Ингибиторы диацилглицеролацилтрансферазы и фармацевтическая композиция, включающая их
Номер патента: 20128
Опубликовано: 29.08.2014
Авторы: Риззо Джон Роберт, Бьючемп Томас Джеймс, Арнольд Маклин Брайан, Ши Цин, Шаус Джон Менерт, Люй Цзяньлян, Кэнада Эмили Джейн, Хембр Эрик Джеймс


























Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль;
где R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -CH3, -ОН, -O(C1-C4 алкила), -О-циклопропила, -О-СН2-фенила, -OC(H)F2 и -OCF3;
R2 выбран из группы, состоящей из Н и -СН3;
R3 выбран из группы, состоящей из Н и -СН3, при условии, что по меньшей мере один из группы, состоящей из R2 и R3, представляет собой Н; и
n равен 1, 2 или 3.
2. Соединение или его соль по п.1, где n равен 1.
3. Соединение или его соль по любому из пп.1 или 2, где R2 представляет собой Н и R3 представляет собой Н.
4. Соединение или его соль по любому из пп.1-3, где R1 выбран из группы, состоящей из Cl и -О-(C1-C4 алкила).
5. Соединение или его соль по любому из пп.1-4, где указанное соединение представляет собой R-изомер.
6. Соединение по любому из пп.1-5 формулы

или его фармацевтически приемлемая соль.
7. Соединение по любому из пп.1-5 формулы

или его фармацевтически приемлемая соль.
8. Фармацевтическая композиция, ингибирующая диацилглицеролацилтрансферазу 1, содержащая соединение по любому из пп.1-7 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
9. Применение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли в качестве лекарственного средства для лечения диабета и/или ожирения.
Текст
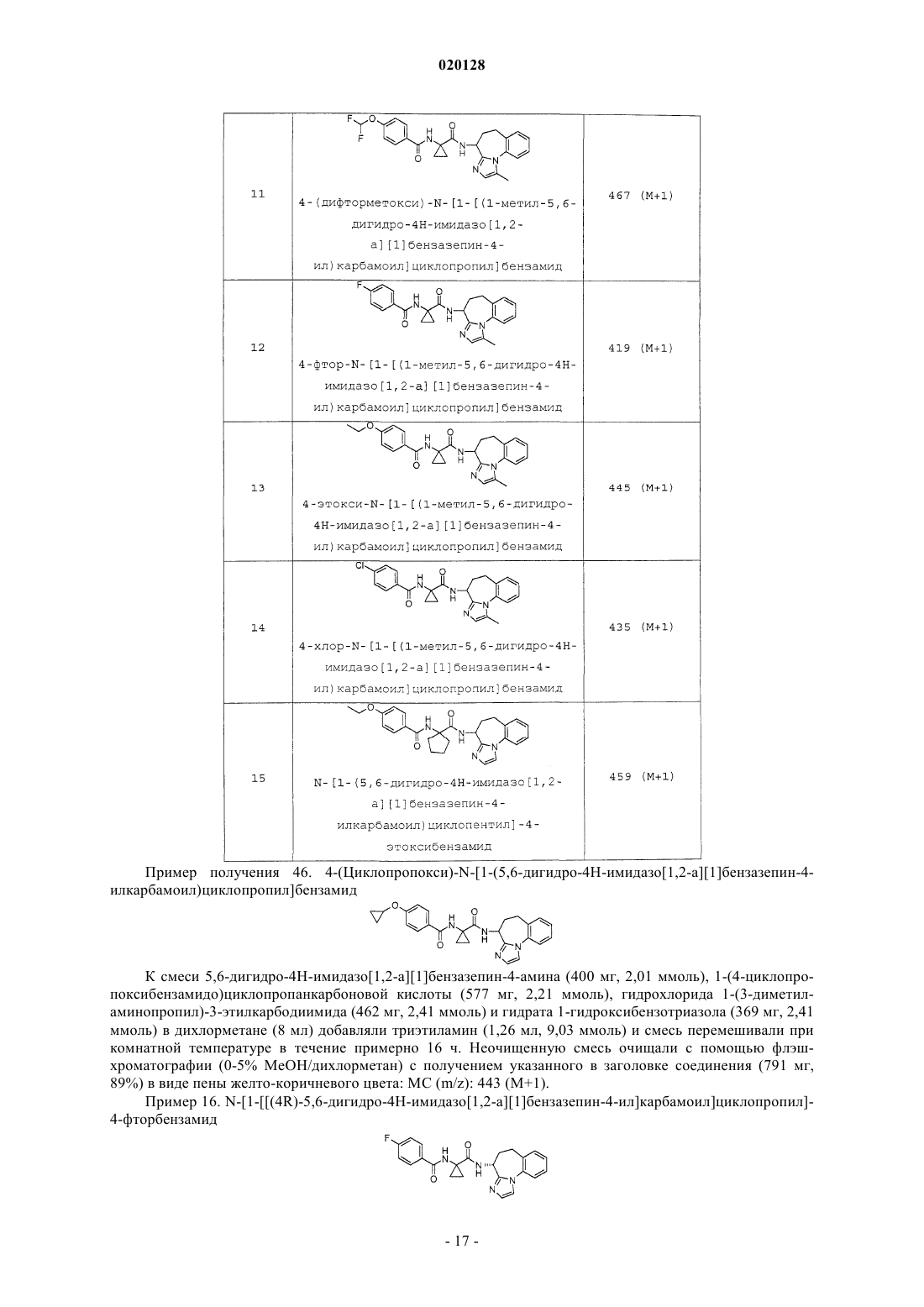
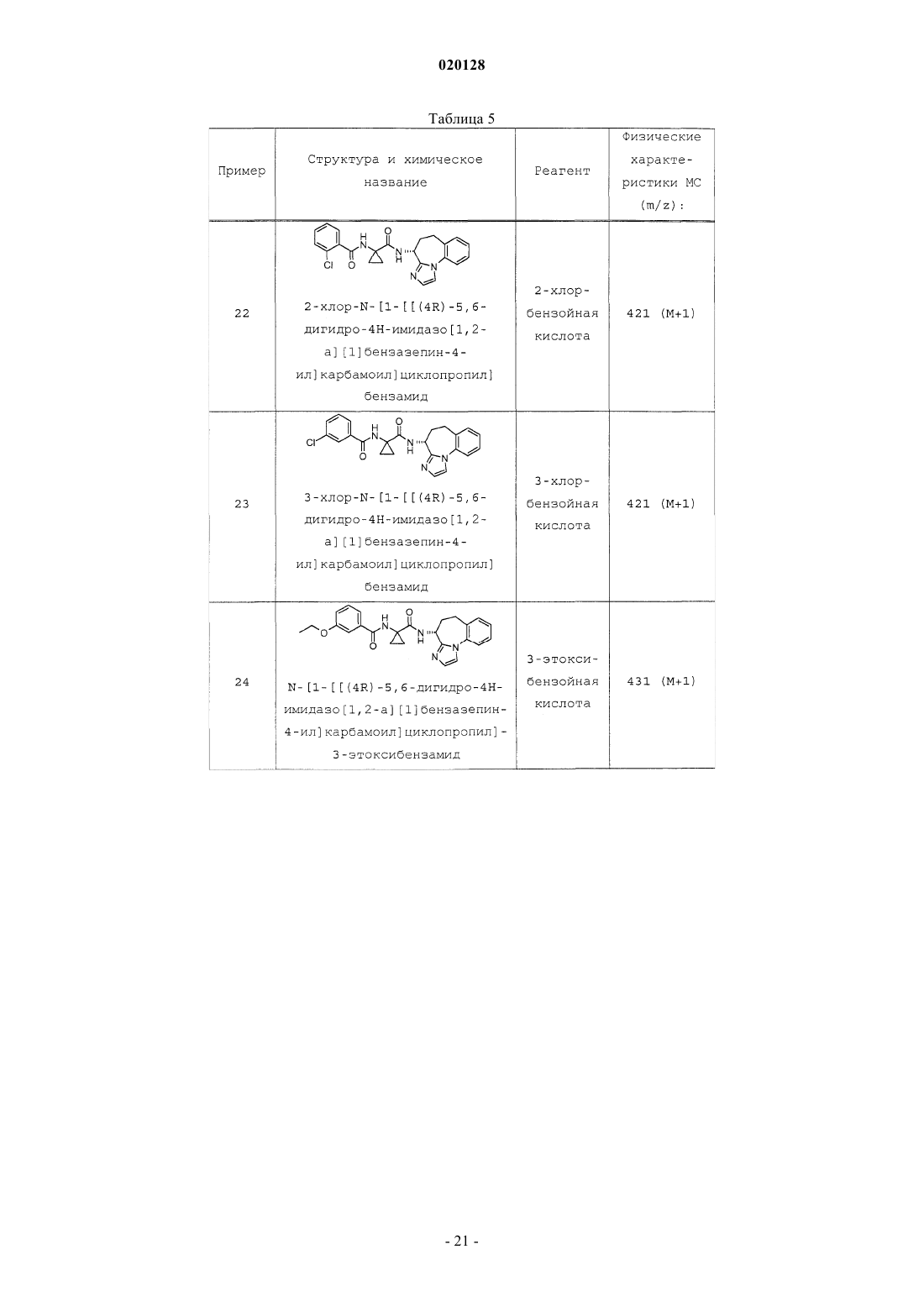
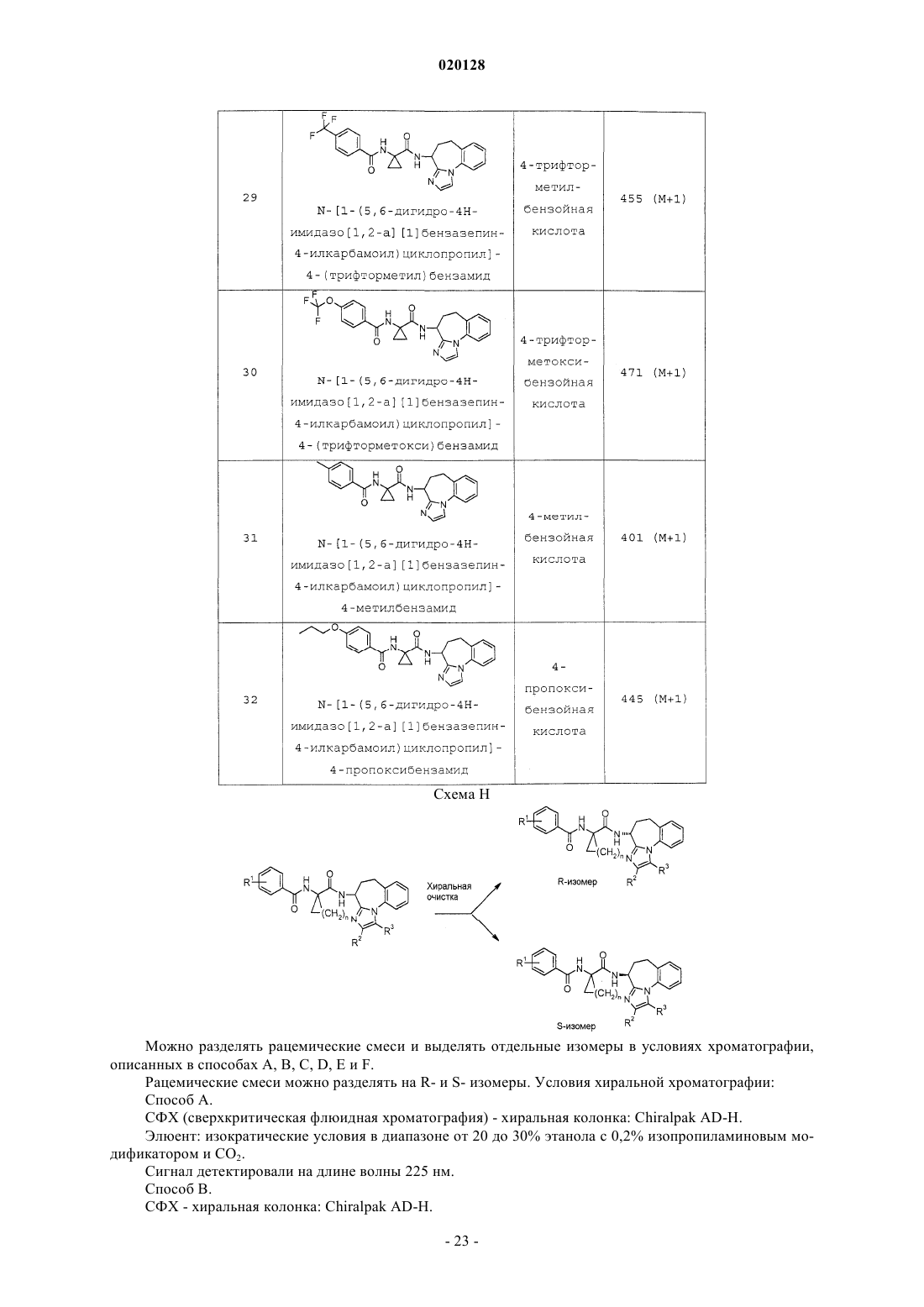
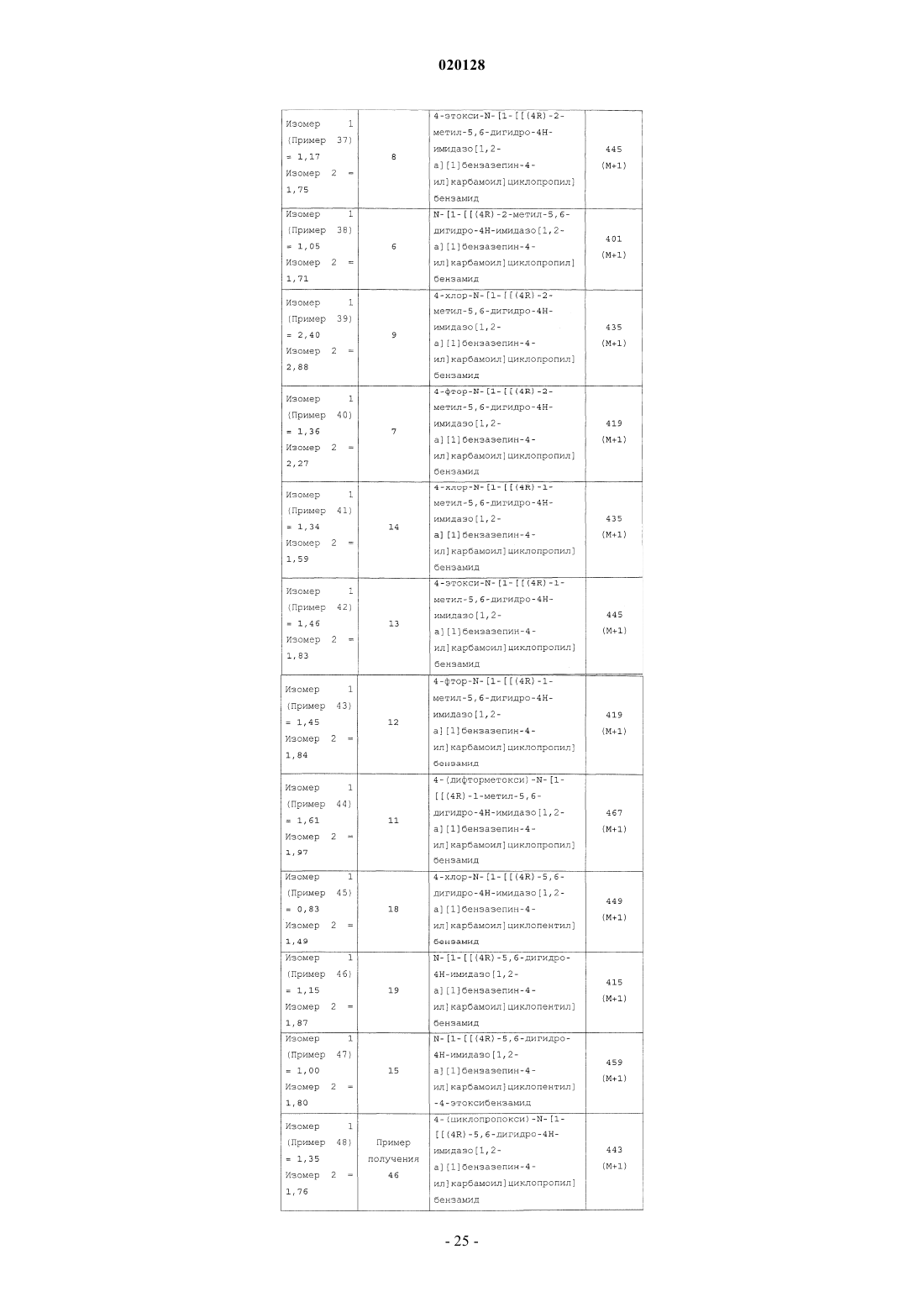
или ее фармацевтически приемлемой соли, где R1 выбран из группы, состоящей из Н, F, Cl, -CF3,-CH3, -ОН, -O(C1-C4 алкила) , -О-циклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3, R2 выбран из группы, состоящей из Н и -СН 3, R3 выбран из группы, состоящей из Н и -СН 3, при условии что по меньшей мере один из группы, состоящей из R2 и R3, представляет собой Н, и n равен 1, 2 или 3, в качестве ингибиторов диацилглицеролацилтрансферазы 1 (DGAT-1). Также настоящее изобретение относится к фармацевтической композиции, содержащей данное соединение, и к применению соединения в качестве лекарственного средства для лечения диабета и/или ожирения.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Согласно настоящему изобретению предложены соединения, которые ингибируют диацилглицеролацилтрансферазу 1 (DGAT-1). Было высказано предположение, что ингибирование DGAT-1, ключевого фермента в синтезе триглицеридов, может представлять собой новый подход к лечению ожирения и/или улучшению чувствительности к инсулину. Несмотря на имеющиеся сообщения о том, что DGAT является превосходной мишенью для низкомолекулярных ингибиторов, сообщалось лишь об ограниченном числе соединений, подавляющих активность DGAT-1. Не существует известных ингибиторов DGAT-1, одобренных для фармацевтического применения. Таким образом, существует потребность в новых малых молекулах, ингибирующих DGAT-1. Существует особая потребность в соединениях, представляющих собой ингибиторыDGAT-1, имеющих желаемые фармакологические характеристики. В заявке на патент США 2008/0090876 (Cheng et.al.) предложены соединения, которые, как сообщается, являются модуляторами DGAT. Соединения, описанные Ченгом (Cheng), представляют собой тианкарбоксамиды. В WO 98/28268 (We et.al.) описаны циклоалкилы, лактамы, лактоны и родственные соединения в качестве ингибиторов высвобождения -амилоидного пептида для применения в лечении болезни Альцгеймера. В патенте США 5696111 (Balwin et.al.) предложены ацилбензазепины в качестве соединений, которые могут применяться для лечения аритмий. В противоположность этому, имидазобензазепины согласно настоящему изобретению структурно отличны от соединений, описанных Ченгом(Cheng), Уи (We) и Балвином (Balwin). Кроме того, не сообщалось и не высказывалось предположений о том, что соединения, описанные Уи или Балвином, могут применяться в качестве ингибиторов DGAT-1. В WO 2010/056496, опубликованной 20 мая 2010 года, сообщается об азепиноновых соединениях, являющихся модуляторами DGAT. Азепиноновые соединения, о которых сообщается в WO 2010/056496,структурно отличны от имидазобензазепинов согласно настоящему изобретению. Соединения согласно настоящему изобретению являются высокоактивными ингибиторами DGAT1. Согласно настоящему изобретению предложен желаемый новый способ лечения, действующий через фармакологический механизм, являющийся уникальным по сравнению с коммерчески доступными способами лечения. Кроме того, некоторые соединения согласно настоящему изобретению селективно ингибируют DGAT-1 по сравнению с DGAT-2. Фармакологический профиль соединений согласно настоящему изобретению в качестве селективных ингибиторов DGAT-1 может являться особенно желательным для применения для лечения ожирения и/или улучшения чувствительности к инсулину. Согласно настоящему изобретению предложены соединения формулы I или их фармацевтически приемлемая соль; где R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -CH3, -ОН, -O(C1-C4 алкила), -Оциклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3;R3 выбран из группы, состоящей из Н и -СН 3; при условии что по меньшей мере один из группы,состоящей из R2 и R3, представляет собой Н; иn равен 1, 2 или 3. Предпочтительно согласно настоящему изобретению предложены соединения формулы I,где R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -CH3, -ОН, -ОСН 3, -ОСН 2 СН 3, -ОСН 2 СН 2 СН 3,-ОСН 2 СН(СН 3)2, -О-циклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3; n равен 1 или 3; и каждый из R2 иR3 представляет собой Н. Предпочтительно согласно настоящему изобретению предложены соединения формулы I или их фармацевтически приемлемые соли; где R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -CH3, -ОН, -ОСН 3, -ОСН 2 СН 3, -ОСН 2 СН 2 СН 3,-ОСН 2 СН(СН 3)2, -О-циклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3; иn равен 1. Предпочтительно согласно настоящему изобретению предложены соединения формулы I или их фармацевтически приемлемые соли; где R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -CH3, -ОН, -O(C1-C4 алкила), -Оциклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3; и n равен 3. Предпочтительно согласно настоящему изобретению предложено соединение формулы I или его фармацевтически приемлемая соль; где R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -CH3, -ОН, -O(C1-C4 алкила), -Оциклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3; n равен 3; и R3 представляет собой Н. Предпочтительно согласно настоящему изобретению предложено соединение формулы I или его фармацевтически приемлемая соль,-1 020128 где R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -CH3, -ОН, -O(C1-C4 алкила), -Оциклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3; n равен 1; и R3 представляет собой Н. Следующее может являться предпочтительным: соединения формулы I или их соли, где R3 представляет собой Н, являются предпочтительными; соединения формулы I или их соли, где n равен 1 или 3, являются предпочтительными; соединения формулы I или их соли, где n равен 1, являются предпочтительными; соединения формулы I или их соли, где R2 представляет собой Н, и R3 представляет собой Н, также являются предпочтительными. Соединения формулы I или их соли, где соединение представляет собой R-изомер, являются предпочтительными. Соединения формулы I или их соли, где R1 выбран из группы, состоящей из F, Cl, -CF3, -СН 3, -ОН,-ОСН 3, -ОСН 2 СН 3, -ОСН 2 СН 2 СН 3, -ОСН 2 СН(СН 3)2, -О-циклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3,являются предпочтительными. Соединения формулы I или их соли, где R1 выбран из группы, состоящей из F, Cl и -O(C1-C4 алкила), являются предпочтительными. Соединения формулы I или их соли, где R1 выбран из группы, состоящей из Cl и -O(C1-C4 алкила),являются предпочтительными. Соединения формулы I или их соли, где R1 представляет собой -ОСН 2 СН 3, являются предпочтительными. Соединения формулы I или их соли, где R1 представляет собой Cl, являются предпочтительными. Соединения формулы I или их соли, где R2 представляет собой СН 3, и R3 представляет собой Н,также являются предпочтительными. Соединения, где n равен 3, являются предпочтительными. Согласно предпочтительному варианту реализации изобретения предложено соединение формулы или его фармацевтически приемлемая соль. Согласно предпочтительному варианту реализации изобретения предложено соединение формулы или его фармацевтически приемлемая соль. Соединения формулы I могут находиться в форме рацемической смеси. Предпочтительный изомер представляет собой "R"-стереоформу, представленную далее в настоящей заявке формулой II. Предпочтительно согласно настоящему изобретению предложены соединения формулы I, имеющие следующую изомерную конформацию, представленную формулой II или их фармацевтически приемлемые соли. Соединения формулы I можно синтезировать и затем разделить R- и S- изомеры с помощью хиральной хроматографии. Соединения формулы I могут быть синтезированы с помощью хирального синтеза для специфичного синтеза предпочтительного R-изомера. Согласно другому варианту реализации настоящего изобретения предложено применение соединения, заявленного согласно настоящему изобретению, или его фармацевтически приемлемых солей для получения лекарственного средства для лечения диабета и/или ожирения. Согласно другому варианту реализации изобретения предложено соединение, заявленное согласно настоящему изобретению, или его соли для применения для лечения ожирения. Согласно другому варианту реализации настоящего изобретения предложено соединение, заявленное в настоящей заявке, или его фармацевтически приемлемые соли для применения в терапии. Согласно другому варианту реализации настоящего изобретения предложено соединение, заявленное согласно настоящему изобретению, или его фармацевтически приемлемые соли, для применения для лечения диабета. Кроме того, изобретение относится к соединению, заявленному согласно настоящему изобретению, для применения для улучшения чувствительности к инсулину у млекопитающего. Согласно настоящему изобретению предложен способ лечения ожирения у млекопитающего, включающий этап введения указанному млекопитающему соединения, заявленного согласно настоящему изобретению, или его фармацевтически приемлемых солей. Согласно настоящему изобретению предложен способ лечения диабета и/или улучшения чувствительности к инсулину у млекопитающего, включающий этап введения указанному млекопитающему соединения, заявленного согласно настоящему изобретению, или его фармацевтически приемлемых солей. Согласно другому варианту реализации настоящего изобретения предложен способ лечения дислипидермии у млекопитающего, включающий этап введения указанному млекопитающему соединения, заявленного согласно настоящему изобретению, или его фармацевтически приемлемых солей. Согласно другому варианту реализации настоящего изобретения предложен способ лечения акне у млекопитающего, включающий этап введения указанному млекопитающему соединения, заявленного согласно настоящему изобретению, или его фармацевтически приемлемых солей. Согласно другому варианту реализации настоящего изобретения, предложен способ лечения вирусного гепатита С (ВГС, HCV), включающий этап введения указанному млекопитающему соединения,заявленного согласно настоящему изобретению, или его фармацевтически приемлемых солей. Согласно другому варианту реализации настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение согласно настоящему изобретению или его фармацевтически приемлемые соли и фармацевтически приемлемый носитель. Согласно другому варианту реализации предложена фармацевтическая композиция согласно настоящему изобретению, дополнительно содержащая второй фармацевтический агент. Согласно другому варианту реализации настоящее изобретение относится к лекарственным формам, содержащим соединение, заявленное согласно настоящему изобретению, или его фармацевтически приемлемые соли, фармацевтически приемлемый носитель и гидроксипропилметилцеллюлозу. Согласно другому варианту реализации изобретения предложена фармацевтическая лекарственная форма, где гидроксипропилметилцеллюлоза представоляет собой ацетат сукцинат гидроксипропилметилцеллюлозы(НРМС-AS-L). Согласно другому варианту реализации изобретения предложен способ получения лекарственной формы, включающий приведение соединения, заявленного согласно настоящему изобретению,или его фармацевтически приемлемых солей, в контакт с гидроксипропилметилцеллюлозой. Согласно другому варианту реализации изобретения предложен способ получения фармацевтической лекарственной формы, где гидроксиметилцеллюлоза представляет собой ацетат сукцинат гидроксипропилметилцеллюлозы (НРМС-AS-L). Термин "фармацевтически приемлемая соль" относится к солям соединений согласно настоящему изобретению, которые считаются приемлемыми для клинического применения и/или применения в области ветеринарии. Указанные соли могут быть получены с помощью способов, известных специалистам в данной области техники. Фармацевтически приемлемые соли и общий метод их получения хорошо известны в данной области техники. См., например, P. Stahl, et al. , HANDBOOK OF PHARMACEUTICALSALTS: PROPERTIES, SELECTION AND USE, (VCHA/Wiley-VCH, 2002); S.M. Berge, et al., "Pharmaceutical Salts," Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977. Соединения согласно настоящему изобретению предпочтительно получают в виде фармацевтических композиций, вводимых различными способами. Термин "фармацевтически приемлемый носитель" означает, что указанный носитель,разбавитель, наполнители и соль является фармацевтически совместимой с другими ингредиентами композиции. Наиболее предпочтительно указанные композиции представляют собой композиции для перорального введения. Фармацевтически приемлемые композиции и способы их получения хорошо известны в данной области техники. См., например, REMINGTON: THE SCIENCE AND PRACTICE OFPHARMACY (A. Gennaro, et al. , eds. , 19 th ed., Mack Publishing Co., 1995). Соединения согласно настоящему изобретению могут быть получены с помощью способов, показанных на схемах А, В, С, D, Е, F, G, Н, I и J, и так, как описано в примерах получения и примерах. Соединения в примерах получения и примерах названы с использованием программы Symyx Draw, версии 3,1. Термины и аббревиатуры, используемые в схемах, примерах получения, примерах и способах в настоящей заявке, имеют общепринятые значения, если иное не указано. Термины и аббревиатуры, используемые в настоящих схемах, примерах получения, примерах и способах, имеют общепринятые значения, если не указано иное. Например, при использовании в настоящей заявке следующие термины имеют указанные значения: N,N-диметилформамид (ДМФА); метил-трет-бутиловый эфир (МТБЭ); тетрагидрофуран (ТГФ); хроматография в псевдоожиженном слое На схеме выше R1 предпочтительно выбран из группы, состоящей из Н, F, Cl, -CF3, -СН 3, -ОН,-O(C1-C4 алкила), -O-циклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3. Более предпочтительно R1 представляет собой -O(C1-C4 алкил) или Cl. Наиболее предпочтительно R1 представляет собой -ОСН 2 СН 3 или(3 л) добавляли оксалилхлорид (189 мл, 2,17 моль). Через 1 ч при температуре 25-26 С смесь концентрировали при пониженном давлении до постоянной массы. Гидрохлорид этил-1-аминоциклопропанкарбоксилата (300 г, 1,81 моль) и дихлорметан (2,5 л) перемешивали на ледяной/водяной бане. Добавляли триэтиламин (631 мл, 4,53 моль) и затем добавляли раствор хлорангидрида (примерно 380 г) в дихлорметане (500 мл). Реакционную смесь перемешивали в течение примерно 16 ч при комнатной температуре. Реакционную смесь разбавляли 1 н. водным раствором соляной кислоты (1 л). Водный слой экстрагировали дихлорметаном (1 л) . Органический слой промывали водой (2 л) и соляным раствором (2 л). Раствор сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок суспендировали в гексанах и фильтровали с получением указанного в заголовке соединения (478 г, 95%) в виде порошка цвета слоновой кости: МС (масс-спектрометрия) (m/z): 278 (М+1). Пример получения 2. 1-[(4-этоксибензоил)амино]циклопропанкарбоновая кислота. Этил-1-[(4-этоксибензоил)амино]циклопропанкарбоксилат (478 г, 1,72 моль) добавляли к тетрагидрофурану (2 л) и метанолу (1 л). Гидроксид лития (LiOH) растворяли (144,7 г, 3,45 моль) в воде (1 л) и добавляли его к полученной смеси. Реакционную смесь перемешивали по мере ее нагревания до 55 С. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. После удаления 2,5 л растворителя смесь разбавляли водой (2 л) и помещали на ледяную/водяную баню. Значение рН доводили до 2 водным 5 н. раствором соляной кислоты (750 мл). Осадок фильтровали,отфильтрованный осадок промывали водой (22 л) и сушили с получением указанного в заголовке соединения (425 г, 99%) в виде белого порошка: МС (m/z): 250 (М+1). Пример получения 3. Этил-1-[(4-хлорбензоил)амино]циклопропанкарбоксилат К диизопропилэтиламину (187,86 мл, 1,08 моль) в дихлорметане (900 мл) добавляли этил-1 аминоциклопропанкарбоксилат (89,2 г, 0,54 моль). Колбу помещали на ледяную/водяную баню и медленно добавляли хлорид 4-хлорбензойной кислоты (75,46 мл, 0,59 моль). После завершения добавления баню удаляли, температуре реакционной смеси позволяли установиться при 20 С и перемешивали в течение примерно 2 ч. Промывали 1 н. раствором соляной кислоты (HCl) с последующей промывкой насыщенным раствором бикарбоната натрия (NaHCO3), водой и затем соляным раствором. Органический слой сушили над сульфатом натрия (Na2SO4), фильтровали и концентрировали при пониженном давлении. Осадок суспендирвоали с гептаном, фильтровали и промывали гептаном с получением указанного в заголовке соединения (142 г, 98%) в виде белого порошка: МС (m/z): 268 (М+1). Пример получения 4. 1-[(4-Хлорбензоил)амино]циклопропанкарбоновая кислота К этил-1-[(4-хлорбензоил)амино]циклопропанкарбоксилату (157 г, 0,58 моль) в 1,4-диоксане (2,4 л) добавляли раствор гидроксида лития (123,05 г, 2,93 моль) в воде (1,2 л). Перемешивали реакционную смесь при комнатной температуре в течение 4 ч и затем указанную смесь концентрировали при пониженном давлении. Смесь разбавляли водой (1 л) и помещали указанную смесь на ледяную/водяную баню. Значение рН доводили до примерно 2 водным раствором 5 н. HCl. Осадок фильтровали, отфильтрованный осадок промывали водой (31 л) и сушили (50 С, вакуумная печь) с получением (132 г, 94%) указанного в заголовке соединения в виде белого порошка: МС (m/z): 240 (М+1). Пример получения 5. Бензил-N-[(3R)-2-оксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамат Метанол (5000 мл) объединяли с 5 н. водным раствором соляной кислоты (1393 мл, 5,57 моль) и нагревали до 35 С в течение 15 мин. Добавляли порциями трет-бутил-N-[(3R)-2-оксо-1,3,4,5-тетрагидро-1 бензазепин-3-ил]карбамат (Armstrong III, Joseph D., et al., Tetrahedron Let. 35(20) pp. 3239-3242 (1994) (77 0 г, ммоль, коммерчески доступный) в течение 1 ч. Смесь концентрировали при пониженном давлении до получения твердого вещества белого цвета. Указанное твердое вещество объединяли с дополнительной порцией (3R)-3-амино-1,3,4,5-тетрагидро-1-бензазепин-2-она (другая партия, такой же способ; 660 г,3,78 моль) и растворяли в воде (2,5 л). К полученному раствору добавляли карбонат натрия (3,17 кг, 3,18 моль) и ацетонитрил (7,5 л). Добавляли по каплям бензилхлорформат (7,14 моль, 1,21 кг) в течение 2 ч. Через 2 ч добавляли этилацетат (8 л) при комнатной температуре и фильтровали. Влажный осадок промывали водой (4,0 л), ацетонитрилом (2,0 л) и этилацетатом (3,0 л). Твердое вещество сушили при пониженном давлении в течение 48 ч с получением указанного в заголовке соединения (2,14 кг, 105%): МС К N-[(3R)-2-оксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамату (2,13 кг, 6,88 моль) в 2-метилтетрагидрофуране (15 л) добавляли пентасульфид фосфора (1,68 кг, 7,57 моль) в течение 30 мин и нагревали до 50 С в течение 24ч. Охлаждали до 29 С, добавляли силикагель (2,0 кг), фильтровали и концентрировали при пониженном давлении. Растворяли полученный осадок в хлороформе (4,0 л) и очищали с помощью хроматографии (смесь этилацетат/гексаны, 1:1) с получением указанного в заголовке соединения (1,65 кг, 73%) в виде твердого вещества красного цвета: МС (m/z): 327 (М+1). Пример получения 7. Бензил-N-[2-(2,2-диметоксиэтиламино)-4,5-дигидро-3 Н-1-бензазепин-3-ил] карбамат Бензил-N-(2-тиоксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил)карбамат (1,78 кг, 5,13 моль) и моногидрат пара-толуолсульфоновой кислоты (48,8 г, 256 ммоль) растворяли в дихлорметане (12 л). Диметилацеталь аминоацетальдегида (2,70 кг, 25 моль) добавляли по каплям в течение 1 ч и перемешивали в течение 36 ч при комнатной температуре. Добавляли дополнительную порцию диметилацеталя аминоацетальдегида (1,40 кг, 13 моль) и перемешивали в течение 12 ч при комнатной температуре. Добавляли воду (5,0 л), отделяли органический слой и экстрагировали водным раствором дихлорметана (2,0 л). Органические слои объединяли и промывали водой, насыщенным раствором хлорида аммиака (6,0 л) и соляным раствором (4,0 л). Органическую фазу фильтровали через кизельгур и концентрировали при пониженном давлении с получением указанного в заголовке соединения (2,30 кг, 118%) в виде желтого масла: МС Смесь бензил-N-[2-(2,2-диметоксиэтиламино)-4,5-дигидро-ЗН-1-бензазепин-3-ил]карбамата (1,80 кг, 4,1 моль) и муравьиной кислоты (6,4 л) нагревали при 95 С в течение 80 мин. Смесь концентрировали при пониженном давлении с получением черного масла и распределяли указанную смесь между дихлорметаном (7,0 л) и водой (5 л). Добавляли карбонат натрия до достижения значения рН водного слоя, равного 7. Органический слой удаляли и промывали водой (3 Х). Водные слои экстрагировали дихлорметаном. Органические слои объединяли и концентрировали при пониженном давлении с получением черного масла. 2,297 кг черного масла разделяли на части примерно по 250 г и каждую часть растворяли в дихлорметане (250 мл). Добавляли к 3,5 кг силикагеля (Merck 9385, 60 А, 230-400 меш). Желаемое соединение вымывали смесью этилацетат/гептан (75:25), начиная с шести объемов колонки (более быстро элюирующиеся примеси вымываются первыми). Продолжали вымывать смесью метанол/этилацетат(5:95) для удаления оставшегося продукта (1082 г восстановленного продукта). Анализ смеси продукта проводили с использованием следующих условий хиральной ВЭЖХ: колонка Chiralpak AD-H, 4,6x150 мм, подвижная фаза ацетонитрил/3 А этанол/диметилэтиламин (15:85:0,2), скорость потока 0,6 мл/мин,УФ-детекция на 250 нм. Наблюдали отношение желаемый/нежелаемый изомер 3:2 в указанных условиях. Смесь изомеров растворяли в концентрации 30 мг/мл в смеси ацетонитрил/3 А этанол (20:80). Изомеры очищали с использованием следующих условий препаративной хиральной ВЭЖХ: колонка Chiralpak ADstate recycle and batch chromatographic techniques, Journal of Chromatography A, 1046 p55-60 (2004. Сбор изомера 1 проводили через 0,4 до 0,9 мин и сбор изомера 2 проводили через 2,4 до 4,2 мин. Желаемый изомер (изомер 2; R-изомер) являлся вторым вымывающимся изомером: бензил-N-[(4R)-5,6-дигидро-4 Нимидазо[1,2-а][1]бензазепин-4-ил]карбамат. Фракции, содержащие продукт, концентрировали при пониженном давлении с получением указанного в заголовке соединения (615 г, 73%) в виде твердого вещества белого цвета: МС (m/z): 334 (М+1). Пример получения 9. Дигидройодид (4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амина К дихлорметану (800 мл) добавляли йодтриметилсилан (783 мл, 5,49 моль) и охлаждали на ледяной/водяной бане. Бензил-N-[(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил]карбамат (610 г,1,83 моль) растворяли в дихлорметане (1 л) и добавляли к раствору йодтриметилсилана. После завершения добавления удаляли ледяную/водяную баню и позволяли реакционной смеси перемешиваться в течение примерно 16 ч при комнатной температуре. Смесь гексанов (6 л) и этанола (1 л) перемешивали на ледяной/водяной бане. К перемешиваемой смеси гексан/этанол осторожно добавляли реакционную смесь для избежания пенообразования. Полученную смесь перемешивали при комнатной температуре в течение 10 мин. Полученное твердое вещество извлекали посредством фильтрования, и отфильтрованный осадок промывали гексанами (32 л) с получением указанного в заголовке соединения (817,1 г, 98 %) в виде легкосыпучего твердого вещества оранжевого цвета: МС (m/z): 200 (М+1). Пример получения 10. (4R)-5,6-Дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амин Бензил-N-[(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил]карбамат (120,0 г, 359,9 ммоль) объединяли с этанолом (2,0 л) в гидрогенизаторе Bchi и добавляли 10% палладий на углеродном носителе (Pd/C) (10,0 г, 9,4 ммоль), суспендированный в толуоле (200 мл). Смесь гидрогенизировали (60 фунтов/кв. дюйм, комнатная температура) в течение примерно 64 ч. Указанную смесь фильтровали через кизельгур и отфильтрованный осадок промывали ТГФ (300 мл). Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (60 г, 84%) в виде белой пены: МС Дигидройодид (4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амина (650 г, 1,43 моль), 1-[(4 этоксибензоил)амино]циклопропанкарбоновую кислоту (427 г, 1,71 моль) и гидрат 1-гидроксибензотриазола (262,5 г, 1,71 моль) добавляли к дихлорметану (3 л). Смесь перемешивали на ледяной/водяной бане и медленно добавляли диизопропилэтиламин (797 мл, 4,57 моль). Добавляли гидро хлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (333,6 г, 1,71 моль) в течение 10 мин. Ледяную/водяную баню удаляли и позволяли смеси перемешиваться в течение примерно 16 ч при комнатной температуре. Реакционную смесь промывали водой (34 л). Органическую фазу сушили над сульфатом натрия. Смесь фильтровали и концентрировали при пониженном давлении до получения объема примерно 1 л. Осадок разбавляли этилацетатом (4 л). 2 л растворителя удаляли при пониженном давлении. Смеси позволяли отстаиваться 1 час при комнатной температуре. Полученный осадок отделяли посредством фильтрования и отфильтрованный осадок промывали гексанами (22 л) с получением указанного в заголовке соединения (569,9 г, 93%) в виде твердого вещества бледно-рыжеватого цвета: МС К суспензии (4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амина (30 г, 151 ммоль), 1-гидроксибензотриазола (20 г, 151 ммоль), триэтиламина (41,97 мл, 301 ммоль) и 1-[(4-хлорбензоил)амино] циклопропанкарбоновой кислоты (40 г, 166 ммоль) в ТГФ (300 мл) при температуре 10 С добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (29 г, 151 ммоль). Ледяную баню удаляли,смесь нагревали до комнатной температуры и указанную смесь перемешивали в течение 4 ч. Смесь выливали в воду (1,0 л) и экстрагировали этилацетатом (1,0 л). Органическую фазу промывали насыщенным раствором бикарбоната натрия (500 мл), сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Осадок суспендировали в этилацетате (300 мл) и нагревали до 71 С. Охлаждали до комнатной температуры, осадок фильтровали и промывали этилацетатом (100 мл) с получением указанного в заголовке соединения (38,8 г, 61%): МС (m/z): 421 (M+1). Схема В Способ хирального синтеза на схеме В приводил к получению "R"-изомера. Трет-бутил-N-[(3R)-2-оксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамат, (полученный, по существу, согласно способу, описанному в источнике Armstrong III, Joseph D., et al, Tetrahedron Let. 35(20) pp. 3239-3242 (1994), 15 г, 54,28 ммоль) суспендировали в 1,4-диоксане (100 мл, 1,17 моль) и охлаждали на ледяной бане. Быстро добавляли HCl в диоксане (100 мл, 4,0 М) и после завершения добавления баню удаляли. Через 3 ч добавляли дополнительную порцию HCl в диоксане (100 мл, 4,0 М). Реакционную смесь перемешивали в течение примерно 16 ч. Твердое вещество собирали посредством фильтрования,промывали малым объемом эфира и сушили указанное твердое вещество в печи с получением указанного в заголовке соединения (11,2 г, 99%) в виде твердого вещества белого цвета: МС (m/z): 177 (М+1). Пример получения 12. Бензил-N-[(3R)-2-оксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамат(270 мл). Добавляли бензилхлорформат (6,24 мл, 42,32 ммоль) и реакционную смесь интенсивно перемешивали при комнатной температуре в течение примерно 16 ч. Осадок собирали посредством фильтрования, промывали водой и эфиром и сушили указанный осадок в вакуумной печи с получением указанного в заголовке соединения (7,2 г, 82%) в виде твердого вещества белого цвета: МС (m/z): 311 (М+1). Пример получения 13. Бензил-N-[(3R)-2-тиоксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамат К раствору бензил-N-[(3R)-2-оксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамата (9,4 г, 30,29 ммоль) в тетрагидрофуране (470 мл) добавляли реагент Лавессона (12,25 г, 30,29 ммоль) и реакционную смесь перемешивали в атмосфере азота в течение примерно 16 ч при комнатной температуре. Осадок удаляли посредством фильтрования и сбрасывали. Фильтрат концентрировали при пониженном давлении и полученный осадок растворяли в этилацетате. Промывали водой и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении с получением вязкого желтого масла. К указанному вязкому маслу добавляли эфир и соскребали его с образованием твердого вещества. Твердое вещество собирали посредством фильтрования и промывали эфиром. Указанное твердое вещество растирали с эфиром и собирали посредством фильтрования с получением указанного в заголовке соединения (7,28 г, 74%) в виде твердого вещества белого цвета: МС (m/z): 349 Бензил-N-[(3R)-2-тиоксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамат (8,71 г, 26,68 ммоль), дихлорид ртути (HgCl2) (9,42 г, 34,69 ммоль) и диметилацеталь аминоацетальдегида (11,65 мл, 106,73 ммоль) объединяли в тетрагидрофуране (250 мл) и нагревали до 55 С в течение 20 мин. Смесь охлаждали до комнатной температуры и фильтровали указанную смесь через кизельгур. Отфильтрованный осадок промывали ТГФ и этилацетатом и фильтрат концентрировали при пониженном давлении. Полученное масло растворяли в дихлорметане, промывали теплой водой (4) и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (9,78 г, 92%) в виде твердого вещества: МС (m/z): 398 (М+1). Пример получения 15. Бензил-N-[(4R)-5,6-дигидро-4 Н-имидазо[1,2-а] [1]бензазепин-4-ил]карбамат Бензил-N-[(3R)-2-(2,2-диметоксиэтиламино)-4,5-дигидро-3 Н-1-бензазепин-3-ил]карбамат (9,78 г,24,61 ммоль) растворяли в муравьиной кислоте (60 мл, 96%) и нагревали при 100 С в течение 1,5 ч. Смеси позволяли охлаждаться и черный осадок удаляли посредством фильтрования через пробку из стекло-8 020128 ваты. Фильтрат концентрировали при пониженном давлении, выливали указанный фильтрат в воду и подщелачивали 1 М NaOH. К полученному осадку добавляли этилацетат и органический слой отделяли. Водный слой экстрагировали этилацетатом. Органические слои объединяли, промывали (соляным раствором), сушили над сульфатом натрия и концентрировали при пониженном давлении с получением указанного в заголовке соединения (6,95 г, 85%): МС (m/z): 334 (М+1), энантиомерный избыток 97,8%, время удерживания для изомера 1 (R-изомера) - 4,58 мин; для изомера 2 (S-изомера) - 3,20 мин (колонка Chiralpak AD-H; 100% EtOH с 0,2% диметилэтиламином; скорость потока 1,0 мл/мин; 225 нм). Пример получения 16. (4R)-5,6-Дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амин Бензил-N-[(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил]карбамат (6,95 г, 20,85 ммоль) растворяли в абсолютном этаноле (375 мл, 6,44 моль) и добавляли 10% палладий на углеродном носителе(Pd/C) (0,70 г, 6,57 ммоль). Смесь помещали на шейкер Parr в атмосфере водорода (Н 2) на 7 ч (60 фунтов/кв. дюйм, комнатная температура). Катализатор удаляли посредством фильтрования и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (4,17 г, 100%) в виде твердого вещества желтого цвета: МС (m/z): 200 (М+1). Схема С Способ синтеза на схеме С приводит к получению рацемической смеси. Пример получения 17. Бензил-N-(2-тиоксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил)карбамат К раствору бензил-N-[(3R)-2-оксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамата (10,0 г, 32,22 ммоль) в толуоле (120 мл) добавляли реагент Лавессона (6,52 г, 16,11 ммоль) и реакционную смесь нагревали в атмосфере азота при 100 С в течение 2,5 ч. Реакционной смеси позволяли охлаждаться и осадок собирали посредством фильтрования. Отфильтрованный осадок промывали (эфиром) и сушили в вакуумной печи с получением указанного в заголовке соединения (5,5 г, 16,8 ммоль). Фильтрат концентрировали при пониженном давлении и очищали с помощью флэш-хроматографии (от 30% гексан/CH2Cl2 до 100% CH2Cl2) с получением указанного в заголовке соединения (3,48 г, 10,7 ммоль). Соединение являлось частично рацемизированным. Общий выход указанного в заголовке соединения составлял 85%: МС (m/z): 327 (М+1). Пример получения 18. Бензил-N-[2-(2,2-диметоксиэтиламино)-4,5-дигидро-3 Н-1-бензазепин-3 ил]карбамат Использовали способ согласно примеру получения 14, где в качестве исходного вещества использовали бензил-N-(2-тиоксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил)карбамат (4,2 г, 12,87 ммоль), с получением указанного в заголовке соединения (4,68 г, 92%) в виде твердого вещества рыжеватого цвета: МС Использовали способ примера получения 15, где в качестве исходного вещества использовали бензил-N-[2-(2,2-диметоксиэтиламино)-4,5-дигидро-3 Н-1-бензазепин-3-ил]карбамат (4,6 г, 11,57 ммоль), с получением указанного в заголовке соединения (3,8 г, 88%): МС (m/z): 334 (М+1). Пример получения 20. 5,6-Дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амин Использовали способ примера получения 16, где в качестве исходного вещества использовали бензил-N-(5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил)карбамат (3,36 г, 10,08 ммоль), с получением указанного в заголовке соединения (2,2 г, 100%) в виде желтого масла: МС (m/z): 200 (М+1). Схема D К раствору бензил-N-[(3R)-2-оксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамата (2,0 г, 6,44 ммоль), хлорацетона (1,19 г, 12,89 ммоль) и фторида тетрабутиламмония (1,29 мл, 1,29 ммоль, 1 М раствор в ТГФ) в тетрагидрофуране (65 мл) добавляли порошкообразный гидроксид калия (КОН) (0,72 г,12,89 ммоль). Смесь перемешивали в течение примерно 16 ч при комнатной температуре. К реакционной смеси добавляли дополнительную порцию гидроксида калия (0,361 г, 6,44 ммоль), хлорацетона (0,59 г,6,44 ммоль), фторида тетрабутиламмония (0,65 мл, 0,65 ммоль, 1 М раствор в ТГФ) и ТГФ (20 мл) и перемешивали в течение примерно 48 ч при комнатной температуре. Реакционную смесь фильтровали через кизельгур и концентрировали фильтрат при пониженном давлении. Осадок очищали с помощью флэш-хроматографии (смесь 10% этилацетат/гексан - 100% этилацетат) с получением указанного в заголовке соединения (1,64 г, 65%): МС (m/z): 389 (М+23). Пример получения 22. Бензил-N-(2-метил-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил)карбамат Бензил-N-[(3R)-1-ацетонил-2-оксо-4,5-дигидро-3 Н-1-бензазепин-3-ил]карбамат (0,7 г, 1,91 ммоль) объединяли с ацетатом аммония (2,94 г, 38,2 ммоль) в уксусной кислоте (20,0 мл) и нагревали при 115 С в течение 24 ч и затем смесь перемешивали при комнатной температуре в течение примерно 16 ч. Реакционную смесь выливали в смесь лед/вода, ощелачивали гидроксидом аммония (28-30%) и экстрагировали дихлорметаном. Промывали водой и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении. Осадок очищали с помощью флэшхроматографии (10% этилацетат/гексан - 50% этилацетат/гексан), затем с помощью сильной катионооб- 10020128 менной колонки (метанол, затем 10% 2 н. раствор аммиака в смеси метанол/CH2Cl2) с получением указанного в заголовке соединения (0,21 г, 32%) в виде твердого вещества белого цвета: МС (m/z): 348(М+1). (По результатам анализа конечного соединения, полученного из указанного промежуточного соединения, указанное в заголовке соединение является частично рацемизированным). Пример получения 23. 2-Метил-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амин Использовали способ из примера получения 16, где в качестве исходного вещества использовали бензил-N-(2-метил-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил)карбамат (0,19 г, 0,55 ммоль), с получением указанного в заголовке соединения (0,082 г, 69%): МС (m/z): 214 (М+1). Схема Е Бензил-N-[(3R)-2-тиоксо-1,3,4,5-тетрагидро-1-бензазепин-3-ил]карбамат (1,5 г, 4,60 ммоль) растворяли в ТГФ (40 мл), охлаждали до 0 С и добавляли одной порцией гидрид натрия (NaH) (192,9 мг, 4,83 ммоль, 60% в минеральном масле). Баню удаляли и реакционную смесь перемешивали в течение примерно 1 ч в атмосфере азота. Добавляли метилйодид (300 мкл, 4,83 ммоль) и перемешивали в течение примерно 1 ч при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении для удаления основной части ТГФ, осадок разбавляли этилацетатом и промывали насыщенным раствором бикарбоната натрия и соляным раствором, сушили над сульфатом натрия и органические слои концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,5 г,96%) в виде твердого вещества белого цвета: МС (m/z): 341 (М+1). Пример получения 25. Бензил-N-(1-метил-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил)карбамат Бензил-N-[(3R)-2-метилсульфанил-4,5-дигидро-3 Н-1-бензазепин-3-ил]карбамат (500 мг, 1,47 ммоль), пропаргиламин (197,28 мкл, 2,94 ммоль) и моногидрат п-толуолсульфоновой кислоты (30 мг,157,71 мкмоль) объединяли в смеси фенилэфир/бифенил (2 мл, 6,53 ммоль) (Dowtherm А) и полученную смесь перемешивали в течение примерно 45 мин при 180 С. Реакционную смесь охлаждали и добавляли эфир (20 мл), затем 2 М HCl (20 мл). Перемешивали в течение нескольких минут, водный слой удаляли и органический слой экстрагировали 2 н. раствором HCl (2). Водные слои объединяли и ощелачивали гидроксидом аммония (28%) до получения щелочного раствора. Осадок собирали посредством фильтрования, промывали водой и сушили в вакуумной печи. Указанный осадок очищали с помощью флэшхроматографии (от 50% 5% раствор аммиака/метанола (2 М) в дихлорметане до 90% 5% раствора) с получением указанного в заголовке соединения (358 мг, 70%) в виде твердого вещества не совсем белого цвета: МС (m/z): 348 (М+1) . Время удерживания для изомера 1 (S-изомера)=3,3 мин; для изомера 2 (Rизомера)=3,7 мин.(колонка Chiralpak AD-H; 100% EtOH с 0,2% диметилэтиламином; скорость потока 1,0 мл/мин; 225 нм). По результатам хирального анализа показана рацемизация примерно на 20%. Пример получения 26. 1-Метил-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амин Бензил-N-(1-метил-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил)карбамат (0,99 г, 2,85 ммоль) и 10% Pd/C (200 мг, 1,88 ммоль) объединяли в абсолютном этаноле (50 мл). Смесь помещали на шейкерParr на 7 ч (водород (Н 2), 60 фунтов/кв. дюйм, комнатная температура). Катализатор удаляли посредством фильтрования, к фильтрату добавляли 10% Pd/C (200 мг, 1,88 ммоль) и полученную смесь помещали на шейкер Parr еще на 7 ч (60 фунтов/кв. дюйм, комнатная температура). Через 7 ч добавляли дополнительную порцию 10% Pd/C (189 мг, 1,78 ммоль) и встряхивали в течение еще 7 ч. Катализатор удаляли посредством фильтрования и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (0,53 г, 87%): МС (m/z): 214 (М+1). Схема F На схеме F R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -СН 3, -ОН, -O(C1-C4 алкила), -Оциклопропила, -O-СН 2-фенила, -OC(H)F2 и -OCF3; R2 выбран из группы, состоящей из Н и СН 3; R3 выбран из группы, состоящей из Н и СН 3, при условии что по меньшей мере один из группы, состоящей изR2 и R3 представляет собой Н; n=1, 2 или 3; и R4 может представлять собой СН 3 или СН 2 СН 3. Предпочтительно, R4 представляет собой СН 2 СН 3. Пример получения 27. 1-Бром-4-винилоксибензол К смеси 4-бромфенола (75 г, 433,5 ммоль), этенилового эфира уксусной кислоты (80,5 мл, 867,0 ммоль) и карбоната натрия (27,6 г, 260,10 ммоль) в толуоле (430 мл) добавляли ди-мю-хлорбис 1,2,5,6 эта)-1,5-циклооктадиен)дииридий (2,9 г, 4,34 ммоль) и смесь нагревали при 100 С в течение 2,5 ч. Указанную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (EtOAc) (300 мл) и промывали один раз водой (400 мл). Органическую фазу отделяли и промывали один раз солевым раствором(250 мл). Полученный материал сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенную смесь очищали с помощью флэш-хроматографии (от 0% до 5% К раствору 1-бром-4-винилокси-бензола (31,75 г, 159,51 ммоль) и хлорйодметана (101,3 г, 574,2 ммоль) в 1,2-дихлорэтане (410 мл) при температуре 0 С медленно добавляли диэтилцинк (1 М раствор в гептане, 380 мл) в течение 1 ч при поддержании температуры ниже 5 С. Смесь белого цвета перемешивали при 0-5 С в течение 2 ч. Реакцию медленно гасили водным насыщенным раствором хлорида аммония (400 мл, выделение тепла) при 5 С и слои разделяли. Водный слой экстрагировали диэтиловым эфиром (2200 мл). Органические слои объединяли, промывали насыщенным раствором хлорида аммония(400 мл), сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (33 г, 97%) в виде масла: ГХ/МС (газовая хроматография/масс-спектрометрия): 212. Пример получения 29. 4-(Циклопропокси)бензойная кислота К раствору (-78 С) 1-бром-4-(циклопропокси)бензола (33 г, 154,9 ммоль) в тетрагидрофуране (500 мл) добавляли 1,6 М раствор бутиллития в гексане (96,8 мл, 154,9 ммоль) при поддержании температуры ниже -70 С. Смесь перемешивали при -78 С в течение 20 мин после завершения добавления. Добавляли сухой лед (464,6 ммоль) 3 порциями с интервалом в 5 мин. Смесь перемешивали при -78 С в течение 30 мин. Охлаждающую баню удаляли и реакцию гасили 10% раствором гидросульфата натрия (NaHSO4)(200 мл). Смесь нагревали до комнатной температуры и экстрагировали 3 Х EtOAc (300 мл). Органические фазы объединяли и промывали один раз соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Осадок суспендировали в диэтиловом эфире и фильтровали. Твердое вещество сушили в вакуумной печи с получением указанного в заголовке соединения (12,9 г, 47%): МС (m/z): 179 (М+1). Пример получения 30. Этил-1-4-(циклопропокси)бензоил]амино]циклопропанкарбоксилат К суспензии (0 С) 4-циклопропоксибензойной кислоты (24,5 г, 137,3 ммоль) в дихлорметане (350 мл), содержащей 2 капли ДМФА, медленно добавляли оксалилхлорид (23,8 мл, 274,7 ммоль). После завершения добавления охлаждающую баню удаляли и полученную смесь перемешивали в течение 2 ч. Полученный материал концентрировали до высыхания при пониженном давлении. Осадок растворяли в дихлорметане (500 мл) и затем добавляли гидрохлорид этил-1-аминоциклопропанкарбоксилата (Indofine 04-265, 24,9 г, 150,5 ммоль) и триэтиламин (57,2 мл, 410,4 ммоль). Смесь перемешивали при комнатной температуре в течение примерно 16 ч. Указанную смесь промывали один раз 10% раствором NaHSO4(250 мл). Органическую фазу отделяли и водную фазу экстрагировали 2 Х дихлорметаном (200 мл). Органические фазы объединяли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенную смесь очищали с помощью флэш-хроматографии (от 0 до 45% 4-Этоксибензойную кислоту (10,0 г, 60,18 ммоль), гидрохлорид этил-1-аминоциклопропанкарбоксилата (коммерчески доступный, 10,96 г, 66,19 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3 этилкарбодиимида (EDC) (13,84 г, 72,21 ммоль) и 1-гидроксибензотриазол (НОВТ) (9,76 г, 72,21 ммоль) объединяли в тетрагидрофуране (500 мл). Добавляли диизопропилэтиламин (DIEA) (23,09 мл, 132,39 ммоль) в течение 3-5 мин и реакционную смесь перемешивали в течение примерно 16 ч при комнатной температуре в атмосфере азота. Смесь разбавляли этилацетатом, промывали разбавленным растворомHCl, разбавляли NaHCO3 и затем соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении. Осадок очищали посредством перекристаллизации из этанола с получением указанного в заголовке соединения (13,93 г, 84%) в виде твердого вещества белого цвета: МС (m/z): 278 (М+1). Соединения, представленные в табл. 1, могут быть получены, по существу, так же, как описано согласно способу в примере получения 31, с использованием реагента в 3 колонке вместо 4-этоксибензойной кислоты и затем могут использоваться без очистки или подвергаться очистке посредством перекристаллизации (из этанола или метанола) или флэш-хроматографии (EtOAc/гексаны, градиент в диапазоне 10-50%). Для примеров получения 36 и 37 использовали гидрохлорид метил-1-аминоциклопентанкарбоксилата (коммерчески доступный) вместо гидрохлорида этил-1-аминоциклопропанкарбоксилата. Соединения согласно примерам получения 32 и 35 очищали путем перекристаллизации из этанола. Соединения согласно примерам получения 33 и 34 очищали с помощью флэш-хроматографии. Соединение согласно примеру получения 36 может использоваться без очистки. Соединение согласно примеру получения 37 очищали путем перекристаллизации из метанола. К раствору этил-1-(4-циклопропоксибензамидо)циклопропанкарбоксилата (33,7 г, 6,48 ммоль) в этаноле (380 мл) добавляли гидроксид натрия (174,7 мл, 174,7 ммоль) и смесь перемешивали при 65 С в течение 3 ч. Полученный материал охлаждали до комнатной температуры и концентрировали при пониженном давлении для удаления этанола. Осадок окисляли 10% раствором NaHSO4. Полученное твердое вещество фильтровали и промывали водой (3 Х) и диэтиловым эфиром (2 Х). Полученный материал сушили в вакуумной печи с получением указанного в заголовке соединения (29,9 г, 98%) в виде твердого вещества белого цвета: МС (m/z): 262 (М+1). Пример получения 39. 1-[(4-Этоксибензоил)амино]циклопропанкарбоновая кислота Этил-1-[(4-этоксибензоил)амино]циклопропанкарбоксилат (4,25 г, 15,33 ммоль) растворяли в 1,4 диоксане (60 мл) и добавляли гидроксид лития (LiOH) (3,22 г, 76,63 ммоль), растворенный в воде (30 мл). Реакционную смесь перемешивали в течение примерно 16 ч при комнатной температуре. Указанную смесь концентрировали при пониженном давлении, добавляли воду, окисляли 5 н. раствором HCl и осадок собирали посредством фильтрования с получением указанного в заголовке соединения (2,8 г, 73%) в виде твердого вещества белого цвета: МС (m/z): 248 (М+1). Соединения согласно примерам получения, представленным в табл. 2, могут быть получены, по существу, так же, как описано согласно способу удаления защитной группы примера получения 39. Таблица 2(4R)-5,6-Дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амин (0,50 г, 2,51 ммоль), 1-[(4-этоксибензоил) амино]циклопропанкарбоновую кислоту (0,75 мг, 3,01 ммоль), 1-гидроксибензотриазол (407 мг, 3,01 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (577 мг, 3,01 ммоль) объединяли в тетрагидрофуране (40 мл). Добавляли диизопропилэтиламин (0,53 мл, 3,01 ммоль) и реакционную смесь перемешивали в течение примерно 16 ч при комнатной температуре в атмосфере азота. Реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении. Осадок очищали с помощью флэш-хроматографии (от 25% EtOAc/гексаны до 100% EtOAc) с по- 15020128 лучением указанного в заголовке соединения (860 мг, 80%): МС (m/z): 431 (М+1). Соединения согласно примерам, представленным в табл. 3, могут быть получены, по существу, так же, как описано согласно способу из примера 3. Таблица 3 К смеси 5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амина (400 мг, 2,01 ммоль), 1-(4-циклопропоксибензамидо)циклопропанкарбоновой кислоты (577 мг, 2,21 ммоль), гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (462 мг, 2,41 ммоль) и гидрата 1-гидроксибензотриазола (369 мг, 2,41 ммоль) в дихлорметане (8 мл) добавляли триэтиламин (1,26 мл, 9,03 ммоль) и смесь перемешивали при комнатной температуре в течение примерно 16 ч. Неочищенную смесь очищали с помощью флэшхроматографии (0-5% МеОН/дихлорметан) с получением указанного в заголовке соединения (791 мг,89%) в виде пены желто-коричневого цвета: МС (m/z): 443 (М+1). Пример 16. N-[1-(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил]карбамоил]циклопропил]4-фторбензамид(4R)-5,6-Дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амин (95,0 мг, 0,48 ммоль), 1-[(4-фторбензоил) амино]циклопропанкарбоновую кислоту (128,0 мг, 0,57 ммоль) и гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) (218,0 мг, 0,57 ммоль) объединяли в N,Nдиметилформамиде (4 мл). Добавляли диизопропилэтиламин (0,21 мл, 1,19 ммоль) и реакционную смесь перемешивали в течение примерно 16 ч при комнатной температуре в атмосфере азота. Реакционную смесь разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении. Осадок очищали с помощью флэш-хроматографии (3% (2 М) раствор аммиака в смеси метанол/дихлорметан) с получением указанного в заголовке соединения (131,0 мг, 68%): МС (m/z): 405 Пример 17 может быть получен, по существу, так же, как описано согласно способу в примере 17. МС (m/z): 387 (М+1). Выход 63%. Схема G В схеме G R1 выбран из группы, состоящей из Н, F, Cl, -CF3, -СН 3, -ОН, -O(С 1-С 4 алкила), -Оциклопропила, -O-СН 2-фенила, OC(H)F2 и -OCF3; R2 выбран из группы, состоящей из Н и СН 3; R3 выбран из группы, состоящей из Н и СН 3, при условии, что по меньшей мере один из группы, состоящей из R2 и(599 мкл, 3,43 ммоль) и реакционную смесь перемешивали в течение примерно 16 ч при комнатной температуре в атмосфере азота. Реакционную смесь разбавляли этилацетатом, промывали разбавленным раствором бикарбоната натрия и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении. Осадок очищали с помощью флэш-хроматографии (от 25%EtOAc/гексаны до 85% EtO/Ac/гексаны) с получением указанного в заголовке соединения (860 мг, 79%) в виде твердого вещества белого цвета: МС (m/z): 383 (М+1). Соединение согласно примеру получения 48 может быть получено, по существу, так же, как описано в способе из примера получения 47, но с использованием 5,6-дигидро-4 Н-имидазо[1,2-а] Соединение согласно примеру получения 49 может быть получено, по существу, так же, как описано в способе из примера получения 47, но с использованием 1-(трет-бутоксикарбониламино) циклопентанкарбоновой кислоты (коммерчески доступной). Очистку проводили с помощью флэш-хроматографии с использованием градиента в диапазоне 20-90% этилацетат/гексан. МС (m/z): 411 (М+1). Пример получения 50. 1-Амино-N-[(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил]циклопропанкарбоксамид трет-Бутил N-[1-(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил]карбамоил]циклопропил] карбамат (856 мг, 2,24 ммоль) растворяли в дихлорметане (20 мл) и охлаждали на ледяной бане. Добавляли трифторуксусную кислоту (10 мл, 132,25 ммоль). Баню удаляли и реакционную смесь перемешивали в течение 1 ч. Указанную смесь концентрировали при пониженном давлении и добавляли этилацетат. Промывали 1 н. раствором NaOH и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения (470 мг, 74%) в виде твердого вещества белого цвета: МС (m/z): 283 (М+1). Пример получения 51. 1-Амино-N-(5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил)циклопентанкарбоксамид Соединение согласно примеру получения 51 может быть получено, по существу, так же, как описано в способе из примера получения 50, но с использованием трет-бутил-N-[1- (5,6-дигидро-4 Нимидазо[1,2-а][1]бензазепин-4-илкарбамоил)циклопентил]карбамата. МС (m/z): 311 (М-1). Пример получения 52. 1-Амино-N-(5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил)циклопропанкарбоксамид Соединение согласно примеру получения 52 может быть получено, по существу, так же, как описано в способе из примера получения 50, но с использованием трет-бутил-N-[1-(5,6-дигидро-4 Нимидазо[1,2-а] [1]бензазепин-4-илкарбамоил)циклопропил]карбамата. МС (m/z): 283 (М+1). Пример 18. 4-Хлор-N-[1-(5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-илкарбамоил)циклопентил]бензамид 1-амино-N-(5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил)циклопентанкарбоксамид (158 мг,509,02 мкмоль), 4-хлорбензойную кислоту (95,64 мг, 610,83 мкмоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (117,10 мг, 610,83 мкмоль) и 1-гидроксибензотриазол (82,54 мг,610,83 мкмоль) объединяли в тетрагидрофуране (12 мл). Добавляли диизопропилэтиламин (106,53 мкл,610,83 мкмоль) и реакционную смесь перемешивали при комнатной температуре в течение примерно 16 ч. Реакционную смесь разбавляли EtOAc, промывали разбавленным раствором бикарбоната натрия и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении. Осадок очищали с помощью флэш-хроматографии (0-40% 5% раствор аммиака/метанол/дихлорметан (2 н.)/дихлорметан) с получением указанного в заголовке соединения (101,0 мг,44%): МС (m/z): 449 (М+1). Соединения согласно примерам, представленным в табл. 4, могут быть получены, по существу, так же, как описано согласно способу из примера 18, но с использованием реагентов, перечисленных в колонке 3. Для очистки использовали флэш-хроматоргафию с использованием 5% раствора аммиака (2 М) в метаноле с градиентом дихлорметан/дихлорметан в диапазоне 0-80%. Таблица 4 1-Амино-N-[(4R)-5,6-дигидро-4 Н-имидазо[1,2-a][1]бензазепин-4-ил]циклопропанкарбоксамид (160 мг, 0,57 ммоль), 4-хлорбензойную кислоту (106 мг, 0,68 ммоль) и гексафторфосфат О-(7 азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) (259,0 мг, 0,68 ммоль) объединяли в N,Nдиметилформамиде (4 мл). Добавляли диизопропилэтиламин (DIEA) (0,25 мл, 1,42 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение примерно 5 ч в атмосфере азота. Реакционную смесь разбавляли этилацетатом, промывали раствором бикарбоната натрия, водой (2 Х) и соляным раствором, сушили над сульфатом натрия и органический слой концентрировали при пониженном давлении. Осадок очищали с помощью флэш-хроматографии (40% EtOAc/гексаны - 100% EtOAc) с получением указанного в заголовке соединения (190,0 мг, 80%): МС (m/z): 421 (М+1). Соединения согласно примерам, представленным в табл. 5, могут быть получены, по существу, так же, как описано согласно способу из примера 21, при использовании реагента из 3 колонки вместо 4 хлорбензойной кислоты и при времени реакции примерно 16 ч с использованием хиральных и рацемических аминов. Очищение осуществляли с помощью флэш-хроматографии с использованием либо (способ 1; примеры 22, 23, 24, 25, 27, 28, 29, 30, 31 и 32): 3-5% раствора аммиака (2 М) в метаноле в градиенте дихлорметан/дихлорметан в диапазоне 0-100%, либо (способ 2; примеры 26): градиента этилацетат/гексан в диапазоне 25-90%. Можно разделять рацемические смеси и выделять отдельные изомеры в условиях хроматографии,описанных в способах А, В, С, D, Е и F. Рацемические смеси можно разделять на R- и S- изомеры. Условия хиральной хроматографии: Способ А. СФХ (сверхкритическая флюидная хроматография) - хиральная колонка: Chiralpak AD-H. Элюент: изократические условия в диапазоне от 20 до 30% этанола с 0,2% изопропиламиновым модификатором и CO2. Сигнал детектировали на длине волны 225 нм. Способ В. СФХ - хиральная колонка: Chiralpak AD-H. Элюент: изократические условия в диапазоне от 25 до 40% изопропанола с 0,2% изопропиламиновым модификатором и CO2. Сигнал детектировали на длине волны 225 нм. Способ С. СФХ - хиральная колонка: Chiralpak OD-H. Элюент: изократические условия с 20% этанолом с 0,2% изопропиламиновым модификатором иCO2. Сигнал детектировали на длине волны 225 мн. Способ D. СФХ - хиральная колонка: Chiralpak OD-H. Элюент: изократические условия в диапазоне от 15 до 20%. метанола с 0,2% изопропиламиновым модификатором и CO2. Сигнал детектировали на длине волны 225 нм. Способ Е. ЖХ - Хиральная колонка: Chiralpak AD-H. Элюент: изократические условия с 100% метанолом с 0,2% изопропиламиновым модификатором. Сигнал детектировали на длине волны 225 нм. Способ F. СФХ (сверхкритическая флюидная хроматография) - хиральная колонка: Chiralcel OJ-H. Элюент: изократические условия с 10% изопропанолом с 0,2% изопропиламиновым модификатором и CO2. Сигнал детектировали на длине волны 225 нм. Соединения согласно примерам, приведенным в табл. 6 (вторая колонка), разделяли на изомеры 1(R-изомер) и 2 (S-изомер) с использованием хроматографических способов А, В, С, D, Е и F. Соединения согласно примерам 29, 30, 20, 8 и 6 разделяли с использованием способа А; соединения согласно примерам 10, 13, 18, 19 и 15 разделяли с использованием способа В; соединения согласно примерам 14, 12 и 11 разделяли с использованием способа С; соединение согласно примеру 7 разделяли с использованием способа D; пример 9 разделяли с использованием способа Е; соединение согласно примеру получения 47 разделяли с использованием способа F. Изомеры 1 (R-изомер) и 2 (S-изомер), номера примеров указанных изомеров и время удерживания для указанных изомеров показаны в колонке 1. Таблица 6 4-Хлор-N-[1-[(4R)-(5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-илкарбамоил]циклопропил] бензамид (439 мг, 1,04 ммоль) растворяли в изопропиловом спирте (13,9 мл) и медленно добавляли моногидрат 4-метилбензолсульфоновой кислоты (198,40 мг, 1,04 ммоль) в изопропиловом спирте (1,60 мл) при комнатной температуре. Смесь перемешивали в течение примерно 2 ч и твердое вещество собирали посредством фильтрования. Указанное твердое вещество сушили в вакуумной печи с получением указанного в заголовке соединения (487 мг, 79%) в виде твердого вещества белого цвета: МС (m/z): 421(М+23). Примеры в табл. 7 могут быть получены, по существу, так же, как описано согласно способу примера 49. Таблица 7 4-Хлор-N-[1-[(4R)-(5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-илкарбамоил]циклопропил]бензамид (350,0 мг, 0,83 ммоль) растворяли в метиловом спирте (12,0 мл) и полученный раствор охлаждали на ледяной бане. Добавляли по каплям раствор соляной кислоты (1 н) в эфире (3,33 мл, 3,33 ммоль). Баню удаляли, смесь перемешивали в течение 30 мин и концентрировали при пониженном давлении с получением указанного в заголовке соединения (323 мг, 85%) в виде твердого вещества белого цвета: МС (m/z): 421 (М+1). Пример 55. Гидрохлорид 4-хлор-N-[1-(5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-илкарбамоил) циклопропил]бензамида Соединение согласно примеру 55 получали, по существу, так же, как описано согласно способу из примера 54. МС (m/z): 421 (M+l). Пример 56. N-[1-(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил]карбамоил]циклопропил]4-гидроксибензамид 4-Бензилокси-N-[1-(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-ил]карбамоил]циклопропил]бензамид (145 мг, 294,37 мкмоль) растворяли в этаноле (20 мл) и гидрогенизировали на аппарате для встряхивани Parr с 10% Pd/C (22,8 мг, 10,71 мкмоль) в течение 18 ч (60 фунтов/кв. дюйм, комнатная температура). Катализатор отфильтровывали и фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения (107 мг, 90%) в виде твердого вещества белого цвета: МС(m/z): 403 (М+1). На схеме J показан альтернативный способ синтеза N-[1-(4R)-5,6-дигидро-4 Н-имидазо[1,2-а][1] бензазепин-4-ил]карбамоил]циклопропил]-4-этоксибензамида. Схема J В реактор загружали 630 кг ДМФА и перемешивали содержимое в течение от 1 до 2 ч под током азота. Загружали 171 кг карбоната цезия и 45,0 кг 3-(2-бромфенил)пропионовой кислоты, полученную смесь перемешивали в течение от 1 до 2 ч при температуре от 80 до 85 С и затем снова охлаждали до температуры от 20 до 25 С. Реактор наполняли 27,0 кг имидазола и 4,4 кг йодида меди и полученную смесь перемешивали в течение от 1 до 2 ч под током азота при температуре от 20 до 25 С. Температуру доводили до температуры от 115 до 125 С и интенсивно перемешивали в течение от 20 до 24 ч, [ВЭЖХ 3%, 3-(2-бромфенил)пропионовой кислоты]. Реакционную смесь охлаждали до 60 С и медленно загружали 181 кг этанола. Реакционную смесь охлаждали до температуры от 0 до 15 С и перемешивали в течение от 8 до 10 ч. Выделяли производное продукта в виде твердого вещества посредством центрифугирования. В реактор загружали влажный осадок, 184 кг этанола и 36 кг ДМФА и перемешивали в течение от 0,5 до 1 ч при температуре от 0 до 15 С. Выделяли производное продукта в виде твердого вещества посредством центрифугирования. Фильтраты объединяли и переносили в реактор с помощью трубчатого фильтра для удаления твердых частиц. Смесь концентрировали до объема от 675 до 720 л при пониженном давлении при поддержании внутренней температуры ниже 65 С. Охлаждали до температуры от 25 до 35 С и загружали в реактор 174 кг этанола. Перемешивали в течение от 10 до 15 мин и проверяли содержание воды в растворе (значение KF0,5%). В вышеуказанный раствор медленно загружали 95 кг тионилхлорида при поддержании внутренней температуры между 25 и 40 С. Смесь нагревали с обратным холодильником в течение от 7 до 10 ч[ВЭЖХ:3% 3-(2-имидазол-1-ил-фенила)-пропионовой кислоты]. Охлаждали до температуры от 20 до 35 С. Смесь фильтровали и отфильтрованный осадок промывали 189 кг этанола. Объединенные фильтраты загружали в реактор и концентрировали при перемешивании при пониженном давлении до объема от 675 до 720 л при поддержании внутренней температуры при 65 С. Доводили температуру до 20-30 С. Медленно добавляли 450 кг воды при поддержании внутренней температуры при 20-30 С и перемешивали в течение 10-15 мин. Полученную смесь промывали гептаном (3153 кг) и доводили рН водного слоя до значения 8,0 с использованием 25% водного Na2CO3 (45 кг) при поддержании внутренней температуры менее 30 С. Полученную смесь экстрагировали МТБЭ (3450 кг), объединенные слои промывали МТБЭ (1369 кг 2%NH4OH; 2362 кг H2O). Органический слой концентрировали до объема от 90 до 180 л при пониженном давлении при поддержании внутренней температуры ниже 40 С. Через трубчатый фильтр загружали 220 кг 2-МеТГФ для удаления твердых частиц, концентрировали до объема от 90 до 180 л при пониженном давлении при поддержании внутренней температуры ниже 40 С. Загружали 214 кг 2-МеТГФ через трубчатый фильтр для удаления твердых частиц (в ходе процесса значение KF0,2%) и переносили раствор в чистые пластиковые пробирки с получением 195,8 кг раствора этил-3-(2-(1 Н-имидазол-1-ил)фенил)пропаноата в 2-МеТГФ. Реактор наполняли 281 кг 2-МеТГФ и 31 кг диизопропиламина. Охлаждали до температуры от -80 до -70 С и медленно загружали 86 кг н-бутиллития в н-гексане при поддержании температуры от -80 до 70 С и перемешивали в течение от 1 до 3 ч. В реактор медленно загружали 195,8 кг раствора этил-3-(2(1 Н-имидазол-1-ил)фенила)пропаноата в 2-МеТГФ при температуре от -80 до -70 С и перемешивали в течение от 2 до 4 ч при от температуре -80 до -70 С (по результатам ВЭЖХ в ходе процесса наблюдали 3% этил-3-(2-(1 Н-имидазол-1-ил)фенила)пропаноата). В реактор медленно загружали 47,8 кг этанола при температуре от -80 до-70 С и перемешивали в течение от 0,5 до 2 ч при температуре от -80 до 70 С. В реактор медленно загружали 310 кг МТБЭ при поддержании внутренней температуры ниже -25 С. Доводили рН до 7,5 292 кг 10% водного раствора лимонной кислоты. Доводили температуру до примерно 0 С. Водный слой отделяли и органический слой промывали 202 кг 25% водным раствором NaCl. Слои разделяли, органический слой концентрировали при пониженном давлении до объема от 41 до 82 л при поддержании внутренней температуры ниже 40 С. Водный слой экстрагировали 200 кг дихлорметана. Органические слои объединяли и перемешивали в течение от 0,5 до 1 ч при температуре от 15 до 25 С. Фильтровали через пластину с диоксидом кремния (20 кг) и промывали пластину 60 кг дихлорметана. Фильтрат концентрировали при пониженном давлении до объема от 82 до 123 л при поддержании внутренней температуры ниже 40 С. 170 кг МТБЭ загружали к осадку и концентрировали при пониженном давлении до объема от 82 до 123 л при поддержании внутренней температуры ниже 50 С. Загружали 208 кг МТБЭ и перемешивали в течение от 10 до 15 мин (контроль в ходе процесса: 10,8% 2-МеТГФ и 4,6% ДХМ) . Загружали 279 кг МТБЭ и концентрировали при пониженном давлении до объема от 82 до 123 л при поддержании внутренней температуры ниже 50 С (остаточный 2-МеТГФ%=1,6%, остаточный ДХМ%=2,7%). 14,5 кг метилацетата загружали к осадку и смесь нагревали с обратным холодильником в течение от 30 до 60 мин. Указанную смесь охлаждали до температуры от 0 до 15 С и перемешивали в течение от 6 до 8 ч при температуре от 0 до 15 С. Твердое вещество выделяли посредством центрифугирования и отфильтрованнвый осадок промывали 35 кг МТБЭ. Полученное твердое вещество сушили с получением 15,8 кг 5,6-дигидроимидазо[1,2-а][1]бензазепин-4-она. ЖХ-МС=199 (М+1), 419 (2 М+23). Пример получения 54. Оксим 5,6-дигидро-4 Н-бензо[f]имидазо[1,2-а]азепин-4-она В реактор загружали в атмосфере азота 42 кг 5,6-дигидроимидазо[1,2-а][1]бензазепин-4-она, 15,5 кг гидрохлорида гидроксиламина, 21,5 кг безводного ацетата натрия и 171 кг метанола. Полученную суспензию коричневого цвета нагревали до температуры от 60 до 70 С в течение 20 ч (с помощью ВЭЖХ в ходе процесса наблюдали 3% 5,6-дигидроимидазо[1,2-а][1]бензазепин-4-она). Реакционную смесь охлаждали до температуры от 23 до 25 С и перемешивали в течение 8 ч. Твердое вещество выделяли посредством центрифугирования и промывали 33 кг холодного метанола. Полученное твердое вещество перемещали в реактор и суспендировали с 210 кг воды при температуре от 85 до 90 С в течение 2 ч. Твердое вещество выделяли посредством центрифугирования и промывали указанное твердое вещество 85 кг воды. Полученное твердое вещество сушили с получением 39,65 кг оксима 5,6-дигидро-4 Нбензо[f]имидазо[1,2-а]азепин-4-она. МС=214 (М+1). Пример получения 55. Дигидрохлорид 5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амина В реактор с инертной атмосферой загружали 153 кг метанола, 9,6 кг оксима 5,6-дигидро-4 Нбензо[f]имидазо[1,2-а]азепин-4-она, 1,9 кг твердого гидроксида натрия, 10 кг воды и 10 кг никеля Ренея(Suzhou Tailida Sci-tech Co., Ltd.). Закачивали в реактор газообразный водород до повышения давления до 58-65 фунтов/кв.дюйм и нагревали до температуры от 60 до 65 С. Через 40 ч охлаждали до 10-20 С и фильтровали через пластину с 8 кг диатомитовой земли для удаления никелевого катализатора, промывая 33 кг метанола. Фильтрат концентрировали в вакууме до достижения объема между 1 и 2 объемами при поддержании температуры реактора ниже 50 С. Загружали 130 кг дихлорметана и концентрировали до получения от 1 до 2 объемов, загружали дополнительные 130 кг дихлорметана и 104 кг воды. Перемешивали при температуре от 10 до 20 С в течение от 10 до 15 мин и оставляли без перемешивания в течение от 30 до 35 мин. Разделяли слои и органический и водный слой переносили в отдельные емкости. Органический слой промывали 104 кг очищенной воды и 104 кг 25% раствора хлорида натрия. Органический слой отделяли от водного слоя. Исходный реактор, используемый для разделения фаз, необходимо промывать минимальным количеством этанола (20 кг), и промывочную порцию этанола добавляли к органическому слою. Органический слой концентрировали в вакууме до объема от 1 до 2 объемов при поддержании внутренней температуры ниже 40 С. Загружали от 57 до 58 кг этанола и концентрировали от до объема от 1 до 2 объемов при поддержании внутренней температуры ниже 40 С и повторяли до тех пор, пока дихлорметан не переставал выявляться по результатам анализа остаточного растворителя (ГХ). Загружали 3 объема этанола и анализировали для выявления остаточного дихлорметана (ГХ). Если дихлормтан не выявлялся, раствор охлаждали до температуры от 10 до 15 С и медленно загружали 100 кг 4 н. HCl в этилацетате. Перемешивали в течение от 3 до 8 ч при температуре от 10 до 20 С, фильтровали и отфильтрованный осадок промывали 25 кг этанола. Указанный отфильтрованный осадок сушили при температуре от 50 до 55 С в течение от 12 до 16 ч с получением 11,1 кг рацемической соли бисHCl амина (1,5-2,0 соль HCl 5,6-дигидро-4 Н-бензо[f]имидазо[1,2-а]азепин-4-амина) МС=200 (М+1). Пример получения 56. (4R)-5,6-Дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амин В реактор загружали 35,9 кг дигидрохлорида 5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амина и 453 мл воды. Медленно добавляли 29,7 л 25% раствора NH4OH в течение 30 мин при поддержании температуры ниже 25 С. После завершения добавления рН составлял 9 и полученный раствор представлял собой чистый раствор коричневого цвета. Кристаллизацию вызывали путем добавления 31,5 г зародышей кристаллизации при температуре 22 С. Перемешивали суспензию в течение 8 ч и охлаждали до 5 С. Отфильтрованный осадок фильтровали и промывали 54 л холодной воды и сушили полученный отфильтрованный осадок в фильтре с использованием N2/вакуума в течение 31 ч, что приводило к получению 5,6-дигидро-4 Н-имидазо[1,2-а][1]бензазепин-4-амина. 29 кг неочищенного отфильтрованного осадка порциями растворяли в 145 л этанола. Удаляли 73 л растворителя посредством перегонки в вакууме. Добавляли 108 л этанола и удаляли и перегоняли 133 л растворителя до тех пор, пока конечная концентрация не составляла 2 л/кг. Проводили анализ по методу Карла-Фишера в ходе процесса для выявления конечного содержания воды 0,1 мас.%.
МПК / Метки
МПК: A61K 31/5517, C07D 487/04, A61P 3/00
Метки: диацилглицеролацилтрансферазы, ингибиторы, композиция, фармацевтическая, включающая
Код ссылки
<a href="https://eas.patents.su/30-20128-ingibitory-diacilglicerolaciltransferazy-i-farmacevticheskaya-kompoziciya-vklyuchayushhaya-ih.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы диацилглицеролацилтрансферазы и фармацевтическая композиция, включающая их</a>
Твердая фармацевтическая композиция, включающая производное бензимидазол-7-карбоксилата и ph регулирующий агент

Номер патента: 16593
Опубликовано: 30.06.2012
Авторы: Йонеяма Судзи, Таноуе Ютака
МПК: A61K 31/00, A61K 9/16, A61K 9/20...
Метки: твердая, бензимидазол-7-карбоксилата, фармацевтическая, регулирующий, агент, включающая, производное, композиция
Формула / Реферат:
1. Твердая фармацевтическая композиция, включающая (5-метил-2-оксо-1,3-диоксол-4-ил)метил-2-этокси-1-{[2'-(5-оксо-4,5-дигидро-1,2,4-оксадиазол-3-ил)бифенил-4-ил]метил}-1Н-бензимидазол-7-карбоксилат или его соль и рН регулирующий агент, имеющий рН от 2 до 5 в воде, в концентрации 1% мас./об. при 25°С.2. Твердая фармацевтическая композиция, включающая калиевую соль...
Твёрдая фармацевтическая композиция, включающая миртазапин

Номер патента: 13739
Опубликовано: 30.06.2010
Авторы: Ферлан Андрей, Церноса Лидия, Пунчух-Колар Алёша, Райер Тадея, Турк Уршка, Гартнар Барбара, Кинцл Майя
МПК: A61K 9/16, A61K 31/55, A61K 9/20...
Метки: композиция, твёрдая, включающая, фармацевтическая, миртазапин
Формула / Реферат:
1. Твердая лекарственная форма для перорального применения, которая содержит:(A) миртазапин или его соль или сольват,(Б) этилцеллюлозу или метилцеллюлозу и(B) по меньшей мере один другой эксципиент.2. Твердая лекарственная форма по п.1, которая содержит производное целлюлозы (Б) в количестве от 0,01 до 25,0 мас.%.3. Твердая лекарственная форма по любому из пп.1, 2, отличающаяся тем, что по меньшей мере часть компонента (А) находится в контакте с...
Фармацевтическая композиция с замедленным высвобождением, включающая венлафаксин

Номер патента: 9957
Опубликовано: 28.04.2008
Авторы: Сегула Закель Мойка, Зупанциц Сильво, Писек Роберт
МПК: A61K 31/137, A61K 9/50, A61K 9/16...
Метки: замедленным, включающая, композиция, венлафаксин, фармацевтическая, высвобождением
Формула / Реферат:
1. Способ получения фармацевтической композиции, содержащей венлафаксин или его фармацевтически приемлемую соль, который включает следующие стадии: (1) получение дисперсии венлафаксина или соли венлафаксина в растворителе, в котором венлафаксин или соль венлафаксина имеют растворимость менее 25 г/л при комнатной температуре; (2) преобразование дисперсии, полученной на стадии (1), в форму частиц; (3) покрывание частиц, полученных на стадии (2),...
Фармацевтическая композиция, включающая производные таксанов, и способ ее получения

Номер патента: 12561
Опубликовано: 30.10.2009
Авторы: Кузнецов Олег Олегович, Забудкин Александр Фридрихович, Чибиляев Тимур Хайдарович, Морозов Павел Игоревич
МПК: A61K 47/10, A61K 31/337, A61J 3/00...
Метки: получения, производные, включающая, способ, таксанов, композиция, фармацевтическая
Формула / Реферат:
- формула изобретения, действующая на территории Договаривающихся государств, для которых ниже не указана особая редакция формулы1. Фармацевтическая композиция, включающая активное вещество, выбранное из группы таксанов, поверхностно-активное вещество и этиловый спирт, отличающаяся тем, что она содержит подкисляющий агент, обеспечивающий рН среды в пределах от 3,0 до 4,5.2. Композиция по п.1, отличающаяся тем, что подкисляющим агентом является...
Фармацевтическая композиция, включающая 1-(3-хлорфенил)-3-алкилпиперазин, для лечения расстройств аппетита

Номер патента: 12443
Опубликовано: 30.10.2009
Авторы: Гульельмотти Анджело, Гарроне Беатриче, Каццолла Никола, Фурлотти Гвидо, Маньяни Маурицио
МПК: A61P 3/04, A61K 31/495
Метки: фармацевтическая, расстройств, композиция, включающая, лечения, аппетита, 1-(3-хлорфенил)-3-алкилпиперазин
Формула / Реферат:
1. Применение 1-(3-хлорфенил)-3-алкилпиперазина формулы (I) в рацемической (R,S)-форме или в форме (S)-энантиомера в которой R является линейной или разветвленной алкильной группой, имеющей от 1 до 3 атомов углерода, или его соли присоединения фармацевтически приемлемой органической или неорганической кислоты в качестве лекарственного средства. 2. Применение согласно п.1 для лечения расстройств аппетита. 3. Применение согласно п.2, отличающееся...
Предыдущий патент: Способ изготовления охлаждающего элемента для пирометаллургического реактора и охлаждающий элемент
Следующий патент: Твердая лекарственная форма, обладающая анорексигенным действием (варианты)
Случайный патент: Способ и устройство скважинной спектрометрии с использованием перестраиваемых оптических фильтров