Высокоэффективные полные и частичные агонисты и антагонисты рецептора ноцицептина/орфанина fq
Номер патента: 11325
Опубликовано: 27.02.2009
Авторы: Гуеррини Ремо, Кало Джироламо, Реголи Доменико, Сальвадори Северо







Формула / Реферат
1. Пептид, имеющий общую формулу (I)

в которой
Хаа1 выбран из группы, состоящей из Phe или N-бензил-глицина (Nphe);
y представляет собой связь между первыми двумя аминокислотными остатками и выбрана из группы, состоящей из CO-NH, и CH2-NH, и СН2-О;
Xbb4 является Phe или pXPhe, где "X" выбран из группы, состоящей из Н, Cl, Br, I, F, NO2 и CN и "р" показывает пара-положение в фенильном кольце Phe;
Хсс7 выбран из группы, состоящей из 2-амино-2-метилпропионовой кислоты (Aib); 2-амино-2-метилмасляной кислоты (Iva); 2-амино-2-этилмасляной кислоты (Deg); 2-амино-2-пропилпентановой кислоты (Dpg); (CaCH3)Leu; (CaCH3)Val; 1-аминоциклопропанкарбоновой кислоты (Ас3с); 1-аминоциклопентанкарбоновой кислоты (Ас5с) и 1-аминоциклогексанкарбоновой кислоты (Ас6с);
Xdd11 выбран из группы, состоящей из Ala; 2-амино-2-метилпропионовой кислоты (Aib); 2-амино-2-метилмасляной кислоты (Iva); 2-амино-2-этилмасляной кислоты (Deg); 2-амино-2-пропилпентановой кислоты (Dpg); (CaCH3)Leu; (CaCH3)Val; 1-аминоциклопропанкарбоновой кислоты (Ас3с); 1-аминоциклопентанкарбоновой кислоты (Ас5с) и 1-аминоциклогексанкарбоновой кислоты (Ac6c);
Хее14 и Xff15 выбраны из группы, состоящей из Arg; Lys; Orn; omoArg; диаминомасляной кислоты; диаминопропионовой кислоты и Trp;
R представляет собой дипептид Asn-Gln-NH2 или Asn-Gln-OH или аминокислоту Asn или с амидо
(-NH2), или с карбоксильной (-ОН) терминальной группой или амино (-NH2) или гидроксильной (-ОН) терминальной группой;
и его фармацевтически приемлемые соли.
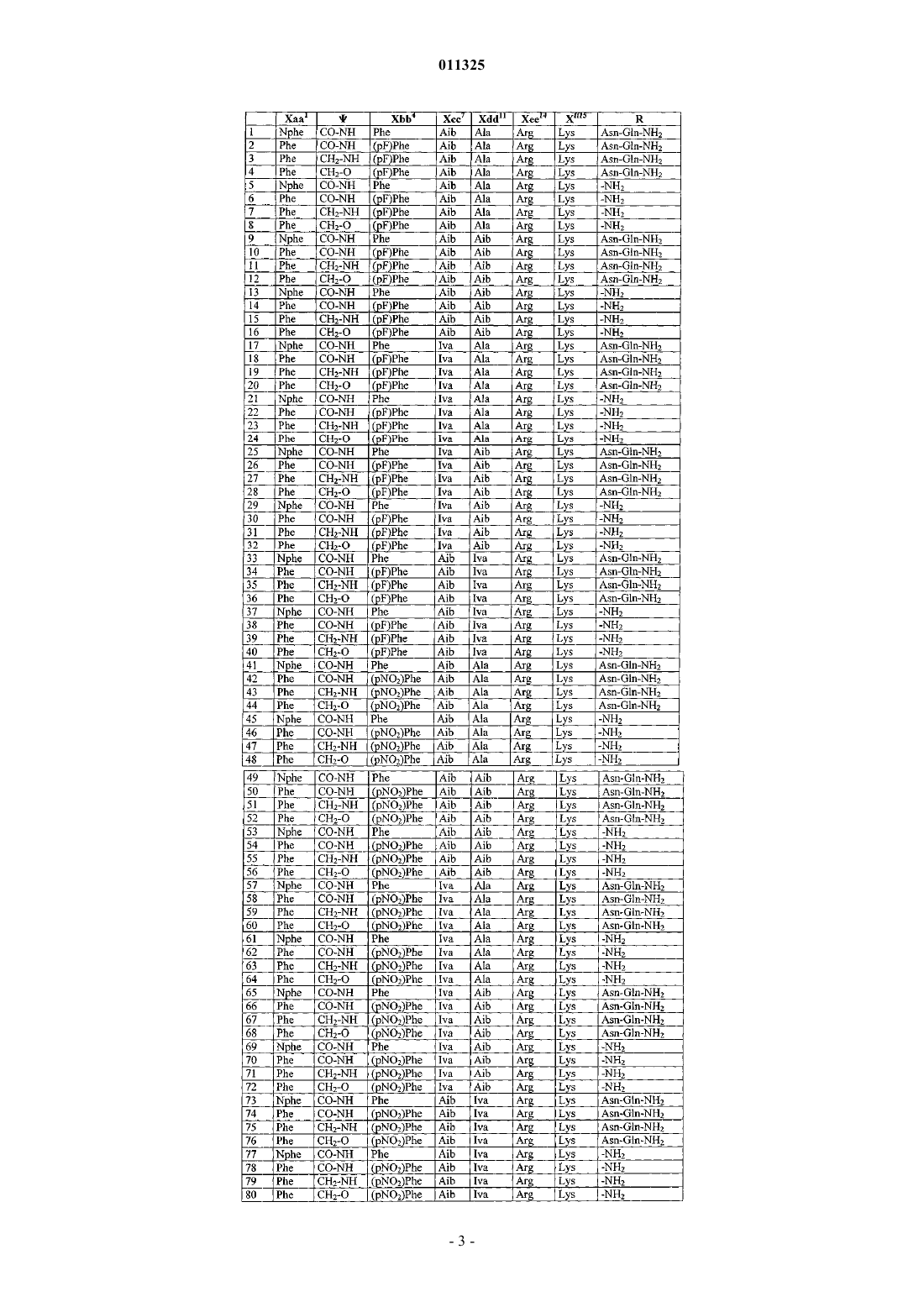
2. Пептид по п.1, выбранный из группы, состоящей из



и его фармацевтически приемлемые соли.
3. Пептид по п.1, в котором
Хаа1 является Phe;
Xbb4 является (pX)Phe, где "X" выбран из группы, состоящей из Н, F, NO2, и "р" указывает пара-положение в фенильном кольце Phe;
Хсс7 выбран из группы, состоящей из 2-амино-2-метилпропионовой кислоты (Aib), 1-аминоциклопентанкарбоновой кислоты (Ac5c) и 2-амино-2-метилмасляной кислоты (Iva);
Xdd11 выбран из группы, состоящей из Ala; 2-амино-2-метилпропионовой кислоты (Aib); 1-аминоциклопентанкарбоновой кислоты (Ас5с), 2-амино-2-метилмасляной кислоты (Iva);
Хее14 является Arg;
Xff15 является Lys и
R представляет собой дипептид Asn-Gln-NH2, или Asn-NH2, или амино(-NH2)группу;
и его фармацевтически приемлемые соли.
4. Пептид по п.3, в котором
y является CO-NH;
"X" является F;
Хсс7 является 2-амино-2-метилпропионовой кислотой (Aib);
Xdd11 является Ala и
R представляет собой дипептид Asn-Gln-NH2;
и его фармацевтически приемлемые соли.
5. Пептид по п.3, в котором
y является CH2-NH;
"X" является F;
Хсс7 является 2-амино-2-метилпропионовой кислотой (Aib);
Xdd11 является Ala и
R представляет собой дипептид Asn-Gln-NH2;
и его фармацевтически приемлемые соли.
6. Пептид по п.2, в котором
Хаа1 является N-бензилглицином (Nphe);
y является CO-NH;
Xbb4 является Phe;
Хсс7 выбран из группы, состоящей из 2-амино-2-метилпропионовой кислоты (Aib), или 2-амино-2-метилмасляной кислоты (Iva);
Xdd11 выбран из группы, состоящей из Ala, или 2-амино-2-метилпропионовой кислоты (Aib), или 2-амино-2-метилмасляной кислоты (Iva) и
R представляет собой дипептид Asn-Gln-NH2 или амино (-NH2) группу;
и его фармацевтически приемлемые соли.
7. Пептид по п.6, в котором
Хсс7 является 2-амино-2-метилпропионовой кислотой (Aib);
Xdd11 является Ala и
R представляет собой дипептид Asn-Gln-NH2;
и его фармацевтически приемлемые соли.
8. Композиция, содержащая пептиды по пп.1-7.
9. Фармацевтическая композиция, содержащая в качестве активного начала пептиды по пп.1-7, объединенные с фармацевтически приемлемыми носителями и/или эксципиентами.
10. Фармацевтическая композиция по п.9, предназначенная для введения через оральный, местный, респираторный, ректальный, интраспинальный, интратекальный, внутрипузырный или парентеральный путь.
11. Фармацевтическая композиция по п.10, где введение производится через интратекальный и парентеральный пути.
12. Применение пептида по пп.1-7 для приготовления лекарственного средства, предназначенного для лечения или предупреждения неврологических и нейросенсорных дисфункций.
13. Применение пептида по пп.3-5 для приготовления лекарственного средства для лечения или предупреждения гипертензии, тахикардии, расстройств задержки воды, гипонатриемии, сердечной недостаточности, гладкомышечных двигательных дисфункций в желудочно-кишечном, респираторном и мочеполовом трактах, воспалительных состояний, периферического или спинального обезболивания, ведения хронической боли и ослабления кашля.
14. Применение по п.13, в котором указанная гладкомышечная двигательная дисфункция включает нейрогенное недержание мочевого пузыря или гиперактивность мочевого пузыря, респираторную дисфункцию.
15. Применение по п.12 для приготовления транквилизатора или лекарственного средства для лечения или предупреждения анорексии.
16. Применение пептида по пп.6-7 для приготовления лекарства для лечения нарушений памяти и настроения, двигательной активности и расстройств потребления пищи или для лечения ожирения.
Текст
011325 Область техники Настоящее изобретение относится к пептидным аналогам ноцицептина/орфанина FQ (N/OFQ), способным регулировать активность рецептора пептида N/OFQ (рецептора NOP), фармацевтическим композициям, содержащим указанные пептидные аналоги, и их применению для лечения дисфункций, патологических условий или патологических состояний, затрагивающих указанный рецептор. Уровень техники В 1994 г. был клонирован новый рецептор, названный ORL1, структурно подобный опиоидным рецепторам; согласно последним рекомендациям Международного союза фармакологов (IUPHAR) NOP является наиболее подходящим названием для этого рецептора. Его эндогенным лигандом (N/OFQ),идентифицированным в конце 1995 г., является гептадекапептид, подобный некоторым опиоидным пептидам (например, динорфину А), который, однако, не связывается с классическими опиоидными рецепторами мю (MOP), дельта (DOP) или каппа (KOP) типов. Клеточные эффекты, опосредованные рецептором NOP, сходны с эффектами, вызываемыми классическими опиоидными рецепторами. С точки зрения структуры и с точки зрения сигнальной трансдукции, система пептид/рецептор N/OFQ-NOP принадлежит к опиоидному семейству, хотя она представляет собой фармакологически отдельную ветвь. Некоторые исследования, выполненные между 1996 и 1998 гг., показали, что N/OFQ может регулировать некоторые функции как в центральной нервной системе (боль, страх, обучение, память, злоупотребление наркотиками, аппетит), так и на периферическом уровне (кровяное давление, ритм сердца, почечные, желудочно-кишечные, мочеполовые периферические и респираторные функции) (более подробно см. Massi etal., Peptides 21, 2000). Начиная с 1996 г., изобретатели настоящего изобретения выполняли исследования на системеN/OFQ-NOP, приведшие к идентификации отдельных лигандов рецептора NOP, таких как: i) N/OFQ(113)-NH2, который представляет собой минимальный функциональный фрагмент с активностью, подобной активности природного лиганда N/OFQ (Calo et al., Eur. J. Pharmacol. 311, R3-5, 1996), ii) N/OFQNH2, который вызывает, особенно in vivo, более сильные и длительные эффекты по сравнению с N/OFQ(Rizzi et al., Naunyn Schmiedebergs Arch. Pharmacol. 363, 161-165, 2001), iii) [Tyr1]N/OFQ(1-13)-NH2, смешанный агонист, который действует на NOP и на классические опиоидные рецепторы (Calo et al., Can. J.Physiol. Pharmacol. 75, 713-8, 1997; Varani et al., Naunyn Schmiedebergs Arch. Pharmacol. 360, 270-7, 1999),iv) [Phe1(CH2-NH)Gly2]N/OFQ(1-13)-NH2, селективный лиганд рецептора NOP, который ведет себя как чистый антагонист, частичный агонист или даже полный агонист, в зависимости от способа получения/ анализа при исследовании (Guerrini et al., Br. J. Pharmacol. 123, 163-5, 1998; Okawa et al., Br. J. Pharmacol. 127, 123-30, 1999) - исходя из подробного анализа фармацевтического действия [Phe1 СН 2NH)Gly2]N/OFQ(1-13)-NH2, сообщенного Calo et al. (Peptides 21, 935-47, 2000), оказывается, что данное соединение, в самом деле, является частичным агонистом NOP, v) [Nphe1]N/OFQ(1-13)-NH2, первый чистый конкурентный антагонист рецептора NOP (Calo et al., Br. J. Pharmacol. 129, 1183-93, 2000; Guerrini etal., J. Med. Chem. 15, 2805-13, 2000). Действие этих лигандов охарактеризовано в некоторых исследованиях in vitro и in vivo (см. Calo et al., Br. J. Pharmacol. 129, 1261-83, 2000). Позднее остаток Phe4 был замещен (pF)Phe или (pNO2)Phe, таким образом были получены сильные селективные агонисты NOP(Guerrini et al., J. Med. Chem. 44, 3956-64, 2001). Другое интересное соединение, [Arg14,Lys15]N/OFQ, было идентифицировано в качестве высокоэффективного агониста (в 17 раз более эффективное, чемN/OFQ), селективного для человеческих рекомбинантных рецепторов NOP, экспрессируемых в клеткахHEK293 (Okada et al., Biochem. Biophys. Res. Commun. 278, 493-8, 2000). Действия этого лиганда были,кроме того, охарактеризованы in vitro с использованием изолированных тканей, чувствительных к(Zhang et al., J. Med. Chem., 45, 5280-5286, 2002) описали аналоги N/OFQ, отличающиеся остатком 2-амино-2-метилпропионовой кислоты (Aib) в положении 7 и/или 11, замещающим остатки Ala и вызывающим увеличение аффинности и силы лиганда. Аналоги N/OFQ были описаны в WO 99/07212, WO 97/07208, WO 99/03491, WO 99/03880 и ЕР 1422240. Полезность данного лиганда была описана в лечении/предупреждении болезней, связанных с гипералгезией, нейроэндокринными функциями, стрессом,двигательной активностью и страхом. В дальнейшем исходной последовательностью пептида N/OFQ является следующая:H-Phe-Gly-Gly-Phe-Thr-Gly-Ala-Arg-Lys-Ser-Ala-Arg-Lys-Leu-Ala-Asn-Gln-OH Описание чертежей Фиг. 1. Эффект интрацеребровентрикулярного (и.ц.в., верхние панели) или интратекального (и.т.,нижние панели) введения N/OFQ (10 нмоль/мышь) и введения в хвост UFP-112 (0,1 нмоль/мышь) в исследовании отдергивания хвоста (ссылка Calo et al., Br. J. Pharmacol. 125, 375-378, 1998). Контрольные животные получили одну и.ц.в. инъекцию физиологического раствора (2 мкл/мышь). Каждая точка представляет собой среднее значениестандартная ошибка измерения из по меньшей мере 4 экспериментов. Фиг. 2. Продолжительность эффектов N/OFQ (10 нмоль/мышь) и UFP-112 (0,1 нмоль/мышь), введенного интрацеребровентрикулярно (и.ц.в.), на самопроизвольную двигательную активность у мышей-1 011325 получали одну и.ц.в. инъекцию физиологического раствора (2 мкл/мышь). Каждая точка представляет собой среднее значениестандартная ошибка измерения из по меньшей мере 4 экспериментов. Фиг. 3. Кинетика действия и обратимости эффектов равноэффективных концентраций N/OFQ иUFP-112 vas deferens мыши после электрической стимуляции. Сокращение vas deferens, вызванное электрической стимуляцией, ингибируется в присутствии UFP-112 или N/OFQ. Подробное описание изобретения Термины, использованные в данном патенте, имеют значение, известное в данной области техники,как, например, в Международном союзе фармакологов (IUPHAR) on Receptor Nomenclature and DragClassification, Pharm. Rev. (2003) Vol. 55, No. 4, p.597, как сообщено здесь: Эффективность - понятие, которое выражает степень, с которой различные агонисты вызывают отличающиеся ответы, даже когда занимают одинаковую долю рецепторов. Активность - выражение активности соединения, определенной в показателях концентрации или количества, необходимого для получения определенного эффекта. Активность измеряется как рЕС 50 для агонистов и как рА 2 для антагонистов. Целями настоящего изобретения являются пептидные аналоги N/OFQ, общей формулы (I) где Хаа 1 является Phe или N-бензилглицин (Nphe);представляет собой связь между первыми двумя аминокислотными остатками и выбрана из CO-NH, и CH2-NH, и СН 2-О; Xbb4 является Phe или (pX)Phe,где "X" представляет собой Н, Cl, Br, I, F, NO2, CN и "р" указывает пара-положение в фенильном кольце(CaCH3)Leu; (CaCH3)Val; 1-аминоциклопропанкарбоновой кислоты (Ас 3 с); 1-аминоциклопентанкарбоновой кислоты (Ac5c) и 1-аминоциклогексанкарбоновой кислоты (Ас 6 с); Хее 14 и Xff15 выбраны из Arg, Lys,Orn, omoArg, диаминомасляной кислоты или Trp; R представляет собой дипептид Asn-Gln-NH2 или AsnGln-OH или аминокислоту Asn или с амидной (-NH2), или с карбоксильной (-ОН) терминальной группой или амино (-NH2) или гидроксильной (-ОН) терминальной группой. Кроме того, изобретение включает фармацевтически приемлемые соли этих соединений (I), особенно соли органических и неорганических кислот, такие как гидрохлорид, гидробромид, фосфаты,сульфаты, ацетаты, сукцинаты, аскорбаты, глюконаты, бензоаты, малеаты, фумараты и стеараты. Соединения согласно изобретению, которые подпадают под формулу I, имеют доказанную фармакологическую активность, даже в 100 раз выше, чем пептидные лиганды, известные в данной области техники. Следовательно, возможно предположить синергический эффект перестановок согласно формуле I: для положений 1, 4, 7, 11, 14, и 15 и связи между первыми двумя аминокислотными остатками. Более высокая активность соединений формулы I и особенно предпочтительных соединений, предпочтительно агонистов и более предпочтительно [(pF)Phe4,Aib7,Arg14,Lys15]N/OFQ-NH2 продемонстрирована в отношении аффинности, активности устойчивости к протеазам, кинетики действия in vitro и, прежде всего, продолжительности их действия in vivo. Предпочтительными соединениями являются соединения формулы (I), в которойпредставляет собой CO-NH, или CH2-NH, или СН 2-О, Хаа 1 является Phe или Nphe, Xbb4 является Phe или (pX)Phe, где"(рХ)" является, как определено выше, Хсс 7 и Xdd11 являются, как определено выше, Хее 14 и Xff15 являются Arg, Lys, Orn, omoArg или Trp; R является -NH2, или -ОН, или Asn-NH2, или Asn-OH, или Asn-GlnNH2, или Asn-Gln-OH. Более предпочтительными являются те соединения формулы (I), в которыхпредставляет собой(pNO2)Phe; Хсс 7 и Xdd11 являются Ala; 2-амино-2-метилпропионовой кислотой (Aib); 2-амино-2-метилмасляной кислотой (Iva); 2-амино-2-этилмасляной (Deg) кислотой; 2-амино-2-пропилпентановой кислотой (Dpg); (CaCH3)Leu; (CaCH3)Val; 1-аминоциклопропанкарбоновой кислотой (Ас 3 с); 1-аминоциклопентанкарбоновой кислотой (Ac5c) и 1-аминоциклогексанкарбоновой кислотой (Ас 6 с); Хее 14 и Xff15 являются Arg или Lys; R представляет собой Asn-Gln-NH2 или -NH2. Даже более предпочтительными являются пептидные аналоги, имеющие формулу (I), в которой вариабельные остатки имеют значение, приведенное в следующей таблице.-3 011325 Среди этого даже более предпочтительными являются соединения, в которыхявляется CO-NH,или CH2-NH, или СН 2-О; Хаа 1 является Phe или Nphe; Xbb4 является Phe, или (pF)Phe, или (pNO2)Phe; Пептидные аналоги согласно изобретению могут быть синтезированы с помощью различных способов, известных из литературы, например Schroeder et al. "The Peptides", vol. 1, Academic Press, 1965;Bodanszky et al. "Peptide Synthesis", Interscience Publisher, 1966; BaranyMerrifield, "The peptides; Analysis,Synthesis, Biology", 2, Academic Press, 1980; E. AtheronR.C. Sheppard, "Solid Phase Peptide Synthesis",IRL Press at Oxford University Press 1989; J. Jones, "The Chemical Synthesis of Peptides", Claredon Press,Oxford 1994. Эти способы включают синтез пептидов на твердой фазе или синтез пептидов в растворимой фазе, синтетические методы органической химии или любую комбинацию вышеупомянутых. Выбор плана синтеза, очевидно, зависит от состава данного пептида. Предпочтительно используются синтетические методы, основанные на соответствующих комбинациях твердофазного синтеза и классических способов в растворимой фазе, включая низкую себестоимость производства, особенно в промышленном масштабе. Подробно указанные методы включают:i) синтез в растворе фрагментов пептидной цепи последовательным сочетанием N-защищенных аминокислот, соответствующим образом активированных, с аминокислотой или С-защищенной пептидной цепью с выделением промежуточных продуктов, последующим селективным снятием защиты N- и С-терминальных концов указанных фрагментов и их повторным сочетанием до тех пор, пока не будет-5 011325 получен необходимый пептид. При необходимости боковые цепи являются незащищенными;ii) твердофазный синтез пептидной цепи от С-терминального конца к N-терминальному концу на нерастворимой полимерной подложке. Пептид убирается из смолы гидролизом с помощью безводной плавиковой кислоты или трифторуксусной кислоты с одновременным снятием защиты с боковых цепей. В конце синтеза пептиды могут быть очищены и выделены обработкой подходящими растворителями и с помощью хроматографических методов, таких как высокоэффективная жидкостная хроматография (ВЭЖХ). Пептидные аналоги согласно изобретению действуют на рецептор NOP как: i) полные агонисты, когда они представляют собой структуру [Phe1(CO-NH)Gly2], ii) частичные агонисты, когда они представляют собой структуру [Phel(CH2-NH)Gly2] или [Phe1(CH2-O)Gly2], и iii) чистые антагонисты, когда они представляют собой структуру [Nphe1(CO-NH)Gly2]. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим пептидные аналоги, описанные здесь, возможно в комбинации с фармацевтически приемлемыми переносчиками и эксципиентами. Композиции данного изобретения могут быть введены оральным или парентеральным путем или через респираторный, ректальный, интраспинальный, интратекальный, внутрипузырный или местный путь в качестве инъецируемого препарата, капсулы, таблетки, гранулы, раствора,суспензии, сиропа, суппозитория, назального спрея, крема, мази, геля, контролирующих высвобождение препарата или другого. Принципы и способы приготовления фармацевтической композиции хорошо известны специалистам в данной области и описаны, например, в Remington's Pharmaceutical Sciences, 18thEdition, Mack Publishing Company, Easton, Pa, 1990. Фармацевтические композиции согласно изобретению будут содержать эффективные количества пептидов (или их производных), как правило, содержащихся в пределах между 0,001 и 100 мг, предпочтительно между 0,01 и 10 мг. Ежедневные дозы будут изменяться в зависимости от типа патологии/дисфункции, возраста, пола и веса тела пациента, общего состояния здоровья и других переменных, которые необходимо оценить по обстоятельствам. Учитывая профиль активности, показанный пептидами изобретения в биологических тестах, фармацевтические композиции, содержащие указанные пептиды, могут применяться для лечения дисфункций, условий или патологических состояний, включая неврологические и нейросенсорные дисфункции. Желательно получить сильную и длительную активацию рецептора NOP для лечения страха, анорексии,гипертензии, тахикардии, расстройств задержки воды, гипонатриемии, застойной сердечной недостаточности, гладкомышечных двигательных дисфункций в желудочно-кишечном, респираторном и мочеполовом трактах (особенно недержания мочи, следующего за неврогенной дисфункцией мочевого пузыря),воспалительных состояний или периферического или спинального обезболивания, особенно для лечения хронической боли или даже более в контроле кашля. Кроме того, возможно использовать антагонисты для лечения памяти, настроения, двигательной активности (например, болезни Паркинсона), расстройств потребления пищи (например, булимии), или, более обобщенно, для лечения пациентов, страдающих ожирением. Высокая молекулярная масса этих соединений и присутствие среди них остатков, которые могут быть положительно заряжены при физиологическом значении рН, делают невероятным то, что они могут преодолевать гематоэнцефалический барьер. Указанные соединения могут оказывать центральное действие после местного введения, даже если они показывают преимущественно периферическое распределение. Например, агонистические соединения могут вызывать обезболивание на уровне центральной нервной системы после интратекального или интраспинального введения. Экспериментальная часть 1. Пептидный синтез. 1.1. Общая схема синтеза. Пептиды изобретения были получены твердофазным синтезом с использованием смолы 4-(2',4'-диметоксифенил-Fmoc-аминометилфеноксиацетамидонорлейцил-смолы (смола Rink-Amide MBHA). Fmoc аминокислоты (флуоренилметоксикарбонил) конденсировали с использованием [О-(7-азабензотриазол-1 ил)-1,1,3,3-тетраметилурониумэкзафторфосфата] (HATU) в качестве реактива для активации карбоксильной функции. Fmoc группы удаляли применением 20% пиперидина в DMF (диметилформамид) и смолу, связанную с защищенным пептидом, обрабатывали реактивом K для того, чтобы получить необработанный пептид. Соединения, содержащие модифицированную пептидную связь между первыми двумя аминокислотными остатками [Phe1(CH2-NH)Gly2] или [Phe1(CH2-O)Gly2], получали конденсацией Boc-Phe-СНО на защищенном пептиде (2-17), или (2-16), или (2-15), связанном со смолой в течение последней стадии синтеза, таким образом восстанавливая in situ промежуточное "имино"-производное сNaBH3CN или конденсируя фрагмент Boc-[Phe1(CH2-O)Gly2]-OH (который был получен следующими способами, сообщенными в литературе: Balboni et al., J. Chem. Soc. Perkin Trans I, 1998, pg 1645-1651) на защищенном пептиде (3-17), или (3-16), или (3-15), связанном со смолой в течение последней стадии синтеза с использованием HATU в качестве конденсирующего вещества. Аналитический контроль как неочищенного, так и конечного продукта был сделан посредством аналитической высокоэффективной жидкостной хроматографии (ВЭЖХ) на приборе Beckmann System-6 011325 применением бинарной элюирующей системы, состоящей из растворителя А: 35 мМ NaH2PO4 (pH 2,1), и растворителя В: 59 мМ NaH2PO4 (рН 2,1)-ацетонитрил (60:40 об./об.), программируя градиент согласно физико-химическим свойствам анализируемых соединений, при скорости потока 1 мл/мин и при длине волны 220 нм. Нечищеный пептид очищали с использованием системы препаративной ВЭЖХ WaterDelta Prep 4000 с колонкой радиального наполнения Water Delta-LC 40 мм (3040 см, С 18, 300 А, 15 мкм),которую элюировали той же подвижной фазой, которую использовали для аналитической ВЭЖХ, и с градиентом, программируемым согласно аналитическому профилю неочищенных продуктов реакции. Молекулярная масса конечного соединения была получена электрораспылительной масс-спектрометрией с применением ZMD2000 микромасс-спектрометра. Для промежуточных соединений некоторых пептидов был выполнен спектроскопический анализ ЯМР 1 Н на приборе Bruker 200 МГц. 1.2. Методика. Пептидные аналоги b), с) и d), описанные выше, были получены согласно методикам, описанным здесь ниже. Смолу Rink-Amide MBHA (0,65 ммоль/г, 0,2 г) обработали пиперидином (20%) в DMF и конденсировали с Fmoc-Gln(Trt)-OH, активируя карбоксильную функцию с HATU. Следующие Fmoc аминокислоты были последовательно соединены в удлиняющуюся пептидную цепь: Fmoc-Asn(Trt)-OH, FmocLys(Boc)-OH, Fmoc-Arg(Pmc)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Arg(Pmc)-OH, Fmoc-Ala-OH, Fmoc-Ser(tBu)OH, Fmoc-Lys(Boc)-OH, Fmoc-Arg(Pmc)-OH, Fmoc-Aib-OH, Fmoc-Gly-OH, Fmoc-Thr(tBu)-OH, Fmoc(pF)Phe-OH, Fmoc-Gly-OH, Fmoc-Gly-OH, Fmoc-Phe-OH. Bce Fmoc аминокислоты (4 экв.) были соединены в удлиняющуюся пептидную цепь с применением HATU (4 экв.) и диизопропилэтиламина (4 экв.) вDMF; реакция сочетания была выполнена за 1 ч. Для того, чтобы оптимизировать выход синтеза и облегчить очистку соединений, требовалось двойное сочетание с ацилированием в течение 1 ч Aib остатка. Для удаления групп Fmoc на каждой стадии использовали пиперидин (20%) в DMF. После снятия защиты последней группы N-Fmoc пептидную смолу отмывали метанолом и высушивали под вакуумом,чтобы получить [(pF)Phe4,Aib7,Arg14,Lys15]-N/OFQ(1-17)-Rink-Amide МВНА-защищенную смолу. Эту защищенную пептидную смолу обрабатывали реактивом K (TFA/H2O/фенол/этандитиол/тиоанизол 82,5:5:5:2,5:5; об./об.; 10 мл/0,2 г смолы) в течение 1 ч при комнатной температуре. После фильтрования использованной смолы растворитель концентрировали под вакуумом и остаток поместили в эфир. Необработанный пептид очищали посредством предварительной ВЭЖХ с обращенной фазой и после лиофилизации получали белый порошок. Синтез [Phe1(CH2-NH)Gly2,(pF)Phe4,Aib7,Arg14,Lys15]-N/OFQ-NH2 (пептид с) выполняли, начиная с промежуточного соединения [(pF)Phe4,Aib7,Arg14,Lys15]-N/OFQ-(2-17)-смолы, синтезированного, как описано выше. Это промежуточное соединение (0,2 г, 0,65 ммоль/г, 0,13 ммоль) ресуспендировали и подвергали набуханию в метаноле, содержащем 1% (об./об.) уксусную кислоту (2 мл). Через 20 мин добавляли раствор, содержащий Boc-Phe-CHO (0,065 г, 0,26 ммоль) и NaBH3CN (0,033 г, 0,52 ммоль), солюбилизированные в метаноле (0,8 мл), и реакционную смесь перемешивали в течение 1,5 ч. Затем смолу отмывали метанолом и обрабатывали реактивом K, как описано выше. Синтез [Phe1(CH2-O)Gly2,(pF)Phe4,Aib7,Arg14,Lys15]N/OFQ-NH2 (пептид d) выполняли, начиная с промежуточного соединения [(pF)Phe4,Aib7,Arg14,Lys15]-N/OFQ(3-17)-смолы, синтезированного, как описано выше. Этот полупродукт (0,2 г, 0,65 ммоль/г,0,13 ммоль) ацилировали на последней стадии Boc-Phe[(CH2-O)]Gly-OH (4 экв., 0,16 г, 0,52 ммоль),активируя карбоксильную функцию с помощью HATU при тех же самых условиях, описанных для нормальных стадий ацилирования. Впоследствии смолу отмывали метанолом и обрабатывали реактивом K,как описано выше. 2. Фармакологические тесты. 2.1. Материалы и методы. Соединения были проверены in vitro на мембранах ооцитов хомяка, экспрессирующих человеческий рекомбинантный рецептор NOP (CHOhNOP) (эксперименты по связыванию с рецептором и эксперименты по стимуляции связывания GTPS), и на vas deferens мыши после электрической стимуляции. Условия, использованные для изучения эффектов соединений в биологических экспериментах (vas deferens мыши), описаны у Bigoni et al. (Naunyn Schmiedebergs Arch. Pharmacol. 359, 160-7, 1999), тогда как условия, использованные для изучения эффектов на CHOhNOP клетках, описаны у Mc Donald et al. (NaunynSchmiedebergs Arch. Pharmacol, 367, 183-187, 2003). В каждой серии экспериментов активность новых соединений сравнивали с активностью природных пептидов N/OFQ. 2.2. Результаты. В экспериментах по связыванию с рецептором доказано, что все проверенные соединения полностью способны к замещению меченного тритием N/OFQ из человеческих рекомбинантных рецепторовNOP. Соединения показали очень разную аффинность к рецептору (pKi) в зависимости от различных химических модификаций. Вообще, соединения со структурой [Phe1(CO-NH)Gly2] показали более высокую аффинность, чем те, которые имеют структуру [Phe1(CH2-NH)Gly2], и чрезвычайно более высокую аффинность, чем имеющие структуру [Nphe1(CO-NH)Gly2]. Кроме того, соединения, имеющие комби-7 011325 нированные модификации [(pF)Phe4,Aib7,Arg14,Lys15], показали аффинность больше, чем те, которые имеют единственные модификации. В функциональных тестах со стимуляцией связывания GTPS и в тестах с ингибированием судороги, вызванной электрической стимуляцией vas deferens мыши, соединения, имеющие структуру[Phe1(CO-NH)Gly2], повторяли эффекты N/OFQ, и в частности вызывали похожие максимальные эффекты, следовательно, действовали как полные агонисты, тогда как соединения, имеющие структуру[Phe1(CH2-NH)Gly2], действовали как частичные агонисты, так как их максимальные эффекты были ниже, чем с N/OFQ. Наконец, соединения, имеющие структуру [Nphe1(CO-NH)Gly2], не вызывали какого-либо эффекта сами по себе, но действовали как конкурентные антагонисты N/OFQ. Для упрощения, табл. 1 представляет результаты, полученные с соединениями [(pF)Phe4,Aib7,Arg14,15 Результаты представляют собой среднее значение из 4-6 определений. ND: не определено, так как соединение дает агонистические эффекты. Как подчеркнуто в табл. 1, соединение UFP-113 ведет себя как частичный агонист рецептора NOP,вызывая максимальные эффекты, которые ниже, чем N/OFQ, и в анализе GTPS, и в исследовании ингибирования судороги, вызванной электрической стимуляцией vas deferens мыши. Доказано, что UFP-111 является прямым и сильным антагонистом, селективным для рецептора NOP. Анализ Schild (выполненный и в экспериментах GTPS, и на системе vas deferens мыши) показывает, что соединение действует как конкурентный антагонист рецептора NOP с величиной активности (выраженной как рА 2) 8,68 и 7,46,соответственно (см. табл. 1). 2.3. Селективность соединения UFP-112. Эффекты UFP-112 опосредованы активацией рецептора NOP, как показано тем фактом, что действие данного пептида на vas deferens мыши не изменялось в присутствии налоксона (неселективного антагониста классических опиоидных рецепторов, но не рецептора NOP), но, оказывается, эффективно антагонизировалось посредством UFP-101, который является селективным антагонистом рецептора NOP([Nphe1,Arg14,Lys15]N/OFQ-NH2, Calo et al., Br. J. Pharmacol. 136, 303-311, 2002). UFP-101, использованный в конкуренции с UFP-112, показал значение силы (рА 2 6,81), сходное с полученным, когда он использовался в конкуренции с эндогенным агонистом N/OFQ (рА 2 6,91). Это показывает, что 3 молекулы(N/OFQ, UFP-112 и UFP-101) взаимодействуют с одним и тем же рецептором: рецептором NOP. Затем,это показано результатами, полученными на тканях нокаутной мыши (Ref. Nishi, M. et al., UnrestrainedEmbo. J. 16 (8): 1858-64, 1997) по гену рецептора NOP (NOP-/-) (см. табл. 2). Таблица 2 Эффекты агониста N/OFQ и UFP112 и агониста DOP, D-Pen2,D-Pen5 энкефалина (DPDPE) на vas deferens дикого типа (NOP+/+) и нокаутной мыши по рецептору NOP (NOP-/-) Ингибирующее действие на сокращение, вызванное электрической стимуляцией, вызываемое UFP112 (сходное с тем, которое найдено с N/OFQ), исчезало в vas deferens, выделенных из мыши NOP-/-, подтверждая, что биологические действия UFP-112 существуют только вследствие взаимодействия с рецептором NOP. Соединение [D-Pen2,D-Pen5]энкефалин, DPDPE (Ref. Life Sci. 1983; 33 Suppl. 1: 447-50), селектив-8 011325 ный агонист DOP, использовали в качестве положительного контроля. Этот контроль показывает, какие ткани, полученные от нокаутной мыши по рецептору NOP, нормально реагируют на ингибирующий стимул, который не использует рецептор NOP. 2.4. Фармакологические тесты на селективность соединений согласно изобретению. Соединения были проверены in vitro на мембранах ооцитов хомяка (СНО), экспрессирующих человеческий рекомбинантный рецептор NOP (CHOhNOP), как в параграфе 2.1, согласно Me Donald et al.(Naunyn Schmiedebergs Arch. Pharmacol. 367, 183-187, 2003). Исследования на селективность этих соединений для рецептора NOP были выполнены посредством исследований рецепторного связывания на мембранах клеток СНО, трансфицированных человеческими рекомбинантными опиоидными рецепторами мю (МОР), дельта (DOP) и каппа (KOP) типов, с использованием того же самого метода, как для CHOhNOP. Исследования селективности были выполнены посредством конкурентных экспериментов согласно методам, описанным у Mc Donald et al. (Naunyn SchmiedebergsArch. Pharmacol. 367, 183-187, 2003). Чтобы измерить pKi для N/OFQ, в качестве радиолиганда был использован меченный тритием N/OFQ, тогда как [3 Н]-дипренорфин был использован для классических опиоидных рецепторов. Активность новых соединений сравнили с активностью природного пептидаN/OFQ. В экспериментах по рецепторному связыванию, выполненному на мембранах трансфицированных клеток СНО, UFP-111, UFP-112 и UFP-113 показали более высокую селективность (100 раз) для рецептора NOP, чем рецепторов МОР, KOP и DOP (см. табл. 3). Таблица 3 Аффинность (pKi) UFP-112, UFP-113 и UFP-111 для рецепторов NOP, MOP, DOP и KOP,трансфицированных в клетки СНО Данные представляют собой среднее значение 4 экспериментов. В качестве меченного тритием лиганда использовали [3H]N/OFQ. 2DAMGO означает [D-Ala(2),N-MePne(4),Gly-ol(5)]энкефалин. 3 В качестве меченного тритием лиганда использовали [3 Н]дипренорфин. 1 3. Исследования эффективности in vivo полного агониста соединения UFP-112. Соединение UFP-112, которое является полным агонистом, было протестировано на мыши in vivo в различных исследованиях: 1) тест на отдергивание хвоста согласно экспериментальным протоколам, описанным в Calo et al.,(Br. J. Pharmacol. 125, 373-378, 1998) and Rizzi et al. (Clin. Pharmacol. 18, 56, 2004); 2) измерение потребления пищи у сытых животных, как описано Rizzi et al. (Naitional Congress of theItalian Society of Neuroscience and joint Italian-Swedish Neuroscience Meetings, Ischia (Napoli) 1-4 October 2005); 3) тест для измерения спонтанной двигательной активности, как описано Rizzi et al. (NaunynSchmiedebergs Arch. Pharmacol. 363, 161-165, 2001. В каждом исследовании активности UFP-112 и N/OFQ были измерены как эквиэффективные дозы. Так как UFP-112 показывает активность выше приблизительно в 100 раз, пептид UFP-112 использовали в дозах, содержащихся в интервале между 0,001 и 0,1 нмоль, и N/OFQ использовали в дозах, содержащихся в интервале между 0,1 и 10 нмоль. В аналгезиометрическом тесте на отдергивание хвоста у мышей UFP-112 в эквиэффективных дозах повторяет эффекты природного лиганда N/OFQ, хотя он показывает свое действие в течение более длительного периода (120 мин).UFP-112 в интервале доз между 0,001-0,1 нмоль вызывает проноцицептивные эффекты, если вводится через интрацеребровентрикулярный (и.ц.в.) путь, тогда как он вызывает антиноцицептивные эффекты при введении интратекально (и.т.) (см. фиг. 1). Указанные эффекты (сходные с теми, которые най-9 011325 дены с N/OFQ) опосредуются активацией рецептора NOP, так как они отсутствуют у NOP-/- мышей.N/OFQ и UFP-112 в эквиэффективных дозах были проверены в тесте на потребление пищи накормленными животными. Оба соединения вызывали значительное увеличение потребления пищи, и также в данных анализах доказано, что UFP-112 является в 100 раз более сильным, чем N/OFQ. В этом тесте гиперфагические эффекты N/OFQ и UFP-112 существуют исключительно благодаря активации рецептораNOP, потому что такие эффекты присутствуют у NOP+/+ мышей, но отсутствуют у NOP-/- мышей. Для того, чтобы исследовать продолжительность действия UFP-112 in vivo, были выполнены эксперименты на мышах, которые сравнили с продолжительностью (от 17 ч 30 мин до 7 ч 30 мин следующего дня) эффекта эквиэффективных доз N/OFQ (10 нмоль) и UFP-112 (0,1 нмоль), при введении обоих соединений и.ц.в., на спонтанную двигательную активность. Оба пептида ингибировали двигательную активность, но эффект N/OFQ заканчивался через 60 мин после и.ц.в. инъекции, тогда как эффект, вызванныйUFP-112, заканчивался после приблизительно 6 ч (см. фиг. 2). 4. Метаболическая стабильность N/OFQ и новых производных соединений UFP-111, UFP-112 иUFP-113 в гомогенатах мозга и в плазме. Образцы плазмы и ткани мозга были получены от самцов мышей Swiss (Morini, Reggio Emilia, Italy,25-30 г). Животное, умерщвленное с помощью анестезии эфиром, перфузировали физиологическим раствором гепарина, введенного через иглу, помещенную в левый желудочек. Затем кровь извлекали и центрифугировали при 14000g в течение 2 мин при комнатной температуре. После отделения от осадка отбирали аликвоты плазмы и хранили при -80 С. После изъятия крови животное далее перфузировали с физиологическим раствором в течение 2 мин перед выемкой мозга. Ткань мозга гомогенизировали в 5 объемах (в./об.) Tris/HCl (50 мМ, рН 7,4, 0 С) с ультра-Turrax (Janke Kunkel, Staufen, FRG) 3 раза в течение 15 с каждый. Супернатант, полученный центрифугированием (3000g в течение 15 мин при 4 С),сливали и затем хранили при -80 С. Содержание белка в препаратах, определенное способом Брэдфорд, как описано в Anal. Biochem.,72, 248-254, 1976, было приблизительно 8 мкг/мкл для гомогената мозга и 17 мкг/мкл для плазмы. Аликвоту 100 мкл раствора каждого пептида (3 мг/500 мкл Tris) инкубировали (при конечной концентрации 6 мкг/мкл) с гомогенатом мозга или плазмой (450 мкл) в общем объеме 1 мл, содержащем буфер Tris/HCl 50 мМ рН 7,4. Инкубацию аликвот выполняли при 37 С в течение различных периодов вплоть до 240 мин. В различные периоды инкубации отбирали аликвоту раствора (100 мкл), а деградацию блокировали добавлением 4,5% TFA раствора (200 мкл). После центрифугирования (3000 об./мин в течение 15 мин) аликвоту (100 мкл) супернатанта вводили в хроматограф ВЭЖХ с обращенной фазой. Анализ ВЭЖХ был выполнен на колонке Kromasil 100-5C18 (4,6250 мм) с использованием хроматографической системы Beckman System Gold, оснащенной УФ-детектором с переменной длиной волны. Экспериментальные условия для элюирования включали градиентный анализ с водой (растворитель А) и ацетонитрилом (растворитель В), оба содержащие 0,1% TFA, при скорости потока 0,7 мл/мин. Следующий протокол был использован для градиентного анализа, выбранного на основе физико-химических характеристик аналита: линейный градиент от 5 до 40% В за 20 мин; линейный градиент от 40 до 60% В за 5 мин; линейный градиент от 60 до 5% В за 5 мин. Элюат проверяли при 220 нм. Период полувыведения (Т 1/2) получили посредством линейной регрессии по методу наименьших квадратов, изображая на диаграммах площади пиков каждого производного как функцию времен инкубации, с использованием по меньшей мере 5 точек для каждого анализа. Данные показаны в табл. 4 как среднеестандартное отклонение и получены по меньшей мере из 3 отдельных экспериментов. Таблица 4 Т 1/2 (мин) N/OFQ и производных в плазме и ткани мозга мышиN/OFQ показал полупериод выведения из плазмы около 1 ч, который очень отличался от полупериода выведения, полученного с гомогенатом мозга, который составлял около 3 мин. Все исследованные пептиды согласно изобретению показали значительно более длинный период полувыведения по сравнению с природным пептидом. В частности, плазменный Т 1/2 UFP-111 и UFP-113 приблизительно вдвое длиннее, чем N/OFQ, тогда как Т 1/2 UFP-112 почти в 3 раза длиннее, чем N/OFQ. Более длинные полупериоды выведения, показанные производными, по сравнению с N/OFQ были более выражены в гомогенате мозга, чем в плазме. Фактически, Т 1/2 всех производных были более чем в 3 раза длиннее, чем значение, показанное N/OFQ (3 мин) в ткани мозга. Эти данные показали, что химические модификации последовательностей UFP-111, 112 и 113 уве- 10011325 личивают их активность как агонистов или антагонистов по сравнению с N/OFQ: такие модификации изменяют их эффективность на рецептор NOP и определяют значительное уменьшение чувствительности к деградации пептидазами, присутствующими и в плазме и в ткани мозга. Эта важная характеристика является, безусловно, определяющей в пролонгировании действия данных молекул in vivo, как хорошо показано для UFP-112 в серии экспериментов, суммированных в разделе 3 (исследования in vivo). 5. Кинетика ингибирующего эффекта UFP-112 на vas deferens мыши. При электрическом стимулировании vas deferens мыши как кинетика действия UFP-112, так и обратимость эффектов после отмывки были намного медленнее, чем с N/OFQ (см. фиг. 3). Это было показано посредством ингибирующего эффекта на сокращение vas deferens, вызванное электрической стимуляцией. Вместе с данными метаболической стабильности это может объяснить более длительное действие invivo UFP-112 по сравнению с эндогенным лигандом N/OFQ. 6. Биологическая активность некоторых соединений формулы I у мыши vas deferens после электрической стимуляции. Табл. 5 суммирует результаты, полученные на vas deferens мыши после электрической стимуляции,в присутствии серии соединений формулы I, несущих различные химические модификации в положении 7 и 11, модели агониста N/OFQ-NH2. Эти данные показывают, что различные аминокислотные замещения не изменяют эффективность соединений, которые действуют как полные агонисты, но в некоторых случаях (например, [Ac5c11]N/OFQ-NH2 и [D/L-Iva11]N/OFQ-NH2) увеличивают активность по сравнению с исходной последовательностью. Следует заметить, что увеличение активности, полученное как результат этих отдельных модификаций (в 2 раза по сравнению с исходной последовательностью), ниже, чем увеличение активности, полученной в результате комбинированной модификации в различных положениях, как в соединении UFP-112 (табл. 1), активность которого возрастает более чем в 100 раз. Таблица 5 Активность ряда соединений с общей формулой I, полученных с различными химическими модификациями в положении 7 и 11, при измерении на vas deferens мыши при электрической стимуляции ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Пептид, имеющий общую формулу (I) в которой Хаа 1 выбран из группы, состоящей из Phe или N-бензил-глицина (Nphe);представляет собой связь между первыми двумя аминокислотными остатками и выбрана из группы, состоящей из CO-NH, и CH2-NH, и СН 2-О;R представляет собой дипептид Asn-Gln-NH2 или Asn-Gln-OH или аминокислоту Asn или с амидо(-NH2), или с карбоксильной (-ОН) терминальной группой или амино (-NH2) или гидроксильной (-ОН) терминальной группой; и его фармацевтически приемлемые соли. 2. Пептид по п.1, выбранный из группы, состоящей из и его фармацевтически приемлемые соли. 3. Пептид по п.1, в котором Хаа 1 является Phe;R представляет собой дипептид Asn-Gln-NH2, или Asn-NH2, или амино(-NH2)группу; и его фармацевтически приемлемые соли. 4. Пептид по п.3, в которомявляется CO-NH;R представляет собой дипептид Asn-Gln-NH2; и его фармацевтически приемлемые соли. 5. Пептид по п.3, в которомявляется CH2-NH;R представляет собой дипептид Asn-Gln-NH2; и его фармацевтически приемлемые соли. 6. Пептид по п.2, в котором Хаа 1 является N-бензилглицином (Nphe);является CO-NH;R представляет собой дипептид Asn-Gln-NH2 или амино (-NH2) группу; и его фармацевтически приемлемые соли. 7. Пептид по п.6, в котором Хсс 7 является 2-амино-2-метилпропионовой кислотой (Aib);R представляет собой дипептид Asn-Gln-NH2; и его фармацевтически приемлемые соли.- 13011325 8. Композиция, содержащая пептиды по пп.1-7. 9. Фармацевтическая композиция, содержащая в качестве активного начала пептиды по пп.1-7, объединенные с фармацевтически приемлемыми носителями и/или эксципиентами. 10. Фармацевтическая композиция по п.9, предназначенная для введения через оральный, местный,респираторный, ректальный, интраспинальный, интратекальный, внутрипузырный или парентеральный путь. 11. Фармацевтическая композиция по п.10, где введение производится через интратекальный и парентеральный пути. 12. Применение пептида по пп.1-7 для приготовления лекарственного средства, предназначенного для лечения или предупреждения неврологических и нейросенсорных дисфункций. 13. Применение пептида по пп.3-5 для приготовления лекарственного средства для лечения или предупреждения гипертензии, тахикардии, расстройств задержки воды, гипонатриемии, сердечной недостаточности, гладкомышечных двигательных дисфункций в желудочно-кишечном, респираторном и мочеполовом трактах, воспалительных состояний, периферического или спинального обезболивания,ведения хронической боли и ослабления кашля. 14. Применение по п.13, в котором указанная гладкомышечная двигательная дисфункция включает нейрогенное недержание мочевого пузыря или гиперактивность мочевого пузыря, респираторную дисфункцию. 15. Применение по п.12 для приготовления транквилизатора или лекарственного средства для лечения или предупреждения анорексии. 16. Применение пептида по пп.6-7 для приготовления лекарства для лечения нарушений памяти и настроения, двигательной активности и расстройств потребления пищи или для лечения ожирения.
МПК / Метки
МПК: A61K 38/33, C07K 14/665
Метки: частичные, рецептора, антагонисты, высокоэффективные, агонисты, полные
Код ссылки
<a href="https://eas.patents.su/30-11325-vysokoeffektivnye-polnye-i-chastichnye-agonisty-i-antagonisty-receptora-nociceptina-orfanina-fq.html" rel="bookmark" title="База патентов Евразийского Союза">Высокоэффективные полные и частичные агонисты и антагонисты рецептора ноцицептина/орфанина fq</a>
Агонисты и антагонисты рецепторов 5-ht2c

Номер патента: 5820
Опубликовано: 30.06.2005
Авторы: Йенссон Маттиас, Тор Маркус, Рингберг Эрик, Пелькман Беньямин, Нильссон Йонас, Нильссон Бьерн, Тейбрант Ян
МПК: A61K 31/497, A61P 25/00, C07D 241/18...
Метки: антагонисты, 5-ht2c, рецепторов, агонисты
Формула / Реферат:
1. Соединение общей формулы (Ib) где X1 представляет -O-, -S- или -N(R27)-; Y1 представляет -O-, -S-, -N(R27)- или -CH2-; Ra представляет C3-8-циклоалкил, арил или гетероарил, каждый из которых может быть, необязательно, замещен в одном или в нескольких положениях независимо друг от друга C1-6-алкилом, C1-6-алкокси, фтор-C1-6-алкокси, 2,2,2-трифторэтокси, C3-5-алкинилокси, C3-5-алкенилокси, диметиламино-C1-6-алкокси, метиламино-C1-6-алкокси,...
Агонисты в2-адренергического рецептора

Номер патента: 3453
Опубликовано: 26.06.2003
Авторы: Чои Сеок-Ки, Моран Эдмунд Дж.
МПК: A61K 38/00, A61K 39/00, A61K 39/395...
Метки: в2-адренергического, агонисты, рецептора
Формула / Реферат:
1. Соединение формулы (II) где Ar1 и Ar3 независимо выбирают из группы, включающей арил, гетероарил и гетероциклил; Ar2 выбирают из группы, включающей фенилен, где группы W и X присоединены в 1,2-, 1,3 и 1,4 положениях фениленового кольца; циклогексилен, необязательно замещенный метилом, где группы W и X присоединены в 1,3 и 1,4 положениях циклогексиленового кольца, и пиперазинилен, где группы W и X присоединены в 1,4 положениях...
Агонисты β2-адренергического рецептора

Номер патента: 4263
Опубликовано: 26.02.2004
Авторы: Чои Сеок-Ки, Моран Эдмунд Дж.
МПК: A61K 31/135, A61K 31/165, A61P 11/00...
Метки: рецептора, агонисты, beta;2-адренергического
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль. 2. Соединение формулы где стереохимия при *C и **C представляет (RS) и (RS); (R) и (R); (R) и (S); (S) и (R) или (S) и (S); или его фармацевтически приемлемая соль. 3. Соединение по п.2, где стереохимия при *C представляет (R) и стереохимия при **C представляет (R). 4. Соединение по п.2, где стереохимия при *C представляет (R) и стереохимия при **C представляет (S). 5....
Агонисты гуанилатциклазного рецептора для лечения тканевого воспаления и канцерогенеза

Номер патента: 6651
Опубликовано: 24.02.2006
Авторы: Джэкоб Гэри С., Шаилубхай Кунвар, Никифорович Грегори
МПК: A61K 38/12, A61K 45/06, A61K 38/10...
Метки: канцерогенеза, агонисты, лечения, гуанилатциклазного, тканевого, воспаления, рецептора
Формула / Реферат:
1. Пептид, состоящий, по существу, из аминокислотной последовательности, выбранной из SEQ ID NO: 2 - SEQ ID NO: 21. 2. Пептид по п.1, где указанный пептид представляет собой (4,12; 7,15) бицикл, имеющий последовательность SEQ ID NO: 20. 3. Пептид по п.1 или 2, где указанный пептид состоит из аминокислотной последовательности, выбранной из SEQ ID NO: 2 - SEQ ID NO: 21. 4. Способ профилактики или лечения первичной или метастатической...
Антагонисты рецептора il-8

Номер патента: 1436
Опубликовано: 26.02.2001
Авторы: Виддаусон Кетрин Луиза, Ратледж Мельвин Кларенс Мл., Вебер Дэниел Франк, Юревич Энтони Джозеф, Херцберг Роберт Филип
МПК: A61K 31/17, A61P 11/06
Метки: рецептора, антагонисты
Формула / Реферат:
1. Способ лечения болезненного состояния, опосредованного хемокином, где хемокин связывается у млекопитающих, нуждающихся в таком лечении, с IL-8 а- или b-рецептором, включающий введение млекопитающему эффективного количества соединения формулы где Х является кислородом или серой; R является любой функциональной группой, имеющей ионизируемый водород и рКа, равный 10 или менее; R1 независимо выбирают из водорода; галогена; нитро; циано;...
Предыдущий патент: Термостабилизированная пресс-композиция, ее применение, формованные детали, состоящие из указанной композиции, способ получения формованных деталей и их применение
Следующий патент: Способ и устройство для определения местоположения и величины скорости течи радиоактивного вещества из емкости, находящейся под давлением
Случайный патент: Имплантант с прикрепленным к нему элементом и способ изготовления такого имплантанта