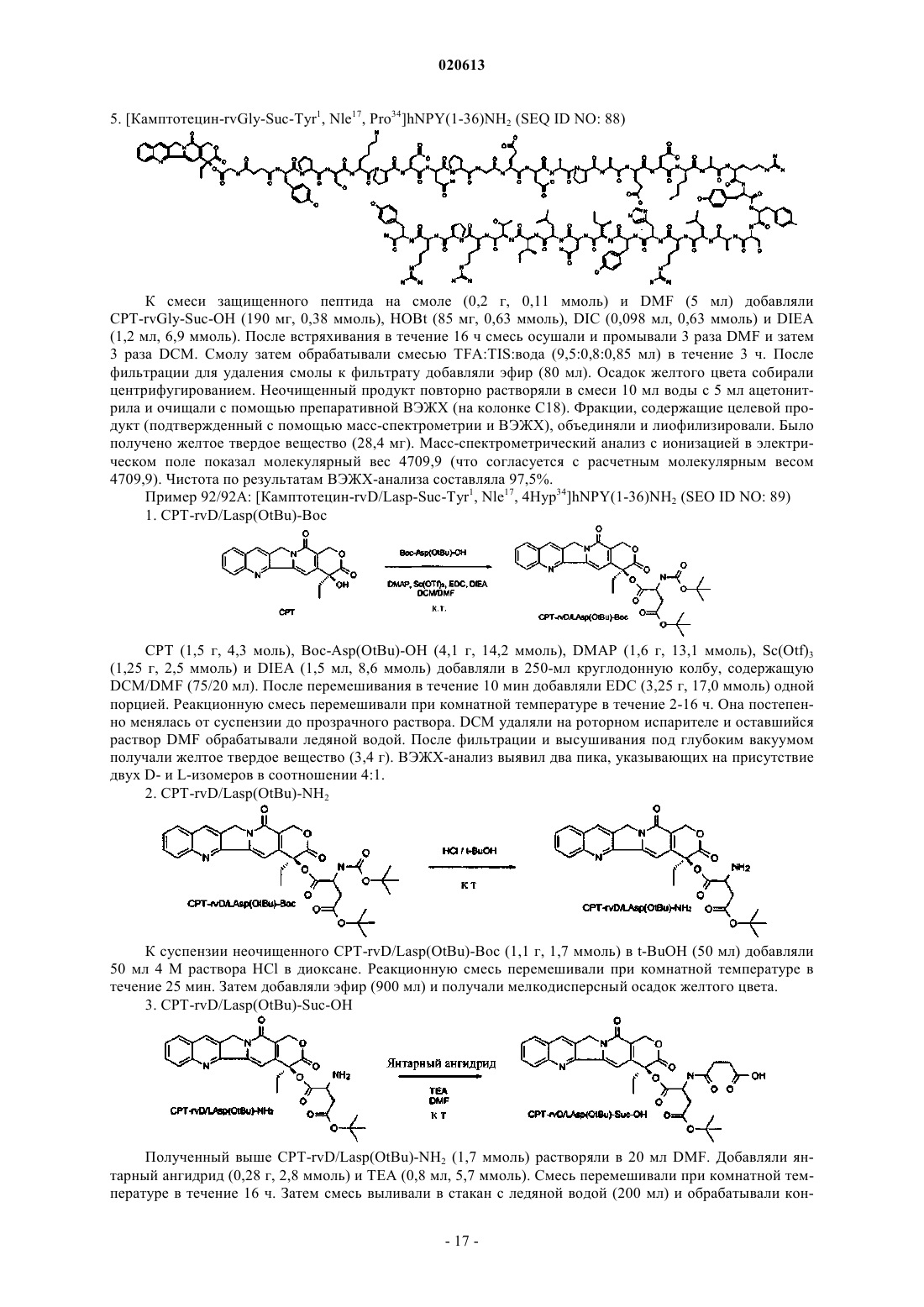
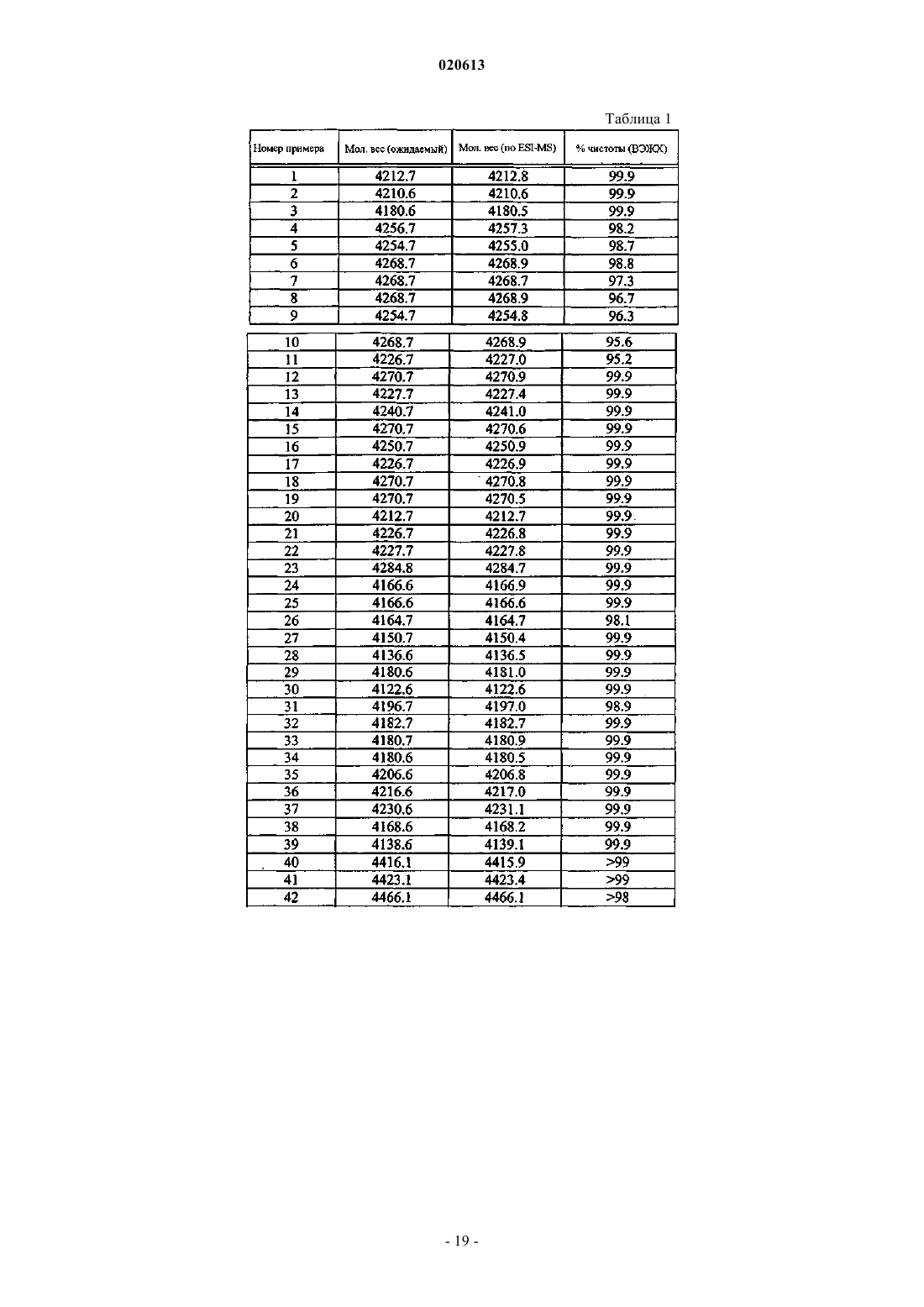
Цитотоксические конъюгаты с соединением, связывающим рецептор нейропептида y
Номер патента: 20613
Опубликовано: 30.12.2014
Авторы: Дун Чжэн Синь, Деоливейра Дэниел Б., Чжоу Кевин Л.
















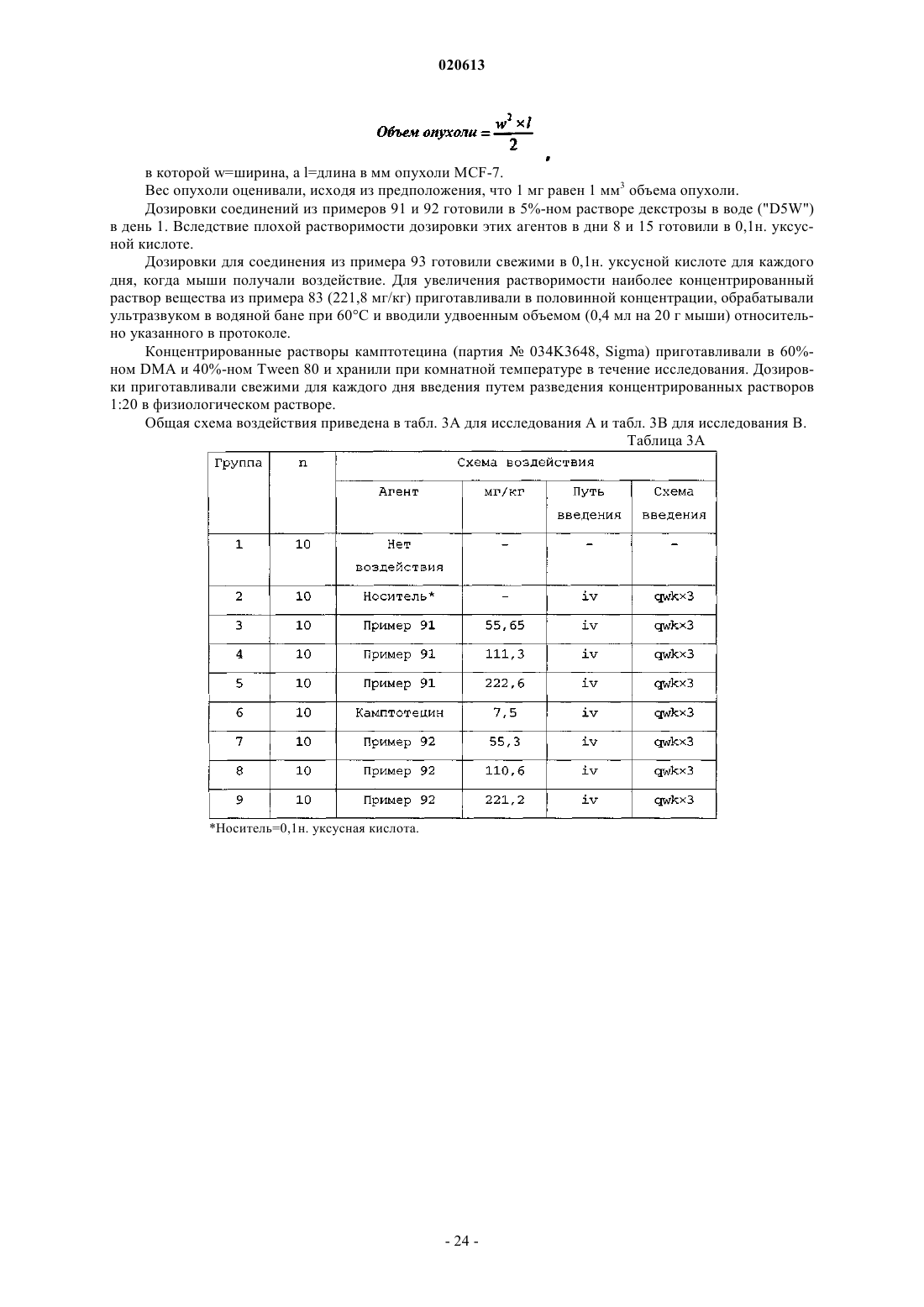
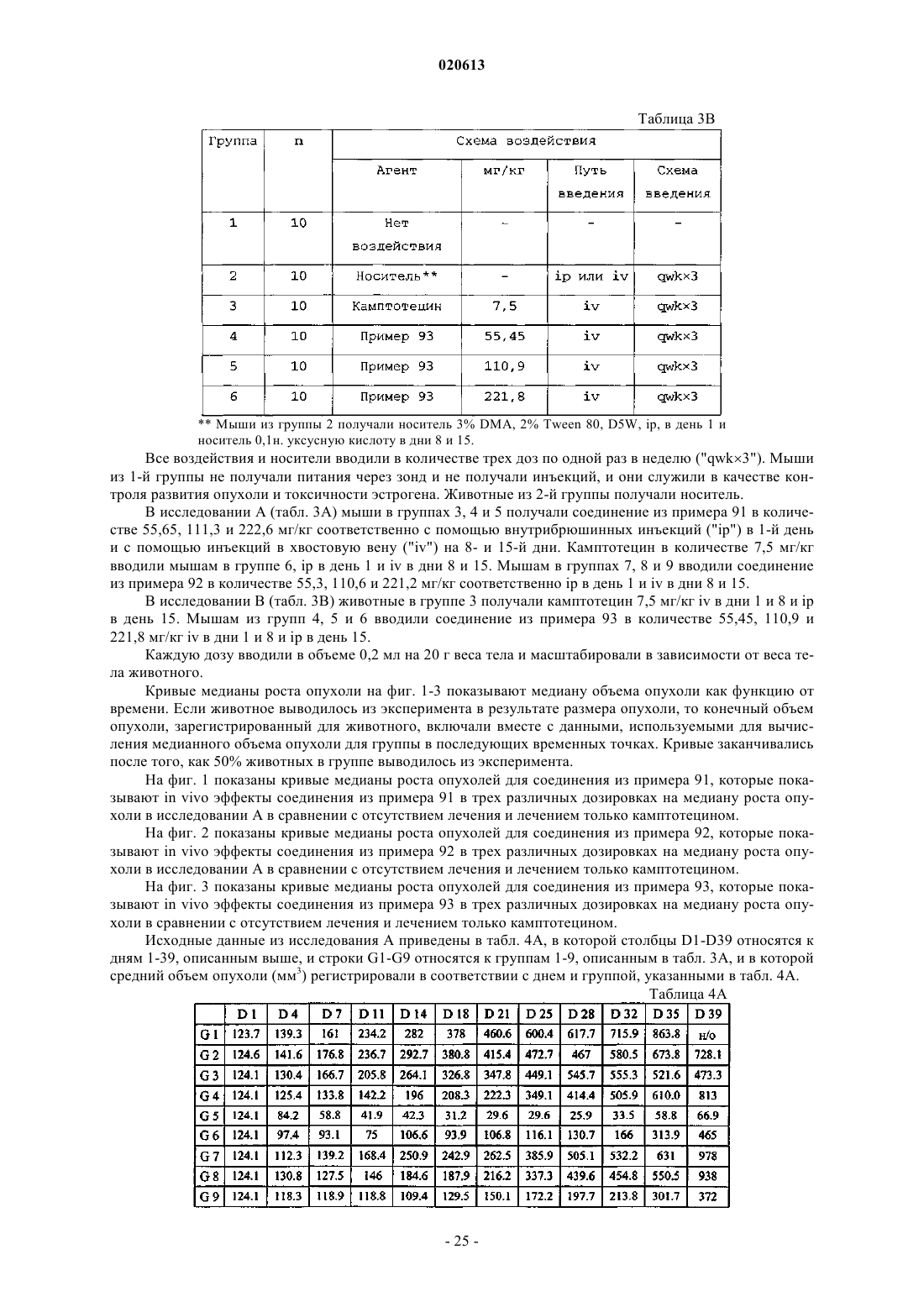
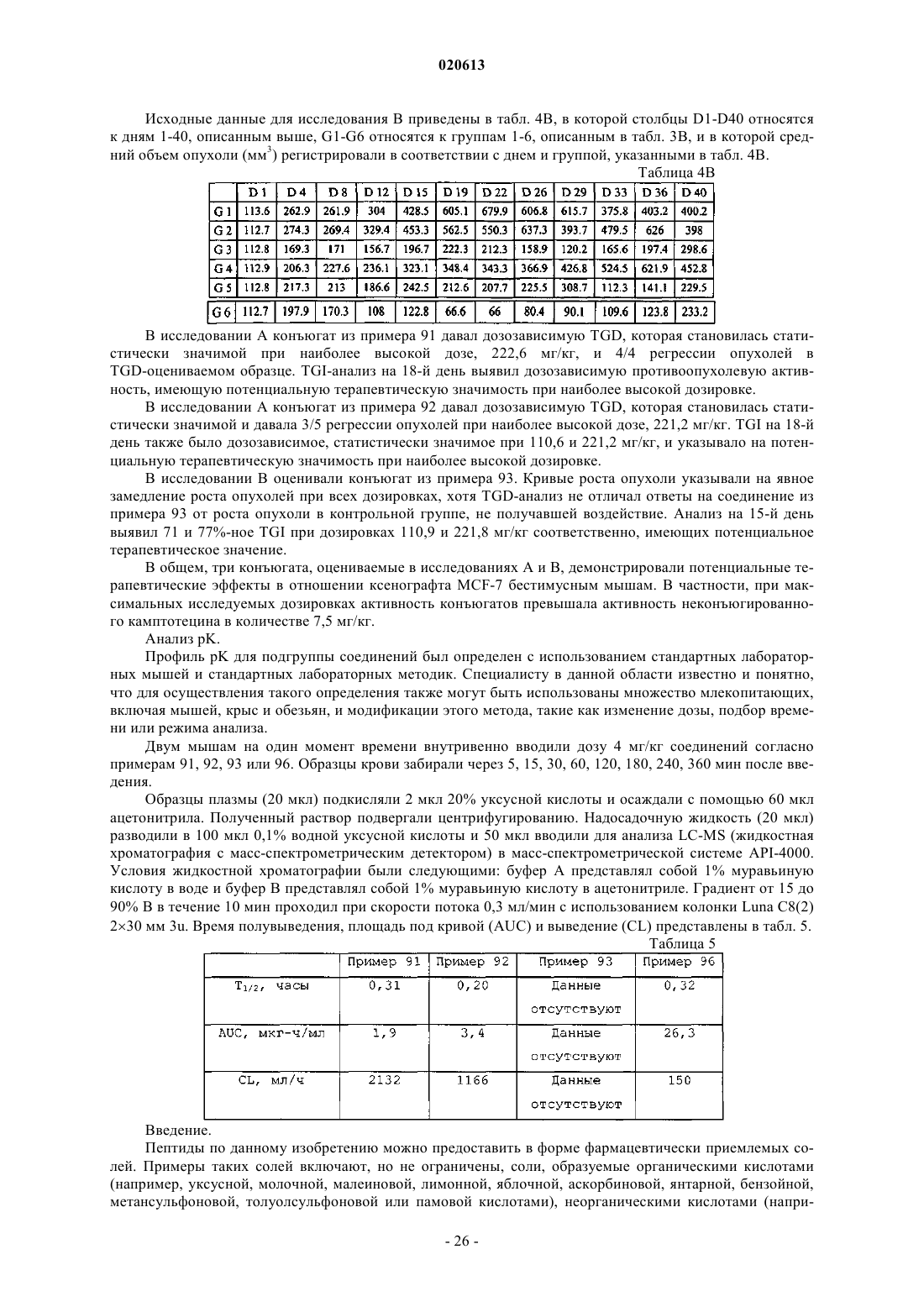



Формула / Реферат
1. Соединение формулы (I)

в которой X является цитотоксическим или цитостатическим средством;
В1 представляет собой rv (аминокислоту);
каждый из В2, В3 и В4, независимо для каждого случая, представляет собой (Doc)m, (Aepa)n или -C(O)-W1-W2-W3-W4-W5-C(O)- или отсутствует;
Z представляет собой аналог hNPY, соответствующий формуле

в которой А1 представляет собой Tyr или HN-CH((CH2)q-N(R2R3))-C(O);
А2 представляет собой Pro;
А3 представляет собой Ser, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А4 представляет собой Lys или HN-CH((CH2)q-N(R2R3))-C(O);
А5 представляет собой Pro;
А6 представляет собой Asp, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А7 представляет собой Asn, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А8 представляет собой Pro;
А9 представляет собой Gly, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А10 представляет собой Glu, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А11 представляет собой Asp, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А12 представляет собой Ala, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А13 представляет собой Pro;
А14 представляет собой Ala, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А15 представляет собой Glu, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А16 представляет собой Asp, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А17 представляет собой Met, А6с, Aib, Nle или HN-CH((CH2)q-N(R2R3))-C(O);
А18 представляет собой Ala, Aib или HN-CH((CH2)q-N(R2R3))-C(О);
А19 представляет собой Arg или HN-CH((CH2)q-N(R2R3))-C(O);
А20 представляет собой Tyr или HN-CH(CH2)q-N(R2R3))-C(O);
А21 представляет собой Tyr или HN-CH((CH2)q-N(R2R3))-C(O);
А22 представляет собой Ser, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А23 представляет собой Ala, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А24 представляет собой Leu, A6c или HN-CH((CH2)q-N(R2R3))-C(O);
А25 представляет собой Arg или HN-CH((CH2)q-N(R2R3))-C(O);
А26 представляет собой His или HN-CH((CH2)q-N(R2R3))-C(O);
А27 представляет собой Tyr или HN-CH((CH2)q-N(R2R3))-C(O);
А28 представляет собой Ile, А6с или HN-CH((CH2)q-N(R2R3))C(O);
А29 представляет собой Asn, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А30 представляет собой Leu, А6с или HN-CH((CH2)q-N(R2R3))-C(O);
А31 представляет собой Ile, Leu или HN-CH((CH2)q-N(R2R3))-C(O);
А32 представляет собой Thr, Aib или HN-CH((CH2)q-N(R2R3))-C(O);
А33 представляет собой Arg, hArg или HN-CH((CH2)q-N(R2R3))-C(O);
А34 представляет собой 4Нур;
А35 представляет собой Arg, Aic, Apc, Lys, 4NH2Phe, 4NH2CH2Phe или HN-CH((CH2)q-N(R2R3))-C(O);
А36 представляет собой Tyr, Aic, HN-CH((CH2)q-N(R2R3))-C(O) или отсутствует;
А37 представляет собой HN-CH((CH2)q-N(R2R3))-C(О) или отсутствует;
R1 представляет собой OH, NH2, (C1-30)алкокси или NH-Х6-CH2-X7, где X6 представляет собой (С1-40)алкил или (С2-40)алкенил и X7 представляет собой H, OH, CO2H или C(O)-NH2;
каждый из W1 и W5, независимо для каждого случая, представляет собой CR4R5;
каждый из R4 и R5, независимо для каждого случая, представляет собой Н, F, Br, Cl, I, (C1-30)алкил, (С2-30)алкенил, замещенный (C1-30)алкил, замещенный (С2-30)алкенил, SR6, S(O)R7 или S(O)2R8 или R4 и R5 вместе образуют (С3-30)циклоалкильное, (С3-30)гетероциклическое или (С5-30)арильное кольцо;
каждый из R6, R7 и R8, независимо для каждого случая, представляет собой (C1-30)алкил, (С2-30)алкенил, замещенный (C1-30)алкил или замещенный (С2-30)алкенил;
каждый из W2, W3 и W4, независимо для каждого случая, представляет собой CR9R10, О, S, (CH2)t или отсутствует;
каждый из R9 и R10, независимо для каждого случая, представляет собой Н, F, Br, Cl, I, (C1-30)алкил, (С2-30)алкенил, замещенный (С1-30)алкил, замещенный (С2-30)алкенил, SR6, S(O)R7 или S(O)2R8 или R9 и R10 вместе образуют кольцевую систему;
m, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10;
n, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10;
q, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4 или 5;
t, независимо для каждого случая, представляет собой 0, 1, 2 или 3;
каждый из R2 и R3, независимо для каждого случая, представляет собой Н, (С1-40)алкил, (C1-40)гетероалкил, (С1-40)ацил, (С2-40)алкенил, (С2-40)алкинил, арил(C1-40)алкил, арил(С1-40)ацил, замещенный (C1-40)алкил, замещенный (C1-40)гетероалкил, замещенный (С1-40)ацил, замещенный (С2-40)алкенил, замещенный (С2-40)алкинил, замещенный арил(C1-40)алкил, замещенный арил(С1-40)ацил, (С1-40)алкилсульфонил или C(NH)-NH2, причем когда R2 представляет собой (С1-40)ацил, арил(C1-40)ацил, замещенный (C1-40)ацил, замещенный арил(С1-40)ацил, (C1-40)алкилсульфонил или C(NH)-NH2, R3 представляет собой Н или (C1-40)алкил, (C1-40)гетероалкил, (С2-40)алкенил, (С2-40)алкинил, арил(C1-40)алкил, замещенный (C1-40)алкил, замещенный (C1-40)гетероалкил, замещенный (С2-40)алкенил, замещенный (С2-40)алкинил или замещенный арил(С1-40)алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором
X представляет собой антрациклин, камптотецин или производное камптотецина, паклитаксел или производное паклитаксела или доксорубицин или производное доксорубицина;
B1 представляет собой rvAsp, rvD-Asp, rvCha, rvD-Cha или rvGly;
B2 представляет собой Suc;
каждый из В3 и В4, независимо для каждого случая, представляет собой (Doc)m, (Aepa)n или отсутствует;
R1 представляет собой NH2;
каждый из R2 и R3, независимо для каждого случая, представляет собой Н или (С1-30)ацил;
при условии, что когда R2 является (C1-30)ацилом, R3 является Н;
каждый из R4 и R5, независимо для каждого случая, представляет собой Н или (С1-40)ацил;
q представляет собой 4; и
каждый из X1, X2, X3, X4 и X5, независимо для каждого случая, представляет собой Н, CH2NH2 или NH2;
или его фармацевтически приемлемая соль.
3. Соединение по п.2, в котором X представляет собой антрациклин, или его фармацевтически приемлемая соль.
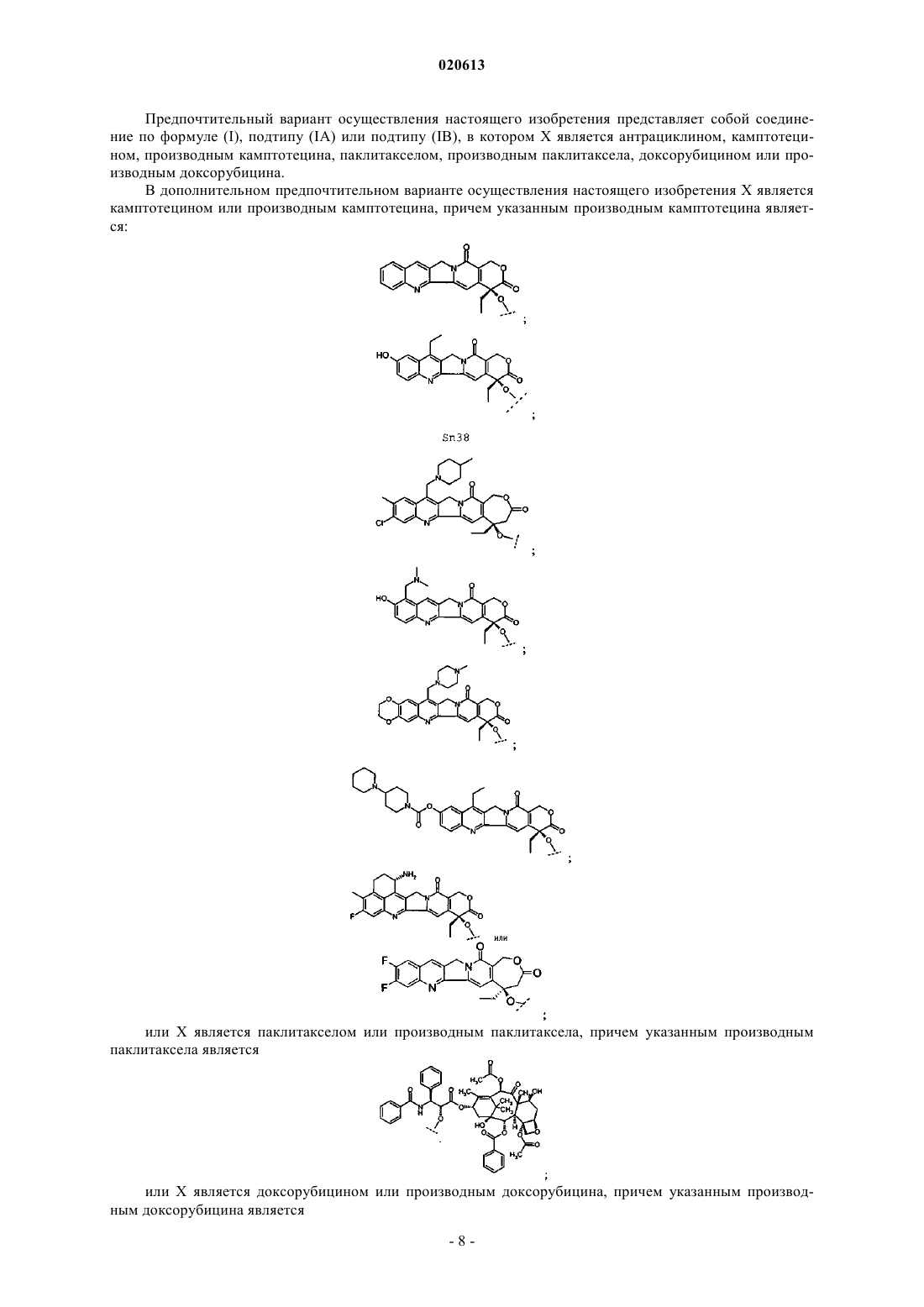
4. Соединение по п.2, в котором X представляет собой камптотецин или производное камптотецина, где указанное производное камптотецина представляет собой


или его фармацевтически приемлемая соль.
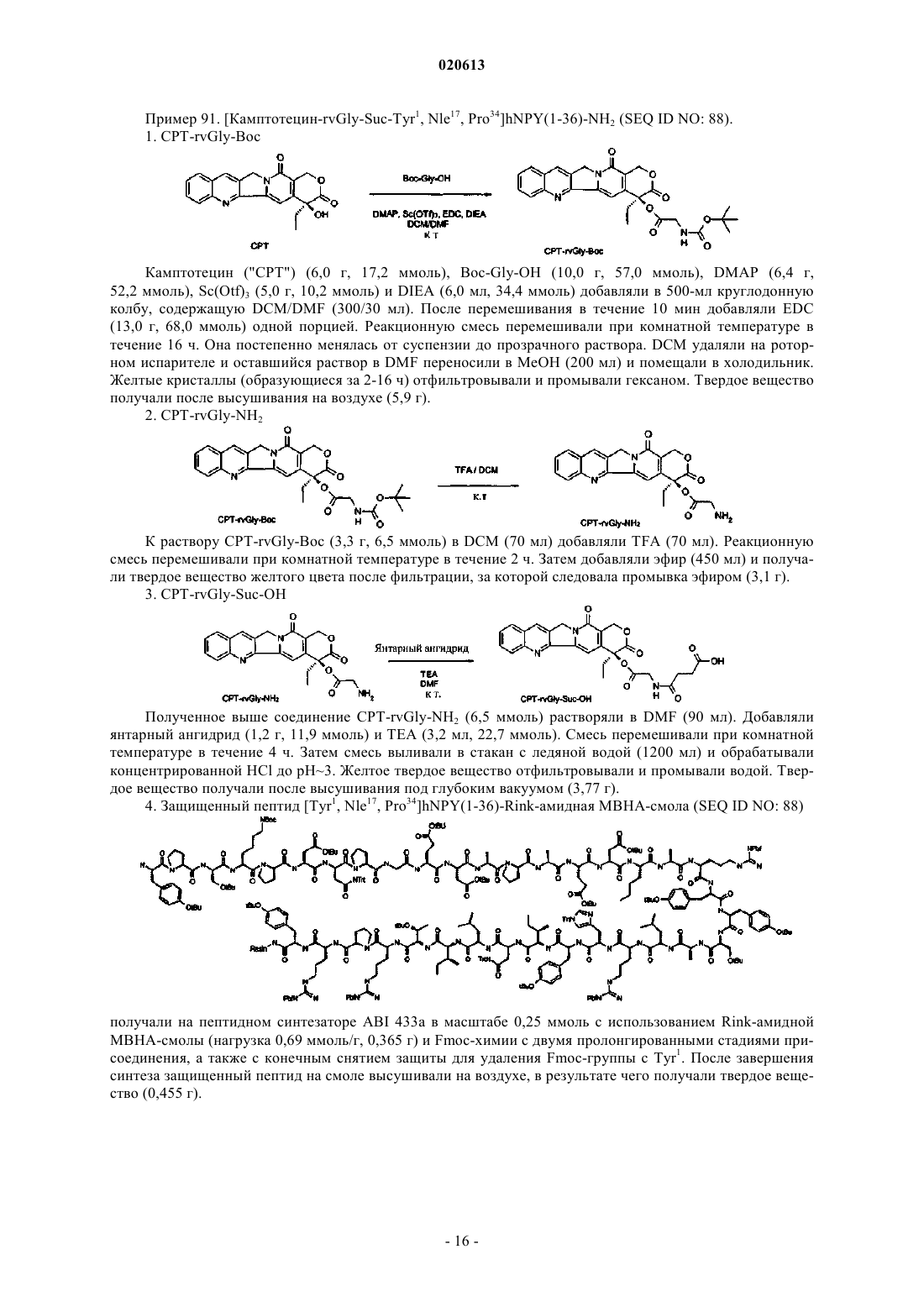
5. Соединение по п.2, в котором X представляет собой паклитаксел или производное паклитаксела, где указанное производное паклитаксела представляет собой

или его фармацевтически приемлемая соль.
6. Соединение по п.2, в котором X представляет собой доксорубицин или производное доксорубицина, где указанное производное доксорубицина представляет собой

или его фармацевтически приемлемая соль.
7. Соединение по любому из пп.2-6, в котором
А1 представляет собой Tyr;
А3 представляет собой Ser;
А4 представляет собой Lys;
А6 представляет собой Asp;
А7 представляет собой Asn;
А9 представляет собой Gly;
А10 представляет собой Glu;
А11 представляет собой Asp;
А12 представляет собой Ala;
А14 представляет собой Ala;
А15 представляет собой Glu;
А16 представляет собой Asp;
А17 представляет собой Aib или Nle;
А18 представляет собой Ala;
А19 представляет собой Arg;
А20 представляет собой Tyr;
А21 представляет собой Tyr;
А22 представляет собой Ser;
А23 представляет собой Ala;
А24 представляет собой Leu;
А25 представляет собой Arg;
А26 представляет собой His;
А27 представляет собой Tyr;
А28 представляет собой Ile;
А29 представляет собой Asn;
А30 представляет собой Leu;
А31 представляет собой Ile или А6с;
А32 представляет собой Thr;
А33 представляет собой Arg;
А35 представляет собой Arg или Aic;
А36 представляет собой Tyr, Aic или отсутствует;
А37 отсутствует,
или его фармацевтически приемлемая соль.
8. Соединение по п.7, в котором X представляет собой камптотецин, или его фармацевтически приемлемая соль.
9. Соединение по п.8, где указанное соединение представляет собой


или его фармацевтически приемлемая соль.
10. Смесь, содержащая [камптотецин-rvAsp-Suc-Tyr1, Nle17, 4Hyp34]hNPY(1-36)-NH2 (SEQ ID NO: 89) и [камптотецин-rvD-Asp-Suc-Tyr1, Nle17, 4Hyp34]hNPY(1-36)-NH2, или ее фармацевтически приемлемая соль.
11. Смесь, содержащая [камптотецин-rvAsp-Suc-Tyr1, Nle17, A6c31, 4Hyp34]hNPY(1-36)-NH2 (SEQ ID NO: 90) и [кампотецин-rvD-Asp-Suc-Tyr1, Nle17, A6c31, 4Hyp34]hNPY(1-36)-NH2, или ее фармацевтически приемлемая соль.
12. Смесь по любому из пп.10, 11, содержащая соотношение (вес.:вес.) примерно 2:98, примерно 5:95, примерно 10:90, примерно 15:85, примерно 20:80, примерно 25:75, примерно 30:70, примерно 35:65, примерно 40:60, примерно 45:50, примерно 50:50, примерно 55:45, примерно 60:40, примерно 65:25, примерно 70:30, примерно 75:25, примерно 80:20, примерно 85:15, примерно 87:13, примерно 88:12, примерно 90:10, примерно 95:5, примерно 97:3 или примерно 98:2 указанной пары соединений, или ее фармацевтически приемлемая соль.
13. Соединение по п.1, в котором Z соответствует:


или его фармацевтически приемлемая соль.
14. Соединение по п.2, в котором А37 отсутствует, или его фармацевтически приемлемая соль.
15. Соединение по п.14, в котором Z соответствует:


или его фармацевтически приемлемая соль.
16. Соединение по п.1 или 2, в котором пептидная связь между А35 и А36 заменена псевдопептидной связью, или его фармацевтически приемлемая соль.
17. Соединение по п.16, в котором А35-А36 представляет собой Lys-y(CH2-NH)Tyr или Lys-y(CH2-N(Ac))Tyr, или его фармацевтически приемлемая соль.
18. Соединение по п.17, в котором Z соответствует

или его фармацевтически приемлемая соль.
19. Фармацевтическая композиция, содержащая эффективное количество соединения по любому из пп.1-9 и 13-18, смесь по любому из пп.10, 11 или их фармацевтически приемлемую соль.
20. Фармацевтическая композиция по п.19, дополнительно содержащая фармацевтически приемлемый носитель.
21. Способ лечения заболевания у субъекта, нуждающегося в этом, включающий введение указанному субъекту терапевтически эффективного количества соединения по любому из пп.1-9 и 13-18, смеси по любому из пп.10, 11 или фармацевтически приемлемой композиции по п.19 или 20, причем указанное заболевание характеризуется нарушенной или нежелательной пролиферацией клеток, экспрессирующих рецептор NPY-Y1.
22. Способ лечения заболевания у субъекта, нуждающегося в этом, по п.21, в котором указанное заболевание выбрано из группы, состоящей из рака молочных желез, рака яичников, глиальных опухолей, почечно-клеточных карцином, нефробластомы и внутриопухолевых кровеносных сосудов.

Текст
ЦИТОТОКСИЧЕСКИЕ КОНЪЮГАТЫ С СОЕДИНЕНИЕМ, СВЯЗЫВАЮЩИМ РЕЦЕПТОР НЕЙРОПЕПТИДА Y Изобретение относится к группам новых конъюгатов нейропептида Y и цитотоксических соединений, к содержащим их композициям и способам, относящимся к их терапевтическому применению для лечения заболевания или состояния, ассоциированных с нарушенной или нежелательной пролиферацией клеток, которые экспрессируют рецепторы NPY-Y1. Область техники Настоящее изобретение относится к направленным цитотоксическим соединениям, содержащим цитотоксическую часть, связанную с направляющей частью, т.е. обеспечивающей направленное действие соединения. Более конкретно, настоящее изобретение относится к цитотоксическим конъюгатам с нейропептидом Y, содержащим их композициям и способам, связанным с их терапевтическим применением для лечения заболеваний или патологических состояний, ассоциированных с нарушенной или нежелательной клеточной пролиферацией, миграцией и/или физиологической активностью. Предпосылки изобретения Большинство цитотоксических лекарственных соединений проявляют нежелательные токсические побочные эффекты вследствие отсутствия избирательности в отношении тканей или клеток, которым необходим терапевтический эффект. Для селективной доставки цитотоксических агентов в целевой тип клеток предпринимались различные подходы. Использование лигандов биологических рецепторов в качестве носителей лекарственных соединений для направленной доставки этих лекарственных соединений в целевые клетки может снизить токсические побочные эффекты и значительно увеличить эффективность доставки лекарственных соединений. Например, в публикации WO 97/19954 по договору о патентной кооперации (РСТ) раскрыты конъюгаты антрациклинового цитотоксического агента, такого как доксорубицин, с пептидным гормоном, таким как LHRH, бомбезин или соматостатин. Цитотоксический агент ковалентно присоединен к пептиду через линкер, имеющий структуру -C(O)-(CH2)n-C(О)-, в которой n=0-7. Аналогично, в европейской патентной заявкеЕР 1118336 раскрыты конъюгаты соматостатиновых аналогов, например октреотида, ланреотида и вапреотида, и цитотоксическое лекарственное соединение, такое как паклитаксел, доксорубицин или камптотецин, соединенные через спейсер, причем указано, что спейсер имеет структуру -C(O)-(CH2)n-C(O)-, в которой n=0-7. В публикации патентной заявки США 2002/0115596 раскрыты конъюгаты цитотоксических агентов и олигопептидов, в которых аминокислотные последовательности пептидов расщепляются предпочтительно свободным простатическим специфическим антигеном. Утверждают, что такие конъюгаты пригодны для лечения рака простаты и доброкачественной гиперплазии простаты. В публикации патентной заявки США 2003/0064984 раскрыты конъюгаты цитотоксических аналогов CC-1065 и дуокармицинов с расщепляемыми линкерными плечами и направляющим агентом, таким как антитело или пептид. Цитотоксические аналоги высвобождаются при расщеплении линкера. В РСТ публикации WO 02/34237 раскрыты конъюгаты активных агентов, ковалентно присоединенных напрямую к полипептиду. Утверждают, что полипептиды стабилизирует активный агент, например,в желудке, посредством конформационной защиты. Однако все еще остро необходимы направленные цитотоксические лекарственные соединения с улучшенными свойствами в отношении направленной специфичности, системной токсичности и фармакокинетики. Предполагается, что применение направленных цитотоксических соединений помогает лечению ряда раковых заболеваний или патологических состояний. Например, предполагается, что при лечении опухолевых или раковых заболеваний, которые сверхэкспрессируют рецепторы нейропептида Y("NPY"), используют направляющее действие и лекарственное действие нативного нейропептида Y человека ("hNPY"), а именно Н-Tyr-Pro-Ser-Lys-Pro-Asp-Asn-Pro-Gly-Glu-Asp-Ala-Pro-Ala-Glu-Asp-MetAla-Arg-Tyr-Tyr-Ser-Ala-Leu-Arg-His-Tyr-Ile-Asn-Leu-Ile-Thr-Arg-Gln-Arg-Tyr-NH2 (SEQ ID NO: 1), или его фрагмента или аналога, в комплексе с цитотоксической частью. Эффект NPY может быть опосредован через несколько подтипов NPY-рецепторов, названныхY1-Y6, из которых хорошо описаны Y1, Y2, Y4 и Y5. Обзор NPY и NPY-рецепторов см., например, в С. Wahlestedt and D. Reis, Annual Review of Pharmacology and Toxicology, 33:309-352 (1993). Исходя из высокой плотности и высокой встречаемости рецептора NPY-Y1 в образцах опухолей молочных желез и метастаз, как описано в РСТ публикации WO 02/43776, рак молочных желез представляет собой важную мишень для NPY-связанных лекарственных соединений. Также было найдено (описано в РСТ публикации WO 02/43776) рецептор нейропептида Y1 экспрессируется исключительно в опухолевой ткани либо в комбинации с Y2-рецептором, либо отдельно, в то время как в здоровой ткани экспрессируется только Y2-рецептор. Соединение, связывающее Y1-рецептор, раскрытое в РСТ публикации WO 02/43776, выбрано из группы, состоящей из следующих соединений: Таким образом, способность соединения специфически взаимодействовать с рецептором NPY-Y1 с помощью селективных к Y1 аналогов NPY, конъюгированных с цитотоксической частью, будет способствовать лечению раковых заболеваний или состояний. Такие раковые заболевания включают, но не ограничены этим, рак молочных желез, рак яичников, глиальные опухоли, почечно-клеточные карциномы,нефробластому и внутриопухолевые кровеносные сосуды. Особые преимущества соединений по настоящему изобретению и их применения в качестве способов лечения опухолей и раковых заболеваний, которые представляют собой важную мишеньNPY-направленных лекарственных соединений, включают, но не ограничены этим, сниженные токсические побочные эффекты, увеличенную эффективность лечения и/или меньшие осложнения в результате мультилекарственной резистентности. Сущность изобретения В одном аспекте изобретение относится к направленным цитотоксическим соединениям со следующей формулой (I): в которой X является цитотоксическим или цитостатическим средством; В 1 представляет собой rv (аминокислоту); каждый из В 2, В 3 и В 4, независимо для каждого случая, представляет собой (Doc)m, (Aepa)n илиZ представляет собой молекулу, которая связывает один или несколько подтипов NPY-рецептора. В формуле (I) Z предпочтительно представляет собой аналог hNPY, соответствующий формуле(С 1-40)алкил или (С 2-40)алкенил и X7 представляет собой H, OH, CO2H или C(O)-NH2; каждый из W1 и W5, независимо для каждого случая, представляет собой CR4R5; каждый из R4 и R5, независимо для каждого случая, представляет собой Н, F, Br, Cl, I, (C1-30)алкил,(С 2-30)алкенил, замещенный (С 1-30)алкил, замещенный (С 2-30)алкенил, SR6, S(O)R7 или S(O)2R8 или R4 и R5 вместе образуют (С 3-30)циклоалкильное, (С 3-30)гетероциклическое или (С 5-30)арильное кольцо; каждый из R6, R7 и R8, независимо для каждого случая, представляет собой (C1-30)алкил,(С 2-30)алкенил, замещенный (C1-30)алкил или замещенный (С 2-30)алкенил; каждый из W2, W3 и W4, независимо для каждого случая, представляет собой CR9R10, О, S, (CH2)t или отсутствует; каждый из R9 и R10, независимо для каждого случая, представляет собой Н, F, Br, Cl, I, (C1-30)алкил,(С 2-30)алкенил, замещенный (C1-30)алкил, замещенный (С 2-30)алкенил, SR6, S(O)R7 или S(O)2R8 или R9 иR10 вместе образуют кольцевую систему;m, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10;n, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10;q, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4 или 5;t, независимо для каждого случая, представляет собой 0, 1, 2 или 3; каждый из X1, X2, X3, X4 и X5, независимо для каждого случая, представляет собой Н, F, Br, Cl, I,(С 1-10)алкил, замещенный (C1-10)алкил, арил, замещенный арил, OH, ОМе, NH2, NO2 или CN; каждый из R2 и R3, независимо для каждого случая, представляет собой Н, (C1-40)алкил,(C1-40)гетероалкил, (C1-40)ацил, (С 2-40)алкенил, (С 2-40)алкинил, арил(С 1-40)алкил, арил(C1-40)ацил, замещенный (С 1-40)алкил, замещенный (C1-40)гетероалкил, замещенный (C1-40)ацил, замещенный (С 2-40)алкенил,замещенный (С 2-40)алкинил, замещенный арил (С 1-40)алкил, замещенный арил(С 1-40)ацил,(C1-40)алкилсульфонил или C(NH)-NH2, причем когда R2 представляет собой (С 1-40)ацил, арил(С 1-40)ацил,замещенный (C1-40)ацил, замещенный арил(С 1-40)ацил, (C1-40)алкилсульфонил или C(NH)-NH2, R3 представляет собой Н или (С 1-40)алкил, (C1-40)гетероалкил, (С 2-40)алкенил, (С 2-40)алкинил, арил(C1-40)алкил,замещенный (С 1-40)алкил, замещенный (C1-40)гетероалкил, замещенный (С 2-40)алкенил, замещенный К подтипу (IA) соединений, описываемых вышеупомянутой формулой (I), относятся соединения, в которыхB2 является Suc; каждый из В 3 и В 4, независимо для каждого случая, представляет собой (Doc)m, (Aepa)n или отсутствует; А 1 представляет собой Tyr или HN-CHCH2)q-N(R2R3-C(O); А 2 представляет собой Pro; А 3 представляет собой Ser, Aib или HN-CHCH2)q-N(R2R3-C(O); А 4 представляет собой Lys или HN-CHCH2)q-N(R2R3-C(O); А 5 представляет собой Pro; А 6 представляет собой Asp, Aib или HN-CHCH2)q-N(R2R3-C(O); А 7 представляет собой Asn, Aib или HN-CHCH2)q-N(R2R3-C(O); А 8 представляет собой Pro; А 9 представляет собой Gly, Aib или HN-CHCH2)q-N(R2R3-С(O); А 10 представляет собой Glu, Aib или HN-CHCH2)q-N(R2R3-C(O); А 11 представляет собой Asp, Aib или HN-CHCH2)q-N(R2R3-C(O); А 12 представляет собой Ala, Aib или HN-CHCH2)q-N(R2R3-C(O); А 13 представляет собой Pro; А 14 представляет собой Ala, Aib или HN-CHCH2)q-N(R2R3-C(O); А 15 представляет собой Glu, Aib или HN-CHCH2)q-N(R2R3-C(О); А 16 представляет собой Asp, Aib или HN-CHCH2)q-N(R2R3-C(O); А 17 представляет собой Met, A6c, Aib, Nle или HN-CHCH2)q-N(R2R3-C(O); А 18 представляет собой Ala, Aib или HN-CHCH2)q-N(R2R3-C(O); А 19 представляет собой Arg или HN-CHCH2)q-N(R2R3-C(O); А 20 представляет собой Tyr или HN-CHCH2)q-N(R2R3-C(O); А 21 Tyr или HN-CHCH2)q-N(R2R3-C(O); А 22 представляет собой Ser, Aib или HN-CHCH2)q-N(R2R3-C(О); А 23 представляет собой Ala, Aib или HN-CHCH2)q-N(R2R3-C(О); А 24 представляет собой Leu, A6c или HN-CHCH2)q-N(R2R3-C(O); А 25 представляет собой Arg или HN-CHCH2)q-N(R2R3-C(O); А 26 представляет собой His или HN-CHCH2)q-N(R2R3-C(О); А 27 представляет собой Tyr или HN-CHCH2)q-N(R2R3-C(O); А 28 представляет собой Ile, А 6 с или HN-CHCH2)q-N(R2R3-C(O); А 29 представляет собой Asn, Aib или HN-CHCH2)q-N(R2R3-C(O); А 30 представляет собой Leu, А 6 с или HN-CHCH2)q-N(R2R3-C(O); А 31 представляет собой Ile, А 6 с, Leu или HN-CHCH2)q-N(R2R3-C(O); А 32 представляет собой Thr, Aib или HN-CHCH2)q-N(R2R3-C(O); А 33 представляет собой Arg или HN-CHCH2)q-N(R2R3-C(O); А 34 представляет собой Tic, Dhp, 4Hyp, Inp, Nip, Pro, hPro или HN-CHCH2)q-N(R2R3-C(O); А 35 представляет собой Arg, Aic, Apc, Lys, 4NH2Phe, 4NH2CH2Phe или HN-CHCH2)q-N(R2R3-C(O); А 36 представляет собой Tyr, Aic, HN-CHCH2)q-N(R2R3-C(O) или отсутствует; А 37 представляет собой HN-CHCH2)q-N(R2R3-С(O) или отсутствует;R1 представляет собой NH2; каждый из R2 и R3, независимо для каждого случая, представляет собой Н или (С 1-30)ацил при условии, что, когда R2 является (C1-30)ацилом, R3 является Н; каждый из R4 и R5, независимо для каждого случая, представляет собой Н или (C1-40)ацил;q представляет собой 4; каждый из X1, X2, X3, X4 и X5, независимо для каждого случая, представляет собой H, CH2NH2 илиNH2. В формуле (I) или подтипе (IA) пептидная связь между А 35 и А 36 может быть замещена псевдопептидной связью, в которой А 35-А 36 могут представлять собой Lys- (CH2-NH)Tyr или В предпочтительном варианте формулы (I) или подтипа (IA) Z соответствует: Предпочтительный вариант осуществления настоящего изобретения представляет собой соединение по формуле (I), подтипу (IA) или подтипу (IB), в котором X является антрациклином, камптотецином, производным камптотецина, паклитакселом, производным паклитаксела, доксорубицином или производным доксорубицина. В дополнительном предпочтительном варианте осуществления настоящего изобретения X является камптотецином или производным камптотецина, причем указанным производным камптотецина является: или X является паклитакселом или производным паклитаксела, причем указанным производным паклитаксела является или X является доксорубицином или производным доксорубицина, причем указанным производным доксорубицина является Дополнительный предпочтительный вариант осуществления изобретения представляет собой любое одно из следующих соединений подтипа (IB): или его фармацевтически приемлемую соль. В дополнительном аспекте изобретение относится к смеси соединений формулы (I), подтипа (IA) или подтипа (IB), в которой линкер rv (аминокислота) находится в D-форме в некоторых соединениях в смеси и в L-форме в некоторых соединениях в смеси. Смесь включает в себя (по весу) примерно 2:98,примерно 5:95, примерно 10:90, примерно 15:85, примерно 20:80, примерно 25:75, примерно 30:70,примерно 35:65, примерно 40:60, примерно 45:50, примерно 50:50, примерно 55:45, примерно 60:40,примерно 65:25, примерно 70:30, примерно 75:25, примерно 80:20, примерно 85:15, примерно 90:10,примерно 95:5, примерно 97:3 или даже примерно 98:2 соединений в смеси, в которых линкер rv (аминокислота) находится в D-форме и в L-форме соответственно. Краткое описание чертежей На фиг. 1 показаны кривые медианы роста опухолей для соединений из примера 91, которые показывают in vivo эффекты соединения из примера 91 в трех различных дозировках на медиану роста опухоли в исследовании А по сравнению с отсутствием лечения или лечением только камптотецином. На фиг. 2 показаны кривые медианы роста опухолей для соединений из примера 92, которые показывают in vivo эффекты соединения из примера 92 в трех различных дозировках на медиану роста опухоли в исследовании А по сравнению с отсутствием лечения или лечением только камптотецином. На фиг. 3 показаны кривые медианы роста опухолей для соединений из примера 93, которые показывают in vivo эффекты соединения из примера 93 в трех различных дозировках на медиану роста опухоли в исследовании В по сравнению с отсутствием лечения или лечением только камптотецином. Подробное описание изобретения Используемый в настоящем описании термин "аминокислота" относится к любой природной или неприродной аминокислоте, включая, но не ограничиваясь, -аминокислоты, -аминокислоты или-аминокислоты, и он может относиться либо к D-, либо к L-аминокислотам, если не указано иное. За исключением N-концевой аминокислоты, все аминокислотные сокращения (например, Ala) в этом описании имеют структуру -NH-Cl(R')-CO-, в которой R и R', каждый независимо, представляют собой водород или боковую цепь аминокислоты (например, R=CH3 и R'=H для Ala), или R и R' могут быть соединены, образуя кольцевую систему. Пептид по этому изобретению также обозначают, используя другой формат, например[Pro34]hNPY(1-36)-NH2 (SEQ ID NO: 83), с замещенными аминокислотами из природной последовательности, помещенными в квадратные скобки, например Pro вместо Gln в hNPY. Обозначение "NH2" вhNPY(1-36)-OH (SEQ ID NO: 1) указывает на свободную кислоту. Для удобства ниже представлен список некоторых сокращений, используемых в настоящей заявке,однако любое сокращение, используемое в настоящей заявке, определение которого не приведено в ней,не используется в значении, противоречащем общепринятому. в которой паралApc - 4-амино-4-карбоксипиперидин, представленный структурой лельные линии "=" указывают на точки присоединения молекулы к другой молекуле или последовательности;N указывает, что структура в круглых скобках присоединена к эпсилон-азоту боковой цепи Lys;Val или Vвалин. Ниже приведено определение других сокращений, используемых в настоящем документе: Ас - ацетил;EMEM - минимальная поддерживающая среда Игла;qwk3 - три дозы, по одной дозе раз в неделю;Lys- (CH2-NH)Tyr имеет структуру Греческая буква используется в настоящем описании для указания того, что пептидная связь заменена псевдопептидной связью. При обозначении аминокислотной последовательности термин используется в формате -А- (Х-X')-В, причем А представляет собой аминоацильный радикал, карбонильная группа которого модифицирована до X, а В представляет собой аминоацильный радикал,-аминогруппа которого была модифицирована до X'. X и X' показаны в виде нитки из символов элементов структуры, разделенных связью, например Lys- (CH2-NH)-Tyr. Указанная аминокислота в обозначении "rv (аминокислота)" присоединена к соединению в "обратной" ориентации. Указанная аминокислота в обозначении "rv (аминокислота)" может иметь либо L-, либо D-конфигурацию. Например, молекула "камптотецин-rvAsp-Suc" имеет структуру: Термин "алкил" относится к углеводородной группе, содержащей один или несколько атомов углерода, в которой атомы углерода, если их несколько, соединены простыми связями, примеры включают,но не ограничены этим, метил, этил, пропил и бутил. Алкильная углеводородная группа может представлять собой прямую цепь или содержать одно или несколько ответвлений или циклические группы, примеры которых включают, но не ограничены этим, изопропил и трет-бутил. Термин "замещенный алкил" относится к алкилу, у которого один или несколько атомов водорода углеводородной группы замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена (т.е. фтора, хлора, брома и йода), OH, CN, SH, NH2, NHCH3, NO2, (С 1-2)алкила, замещенного 1-6 атомами галогена, CF3, OCH3, OCF3 и (CH2)0-4-COOH. В различных вариантах осуществления изобретения присутствуют 1, 2, 3 или 4 заместителя. Присутствие (CH2)0-4-COOH приводит к получению алкилсодержащей кислоты. Примеры алкилсодержащих кислот, содержащих (CH2)0-4-COOH,включают в себя 2-норборнануксусную кислоту, трет-масляную кислоту и 3-циклопентилпропионовую кислоту. Термин "гетероалкил" относится к алкилу, у которого один или несколько атомов углерода в углеводородной группе замещены одним или несколькими из следующих атомов или групп: амино, амидо, О,S, N и карбонил. В различных вариантах осуществления изобретения присутствуют 1 или 2 гетероатома. Термин "замещенный гетероалкил" относится к гетероалкилу, у которого один или несколько атомов углерода углеводородной группы замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена (т.е. фтора, хлора, брома и йода), OH, CN, SH, NH2, NHCH3, NO2,(C1-2)алкила, замещенного 1-6 атомами галогена, CF3, OCH3, OCF3 и (CH2)0-4-COOH. В различных вариантах осуществления изобретения присутствуют 1, 2, 3 или 4 заместителя. Термин "алкенил" относится к углеводородной группе, образованной двумя или несколькими атомами углерода, в которой присутствуют одна или несколько двойных связей углерод-углерод, примеры которой включают, но не ограничены этим, винил, аллил, бутенил и пропенил. Алкенильная углеводородная группа может представлять собой прямую цепь или содержать одно или несколько ответвлений или циклические группы, примеры которых включают, но не ограничены этим, н-бутенил относительно т-бутенила и н-пентенил в сравнении с циклопентенилом. Термин "замещенный алкенил" относится к алкенилу, у которого один или несколько атомов водорода замещены одним или несколькими заместителями, выбранными из группы, состоящей из галогена(т.е. фтора, хлора, брома и йода), OH, CN, SH, NH2, NHCH3, NO2, (C1-2)алкила, замещенного 1-6 атомами галогена, CF3, OCH3, OCF3 и (CH2)0-4-COOH. В различных вариантах осуществления изобретения присутствуют 1, 2, 3 или 4 заместителя. Термин "арил" относится к необязательно замещенной ароматической группе по меньшей мере с одним кольцом, имеющим конъюгированную -электронную систему, содержащую до двух конъюгированных или конденсированных кольцевых систем. Арил включает в себя, но не ограничен этим, карбоциклическую арильную, гетероциклическую арильную и биарильную группы. Предпочтительно, арил является 5- или 6-членным кольцом. Предпочтительные атомы для гетероциклического арила включают,но без ограничения, один или несколько из атомов серы, кислорода и азота. Примеры арила включают,но не ограничены этим, фенил, 1-нафтил, 2-нафтил, индол, хинолин, 2-имидазол и 9-антрацен. Заместители арила выбраны из группы, состоящей из (С 1-4)алкила, (С 1-4)алкокси, галогена (т.е. фтора, хлора,брома и йода), OH, CN, SH, NH2, NO2, (C1-2)алкила, замещенного 1-5 атомами галогена, CF3, OCF3 и(CH2)0-4-COOH. В различных вариантах осуществления изобретения арил содержит 0, 1, 2, 3 или 4 заместителя. Термин "алкиларил" относится к "алкилу", соединенному с "арилом", как описано выше. Предполагается, что термин "циклоалкил" включает моноциклоалкильную или бициклоалкильную группу с указанным числом атомов углерода, известным опытным специалистам в данной области. Термин "гетероцикл" включает моноциклические и бициклические системы с одним или несколькими гетероатомами, такими как кислород, азот и сера. Кольцевая система может быть ароматической,например пиридин, индол, хинолин, пиримидин, тиофен (также известный как тиенил), фуран, бензотиофен, тетразол, дигидроиндол, индазол, N-формилиндол, бензимидазол, тиазол и тиадиазол. Кольце- 13020613 вые системы также могут быть неароматическими, как, например, но не ограниченные этим, пирролидин, пиперидин, морфолин и т.п. Синтез. Соединения по данному изобретению можно получить с использованием методик, описанных в примерах настоящего документа, а также с помощью методик, хорошо известных в данной области. Например, полипептидную область аналога NPY можно синтезировать и/или модифицировать химическим или биохимическим способом. Примеры методик биохимического синтеза, включающие введение нуклеиновой кислоты в клетку и экспрессию нуклеиновых кислот, можно найти, например, в Stewart, J.M., etal., Solid Phase Synthesis, Pierce Chemical Co., 2nd ed. (1984) и, например, в Sambrook et al., Molecular Cloning: A Laboratory Manual, 2nd Edition, Cold Spring Harbor Laboratory Press (1989). Нижеследующие примеры также иллюстрируют способы синтеза направленных цитотоксических соединений по настоящему изобретению. Примеры приведены в иллюстративных целях и никоим образом не подразумевают ограничение объема изобретения. Пример 1. [Aib10, 4Hyp34]hNPY(1-36)-NH2 (SEQ ID NO: 3). Указанный в заголовке пептид синтезировали с помощью Fmoc-химии. С-концевую часть пептида(остатки 18-36) синтезировали на пептидном синтезаторе ABI 433A (Applied Biosystems, Foster City, CA,USA) в масштабе синтеза 1,0 ммоль. 1,37 г Rink-амидной МВНА-смолы с замещением 0,73 ммоль(Novabiochem, San Diego, CA, USA) помещали в реакционный сосуд. Смолу обрабатывали 10 мл NMP в течение 15 мин для набухания смолы. Для синтеза пептида использовали протокол ABI FastMoc 1.0. Каждый цикл состоял из удаления N-концевой Fmoc-группы с помощью 20%-ного пиперидина с последующей многократной промывкой NMP. Готовые картриджи для каждой аминокислоты (1 ммоль) затем растворяли в 0,45 M HOBT/HBTU. После времени, достаточного для растворения аминокислоты,ее автоматически переносили в активационный сосуд. Еще два аминокислотных картриджа (1 ммоль) растворяли и переносили в сосуд для активации до общего количества 3 экв. аминокислоты, используемых на стадию присоединения. Затем в реакционный сосуд добавляли DIPEA (3 мл 2 М раствора) до общего количества 6 экв. DIPEA. Все эту смесь затем добавляли к смоле и оставляли для перемешивания на 15 мин. Затем освобождали реакционный сосуд, промывали NMP и затем проводили вторую стадию присоединения. После второй стадии присоединения смолу снова тщательно промывали. Каждую аминокислоту присоединяли два раза аналогичным образом. После стадии присоединения первого остатка Tyr для каждой из следующих 4 стадий присоединения и каждой стадии присоединения Arg смолу блокируют 5 мл блокирующего раствора (0,5 М уксусного ангидрида/0,13 М DIPEA/0,01 М НОВТ) для блокирования любых неацилированных участков смолы. Для стадий присоединения использовали следующие аминокислотные картриджи: цикл 1) Fmoc-Tyr(tBu)-OH; цикл 2) Fmoc-Arg(Pbf)-OH; цикл 3) Fmoc-4Hyp-OH; цикл 4) Fmoc-Arg(Pbf)-OH; цикл 5) Fmoc-Thr(tBu)-OH; цикл 6) Fmoc-Ile-OH; цикл 7) Fmoc-Leu-OH; цикл 8) Fmoc-Asn(Trt)-OH; цикл 9) Fmoc-Ile-OH; цикл 10) Fmoc-Tyr(tBu)-OH; цикл 11) Fmoc-His(Trt)-OH; цикл 12) Fmoc-Arg(Pbf)-OH; цикл 13) Fmoc-Leu-OH; цикл 14) Fmoc-Ala-OH; цикл 15) Fmoc-Ser(tBu)-OH; цикл 16) Fmoc-Tyr(tBu)-OH; цикл 17) Fmoc-Tyr(tBu)-OH; цикл 18) Fmoc-Arg(Pbf)-OH и цикл 19) Fmoc-Ala-OH. После последней стадии присоединения смолу промывали NMP, затем следовало стандартное удаление N-концевой Fmoc-группы, промывка NMP с последующей промывкой DCM. После сборки С-концевой части пептидной цепи (остатки 18-36) 0,1 смолы (0,1 ммоль) использовали для конструирования N-концевой части пептида, при этом остаток сохраняли. N-концевую часть указанного в заглавии пептида (остатки 1-17) синтезировали, используя Fmoc-химию с помощью микроволнового излучения,на пептидном синтезаторе Liberty (СЕМ, Matthews, NC, USA) в масштабе синтеза 0,1 ммоль. Смолу из предшествующего синтеза помещали в коническую пробирку объемом 50 мл вместе с 15 мл DMF и загружали в позицию для смолы на синтезаторе. Смола затем количественно переносилась в реакционный сосуд автоматически. Использовали стандартный протокол синтеза Liberty для масштаба 0,1 ммоль. Этот протокол включает в себя снятие N-концевой Fmoc-группы первой обработкой 7 мл 20%-ного пиперидина, содержащего 0,1 М НОВТ, в DMF. Первое снятие защиты проводили в течение 30 с с помощью микроволн (45 Вт, максимальная температура 75 С) и продувки азотом (3 с продувка/7 с перерыв). Затем реакционный сосуд осушали и проводили вторую обработку пиперидином, аналогичную первой обработке, за исключением того, что она длилась 3 мин. Затем смолу осушали и тщательно промывали несколько раз DMF. Затем добавляли защищенную аминокислоту, Fmoc-Met-OH в виде 0,2 М концентрированного раствора в DMF (2,5 мл, 5 экв.) с последующим добавлением 1,0 мл 0,45 M (4,5 экв.) HBTU в DMF. После этого добавляли 0,5 мл 2 M (10 экв.)DIPEA в NMP. Стадию присоединения проводили в течение 5 мин с использованием микроволнового излучения мощностью 20 Вт при максимальной температуре 75 С, и таким же соотношением времени продувки азотом и времени перерыва. После первой стадии присоединения содержимое реакционного сосуда удаляли в отходы и повторяли стадию присоединения. Затем начинали цикл 2 аналогично циклу 1. Все аминокислоты вводили аналогичным образом и использовали стратегию двукратного присоединения для всей последовательности. При присоединении остатков 9-10 (Gly-Aib) использовали процедуру блокирования немедленно после стадии присоединения. Блокирование выполняли, добавляя 7 мл 0,5 М уксусного ангидрида, содержащего 0,015 М НОВТ вNMP, совместно с 2 мл 2 М раствора DIPEA, используя многостадийный протокол обработки микроволновым излучением: при мощности 50 Вт в течение 30 с (максимальная температура - 65 С) с последующим перерывом в течение 30 с и следующим раундом 30-секундной обработки микроволновым излучением (50 Вт), и последующим 30-секундным перерывом. Смолу осушали и тщательно промывали DMF. Использовали следующие аминокислоты (Advanced Chemtech, Louisville, KY, USA): цикл 20) Fmoc-Met-OH; цикл 21) Fmoc-Asp(OtBu)-OH; цикл 22) Fmoc-Glu(OtBu)-OH; цикл 23) Fmoc-Ala-OH; цикл 24) Fmoc-Pro-OH; цикл 25) Fmoc-Ala-OH; цикл 26) Fmoc-Asp(OtBu)-OH; цикл 27) Fmoc-Aib-OH; цикл 28) Fmoc-Gly-OH; цикл 29) Fmoc-Pro-OH; цикл 30) Fmoc-Asn(Trt)-OH; цикл 31) Fmoc-Asp(OtBu)-OH; цикл 32) Fmoc-Pro-OH; цикл 33) Fmoc-Lys(Boc)-OH; цикл 34) Fmoc-Ser(tBu)-OH; цикл 35) Fmoc-Pro-OH; цикл 36) Fmoc-Tyr(tBu)-OH. После завершения синтеза пептидной цепи для удаления N-концевой Fmoc-группы использовали стандартную обработку пиперидином с помощью стандартной процедуры снятия защиты, описанной выше. Смолу тщательно промывали DMF и затем помещали обратно в 50-мл коническую пробирку, используя DMF в качестве растворителя для переноса. С пептида удаляли защиту и отщепляли от смолы с помощью обработки 5 мл следующего реагента: 5% TIS, 2% воды, 5% (вес./об.) DTT и 88% TFA и перемешивания в течение 3,5 ч. Фильтрат собирали в 45 мл холодного безводного этилового эфира. Осадок осаждали в течение 10 мин при 3500 об/мин в охлаждаемой центрифуге. Эфир декантировали и пептид растворяли в свежем эфире. Обработку этиловым эфиром проводили всего 2 раза. После последней промывки эфиром пептид оставляли сушиться на воздухе для удаления остатков эфира. Осадок пептида ресуспендировали в 8 мл ацетонитрила с последующим добавлением 8 мл деионизированной воды и оставляли до полного растворения. Раствор пептида затем анализировали с помощью масс-спектрометрии. Масс-спектрометрический анализ с использованием ионизации распылением в электрическом поле идентифицировал основной продукт с массой 4212,1, соответствующей целевому продукту. Анализ с помощью аналитической ВЭЖХ на колонке С 18 2504,6 мм (Phenomenex, Torrance, CA, USA) в градиенте ацетонитрила 2-60%(0,1% TFA) за 30 мин идентифицировал основной продукт с чистотой 45%. Неочищенный пептид затем очищали с помощью препаративной ВЭЖХ на обращенно-фазовой колонке С 18 в градиенте ацетонитрила 10-60% (0,1% TFA) за 50 мин при скорости потока 10 мл/мин. Очищенный продукт анализировали с помощью ВЭЖХ на чистоту (99%) и с помощью масс-спектрометрии для определения массы(4212,8 Да), причем экспериментально определенная масса хорошо соответствовала ожидаемой массе 4212,7. После чего пептид лиофилизировали, получая 39 мг очищенного продукта с выходом 9%.(13,0 г, 68,0 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Она постепенно менялась от суспензии до прозрачного раствора. DCM удаляли на роторном испарителе и оставшийся раствор в DMF переносили в MeOH (200 мл) и помещали в холодильник. Желтые кристаллы (образующиеся за 2-16 ч) отфильтровывали и промывали гексаном. Твердое вещество получали после высушивания на воздухе (5,9 г). 2. CPT-rvGly-NH2 К раствору CPT-rvGly-Boc (3,3 г, 6,5 ммоль) в DCM (70 мл) добавляли TFA (70 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем добавляли эфир (450 мл) и получали твердое вещество желтого цвета после фильтрации, за которой следовала промывка эфиром (3,1 г). 3. CPT-rvGly-Suc-OH Полученное выше соединение CPT-rvGly-NH2 (6,5 ммоль) растворяли в DMF (90 мл). Добавляли янтарный ангидрид (1,2 г, 11,9 ммоль) и TEA (3,2 мл, 22,7 ммоль). Смесь перемешивали при комнатной температуре в течение 4 ч. Затем смесь выливали в стакан с ледяной водой (1200 мл) и обрабатывали концентрированной HCl до рН 3. Желтое твердое вещество отфильтровывали и промывали водой. Твердое вещество получали после высушивания под глубоким вакуумом (3,77 г). 4. Защищенный пептид [Tyr1, Nle17, Pro34]hNPY(1-36)-Rink-амидная МВНА-смола (SEQ ID NO: 88) получали на пептидном синтезаторе ABI 433 а в масштабе 0,25 ммоль с использованием Rink-амидной МВНА-смолы (нагрузка 0,69 ммоль/г, 0,365 г) и Fmoc-химии с двумя пролонгированными стадиями присоединения, а также с конечным снятием защиты для удаления Fmoc-группы с Tyr1. После завершения синтеза защищенный пептид на смоле высушивали на воздухе, в результате чего получали твердое вещество (0,455 г). К смеси защищенного пептида на смоле (0,2 г, 0,11 ммоль) и DMF (5 мл) добавляли(1,2 мл, 6,9 ммоль). После встряхивания в течение 16 ч смесь осушали и промывали 3 раза DMF и затем 3 раза DCM. Смолу затем обрабатывали смесью TFA:TIS:вода (9,5:0,8:0,85 мл) в течение 3 ч. После фильтрации для удаления смолы к фильтрату добавляли эфир (80 мл). Осадок желтого цвета собирали центрифугированием. Неочищенный продукт повторно растворяли в смеси 10 мл воды с 5 мл ацетонитрила и очищали с помощью препаративной ВЭЖХ (на колонке С 18). Фракции, содержащие целевой продукт (подтвержденный с помощью масс-спектрометрии и ВЭЖХ), объединяли и лиофилизировали. Было получено желтое твердое вещество (28,4 мг). Масс-спектрометрический анализ с ионизацией в электрическом поле показал молекулярный вес 4709,9 (что согласуется с расчетным молекулярным весом 4709,9). Чистота по результатам ВЭЖХ-анализа составляла 97,5%. Пример 92/92 А: [Камптотецин-rvD/Lasp-Suc-Tyr1, Nle17, 4Hyp34]hNPY(1-36)NH2 (SEO ID NO: 89) 1. CPT-rvD/Lasp(OtBu)-BocDCM/DMF (75/20 мл). После перемешивания в течение 10 мин добавляли EDC (3,25 г, 17,0 ммоль) одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 2-16 ч. Она постепенно менялась от суспензии до прозрачного раствора. DCM удаляли на роторном испарителе и оставшийся раствор DMF обрабатывали ледяной водой. После фильтрации и высушивания под глубоким вакуумом получали желтое твердое вещество (3,4 г). ВЭЖХ-анализ выявил два пика, указывающих на присутствие двух D- и L-изомеров в соотношении 4:1. 2. CPT-rvD/Lasp(OtBu)-NH2 К суспензии неочищенного CPT-rvD/Lasp(OtBu)-Boc (1,1 г, 1,7 ммоль) в t-BuOH (50 мл) добавляли 50 мл 4 М раствора HCl в диоксане. Реакционную смесь перемешивали при комнатной температуре в течение 25 мин. Затем добавляли эфир (900 мл) и получали мелкодисперсный осадок желтого цвета. 3. CPT-rvD/Lasp(OtBu)-Suc-OH Полученный выше CPT-rvD/Lasp(OtBu)-NH2 (1,7 ммоль) растворяли в 20 мл DMF. Добавляли янтарный ангидрид (0,28 г, 2,8 ммоль) и TEA (0,8 мл, 5,7 ммоль). Смесь перемешивали при комнатной температуре в течение 16 ч. Затем смесь выливали в стакан с ледяной водой (200 мл) и обрабатывали кон- 17020613 центрированной НС 1 до рН 3. Получали мелкодисперсное твердое вещество желтого цвета (0,72 г). ВЭЖХ-анализ выявил два пика в соотношении 4:1. 4. Защищенный пептид [Tyr1, Nle17, 4Hyp34]hNPY(1-36)-Rink-амидная МВНА-смола(SEQ ID NO: 89). Указанный пептид получали с помощью процедуры, аналогичной процедуре в примере 81. 5. [Камптотецин-rvD/Lasp-Suc-Tyr1, Nle17, 4 Нур 34]hNPY(1-36)NH2 (SEQ ID NO: 89). К смеси защищенного пептида [Tyr1, Nle17, 4Hyp34]hNPY(1-36)-Rink-амидная МВНА-смола(0,125 ммоль) и DMF (5 мл) добавляли CPT-rvD/Lasp(OtBu)-Suc-OH (230 мг, 0,37 ммоль), HOBt (84 мг,0,63 ммоль), DIC (0,097 мл, 0,63 ммоль) и DIEA (1,3 мл, 7,5 ммоль). Следовали процедуре, аналогичной процедуре из примера 81. Количество очищенного полученного вещества составляло 31,0 мг. Вычисленный с помощью масс-спектрометрического анализа с ионизацией в электрическом поле молекулярный вес составлял 4783,8 (что согласуется с расчетным молекулярным весом 4784,2). Чистота по результатам аналитической ВЭЖХ составляла 100%. Пример 93.[CPT-rvD/Lasp-Suc-Tyr1, Nle17, A6c31, 4Hyp34]hNPY(1-36)-NH2 получали аналогично примеру 82 изCPT-rvD/Lasp(OtBu)-Suc-OH и соответствующего пептида [Tyr1, Nle17, А 6 с 31, 4 Нур 34]hNPY(1-36)-Rinkамидная МВНА-смола. Количество очищенного полученного вещества составляло 23,0 мг. Массспектрометрический анализ с ионизацией в электрическом поле показал, что молекулярный вес составлял 4796,0 (что согласуется с расчетным молекулярным весом 4796,2). Чистота по результатам аналитической ВЭЖХ составляла 100%. Другие соединения по изобретению средний специалист в данной области может получить, используя процедуры синтеза, аналогичные описанным в приведенных выше примерах. Физические параметры соединений из примеров 1-77 и 91-107 приведены в табл. 1.In vitro радиолигандный анализ связывания NPY-Y1 и NPY-Y2 рецептора. Клеточные линии нейробластомы человека, SK-N-MC и SK-N-BE2 (Американская коллекция типовых культур, Rockville, MD, USA), экспрессирующие рецепторы NPY-Y1 и NPY-Y2 соответственно,культивировали в среде ЕМЕМ, содержащей 10% фетальной бычьей сыворотки и 5% экстракта куриных эмбрионов, при 37 С во влажной атмосфере в 95% воздуха и 5% CO2. Для радиолигандного анализа связывания in vitro NPY-Y1 и NPY-Y2 соответствующие клетки вали дважды с помощью центрифугирования (39000g в течение 10 мин) и конечные осадки ресуспендировали в 50 мМ Tris-HCl, содержащем 2,5 мМ MgCl2, 0,1 мг/мл бацитрацина (Sigma Chemical, St. Louis,MO, USA) и 0,1% BSA. Для анализа аликвоты (0,4 мл) приготовленных суспензий инкубировали с 0,05 нМ [125I]PYY(2200 Ки/ммоль, Perkin-Elmer, Boston, MA) в присутствии или без 0,05 мл немеченного конкурентного тестируемого пептида или тестируемого направленного цитотоксического соединения. После 100-минутной инкубации (при 25 С) связавшийся [125I]PYY отделяли от свободного быстрой фильтрацией через GF/C-фильтры (Brandel, Gaithersburg, MD, USA), которые предварительно были вымочены в 0,3%-ном растворе полиэтиленимина. Фильтры затем промывали три раза 5-мл аликвотами ледяного 50 мМ Tris-HCl и связанную радиоактивность, задержанную на фильтрах, измеряли с помощью гаммаспектрометрии (Wallac LKB, Gaithersburg, MD, USA). Удельное связывание определяли как общее количество связавшегося [125I]PYY минус количество,связавшееся в присутствии 1000 нМ PYY (Bachem, Torrence, CA, USA). Константы ингибирования (Ki) вычисляли с помощью известного уравнения Ченга-Прусова, и эти данные совместно с селективностью указанных соединений относительно NPY-Y1 и NPY-Y2 приведены в табл. 2. Каждое из соединений из примеров 1-38, 40-61, 64-77 и 91-103 тестировали в приведенных выше радиолигандных анализах, и было найдено, что практически все из указанных соединений имели Ki ниже 100 нМ, а также некоторые из соединений из примеров имели Ki в субнаномолярном диапазоне. Также было найдено, что практически все из указанных соединений высокоизбирательно связывали NPY-Y1 по сравнению с NPY-Y2. Таблица 2 Анализ роста опухоли in vivo. Соединения из примеров 91, 92 и 93 оценивали в двух исследованиях: "Исследование А" и "Исследование В", в каждом из которых использовали одну группу (n=10) не получавших воздействие животных, одну группу получавших носитель животных и одну группу животных, получавших неконъюгированный камптотецин в количестве 7,5 мг/кг. Введение проводили один раз в неделю в течение трех недель с помощью инъекций в хвостовую вену, за исключением случаев, когда плохая растворимость лекарственного соединения или некроз хвоста требовали внутрибрюшинных инъекций. В исследовании А оценивали группы мышей, получавших соединение из примера 91 в количестве 55,65, 111,3 и 222,6 мг/кг и из примера 92 в количестве 55,3, 110,6 и 221,2 мг/кг. Соединение из примера 93 оценивали в исследовании В в количестве 55,45, 110,9 и 221,8 мг/кг. Противоопухолевую активность оценивали по задержке роста опухоли ("TGD"), определяемой как разница медианного времени для достижения конечного размера опухоли в группе, получавшей исследуемое воздействие, относительно контрольной группы, и по ингибированию роста опухоли ("TGI"), определяемому как разница между медианными объемами опухолей в группах, получавших исследуемое воздействие, и контрольной группой в такой день исследования, который обеспечивает баланс между поддающимися измерению ответами в группе и большими размерами групп. Токсичность оценивали по весу тела и клиническим наблюдениям. В день 1 (определение "Дня 1" дано ниже) возраст самок бестимусных мышей (nu/nu, Harlan) составлял 10-11 недель, а их вес находился в диапазоне 20,2-31,1 г и 21,2-29,8 г. Животные получали adlibitum воду (очищенную с помощью обратного осмоса, содержащую 1 ppm Cl) и стерилизованное с помощью радиации питание "NIH 31 Modified and Irradiated Lab Diet", состоящее из 18% сырого белка,5% сырого жира и 5% сырой клетчатки. Мышей содержали на стерилизованной с помощью радиации подстилке для лабораторных животных "ALPHA-dribed-o'cobs" в неподвижных микроизоляторах на 12-часовом световом цикле при 21-22 С (70-72F) и 40-60%-ной влажности. Клетки карциномы молочных желез человека MCF-7 растили в среде R3MI 1640, содержащей 100 ед/мл натриевой соли пенициллина G, 100 мкг/мл стрептомицина сульфата и 25 мкг/мл гентамицина. В среду добавляли 10% инактивированной нагревом фетальной бычьей сыворотки и 2 мМ глутамина. Дополнительную буферную емкость обеспечивали с помощью 10 мМ HEPES и 0,075%-ного бикарбоната натрия. Опухолевые клетки культивировали в флаконах для культуры тканей в увлажняемом инкубаторе при 37 С в атмосфере, состоящей из 95% воздуха и 5% CO2. За 3-7 дней до инъекции клеток каждой мыши между лопатками имплантировали капсулу, содержащую 17 эстрадиол (0,36 мг, высвобождение в течение 60 дней, Innovative Research of America). Используемые для имплантации опухолевые клетки MCF-7 собирали в течение логарифмической фазы роста и ресуспендировали в фосфатно-солевом буфере в количестве 5107 клеток/мл. Каждой мыши вводили подкожно с помощью инъекций в правый бок 1107 клеток. За развитием опухолей наблюдали дважды в неделю и затем ежедневно после достижения опухолью объема 80-120 мм 3. На 18-й день,который в исследовании А обозначали как "День 1", животных сортировали по девяти группам (число животных в группе, n=10) с индивидуальными размерами опухолей, варьирующими в диапазоне от 63 до 172 мм 3 и средним объемом опухоли в группе, составляющим 123,7-124,6 мм 3. Вследствие плохой приживаемости опухолей число животных было недостаточным для дополнительных трех групп, включенных в исходный протокол. Поэтому было проведено отдельное исследование ("Исследование В"), которое включало шесть групп (n=10) животных. В этом исследовании между имплантацией клеток и разделением по группам прошло 17 дней. Индивидуальные размеры опухолей на день 1 варьировали от 63 до 221 мм 3, а средний объем опухоли для групп варьировал от 112,7 до 113,6 мм 3. Размер опухоли в мм 3 вычисляли по формуле в которой w=ширина, а l=длина в мм опухоли MCF-7. Вес опухоли оценивали, исходя из предположения, что 1 мг равен 1 мм 3 объема опухоли. Дозировки соединений из примеров 91 и 92 готовили в 5%-ном растворе декстрозы в воде ("D5W") в день 1. Вследствие плохой растворимости дозировки этих агентов в дни 8 и 15 готовили в 0,1 н. уксусной кислоте. Дозировки для соединения из примера 93 готовили свежими в 0,1 н. уксусной кислоте для каждого дня, когда мыши получали воздействие. Для увеличения растворимости наиболее концентрированный раствор вещества из примера 83 (221,8 мг/кг) приготавливали в половинной концентрации, обрабатывали ультразвуком в водяной бане при 60 С и вводили удвоенным объемом (0,4 мл на 20 г мыши) относительно указанного в протоколе. Концентрированные растворы камптотецина (партия 034K3648, Sigma) приготавливали в 60%ном DMA и 40%-ном Tween 80 и хранили при комнатной температуре в течение исследования. Дозировки приготавливали свежими для каждого дня введения путем разведения концентрированных растворов 1:20 в физиологическом растворе. Общая схема воздействия приведена в табл. 3 А для исследования А и табл. 3 В для исследования В. Таблица 3 А Мыши из группы 2 получали носитель 3% DMA, 2% Tween 80, D5W, ip, в день 1 и носитель 0,1 н. уксусную кислоту в дни 8 и 15. Все воздействия и носители вводили в количестве трех доз по одной раз в неделю ("qwk3"). Мыши из 1-й группы не получали питания через зонд и не получали инъекций, и они служили в качестве контроля развития опухоли и токсичности эстрогена. Животные из 2-й группы получали носитель. В исследовании А (табл. 3 А) мыши в группах 3, 4 и 5 получали соединение из примера 91 в количестве 55,65, 111,3 и 222,6 мг/кг соответственно с помощью внутрибрюшинных инъекций ("ip") в 1-й день и с помощью инъекций в хвостовую вену ("iv") на 8- и 15-й дни. Камптотецин в количестве 7,5 мг/кг вводили мышам в группе 6, ip в день 1 и iv в дни 8 и 15. Мышам в группах 7, 8 и 9 вводили соединение из примера 92 в количестве 55,3, 110,6 и 221,2 мг/кг соответственно ip в день 1 и iv в дни 8 и 15. В исследовании В (табл. 3B) животные в группе 3 получали камптотецин 7,5 мг/кг iv в дни 1 и 8 и ip в день 15. Мышам из групп 4, 5 и 6 вводили соединение из примера 93 в количестве 55,45, 110,9 и 221,8 мг/кг iv в дни 1 и 8 и ip в день 15. Каждую дозу вводили в объеме 0,2 мл на 20 г веса тела и масштабировали в зависимости от веса тела животного. Кривые медианы роста опухоли на фиг. 1-3 показывают медиану объема опухоли как функцию от времени. Если животное выводилось из эксперимента в результате размера опухоли, то конечный объем опухоли, зарегистрированный для животного, включали вместе с данными, используемыми для вычисления медианного объема опухоли для группы в последующих временных точках. Кривые заканчивались после того, как 50% животных в группе выводилось из эксперимента. На фиг. 1 показаны кривые медианы роста опухолей для соединения из примера 91, которые показывают in vivo эффекты соединения из примера 91 в трех различных дозировках на медиану роста опухоли в исследовании А в сравнении с отсутствием лечения и лечением только камптотецином. На фиг. 2 показаны кривые медианы роста опухолей для соединения из примера 92, которые показывают in vivo эффекты соединения из примера 92 в трех различных дозировках на медиану роста опухоли в исследовании А в сравнении с отсутствием лечения и лечением только камптотецином. На фиг. 3 показаны кривые медианы роста опухолей для соединения из примера 93, которые показывают in vivo эффекты соединения из примера 93 в трех различных дозировках на медиану роста опухоли в сравнении с отсутствием лечения и лечением только камптотецином. Исходные данные из исследования А приведены в табл. 4 А, в которой столбцы D1-D39 относятся к дням 1-39, описанным выше, и строки G1-G9 относятся к группам 1-9, описанным в табл. 3 А, и в которой средний объем опухоли (мм 3) регистрировали в соответствии с днем и группой, указанными в табл. 4 А. Таблица 4 А Исходные данные для исследования В приведены в табл. 4 В, в которой столбцы D1-D40 относятся к дням 1-40, описанным выше, G1-G6 относятся к группам 1-6, описанным в табл. 3 В, и в которой средний объем опухоли (мм 3) регистрировали в соответствии с днем и группой, указанными в табл. 4 В. Таблица 4 В В исследовании А конъюгат из примера 91 давал дозозависимую TGD, которая становилась статистически значимой при наиболее высокой дозе, 222,6 мг/кг, и 4/4 регрессии опухолей вTGD-оцениваемом образце. TGI-анализ на 18-й день выявил дозозависимую противоопухолевую активность, имеющую потенциальную терапевтическую значимость при наиболее высокой дозировке. В исследовании А конъюгат из примера 92 давал дозозависимую TGD, которая становилась статистически значимой и давала 3/5 регрессии опухолей при наиболее высокой дозе, 221,2 мг/кг. TGI на 18-й день также было дозозависимое, статистически значимое при 110,6 и 221,2 мг/кг, и указывало на потенциальную терапевтическую значимость при наиболее высокой дозировке. В исследовании В оценивали конъюгат из примера 93. Кривые роста опухоли указывали на явное замедление роста опухолей при всех дозировках, хотя TGD-анализ не отличал ответы на соединение из примера 93 от роста опухоли в контрольной группе, не получавшей воздействие. Анализ на 15-й день выявил 71 и 77%-ное TGI при дозировках 110,9 и 221,8 мг/кг соответственно, имеющих потенциальное терапевтическое значение. В общем, три конъюгата, оцениваемые в исследованиях А и В, демонстрировали потенциальные терапевтические эффекты в отношении ксенографта MCF-7 бестимусным мышам. В частности, при максимальных исследуемых дозировках активность конъюгатов превышала активность неконъюгированного камптотецина в количестве 7,5 мг/кг. Анализ pK. Профиль pK для подгруппы соединений был определен с использованием стандартных лабораторных мышей и стандартных лабораторных методик. Специалисту в данной области известно и понятно,что для осуществления такого определения также могут быть использованы множество млекопитающих,включая мышей, крыс и обезьян, и модификации этого метода, такие как изменение дозы, подбор времени или режима анализа. Двум мышам на один момент времени внутривенно вводили дозу 4 мг/кг соединений согласно примерам 91, 92, 93 или 96. Образцы крови забирали через 5, 15, 30, 60, 120, 180, 240, 360 мин после введения. Образцы плазмы (20 мкл) подкисляли 2 мкл 20% уксусной кислоты и осаждали с помощью 60 мкл ацетонитрила. Полученный раствор подвергали центрифугированию. Надосадочную жидкость (20 мкл) разводили в 100 мкл 0,1% водной уксусной кислоты и 50 мкл вводили для анализа LC-MS (жидкостная хроматография с масс-спектрометрическим детектором) в масс-спектрометрической системе API-4000. Условия жидкостной хроматографии были следующими: буфер А представлял собой 1% муравьиную кислоту в воде и буфер В представлял собой 1% муравьиную кислоту в ацетонитриле. Градиент от 15 до 90% В в течение 10 мин проходил при скорости потока 0,3 мл/мин с использованием колонки Luna С 8(2) 230 мм 3u. Время полувыведения, площадь под кривой (AUC) и выведение (CL) представлены в табл. 5. Таблица 5 Введение. Пептиды по данному изобретению можно предоставить в форме фармацевтически приемлемых солей. Примеры таких солей включают, но не ограничены, соли, образуемые органическими кислотами(например, уксусной, молочной, малеиновой, лимонной, яблочной, аскорбиновой, янтарной, бензойной,метансульфоновой, толуолсульфоновой или памовой кислотами), неорганическими кислотами (напри- 26020613 мер, хлористо-водородной, серной или фосфорной кислотами) и полимерными кислотами (например,дубильной кислотой, карбоксиметилцеллюлозой, полимолочной, полигликолевой кислотами или сополимерами полимолочной-гликолевой кислот). Обычный способ изготовления соли пептида по настоящему изобретению хорошо известен в данной области и может быть выполнен стандартными способами обмена солей. Соответственно, соль трифторуксусной кислоты и пептида по настоящему изобретению(TFA соль образуется в результате очистки пептида с использованием препаративной ВЭЖХ с элюцией буферными растворами, содержащими TFA) можно превратить в другую соль, такую как ацетатная соль,путем растворения пептида в небольшом количестве 0,25 н. водного раствора уксусной кислоты. Полученный в результате раствор наносят на полупрепаративную ВЭЖХ-колонку (Zorbax, 300 SB, C-8). Колонку элюируют (1) 0,1 н. водным раствором ацетата аммония в течение 0,5 ч, (2) 0,25 н. водным раствором уксусной кислоты в течение 0,5 ч и (3) линейным градиентом (от 20 до 100% раствора В за 30 мин) при скорости потока 4 мл/мин (раствором А является 0,25 н. водный раствор уксусной кислоты; раствором В является 0,25 н. раствор уксусной кислоты в смеси ацетонитрил/вода, 80/20). Содержащие пептид фракции собирают и лиофилизируют. Дозировка действующего ингредиента в композициях по данному изобретению может варьировать,однако необходимо, чтобы количество действующего ингредиента было достаточным для получения соответствующей лекарственной формы. Конкретная дозировка зависит от желаемого терапевтического эффекта, пути введения и продолжительности лечения. В общем, эффективная дозировка для активности по настоящему изобретению находится в диапазоне от 110-7 до 200 мг/кг/день, предпочтительно от 110-4 до 100 мг/кг/день, которую можно вводить одной дозой или разделенной на несколько доз. Соединения по настоящему изобретению можно вводить перорально, парентерально (например, с помощью внутримышечных, внутрибрюшинных, внутривенных или подкожных инъекций или имплантов), назально, вагинально, ректально, сублингвально или местно, и соединения можно ввести в фармацевтический состав совместно с фармацевтически приемлемыми носителями для получения лекарственных форм, подходящих для каждого пути введения. Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли,порошки и гранулы. В таких твердых лекарственных формах действующее соединение смешано по меньшей мере с одним инертным фармацевтически приемлемым носителем, таким как сахароза, лактоза или крахмал. Такие лекарственные формы также обычно содержат дополнительные вещества, помимо таких инертных разбавителей, например смазывающие вещества, такие как стеарат магния. В случае капсул, таблеток и пилюль лекарственные формы могут также содержать буферные агенты. Таблетки и пилюли можно дополнительно приготовить с кишечно-растворимой оболочкой. Жидкие лекарственные формы для перорального введения включают, без ограничения, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и т.п., содержащие инертные разбавители, обычно используемые в данной области, такие как вода. Помимо таких инертных разбавителей, композиции могут также включать в себя адъюванты, такие как увлажняющие агенты, эмульгирующие и суспендирующие агенты, и подсластители, вкусовые и ароматические агенты. Препараты по настоящему изобретению для парентерального введения включают, без ограничения,стерильные водные или неводные растворы, суспензии, эмульсии и т.п. Примеры неводных растворителей или носителей включают пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин, и подходящие для инъекций органические сложные эфиры, такие как этилолеат. Такие лекарственные формы могут также содержать адъюванты, такие как консерванты, увлажняющие, эмульгирующие и диспергирующие агенты. Их можно стерилизовать, например, фильтрацией композиций через задерживающие бактерии фильтры, включением в композиции стерилизующих агентов, обработкой композиций радиоактивным излучением или нагревом. Их также можно изготовить в форме стерильных твердых композиций, которые можно растворить в стерильной воде или какой-либо другой стерильной среде для инъекций, непосредственно перед использованием. Композициями для ректального или вагинального введения предпочтительно являются суппозитории, которые могут содержать кроме действующего вещества вспомогательные вещества, такие как масло какао или суппозиторный воск. Композиции для назального или сублингвального введения также приготавливают со стандартными вспомогательными веществами, хорошо известными в данной области. Кроме того, соединение по настоящему изобретению можно вводить в композиции с замедленным высвобождением, такие как композиции, описанные в следующих патентах и патентных заявках. В патенте США 5672659 описаны композиции с замедленным высвобождением, содержащие биологически активный агент и полиэфир. В патенте США 5595760 описаны композиции с замедленным высвобождением, содержащие биологически активный агент в желируемой форме. В патенте США 5821221 описаны полимерные композиции с замедленным высвобождением, содержащие биологически активный агент и хитозан. В патенте США 5916883 описаны композиции с замедленным высвобождением, содержащие биологически активный агент и циклодекстрин. В РСТ публикации WO 99/38536 описаны легко всасываемые композиции биологически активного агента с замедленным высвобождением. В РСТ публикации WO 00/04916 описан способ изготовления микрочастиц, содержащих терапевтический агент,такой как пептид, в системе масло в воде. В РСТ публикации WO 00/09166 описаны комплексы, содержащие терапевтический агент, такой как пептид, и фосфорилированный полимер. В РСТ публикацииWO 00/25826 описаны комплексы, содержащие терапевтический агент, такой как пептид, и полимер, несущий неполимеризующийся лактон. Если не указано иное, то все используемые в настоящем описании технические и научные термины имеют такое же значение, какое под ними понимает средний специалист в области, к которой принадлежит изобретение. Кроме того, все публикации, патентные заявки, патенты и другие ссылки, упомянутые в настоящем описании, тем самым полностью включены в него путем ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) в которой X является цитотоксическим или цитостатическим средством; В 1 представляет собой rv (аминокислоту); каждый из В 2, В 3 и В 4, независимо для каждого случая, представляет собой (Doc)m, (Aepa)n илиZ представляет собой аналог hNPY, соответствующий формуле(С 1-40)алкил или (С 2-40)алкенил и X7 представляет собой H, OH, CO2H или C(O)-NH2; каждый из W1 и W5, независимо для каждого случая, представляет собой CR4R5; каждый из R4 и R5, независимо для каждого случая, представляет собой Н, F, Br, Cl, I, (C1-30)алкил,(С 2-30)алкенил, замещенный (C1-30)алкил, замещенный (С 2-30)алкенил, SR6, S(O)R7 или S(O)2R8 или R4 и R5 вместе образуют (С 3-30)циклоалкильное, (С 3-30)гетероциклическое или (С 5-30)арильное кольцо; каждый из R6, R7 и R8, независимо для каждого случая, представляет собой (C1-30)алкил,(С 2-30)алкенил, замещенный (C1-30)алкил или замещенный (С 2-30)алкенил; каждый из W2, W3 и W4, независимо для каждого случая, представляет собой CR9R10, О, S, (CH2)t или отсутствует; каждый из R9 и R10, независимо для каждого случая, представляет собой Н, F, Br, Cl, I, (C1-30)алкил,(С 2-30)алкенил, замещенный (С 1-30)алкил, замещенный (С 2-30)алкенил, SR6, S(O)R7 или S(O)2R8 или R9 иR10 вместе образуют кольцевую систему;m, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10;n, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10;q, независимо для каждого случая, представляет собой 0, 1, 2, 3, 4 или 5;t, независимо для каждого случая, представляет собой 0, 1, 2 или 3; каждый из R2 и R3, независимо для каждого случая, представляет собой Н, (С 1-40)алкил,(C1-40)гетероалкил, (С 1-40)ацил, (С 2-40)алкенил, (С 2-40)алкинил, арил(C1-40)алкил, арил(С 1-40)ацил, замещенный (C1-40)алкил, замещенный (C1-40)гетероалкил, замещенный (С 1-40)ацил, замещенный (С 2-40)алкенил,замещенный(С 2-40)алкинил или замещенный арил(С 1-40)алкил; или его фармацевтически приемлемая соль. 2. Соединение по п.1, в которомX представляет собой антрациклин, камптотецин или производное камптотецина, паклитаксел или производное паклитаксела или доксорубицин или производное доксорубицина;B2 представляет собой Suc; каждый из В 3 и В 4, независимо для каждого случая, представляет собой (Doc)m, (Aepa)n или отсутствует;R1 представляет собой NH2; каждый из R2 и R3, независимо для каждого случая, представляет собой Н или (С 1-30)ацил; при условии, что когда R2 является (C1-30)ацилом, R3 является Н; каждый из R4 и R5, независимо для каждого случая, представляет собой Н или (С 1-40)ацил;q представляет собой 4; и каждый из X1, X2, X3, X4 и X5, независимо для каждого случая, представляет собой Н, CH2NH2 илиNH2; или его фармацевтически приемлемая соль. 3. Соединение по п.2, в котором X представляет собой антрациклин, или его фармацевтически приемлемая соль. 4. Соединение по п.2, в котором X представляет собой камптотецин или производное камптотецина,где указанное производное камптотецина представляет собой
МПК / Метки
МПК: A61K 38/16
Метки: рецептор, связывающим, конъюгаты, соединением, нейропептида, цитотоксические
Код ссылки
<a href="https://eas.patents.su/30-20613-citotoksicheskie-konyugaty-s-soedineniem-svyazyvayushhim-receptor-nejjropeptida-y.html" rel="bookmark" title="База патентов Евразийского Союза">Цитотоксические конъюгаты с соединением, связывающим рецептор нейропептида y</a>
Цитотоксические агенты, включающие новые производные томаймицина, конъюгаты, способы получения конъюгатов, фармацевтическая композиция, включающая конъюгат, и их терапевтическое применение

Номер патента: 17196
Опубликовано: 30.10.2012
Авторы: Чари Рави В.Дж., Дэн Юнхун, Ли Вэй, Гози Лоранс, Чжао Роберт, Бушар Эрве, Коммерсон Ален
МПК: C07D 487/04
Метки: конюгатов, способы, конъюгат, фармацевтическая, производные, конъюгаты, композиция, агенты, получения, терапевтическое, включающие, включающая, применение, новые, цитотоксические, томаймицина
Формула / Реферат:
1. Соединения томаймицина формулы (I)гдепредставляет собой необязательную одинарную связь;представляет собой либо одинарную связь, либо двойную связь;при условии, что когдапредставляет собой одинарную связь, то U и U', одинаковые или разные, независимо представляют собой Н и W и W', одинаковые или разные, независимо выбраны из группы, состоящей из OH, -OR, -OCOR, -OCOOR, -OCONRR', -NRCONRR', -OCSNHR, -SH, -SR, -SOR, -SOOR, -SO3-, -NRSOOR,...
Аналоги нейропептида y, содержащие по меньшей мере одну замену на синтетическую аминокислоту

Номер патента: 19498
Опубликовано: 30.04.2014
Авторы: Чжоу Кевин Л., Дун Чжэн Синь, Деоливейра Дэниел Б.
МПК: A61K 38/00
Метки: синтетическую, аминокислоту, замену, содержащие, аналоги, одну, нейропептида, мере, меньшей
Формула / Реферат:
1. Соединение формулы (I) (SEQ ID NO: 2)где А1 представляет собой Tyr, (X1,X2,X3,X4,X5)Phe или HN-СН((CH2)n-N(R4R5))-С(O);А2 представляет собой Pro, 3Hyp, цис-3Hyp, 4Hyp или цис-4Hyp;А3 представляет собой Ser, Abu, Aib, Ala, Thr или HN-CH((CH2)n-N(R4R5))-C(O);А4 представляет собой Lys, Arg, hArg, Dab, Dap, Orn или HN-CH((CH2)n-N(R4R5))-C(O);А5 представляет собой Pro, 3Hyp, цис-3Hyp, 4Hyp или цис-4Hyp;А6 представляет собой Asp, Aib, Asn, Gln, Glu...
Цитотоксические депсипептиды
Номер патента: 10585
Опубликовано: 30.10.2008
Авторы: Куэвас Кармен, Малет Лейре, Каньедо Либрада Мария, Ромеро Пако, Фернандо Рейес Хосе
МПК: C07K 5/097, A61P 35/00, A61K 38/12...
Метки: депсипептиды, цитотоксические
Формула / Реферат:
1. Соединение общей формулы I где каждая из групп R1 независимо выбрана из группы, состоящей из водорода, незамещенного С1-С4алкилидена, замещенного или незамещенного С1-С4алкила, где, в случае замещенных производных, заместитель представляет собой OR', где R' представляет собой С(=O)С1-С4алкил или С1-С4алкил; R2 представляет собой водород; каждая из групп R3 независимо выбрана из группы, состоящей из водорода, галогена, гидроксила, замещенного...
Усовершенствованные цитотоксические агенты, содержащие новые мэйтансиноиды

Номер патента: 10909
Опубликовано: 30.12.2008
Авторы: Уиддисон Уэйн К., Чари Рави В.Дж.
МПК: C07D 491/12, A61K 31/5365
Метки: агенты, содержащие, цитотоксические, новые, мэйтансиноиды, усовершенствованные
Формула / Реферат:
1. Мэйтансиноид, имеющий в положении С-3, С-14-гидроксиметил, С-15-гидрокси или С-20-десметил боковую цепочку ацилированной аминокислоты с ацильной группой, несущей затрудненную сульфгидрильную группу, в которой атом углерода ацильной группы, несущей тиоловую функциональную группу, имеет один или два заместителя, причем указанные заместители представляют собой линейный алкил или алкенил, имеющий от 1 до 10 атомов углерода, разветвленный или...
Цитотоксические агенты, содержащие новые таксаны

Номер патента: 12625
Опубликовано: 30.10.2009
Авторы: Миллер Майкл Л., Чари Рави В Дж., Коммерсон Ален, Балоглу Эркан
МПК: A61P 35/00, C07D 493/06, A61K 39/395...
Метки: таксаны, содержащие, новые, цитотоксические, агенты
Формула / Реферат:
1. Таксаны, имеющие следующую формулу (I): где Z означает радикал формулы II; R1 представляет собой трет-бутоксигруппу; R3 представляет собой изобутенил или фурил; R4 представляет собой связующую группу, выбранную из дисульфидной группы и тиоэфирной группы, гидроксигруппу, ацетоксигруппу, (С2-С4)алкилоксикарбонилоксигруппу, (С1-С2)диалкилкарбамоилоксигруппу, причем ацетоксигруппа, (С2-С4)алкилоксикарбонилоксигруппа и...
Предыдущий патент: Применение 7-хлор-n,n,5-триметил-4-оксо-3-фенил-3,5-дигидро-4н-пиридазин[4,5-b]индол-1-ацетамида в качестве биомаркера уровней периферического бензодиазепинового рецептора
Следующий патент: Натягиваемое абсорбирующее изделие
Случайный патент: Наклонная кровельная система и изоляционная плита для наклонных кровельных систем