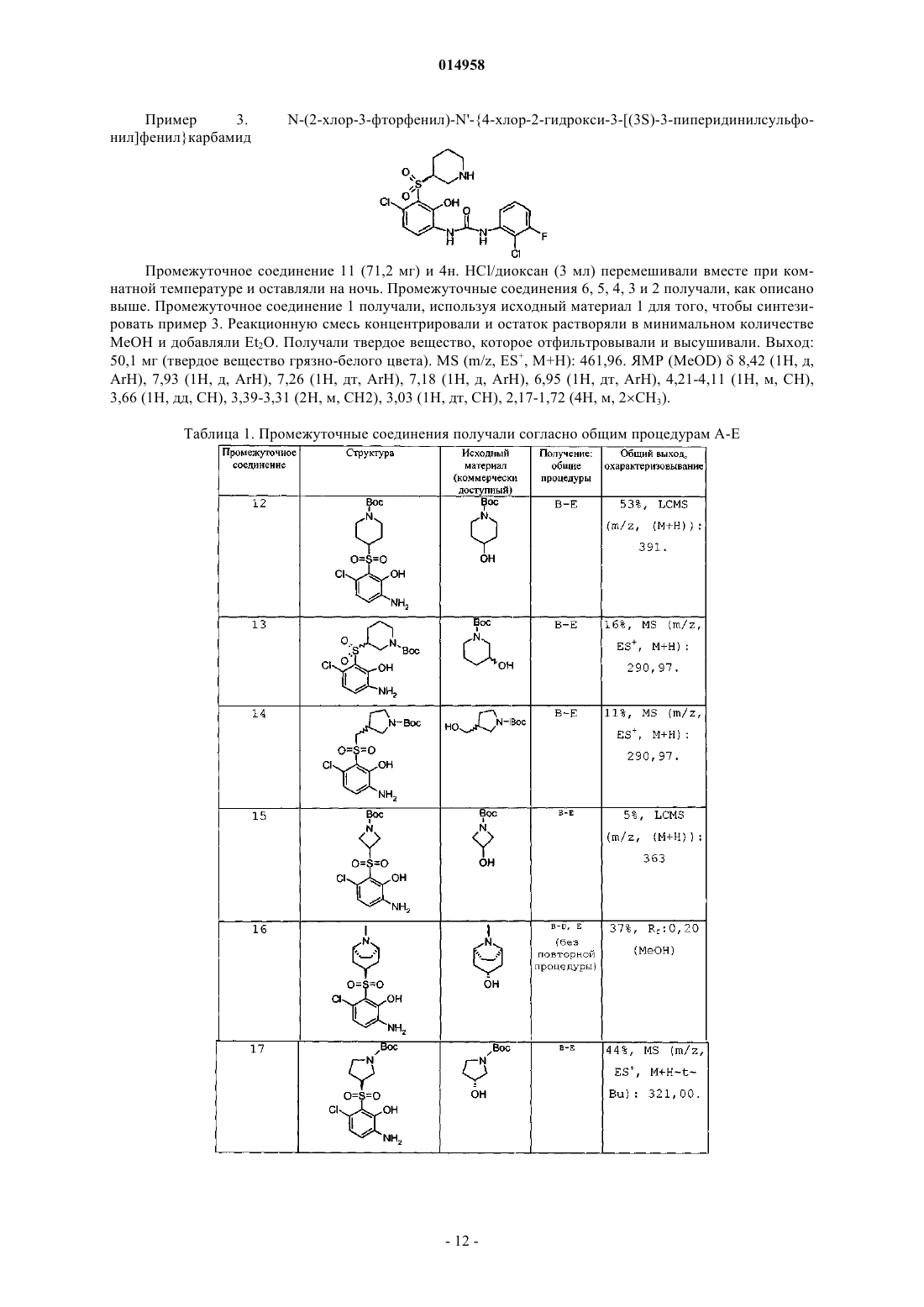
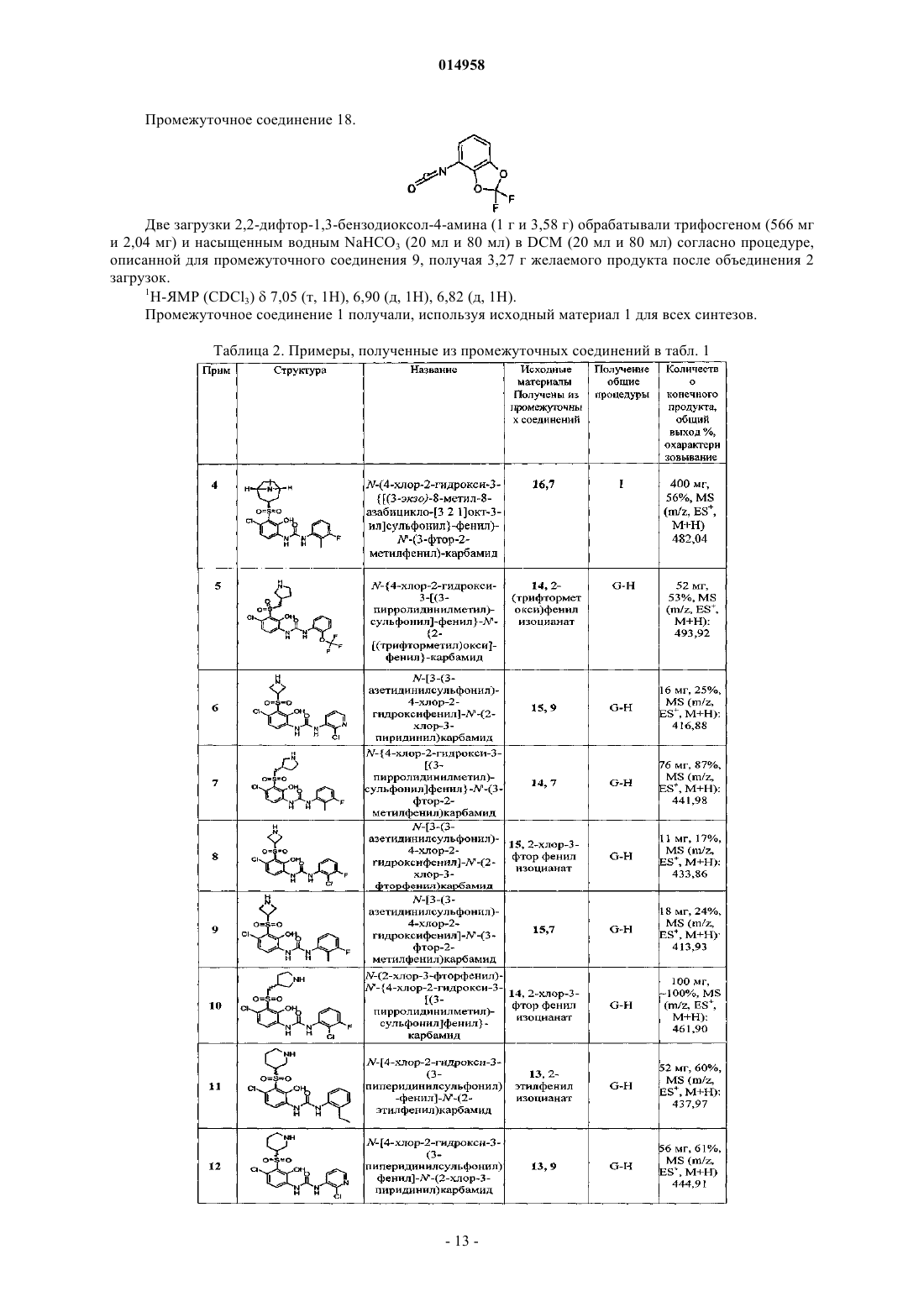
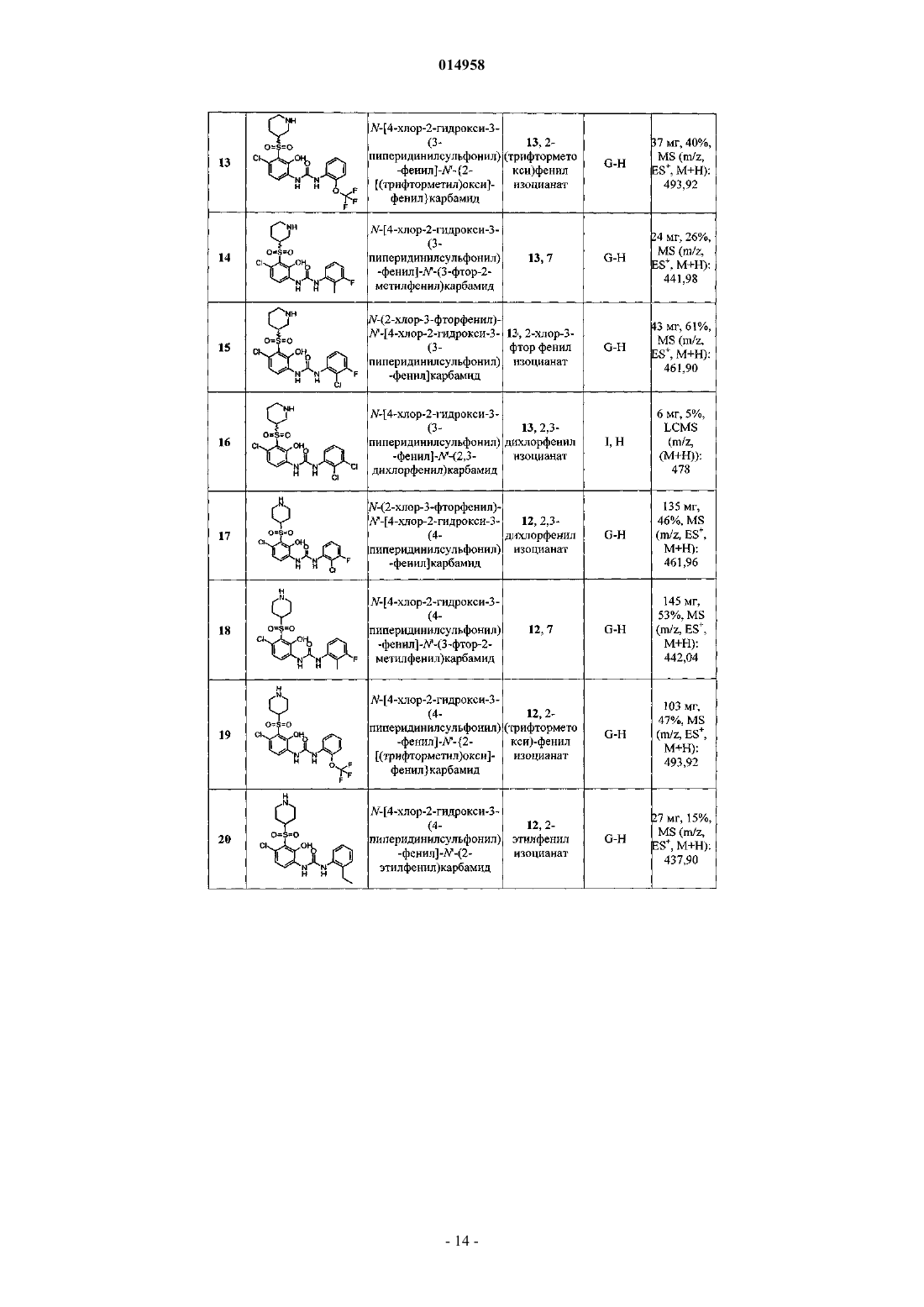
Антагонисты рецептора il – 8



























Формула / Реферат
1. Соединение согласно формуле (I)

в которой
X выбран из группы, состоящей из галогена, C1-3алкила, C1-3алкокси, циано, CF3 и OCF3;
R2 выбран из группы, состоящей из C3-6циклоалкила, фенила и гетероарила, причем фенил или гетероарил в случае необходимости замещены один или два раза независимо заместителем, выбранным из группы, состоящей из C1-3алкила, галогена, CF3, OCF3, фенилокси и бензилокси; или
R2 обозначает фенил, замещенный метилендиокси, или фенил, замещенный (ди-галоген-замещенным)метилендиокси;
R1 обозначает C4-8гетероцикл (CH2)n-, в котором гетероцикл представляет собой пирролидин-2-ил, пирролидин-3-ил, пиперидин-4-ил, пиперидин-3-ил или азетидин-3-ил, причем каждая группа в случае необходимости замещена независимо один или два раза заместителем, выбранным из группы, состоящей из C1-3алкила, C(O)OR4 и C(O)R5;
или
R1 выбран из следующих кольцевых систем (a-k)

R3 обозначает H или C1-3алкил;
R4 и R5 независимо обозначают C1-3алкил;
n = 0 или 1,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором X обозначает галоген.
3. Соединение по п.1, в котором X обозначает хлор.
4. Соединение по п.1, в котором R2 обозначает фенил, в случае необходимости замещенный независимо один или два раза заместителем, выбранным из группы, состоящей из C1-3алкила, галогена, OCF3 и фенилокси.
5. Соединение по п.4, в котором R2 выбран из группы, состоящей из 3-фтор-2-метилфенила, 2-трифторметилоксифенила, 2-хлор-3-фторфенила, 2-этилфенила или 2-фенилоксифенила.
6. Соединение по п.5, в котором R2 обозначает 3-фтор-2-метилфенил или 2-хлор-3-фторфенил.
7. Соединение по п.1, в котором R2 обозначает пиридил, в случае необходимости замещенный один раз галогеном.
8. Соединение по п.7, в котором R2 обозначает 2-хлор-3-пиридил.
9. Соединение по п.1, в котором n = 0.
10. Соединение по п.9, в котором R1 обозначает пиперидин-3-ил.
11. Соединение по п.9 или 10, в котором X обозначает галоген.
12. Соединение по п.1, в котором R1 обозначает 3-экзо-8-метил-8-азабицикло[3.2.1]окт-3-ил или 3-экзо-8-азабицикло[3.2.1]окт-3-ил.
13. Соединение по пп.9, 10 или 11, в котором R2 выбран из группы, состоящей из 3-фтор-2-метилфенила, 2-трифторметилоксифенила, 2-хлор-3-фторфенила, 2-этилфенила или 2-фенилоксифенила.
14. Соединение по п.1, представляющее собой
N-(4-хлор-2-гидрокси-3-{[(3-экзо)-8-метил-8-азабицикло[3.2.1]окт-3-ил]сульфонил}фенил)-N'-(3-фтор-2-метилфенил)карбамид;
N-[3-(3-азетидинилсульфонил)-4-хлор-2-гидроксифенил]-N'-(2-хлор-3-пиридинил)карбамид;
N-[3-(3-азетидинилсульфонил)-4-хлор-2-гидроксифенил]-N' -(2-хлор-3-фторфенил)карбамид; или
N-[3-(3-азетидинилсульфонил)-4-хлор-2-гидроксифенил]-N'-(3-фтор-2-метилфенил)карбамид; или его фармацевтически приемлемая соль.
15. Соединение по п.1, представляющее собой N-{3-[(3-экзо)-8-азабицикло[3.2.1]окт-3-илсульфонил]-4-хлор-2-гидроксифенил}-N'-(2-хлор-3-пиридинил)карбамид;
N-{3-[(3-экзо)-8-азабицикло[3.2.1]окт-3-илсульфонил]-4-хлор-2-гидроксифенил}-N'-(3-фтор-2-метилфенил) карбамид;
N-{3-[(3-экзо)-8-азабицикло[3.2.1]окт-3-илсульфонил]-4-хлор-2-гидроксифенил}-N'-[2-(фенилокси)фенил] карбамид или
N-{3-[(3-экзо)-8-азабицикло[3.2.1]окт-3-илсульфонил]-4-хлор-2-гидроксифенил}-N'-(2-хлор-3-фторфенил) карбамид,
или его фармацевтически приемлемая соль.
16. Соединение по п.1, представляющее собой
N-{4-хлор-2-гидрокси-3-[(3-пирролидинилметил)сульфонил]фенил}-N'-(3-фтор-2-метилфенил)карбамид;
N-{4-хлор-2-гидрокси-3-[(3-пирролидинилметил)сульфонил]фенил}-N'-{2-[(трифторметил)окси]фенил}карбамид;
N-(2-хлор-3-фторфенил)-N'-{4-хлор-2-гидрокси-3-[(3-пирролидинилметил)сульфонил]фенил}карбамид;
N-{4-хлор-2-гидрокси-3-[(3R)-3-пирролидинилсульфонил]фенил}-N'-(2-этилфенил)карбамид;
N-{4-хлор-2-гидрокси-3-[(3R)-3-пирролидинилсульфонил]фенил}-N'-(2,2-дифтор-1,3-бензодиоксол-4-ил)карбамид;
N-{4-хлор-2-гидрокси-3-[(3R)-3-пирролидинилсульфонил]фенил}-N'-{2-[(трифторметил)окси]фенил}карбамид;
N-{4-хлор-2-гидрокси-3-[(3S)-3-пирролидинилсульфонил]фенил}-N'-(2-хлор-3-пиридинил)карбамид;
N-{4-хлор-2-гидрокси-3-[(3S)-3-пирролидинилсульфонил]фенил}-N'-(3-фтор-2-метилфенил)карбамид;
N-{4-хлор-2-гидрокси-3-[(3R)-3-пирролидинилсульфонил]фенил}-N'-[2-(фенилокси)фенил]карбамид;
N-(2-хлор-3-фторфенил)-N'-{4-хлор-2-гидрокси-3-[(3S)-3-пирролидинилсульфонил]фенил}карбамид или
N-(2-хлор-3-фторфенил)-N'-{4-хлор-2-гидрокси-3-[(3R)-3-пирролидинилсульфонил]фенил}карбамид;
или его фармацевтически приемлемая соль.
17. Соединение по п.1, представляющее собой
N-[4-хлор-2-гидрокси-3-(4-пиперидинилсульфонил)фенил]-N'-(2,2-дифтор-1,3-бензодиоксол-4-ил)карбамид;
N-[4-хлор-2-гидрокси-3-(4-пиперидинилсульфонил)фенил]-N'-(2-хлор-3-пиридинил)карбамид;
N-[4-хлор-2-гидрокси-3-(4-пиперидинилсульфонил)фенил]-N'-(2-этилфенил)карбамид;
N-[4-хлор-2-гидрокси-3-(4-пиперидинилсульфонил)фенил]-N'-{2-[(трифторметил)окси]фенил}карбамид;
N-[4-хлор-2-гидрокси-3-(4-пиперидинилсульфонил)фенил]-N'-(3-фтор-2-метилфенил)карбамид;
N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(2-этилфенил)карбамид;
N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(2-хлор-3-пиридинил)карбамид;
N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-{2-[(трифторметил)окси]фенил}карбамид;
N-[4-хлор-2-гидрокси-3-(4-пиперидинилсульфонил)фенил]-N'-[2-(фенилокси)фенил]карбамид;
4-{[6-хлор-3-({[(3-фтор-2-метилфенил)амино]карбонил}амино)-2-гидроксифенил]сульфонил}-1-пиперидинэтилкарбоксилат;
4-({6-хлор-2-гидрокси-3-({[2-(фенилокси)фенил]амино}карбонил)амино]фенил}сульфонил)-1-пиперидинэтилкарбоксилат;
N-(2-хлор-3-фторфенил)-N'-[4-хлор-2-гидрокси-3-(4-пиперидинилсульфонил)фенил]карбамид;
4-{[6-хлор-3-({[(2-хлор-3-фторфенил)амино]карбонил}амино)-2-гидроксифенил]сульфонил}-1-пиперидинэтилкарбоксилат;
N-{4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенил}-N'-(3-фтор-2-метилфенил)карбамид;
N-{4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенил}-N'-(2-хлор-3-пиридинил)карбамид;
N-(2-хлор-3-фторфенил)-N'-{4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенил}карбамид;
N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(3-фтор-2-метилфенил)карбамид;
N-(2-хлор-3-фторфенил)-N'-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]карбамид; или
N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(2,3-дихлорфенил)карбамид;
или его фармацевтически приемлемая соль.
18. Соединение по п.1, представляющее собой
N-{4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенил}-N'-(3-фтор-2-метилфенил)карбамид;
N-{4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенил}-N'-(2-хлор-3-пиридинил)карбамид;
N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(3-фтор-2-метилфенил)карбамид и
N-(2-хлор-3-фторфенил)-N'-{4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенил}карбамид;
или его фармацевтически приемлемая соль.
19. Соединение по п.1, представляющее собой
N-{4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенил}-N'-(3-фтор-2-метилфенил)карбамид;
N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(3-фтор-2-метилфенил)карбамид;
или его фармацевтически приемлемая соль.
20. Соединение по п.19, представляющее собой гидрохлорид.
21. Соединение по п.1, которое является N-(2-хлор-3-фторфенил)-N'-{4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенил}карбамидом, или его фармацевтически приемлемая соль.
22. Фармацевтическая композиция, содержащая соединение по любому из пп.1 или 14-21 и фармацевтически приемлемый носитель или разбавитель.
23. Применение соединения по любому из пп.1 или 14-21 в качестве активного терапевтического вещества.
24. Применение соединения по любому из пп.1 или 14-21 для лечения опосредуемого хемокинами заболевания, причем хемокин связывается с IL-8a или b рецептором у млекопитающего.
25. Применение соединения по любому из пп.1 или 14-21 для лечения заболевания, выбранного из группы, состоящей из астмы, хронического обструктивного заболевания легких и респираторного дистресс-синдрома взрослых.
26. Применение соединения по любому из пп.1 или 14-21 для получения лекарственного средства, пригодного для лечения опосредуемого хемокинами заболевания.
27. Применение соединения по любому из пп.1 или 14-21 для получения лекарственного средства, пригодного для лечения опосредуемого хемокинами заболевания, причем заболевание выбрано из группы, состоящей из астмы, хронического обструктивного заболевания легких, респираторного дистресс-синдрома взрослых, псориаза, воспалительного заболевания кишечника, болезни Крона и неспецифического язвенного колита.
28. Применение соединения по любому из пп.1 или 14-21 для получения лекарственного средства, пригодного для лечения заболевания, выбранного из группы, состоящей из псориаза, аллергического дерматита, остеоартрита, ревматоидного артрита, астмы, хронического обструктивного заболевания легких, респираторного дистресс-синдрома взрослых, воспалительного заболевания кишечника, болезни Крона, неспецифического язвенного колита, инсульта, септического шока, эндотоксического шока, грамотрицательного сепсиса, синдрома токсического шока, реперфузионного повреждения сердца и почек, гломерулонефрита, тромбоза, реакции трансплантат против хозяина, болезни Альцгеймера, отторжения аллотрансплантанта, малярии, рестиноза, ангиогенеза, атеросклероза, остеопороза, гингивита, вирусных заболеваний, таких как риновирусное заболевание, и нежелательного высвобождения гематопоэтических стволовых клеток.
29. Фармацевтическая композиция, содержащая соединение по любому из пп.1 или 14-21 и один или более дополнительных терапевтических ингредиентов.
30. Применение композиции по п.29, в которой дополнительным терапевтическим ингредиентом является антагонист рецептора CXCR3 или антагонист рецептора CCR5.
31. Способ синтеза соединения по любому из пп.1 или 14-21, включающий стадии:
a) окисления сульфида согласно формуле (II)

в которой R1 и X имеют значения, определенные согласно формуле (I) в п.1, до сульфона согласно формуле (III)

b) гидролиза этого сульфона до аминофенола согласно формуле (IV)

и
c) действия на этот аминсфенол изоцианатом или предшественником изоцианата, имеющим формулу R2C=N=O или R2CON3, с получением конечного продукта, и причем R2 имеет значение, определенное для формулы (I); и присоединение к группе R1 и удаление от группы R1 кислотно-лабильной защитной группы по мере необходимости.
32. Промежуточное соединение, выбранное из группы, состоящей из формулы (II)

и формулы (III)

в которых R1 и X имеют значения, определенные в формуле (I) по п.1.
33. Промежуточное соединение, имеющее формулу (IV)

в которой R1 и X имеют значения, определенные в формуле (I) по п.1.
34. Способ по п.31, в котором кислотно-лабильная защитная группа представляет собой группу t-Boc.
35. Соединение по п.32, выбранное из группы, состоящей из

36. Соединение по п.32, имеющее формулу

37. Способ по п.31, в котором соединение формулы (I) по п.1 имеет группу R1, защищенную кислотно-лабильной защитной группой t-Boc, и выбрано из

Текст
Это изобретение относится к новым соединениям формулы (I) и композициям, которые могут быть использованы в лечении болезненных состояний, опосредуемых хемокином Интерлейкином 8 (IL-8). 014958 Это изобретение относится к новым сульфоновым соединениям, фармацевтическим композициям,способам их получения и их применению в лечении заболеваний, опосредуемых IL-8, GRO, GRO,GRO, NAP-2 и ENA-78. Уровень техники По отношению к интерлейкину-8 (IL-8) использовали много различных названий, таких как белок-1 аттрактант/активатор нейтрофилов (NAP-1), моноцитарный хемотаксический фактор нейтрофилов(MDNCF), нейтрофил-активизирующий фактор (NAF) и T-лимфоцитарный хемотаксический фактор. Интерлейкин-8 является хемоаттрактантом для нейтрофилов, базофилов и субпопуляции T-клеток. Он продуцируется большинством ядерных клеток, включая макрофаги, фибробласты, эндотелиальные и эпителиальные клетки, подвергнутые действию TNF, IL-1, IL-1 или LPS, и самими нейтрофилами под действием LPS или хемотаксических факторов, таких как FMLP. М. Baggiolini et al., J. Clin. Invest. 84,1045 (1989); J. Schroder et al., J. Immunol. 139, 3474 (1987) and J. Immunol. 144, 2223 (1990); Strieter, et al.,Science 243, 1467 (1989) and J. Biol. Chem. 264, 10621, 1989); Cassatella et al., J. Immunol. 148, 3216 (1992).GRO, GRO, GRO и NAP-2 также принадлежат к семейству хемокинов . Как и IL-8, эти хемокины также упоминаются под различными названиями. Например, GRO называют MGSA соответственно (Melanoma Growth Stimulating Activity), см. Richmond et al., J. Cell Physiology 129, 375ELR, непосредственно предшествующее звену CXC, связываются с IL-8 В рецептором (CXCR2).IL-8, GRO, GRO, GRO, NAP-2 и ENA-78 стимулируют множество функций in vitro. Было показано, что они все имеют свойства хемоаттрактантов в отношении нейтрофилов, в то время как IL-8 иGRO демонстрировали хемотактическую активность в отношении T-лимфоцитов и базофилов. Кроме того, IL-8 может индуцировать высвобождение гистамина из базофилов и как у здоровых, так и страдающих аллергией людей. GRO- и IL-8 могут, кроме того, индуцировать высвобождение лизосомальных ферментов и окислительный взрыв из нейтрофилов. Также было показано, что IL-8 увеличивает поверхностную экспрессию Mac-1 (CD11b/CD18) на нейтрофилах без синтеза белка de novo. Это может способствовать усилению адгезии нейтрофилов к клеткам сосудистого эндотелия. Многие известные заболевания характеризуются массивной инфильтрацией нейтрофилов. Как и IL-8, GRO, GRO, GRO и NAP-2 промотируют аккумуляцию и активацию нейтрофилов, эти хемокины участвуют в механизме разнообразных острых и хронических воспалительных нарушений, включая псориаз и ревматоидный артрит, Baggiolini et al., FEBS Lett. 307, 97 (1992); Miller et al., Crit. Rev. Immunol. 12, 17 (1992); Oppenheim et al., Annu. Rev. Immunol. 9, 617 (1991); Seitz et al., J. Clin. Invest. 87, 463 (1991); Miller et al., Am.Rev. Respir. Dis. 146, 427 (1992); Donnely et al., Lancet 341, 643 (1993). Кроме того, хемокины ELR (те,которые содержат аминокислотное звено ELR непосредственно перед звеном CXC) также участвуют в ангиостазе, Strieter et al., Science 258, 1798 (1992).In vitro, IL-8, GRO, GRO, GRO и NAP-2 индуцируют изменение формы нейтрофилов, хемотаксис, высвобождение гранул и окислительный взрыв, связываясь с рецепторами семитрансмембранного,G-белок-связанного семейства и активируя их, в особенности, связываясь с рецепторами IL-8, особенно с рецептором IL-8 (CXCR2). Thomas et al., J. Biol. Chem. 266, 14839 (1991); и Holmes et al., Science 253,1278 (1991). Имеет прецедент разработка непептидных маломолекулярных антагонистов членов этого семейства рецепторов. Для обзора см. R. Freidinger в: Progress in Drug Research, Vol. 40, pp. 33-98, Birkhauser Verlag, Basel 1993. Следовательно, рецептор IL-8 представляет собой многообещающую мишень для разработки новых противовоспалительных средств. Были охарактеризованы два высокоаффинных рецептора IL-8 человека (77% гомологии): CXCR1,который связывается только с IL-8 с высоким сродством, и CXCR2, который имеет высокое сродство в отношении IL-8, а также для GRO, GRO, GRO и NAP-2. См. Holmes et al., выше; Murphy et al., Science 253, 1280 (1991); Lee et al., J. Biol. Chem. 267, 16283 (1992); LaRosa et al., J. Biol. Chem. 261, 25402 (1992);and Gayle et al., J. Biol. Chem. 268, 7283 (1993). По-прежнему существует потребностью в лечении, в этой области, в соединениях, которые являются способными к связыванию с рецепторами CXCR1 и/или CXCR2. Поэтому состояния, связанные с увеличением продукции IL-8 (который является ответственным за хемотаксис нейтрофилов и субпопуляцииT-лимфоцитов в область воспаления), по-видимому, могут излечиваться при помощи соединений, которые являются ингибиторами связывания рецептора IL-8. Сущность изобретения Настоящее изобретение включает новые антагонисты рецептора IL-8, представленные формулой(I), и композиции, содержащие соединения по изобретению и фармацевтически приемлемый носитель или разбавитель. Настоящее изобретение также включает новые антагонисты рецептора IL-8, представленные формулой (I), и комбинации, включающие соединения по изобретению и один или более дополнительных терапевтических ингредиентов. Настоящее изобретение также включает способ лечения опосредуемых хемокинами заболеваний,причем, хемокином является хемокин, который связывается с рецептором IL-8 или , и этот способ-1 014958 включает введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Настоящее изобретение также относится к способу ингибирования связывания IL-8 на его рецепторах у млекопитающего, особенно у человека, который включает введение эффективного количества соединения формулы (I). Подробное описание изобретения Соединения формулы (I), которые могут быть использованы в настоящем изобретении, представлены структурой:R2 выбран из группы, состоящей из C3-6 циклоалкила, фенила и гетероарила, причем фенил или гетероарил в случае необходимости замещены, один или два раза, независимо, заместителем, выбранным из группы, состоящей из C1-3 алкила, галогена, CF3, OCF3, фенилокси и бензилокси; илиR2 обозначает фенил, замещенный метилендиокси или (дигалогензамещенным)-метилендиокси;R1 обозначает C4-8 гетероцикл (CH2)n, причем гетероцикл в случае необходимости замещен, независимо, один или два раза, заместителем, выбранным из группы, состоящей из C1-3 алкила, C(O)OR4 иR1 выбран из следующих кольцевых систем (a-k): в которых R3 обозначает H или C1-3 алкил; или их фармацевтически приемлемая соль. В рамках изобретения "C1-3 алкил" относится к прямой или разветвленной насыщенной углеводородной группе, содержащей 1-3 атома углерода. Примеры таких групп включают метил, этил, н-пропил,или изопропил. В рамках изобретения "циклоалкил" относится к насыщенному моноциклическому углеводородному кольцу из 3-6 атомов углерода. Примеры таких групп включают циклопропил, циклобутил, циклопентил, циклогексил и т.п. В рамках изобретения "гетероцикл" относится к 4-8-членному насыщенному или неароматическому ненасыщенному моноциклическому кольцу, содержащему 1-3 гетероатома, выбранных из кислорода,азота или серы. Примеры таких колец включают пирролидинил, азетидинил, пиразолидинил, пиперидинил, пиперазинил и т.п. В рамках изобретения "гетероарил" относится к 5-6 членному моноциклическому ароматическому кольцу, содержащему 1-3 гетероатома, выбранных из кислорода, азота и серы. Примеры таких моноциклических ароматических колец включают пиридил, пирролил, пиранил, пиразолил, пиримидил, пиридазинил, пиразинил и т.п. В рамках изобретения "галоген" относится к F, Cl, Br или I.-2 014958 В рамках изобретения "C1-3 алкокси" относится к прямой или разветвленной алкоксигруппе, содержащей 1-3 атома углерода. Примерами алкокси, включенных в рамки изобретения, являются метокси,этокси, пропокси и проп-2-окси и т.п. В рамках изобретения "в случае необходимости замещенный", если не определено специфически,означает замещенный, независимо в каждом случае, от одного до трех раз такими группами, как галоген,C1-3 алкил, C1-3 алкокси, гетероцикл, арил и гетероарил, так что возможные заместители могут быть также замещены, за исключением галогена, от одного до трех раз, независимо, галогеном или C1-2 алкилом. Соединения согласно настоящему изобретению могут содержать один или более асимметрических атомов углерода и могут существовать в рацемических и оптически активных формах. Все эти соединения и диастереомеры находятся в рамках настоящего изобретения. Предпочтительно, X выбирают из группы, состоящей из галогена, C1-3 алкила, C1-3 алкокси, циано,CF3 и OCF3. В одном варианте осуществления X обозначает галоген. В другом варианте осуществления X выбирают из группы, состоящей из F, Cl и Br. В другом варианте осуществления X обозначает Cl. Предпочтительно R2 выбирают из группы, состоящей из C3-6 циклоалкила, фенила и гетероарила,причем фенил или гетероарил в случае необходимости замещены, один или два раза, независимо, заместителем, выбранным из группы, состоящей из C1-3 алкила, галогена, CF3, OCF3, фенилокси и бензилокси; илиR2 обозначает фенил, замещенный метилендиокси или (дигалогензамещенным)метилендиокси. В одном варианте осуществления R2 обозначает фенил, в случае необходимости замещенный, независимо, один или два раза заместителем, выбранным из группы, состоящей из C1-3 алкила, галогена, OCF3 или фенилокси. В одном варианте осуществления R2 обозначает пиридил, в случае необходимости замещенный один раз галогеном. В другом варианте осуществления R2 обозначает пиридил, замещенный хлором. В другом варианте осуществления R2 обозначает фенил, замещенный дифторметилендиокси. В другом варианте осуществления R2 обозначает C3-6 циклоалкил. В другом варианте осуществления R2 обозначает галогенметилфенил, тригалогенметилоксифенил,дигалогенфенил, этилфенил или фенилоксифенил. В другом варианте осуществления R2 обозначает 3-фтор-2-метилфенил, 2-трифторметилоксифенил,2-хлор-3-фторфенил, 2-этилфенил или 2-фенилоксифенил. В другом варианте осуществления R2 обозначает галогенпиридил. В другом варианте осуществления R2 обозначает 2-хлор-3-пиридил. Предпочтительно R1 обозначает C4-8 гетероцикл (CH2)n, причем гетероцикл в случае необходимости замещен, независимо, несколько раз заместителем, выбранным из группы, состоящей из C1-3 алкила,C(O)OR4 и C(O)R5; и в котором R4 и R5 обозначают независимо C1-3 алкил и n = 0 или 1; илиR1 выбран из группы, состоящей из следующих кольцевых систем (a-k) в которых R3 обозначает H или C1-3 алкил. В одном варианте осуществления R1 гетероцикл обозначает пирролидинил. В одном варианте осуществления R1 гетероцикл выбран из группы, состоящей из азетидинила, пиперидинила и пирролидинила. В другом варианте осуществления R1 выбран из группы, состоящей из пирролидинилметила, азе-3 014958 тидинила, пиперидинила, пирролидинила и этил-1-пиперидинилкарбоксилата. В другом варианте осуществления R1 выбран из группы, состоящей из 3-пирролидинилметила, 3 азетидинила, 4-пиперидинила, 3-пиперидинила, 3-пирролидинила и этил-1-пиперидинилкарбоксилата. Предпочтительно n = 0 или 1. В одном варианте осуществления n = 0. В другом варианте осуществления n = 1. В одном варианте осуществления R1 обозначает гетероцикл, выбранный из группы, состоящей из 23 азабицикло[2.2.1]гептила,8-азабицикло[3.2.1]октила,8-метил-8-азабицикло[3.2.1]октила,азабицикло[3.2.1]октила, 2-азабицикло[2.2.2]октила и 2-окса-5-азабицикло[2.2.2]октила. В другом варианте осуществления R1 обозначает 3-экзо-8-метил-8-азабицикло[3.2.1]окт-3-ил или 3 экзо-8-азабицикло[3.2.1]окт-3-ил. Подходящие фармацевтически приемлемые соли известны специалисту и включают основные соли неорганических и органических кислот, таких как соляная кислота, бромисто-водородная кислота, серная кислота, фосфорная кислота, метансульфокислота, этансульфокислота, уксусная кислота, яблочная кислота, винная кислота, лимонная кислота, молочная кислота, щавелевая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, бензойная кислота, салициловая кислота, фенилуксусная кислота и миндальная кислота. Кроме того, фармацевтически приемлемые соли соединений формулы (I) можно также образовать с фармацевтически приемлемым катионом, например, если заместитель включает карбоксильную группу. Подходящие фармацевтически приемлемые катионы известны специалисту и включают щелочные, щелочно-земельные, аммониевые и четвертичноаммониевые катионы. Примеры соединений согласно настоящему изобретению включают, но не ограничены ими:N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(2,3-дихлорфенил)карбамид; или их фармацевтически приемлемую соль. Предпочтительно соль является гидрохлоридом. В одном варианте осуществления соединения согласно настоящему изобретению включают, но не ограничены имиN-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(2,3-дихлорфенил)карбамид; или их фармацевтически приемлемую соль. В другом варианте осуществления соединения согласно настоящему изобретению включают, но не ограничены ими:N-[4-хлор-2-гидрокси-3-(3-пиперидинилсульфонил)фенил]-N'-(2,3-дихлорфенил)карбамид; или их фармацевтически приемлемую соль. В другом варианте осуществления соединения согласно настоящему изобретению включают, но не ограничены имиN-(2-хлор-3-фторфенил)-N'-4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфонил]фенилкарбамид; или их фармацевтически приемлемую соль. Способы получения Соединения по изобретению могут синтезироваться способом, включающим стадии:A) окисление сульфида согласно формуле (II) до сульфона согласно формуле (III):B) гидролиз этого сульфона до аминофенола согласно формуле (IV)C) действие на этот аминофенол изоцианатом или предшественником изоцианата с получением конечного продукта. Настоящее изобретение также включает новые промежуточные соединения, выбранные из группы,состоящей из формул (II) Соединения формулы (I) могут быть получены с использованием процедур синтеза, некоторые из-6 014958 которых проиллюстрированы ниже в схеме 1. Синтез, предусмотренный в этих схемах, может быть использован для получения соединений формулы (I), имеющих различные группы, которые вводят в реакцию, используя дополнительные заместители, которые соответственно защищают, чтобы добиться совместимости с реакциями, описанными здесь. Последующее удаление защитных групп, в этих случаях,затем приводит к соединениям, имеющим раскрытую здесь природу. После формирования карбамидного ядра другие соединения этих формул могут быть получены с использованием стандартных методик взаимного превращения функциональных групп, известных из уровня техники. Схема 1HCl; e) R2C=N=O или R2CON3. В примерах, где R1 содержат 3 или группу или группы ароматического амина, соединения формулы (I) могут быть получены согласно схеме 1. Сульфонил хлорид 1 (полученный согласно WO 01/68033 A2, включенной в настоящее описание в отношении раскрытия получения соединений согласно схеме 1, 1) восстанавливают до соответствующего тиола 2 в таких условиях, как трифенилфосфин в DMF. Альтернативно, соединение 2 может быть получено гашением соответствующего аниона лития (см. WO 01/68033 A2, включенную в настоящее описание в отношении раскрытия получения соединений согласно схеме 1, 1) серой (S8). Алкилирование 2 затем осуществляют путем обработки с использованием R1-LG, где LG обозначает удаляемую группу,такую как хлор, бром, йод или сульфонат, в присутствии основания. Полученные сульфиды 3 затем окисляют до сульфонов 4 действием окислителя, такого как перекись водорода. Гидролиз бензоксазольной группы приводит к аминофенолам 5, которые, в свою очередь, подвергают действию изоцианата или подходящего предшественника изоцианата, который может быть преобразован в изоцианат in situ. Эта реакция приводит к конечному карбамиду 6. В случаях, когда R1 содержит 1 или 2 амин, защищенный группой Boc или подобной кислотнолабильной защитной группой, соединения могут быть получены, как показано в схеме 2. Формирование сульфона и гидролиз выполняют, как описано выше, однако, так как последний приводит к удалению группы Boc, она должна быть введена повторно, что может быть сделано в подходящих реакционных условиях, таких как Boc ангидрид и гидроксид натрия, приводя к соединению 5a. После формирования карбамида группу Boc удаляют, получая желаемый продукт 7 в кислых условиях, таких как 4 н. соляная кислота в 1,4-диоксане. Наконец, полученный амин может быть далее переработан с помощью операции,известной специалисту как восстановительное аминирование, с получением 2 или 3 аминов 8. Схема 2 где: a) i) H2SO4/водн. 1,4-диоксан; ii) NaOH, Boc2O; b) R2C=N=O или R2CON3; c) 4 н. HCl, 1,4-7 014958 диоксан; d) NaCNBH3, альдегид или кетон. Промежуточные соединения 4a, 5a, 6a, 7 и 8 представляют собой новые промежуточные соединения в схеме 2. Примеры синтеза Следующие примеры иллюстрируют изобретение. Эти примеры не предназначены для ограничения объема настоящего изобретения, а скорее представляют собой руководство для специалиста по получению и использованию соединений, композиций и способов согласно настоящему изобретению. Хотя описаны конкретные варианты осуществления настоящего изобретения, специалисту понятно, что различные изменения и модификации могут быть сделаны без отхода от духа и объема изобретения. Все ссылки на эфир относятся к простому диэтиловому эфиру, солевой раствор относится к насыщенному водному раствору NaCl, DCM относится к дихлорметану, THF относится к тетрагидрофурану,EtOAc относится к этилацетату, Hex и Hx относятся к гексану, IMS относится к промышленному метиловому спирту, ТВМЕ относится к простому трет-бутилметиловому эфиру, DMF относится к диметилформамиду, BOC и Boc относятся к трет-бутилоксикарбонилу. Если не указано иное, все температуры выражены в C (градусах по шкале Цельсия). Все реакции проводят в инертной атмосфере при комнатной температуре, если не указано иное. Спектры 1H ЯМР регистрировали на спектрометре Jeol Delta2 (при 300 МГц). Химические сдвиги выражены в частях на миллион (ррm, единицы ). Структуры расщепления описывают выдимые мультиплетности и определяются как s (синглет), d (дублет), t (триплет), q (квартет), quint (квинтет), m (мультиплет), br (уширенный). Если не указано иное, "флэш" и "хроматография на колонках" относятся к флэш-хроматографии на колонке с силикагелем с использованием указанных систем растворителей. Данные LC-MS были получены либо на PE Sciex Single Quadrupole LC/MS API-150, объединенном с системой LC Shimadzu (Контроллер SCL-10A и двойной УФ-детектор), либо на Waters micromass ZQ,объединенном с модулем разделения Waters 2695. Исходный материал 1. N-(3,4-дихлорфенил)-2,2-диметилпропанамид 3,4-дихлоранилина (150 г) растворяли в 1,0 л TBME, и раствор охлаждали до 10C. Гидроксид натрия (140,7 г 30%-ого водного раствора) добавляли при механическом перемешивании. Пивалоилхлорид(125,9 мл) добавляли по каплям, поддерживая внутреннюю температуру 35C. После добавления температуру реакции поддерживали равной 30-35C еще 30 мин. Реакционной смеси затем давали охладиться до комнатной температуры и затем выдерживали при 0-5C в течение 1 ч. Полученный осадок фильтровали и промывали 600 мл смеси вода/MeOH (90/10) и затем 900 мл воды. Полученное твердое вещество высушивали в вакуумном сушильном шкафу при 55C в течение 4 дней. Выход: 162 г. 1N-(3,4-дихлорфенил)-2,2-диметилпропанамид (121 г) растворяли в 720 мл THF, и раствор охлаждали до -50C. Добавляли бутиллитий (433 мл, 2,5 н. в гексане), поддерживая внутреннюю температуру от 45 до -35C (конечная темп. -35C). Выдерживали при -25C в течение 40 мин. Проверка ВЭЖХ реакционной смеси показала, что осталось 5-10% исходного материала. Дополнительные 35 мл бутиллития добавляли при -30C, и реакционную смесь выдерживали при температуре от -30 до -25C в течение дальнейших 30 мин (ВЭЖХ: никаких значительных изменений). Реакционную смесь охлаждали до -45C и барботировали SO2 через раствор до достижения насыщенности. Затем реакционную смесь перемешивали при температуре от -10 до 0C в течение 45 мин. Через раствор барботировали аргон (2 объема двойного баллона), после чего реакционную смесь охлаждали до -5C. Добавляли сульфурилхлорид (58,8 мл),поддерживая температуру ниже 22C. Затем реакционную смесь выдерживали при 10-15C в течение 1 ч(ВЭЖХ: полное завершение реакции). Добавляли EtOAc и смесь концентрировали, промывали водой,насыщенным водным бикарбонатом натрия и солевым раствором, высушивали над MgSO4, и растворитель выпаривали в вакууме. Сырой материал кристаллизовали и растирали с горячим гексаном. Выход: 87,2 г 1 Исходный материал 1, N-(3,4-дихлорфенил)-2,2-диметилпропанамид (полученный согласно WO 01/68033 A2, включенной в настоящее описание путем ссылки в отношении раскрытия синтеза Исходного материала 1, также описанного выше) растворяли в сухом THF (400 мл), затем охлаждали до -75C в атмосфере аргона. n-BuLi (160 мл, 2,5 М в гексане, 5 экв.) добавляли по каплям, поддерживая температуру ниже -60C. После добавления всего n-BuLi реакционную смесь перемешивали при -5C в течение 1,5-8 014958 ч, затем охлаждали до -70C и добавляли серу ("цветки серы") (13 г) с последующим перемешиванием при температуре от -70C до комнатной температуры в течение ночи. После перемешивания реакционной смеси при -10C раствор менял цвет с желтого на коричнево/оранжевый. Реакционную смесь охлаждали до 0C, гасили 2 н. раствором HCl (200 мл) и перемешивали в течение 10 мин. Органический слой отделяли и подщелачивали 2 н. раствором NaOH до pH 12-13, затем промывали EtOAc. Водный слой повторно подкисляли 2 М раствором HCl до pH приблизительно 1 и экстрагировали дихлорметаном (2X),который промывали водой, высушивали над Na2SO4 и концентрировали. Сырой продукт очищали с использованием хроматографии на колонках 1:5 (EtOAc/Hex). Выход: 6 г (30%, оранжевое масло). 1H-ЯМР (CDCl3)7,39-7,30 (м, 2H), 4,08 (с, 1H), 1,50 (9H, c). Альтернативно, промежуточное соединение 1 получали следующим образом. Трифенилфосфин (89 г) растворяли в DCM (200 мл) и DMF (2,2 мл). Раствор охлаждали в ванне лед/метанол до -1C. К этому добавляли раствор 6-хлор-2-(1,1-диметилэтил)-1,3-бензоксазол-7 сульфонилхлорида, исходного материала 2 (полученного согласно WO 01/68033 A2, включенной в настоящее описание путем ссылки в отношении раскрытия синтеза исходного материала 2, также описанного выше) (35 г) в DCM (100 мл за 30 мин, поддерживая температуру ниже 15C. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 18 ч. Реакционную смесь гасили,используя 2 н. соляную кислоту (200 мл). Фазы разделяли и органическую фазу упаривали в вакууме. Остаток суспендировали в 2 н. гидроксиде натрия (400 мл) и быстро перемешивали в течение 3 ч. Твердое вещество удаляли фильтрацией и промывали водой. Объединенные фильтрат и смывы охлаждали в ванне со льдом/водой и подкисляли с использованием 5 н. соляной кислоты до рН 1. Экстрагировали, используя TBME (400 мл). Органическую фазу высушивали над сульфатом магния и упаривали в вакууме,получая Промежуточное соединение 1 (22,85 г) в форме твердого вещества коричневого цвета. Промежуточное соединение 2 (общая процедура A).DCM (100 мл). Объединенные органические фазы промывали водой (250 мл), затем солевым раствором(50 мл), высушивали (Na2SO4) и концентрировали. Остаток хроматографировали на колонке (флэш,элюирование с градиентом 0-10% MeOH/DCM). Выход: 1,55 г. 1 К раствору промежуточного соединения 2 (1 г) в DCM (10 мл) добавляли Et3N (1,38 мл), затем по каплям при 0C MsCl (0,46 мл). После перемешивания при 0C в течение 1 ч реакционную смесь нагревали при комнатной температуре, гасили водой (10 мл) и разделяли. Водный слой экстрагировали DCM(220 мл). Объединенные органические фазы промывали водой (40 мл), добавляли лопатку кремнезема,высушивали (NaSO4) и концентрировали. Выход: 1,4148 г. 1 К суспензии NaH (0,30 г) в THF (20 мл) добавляли по каплям промежуточное соединение 1 (используя Исходный материал 1) (1,22 г). После перемешивания в течение 1 ч добавляли промежуточное соединение 3 (1,41 г) в THF, реакционную смесь нагревали при 80C и оставляли в течение ночи. Реакционную смесь охлаждали до комнатной температуры, затем гасили водным насыщенным NaHCO3 (50 мл). Реакционную смесь экстрагировали DCM (250 мл). Объединенные органические фазы промывали водой (100 мл), высушивали (NaSO4) и концентрировали. Остаток хроматографировали на колонке (флэш,20% EtOAC/Hx, силикагель). Выход: 946,9 мг. К раствору промежуточного соединения 4 (946,9 мг) в DCM (10 мл) добавляли при -10C mCPBA(2,31 г) в DCM (10 мл). Реакционную смесь перемешивали при -10C в течение 1 ч, затем нагревали до комнатной температуры. Реакционную смесь гасили водным насыщенным NaHCO3 (50 мл), затем экстрагировали DCM (270 мл). Объединенные органические фазы промывали водой (50 мл), высушивали(Na2SO4) и концентрировали. Остаток хроматографировали на колонке (флэш, 30% EtOAc/Hx, силикагель). Выход: 353,6 мг (35%, желтое масло). MS (m/z, ES+, M+H): 457,08. Промежуточное соединение 6 (общая процедура E). К раствору промежуточного соединения 5 (353 мг) в IMS (5 мл) добавляли водный концентрированный раствор HCl (5 мл). Реакционную смесь нагревали до 80C и оставляли на ночь. Реакционную смесь охлаждали до комнатной температуры и концентрировали, чтобы удалить IMS. Остаток подщелачивали до pH 12 водным насыщенным NaOH, добавляли при 0C EtOAc (30 мл), BOC2O (1 экв., 0,17 г) и оставляли на ночь. Реакционную смесь разделяли, и водный слой экстрагировали EtOAc (230 мл). Объединенные органические фазы высушивали (Na2SO4) и концентрировали. Остаток хроматографировали на колонке (флэш, элюирование с градиентом 10-30% EA/Hx). Выход: Выделяли две фракции, содержащие продукт: 58,0 мг и 180,9 мг. MS (m/z, ES+, M+H): 291,01. Промежуточное соединение 7 (общая процедура F). 3-фтор-2-метиланилин (7,4 г) растворяли в DCM (220 мл) при комнатной температуре в атмосфере аргона. После охлаждения до 0C добавляли водный насыщенный NaHCO3 (220 мл), затем трифосген(5,85 г). Реакционную смесь перемешивали при 0C в течение 1 ч. После этого времени продукт экстрагировали DCM (250 мл). Органические фракции объединяли, высушивали над MgSO4 и растворитель удаляли в вакууме, получая желтое масло. Добавление гексана приводило к осаждению белой соли, которую отфильтровывали. Удаление гексана в вакууме привело к желтому маслу (7,69 г, 86 %). 1H-ЯМР К раствору промежуточного соединения 6 (60 мг) в DCM (3 мл) добавляли промежуточное соединение 7 (70 мг) и реакционную смесь оставляли на выходные. Реакционную смесь концентрировали и остаток хроматографировали на колонке (флэш, элюирование с градиентом 20%-30% EtOAc/Hx). Выход: 56,2 мг. MS (m/z, ES+, M+H): 542,01. Пример 1. Промежуточное соединение 8 (56,2 мг) и 4 н. HCl/диоксан (3 мл) перемешивали вместе при комнатной температуре и оставляли на ночь. Промежуточные соединения 6, 5, 4, 3 и 2 получали, как описано- 10014958 выше. Промежуточное соединение 1 получали, используя исходный материал 1, для того, чтобы синтезировать пример 1. Реакционную смесь концентрировали и остаток растворяли в минимальном количестве MeOH, и добавляли Et2O. Получали твердое вещество, которое отфильтровывали и высушивали. Сырой выход: 28,4 мг. Сырой продукт растворяли в минимальном количестве MeOH и добавляли Et2O. Получали твердое вещество, растворитель декантировали и твердое вещество высушивали. Выход: 18,8 мг. MS (m/z, ES+, M+H): 441,98. ЯМР (MeOD)8,40 (1H, д, ArH), 7,46 (1H, д, ArH), 7,19-7,15 (2H, м,ArH), 6,85 (1H, т, ArH), 4,14 (1H, дт, CH), 3,66 (1H, дд, CH), 3,37 (2H, д, CH2), 3,04 (1H, дт, CH) , 2,19 (3H,С, ArCH3), 2,14-1,69 (4 Н, м, 2CH2). Промежуточное соединение 9. Трифосген (7,7 г) в DCM (40 мл) добавляли при 0C к 3-амино-2-хлорпиридину (10 г) в DCM (200 мл) и насыщенном водном NaCHO3 (200 мл) . Реакционную смесь перемешивали в течение 1 ч. Затем продукт экстрагировали DCM (250 мл), высушивали над Na2SO4 и растворитель удаляли в вакууме, получая твердое вещество грязно-белого цвета. Растирание с гексаном с последующей фильтрацией твердых частиц и удалением растворителя из элюента привело к продукту в форме бесцветного масла. После промывки продукта аргоном и помещения его в рефрижератор, получали кристаллическое твердое вещество белого цвета (6 г). 1H-ЯМР (CDCl3)8,22 (с ушир., 1H), 7,45 (д, 1H), 7,27-7,20 (м, 1H). Промежуточное соединение 10. К раствору промежуточного соединения 6 (60 мг) в DCM (3 мл) добавляли промежуточное соединение 9 (71 мг) и реакционную смесь оставляли на выходные. Реакционную смесь концентрировали и остаток хроматографировали на колонке (флэш, 20-30 % EtOAc/Hx, силикагель). Выход: 60,0 мг. MS Промежуточное соединение 10 (60 мг) и 4 н. HCl/диоксан (3 мл) перемешивали вместе при комнатной температуре и оставляли на ночь. Промежуточные соединения 6, 5, 4, 3 и 2 получали, как описано выше. Промежуточное соединение 1 получали, используя исходный материал 1 для того, чтобы синтезировать пример 2. Реакционную смесь концентрировали, и остаток растворяли в минимальном количестве К раствору промежуточного соединения 6 (60 мг) в DCM (3 мл) добавляли 2-хлор-3 фторфенилизоцианат (79 мг) и реакционную смесь оставляли на выходные. Реакционную смесь концентрировали и остаток хроматографировали на колонке (флэш, элюирование с градиентом 0-30%N-(2-хлор-3-фторфенил)-N'-4-хлор-2-гидрокси-3-[(3S)-3-пиперидинилсульфо Промежуточное соединение 11 (71,2 мг) и 4 н. HCl/диоксан (3 мл) перемешивали вместе при комнатной температуре и оставляли на ночь. Промежуточные соединения 6, 5, 4, 3 и 2 получали, как описано выше. Промежуточное соединение 1 получали, используя исходный материал 1 для того, чтобы синтезировать пример 3. Реакционную смесь концентрировали и остаток растворяли в минимальном количествеMeOH и добавляли Et2O. Получали твердое вещество, которое отфильтровывали и высушивали. Выход: 50,1 мг (твердое вещество грязно-белого цвета). MS (m/z, ES+, M+H): 461,96. ЯМР (MeOD)8,42 (1H, д,ArH), 7,93 (1H, д, ArH), 7,26 (1H, дт, ArH), 7,18 (1H, д, ArH), 6,95 (1H, дт, ArH), 4,21-4,11 (1H, м, CH),3,66 (1H, дд, CH), 3,39-3,31 (2H, м, CH2), 3,03 (1H, дт, CH), 2,17-1,72 (4 Н, м, 2CH3). Таблица 1. Промежуточные соединения получали согласно общим процедурам A-E Две загрузки 2,2-дифтор-1,3-бензодиоксол-4-амина (1 г и 3,58 г) обрабатывали трифосгеном (566 мг и 2,04 мг) и насыщенным водным NaHCO3 (20 мл и 80 мл) в DCM (20 мл и 80 мл) согласно процедуре,описанной для промежуточного соединения 9, получая 3,27 г желаемого продукта после объединения 2 загрузок. 1H-ЯМР (CDCl3)7,05 (т, 1H), 6,90 (д, 1H), 6,82 (д, 1H). Промежуточное соединение 1 получали, используя исходный материал 1 для всех синтезов. Таблица 2. Примеры, полученные из промежуточных соединений в табл. 1N-BOC-нортропинон (6,3 г) растворяли в MeOH (150 мл) и охлаждали до 0C. NaBH4 добавляли частями, и реакционную смесь перемешивали при 0C в течение 2,5 ч. Реакционную смесь гасили водой,подкисляли до pH 3 и экстрагировали DCM (3). Объединенные органические слои высушивали надNa2SO4 и концентрировали, получая желаемый продукт (5,0 г, 79%, твердое вещество белого цвета). Промежуточное соединение 19 получали из промежуточного соединения 18 и промежуточного соединения 1 (с использованием исходного материала 1) согласно общим процедурам B, C, D и E. Общий выход: 29%, MS (m/z, ES+, M+H): 316,99. В круглодонную колбу на 100 мл в атмосфере аргона добавляли (R)-(-)-N-BOC-3-пирролидинол (1,5 г). Затем добавляли CBr4 (1,5 г), затем сухой THF (50 мл), и смесь охлаждали до 5C. Затем добавлялиPPh3 за 5 мин и развитие реакции отслеживали с помощью TLC. Твердое вещество удаляли фильтрацией и промывали эфиром. Фильтрат концентрировали и очищали хроматографией на колонках с элюированием 1:3 EtOAc:Hx. Выход: 2,3 г ( 100%, используемых без дальнейшей очистки). 1 Промежуточное соединение 1 (1,0 г), промежуточное соединение 21 (1,24 г) и K2CO3 (0,49 г) в метилэтилкетоне (10 мл) перемешивали при комнатной температуре в течение ночи. Промежуточное соединение 1 получали, используя исходный материал 1. Реакционную смесь гасили водой, экстрагировалиEtOAc, и органические слои высушивали над Na2SO4 и концентрировали. Сырой продукт очищали хроматографией на колонках с элюированием 1:4 EtOAc:Нх. Выход: 0,60 г (36%, коричневое масло). 1(0,68 г). Полученную смесь перемешивали при -10C в течение 30 мин. Реакционную смесь гасили раствором NaHCO3, экстрагировали DCM, промывали водой. Органические слои высушивали над Na2SO4,концентрировали и очищали хроматографией на колонках с элюированием 1:3 EtOAc:Hx. Выход: 0,30 г(69%, твердое вещество белого цвета). LC-MS (m/z, M+H): 443. Промежуточное соединение 24 (общая процедура J). Промежуточное соединение 23 (0,30 г) перемешивали в THF (2 мл), затем добавляли конц. HCl и нагревали при 80C в течение ночи. Реакционную смесь охлаждали, растворитель удаляли и оставшуюся смесь подщелачивали 50%-ным водным раствором NaOH. Смесь охлаждали до 0C и добавляли EtOAc,затем BOC2O (0,155 г) и полученную смесь перемешивали при комнатной температуре в течение ночи. Смесь разбавляли EtOAc, и органическую фазу отделяли, высушивали над Na2SO4 и концентрировали. Сырой продукт очищали хроматографией на колонках с элюированием с градиентом 33-75% EtOAc в гексане (количественный) 0,25 г. LC-MS (m/z, M+H-tBu): 321,00. Промежуточное соединение 25. Промежуточное соединение 25 получали из промежуточного соединения 1 (0,5 г) и N-BOC-4 бромпиперидина (0,59 г) согласно общим процедурам I, D и J. Выход: 0,50 г (58% в целом). Промежуточное соединение 1 получали, используя исходный материал 1. Промежуточное соединение 25 (0,25 г) и 2-феноксифенилизоцианат (0,175 г) растворяли в DMF (3 мл) и реакционные смеси перемешивали при комнатной температуре в течение ночи. Избыток DMF выпаривали и оставшуюся смесь загружали на колонку и элюировали смесью 1:3 EtOAc:Нх. Выход: 400 мг(количественный, твердое вещество белого цвета) MS (m/z, ES+, M+H): 574,17. Пример 28. N-[4-хлор-2 тидрокси-3-(4-пиперидинилсульфонил)фенил]-N'-[2-(фенилокси)фенил]карбамид Пример 27 (0,20 г) перемешивали в CHCl3 (2 мл) при комнатной температуре в атмосфере аргона. Добавляли йодтриметилсилан (0,21 г) и смесь нагревали с обратным холодильником в течение ночи. Реакция, однако, не начиналась, пока не были добавлены две загрузки хлортриметилсилана (0,037 г за загрузку) и NaI (0,051 г за загрузку), и реакционную смесь не нагревали при 75C в течение еще 6 ч. В это время добавляли MeOH и смесь перемешивали в течение еще 30 мин, после чего растворители удаляли и добавляли раствор аммиака в метаноле. Смесь экстрагировали DCM, органический слой промывали водой, высушивали над Na2SO4 и концентрировали. Сырой продукт растирали со смесью DCM/эфир и затем очищали хроматографией на колонках с элюированием метанолом. Выход: 10 мг (6%, твердое вещество светло-коричневого цвета) MS (m/z, ES+, M+H): 502,06. Промежуточные соединения 4-6, 8, 10-17, 20, 22-25 являются новыми промежуточными соединениями. Таблица 3. Примеры, полученные из промежуточных соединений 20, 24 и 25.- 18014958 Способ лечения Соединения формулы (I) или их фармацевтически приемлемая соль могут использоваться в получении лекарственного средства для профилактического или терапевтического лечения любого болезненного состояния у человека или другого млекопитающего, которое усилено или вызвано чрезмерной или нерегулируемой продукцией цитокина IL-8 клетками такого млекопитающего, такими как, но не ограничиваясь ими, моноциты и/или макрофаги, или других хемокинов, которые связываются с IL-8 илирецептором, также называемым рецептором типа I или II. Соответственно, настоящее изобретение относится к способу лечения заболевания, опосредуемого хемокином, причем хемокином является хемокин, который связывается с IL-8 илирецептором, и этот способ включает введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В частности, хемокинами являются IL-8, GRO, GRO, GRO, NAP-2 или ENA-78. Соединения формулы (I) вводят в количестве, достаточном для ингибирования функций цитокина,в особенности IL-8, GRO, GRO, GRO, NAP-2 или ENA-78, такой, что они биологически даунрегулируются до нормальных уровней физиологической функции, или в некотором случае до недостаточных уровней, что приводит к улучшению в случае болезненного состояния. Патологические уровниIL-8, GRO, GRO, GRO, NAP-2 или ENA-78, например, в контексте настоящего изобретения, составляют: (i) уровни свободного IL-8, больше или равные 1 пикограм на 1 мл; (ii) любые связанные с клетками IL-8, GRO, GRO, GRO, NAP-2 или ENA-78 выше нормальных физиологических уровней; или (iii) присутствие IL-8, GRO, GRO, GRO, NAP-2 или ENA-78 выше основных уровней в клетках или тканях, в которых IL-8, GRO, GRO, GRO, NAP-2 или ENA-78, соответственно, продуцируются. Существует много болезненных состояний, при которых чрезмерная или нерегулируемая продукция IL-8 участвует в усилении и/или порождении заболевания. Хемокин-опосредуемые заболевания включают псориаз, аллергический дерматит, остеоартрит, ревматоидный артрит, астму, хроническое обструктивное заболевание легких, респираторный дистресс-синдром взрослых, воспалительное заболевание кишечника, болезнь Крона, неспецифический язвенный колит, инсульт, септический шок, эндотоксический шок, грамотрицательный сепсис, синдром токсического шока, реперфузионное повреждение сердца и почек, гломерулонефрит, тромбоз, реакцию трансплантат против хозяина, болезнь Альцгеймера, отторжение аллотрансплантата, малярию, рестиноз, ангиогенез, атеросклероз, остеопороз, гингивит,вирусные заболевания, такие как риновирусные заболевания, или нежелательное высвобождение гематопоэтических стволовых клеток. Эти заболевания прежде всего характеризуются массивной инфильтрацией нейтрофилов, инфильтрацией T-клеток или ростом новых сосудов и связаны с увеличенной продукцией IL-8, GRO, GRO,GRO, NAP-2 или ENA-78, которая является ответственной за хемотаксис нейтрофилов в воспалительный участок или направленный рост эндотелиальных клеток. В отличие от других воспалительных цитокинов (IL-1, TNF и IL-6), IL-8, GRO, GRO, GRO, NAP-2 или ENA-78 имеют уникальное свойство в том, что они промотируют хемотаксис нейтрофилов, высвобождение ферментов, включая, но не ограничиваясь им, высвобождение эластазы, а также продукцию и активацию супероксидов. -Хемокины, но особенно GRO, GRO, GRO, NAP-2 или ENA-78, работающие через рецептор IL-8 типа I или II, могут промотировать неоваскуляризацию опухолей, промотируя направленный рост эндотелиальных клеток. Поэтому ингибирование индуцированных IL-8 хемотаксиса или активации приведет к непосредственному снижению инфильтрации нейтрофилов. Недавние исследования также демонстрируют роль хемокинов в лечении ВИЧ инфекций, Littlemanet al., Nature 381, pp. 661 (1996) and Koup et al., Nature 381, pp. 667 (1996). Настоящее исследование также показывает использование ингибиторов IL-8 в лечении атеросклероза. Первая ссылка, Boisvert et al., J. Clin. Invest, 1998, 101:353-363, демонстрирует, через пересадку костного мозга, что отсутствие рецепторов IL-8 на стволовых клетках (и, поэтому, на моноцитах/макрофагах) приводит к сокращению развития атеросклеротических бляшек у мышей с дефицитом рецепторов ЛПНП. Дополнительные ссылки: Apostolopoulos, et al., Arterioscler. Thromb. Vasc. Biol. 1996,16:1007-1012; Liu, et al., Arterioscler. Thromb. Vasc. Biol, 1997, 17:317-323; Rus, et al., Atherosclerosis. 1996,127:263-271.; Wang et al., J. Biol. Chem. 1996, 271:8837-8842; Yue, et al., Eur. J. Pharmacol. 1993, 240:81-84;et al., Arterioscler. Thromb., 1994, 14:47-53. Настоящее изобретение также относится к средству для лечения повреждений ЦНС с помощью соединений-антагонистов рецептора хемокина формулы (I). Такое лечение обеспечивается в острый период, а также для профилактики повреждения у лиц, считающихся предрасположенными к такому повреждению. Повреждения ЦНС, как определено здесь, включают как открытые, так и проникающие травмы головы, например, при хирургическом вмешательстве, или закрытые травматические повреждения головы,такие как повреждение области головы. Также в рамки этого определения входят ишемический инсульт,особенно в области мозга. Ишемический инсульт может быть определен как очаговое неврологическое нарушение, которое- 19014958 следует из недостаточного кровоснабжения специфической области мозга, обычно как следствие эмболии, тромбов или местного атероматозного смыкания кровеносного сосуда. Роль воспалительных цитокинов в этой области выявлена, и настоящее изобретение обеспечивает средства для потенциального лечения этих повреждений. Ранее были доступны относительно ограниченные виды лечения острого повреждения, такого как описано.TNF- представляет собой цитокин провоспалительного действия, включая экспрессию молекулы адгезии лейкоцита к эндотелию. Лейкоциты инфильтруются в области ишемического повреждения мозга, и, следовательно, соединения, которые ингибируют или снижают уровни TNF, по-видимому, могут быть использованы для лечения ишемического повреждения головного мозга. См. Liu et al., Stroke, Vol. 25.,7, pp. 1481-88 (1994), раскрытие которого включено в настоящее описание путем ссылки. Модели закрытых черепно-мозговых повреждений и лечения смешанными средствами 5-LO/CO обсуждаются в Shohami et al., J. of VaiscClinical Physiology and Pharmacology, vol. 3,2, pp. 99-107(1992). Было обнаружено, что лечение, которое уменьшает формирование отека, улучшает функциональный результат у подвергаемых лечению животных. Соединения формулы (I) вводят в количестве, достаточном для ингибирования IL-8, связывание с рецепторами IL-8 или , что доказывается уменьшением хемотаксиса и активации нейтрофилов. Открытие того, что соединения формулы (I) являются ингибиторами связывания IL-8, основано на эффектах соединений формулы (I) в тестах in vitro на связывание с рецептором, которые описаны здесь. Было показано, что соединения формулы (I) являются ингибиторами рецепторов IL-8 типа II. В рамках изобретения термин "опосредуемое IL-8 заболевание или болезненное состояние" относится к любому и всем болезненным состояниям, в которых IL-8, GRO, GRO, GRO, NAP-2 или ENA78 играет роль либо за счет продукции IL-8, GRO, GRO, GRO, NAP-2 или ENA-78 как таковых, либо за счет того, что IL-8, GRO, GRO, GRO, NAP-2 или ENA-78 вызывают высвобождение других монокинов, таких как, но не ограничиваясь ими, IL-1, IL-6 или TNF. Болезненное состояние, в котором основной составляющей является, например, IL-1, и чья продукция или действие усиливается или секретируется в ответ на IL-8, поэтому следует считать болезненным состоянием, опосредуемым IL-8. В рамках изобретения термин "опосредуемое хемокином заболевание или болезненное состояние" относится к любому и всем болезненным состояниям, в которых играет роль хемокин, который связывается с IL-8 илирецептором, такой как, но не ограничиваясь ими, IL-8, GRO, GRO, GRO, NAP-2 или ENA-78. Оно включает болезненное состояние, в котором IL-8 играет роль либо за счет продукцииIL-8 как такового, либо за счет того, что IL-8 вызывает высвобождение других монокинов, таких как, но не ограничиваясь ими, IL-1, IL-6 или TNF. Болезненное состояние, в котором основной составляющей является, например, IL-1, и чья продукция или действие усиливается или секретируется в ответ на IL-8,поэтому следует считать болезненным состоянием, опосредуемым IL-8. В рамках изобретения термин "цитокин" относится к любому секретируемому полипептиду, который воздействует на функции клеток и представляет собой молекулу, которая модулирует взаимодействия между клетками в иммунной, воспалительной или гематопоэтической реакции. Цитокин включает,но не ограничен ими, монокины и лимфокины, независимо от того, какие клетки их продуцируют. Например, монокин обычно упоминается как продуцируемый и секретируемый одноядерной клеткой, такой как макрофаг и/или моноцит. Многие другие клетки, однако, также продуцируют монокины, такие какNK-клетки, фибробласты, базофилы, нейтрофилы, зндотелиальные клетки, мозговые астроциты, клетки стромы костного мозга, кератиноциты эпидермиса и B-лимфоциты. Лимфокины обычно упоминаются как продуцируемые лимфоцитами. Примеры цитокинов включают, но не ограничены ими, интерлейкин 1 (IL-1), интерлейкин-6 (IL-6), интерлейкин-8 (IL-8), фактор некроза опухоли(TNF-) и фактор некроза опухоли(TNF-). В рамках изобретения термин "хемокин" относится к любому секретируемому полипептиду, который воздействует на функции клеток и представляет собой молекулу, которая модулирует взаимодействия между клетками в иммунной, воспалительной или гематопоэтической реакции, подобно термину"цитокин", раскрытому выше. Хемокин прежде всего секретируется трансмембранно из клеток и вызывает хемотаксис и активацию специфических белых кровяных телец и лейкоцитов, нейтрофилов, моноцитов, макрофагов, T-клеток, B-клеток, эндотелиальных клеток и гладкомышечных клеток. Примеры хемокинов включают, но не ограничены ими, IL-8, GRO, GRO, GRO, NAP-2, ENA-78, IP-10, MIP-1,MIP-, PF4 и MCP 1, 2 и 3. Для использования соединения формулы (I) или его фармацевтически приемлемой соли в терапии его обычно составляют в фармацевтическую композицию в соответствии с обычной фармацевтической практикой. Это изобретение, поэтому, также относится к фармацевтической композиции, содержащей эффективное, нетоксичное количество соединения формулы (I) и фармацевтически приемлемый носитель или разбавитель. Соединения формулы (I), их фармацевтически приемлемые соли и содержащие их фармацевтические композиции могут быть введены любым из путей, обычно используемых для введения лекарственного средства, например перорально, топически, парентерально или ингаляцией. Соединения формулы(I) могут вводиться в обычных лекарственных формах, полученных путем комбинации соединения формулы (I) со стандартными фармацевтическими носителями согласно обычным процедурам. Соединения формулы (I) могут также вводиться в обычных дозах в комбинации с известным вторым терапевтически активным соединением. Эти процедуры могут включать смешивание, грануляцию и прессование или растворение ингредиентов, в зависимости от желаемого препарата. Следует понимать, что форма и характер фармацевтически приемлемого характера или разбавителя диктуется количеством активного ингредиента, с которым он должен быть скомбинирован, путем введения и другими известными переменными. Носитель(и) должен быть "приемлемым" в смысле совместимости с другими ингредиентами состава и не быть вредным для реципиента. Фармацевтический носитель может быть, например, твердым или жидким. Примерами твердых носителей являются лактоза, фарфоровая глина, сахароза, тальк, желатин, агар-агар, пектин, гуммиарабик,стеарат магния, стеариновая кислота и т.п. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло, вода и т.п. Точно так же носитель или разбавитель могут включать материал для задержки высвобождения, известный из уровня техники, такой как глицерилмоностеарат или глицерилдистеарат, индивидуально или с воском. Могут быть использованы самые различные фармацевтические формы. Так, если используется твердый носитель, препарат может быть таблетирован, помещен в твердую желатиновую капсулу в форме порошка или гранулы или находиться в форме пастилок или леденцов. Количество твердого носителя широко варьирует, но предпочтительно составляет от приблизительно 25 мг до приблизительно 1 г. Когда используется жидкий носитель, препарат может быть в форме сиропа, эмульсии, мягкой желатиновой капсулы, стерильной жидкости для инъекций, такой как ампула или неводная жидкая суспензия. Соединения формулы (I) могут вводиться топически, что является несистемным введением. Оно включает нанесение соединения формулы (I) наружно на эпидермис или полость рта и инстилляцию такого соединения в ухо, глаз и нос таким образом, что соединение в основном не поступает в кровоток. Напротив, системное введение относится к пероральному, внутривенному, внутрибрюшинному и внутримышечному введению. Составы, подходящие для топического введения, включают жидкие или полужидкие препараты,подходящие для проникновения через кожу в зону воспаления, такие как линименты, лосьоны, кремы,мази или пасты и капли, подходящие для введения в глаз, ухо или нос. Активный ингредиент может составлять, для топического введения, от 0,001 до 10 вес./вес.%, например от 1 до 2 вес.% от массы состава. Он может, однако, составлять 10 вес./вес.%, но предпочтительно составляет менее 5 вес./вес.%, более предпочтительно от 0,1 до 1 вес./вес. % от массы состава. Лосьоны согласно настоящему изобретению включают подходящие для нанесения на кожу или глаз. Лосьон для глаз может включать стерильный водный раствор, в случае необходимости содержащий бактерицид, и может быть получен способами, подобными способам для получения капель. Лосьоны или линименты для нанесения на кожу могут также включать средство для ускорения высыхания и охлаждения кожи, такие как спирт или ацетон, и/или увлажнитель, такой как глицерин или масло, такое как касторовое масло или арахисовое масло. Кремы, мази или пасты согласно настоящему изобретению представляют собой полутвердые составы активного ингредиента для наружного нанесения. Они могут быть получены путем смешивания активного ингредиента в тонко раздробленной или порошковой форме, индивидуально или в растворе или суспензии в водной или неводной жидкости, при помощи подходящих машин, с сальной или несальной основой. Основа может включать углеводороды, такие как твердый, мягкий или жидкий парафин, глицерин, пчелиный воск, металлическое мыло; мазь на основе камеди; масло природного происхождения,такое как миндальное, кукурузное, арахисовое, касторовое или оливковое масло; ланолин или его производные, или жирную кислоту такую как стеариновая или олеиновая кислота, вместе со спиртом, таким как пропиленгликоль или макрогель. Состав может включать любое подходящее поверхностно-активное средство, такое как анионное, катионное или неионогенное поверхностно-активное вещество, такое как сложный эфир сорбитана или его полиоксиэтиленовое производное. Могут также быть включены суспендирующие агенты, такие как натуральные смолы, производные целлюлозы или неорганические материалы, такие как кремниевые кремнеземы, и другие ингредиенты, такие как ланолин. Капли согласно настоящему изобретению могут включать стерильные водные или масляные растворы или суспензии и могут быть получены путем растворения активного ингредиента в подходящем водном растворе бактерицидного и/или фунгицидного средства и/или любого другого подходящего консерванта, и предпочтительно включают поверхностно-активное средство. Полученный раствор может быть затем осветлен фильтрацией, перенесен в подходящий контейнер, который затем запечатывают и стерилизуют, обрабатывая в автоклаве или поддерживая в 98-100C в течение получаса. Альтернативно,раствор может быть стерилизован фильтрацией и помещен в контейнер асептической методикой. Примерами бактерицидных и фунгицидных средств, подходящих для включения в капли, являются нитрат или ацетат фенилртути (0,002%), хлорид бензалкония (0,01%) и хлоргексидинацетат (0,01%). Подходящие растворители для получения масляного раствора включают глицерин, разбавленный спирт и пропиленгликоль.- 21014958 Соединения формулы (I) могут вводиться парентерально, т.е. внутривенным, внутримышечным,подкожным, внутриносовым, внутриректальным, внутривлагалищным или внутрибрюшинным введением. Подкожные и внутримышечные формы парентерального введения обычно являются предпочтительными. Подходящие лекарственные формы для такого введения могут быть получены обычными методиками. Соединения формулы (I) могут также вводиться ингаляцией, т.е. внутриносовым и пероральным ингаляционным введением. Подходящие лекарственные формы для такого введения, такие как состав аэрозоля или ингалятор с отмеренными дозами, могут быть получены обычными методиками. Для всех способов использования, раскрытого здесь для соединений формулы (I), суточная доза при пероральном режиме предпочтительно составляет от приблизительно 0,01 до приблизительно 80 мг/кг полной массы тела. Суточная доза при парентеральном режиме предпочтительно составляет от приблизительно 0,001 до приблизительно 80 мг/кг полной массы тела. Суточная доза при топическом режиме предпочтительно составляет от 0,1 до 150 мг, вводимых 1-4, предпочтительно 2 или 3 раза в день. Суточная доза при режиме ингаляции предпочтительно составляет от приблизительно 0,01 до приблизительно 1 мг/кг в сутки. Специалисту также будет понятно, что оптимальное количество и интервал введения индивидуальных доз соединения формулы (I) или его фармацевтически приемлемой соли будут определены в зависимости от природы и степени серьезности подвергаемого лечению состояния, формы, пути и места введения, и конкретного получающего лечение пациента, и что такие оптимумы могут быть определены обычными методиками. Специалисту также будет понятно, что оптимальный курс лечения, то есть число доз соединения формулы (I) или его фармацевтически приемлемой соли, вводимое в сутки в течение определенного числа дней, может быть установлен специалистом с использованием обычных тестов для определения курса лечения. Комбинации Соединение и фармацевтические составы согласно изобретению могут использоваться в комбинации с или включать одно или более других терапевтических средств, например, выбранных из противовоспалительных средств, антихолинергических средств (особенно антагонистов рецептора M1/M2/M3),агонистов 2-адренорецептора, антиинфекционных средств, таких как антибиотики, противовирусные средства или антигистамины. Изобретение, таким образом, относится, в дальнейшем аспекте, к комбинации, включающей соединение формулы (I) или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное, вместе с одним или более другими терапевтически активными средствами, например, выбранными из противовоспалительного средства, такого как кортикостероид или NSAID, антихолинергического средства, агониста 2-адренорецептора, противоинфекционного средства, такого как антибиотик или противовирусное средство, или антигистамин. Один вариант осуществления изобретения охватывает комбинации, включающие соединение формулы (I) или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с агонистом 2-адренорецептора и/или антихолинергическим средством, и/или ингибитором PDE-4, и/или антигистамином. Специалисту будет понятно, что, в случае необходимости, другой терапевтический ингредиент(ингредиенты) может использоваться в форме солей, например солей щелочного металла или солей амина, или солей присоединения с кислотой, или пролекарств, или в форме сложных эфиров, например,сложных эфиров низшего алкила, или в форме сольватов, например, гидратов, чтобы оптимизировать активность и/или стабильность, и/или физические свойства, такие как растворимость, терапевтического ингредиента. Также понятно, что, в случае необходимости, терапевтические ингредиенты могут использоваться в оптически чистой форме. В одном варианте осуществления изобретение охватывает комбинацию, включающую соединение по изобретению вместе с агонистом 2-адренорецептора. Примеры агонистов 2-адренорецептора включают сальметерол (который может быть рацематом или единственным энантиомером, таким как Rэнантиомер), сальбутамол (который может быть рацематом или единственным энантиомером, таким какR-энантиомер), формотерол (который может быть рацематом или единственным диастереомером, таким как R,R-диастереомер), сальмефамол, фенотерол, кармотерол, этантерол, наминтерол, кленбутерол, пирбутерол, флербутерол, репротерол, бамбутерол, индакатерол, тербуталин, и их соли, например ксинафоат(1-гидрокси-2-нафталинкарбоксилат) сальметерола, сульфат или свободное основание сальбутамола, или фумарат формотерола. В одном варианте осуществления агонисты 2-адренорецептора являются агонистами 2-адренорецептора длительного действия, например, соединениями, которые обеспечивают эффективную бронходилатацию в течение приблизительно 12 ч или дольше. Другие агонисты 2 адренорецептора включают описанные в WO 2002/066422, WO 2002/070490, WO 2002/076933, WO 2002/024439, WO 2003/072539, WO 2003/091204, WO 2004/016578, WO 2004/022547, WO 2004/037807,WO 2004/037773, WO 2004/037768, WO 2004/039762, WO 2004/039766, WO 2001/42193 и WO 2003/042160. Другие примеры агонистов 2-адренорецептора включают 3-(4-[6-2R)-2-гидрокси-2-[4-гидрокси-3-(гидроксиметил)фенил]этиламино)гексил]оксибутил)бензолсульфонамид;N-22-[4-(3-фенил-4-метоксифенил)аминофенил]этил-2-гидрокси-2-(8-гидрокси-2(1H)-хинолинон 5-ил)этиламин и 5-[(R)-2-(2-4-[4-(2-амино-2-метилпропокси)фениламино]фенилэтиламино)-1-гидроксиэтил]-8 гидрокси-1H-хинолин-2-он. Агонист 2-адренорецептора может быть в форме соли, образованной с фармацевтически приемлемой кислотой, выбранной из серной, хлористо-водородной, фумаровой, гидроксинафтоевой (например,1- или 3-гидрокси-2-нафтоевой), коричной, замещенной коричной, трифенилуксусной, сульфамовой,сульфаниловой, нафталинакриловой, бензойной, 4-метоксибензойной, 2- или 4-гидроксибензойной, 4 хлорбензойной и 4-фенилбензойной кислотой. Подходящие противовоспалительные средства включают кортикостероиды. Примерами кортикостероидов, которые могут использоваться в комбинации с соединениями по изобретению, являются пероральные и ингаляционные кортикостероиды и их пролекарства, которые имеют противовоспалительную активность. Примеры включают метилпреднизолон, преднизолон, дексаметазон, флутиказонпропионат, Sфторметиловый эфир 6,9-дифтор-11-гидрокси-16-метил-17-[(4-метил-1,3-тиазол-5-карбонил)окси]-3-оксоандроста-1,4-диен-17-тиокарбоновой кислоты, S-фторметиловый эфир 6,9-дифтор 17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-тиокарбоновой кислоты (флутиказон фуроат), S-(2-оксотетрагидрофуран-3S-ил)овый эфир 6,9-дифтор-11-гидрокси 16-метил-3-оксо-17-пропионилоксиандроста-1,4-диен-17-тиокарбоновой кислоты, S-цианометиловый эфир 6,9-дифтор-11-гидрокси-16-метил-3-оксо-17-(2,2,3,3-тетраметилциклопропилкарбонил)оксиандроста-1,4-диен-17-тиокарбоновой кислоты и S-фторметиловый эфир 6,9-дифтор-11 гидрокси-16-метил-17-(1-метилциклопропилкарбонил)окси-3-оксоандроста-1,4-диен-17-тиокарбоновой кислоты, сложные эфиры беклометазона (например, 17-пропионат или 17,21-дипропионат), будезонид, флунисолид, сложные эфиры мометазона (например, мометазон фуроат), триамцинолон ацетонид,рофлепонид, циклезонид (16,17-(R)-циклогексилметилен]бис(окси)]-11,21-дигидроксипрегна-1,4 диен-3,20-дион), бутиксокорт пропионат, RPR-106541 и ST-126. В одном варианте осуществления кортикостероиды включают флутиказон пропионат, S-фторметиловый эфир 6,9-дифтор-11-гидрокси-16 метил-17-[(4-метил-1,3-тиазол-5-карбонил)окси]-3-оксоандроста-1,4-диен-17-тиокарбоновой кислоты,S-фторметиловый эфир 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-тиокарбоновой кислоты, S-цианометиловый эфир 6,9-дифтор-11-гидрокси 16-метил-3-оксо-17-(2,2,3,3-тетраметилциклопропилкарбонил)окси-андроста-1,4-диен-17 тиокарбоновой кислоты и S-фторметиловый эфир 6,9-дифтор-11-гидрокси-16-метил-17-(1 метилциклопропилкарбонил)окси-3-оксоандроста-1,4-диен-17-тиокарбоновой кислоты. В одном варианте осуществления кортикостероид представляет собой S-фторметиловый эфир 6,9-дифтор-17-[(2 фуранипкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-тиокарбоновой кислоты. Примеры кортикостероидов также включают описанные в WO 2002/088167, WO 2002/100879, WO 2002/12265, WO 2002/12266, WO 2005/005451, WO 2005/005452, WO 2006/072599 и WO 2006/072600. Нестероидные соединения, имеющее агонизм в отношении глюкокортикоидов, которые могут обладать селективностью в плане трансрепрессии над трансактивацией и могут быть использованы в комбинированной терапии, включают описанные в следующих опубликованных заявки на патенты и патентах: WO 2003/082827, WO 1998/54159, WO 2004/005229, WO 2004/009017, WO 2004/018429, WO 2003/104195, WO 2003/082787, WO 2003/082280, WO 2003/059899, WO 2003/101932, WO 2002/02565,WO 2001/16128, WO 2000/66590, WO 2003/086294, WO 2004/026248, WO 2003/061651, WO 2003/08277,WO 2006/000401, WO 2006/000398 и WO 2006/015870. Нестероидные соединения, имеющие агонизм в отношении глюкокортикоидов, которые могут обладать селективностью в плане трансрепрессии над трансактивацией и могут быть использованы в комбинированной терапии, включают описанные в следующих патентах: WO 2003/082827, WO 1998/54159,WO 2004/005229, WO 2004/009017, WO 2004/018429, WO 2003/104195, WO 2003/082787, WO 2003/082280, WO 2003/059899, WO 2003/101932, WO 2002/02565, WO 2001/16128, WO 2000/66590, WO 2003/086294, WO 2004/026248, WO 2003/061651 и WO 2003/08277. Примеры противовоспалительных средств включают нестероидные противовоспалительные препараты (NSAID).(PDE) (например, теофиллин, ингибиторы PDE4 или смешанные ингибиторы PDE3/PDE4), антагонисты лейкотриена, ингибиторы синтеза лейкотриена (например монтелукаст), ингибиторы iNOS, ингибиторы триптазы и эластазы, антагонисты бета-2 интегрина и агонисты или антагонисты рецептора аденозина(например, агонисты аденозина 2a), антагонисты цитокина (например, антагонисты хемокина, такие как антагонист CCR3) или ингибиторы синтеза цитокина, или ингибиторы 5-липооксигеназы. В одном варианте осуществления изобретение охватывает ингибиторы iNOS (индуцируемой синтазы оксида азота) для перорального введения. Примеры ингибиторов iNOS включают раскрытые в WO 1993/13055, WO 1998/30537, WO 2002/50021, WO 1995/34534 и WO 1999/62875. Примеры ингибиторов CCR3 включают раскрытые в WO 2002/26722. В одном варианте осуществления изобретение относится к применению соединений Формулы (I) в комбинации с ингибитором фосфодиэстеразы 4 (PDE4), например, в случае состава, адаптированного для ингаляции. Ингибитор PDE4, который может быть использован в этом аспекте изобретения, может быть любым соединением, которое известно как или которое действует как ингибитор PDE4, например, как ингибитор PDE4B и/или PDE4D. РВЕ 4-ингибирующие соединения включают цис-4-циано-4-(3-циклопентилокси-4-метоксифенил)циклогексан-1-карбоновую кислоту,2-карбометокси-4-циано-4-(3-циклопропилметокси-4 дифторметоксифенил)циклогексан-1-он и цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-ол]. Кроме того, цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновая кислота (также известная как циломиласт) и ее соли, сложные эфиры, пролекарства или физические формы, которые описаны в патенте США 5552438, выданном 3 сентября 1996. Другие PDE4-ингибирующие соединения включают AWD-12-281 (N-(3,5-дихлор-4-пиридинил)-1[4-фторфенил)метил]-5-гидроксиоксо-1H-индол-3-ацетамид) от Elbion (Hofgen, N. et al. 15th EFMC Intal. Eur Resp J. [Annu Cong Eur Resp Soc (Sept 19-23, Geneva) 1998] 1998, 12 (Suppl. 28): Abst P2393); рофлумиласт (3-(циклопропилметокси)-N-(3,5-дихлор-4-пиридинил)-4-(дифторметокси)бензамид) (см. EP 0706513 B1 на имя Byk Gulden Lomberg, например см. пример 5); фталазинон (WO1999/47505) от BykGulden; Пумафентрин,(-)-p-[(4aR,10bS)-9-этокси-1,2,3,4,4a,10b-гексагидро-8-метокси-2 метилбензо[c][1,6]нафтиридин-6-ил]-N,N-диизопропилбензамид, который является смешанным ингибитором PDE3/PDE4, который был получен и опубликован Byk-Gulden, ныне Altana; арофиллин, разработанный Almirall-Prodesfarma; VM554/UM565 от Vernalis; или T-440 (Tanabe Seiyaku; Fuji, К. et al. J Pharmacol Exp Ther,1998, 284(1): 162), ИТ 2585. Другие PDE4-ингибирующие соединения раскрыты в опубликованных международных заявках на патенты WO 2004/024728, WO 2004/056823, WO 2004/103998 (например, пример 399 или 544), WO 2005/058892, WO 2005/090348, WO 2005/090353 и WO 2005/090354, все на имя Glaxo Group Limited. Примерами антихолинергических средств являются соединения, которые действуют как антагонисты на мускариновых рецепторах, в особенности соединения, которые являются антагонистами рецепторов M1 или M3, двойными антагонистами рецепторов M1/M3 или M2/M3, или пан-антагонистами рецепторов M1/M2/M3. Примеры соединений для введения путем ингаляции включают ипратопиум (например,бромид CAS 22254-24-6, выпускаемый под названием Atrovent), окситропиум (например, бромид CAS 30286-75-0) и тиотропиум (например, бромид CAS 136310-93-5, выпускаемый под названием Spiriva). Также интерес представляют реватропат (например, гидробромид, CAS 262586-79-8) и LAS-34273, который раскрыт в WO2001/04118. Примеры соединений для перорального введения включают пирензепин(CAS 28797-61-7), дарифенацин (CAS 133099-04-4, или CAS 133099-07-7 для гидробромида, выпускаемый под названием Enablex), оксибутинин (CAS 5633-20-5, выпускаемый под названием Ditropan), теродилин (CAS 15793-40-5), тольтеродин (CAS 124937-51-5, или CAS 124937-52-6 для тартрата, выпускаемый под названием Detrol), отилониум (например, бромид CAS 26095-59-0, выпускаемый под названиемSpasmomen), троспиум хлорид (CAS 10405-02-4) и солифенацин (CAS 242478-37-1, или CAS 242478-38-2 для сукцината, также известный как YM-905 и выпускаемый под названием Vesicare). Дополнительные соединения раскрыты в WO 2005/037280, WO 2005/046586 и WO 2005/104745. Комбинации по изобретению включают, но не ограничены ими(1R,5S)-3-(2-циано-2,2-дифенилэтил)-8-метил-8-2-[(фенилметил)окси]этил-8-азониабицикло[3.2.1]октан бромид. Другие антихолинергические средства включают соединения, которые раскрыты в заявке на патент США 60/487981, полностью включенной в настоящее описание путем ссылки. Они включают, например:(эндо)-3-2,2-дифенил-3-[(1-фенилметаноил)амино]пропил-8,8-диметил-8-азониабицикло[3.2.1]октан бромид. В одном варианте осуществления изобретение относится к комбинации, включающей соединение формулы (I) или его фармацевтически приемлемую соль вместе с антагонистом H1. Примеры антагонистов H1 включают, без ограничения, амелексанокс, астемизол, азатадин, азеластин, акривастин, бромфенирамин, цетиризин, левоцетиризин, эфлетиризин, хлорфенирамин, клемастин,циклизин, каребастин, ципрогептадин, карбиноксамин, дескарбоэтоксилоратадин, доксиламин, диметинден, эбастин, эпинастин, эфлетиризин, фексофенадин, гидроксизин, кетотифен, лоратадин, леовкабастин,мизоластин, меквитазин, миансерин, ноберастин, меклизин, норастемизол, клопатадин, пикумаст, пириламин, прометазин, терфенадин, трипеленнамин, темеластин, тримепразин и трипролидин, особенно цетиризин, левоцетиризин, эфлетиризин и фексофенадин. В другом варианте осуществления изобретение относится к комбинации, включающей соединение формулы (I) или его фармацевтически приемлемую соль вместе с антагонистом H3 (и/или обратным агонистом). Примеры антагонистов H3 включают, например, соединения, раскрытые в WO 2004/035556 и в WO 2006/045416. Другие антагонисты рецептора гистамина, которые могут использоваться в комбинации с соединениями согласно настоящему изобретению, включают антагонисты (и/или обратные агонисты) рецептора H4, например, соединения, раскрытые в Jablonowski et al., J. Med. Chem. 46:3957-3960 (2003). В одном варианте осуществления изобретение относится к комбинации, включающей соединение формулы (I) или его фармацевтически приемлемую соль вместе с антагонистом рецептора CCR5, таким как 4,4-дифтор-N-1S)-3-3-[3-метил-5-(1-метилэтил)-4H-1,2,4-триазол-4-ил]-8-азабицикло[3.2.1]окт-8 ил-1-фенилпропил)циклогексанкарбоксамид В одном варианте осуществления изобретение относится к комбинации, включающей соединение формулы (I) или его фармацевтически приемлемую соль этого вместе с антагонистом рецептора CXCR3,таким как N-1R)-1-3-[4-(этилокси)фенил]-4-оксо-3,4-дигидропиридо[2,3-d]пиримидин-2-илэтил)-N(3-пиридинилметил)-2-4-[(трифторметил)окси]фенилацетамид: Изобретение, таким образом, относится, в следующем аспекте, к комбинации, включающей соединение формулы (I) и/или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с ингибитором PDE4. Изобретение, таким образом, относится, в следующем аспекте, к комбинации, включающей соединение формулы (I) и/или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с агонистом 2-адренорецептора. Изобретение, таким образом, относится, в следующем аспекте, к комбинации, включающей соединение формулы (I) и/или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с кортикостероидом. Изобретение, таким образом, относится, в следующем аспекте, к комбинации, включающей соединение формулы (I) и/или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с нестероидным агонистом GR. Изобретение, таким образом, относится, в следующем аспекте, к комбинации, включающей соединение формулы (I) и/или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с антихолинергическим средством. Изобретение, таким образом, относится, в следующем аспекте, к комбинации, включающей соединение формулы (I) и/или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с антигистамином. Изобретение, таким образом, относится, в следующем аспекте, к комбинации, включающей соединение формулы (I) и/или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с ингибитором PDE4 и агонистом 2-адренорецептора. Изобретение, таким образом, относится, в следующем аспекте, к комбинации, включающей соединение формулы (I) и/или его фармацевтически приемлемую соль, сольват или физиологически функциональное производное вместе с антихолинергическим средством и ингибитором PDE-4. Комбинации, упомянутые выше, могут быть представлены для использования в форме фармацевтического состава, и таким образом, фармацевтические составы, включающие комбинацию, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем представляют собой дальнейший аспект изобретения. Индивидуальные соединения таких комбинаций могут вводиться последовательно или одновременно в отдельных или комбинированных фармацевтических составах. В одном варианте осуществления индивидуальные соединения вводят одновременно в комбинированном фармацевтическом составе. Подходящие дозы известных терапевтических средств могут быть легко определены специалистом. Изобретение, таким образом, относится в следующем аспекте к фармацевтической композиции, содержащей комбинацию соединения по изобретению вместе с другим терапевтически активным средством. Изобретение, таким образом, относится в следующем аспекте к фармацевтической композиции, содержащей комбинацию соединения по изобретению вместе с ингибитором PDE4. Изобретение, таким образом, относится в следующем аспекте к фармацевтической композиции, содержащей комбинацию соединения по изобретению вместе с агонистом 2-адренорецептора. Изобретение, таким образом, относится, в следующем аспекте, к фармацевтической композиции,содержащей комбинацию соединения по изобретению вместе с кортикостероидом. Изобретение, таким образом, относится, в следующем аспекте, к фармацевтической композиции,содержащей комбинацию соединения по изобретению вместе с нестероидным агонистом GR. Изобретение, таким образом, относится, в следующем аспекте, к фармацевтической композиции,содержащей комбинацию соединения по изобретению вместе с антихолинергическим средством. Изобретение, таким образом, относится, в следующем аспекте, к фармацевтической композиции,содержащей комбинацию соединения по изобретению вместе с антигистамином. Изобретение, таким образом, относится, в следующем аспекте, к фармацевтической композиции,содержащей комбинацию соединения по изобретению вместе с антагонистом рецептора CXCR3. Изобретение, таким образом, относится, в следующем аспекте, к фармацевтической комбинации по изобретению вместе с антагонистом рецептора CCR5. Изобретение будет далее описано при помощи следующих биологических примеров, которые являются просто иллюстративными и не должны быть рассмотрены как ограничивающие объем настояще- 26014958 го изобретения. Биологические примерыIL-8 и GRO- хемокин-ингибирующие эффекты соединений согласно настоящему изобретению определяли с помощью следующих тестов in vitro: Тест связывания с рецептором:[125I] IL-8 (рекомбинантный человеческий) был получен от GE Healthcare, с удельной активностью 2000 Ci/ммоль. Все другие химические соединения имели степень чистоты, подходящую для проведения анализов. Высокие уровни рекомбинантного человеческого CXCR1 (IL-8 типа ) и CXCR2 (IL-8 типа ) индивидуально экспрессировали в неприлипших клетках Яичника Китайского хомячка (CHO), как описано ранее (Holmes, et al., Science, 1991, 253, 1278). Мембраны получали согласно ранее описанному протоколу, Haour, et al., J. Biol. Chem., 249, pp. 2195-2205 (1974, включенному в настоящее описание в отношении получения мембран, за исключением того, что буфер гомогенизации был модифицирован и содержал 40 мМ Tris-HCl (pH 7,5), 1 мМ MgSO4, 0,5 мМ EGTA (этиленгликоль-бис(2-аминоэтиловый эфир)-N, N, N', N'-тетра-уксусная кислота), 1 мМ PMSF (-толуолсульфонилфторид), 2,5 мг/л лейпептина и 0,1 мг/мл апротинина. Клетки гомогенизировали и центрифугировали при 2000 об/мин в течение 10 мин. Супернатант центрифугировали при 100000g в течение 1 ч. Супернатант отбрасывали и мембраны сохраняли при -80C. Мембранную концентрацию белка определяли, используя реактив BioRad согласно протоколу производителя, используя бычий сывороточный альбумин (BSA) в качестве стандарта. Все связывание IL-8 проводили, используя тест сцинтилляционной близости (SPA), используя шарики агглютинина проростков пшеницы в формате планшета с 96 лунками. Мембраны CHO-CXCR1 илиNaCl. Соединения разбавляли в диметилсульфоксиде с 20X конечным разведением (конечная концентрация соединения от 1 нМ до 30 мкМ и конечная концентрация ДМСО 5%). Тест проводили на планшетах с 96 лунками (optiplate 96, Packard) при комнатной температуре, в 0,1 мл связывающего буфера с мембранами и 0,04% CHAPS (3-[(3-холамидопропил)диметиламмонио]-1-пропанесульфонат), 0,0025% BSA и 0,23 нМ [125I] IL-8. Планшеты встряхивали на платформе в течение 1 ч, в конце инкубации планшеты центрифугировали при 2000 об./мин в течение 5 мин и считывали в счетчике Top Count. Рекомбинантный IL-8 R, CXCR1 или Типа I, рецептор также упомянут здесь как непермиссивный рецептор, а рекомбинантный IL-8 R, CXCR2 или Типа II рецептор упоминается как пермиссивный рецептор. Использованные в примерах соединения формулы (I), примеры 1-38, показали положительную ингибирующую активность в этом тесте с уровнями IC50 30 мкМ и будут считаться активными. Тест Хемотаксиса: Ингибирующие свойства in vitro этих соединений были определены в тесте хемотаксиса нейтрофилов. Первичные человеческие нейтрофилы выделяли из периферической цельной крови, используя центрифугирование в прерывистом градиенте Перколла, осаждение декстраном и гипотонический лизис. Хемоаттрактанты IL-8 (CXCL8) или GRO- (CXCL1) помещали в нижнюю камеру 96-многолуночной камеры (ChemoTx System, Neuro Probe, Gaithersburg, MD). Используемая концентрация агониста была концентрацией EC80. Эти две камеры были разделены 5 мкм мембраной из поликарбоната. Когда тестировали соединения по изобретению, их предварительно инкубировали с клетками, после чего помещали в верхнюю часть фильтра. Хемотаксис проводили в течение 45 мин в увлажненном термостате при 37C с 5%-ным CO2. В конце инкубационного периода мембрану удаляли, и мигрировавшие клетки в нижней камере переносили на планшет с 96 лунками. Эти клетки измеряли, используя люминесцентный тест жизнеспособности клеток (Celltiter-Glo, Promega, Madison, WI). Каждый образец проверяли в двойном экземпляре, и каждое соединение повторяло по крайней мере три раза. Клетки положительного контроля были клетками без добавленного соединения и представляют максимальный хемотактический ответ. Отрицательный контроль (нестимулированный) был без добавления хемокина к нижней камере. Различие между положительным контролем и отрицательным контролем представляет собой хемотактическую активность клеток. В этом тесте были проверены примеры 1-3. Соединение считали активным, если значения IC50 составляли 5 мкМ. Тест Цельной крови человека CD11b Обозначенные соединения были проверены на их способность ингибировать GROиндуцированную экспрессию интегрина CD11b на нейтрофилах в цельной крови человека. Забор крови производили (9 мл), используя катетер Бабочкаи шприц на 10 мл, содержащий 0,2 мл рабочего Гепарина Натрия. Кровь сохраняли при 37C до помещения на лед на стадии 5, описанной ниже. Сток-растворы соединения затем разбавляли в 12 раз до максимальной конечной концентрации 120 мкМ. Затем готовили 1/2 Log серийные разведения в носителе. 10 мкл разведений соединения или носителя добавляли в подходящие полипропиленовые пробирки 1275. Сто микролитров цельной крови добавляли в пробирку и инкубировали в течение 10 мин при 37C на водяной бане с начальным (аккуратным) взбалтыванием и повторным в момент времени 5 мин. Сток GRO разбавляли 1:166,66 в 0,1 %BSA-DPBS до "12" концентрации 120 нМ и 10 мкл разведения GRO или 0,1% BSA-DPBS добавляли в подходящие пробирки так, чтобы заключительная концентрация GRO равнялась 10 нМ. Пробирки инкубировали в течение 10 мин при 37C при аккуратном взбалтывании вручную, повторяемом в момент времени 5 мин. Образцы затем помещали на лед и добавляли 250 мкл охлажденного на льду рабочего разведения CellFix с последующей минутной инкубацией на льду. В ходе инкубации GRO подготавливали пробирки Eppendorf на 1,5 мл, добавляя подходящие антитела. Каждая пробирка получила 10 мклCD11b-FITC и 5 мкл CD16-PE, за исключением изотипического контроля, который получил 10 мклIgG2a-FITC вместо CD11b. Дополнительно 50 мкл фиксированной крови из каждой пробирки добавляли в подходящую пробирку Eppendorf. Образцы инкубировали в течение 20 мин при 4C в темноте. Дополнительно смеси кровь/антитела в 500 мкл холодного DPBS добавляли в соответствующую маркированную полистирольную пробирку 1275. Полученную смесь сохраняли на льду. Добавляли сток LDS (10 мкл), и смесь инкубировали в течение 10 мин при 4C до анализа потока. Образцы сохраняли в затемненной среде. Добавление LDS заканчивали, когда образцы собирали на счетной камере потока так, что все образцы разгоняли 10-20 мин после добавления LDS. Объемная скорость потока среды использовалась для аккумуляции потока, и порог FL3 увеличивали, чтобы удалить красные кровяные клетки из анализа, используя сигнал LDS. Цветную компенсацию устанавливали должным образом, используя немаркированные образцы и одноцветные образцы, чтобы вычесть пятно LDS в РЕ и пятно PE в FITC и FITC в PE. Для цитометра BD LSR LDS=FL3, PE=FL2,FITC=FL1. Были получены минимум 2000-3000 событий, которые удовлетворяют воротам гранулоцитаSSC vs. FSC и были CD16 позитивными сигналом FL2. Использованные в примерах соединения формулы (I) примеры 1-3, 12, 17, 18, 23 и 26 показали положительную ингибирующую активность в этом тесте со значениями IC50 5 мкМ и будут считаться активными. Соединения примеров 1-3, 12, 17, 18, 23 и 26, проверенные в вышеописанном тесте, имели значение IC50 от приблизительно 2 до приблизительно 0,5 мкМ. Мобилизация кальция в клетках CHO-K1, стабильно экспрессирукдцих CXCR2 и G16:DMEM/FL2 (HAM) 1:1, w/10% FCS (инактивированной высокой температурой), w/2 мМ L-глутамина,w/0,4 мг/мл G418, поддерживая при 37C в инкубаторе с 5% CO2. За 24 ч до проведения теста клетки собирали и высеивали в плотности 40000 клеток на лунку в 96-луночные планшеты с черными стенками и прозрачным дном (Packard View) и возвращали в инкубатор с CO2. В день проведения теста соединения последовательно разбавляли в 100% ДМСО в 300X до желаемой тестируемой концентрации. Среды для выращивания отсасывали от клеток и заменяли 100 мкл среды загрузки (ЕМЕМ с солями Earl w/Lглутамин, 0,1% BSA, (Bovuminar Cohen Fraction V от Seriologicals Corp.), 4 мкМ флуоресцентного индикатора сложного Флуо-4-ацетоксиметилового эфира (Fluo-4, от Molecular Probes), и 2,5 мМ пробенцида),и инкубировали в течение 1 ч при 37 С в инкубаторе с CO2. Среду загрузки отсасывали и заменяли 100 мкл ЕМЕМ с солями Earl w/L-глутамин, 0,1% желатина, и 2,5 мМ пробенцида и инкубировали еще 10 мин. Последовательно разбавленное соединение (3 мкл) в ДМСО (300X) переносили на 96-луночные планшеты, содержащие 297 мкл KRH (120 мМ NaCl, 4,6 мМ KCl, 1,03 мМ KH2PO4, 25 мМ NaHCO3, 1,0 мМ CaCl2, 1,1 мМ MgCl2, 11 мМ глюкозы, 20 мМ HEPES (рН 7,4 w/2,5 мМ пробенцида и 0,1% желатина(соединение теперь в 3X). Среду отсасывали от клеток, и клетки промывали 3 раза KRH w/2,5 мМ пробенцида, w/0,1% желатина. К лунке добавляли KRH (100 мкл) w/2,5 мМ пробенцида с 0,1% желатина,затем к лунке добавляли 50 мкл 3X соединения в KRH w/2,5 мМ пробенцида и 0,1% желатина (соединение теперь в 1X) и инкубировали при 37C в инкубаторе с CO2 в течение 10 мин. Лунки помещали наFLIPR (Fluorometric Imaging Plate Reader, Molecular Devices, Sunnyvale CA) для анализа, как описано ранее (Sarau et al., 1999). Процент максимальной индуцируемой человеческим IL-8 мобилизации Ca2+, индуцированной 1,0 нМ IL-8, концентрацию EC80 для CXCR2, определяли для каждой концентрации соединения и IC50 вычисляли как концентрацию тестируемого соединения, которая ингибирует 50% максимального ответа, индуцированного 1,0 нМ IL-8. Примеры 1-38 показали положительную ингибирующую активность в этом тесте со значениями IC50 10 мкМ и будут считаться активными. Соединения 138, проверенные с помощью вышеописанного теста, имели IC50 от приблизительно 6000 нМ до приблизительно 5 нМ. Приведенное выше описание полностью раскрывает изобретение, включая его предпочтительные варианты осуществления. Модификации и улучшения вариантов осуществления, специфически раскрытых здесь, находятся в рамках следующей далее формулы изобретения. Без дальнейших уточнений понятно, что специалист, используя предыдущее описание, может использовать настоящее изобретение в наиболее полной степени. Поэтому примеры, приведенные здесь, должны рассматриваться как просто иллюстративные и никоим образом не ограничивающие объем настоящего изобретения. Варианты осуществления изобретения, на которое испрашиваются исключительные права собственности или привилегии, определены следующим образом.- 28014958 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение согласно формуле (I)R2 выбран из группы, состоящей из C3-6 циклоалкила, фенила и гетероарила, причем фенил или гетероарил в случае необходимости замещены, один или два раза, независимо заместителем, выбранным из группы, состоящей из C1-3 алкила, галогена, CF3, OCF3, фенилокси и бензилокси; илиR1 обозначает C4-8 гетероцикл (CH2)n-, в котором гетероцикл представляет собой пирролидин-2-ил,пирролидин-3-ил, пиперидин-4-ил, пиперидин-3-ил или азетидин-3-ил, причем каждая группа в случае необходимости замещена независимо один или два раза заместителем, выбранным из группы, состоящей из C1-3 алкила, C(O)OR4 и C(O)R5; илиR1 выбран из следующих кольцевых систем (a-k)n = 0 или 1,или его фармацевтически приемлемая соль. 2. Соединение по п.1, в котором X обозначает галоген. 3. Соединение по п.1, в котором X обозначает хлор. 4. Соединение по п.1, в котором R2 обозначает фенил, в случае необходимости замещенный, независимо один или два раза, заместителем, выбранным из группы, состоящей из C1-3 алкила, галогена, OCF3 и фенилокси. 5. Соединение по п.4, в котором R2 выбран из группы, состоящей из 3-фтор-2-метилфенила, 2 трифторметилоксифенила, 2-хлор-3-фторфенила, 2-этилфенила или 2-фенилоксифенила. 6. Соединение по п.5, в котором R2 обозначает 3-фтор-2-метилфенил или 2-хлор-3-фторфенил. 7. Соединение по п.1, в котором R2 обозначает пиридил, в случае необходимости замещенный один раз галогеном. 8. Соединение по п.7, в котором R2 обозначает 2-хлор-3-пиридил. 9. Соединение по п.1, в котором n = 0. 10. Соединение по п.9, в котором R1 обозначает пиперидин-3-ил. 11. Соединение по п.9 или 10, в котором X обозначает галоген. 12. Соединение по п.1, в котором R1 обозначает 3-экзо-8-метил-8-азабицикло[3.2.1]окт-3-ил или 3 экзо-8-азабицикло[3.2.1]окт-3-ил. 13. Соединение по пп.9, 10 или 11, в котором R2 выбран из группы, состоящей из 3-фтор-2 метилфенила, 2-трифторметилоксифенила, 2-хлор-3-фторфенила, 2-этилфенила или 2-фенилоксифенила. 14. Соединение по п.1, представляющее собой N-(4-хлор-2-гидрокси-3-[(3-экзо)-8-метил-8 азабицикло[3.2.1]окт-3-ил]сульфонилфенил)-N'-(3-фтор-2-метилфенил)карбамид;N-[3-(3-азетидинилсульфонил)-4-хлор-2-гидроксифенил]-N'-(3-фтор-2-метилфенил)карбамид; или его фармацевтически приемлемая соль.
МПК / Метки
МПК: C07D 211/84, A01N 43/40, A61K 31/42, A61K 31/44, C07D 413/00, A01N 43/76, C07D 213/63, C07D 211/72, C07D 263/52, C07D 213/70, C07D 263/60
Метки: антагонисты, рецептора
Код ссылки
<a href="https://eas.patents.su/30-14958-antagonisty-receptora-il-8.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты рецептора il – 8</a>
Производные индола как антагонисты рецептора 5-нт

Номер патента: 304
Опубликовано: 29.04.1999
Авторы: Форбес Ян Томсон, Джоунз Грэхэм Элджин, Вайман Пол Эдриан, Дакворт Дэвид Малькольм, Гэстер Лэрэми Мэри, Малхоллэнд Кит Раймонд, Дэвис Дэвид Томас
МПК: C07D 401/12, A61K 31/44
Метки: антагонисты, производные, рецептора, 5-нт, индола
Формула / Реферат:
1. Соединение формулы (I) или его соль где Р1 и Р2 независимо представляет собой фенил, ароматические или частично насыщенные моноциклические или бициклические гетероциклические кольца, содержащие до трех гетероатомов, выбранных из азота, кислорода или серы; А представляет собой связь, цепь из 1-5 атомов, необязательно замещенных С1-6алкилом, или А представляет собой необязательно замещенный фенил или необязательно замещенное 5-7-членное...
Антагонисты рецептора il-8

Номер патента: 1436
Опубликовано: 26.02.2001
Авторы: Виддаусон Кетрин Луиза, Херцберг Роберт Филип, Вебер Дэниел Франк, Юревич Энтони Джозеф, Ратледж Мельвин Кларенс Мл.
МПК: A61K 31/17, A61P 11/06
Метки: антагонисты, рецептора
Формула / Реферат:
1. Способ лечения болезненного состояния, опосредованного хемокином, где хемокин связывается у млекопитающих, нуждающихся в таком лечении, с IL-8 а- или b-рецептором, включающий введение млекопитающему эффективного количества соединения формулы где Х является кислородом или серой; R является любой функциональной группой, имеющей ионизируемый водород и рКа, равный 10 или менее; R1 независимо выбирают из водорода; галогена; нитро; циано;...
Бензоксазиноновые антагонисты рецептора допамина d4.

Номер патента: 1486
Опубликовано: 23.04.2001
Авторы: Беллиотти Томес, Вайс Лауренс Дейвид, Вустров Дейвид Юрген
МПК: C07D 265/36, A61K 31/535
Метки: антагонисты, бензоксазиноновые, рецептора, допамина
Формула / Реферат:
1. Соединение формулы (I) где R1 и R2 означают независимо водород или алкил с числом углеродных атомов от 1 до 6, X означает С, N или СН, при этом в случае X=С штрихованная линия означает связь, а в других случаях отсутствует; R3 означает фенил, нафтил, гетероарил, замещенный фенил, замещенный нафтил или замещенный гетероарил, при этом каждый заместитель независимо выбран из группы, включающей галоген, алкоксигруппу с числом углеродных...
Антагонисты рецептора гистамина-3

Номер патента: 13602
Опубликовано: 30.06.2010
Авторы: Чандрасекаран Рамалакшми Йегна, Вагер Тревис Т., Батлер Тодд Уилльям
МПК: C07D 207/04, C07C 237/24, A61K 31/165...
Метки: гистамина-3, рецептора, антагонисты
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль,где R1и R2, каждый независимо, выбран из группы, включающейводород;С1-С8-алкил, необязательно замещенный 1-4 галогенами;С1-С4-алкильную группу, необязательно замещенную заместителем, выбранным из группы, состоящей из ОН, одного до четырех С1-С4-алкила, С3-С7-циклоалкила, С1-С4-диалкиламино, С6-С14-арила, необязательно замещенного галогеном и необязательно замещенного группой...
Изоксазолиновые антагонисты рецептора фибриногена.

Номер патента: 924
Опубликовано: 26.06.2000
Авторы: Батт Дуглас Гай, Хуссейн Минур Алван, Моуса Шакер Ахмед, Деградо Уильям Фрэнк, Ксу Чу-Биао, Витьяк Джон, Силеки-Дзурдз Таис Мотриа, Олсон Ричард Эрик, Кайн Гари Авонн, Пинто Дональд
МПК: A61K 31/42, A61P 7/02, C07D 261/04...
Метки: рецептора, изоксазолиновые, антагонисты, фибриногена
Формула / Реферат:
1. Соединение формулы I или их формы фармацевтически приемлемых солей, где R1 выбран из R2(R3)N(CH2)qZ-, R2(R3)N(R2N=)CN(R2)(CH2)qZ-, группы пиперазинил-(СН2)qZ- или Z выбран из О, S, S(=О) или S(=O)2; R2 и R3 независимо выбраны из: Н, C1-С10алкила, С2-С6алкенила, C3-С11циклоалкила, С4-С11циклоалкилалкила, С6-С10арила, С7-С11арилалкила, С2-С7алкилкарбонила, С6-С10арилкарбонила, С2-С10алкоксикарбонила, С4-С11циклоалкоксикарбонила,...
Предыдущий патент: Дикетопиперазиновые и пиперидиновые производные в качестве противовирусных агентов
Следующий патент: Ингибиторы глицинового переносчика-1
Случайный патент: Устройство дозатора напитка и контейнер, снабженный резервуаром среды под давлением