Селективный модулятор рецепторов эстрогена
Номер патента: 20742
Опубликовано: 30.01.2015
Авторы: Кларк Кристиан Александер, Ричардсон Тимоти Иво, Хаммел Конрад Уилсон, Джоунз Скотт Алан, Льюис Джордж Сал, Додж Джеффри Алан, Хинклин Рональд Джей







Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль,
где R1 выбран из группы, состоящей из Н, -C1-C4-алкила, F, Cl, -CN, -C(O)R3, -(С1-С3-алкил)ОН, -ОСН3,
-S(O)2R4, -S(O)CH3, -CF3 и -S(C1-C3-алкил);
R2 выбран из группы, состоящей из Н, F и СН3;
R3 выбран из группы, состоящей из ОН, -ОСН3, СН3, -N(CH3)2;
R4 выбран из группы, состоящей из -C1-C4-алкила, -N(CH3)2 и -CF3; и
R5 представляет собой Н.
2. Соединение по п.1 формулы

или его фармацевтически приемлемая соль,
где R1 выбран из группы, состоящей из Н, -C1-C4-алкила, F, Cl, -CN, -C(O)R3, -(C1-С3-алкил)ОН, -ОСН3,
-S(O)2R4, -S(O)CH3 и -S(C1-С3-алкил);
R2 выбран из группы, состоящей из Н, F и СН3;
R3 выбран из группы, состоящей из ОН, -ОСН3, СН3, -N(CH3)2;
R4 выбран из группы, состоящей из -C1-C4-алкила, -N(CH3)2 и -CF3; и
R5 представляет собой Н.
3. Соединение по любому из пп.1 или 2, где R2 выбран из Н и СН3.
4. Соединение по п.3, где R2 представляет собой Н.
5. Соединение по любому из пп.1-4, где R1 выбран из группы, состоящей из Н, C1-C4-алкила, F, Cl, CF3, -CN, -C(O)R3, -S(O)2R4, -S(O)CH3 и -SCH3.
6. Соединение по п.5, где R1 выбран из группы, состоящей из Н, -СН3, -СН2СН3, -СН(СН3)2, -СН2СН(СН3)2, F, Cl, -CF3, -CN, -С(О)СН3, -C(O)N(CH3)2, -S(O)2CH(CH3)2, -S(O)2CH2CH3, -S(O)2CH3, -S(O)CH3 и -SCH3.
7. Соединение по п.6, где R1 представляет собой -СН3.
8. Соединение по любому из пп.1-7, представляющее собой соединение формулы

или его фармацевтически приемлемая соль.
9. Соединение по любому из пп.1-7, представляющее собой соединение формулы

или его фармацевтически приемлемая соль.
10. Соединение по п.1, представляющее собой соединение формулы

или его фармацевтически приемлемая соль.
11. Соединение по любому из пп.1-10, где указанное соединение представляет собой фармацевтически приемлемую соль.
12. Соединение по п.11, где соль представляет собой гидрохлорид.
13. Фармацевтическая композиция, содержащая по меньшей мере одно соединение по любому из пп.1-12 и фармацевтически приемлемый носитель.
14. Способ лечения остеоартрита у млекопитающего, включающий введение млекопитающему соединения по любому из пп.1-12.
15. Применение соединения по любому из пп.1-12 в качестве лекарственного средства для лечения остеоартрита.
16. Применение соединения по любому из пп.1-12 для лечения остеоартрита.
17. Соединение формулы

или его фармацевтически приемлемая соль.
18. Соединение по п.17, представляющее собой гидрохлоридную соль.
Текст
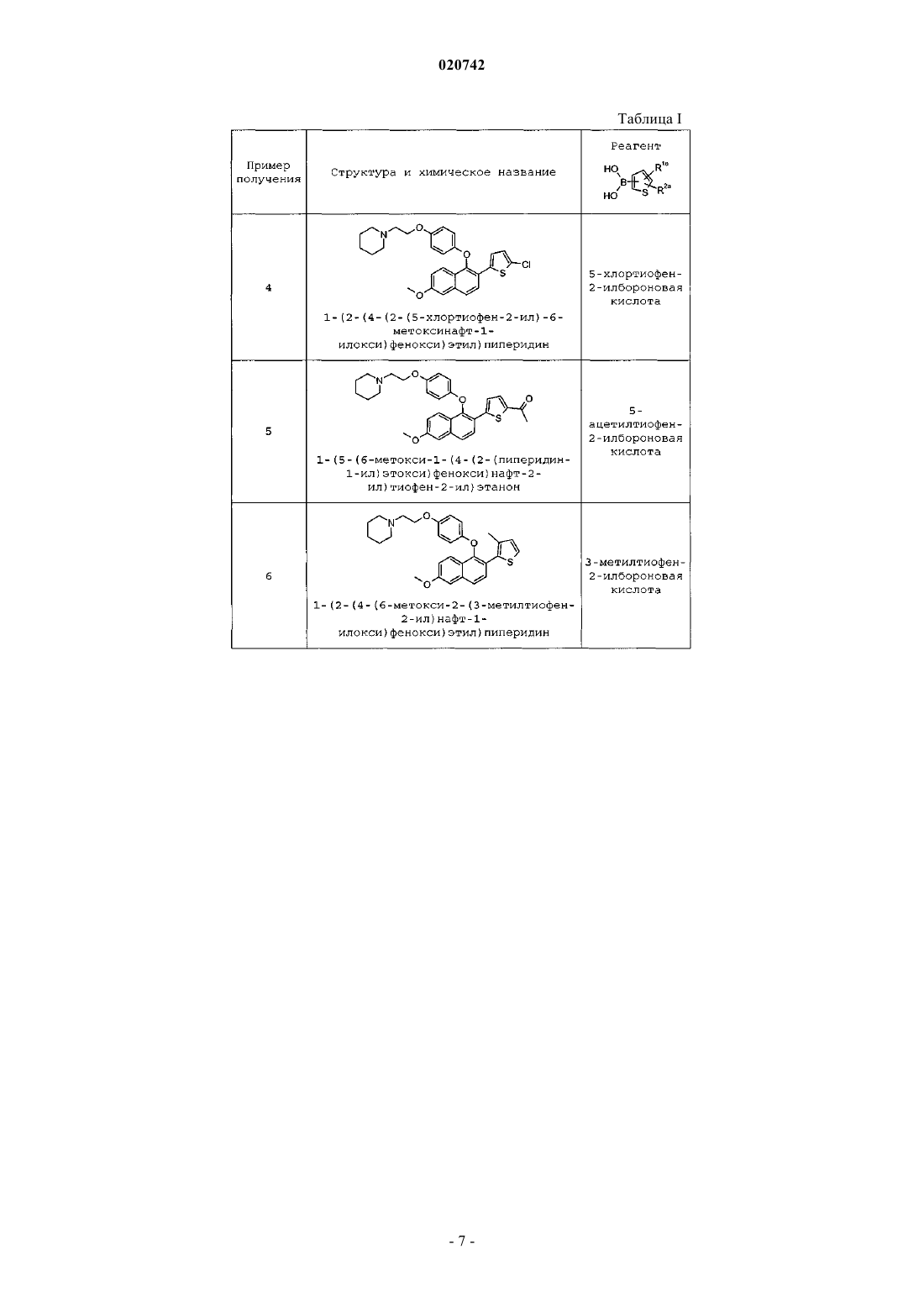
СЕЛЕКТИВНЫЙ МОДУЛЯТОР РЕЦЕПТОРОВ ЭСТРОГЕНА или его фармацевтически приемлемой соли, а также к соединению формулы (I) или его фармацевтически приемлемой соли, представляющим собой селективные модуляторы рецепторов эстрогена, которые полезны при лечении остеоартрита.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Большинство коммерчески доступных терапевтических средств для лечения остеоартрита ("ОА") направлены на уменьшение воспаления и облегчение боли, связанных с ОА. Одобренные способы лечения ОА могут представлять собой инвазивные способы, терять эффективность при длительном применении и могут подходить не для всех пациентов. Таким образом, требуются новые способы лечения пациентов, страдающих ОА. Сообщалось о преклинических исследованиях эстрогенов и различных селективных модуляторов рецепторов эстрогена (SERM) и выявленной активности в отношении подавления заболевания в моделях остеоартрита суставов. Специалистам известно множество молекул SERM. Было обнаружено, что многие известные SERM обладают агонистической активностью по отношению к эстрогену в кости; тем не менее, в настоящее время ни один из SERM не одобрен для лечения ОА. Селективная агонистическая или антагонистическая активность молекулы SERM в различных тканях может приводить к изменению показателей безопасности и эффективности SERM. Например, молекулы SERM, обладающие агонистической активностью в матке, могут быть связаны с нежелательным вагинальным кровотечением. МолекулыSERM могут обладать селективной агонистической и/или антагонистической активностью, например, в кости, груди и/или матке. Заявитель полагает, что соединения, являющиеся SERM, проявляющие выраженную антагонистическую активность в матке и при этом обладающие агонистической активностьюSERM в кости, могут быть особенно подходящими для применения для лечения ОА. Соединения SERM,оказывающие положительное воздействие на проявления или симптомы ОА и при этом обладающие приемлемыми показателями безопасности, могли бы стать особенно подходящей дополнительной терапевтической возможностью. В патенте США 5728724 ("патент '724") предложены соединения SERM, имеющие структуру [2(2-тиенил)-6-гидроксибензотиен-3-ил][4-[2-(1-пиперидинил)этокси]фенил]метанона. При этом указано,что эти соединения подходят для лечения остеопороза, постменопаузального синдрома, эндометриоза и ряда других состояний, связанных с активностью SERM. Тем не менее, в патенте не сообщается, что указанные соединения SERM подходят для лечения ОА. Кроме того, молекулы SERM согласно патенту '724 по своей структуре отличны от соединений, предложенных в настоящем изобретении. В отличие от патента '724 согласно настоящему изобретению необходимо соединение, содержащее линкерную оксигруппу, присоединенную к нафталиновому остову молекулы. В настоящем изобретении предложено новое высокоактивное соединение, являющееся SERM. При этом указанная молекула SERM проявляет селективный антагонизм по отношению к эстрогену в матке с обеспечением фармакологических характеристик, особенно желательных при применении для лечения ОА. Кроме того, указанный селективный SERM может обеспечивать желаемые показатели безопасности для применения для лечения ОА. В настоящем изобретении предложены соединения формулы (I) или их фармацевтически приемлемая соль,где R1 выбран из группы, состоящей из Н, -C1-C4-алкила, F, Cl, -CN, -C(O)R3, -(С 1-С 3-алкил)ОН,-ОСН 3, -S(O)2R4, -S(O)CH3, -CF3 И -S(C1-C3-алкил);R5 представляет собой Н. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемая соль,где R1 выбран из группы, состоящей из Н, -C1-C4-алкила, F, Cl, -CN, -C(O)R3, -(C1-C3-алкил)ОН,-ОСН 3, -S(O)2R4, -S(O)CH3 и -S(C1-С 3-алкил);R5 представляет собой Н. В другом варианте реализации настоящего изобретения предложены соединения формулы (I), где В другом варианте реализации настоящего изобретения предложены соединения формулы (I), гдеR2 представляет собой Н. В другом варианте реализации настоящего изобретения предложены соединения формулы (I), гдеR1 выбран из группы, состоящей из Н, С 1-С 4-алкила, F, Cl, CF3, -CN, -C(O)R3, -S(O)2R4, -S(O)CH3 и -SCH3. В следующем варианте реализации настоящего изобретения предложены соединения формулы (I),где R1 выбран из группы, состоящей из Н, -СН 3, -СН 2 СН 3, -СН(СН 3)2, -СН 2 СН (СН 3)2, F, Cl, -CF3, -CN,-С(О)СН 3, -C(O)N(CH3)2, -S(О)2 СН (СН 3)2, -S(О)2 СН 2 СН 3, -S(O)2CH3, -S(O)CH3 и -SCH3. В другом варианте реализации настоящего изобретения предложены соединения формулы (I), гдеR1 представляет собой -СН 3. В другом варианте реализации настоящего изобретения предложены соединения формулы (I), которые представляют собой 2-тиофен или его фармацевтически приемлемую соль. В другом варианте реализации настоящего изобретения предложены соединения формулы (I), которые представляют собой 3-тиофен или его фармацевтически приемлемую соль. В настоящем изобретении предложено также соединение формулы (А) или его фармацевтически приемлемая соль. Предпочтительно соединение формулы (А) представляет собой гидрохлоридную соль. В следующем варианте реализации настоящего изобретения соединение формулы (I) представляет собой фармацевтически приемлемую соль. Предпочтительно соль представляет собой гидрохлорид. Согласно другому варианту реализации настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В другом варианте реализации настоящего изобретения предложен способ лечения остеоартрита у млекопитающего, включающий введение млекопитающему соединения согласно настоящему изобретению. Кроме того, настоящее изобретение относится к применению соединения, предложенного в настоящем изобретении, в качестве лекарственного средства для лечения остеоартрита. Кроме того, настоящее изобретение относится к применению соединения, предложенного в настоящем изобретении, для лечения остеоартрита."Фармацевтически приемлемая соль" относится к солям соединений согласно настоящему изобретению, которые рассматривают в качестве приемлемых для применения в клинических целях и/или ветеринарии. Указанные соли могут быть получены по способам, известным специалисту в данной области техники. Фармацевтически приемлемые соли и общая методология их получения хорошо известны в данной области техники; см., например, работу П. Сталя с соавт. (P. Stahl et al.), HANDBOOK OFPHARMACEUTICAL SALTS: PROPERTIES, SELECTION AND USE, (VCHA/Wiley-VCH, 2002); С.М. Берджа с соавт. (S.M. Berge, et al.), "Pharmaceutical Salts," Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977. Соединения согласно настоящему изобретению предпочтительно получают в виде фармацевтических композиций, вводимых различными способами. Термин "фармацевтически приемлемый носитель" означает, что носитель, разбавитель, эксципиенты и соль являются фармацевтически совместимыми с другими ингредиентами композиции. Более предпочтительно указанные композиции подходят для перорального введения. Фармацевтически приемлемые композиции и способы их получения хорошо известны в данной области техники; см., например, REMINGTON: THE SCIENCE AND PRACTICE OF почтительные фармацевтически приемлемые соли включают гидрохлорид, метансульфонат и метилбензосульфонат. Гидрохлоридная и метилбензосульфонатная соли являются особенно предпочтительными фармацевтически приемлемыми солями. Остеоартрит (далее "ОА") представляет собой хроническое дегенеративное заболевание суставов. Симптомы ОА представляют собой, например, сильную боль, ограниченную подвижность и утрату работоспособности пораженных суставов. До настоящего времени применение ралоксифена, являющегося известным SERM, для лечения ОА изучали в ходе двух исследований. Первое исследование представляло собой проспективное плацебонеконтролируемое обсервационное исследование, в котором было показано, что применение ралоксифена значительно снижало частоту сообщений о боли в различных частях скелета, уменьшало употребление анальгетиков и улучшало качество сна. Во втором исследовании лечение ралоксифеном приводило к небольшому, но значимому улучшению клинического исхода (15% снижению индекса WOMAC) после одного года лечения. Также было показано, что как ралоксифен, так и другой SERM, левормелоксифен,приводили к значительному снижению уровней CTX-II, являющегося маркером хрящевого обмена коллагена II типа, в моче. Необходимы новые соединения SERM, проявляющие особенную селективность,которые можно применять для лечения ОА. Соединения согласно настоящему изобретению могут быть получены с применением способов,представленных на схеме А и описанных в примерах. Названия соединений в примерах получения и примерах даны с помощью ChemDraw Ultra version 10.0 и Symyx Draw Version 3.1. Термины и сокращения, используемые на схемах, в примерах получения, примерах и методиках в настоящем описании имеют свои обычные значения, если не указано иное. Например, в настоящем описании следующие термины имеют указанные значения: "NBS" означает N-бромсукцинимид, "BnBr" означает бензилбромид, "Cs2CO3" означает карбонат цезия, "CuOTf" означает трифлат меди, "Tf2O" означает ангидрид трифторметансульфокислоты, "Pd(OH)2/C" означает Pd(OH)2 на угле, "Pd(Ac)2" означает ацетат палладия(II), "PCy3" означает трихлоргексилфосфин, "ACN" означает ацетонитрил, "CsF" означает фторид цезия, "BBr3" означает бромид бора, "ДМФ" означает диметилформамид, "nBuLi" означает нбутиллитий, "ТГФ" означает тетрагидрофуран, "Et3SiH" означает триэтилсилан, "TFA" означает трифторуксусную кислоту, "POCl3" означает фосфорилхлорид, "MgSO4" означает сульфат магния, "NH4OH" означает гидроксид аммония, "Na2SO4" означает сульфат натрия, "Pd(dppf)Cl2" означает хлорид (1,1-бис(дифенилфосфино)ферроцен)палладия(II), "dppf" означает 1,1-бис-(дифенилфосфино)ферроцен, "КОАс" означает ацетат калия, "DCM" означает дихлорметан, "DIPEA" означает диизопропилэтиламин, "TFAA" означает ангидрид 2,2,2-трифторуксусной кислоты, "n-BuLi" означает н-бутиллитий, "mCPBA" означает мета-хлорпербензойную кислоту, "Pd(PPh3)4" означает тетракис(трифенилфосфин)палладий, "TEA" означает триэтиламин, "ДМАА" означает N,N-диметилацетамид, "TBAI" означает йодид тетра-Dбутиламмония, "ДМАП" означает 4-диметиламинопиридин, "NaClO2" означает хлорит натрия, "DDQ" означает 2,3-дихлор-5,6-дицианобензохинон, "ДМСО" означает диметилсульфон, "SCX" означает сильный катионный обмен, "LRMS" означает масс-спектрометрию низкого разрешения, "ДСК" означает дифференциальную сканирующую калориметрию, а "ТП" означает температуру плавления. К диметилформамиду (ДМФ, 250 мл) при температуре окружающей среды добавляют 6 метоксинафт-2-ол (20 г, 114,8 ммоль) с последующим добавлением N-бромсукцинимида (NBS, 21,5 г,120 ммоль) через 30 мин. Через 45 мин разбавляют водой (800 мл), собирают и сушат осадок с получением 25,5 г (87%) 1-бром-6-метоксинафт-2-ола. К ДМФ (800 мл) добавляют 1-бром-6-метоксинафт-2-ол (66,7 г, 264 ммоль), карбонат калия (K2CO3,40,0 г, 290 ммоль) и бензилбромид (49,6 г, 290 ммоль). Перемешивают смесь при температуре окружающей среды в течение 1 ч. Для осаждения продукта добавляют воду (400 мл). Собирают осадок и промывают осадок на фильтре гептаном(3125 мл), затем сушат с получением 2-бензилокси-1-бром-6 метоксинафталина (83,7 г, 98,9 ммоль). Смешивают толуол (200 мл), 2-бензилокси-1-бром-6-метоксинафталин (30 г, 87,4 ммоль), 4-(2 пиперидин-1-илэтокси)фенол (23,2 г, 105 ммоль) и карбонат цезия (34,4 г, 105 ммоль). Смесь кипятят с обратным холодильником. Удаляют часть толуола (100 мл) отгонкой. К реакционной смеси добавляют этилацетат (390 мг, 4,37 ммоль) и бензольный комплекс трифлата меди (2,20 г, 4,37 ммоль) и перемешивают в течение 5 мин. Удаляют растворитель перегонкой и нагревают полученный остаток до 174 С в течение 1,5 ч. Растворяют остаток в смеси этилацетата (200 мл) и водного раствора HCl (1 н., 90 мл). Выделяют и концентрируют органический раствор с получением остатка. Очищают остаток хроматографией на силикагеле с получением 1-2-[4-(2-бензилокси-6-метоксинафт-1-илокси)фенокси]этилпиперидина (12,4 г, 26,2 ммоль). В смесь метанол/этилацетат (1:1, 490 мл) добавляют 1-2-[4-(2-бензилокси-6-метоксинафт-1 илокси)фенокси]этилпиперидин (12,4 г, 25,5 моль) и нагревают для получения раствора. Удаляют источник нагрева и добавляют формиат аммония (4,83 г, 76,6 ммоль) и Pd(OH)2 на угле (20 мас.%, 1,58 г,1,12 ммоль). Кипятят раствор с обратным холодильником в течение 50 мин, затем фильтруют. Концентрируют фильтрат с получением 6-метокси-1-[4-(2-пиперидин-1-илэтокси)фенокси]нафт-2-ола (9,9 г,25,1 ммоль). Охлаждают дихлорметан (290 мл), триэтиламин (3,08 г, 30,4 ммоль) и 6-метокси-1-[4-(2-пиперидин 1-илэтокси)фенокси]нафт-2-ол (9,2 г, 23,4 ммоль) до -50 С и добавляют ангидрид трифторметансульфоновой кислоты (7,26 г, 25,7 ммоль). Перемешивают полученную смесь при -50 С в течение 2 ч, затем оставляют смесь для нагревания до температуры окружающей среды с последующим дополнительным перемешиванием в течение 1 ч. Добавляют солевой раствор (150 мл) и затем отделяют органический рас-4 020742 твор. Промывают органический раствор насыщенным водным раствором NaHCO3, затем сушат перед концентрированием с получением остатка. Кристаллизуют остаток с помощью смеси диэтиловый эфиргексан с получением указанного в заголовке соединения (11,2 г, 21,27 ммоль). LRMS (m/z): 526 (М+1). Пример получения 2. Синтез гидрохлорида 1-(2-(4-(6-метокси-2-(5-метилтиофен-2-ил)нафт-1 илокси)фенокси)этил)пиперидина. Суспендируют трифторметансульфонат 6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2 ила (398,8 г, 758,8 ммоль), 5-метил-2-тиофенбороновую кислоту (224 г, 1,58 моль) и фторид цезия (300 г,1,97 моль) в ацетонитриле (12 л). Дегазируют полученную суспензию через газоотводную трубку в течение 30 мин при нагревании до 50 С. Обрабатывают смесь трициклогексилфосфином (8 г, 28,5 ммоль) и дегазируют в течение 10 мин, затем обрабатывают ацетатом палладия(II) (4 г, 17,8 ммоль), дополнительно дегазируют в течение 5 мин и оставляют перемешиваться на ночь при 80 С. Добавляют трициклогексилфосфин (8 г, 28,5 ммоль) и ацетат палладия(II) (4 г, 17,8 ммоль) и дополнительно нагревают смесь в течение 8 ч, затем оставляют раствор на ночь для медленного охлаждения. Фильтруют охлажденный раствор через большую пластину из стеклокерамики и концентрируют фильтрат. Суспендируют остаток и осадок на фильтре в воде (4 л) и этилацетате (8 л), затем переносят в делительную воронку и обрабатывают насыщенным водным раствором бикарбоната натрия (300 г, 3,57 моль). Тщательно перемешивают в течение 10 мин, затем разделяют слои. Промывают органический слой насыщенным водным раствором бикарбоната натрия, солевым раствором и сушат над MgSO4 в течение ночи. Фильтруют смесь и обрабатывают фильтрат HCl (4 н. в диоксане, 200 мл, 800 ммоль). Перемешивают полученную суспензию в течение 60 мин, затем фильтруют с получением указанного в заголовке соединения (352 г, 690,06 ммоль). Готовят раствор гидрохлорида 1-(2-(4-(6-метокси-2-(5-метилтиофен-2-ил)нафт-1 илокси)фенокси)этил)пиперидина (346, 9 г, 680,07 ммоль) в дихлорметане (12 л) и охлаждают до 3 С. Обрабатывают полученный раствор трибромидом бора (1 М в дихлорметане, 301,4 мл, 3,19 моль) через канюлю в течение 20 мин. Перемешивают полученную смесь при 0 С в течение 90 мин. Выливают смесь на колотый лед и обрабатывают порциями бикарбоната натрия (1 кг, 11,90 моль) в течение 30 мин. Фильтруют полученную суспензию с получением темного осадка. Переносят фильтрат в делительную воронку. Разделяют растворы и концентрируют органический раствор. Объединяют остаток с осадком на фильтре и суспендируют в 20% растворе изопропанола в хлороформе (6 л) и перемешивают с водой (4 л), содержащей 200 г бикарбоната натрия, в течение 3 ч. Разделяют слои и промывают органический слой солевым раствором, сушат (MgSO4), фильтруют и выпаривают с получением 324 г грязно-белого твердого вещества. Очищают полученное твердое вещество с помощью хроматографии на силикагеле с получением 281 г бледно-желтого порошка. Перемешивают твердое вещество в тетрагидрофуране (11 л,135,18 моль) и обрабатывают S-триамином (446 г, 2,05 моль). После перемешивания в течение ночи фильтруют через целит и концентрируют фильтрат с получением белого порошка. Перемешивают порошок в тетрагидрофуране (11 л, 135,18 моль) и снова обрабатывают S-триамином (357 г, 1,64 моль). После перемешивания в течение выходных фильтруют через целит и концентрируют фильтрат с получением белого порошка. Суспендируют полученный белый порошок в метаноле (5 л) при комнатной температуре. В отдельном сосуде к метанолу (2 л) добавляют за один прием HCl (12 н. в воде, 85 мл, 1,04 моль). Добавляют указанный раствор к суспензии и перемешивают в течение 1 ч. Охлаждают смесь до 2 С и перемешивают в течение 1 ч. Фильтруют полученную смесь с получением порошка кремового цвета. Сушат полученный порошок в вакууме при 85 С в течение ночи с получением указанного в заголовке соединения (258 г, 520,0 ммоль). LRMS (m/z): 460 (M+1-HCl). ТП (ДСК)=248,82 С. Пример получения 66. Синтез 6-(5-метилтиофен-2-ил)-5-(4-(2-(пиперидин-2 ил)этокси)фенокси)нафт-2-ола. Растворяют гидрохлорид 1-(2-(4-(6-метокси-2-(5-метилтиофен-2-ил)нафт-1-илокси)фенокси)этил)пиперидина (5,15 г, 10,1 ммоль) в дихлорметане (340 мл). Охлаждают полученный раствор до 0 С. В отдельном сосуде в атмосфере азота смешивают чистый трибромид бора (3,9 мл, 40,4 ммоль) и дихлорметан (36,5 мл). Добавляют полученный раствор трибромида бора к охлажденному раствору. Перемешивают полученную смесь при 0 С в течение 2,5 ч. Гасят смесь насыщенным водным раствором бикарбоната натрия и оставляют для нагревания до комнатной температуры. Разделяют слои и экстрагируют водный слой 20% раствором метанола в дихлорметане (10 мл 3). Концентрируют объединенные органические растворы с получением твердого вещества. Суспендируют твердое вещество в метаноле(234 мл) и добавляют HCl (1 н. в воде, 10,8 мл). Помещают полученный раствор на ионообменную колон-5 020742 ку с кислым SCX. Промывают колонку 40% раствором метанола в дихлорметане, затем элюируют целевое вещество 40% раствором 7 М раствора аммиака в метаноле в дихлорметане. Концентрируют аммиаксодержащий элюент с получением указанного в заголовке соединения (4,54 г, 9,88 моль). LRMS (m/z): 460 (М+1). Пример получения 67. Синтез метансульфоната 6-(5-метилтиофен-2-ил)-5-(4-(2-(пиперидин-1 ил)этокси)фенокси)нафт-2-ола. Помещают 6-(5-метилтиофен-2-ил)-5-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ол (105,7 мкг,0,23 ммоль) в пробирку. Добавляют этилацетат (2 мл) и метансульфокислоту (18 мкл). Нагревают полученную смесь до 60 С при перемешивании. К полученному ярко-оранжевому раствору с небольшим количеством темно-коричневого смолистого осадка добавляют тетрагидрофуран (1 мл). Обрабатывают полученный раствор ультразвуком в течение 15 мин. Осторожно отделяют суспензию от смолистого осадка, образовавшегося на стенках сосуда, декантированием. Осторожно собирают полученное твердое вещество посредством вакуумного фильтрования и промывают полученный осадок пентаном (22 мл). Сушат полученное твердое вещество при 40 С в вакуум-сушильном шкафу в течение ночи с получением указанного в заголовке соединения. ТП (ДСК)=161,90 С. Пример получения 68. Синтез 4-метилбензолсульфоната 6-(5-метилтиофен-2-ил)-5-(4-(2(пиперидин-1-ил)этокси)фенокси)нафт-2-ола. Помещают 6-(5-метилтиофен-2-ил)-5-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ол (103,4 мг,0,22 ммоль) в пробирку. Добавляют этилацетат (4 мл) и п-толуолсульфокислоту (52 мг, 0,30 ммоль) и перемешивают при 60 С. Добавляют тетрагидрофуран до получения прозрачного раствора и обрабатывают полученный раствор ультразвуком в течение 15 мин. Осторожно отделяют суспензию от смолистого осадка, образовавшегося на стенках сосуда, декантированием. Фильтруют суспензию с получением осадка в виде светло-розового твердого вещества. Промывают осадок пентаном (22 мл) и сушат при 40 С в вакуум-сушильном шкафу в течение ночи с получением указанного в заголовке соединения. ТП Согласно схеме В необязательно замещенную тиофенбороновую кислоту подвергают сочетанию с соединениями формулы 1 с образованием соединений формулы 2, где R1a выбран из группы, состоящей из Н, -C1-C4-алкила, F, Cl, -CN и -C(O)R3, a R2a выбран из группы, состоящей из Н, F и СН 3. Предпочтительно R1a представляет собой Н, -Cl, -С(О)СН 3, СН 3, CN, a R2a представляет собой Н или СН 3. Пример получения 3. 1-(2-(4-(6-Метокси-2-(тиофен-2-ил)нафт-1-илокси)фенокси)этил)пиперидин Смешивают трифторметансульфонат 6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2 ила (6,0 г, 11,4 ммоль), тиофен-2-бороновую кислоту (4,4 г, 34,3 ммоль), трициклогексилфосфин (1,1 г,4,0 ммоль), ацетат палладия(II) (0,51 г, 2,3 ммоль), фторид цезия (15,6 г, 102,8 ммоль) и ацетонитрил (150 мл, дегазированный азотом в течение 30 мин) в круглодонной колбе в атмосфере азота. Нагревают полученную смесь до 80 С и перемешивают в течение 40 мин. Отфильтровывают твердое вещество и промывают ацетонитрилом. Объединяют фильтрат и промывные воды и концентрируют в вакууме. Растворяют остаток в метаноле и помещают на ионообменную колонку с кислым SCX. Промывают колонку метанолом, затем элюируют 2 М раствором аммиака в метаноле. Концентрируют аммиаксодержащий элюент в вакууме с получением светло-коричневого твердого вещества. Очищают твердое вещество посредством флэш-хроматографии на силикагеле с получением указанного в заголовке соединения (3,88 г, 8,4 ммоль). 1 Н ЯМР (d6-ДМСО)1,29 (дд, J=5,9, 11,2 Гц, 2 Н), 1,43-1,37 (м, 5 Н), 2,32 (с, 4 Н), 2,52 (т, J=5,9 Гц, 2 Н),3,87 (т, J=6,1 Гц, 2 Н), 6,62-6,60 (м, 2 Н), 6,76-6,74 (м, 2 Н), 7,09-7,03 (м, 2 Н), 7,37 (д, J=2,6 Гц, 1 Н), 7,487,47 (м, 1 Н), 7,62-7,59 (м, 2 Н), 7,76 (д, J=8,8 Гц, 1 Н), 7,96 (д, J=8,8 Гц, 1 Н). Соединения примеров получения, представленные в табл. I, могут быть получены, по существу, в соответствии с описанием примера получения 3 с применением реагента (столбец 3), который используют вместо тиофен-2-бороновой кислоты. Согласно схеме С соединения формулы 3 сначала превращают в литийорганическое соединение, а затем добавляют к электрофилу ([Е+]) с образованием соединений формулы 4, где Е представляет собой 1-(2-(4-(2-(5-Фтортиофен-2-ил)-6-метоксинафт-1 В пробирку с закручивающейся крышкой в атмосфере аргона добавляют 1-(2-(4-(6-метокси-2(тиофен-2-ил)нафт-1-илокси)фенокси)этил)пиперидин (153 мг, 0,4 ммоль) и тетрагидрофуран (3,3 мл). Охлаждают полученный раствор до 78 С. По каплям добавляют н-бутиллитий (1,6 М раствор в гексане,230 мкл, 0,4 ммоль). Нагревают полученный раствор до 0 С и перемешивают в течение 30 мин. Добавляют раствор N-фторбензолсульфонимида (210,0 мг, 0,6 ммоль) в тетрагидрофуране (500 мкл). Оставляют полученную смесь нагреваться до комнатной температуры и перемешивают в течение 2 ч. Добавляют 1 М раствор HCl, разбавляют диэтиловым эфиром и пропускают через ионообменную колонку с кислымSCX. Промывают колонку метанолом, затем элюируют целевое вещество 2 М раствором аммиака в метаноле. Концентрируют аммиаксодержащий элюент с получением желтого твердого вещества. Очищают желтое твердое вещество с помощью флэш-хроматографии на силикагеле с получением указанного в заголовке соединения (59,0 мг, 0,1 ммоль): масс-спектрометрия (m/z): 478 (М+1). Соединения примеров получения, представленные в табл. II, могут быть получены, по существу, в соответствии с описанием примера получения 13 с применением реагента (столбец 3), который используют вместо N-фторбензолсульфонимида. Таблица II Согласно схеме D соединения формулы 5 окисляют двумя эквивалентами пероксимоносульфата сульфата калия с образованием соединений формулы 6, где R4a представляет собой -СН 3, -СН 2 СН 3 или В круглодонную колбу в атмосфере азота добавляют 1-(2-(4-(2-(5-(этилтио)тиофен-2-ил)-6 метоксинафт-1-илокси)фенокси)этил)пиперидин (226 мг, 0,43 ммоль), тетрагидрофуран (8,7 мл) и метанол (8,7 мл). Охлаждают полученный раствор до 0 С и добавляют раствор пероксимоносульфата сульфата калия (535 мг, 0,87 ммоль) в воде (4,5 мл). Перемешивают полученную смесь при 0 С в течение 30 мин. Оставляют смесь для нагревания до комнатной температуры и перемешивают в течение 1 ч. Помещают смесь на ионообменную колонку с кислым SCX. Промывают колонку смесью 1:1 метанола и дихлорметана, затем элюируют смесью 1:1 2 М раствора аммиака в метаноле и дихлорметана. Концентрируют аммиаксодержащий элюент в вакууме с получением грязно-белой пены. Растворяют пену в дихлорметане и обрабатывают HCl (1 М раствор в диэтиловом эфире, 900 мкл). Концентрируют полученную смесь в вакууме с получением указанного в заголовке соединения (142 мг, 0,24 ммоль): массспектрометрия (m/z): 552 (М+1-HCl). Соединения примеров получения, представленные в табл. III, могут быть получены, по существу, в соответствии с описанием примера получения 22 с применением реагента (столбец 3), который используют вместо 1-(2-(4-(2-(5-(этилтио)тиофен-2-ил)-6-метоксинафт-1-илокси)фенокси)этил)пиперидина. Таблица III Согласно схеме Е соединение формулы 7 окисляют одним эквивалентом пероксимоносульфата сульфата калия с образованием соединения формулы 8. Пример получения 25. 1-(2-(4-(6-Метокси-2-(5-(метилсульфинил)тиофен-2-ил)нафт-1 илокси)фенокси)этил)пиперидин В круглодонную колбу в атмосфере азота добавляют 1-(2-(4-(6-метокси-2-(5(метилсульфинил)тиофен-2-ил)нафт-1-илокси)фенокси)этил)пиперидин (84 мг, 0,17 ммоль), тетрагидрофуран (3,3 мл) и метанол (3,3 мл). Охлаждают полученный раствор до 0 С и добавляют раствор перок- 11020742 симоносульфата сульфата калия (102 мг, 0,17 ммоль) в воде (1,7 мл). Перемешивают полученную смесь при 0 С в течение 30 мин. Помещают смесь на ионообменную колонку с кислым SCX. Промывают колонку 1:1 смесью метанол/дихлорметан и элюируют 1:1 смесью 2 М раствор аммиака в метаноле/дихлорметан. Концентрируют аммиаксодержащий элюент в вакууме с получением указанного в заголовке соединения (87 мг, 0,17 ммоль): масс-спектрометрия (m/z): 522 (М+1). Схема F Согласно схеме F соединения формулы 9 восстанавливают до соединений формулы 10. Пример получения 26. 1-(2-(4-(2-(5-Изопропилтиофен-2-ил)-6-метоксинафт-1 илокси)фенокси)этил)пиперидин В колбу в атмосфере аргона добавляют 2-(5-(6-метокси-1-(4-(2-(пиперидин-1 ил)этокси)фенокси)нафт-2-ил)тиофен-2-ил)пропан-2-ол (84 мг, 0,16 ммоль), триэтилсилан (1,7 мл) и дихлорметан (0,85 мл). Добавляют трифторуксусную кислоту (0,81 мл) и перемешивают полученную смесь в течение 45 мин. Помещают смесь на ионообменную колонку с кислым SCX. Промывают колонку смесью 1:2 метанол/дихлорметан и элюируют смесью 1:2 2 М раствор аммиака в метаноле/дихлорметан. Концентрируют аммиаксодержащий элюент в вакууме с получением указанного в заголовке соединения Согласно схеме G соединение формулы 11 сначала превращают в хлорангидрид, затем подвергают взаимодействию с гидроксидом аммония с получением амида соединения формулы 12. Соединение формулы 12 затем дегидратируют, а затем образуется гидрохлоридная соль, давая соединение формулы 13. Пример получения 27. 5-(6-Метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-2 карбоксамид В круглодонную колбу в атмосфере азота добавляют 5-(6-метокси-1-(4-(2-(пиперидин-1 ил)этокси)фенокси)нафт-2-ил)тиофен-2-карбоновую кислоту (86 мг, 0,2 ммоль) и дихлорметан (2,4 мл). Охлаждают суспензию до 0 С и последовательно добавляют оксалилхлорид (22,2 мкл, 0,3 ммоль) и диметилформамид (30,0 мкл). Нагревают полученную смесь до комнатной температуры и перемешивают в течение 30 мин. Концентрируют в вакууме с получением желтого твердого вещества. К твердому веществу добавляют тетрагидрофуран (2,5 мл). К полученной суспензии добавляют раствор аммиака (12,1 М в воде, 3 мл, 36,4 ммоль) в тетрагидрофуране (5,7 мл). Перемешивают полученный раствор в течение 30 мин, затем разбавляют диэтиловым эфиром. Разделяют слои и сушат органический слой (Na2SO4), фильтруют и концентрируют в вакууме с получением желтовато-коричневого твердого вещества. Растворяют полученное твердое вещество в метаноле и помещают на ионообменную колонку с кислым SCX. Промывают колонку метанолом, затем элюируют целевое вещество 2 М раствором аммиака в метаноле. Концентрируют аммиаксодержащий элюент с получением указанного в заголовке соединения (79 мг,0,16 ммоль). 1 Н ЯМР (d6-ДМСО)1,32-1,27 (м, 2 Н), 1,44-1,38 (м, 4 Н), 2,33 (с, 4 Н), 2,53 (т, J=5,9 Гц, 2 Н),- 12020742 В круглодонную колбу в атмосфере азота добавляют 5-(6-метокси-1-(4-(2-(пиперидин-1 ил)этокси)фенокси)нафт-2-ил)тиофен-2-карбоксамид (79 мг, 0,16 ммоль) и фосфорилхлорид (13 мл, 139,9 ммоль). Нагревают полученную смесь до 100 С и перемешивают в течение 15 мин. Охлаждают смесь до комнатной температуры и концентрируют в вакууме с получением желтого остатка. Осторожно гасят полученный остаток метанолом и помещают на ионообменную колонку с кислым SCX. Промывают колонку метанолом, затем элюируют целевое вещество 2 М раствором аммиака в метаноле. Концентрируют аммиаксодержащий элюент в вакууме с получением бледно-желтого твердого вещества. Растворяют полученное твердое вещество в дихлорметане и обрабатывают HCl (1 M раствор в диэтиловом эфире, 5 мл). Концентрируют в вакууме с получением указанного в заголовке соединения (81,5 мг, 0,16 ммоль). 1 Н ЯМР (d6-ДМСО)0,82-0,73 (м, 1 Н), 1,33-1,23 (м, 4 Н), 2,88-2,80 (м, 2 Н), 3,36-3,31 (м, 5 Н), 3,81 (с, 3 Н),4,23-4,19 (м, 2 Н), 6,73-6,63 (м, 2 Н), 6,87-6,77 (м, 2 Н), 7,15-7,09 (м, 1 Н), 7,41-7,36 (м, 1 Н), 7,62-7,54 (м,1 Н), 7,87-7,77 (м, 3 Н), 8,09-8,05 (м, 1 Н), 10,58-10,45 (м, 1 Н). Схема Н Удаляют защитную(ые) группу(группы) из соединений формулы 14 с образованием гидрохлоридной соли соединений формулы (I). Пример 5. Гидрохлорид 5-(4-(2-(пиперидин-1-ил)этокси)фенокси)-6-(тиофен-2-ил)нафт-2-ола Растворяют 1-(2-(4-(6-метокси-2-(тиофен-2-ил)нафт-1-илокси)фенокси)этил)пиперидин (100 мг,0,22 ммоль) в дихлорметане и обрабатывают HCl (1 М раствор в диэтиловом эфире, 220 мкл, 0,22 ммоль). Концентрируют в вакууме с получением желтого твердого вещества и добавляют дихлорметан (7,3 мл). Охлаждают полученный раствор до 0 С и добавляют трибромид бора (1 М раствор в дихлорметане, 870 мкл, 0,87 ммоль). Перемешивают полученную смесь при 0 С в течение 2,5 ч. Гасят смесь насыщенным водным раствором бикарбоната натрия и оставляют для нагревания до комнатной температуры. Разделяют слои и экстрагируют водный слой 20% раствором метанола в дихлорметане (10 мл 3). Осушают объединенные органические слои (Na2SO4), фильтруют и концентрируют в вакууме с получением желтого осадка. Очищают остаток посредством флэш-хроматографии на силикагеле с получением грязнобелой пены. Суспендируют пену в ACN и добавляют HCl (5 М раствор в воде, 1 мл). Замораживают полученный раствор и лиофилизируют с получением указанного в заголовке соединения (50,5 мг, 0,12 ммоль): масс-спектрометрия (m/z): 446(М+1-HCl). Соединения примеров в табл. IV могут быть получены, по существу, путем удаления защитной(ых) группы(групп) в соответствии с примером 5. В круглодонную колбу добавляют 3-бром-4-метилтиофен (0,885 г, 5,00 ммоль), хлорид (1,1'-бис(дифенилфосфино)ферроцен)палладия(II) (408 мг, 499,82 мкмоль), 1,1'-бис-(дифенилфосфино)ферроцен(277,09 мг, 499,82 мкмоль), бис-(пинаколато)дибор (2,54 г, 10 ммоль), ацетат калия (1,47 г, 14,99 ммоль) и 1,4-диоксан (50 мл). Продувают реакционный сосуд аргоном. Перемешивают смесь при 85 С в течение 16 ч. Охлаждают смесь до комнатной температуры. Фильтруют смесь и промывают твердое вещество 1,4-диоксаном. Концентрируют фильтрат. Очищают полученный остаток с помощью флэшхроматографии на силикагеле с получением указанного в заголовке соединения (0,62 г, 2,5 ммоль). Соединения примеров получения в табл. V могут быть получены, по существу, в соответствии с описанием примера получения 29 с применением реагента (столбец 3), используемого вместо 3-бром-4 метилтиофена. Таблица V В круглодонную колбу добавляют ацетонитрил (10 мл), 6-метокси-1-(4-(2-(пиперидин-1 ил)этокси)фенокси)нафт-2-илтрифторметансульфонат (150 мг, 285,4 мкмоль), 4-(4,4,5,5-тетраметил 1,3,2-диоксаборолан-2-ил)тиофен-3-карбонитрил (134,2 мг, 570,8 мкмоль), трициклогексилфосфин (24,0 мг, 85,6 мкмоль), ацетат палладия(II) (12,8 мг, 57,1 мкмоль) и фторид цезия (390,2 мг, 2,6 ммоль). Трижды продувают реакционный сосуд аргоном. Перемешивают смесь при 80 С в течение 1,5 ч. Фильтруют смесь и промывают твердое вещество ацетонитрилом. Концентрируют фильтрат. Очищают полученный остаток ионообменной смолой DOWEX с получением указанного в заголовке соединения (87 мг, 179,8 мкмоль). Соединения примеров получения в табл. VI могут быть получены, по существу, в соответствии с описанием примера получения 31 с применением реагента (столбец 3), используемого вместо 4-(4,4,5,5 тетраметил-1,3,2-диоксаборолан-2-ил)тиофен-3-карбонитрила. Добавляют раствор хлороводорода в диэтиловом эфире (1 М, 180 мкл, 180,0 мкмоль) к приготовленному раствору 4-(6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-3 карбонитрила (87 мг, 17 9,5 мкмоль) в дихлорметане (2 мл). Перемешивают смесь в течение 10 мин. Концентрируют смесь с получением указанного в заголовке соединения (95 мг, 179 мкмоль). Соединения примеров получения в табл. VII могут быть получены, по существу, в соответствии с описанием примера получения 35 с применением реагента (столбец 3), используемого вместо 4-(6 метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-3-карбонитрила. Таблица VII Медленно добавляют трибромид бора в дихлорметане (4 М раствор, 182 мкл, 728,0 мкмоль) к раствору гидрохлорида 4-(6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-3 карбонитрила (95 мг, 182,3 мкмоль) и дихлорметана (4 мл), перемешиваемому при 0 С. Перемешивают смесь при 0 С в течение 1,5 ч. Гасят смесь водным раствором бикарбоната натрия. Трижды экстрагируют смесь 20% метанол/дихлорметан. Промывают объединенные органические слои водой. Сушат полученный раствор над сульфатом натрия, фильтруют и концентрируют досуха. Очищают остаток с помощью флэш-хроматографии на силикагеле с получением указанного в заголовке соединения (52 мг, 111,2 мкмоль). Соединения примеров получения в табл. VIII могут быть получены, по существу, в соответствии с описанием примера получения 39 с применением реагента (колонка 3), используемого вместо гидрохлорида 4-(6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-3-карбонитрила. Таблица VIII Альтернативный синтез соединения согласно примеру 14. Гидрохлорид 4-(6-гидрокси-1-(4-(2(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-3-карбонитрила Добавляют раствор хлороводорода в диэтиловом эфире (1 М, 122 мкл, 122,0 ммоль) к раствору 4-(6 гидрокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-3-карбонитрила (52 мг, 110,5 мкмоль) и дихлорметана (4 мл). Обрабатывают смесь ультразвуком в течение 5 мин, а затем концентрируют с получением указанного в заголовке соединения (57 мг, 110,5 мкмоль). МС (m/z): 471 (М+1-HCl). Соединения примеров в табл. IX могут быть получены, по существу, в соответствии с описанием альтернативного способа синтеза соединения согласно примеру 14 с применением реагента (столбец 3),используемого вместо 4-(6-гидрокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-3 карбонитрила. К раствору 2,3-диметилтиофена (0,55 г, 4,90 ммоль) в дихлорметане (20 мл) добавляют Nбромсукцинимид (0,96 мг, 5,39 ммоль). Перемешивают реакционную смесь в течение ночи. Концентрируют смесь. Очищают остаток с помощью флэш-хроматографии на силикагеле с получением указанного в заголовке соединения (704 мг, 3,68 ммоль). Пример получения 44. 2-Бром-3,5-диметилтиофен. Соединение примера получения 44 может быть получено, по существу, в соответствии с описанием примера получения 43 с применением в качестве реагента 2,4-диметилтиофена. Схема K При перемешивании к раствору 2-бромтиофен-3-карбоновой кислоты (500 мг, 2,41 ммоль) и толуола (20 мл) добавляют тионилхлорид (0,35 мл, 4,80 ммоль). Продувают реакционный сосуд азотом. Смесь кипятят с обратным холодильником и перемешивают в течение 3 ч. Концентрируют смесь с получением указанного в заголовке соединения (540 мг, 2,41 ммоль). Применяют соединение в следующей процедуре без дополнительной очистки. Пример получения 46. 5-Бромтиофен-3-карбонилхлорид. Соединение примера получения 46 может быть получено, по существу, в соответствии с описанием примера получения 45 с применением в качестве реагента 5-бромтиофен-3-карбоновой кислоты. К водному раствору аммиака (25%, 3 мл, 17,44 ммоль) при перемешивании добавляют 2 бромтиофен-3-карбонилхлорид (540 мг, 2,39 ммоль). Перемешивают смесь в течение 30 мин. Концентрируют смесь. Собирают полученный осадок посредством фильтрования и промывают твердое вещество водой. Сушат белый осадок в вакууме с получением указанного в заголовке соединения (450 мг, 2,17 ммоль). Пример получения 48. 5-Бромтиофен-3-карбоксамид. Соединение примера получения 48 может быть получено, по существу, в соответствии с примером получения 47 с применением в качестве реагента 5-бромтиофен-3-карбонилхлорида. Пример получения 49. 2-Бромтиофен-3-карбонитрил К раствору 2-бромтиофен-3-карбоксамида (200 мг, 970,58 мкмоль) и триэтиламина (0,34 мл, 2,44 ммоль) в ТГФ (10 мл) при перемешивании при 5 С с помощью шприца добавляют ангидрид 2,2,2 трифторуксусной кислоты. Убирают охлаждающую баню, нагревают смесь до комнатной температуры и перемешивают в течение 16 ч. Концентрируют смесь. Добавляют воду и дихлорметан. Трижды экстрагируют смесь дихлорметаном. Сушат объединенные экстракты над сульфатом натрия, фильтруют и концентрируют досуха. Очищают остаток с помощью флэш-хроматографии на силикагеле с получением указанного в заголовке соединения (113 мг, 601,76 мкмоль). Пример получения 50. 5-Бромтиофен-3-карбонитрил. Соединение примера получения 50 может быть получено, по существу, в соответствии с примером получения 49 с применением в качестве реагента 5-бромтиофен-3-карбоксамида. Схема L(2,62 г, 4,99 ммоль) и дихлорметана (20 мл) добавляют раствор хлороводорода в диэтиловом эфире (1,0 М, 5,5 мл, 5,50 ммоль). Перемешивают смесь в течение 10 мин и затем концентрируют с получением указанного в заголовке соединения (2,8 г, 4,99 ммоль). Пример получения 52. 6-Гидрокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2 илтрифторметансульфонат. К раствору гидрохлорида 6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2 илтрифторметансульфоната (2,8 г, 4,98 ммоль) и дихлорметана (100 мл) добавляют раствор трибромида бора в дихлорметане (4 М, 5 мл, 20,00 ммоль). Перемешивают реакционную смесь в течение 2 ч, а затем гасят водным раствором бикарбоната натрия. Трижды экстрагируют смесь дихлорметаном. Объединяют экстракты, сушат над сульфатом натрия, фильтруют и концентрируют с получением указанного в заголовке соединения (2,4 г, 4,68 ммоль). Пример получения 53. 5-(4-(2-(Пиперидин-1-ил)этокси)фенокси)-6-(трифторметилсульфонилокси)нафт-2-илацетат. К раствору 6-гидрокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-илтрифторметансульфоната(2,4 г, 4,69 ммоль) и диизопропилэтиламина (2,45 мл, 14,05 ммоль) добавляют ацетилхлорид (0,67 мл,9,41 ммоль). Перемешивают смесь в течение 1 ч, а затем гасят водным раствором бикарбоната натрия. Трижды экстрагируют смесь дихлорметаном. Объединяют экстракты, сушат над сульфатом натрия,фильтруют и концентрируют с получением указанного в заголовке соединения (2,33 г, 4,22 ммоль). Пример получения 54. Гидрохлорид 6-гидрокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2 илбороновой кислоты В круглодонную колбу добавляют бис-(неопентилгликолято)дибор (4,2 г, 18,59 ммоль), 5-(4-(2(пиперидин-1-ил)этокси)фенокси)-6-(трифторметилсульфонилокси)нафт-2-илацетат (2,33 г, 4,21 моль),ацетат палладия(II) (0,199 г, 886,38 мкмоль), фторид цезия (5,21 г, 34,30 ммоль), трициклогексилфосфин(0,41 г, 1,40 ммоль) и ацетонитрил (50 мл). Смесь кипятят с обратным холодильником при перемешивании в течение 1 ч в атмосфере азота. Охлаждают смесь, фильтруют и промывают твердое вещество ацетонитрилом. Концентрируют объединенные фильтраты и промывные воды. Суспендируют полученный остаток в диэтиловом эфире (40 мл) и обрабатывают ультразвуком в течение 30 мин. Удаляют осадок фильтрованием и концентрируют фильтрат с получением неочищенного промежуточного продукта. К раствору неочищенного промежуточного продукта в диэтиловом эфире (50 мл) добавляют диэтаноламин(405,25 мкл, 4,21 ммоль). Перемешивают смесь в течение 1 ч. Декантируют органический слой и растворяют оставшийся остаток в метаноле (20 мл) и 10 мл воды. Добавляют концентрированную HCl и перемешивают полученную смесь в течение 16 ч. Концентрируют смесь для удаления МеОН. Трижды экстрагируют водный остаток дихлорметаном. Объединяют экстракты, сушат над сульфатом натрия, фильтруют и концентрируют досуха. Очищают остаток с помощью флэш-хроматографии на силикагеле с получением указанного в заголовке соединения (0,6 г, 1,47 ммоль). Пример получения 55. 3-(6-Гидрокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-2 карбонитрил К раствору гидрохлорида 6-гидрокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-илбороновой кислоты (100 мг, 245,5 мкмоль) в этаноле (2 мл) и ацетонитриле (8 мл) добавляют 3-бромтиофен-2 карбонитрил (93 мг, 494,6 мкмоль), ацетат палладия(II) (12 мг, 53,4 мкмоль), трициклогексилфосфин (21 мг, 74,9 мкмоль), фторид цезия (336 мг, 2,2 ммоль). Трижды продувают реакционный сосуд азотом. Перемешивают смесь при 85 С в течение 2 ч. Охлаждают и фильтруют смесь. Промывают полученное твердое вещество ацетонитрилом. Концентрируют объединенные фильтрат и промывные воды. Очищают остаток на ионообменной DOWEX смоле с получением неочищенного продукта. Очищают неочищенный продукт с помощью препаративной ВЭЖХ с получением указанного в заголовке соединения (12 мг, 24,55 мкмоль). Соединения примеров получения в табл. X могут быть получены, по существу, в соответствии с примером получения 55 с применением реагента (столбец 3), используемого вместо 3-бромтиофен-2 карбонитрила. К раствору 3-(6-гидрокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)нафт-2-ил)тиофен-2 карбонитрила (12 мг, 24,5 мкмоль) и дихлорметана (4 мл) добавляют раствор хлороводорода в диэтиловом эфире (1 М, 28 мкл, 28,0 мкмоль). Обрабатывают смесь ультразвуком в течение 5 мин, а затем концентрируют с получением указанного в заголовке соединения (13 мг, 24,5 мкмоль). Масс-спектр (m/z): 471 (M+1-HCl). Соединения примеров в табл. XI могут быть получены, по существу, в соответствии с примером 26 с применением реагента (столбец 3), используемого вместо 3-(6-гидрокси-1-(4-(2-(пиперидин-1 ил)этокси)фенокси)нафт-2-ил)тиофен-2-карбонитрила. Таблица XI Пример получения 64. Триметил-(5-метилсульфонил-3-тиенил)станнан. К раствору 2,4-дибромтиофена (7 мл, 62 ммоль) в диэтиловом эфире (240 мл) при -78 С добавляют н-бутиллитий (1,6 М раствор в гексане, 39 мл, 62 ммоль). Через 0,5 ч добавляют н-бутиллитий (1,6 М раствор в гексане, 15,6 мл, 25 ммоль) и перемешивают смесь при -78 С в течение дополнительных 15 мин. Добавляют метилдисульфанилметан (6 мл, 68 ммоль) и перемешивают реакционную смесь до нагревания ее до комнатной температуры. Выливают реакционную смесь в смесь льда и насыщенного водного раствора хлорида аммония. Разделяют слои и экстрагируют водный слой диэтиловым эфиром. Объединяют органические слои и промывают насыщенным водным раствором хлорида аммония, водой и солевым раствором, затем сушат сульфатом натрия. Концентрируют полученный раствор с получением 4-бром-2-метилсульфанилтиофена (9,3 г, 44,4 ммоль). Растворяют 4-бром-2-метилсульфанилтиофен (9,3 г, 44,4 ммоль) в дихлорметане (230 мл) и охлаждают до 0 С. В виде 3 порций с 5-минутными интервалами добавляют мета-хлорпербензойную кислоту(28 г, 162 ммоль). Перемешивают полученную смесь до нагревания ее до комнатной температуры. Разбавляют реакционную смесь диэтиловым эфиром и промывают 5% водным раствором сульфита натрия,насыщенным водным раствором бикарбоната натрия и солевым раствором, затем сушат сульфатом натрия. Концентрируют полученный раствор и очищают остаток с помощью флэш-хроматографии (10-20% этилацетат в гексане) с получением 4-бром-2-метилсульфонилтиофена (5 г, 20,73 ммоль). Растворяют 4-бром-2-метилсульфонилтиофен (0,6 г, 2,5 ммоль) и толуоле (18 мл). Добавляют гексаметилдиолово (1,8 мл, 3,8 ммоль) и тетракис(трифенилфосфино)палладий(0) (0,05 г, 0,04 ммоль). Нагревают полученную смесь до 85 С и перемешивают в течение 4 ч. Охлаждают смесь до комнатной температуры и проводят разделение солевым раствором. Концентрируют органический раствор и очищают с помощью флэш-хроматографии (0-10% этилацетат в гексане) с получением триметил-(5 метилсульфонил-3-тиенил)станнана (0,5 г, 1,53 ммоль). Пример получения 65. 1-[2-[4-6-Метокси-2-(5-метилсульфонил-3-тиенил)-1 нафтил]окси]фенокси]этил]пиперидин Смешивают ацетат палладия(II) (0,043 г, 0,19 ммоль) и трициклогексилфосфин (0,081 г, 0,29 ммоль) в ацетонитриле (15 мл) и обрабатывают ультразвуком в течение 10 мин. К суспензии фторида цезия (0,5 г, 3,3 ммоль) в ацетонитриле (40 мл) добавляют смесь, содержащую [6-метокси-1-[4-[2-(1 пиперидил)этокси]фенокси]-2-нафтил]трифторметансульфонат (0,5 г, 0,95 ммоль), триметил-(5 метилсульфонил-3-тиенил)станнан (0,94 г, 2,9 ммоль) и палладий. Нагревают полученную смесь до 90 С и перемешивают в течение 18 ч. Охлаждают смесь до комнатной температуры и концентрируют. Разделяют полученный остаток между этилацетатом и насыщенным водным раствором бикарбоната натрия. Разделяют слои и промывают органический слой насыщенным водным раствором хлорида аммония и солевым раствором, затем сушат сульфатом натрия. Концентрируют полученный раствор и очищают с помощью флэш-хроматографии (0-5% метанола в дихлорметане) с получением указанного в заголовке соединения (0,3 г, 0,55 ммоль). Пример 35. Трифторацетат 6-(5-метилсульфонил-3-тиенил)-5-[4-[2-(1 пиперидил)этокси]фенокси]нафт-2-ола Растворяют 1-[2-[4-6-метокси-2-(5-метилсульфонил-3-тиенил)-1 нафтил]окси]фенокси]этил]пиперидин (0,3 г, 0,55 ммоль) в этилацетате. Добавляют диэтиловый эфир (10 мл) и охлаждают до 0 С. Добавляют соляную кислоту (2 М раствор в диэтиловом эфире, 0,4 мл, 0,84 ммоль) и собирают полученный осадок фильтрованием. Растворяют твердое вещество в этилацетате и концентрируют с получением гидрохлорида 1-[2-[4-6-метокси-2-(5-метилсульфонил-3-тиенил)-1 нафтил]окси]фенокси]этил]пиперидина (0,35 г, 0,55 ммоль). Растворяют гидрохлорид 1-[2-[4-6-метокси-2-(5-метилсульфонил-3-тиенил)-1-нафтил]окси]фенокси]этил]пиперидина (0,35 г, 0,55 ммоль) в дихлорметане (15 мл) и охлаждают до 0 С. К полученному охлажденному раствору добавляют трибромид бора (0,3 мл, 3,1 ммоль). Перемешивают полученную смесь в течение 1 ч при 0 С. Разделяют реакционную смесь на порции между этилацетатом и насыщенным водным раствором бикарбоната натрия. Разделяют слои и экстрагируют водный слой этилацетатом(2). Промывают объединенные органические слои солевым раствором и сушат сульфатом натрия. Концентрируют полученный раствор и очищают с помощью жидкостной хроматографии высокого давления с получением указанного в заголовке соединения (24 мг, 0,04 ммоль). Масс-спектр (m/z): 524 (M+1-TFA). Схема N Согласно схеме N трифторметансульфонатное соединение превращают в соединение бороновой кислоты. Гидрохлоридная соль 6-бороновая кислота-5-[4-(2-пиперидин-1-илэтокси)фенокси]нафт-2-ола может быть получена в соответствии со способом, предложенным в публикации РСТ WO2005073204. Соединение бороновой кислоты превращают в замещенное тиофеновое соединение с применением в качестве реагента 3-бром-2-хлортиофена или 2-бром-5-метилтиофена с применением способа, по существу соответствующего способу, описанному в WO2005073204. Пример 36. Трифторацетат 6-(2-хлор-3-тиенил)-5-[4-[2-(1-пиперидил)этокси]фенокси]нафт-2-ола Трифторацетат 6-(2-хлор-3-тиенил)-5-[4-[2-(1-пиперидил)этокси]фенокси]нафт-2-ола получают, по существу, согласно способу, аналогичному способу получения трифторацетатной соли 5-[4-[2-(пиперид 1-ил)этокси]фенокси]-6-(2,3,4-трифторфенил)нафт-2-ола, приведенному в WO2005073204, с применением 3-бром-2-хлортиофена. Масс-спектр (m/z): 480 (М+1-TFA). Схема О Альтернативный способ синтеза соединения согласно примеру 1 в форме свободного основания Пример получения 69. 1-Хлор-6-метокси-3,4-дигидронафталин-2-карбальдегид. В круглодонную колбу добавляют соединение 1 (на схеме О; 6-метокситетралин-1-он) (6,11 г, 34,7 ммоль) и N,N-диметилацетамид (20 мл, 2 59,5 ммоль). 5 раз продувают реакционный сосуд аргоном. К реакционной смеси по каплям добавляют POCl3 (8 мл, 148,9 ммоль). При перемешивании нагревают реакционную смесь до 105 С и поддерживают эту температуру в течение 4 ч. Гасят реакционную смесь смесью лед-вода. Трижды экстрагируют смесь этилацетатом и отбрасывают водную фазу. Объединяют органические слои и сушат над MgSO4, фильтруют и концентрируют досуха с получением 1-хлор-6 метокси-3,4-дигидронафталин-2-карбальдегида в виде коричневого твердого вещества (5,94 г, 70% выход). Пример получения 70. 6-Метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)-3,4-дигидронафталин 2-карбальдегид. В трехгорлую круглодонную колбу добавляют 1-хлор-6-метокси-3,4-дигидронафталин-2 карбальдегид (2,25 г, 10,1 ммоль), 4-(2-(пиперидин-1-ил)этокси)фенол (1,8 г, 8,24 ммоль), йодид тетра-Nбутиламмония (50 мг, 0,14 ммоль), карбонат калия (4,1 г, 29,8 ммоль) и 4-диметиламинопиридин (120 мг,0,99 ммоль). Продувают реакционный сосуд азотом. К реакционной смеси медленно добавляют диметилформамид (30 мл). Нагревают смесь до 100 С при перемешивании и поддерживают температуру в течение 4 ч. Охлаждают реакционную смесь до комнатной температуры и гасят смесью лед-вода. Трижды экстрагируют смесь этилацетатом. Сушат объединенные органические слои над MgSO4, фильтруют и концентрируют с получением 6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)-3,4-дигидронафталин 2-карбальдегида (4,11 г). Пример получения 71. 6-Метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)-3,4-дигидронафталин 2-карбоновая кислота. В круглодонную колбу добавляют 6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)-3,4 дигидронафталин-2-карбальдегид (3,0 г, 4,34 ммоль), резорцин (531 мг, 4,8 ммоль), ТГФ (8 мл), этанол (8 мл) и уксусную кислоту (0,9 мл). Перемешивают смесь при 25 С в течение 5 мин. К реакционной смеси медленно добавляют раствор хлорита натрия (1,3 г, 11,2 ммоль) в воде (8 мл). Перемешивают смесь при 80 С в течение 2 ч. Гасят реакционную смесь смесью лед-вода. При перемешивании добавляют в сосуд этилацетат. Трижды промывают смесь разбавленным раствором NaOH и отбрасывают органическую фазу. В водный слой добавляют воду до достижения рН, равного 5-6. Водный слой экстрагируют 5 раз этилацетатом и отбрасывают водную фазу. Сушат органический слой над MgSO4, фильтруют и концентрируют досуха, дополнительно очищают с получением 400 мг 6-метокси-1-(4-(2-(пиперидин-1 ил)этокси)фенокси)-3,4-дигидронафталин-2-карбоновой кислоты. 1 Н ЯМР (d-ДМСО, 300 МГц): 9,97 (1 Н,с) 7,12 (1 Н, м), 6,86 (5 Н, м), 6,71 (1 Н, м), 4,27 (2 Н, м), 3,73 (3 Н, с), 4,26 (2 Н, с), 3,41 (3 Н, м), 2,82 (2 Н, м),2,59 (2 Н, м), 1,70 (6 Н, м). Пример получения 72. 1-(2-(4-(2-Бром-6-метокси-3,4-дигидронафт-1-илокси)фенокси)этил)пиперидин. В колбу добавляют 6-метокси-1-(4-(2-(пиперидин-1-ил)этокси)фенокси)-3,4-дигидронафталин-2 карбоновую кислоту (0,4 г, 0,68 ммоль), дихлорметан (10 мл) и триэтиламин (0,2 мл, 1,43 ммоль). Перемешивают смесь при 25 С в течение 10 мин. Порциями добавляют N-бромсукцинимид (0,5 г, 4,13 экв.,2,81 ммоль). Охлаждают реакционную смесь до комнатной температуры и гасят смесью лед-вода. Триж- 28020742 ды экстрагируют смесь этилацетатом и отбрасывают водную фазу. Сушат объединенные органические слои над MgSO4, фильтруют и концентрируют досуха с получением 1-(2-(4-(2-бром-6-метокси-3,4 дигидронафт-1-илокси)фенокси)этил)пиперидина в виде желтого масла (300 мг). Пример получения 73. 1-(2-(4-(2-Бром-6-метоксинафт-1-илокси)фенокси)этил)пиперидин. В круглодонную колбу добавляют 1-(2-(4-(2-бром-6-метокси-3,4-дигидронафт-1 илокси)фенокси)этил)пиперидин (6,7 г, 6,6 ммоль) и ацетонитрил (50 мл). Перемешивают смесь при 25 С в течение 5 мин. К реакционной смеси медленно добавляют 2,3-дихлор-5,6-дицианобензохинон (DDQ)(3,2 г, 14,1 ммоль). Нагревают смесь до 80 С и перемешивают при указанной температуре в течение 14 ч. Охлаждают реакционную смесь до комнатной температуры и гасят смесью лед-вода. Трижды экстрагируют смесь этилацетатом и отбрасывают водную фазу. Сушат объединенные органические слои(MgSO4), фильтруют и концентрируют досуха. Очищают с помощью флэш-хроматографии с получением 1-(2-(4-(2-бром-6-метоксинафт-1-илокси)фенокси)этил)пиперидина в виде коричневого твердого вещества (817 мг). 1H ЯМР (d-ДМСО, 300 МГц): 7,68 (3 Н, м), 7,43 (1 Н, с), 7,15 (1 Н, м), 6,97 (2 Н, д), 6,72 (2 Н, д),4,26 (2 Н, с), 3,85 (3 Н, с), 3,48 (4 Н, м), 2,96 (2 Н, м), 1,70 (6 Н, м). Пример получения 74. 1-(2-(4-(6-Метокси-2-(5-метилтиофен-2-ил)нафт-1-илокси)фенокси)этил)пиперидин. В трехгорлую круглодонную колбу добавляют 1-(2-(4-(2-бром-6-метоксинафт-1 илокси)фенокси)этил)пиперидин (25 мг, 75,9 мкмоль), 5-метилтиофен-2-илбороновую кислоту (20 мг,140,86 мкмоль), тетракис(трифенилфосфин)палладий (20 мг, 17,3 мкмоль) и диметилсульфон (ДМСО) (2 мл). 5 раз продувают реакционную смесь азотом. Нагревают смесь при 100 С в течение 6 ч, а затем при 80 С в течение 40 ч. Пример получения 75. 6-(5-Метилтиофен-2-ил)-5-(4-(2-(пиперидин-2-ил)этокси)фенокси)нафт-2-ол Удаляют защитную(ые) группу(ы) из 1-(2-(4-(6-метокси-2-(5-метилтиофен-2-ил)нафт-1 илокси)фенокси)этил)пиперидина с применением способа, описанного в примере 1. Исследование биологической активности. Анализ связывания рецептора эстрогена. Соединения испытывали на предмет аффинности связывания с обоими типами рецептора эстрогена(ER и ER) с помощью исследования конкурентного связывания, которое является мерой способности соединения замещать 3 Н-эстрадиол из рецепторов. Значения IC50 и Ki могут быть рассчитаны для обоих типов рецепторов. Исследование конкурентного связывания проводили в буферном растворе, содержащем 50 мМHEPES буферный агент, рН 7,5, 1,5 мМ этилендиаминтетрауксусную кислоту (ЭДТА), 150 мМ NaCl,10% глицерин, 1 мг/мл овальбумина и 5 мМ дитиотреитола (DTT) с применением 0,025 мкКи на лунку 3 Н-эстрадиола (NEN NET517 при 118 Ки/ммоль, 1 мкКи/мл), 10 нг/лунка ER или ER рецептора. Анализируемое соединение добавляли в 10 различных концентрациях. Неспецифическое связывание определяли в присутствии 1 мкМ эстрадиола (17-эстрадиола). Реакционную смесь (140 мкл) инкубировали в течение 4 ч при комнатной температуре, затем к каждой реакционной смеси добавляли 70 мкл охлажденного DCC буферного раствора (в расчете на 50 мкл исследуемого буферного раствора DCC буферный раствор содержит 750 мг угля и 250 мг декстрана). Содержимое планшетов перемешивали в течение 8 мин в орбитальном встряхивателе при 4 С. Затем планшеты центрифугировали при 3000 об/мин при 4 С в течение 10 мин. Аликвоту смеси, равную 120 мкл, переносили в другой 96-луночный чистый плоскодонный планшет, и в каждую лунку добавляли 175 мкл сцинтилляцинной жидкости. Планшеты закрывали и интенсивно встряхивали в орбитальном встряхивателе. После инкубирования в течение 2,5 ч планшеты помещали в считывающее устройство. Данные использовали для вычисления IC50 и процент ингибирования при 10 мкМ. Kd 3 Н-эстрадиола определяли как связывание ER или ER рецепторов при насыщении. Значения IC50 анализируемых соединений переводили в значения Ki с применением уравнения Ченга-Прусова, а Kd определяли с помощью исследования связывания при насыщении. Все соединения согласно примерам, описанные в настоящей заявке, демонстрировали активность в ходе исследования связывания, при этом измеренные значения Ki- составляли менее чем 20 нМ для ER рецептора, а значения Ki- составляли менее чем 200 нМ для ER рецептора. Для соединения согласно примеру 1 определяли, что измеренное значение Ki- составляет 0,150,22 нМ (среднее геометрическоестандартное отклонение), при этом измеренная аффинность к ER рецептору составляла Ki-=0,200,20 нМ (среднее геометрическоестандартное отклонение). Таким образом, показано связывание с высокой аффинностью соединения согласно настоящему изобретению с обоими ER рецепторами. Исследование пролиферации клеток Исикавы. В данном исследовании измеряли пролиферацию клеток (с помощью измерения активности щелоч- 29
МПК / Метки
МПК: C07D 333/08, C07D 333/28, C07D 333/38, C07D 333/22, C07D 333/34, A61P 19/00, A61K 31/381
Метки: модулятор, эстрогена, селективный, рецепторов
Код ссылки
<a href="https://eas.patents.su/30-20742-selektivnyjj-modulyator-receptorov-estrogena.html" rel="bookmark" title="База патентов Евразийского Союза">Селективный модулятор рецепторов эстрогена</a>
Селективные модуляторы рецепторов эстрогена для лечения вазомоторных симптомов

Номер патента: 16613
Опубликовано: 30.06.2012
Авторы: Фрэнк Скотт Алан, Додж Джеффри Алан, Уоллас Оуэн Брендан, Дэлли Роберт Дин, Хинклин Рональд Джей, Шеферд Тимоти Алан
МПК: A61K 31/55, A61K 31/4523, A61K 31/4453...
Метки: модуляторы, симптомов, эстрогена, рецепторов, вазомоторных, селективные, лечения
Формула / Реферат:
1. Соединение формулы (I)где m равно 0, 1 или 2;n равно 1, 2, 3 или 4;R представляет собой Н или метил при условии, что если m равно 1 или 2, то R должен представлять собой Н, и если m равно 0, то R должен представлять собой метил;R1 представляет собой Н, SO2(н-С4-С6-алкил) или COR2;X представляет собой О или NR3;X1 представляет собой О, СН2 или С=O;R6 представляет собой Н или F,R2 представляет собой C1-С6-алкил; C1-C6-алкокси; NR5R5a; фенокси...
Селективные модуляторы рецепторов эстрогена для лечения вазомоторных симптомов

Номер патента: 12262
Опубликовано: 28.08.2009
Авторы: Хинклин Рональд Джей, Фрэнк Скотт Алан, Уоллас Оуэн Брендан, Додж Джеффри Алан, Шеферд Тимоти Алан, Дэлли Роберт Дин
МПК: A61K 31/4523, A61K 31/55, A61K 31/4453...
Метки: рецепторов, симптомов, вазомоторных, селективные, модуляторы, эстрогена, лечения
Формула / Реферат:
1. Соединение формулы Ia где m равно 0, 1 или 2; n равно 1, 2, 3 или 4; R представляет собой Н или метил, при условии, что если m равно 1 или 2, то R должен представлять собой Н, и если m равно 0, то R должен представлять собой метил; R1 представляет собой Н, SO2(н-С4-С6-алкил) или COR2; X представляет собой О или NR3; Y представляет собой О, S, SO или NR4; R2 представляет собой C1-C6-алкил, C1-C6-алкокси, NR5R5a, фенокси или фенил,...
Селективный антагонист опиоидных рецепторов каппа

Номер патента: 17484
Опубликовано: 28.12.2012
Авторы: Митч Чарльз Говард, Диас Буэсо Нурия, Маккинзи Дэвид Ли, Педрегал-Терсеро Консепсьон
МПК: A61P 25/32, A61K 31/40, C07D 207/08...
Метки: селективный, антагонист, опиоидных, рецепторов, каппа
Формула / Реферат:
1. 3-Фтор-4-[4-[2-(3,5-диметилфенил)пирролидин-1-илметил]фенокси]бензамид или его фармацевтически приемлемая соль.2. Соединение по п.1, представляющее собой (S)-3-фтор-4-[4-[2-(3,5-диметилфенил)пирролидин-1-илметил]фенокси]бензамид или его фармацевтически приемлемую соль.3. Фармацевтическая композиция, обладающая антагонистической активностью в отношении опиоидного рецептора каппа, содержащая соединение по п.1 или 2 или фармацевтически...
Модулятор рецепторов глюкокортикостероидов и его применение

Номер патента: 14695
Опубликовано: 30.12.2010
Авторы: Карсон Мэттью Уилльям, Коулан Майкл Джозеф
МПК: A61P 19/02, C07D 491/044, A61K 31/4745...
Метки: модулятор, рецепторов, глюкокортикостероидов, применение
Формула / Реферат:
1. Соединение, которое представляет собой (Е)-N-{3-[1-(8-хлор-11Н-10-окса-1-азадибензо[a,d] циклогептен-5-илиден)пропил]фенил}метансульфонамид, или его фармацевтически приемлемая соль.2. Соединение по п.1, которое представляет собой (E)-N-{3-[1-(8-хлор-11Н-10-окса-1-азадибензо[a,d] циклогептен-5-илиден)пропил]фенил}метансульфонамид.3. Соль по п.1, которая представляет собой...
Лечение нарушений центральной нервной системы селективными модуляторами рецепторов эстрогена

Номер патента: 2360
Опубликовано: 25.04.2002
Авторы: Нэдлер Мэри Патриция, Брайант Генри Ульман, Бэйлес Келли Рени, Пол Стивен Марк
МПК: A61K 31/381, C07D 333/52, A61P 25/24...
Метки: нервной, эстрогена, рецепторов, системы, лечение, модуляторами, центральной, селективными, нарушений
Формула / Реферат:
1. Способ лечения у пациента, нуждающегося в таком лечении, нарушения центральной нервной системы, выбранного из депрессии, смены настроений и болезни Альцгеймера, включающий введение терапевтически эффективного количества соединения, имеющего структуру или его фармацевтически приемлемой соли или пролекарства R1 и R2 независимо выбирают из группы, состоящей из гидрокси и алкокси, содержащего от одного до четырех атомов углерода ; и R3 и R4...
Предыдущий патент: Способ диагностики деталей двигателя внутреннего сгорания
Следующий патент: Способ извлечения диоксида углерода из газа с использованием нагреваемого технологическим газом ребойлера для извлечения диоксида углерода в отпарном аппарате
Случайный патент: Опорный поручень для лиц с ограниченными возможностями для использования в санузлах