4-арилпиперидины
Номер патента: 11029
Опубликовано: 30.12.2008
Авторы: Марзабади Мохаммад Р., Делеон Джон Э., Чэнь Чень-Ань, Ветцель Джон М., Лу Кай, Цзян Юй
























Формула / Реферат
1. Соединение со структурой

где каждый X является независимо CR1;
где каждый R1 является независимо -Н, -F, -Cl, -Br, -I, -CN, -NO2, линейным или разветвленным C1-C7 алкилом, C1-C7 алкилокси, монофторалкилом или полифторалкилом, или C3-C6 циклоалкил-C1-C7 алкилом;
каждый R2 является независимо -Н, -F, -Cl или линейным или разветвленным C1-C4 алкилом, монофторалкилом или полифторалкилом;
n является целым числом от 2 до 6 включительно;
В является CH2, CHOH, О или CO;
Y является С или N;
Z является О, CH2, CO, CHOH или нулем;
и А является фенилом или гетероарилом, где фенил или гетероарил являются необязательно замещенными тремя или менее R2;
причем гетероарил выбирают из группы, включающей фуранил, тиенил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, оксадиазолил, триазолил, тиадиазолил, пиридил, пиридинил, пиридазинил, пиримидинил, пиразинил и триазинил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где n равняется 2 или 3.
3. Соединение по п.2, где каждый R1 является независимо -Н, -F, -Cl, линейным или разветвленным C1-C4 алкилом или алкокси или C3-C6 циклоалкил-C1-C4 алкилом.
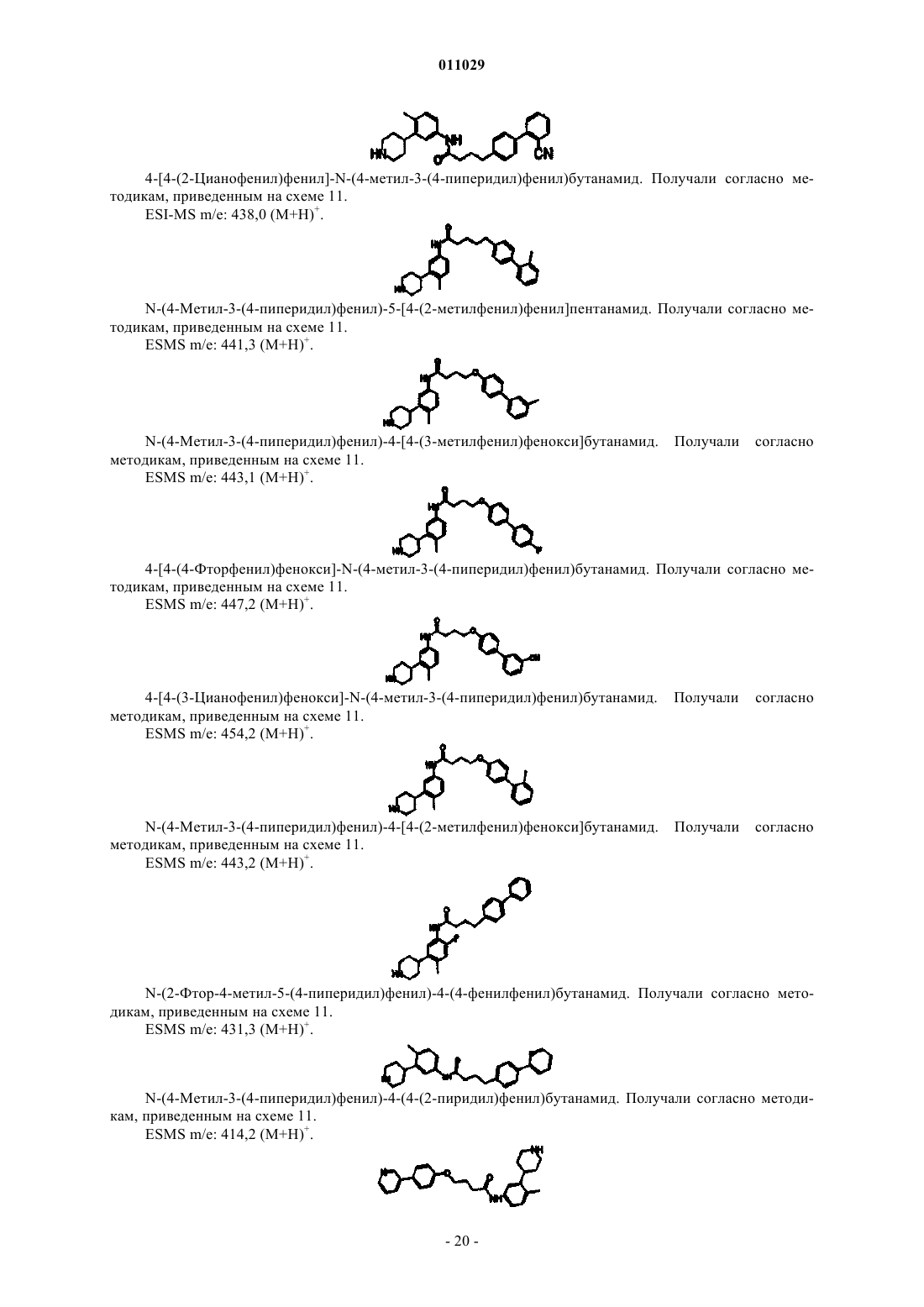
4. Соединение по п.3, где каждый R2 является независимо -Н, -F, -Cl или линейным или разветвленным C1-C4 алкилом, монофторалкилом или полифторалкилом.
5. Соединение по п.4, где А является пиридинилом, необязательно замещенным тремя или менее R2.
6. Соединение по п.4, где А является тиенилом, необязательно замещенным тремя или менее R2.
7. Соединение по п.4, где А является фуранилом, необязательно замещенным тремя или менее R2.
8. Соединение по п.4, где А является тиазолилом, необязательно замещенным тремя или менее R2.
9. Соединение по п.4, где А является имидазолилом, необязательно замещенным тремя или менее R2.
10. Соединение по п.4, где А является пиразолилом, необязательно замещенным тремя или менее R2.
11. Соединение по п.4, где А является оксазолилом, необязательно замещенным тремя или менее R2.
12. Соединение по п.4, где А является триазинилом, необязательно замещенным тремя или менее R2.
13. Соединение по п.4, где А является фенилом, необязательно замещенным тремя или менее R2.
14. Соединение по п.13, где Z является CH2, CO или CHOH.
15. Соединение по п.13, где Z является O или нулем.
16. Соединение по п.15, где В является CH2.
17. Соединение по п.16, где Z является О.
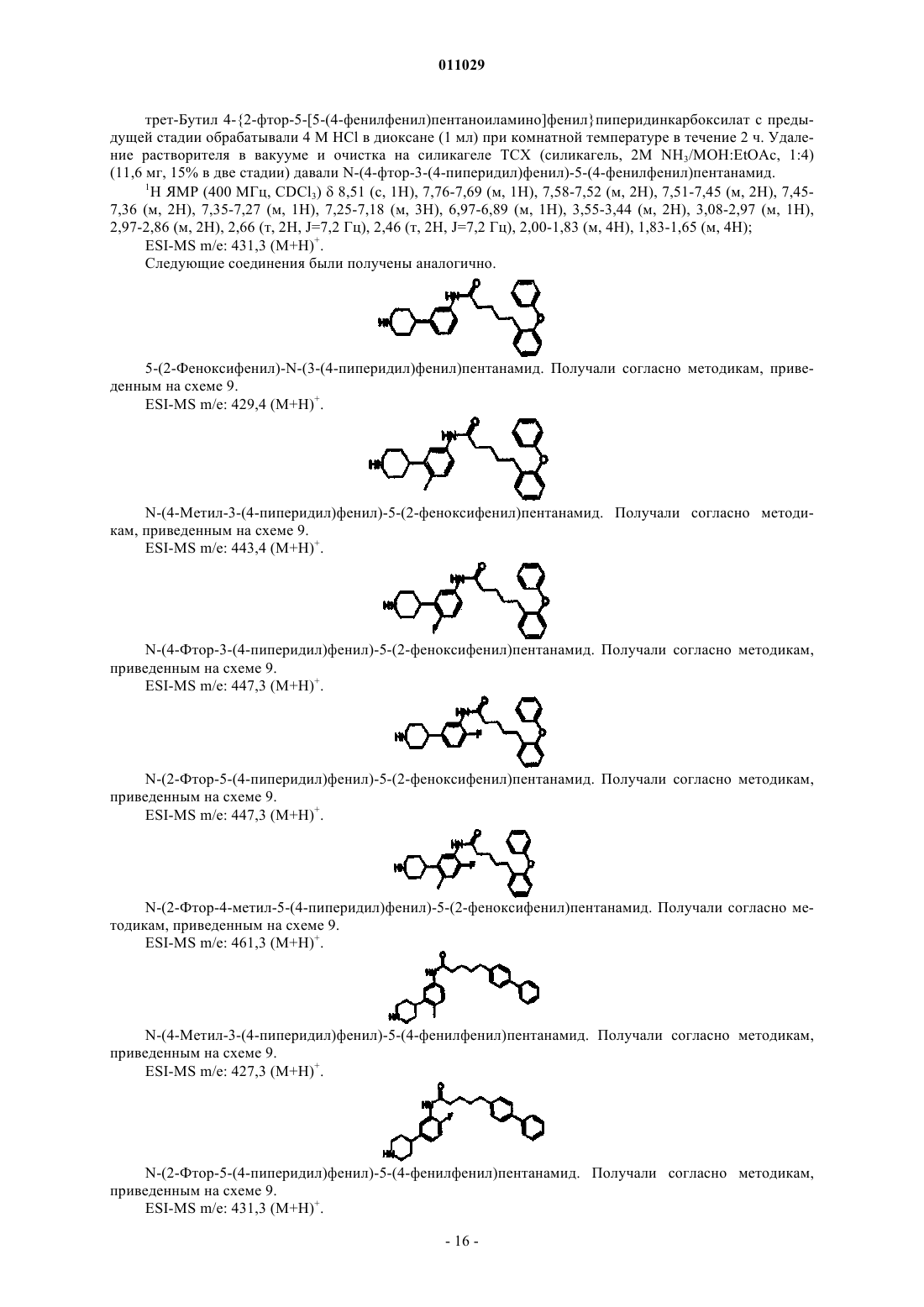
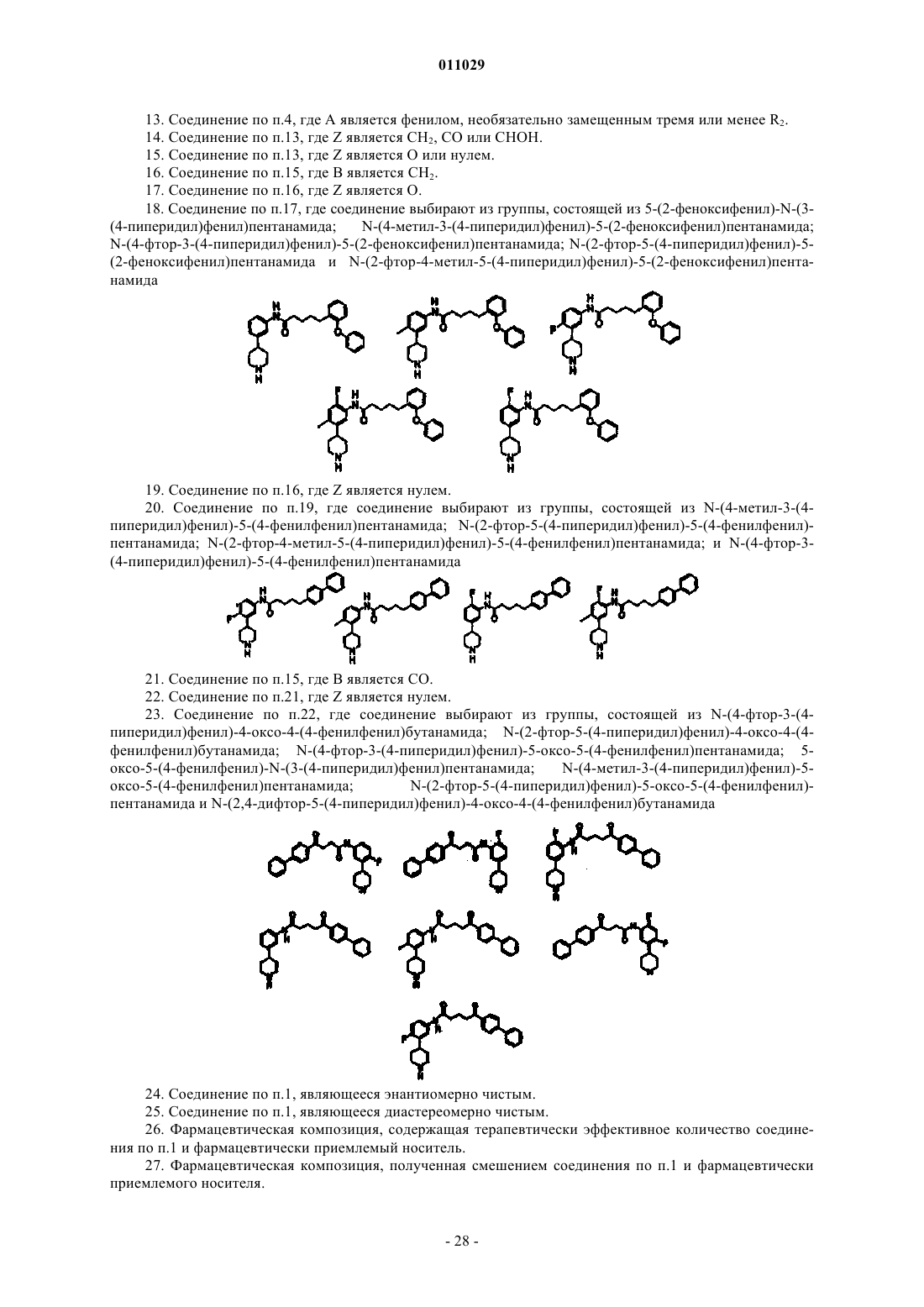
18. Соединение по п.17, где соединение выбирают из группы, состоящей из 5-(2-феноксифенил)-N-(3-(4-пиперидил)фенил)пентанамида; N-(4-метил-3-(4-пиперидил)фенил)-5-(2-феноксифенил)пентанамида;
N-(4-фтор-3-(4-пиперидил)фенил)-5-(2-феноксифенил)пентанамида; N-(2-фтор-5-(4-пиперидил)фенил)-5-(2-феноксифенил)пентанамида и N-(2-фтор-4-метил-5-(4-пиперидил)фенил)-5-(2-феноксифенил)пентанамида

19. Соединение по п.16, где Z является нулем.
20. Соединение по п.19, где соединение выбирают из группы, состоящей из N-(4-метил-3-(4-пиперидил)фенил)-5-(4-фенилфенил)пентанамида; N-(2-фтор-5-(4-пиперидил)фенил)-5-(4-фенилфенил)пентанамида; N-(2-фтор-4-метил-5-(4-пиперидил)фенил)-5-(4-фенилфенил)пентанамида; и N-(4-фтор-3-(4-пиперидил)фенил)-5-(4-фенилфенил)пентанамида

21. Соединение по п.15, где В является CO.
22. Соединение по п.21, где Z является нулем.
23. Соединение по п.22, где соединение выбирают из группы, состоящей из N-(4-фтор-3-(4-пиперидил)фенил)-4-оксо-4-(4-фенилфенил)бутанамида; N-(2-фтор-5-(4-пиперидил)фенил)-4-оксо-4-(4-фенилфенил)бутанамида; N-(4-фтор-3-(4-пиперидил)фенил)-5-оксо-5-(4-фенилфенил)пентанамида; 5-оксо-5-(4-фенилфенил)-N-(3-(4-пиперидил)фенил)пентанамида; N-(4-метил-3-(4-пиперидил)фенил)-5-оксо-5-(4-фенилфенил)пентанамида; N-(2-фтор-5-(4-пиперидил)фенил)-5-оксо-5-(4-фенилфенил)пентанамида и N-(2,4-дифтор-5-(4-пиперидил)фенил)-4-оксо-4-(4-фенилфенил)бутанамида

24. Соединение по п.1, являющееся энантиомерно чистым.
25. Соединение по п.1, являющееся диастереомерно чистым.
26. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
27. Фармацевтическая композиция, полученная смешением соединения по п.1 и фармацевтически приемлемого носителя.
28. Способ получения фармацевтической композиции, включающий смешение соединения по п.1 и фармацевтически приемлемый носитель.
29. Применение соединения по пп.1-25 для изготовления лекарственного средства для лечения пациента, страдающего от аффективного расстройства, выбранного из группы, состоящей из депрессии, глубокой депрессии, маниакально-депрессивного психоза, агорафобии, специфической фобии, социофобии, навязчивого состояния, посттравматического стресса, острого стрессового расстройства и состояния тревоги, включающее введение пациенту терапевтически эффективного количества соединения по п.1.
30. Применение соединения по пп.1-25 для изготовления лекарственного средства для лечения пациента, страдающего от нарушения мочеиспускания, выбранного из группы, состоящей из непроизвольного мочеиспускания, позыва к мочеиспусканию, частого мочеиспускания, недержания мочи, никтурии и энуреза, включающее введение пациенту терапевтически эффективного количества соединения по п.1.
31. Применение соединения по пп.1-25 для изготовления лекарственного средства для лечения пациента, страдающего от расстройства питания, выбранного из группы, состоящей из ожирения, булимии, нейрогенной булимии и нейрогенной анорексии, включающее введение пациенту терапевтически эффективного количества соединения по п.1.
Текст
011029 Настоящее изобретение относится к 4-арилпиперидинам и родственным гетероциклическим соединениям, применяемым в качестве терапевтических средств для лечения физиологических недугов, таких как определенные психические состояния, включающие, но этим не ограничиваясь, депрессию и состояние тревоги. Кроме того, терапевтическое средство настоящего изобретения может применяться для лечения ожирения или неотложного недержания мочи. Меланинконцентрирующий гормон (MCH) является циклическим нейропептидом, первоначально выделенным из гипофизов лососевых (костистых рыб) (Kawauchi et al., 1983). Была опубликована идентификация связанного с G-белком рецептора для MCH (Chambers et al., 1999; Saito et al., 1999). Эти группы идентифицировали MCH как эндогенный лиганд для человеческого рецептора SLC-1, связанного "сиротским" G-белком (Lakaye et al.,1998). После этого открытия было обнаружено, что MCH млекопитающих (19 аминокислот) является высококонсервативным у крысы, мыши и человека, проявляя 100% аминокислотную идентичность. Сообщалось, что гомолог этого рецептора у крысы, называемый сейчас MCH1, локализован в областях мозга крысы. В наших собственных исследованиях были оценены антагонисты MCH1 на различных животных моделях, которые хорошо известны как позволяющие предсказывать эффективность соединений для человека, см. Borowsky, В., et al. (2002). Эти эксперименты наводят на мысль, что MCH1 антагонисты могут применяться для лечения депрессии и/или состояния тревоги. После картирования мест связывания для [(3)Н]SNAP-7941, селективного и эффективного MCH1 антагониста в мозге крыс, авторы оценивали его влияние в серии поведенческих моделей. SNAP-7941 оказывал воздействия, аналогичные воздействию антидепрессантов и анксиолитиков, используемых в клинике, на трех животных моделях депрессия/тревога: тест принудительного плавания на крысах, тест социального взаимодействия на крысах и тест на вокализацию при отделении от матери на морских свинках. Эти наблюдения предполагают, чтоMCH1 антагонист может применяться для лечения депрессии и/или состояния тревоги. Кроме того, в недавних сообщениях по фенотипу MCH1 генетически модифицированных мышей было высказано предположение о связи между MCH1 и воздействиями MCH на питание. Две группы независимо показали, что целевой разрыв гена MCH1 рецептора (MCH1 генетически модифицированный) у мышей дает в результате животных, которые, будучи гиперфагическими, являются худыми и имеют пониженную массу тела по отношению к однопометным животным дикого типа (Marsh et al.,2002; Chen et al., 2002). Снижение массы тела приписывают повышению метаболизма. Каждая группа продемонстрировала, что MCH1 генетически модифицированные мыши устойчивы к вызываемому питанием ожирению, и обычно характеризуются весами, аналогичными однопометным животным, содержавшимся на привычном питании. В литературе описаны молекулы искусственного антагониста для MCH1 рецептора. Bednarek et al.(2002) сообщил о синтезе антагонистов MCH1 с высоким сродством к пептиду. Небольшая молекула антагониста MCH1 описана Takekawa et al. (2002). В лабораториях авторов авторы открыли маленькие молекулы, которые являются антагонистамиMCH1 рецептора. Соответственно, соединения настоящего изобретения могут применяться для лечения состояний, перечисленных выше. Настоящее изобретение относится к соединениям со структурой где каждый X является независимо CR1 или N, при условии, что если один X является N, то оставшиесяC1-C7 алкилом, алкилокси, монофторалкилом или полифторалкилом, или C3-C6 циклоалкил-C1-C7 алкилом; каждый R2 является независимо -H, -F, -Cl, -Br, -I, -CN, -NO2 или линейным или разветвленным C1C7 алкилом, монофторалкилом или полифторалкилом;n является целым числом от 2 до 6 включительно;Z является О, S, SO, SO2, CH2, CO, CHOH или нулем; А является фенилом или гетероарилом, где фенил или гетероарил являются необязательно замещенными тремя или менее R2; или к его фармацевтически приемлемой соли. В одном из вариантов осуществления изобретения соединение выбирают из одного из конкретных соединений, раскрытых в подробном описании изобретения. В варианте осуществления настоящего изобретения соединение является энантиомерно чистым. В-1 011029 другом варианте осуществления изобретения соединение является диастереомерно чистым. В дополнительном варианте осуществления изобретения соединение является энантиомерно и диастереомерно чистым. Настоящее изобретение дополнительно относится к фармацевтической композиции, которая содержит терапевтически эффективное количество соединения настоящего изобретения и фармацевтически приемлемый носитель. Настоящее изобретение дополнительно относится к фармацевтической композиции, получаемой смешением соединения настоящего изобретения и фармацевтически приемлемого носителя. Настоящее изобретение также относится к способу получения фармацевтической композиции, содержащему смешение соединения настоящего изобретения с фармацевтически приемлемым носителем. Кроме того, изобретение относится к способу лечения пациента, страдающего от аффективных расстройств, выбранных из группы, состоящей из депрессии, глубокой депрессии, маниакальнодепрессивного психоза, агорафобии, специфической фобии, социофобии, навязчивого состояния, посттравматического синдрома, острого стрессового расстройства и беспокойства, включающему введение пациенту терапевтически эффективного количества соединения изобретения. В отдельном варианте осуществления изобретения заболеванием является депрессия или состояние тревоги. Кроме того, изобретение также относится к способу лечения пациента, страдающего от нарушений мочеиспускания, выбранного из группы, состоящей из энуреза, позыва к мочеиспусканию, частого мочеиспускания, недержания мочи, никтурии и энуреза, включающему введение пациенту терапевтически эффективного количества соединения изобретения. В отдельном варианте осуществления изобретения заболеванием является позыв к мочеиспусканию. Изобретение относится также к способу лечения пациента, страдающего расстройством питания,выбранным из группы, состоящей из ожирения, булимии, нейрогенной булимии и нейрогенной анорексии, включающему введение пациенту терапевтически эффективного количества соединения изобретения. В отдельном варианте осуществления изобретения заболеванием является ожирение. В настоящем изобретении термин линейный или разветвленный C1-C7 алкил относится к насыщенному углеводороду, имеющему от одного до семи углеродных атомов включительно. Примерами таких заместителей являются, но этим не ограничиваясь, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2 бутил, 2-метил-2-пропил и 2-метил-1-пропил. Термин линейный или разветвленный C1-C7 алкилокси относится к насыщенной алкоксигруппе, имеющей от одного до семи углеродных атомов включительно,со свободной валентностью на кислороде. Примеры таких заместителей включают, но этим не ограничиваясь, метокси, этокси, н-бутокси и так далее. Термин C3-C6 циклоалкил-C1-C7 алкил обозначает насыщенный алкилуглеводород, замещенный моноциклическим карбоциклическим кольцом, имеющий от трех до семи углеродных атомов, присоединенных к C1-C7 алкильному фрагменту. Примеры таких заместителей включают, но этим не ограничиваясь, циклопропилметил, циклопентилэтил, циклогексил-нпропил и так далее. Используемый в настоящем изобретении термин "гетероарил относится к 5- или 6-членным ненасыщенным циклам, которые содержат один или более атомов кислорода, серы или азота. Примеры гетероарильных групп включают, но этим не ограничиваясь, фуранил, тиенил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, оксадиазолил, триазолил, тиадиазолил, пиридил,пиридинил, пиридазинил, пиримидинил, пиразинил и триазинил. Изобретение предлагает каждый чистый стереоизомер любого описанного здесь соединения. Такие стереоизомеры могут включать энантиомеры, диастереомеры, или Е- или Z-изомеры алкенов или иминов. Изобретение также относится к стереоизомерным смесям, включая рацемические смеси, диастереомерные смеси или E/Z изомерные смеси. Стереоизомеры могут быть синтезированы в чистой форме(Nogradi, M.; Stereoselective Synthesis, (1987) VCH Editor Ebel, H. and Asymmetric Synthesis, Volumes 3 В 5, (1983) Academic Press, Editor Morrison, J.), или они могут быть выделены различными способами, такими как кристаллизация и хроматографические методы (Jaques, J.; Collet, A.; Wilen, S.; Enantiomer, Racemates. and Resolutions, 1981, John Wiley and Sons and Asymmetric Synthesis, Vol. 2, 1983, Academic Press,Editor Morrison, J). Кроме того, соединения настоящего изобретения могут присутствовать в виде смеси энантиомеров, диастереомеров или изомеров. Более того, два или более соединений могут присутствовать в виде рацемической или диастереомерной смеси. Предпочтительно, чтобы чистота соединения настоящего изобретения достигала 80%, более предпочтительно 90% и наиболее предпочтительно 95%. Это изобретение включает фармацевтически приемлемые соли и комплексы всех описанных здесь соединений. Кислоты и основания, из которых эти соли получают, включают, но этим не ограничиваясь, перечисленные здесь кислоты и основания. Кислоты включают, но этим не ограничиваясь, следующие неорганические кислоты: соляную кислоту, бромистоводородную кислоту, йодисто-водородную кислоту, серную кислоту и борную кислоту. Кислоты включают, но этим не ограничиваясь, следующие органические кислоты: уксусную кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, малеиновую кислоту, лимонную кислоту, метансульфоновую кислоту, бензойную кислоту, гликолевую кислоту, молочную кислоту и миндальную кислоту. Основания включают, но этим не ограничиваясь, аммиак, метиламин, этиламин, пропила-2 011029 мин, диметиламин, диэтиламин, триметиламин, триэтиламин, этилендиамин, гидроксиэтиламин, морфолин, пиперазин и гуанидин. Это изобретение дополнительно предлагает гидраты и полиморфы всех описанных здесь соединений. Объем настоящего изобретения включает пролекарства соединений изобретения. Обычно такие пролекарства являются функциональными производными соединений изобретения, которые легко превращаются in vivo в требуемое соединение. Таким образом, в настоящем изобретении термин "введение" обозначает лечение различных описанных состояний с помощью конкретно раскрытого соединения, или соединения, которое не может быть конкретно раскрыто, но которое превращается в конкретное соединение in vivo после введения пациенту. Традиционные методики для выбора и приготовления соответствующих производных пролекарств описаны, например, в Design of Prodrugs, ed. H. Bundgaard, Elsevier,1985. Настоящее изобретение дополнительно включает метаболиты соединений настоящего изобретения. Метаболиты включают активные вещества, продуцируемые при введении соединений этого изобретения в биологическую среду. Как уже упоминалось в сущности изобретения, настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения изобретения и фармацевтически приемлемый носитель. В одном из вариантов осуществления изобретения количество соединения составляет от около 0,01 до около 800 мг. В другом варианте осуществления изобретения количество соединения составляет от около 0,01 до около 500 мг. В еще одном варианте осуществления изобретения количество соединения составляет от около 0,1 до около 250 мг. В другом варианте осуществления изобретения количество соединения составляет от около 0,1 до около 60 мг. В еще одном варианте осуществления изобретения количество соединения составляет от около 1 до около 20 мг. В дополнительном варианте осуществления изобретения носителем является жидкость и композицией является раствор. В другом варианте осуществления изобретения носителем является твердое вещество и композицией является таблетка. В другом варианте осуществления изобретения носителем является гель и композиция представляет капсулу, суппозиторий или крем. В дополнительном варианте осуществления изобретения соединение может входить в рецептуру как часть фармацевтически приемлемого трансдермального пластыря. В еще одном варианте осуществления изобретения соединение может вводиться пациенту посредством распыления или ингаляции. Настоящее изобретение также относится к фармацевтической композиции, полученной смешением терапевтически эффективного количества соединения изобретения и фармацевтически приемлемого носителя. Изобретение относится к способу получения фармацевтической композиции, включающей смешение терапевтически эффективного количества соединения изобретения и фармацевтически приемлемого носителя. Твердый носитель может включать одно или более веществ, которые могут также действовать как эндогенные носители (например, носители в виде питательного вещества или питательного микроэлемента), ароматизирующие вещества, скользящие вещества, солюбилизаторы, суспендирующие вещества,наполнители, вещества, способствующие скольжению, вещества, способствующие прессованию, связующие или средства, разлагающие таблетку; это может быть также инкапсулированный материал. В случае порошков носителем является тонко измельченное твердое вещество, которое смешивают с тонко измельченным действующим ингредиентом. В случае таблеток действующий ингредиент смешивают с носителем, обладающим необходимыми свойствами спрессовываться в соответствующих пропорциях и спрессовываться в желаемую форму и размер. Предпочтительно, чтобы порошки и таблетки содержали до 99% активного ингредиента. Подходящие твердые носители включают, например, фосфат кальция,стеарат магния, тальк, сахара, лактозу, декстрин, крахмал, желатин, целлюлозу, поливинилпирролидон,низкоплавкие воски и ионообменные смолы. Жидкие носители применяют для получения растворов, суспензий, эмульсий, сиропов, эликсиров и композиций под давлением. Активный ингредиент может быть растворен или суспендирован в фармацевтически приемлемом жидком носителе, таком как вода, органический растворитель, смесь фармацевтически приемлемых масел или жиров. Жидкий носитель может содержать другие подходящие фармацевтические добавки, такие как солюбилизаторы, эмульгаторы, буферы, консерванты, подслащивающие вещества, ароматизирующие вещества, суспендирующие вещества, загустители, окрашивающие вещества, регуляторы вязкости, стабилизаторы или осморегуляторы. Примеры подходящих жидких носителей для перорального и парентерального введения включают воду (частично содержащую вышеназванные добавки, например производные целлюлозы, предпочтительно раствор натрий-карбоксиметилцеллюлозы), спирты (включая одноатомные и полиатомные спирты, например гликоли) и их производные, и масла (например, фракционированные кокосовое масло и арахисовое масло). Для парентерального введения носителем может также быть масляный эфир, такой как этилолеат или изопропилмиристат. Стерильные жидкие носители применяют в стерильных жидких формах композиций для парентерального введения. Жидкий носитель для композиций, находящихся под давлением, может быть галогенированным углеводородом или другим фармацевтически приемлемым пропеллентом. Жидкие фармацевтические композиции, которые являются стерильными растворами или суспензиями, могут применяться, например, путем внутримышечных, интратекальных, эпидуральных, интраперитонеальных или подкожных инъекций. Стерильные растворы могут также быть введены внутривенно.-3 011029 Соединения могут быть приготовлены как стерильная твердая композиция, которая может быть растворена или суспендирована в момент введения с использованием стерильной воды, физиологического раствора или других соответствующих стерильных сред для инъекций. Подразумевается, что носители включают необходимые инертные связующие, суспендирующие вещества, вещества, способствующие скольжению, ароматизирующие вещества, подслащивающие вещества, консерванты, красители и покрытия. Соединение может быть введено перорально в виде стерильного раствора или суспензии, содержащего другие растворенные вещества или суспендирующие вещества (например, достаточное количество физиологического раствора или глюкозы, чтобы сделать раствор изотоническим), соли желчных кислот,гуммиарабик, желатин, сорбитмоноолеат, полисорбат 80 (олеатные эфиры сорбита и его ангидриды, сополимеризованные с окисью этилена) и другие подобные. Соединение может быть также введено перорально в виде или жидкой, или твердой композиции. Композиции, подходящие для перорального введения, включают твердые формы, такие как пилюли, капсулы, гранулы, таблетки и порошки, и жидкие формы, такие как растворы, сиропы, эликсиры и суспензии. Формы, подходящие для парентерального введения, включают стерильные растворы, эмульсии и суспензии. Оптимальные дозы для введения могут быть определены специалистами в этой области, и они будут меняться в зависимости от того, какое соединение используется, силы препарата, способа введения и развития болезненного состояния. Дополнительные факторы, зависящие от подвергаемого лечению конкретного пациента, будут приводить к необходимости корректировки дозы, включая возраст пациента, вес, пол, питание и время введения. Применительно к пациенту, "терапевтически эффективное количество" означает любое количество соединения, которое при введении пациенту, страдающему от заболевания, против которого соединения являются эффективными, вызывает восстановление, ремиссию или регрессию заболевания. Применительно к пациенту, "пациент" означает позвоночное, млекопитающее или человек. Настоящее изобретение относится к способу лечения повышенной активности мочевого пузыря с симптомами позыва к непроизвольному мочеиспусканию, недержания и/или частоты мочеиспускания у пациента, включающему введение пациенту количество соединения изобретения, которое является эффективным для лечения повышенной активности мочевого пузыря у пациента. Это изобретение также предлагает способ облегчения позыва к непроизвольному мочеиспусканию у пациента, страдающего повышенной активностью мочевого пузыря, включающий введение пациенту количество соединения изобретения, которое является эффективным для облегчения состояния позыва к непроизвольному мочеиспусканию у пациента. Это изобретение дополнительно предлагает способ облегчения состояния недержания мочи у пациента, страдающего повышенной активностью мочевого пузыря, который содержит введение пациенту количества соединения изобретения, которое является эффективным для облегчения состояния недержания мочи у пациента. Кроме того, это изобретение предлагает способ облегчения состояния повышенной частоты мочеиспускания у пациента, страдающего повышенной активностью мочевого пузыря, который содержит введение пациенту количества соединения изобретения, которое является эффективным для облегчения состояния повышенной частоты мочеиспускания у пациента. Настоящее изобретение также относится к способу лечения пациента, страдающего от нарушений мочеиспускания, который включает введение пациенту количества соединения изобретения, которое является эффективным для лечения нарушений мочеиспускания у пациента. В некоторых вариантах осуществления изобретения нарушением мочеиспускания является непроизвольное мочеиспускание, гиперактивный мочевой пузырь, неопределимый позыв к мочеиспусканию, частое мочеиспускание, недержание мочи,никтурия или энурез. Гиперактивный мочевой пузырь и недержание мочи могут или не могут быть связаны с доброкачественной гиперплазией предстательной железы. Настоящее изобретение относится к способу облегчения симптомов заболевания у пациента, которое восприимчиво к лечению антагонистомMCH1 рецептора, включающему введение пациенту количества MCH1 антагониста, которое эффективно для облегчения симптомов, в котором MCH1 антагонистом является любое из соединений изобретения. В варианте осуществления изобретения пациентом является позвоночное, млекопитающее, человек или собака. В другом варианте осуществления изобретения соединение вводят перорально. В еще одном варианте осуществления изобретения соединение вводят в комбинации с пищей. Это изобретение относится к способу регулирования режима питания пациента, включающему введение пациенту количества соединения изобретения, эффективного для снижения потребления пищи пациентом. Это изобретение также относится к способу лечения расстройства питания у пациента, включающему введение пациенту количества соединения этого изобретения, эффективного для снижения потребления пищи пациентом. В варианте осуществления настоящего изобретения расстройством питания является булимия, ожирение или нейрогенная булимия. В варианте осуществления настоящего изобретения пациентом является позвоночное, млекопитающее, человек или собака. В дополнительном варианте осуществления изобретения соединение вводят в комбинации с пищей. Настоящее изобретение дополнительно предлагает способ уменьшения массы тела пациента, включающий введение пациенту количества соединения изобретения,эффективного для уменьшения массы тела пациента. Настоящее изобретение также относится к способу лечения пациента, страдающего от глубокой депрессии, психической депрессии, маниакально-депрессивного психоза I и II, шизоаффективного психоза,-4 011029 когнитивных заболеваний с депрессивным настроением, расстройства личности, бессонницы, сонливости, нарколепсии, циркадного ритма сна, ночных кошмаров, нарушения сна, связанного со страхом, лунатизма, навязчивого состояния, расстройства панического типа, с или без агорафобии, посттравматического стресса, социальной тревоги, социофобии и генерализованного тревожного расстройства. Настоящее изобретение также относится к способу лечения пациента, страдающего депрессией, включающему введение пациенту количества соединения этого изобретения, эффективного для лечения депрессии у пациента. Настоящее изобретение дополнительно относится к способу лечения пациента, страдающего от состояния тревоги, включающему введение пациенту количества соединения этого изобретения, эффективного для лечения состояния тревоги у пациента. Настоящее изобретение также относится к способу лечения пациента, страдающего от депрессии и состояния тревоги, включающему введение пациенту количества соединения этого изобретения, эффективного для лечения депрессии и состояния тревоги у пациента. Кроме того, изобретение относится к конкретным вариантам осуществления настоящего изобретения. В одном варианте осуществления изобретения n является целым числом от 2 до 5 включительно. В другом варианте осуществления n равно 2 или 3. В одном варианте осуществления изобретения каждый R1 является независимо -Н, -F, -Cl, линейным или разветвленным C1-C4 алкилом и -алкокси или C3-C6 циклоалкил-C1-C4 алкилом. В одном варианте осуществления изобретения каждый R2 является независимо -Н, -F, -Cl или линейным или разветвленным C1-C4 алкилом, монофторалкилом или полифторалкилом. В одном варианте осуществления изобретения один X является N. В одном варианте осуществления изобретения каждый X является С. В одном варианте осуществления изобретения А является пиридинилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения А является тиенилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения А является фуранилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения А является триазолилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения является имидазолилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения А является пиразолилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения А является оксазолилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения А является триазинилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения А является фенилом, необязательно замещенным тремя или менее R2. В одном варианте осуществления изобретения Z является S, SO или SO2. В одном варианте осуществления изобретения Z является CH2, CO или CHOH. В одном варианте осуществления изобретения Z является O или нулем. В одном варианте осуществления изобретения В является CH2. В одном варианте осуществления изобретения Z является O. В одном варианте осуществления изобретения Z является нулем. В одном варианте осуществления изобретения Z является CO. В одном варианте осуществления изобретения Z является нулем. Изобретение будет легче понять из приводимых далее деталей эксперимента. Однако специалист в этой области отчетливо осознает, что конкретные способы и обсуждаемые в описании результаты просто иллюстрируют изобретение, описанное более полно в формуле изобретения, которая следует далее.I. Схемы синтезов Схема 1(b) Сочетание на палладии или Cu. Для синтеза бифенила, простых эфиров диарила и тиоэфира диарила исходные реагенты коммерчески доступны или в качестве альтернативы могут быть получены из различных промежуточных соединений, известных специалистам в этой области. Дополнительную информацию можно найти в следующих источниках: Suzuki, 1995, Chem. Rev. 95, 2457; Suzuki, 1999, J. Organomet. Chem. 576 (1-2), 147-168; Для цинкорганических реагентов исходные вещества коммерчески доступны или в качестве альтернативы могут быть получены из различных промежуточных соединений, известных специалистам в этой области. Дополнительную информацию можно найти в следующих источниках: Rieke, 1991, J. Org.Chem. 56, 1445; Rieke, 1997, Tetrahedron 53, 1925 и приведенных здесь ссылках. Для реакции Виттига исходные реагенты коммерчески доступны или в качестве альтернативы могут быть получены из различных промежуточных соединений, известных специалистам в этой области. Дополнительную информацию можно найти в следующих источниках: Fesik, 1997, J. Med. Chem. 40, 3144-3150 и приведенных здесь ссылках. Схема 7-7 011029 Для синтезов бифенила, простых эфиров диарила и диарилтиоэфира исходные реагенты коммерчески доступны или в качестве альтернативы могут быть получены из различных промежуточных соединений, известных специалистам в этой области. Дополнительную информацию можно найти в следующих источниках: Suzuki, 1995, Chem. Rev. 95, 2457; Suzuki, 1999, J.Organomet. Chem. 576(1-2), 147-168;Schopfer, 2001, Tetrahedron, 57, 3069- 3073; Venkataraman, 2002, Org. Lett. 16, 2803-2806; Hartwig, 1998,Angew. Chem. Int. Ed. 37, 2046-2067 и приведенных здесь ссылках. Для синтезов сочетанием по Ульману исходные реагенты коммерчески доступны или в качестве альтернативы могут быть получены из различных промежуточных соединений, известных специалистам в этой области. Дополнительную информацию можно найти в следующих источниках: Song, 2002, Org. Промежуточные соединения биарила, используемые в схеме 9, могут в качестве альтернативы быть синтезированы по реакции Фриделя-Крафтса, используя активированные карбоновые кислоты и биарильные системы, как изображено на схеме 10. Схема 11Y1=B(OH)2 или боронат; Y2=Br, I, OTf Биарилы, изображенные на схеме 11, могут быть синтезированы по схеме Сузуки, используя бороновые кислоты/боронаты и арилгалогениды/трифлаты, как описано в схеме 11.II. Подробные примеры синтезов Следующие примеры иллюстрируют способы, применяемые для получения соединений изобретения. Общие методы. Все реакции проводили в атмосфере азота, и реагенты, неразбавленные или в соответствующих растворителях, вводили в реакционный сосуд с помощью шприца и канюли. Безводные растворители приобретали у фирмы Aldrich Chemical Company и использовали без дополнительной обработки. Описанные в патенте примеры называли с использованием программы ACD/Name Program (version 4.01, Advanced Chemistry Development Inc., Торонто, Онтарио, M5H2L3, Канада). ЯМР спектры на 1H и 13 С регистрировали при 300 и 75 МГц соответственно (GE QE Plus) или при 400 и 100 МГц соответственно(Bruker Avance) в CDCl3 в качестве растворителя и с тетраметилсиланом в качестве внутреннего стандарта, если не указано иначе. Химические сдвигивыражали в ppm, константу взаимодействия (J) выражали в Гц и диаграммы расщепления описывали следующим образом: s=синглет; d=дуплет; t=триплет;q=квартет; квинтет; секстет; септет; br=уширенный; m=мультиплет; dd=дуплет дуплетов; =двойной дуплет дуплетов; dt=дуплет триплетов; td=триплет дуплетов; dm=дуплет мультиплетов. Элементный анализ проводили при помощи Robertson Microlit Laboratories, Inc. Если не указано иначе, масс-спектры получали с использованием ионизации электрораспылением (ESMS, Micromass Platform II или Quattro Micro) и молекулярная масса приводятся в виде (М+Н)+ или (М-Н)-. Тонкослойную хроматографию (ТСХ) проводили на стеклянных пластинках с предварительно нанесенным силикагелем 60 F254 (0,25 мм, ЕМSeparations Tech.). Препаративную ТСХ проводили на стеклянных пластинках с предварительно нанесенным силикагелем марки GF (2 мм, Analtech). Колоночную флэш-хроматографию проводили на силикагеле марки Merck silicagel 60 (230-400 меш). Температуры плавления (т.пл.) определяли в открытых капиллярных трубках на приборе Mel-Temp, и эти значения не скорректированы. Эксперименты с СВЧизлучением проводили с использованием аппаратуры Biotage Emyrs Optimize или Smithcreator. Патрон"Bond elute". Патрон, заполненный двуокисью кремния марки Isolute, для твердофазной экстракции с силанольными группами на поверхности частиц двуокиси кремния поставляет фирма Argonaut Technologies. трет-Бутил 4-[(трифторметил)сульфонил]окси-3,6-дигидро-1(2 Н)-пиридинкарбоксилат. н-Бутиллитий (17,6 мл, 44,2 ммоль, 2,5 М в гексанах) добавляли к раствору диизопропиламина (96,2 мл, 44,2 ммоль) в безводном ТГФ (40,0 мл) при 0 С и образующуюся смесь перемешивали в течение 20 мин. Реакционную смесь охлаждали до -78 С и добавляли по каплям трет-бутил 4-оксо-1-пиперидинкарбоксилат(Aldrich Chemical Company, 7,97 г, 40,0 ммоль) в ТГФ (40,0 мл) к реакционной смеси, которую затем перемешивали в течение 30 мин. Добавляли по каплям к реакционной смеси Tf2NPh (42,0 ммоль, 15,0 г) в ТГФ (40,0 мл) и реакционную смесь перемешивали при 0 С в течение ночи. Реакционную смесь концентрировали в вакууме, повторно растворяли в смеси гексан:EtOAc (9:1), пропускали через слой оксида алюминия и слой оксида алюминия промывали смесью гексан:EtOAc (9:1). Объединенные экстракты концентрировали в вакууме с получением целевого продукта (16,5 г), который был загрязнен исходным реагентом Tf2NPh. 1 Н ЯМР (400 МГц, CDCl3)5,77 (с,1 Н), 4,05 (дм, 2 Н, J=3,0 Гц), 3,63 (т, 2 Н, J=5,7 Гц), 2,45 (м, 2 Н),1,47 (с, 9 Н). трет-Бутил 4-(3-аминофенил)-3,6-дигидро-1(2 Н)-пиридинкарбоксилат. Дегазированную смесь 2,0 М водного раствора Na2CO3 (4,20 мл), трет-бутил 4-[(трифторметил)сульфонил]окси-3,6-дигидро-1(2 Н)пиридинкарбоксилата (0,500 г, 1,51 ммоль), 3-аминофенилбороновой кислоты полусульфата (0,393 г,2,11 ммоль), хлорида лития (0,191 г, 4,50 ммоль) и тетракис-трифенилфосфин паладия (0,080 г, 0,075 ммоль) в диметоксиэтане (5,00 мл) нагревали до кипения с обратным холодильником в течение 3 ч в атмосфере аргона. Органический слой охлажденной реакционной смеси отделяли и водный слой промывали этилацетатом (350 мл). Объединенные органические растворы сушили и концентрировали в вакууме. Неочищенный продукт подвергали хроматографии (двуокись кремния, гексаны:EtOAc:дихлорметан 6:1:1 с 1% изопропиламина) с получением требуемого продукта (0,330 г, 81%). 1 Н ЯМР (400 МГц, CDCl3)7,12 (т, 1 Н, J=7,60 Гц), 6,78 (д, 1 Н, J=8,4 Гц), 6,69 (т, 1 Н, J=2,0 Гц), 6,59 трет-Бутил 4-[3-(амино)фенил]-1-пиперидинкарбоксилат. Смесь трет-бутил 4-(3-аминофенил)-3,6 дигидро-1(2 Н)-пиридинкарбоксилата (3,10 г, 11,3 ммоль) и 10% Pd/C (1,00 г) в этаноле (100 мл) гидрировали при комнатной температуре при использовании метода баллона с водородом в течение 2 дней. Реакционную смесь фильтровали через целит и промывали этанолом. Объединенные этанольные экстракты концентрировали в вакууме и остаток подвергали хроматографии на двуокиси кремния (дихлорметан:метанол:изопропиламин 95:5:1) с получением требуемого продукта (2,63 г, 84%). 1 Н ЯМР (400 МГц, CDCl3)7,10 (т, 1 Н, J=7,6 Гц), 6,62 (д, 1 Н, J=8,4 Гц), 6,60-6,59 (м, 2 Н), 4,27-4,18 1-Бром-2,4-дифтор-5-нитробензол. При 0 С к смеси 1-бром-2,4-дифторбензола (20,0 г; 11,7 мл; 0,100 моль) и H2SO4 (76,8 мл) добавляли HNO3 (68,0 мл) в течение 45 мин с такой скоростью, чтобы внутренняя температура была 7 С. Образующуюся смесь перемешивали в течение 1 ч при 0 С, переливали в ледяную воду (400 мл), интенсивно перемешивали в течение 2-3 мин и экстрагировали CH2Cl2(400 мл). CH2Cl2 экстракт промывали рассолом (1500 мл), сушили над Na2SO4, фильтровали и испаряли с получением продукта в виде желтого масла (23,5 г, 95%). 1 Н ЯМР (300 МГц, CDCl3)7,14 (ддд, J=0,3, 7,8, 9,9 Гц, 1 Н), 8,39 (т, J=7,2 Гц, 1 Н). 2-Бром-5-фтор-4-нитротолуол. К нагреваемой с обратным холодильником смеси нитрониума тетрафторбората (11,6 г; 87,0 ммоль) и CH2Cl2 (60,0 мл) добавляли 2-бром-5-фтортолуол (15,0 г, 10,0 мл,79,0 ммоль) в течение 5 мин. Смесь перемешивали при нагревании с обратным холодильником в течение 4,5 ч, охлаждали и переливали в ледяную воду (150 мл). Водную порцию экстрагировали CH2Cl2(1150 мл). Объединенные CH2Cl2 экстракты промывали рассолом (100 мл), сушили над Na2SO4, фильтровали и испаряли с получением 18,3 г неочищенного продукта, который обрабатывали гексаном и испаряли до появления кристаллов. Смесь охлаждали до -70 С и гексан отделяли от образующегося твердого вещества декантацией. Оставшийся гексан удаляли испарением с получением 9,77 г продукта в полутвердом виде (53%). Маточные растворы испаряли и очищали колоночной хроматографией (силикагель,2% EtOAc в гексане). Испарение соответствующих фракций давало 1,0 г продукта (суммарно 59%). 1 Н ЯМР (300 МГц, CDCl3)2,48 (с, 3 Н), 7,20 (д, J=11,7 Гц, 1 Н), 8,26 (д, J=6,9 Гц, 1 Н).(444 мг, 4,53 ммоль), PdCl2dppf (37,0 мг, 3,00 мол.%) и dppf (25,0 мг, 3,00 мол.%), добавляли раствор трет-бутил 4-[(трифторметил)сульфонил]окси-3,6-дигидро-1(2 Н)-пиридинкарбоксилата (500 мг,1,51 ммоль) в 1,4-диоксане (10,0 мл) при комнатной температуре в атмосфере аргона. Смесь нагревали при 80 С в течение ночи. После охлаждения до комнатной температуры смесь фильтровали через целит,и целит промывали EtOAc (320 мл). Фильтраты концентрировали в вакууме. Образующийся остаток растворяли в EtOAc и промывали H2O и рассолом, сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью флэш-хроматографии (1:9 EtOAc:гексан) с получением трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидро-1(2 Н)-пиридинкарбоксилата (355 мг, 76%). 1 трет-Бутил 4-(5-нитро-2-метилфенил)-3,6-дигидро-1(2 Н)-пиридинкарбоксилат. К раствору 2-бром- 10011029 1-метил-4-нитробензола (14,0 г, 64,8 ммоль) и ДМФ (400 мл) добавляли трет-бутил 4-(4,4,5,5-тетраметил 1,3,2-диоксаборолан-2-ил)-3,6-дигидро-1(2 Н)-пиридинкарбоксилат (20,0 г; 64,8 ммоль), K2CO3 (27,0 г; 194 ммоль) и PdCl2dppfCH2Cl2 (3,20 г; 3,90 ммоль; загрузка катализатора 6 мол.%). Образующуюся смесь нагревали при 80 С в атмосфере азота в течение 6 ч, охлаждали до комнатной температуры, давали возможность отстояться в течение 18 ч, затем охлаждали до 4 С. В течение 10 мин добавляли воду (400 мл) с такой скоростью, чтобы температура была 35 С. Добавляли EtOAc (400 мл), смесь перемешивали в течение 15 мин и слой EtOAc удаляли. Повторяли эту процедуру экстракции EtOAc (2400 мл). Органические экстракты объединяли, промывали водой (800 мл) и насыщенным водным раствором NaCl(320 мл), фильтровали через Celite, сушили над MgSO4, фильтровали и испаряли с получением темного остатка, который очищали колоночной хроматографией (силикагель, 70:30 гексан/EtOAc). Испарение соответствующих фракций давало 22,0 г продукта в виде твердого вещества, который использовали на следующей стадии. 1 трет-Бутил 4-(5-амин-2-метилфенил)-1-пиперидинкарбоксилат. Смесь трет-бутил 4-(5-нитро-2-метилфенил)-3,6-дигидро-1(2 Н)-пиридинкарбоксилата (22,0 г), этанола (абсолютный, 300 мл) и 10% Pd-C (2,00 г) выдерживали при 55-60 psi в атмосфере водорода в течение 66 ч. Смесь фильтровали и концентрировали с получением неочищенного зеленого масла (55% конверсии согласно 1 Н ЯМР). К раствору масла и этанола (300 мл) добавляли 10% Pd-C (2,00 г) и образующуюся смесь выдерживали при 55-60 psi в атмосфере водорода в течение 20 ч 15 мин. Смесь фильтровали через Celite и осадок промывали этанолом (200 мл) и EtOAc (100 мл). Фильтрат концентрировали,разбавляли EtOAc (500 мл), сушили над MgSO4, фильтровали и испаряли с получением 18,4 г масла, которое растворяли в EtOAc (10 мл) и гексане (350 мл) и давали возможность отстояться при 5 С в течение 18 ч. Образующуюся смесь фильтровали с получением 13,4 г продукта в виде твердого вещества (выход 71% за две стадии). 1Pd-C (50% воды по весу, 3,20 г) выдерживали при 60 psi в атмосфере водорода в течение 16 ч. Смесь фильтровали через слои Celite и Celite промывали этанолом (425 мл). Фильтрат концентрировали с получением 17,0 г вязкого масла, которое далее сушили под высоким вакуумом в течение нескольких часов. Добавляли гексаны (125 мл) и смесь охлаждали до 5 С в течение 30 мин при интенсивном перемешивании. Образующееся твердое вещество отфильтровывали и твердый осадок промывали маточными растворами, затем холодными гексанами (50 мл) и сушили с получением 15,5 г продукта в виде бежевого твердого вещества (выход 94%). 1(30,0 г; 142 ммоль) и H2SO4 (115 мл) добавляли HNO3 (68%; 102 мл) в течение 1 ч 25 мин с такой скоростью, чтобы внутренняя температура была 8 С. Образующуюся смесь перемешивали в течение 1 ч 50 мин при 0 С (температура в течение 1 ч 50 мин=4,6 С), переливали на лед (1200 мл) и воду (650 мл), интенсивно перемешивали в течение 30 мин и экстрагировали CH2Cl2 (3600 мл). CH2Cl2 экстракты объединяли, промывали водой (2600 мл), сушили над MgSO4, фильтровали и испаряли с получением продукта в виде прозрачного желтого масла (35,0 г, выход 99%). 1 Н ЯМР (300 МГц, CDCl3)7,01 (ддд, J=2,4, 7,8, 9,3 Гц, 1 Н); 19 3-Амино-1-бром-2,4,6-трифторбензол. Смесь 1-бром-3-нитро-2,4,6-трифторбензола (15,1 г; 59,0 ммоль), этанола (590 мл), циклогексана (177 мл) и Pd(OH)2 (5,90 г) нагревали при 80-85 С (внешняя температура) в атмосфере азота в течение 2 ч 5 мин (Твнутренняя=70 С). Смесь охлаждали до 30 С, фильтровали через Celite и промывали EtOAc (200 мл) и этанолом (200 мл). Желтый фильтрат концентрировали и сушили под высоким вакуумом при 60 С с получением 11 г продукта в виде твердого вещества Бензил 5-бром-3-пиридинилкарбамат. К суспензии 5-бромникотиновой кислоты (20,0 г, 99,0 ммоль) в толуоле (200 мл) добавляли дифенилфосфорилазид (25,6 мл, 118,8 ммоль) и Et3N (16,6 мл,118,8 ммоль). После перемешивания при комнатной температуре в течение 30 мин, добавляли бензиловый спирт (15,4 мл, 148,5 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч, затем нагревали с обратным холодильником в течение ночи. После охлаждения до комнатной температуры реакционную смесь промывали H2O, NaHCO3 и рассолом, сушили над MgSO4 и концентрировали. Очищали флэш-хроматографией (15-50% EtOAc/гексан) с получением 22,2 г (72,5 ммоль, 73%) бензил 5 бром-3-пиридинилкарбамата. 1 Н ЯМР (400 МГц, CDCl3)8,39-8,32 (м, 2 Н), 8,29 (с, 1 Н), 7,45-7,32 (м, 5 Н), 6,94 (с, 1 Н), 5,22 (с,2 Н); 5-(2-Феноксифенил)валериановая кислота. К раствору 5-(2-феноксифенил)-5-оксовалериановой кислоты (Rieke Metals, Inc.) (500 мг, 1,8 ммоль) в уксусной кислоте (4 мл) при комнатной температуре добавляли Pd/C (10%, 50 мг) и 0,2 мл HCl (концентрированная). Образующуюся смесь затем гидрировали(комнатная температура, 100 psi, в течение ночи). Реакционную смесь фильтровали через целит и растворитель удаляли в вакууме. Остаток подвергали воздействию глубокого вакуума для удаления следовых количеств уксусной кислоты. Неочищенный продукт использовали на следующей стадии без какой-либо дополнительной очистки (500 мг, 1,8 ммоль, количественный выход). 1 Н ЯМР (400 МГц, CDCl3)7,35-7,21 (м, 3 Н), 7,19-7,12 (м, 1 Н), 7,11-7,01 (м, 2 Н), 6,96-6,83 (м, 3 Н),2,65 (т, 2 Н, J=7,2 Гц), 2,34 (т, 2 Н, J=7,2 Гц), 1,73-1,59 (м, 4 Н);(54,1 мг, 0,550 ммоль) и 1,1'-бис-(дифенилфосфино)ферроцендихлорпалладий (5,60 мг, 5 мол.%) в диметилсульфоксиде (1,50 мл) нагревали при 100 С в течение 18 ч. Реакционную смесь концентрировали в вакууме до получения темного масла. Остаток разбавляли EtOAc, фильтровали и фильтрат концентрировали в вакууме до получения неочищенной кислоты (38,0 мг, 95%).[4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]масляная кислота. Смесь 4-(4,4,5,5 тетраметил-1,3,2-диоксаборолан-2-ил)фенола (2,00 г, 10,0 ммоль), этил 4-бромбутаноата (2,00 мл, 14,9 ммоль), K2CO3 (2,40 г, 17,0 ммоль), NH4I (1,00 г, 7,00 ммоль) и ацетонитрила (14 мл) нагревали при 140 С в целом в течение 90 мин с периодическим контролем. Реакционную смесь распределяли между водой (100 мл) и диэтиловым эфиром (50 мл), отделяли и промывали диэтиловым эфиром (50 мл). Объединенные органические экстракты сушили над сульфатом натрия, затем концентрировали в вакууме с получением желтого масла. Неочищенный продукт очищали с помощью патрона "Bond elute" (70 г, проводили дважды), элюируя пентаном, затем смесью пентан:диэтиловый эфир 4:1, затем смесью пентан:диэтиловый эфир 1:1 с получением этил 4-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)фенокси]бутаноата в виде бесцветного масла, (1,17 г, 35%). 1H ЯМР (CDCl3)7,73 (м, 2 Н), 6,87 (м, 2 Н), 4,14 (кв, J=7,1 Гц, 2 Н), 4,03 (т, J=6,1 Гц, 2 Н), 2,51 (т,J=7,3 Гц, 2 Н), 2,11 (м, 2 Н), 1,33 (с, 12 Н), 1,25 (т, J=7,1 Гц, 3 Н). Этил 4-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]бутаноат (1,17 г, 3,50 ммоль) растворяли в этаноле (10 мл) и добавляли гидроксид лития (1 М, 10 мл). Образующуюся смесь перемешивали при температуре окружающей среды в течение 1 ч, затем подкисляли соляной кислотой (1 М, 25,0 мл). Водную смесь экстрагировали EtOAc (320 мл) и объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме с получением 4-[4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)фенокси]масляной кислоты в виде белого твердого вещества (0,906 г, 96%). 1 Н ЯМР (CDCl3)7,65 (м, 2 Н), 6,89 (м, 2 Н), 4,04 (т, J=6,2 Гц, 2 Н), 2,48 (т, J=7,3 Гц, 2 Н), 2,06 (м, 2 Н),1,32 (с, 12 Н). 4-(4-Йодфенокси)масляную кислоту получали из 4-йодфенола и этил 4-бромбутаноата по аналогичной методике, использованной для получения 4-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)фенокси]масляной кислоты. 1 Н ЯМР (CD3OD)7,54 (м, 2 Н), 6,73 (м, 2 Н), 3,99 (т, J=6,3 Гц, 2 Н), 2,47 (т, J=7,3 Гц, 2 Н), 2,04 (м,2 Н).(15 мл). Трихлорид алюминия (2,99 г, 22,4 ммоль) добавляли порциями к реакционной смеси, перемешивание продолжалось в течение 16 ч при температуре окружающей среды, и затем смесь выливали на измельченный лед (50 мл). Реакционной смеси давали возможность постоять в течение 1 ч, двухфазную смесь разделяли и водную фазу экстрагировали дихлорметаном. Объединенные органические экстракты промывали соляной кислотой (2, 0,1 М), затем раствором карбоната натрия (2, 5%) и сушили над сульфатом магния и концентрировали в вакууме с получением твердого вещества бледно-желто-коричневого цвета. Неочищенный продукт растворяли в тетрагидрофуране (10 мл) и гидроксид лития (0,82 М, 5 мл) добавляли и смесь перемешивали в течение 2 ч. Реакционную смесь концентрировали в вакууме и водный остаток экстрагировали дихлорметаном, затем EtOAc, затем подкисляли соляной кислотой (0,1M). Водную фазу дополнительно экстрагировали EtOAc (3), объединенные органические экстракты сушили над сульфатом магния и концентрировали в вакууме с получением 6-оксо-6-(4-фенилфенил)гексановой кислоты в виде твердого вещества кремового цвета (0,220 г, 21%).ESMS m/e: 283,2 (M-Н)-; 1 Н ЯМР (CDCl3)8,03 (м, 2 Н), 7,68 (м, 2 Н), 7,63 (м, 2 Н), 7,47 (м, 2 Н) 7,40 (м, 1 Н), 3,04 (т, J=7,0 Гц,2 Н), 2,44 (т, J=7,2 Гц, 2 Н), 1,84 (м, 2 Н), 1,77 (м, 2 Н). 6-Оксо-6-(4-фенилфенил)гексановую кислоту (0,220 г, 0,780 ммоль) растворяли в трифторуксусной кислоте (5 мл) и добавляли по каплям триэтилсилан (0,310 мл, 1,25 ммоль). Реакционную смесь нагревали при 55 С, в атмосфере азота в течение 72 ч, затем охлаждали до температуры окружающей среды и концентрировали в вакууме с получением маслянистого твердого вещества. Неочищенный продукт распределяли между насыщенным водным раствором карбоната натрия и диэтиловым эфиром. Водный слой подкисляли соляной кислотой, экстрагировали диэтиловым эфиром (310 мл), объединенные органические экстракты сушили над сульфатом магния и концентрировали в вакууме с получением 6-(4 фенилфенил)гексановой кислоты в виде твердого вещества бледно-желто-коричневого цвета (105 мг,50%). 5-4-[4-(Трифторметил)фенил]фенилпентановая кислота. Смесь 5-(4-бромфенил)пентановой кислоты (300 мг, 1,17 ммоль), 4-(трифторметил)бензолбороновой кислоты (243 мг, 1,28 ммоль), карбоната натрия (154 мг, 1,41 ммоль), воды (1,55 мл) и изопропанола (0,220 мл) нагревали при 150 С в течение 10 мин. Реакционную смесь распределяли между водой и EtOAc, органическую фазу отделяли, сушили над сульфатом магния и концентрировали в вакууме с получением 5-4-[4-(трифторметил)фенил]фенилпентановой кислоты в виде твердого вещества (120 мг, 32%).CH2Cl2/ДМФ (10:1) перемешивали в течение 15 мин с последующим добавлением трет-бутил 4-(5-амино 2-фторфенил)-1-пиперидинкарбоксилата (0,17 ммоль, 50 мг). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Реакционная смесь непосредственно (без какой-либо обработки) была подвергнута препаративной ТСХ (силикагель, 1:1 гексан/EtOAc) с получением трет-бутил 4-2 фтор-5-[5-(4-фенилфенил)пентаноиламино]фенилпиперидинкарбоксилата. 1 Н ЯМР (400 МГц, CDCl3)7,60-7,55 (м, 2 Н), 7,54-7,48 (м, 2 Н), 7,45-7,39 (м, 2 Н), 7,39-7,28 (м, 4 Н),7,27-7,22 (м, 2 Н), 6,98-6,91 (м, 1 Н), 4,33-4,13 (ушир., 2 Н), 3,01-2,90 (м, 1 Н), 2,89-2,60 (м, 2 Н), 2,69 (т, 2 Н,J=7,2 Гц), 2,37 (т, 2 Н, J=7,2 Гц), 1,85-1,04 (м, 9 Н);- 15011029 трет-Бутил 4-2-фтор-5-[5-(4-фенилфенил)пентаноиламино]фенилпиперидинкарбоксилат с предыдущей стадии обрабатывали 4 М HCl в диоксане (1 мл) при комнатной температуре в течение 2 ч. Удаление растворителя в вакууме и очистка на силикагеле ТСХ (силикагель, 2 М NH3/MOH:EtOAc, 1:4)ESI-MS m/e: 431,3 (М+Н)+. Следующие соединения были получены аналогично. 5-(2-Феноксифенил)-N-(3-(4-пиперидил)фенил)пентанамид. Получали согласно методикам, приведенным на схеме 9.N-(4-Метил-3-(4-пиперидил)фенил)-5-(2-феноксифенил)пентанамид. Получали согласно методикам, приведенным на схеме 9.N-(2-Фтор-4-метил-5-(4-пиперидил)фенил)-5-(2-феноксифенил)пентанамид. Получали согласно методикам, приведенным на схеме 9.N-(2-Фтор-4-метил-5-(4-пиперидил)фенил)-5-(4-фенилфенил)пентанамид. Получали согласно методикам, приведенным на схеме 9.N-(4-Фтор-3-(4-пиперидил)фенил)-4-оксо-4-(4-фенилфенил)бутанамид. Получали согласно методикам, приведенным на схеме 9.N-(2-Фтор-5-(4-пиперидил)фенил)-4-оксо-4-(4-фенилфенил)бутанамид. Получали согласно методикам, приведенным на схеме 9.N-(4-Фтор-3-(4-пиперидил)фенил)-5-оксо-5-(4-фенилфенил)пентанамид. Получали согласно методикам, приведенным на схеме 9.N-(4-Метил-3-(4-пиперидил)фенил)-5-оксо-5-(4-фенилфенил)пентанамид. Получали согласно методикам, приведенным на схеме 9.N-(2-Фтор-5-(4-пиперидил)фенил)-5-оксо-5-(4-фенилфенил)пентанамид. Получали согласно методикам, приведенным на схеме 9.N-(2,4-Дифтор-5-(4-пиперидил)фенил)-4-оксо-4-(4-фенилфенил)бутанамид. Получали согласно методикам, приведенным на схеме 9.N-(4-Метил-3-(4-пиперидил)фенил)-5-4-[4-(трифторметил)фенил]фенилпентанамид. согласно методикам, приведенным на схеме 9. 4-[4-(3-Фторфенил)фенил]-N-(4-метил-3-(4-пиперидил)фенил)бутанамид. Смесь трет-бутил 4-(3 амино-6-метилфенил)пиперидинкарбоксилата (39,6 мг, 0,130 ммоль), 4-[4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)фенил]масляной кислоты (38,0 мг, 0,130 ммоль), [диметиламино-([1,2,3]триазоло[4,5-b]пиридин-3-илокси)метилен]диметиламмонийгексафторфосфата (HATU, 54,3 мг, 0,140 ммоль) и диизопропилэтиламина (68,0 мкл, 0,390 ммоль) в диметилформамиде (2 мл) перемешивали при температуре окружающей среды в течение 18 ч. Реакционную смесь концентрировали в вакууме, растворяли вEtOAc (20 мл) и промывали бикарбонатом натрия (насыщенным, 10 мл) и водой (210 мл), затем сушили над сульфатом натрия и концентрировали в вакууме с получением смолы. Очистка на силикагеле (силикагель 60, 40 мл) с элюированием смесью циклогексан: EtOAc, 8:1, затем 4:1 давала трет-бутил 4-(2 метил-5-4-[4-(4,4,5,5-тетраметил(1,3,2-диоксаборолан-2-илфенил]бутаноиламинофенил)пиперидинкарбоксилат в виде желтого масла (39,0 мг, 53%).(0,8 мл) добавляли к смеси 3-фторбромбензола (6,55 мг, 0,370 ммоль), 1,1'-бис-(дифенилфосфино)ферроцендихлорпалладия (3,30 мг, 8 мол.%) и раствора карбоната цезия (2 М, 50 мкл) и полученную смесь нагревали микроволнами при 110 С в течение 25 мин. Реакционную смесь концентрировали в вакууме, затем распределяли между водой (20 мл) и EtOAc (210 мл). Органическую фазу отделяли, промывали водой (220 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением смолы. Очистка неочищенного продукта на силикагеле (силикагель 60, 20 мл) с элюированием смесью циклогексан:EtOAc, 85:15, затем 4:1 давала трет-бутил 4-(5-4-[4-(3-фторфенил)фенил]бутаноиламино-2 метилфенил)пиперидинкарбоксилат (13,0 мг, 73%).ESI-MS m/e: 431,3 (М-С 5 Н 8 О 2+Н)+. Полученный трет-бутил 4-(5-4-[4-(3-фторфенил)фенил]бутаноиламино-2-метилфенил)пиперидинкарбоксилат растворяли в дихлорметане (0,8 мл) и трифторуксусную кислоту (95%, 0,8 мл) добавляли и реакционную смесь перемешивали в течение 2 ч, затем концентрировали в вакууме. Остаток по- 18011029 вторно растворяли в ацетонитриле (1 мл), соляную кислоту (1 М, 1 мл) добавляли, и реакционную смесь концентрировали в вакууме с получением гидрохлорида 4-[4-(3-фторфенил)фенил]-N-(4-метил-3-(4 пиперидинил)фенил)бутанамида в виде белого твердого вещества (11,2 мг, 98%).(460 мг, 1,50 ммоль), гидрохлорида 1-[3-диметиламинопропил]-3-этилкарбодиимида (370 мг, 1,90 ммоль) и HOBt (320 мг, 2,30 ммоль) в диметилформамиде (10 мл) перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь распределяли между водой (80 мл) и диэтиловым эфиром(320 мл). Органическую фазу сушили над сульфатом натрия и концентрировали в вакууме с получением желтого масла. Очистка неочищенного продукта с помощью патрона для твердофазной экстракции(5 г) с элюированием пентаном, затем смесью пентан:диэтиловый эфир 1:1, затем смесью 1:4, затем 100% диэтиловым эфиром давала трет-бутил 4-5-[4-(4-йодфенокси)бутаноиламино]-2-метилфенилпиперидинкарбоксилат в виде пены кремового цвета (514 мг, 59%).ESMS m/e: 579,2 (M+Н)+; 1 Н ЯМР (CDCl3)7,54 (м, 2 Н), 7,30 (д, J=2,1 Гц, 1 Н), 7,23 (дд, J=8,2, 2,1 Гц, 1 Н), 7,17 (ушир.с, 1 Н),7,09 (д, J=8,2 Гц, 1 Н), 6,67 (м, 2 Н), 4,26 (ушир.с, 2 Н), 4,03 (т, J=5,9 Гц, 2 Н), 2,80 (м, 3 Н), 2,55 (т, J=7,1 Гц,2 Н), 2,30 (с, 3 Н), 2,20 (м, 2 Н), 1,73 (ушир.д, J=13,6 Гц, 2 Н), 1,58 (м, 2 Н), 1,49 (с, 9 Н). трет-Бутил 4-5-[4-(4-йодфенокси)бутаноиламино]-2-метилфенилпиперидинкарбоксилат и 2 фторбензолбороновая кислота (35 мг, 0,250 ммоль), тетракис-трифенилфосфинпалладий(0) (12,0 мг,10 мол.%), водный раствор карбоната натрия (2M, 1 мл) и диметоксиэтан (2 мл) нагревали СВЧизлучением при 120 С в течение 15 мин. Реакционную смесь разбавляли водой (2 мл), экстрагировали этилацетатом (30,5 мл) и объединенные органические фазы испаряли в токе азота. Полученную смолу очищали в патроне для твердофазной экстракции (5 г) с элюированием смесью циклогексан:этилацетат 9:1, затем смесью 3:2 с получением трет-бутил 4-(5-4-[4-(2-фторфенил)фенокси]бутаноиламино-2 метилфенил)пиперидинкарбоксилата (60,0 мг, количественно).(2 Н, ушир.д, J=12,3 Гц), 1,60 (2 Н, кв. д, J=12,3, 3,8 Гц), 1,48 (9 Н, с). Амид растворяли в дихлорметане (1 мл) и трифторуксусную кислоту (1 мл) добавляли. Полученный раствор взбалтывали при вращении в течение 1 ч, затем испаряли в токе азота с получением масла. Очистка в амино SPE патроне (2 г) с элюированием диэтиловым эфиром, затем этилацетатом, затем 10% метанолом в этилацетате давала 4-[4-(2-фторфенил)фенокси]-N-(4-метил-3-(4-пиперидил)фенил)бутанамид. Его растворяли в метаноле (2 мл) и добавляли соляную кислоту (1M, 3 мл) и полученную смесь концентрировали в вакууме и превращали в порошок с помощью диэтилового эфира, получая 4-[4-(2 фторфенил)фенокси]-N-(4-метил-3-(4-пиперидил)фенил)бутанамид.(д, J=2,0 Гц, 1 Н), 7,49 (м, 3 Н), 7,37 (м, 1 Н), 7,28 (м, 3 Н), 7,07 (д, J=8,6 Гц, 1 Н), 7,04 (м, 2 Н), 4,07 (т, J=6,4 Гц, 2 Н), 3,36 (4 Н, под пиком воды), 3,02 (м, 3 Н), 2,25 (с, 3 Н), 2,05 (квинтет, 2 Н), 1,81 (м, 4 Н). Следующие соединения получали аналогично. 4-[4-(4-Цианофенил)фенил]-N-(4-метил-3-(4-пиперидил)фенил)бутанамид. Получали согласно методикам, приведенным на схеме 11. 4-[4-(2-Цианофенил)фенил]-N-(4-метил-3-(4-пиперидил)фенил)бутанамид. Получали согласно методикам, приведенным на схеме 11.N-(4-Метил-3-(4-пиперидил)фенил)-5-[4-(2-метилфенил)фенил]пентанамид. Получали согласно методикам, приведенным на схеме 11.N-(4-Метил-3-(4-пиперидил)фенил)-4-[4-(3-метилфенил)фенокси]бутанамид. методикам, приведенным на схеме 11. 4-[4-(4-Фторфенил)фенокси]-N-(4-метил-3-(4-пиперидил)фенил)бутанамид. Получали согласно методикам, приведенным на схеме 11. 4-[4-(3-Цианофенил)фенокси]-N-(4-метил-3-(4-пиперидил)фенил)бутанамид. методикам, приведенным на схеме 11.N-(4-Метил-3-(4-пиперидил)фенил)-4-[4-(2-метилфенил)фенокси]бутанамид. методикам, приведенным на схеме 11.N-(2-Фтор-4-метил-5-(4-пиперидил)фенил)-4-(4-фенилфенил)бутанамид. Получали согласно методикам, приведенным на схеме 11.N-(4-Метил-3-(4-пиперидил)фенил)-4-(4-(2-пиридил)фенил)бутанамид. Получали согласно методикам, приведенным на схеме 11.N-(4-Метил-3-(4-пиперидил)фенил)-4-(4-(3-пиридил)фенокси)бутанамид. Получали согласно методикам, приведенным на схеме 11.N-(4-Метил-3-(4-пиперидил)фенил)-5-(4-(4-пиридил)фенил)пентамид. Получали согласно методикам, приведенным на схеме 11.N-(4-Метил-3-(4-пиперидил)фенил)-4-(4-(2-пиридил)фенокси)бутанамид. Получали согласно методикам, приведенным на схеме 11.III. Композиции для перорального введения В качестве конкретного варианта осуществления композиции для перорального введения соединения настоящего изобретения 100 мг одного из описанных соединений смешивают с достаточно тонко измельченной лактозой до достижения суммарного количества от 580 до 590 мг для заполнения твердых желатиновых капсул размера 0.IV. Фармакологическая оценка соединений на клонированном MCH1 рецепторе крыс Фармакологические свойства соединений настоящего изобретения оценивали на клонированномMCH1 рецепторе крыс с использованием описанных ниже протоколов. Клетки-хозяева. Для изучения гетерогенно экспрессивных белков может быть использовано большое разнообразие клеток-хозяев. Эти клетки включают, но этим не ограничиваясь, соответствующие линии млекопитающих, такие как Cos-7, CHO, LM(tk-), HEK293 и Peak rapid 293; клеточные линии насекомых, такие какSf9 и Sf21; клетки земноводных, такие как xenopus oocytes (яйцеклетки лягушки); и другие. Клетки COS 7 выращивают в 150-мм чашках в среде DMEM с добавками (среда Игла в модификации Дульбекко с 10% телячьей сывороткой, 4 мМ глутамина, 100 единиц/мл пеницилина/ 100 фг/мл стрептомицина) при 37 С, 5% CO2. Исходные чашки клеток COS-7 трипсинизируют и разводят 1:6 каждые 3-4 дня. Клетки почки человеческого эмбриона 293 выращивают в 150-мм чашках в среде DMEM с дополнениями (10% телячьей сыворотки, 4 ммоль глутамина, 100 единиц/мл пеницилина/100 фг/мл стрептомицина) при 37 С,5% CO2. Исходные чашки клеток 293 трипсинизируют и разводят 1:6 каждые 3-4 дня. Клетки почки человеческого эмбриона Peak rapid 293 (Peakr293) выращивают на 150-мм чашках в среде DMEM с дополнениями (10% телячьей сыворотки, 10% L-глутамина, 50 фг/мл гентамицина) при 37 С, 5% CO2. Исходные чашки клеток Peak rapid 293 трипсинизируют и разводят 1:12 каждые 3-4 дня. Клетки фибробластов мыши LM (tk-) выращивают в 150-мм чашках в среде DMEM с дополнениями(среда Игла в модификации Дульбекко с 10% телячьей сывороткой, 4 ммоль глутамина, 100 единиц/мл пенициллина/100 фг/мл стрептомицина) при 37 С, 5% CO2. Исходные чашки клеток LM (tk-) трипсинизируют и разводят 1:10 каждые 3-4 дня. Клетки яичников китайского хомячка (CHO) выращивают на 150-мм чашках в среде Хэма F-12 с дополнениями (10% телячья сыворотка, 4 мМ L-глутамина и 100 единиц/мл пеницилина/100 фг/мл стрептомицина) при 37 С, 5% CO2. Исходные чашки клеток CHO трипсинизируют и разводят 1:8 каждые 3-4 дня. Клетки-фибробласты мышиного эмбриона NIH-3T3 выращивают в 150-мм чашках в среде Игла в модификации Дульбекко (DMEM) с дополнениями (с 10% телячьей сывороткой, 4 мМ глутамина, 100 единиц/мл пенициллина/100 фг/мл стрептомицина) при 37 С,5% CO2. Исходные чашки клеток NIH-3T3 трипсинизируют и разводят 1:15 каждые 3-4 дня. Клетки Sf9 иSf21 выращивают в монослоях на 150-мм чашках для тканевых культур в среде TMN-FH, дополненной 10% телячьей сывороткой, при 27 С, без CO2. Клетки насекомых High Five insect выращивают на 150-мм чашках для тканевых культур в среде Ex-Cell 400, дополненной L-глутамином, также при 27 С, безCO2. В некоторых случаях клеточные линии, которые растут в виде сросшихся монослоев, могут быть превращены в суспензионную культуру для увеличения выхода клеток и обеспечения больших партий единообразно испытываемого материала для стандартных проектов скрининга рецептора. Транзиторная экспрессия. Исследуемые белки, кодируемые ДНК, могут быть транзиторно экспрессированы в различных клеточных линиях млекопитающих, насекомых, земноводных и других видов различными методами, включающими, но этим не ограничиваясь, кальций-фосфатный, ДЭАЭ-декстрановый, липосомный, вирусный,электропорации и микроинъекционного введения. Каждый из этих методов может требовать оптимизации соответствующих экспериментальных параметров, зависящих от ДНК, клеточной линии и типа анализа, который будет далее использоваться. Типичный протокол для кальций-фосфатного метода приме- 21011029 нительно к клеткам Peak rapid 293 описывается следующим образом. Сросшиеся клетки собирают приблизительно за двадцать четыре часа до трансфекции и пересевают при плотности 3,5106 клеток/чашка в 150-мм чашку для тканевых культур и инкубируют в течение ночи при 37 С при 5% CO2. 250 фл смесиCaCl2 и ДНК (15 фг ДНК в 250 мМ CaCl2) добавляют в 5 мл пластиковую пробирку и медленно добавляют при несильном перемешивании 500 фл 2 Х HBS (280 мМ NaCl, 10 ммоль KCl, 1,5 мМ Na2HPO4, 12 мМ декстрозы, 50 мМ HEPES). Смесь инкубируют в течение 20 мин при комнатной температуре для образования осадка ДНК. Смесь осадка ДНК затем добавляют в культуральную среду в каждой чашке и инкубируют в течение 5 ч при 37 С, 5% CO2. После инкубации в каждую чашку добавляют 5 мл культуральной среды (DMEM, 10% FBS, 10% L-глутамина и 50 мкг/мл гентамицина). Клетки затем инкубируют в течение от 24 до 48 ч при 37 С, 5% CO2. Типичный протокол для ДЭАЭ-декстранового метода применительно к клеткам Cos-7 описывается следующим образом. Клетки, которые будут использованы для трансфекции, разбавляют за 24 ч до трансфекции для приготовления флаконов, которые заполняются во время трансфекции на 70-80%. Вкратце, 8 фг рецептора ДНК плюс 8 фг любой необходимой дополнительной ДНК (например, G вектора экспрессии белка, репортерной конструкции, маркера устойчивости к антибиотику, пустого вектора и так далее) добавляют к 9 мл полной среды DMEM плюс смесь ДЭАЭдекстран (10 мг/мл в PBS). Клетки Cos-7, высеянные во флакон Т 225 (заполняемый наполовину), промывают один раз PBS и добавляют в каждый флакон смесь ДНК. Клетки инкубируют в течение 30 мин при 37 С, 5% CO2. Вслед за инкубацией, добавляют в каждую колбу 36 мл полной среды DMEM с 80 фМ хлорохина и инкубируют дополнительно в течение 3 ч. Среду затем аспирируют и добавляют 24 мл полной среды, содержащей 10% ДМСО, в течение ровно 2 мин и затем аспирируют. Клетки затем промывают 2 раза PBS и добавляют в каждую колбу 30 мл полной среды DMEM. Клетки затем инкубируют в течение ночи. На следующий день клетки собирают путем трипсинизации и пересеивают при необходимости, в зависимости от типа анализа, который будет применяться. Типичный протокол для липосомного метода трансфекции применительно к клеткам CHO описывается следующим образом. Клетки, которые будут использованы для трансфекции, разводят за 24 ч до трансфекции для приготовления флаконов, которые заполняются во время трансфекции на 70-80%. Суммарно 10 фг ДНК, которое может включать меняющиеся отношения рецептора ДНК плюс любую необходимую дополнительную ДНК (например, G вектора экспрессии белка, репортерной конструкции,маркера устойчивости к антибиотику, пустого вектора и так далее) используют для трансфекции каждого 75 см 2 флакона клеток. Липосомную трансфекцию проводят согласно рекомендациям производителей(LipofectAMINE, GibcoBRL, Bethesda, MD). Подвергнутые трансфекции клетки собирают через 24 ч после трансфекции и используют или пересевают согласно требованиям анализа, который будет использоваться. Типичный протокол для метода электропорации применительно к клеткам Cos-7 описывается следующим образом. Клетки, которые будут использованы для трансфекции, разводят за 24 ч до трансфекции для приготовления флаконов, которые заполняются во время трансфекции наполовину. Клетки собирают путем трипсинизации, ресуспендируют в их среде для выращивания и подсчитывают. 4106 клеток суспендируют в 300 фл среды DMEM и помещают в кювету для электропорации. 8 фг рецептора ДНК плюс 8 фг любой необходимой дополнительной ДНК (например, G вектора экспрессии белка, репортерной конструкции, маркера устойчивости к антибиотику, пустого вектора и так далее) добавляют к клеточной суспензии, кювету помещают в BioRad Gene Pulser и подвергают электрическому импульсу(Gene Pulser установки: 0,25 кВ напряжение, 950 фФ емкость). Вслед за импульсом в каждую кювету добавляют 800 фл полной среды DMEM и суспензию переносят в стерильную пробирку. Полную среду добавляют в каждую пробирку, чтобы довести конечную концентрацию клеток до 1105 клеток/100 фл. Клетки затем при необходимости высевают в зависимости от типа анализа, который будет проводиться. Типичный протокол для вирусной экспрессии гетерологичных белков описывается следующим образом для клеток насекомых Sf9. Кодирующая область ДНК, кодирующая рецептор, раскрытая здесь,может быть субклонирована в pBlueBacIII в существующие сайты рестрикции или сайты, заданные в последовательности 5' и 3' к кодирующей области полипептидов. Для генерации бакуловируса, 0,5 фг вирусной ДНК (BaculoGold) и 3 фг структурного компонента ДНК, кодирующего полипептид, могут быть со-трансфекцированы в 2106 Spodoptera frugiperda клетки насекомых Sf9 методом соосаждения фосфатом кальция, как описано фирмой Pharmingen (в "Baculovirus Expression Vector System: Proceduresand Methods Manual"). Клетки затем инкубируют в течение 5 дней при 27 С. Супернатант посева сотрансфекции может быть собран центрифугированием, и рекомбинационная вирусная бляшка может быть очищена. Методика инфицирования клеток вирусом, приготовления запасов вируса и определение титра запасов вируса является такой, как описано в руководстве фирмы Pharmingen. Аналогичные принципы обычно можно было бы применять к экспрессии клеток млекопитающих с помощью ретровирусов,вируса леса Семлики и двухспиральных ДНК-вирусов, таких как адено-, герпес- и vacinia-вирусы, и другие подобные. Стабильная экспрессия. Гетерологическая ДНК может быть устойчиво введена в клетки-хозяева, заставляя клетку постоянно экспрессировать чужеродный белок. Методы доставки ДНК в клетку являются аналогичными тем,- 22011029 которые описаны выше для транзиторной экспрессии, но требуют сотрансфекции вспомогательного гена,чтобы придать лекарственную устойчивость в заданной клетке-хозяине. Достигаемая лекарственная устойчивость может быть использована для выбора и поддержания клеток, которые приняли гетерологическую ДНК. Ассортимент генов устойчивости является доступным, включая, но этим не ограничиваясь,неомицин, канамицин и гигромицин. Для целей исследований рецептора стабильную экспрессию гетерологического рецептора белка проводят в клетках, но без ограничения, млекопитающих, включая CHO,HEK293, LM (tk-) и так далее. Приготовление клеточной мембраны. Для исследования связывания суспендируют осадки трансфекцированных клеток в буфере, охлажденном льдом (20 мМ TrisHCl, 5 мМ EDTA, pH 7,4) и гомогенизируют с помощью ультразвука в течение 7 с. Клеточные лизаты центрифугируют при 200g в течение 5 мин при 4 С. Супернатанты центрифугируют затем при 40000g в течение 20 мин при 4 С. Образующиеся осадки промывают один раз в буфере для гомогенизации и суспендируют в связывающем буфере (см. методы связывания радиолиганда). Концентрации белков определяют методом Брэдфорда (Bradford, 1976), используя бычий сывороточный альбумин в качестве стандарта. Исследования связывания обычно проводят немедленно, однако можно приготавливать мембраны периодически и хранить их замороженными в жидком азоте для использования в будущем. Исследования по связыванию радиолиганда. Исследования по связыванию радиолиганда для MCH1 рецептора у крыс проводили с использованием плазмиды pcDNA3.1-rMCH1-f (АТСС Patent Deposit Designation No. PTA-3505). ПлазмидаpcDNA3.1-rMCH1-f содержит регулятивные элементы, необходимые для экспрессии ДНК в клетке млекопитающих, оперативно связанные с ДНК, кодирующая MCH1 рецептор крыс так, что приводит к его экспрессии. Плазмида pcDNA3.1-rMCH1-f была депонирована 5 июля 2001 г. в Американской коллекции типовых культур (American Type Culture Collection (ATCC), 12301 Parklawn Drive, Rockville, Maryland 20852, U.S.A.) в соответствии со статьями Будапештского договора о международном признании депонирования микроорганизмов для целей патентной процедуры и ей был присвоен идентификационный номер АТСС Patent Deposit Designation No. PTA-3505. Исследования по связыванию могут также быть проведены, как описано ниже, с использованием плазмиды pEXJ.HR-TL231 (АТСС Accession No. 203197). Плазмида pEXJ.HR-TL231 кодирует MCH1 рецептор человека и была депонирована 17 сентября 1998 г. в Американской коллекции типовых культур (American Type Culture Collection (АТСС), 12301Parklawn Drive, Rockville, Maryland 20852, U.S.A.) в соответствии со статьями Будапештского договора о международном признании депонирования микроорганизмов для целей патентной процедуры и ей был присвоен идентификационный номер АТСС Accession No. 203197. Клетки почки человеческого эмбриона Peak rapid 293 (Peakr293 клетки) кратковременно подвергали трансфекции с ДНК, кодирующей MCH1 рецептор, с использованием кальций-фосфатного метода, и клеточные мембраны готовили так, как описано выше. Эксперименты по связыванию с мембранами клеток Peakr293, подвергнутых трансфекцииMCH1 рецептором крыс, проводили с 0,08 нМ [3 Н]соединения А, используя инкубационный буфер, состоящий из 50 мМ Tris pH 7,4, 10 мМ MgCl2, 0,16 мМ PMSF, 1 мМ 1,10-фенантролина и 0,2% BSA. Связывание проводили при 25 С в течение 90 мин. Инкубации прерывали быстрой фильтрацией в вакууме через фильтры из стекловолокна GF/C, предварительно вымоченные в 5% PEI, используя 50 нМ Tris pH 7,4 в качестве промывного буфера. Во всех экспериментах неспецифическое связывание определяется с использованием 10 пМ меченного тритием метил (4S)-3-[(3-4-[3-(ацетиламино)фенил]-1 пиперидинилпропил)амино]карбонил-4-(3,4-дифторфенил)-6-(метоксиметил)-2-оксо-1,2,3,4 тетрагидро-5-пиримидинкарбоксилата. Синтез этого меченного изотопом соединения описан в патентной заявке WO 03/04027. Данные по результатам связывания. Обозначенный выше метод использовали для идентификации соединений настоящего изобретения в качестве сильнодействующих ингибиторов MCH1 рецептора. Значения Ki для раскрытого соединения изменяются в интервале от 0,1 нМ до 1000 нМ. В одном варианте осуществления изобретения сродство связывания для соединений в отношении MCH1 рецептора составляет от 0,5 нМ до 500 нМ. В одном варианте осуществления изобретения сродство связывания для соединений в отношении MCH1 рецептора составляет от 1,0 нМ до 100 нМ. В другом варианте осуществления изобретения сродство связывания для соединений в отношении MCH1 рецептора составляет от 1,0 до 75 нМ.V. Методы in vivo А. Ожирение. Следующие два (2) метода описывают протоколы, которые могут быть использованы для предсказания эффективности MCH1 антагонистов для лечения ожирения. 1. Влияние MCH1 антагонистов на вес тела (3 дня). Самцов крыс линии Лонг-Эванс (Charles River), весящих 180-200 г, помещают в клетки в группах по четыре штуки на 12-часовой цикл свет/темнота со свободным доступом к пище и воде. Тестируемые соединения вводят дважды в день путем подкожной инъекции за 1 ч до начала темного цикла и через 2 ч после того, как зажигают свет, в течение трех дней. Всех крыс взвешивают ежедневно после каждой утренней инъекции. Суммарные результаты выражают как вес тела (граммы), прибавленный за день (стандартная ошибка среднего), и подвергают двухфакторному дисперсионному анализу ANOVA. Данные для каждой временной точки подвергают однофакторному дисперсионному анализу ANOVA с апостериорным вычислением с использованием критерия Ньюмена-Кеулса. Данные затем анализируют с использованием программы GraphPad Prism (v2.01) (GraphPad Software, Inc., San Diego, CA). 2. Влияние MCH1 антагонистов на потребление подслащенного сгущенного молока. Самцов мышей линии C57BL/6 (Charles River), весящих 17-19 г, в начале экспериментов помещают в клетки в группах по четыре или пять на 12-часовой цикл свет/темнота со свободным доступом к пище и воде. В течение 7 дней мышей взвешивают, помещают в индивидуальные клетки и дают возможность пить подслащенное сгущенное молоко (Nestle, разбавленное 1:3 водой) в течение 1 ч, 2-4 ч во время цикла с освещением. Количество потребленного молока определяют взвешиванием бутылки с молоком до и после каждого приема жидкости. В день теста мышам подкожно вводили тестируемое соединение (3, 10 или 30 мг/кг в 0,01% молочной кислоте), носитель (0,01% молочная кислота), d-фенфлурамина (10 мг/кг в 0,01% молочной кислоты) за 30 мин до доступа к молоку. Количество молока, потребленного в день теста (в мл молока/кг веса тела), сравнивают с базисным потреблением для каждой мыши, определенным в течение предыдущих 2 дней. Данные для каждой временной точки подвергают однофакторному дисперсионному анализу ANOVA. В. Депрессия. Следующий метод описывает протокол, который может быть использован для предсказания эффективности MCH1 антагонистов для лечения депрессии. 1. Тест принудительного плавания (ППТ) на крысах. В качестве животных во всех экспериментах используют самцов крыс линии Sprague Dawley (Taconic Farms, NY). Крыс помещают по 5 штук в клетку и выдерживают на 12:12-часовом цикле светтемнота. Крыс приручали в течение 1 мин каждый день на протяжении 4 дней до поведенческого тестирования. Введение лекарственного препарата. Животных распределяют случайным образом для подкожного введения только носителя (2,5%EtOH/2,5% Tween-80), имипрамина (положительный контроль; 60 мг/кг) или тестируемого соединения за 60 мин до начала 5-минутного периода теста. Все инъекции делаются с использованием 1 см 3 туберкулинового шприца с размерами игл 263/8 (Becton-Dickinson, VWR Scientific, Bridgeport, NJ). Объем инъекции был 1 мл/кг. Экспериментальное проектирование. Методика, используемая в этом исследовании, аналогична той, которая описана ранее (Porsolt, et al.,1978), за исключением того, что глубина воды по этой методике составляет 31 см. Большая глубина в этом тесте не позволяет крысам поддерживать себя за счет касания низа цилиндра лапами. Сеансы плавания проводили путем помещения крыс в индивидуальные цилиндры из плексигласа (46 см высотой 20 см в диаметре), содержащие 23-25 С воду глубиной 31 см. Тесты на плавание проводят всегда между 9.00 и 17.00 часами, и они состоят из исходного 15-минутного установочного теста с последующим на 24 ч позже 5-минутным тестом. Лекарственные препараты вводят за 60 мин до 5-минутного периода теста. После всех сеансов плавания крыс вынимают из цилиндров, сушат бумажными полотенцами и помещают в нагретую клетку в течение 15 мин и возвращают в их персональные клетки. Все сеансы теста снимают и записывают на видеопленку с помощью цветной видеокамеры для дальнейшего скоринга. Поведенческий скоринг. Поведение крыс оценивает с 5-секундными интервалами во время 5-минутного теста специальный человек, которому не известно условие лечения. Подсчитываемыми видами поведения являются: 1) неподвижность - крыса остается плавающей в воде без борьбы и только делает те движения, которые необходимы для поддержания головы над водой; 2) выкарабкивание - крыса делает активные движения передними лапами в направлении в воду и из воды, обычно направленные к стенкам; 3) плавание - крыса делает активные плавательные движения, большие, чем необходимо для того,чтобы просто держать голову над водой, например плавание кругами в цилиндре; и 4) ныряние - тело крысы полностью погружено.- 24011029 Анализ данных. Данные теста принудительного плавания (неподвижность, плавание, выкарабкивание, ныряние) подвергают рандомизированному однофакторному дисперсионному анализу ANOVA с апостериорным вычислением с использованием критерия Ньюмена-Кеулса. Данные анализируют с использованием программы GraphPad Prism (v2.01) (GraphPad Software,Inc., San Diego, CA). 2. Тест принудительного плавания (ППТ) на мышах. Во всех экспериментах в качестве животных используют мышей линии DBA/2 (Taconic Farms, NY). Животных размещают по 5 штук в клетке в регулируемой среде с 12:12-часовым циклом свет:темнота. Животных приручают 1 мин каждый день в течение 4 дней до эксперимента. Это методика включает имитацию принудительного кормления через зонд 1,5 дюйма. Введение лекарственного препарата. Животных распределяют случайным образом для введения только носителя (5% EtOH/5% Tween80), только тестируемого соединения, или имипрамина (60 мг/кг) путем перорального принудительного кормления за 1 ч до плавательного теста. Экспериментальное проектирование. Методика теста принудительного плавания для мышей является аналогичной методике, описанной выше для крыс, с некоторыми модификациями. Цилиндром, используемым для теста, является лабораторный стакан емкостью 1 л (10,5 см диаметр 15 см высота), наполненный до 800 мл (10 см глубина) 2325 С водой. Для каждой мыши проводят только один 5-минутный плавательный тест между 13.00 и 17.00 ч. Лекарственный препарат вводят за 30-60 мин до 5-минутного периода теста. После всех сеансов плавания мышей удаляют из цилиндров, сушат бумажными полотенцами и помещают в нагретую клетку на 15 мин. Все сеансы теста снимают и записывают на видеопленку с помощью цветной видеокамерыSony для дальнейшего скоринга. Поведенческий скоринг. Поведение во времени от 2 до 5 мин теста воспроизводится на мониторе телевизора и обсчитывается исследователем. Суммарное время, потраченное на неподвижность (животное, плавающее только с минимальными движениями для того, чтобы остаться на плаву) и подвижность (плавание и движения выше тех, которые необходимы для того, чтобы оставаться на плаву), записывается. Анализ данных. Данные теста принудительного плавания (время проявления неподвижности, подвижности; секунды) подвергают рандомизированному однофакторному дисперсионному анализу ANOVA с апостериорным вычислением с использованием критерия Ньюмена-Кеулса. Данные анализируют с использованием программы GraphPad Prism (v2.01) (GraphPad Software,Inc., San Diego, CA) С. Состояние тревоги. Следующий метод описывает протокол, который может быть использован для предсказания эффективности MCH1 антагонистов для лечения состоянии тревоги. Тест социального взаимодействия (СВТ). Крысам дают возможность привыкнуть к средствам ухода за животными в течение 5 дней и размещают по одной в клетку за 5 дней до теста. Животных приручают в течение 5 мин в день. Проектирование и методику теста "социального взаимодействия" проводят, как описано ранее Kennett, et al. (1997). В день теста к подобранной по весу паре крыс (5%), незнакомых друг с другом, применяют одинаковое лечение и возвращают в их персональные клетки. Животных случайным образом подразделяют на 5 групп, подвергаемых лечению, по 5 пар в каждой группе, и чрескожно вводят тестируемое соединение(10, 30 или 100 мг/кг), носитель (1 мл/кг) или хлордиазепоксид (5 мг/кг). Дозирование проводят за 1 ч до тестирования. Крыс затем помещают на 15 мин в белый ящик для теста из оргстекла марки Perspex или арену (543726 см), в которых пол разделен на 24 равных квадрата. Используют воздушный кондиционер для генерирования фонового шума и поддержания температуры в комнате около 74F. Все сессии записывают на видеопленку с использованием портативной видеокамеры JVC (модель GR-SZ1, ElmwoodPark, NJ) с видеокассетами длительностью 30 мин или TDK (HG ultimate brand) или Sony. Все сессии проводят между 13.00-16.30 часами. Активное социальное взаимодействие, определяемое как чистка,обнюхивание, покусывание, боксирование, борьба, слежение и ползанье над или под, подсчитывают с использованием секундомера (Sportsline model no. 226, 1/100 с точностью). Подсчитывают число эпизодов вертикальной стойки (животное полностью поднимает свое тело на задние конечности), чистки (облизывание, покусывание, почесывание тела), и умывания мордочки (т.е. лапки двигаются неоднократно над мордочкой), и число пересеченных квадратов. Пассивное социальное взаимодействие (животные лежат рядом друг с другом или одно сверху другого) не подсчитывали. Все поведения оцениваются позже наблюдателем, которому неизвестно, какое лечение применялось к каждой паре. В конце каждого теста ящик тщательно вытирают увлажненными бумажными полотенцами. Животные. Самцов крыс альбиносов линии Sprague Dawley (Taconic Farms, NY) помещают в клетки попарно с 12-часовым циклом свет/темнота (свет зажигают в 07.00 часов) со свободным доступом к пище и воде.- 25011029 Введение лекарственного препарата. Тестируемое соединение растворяют или в 100% ДМСО или 5% молочной кислоте, об./об. (SigmaChemical Co., St. Louis, MO). Хлордиазепоксид (Sigma Chemical Co., St. Louis, МО) растворяют в дважды дистиллированной воде. Носитель состоит из 50% ДМСО (об./об.) или 100% диметилацетамида (ДМА). Все растворы лекарственного препарата готовят за 10 мин до инъекции и растворы выбрасывают в конце дня теста. Объем вводимого раствора лекарственного средства составляет 1 мл/кг. Анализ данных. Данные по социальному взаимодействию (время взаимодействия, вертикальная стойка и пересеченные квадраты) подвергают рандомизированному однофакторному дисперсионному анализу ANOVA с апостериорным вычислением с использованием критерия Стьюдента-Ньюмена-Кеулса. Данные подвергают проверке на нормальность распределения (критерий Шапиро-Уилка). Данные анализируют с помощью программы GBSTAT, версии 6.5 (Dynamics Microsystems, Inc., Silver Spring, MD, 1997).D. Мочевые расстройства. Воздействие соединений на рефлекс мочеиспускания может быть оценено путем использования модели "вызываемого растяжением ритмического сокращения" (ВРРС), как описано в предшествующих публикациях (например, Maggi et al., 1987; Morikawa et al., 1992), и/или модели непрерывной медленной чреспузырной инфузии (НМЧИ). 1. Модель DIRC. Самок крыс линии Sprague Dawley, весящих примерно 300 г, анестезируют путем введения подкожно уретана (1,2 г/кг). В трахею вводят катетер PE240 для обеспечения чистоты дыхательных путей на протяжении эксперимента. Делают разрез по средней линии брюшной стенки и выделяют левый и правый мочеточники. Мочеточники перевязывают дистально (чтобы предотвратить утечку мочи из мочевого пузыря) и проксимально устанавливают катетер PE10. Разрез закрывают с использованием шелковых нитей размером 4-0, оставляя PE10 трубки, направленные наружу для удаления мочи. В мочевой пузырь вставляют в трансуретральном направлении катетер PE50 на 2,5 см над отверстием уретры. Этот катетер прикрепляют к хвосту, используя тесьму, и подсоединяют к датчику давления. Для предотвращения утечки из мочевого пузыря катетер плотно привязывают к наружному отверстию уретры, используя шелк размером 4-0. Для стимуляции рефлекса мочеиспускания мочевой пузырь сначала опорожняют путем надавливания на нижнюю часть живота и затем наполняют физиологическим раствором до увеличения его объема в 100 раз (максимум=2 мл) до тех пор, пока не происходит спонтанное сокращение мочевого пузыря (обычно 20-40 мм рт.ст.) с частотой одно сокращение каждые 2-3 мин. Как только устанавливается регулярный ритм, вводят внутривенно или подкожно носитель (физиологический раствор) или тестируемые соединения для исследования их влияния на активность мочевого пузыря. В качестве положительного контроля часто используют 5-HT1A антагонист WAY-100635. Данные выражают как интервал сокращения (в секундах) до введения лекарственного препарата (базальный) или после введения носителя или тестируемого соединения. 2. Модель непрерывной медленной чреспузырной инфузии (НМЧИ) на крысах. Для исследования используют самцов крыс линии Sprague Dawley, весящих примерно 300 г. Крыс анестезируют пентобарбитоном натрия (50 мг/кг, подкожно). Через разрез по средней линии брюшной стенки обнажают мочевой пузырь и полиэтиленовый катетер (РЕ 50) вводят в мочевой пузырь через небольшой разрез на куполе мочевого пузыря и катетер прикрепляют с помощью шовного материала. Другой конец катетера выводят подкожно на дорсальную область шеи. Аналогично другой катетер (РЕ 50) вводят в желудок через околосрединный разрез брюшной стенки со свободным концом, выведенным подкожно в области шеи. Хирургические раны закрывают шелковым 4-0 швом и животному дают возможность восстановиться при соответствующем послеоперационном уходе. На следующий день животное помещают в фиксирующее устройство для крыс. Открытый конец катетера в мочевом пузыре соединяют с датчиком давления, так же как и с инфузионным насосом, через трехходовой кран. Циклы опорожнения мочевого пузыря инициируют непрерывной инфузией физиологического раствора при скорости 100 мкл/мин. Повторяющиеся опорожняющие сокращения записывают с помощью программы сбора данных в режиме реального времени Power Lab. После записи базального образца опорожнения вводят тестируемый лекарственный препарат или носитель в течение часа непосредственно в желудок через внутрижелудочный катетер, и циклы опорожнения регистрируют в течение 5 ч. Давление мочеиспускания и частоту вычисляют до и после лечения (с интервалом каждые 30 мин) для каждого животного. Объем мочевого пузыря вычисляют из частоты мочеиспускания, исходя из постоянной инфузии 100 мкл/мин. Влияние тестируемого лекарственного препарата выражают как процент от базального,объема мочевого пузыря до объема после применения лекарственного препарата. Для сравнения в качестве положительного контроля часто используют WAY 100635. Ссылки где каждый X является независимо CR1; где каждый R1 является независимо -Н, -F, -Cl, -Br, -I, -CN, -NO2, линейным или разветвленным C1C7 алкилом, C1-C7 алкилокси, монофторалкилом или полифторалкилом, или C3-C6 циклоалкил-C1-C7 алкилом; каждый R2 является независимо -Н, -F, -Cl или линейным или разветвленным C1-C4 алкилом, монофторалкилом или полифторалкилом;n является целым числом от 2 до 6 включительно; В является CH2, CHOH, О или CO;Z является О, CH2, CO, CHOH или нулем; и А является фенилом или гетероарилом, где фенил или гетероарил являются необязательно замещенными тремя или менее R2; причем гетероарил выбирают из группы, включающей фуранил, тиенил, пирролил, оксазолил, тиазолил, имидазолил, пиразолил, изоксазолил, изотиазолил, оксадиазолил, триазолил, тиадиазолил, пиридил, пиридинил, пиридазинил, пиримидинил, пиразинил и триазинил; или его фармацевтически приемлемая соль. 2. Соединение по п.1, где n равняется 2 или 3. 3. Соединение по п.2, где каждый R1 является независимо -Н, -F, -Cl, линейным или разветвленнымC1-C4 алкилом или алкокси или C3-C6 циклоалкил-C1-C4 алкилом. 4. Соединение по п.3, где каждый R2 является независимо -Н, -F, -Cl или линейным или разветвленным C1-C4 алкилом, монофторалкилом или полифторалкилом. 5. Соединение по п.4, где А является пиридинилом, необязательно замещенным тремя или менее R2. 6. Соединение по п.4, где А является тиенилом, необязательно замещенным тремя или менее R2. 7. Соединение по п.4, где А является фуранилом, необязательно замещенным тремя или менее R2. 8. Соединение по п.4, где А является тиазолилом, необязательно замещенным тремя или менее R2. 9. Соединение по п.4, где А является имидазолилом, необязательно замещенным тремя или менееR2. 10. Соединение по п.4, где А является пиразолилом, необязательно замещенным тремя или менееR2. 11. Соединение по п.4, где А является оксазолилом, необязательно замещенным тремя или менееR2. 12. Соединение по п.4, где А является триазинилом, необязательно замещенным тремя или менее- 27011029 13. Соединение по п.4, где А является фенилом, необязательно замещенным тремя или менее R2. 14. Соединение по п.13, где Z является CH2, CO или CHOH. 15. Соединение по п.13, где Z является O или нулем. 16. Соединение по п.15, где В является CH2. 17. Соединение по п.16, где Z является О. 18. Соединение по п.17, где соединение выбирают из группы, состоящей из 5-(2-феноксифенил)-N-(3(4-пиперидил)фенил)пентанамида; 19. Соединение по п.16, где Z является нулем. 20. Соединение по п.19, где соединение выбирают из группы, состоящей из N-(4-метил-3-(4 пиперидил)фенил)-5-(4-фенилфенил)пентанамида; N-(2-фтор-5-(4-пиперидил)фенил)-5-(4-фенилфенил)пентанамида; N-(2-фтор-4-метил-5-(4-пиперидил)фенил)-5-(4-фенилфенил)пентанамида; и N-(4-фтор-3(4-пиперидил)фенил)-5-(4-фенилфенил)пентанамида 21. Соединение по п.15, где В является CO. 22. Соединение по п.21, где Z является нулем. 23. Соединение по п.22, где соединение выбирают из группы, состоящей из N-(4-фтор-3-(4 пиперидил)фенил)-4-оксо-4-(4-фенилфенил)бутанамида; N-(2-фтор-5-(4-пиперидил)фенил)-4-оксо-4-(4 фенилфенил)бутанамида; N-(4-фтор-3-(4-пиперидил)фенил)-5-оксо-5-(4-фенилфенил)пентанамида; 5 оксо-5-(4-фенилфенил)-N-(3-(4-пиперидил)фенил)пентанамида; 24. Соединение по п.1, являющееся энантиомерно чистым. 25. Соединение по п.1, являющееся диастереомерно чистым. 26. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. 27. Фармацевтическая композиция, полученная смешением соединения по п.1 и фармацевтически приемлемого носителя.- 28011029 28. Способ получения фармацевтической композиции, включающий смешение соединения по п.1 и фармацевтически приемлемый носитель. 29. Применение соединения по пп.1-25 для изготовления лекарственного средства для лечения пациента, страдающего от аффективного расстройства, выбранного из группы, состоящей из депрессии,глубокой депрессии, маниакально-депрессивного психоза, агорафобии, специфической фобии, социофобии, навязчивого состояния, посттравматического стресса, острого стрессового расстройства и состояния тревоги, включающее введение пациенту терапевтически эффективного количества соединения по п.1. 30. Применение соединения по пп.1-25 для изготовления лекарственного средства для лечения пациента, страдающего от нарушения мочеиспускания, выбранного из группы, состоящей из непроизвольного мочеиспускания, позыва к мочеиспусканию, частого мочеиспускания, недержания мочи, никтурии и энуреза, включающее введение пациенту терапевтически эффективного количества соединения по п.1. 31. Применение соединения по пп.1-25 для изготовления лекарственного средства для лечения пациента, страдающего от расстройства питания, выбранного из группы, состоящей из ожирения, булимии,нейрогенной булимии и нейрогенной анорексии, включающее введение пациенту терапевтически эффективного количества соединения по п.1.
МПК / Метки
МПК: A61K 31/445, C07D 401/14, C07D 401/04, C07D 401/10, C07D 211/68
Метки: 4-арилпиперидины
Код ссылки
<a href="https://eas.patents.su/30-11029-4-arilpiperidiny.html" rel="bookmark" title="База патентов Евразийского Союза">4-арилпиперидины</a>
Ингибирующие mtp арилпиперидины или пиперазины, замещенные 5-членными гетероциклами

Номер патента: 9455
Опубликовано: 28.12.2007
Авторы: Ван Дер Векен Луи Йозеф Элизабет, Вьейевуа Марсель, Мерпул Ливен, Бакс Лео Якобус Йозеф, Линдерс Йоаннес Теодорус Мария, Яроскова Либуше, Рувенс Петер Вальтер Мария
МПК: C07D 233/54, C07D 249/08, A61P 19/00...
Метки: ингибирующие, замещенные, 5-членными, гетероциклами, арилпиперидины, пиперазины
Формула / Реферат:
1. Соединение формулы (I) его N-оксиды, фармацевтически приемлемые кислотно-аддитивные соли и стереохимически изомерные формы, где пунктирная линия представляет необязательную связь, и она отсутствует, когда X2 представляет атом азота; радикал -Y1-Y2- представляет радикал формулы где в двухвалентных радикалах формулы (а-1) или (а-2) атом водорода может быть необязательно заменен C1-6алкилом или фенилом; или в двухвалентных радикалах формулы...
Предыдущий патент: Жаропрочный сплав и способ получения минеральной ваты
Следующий патент: Новая кристаллическая форма v агомелатина, способ её получения и фармацевтические композиции, которые её содержат
Случайный патент: Чистящие средства для твердых поверхностей на основе смесей, полученных в результате метатезиса натуральных масел