (30 мл) добавляли нBuLi (2,5 М в гексане, 1,2 экв., 1,69 мл) по каплям в течение 10 мин при -78 С в атмосфере азота. Смесь перемешивали в течение 1 ч при этой температуре перед добавлением по каплям раствора соединения 1 с(0,72 г, 1 экв.) в сухом ТГФ (10 мл) и перемешивали в течение 1 ч при этой температуре. Реакционную смесь оставляли медленно нагреваться до кг и перемешивали в течение ночи. Реакцию останавливали раствором насыщенного раствора хлорида аммония (50 мл) и ЭА (250 мл). Отделяли органический слой,и водный слой промывали ЭА (2100 мл). Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта в виде коричневого масла. Продукт далее выделяли посредством КХ с ПЭ/ЭА (от 19:1 до 3:1). Реакцию и очистку повторяли дважды в том же масштабе и объединенный продукт (3,13 г) очищали повторно в тех же условиях с получением конечного продукта 1 (1,7 г, 19%). 1 ствии этого времени ТГФ удаляли при пониженном давлении. Оставшийся водный раствор разбавляли водой (25 мл) и промывали Et2O (250 мл). Затем водный слой переносили в круглодонную колбу и подкисляли до рН 6 с помощью 1N HCl. Образовавшийся белый осадок отфильтровывали и сушили при пониженном давлении при 50 С с получением указанного в заголовке соединения 2 (1,3 г, 78%, индивидуальный изомер по данным 1H ЯМР и ЖХ-МС) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, CD3OD) : 7,98 (с, 1H), 7,86 (д, J = 7,6 Гц, 1 Н), 7,58-7,46 (м, 5 Н), 7,41 (т, J = 7,6 Гц,1 Н), 6,91 (д, J = 2,4 Гц, 1H), 6,80 (дд, J = 8,8, 2,4 Гц, 1H), 4,95 (с, 2 Н), 3,29-3,25 (м, 2 Н), 2,96 (м, 1H), 2,55-2,49(м, 2 Н), 2,37 (м, 1H), 1,24-1,22 (м, 4 Н). МС (ионизация электроспреем, ESI-) m/z: 584 (582) [М-1]-. Соответствующие интенсивные ядерные эффекты Оверхаузера (ЯЭО) (получены из спектровROESY (спектроскопия ЯЭО во вращающейся системе координат); показаны ниже с помощью стрелок) указывают на то, что два ароматических фрагмента соединения согласно примеру 2 находятся в 1,3 транс-ориентации Альтернативный путь для примера 2. Стадия 1: 3-(3-бромфенил)циклобутанон (2 а).N,N-Диметилацетамид (9,0 г, 103 ммоль) растворяли в 1,2-дихлорэтане (200 мл). Раствор охлаждали до 0 С перед добавлением ангидрида трифторметансульфоновой кислоты (63 г, 223 ммоль). Реакционную смесь перемешивали дополнительно в течение 60 мин при 0 С. Затем добавляли 1-бром-3 винилбензол (15 г, 81,9 ммоль) и 2,4,6-коллидин (10,5 г, 86,6 ммоль). Реакционную смесь нагревали при температуре кипения в течение ночи, реакцию останавливали добавлением воды (300 мл) и перемешивали в течение 2 ч при кт. Смесь экстрагировали ДХМ (300 мл 3). Объединенные органические слои сушили над Na2SO4 и концентрировали в вакууме. Очистка посредством КХ (ЭА/ПЭ = 1:20) приводила к получению указанного в заголовке соединения 2 а (5,0 г, 27%) в виде бледно-желтого твердого вещества. Стадия 2: 3-(3-бромфенил)-1-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4 ил)метокси)фенил)циклобутанол (2b). К раствору соединения 1 а (14 г, 29,6 ммоль) в сухом ТГФ (500 мл) добавляли по каплям при -78 С н-BuLi (18,5 мл, 1,6 М в гексане, 29,6 ммоль). Смесь перемешивали дополнительно в течение 1 ч при-78 С и добавляли по каплям раствор соединения 2 а (6,5 г, 28,9 ммоль) в сухом ТГФ (50 мл). Полученную смесь перемешивали при -78 С в течение 1 ч и затем нагревали до кт, реакцию останавливали насыщенным водн. NH4Cl (500 мл). Смесь экстрагировали ЭА (500 мл 2), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток очищали посредством КХ (ЭА/ПЭ = 1:5) с получением указанного в заголовке соединения 2b (6,5 г, 37%) в виде белого твердого вещества. Стадия 3: 3-(3-цианофенил)-1-(2-хлор-4-5-циклопропил-3-(2.6-дихлорфенил)изоксазол-4 ил)метокси)фенил)циклобутанол (2 с). К раствору соединения 2b (3,1 г, 5 ммоль) в ДМФА (50 мл) добавляли в атмосфере аргона Zn(CN)2(500 мг, 4,3 ммоль), Pd2(dba)3 (300 мг, 0,33 ммоль) и реагент Ксантфос (Xantphos) (150 мг, 0,31 ммоль). Смесь перемешивали в течение 10 ч при 115 С при микроволновом облучении. После охлаждения до кт реакционную смесь разбавляли водой (250 мл) и экстрагировали ЭА (250 мл 2). Объединенные органические слои промывали солевым раствором (100 мл 3) и сушили над Na2SO4. Остаток очищали посредством КХ (ЭА/ПЭ) с получением указанного в заголовке соединения 2 с (1,2 г, 42%) в виде бледножелтого твердого вещества. Стадия 4: 3-1s,3s)-3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси) фенил)-3-гидроксициклобутил)бензойная кислота (2). К раствору соединения 2 с (15 г, 24,2 ммоль) в EtOH (750 мл) добавляли водн. NaOH (40 г в 100 мл воды). Полученную смесь нагревали до кипения в течение ночи и затем охлаждали до кт. Реакционную смесь концентрировали в вакууме для удаления летучего растворителя, разбавляли водой (1000 мл) и доводили рН до 2 с помощью разбавленной водн. HCl (1N). Образовавшийся осадок собирали фильтрованием с получением неочищенного продукта в виде желтого твердого вещества (13,8 г). Очистка посредством препаративной обращенно-фазовой ВЭЖХ (ОФ-ВЭЖХ) приводила к получению указанного в заголовке соединения 2 (8,0 г, 56%, индивидуальный изомер по данным 1H ЯМР) в виде белого твердого вещества. Препаративный пример 3 Стадия 1: метиловый эфир 3-(3-гидроксиазетидин-1-ил)бензойной кислоты (3 а). К раствору метил-3-иодбензоата (4,5 г, 17,2 ммоль) в ДМСО (30 мл) добавляли хлороводородную соль 3-азетидин-3-ола (1,3 г, 11,8 ммоль), CS2CO3 (9,5 г, 29,2 ммоль), CuI (446 мг, 2,3 ммоль) и L-пролин(540 мг, 4,7 ммоль) и смесь далее нагревали при 90 С в течение 18 ч в атмосфере аргона. Раствор разбавляли ЭА и водой, органический слой три раза промывали солевым раствором, концентрировали при пониженном давлении и очищали посредством КХ (ПЭ/ЭА = 2:1) с получением соединения 3 а (1,6 г, 66%) в виде желтого твердого вещества. Стадия 2: метиловый эфир 3-(3-оксоазетидин-1-ил)бензойной кислоты (3). К раствору соединения 3 а (1,60 г, 7,7 ммоль) в сухом ДХМ (30 мл) добавляли периодинан ДессаМартина (6,5 г, 15,4 ммоль) при 0 С, смесь перемешивали при кт в течение 2 ч в атмосфере N2. Реакцию останавливали насыщенным раствором бикарбоната натрия и смесь разбавляли ЭА. Органическую часть промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали посредством КХ (ПЭ/ЭА = 4:1) с получением соединения 3 (1,2 г, 75%) в виде белого твердого вещества. Пример 4: 3-1s,3s)-3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)-3-гидроксициклобутил)-N-(метилсульфонил)бензамид (4) К раствору соединения 2 (100 мг, 0,17 ммоль) в ДХМ (5 мл) добавляли EDCIHCl (100 мг, 0,52 ммоль), ДМАП (100 мг, 0,81 ммоль) и MeSO2NH2 (40 мг, 0,42 ммоль). Смесь перемешивали при 30 С в течение ночи и затем разбавляли ЭА и промывали Н 2 О, солевым раствором и сушили над Na2SO4. Концентрирование в вакууме и очистка посредством препаративной ТСХ приводили к получению неочищенного целевого соединения в виде светло-желтого твердого вещества. После очистки посредством ОФ-ВЭЖХ получали указанное в заголовке соединение 4 (38 мг, 33%) в виде белого твердого вещества. 1(276 мг, 2 ммоль), фенилметантиол (125 мг, 1 ммоль), Pd2(dba)3 (200 мг, 0,22 ммоль) и Ксантфос (75 мг,0,16 ммоль). Далее смесь перемешивали при 115 С в течение 4 ч. После охлаждения до кт реакционную смесь разбавляли водой (100 мл) и экстрагировали ЭА (100 мл 2). Объединенные органические слои промывали солевым раствором (100 мл 2), сушили над Na2SO4 и концентрировали досуха. Очистка посредством КХ привела к получению соединения 5 а (200 мг, 30%) в виде бледно-желтого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3) : 7,36-7,32 (м, 3 Н), 7,28-7,07 (м, 9 Н), 7,01 (д, J = 7,2 Гц, 1 Н), 6,82 (д, J = 2,0 Гц, 1H), 6,66 (дд, J = 8,8, 2,0 Гц, 1H), 4,75 (с, 2 Н), 4,04 (с, 2 Н), 3,06-3,00 (м, 2 Н), 2,84-2,78 (м, 2 Н), 2,442,38 (м, 3 Н), 2,09 (м, 1H), 1,24-1,18 (м, 2 Н), 1,11-1,08 (м, 2 Н). МС (ESI+) m/z: 662 [M+1]+. Стадия 2: 3-(3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)-3 гидроксициклобутил)бензол-1-сульфонилхлорид (5b). К раствору соединения 5 а (34 мг, 0,05 ммоль) в CH3CN/HOAC/H2O (1 мл/37 мкл/25 мкл) добавляли 2,4-дихлор-5,5-диметилгидантоин (20 мг, 0,1 ммоль). Смесь перемешивали при 0-5 С в течение 2 ч. Ре- 17024843 акционную смесь разбавляли водой и экстрагировали CH2Cl2. Объединенные органические слои промывали 5% раствором NaHCO3, солевым раствором и сушили над Na2SO4. Концентрирование досуха привело к получению неочищенного продукта 5b (30 мг) в виде бесцветного масла, которое использовали непосредственно для следующей стадии. Стадия 3: 3-(3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)-3 гидроксициклобутил)бензолсульфонамид (5). К раствору соединения 5b (30 мг) в CH3CN (2 мл) добавляли NH4OH (0,3 мл). Смесь перемешивали при кт в течение 1 ч. Концентрирование досуха и очистка посредством препаративной ОФ-ВЭЖХ привели к получению указанного в заголовке соединения 5 (3,5 мг, 10% на две стадии) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3) : 7,85 (с, 1H), 7,77 (д, J = 7,6 Гц, 1H), 7,54-7,41 (м, 5 Н), 7,35 (д, J = 8,4 Гц,1H), 6,90 (с, 1H), 6,75 (д, J = 8,4 Гц, 1H), 4,83 (с, 2 Н), 4,77 (с, уширенный, 2 Н), 3,20 (т, J = 10,4 Гц, 2 Н),3,04 (м, 1 Н), 2,58 (т, J = 10,6 Гц, 2 Н), 2,17 (м, 1H), 1,31-1,30 (м, 2 Н), 1,20-1,16 (м, 2 Н). МС (ESI-) m/z: 617 К раствору соединения 2b (200 мг, 0,32 ммоль) в ДМСО добавляли метансульфинат натрия (50 мг,0,46 ммоль), CuI (20 мг, 0,1 ммоль), L-пролин (37 мг, 0,32 ммоль) и диизопропилэтиламин (ДИЭА) (41 мг, 0,32 ммоль). Смесь перемешивали при 95 С в течение ночи и затем разбавляли водой и экстрагировали ЭА. Объединенные органические слои промывали водой и сушили над Na2SO4. Концентрирование досуха при пониженном давлении и очистка посредством препаративной ОФ-ВЭЖХ приводили к получению указанного в заголовке соединения 6 в виде белого твердого вещества (35 мг, 21%, индивидуальный изомер по данным 1 Н ЯМР и ЖХ-МС). 1 Н ЯМР (400 МГц, CDCl3) : 7,84 (с, 1H), 7,79 (д, J = 7,6 Гц, 1H), 7,60 (д, J = 7,6 Гц, 1 Н), 7,53 (т, J = 7,6 Гц, 1 Н), 7,44-7,41 (м, 3 Н), 7,34 (т, J = 7,2 Гц, 1 Н), 6,90 (д, J = 2,8 Гц, 1 Н), 6,75 (дд, J = 8,4, 2,0 Гц, 1H),4,83 (с, 2 Н), 3,24-3,19 (м, 2 Н), 3,08-3,04 (м, 4 Н), 2,62-2,56 (м, 2 Н), 2,17 (м, 1 Н), 1,31-1,29 (м, 2 Н), 1,20-1,16 Стадия 1: метиловый эфир 1-изопропил-5-винил-1H-пиразол-3-карбоновой кислоты (7 а). Суспензию метилтрифенилфосфонийбромида (2,69 г, 7,52 ммоль) в сухом ТГФ (40 мл) охлаждали до -78 С и добавляли по каплям н-бутиллитий (1,6 М раствор в гексане, 3,7 мл, 5,91 ммоль). Желтооранжевую суспензию перемешивали при -78 С в течение 50 мин и затем добавляли по каплям раствор метилового эфира 5-формил-1-изопропил-1H-пиразол-3-карбоновой кислоты (полученного как описано вWO 2011/020615, 1,05 г, 5,37 ммоль) в сухом ТГФ (10 мл). Смесь перемешивали при -78 С в течение 1,75 ч, охлаждающую баню удаляли и смесь (грязно-белую суспензию) перемешивали при кт в течение 1 ч. Затем смесь распределяли между разбавленным водн. раствором NaHCO3 (150 мл) и ЭА (150 мл). Водный слой дважды экстрагировали ЭА (по 50 мл каждый раз) и объединенный органический слой дважды промывали водой (по 50 мл каждый раз) и концентрировали без высушивания с получением 2,74 г желтого медленно кристаллизующегося масла. Неочищенный продукт очищали посредством КХ (предадсорбция CH2Cl2, гексан/ЭА 4:1) с получением алкена 7 а (590 мг, 57%) в виде бесцветного масла. 1 Н ЯМР (ДМСО-d6) : 7,02 (с, 1H), 6,87 (дд, J = 17,3, 11,2 Гц, 1H), 5,94 (дд, J = 17,3, 1,3 Гц, 1H), 5,45(7b). Реакцию проводили в двух сухих запаянных трубках (две равные по количеству порции). Порции объединяли для обработки и очистки. Методика для одной порции: к раствору N,N-диметилацетамида(0,22 мл, 2,34 ммоль) в 1,2-дихлорэтане (12 мл) добавляли по каплям в атмосфере азота при температуре от -15 до -20 С ангидрид трифторметансульфоновой кислоты (0,43 мл, 2,57 ммоль) с образованием непрозрачной суспензии. Смесь перемешивали при -15 С в течение 10 мин и добавляли по каплям раствор алкена 7 а (151 мг, 0,78 ммоль) и симм-коллидин (0,42 мл, 3,12 ммоль) в 1,2-дихлорэтане (3 мл) (образовался желтый раствор). По окончании добавления охлаждающую баню удаляли, смесь оставляли нагреваться до кт (оранжевый мутный раствор) и трубку запаивали. Далее смесь перемешивали при 90 С в течение 15 ч (коричневые смеси). Добавляли воду (5 мл) при кт и смеси перемешивали при 100 С в течение 2 ч (мутные двухфазные растворы). После охлаждения до кт смеси объединяли и распределяли между разбавленным водн. раствором NaHCO3 и CH2Cl2 и водный слой экстрагировали три раза CH2Cl2 (по 30 мл каждый раз). Объединенный органический слой сушили (Na2SO4), фильтровали и концентрировали с получением коричневого масла (2,2 г). Очистка посредством КХ (613 см, предадсорбция CH2Cl2, толуол/ЭА 3:1) приводила к получению циклобутанона 7b (115,5 мг, 31%) в виде желтого масла. 1 Н ЯМР (ДМСО-d6) : 6,81 (с, 1 Н), 4,58 (септ, J = 6,5 Гц, 1 Н), 3,78 (с, ЗН), 3,85-3,73 (м, 1 Н), 3,593,45 (м, 2 Н), 3,37-3,24 (м, 2 Н, частично перекрывается сигналом воды), 1,39 (д, J = 6,6 Гц, 6 Н).C12H16N2O3 (236,27). ЖХ-МС (ESI): 237 [М+Н]+. Стадия 3: метиловый эфир 5-1s,3s)-3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4 ил)метокси)фенил)-3-гидроксициклобутил)-1-изопропил-1H-пиразол-3-карбоновой кислоты (7). Раствор бромида 1 а (368 мг, 0,78 ммоль) в сухом ТГФ (6 мл) охлаждали до -78 С и добавляли по каплям 1,6 М раствор н-бутиллития в гексане (0,48 мл, 0,76 ммоль). Смесь перемешивали при -78 С в течение 20 мин и добавляли по каплям раствор циклобутанона 7b (164 мг, 0,69 ммоль) в сухом ТГФ (4 мл). Смесь перемешивали при -78 С в течение 2,5 ч и насыщенный водн. раствор NH4Cl (1 мл) добавляли по каплям при этой температуре. Охлаждающую баню удаляли и смесь оставляли нагреваться до кт и перемешивали при кт в течение 0,5 ч. Смесь затем добавляли в разбавленный водн. раствор NH4Cl и экстрагировали три раза ЭА. Объединенный органический слой сушили (Na2SO4), фильтровали и концентрировали с получением 516 мг почти бесцветного масла. Очистка посредством КХ (4,523 см, предадсорбция CH2Cl2, элюент гексан/ацетон = 2:1) приводила к получению регенерированного циклобутанона 7b (31,3 мг, 19%, слегка желтоватое масло) и неочищенного продукта (333 мг). Повторная очистка посредством КХ (422 см, гексан/ЭА =1:1) или препаративной ТСХ приводила к получению чистого продукта 7 (210 мг, 48%) в виде белой пены. 1H ЯМР (ДМСО-d6) : 7,65 (д, J = 2,1 Гц, 1H), 7,62 (с, 1 Н), 7,59-7,48 (м, 2 Н), 6,92 (д, J = 2,4 Гц, 1H),6,76 (дд, J = 8,6, 2,6 Гц, 1H), 6,66 (с, 1H), 5,49 (с, 1H), 4,92 (с, 2 Н), 4,42 (квинтетоподобный м, J = 6,5 Гц,1 Н), 3,78 (с, 3 Н), 3,24-3,11 (м, 2 Н, частично перекрывается сигналом воды), 3,04-2,90 (м, 1 Н), 2,54-2,33(м, 3 Н, частично перекрывается сигналом ДМСО), 1,32 (д, J = 6,5 Гц, 6 Н), 1,26-1,08 (м, 4 Н). Сложный эфир 7 (98,3 мг, 0,156 ммоль) растворяли в смеси ТГФ (7,5 мл), МеОН (2,5 мл) и воды (2,5 мл) и добавляли при кт LiOHН 2 О (65 мг, 1,56 ммоль). Смесь перемешивали при кт в течение 18 ч. Смесь распределяли между разбавленным водн. раствором NH4Cl и ЭА, и органический слой один раз промывали водой. Объединенный водный слой дважды экстрагировали ЭА. Объединенный органический слой сушили (Na2SO4), фильтровали и концентрировали с получением 103 мг почти белого твердого вещества. Продукт очищали посредством КХ (33,5 см, ЭА/EtOH = от 10:1 до 1:4) с получением соединения 8(94,8 мг, 99%) в виде белого твердого вещества. 1(дд, J = 8,6, 2,4 Гц, 1 Н), 6,38 (с, 1 Н), 5,51 (с, 1H, обменивается с D2O), 4,92 (с, 2 Н), 4,31 (квинтетоподобный м, J = 6,5 Гц, 1 Н), 3,25-3,08 (м, 2 Н, частично перекрывается сигналом воды), 2,93-2,77 (м, 1H), 2,572,43 (м, 1H, скрыт сигналом ДМСО), 2,43-2,29 (м, 2 Н, частично перекрывается сигналом ДМСО), 1,29 (д,J = 6,5 Гц, 6 Н), 1,26-1,08 (м, 4 Н). Сигнал СО 2 Н в спектре не виден. C30H28Cl3N3O5 (616,92). ЖХ-МС (ESI): 616, 618 [М+Н]+. Альтернативный путь для примера 8. Стадия 1: 1-(3-метиленциклобутил)этанон (8 а). Метиленциклобутанкарбонитрил (5,0 г, 53,7 ммоль) растворяли в сухом диэтиловом эфире (25 мл),охлаждали в ледяной бане и добавляли по каплям MeMgBr (26,8 мл, 80,5 ммоль, 3 М в эфире). Смесь оставляли перемешиваться в течение ночи при кт, охлаждали до 0 С, осторожно завершали реакцию 15% водн. раств. NaHSO4 (100 мл). Смесь перемешивали при кт в течение 30 мин и слои разделяли. Водную фазу экстрагировали пентаном (50 мл) и диэтиловым эфиром (50 мл). Объединенные органические слои промывали солевым раствором и сушили над Na2SO4. Растворители удаляли под вакуумом при кт и получали неочищенный продукт в виде желтоватой жидкости. Стадия 2: этиловый эфир 4-(3-метиленциклобутил)-2,4-диоксобутановой кислоты (8b). Натрий (1,15 г, 49,9 ммоль) растворяли в сухом EtOH (30 мл, денатурированного 5% диэтиловым эфиром). Соединение 8 а (5,5 г, 49,9 ммоль, неочищенное) растворяли в сухом EtOH (45 мл) и добавляли полученный выше раствор этоксида натрия. Эту смесь перемешивали при кт в течение 15 мин и затем добавляли по каплям диэтилоксалат (6,8 мл, 49,9 ммоль). Реакционную смесь помещали в предварительно нагретую (до 67 С) масляную баню и перемешивали при этой температуре в течение 4,5 ч. Смесь оставляли при кт в течение ночи. Растворитель удаляли, добавляли ЭА (100 мл) и 1 М HCl (70 мл) и отделяли органическую фазу. Водную фазу повторно экстрагировали ЭА (50 мл). Объединенные органические фазы промывали водой, солевым раствором и сушили над безв. Na2SO4. Растворитель удаляли при пониженном давлении и остаток очищали на силикагеле с применением смеси гексан/МТБЭ 9:1 в качестве элюента с получением чистого продукта 8b. Выход: 6,29 г, 56% на две стадии. 1 Н ЯМР (CDCl3),(м.д.): 6,36 (с, 1H), 4,85-4,80 (м, 2 Н), 4,34 (д, J = 8,0 Гц, 2 Н), 3,35-3,25 (м, 1H),3,05-2,85 (м, 4 Н), 1,36 (т, J = 8,0 Гц, 3 Н). Стадия 3: этиловый эфир 1-изопропил-5-(3-метиленциклобутил)-1H-пиразол-3-карбоновой кислоты(8 с). Соединение 8b (6,29 г, 29,9 ммоль) растворяли в сухом EtOH (65 мл, денатурированного 5% МеОН) и добавляли гидрохлорид изопропилгидразина (3,97 г, 35,9 ммоль). Реакционную смесь перемешивали в течение 3 ч при кт. Растворитель удаляли и к маслянистому остатку последовательно добавляли ЭА (100 мл), воду (50 мл) и насыщ. NaHCO3 (50 мл). Слои разделяли и водную фазу повторно экстрагировали ЭА(50 мл). Объединенные органические фазы промывали солевым раствором (70 мл) и сушили над безв.Na2SO4. Растворитель удаляли под вакуумом и остаток сушили при пониженном давлении. Выход: 7,23 г(содержит 3,4% EtOAc по данным ЯМР, в пересчете на чистое вещество выход: 6,98 г, 94%). Неочищенный продукт 8 с характеризовался 98% чистотой по данным ВЭЖХ и ЯМР. 1 Н ЯМР (CDCl3),(м.д.): 6,62 (с, 1H), 4,88-4,82 (м, 2 Н), 4,42-4,32 (м, 3 Н), 3,56-3,45 (м, 1H), 3,17-3,07(8d). Соединение 8 с (6,45 г, 26,0 ммоль) растворяли в смеси MeCN (77 мл) и воды (13 мл) и охлаждали в ледяной бане. К этому раствору добавляли RuCl3 Н 2 О (0,19 г, 0,86 ммоль) с последующим добавлением порциями NaIO4 (19,35 г, 90,9 ммоль). Во время этого добавления наблюдали экзотермический эффект. Полученную вязкую суспензию перемешивали при кт в течение 45 мин. Реакционную смесь разбавляли водн. раств. Na2S2O3 (10%, 260 мл), водой (50 мл) и ДХМ (100 мл). Фазы разделяли и водную фазу экстрагировали ДХМ (270 мл). Объединенные органические фазы промывали водн. раств. Na2S2O3 (10%,50 мл), водой (100 мл), солевым раствором (100 мл) и сушили над безв. Na2SO4. Неочищенный продукт(6,5 г) очищали на силикагеле, элюируя смесью гексан/МТБЭ с получением чистого продукта в виде масла, затвердевающего при хранении при -20 С. Выход: 5,8 г (78% на две стадии). 1 Н ЯМР (ДМСО-d6),(м.д.): 6,78 (с, 1H), 4,57 (h, J = 8,0 Гц, 1H), 4,26 (д, J = 8,0 Гц, 2 Н), 3,85-3,75 (м,1H), 3,58-3,45 (м, 2 Н), 3,35-3,25 (м, 2 Н), 1,39 (д, J = 8,0 Гц, 6 Н), 1,28 (т, J = 8,0 Гц, 3 Н). Стадия 5: 4-4-бром-3-хлорфенокси)метил)-5-циклопропил-3-(2,6-дихлорфенил)изоксазол (8 е). 3-Хлор-4-бромфенол (3,8 г, 18,3 ммоль) смешивали с (5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метанолом (3,47 г, 12,2 ммоль) и трифенилфосфином (6,41 г, 24,4 ммоль) в толуоле (150 мл). Смесь охлаждали в ледяной бане и добавляли по каплям DIAD (4,8 мл, 24,4 ммоль) в виде раствора в толуоле (10 мл). Реакционную смесь перемешивали при кт в течение 21 ч и растворители удаляли на роторном испарителе с получением желтого маслянистого остатка. Остаток растворяли в ДХМ (200 мл),добавляли силикагель (20 г) и смесь упаривали досуха. Этот материал наносили в верхнюю часть колонки с силикагелем и очищали элюированием смесью гексан/МТБЭ 9:1. Фракции, содержащие продукт,объединяли и растворитель удаляли при пониженном давлении с получением чистого продукта 8 е в виде бесцветного масла, кристаллизовавшегося при сушке в вакууме в течение ночи. Выход: 5,07 г (88%). 1 Н ЯМР (CDCl3),(м.д.): 7,45-7,30 (м, 4 Н), 6,90 (с, 1 Н), 6,60-6,55 (м, 1H), 2,15-2,07 (м, 1 Н), 1,32-1,25LiCl (0,684 г, 16,15 ммоль) растворяли в ТГФ (20 мл) при кт и добавляли /PrMgCl (2,0 М в ТГФ, 8,1 мл, 16,15 ммоль). Смесь перемешивали в течение 10 мин при кт, охлаждали на ледяной бане и добавляли в течение 5 мин раствор соединения 8 е (2,55 г, 5,38 ммоль) в ТГФ (20 мл). Охлаждающую баню удаляли и смесь перемешивали при кт в течение 4 ч. Смесь охлаждали до -10 С и быстро добавляли раствор соединения 8d (1,48 г, 5,92 ммоль) в ТГФ (16 мл). Смесь перемешивали при кт в течение 90 мин и затем добавляли водн. 0,5 М NaHSO4 (35 мл) и ЭА (50 мл). Полученную смесь перемешивали в течение 10 мин,- 20024843 слои разделяли и водный слой экстрагировали ЭА (30 мл). Объединенные органические фазы промывали водн. NaHCO3 (50 мл), солевым раствором (50 мл) и сушили над безв. Na2SO4. После удаления растворителя получали неочищенный продукт (3,79 г) в виде белой пены. 3,6 г этого продукта очищали на колонке с силикагелем, элюируя смесью гексан/ЭА 3:2 с получением чистого продукта 8f в виде твердой пены. Выход: 1,62 г (49%). 1 Н ЯМР (ДМСО-d6),(м.д.): 7,65-7,47 (м, 4 Н), 6,93-6,91 (м, 1H), 6,79-6,72 (м, 1H), 6,65 (с, 1H), 5,48(с, 1H), 4,92 (с, 2 Н), 4,42 (h, J = 8,0 Гц, 1H), 4,26 (д, J = 8,0 Гц, 2 Н), 3,32 (с, 2 Н), 3,22-3,14 (м, 2 Н), 3,052,90 (м, 1H), 2,45-2,35 (м, 2 Н), 1,35-1,10 (м, 14 Н). Стадия 7: 5-1s,3s)-3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)-3-гидроксициклобутил)-1-изопропил-1H-пиразол-3-карбоновая кислота (8). Соединение 8f (1,60 г, 2,48 ммоль) растворяли в ТГФ (100 мл), затем добавляли последовательно МеОН (50 мл), воду (50 мл) и LiOHН 2 О (1,04 г, 24,8 ммоль). Смесь перемешивали в течение 4,5 ч при кт и затем концентрировали при пониженном давлении для удаления МеОН и ТГФ. Оставшийся водный раствор подкисляли добавлением водн. 1 М HCl (24 мл) до рН 4,05 (контроль с помощью рН-электрода). Уже при рН, приблизительно равном 7, началось образование осадка. Полученное твердое вещество отфильтровывали, промывали на фильтре водой и сушили в вакууме при кт с получением продукта 8 в виде белого порошка. Выход: 1,40 г (92%). 1 Н ЯМР (CDCl3),(м.д.): 7,44-7,32 (м, 4 Н), 6,91 (д, J = 4,0 Гц, 1H), 6,78 (с, 1H), 6,75 (дд, J = 4,0 Гц, J = 8,0 Гц, 1H), 4,83 (с, 2 Н), 4,35-4,20 (м, 1H), 3,25-3,14 (м, 2 Н), 3,04-2,90 (м, 1 Н), 2,62-2,54 (м, 2 Н), 2,21-2,11 Соединение согласно примеру 8 А может быть получено путем гидролиза сложноэфирной группы неочищенного продукта 8f, как описано для примера 8, и выделения из неочищенного продукта 8 в качестве минорного изомера посредством препаративной ОФ-ВЭЖХ. 1 Н ЯМР (CDCl3),(м.д.): 7,42-7,30 (м, 2 Н), 7,11 (д, J = 8,0 Гц, 1H), 6,75-6,65 (м, 1H), 6,57 (с, 1 Н),4,79 (с, 2 Н), 4,50-4,41 (м, 1H), 3,96-3,85 (м, 1H), 2,98-2,90 (м, 2 Н), 2,67-2,57 (м, 2 Н), 2,20-2,09 (м, 1 Н), 1,51(д, J = 8,0 Гц, 6 Н), 1,32-1,14 (м, 4 Н). 13 С ЯМР (CDCl3),(м,д,): 172,6, 166,2, 159,2, 158,4, 147,4, 141,2, 135,7, 134,6, 132,8, 131,3, 128,1,127,7, 127,5, 116,8, 113,5, 110,0, 105,8, 75,1, 59,8, 51,2, 41,8, 25,4, 22,6, 8,5, 7,8. МС (ESI+) m/z: 616,3 [M+1]+. Конфигурация противолежащих атомов кольца основного изомера (соединение 8) и минорного изомера (соединение 8 А) была подтверждена с помощью экспериментов по измерению ЯЭО. Обнаруженные характерные ЯЭО между протонами указаны на следующих чертежах двойными стрелками: ЯЭО, обнаруженные для соединения согласно примеру 8 с 1,3-транс-конфигурацией противолежащих ароматических фрагментов ЯЭО, обнаруженные для соединения согласно примеру 8 А с 1,3-цис-конфигурацией противолежащих ароматических фрагментов Пример 9: метиловый эфир 6-(3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4 ил)метокси)фенил)-3-гидроксициклобутил)-1-метил-1H-индазол-3-карбоновой кислоты (9) Стадия 1: метиловый эфир 1-метил-6-винил-1H-индазол-3-карбоновой кислоты (9 а). К раствору метилового эфира 6-бром-1-метил-1H-индазол-3-карбоновой кислоты (60 мг, 0,22 ммоль) в ДМФА (10 мл) добавляли трибутил(винил)олово (99 мкл, 0,34 ммоль), Pd(Ph3)4 (11 мг, 9 мкмоль). После того как добавление было завершено, смесь перемешивали при 90 С в течение 4 ч в атмосфере Ar. Далее растворитель удаляли при пониженном давлении. Очистка посредством КХ приводила к получению соединения 9 а (52 мг, 88%). Стадия 2: метиловый эфир 1-метил-6-(3-оксоциклобутил)-1H-индазол-3-карбоновой кислоты (9b). В соответствии с методикой, описанной в примере 7, стадия 2, соединение 9b получали из соединения 9 а с выходом 57%. 1 Н ЯМР (400 МГц, CDCl3),(м.д): 8,14 (д, J= 8,4 Гц, 1H), 7,31 (с, 1H), 7,23 (д, J = 8,8 Гц, 1H), 4,13(с, 3 Н), 3,99 (с, 3 Н), 3,87-3,79 (м, 1H), 3,58-3,51 (м, 2 Н), 3,33-3,26 (м, 2 Н). m/z: 259 [М+1]+. Стадия 3: метиловй эфир 6-(3-(2-хлор-4-5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)-3-гидроксициклобутил)-1-метил-1H-индазол-3-карбоновой кислоты (9). В соответствии с методикой, описанной в примере 7. стадия 3, соединение 9 получали из соединения 9b с выходом 40%. Пример 10: 6-(3-(2-хлор-4-5-циклопропил-3-(2.6-дихлорфенил)изоксазол-4-ил)метокси)фенил)-3 гидроксициклобутил)-1-метил-1H-индазол-3-карбоновая кислота (10) В соответствии с методикой, описанной в примере 8, соединение 10 получали из соединения 9 с выходом 45% в виде белого твердого вещества. 1 Н ЯМР (400 МГц, CDCl3 : 8,14 (д, J = 8,0 Гц, 1 Н), 7,48 (д, J = 8,8 Гц, 1H), 7,43-7,32 (м, 4 Н), 7,29 (м,1 Н), 6,92 (д, J = 2,4 Гц, 1 Н), 6,76 (дд, J = 7,2 Гц, 2,4 Гц, 1H), 4,84 (с, 2 Н), 4,18 (с, 3 Н), 3,45-3,40 (м, 1H),3,28-3,23 (м, 2 Н), 3,19-3,10 (м, 1H), 2,68-2,63 (м, 2 Н), 2,21-2,14 (м, 1H), 1,33-1,29 (м, 2 Н), 1,20-1,15 (м,2 Н). m/z: 638 [М+1]+. Препаративный пример 11 Стадия 1: метиловый эфир 5-(3-гидроксиазетидин-1-ил)никотиновой кислоты (11 а). Смесь метилового эфира 5-бромникотиновой кислоты (2,00 г, 9,26 ммоль), азетидин-3-ола (1,01 г,9,26 ммоль), Cs2CO3 (9,06 г, 27,8 ммоль), BINAP (1,15 г, 1,85 ммоль) и Pd(OAc)2 (0,44 г, 1,85 ммоль) в сухом диоксане (115 мл) нагревали в течение ночи при 85 С в атмосфере N2. Полученную смесь фильтровали, концентрировали при пониженном давлении и очищали посредством препаративной ВЭЖХ с получением соединения 11 а (250 мг, 13%) в виде желтого твердого вещества. Стадия 2: метиловый эфир 5-(3-оксоазетидин-1-ил)никотиновой кислоты (11). К раствору соединения 11 а (250 мг, 1,20 ммоль) в сухом ДХМ (15 мл) добавляли периодинан ДессаМартина (1,014 г, 2,40 ммоль) при 0 С в атмосфере N2 и раствор перемешивали при кт в течение 2 ч. В полученный раствор для остановки реакции добавляли насыщенный раствор бикарбоната натрия и разбавляли ЭА. Органическую часть промывали солевым раствором, сушили над Na2SO4, фильтровали,концентрировали при пониженном давлении и очищали посредством КХ (ДХМ/МеОН = 150:1) с получением соединения 11 (140 мг, 57%) в виде желтого твердого вещества. Препаративный пример 12. С помощью методики, аналогичной описанной в препаративном примере 11, получали следующее соединение: Примеры 13/1-13/9. В следующей таблице приведены дополнительные примеры соединений, полученных в соответствии с упомянутыми выше препаративными примерами и примерами. Все перечисленные соединения были получены в виде индивидуальных изомеров. Примеры 14/1 и 14/2. С помощью методики, аналогичной описанной выше в примерах 1-13 и на схемах, получали следующие соединения, используя соответствующие структурные элементы. Следующее соединение может быть получено тем же образом путем применения методик, аналогичных описанным выше Анализ Анализ активности методом резонансного переноса энергии флуоресценции (FRET). Определение опосредованного лигандом взаимодействия пептида-кофактора для количественной оценки связывания лиганда с ядерным рецептором FXR проводили следующим образом. Получение лигандсвязывающего домена FXR- человека: ЛСД FXR- человека экспрессировали в Е. coli штамм BL21(DE3) в виде слитого белка, содержащего на N-конце последовательность глутатионS-трансферазы (GST). ДНК, кодирующую лигандсвязывающий домен FXR, клонировали в векторpDEST15 (Invitrogen). Экспрессия была под контролем индуцируемого ИПТГ Т 7-промотора. Аминокислоты, составляющие границы лигандсвязывающего домена, представляли собой аминокислоты 187-472 из файла базы данных NM005123 (RefSeq). Экспрессия и очистка ЛСД FXR: ночную прекультуру трансформированного штамма Е. coli разбав- 24024843CD600 = 0,4-0,6. Далее индуцировали экспрессию гена добавлением 0,5 мМ ИПТГ. Клетки инкубировали дополнительно в течение 6 ч при 30 С и скорости перемешивания 180 об/мин. Клетки собирали центрифугированием (7000g, 7 мин, кт). На 1 л исходной клеточной культуры клетки ресуспендировали в 10 мл буфера для лизиса (50 мМ глюкозы, 50 мМ Трис рН 7,9, 1 мМ ЭДТА и 4 мг/мл лизоцима) и оставляли на льду в течение 30 мин. Затем клетки подвергали обработке ультразвуком и удаляли остатки клеток посредством центрифугирования (22000g, 30 мин, 4 С). На 10 мл супернатанта добавляли 0,5 мл суспензии предварительно промытой глутатионсефарозы 4 В (Qiagen) и полученную суспензию медленно перемешивали вращением в течение 1 ч при 4 С. Гранулы глутатионсефарозы 4 В осаждали центрифугированием (2000g, 15 с, 4 С) и дважды промывали промывочным буфером (25 мМ Трис, 50 мМ KCl, 4 мМ MgCl2 и 1 М NaCl). Осадок ресуспендировали в 3 мл элюирующего буфера на 1 л исходной культуры(элюирующий буфер: 20 мМ Трис, 60 мМ KCl, 5 мМ MgCl2 и 80 мМ глутатиона, добавленного в виде порошка непосредственно перед использованием). Суспензию оставляли перемешиваться вращением в течение 15 мин при 4 С, гранулы осаждали и проводили повторное элюирование половиной объема элюирующего буфера по сравнению с первой элюцией. Элюаты объединяли и подвергали диализу в течение ночи против буфера 20 мМ Hepes (рН 7,5), содержащего 60 мМ KCl, 5 мМ MgCl2, а также 1 мМ дитиотреитола и 10% (об/об) глицерина. Содержание белка анализировали посредством электрофореза в полиакриламидном геле в присутствии ДСН (SDS-PAGE). Метод позволяет измерять способность предполагаемых лигандов модулировать взаимодействие между очищенным лигандсвязывающим доменом (ЛСД) FXR, экспрессируемым в бактериях, и синтетическим биотинилированным пептидом на основе остатков 676-700 из SRC-1 (LCD2, 676-700). Последовательность использованного пептида представляла собой B-CPSSHSSLTERHKILHRLLQEGSPS-COOH,где N-конец был биотинилирован (В). Лигандсвязывающий домен (ЛСД) FXR экспрессировали в виде белка, слитого с GST, в клетках BL-21 с применением вектора pDEST15. Клетки лизировали посредством обработки ультразвуком и слитые белки очищали на глутатионсефарозе (Pharmacia) в соответствии с инструкциями производителя. Для скрининга соединений на их способность влиять на взаимодействиеFXR и пептида применяли технологию LANCE компании Perkin Elmer. Этот метод основан на зависимом от связывания переноса энергии от донора к флуорофору-акцептору, присоединенному к связывающумуся исследуемому партнеру. Для простоты обработки и уменьшения фона от соединения флуоресцентная технология LANCE включает применение стандартных флуорофорных меток и детектирования с временным разрешением. Анализ проводили в конечном объеме 25 мкл в 384-луночном планшете, в буфере на основе Трис (20 мМ Трис-HCl, рН 7,5; 60 мМ KCl, 5 мМ MgCl2; 35 нг/мкл БСА), содержащем 20-60 нг/лунка рекомбинантного ЛСД FXR, слитого с GST, 200-600 нМ N-терминально биотинилированного пептида, представляющего собой аминокислоты 676-700 SRC-1, 200 нг/лунка конъюгата стрептавидин-xIAPC (Prozyme) и 6-10 нг/лунка конъюгата реактива Eu W1024 с антителами к GST (Perkin Elmer). Содержание ДМСО в образцах поддерживали на уровне 1%. После приготовления смеси для анализа и разбавления потенциальных FXR-модулирующих лигандов аналитические пробы доводили до состояния равновесия в течение 1 ч в темноте при кт в черных 384-луночных планшетах для флуоресцентного иммуноанализа (ФИА) (Greiner). Детекцию сигнала LANCE осуществляли с помощью прибора для регистрации флуоресценции Perkin Elmer VICTOR2VTM Multilabel Counter. Результаты были визуализированы путем нанесения на график соотношения между излучаемым светом при 665 и 615 нм. В отсутствие добавленного лиганда наблюдается образование комплекса FXR-пептид на базовом уровне. Лиганды, способствующие образованию комплекса, вызывают зависимое от концентрации увеличение разрешенного по времени сигнала флуоресценции. Следовало ожидать, что соединения, которые связываются одинаково хорошо и с мономерным FXR, и с комплексом FXR-пептид, не должны были давать каких-либо изменений сигнала, тогда как лиганды, которые связываются преимущественно с мономерным рецептором,должны были вызывать зависимое от концентрации уменьшение наблюдаемого сигнала. Для оценки ингибиторной способности соединений определяли величины EC50, например для соединений, представленных ниже в табл. 1 (А = ЕС 50 25 нМ; В = 25 ЕС 50 100 нМ; С = ЕС 50 100 нМ). Таблица 1 Одногибридное исследование на клетках млекопитающих (М 1 Н). Определение опосредованной лигандом трансактивации под управлением GaW-промотора для количественной оценки обусловленной связыванием лиганда активации FXR проводили следующим образом: фрагмент кДНК, кодирующий лигандсвязывающий домен FXR, клонировали в вектор pCMV-BD(Stratagene) в виде гибрида с ДНК-связывающим доменом транскрипционного фактора GAL4 дрожжей под контролем CMV-промотора. Аминокислоты, составляющие границы лигандсвязывающего домена,- 25024843 представляли собой аминокислоты 187-472 из файла базы данных NM005123 (RefSeq). В качестве репортерной плазмиды использовали плазмиду pFR-Luc (Stratagene), содержащую синтетический промотор с пятью тандемными повторами дрожжи сайтов связывания GAL4 дрожжей, контролирующий экспрессию гена люциферазы Photinus pyralis (американского светлячка) в качестве репортерного гена. В целях повышения точности эксперимента клетки котрансфицировали плазмидой pRL-CMV (Promega). pRLCMV содержит конститутивный CMV-промотор, контролирующий экспрессию люциферазы Renilla reniformis. Все анализы с применением репортерного гена Gal4 проводили на клетках HEK293 (полученных от DSMZ, Braunschweig, Germany), которые выращивали в культуральной среде MEM (модифицированная среда Игла) с L-глутамином и сбалансированным солевым раствором (BSS) Эрла с добавлением 10% фетальной бычьей сыворотки, 0,1 мМ заменимых аминокислот, 1 мМ пирувата натрия и 100 единиц/мл пенициллина/стрептавидина при 37 С в атмосфере с 5% СО 2. Среда и добавки были приобретены в Invitrogen. Для анализа клетки высевали в 96-луночные планшеты в концентрации 5105 клеток/лунка в 100 мкл на лунку среды MEM без фенолового красного и L-глутамина и с BSS Эрла с добавкой 10% смеси уголь/декстран, обработанной ФБС (Hyclone, South Logan, Utah), 0,1 мМ заменимых аминокислот, 2 мМ глутамина, 1 мМ пирувата натрия и 100 единиц/мл пенициллина/стрептавидина, инкубировали при 37 С в атмосфере с 5% СО 2. На следующий день клетки характеризовались слиянием 90%. Среду удаляли и клетки подвергали временной трансфекции тремя вышеописанными плазмидами с применением реагента для трансфекции OptiMEM на основе полиэтиленимина (OptiMEM, Invitrogen; Polyethyleneimine,по каталогу Aldrich 40,827-7) в количестве 20 мкл на лунку. Через 2-4 ч после добавления трансфицирующей смеси добавляли MEM того же состава, который использовали для посева клеток. Затем добавляли стоковые растворы соединений, предварительно разбавленные в MEM (конечная концентрация переносящей среды не превышала 0,1%). Клетки дополнительно инкубировали в течение 16 ч перед последовательным измерением активности люциферазы светлячка и Renilla в одном и том же клеточном экстракте с применением системы для анализа люциферазной активности Dual Light (Dyer et al., Anal. Biochem. 2000, 282, 158-161). Все эксперименты проводили в трех повторностях. Для оценки способности типовых соединений выступать в качестве агонистов FXR определяли диапазоны эффективных концентраций в анализе М 1 Н, которые представлены ниже в табл. 2 (А = ЕС 50 25 нМ; В = 25EC50100 нМ; С = ЕС 50100 нМ). Таблица 2 Анализ растворимости в воде. Растворимость в воде в среде ФСБ, рН 7,4 определяли следующим образом. 10 мМ стоковый раствор соединения в ДМСО добавляли в ФСБ (рН 7,4) до достижения теоретической конечной концентрации 200 мкМ. Полученный раствор/суспензию встряхивали при 1250 об/мин в течение 1 ч и затем оставляли в темноте при кт на 23 ч. По истечение этого периода времени все осадки отделяли от раствора посредством центрифугирования при 3900 об/мин в течение 30 мин. Растворимость в воде определяли путем сравнения площади основного пика калибровочного стандарта (200 мкМ) в органическом растворителе (метанол/вода 60:40, об/об) с площадью соответствующего пика образца в буфере. В качестве метода детектирования применяли ВЭЖХ-УФ/ВИД при 230 нм. Анализ проникающей способности с параллельным применением искусственных мембран(РАМРА). Для анализа РАМРА готовили стоковые растворы исследуемых соединений в ДМСО с концентрацией 5 мМ. Стоковые растворы эталонных соединений с концентрацией 5 мМ готовили в EtOH (карбамазепин, гуанабенз) или в EtOH:Н 2 О 1:1 (об/об) (цефтриаксон) соответственно. Соединения разбавляли ФСБ (рН 7,4) для получения исходных растворов, содержащих 5% соответствующего органического растворителя и 250 мкМ эталонных соединений или 10 мкМ исследуемых соединений соответственно. Для анализа использовали модифицированную методику РАМРА, описанную Kansy et al. (J. Med. Chem. 1998, 41, 1007). Эталонные соединения с низкой (цефтриаксон), средней (гуанабенз) и высокой (карбамазепин) проникающей способностью были включены в качестве внутренних контролей. Эксперименты по определению проникающей способности проводили в 96-луночном планшетеMultiscreen (донор), покрытой 96-луночным планшетом с фильтром Multiscreen Immobilon (акцептор). Гидрофобный материал фильтра планшета Immobilon предварительно смачивали 70% этанолом и обрабатывали раствором липидов (лецитина, растворенного в додекане). Донорный планшет заполняли исследуемыми соединениями и эталонными соединениями, и оба планшета вставляли друг в друга и помещали на орбитальный шейкер на 15 мин при 100 об/мин. Исследование переноса начинали путем внесения 150 мкл буфера ФСБ, содержащего исследуемые и эталонные соединения, в донорный планшет. Че- 26024843 рез 15-16 ч диффузии при кт собирали содержимое акцепторного и донорного планшетов и количественно оценивали с помощью детектирования ЖХ/МС (исследуемые соединения) или посредством УФспектроскопии с использованием спектрофотометра Spectramax Plus384 (Molecular Devices) (эталонные соединения). Максимумы поглощения для эталонных соединений цефтриаксона, гуанабенза и карбамазепина составляли 240, 270 и 286 нм соответственно. Восстановленные образцы готовили, как описано для образцов для анализа проникающей способности, и инкубировали в типовых флаконах в течение периода процесса проникновения в тех же условиях. Для анализа исследуемых соединений методом ЖХ/МС 100 мкл инкубируемых растворов отбирали из акцепторных и донорных компартментов и обрабатывали для осаждения ацетонитрилом (ACN), как описано ниже. Кроме того, образцы исследуемых соединений из липидного слоя экстрагировали путем промывания каждой лунки два раза 150 мкл ЭА. Растворы собирали в реакционные пробирки объемом 1,5 мл и растворитель упаривали. Высушенные остатки ресуспендировали в смеси ФСБ/ДМСО/ACN,отражающей состав акцепторных и донорных образцов (т.е. 100 мкл буфера с добавкой 5% ДМСО, 200 мкл ACN + внутренний стандарт (ВС. Конечное содержание растворителя в каждом образце составляло 66% ACN. Образцы из донорных и акцепторных компартментов и калибровочные стандарты осаждали добавлением 200 мкл ACN/BC или 400 мкл ACN/BC соответственно. После интенсивного встряхивания (10 с) и центрифугирования (5 мин при 4800g, кт) не содержащие частиц супернатанты подвергали анализу методом ЖХ-МС/МС. Мембранные компартменты экстрагировали, как описано выше. После восстановления образцы интенсивно встряхивали (10 с) и осаждали (5 мин при 4800g, кт). Не содержащие частиц супернатанты подвергали анализу методом ЖХ-МС/МС. Для анализа соединений согласно настоящему изобретению использовали систему для ВЭЖХ, состоящую из насоса Accela U-HPLC и автоматического дозатора Accela (Thermo Fisher Scientific, USA). Масс-спектрометрический анализ выполняли с помощью масс-спектрометра Exactive (технология с орбитальной ионной ловушкой и точным определением массы), снабженного подогреваемой поверхностью контакта для ионизации электрораспылением (H-ESI2) (Thermo Fisher Scientific, USA), подключенного к ПК со стандартным программным обеспечением Xcalibur 2.1. ЖХ выполняли в режиме градиента (табл. 3) с применением ACN/0,1% муравьиной кислоты в качестве органической фазы (А) и 10 мМ формиата аммония/0,1% муравьиной кислоты в качестве водной фазы (В); и скорость создаваемого насосом потока устанавливали на 500 мкл/мин. Разделение проводили на аналитической колонке Gemini C6-Phenyl, 3 мкм, 502,0 мм (Phenomenex, Germany) с предколонкой В качестве файла для настройки МС использовали стандартный файл настройки для всех анализируемых соединений с примением режима положительных или отрицательных ионов. В качестве фиксированной массы для внутренней калибровки масс использовали значение [М+Н]+ иона диизооктилфталата (m/z 391,28429), повсеместно присутствующего в системе растворителей. Анализируемое соединение обнаруживали путем сканирования в диапазоне 1 Томсон около ожидаемой массы моноизотопного иона [М+Н]+ или [М-Н]-. Разрешающая способность орбитальной ионной ловушки по массам была установлена на величину 50000. Точную массу каждого анализируемого соединения применяли для интегрирования пиков. Дополнительные настройки приборов были следующими: главный контроллер - газ выключен, автоматическая регулировка усиления (АРУ) - высокий динамический диапазон, макс, время впрыска в ловушку 100 мс, защитный газ 30, вспомогательный газ 8, продувочный газ 2, напряжение на источнике ионизации 4 кВ, температура капилляра 250 С, температура нагревателя ESI2 250 С. Цель настоящего изобретения заключалась в создании агонистов FXR с улучшенными физикохимическими свойствами по сравнению с соединениями, завленными в WO 2011/020615. Она была достигнута путем введения полярной гидроксильной группы в 1,3-циклобутилиденовую или 1,3 азетидинилиденовую группы, заменяющие прежнее 1,2-циклопропилиденовое кольцо Неожиданно было обнаружено, что полученные соединения сохраняли свою активность по отношению к рецептору FXR, но демонстрировали улучшенные физико-химические свойства, такие как более высокая растворимость в воде и/или мембранная проницаемость. Прямое сравнение соответствующих соединений из двух серий представлено в табл. 4. Поток (%) = (с акцепторная лунка)/(с донорная лунка + с акцепторная лунка)1002 н.о. = не определено В каждом случае либо растворимость в воде, либо мембранная проницаемость по данным РАМРА,либо обе характеристики значительно были улучшены путем введения гидроксициклобутильного или гидроксиазетидильного фрагмента. Как и большинство молекул, активных по отношению к ядерным рецепторам, агонисты FXR, как правило, являются очень липофильными (М.L. Crawley, Expert Opin.Ther. Patents 2010, 20, 1047). Таким образом, лучшая растворимость в воде и мембранная проницаемость подразумевают более высокую биодоступность при пероральном введении и в общем случае лучшую пригодность для клинической разработки этих соединений в качестве лекарственных средств (L. Huang,J. Dong, S. Karki in Evaluation of drug candidates for preclinical development (Eds. С. Han, С.В. Davis, В. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, соответствующее следующей формуле (1), его энантиомер, диастереомер или фармацевтически приемлемая соль где R выбран из группы, состоящей из COOR6, CONR7R8, тетразолила, SO2NR7R8, SO2-C1-6 алкила, при этом R6 независимо выбран из группы, состоящей из Н или С 1-6 алкила, a R7 и R8 независимо друг от друга выбраны из группы, состоящей из Н, C1-6 алкилен-R9, SO2-C1-6 алкила, где R9 выбран из группы, состоящей из СООН и SO3H; А выбран из группы, состоящей из фенила, пиридила, пиразолила, индазолила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из О-С 1-6 алкила, С 1-6 алкила;Q выбран из группы, состоящей из фенила, который возможно замещен одной или двумя группами,независимо выбранными из группы, состоящей из галогена; 4. Соединение по любому из пп.1-3, отличающееся тем, что Z представляет собой 5. Соединение по любому из пп.1-4, выбранное из
МПК / Метки
МПК: A61K 31/42, A61P 3/10, C07D 261/08, A61P 1/16, A61P 35/00, C07D 413/14, C07D 413/12
Метки: nr1h4, x-рецептор, соединения, активность, модулирующие, связывающие, фарнезоидный
Код ссылки
<a href="https://eas.patents.su/30-24843-soedineniya-svyazyvayushhie-farnezoidnyjj-x-receptor-nr1h4-i-moduliruyushhie-ego-aktivnost.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения, связывающие фарнезоидный x-рецептор (nr1h4) и модулирующие его активность</a>
Новые соединения, связывающие фарнезоидный х-рецептор (fxr) (nr1h4) и модулирующие его активность

Номер патента: 24083
Опубликовано: 31.08.2016
Авторы: Кинцель Олаф, Абель Ульрих, Штеенек Кристоф, Кремозер Клаус
МПК: A61K 31/443, A61K 31/42, A61K 31/4192...
Метки: новые, модулирующие, х-рецептор, fxr, активность, фарнезоидный, соединения, связывающие, nr1h4
Формула / Реферат:
1. Соединение согласно формуле (1), его энантиомер, диастереомер, таутомер, сольват или фармацевтически приемлемая соль:в которой R выбран из группы, состоящей из COOR6, CONR7R8, тетразолила или Н, где R6 независимо выбран из группы, состоящей из Н или C1-C6-алкила, и R7 и R8 независимо друг от друга выбраны из группы, состоящей из Н, C1-C6-алкила, C1-C6-галоалкила, C1-C6-алкилена-R9, SO2-C1-C6-алкила, R9 выбран из группы, состоящей из COOH, OH...
Димерные тромбопоэтиновые пептидные миметики, связывающие мpl рецептор и имеющие тромбопоэтичеcкую активность

Номер патента: 3998
Опубликовано: 25.12.2003
Авторы: Читхэм Дженет, Фейдже Улрих, Лиу Чуан-Фа
МПК: C07K 14/52, A61K 38/19, A61P 7/06...
Метки: активность, рецептор, димерные, имеющие, пептидные, тромбопоэтиновые, тромбопоэтичеcкую, связывающие, миметики
Формула / Реферат:
1. Соединение, которое связывается с рецептором mpl, имеющее структуру TMP1(L1)n-TMP2, где TMP1 и TMP2, каждый независимо друг от друга, выбран из группы коровых соединений, имеющих структуру X2 - X3 - X4 - X5 - X6 - X7 - X8 - X9 - X10, причем X2 выбран из группы, включающей Glu, Asp, Lys и Val; X3 выбран из группы, включающей Gly и Ala; X4 представляет собой Pro; X5 выбран из группы, включающей Thr и Ser; X6 выбран из группы, включающей...
Соединения, модулирующие активность ppar&gamma

Номер патента: 4887
Опубликовано: 26.08.2004
Авторы: Хагивара Ацуси, Рубенштейн Стивен М., Синкаи Хисаси, Хауз Джонатан Б., Макджи Лоренс Р., Фурукава Нобору
МПК: C07C 311/21, A61K 31/18, C07D 213/65...
Метки: соединения, активность, модулирующие, ppar&gamma
Формула / Реферат:
1. Соединение формулы где Ar1 представляет собой незамещенный 2-бензотиазолил или замещенный 2-бензотиазолил, имеющий от 1 до 3 заместителей, независимо выбранных из группы, включающей галоген, -OR', -OC(O)R', -NR'R", -SR', -R', -CN, -NO2-, -CO2R', -CONR'R", -C(O)R', -NR"C(O)R', -S(O2)R', -S(O)2NR'R", перфтор(C1-C4)алкокси и перфтор(C1-C4)алкил, где R' и R" являются независимо выбранными из группы, включающей водород,...
Соединения, модулирующие активность ppar, и способы их получения

Номер патента: 9518
Опубликовано: 28.02.2008
Авторы: Браттон Лэрри Дон, Филзен Гари Фредерик, Аненгст Пол Чарльз, Гейер Эндрю Джордж, Ауэрбах Брюс Джеффри, Триведи Бхарат Калидас
МПК: A61K 31/19, C07C 317/22, C07C 323/19...
Метки: соединения, способы, модулирующие, ppar, активность, получения
Формула / Реферат:
1. Соединение, выбранное из [4-(бифенил-4-илметилсульфанил)-5-метокси-2-метилфенокси]уксусной кислоты; [5-метокси-2-метил-4-(4'-трифторметилбифенил-4-илметилсульфанил)фенокси]уксусной кислоты; [4-(2',4'-дихлорбифенил-4-илметилсульфанил)-5-метокси-2-метилфенокси]уксусной кислоты; [5-метокси-2-метил-4-(3'-трифторметилбифенил-4-илметилсульфанил)фенокси]уксусной кислоты; [4-(4'-фторбифенил-4-илметилсульфанил-5-метокси-2-метилфенокси]уксусной...
Соединения, модулирующие активность toll-подобных рецепторов

Номер патента: 19768
Опубликовано: 30.06.2014
Авторы: Чэнь Майкл, Коттэм Ховард Б., Карсон Деннис А., Хайяши Томоко
МПК: C07D 473/02, A61K 31/52, A61P 29/00...
Метки: модулирующие, соединения, активность, toll-подобных, рецепторов
Формула / Реферат:
1. Соединение, имеющее структуру согласно формуле Iили его фармацевтически приемлемая соль, включая его гидрат,где X представляет собой N или CR2;R представляет собой -OR1, -SR1 или -NRaRb,X1 представляет собой связь или -О-, -S- или -NRc-;Rc представляет собой водород, C1-C10-алкил или замещенный C1-C10-алкил или Rc и R1 вместе с атомом азота могут образовывать гетероциклическое кольцо или замещенное гетероциклическое кольцо;каждый R1...
Предыдущий патент: Соединения в качестве модуляторов протеинкиназы pi3k
Следующий патент: Иммуноконъюгат и его применение для лечения рака
Случайный патент: Cамофиксирующийся механизм