Пролекарства пиперидинильного производного в качестве модуляторов активности хемокинового рецептора
Номер патента: 22150
Опубликовано: 30.11.2015
Авторы: Сантелла Джозеф В., Картер Перси Х., Наир Сатиш, Корнелиус Линдон А.М., Дар Т.Дж.Мьюрали, Хайнс Джон, Дунсия Джон В., Ву Хонг, Вориер Йакумар С.





























Формула / Реферат
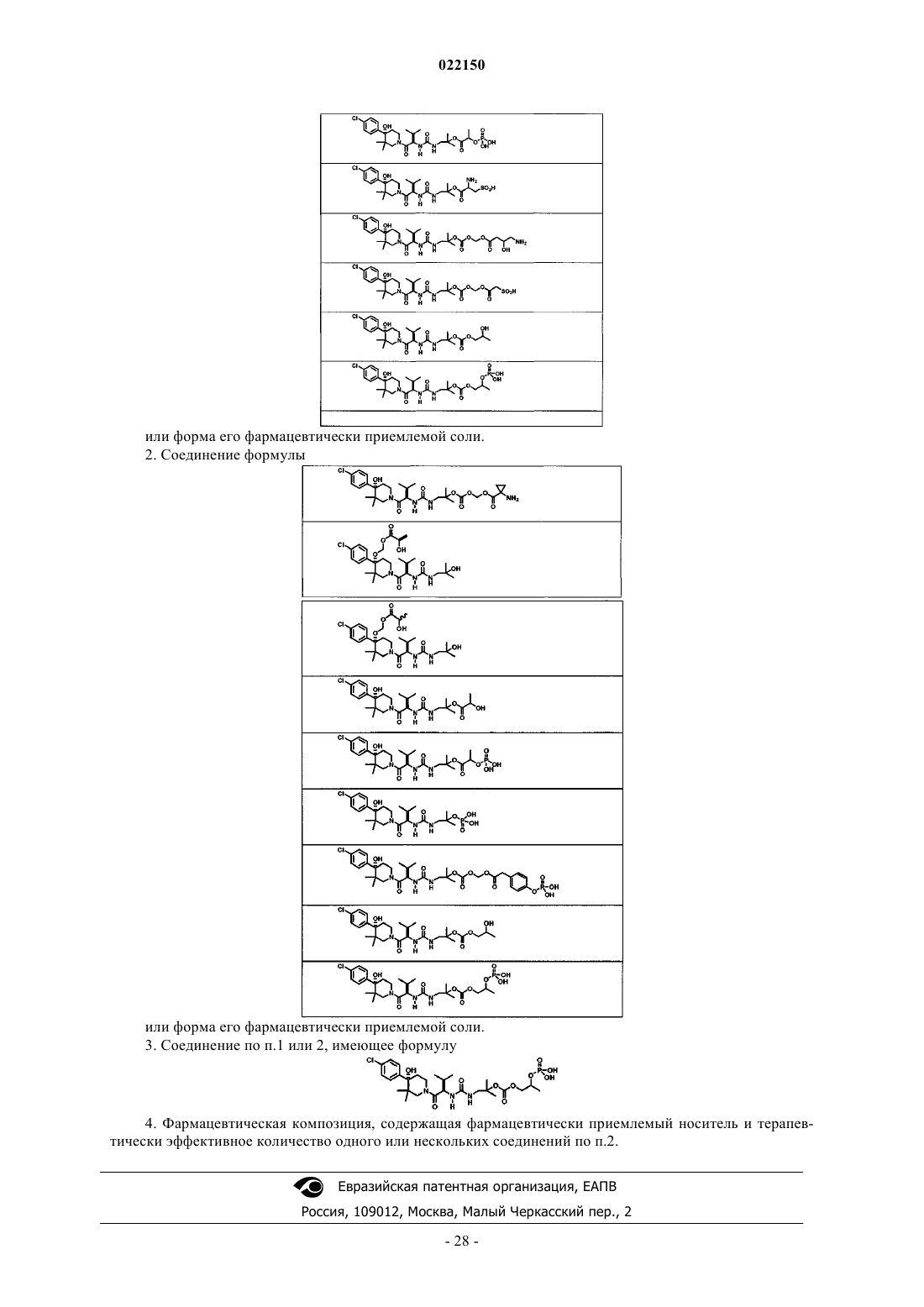
1. Соединение формулы



или форма его фармацевтически приемлемой соли.
2. Соединение формулы


или форма его фармацевтически приемлемой соли.
3. Соединение по п.1 или 2, имеющее формулу

4. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество одного или нескольких соединений по п.2.
Текст
ПРОЛЕКАРСТВА ПИПЕРИДИНИЛЬНОГО ПРОИЗВОДНОГО В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ ХЕМОКИНОВОГО РЕЦЕПТОРА Изобретение относится к пролекарствам соединения формулы (I) или их стереоизомерам или фармацевтически приемлемым солям. Кроме того, раскрыты способы лечения и профилактики воспалительных заболеваний, таких как бронхиальная астма и аллергические заболевания, а также аутоиммунные патологии, такие как ревматоидный артрит, с применением соединений пролекарства. Область техники Изобретение относится, главным образом, к пролекарствам пиперидинильного модулятора активности хемокинового рецептора, содержащим их фармацевтическим композициям и способам их применения в качестве средства для лечения и профилактики воспалительных заболеваний, аллергических и аутоиммунных заболеваний и, в частности, ревматоидного артрита и отторжения трансплантата. Уровень техники Хемокины представляют собой хемотактические цитокины с молекулярной массой 6-15 кДа, которые высвобождаются широким спектром клеток для привлечения и активации, помимо прочих типов клеток, моноцитов, макрофагов, Т- и В-лимфоцитов, эозинофилов, базофилов и нейтрофилов. Хемокины подразделяют на два основных класса, СХС и СС, в зависимости от того, являются ли первые два цистеина в аминокислотной последовательности разделенными одной аминокислотой (СХС) или смежными(СС) СХС-хемокины, такие как интерлейкин-8 (IL-8), нейтрофил-активирующий белок-2 (NAP-2) и белок-стимулятор активности роста меланомы (MGSA), преимущественно обуславливают хемотаксис нейтрофилов и Т-лимфоцитов, тогда как СС-хемокины, такие как RANTES, MIP-1, MIP-1 , моноцитарные хемотактические белки (МСР-1, МСР-2, МСР-3, МСР-4 и МСР-5) и эотаксины (-1 и -2), обуславливают хемотаксис, помимо прочих типов клеток, макрофагов, Т-лимфоцитов, эозинофилов, дендритных клеток и базофилов. Хемокины связываются со специфическими рецепторами поверхности клетки, принадлежащими к семейству сопряженных с G-белком белков с семью трансмембранными доменами, которые называют"хемокиновыми рецепторами". Связываясь с соответствующими им лигандами, хемокиновые рецепторы передают внутриклеточный сигнал через связанные тримерные G-белки, что приводит, помимо прочих ответов, к быстрому повышению концентрации внутриклеточного кальция, изменениях формы клетки,усиленной экспрессии молекул клеточной адгезии, дегрануляции и стимуляции миграции клеток. Существует по меньшей мере десять хемокиновых рецепторов человека, которые связывают или отвечают на СС-хемокины со следующими характерными профилями: CCR-1 (или "CKR-1", или "СС-CKR-1") [MIP1, МСР-3, МСР-4, RANTES] CCR-2A и CCR-2B (или "CKR-2A"/"CKR-2B", или "CC-CKR-2A"/"CCCKR-2B") [МСР-1, МСР-2, МСР-3, МСР-4, МСР-5]; CCR-3 (или "CKR-3", или "CC-CKR-3") [эотаксин-1,эотаксин-2, RANTES, МСР-3, МСР-4]; CCR-4 (или "CKR-4", или "CC-CKR-4") [TARC, MDC]; CCR-5CCR-10 (или "CKR-10", или "CC-CKR-10") [MCP-1, МСР-3]; и CCR-11 [МСР-1, МСР-2 и МСР-4]. Помимо хемокиновых рецепторов млекопитающих было показано, что цитомегаловирусы, герпесвирусы и поксвирусы млекопитающих экспрессируют в инфицированных клетках белки со свойствами связывания хемокиновых рецепторов. СС-хемокины человека, такие как RANTES и МСР-3, могут вызывать быструю мобилизацию кальция посредством этих кодируемых вирусом рецепторов. Экспрессия рецептора может быть пермиссивной для инфекции, обеспечивая нарушение нормального надзора со стороны иммунной системы и ответа на инфекцию. Кроме того, хемокиновые рецепторы человека, такие как CXCR4, CCR2, CCR3, CCR5 и CCR8, могут действовать как корецепторы для инфицирования клеток млекопитающих микробами, как в случае, например, вирусов иммунодефицита человека (HIV, ВИЧ). Было сделано заключение о том, что хемокины и соответствующие им рецепторы являются важными медиаторами воспалительных, инфекционных и иммунорегуляторных нарушений и заболеваний,включая брохиальную астму и аллергические заболевания, а также аутоиммунных патологий, таких как ревматоидный артрит и атеросклероз (обзор представлен в Carter, P.H., Current Opinion in Chemical Biology 2002, 6, 510; Trivedi et al., Ann. Reports Med. Chem. 2000, 35, 191; Saunders et al., Drug Disc. Today 1999, 4, 80; Premack et al., Nature Medicine 1996, 2, 1174). Например, хемокин-макрофагальный воспалительный белок-1 (MIP-1) и его рецептор СС-хемокиновый рецептор 1 (CCR-1) играют ведущую роль в привлечении лейкоцитов в очаги воспаления и в последующей активации этих клеток. Если хемокинMIP-1 связывается с CCR-1, это индуцирует быстрое повышение концентрации внутриклеточного кальция, усиленную экспрессию молекул клеточной адгезии, дегрануляцию клеток и стимуляцию миграции лейкоцитов. К тому же, хемотактические свойства MIP-1 у людей были продемонстрированы экспериментально. Люди после внутрикожной инъекции MIP-1 испытывали быстрый и значительный приток лейкоцитов к месту инъекции (Brummet, M.E., J. Immun. 2000,164, 3392-3401). Важность взаимодействия MIP-1/CCR-1 была продемонстрирована в экспериментах на генномодифицированных мышах. MIP-1 -/- мыши характеризовались нормальными количествами лейкоцитов,но были неспособны рекрутировать моноциты в очаги вирусного воспаления после иммунной стимуляции. Недавно было показано, что MIP-1 -/- мыши устойчивы к индуцируемому антителами против коллагена артриту. Подобным образом, CCR-1 -/- мыши были неспособны рекрутировать нейтрофилы после стимуляции MIP-1 in vivo; более того, нейтрофилы периферической крови CCR-1-нулевых мышей не мигрировали в ответ на MIP-1, демонстрируя таким образом специфичность взаимодействия MIP1/CCR-1. Заслуживают упоминания жизнеспособность и общее нормальное состояние здоровья MIP-1-/- и CCR-1 -/- животных, у которых нарушение взаимодействия MIP-1/CCR-1 не вызывает физиологического кризиса. В совокупности эти данные приводят к выводу, что молекулы, которые блокируют действия MIP-1, были бы применимы для лечения ряда воспалительных и аутоиммунных нарушений. Эта гипотеза теперь подтверждена на ряде описанных ниже различных животных моделей заболеваний. Известно, что содержание MIP-1 повышено в синовиальной жидкости и крови больных ревматоидным артритом. Более того, в нескольких исследованиях была продемонстрирована потенциальная терапевтическая ценность антагонизма взаимодействия MIP-1/CCR1 для лечения ревматоидного артрита. Следует также заметить, что CCR-1 также является рецептором для хемокинов RANTES, МСР-3,НСС-1, Lkn-l/HCC-2, НСС-4 и MPIF-1 (Carter, P.H., Curr. Opin Chem. Bio. 2002, 6, 510-525). Поскольку,предположительно, описанное в настоящем документе новое соединение формулы (I) проявляет антагонизм в отношении MIP-1 путем связывания с рецептором CCR-1, это соединение, возможно, также является эффективным антагонистом воздействий вышеупомянутых лигандов, опосредуемых CCR-1. Соответственно, когда в настоящем документе делается ссылка на "антагонизм в отношении MIP-1", следует допускать, что она является эквивалентной понятию "антагонизм в отношении хемокиновой стимуляции CCR-1". Недавно несколько групп описали разработку низкомолекулярных антагонистов MIP-1 (обзор приведен в Carson, K.G. et al., Ann. Reports Med. Chem. 2004, 39, 149-158). Сущность изобретения Соответственно, настоящее изобретение относится к пролекарствам антагониста или частичного агониста/антагониста активности MIP-1 или рецептора CCR-1 или их фармацевтически приемлемым солям. Настоящее изобретение относится к фармацевтическим композициям, содержащим фармацевтически приемлемый носитель и терапевтически эффективное количество пролекарств соединения согласно настоящему изобретению или его фармацевтически приемлемой солевой формы. Настоящее изобретение относится к способу лечения ревматоидного артрита и отторжения трансплантата, предусматривающему введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества одного или нескольких пролекарств соединения согласно настоящему изобретению или его фармацевтически приемлемой солевой формы. Настоящее изобретение относится к способу лечения воспалительных заболеваний, предусматривающему введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества одного или нескольких пролекарств соединения согласно настоящему изобретению или его фармацевтически приемлемой солевой формы. Настоящее изобретение относится к пролекарствам пиперидинильного производного для применения в терапии. Настоящее изобретение относится к применению пролекарства пиперидинильного производного для производства лекарственного препарата для лечения воспалительных заболеваний. Подробное описание изобретения Согласно одному варианту осуществления настоящее изобретение относится к пролекарствам нового соединения формулы (I) или их стереоизомерам или фармацевтически приемлемым солям. Настоящее изобретение относится к пролекарствам пиперидинильного соединения, которые характеризуются неожиданно предпочтительным профилем по сравнению с активностью известных ингибиторов CCR-1, например, производных пиперидинила, описанных в заявке US2007/0208056 А 1, опубликованной 6 сентября 2007 г., и принадлежащей заявителю. Эти пролекарства получали для обеспечения соединений с улучшенной проникающей способностью, растворимостью и/или стабильностью для целей составления и хранения лекарственных средств. Эти пролекарства или их фармацевтически приемлемые соли характеризуются следующими структурами: Предпочтительные соединения согласно настоящему изобретению включают Согласно другому варианту осуществления настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество одного или нескольких пролекарств соединения формулы (I). При этом соединения согласно изобретению могут использоваться для модуляции активности хемокина или хемокинового рецептора, модуляции активности рецептора CCR-1 или модуляции активности MIP-1, МСР-3, МСР-4, RANTES, предпочтительно модуляции опосредованной рецептором CCR-1 активности MIP-1. При этом пролекарства соединения формулы (I) могут использоваться для лечения нарушений, причем указанное нарушение выбрано из остеоартрита, аневризмы, лихорадки, сердечнососудистых эффектов, болезни Крона, застойной сердечной недостаточности, аутоиммунных заболеваний, ВИЧ-инфекции, ВИЧ-ассоциированной деменции, псориаза, идиопатического легочного фиброза,артериосклероза трансплантата, вызванной физическими или химическими факторами травмы головного мозга, нейропатической боли, воспалительного заболевания кишечника, альвеолита, язвенного колита,системной красной волчанки, нефротоксического сывороточного нефрита, гломерулонефрита, бронхиальной астмы, рассеянного склероза, атеросклероза, ревматоидного артрита, рестеноза, трансплантации органов, псориатического артрита, множественной миеломы, аллергий, например, дегрануляции тучных клеток в коже и конъюктиве глаза, печеночноклеточной карциномы, колоректальной злокачественной опухоли, остеопороза, фиброза почек и других видов злокачественных опухолей, предпочтительно болезни Крона, псориаза, воспалительного заболевания кишечника, системной красной волчанки, рассеянного склероза, ревматоидного артрита, множественной миеломы, аллергий, например, дегрануляции тучных клеток в коже и конъюктиве глаза, печеночноклеточной карциномы, остеопороза и фиброза почек. Одно или нескольких пролекарств соединения формулы (I) могут использоваться для применения для приготовления лекарственного препарата для лечения нарушения, причем указанное нарушение выбрано из остеоартрита, аневризмы, лихорадки, сердечно-сосудистых эффектов, болезни Крона, застойной сердечной недостаточности, аутоиммунных заболеваний, ВИЧ-инфекции, ВИЧ-ассоциированной деменции, псориаза, идиопатического легочного фиброза, артериосклероза трансплантата, вызванной физическими или химическими факторами травмы головного мозга, нейропатической боли, воспалительного заболевания кишечника, альвеолита, язвенного колита, системной красной волчанки, нефротоксического сывороточного нефрита, гломерулонефрита, бронхиальной астмы, рассеянного склероза, атеросклероза,ревматоидного артрита, рестеноза, трансплантации органов, псориатического артрита, множественной миеломы, аллергий, например, дегрануляции тучных клеток в коже и конъюктиве глаза, печеночноклеточной карциномы, колоректальной злокачественной опухоли, остеопороза, фиброза почек и других видов злокачественных опухолей, предпочтительно болезни Крона, псориаза, воспалительного заболевания кишечника, системной красной волчанки, рассеянного склероза, ревматоидного артрита, множественной миеломы, аллергий, например, дегрануляции тучных клеток в коже и конъюктиве глаза, печеночноклеточной карциномы, остеопороза и фиброза почек. Согласно другому варианту осуществления настоящее изобретение относится к фармацевтической композиции, содержащей одно или несколько пролекарств соединения формулы (I) и один или несколько активных ингредиентов. При этом для модуляции активности хемокина или хемокинового рецептора предусмотрено введение нуждающемуся в этом пациенту терапевтически эффективного количества фармацевтической ком-5 022150 позиции, содержащей одно или несколько пролекарств соединения формулы (I) и один или несколько активных ингредиентов. При этом введение нуждающемуся в этом пациенту терапевтически эффективного количества фармацевтической композиции модулярует активность рецептора CCR-1, MIP-1, МСР-3, МСР-4, RANTES,предпочтительно опосредованной рецептором CCR-1 активность MIP-1. Введение нуждающемуся в этом пациенту терапевтически эффективного количества фармацевтической композиции, содержащей одно или несколько пролекарств соединения формулы (I) и один или несколько активных ингредиентов может использоваться для лечения нарушений, причем указанное нарушение выбрано из остеоартрита, аневризмы, лихорадки, сердечно-сосудистых эффектов, болезни Крона, застойной сердечной недостаточности, аутоиммунных заболеваний, ВИЧ-инфекции, ВИЧассоциированной деменции, псориаза, идиопатического легочного фиброза, артериосклероза трансплантата, вызванной физическими или химическими факторами травмы головного мозга, нейропатической боли, воспалительного заболевания кишечника, альвеолита, язвенного колита, системной красной волчанки, нефротоксического сывороточного нефрита, гломерулонефрита, бронхиальной астмы, рассеянного склероза, атеросклероза, ревматоидного артрита, рестеноза, трансплантации органов, псориатического артрита, множественной миеломы, аллергий, например, дегрануляции тучных клеток в коже и конъюктиве глаза, печеночноклеточной карциномы, колоректальной злокачественной опухоли, остеопороза,фиброза почек и других видов злокачественных опухолей, предпочтительно болезни Крона, псориаза,воспалительного заболевания кишечника, системной красной волчанки, рассеянного склероза, ревматоидного артрита, множественной миеломы, аллергий, например, дегрануляции тучных клеток в коже и конъюктиве глаза, печеночноклеточной карциномы, остеопороза и фиброза почек. Настоящее изобретение может быть осуществлено в других конкретных формах без отклонения от его сущности или существенных признаков. Настоящее изобретение также относится ко всем комбинациям упомянутых в настоящем документе альтернативных аспектов настоящего изобретения. Следует понимать, что любые и все варианты осуществления настоящего изобретения могут быть предусмотрены в сочетании с любым другим вариантом осуществления для описания дополнительных вариантов осуществления настоящего изобретения. Кроме того, любые элементы варианта осуществления могут быть объединены с любым и всеми другими элементами любого варианта осуществления для описания дополнительных вариантов осуществления. Определения Описанные в настоящем документе соединения могут содержать центры асимметрии. Содержащие асимметрично замещенный атом соединения согласно настоящему изобретению могут быть выделены в оптически активных или рацемических формах. Из предшествующего уровня техники хорошо известно,как получать оптически активные формы, например, путем разделения рацемических форм или путем синтеза из оптически активных исходных веществ. Среди описанных в настоящем документе соединений также может присутствовать много геометрических изомеров олефинов, двойных связей C=N и т.п., и все подобные стабильные изомеры предусмотрены в соответствии с настоящим изобретением. Описаны цис- и транс-геометрические изомеры соединений согласно настоящему изобретению, и они могут быть выделены в виде смеси изомеров или в виде отдельных изомерных форм. Если конкретные стереохимический вариант или изомерная форма не указаны особо, предусмотрены все хиральные, диастереомерные, рацемические формы и все геометрические изомерные формы структуры. Один энантиомер соединения формулы I может характеризоваться превосходной активностью по сравнению с другим. Таким образом, все стереохимические варианты считаются частью настоящего изобретения. Как известно среднему специалисту в данной области техники, при необходимости разделение рацемического вещества может быть достигнуто при помощи HPLC с применением хиральной колонки или путем разделения с применением средства для разделения. Используемое в настоящем документе выражение "фармацевтически приемлемый" относится к таким соединениям, веществам, композициям и/или лекарственным формам, которые по результатам тщательной медицинской оценки подходят для применения для взаимодействия с тканями человека и животных без избыточной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, в соответствии с приемлемым соотношением польза/риск. Используемое в настоящем документе выражение "фармацевтически приемлемые соли" относится к производным раскрытых соединений, причем исходное соединение модифицировано путем образования его солей с кислотами или основаниями. Примеры фармацевтически приемлемых солей включают без ограничения соли неорганических или органических кислот и основных остатков, таких как амины; щелочные или органические соли кислотных остатков, таких как карбоновые кислоты; и т.п. Фармацевтически приемлемые соли включают традиционные нетоксичные соли или четвертичные аммониевые соли исходного соединения, образованного, например, нетоксичными неорганическими или органическими кислотами. Например, такие традиционные нетоксичные соли включают соли, образуемые неорганическими кислотами, например, хлористо-водородной, бромисто-водородной, серной, сульфамовой,фосфорной, азотной и т.п.; и соли, образуемые органическими кислотами, например уксусной, пропио-6 022150 новой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, памовой, малеиновой, гидроксималеиновой, фенилуксусной, глутаминовой, бензойной, салициловой,сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, оксалиновой, изэтионовой и т.п. Фармацевтически приемлемые соли согласно настоящему изобретению могут быть синтезированы традиционными химическими способами из содержащего основный или кислотный фрагмент исходного соединения. Как правило, такие соли могут быть получены путем осуществления взаимодействия свободных кислотных или основных форм этих соединений со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе или в их смеси; как правило,предпочтительной является неводная среда, такая как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечни подходящих солей представлены в Remington's Pharmaceutical Sciences, 17th ed, MackPublishing Company, Easton, PA, 1985, p. 1418, раскрытие которых таким образом включено в настоящий документ посредством ссылки. Указанные процитированные документы включены в настоящий документ посредством ссылки. Кроме того, соединения формулы I после их получения предпочтительно выделяют и очищают для приготовления композиции, содержащей количество соединения формулы I, по массе равное или больше чем 99% ("по существу чистое" соединение I), которое затем используют или включают в состав согласно описанию в настоящем документе. Такие "по существу чистые" соединения формулы I также предусмотрены как часть настоящего изобретения. Предусмотрены все стереоизомеры соединений согласно настоящему изобретению или в виде смеси, или в чистой, или по существу чистой форме. Соединения согласно настоящему изобретению могут содержать центры асимметрии при любых атомах углерода, включая любой из заместителей R, и/или характеризоваться полиморфизмом. Следовательно, соединения формулы I могут существовать в энантиомерной или диастереомерной формах или в виде их смеси. В способах получения в качестве исходных веществ могут быть использованы рацематы, энантиомеры или диастереомеры. Если получены энантиомерные или диастереомерные продукты, они могут быть разделены традиционными способами,например при помощи хроматографии или фракционной кристаллизацией. Подразумевается, что термины "стабильное соединение" и "стабильная структура" обозначают соединение, достаточно стойкое для того, чтобы его можно было подвергать выделению из реакционной смеси с достаточной степенью чистоты и включению в состав эффективного лекарственного препарата. Подразумевается, что настоящее изобретение относится к стабильным соединениям. Подразумевается, что "терапевтически эффективное количество" включает количество соединения согласно настоящему изобретению в отдельности или количество комбинации заявленных соединений или количество соединения согласно настоящему изобретению в комбинации с другими активными ингредиентами, эффективными для ингибирования MIP-1 или эффективными для лечения или профилактики воспалительных нарушений. Используемые в настоящем документе термины "лечить" или "лечение" охватывают лечение болезненного состояния у млекопитающих, в частности у людей, и включают: (а) профилактику развития болезненного состояния у млекопитающего, в частности, когда такое млекопитающее предрасположено к развитию болезненного состояния, но его наличие еще не было диагностировано; (b) ингибирование болезненного состояния, т.е. остановку его развития; и/или (с) облегчение болезненного состояния, т.е. вызывание регрессии болезненного состояния. Синтез Соединения согласно настоящему изобретению получали, как показано в следующих примерах,схемах реакций и их описаниях, а также в соответствии с известными из релевантной литературы процедурами, которые могут быть использованы специалистом в данной области техники. Далее представлены типичные реагенты и методики для проведения этих реакций. Соединения формулы 1.3 могут быть получены показанным на схеме 1 способом. Начиная с соединения 1, путем осуществления взаимодействия с соответствующим фосфорилирующим средством с последующим окислением, например, пероксидом водорода, может быть получено защищенное фосфатное соединение 1.2. Защитные группы, которые могут быть использованы для этого превращения, представляют собой, например, аллил и бензил, и понятно, что специалист в области органического синтеза сможет использовать в этом способе и другие защитные группы. Удаление защитных групп позволяет получить соединение формулы 1.3. Соединения формул 2.1, 2.3 и 2.4 могут быть получены указанными на схеме 2 способами. Путем связывания соединения 1 с разными карбоновыми кислотами, ангидридами или хлорангидридами могут быть получены соединения общей формулы 2.1, которым может потребоваться дальнейшая модификация путем гидролиза до конечных аналогов. Соединения формулы 2.3 могут быть получены путем стандартного связывания с защищенными аминокислотными производными с последующим снятием защитной группы с аминогруппы с применением стандартных способов. Путем осуществления взаимодействия соединения 1 с функционализированным хлорформиатом, таким как 2.4, может быть получено соединение 2.2, которое далее может взаимодействовать с нуклеофилом, таким как морфолин, с получением соединения 2.5. Схема 2 Соединения общих структур 3.4 и 3.5 могут быть получены указанными на схеме 3 способами. Метилтиометиловый эфир 3.1 может быть получен путем осуществления взаимодействия соединения 3.1 сDMSO и уксусным ангидридом. Получение соединений 3.4 и 3.5 может быть обеспечено путем осуществления взаимодействия соединения 3.3 с таким соответствующим активирующим реагентом, как NBS или CuBr2, с последующим осуществлением взаимодействия с нуклеофилом, таким как соль фосфорной кислоты или молочной кислоты или аминокислоты. Схема 3 Соединения формулы 4.3 могут быть получены указанными на схеме 4 способами. Получение соединения 4.1 может быть обеспечено путем осуществления взаимодействия соединения 1 с хлорметилхлорформиатом в соответствующем растворителе в присутствии основания, такого как пиридин. Полу-8 022150 чение соединения 4.2 может быть обеспечено путем осуществления взаимодействия соединения 4.1 с защищенным аминокислотным производным в присутствии основания, такого как K2CO3. Получение соединения 4.3 может быть обеспечено путем снятия защитных групп с аминокислотного компонента. Также на схеме 4 указаны соединения, которые могут быть получены этим способом. Схема 4 Соединение 4.1 может быть использовано в качестве промежуточного соединения для получения различных дополнительных аналогов пролекарства, таких как показаны на схеме 5. Например, получение соединения 5.1 может быть обеспечено путем осуществления взаимодействия соединения 4.1 с фумаровой кислотой в присутствии основания, такого как TEA. Дополнительно, получение соединения 5.2 может быть обеспечено путем осуществления прямого взаимодействия с фосфорной кислотой и TEA. Соединения формулы 5.3 могут быть получены путем осуществления взаимодействия соединения 4.1 с соответствующим образом защищенной и функционализированной карбоновой кислотой в присутствии основания, с которого, в свою очередь, могут быть сняты защитные группы с получением соединения 5.3. Кроме того, соединения формулы 5.4 могут быть получены путем осуществления взаимодействия соответствующим образом функционализированной фенилуксусной кислоты с соединением 4.1, затем путем снятия защитных групп с фосфата при помощи гидрирования с получением соединения 5.4. Схема 5 Соединение 4.1 также может быть использовано для получения дополнительных соединений, таких как аналоги, показанные на схеме 6. Соединения 6.1 и 6.2 могут быть получены прямым замещением хлорида кислотой с последующим окислением серу содержащего фрагмента и снятием защитных групп. Соединение 6.3 может быть получено путем превращения хлорангидрида 4.1 в более реакционноспособный йодангидрид при помощи Nal с последующим осуществлением взаимодействия с Na2SO3. Схема 6 Кроме того, соединения формулы 7.2 могут быть получены путем осуществления взаимодействия соединения 1 с уксусным ангидридом и DMSO с получением метилтиометилового эфира 7.1, который, в свою очередь, может взаимодействовать с таким активирующим агентом, как NBS или NIS, с последующим взаимодействием с фосфорной кислотой и основанием с получением соединения 7.2. Схема 7 Дополнительные пролекарства соединения 1 могут быть получены указанными на схеме 8 способами. Сложный эфир 8.1 может быть получен путем осуществления взаимодействия соединения 1 с защищенным производным молочной кислоты, таким как бензилмолочная кислота, со связывающим агентом. Путем удаления защитной группы с последующим образованием фосфата на вторичном спирте могут быть получены соответственно спирт 8.2 и фосфат 8.3. Подобным образом, соединение 1 может взаимодействовать с гидроксизащищенной кислотой, такой как 3-О-бензилпропионовая кислота, с получением сложного эфира 8.4. Соединение 8.6 может быть получено в результате снятия защитных групп с образованием спирта 8.5 и превращением в фосфат при стандартных условиях. Схема 8 Образованные сульфоновой кислотой пролекарства соединения 1 могут быть получены указанными на схеме 9 способами. Соединение 9.2 может быть получено путем осуществления взаимодействия соединения 1 с хлорацетилхлоридом в присутствии основания, такого как пиридин. Производное сульфоновой кислоты 9.2 может быть получено путем осуществления взаимодействия с Na2SO3 в соответствующем растворителе. Пролекарства формулы 9.5 могут быть получены путем осуществления первого взаимодействия соединения 1 с акрилоилхлоридом с получением соединения 9.3. Путем осуществления взаимодействия соединения 9.3 с серу содержащим нуклеофилом, таким как тиоуксусная кислота, может быть получено соединение 9.4, которое может быть превращено в сульфоновую кислоту с применением стандартных способов (KOAc, АсОН, Оксон). Схема 9 Указанными на схеме 10 способами могут быть получены дополнительные аналоги пролекарств. Соединение 10.1 может быть получено путем осуществления взаимодействия соединения 1 с надлежащим образом защищенным производным 4-гидроксифенилуксусной кислоты. Путем удаления защитной группы может быть получено фенольное соединение 10.2, которое может быть превращено в фосфат с применением способов, подобных описанным ранее. Схема 10 Дополнительные образованные карбонатом пролекарства соединения 1 могут быть получены указанными на схеме 11 способами. Путем осуществления взаимодействия соединения 1 с аллилхлорформиатом в присутствии основания может быть получено соединение 11.1, которое затем может быть превращено в кетон 11.2 с применением известных способов (например, PdCl2, CuCl2, THF, вода). Затем кетон может быть восстановлен до спирта 11.3, при помощи которого за две дополнительные стадии (фосфорилирование и снятие защитных групп) может быть получено фосфатное пролекарство 11.4. Раствор трет-бутил-4-оксопиперидин-1-карбоксилата (52,47 г, 263 ммоль) в THF (1000 мл) охлаждали до 0 С и обрабатывали 4-мя равными порциями гидрида натрия (60% суспензия в минеральном масле) (22,12 г, 553 ммоль) с 5-минутными интервалами. Полученную суспензию перемешивали при 0 С в течение 45 мин ("мин") и затем обрабатывали добавлением по каплям йодметана (41,2 мл, 658 ммоль). Смесь перемешивали в течение 1 ч ("ч" или "час") и затем оставляли нагреться до комнатной температуры ("к.т."). Через 90 мин ледяную баню удаляли, наблюдали кратковременный экзотермический эффект(20-40 С за 3 мин) и интенсивное выделение газа. Ледяную баню удаляли и смесь оставляли перемешиваться в течение ночи при медленном нагревании до комнатной температуры. Реакцию гасили насыщенным хлоридом аммония (200 мл), затем обрабатывали достаточным количеством воды для растворения солей, которые осаждались. Слои разделяли и органическую фазу концентрировали в вакууме. Водную фазу экстрагировали этилацетатом и этот экстракт объединяли с остатком из первой органической фазы. Полученный раствор разбавляли 500 мл этилацетата и смесь промывали 2 раза водой, один раз насыщенным солевым раствором, сушили над сульфатом натрия и затем концентрировали в вакууме с получением вязкого масла, которое затвердевало при стоянии. Затвердевшую массу растворяли в 100 мл кипящих гексанов и полученный раствор оставляли охлаждаться до комнатной температуры, причем он стоял в течение ночи. По прошествии этого времени осевшие кристаллы собирали путем фильтрования,промывали небольшим количеством очень холодных гексанов и сушили с получением указанного в заголовке соединения в виде порошка (19,5 г, 86 ммоль, выход 32,6%). MS (ES+) = 172, 154. Стадия 2. -трет-Бутил-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-карбоксилат Раствор 4-бромхлорбензола (136,6 г, 0,71 моль) в безводном THF (1000 мл) охлаждали до -78 С и затем обрабатывали добавлением по каплям 1,6 М раствора н-бутиллития в гексанах (466 мл, 0,75 моль) со скоростью, которая обеспечивала поддержание внутренней температуры ниже -60 С. Полученную смесь перемешивали при -78 С в течение 1,5 ч, в ходе чего наблюдали образование осадка. Полученную суспензию обрабатывали добавлением по каплям раствора терт-бутил-3,3-диметил-4-оксопиперидин-1 карбоксилата (73,7 г, 0,32 моль) в безводном THF (400 мл) со скоростью, которая обеспечивала поддержание внутренней температуры ниже -60 С. Смесь перемешивали при -78 С в течение 2 ч, во время которых наблюдали образование прозрачного раствора. Реакционную смесь гасили насыщенным хлоридом аммония (300 мл) и полученную смесь оставляли нагреваться до комнатной температуры. Водные и органические слои разделяли и органическую фазу концентрировали в вакууме с получением остатка. Водную фазу экстрагировали 2 этилацетатом (300 мл). Объединенные экстракты добавляли к остатку из исходной органической фазы и полученную смесь разбавляли до 1200 мл этилацетатом. Полученный раствор промывали 2 водой (300 мл), один раз насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме с получением остатка. Остаток распускали в кипящих гексанах (300 мл) и полученную суспензию охлаждали до комнатной температуры. При достижении предусмотренной температуры белые твердые вещества собирали путем фильтрования и промывали 2 гексанами и затем сушили на воздухе с получением указанного в заголовке соединения в виде белого порошка (93,7 г. выход 85%). Стадия 3. -4-(4-Хлорфенил)-3,3-диметилпиперидин-4-ол Раствор -трет-бутил-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-карбоксилата (93,7 г,0,276 моль) в диоксане (100 мл) обрабатывали 4 М раствором HCl в диоксане (275 мл. 1,1 моль). Полученную смесь перемешивали при комнатной температуре в течение 4 ч. По прошествии этого времени смесь концентрировали в вакууме и затем концентрировали 3 из метиленхлорида (200 мл) с удалением остаточного HCl. Полученный остаток перемешивали в 1 М NaOH (500 мл) и полученную суспензию экстрагировали 4 при помощи 500 мл этилацетата. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения (66,8 г, количественный выход) в виде твердого вещества. Стадия 4. (S)-4-(4-Хлорфенил)-3,3-диметилпиперидин-4-ол Суспензию -4-(4-хлорфенил)-3,3-диметилпиперидин-4-ола (175 г) и L-винной кислоты (0,9 эквив.) в МЕК (3,22 л) нагревали до температуры возгонки. При достижении предусмотренной температуры добавляли воду (100 мл) с получением раствора. Полученный раствор нагревали с обратным холодильником в течение 1 ч и затем оставляли охлаждаться до комнатной температуры и перемешивали в течение 48 ч. По прошествии этого времени полученную взвесь отфильтровывали и собранные твердые вещества сушили под вакуумом с получением 123,4 г соли винной кислоты. Это вещество объединяли с другой серией такого же размера и объединенные твердые вещества суспендировали в МЕК (2,55 л) и воде (0,25 л). Полученный раствор нагревали до температуры возгонки и добавляли дополнительное количество воды (0,2 л) для растворения смеси. Раствор нагревали с обратным холодильником в течение 2 ч и затем оставляли охлаждаться до комнатной температуры и перемешивали в течение выходных. По окончании этого периода полученные твердые вещества собирали путем фильтрования и сушили с получением 219 г соли. Соль разделяли на две равные порции. Каждую порцию суспендировали в воде (2 л) и затем для осаждения свободного основания пиперидина добавляли 50% NaOH. После фильтрования и сушки выделяли 126,3 г указанного в заголовке соединения (выход 72%, 99% э и.). Стадия 5. трет-Бутил-(R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил 1-оксобутан-2-илкарбамат В трехгорлую круглодонную ("RB"') колбу емкостью 3 л добавляли (R)-2-(третбутоксикарбониламино)-3-метилбутановую кислоту (39,8 г, 183 ммоль), CH2Cl2 (1,6 л), (S)-4-(4 хлорфенил)-3,3-диметилпиперидин-4-ол (40,0 г, 167 ммоль), EDC (70,4 г, 367 ммоль) и HOBt (56,2 г, 416 ммоль). По окончании добавления реакционную смесь перемешивали в течение 30 мин при к.т. По прошествии этого времени добавляли триэтиламин (TEA, 93 мл, 668 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 20 ч. По окончании этого периода реакционную смесь промывали Na2CO3 (3 300 мл, примечание: первую промывку Na2CO3 подвергали фильтрованию на вакуумном фильтре и полученный фильтрат обратно экстрагировали CH2Cl2), 1 н. HCl (3300 мл), водой (400 мл) и насыщенным солевым раствором (300 мл). Полученный раствор сушили над Na2SO4 и концентрировали до полутвердого вещества (106 г, теоретический выход равен 73,2 г). На следующей стадии осуществляли взаимодействие полутвердого вещества без дополнительной очистки. Стадия 6. (R)-2-Амино-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метилбутан-1-он, HCl в диоксане, 720 мл, 2880 ммоль). По окончании добавления реакционную смесь перемешивали при к.т в течение 2,5 ч. По прошествии этого времени реакционную смесь концентрировали с получением геля Гель выпаривали совместно с метанолом (8100 мл) и затем CH2Cl2 (7100 мл) с получением твердого вещества (при изначальном взвешивании 57 г, соль HCl). Стадия 7. Фенил-(R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1 оксобутан-2-илкарбамат Синтез карбаматов проводили в двух отдельных колбах. Раскрытые в настоящем документе количества представляют собой общие значения, использованные для проведения экспериментов в двух колбах результаты. (R)-2-Амино-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метилбутан-1-он, HCl. (20 г, 53,3 ммоль) и DIPEA (18,61 мл, 107 ммоль) смешивали в CH2Cl2 (15 мл) при к.т. при перемешивании и затем через капельную воронку по каплям добавляли фенилкарбонилхлорид (6,71 мл,53,3 ммоль) в 10 мл метиленхлорида. По окончании добавления реакционную смесь перемешивали в течение одного часа. По прошествии этого времени добавляли дополнительные 0,2 эквив. DIPEA с последующим добавлением раствора фенилхлорформиата в метиленхлориде. Органический и водный слои разделяли. Органический слой промывали 1 н. HCl, нас. водн. NaHCO3 и насыщенным солевым раствором; сушили и затем десорбировали с получением масла. В масло при перемешивании при к.т. добавляли 25 мл MeCN. По окончании добавления и после перемешивания в течение 10 мин образовывались твердые вещества. Добавляли эфир (50 мл) и полученную смесь перемешивали в течение 5 мин. По прошествии этого времени добавляли дополнительное количество эфира (25 мл) и перемешивание продолжали в течение 15 мин. По окончании этого периода полученные твердые вещества собирали путем фильтрования и затем промывали эфиром с получением 13 г неочищенного твердого вещества, которое использовали без дополнительной очистки. Фильтрат концентрировали с получением остатка. Остаток очищали на силикагеле (9:1 - 3:1 - 1:1 гексанов/EtOAc - 100% EtOAc) с получением дополнительных 6,72 г продукта (выход общей массы 19,7 г, выход 81%). Стадия 8. Соединение формулы I Фенил-(R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1-оксобутан-2 илкарбамат (16,0 г, 34,9 ммоль), 1-амино-2-метилпропан-2-ол (3,42 г, 38,3 ммоль) и DIPEA (6,70 мл, 38.3 ммоль) смешивали в атмосфере азота при перемешивании в MeCN (30 мл) при к.т. Полученную суспензию нагревали до температуры возгонки, во время чего суспензия становилась бесцветным раствором. После перемешивания с обратным холодильником в течение приблизительно 20 мин твердые вещества осаждали. После перемешивания с обратным холодильником в течение 1,5 ч добавляли 20 мл ацетонитрила и другие 0,1 эквив. 1-амино-2-метилпропан-2-ола и DIPEA. Реакционную смесь перемешивали в течение дополнительных 1,5 ч. По прошествии этого времени реакционную смесь прекращали нагревать и оставляли охлаждаться до к.т. При охлаждении до к.т. добавляли воду для осаждения продукта (240 мл) и полученную свободнотекучую суспензию перемешивали в течение ночи. По окончании этого периода полученные твердые вещества собирали путем фильтрования, промывали 2 раза водой и затем сушили в высоком вакууме в течение 6 ч с получением 15,4 г твердых веществ. Эти твердые вещества (и дополнительный 1 г пробной партии) при перемешивании суспендировали в 50 мл ацетона при к.т. и затем добавляли 3-кратный объем воды (150 мл). Свободнотекучую суспензию перемешивали в течение ночи. По прошествии этого времени полученные твердые вещества собирали путем фильтрования, дважды промывали водой и затем сушили в течение 48 ч с получением 15,2 г соединения формулы I в виде твердого вещества. 1 Н ЯМР (500 МГц, метанол-d4, ротамерный)ppm 7,47 (dd, J=15,4, 8,8 Гц, 4 Н), 7,31 Стадия 9. Соединение согласно примеру 1 Раствор соединения 1 (200 мг, 0,441 ммоль), 2-9 Н-флуорен-9-ил)метокси)карбониламино) уксусной кислоты (262 мг, 0,881 ммоль) и DMAP (10,76 мг, 0,088 ммоль) обрабатывали DCC (182 мг, 0,881 ммоль) и смесь перемешивали при комнатной температуре в течение 7 ч. Анализ LC/MS позволил определить только 50% превращения исходного вещества и показал, что в результате полимеризации аминокислоты образовалось значительное количество ангидрида. Смесь обрабатывали дополнительным количеством 2-9 Н-флуорен-9-ил)метокси)карбониламино)уксусной кислоты (262 мг, 0,881 ммоль) иDCC (182 мг, 0,881 ммоль), затем перемешивали в течение ночи при комнатной температуре. Смесь фильтровали и фильтрат очищали на колонке с 24 г силикагеля, элюируя при 40 мл/мин градиентом этилацетата/гексанов, с получением пленки, которая содержала некоторое количество неопределенных примесей. Вещество повторно очищали на колонке с 24 г силикагеля, элюируя при 40 мл/мин 4%, затем 10% метанолом/метиленхлоридом, с получением 1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси 3,3-диметилпиперидин-1-ил)-3-метил-1-оксобутан-2-ил)уреидо)-2-метилпропан-2-ил-2-9 Н-флуорен-9 ил)метокси)карбониламино)ацетата (197 мг, 0,261 ммоль, выход 59,2%) в виде бесцветной пленки. MS(ES+) = 733,3 (М+Н)+. Раствор 1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1-оксобутан-2-ил)уреидо)-2-метилпропан-2-ил-2-9 Н-флуорен-9-ил)метокси)карбониламино)ацетата (180 мг,0,245 ммоль) и 1-октантиола (0,426 мл, 2,455 ммоль) в безводном THF (3 мл) обрабатывали DBU (7,40 мкл, 0,049 ммоль) и смесь перемешивали в течение ночи при комнатной температуре. Растворитель выпаривали потоком азота и остаток очищали на колонке с 12 г силикагеля, элюируя при 30 мл/мин градиентом метанола/метиленхлорида, с получением 1-(3-R)-1-S)-4-(4-хлорфенил)-4 гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1-оксобутан-2-ил)уреидо)-2-метилпропан-2-ил-2 аминоацетата (105 мг, 0,205 ммоль выход 84%) в виде бесцветного стекла MS (ES+) = 511,1 (М+Н)+. Пример 3. Раствор соединения 1 (215 мг, 0,426 ммоль), BOC-L-Валина (231 мг, 1,066 ммоль) и 4(диметиламино)пиридина (130 мг, 1,066 ммоль) в DCM (5 мл) обрабатывали раствором N,N'дициклогексилкарбодиимида (246 мг, 1,193 ммоль) в DCM (2 мл) и смесь перемешивали при к.т. в течение 48 ч. Реакционную смесь фильтровали с удалением N,N-дициклогексилмочевины и фильтрат концентрировали на роторном испарителе. Остаток растворяли в EtO Ас (15 мл), охлаждали до 0 С и отфильтровывали дополнительную N,N-дициклогексилмочевину. Фильтрат промывали 1 М KHSO4 (3 раза) водой и 5% NaHCO3 (3 раза), сушили (Na2SO4) и концентрировали. Неочищенный продукт очищали флэш-хроматографией с применением картриджа с 12 г силикагеля и элюировали 0-75% EtOAc/ гексанами с получением необходимого продукта (S)-1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1-оксобутан-2-ил)уреидо)-2-метилпропан-2-ил-2-(трет-бутоксикарбониламино)-3-метилбутаноата(170 мг, 0,260 ммоль, выход 61,1%) в виде твердого вещества. Раствор(S)-1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1 оксобутан-2-ил)уреидо)-2-метилпропан-2-ил-2-(трет-бутоксикарбониламино)-3-метилбутаноата (170 мг,0,260 ммоль) в DCM (0,5 мл) обрабатывали 4 М хлористо-водородной кислотой (4 мл, 16,00 ммоль) в диоксане и перемешивали при к.т. Через 10 мин при помощи LC-MS было показано, что израсходование начального вещества составило 95%. Реакционную смесь по каплям добавляли к насыщенному раствору NaHCO3 (35 мл) и смесь экстрагировали DCM (350 мл). Экстракт промывали насыщенным солевым раствором, сушили (Na2SO4) и неочищенный продукт очищали хроматографией среднего давления на силикагеле С 18 (картридж с 13 г) и элюировали 0-70% А/В. Скорость потока: 30 мл/мин. Подвижная фаза: А: 10% МеОН - 90% вода - 0,1 % TFA. В: 90% МеОН -10% вода - 0,1% TFA. Содержащую продукт фракцию объединяли, концентрировали на роторном испарителе с удалением большей части метанольного растворителя, остаток обрабатывали насыщенным раствором NaHCO3 до рН 8 и экстрагировали DCM. Экстракт промывали насыщенным солевым раствором, сушили (Na2SO4) и концентрировали на роторном испарителе с получением продукта в виде полукристаллического твердого вещества. Твердое вещество растворяли в ацетонитриле/воде (2 мл/8 мл) и мутный раствор замораживали и лиофилизировали с получением (S)-1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпипери- 15022150 мг, Соединение 1 (0,215 г, 0,474 ммоль) в DCM (3 мл) добавляли в пиридин (0,192 мл, 2,368 ммоль) с последующим добавлением по каплям хлорметилхлорформиата (0,084 мл, 0,947 ммоль) в течение 3 мин при к.т. Исходная гетерогенная смесь становилась гомогенной к концу добавления хлорметилхлорформиата. Содержимое перемешивали при к.т. в течение 1 ч, в это время при помощи LCMS (Luna 3u C18 4,630 мм; растворитель А = 10% МеОН, 90% Н 2 О, 0,1% TFA, растворитель В = 90% МеОН, 10% Н 2 О,0,1% TFA) отмечали присутствие основного пика с желаемой массой. Реакционную смесь концентрировали, остаток растворяли в МеОН (1,5 мл) и подвергали препаративной HPLC (Phenomenex Luna AXIA 5u C18 30100 мм; градиент 8 мин; растворитель А = 10% МеОН, 90% Н 2 О, 0,1% TFA, растворитель В = 90% МеОН, 10% Н 2 О, 0,1% TFA). Необходимые фракции собирали, концентрировали и разделяли междуDCM (10 мл) и насыщенным солевым раствором (10 мл). Слой DCM отделяли, сушили над сульфатом натрия и концентрировали с получением хлорметил-1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3 диметилпиперидин-1-ил)-3-метил-1-оксобутан-2-ил)уреидо)-2-метилпропан-2-илкарбоната (0,19 г, 0,348 ммоль, выход 73,4%). К хлорметил-1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1 оксобутан-2-ил)уреидо)-2-метилпропан-2-илкарбонату (0,19 г, 0,348 ммоль) в DMF (2 мл) последовательно добавляли триэтиламин (0,048 мл, 0,348 ммоль) и фумаровую кислоту (0,020 г, 0,174 ммоль) при к.т. Содержимое нагревали при 70 С (темпер. масляной бани) в течение 20 ч. Реакционную смесь охлаждали до к.т. и подвергали препаративной HPLC (Phenomex Luna AXIA 30 х 100 мм; градиент 10 мин; растворитель А = 10% CH3CN, 90% Н 2 О, 0,1% TFA, растворитель В = 90% CH3CN, 10% Н 2 О, 0,1% TFA). Необходимые фракции собирали и концентрировали. Остаток разделяли между DCM (20 мл) и насыщенным солевым раствором (10 мл). Слой DCM сушили над сульфатом натрия и концентрировали с получением (R,Е)-3-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-карбонил)-2,8,8-триметил 5,10,14-триоксо-9,11,13-триокса-4,6-диазагептадек-15-ен-17-овой кислоты (0,022 г, 0,035 ммоль, выход 10,11%). Полученное полутвердое вещество растворяли в МеОН (0,2 мл) и воде (1,5 мл) и сушили сублимацией в течение ночи до образования твердого вещества. Аналитическая RP-HPLC: tR = 3,40 мин (YMC S5 Стадия 1. Соединение 1 в CH2Cl2 (12 мл) при 0 С добавляли в пиридин (0,534 мл, 6,61 ммоль) с последующим добавлением 2-бромэтилхлорформиата (413 мг, 1,982 ммоль). Реакционную смесь оставляли медленно нагреваться до к.т. и перемешивали в течение 4 ч. Реакционную смесь концентрировали и разделяли между ЕА (40 мл) и водой (20 мл). Слои разделяли и ЕА слой дополнительно промывали 0,5 н.HCl, разбавляли водным NaHCO3, затем насыщенным солевым раствором. ЕА слой сушили (Na2SO4) отфильтровывали и концентрировали. Продукт очищали при помощи флэш-хроматографии на SiO2 (2550% ЕА/гептан - 100% ЕА) и сушили с получением 2-бромэтил-1-(3-R)-1-S)-4-(4-хлорфенил)-4 гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1-оксобутан-2-ил)уреидо)-2-метилпропан-2-илкарбонатаK2CO3 (356 мг, 2,58 ммоль) в DMF (3 мл) добавляли морфолин и реакционную смесь перемешивали при к.т. в течение ночи. После сушки неочищенного продукта в течение ночи в высоком вакууме остаточное стекло очищали при помощи колоночной хроматографии (50% ЕА/гепт. - 100% ЕА - 5% МеОН/ЕА) с получением 1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1-оксобутан-2-ил)уреидо)-2-метилпропан-2-ил-2-морфолинэтилкарбоната (620 мг, 1,014 ммоль, выход 79%) в виде пены. 1 Н ЯМР (CDCl3, 400 МГц, ротамерный) 7,40-7,24 (m, 4 Н), 5,44 (dd, J=21,9, 8,8 Гц, 1H), 5,13(dt, J=21,1, 5,7 Гц, 1H), 4,82 (m, 1H), 4,65 (br d, 0,5 Н), 4,20 (t, J=6,2 Гц, 2 Н), 3,95 (m, 0,5 Н), 3,71 (t, J=4,8 Гц, 4 Н), 3,58 (m, 1 Н), 3,43 (m. 3 Н), 3,15 (dt, J=13,1, 2,6 Гц, 0,5 Н), 3,05 (d, J=12,7 Гц, 0,5 Н), 2,64 (t, J=5,7 Гц, 2 Н), 2,51 (t, J=4,4 Гц, 4 Н), 1,96 (m, 1 Н), 1,54 (m, 1 Н), 1,47-1,43 (m, 6 Н), 1,06 (d, J=6,6 Гц, 1,5 Н), 0,970,76 (m, 10,5 Н). Чистота HPLC: 99%, время удерживания 3,23 мин (Chromalith Speedrod, С 18, 4,650 мм,10% МеОН/вода - 90% МеОН/вода с 0,2% Н 3 РО 4, градиент 4 мин, 4 мл/мин). LCMS: 611,38 (М+). Образование гидрохлорида К раствору 1-(3-R)-1-S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1 оксобутан-2-ил)уреидо)-2-метилпропан-2-ил-2-морфолинэтилкарбоната (222 мг, 0,363 ммоль) в ЕА (4 мл) добавляли HCl в диоксане (0,086 мл, 0,345 ммоль). Смесь недолго облучали ультразвуком до образования в растворе легкого помутнения, затем оставляли быстро перемешиваться в течение ночи. Твердые вещества отфильтровывали и сушили с получением 220 мг твердого вещества. 1 Н ЯМР (DMSO-d6, 400 МГц, ротамерный)7,45 (m, 2 Н), 7,35 (m, 2 Н), 6,44 (m, 0,5 Н, 6,20 (m, 0,5 Н), 5,09 (s, 0,7 Н), 4,58 (m, 0,7 Н),4,39 (m, 2 Н), 3,94 (m, 4 Н), 3,73 (t, J=11,9 Гц, 2 Н), 3,43-2,89 (m, 18 Н), 1,52-1,32 (m, 8 Н), 0,2 (d, J= 6,6 Гц,1 Н), 0,82 (d, J=6,6 Гц, 3 Н), 0,79 (d, J=6,6 Гц, 2 Н), 0,69 (m, 3 Н), 0,61 (m, 4 Н). Чистота HPLC: 99%, время удерживания 3,25 мин (Chromalith Speedrod, С 18, 4,650 мм, 10% МеОН/вода - 90% МеОН/вода с 0,2% Н 3 РО 4, градиент 4 мин, 4 мл/мин). Пример 8(456 мг, 1,322 ммоль) с последующим добавлением 1H-тетразола (93 мг, 1,322 ммоль). Реакционная смесь содержала некоторое количество твердых веществ, которые растворялись через несколько минут. Добавляли Н 2 О 2 (0,020 мл, 0,661 ммоль) и перемешивали несколько часов. Затем реакционную смесь промывали 10% Na2S2O5, 1 г HCl и насыщенным солевым раствором. Органический слой сушили над сульфатом натрия и концентрировали с получением 560 мг стекла, которое очищали на силикагеле в 3:1 1:1 гексанах/EtOAc - 100% EtOAc - 9:1 метиленхлорида/метанола с получением дибензил-1-(3-R)-1S)-4-(4-хлорфенил)-4-гидрокси-3,3-диметилпиперидин-1-ил)-3-метил-1-оксобутан-2-ил)уреидо)-2 метилпропан-2-илфосфата (460 мг, 0,644 ммоль, выход 97%) продукта в виде стекла. Применимость В основном было показано, что пролекарства соединения формулы (I) являются модуляторами активности хемокиновых рецепторов. Ожидается, что соединения будут применимы для лечения заболеваний человека, связанных с хемокинами и соответствующими им рецепторами, поскольку они характеризуются активностью модуляторов активности хемокиновых рецепторов. Пролекарства согласно настоящему изобретению получали для обеспечения свойств улучшенной растворимости и стабильности соединения I, как показано ниже в табл. I. Растворимость соединения 1 составляет 0,047 мг/мл в буфере с рН 6,6, как определено изложенными выше способами. Период полужизни при рН 1 составляет 30 ч, а при рН 7,4 составляет 22 ч в соответствии с изложенными способами в описанных выше анализах. Для таблицы 1 период полужизни в воде относится к превращению пролекарства в соединение 1. Анализ растворимости в воде при условиях термодинамического равновесия Приготовление стандартов Калибровочный стандарт готовят путем точного взвешивания 0,5-0,7 мг образца в 5 мл метанола. Если вещество не является полностью растворимым в метаноле, применяют другие растворители, такие как DMSO, или смешанные растворители. Калибровочный стандарт, как правило, готовят свежим непосредственно перед началом анализа. Для определения концентрации конечного раствора следует применять построенную по двум точкам калибровочную кривую. Последовательное разведение выполняют на стандартном растворе. Приготовление исследуемого образца Конечный насыщенный раствор готовят путем добавления 1,0 мл соответствующего водного растворителя к оставшейся части вещества (1 мг/1 мл) в пузырек емкостью 1 драхма. Раствор подвергают обработке ультразвуком и перемешивают на вортекс-мешалке в течение 30 с. Раствор образца помещают на орбитальный шейкер, который непрерывно покачивает растворы образца в течение 15-24 ч при комнатной температуре. Конечный насыщенный раствор затем переносят в пробирку "Эппендорф" емкостью 1,5 мл и центрифугируют в течение 2 мин при 10000 об./мин. Супернатант из насыщенного раствора переносят в стеклянную пробирку для HPLC без фильтрования, поскольку объем 1,5 мл недостаточен для пропитывания шприцевого фильтра. Эта процедура подготовки образца сводит к нулю эффекты неспецифического связывания фильтровальной установкой. Количественное определение с помощью LC Стандарты и образец анализировали с помощью HPLC (ВЭЖХ) с применением либо диодной матрицы для UV/видимого спектра, либо переменной длины волны определения. Типичные длины волны для количественного определения составляют 210 или 254 нм; длина волны определения может быть подобрана индивидуально для оптимизации чувствительности. Дополнительно к определению в UV рекомендуют масс-спектрометрическое определение, если доступно, с целью подтверждения подлинности интересующего пика при HPLC в UV. Разведения водных исследуемых растворов выполняют, если пик при HPLC в UV находится за пределами линейной части стандартной калибровочной кривой. Типичные разведения включают 100 мкл/900 мкл (X10) или 500 мкл/500 мкл (2 Х), в случае необходимости. Реактивы Используют растворители качества для HPLC. Анализ стабильности раствора Ацетонитриловый маточный раствор: готовят ацетонитриловый маточный раствор пролекарства путем растворения 1,5-2,0 мг точно взвешенного соединения в 5,0 мл ацетонитрила в 5 мл мерной пробирке. Рабочий буферный раствор рН 7,4: готовят рабочий буферный раствор рН 7,4 лекарственного средств путем добавления 1,5 мл ацетонитрилового маточного раствора к 3,5 мл буфера для стабильности в 20 мл сцинтилляционный флакон, очень хорошо перемешивают. С помощью 3 мл шприца удаляют 3 мл раствора и фильтруют с применением шприцевого фильтра Gelman с размером пор 0,45 мкм в чистую 1,5 мл пробирку для LC. Этот профильтрованный раствор применяют для оценки разрушения пролекарства в течение исследования. Целевая концентрация: 90-120 мкг/мл (70% водный: 30% ацетонитриловый). Кислый рабочий раствор рН 1,0: готовят рабочий раствор лекарственного средства с рН 1,0 путем добавления 1,5 мл ацетонитрилового маточного раствора к 3,5 мл буфера для стабильности в 20 мл сцинтилляционный флакон, очень хорошо перемешивают. С помощью 3 мл шприца удаляют 3 мл раствора и фильтруют с применением шприцевого фильтра Gelman с размером пор 0,45 мкм в чистую 1,5 мл пробирку для LC. Этот профильтрованный раствор применяют для оценки разрушения пролекарства в течение исследования. Целевая концентрация: 90-120 мкг/мл (70% водный: 30% ацетонитриловый). Один образец (n=1) готовят для каждого условия рН при следовании описанной выше процедуре приготовления образца. Образцы затем помещают в поддерживаемый при 37 С автодозатор для HPLC и образцы анализируют в течение 24 ч. Оставшееся пролекарство (%) представляют относительно исходной площади пика (t = 0 ч). В случаях, когда можно рассчитать полупериод превращения в материнское соединение, получают значение t1/2. Подтверждение превращения в материнское соединение из пролекарства получают с помощью жидкостной хромато-масс-спектрометрии (LCMS) и анализа HPLC образцов в конечный момент времени. Типичные параметры LC показаны ниже. Система HPLC: серия HP 1100, Hewlett Packard., подогреваемый автодозатор. Аналитическая колонка: Synergi 4u Hydro С 18, 4,6 мм 5,0 см, Phenomenex. Температура колонки: 40 С. Температура автодозатора: 37 С. Скорость потока: 1,0 мл/мин. Объем введения: 10 мкл. Подвижная фаза: А: ацетонитрил. В: 0,1% фосфорная кислота в воде. Время хроматографирования: 14,0 мин. Типичные параметры для жидкостной хромато-масс-спектрометрии (LCMS) показаны ниже: Система для жидкостной хроматографии-масс-спектрометрии (LC-MS): система HPLC Surveyor,ThermoFinnigan LCQ Deca XP Max (ионная ловушка). Аналитическая колонка: Synergi 4u Hydro С 18, 4,6 мм 5,0 см, Phenomenex. Температура колонки: 40 С. Температура автодозатора: 22 С. Скорость потока: 1,0 мл/мин. Объем введения: 5 мкл. Подвижная фаза: А: 95% ацетонитрила/5% 20 мМ ацетата аммония. В: 5% ацетонитрила/95% 20 мМ ацетата аммония. Параметры LCQ Скорость потока высушивающего газа: 81,64. Скорость потока вспомогательного/продувочного газа: 19,01. Сила тока (мкА): 10,89. Напряжение (кВ): 5,00. Капилляр (С): 348,10. Напряжение на капилляре (В): 30,44. Хемокиновые рецепторы млекопитающих обеспечивают мишень для препятствования или стимуляции функции иммунной клетки у млекопитающего, такого как человек. Соединения, которые ингибируют или стимулируют функцию хемокинового рецептора, являются особенно применимыми для модуляции функции иммунной клетки для терапевтических целей. Соответственно, настоящее изобретение относится к одному или нескольким пролекарствам соеди- 20022150 нения формулы (I), которые считаются применимыми для профилактики и/или лечения множества разнообразных воспалительных, инфекционных и иммунорегуляторных нарушений и заболеваний, включая бронхиальную астму и аллергические заболевания, инфекции патогенных микроорганизмов (которые по определению включают вирусы), а также аутоиммунных патологий, таких как ревматоидный артрит и атеросклероз. Например, соединения согласно настоящему изобретению, которые ингибируют одну или несколько функций хемокинового рецептора млекопитающих (например, хемокинового рецептора человека),можно вводить для ингибирования (т.е. ослабления или профилактики) воспаления или инфекционного заболевания. В результате ингибируется один или несколько воспалительных процессов, таких как миграция лейкоцитов, адгезия, хемотаксис, экзоцитоз (например, ферментов, гистамина) или высвобождение медиаторов воспаления. Подобным образом соединения согласно настоящему изобретению, которые стимулируют одну или несколько функций хемокинового рецептора млекопитающего (например, хемокинового рецептора человека), при введении стимулируют (индуцируют или усиливают) иммунную или воспалительную реакцию, такую как миграция лейкоцитов, адгезия, хемотаксис, экзоцитоз (например, ферментов, гистамина) или высвобождения медиаторов воспаления, что приводит к полезной стимуляции воспалительных процессов. Например, эозинофилы могут быть рекрутированы для борьбы с паразитарными инвазиями. К тому же лечение вышеупомянутых воспалительных, аллергических и аутоиммунных заболеваний также может быть предусмотрено для соединения согласно настоящему изобретению, которое стимулирует одну или несколько функций хемокинового рецептора млекопитающего, если предусмотрена доставка достаточного количества соединения, чтобы вызвать прекращение экспрессии рецептора на клетках посредством индукции интернализации хемокинового рецептора, или такая доставка соединения, которая приводит к нарушению направлений миграции клеток. Помимо приматов, таких как люди, могут получать лечение множество других млекопитающих. Например, лечение могут получать млекопитающие, включающие без ограничения коров, овец, коз, лошадей, собак, кошек, морских свинок, крыс или других видов крупного рогатого скота, мелкого рогатого скота, лошадиных, собачьих, кошачьих, грызунов или мышиных. Однако способ также может быть реализован и на других видах, таких как виды птиц. Получающий лечение вышеописанными способами субъект является млекопитающим, самцом или самкой, для которого желательна модуляция активности хемокинового рецептора. Подразумевается, что используемый в настоящем документе термин "модуляция" охватывает антагонизм, агонизм, частичный антагонизм и/или частичный агонизм. Заболевания или состояния человека или другого вида, которые можно лечить ингибиторами функции хемокинового рецептора, включают без ограничения воспалительные или аллергические заболевания и состояния, включая респираторные аллергические заболевания, такие как бронхиальная астма, аллергический ринит, связанные с гиперчувствительностью легочные заболевания, гиперчувствительный пневмонит, эозинофильный целлюлит (например, синдром Велла), эозинофильные пневмонии (например, синдром Леффлера, хроническая эозинофильная пневмония), эозинофильный фасциит (например,синдром Шульмана), гиперчувствительность замедленного типа, интерстициальные заболевания легких(ILD) (например, идиопатический легочный фиброз или ILD, ассоциированные с ревматоидным артритом, системной красной волчанкой, анкилозирующим спондилитом, системным склерозом, синдромом Шегрена, полиомиозитом или дерматомиозитом); общую анафилактическую реакцию, или реакции гиперчувствительности, лекарственные аллергии (например, на пенициллин, цефалоспорины), синдром эозинофилии-миалгии вследствие приема загрязненного триптофана, аллергии на укусы насекомых; аутоиммунные заболевания, такие как ревматоидный артрит, псориатический артрит, рассеянный склероз,системная красная волчанка, тяжелая миастения, ювенильный сахарный диабет; гломерулонефрит, аутоиммунный тиреоидит, болезнь Бехчета; отторжение трансплантата (например, при трансплантации),включая отторжение аллотрансплатата или реакцию "трансплантат против хозяина"; воспалительные заболевания кишечника, такие как болезнь Крона и язвенный колит; спондилоартропатии; склеродермию; псориаз (включая псориаз, опосредованный Т-клетками) и воспалительные дерматозы, такие как дерматит, экзема, атопический дерматит, аллергический контактный дерматит, крапивница; васкулит(например, некротизирующий, кожный и гиперчувствительный васкулит); эозинофильный миозит, эозинофильный фасциит; виды злокачественных опухолей с лейкоцитарной инфильтрацией кожи или органов. Можно лечить и другие заболевания или состояния, при которых следует подавить нежелательные воспалительные реакции, включая без ограничения реперфузионные повреждения, атеросклероз, определенные злокачественные новообразования системы крови, цитокин-индуцируемая токсичность (например, септический шок, эндотоксический шок), полимиозит, дерматомиозит. Инфекционные заболевания или состояния человека или другого вида, которые можно лечить ингибиторами функции хемокинового рецептора, включают без ограничения ВИЧ-инфекцию. Заболевания или состояния людей или других видов, которые можно лечить стимуляторами функции хемокинового рецептора, включают без ограничения иммуносупрессию, такую как имеющая место у индивидов с синдромами иммунодефицита, такими как СПИД (AIDS), или другими вирусными инфекциями, индивидов, подвергающихся лучевой терапии, химиотерапии, терапии аутоиммунного заболева- 21022150 ния или лекарственной терапии (например, кортикостероидами), которая вызывает иммуносупрессию; иммуносупрессию вследствие врожденной недостаточности функции рецептора или другой причины; и инфекционные заболевания, такие как паразитарные заболевания, включая без ограничения инвазию гельминтов, таких как нематоды (круглые черви) (трихуриаз, энтеробиоз, аскаридоз, анкилостомоз,стронгилоидоз, трихинеллез, филяриатоз); трематоды (сосальщики) (шистосомоз, клонорхоз), цестоды(ленточные черви) (эхинококкоз, тениаринхоз, цистицеркоз); инвазия червей во внутренние органы, висцеральный синдром "блуждающей личинки" (например, токсокара), эозинофильный гастроэнтерит (например, Anisaki sp., Phocanema sp.), кожный синдром "блуждающей личинки" (Ancylostona braziliense,Ancylostoma caninum). Соединения согласно настоящему изобретению являются соответственно применимыми для профилактики и лечения множества разнообразных воспалительных, инфекционных и иммунорегуляторных нарушений и заболеваний. К тому же лечение вышеупомянутых воспалительных, аллергических и аутоиммунных заболеваний может также быть предусматрено для стимуляторов функции хемокинового рецептора, если предусмотрена доставка достаточного количества соединения, чтобы вызвать прекращение экспрессии рецептора на клетках посредством индукции интернализации хемокинового рецептора, или такую доставку соединения, которая приводит к нарушению направлений миграции клеток. Согласно другому аспекту настоящее изобретение может быть применено для оценки предполагаемых специфических агонистов или антагонистов сопряженного с G-белком рецептора. Настоящее изобретение относится к применению одного или нескольких пролекарств соединения формулы (I) в подготовке и выполнении скрининг-анализов в отношении соединений, которые модулируют активность хемокиновых рецепторов. Кроме того, соединения согласно настоящему изобретению применимы для установления или определения сайта связывания других соединений с хемокиновыми рецепторами, например, путем конкурентного ингибирования или в качестве эталона при анализе для сравнения их известной активности с соединением с неизвестной активностью. При разработке новых анализов или протоколов соединения согласно настоящему изобретению можно применять для исследования их эффективности. В частности, такие соединения могут быть предусмотрены в коммерческом наборе, например, для применения в фармацевтических исследованиях, касающихся вышеупомянутых заболеваний. Соединения согласно настоящему изобретению также применимы для оценки предполагаемых специфических модуляторов хемокиновых рецепторов. К тому же соединения по настоящему изобретению могут быть использованы для проверки специфичности сопряженных с G-белком рецепторов, которые не предполагаются как хемокиновые рецепторы, либо как служащие примерами соединений, которые не связываются, либо как структурные варианты активных в отношении этих рецепторов соединений, что может помочь определить конкретные участки взаимодействия. Пролекарства соединения формулы (I) могут использоваться для лечения или профилактики нарушений, выбранных из ревматоидного артрита, остеоартрита, септического шока, атеросклероза, аневризмы, лихорадки, сердечно-сосудистых эффектов, гемодинамического шока, септического синдрома, постишемического реперфузионного повреждения, малярии, болезни Крона, воспалительных заболеваний кишечника, микобактериальной инфекции, менингита, псориаза, застойной сердечной недостаточности,фибротических заболеваний, кахексии, отторжения трансплантата, аутоиммунных заболеваний, кожных воспалительных заболеваний, рассеянного склероза, радиационного повреждения, повреждения альвеол при гипероксии, ВИЧ, деменции при ВИЧ, инсулиннезависимого сахарного диабета, бронхиальной астмы, аллергического ринита, атопического дерматита, идиопатического легочного фиброза, буллезного пемфигоида, гельминтных паразитарных инвазий, аллергического колита, экземы, конъюнктивита,трансплантации, семейной эозинофилии, эозинофильного целлюлита, эозинофильных пневмоний, эозинофильного фасциита, эозинофильного гастроэнтерита, лекарственной эозинофилии, муковисцидоза,синдрома Черджа-Стросса, лимфомы, болезни Ходжкина, карциномы ободочной кишки, синдрома Фелти, саркоидоза, увеита, болезни Альцгеймера, гломерулонефрита и системной красной волчанки. Согласно другому аспекту пролекарства соединения формулы (I) могут использоваться для лечения или профилактики воспалительных нарушений, выбранных из ревматоидного артрита, остеоартрита,атеросклероза, аневризмы, лихорадки, сердечно-сосудистых эффектов, болезни Крона, воспалительных заболеваний кишечника, псориаза, застойной сердечной недостаточности, рассеянного склероза, аутоиммунных заболеваний, кожных воспалительных расстройств. Согласно другому аспекту пролекарства соединения могут использоваться для лечения или профилактики воспалительных нарушений, выбранных из ревматоидного артрита, остеоартрита, атеросклероза,болезни Крона, воспалительных заболеваний кишечника и рассеянного склероза. Комбинированная терапия для профилактики и лечения воспалительных, инфекционных и иммунорегуляторных нарушений и заболеваний, включая бронхиальную астму и аллергические заболевания, а также аутоиммунные патологии, такие как ревматоидный артрит и атеросклероз, и приведенных выше патологий иллюстрируется комбинацией соединений согласно настоящему изобретению и других известных для таких применений соединений. Например, при лечении или профилактике воспаления соединения согласно настоящему изобретению могут быть применены совместно с противовоспалительным или анальгезирующим средством, таким как опиатный агонист, ингибитор липоксигеназы, ингиби- 22022150 тор циклооксигеназы-2, ингибитор интерлейкинов, такой как ингибитор интерлейкина-1, ингибитор фактора некроза опухолей, антагонист NMDA, ингибитор оксида азота или ингибитор синтеза оксида азота,нестероидное противовоспалительное средство, ингибитор фосфодиэстеразы или цитокинсупрессирующее противовоспалительное средство, например, с таким соединением, как ацетаминофен,аспирин, кодеин, фентанил, ибупрофен, индометацин, кеторолак, морфин, напроксен, фенацетин, пироксикам, стероидный анальгетик, суфентанил, сулиндак, интерферон-альфа и т.п. Подобным образом, соединения согласно настоящему изобретению могут быть введены с анальгезирующим средством; потенцирующим средством, таким как кофеин, антагонист Н 2-гистаминовых рецепторов, симетикон, гидроксид алюминия или магния; противоотечным средством, таким как фенилэфрин, фенилпропаноламин,псевдоэфедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или леводезоксиэфедрин; и противокашлевым средством, таким как кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; диуретиком; и седативным или неседативным антигистаминным средством. Подобным образом, соединения согласно настоящему изобретению могут быть применены в комбинации с другими лекарственными средствами, которые применяют при лечении/профилактике/подавлении или снижении интенсивности симптомов заболеваний или состояний, при которых применимы соединения согласно настоящему изобретению. Такие другие лекарственные средства можно вводить путем и в количестве, которые обычно используют для этого, одновременно или последовательно с соединением согласно настоящему изобретению. Если соединения согласно настоящему изобретению применяют одновременно с одним или несколькими другими лекарственными средствами, в дополнение к соединениям согласно настоящему изобретению может быть применена содержащая такое другое лекарственное средство фармацевтическая композиция. Соответственно, фармацевтические композиции согласно настоящему изобретению включают таковые, которые также содержат один или несколько других активных ингредиентов дополнительно к соединениям согласно настоящему изобретению. Примеры других активных ингредиентов, которые могут быть объединены с соединениями согласно настоящему изобретению или введены отдельно или в таких же фармацевтических композициях,включают без ограничения (а) антагонисты интегрина, такие как антагонисты селектинов, ICAM и VLA4; (b) стероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон, дексаметазон и гидрокортизон; (с) иммунодепрессанты, такие как циклоспорин, такролимус, рапамицин и другие иммунодепрессанты FK-506 типа; (d) антигистамины (антагонисты Н 1-гистамина), такие как бромфенирамин,хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин,фенирамин, пириламин, астемизол, терфенадин, лоратадин, цетиризин, фексофенадин, дескарбоэтоксилоратадин и т.п.; (е) нестероидные противоастматические средства, такие как b2-агонисты (тербуталин,метапротеренол, фенотерол, изоэтарин, альбутерол, битолтерол и пирбутерол), теофиллин, кромолин натрия, атропин, ипратропия бромид, антагонисты лейкотриена (зафирлукаст, монтелукаст, пранлукаст,иралукаст, побилукаст, SKB-102,203), ингибиторы биосинтеза лейкотриенов (зилейтон, BAY-1005); (f) нестероидные противовоспалительные средства (NSAID), такие как производные пропионовой кислоты(алминопрофен, бенксапрофен, буклоксовая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен,флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен,пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (индометацин, ацеметацин, алклофенак, клиданак, диклофенак, фенклофенак, фенклозиновая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенаминовой кислоты (флуфенамовая кислота, меклофенамовая кислота, мефенамовая кислота, нифлумовая кислота и толфенамовая кислота), производные бифенилкарбоновой кислоты(ацетилсалициловая кислота, сульфасалазин) и пиразолоны (апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (g) ингибиторы циклооксигеназы-2 (СОХ-2); (h) ингибиторы фосфодиэстеразы IV типа (PDE-IV); (i) другие антагонисты хемокиновых рецепторов; (j) средства, понижающие уровень холестерина, такие как ингибиторы HMG-CoA-редуктазы (ловастатин, симвастатин и правастатин, флувастатин, аторвастатин и другие статины), секвестранты (холестирамин и колестипол),никотиновая кислота, производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (k) противодиабетические средства, такие как инсулин, сульфонилмочевины,бигуаниды (метформин), ингибиторы -глюкозидазы (акарбоза) и глитазоны (троглитазон и пиоглитазон); (1) препараты интерферонов (интерферон альфа-2 а, интерферон-2 В, интерферон альфа-N3, интерферон бета-1 а, интерферон бета-1b, интерферон гамма-1b); (m) противовирусные соединения, такие как эфавиренз, невирапин, индинавир, ганцикловир, ламивудин, фамцикловир и залцитабин; (n) другие соединения, такие как 5-аминосалициловая кислота и ее пролекарства, антиметаболиты, такие как азатиоприн и 6-меркаптопурин, и цитотоксические химиотерапевтические средства против злокачественных опухолей. Массовое отношение соединений согласно настоящему изобретению со вторым активным ингредиентом может меняться и будет зависеть от эффективных доз каждого ингредиента. Как правило, используют эффективную дозу каждого. Таким образом, например, если соединения согласно настоящему изобретению объединяют с NSAID, массовое отношение соединения согласно на- 23022150 стоящему изобретению к NSAID, как правило, находится в диапазоне от приблизительно 1000:1 до приблизительно 1:1000, или альтернативно от приблизительно 200:1 до приблизительно 1:200. Как правило,комбинации соединений согласно настоящему изобретению и других активных ингредиентов также находятся в пределах вышеупомянутого диапазона, но в каждом случае следует использовать эффективную дозу каждого активного ингредиента. Млекопитающему соединения могут быть введены в терапевтически эффективном количестве. Под"терапевтически эффективным количеством" подразумевается количество соединения, которое при введении млекопитающему отдельно или в комбинации с дополнительным терапевтическим средством является эффективным для профилактики или облегчения состояния тромбоэмболического заболевания или прогрессирования заболевания. Дозировка и состав Соединения согласно настоящему изобретению могут быть введены в таких пероральных лекарственных формах, как таблетки, капсулы (каждая из которых содержит составы замедленного высвобождения или высвобождения с установленным временем), пилюли, порошки, гранулы, эликсиры, тинктуры,суспензии, сиропы и эмульсии. Они также могут быть введены во внутривенной (болюсной или инфузионной), интраперитонеальной, подкожной или внутримышечной форме, причем все используемые лекарственные формы хорошо известны специалистам в области фармации. Они могут быть введены по отдельности, но обычно их вводят с фармацевтическим носителем, отобранным согласно выбранному пути введения и стандартной фармацевтической практике. Разумеется, схема введения соединений будет изменяться в зависимости от таких известных факторов, как фармакодинамические характеристики конкретного средства и его способа и пути введения, вида, возраста, пола, самочувствия, состояния здоровья и массы тела реципиента; природы и степени выраженности симптомов; вида совместного лечения; частоты введения; пути введения, функции почек и печени пациента и желаемого эффекта. Врач или ветеринар могут определить и прописать эффективное количество лекарственного средства, необходимого для профилактики, противодействия или остановки развития тромбоэмболического нарушения. В соответствии с общим руководством ежедневная пероральная дозировка каждого активного ингредиента при использовании для указанных эффектов находится в диапазоне от приблизительно 0,001 до 1000 мг/кг от массы тела или от приблизительно 0,01 до 100 мг/кг от массы тела в сутки или альтернативно от приблизительно 1,0 до 20 мг/кг/сутки. При внутривенном введении дозировки находятся в диапазоне от приблизительно 1 до приблизительно 10 мг/кг/мин при постоянной скорости инфузии. Соединения согласно настоящему изобретению могут быть введены однократной суточной дозой или суммарная суточная доза может быть введена раздельными дозами за два, три или четыре раза в сутки. Соединения согласно настоящему изобретению могут быть введены в интраназальной форме путем местного применения подходящих интраназальных носителей или чрескожным путем с применением чрескожных пластырей. Разумеется, при введении в форме системы чрескожной доставки введение непрерывно в отличие от прерывистого режима введения. Соединения, как правило, вводят в смеси с подходящими фармацевтическими разбавителями, наполнителями или носителями (в совокупности называемыми в настоящем документе фармацевтическими носителями), соответствующе выбранными согласно предполагаемой форме введения, а именно форме перорально вводимых таблеток, капсул, эликсиров, сиропов и т.п., и как предусмотрено традиционной фармацевтической практикой. Например, для перорального введения в форме таблетки или капсулы активный компонент лекарственного средства может быть объединен с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как лактоза, крахмал, сахароза, глюкоза, метилцеллюлоза, стеарат магния,двузамещенный фосфат кальция, сульфат кальция, маннит, сорбит и т.п.; для перорального введения в жидкой форме компоненты перорального лекарственного средства могут быть объединены с любым пероральным нетоксическим фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Более того, когда желательно или необходимо, в смесь могут быть также включены подходящие связующие, смазывающие вещества, разрыхлители и красители. Подходящие связующие включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические камеди, такие как гуммиарабик, трагакант или альгинат натрия,карбоксиметилцеллюлоза, полиэтиленгликоль, воск и т.п. Используемые в этих лекарственных формах смазывающие вещества включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Разрыхлители содержат без ограничения крахмал, метилцеллюлозу, агар,бентонит, ксантановую камедь и т.п. Соединения согласно настоящему изобретению также могут быть введены в форме таких липосомальных систем доставки, как небольшие моноламеллярные везикулы, большие моноламеллярные везикулы и многослойные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения согласно настоящему изобретению также могут быть соединены с растворимыми полимерами в качестве нацеливаемых носителей лекарственного средства. Такие полимеры могут вклю- 24022150 чать поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенные пальмитоиловыми остатками. Кроме того, соединения согласно настоящему изобретению могут быть соединены с представителями класса биологически разлагаемых полимеров, применимых для достижения контролируемого высвобождения лекарственного средства, например полимолочной кислоты, полигликолиевой кислоты, сополимеров полимолочной и полигликолевой кислоты, полиэпсилонкапролактона, полигидроксимасляной кислоты, сложных полиортоэфиров, полиацеталей, полигидропиранов, полицианоацилатов и сшитых или амфипатических блоксополимеров гидрогелей. Подходящие для введения лекарственные формы (фармацевтические композиции) могут содержать от приблизительно 1 мг до приблизительно 100 млг активного ингредиента на стандартную лекарственную форму. Как правило, в этих фармацевтических композициях активный ингредиент находится в количестве приблизительно 0,5-95 мас.% от общей массы композиции. Желатиновые капсулы могут содержать активный ингредиент и порошкообразные носители, такие как лактоза, крахмал, производные целлюлозы, стеарат магния, стеариновую кислоту и т.п. Подобные разбавители могут быть использованы для получения спрессованных таблеток. Как таблетки, так и капсулы могут быть произведены как продукты замедленного высвобождения для обеспечения непрерывного высвобождения лекарственного средства в течение периода времени, исчисляемого часами. Спрессованные таблетки могут быть покрыты сахаром, или покрыты пленкой для маскировки любого неприятного вкуса и для защиты таблетки от атмосферной среды, или покрыты энтеросолюбильной оболочкой для селективного распада в желудочно-кишечном тракте. Жидкие лекарственные формы для перорального введения могут содержать красители и ароматизаторы для улучшения приемлемости для пациента. Как правило, подходящие носители для парентеральных растворов представляют собой воду, подходящее масло, солевой раствор, водный раствор декстрозы (глюкозы) и растворы сходных сахаров и гликоли, такие как пропиленгликоль или полиэтиленгликоли. Растворы для парентерального введения могут содержать растворимую в воде соль активного ингредиента, подходящие стабилизирующие средства и при необходимости буферные вещества. Подходящими стабилизаторами являются антиоксиданты, такие как бисульфит натрия, сульфит натрия или аскорбиновая кислота, или отдельно, или в комбинации. Также применяют лимонную кислоту и ее соли и EDTA натрия. К тому же парентеральные растворы могут содержать консерванты, как хлорид бензалкония, метил- или пропилпарабен и хлорбутанол. Подходящие фармацевтические носители описаны в Remington's Pharmaceutical Sciences, Mack Publishing Company, стандартном руководстве в этой области техники. Характерные применимые фармацевтические лекарственные формы для введения соединений согласно настоящему изобретению могут быть описаны далее. Капсулы Большое число единичных капсул может быть получено путем заполнения каждой из стандартных состоящих из двух частей твердых желатиновых капсул 100 мг порошкообразного активного ингредиента, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния. Мягкие желатиновые капсулы Смесь активного ингредиента в перевариваемом масле, таком как соевое масло, хлопковое масло или оливковое масло, может быть получена и впрыснута посредством насоса вытесняющего действия в желатин с образованием мягких желатиновых капсул, содержащих 100 мг активного ингредиента. Капсулы должны быть промыты и высушены. Таблетки Таблетки могут быть получены традиционными способами таким образом, чтобы стандартная лекарственная форма содержала 100 мг активного ингредиента, 0,2 мг коллоидного диоксида кремния, 5 мг стеарата магния, 275 мг микрокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы. Для усиления приятного вкуса и замедления всасывания могут быть использованы соответствующие покрытия. Инъекционное лекарственное средство Парентеральная композиция, подходящая для введения путем инъекции, может быть получена перемешиванием 1,5 мас.% активного ингредиента в 10 об.% пропиленгликоля и воды. Раствор должен быть сделан изотоническим при помощи хлорида натрия и стерилизован. Суспензия Для перорального введения может быть получена водная суспензия, чтобы каждые 5 мл содержали 100 мг мелкодисперсного активного ингредиента, 200 мг карбоксиметилцеллюлозы натрия, 5 мг бензоата натрия, 1,0 г раствора сорбита, U.S.P. и 0,025 мл ванилина. Если соединения согласно настоящему изобретению объединены, например, с другими противокоагулирующими средствами, то суточная доза может составлять от приблизительно 0,1 до 100 мг соединений согласно настоящему изобретению и от приблизительно 1 до 7,5 мг второго противокоагулирующего средства на килограмм массы тела пациента. Как правило, в лекарственной форме в виде таблетки соединения согласно настоящему изобретению могут присутствовать в количестве от приблизительно 5 до 10 мг на стандартную лекарственную форму, а второе противокоагулирующее средство в количестве от приблизительно 1 до 5 мг на стандартную лекарственную форму. Как правило, если два или несколько из вышеуказанных вторых терапевтических средств вводят с соединениями согласно настоящему изобретению, ввиду аддитивного или синергетического эффекта терапевтических средств при введении в комбинации количество каждого компонента в типичной суточной дозировке и типичной лекарственной форме может быть уменьшено относительно обычной дозировки средства для раздельного введения. В особенности при объединении в стандартную лекарственную форму возникает возможность для химического взаимодействия между объединенными активными ингредиентами. По этой причине, когда соединения согласно настоящему изобретению и второе терапевтическое средство объединены в одну стандартную лекарственную форму, они включены в состав таким образом, чтобы несмотря на то, что активные ингредиенты объединены в одну стандартную лекарственную форму, физический контакт между активными ингредиентами был минимизирован (т.е. уменьшен). Например, один активный ингредиент может быть покрыт энтеросолюбильной оболочкой. Путем покрытия энтеросолюбильной оболочкой одного из активных ингредиентов может быть не только минимизирован контакт между объединенными активными ингредиентами, также возможен контроль высвобождения одного из этих компонентов в желудочно-кишечном тракте так, чтобы один из этих компонентов высвобождался не в желудке, а в кишечнике. Один из активных ингредиентов также может быть покрыт веществом, которое обеспечивает замедленное высвобождение на протяжении всего желудочно-кишечного тракта и также служит для минимизации физического контакта между объединенными активными ингредиентами. Кроме того, компонент длительного высвобождения может быть дополнительно покрыт энтеросолюбильной оболочкой, чтобы высвобождение этого компонента проходило только в кишечнике. Другая методика будет предусматривать приготовление комбинированного продукта, в котором один компонент покрыт полимером длительного высвобождения и/или полимером, высвобождающимся в кишечнике, а другой компонент также покрыт полимером, как гидроксипропилметилцеллюлоза (НРМС) низкой вязкости, или другими известными из настоящего уровня техники соответствующими веществам для дополнительного разделения активных компонентов. Полимерные покрытия служат для образования дополнительного барьера для взаимодействия с другим компонентом. На основании настоящего описания эти, а также другие пути минимизации контакта между компонентами продуктов комбинации согласно настоящему изобретению, введенные или в виде одной лекарственной формы, или введенные в отдельных формах, но в то же время таким же способом, будут вполне очевидны специалистам в данной области техники. Хотя настоящее изобретение было описано подробно и со ссылкой на конкретные варианты его осуществления, специалисту в данной области техники будет очевидно, что различные изменения и модификации могут быть сделаны без отклонения от его сущности и объема или форма его фармацевтически приемлемой соли. 2. Соединение формулы или форма его фармацевтически приемлемой соли. 3. Соединение по п.1 или 2, имеющее формулу 4. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество одного или нескольких соединений по п.2. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2
МПК / Метки
МПК: C07D 211/52
Метки: модуляторов, производного, хемокинового, активности, пиперидинильного, рецептора, пролекарства, качестве
Код ссылки
<a href="https://eas.patents.su/29-22150-prolekarstva-piperidinilnogo-proizvodnogo-v-kachestve-modulyatorov-aktivnosti-hemokinovogo-receptora.html" rel="bookmark" title="База патентов Евразийского Союза">Пролекарства пиперидинильного производного в качестве модуляторов активности хемокинового рецептора</a>
Пиперидинильные производные в качестве модуляторов активности хемокинового рецептора

Номер патента: 19192
Опубликовано: 30.01.2014
Авторы: Гарднер Дэниел С., Додд Дхармпал С., Хайнс Джон, Кавалларо Куллен Л., Картер Перси Х., Лиу Руи-Кин, Сантелла Джозеф Б., Дунсиа Джон В.
МПК: A61K 31/445, C07D 211/18, A61K 31/454...
Метки: производные, хемокинового, качестве, модуляторов, рецептора, активности, пиперидинильные
Формула / Реферат:
1. Соединение формулы (Ib)или его стереоизомеры, или фармацевтически приемлемые солевые формы, гдеТ представляет собойR1 представляет собой алкил, фенил или циклоалкил, все из которых могут быть необязательно замещены с помощью 0-5 R1a;R1a при каждом появлении независимо выбран из алкила, галогеналкила, арила, галогена, -С(=O)ОН, -C(=O)O(CR8R8)rR10 и -ОН, где арил может быть необязательно замещен с помощью 0-3 R1b;R1b при каждом появлении...
Пиперидинильное производное как модулятор активности хемокинового рецептора

Номер патента: 21224
Опубликовано: 29.05.2015
Автор: Сантелла Джозеф Б.
МПК: A61P 19/02, A61K 31/451, C07D 211/52...
Метки: модулятор, активности, производное, хемокинового, пиперидинильное, рецептора
Формула / Реферат:
1. Соединение формулы (I)или его стереоизомер или фармацевтически приемлемая соль.2. Фармацевтическая композиция для лечения и предупреждения воспалительных заболеваний, аллергических и аутоиммунных заболеваний, содержащая терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.3. Способ модуляции активности хемокина или хемокинового рецептора, заключающийся во введении нуждающемуся в этом пациенту...
Производные пиперазинилпиперидина в качестве антагонистов хемокинового рецептора

Номер патента: 15517
Опубликовано: 31.08.2011
Авторы: Чжан Кэ, Ананд Раджан, Сюэ Чу-Бяо, Цао Ганьфэн, Хуан Тайшэн, Ван Аньлай, Чэнь Лихуа, Мелони Дэвид, Меткаф Брайан, Гленн Джозеф
МПК: A61K 31/495
Метки: хемокинового, пиперазинилпиперидина, качестве, рецептора, антагонистов, производные
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль, в которомR1 представляет собой C1-C20 гетероарил, необязательно замещенный одним или несколькими R6;R2 представляет собой Н, галоген, циано, нитро, C1-С6 алкил, C1-С6галогеналкил, С2-С6 алкенил, С2-С6алкинил, C6-C18 арил, C1-C20 гетероарил, С3-С7 циклоалкил, С3-С20гетероциклоалкил, SOR7, SO2R7, COR8, OR9, SR9, COOR9, NR10R11 или NR10COR8;R3 представляет собой F, Cl, Br, I, C1-C4...
4-феноксиметилпиперидины в качестве модуляторов активности gpr119

Номер патента: 17260
Опубликовано: 30.11.2012
Авторы: Леле Жеральд, Уэсткотт-Бейкер Лукас, Никулин Виктор, Эппле Роберт
МПК: C07D 211/30, A61K 31/4523, A61P 3/10...
Метки: модуляторов, активности, качестве, 4-феноксиметилпиперидины, gpr119
Формула / Реферат:
1. Соединение формулы Iгде R1 выбран из фенила, пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, пиримидин-5-ила, пиразол-4-ила, 1H-пиразол-4-ила, 6-оксо-1,6-дигидропиридин-3-ила, 2-оксо-1,2-дигидропиримидин-5-ила, 2-оксо-1,2-дигидропиридин-4-ила и 2-оксо-2,3-дигидрооксазоло[4,5-b]пиридин-6-ила; где указанный фенил, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил, пиримидин-5-ил, пиразол-4-ил, 1H-пиразол-4-ил, 6-оксо-1,6-дигидропиридин-3-ил,...
Соединения и композиции в качестве модуляторов активности gpr119

Номер патента: 18703
Опубликовано: 30.10.2013
Авторы: Кау Кристофер, Эппле Роберт
МПК: A61K 31/497, A61P 3/10, A61P 3/04...
Метки: gpr119, активности, модуляторов, качестве, соединения, композиции
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, в которомА может содержать вплоть до 2 кольцевых атомов углерода, замещенных оксогруппой;n выбран из 0, 1 и 2;t1 и t2, каждый независимо, выбраны из 0 и 1;R1 выбран из цианогруппы, -S(O)2R6a, -N(R6a)S(O)2R6a, -S(O)2OR6a, -S(O)2C(O)R6a, -S(O)2C(O)OR6a и -S(O)2NR6aR6b, где R6a выбран из C1-6алкила, где указанный алкил в R6a необязательно замещен 1-3 радикалами, независимо...
Предыдущий патент: Абсорбирующее изделие
Случайный патент: Система стабилизации напряжения линий электроснабжения