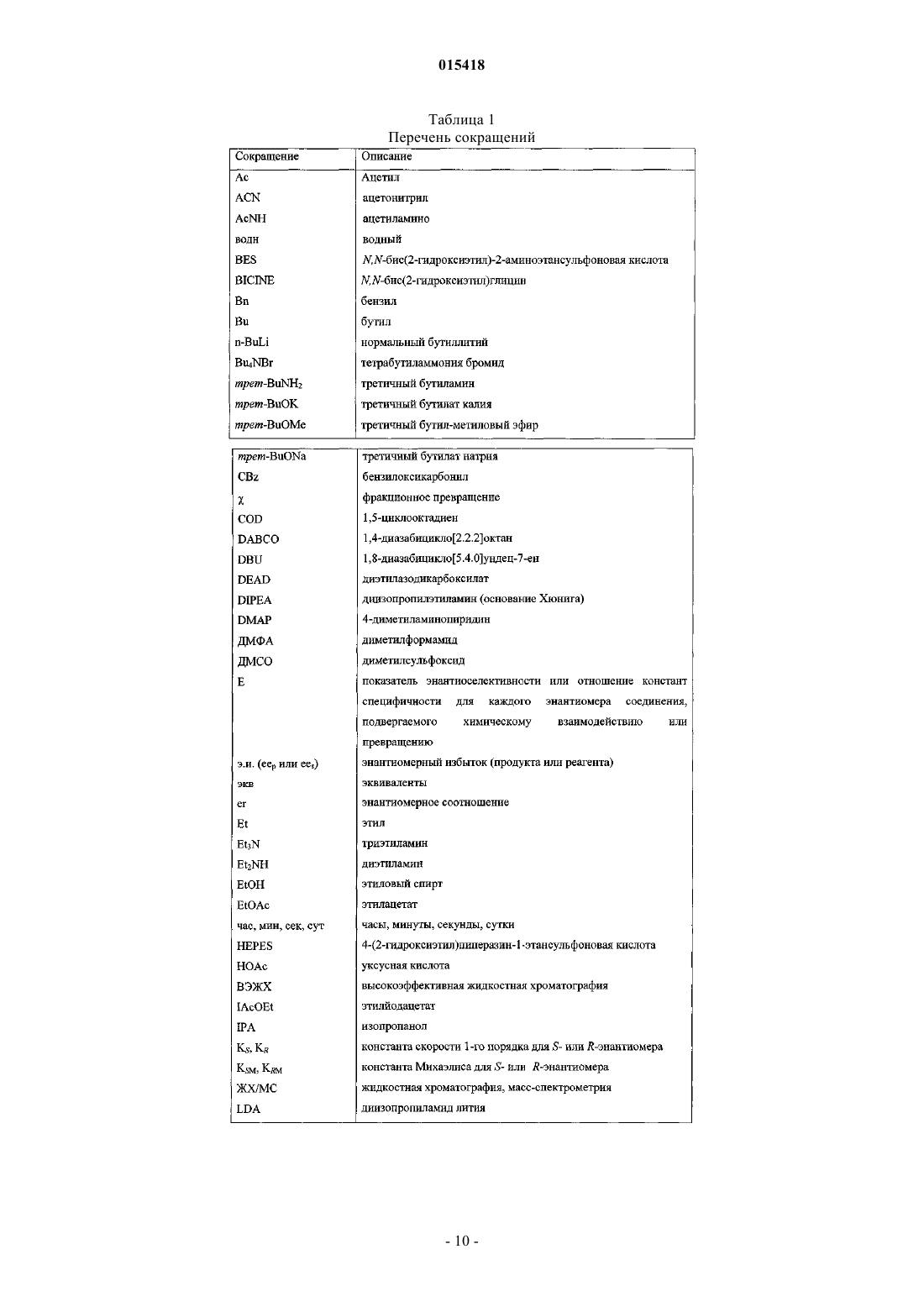
Получение прегабалина и родственных соединений
Номер патента: 15418
Опубликовано: 31.08.2011
Авторы: Келлехер Патрик Жерар Томас, Мартинес Карлос Альберто, Талли Уилльям Юджин, Тао Йунхуа, Дюмон Ив Рене, Ху Шангхуи


























Формула / Реферат
1. Способ получения соединения формулы 1
![]()
или его фармацевтически приемлемых комплекса, соли, сольвата или гидрата, где R1и R2 являются разными, и каждый независимо выбран из атома водорода, C1-12алкила, C3-12циклоалкила и замещенного
C3-12циклоалкила, включающий:
(а) взаимодействие соединения формулы 2

или его соли с кислотой и водой с получением соединения формулы 1 или его соли и
(б) возможно превращение соединения формулы 1 или его соли в фармацевтически приемлемые комплекс, соль, сольват или гидрат, где R1 и R2 в формуле 2 являются такими, как определено в формуле 1.
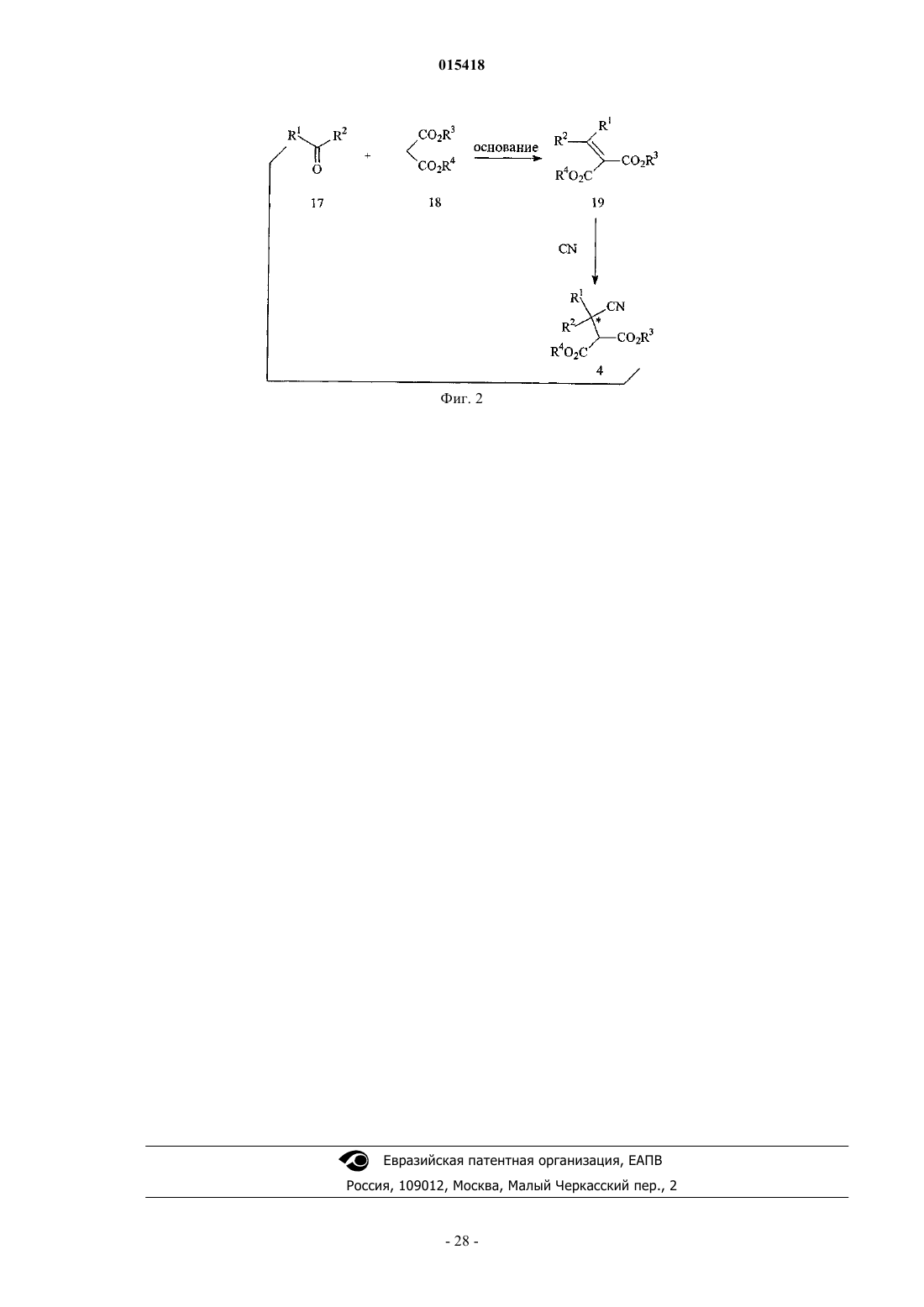
2. Способ по п.1, дополнительно включающий восстановление цианогруппировки соединения формулы 3

или его соли с получением соединения формулы 2 или его соли, где
R1 и R2в формуле 3 являются такими, как определено в формуле 1; и
R3 в формуле 3 представляет собой C1-12алкил, C3-12циклоалкил или арил-C1-6алкил.
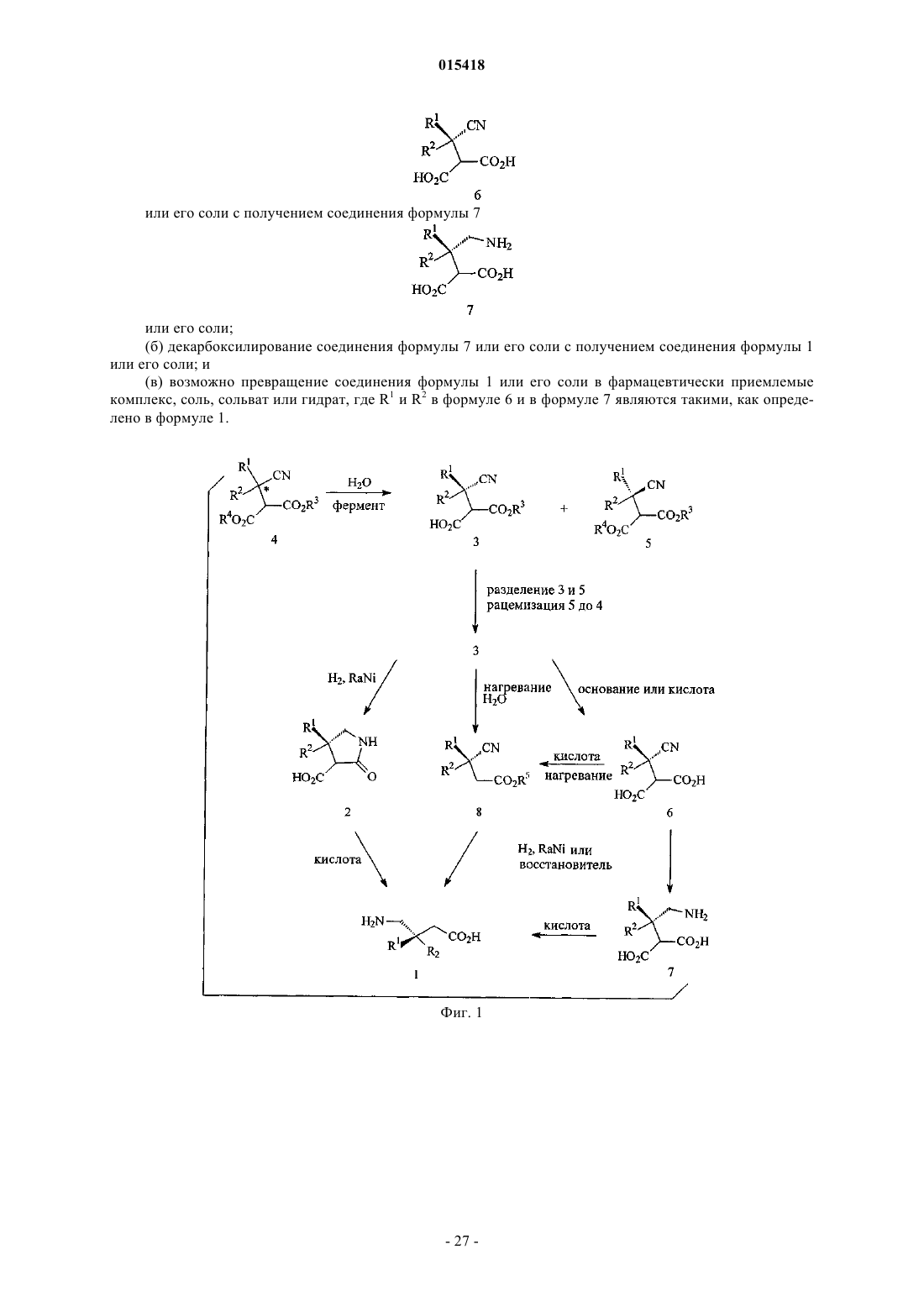
3. Способ получения соединения формулы 1
![]()
или его фармацевтически приемлемых комплекса, соли, сольвата или гидрата, где R1и R2 являются разными, и каждый независимо выбран из атома водорода, C1-12алкила, C3-12циклоалкила и замещенного
C3-12циклоалкила, включающий:
(а) восстановление цианогруппировки соединения формулы 6

или его соли с получением соединения формулы 7

или его соли;
(б) декарбоксилирование соединения формулы 7 или его соли с получением соединения формулы 1 или его соли и
(в) возможно превращение соединения формулы 1 или его соли в фармацевтически приемлемые комплекс, соль, сольват или гидрат, где R1и R2 в формуле 6 и в формуле 7 являются такими, как определено в формуле 1.
Текст
Дата публикации и выдачи патента Номер заявки Раскрыты способы получения энантиомерно обогащенных -аминокислот формулы 1, в том числе(S)-(+)-3-аминометил-5-метилгексановой кислоты (прегабалина), посредством ферментативного кинетического разделения. Ху Шангхуи, Мартинес Карлос Альберто, Тао Йунхуа (US), Талли Уилльям Юджин, Келлехер Патрик Жерар Томас, Дюмон Ив Рене (IE) Поликарпов А.В., Борисова Е.Н. (RU) 015418 Область изобретения Настоящее изобретение относится к способам и веществам для получения энантиомерно обогащенных -аминокислот посредством ферментативного кинетического разделения и особенно полезно для получения -аминокислот, которые обладают аффинностью связывания с человеческой 2 субъединицей кальциевого канала, в том числе прегабалина и родственных соединений. Предшествующий уровень техники Прегабалин, (S)-(+)-3-аминометил-5-метилгексановая кислота, является родственным эндогенному ингибиторному нейромедиатору -аминомасляной кислоте (ГАМК), которая вовлечена в регуляцию активности нейронов головного мозга. Прегабалин проявляет противоэпилептическую активность, как обсуждалось в патенте США 5563175, R. B. Silverman et al., и считается, что он является полезным для лечения, среди других состояний, боли, физиологических состояний, ассоциированных с психомоторными стимуляторами, воспаления, желудочно-кишечного повреждения, алкоголизма, бессонницы и различных психиатрических расстройств, включая манию и биполярное расстройство. Смотри, соответственно, патент США 6242488, L. Bueno et al.; патент США 6326374, L. MagnusC. A. Segal, и патент США 6001876, L.Singh; патент США 6194459, Н. С. Akunne et al.; патент США 6329429, D.Schrier et al.; патент США 6127418, L. Bueno et al.; патент США 6426368, L. Bueno et al.; патент США 6306910, L. MagnusC. A. Segal и патент США 6359005, A. C. Pande, которые включены в данное описание ссылкой во всей их полноте и для любых целей. Прегабалин получали различными путями. Обычно рацемическую смесь 3-аминометил-5 метилгексановой кислоты синтезируют и затем разделяют на R- и S-энантиомеры. В таких способах могут быть использованы азидное промежуточное соединение, малонатное промежуточное соединение или синтез Хоффмана (Hofman). См., соответственно, патент США 5563175, R. B. Silverman et al., патенты США 6046353, 5840956 и 5637767, Т. М. Grote et al.; и патенты США 5629447 и 5616793, B. K.HuckabeeD. M. Sobieray, которые включены в данное описание ссылкой во всей их полноте и для любых целей. В каждом из этих способов рацемат подвергают взаимодействию с хиральной кислотой (разделяющий агент) с образованием пары диастереоизомерных солей, которые разделяют известными методами, такими как фракционная кристаллизация и хроматография. Эти способы, таким образом, включают значительную обработку после получения рацемата, которая, наряду с разделяющим агентом, увеличивает себестоимость производства. Более того, нежелательный R-энантиомер часто отбрасывают, так как он не может быть эффективно переработан, из-за чего эффективная производительность процесса снижается до 50%. Прегабалин также синтезировали непосредственно, используя хиральное вспомогательное вещество(4R,5S)-4-метил-5-фенил-2-оксазолидинон. Смотри, например, патенты США 6359169, 6028214,5847151, 5710304, 5684189, 5608090 и 5599973, все R. B. Silverman et al., которые включены в данное описание ссылкой во всей их полноте и для любых целей. Хотя эти способы обеспечивают получение прегабалина с высокой энантиомерной чистотой, они менее желательны для крупномасштабного синтеза, так как в них используются сравнительно дорогостоящие реагенты (например хиральное вспомогательное вещество), которые сложны в обработке, а также специальное криогенное оборудование для достижения требуемых рабочих температур, которые могут достигать -78C. В недавно опубликованной заявке на патент США обсуждается способ получения прегабалина посредством асимметрического гидрирования циано-замещенного олефина с получением хирального циано-предшественника (S)-3-аминометил-5-метилгексановой кислоты. Смотри переуступленную должным образом заявку на патент США 2003/0212290 Al, Burk et al., опубликованную 13 ноября 2003 г., которая включена в данное описание ссылкой во всей ее полноте для любых целей. Циано-предшественник затем восстанавливают с получением прегабалина. В асимметрическом гидрировании используют хиральный катализатор, который состоит из переходного металла, связанного с бифосфиновым лигандом,такой как (R,R)-Me-DUPHOS -)-1,2-бис 2R,5R)-2,5-диметилфосфолано)бензол). Этот способ приводит в результате к существенному обогащению прегабалином по сравнению с (R)-3-(аминометил)-5 метилгексановой кислотой. Способ, обсуждаемый в заявке на патент США 2003/0212290 A1, представляет собой рентабельный способ получения прегабалина, однако по различным причинам были бы желательны дальнейшие улучшения. Например, бифосфиновые лиганды, включая запатентованный лиганд (R,R)-Me-DUPHOS,часто трудно получать, так как они имеют два хиральных центра, что увеличивает их стоимость. Более того, асимметрическое гидрирование требует использования специального оборудования, способного работать с H2, что увеличивает капитальные затраты. Краткое изложение сущности изобретения В настоящем изобретении предложены способы получения энантиомерно обогащенных аминокислот формулы 1 или его фармацевтически приемлемых комплекса, соли, сольвата или гидрата, где R1 и R2 являются разными, и каждый независимо выбран из атома водорода, C1-12 алкила, C3-12 циклоалкила и замещенного Способы по настоящему изобретению включают кинетическое разделение рацемического цианодиэфирного промежуточного соединения (формула 4 или 12) с использованием фермента, который адаптирован к энантиоселективному гидролизу сложноэфирной группировки данного промежуточного соединения. Полученный в результате моноэфир дикарбоновой кислоты (формула 3 или 11), который является по существу энантиомерно чистым, подвергают дополнительному взаимодействию с получением нужных энантиомерно обогащенных -аминокислот (формула 1 или 9). Необогащенный энантиомер (формула 5 или 13) из данного кинетического разделения может быть повторно использован в ферментативном разделении после рацемизации, благодаря чему увеличивается общий выход. Заявленные способы имеют значительные преимущества перед существующими способами получения энантиомерно обогащенных -аминокислот (формула 1 и 9). Например, оптически активные аминокислоты могут быть получены без использования хиральных вспомогательных веществ или запатентованных катализаторов гидрирования, что приведет к более низкой себестоимости единицы продукции. Поскольку ферментативные процессы могут быть осуществлены при комнатной температуре и атмосферном давлении, заявленные способы будут способствовать минимизации режимных конфликтов,возникающих в результате использования специализированного оборудования, способного работать с высокими давлениями и низкими температурами. Как отмечено в примерах, настоящее изобретение может быть использовано для получения прегабалина, начиная с рацемического цианозамещенного диэфира (формула 12) с хорошим выходом (от 26 до 31%) после переработки одной партии непрореагировавшего энантиомера (формула 13). Это преобразуется в приблизительно 50%-ную экономию по стоимости товара по сравнению с малонатным способом, описанным выше. В одном аспекте настоящего изобретения предложен способ получения соединения формулы 1 или его фармацевтически приемлемых комплекса, соли, сольвата или гидрата, где R1 и R2 являются разными, и каждый независимо выбран из атома водорода, C1-12 алкила, C3-12 циклоалкила и замещенного(а) взаимодействие соединения формулы 2 или его соли с кислотой и водой с получением соединения формулы 1 или его соли; и(б) возможно превращение соединения формулы 1 или его соли в фармацевтически приемлемые комплекс,соль, сольват или гидрат, где R1 и R2 в формуле 2 являются такими, как определено в формуле 1. В предпочтительном воплощении данный способ дополнительно включает восстановление цианогруппировки соединения формулы 3-2 015418 или его соли с получением соединения формулы 2 или его соли, где В другом аспекте настоящего изобретения предложен способ получения соединения формулы 1 или его фармацевтически приемлемых комплекса, соли, сольвата или гидрата, гдеR1 и R2 являются разными, и каждый независимо выбран из атома водорода, C1-12 алкила, C3-12 циклоалкила и замещенного C3-12 циклоалкила, включающий:(а) восстановление цианогруппировки соединения формулы 6 или его соли с получением соединения формулы 7(б) декарбоксилирование соединения формулы 7 или его соли с получением соединения формулы 1 или его соли; и(в) возможно превращение соединения формулы 1 или его соли в фармацевтически приемлемые комплекс, соль, сольват или гидрат, где R1 и R2 в формуле 6 и 7 являются такими, как определено в формуле 1. Соединение формулы 6, указанной выше, может быть получено путем гидролиза соединения формулы 3 или его соли, где R1 и R2 в формуле 3 являются такими, как определено в формуле 1, и R3 представляет собой C1-12 алкил, C3-12 циклоалкил или арил-C1-6 алкил. В еще одном аспекте настоящего изобретения предложен способ получения соединения формулы 1 или его фармацевтически приемлемых комплекса, соли, сольвата или гидрата, гдеR1 и R2 являются разными, и каждый независимо выбран из атома водорода, C1-12 алкила, C3-12 циклоалкила и замещенного C3-12 циклоалкила, включающий:(а) восстановление цианогруппировки соединения формулы 8 или его соли с получением соединения формулы 1 или его соли; и(б) возможно превращение соединения формулы 1 или его соли в фармацевтически приемлемые комплекс, соль, сольват или гидрат, где R1 и R2 в формуле 8 являются такими, как определено в формуле-3 015418 1, и R1 в формуле 8 представляет собой атом водорода, C1-12 алкил,C3-12 циклоалкил или арил-C1-6 алкил. Соединение формулы 8 может быть получено путем декарбоксилирования соединения формулы 3,указанной выше, или его соли, либо путем гидролиза и декарбоксилирования соединения формулы 3 или его соли, с получением соединения формулы 8 или его соли. Соединение формулы 3, указанной выше,или его соль, может быть получено путем:(а) приведения соединения формулы 4 в контакт с ферментом с получением соединения формулы 3 и соединения формулы 5 где данный фермент адаптирован к энантиоселективному гидролизу соединения формулы 4 до соединения формулы 3 или его соли;(б) выделения соединения формулы 3 или его соли; и(в) возможно рацемизации соединения формулы 5 с получением соединения формулы 4, где R1, R2 и R3 в формуле 4 и формуле 5 являются такими, как определено в формуле 1 и формуле 3; и R4 в формуле 4 и формуле 5 является таким же, как R3, или отличается от него и представляет собой C1-12 алкил, C3-12 циклоалкил или арил-C1-6 алкил. Любое количество ферментов может быть использовано для энантиоселективного гидролиза соединения формулы 4 до соединения формулы 3 или его соли. Полезные ферменты включают липазы, например липазы, получаемые из Thermomyces lanuginosus. В еще одном аспекте настоящего изобретения предложен способ получения соединения формулы 11 или его соли, где R3 представляет собой C1-12 алкил, C3-12 циклоалкил или арил-C1-6 алкил, включаю(а) приведение соединения формулы 12 в контакт с ферментом с получением соединения формулы 11 и соединения формулы 13 где данный фермент адаптирован к энантиоселективному гидролизу соединения формулы 12 до соединения формулы 11 или его соли;(б) выделение соединения формулы 11 или его соли; и(в) возможно рацемизацию соединения формулы 13 с получением соединения формулы 12, гдеR4 в формуле 12 и 13 является таким же, как R3, или отличается от него и представляет собой C1-12 алкил, C3-12 циклоалкил или арил-C1-6 алкил. В способе получения соединения формулы 11 соответствующие соли соединения формулы 11 включают соединения, выбранные из солей щелочных металлов, таких как калиевая соль; солей первичных аминов, таких как соль трет-бутиламина; и солей вторичных аминов. Кроме того, полезные ферменты включают липазы, такие как липазы, получаемые из Thermomyces lanuginosus. Настоящее изобретение включает все комплексы и соли, независимо от того, являются они или не являются фармацевтически приемлемыми, сольваты, гидраты и полиморфные формы раскрытых соединений. Некоторые соединения могут содержать алкенильную или циклическую группу, так что цис/транс(или Z/E) стереоизомеры являются возможными, или могут содержать кето или оксимную группу, так что может иметь место таутомерия. В таких случаях, настоящее изобретение в общем случае включает все Z/E изомеры и таутомерные формы, являются ли они чистыми, по существу, чистыми или смесями. Краткое описание графических материалов На фиг. 1 изображена схема получения энантиомерно обогащенных -аминокислот (формула 1). На фиг. 2 изображена схема получения цианозамещенных диэфиров (формула 4). Подробное описание изобретения определения и сокращения Если не указано иное, в данном описании используют определения, предложенные ниже. Некоторые из определений и формул могут включать дефис ("-") для указания связи между атомами или точки присоединения к названному или неназванному атому или группе атомов. Другие определения и формулы могут включать знак равенства ("=") или символ идентичности ("=") для указания двойной связи или тройной связи соответственно. Некоторые формулы могут также включать одну или более чем одну звездочку для указания стереогенных (асимметрических или хиральных) центров, хотя отсутствие звездочки не указывает на то, что у этого соединения отсутствует стереоцентр. Такие формулы могут относиться к рацемату, или к индивидуальным энантиомерам, или к индивидуальным диастереомерам,которые могут быть, либо могут не быть чистыми или, по существу, чистыми."Замещенные" группы представляют собой группы, в которых один или более чем один атом водорода заменен одной или более группами, отличными от водорода, при условии, что соблюдены требования валентности и что результатом этого замещения является химически стабильное соединение."Примерно" или "приблизительно", при использовании в связи с измеримой количественной переменной, относится к указанному значению этой переменной или ко всем значениям этой переменной,которые находятся в пределах экспериментальной ошибки указанного значения (например в пределах 95%-ного доверительного интервала для среднего) или в пределах 10% от указанного значения в зависимости от того, что из них больше."Алкил" относится к неразветвленным и разветвленным насыщенным углеводородным группам,обычно имеющим определенное число атомов углерода (то есть C1-6 алкил относится к алкильной группе,имеющей 1, 2, 3, 4, 5 или 6 атомов углерода, и C1-12 алкил относится к алкильной группе, имеющей 1, 2, 3,4, 5, 6, 7, 8, 9, 10, 11 или 12 атомов углерода). Примеры алкильных групп включают, без ограничения ими, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, пент-1-ил, пент-2-ил,пент-3-ил, 3-метилбут-1-ил, 3-метилбут-2-ил, 2-метилбут-2-ил, 2,2,2-триметилэт-1-ил, н-гексил и тому подобное."Алкенил" относится к неразветвленным и разветвленным углеводородным группам, имеющим одну или более чем одну ненасыщенную углерод-углеродную связь и обычно имеющим определенное число атомов углерода. Примеры алкенильных групп включают, без ограничения ими, этенил, 1-пропен-1 ил, 1-пропен-2-ил, 2-пропен-1-ил, 1-бутен-1-ил, 1-бутен-2-ил, 3-бутен-1-ил, 3-бутен-2-ил, 2-бутен-1-ил, 2 бутен-2-ил, 2-метил-1-пропен-1-ил, 2-метил-2-пропен-1-ил, 1,3-бутадиен-1-ил, 1,3-бутадиен-2-ил и тому подобное."Алкинил" относится к неразветвленным и разветвленным углеводородным группам, имеющим одну или более чем одну тройную углерод-углеродную связь и обычно имеющим определенное число атомов углерода. Примеры алкинильных групп включают, без ограничения ими, этинил, 1-пропин-1-ил, 2 пропин-1-ил, 1-бутин-1-ил, 3-бутин-1-ил, 3-бутин-2-ил, 2-бутин-1-ил и тому подобное."Алканоил" и "алканоиламино" относятся, соответственно, к алкил-C(O)- и алкил-C(O)-NH-, где алкил является таким, как определено выше, и обычно включают определенное число атомов углерода, в том числе атом углерода карбонильной группы. Примеры алканоильных групп включают, без ограничения ими, формил, ацетил, пропионил, бутирил, пентаноил, гексаноил и тому подобное."Алкеноил" и "алкиноил" относятся, соответственно, к алкенил-C(O)- и алкинил-C(O)-, где алкенил и алкинил являются такими, как определено выше. Ссылки на алкеноил и алкиноил обычно включают определенное число атомов углерода, за исключением атома углерода карбонильной группы. Примеры алкеноильных групп, включают, без ограничения ими, пропеноил, 2-метилпропеноил, 2-бутеноил, 3 бутеноил, 2-метил-2-бутеноил, 2-метил-3-бутеноил, 3-метил-3-бутеноил, 2-пентеноил, 3-пентеноил, 4 пентеноил и тому подобное. Примеры алкиноильных групп включают, без ограничения ими, пропиноил,2-бутиноил, 3-бутиноил, 2-пентиноил, 3-пентиноил, 4-пентиноил и тому подобное."Алкокси", "алкоксикарбонил" и "алкоксикарбониламино" относятся, соответственно, к алкил-O-,алкенил-O- и алкинил-O-; к алкил-O-C(O)-, алкенил-O-С(О)-, алкинил-O-C(O)-; и к алкил-O-C(O)-NH-,алкенил-O-C(O)-NH- и алкинил-O-C(O)-NH-, где алкил, алкенил и алкинил являются такими, как определено выше. Примеры алкоксигрупп включают, без ограничения ими, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентокси, втор-пентокси и тому подобное. Примеры алкоксикарбонильных групп включают, без ограничения ими, метоксикарбонил, этоксикарбонил, нпропоксикарбонил,изопропоксикарбонил,н-бутоксикарбонил,втор-бутоксикарбонил,третбутоксикарбонил, н-пентоксикарбонил, втор-пентоксикарбонил и тому подобное."Алкиламино", "алкиламинокарбонил", "диалкиламинокарбонил", "алкилсульфонил", "сульфониламиноалкил" и "алкилсульфониламинокарбонил" относятся, соответственно, к алкил-NH-, алкил-NHC(O)-, алкил 2-N-C(O)-, алкил-S(O2)-, HS(O2)-NH-алкил- и алкил-S(O)-NH-C(O)-, где алкил является таким, как определено выше."Аминоалкил" и "цианоалкил" относятся, соответственно, к группе NH2-алкил и NC-алкил, где алкил является таким, как определено выше."Гало", "галоген" и "галогено" могут быть использованы взаимозаменяемо и относятся к фторо,хлоро, бромо и йодо."Галогеноалкил", "галогеноалкенил", "галогеноалкинил", "галогеноалканоил", "галогеноалкеноил","галогеноалкиноил", "галогеноалкокси" и "галогеноалкоксикарбонил" относятся, соответственно, к алкильным, алкенильным, алкинильным, алканоильным, алкеноильным, алкиноильным, алкоксильным и алкоксикарбонильным группам, замещенным одним или более атомами галогена, где алкил, алкенил,алкинил, алканоил, алкеноил, алкиноил, алкокси и алкоксикарбонил являются такими, как определено выше. Примеры галогеноалкильных групп включают, без ограничения ими, трифторметил, трихлорметил, пентафторэтил, пентахлорэтил и тому подобное."Гидроксиалкил" и "оксоалкил" относятся, соответственно, к группам HO-алкил и O=алкил, где алкил является таким, как определено выше. Примеры гидроксиалкильных и оксоалкильных групп включают, без ограничения ими, гидроксиметил, гидроксиэтил, 3-гидроксипропил, оксометил, оксоэтил, 3 оксопропил и тому подобное."Циклоалкил" относится к насыщенным моноциклическим и бициклическим углеводородным кольцам, обычно имеющим определенное число атомов углерода, составляющих кольцо (то есть C3-7 циклоалкил относится к циклоалкильной группе, имеющей 3, 4, 5, 6 или 7 атомов углерода в качестве кольцевых атомов). Циклоалкил может быть присоединен к родительской группе или к субстрату по любому кольцевому атому, пока такое присоединение не будет нарушать требования валентности. Подобным образом, циклоалкильные группы могут включать один или более чем один заместитель, не являющийся водородом, пока такое замещение не будет нарушать требования валентности. Полезные заместители включают, без ограничения ими, алкил, алкенил, алкинил, галогеноалкил, галогеноалкенил, галогеноалкинил, алкокси, алкоксикарбонил, алканоил и галогено, как определено выше, и гидрокси, меркапто,нитро и амино. Примеры моноциклических циклоалкильных групп включают, без ограничения ими, циклопропил,циклобутил, циклопентил, циклогексил и тому подобное. Примеры бициклических циклоалкильных групп включают, без ограничения ими, бицикло [1.1.0]бутил, бицикло [1.1.1]пентил, бицикло[2.1.0]пентил, бицикло[2.1.1]гексил, бицикло[3.1.0]гексил, бицикло[2.2.1]гептил, бицикло [3.2.0]гептил,бицикло [3.1.1]гептил, бицикло [4.1.0]гептил, бицикло[2.2.2]октил, бицикло[3.2.1]октил, бицикло[4.1.1]октил, бицикло[3.3.0]октил, бицикло[4.2.0]октил, бицикло[3.3.1]нонил, бицикло[4.2.1]нонил,бицикло[4.3.0]нонил,бицикло[3.3.2]децил,бицикло[4.2.2]децил,бицикло[4.3.1]децил,бицикло[4.4.0]децил, бицикло[3.3.3]ундецил, бицикло[4.3.2]ундецил, бицикло[4.3.3]додецил и тому подобное,которые могут быть присоединены к родительской группе или к субстрату по любому из кольцевых атомов, пока такое присоединение не будет нарушать требования валентности."Циклоалкенил" относится к моноциклическим и бициклическим углеводородным кольцам, имеющим одну или более чем одну ненасыщенную углерод-углеродную связь и обычно имеющим определенное число атомов углерода, составляющих кольцо (то есть C3-7 циклоалкенил относится к циклоалкенильной группе, имеющей 3, 4, 5, 6 или 7 атомов углерода в качестве кольцевых атомов). Циклоалкенил может быть присоединен к родительской группе или к субстрату по любому кольцевому атому, пока такое присоединение не будет нарушать требования валентности. Также циклоалкенильные группы могут включать один или более чем один заместитель, не являющийся водородом, пока такое замещение не будет нарушать требования валентности. Полезные заместители включают, без ограничения ими, алкил,алкенил, алкинил, галогеноалкил, галогеноалкенил, галогеноалкинил, алкокси, алкоксикарбонил, алканоил и галогено, как определено выше, и гидрокси, меркапто, нитро и амино."Циклоалканоил" и "циклоалкеноил" относятся к группам циклоалкил-C(O)- и циклоалкенил-C(O)-,соответственно, где циклоалкил и циклоалкенил являются такими, как определено выше. Ссылки на циклоалканоил и циклоалкеноил обычно включают определенное число атомов углерода, за исключением атома углерода карбонильной группы. Примеры циклоалканоильных групп включают, без ограничения ими, циклопропаноил, циклобутаноил, циклопентаноил, циклогексаноил, циклогептаноил, 1-6 015418 циклобутеноил, 2-циклобутеноил, 1-циклопентеноил, 2-циклопентеноил, 3-циклопентеноил, 1 циклогексеноил, 2-циклогексеноил, 3-циклогексеноил и тому подобное."Циклоалкокси" и "циклоалкоксикарбонил" относятся, соответственно, к группам циклоалкил-O- и циклоалкенил-O- и к циклоалкил-O-C(O)- и циклоалкенил-O-C(O)-, где циклоалкил и циклоалкенил являются такими, как определено выше. Ссылки на циклоалкокси и циклоалкоксикарбонил обычно включают определенное число атомов углерода, за исключением атома углерода карбонильной группы. Примеры циклоалкокси групп включают, без ограничения ими циклопропокси, циклобутокси, циклопентокси, циклогексокси, 1-циклобутенокси, 2-циклобутенокси, 1-циклопентенокси, 2-циклопентенокси, 3 циклопентенокси, 1-циклогексенокси, 2-циклогексенокси, 3-циклогексенокси и тому подобное. Примеры циклоалкоксикарбонильных групп включают, без ограничения ими, циклопропоксикарбонил, циклобутоксикарбонил, циклопентоксикарбонил, циклогексоксикарбонил, 1-циклобутеноксикарбонил, 2 циклобутеноксикарбонил, 1-циклопентеноксикарбонил, 2-циклопентеноксикарбонил, 3-циклопентеноксикарбонил, 1-циклогексеноксикарбонил, 2-циклогексеноксикарбонил, 3-циклогексеноксикарбонил и тому подобное."Арил" и "арилен" относятся к моновалентным и дивалентным ароматическим группам, соответственно, включающим 5- и 6-членные моноциклические ароматические группы, содержащие от 0 до 4 гетероатомов, независимо выбранных из азота, кислорода и серы. Примеры моноциклических арильных групп включают, без ограничения ими, фенил, пирролил, фуранил, тиофенил, тиазолил, изотиазолил,имидазолил, триазолил, тетразолил, пиразолил, оксазолил, изоксазолил, пиридинил, пиразинил, пиридазинил, пиримидинил и тому подобное. Арильные и ариленовые группы также включают бициклические группы, трициклические группы и так далее, включающие конденсированные 5- и 6-членные кольца,описанные выше. Примеры многоциклических арильных групп включают, без ограничения ими, нафтил,бифенил, антраценил, пиренил, карбазолил, бензоксазолил, бензодиоксазолил, бензотиазолил, бензоимидазолил, бензотиофенил, хинолил, изохинолил, индолил, бензофуранил, пуринил, индолизинил и тому подобное. Эти арильные и ариленовые группы могут быть присоединены к родительской группе или к субстрату по любому кольцевому атому, пока такое присоединение не будет нарушать требования валентности. Также арильные и ариленовые группы могут включать один или более чем один заместитель,не являющийся водородом, пока такое замещение не будет нарушать требования валентности. Полезные заместители включают, без ограничения ими, алкил, алкенил, алкинил, галогеноалкил, галогеноалкенил,галогеноалкинил, циклоалкил, циклоалкенил, алкокси, циклоалкокси, алканоил, циклоалканоил, циклоалкеноил, алкоксикарбонил, циклоалкоксикарбонил и галогено, как определено выше, и гидрокси, меркапто, нитро, амино и алкиламино."Гетероцикл" и "гетероциклил" относятся к насыщенным, частично ненасыщенным или ненасыщенным моноциклическим или бициклическим кольцам, имеющим от 5 до 7, или от 7 до 11 кольцевых атомов соответственно. Эти группы включают кольцевые атомы, выбранные из атомов углерода и 1-4 гетероатомов, независимо представляющих собой азот, кислород или серу, и могут включать любую бициклическую группу, в которой любой из определенных выше моноциклических гетероциклов конденсированы с бензольным кольцом. Гетероатомы азота и серы возможно могут быть окислены. Гетероциклическое кольцо может быть присоединено к родительской группе или к субстрату по любому гетероатому или атому углерода, пока такое присоединение не будет нарушать требования валентности. Также любые из кольцевых атомов углерода или азота могут включать один или более чем один заместитель, не являющийся водородом, пока такое замещение не будет нарушать требования валентности. Полезные заместители включают, без ограничения ими, алкил, алкенил, алкинил, галогеноалкил, галогеноалкенил,галогеноалкинил, циклоалкил, циклоалкенил, алкокси, циклоалкокси, алканоил, циклоалканоил, циклоалкеноил, алкоксикарбонил, циклоалкоксикарбонил и галогено, как определено выше, и гидрокси, меркапто, нитро, амино и алкиламино. Примеры гетероциклов включают, без ограничения ими, акридинил, азоцинил, бензимидазолил,бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензотиазолил, бензотриазолил, бензотетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хроменил, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуран, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1Hиндазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензофуранил, изохроманил,изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, морфолинил, нафтиридинил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5 оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксазолидинил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатиинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил,пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, 6H-1,2,5 тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил,тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-7 015418 триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил."Гетероарил" и "гетероарилен" относятся, соответственно, к моновалентным и дивалентным гетероциклам или гетероциклическим группам, как определено выше, которые являются ароматическими. Гетероарильные и гетероариленовые группы представляют собой подмножество арильных и ариленовых групп соответственно."Уходящая группа" относится к любой группе, которая покидает молекулу во время процесса фрагментации, включая реакции замещения, реакции элиминации и реакции присоединения-элиминации. Уходящие группы могут быть нуклеофугами, когда группа уходит с парой электронов, которые ранее служили связью между этой уходящей группой и молекулой, или могут быть электрофугами, когда группа уходит без пары электронов. Способность уходящей группы - нуклеофуга уходить зависит от силы образующего ее основания, причем наиболее сильные основания дают наиболее слабые уходящие группы. Типичные уходящие группы - нуклеофуги включают азот (например из солей диазония); сульфонаты, включая алкилсульфонаты (например мезилат), фторалкилсульфонаты (например трифлат, гексафлат, нонафлат и трезилат) и арилсульфонаты (например тозилат, брозилат, клозилат и нозилат). Другие включают карбонаты, галогенид-ионы, карбоксилатные анионы, фенолятные ионы и алкилаты. Некоторые более сильные основания, такие как NH2- и OH-, можно сделать более хорошими уходящими группами путем обработки кислотой. Типичные уходящие группы - электрофуги включают протон, CO2 и металлы."Энантиомерный избыток" или "ee" является характеристикой для данного образца, избытка одного энантиомера относительно рацемического образца хирального соединения и выражается в процентах. Энантиомерный избыток определяют как 100 (er-1)/(er+1), где "er" представляет собой отношение энантиомера, присутствующего в большем количестве, к энантиомеру, присутствующему в меньшем количестве."Диастереомерный избыток" или "д.и." является характеристикой для данного образца, избытка одного диастереомера относительно образца, содержащего равные количества диастереомеров, и выражается в процентах. Диастереомерный избыток определяют как 100(dr-1)/(dr+1), где "dr" представляет собой отношение диастереомера, присутствующего в большем количестве, к диастереомеру, присутствующему в меньшем количестве."Стереоселективный", "энантиоселективный", "диастереоселективный" и их варианты относятся к данному способу (например гидролизу сложного эфира, гидрированию, гидроформилированию, -аллилпалладиевому сочетанию, гидросилилированию, гидроцианированию, метатезису олефинов, гидроацилированию, изомеризации аллиламинов и так далее), при котором получают большее количество одного стереоизомера, энантиомера или диастереоизомера, чем другого соответственно."Высокая степень стереоселективности", "высокая степень энантиоселективности", "высокая степень диастереоселективности" и их варианты относятся к данному способу, при котором получают продукты, имеющие избыток одного стереоизомера, энантиомера или диастереоизомера, составляющий по-8 015418 меньшей мере приблизительно 90% от содержания продуктов. Для пары энантиомеров или диастереомеров высокая степень энантиоселективности или диастереоселективности будет соответствовать э.и. или д.и. по меньшей мере приблизительно 80%."Стереоизомерно обогащенный", "энантиомерно обогащенный", "диастереомерно обогащенный" и их варианты относятся, соответственно, к образцу соединения, который содержит большее количество одного стереоизомера, энантиомера или диастереомера, чем другого. Степень обогащения может быть измерена в % от общего продукта или, для пары энантиомеров или диастереомеров, в э.и. или д.и"По существу чистый стереоизомер", "по существу чистый энантиомер", "по существу чистый диастереомер" и их варианты относятся, соответственно, к образцу, содержащему стереоизомер, энантиомер или диастереомер, составляющий по меньшей мере приблизительно 95% образца. Для пары энантиомеров и диастереомеров по существу чистый энантиомер или диастереомер будет соответствовать образцам, имеющим э.и. или д.и. приблизительно 90% или выше."Чистый стереоизомер", "чистый энантиомер", "чистый диастереомер" и их варианты относятся,соответственно, к образцу, содержащему стереоизомер, энантиомер или диастереомер, составляющий по меньшей мере приблизительно 99,5% образца. Для пары энантиомеров и диастереомеров чистый энантиомер или чистый диастереомер будет соответствовать образцам, имеющим э.и. или д.и. приблизительно 99% или выше."Противоположный энантиомер" относится к молекуле, которая не является суперпозиционным зеркальным отображением сравнительной молекулы, которая может быть получена обращением всех стереогенных центров этой сравнительной молекулы. Например, если сравнительная молекула имеет абсолютную стереохимическую S-конфигурацию, тогда противоположный энантиомер имеет абсолютную стереохимическую R-конфигурацию. Также, если сравнительная молекула имеет абсолютную стереохимическую S,S-конфигурацию, тогда противоположный энантиомер имеет стереохимическую R,Rконфигурацию и так далее."Стереоизомеры" определенного соединения относятся к противоположному энантиомеру соединения и к любым диастереоизомерам или геометрическим изомерам (Z/E) этого соединения. Например,если определенное соединение имеет стереохимическую S,R,Z-конфигурацию, его стереоизомеры будут включать его противоположный энантиомер, имеющий R,S,Z-конфигурацию, его диастереомеры, имеющие S,S,Z-конфигурацию и R,R,Z-конфигурацию, и его геометрические изомеры, имеющие S,R,Eконфигурацию, R,S,E-конфигурацию, S,S,E-конфигурацию и R,R,E-конфигурацию."Показатель энантиоселективности" или "E" относится к отношению констант специфичности для каждого энантиомера соединения, подвергаемого химическому взаимодействию или превращению, и может быть рассчитан (для S-энантиомера) из следующего выражения: где KS и KR представляют собой константы скорости 1-го порядка для превращения S- и Rэнантиомеров соответственно; KSM И KRM представляют собой константы Михаэлиса для S- и Rэнантиомеров соответственно;представляет собой показатель фракционного превращения субстрата;eep и ees представляют собой энантиомерный избыток продукта и субстрата (реагента) соответственно."Липазная единица" или "ЛЕ" относится к количеству фермента (в г), которое высвобождает 1 мкмоль титруемой масляной кислоты/мин при приведении в контакт с трибутирином и эмульгатором"Сольват" относится к молекулярному комплексу, содержащему раскрытое или заявленное соединение и стехиометрическое или нестехиометрическое количество одной или более молекул растворителя"Гидрат" относится к растворителю, содержащему раскрытое или заявленное соединение и стехиометрическое или нестехиометрическое количество воды."Фармацевтически приемлемые комплексы, соли, сольваты или гидраты" относятся к комплексам,солям присоединения кислот или оснований, сольватам или гидратам заявленных и раскрытых соединений, которые, в пределах объема обоснованного медицинского суждения, являются подходящим для применения в контакте с тканями пациентов, без чрезмерной токсичности, раздражения, аллергической реакции и тому подобного, соответствуют приемлемому соотношению польза/риск, и эффективным для их предполагаемого применения."Предкатализатор" или "предшественник катализатора" относится к соединению или набору соединений, которые превращаются в катализатор перед применением."Лечение" относится к изменению течения, облегчению, задержке развития или к предупреждению расстройства или состояния, к которому этот термин применяется, или к предупреждению одного или более симптомов такого расстройства или состояния."Лечение" относится также к акту "лечения", как оно определено непосредственно выше. В табл. 1 перечислены сокращения, используемые в данном описании. В некоторых из реакционных схем и примеров, приведенных ниже, определенные соединения могут быть получены с использованием защитных групп, которые предотвращают нежелательную химическую реакцию на реакционноспособных в иных обстоятельствах сайтах. Защитные группы могут также быть использованы для того, чтобы увеличить растворимость или иным образом модифицировать физические свойства соединения. Для обсуждения стратегий защитных групп, описания материалов и методов введения и удаления защитных групп и компиляции полезных защитных групп для общепринятых функциональных групп, включая амины, карбоновые кислоты, спирты, кетоны, альдегиды и тому подобное, смотри T. W. Greene and P. G. Wuts Protecting Groups in Organic Chemistry (1999) и P. Kocienski Protective Groups (2000), которые включены в данное описание ссылкой во всей их полноте для любых целей. В дополнение, в некоторых из схем и примеров, представленных ниже, могут быть опущены под- 11015418 робности обычных реакций, включая окисление, восстановление и другие, которые известны специалистам обычной квалификации в области органической химии. Подробности таких взаимодействий можно найти в ряде монографий, включая Richard Larock, Comprehensive Organic Transformations (1999) и многотомную серию под редакцией Michael B. Smith и других, Compendium of Organic Synthetic Methods(1974-2003). В общем случае, исходные вещества и реагенты могут быть получены из коммерческих источников или могут быть получены согласно литературным источникам. В общем случае, химические трансформации, описанные в данном описании изобретения, могут быть осуществлены с использованием по существу стехиометрических количеств реагентов, хотя для некоторых реакций может быть полезно использование избытка одного или более реагентов. Кроме того,многие реакции, раскрытые в данном описании изобретения, включая энантиоселективный гидролиз рацемического диэфира (формула 4), подробно описанный ниже, могут быть проведены при приблизительно комнатной температуре (КТ), однако для отдельных реакциях может требоваться использование более высоких или более низких температур, в зависимости от кинетики, выходов реакции и тому подобного. Более того, для многих химических трансформаций может быть использован один или более чем один совместимый растворитель, который может оказывать влияние на скорость и выход реакции. В зависимости от природы реагентов такой один или более чем один растворитель может представлять собой полярный протонный растворитель, полярный апротонный растворитель, неполярный растворитель или некоторую комбинацию. Любые ссылки в данном описании на интервал концентраций, интервал температур, интервал pH, интервал загрузки катализаторов и так далее, независимо от того используется ли специально слово "интервал" или нет, включают указанные граничные значения. В настоящем изобретении предложены способы получения оптически активных -аминокислот(формула 1), включая их фармацевтически приемлемые соли, сложные эфиры, амиды или пролекарства. Соединения формулы 1 включают заместители R1 и R2, которые определены выше. Полезные соединения формулы 1 таким образом включают соединения, в которых R1 представляет собой атом водорода, а R2 представляет собой C1-12 алкил, C3-12 циклоалкил или замещенный C3-12 циклоалкил, или соединения, в которых R2 представляет собой атом водорода, а R1 представляет собой C1-12 алкил, C3-12 циклоалкил или замещенный C3-12 циклоалкил. В частности, полезные соединения формулы 1 включают соединения, в которых R1 представляет собой атом водорода, а R2 представляет собой C1-6 алкил или C3-7 циклоалкил,или соединения, в которых R2 представляет собой атом водорода, а R1 представляет собой C1-6 алкил илиC3-7 циклоалкил. Особенно полезные соединения формулы 1 включают соединения, в которых R1 представляет собой атом водорода, а R2 представляет собой C1-4 алкил, такие как прегабалин (формула 9). На фиг. 1 показан способ получения оптически активных -аминокислот (формула 1). Этот способ включает стадию приведения в контакт или объединения реакционной смеси, которая состоит из цианозамещенного диэфира (формула 4) и воды, с ферментом с получением смеси продукта, которая включает оптически активный моноэфир дикарбоновой кислоты (формула 3) и оптически активный диэфир (формула 5). Цианозамещенный диэфир (формула 4) имеет стереогенный центр, который обозначен звездочкой и, как описано ниже, может быть получен в соответствии со схемой взаимодействий, показанной на фиг. 2. Перед приведением в контакт фермента цианозамещенный диэфир (формула 4) обычно содержит рацемическую (эквимолярную) смесь диэфира формулы 5 и его противоположного энантиомера. Заместители R1, R2 и R3 в формуле 3, формуле 4 и формуле 5 и заместитель R4 в формуле 4 и формуле 5 являются такими, как определено выше в отношении формулы 1. В общем случае и если не определено по-другому, когда конкретный идентификатор заместителя (R1, R2, R3 и так далее) определен в первый раз в связи с какой-либо формулой, такой же идентификатор заместителя, используемый в последующей формуле, будет иметь то же самое значение, как и в более ранней формуле. Фермент (или биокатализатор) может представлять собой любой белок, который, несмотря на то,что он оказывает незначительное влияние на соединение формулы 5 или не оказывает такого влияния,будет катализировать гидролиз его противоположного энантиомера с получением моноэфира дикарбоновой кислоты (формулы 3). Полезные ферменты для энантиоселективного гидролиза соединения формулы 4 в соединения формулы 3 могут, таким образом, включать гидролазы, в том числе липазы, некоторые протеазы и другие энантиоселективные эстеразы. Такие ферменты могут быть получены из ряда природных источников, в том числе органов животных и микроорганизмов. См., например, табл. 2 в отношении неограничивающего перечня имеющихся в продаже гидролаз. Как показано в разделе Примеры, ферменты, полезные для энантиоселективного превращения цианозамещенного диэфира (формула 4 и формулы 12) в нужный оптически активный моноэфир дикарбоновой кислоты (формула 3 и формула 11) включают липазы. Особенно полезные липазы включают ферменты, получаемые из микроорганизмов Thermomyces lanuginosus, например доступные в Novo-NordiskA/S под товарным знаком LIPOLASE (CAS No. 9001-62-1). Ферменты LIPOLASE получают путем глубинной ферментации микроорганизма Aspergillus oryzae, генетически модифицированного с помощью ДНК из Thermomyces lanuginosus DSM 4109, которая кодирует аминокислотную последовательность этой липазы. LIPOLASE 100L и LIPOLASE 100T доступны в виде жидкого раствора и гранулированного твердого вещества соответственно, причем каждая из них имеет номинальную активность 100 кЛЕ/г. Другие формы LIPOLASE включают LIPOLASE 50L, которая имеет половину активностиLIPOLASE 100L, и LIPOZYME 100L, которая имеет такую же активность, как и LIPOLASE 100L,но пищевое качество. Различные методики скрининга могут быть использованы для идентификации подходящих ферментов. Например, большое количество имеющихся в продаже ферментов может быть отобрано с использованием методик высокопроизводительного скрининга, описанных ниже в разделе Примеры. Другие ферменты (или микробиологические источники ферментов) могут быть отобраны с использованием методик выделения с обогащением. Такие методики обычно включают использование сред, лимитированных по углероду или по азоту, дополненных субстратом обогащения, который может представлять собой рацемический субстрат (формула 4) или структурно подобное соединение. Потенциально полезные микроорганизмы выбирают для последующего исследования, основываясь на их способности расти в среде, содержащей субстрат обогащения. Эти микроорганизмы затем оценивают на их способность энантиоселективно катализировать гидролиз сложного эфира путем приведения в контакт суспензий микробных клеток с рацемическим субстратом и тестирования на присутствие нужного оптически активного моноэфира дикарбоновой кислоты (формула 3) с использованием аналитических способов, таких как хиральная высокоэффективная жидкостная хроматография (ВЭЖХ), газожидкостная хроматография, жидкостная хроматография/масс-спектрометрия (ЖХ/МС) и тому подобное. Как только микроорганизм, имеющий необходимую гидролитическую активность, выделен, может быть использована ферментная инженерия для улучшения свойств фермента, продуцируемого им. В качестве примера и без ограничения, ферментная инженерия может быть использована для увеличения выхода и энантиоселективности гидролиза сложного эфира, для расширения рабочих интервалов температуры и pH этого фермента и для улучшения устойчивости фермента к органическим растворителям. Полезные методики ферментной инженерии включают способы рационального конструирования, такие как сайт-направленный мутагенез, и методики in vitro-управляемой эволюции, которые используют успеш- 15015418 ные стадии случайного мутагенеза, экспрессии генов и высокопроизводительный скрининг для оптимизации желаемых свойств. См., например, K. M. KoellerC.-H. Wong, "Enzymes for chemical synthesis",Nature 409:232-240 (11 Jan. 2001), и указанные там источники информации, полные описания которых включены в данное описание посредством ссылки. Фермент может присутствовать в форме целых микробных клеток, пермеабилизованных микробных клеток, экстрактов микробных клеток, частично очищенных ферментов, очищенных ферментов и тому подобного. Фермент может содержать дисперсию частиц, имеющих средний размер частиц, исходя из объема, менее чем приблизительно 0,1 мм (тонкая дисперсия) или приблизительно 0,1 мм или больше(грубая дисперсия). Грубые дисперсии ферментов имеют потенциальные преимущества при обработке перед тонкими дисперсиями. Например, грубые частицы фермента могут быть использованы повторно в периодических процессах или в полунепрерывных либо непрерывных процессах и обычно могут быть отделены (например путем фильтрации) от других компонентов биопревращения легче, чем тонкие дисперсии ферментов. Полезные грубые дисперсии ферментов включают поперечно-сшитые кристаллы ферментов(CLECs) и поперечно-сшитые агломераты ферментов (CLEAs), которые состоят главным образом из фермента. Другие грубые дисперсии могут включать ферменты, иммобилизованные на, или в, нерастворимой подложке. Полезные твердые подложки включают полимерные матрицы, состоящие из альгината кальция, полиакриламида, EUPERGIT, и других полимерных материалов, а также неорганические матрицы, такие как CELITE. Обобщенное описания CLECs и других методик иммобилизации ферментов смотри в патенте США 5618710, М. A. NaviaN. L. St. Clair. Обобщенное описание CLEAs, включая их получение и применение, см. в заявке на патент США 2003/0149172, L. CaoJ. Elzinga et al. Смотри также в А. М. Anderson, Biocat. Biotransform, 16:181 (1998) и P. Lopez-Serrano et al., Biotechnol. Lett. 24:1379-83 (2002) обсуждение применимости CLECs- и CLEAs-технологии к липазе. Полные описания вышеупомянутых источников информации включены в данное описание ссылкой для любых целей. Реакционная смесь может содержать одну фазу или может содержать множество фаз (например двух- или трехфазная система). Так, например, энантиоселективный гидролиз, показанный на фиг. 1, может происходить в единственной водной фазе, которая содержит фермент, первоначально рацемический субстрат (формула 4), нежелательный оптически активный диэфир (формула 5) и нужный оптически активный моноэфир дикарбоновой кислоты (формула 3). Альтернативно, реакционная смесь может содержать многофазную систему, которая включает водную фазу в контакте с твердой фазой (например ферментом или продуктом), водную фазу в контакте с органической фазой, или водную фазу в контакте с органической фазой и твердой фазой. Например, энантиоселективный гидролиз может быть осуществлен в двухфазной системе, состоящей из твердой фазы, которая содержит фермент, и водной фазы, которая содержит первоначально рацемический субстрат, нежелательный оптически активный диэфир и нужный оптически активный моноэфир дикарбоновой кислоты. Альтернативно, энантиоселективный гидролиз может быть осуществлен в трехфазной системе, состоящей из твердой фазы, которая содержит фермент, органической фазы, которая первоначально содержит рацемический субстрат (формула 4), и водной фазы, которая первоначально содержит небольшую фракцию рацемического субстрата. Поскольку нужный оптически активный моноэфир дикарбоновой кислоты (формула 3) имеет более низкий рКа, чем непрореагировавший оптически активный диэфир(формула 5) и, следовательно, обладает более высокой растворимостью в воде, органическая фаза в ходе реакции обогащается непрореагировавшим диэфиром, в то время как водная фаза обогащается нужным моноэфиром дикарбоновой кислоты. Количества рацемического субстрата (формула 4) и биокатализатора, используемые в энантиоселективном гидролизе, среди прочего, будут зависеть от свойств конкретного циано-замещенного диэфира и фермента. В общем случае, однако, в данном взаимодействии может быть использован субстрат,имеющий первоначальную концентрацию от приблизительно 0,1 М до приблизительно 3,0 М, и во многих случаях, имеющий первоначальную концентрацию от приблизительно 1,5 М до приблизительно 3,0 М. Дополнительно, в данном взаимодействии может быть использован фермент, загружаемый в количестве от приблизительно 1 до приблизительно 10%, и во многих случаях, загружаемый в количестве от приблизительно 3 до приблизительно 4% (об./об.). Энантиоселективный гидролиз может быть осуществлен в широком интервале температур и значений pH. Например, взаимодействие может быть осуществлено при температуре от приблизительно 10C до температуре приблизительно 50C, но обычно его осуществляют при приблизительно КТ. Такие температуры в общем случае позволяют осуществить, по существу, полное превращение (например от приблизительно 42% до приблизительно 50%) рацемата (формула 4) в течение приемлемого количества времени (от приблизительно 2 ч до приблизительно 24 ч) без дезактивации фермента. Кроме того, энантиоселективный гидролиз может быть осуществлен при pH от приблизительно 5 до pH приблизительно 10,более типично при pH от приблизительно 6 до pH приблизительно 9, и часто при pH от приблизительно 6,5 до pH приблизительно 7,5. В отсутствие контроля рН, значение pH реакционной смеси будет уменьшаться, по мере того как- 16015418 происходит гидролиз субстрата (формула 4) из-за образования моноэфира дикарбоновой кислоты (формула 3). Чтобы компенсировать это изменение, реакция гидролиза может проходить с внутренним контролем pH (то есть в присутствии подходящего буфера) или может проходить с внешним контролем pH посредством добавления основания. Подходящие буферы включают фосфат калия, фосфат натрия, ацетат натрия, ацетат аммония, ацетат кальция, BES, BICINE, HEPES, MES, MOPS, PIPES, TAPS, TES,TRICINE, Tris, TRIZMA или другие буферы, имеющие pKa от приблизительно 6 до pKa приблизительно 9. Концентрация буфера в общем случае варьируется в интервале от приблизительно 5 мМ до приблизительно 1 мМ, и обычно варьируется в интервале от приблизительно 50 мМ до приблизительно 200 мМ. Подходящие основания включают водные растворы, состоящие из KOH, NaOH, NH4OH и так далее,имеющие концентрации в интервале от приблизительно 0,5 М до приблизительно 15 М, или более типично в интервале от приблизительно 5 М до приблизительно 10 М. Также могут быть использованы другие неорганические добавки, такие как ацетат кальция. После или во время ферментативного превращения рацемата (формула 4) нужный оптически активный моноэфир дикарбоновой кислоты (формула 3) выделяют из смеси продукта, используя стандартные методики. Например, в случае периодического процесса взаимодействия в единственной (водной) фазе, смесь продукта может быть экстрагирована один или более раз неполярным органическим растворителем, таким как гексан или гептан, в результате чего нужный моноэфир дикарбоновой кислоты (формула 2) и непрореагировавший диэфир (формула 5) распределяются в водные и органические фазы соответственно. Альтернативно, в случае взаимодействия в многофазной системе с использованием водной и органической фаз, обогащенных нужным моноэфиром (формула 3) и нежелательным диэфиром (формула 5) соответственно, моноэфир и диэфир могут быть разделены по партиям после взаимодействия или могут быть разделены в полунепрерывном или непрерывном процессе в процессе энантиоселективного гидролиза. Как показано на фиг. 1, непрореагировавший диэфир (формула 5) может быть выделен из органической фазы и рацемизирован с получением рацемического субстрата (формула 4). Полученный рацемат(формула 4) может быть повторно использован или объединен с непрореагировавшим рацемическим субстратом, который затем подвергают ферментативному превращению до соединения формулы 3, как описано выше. Повторное использование непрореагировавшего диэфира (формула 5) увеличивает общий выход энантиоселективного гидролиза выше 50%, благодаря чему повышается экономия вещества и снижаются издержки, связанные с устранением нежелательных энантиомеров. Обработка диэфира (формула 5) основанием, достаточно сильным, чтобы отделить кислотный протон малонатной группировки, обычно приводит к инверсии стереогенного центра и образованию рацемического субстрата (формула 4). Полезные основания включают органические основания, такие как алкилаты (например этилат натрия), линейные алифатические амины и циклические амины и органические основания, такие как KOH, NaOH, NH4OH и тому подобное. Взаимодействие осуществляют в совместимом растворителе, включая полярные протонные растворители, такие как EtOH, или апротонные полярные растворители, такие как МТВЕ. Температуры взаимодействий выше КТ обычно повышают выход в данном способе рацемизации. Как показано на фиг. 1, по существу, энантиомерно чистый моноэфир дикарбоновой кислоты (формула 3) можно превратить в оптически активную -аминокислоту (формула 1), используя по меньшей мере три разных способа. В одном способе моноэфир (формула 3) гидролизуют в присутствии кислотного катализатора или основного катализатора с получением оптически активной цианозамещенной дикарбоновой кислоты (формула 6) или соответствующей соли. Цианогруппировку полученной в результате дикарбоновой кислоты (или ее соли) восстанавливают с получением оптически активной аминодикарбоновой кислоты (формула 7) или соответствующей соли, которую затем декарбоксилируют путем обработки кислотой, путем нагревания, или тем и другим, с получением нужной оптически активной -аминокислоты (формула 1). Цианогруппировка может быть восстановлена посредством взаимодействия с H2 в присутствии каталитических количеств никеля Ренея, палладия, платины и тому подобного, или посредством взаимодействия с восстановителем, таким как LiAlH4, BH3-Me2S и тому подобным. Полезные кислоты для реакций гидролиза и декарбоксилирования включают минеральные кислоты, такие как HClO4, HI, H2SO4, HBr, HCl и тому подобное. Полезные основные катализаторы для реакции гидролиза включают различные гидроксиды и оксиды щелочных и щелочно-земельных металлов,включая LiOH, NaOH, KOH и тому подобное. В другом способе моноэфир дикарбоновой кислоты (формула 3) подвергают восстановительной циклизации с образованием оптически активного циклического 3-карбоксипирролидин-2-она (формула 2), который затем обрабатывают кислотой с получением нужной энантиомерно обогащенной аминокислоты (формула 1). Восстановительная циклизация может быть осуществлена путем взаимодействия моноэфира (формула 3) с H2 в присутствии каталитических количеств никеля Ренея, палладия, платины и тому подобного. Одна или более чем одна кислота может быть использована для гидролиза и декарбоксилирования получающейся в результате лактамной кислоты (формула 2), включая минеральные кислоты, такие как HClO4, HI, H2SO4, HBr и HCl, и органические кислоты, такие как HOAc, ТФУ, пара- 17015418TSA и тому подобное. Концентрация кислот может варьироваться в интервале от приблизительно 1 н. до приблизительно 12 н., и количество кислот может варьироваться в интервале от приблизительно 1 до приблизительно 7 экв. Реакции гидролиза и декарбоксилирования могут быть осуществлены при температуре от приблизительно КТ или выше, или при температуре от приблизительно 60C или выше, или при температуре в интервале от приблизительно 60 до приблизительно 130C. В третьем способе сложноэфирную группировку моноэфира дикарбоновой кислоты (формула 3) сначала гидролизуют с получением цианозамещенной дикарбоновой кислоты (формула 6 или ее соли),как описано выше. Полученную в результате дикарбоновую кислоту (или ее соль) затем декарбоксилируют с получением оптически активной цианозамещенной карбоновой кислоты или ее соли (формула 8, в которой R5 представляет собой атом водорода, хотя R5 может также представлять собой C1-12 алкил, C3-12 циклоалкил или арил-C1-6 алкил, как отмечено выше). Могут быть использованы те же самые условия, которые используются для декарбоксилирования лактамной кислоты (формула 2) или -аминодикарбоновой кислоты (формула 7). Вместо первого гидролиза сложноэфирной группировки, моноэфир дикарбоновой кислоты (формула 3) может быть сначала декарбоксилирован непосредственно до цианозамещенного моноэфира (формула 8) путем нагревания водного раствора моноэфира дикарбоновой кислоты (в виде соли) до температуры от приблизительно 50C до температуры дефлегмации. Могут быть использованы также условия Крапчо (ДМСО/NaCl/вода). В любом случае, цианогруппировку соединения формулы 8 затем восстанавливают с получением оптически активной 7-аминокислоты (формула 1). В дополнение к никелю Ренея ряд других катализаторов может быть использован для восстановления цианогруппировки соединений формулы 3, 6 и 8. Они включают, без ограничения ими, гетерогенные катализаторы, содержащие от приблизительно 0,1% до приблизительно 20% и, более типично, от приблизительно 1 до приблизительно 5 мас.% переходных металлов, таких как Ni, Pd, Pt, Rh. Re, Ru и Ir, включая их оксиды и комбинации, которые обычно наносят на подложку из различных материалов, включая Al2O3, C, CaCO3, SrCO3, BaSO4,MgO, SiO2, TiO2, ZrO2 и тому подобное. Многие из этих металлов, включая Pt, могут содержать добавку амина, сульфида или еще одного металла, такого как Pb, Cu и Zn. Полезные катализаторы, таким образом, включают палладиевые катализаторы, такие как Pd/C, Pd/SrCCb, Pd/Al2O3, Pd/MgO, Pd/CaCO3,Pd/BaSO4, PdO, черный Pd, PdCl2 и тому подобные, содержащие от приблизительно 1 до приблизительно 5 мас.% Pd. Другие полезные катализаторы включают Rh/C, Ru/C, Re/C, PtO2, Rh/C, RuO2 и тому подобное. Каталитическое восстановление цианогруппировки обычно осуществляют в присутствии одного или более полярных растворителей, включая, без ограничения ими, воду, спирты, простые эфиры, сложные эфиры и кислоты, такие как MeOH, EtOH, IPA, ТГФ, EtAc и HOAc. Это взаимодействие может быть осуществлено при температурах в интервале от приблизительно 5 до приблизительно 100C, хотя взаимодействия при КТ являются более общепринятыми. В общем случае, отношение субстрата к катализатору может варьироваться в интервале от приблизительно 1:1 до приблизительно 1000:1, по массе, и давление H2 может варьироваться в интервале от приблизительно атмосферного давления, т.е. избыточного давления 0 фунт/кв. дюйм, до избыточного давления приблизительно 1500 фунт/дюйм 2 (10,34 МПа). Более типично, отношения субстрата к катализатору варьируются в интервале от приблизительно 4:1 до приблизительно 20:1, и давление H2 варьируется в интервале от приблизительно 25 фунт/дюйм 2 (172,4 кПа) до приблизительно 150 фунт/дюйм 2 (1034 КПа). Все предыдущие способы могут быть использованы для превращения, по существу, энантиомерно чистого моноэфира (формула 3) в оптически активную -аминокислоту (формула 1), но каждый может иметь определенные преимущества перед другими. Например, после кислотной обработки в способе с использованием восстановительной циклизации, лактамная кислота (формула 2) может быть выделена и очищена путем экстрагирования ее в органический растворитель, в то время как циано-замещенная дикарбоновая кислота (формула 6) может быть более сложной для выделения ввиду ее сравнительно более высокой растворимости в воде. Выделение лактамной кислоты (формула 2) снижает остаток водорастворимых примесей в конечной смеси продукта и обеспечивает более высокую концентрацию реагента (например от приблизительно 1 до приблизительно 2 М) во время гидролиза и декарбоксилирования, благодаря чему увеличивается производительность способа. Кроме того, непосредственное декарбоксилирование путем нагревания водного раствора моноэфира дикарбоновой кислоты (формула 3) дает цианомоноэфир (формула 8) с высокой энантиомерной чистотой. Это соединение может быть выделено из реакционной среды путем экстракции в органический растворитель или путем прямого разделения фаз, обеспечивающими эффективное удаление неорганических примесей с помощью водной фазы. Двумя преимуществами этого подхода являются высокая производительность взаимодействия и исключение сильно кислотных условий. На фиг. 2 показан способ получения цианозамещенных диэфиров (формула 4), которые могут служить в качестве субстратов для ферментативного энантиоселективного гидролиза, показанного на фиг. 1. Способ включает перекрестную альдольную конденсацию, которая включает взаимодействие асимметрического кетона или альдегида (формула 17) с диэфиром малоновой кислоты (формула 18) в присутствии каталитических количеств основания с получением диэфира ,-ненасыщенной малоновой кислоты(формула 19), в которой R1, R2, R3 и R4 являются такими, как определено выше в связи с формулой 1. Этот тип реакции перекрестной альдольной конденсации известен как конденсация Кновенагеля, которая описана в ряде литературных обзоров. Смотри, например, B. K. Wilk, Tetrahedron 53:7097-7100 (1997) и цитированные там источники информации, полные описания которых включены в данное описание ссылкой для любых целей. В общем случае может быть использовано любое основание, способное образовывать енолят-ион из диэфира малоновой кислоты (формула 18), включая вторичные амины, такие как ди-н-пропиламин, диизопропиламин, пирролидин и другие, и их соли. Это взаимодействие может включать карбоновую кислоту, такую как HOAc, для нейтрализации продукта и для сведения к минимуму енолизации асимметрического кетона или альдегида (формула 17). Во взаимодействиях, включающих асимметрические кетоны, для облегчения взаимодействия могут также быть использованы кислоты Льюиса, такие как тетрахлорид титана, хлорид цинка, ацетат цинка и тому подобное. Это взаимодействие обычно протекает в углеводородном растворителе в условиях кипячения с обратным холодильником. Полезные растворители включают гексан, гептан, циклогексан, толуол, метил-трет-бутиловый эфир и тому подобное, с азеотропным удалением воды. На следующей стадии источник цианида, такой как HCN, цианогидрин ацетона, цианид щелочного металла (NaCN, KCN и другие) или цианид щелочно-земельного металла (цианид магния и другие), подвергают коньюгатному присоединению по -углероду диэфира ,-ненасыщенной малоновой кислоты(формула 19). Эту реакцию обычно осуществляют в одном или более полярных протонных растворителях, включая EtOH, MeOH, н-пропанол, изопропанол, или полярных апротонных растворителях, таких как ДМСО и тому подобное. Последующая кислотная обработка дает цианозамещенный диэфир (формула 4). Применение способа, изображенного на фиг. 2, с получением предшественника прегабалина (формула 12), см. в патенте США 5637767, Grote et al., который включен в данное описание ссылкой во всей его полноте и для любых целей. Нужные (S)- или (R)-энантиомеры любых из соединений, раскрытых в данном описании, могут быть дополнительно обогащены посредством классического разделения, хиральной хроматографии или перекристаллизации. Например, оптически активные -аминокислоты (формула 1), в частности прегабалин (формула 9), могут быть подвергнуты взаимодействию с энантиомерно чистым соединением (например кислотой или основанием) с получением пары диастереоизомеров, каждый из которых состоит из индивидуального энантиомера, которые разделяют посредством фракционной перекристаллизации или хроматографии. Нужный энантиомер затем регенерируют из соответствующего диастереоизомера. Дополнительно, нужный энантиомер часто может быть дополнительно обогащен путем перекристаллизации в подходящем растворителе, когда он имеется в достаточном количестве (например обычно не менее чем приблизительно 85% э.и., и в некоторых случаях не менее чем приблизительно 90% э.и.). Как изложено в данном описании, многие из раскрытых соединений имеют стереоизомеры. Некоторые из этих соединений могут существовать в виде индивидуальных энантиомеров (энантиомерно чистые соединения) или смесей энантиомеров (обогащенных и рацемических образцов), что зависит от относительного избытка одного энантиомера над другим в образце, могут проявлять оптическую активность. Такие стереоизомеры, которые не являются зеркальными отображениями при наложении друг на друга, имеют стереогенную ось или один или более чем один стереогенный центр (то есть хиральность). Другие раскрытые соединения могут быть стереоизомерами, которые не являются зеркальными отображениями. Такие стереоизомеры, которые известны как диастереоизомеры, могут быть хиральными или ахиральными (не содержат стереогенных центров). Они включают молекулы, содержащие алкенильную или циклическую группу, так что возможными являются цис/транс- (или Z/E-)стереоизомеры, или молекулы, содержащие два или более чем два стереогенных центра, в которых инверсия единственного стереогенного центра образует соответствующий диастереоизомер. Если не указано или не очевидно другое(например при использования изображения стереосвязей, указателя стереоцентра и так далее), объем настоящего изобретения в общем случае включает указанное соединение и его стереоизомеры, независимо от того, является ли каждый из них чистым (например энантиомерно чистым) или они являются смесями (например энантиомерно обогащенными или рацемическими). Некоторые из соединений могут также содержать кето- или оксимную группу, так что может иметь место таутомерия. В таких случаях настоящее изобретение в общем случае включает таутомерные формы, независимо от того, является ли каждая из них чистой или они являются смесями. Многие из соединений, описанных в данном описании, включая соединения, представленные формулой 1 и 9, способны к образованию фармацевтически приемлемых солей. Эти соли включают, без ограничения ими, соли присоединения кислот (в том числе дикислот) и соли основания. Фармацевтически приемлемые соли присоединения кислот включают нетоксичные соли, получаемые из органических кислот, таких как соляная, азотная, фосфорная, серная, бромисто-водородная, иодисто-водородная, фтористо-водородная, фосфористая и тому подобное, а также нетоксичные соли, получаемые из органических кислот, таких как алифатические моно- и дикарбоновые кислоты, фенилзамешенные алкановые кислоты,гидроксиалкановые кислоты, алкендионовые кислоты, ароматические кислоты, алифатические и арома- 19015418 тические сульфоновые кислоты и так далее. Такие соли, таким образом, включают сульфат, пиросульфат,бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, трифторацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, манделат, бензоат, хлорбензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малат, тартрат, метансульфонат и тому подобное. Фармацевтически приемлемые соли основания включают нетоксичные соли, получаемые из оснований, включая катионы металлов, такие как катион щелочного или щелочно-земельного металла, а также амины. Примеры подходящих катионов металла включают, без ограничения ими, катионы натрия(Na+), катионы калия (K+), катионы магния (Mg2+), катионы кальция (Ca2+) и тому подобное. Примеры подходящих аминов включают, без ограничения ими, N,N'-дибензилэтилендиамин, хлорпрокаин, холин,диэтаноламин, дициклогексиламин, этилендиамин, N-метилглюкамин, прокаин и трет-бутиламин. Обсуждение полезных солей присоединения кислот и оснований см. в S. M. Berge et al., "Pharmaceutical Salts",66 J. of Pharm. Sci, 1-19 (1977); см. также Stahl and Wermuth, Handbook of Pharmaceutical Salts. Properties,Selection, and Use (2002). Фармацевтически приемлемую соль присоединения кислоты (или соль основания) можно получать путем приведения в контакт свободного основания (или свободной кислоты) или цвиттер-иона соединения с достаточным количеством нужной кислоты (или основания) с образованием нетоксичной соли. Если соль осаждается из раствора, ее можно выделить путем фильтрации, иначе соль может быть выделена путем выпаривания растворителя. Свободное основание (или свободную кислоту) можно также регенерировать путем приведения в контакт соли присоединения кислоты с основанием (или соли основания с кислотой). Хотя некоторые физические свойства свободного основания (или свободной кислоты) и его соответствующей соли присоединения кислоты (или соли основания) могут отличаться (например растворимость, кристаллическая структура, гигроскопичность и так далее), свободное основание соединения и соль присоединения кислоты (или его свободная кислота и соль основания) являются в остальном такими же для целей этого описания. Раскрытые и заявленные соединения могут существовать как в несольватированных, так и в сольватированных формах, а также в виде других типов комплексов помимо солей. Полезные комплексы включают клатраты или комплексы включения соединение-хозяин, где эти соединение и хозяин присутствуют в стехиометрических или нестехиометрических количествах. Полезные комплексы могут также содержать два или более органических, неорганических, или органических и неорганических компонента в стехиометрических или нестехиометрических количествах. Полученные в результате комплексы могут быть ионизированными, частично ионизированными или неионизированными. Обзор таких комплексов см. в J. K. Haleblian, J. Pharm. Sci. 64(8):1269-88 (1975). Фармацевтически приемлемые сольваты также включают гидраты и сольваты, в которых кристаллизационный растворитель может быть замещен изотопами, например D2O, d6-ацетон, d6-ДМСО и так далее. В общем случае, для целей данного описания,ссылки на несольватированную форму соединения также включают соответствующую сольватированную или гидратированную форму этого соединения. Раскрытые соединения также включают все фармацевтически приемлемые изотопные варианты, в которых по меньшей мере один атом заменен атомом, имеющим тот же самый атомный номер, но атомную массу, отличную от атомной массы, обычно обнаруживаемой в природе. Примеры изотопов, подходящих для включения в раскрытые соединения, включают, без ограничения ими, изотопы водорода, такие как 2H и 3H; изотопы углерода, такие как 13C и 14C; изотопы азота, такие как 15N; изотопы кислорода,такие как 17O и 18O; изотопы фосфора, такие как 31P и 32P; изотопы серы, такие как 35S; изотопы фтора,такие как 18F; и изотопы хлора, такие как 36Cl. Применение изотопных вариантов (например дейтерия 2H) может иметь некоторые терапевтические преимущества, вытекающие из большей метаболической стабильности, например увеличенный период полувыведения in vivo или сниженные требования в отношении дозировки. Дополнительно, некоторые изотопные варианты раскрытых соединений могут включать радиоактивные изотопы (например тритий 3H, или 14C), которые могут быть полезны в исследованиях распределения лекарственного средства и/или субстрата в тканях. Примеры Следующие примеры предназначены для иллюстрации, не являются ограничивающими и представляют собой конкретные воплощения настоящего изобретения. Общие материалы и методы Скрининг ферментов осуществляли, используя 96-луночный планшет, как описано в D. Yazbeck etal., Synth. Catal. 345:524-32 (2003), полное описание которой включено ссылкой для любых целей. Все ферменты, используемые в планшете для скрининга (см. табл. 2), были получены от коммерческих поставщиков ферментов, включая Amano (Nagoya, Japan), Roche (Basel, Switzerland), Novo Nordisk (Bagsvaerd, Denmark), Altus Biologies Inc. (Cambridge, MA), Biocatalytics (Pasadena, CA), Toyobo (Osaka, Japan),Sigma-Aldrich (St. Louis, MO) и Fluka (Buchs, Switzerland). Реакции скрининга осуществляли в EppendorfThermomixer-R (VWR). В последующих крупномасштабных ферментативных разделениях использовали- 20015418 Ядерный магнитный резонанс Спектры 1H-ЯМР при 300 МГц и 13C-ЯМР при 75 МГц получали на спектрометре BRUKER 300 UltraShield, оборудованном 5-мм автоматически переключаемым датчиком PHQNP. Спектры обычно регистрировали при температуре, близкой к КТ, и использовали стандартные методики автоблокировки,авторегулировки и автоусиления. Образцы обычно пропускали при 20 Гц для 1D-экспериментов. Спектры 1H-ЯМР получали, используя импульсы под наклоном угла 30, 1,0-секундный период рециркуляции и 16 сканирований с разрешением 0,25 Гц/точку. Охват данных составлял обычно 8000 Гц, от +18 до -2 м.д. (стандарт ТМС при 0 миллионных долей (м.д., и измерение осуществляли с 0,3 Гц уширением линии. Обычное время сбора данных составляло 5-10 с. Стандартные спектры 13C-ЯМР получали, используя импульсы под наклоном угла 30, 2,0-секундный период рециркуляции и 2048 сканирований с разрешением 1 Гц/точку. Широта спектра составляла обычно 25 КГц от +235 до -15 м.д. (стандарт ТМС при 0 м.д.). Непрерывно применяли расщепление протона и 1 Гц уширение линии во время измерения. Обычное время сбора данных составляло 102 мин. Масс-спектрометрия Масс-спектрометрию осуществляли на HEWLETT PACKARD 1100MSD, используя программное обеспечение HP Chemstation Plus. Жидкостную хроматографию (ЖХ) осуществляли на системе Agilent 1100 quaternary LC с жидкостным манипулятором Agilent в качестве пробоотборника. Данные получали при ионизации электрораспылением, используя ACN/воду (содержащую 0,1% муравьиной кислоты) в качестве растворителя (от 10% CAN до 90%, 7 мин). Температура образца составляла 350C, источника 150C. Коронный разряд составлял 3000 В для положительного иона и 3000 В для отрицательного иона. Высокоэффективная жидкостная хроматография Высокоэффективную жидкостную хроматографию (ВЭЖХ) осуществляли на аппаратуре серии 1100 AGILENT TECHNOLOGIES, оборудованной автоматическим пробоотборником Agilent 220 HPLC,четверным насосом и УФ-детектором. ЖХ контролировали компьютерно, используя программное обеспечение HP Chemstation Plus. Хиральную ВЭЖХ на нормальной фазе осуществляли, используя колонки для хиральной ВЭЖХ, полученные от Chiral Technologies (Exton, PA) и Phenomenex (Torrance, CA). Газовая хроматография Газовую хроматографию (ГХ) осуществляли в системе Agilent 6890N network GC 110 В, оборудованной FID-детектором с электрометром, капиллярным инжектором 7683 Series split/splitless, релейной платой, контролирующей четыре внешних события, и встроенным принтером/самописцем. Энантиомерный избыток диэфира (формула 13, R3 =R4 =Et) и моноэфира (формула 11, R3 =Et) получали, используя колонку CHIRALDEX G-TA (30 м 0,25 мм) с газом-носителем гелием и при 135C. В таких условиях моноэфир разлагался с получением этилового эфира (S)-3-циано-5-метилгексановой кислоты, и э.и. определяли исходя из продукта разложения. Колонки для хиральной ГХ, используемые в анализе, получали от Astec, Inc. (Whippany, NJ). Пример 1. Скрининг ферментов посредством ферментативного гидролиза этилового эфира (R/S)-3 циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 20) с получением (3S)-3-циано-2 этоксикарбонил-5-метилгексановой кислоты (формула 21) Скрининг ферментов осуществляли, используя набор для скрининга, состоящий из индивидуальных ферментов, помещенных в отдельные лунки 96-луночного планшета, который готовили заранее в соответствии со способом, описанным в D. Yazbeck et al., Synth. Catal. 345:524-32 (2003). Каждая из лунок имела свободный объем 0,3 мл (планшет с неглубокими лунками). Одна лунка 96-луночного планшета содержала только фосфатный буфер (10 мкл, 0,1 М, pH 7,2), другая лунка содержала только ACN (10 мкл), и каждая из оставшихся лунок содержала один из 94 ферментов, перечисленных в табл.2 (10 мкл,100 мг/мл). Перед использованием набор для скрининга извлекали из места хранения при -80C, и ферменты оставляли размораживаться при КТ в течение приблизительно 5 мин. Фосфатно-калиевый буфер(85 мкл, 0,1 М, pH 7,2) распределяли в каждую из лунок, используя многоканальную пипетку. Затем к каждой лунке добавляли концентрированный субстрат (формула 20, 5 мкл) с помощью многоканальной пипетки, и эти 96 реакционные смеси инкубировали при 30C и 750 об/мин. Реакции гасили и образцы отбирали через 24 ч путем переноса каждой из этих реакционных смесей в отдельные лунки второго 96 луночного планшета. Каждая из лунок имела свободный объем 2 мл (планшет с глубокими лунками) и содержала EtOAc (1 мл) и HCl (1 н., 100 мкл). Компоненты каждой лунки смешивали путем отсасывания содержимого лунки пипеткой. Второй планшет центрифугировали и 100 мкл органического супернатанта переносили из каждой лунки в отдельные лунки третьего 96-луночного планшета (плоского планшета). Лунки третьего планшета затем герметично закрывали, используя проницаемое матовое покрытие.- 21015418 Как только лунки были герметично закрыты, третий планшет переносили в ГХ-систему для определения оптической чистоты (э.и.). В табл. 3 приведены фермент, товарный знак, поставщик и значение E для некоторых ферментов,которые были подвергнуты скринингу. Для данного фермента значение E можно интерпретировать как относительную реакционную способность пары энантиомеров (субстратов). Значения E, приведенные в табл.3, рассчитывали на основании данных ВЭЖХ (фракционная конверсия, , и э.и.), используя компьютерную программу под названием Ee2, доступную от University of Graz. В общем случае, ферменты,обладающие S-селективностью и значением E приблизительно 35 или выше, подходят для крупномасштабного производства. Таблица 3. Результаты скрининговых взаимодействий примера 1 Пример 2. Ферментативное разделение этилового эфира (R/S)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 20) с получением калиевой соли (3S)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 23) и этилового эфира (R)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 22) В реактор (392 л), оборудованный верхней мешалкой, загружали фосфатно-калиевый буфер (292,2 л, 10 мМ, pH 8,0) и LIPOLASE 100L, type EX (3,9 л). Эту смесь перемешивали при 800 об/мин в течение 1 мин и добавляли KOH (2 М), чтобы довести pH до 8,0. Добавляли этиловый эфир (R/S)-3-циано-2 этоксикарбонил-5-метилгексановой кислоты (формула 20, 100 кг), и полученную смесь растирали с водным NaOH (50%) во время гидролиза для поддержания pH на уровне 8,0. Ход реакции контролировали посредством ВЭЖХ (колонка C18, 4,6 мм 150 мм, детекция при 200 нм). После достижения приблизительно 40-45%-ного превращения (например через приблизительно 24 ч) эту реакционную смесь переносили на разделительную воронку. Водную смесь экстрагировали гептаном (205 л). Добавляли EtOH (абсолютный) (вплоть до приблизительно 5% об./об.) для разрушения образовавшейся легкой эмульсии и водный и органический слои разделяли. Стадию экстракции повторяли дважды, и водный слой, содержащий калиевую соль (3S)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 23), можно было дополнительно концентрировать в вакууме (например до 25-50% от его первоначального объема). Органические слои, содержащие этиловый эфир (R)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 22), объединяли, сушили и концентрировали. Полученный в результате диэтиловый эфир затем рацемизировали в соответствии с примером 6. MC m/z [M+H]+227. 1 Н ЯМР (300 МГц, D2O):2,35 (dd, 6H), 2,70 (t, 3H), 2,85 (m, 1H), 2,99 (m, 1H), 3,25 (m, 1H), 4,75 (m,- 22015418 1H), 5,60 (q, 2H). 13C ЯМР (75 м.д., D2O):172,19, 171,48, 122,85, 62,70, 59,49, 40,59, 31,83, 27,91, 23,94, 21,74, 14,77. Пример 3. Ферментативное разделение этилового эфира (R/S)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 20) с получением калиевой соли (3S)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 23) и этилового эфира (R)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 22) В реактор (3,92 л), оборудованный верхней мешалкой, загружали кальций-ацетатный буфер (1,47 л,100 мМ, pH 7,0) и этиловый эфир (R/S)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 20, 1 кг). Эту смесь перемешивали при 1100 об/мин в течение 5 мин и добавляли KOH (5 М) для доведения pH до 7,0. Добавляли LIPOLASE 100L, type EX (75 мл) и полученную смесь растирали с KOH (5 М) во время гидролиза для поддержания pH на уровне 7,0. Ход реакции контролировали посредством ВЭЖХ (колонка C18, 4,6 мм 150 мм, детекция при 200 нм). После достижения приблизительно 42-45%ного превращения (например через приблизительно 20-25 ч), реакционную смесь переносили на разделительную воронку. Водную смесь экстрагировали гексаном (100% об./об.). Добавляли EtOH (абсолютный)(вплоть до приблизительно 5% об./об.) для разрушения образовавшейся легкой эмульсии, и водный и органический слои разделяли. Стадию экстракции повторяли дважды с получением водного слоя, содержащего калиевую соль (3S)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 23), которая может быть использована в следующих превращениях без выделения. Органические слои, содержащие этиловый эфир (R)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 22), объединяли, сушили и концентрировали. Полученный в результате диэтиловый эфир затем рацемизировали в соответствии с примером 6. Пример 4. Получение (S)-4-изобутил-2-оксопирролидин-3-карбоновой кислоты (формула 10) из калиевой соли (3S)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 23) В сосуд загружали водный раствор, содержащий калиевую соль (3S)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 23, 411 л, из примера 2). К этой смеси добавляли никель Ренея (50%ный водный раствор, Sigma-Aldrich) и в этот сосуд вводили газообразный водород в течение 20-часового промежутка времени для поддержания избыточного давления 50 фунт/дюйм 2 (344,74 кПа) в свободном пространстве сосуда во время реакции. Реакцию гидрирования контролировали по поглощению H2 и ВЭЖХ-анализу (колонка C18, 4,6 мм 150 мм, детекция при 200 нм) содержимого сосуда. После завершения реакции водную смесь фильтровали для удаления катализатора Ni Ренея. Значение pH концентрированного раствора доводили до 3,0, используя 37%-ную HCl (приблизительно 14 л). Полученный в результате раствор экстрагировали три раза EtOAc (50% об./об.). Объединенные органические слои концентрировали в вакууме с получением (S)-4-изобутил-2-оксопирролидин-3-карбоновой кислоты (формула 10). MC m/z [M+H]+186.1130. 13 С ЯМР (75 м.д., CDCl3):175,67, 172,23, 54,09, 47,62, 43,69, 37,22, 26,31, 23,34, 22,54. Выход 40-42%; 97% э.и Пример 5. Получение прегабалина (формула 9) из (S)-4-изобутил-2-оксопирролидин-3-карбоновой кислоты (формула 10)(формула 10), HCl (36-38%, 30 л) и воду (29 л). К этому раствору добавляли НОАс (1 л) и полученную суспензию нагревали в течение 36-38 ч при 80C и в течение еще 6 ч при 110C. Степень реакции контролировали посредством ВЭЖХ (колонка C18, 4,6 мм 150 мм, детекция при 200 нм). Воду и избытокHCl выпаривали с получением масла, которое промывали МТВЕ (215 л). К этому маслу добавляли воду,и смесь перемешивали до тех пор, пока раствор не становился прозрачным. Значение pH этого раствора доводили до 5,2-5,5, используя KOH (приблизительно 6 кг), что привело в результате к осаждению прегабалина. Эту смесь нагревали до 80C и затем охлаждали до 4C. Через 10 ч кристаллический прегабалин отфильтровывали и промывали IPA (12 л). Фильтрат концентрировали в вакууме с получением остаточного масла. К этому остаточному маслу добавляли воду (7,5 л) и EtOH (5,0 л) и полученную смесь- 23015418 нагревали до 80C и затем охлаждали до 4C. Через 10 ч вторую партию кристаллов прегабалина отфильтровывали и промывали IPA (1 л). Объединенные кристаллы прегабалина сушили в вакуумной печи при 45C в течение 24 ч. MC m/z [M+H]+160,1340. 1 Н ЯМР (300 МГц, D2O):2,97 (dd, J=5,4, 12,9 Гц, 1H), 2,89 (dd, J=6,6, 12,9 Гц, 1H), 2,05-2,34 (m,2H), 1,50-1,70 (септет, J=6,9 Гц, 1H), 1,17 (t, J=7,0 Гц, 2H), 0,85 (dd, J=2,2, 6,6 Гц, 6H). 13 С ЯМР (75 м.д., D2O):181,54, 44,32, 41,28, 32,20, 24,94, 22,55, 22,09. Выход 80-85%; э.и. 99,5%. Пример 6. Получение этилового эфира (R/S)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты(формула 20) посредством рацемизации этилового эфира (R)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 22) В сосуд загружали этиловый эфир (R)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты(формула 22, 49,5 кг) и EtOH (250 л). К этой смеси добавляли этилат натрия (21 мас./мас.%, в EtOH, 79,0 л, 1,1 экв.) и ее нагревали до 80C в течение 20 ч. После завершения реакции смесь оставляли охлаждаться до КТ и нейтрализовали добавлением HOAc (12,2 л). После выпаривания EtOH к этой смеси добавляли МТВЕ (150 л) и полученный в результате раствор фильтровали и упаривали с получением этилового эфира (R/S)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 20) с количественным выходом. Пример 7. Получение этилового эфира (S)-3-циано-5-метилгексановой кислоты (формула 24) из В 50-мл круглодонную колбу загружали (3S)-3-циано-2-этоксикарбонил-5-метилгексановую кислоту (формула 21, 3,138 г, 13,79 моль), NaCl (927 мг, 1,15 экв.), деионизованную воду (477 мкл, 1,92 экв.) и ДМСО (9,5 мл). Полученную смесь нагревали до 88C и выдерживали при этой температуре в течение 17 ч. Отбирали образец для ЖХ и ЖХ/МС анализов, которые показали присутствие исходного вещества(формула 21) и продуктов (формула 24 и формулы 25). Температуру этой смеси затем повышали до 135C и оставляли взаимодействовать в течение еще 3,5 ч. Отбирали второй образец для ЖХ и ЖХ/МС анализов, которые показали отсутствие исходного вещества (формула 21) и показали, помимо нужных продуктов (формула 24 и формула 25), присутствие неидентифицированных побочных продуктов. Этиловый эфир (S)-3-циано-5-метилгексановой кислоты (формула 24): 97,4% э.и. после 88C; 97,5% э.и. после 135C. Пример 8. Определение оптической чистоты (э.и.) (S)-4-изобутил-2-оксопирролидин-3-карбоновой кислоты (формула 10) Оптическую чистоту (S)-4-изобутил-2-оксопирролидин-3-карбоновой кислоты (формула 10) определяли способом дериватизации. Образец (S)-4-изобутил-2-оксопирролидин-3-карбоновой кислоты этерифицировали, используя EtOH, в присутствии каталитического количества безводной HCl в диоксане при 70C. Полученный в результате лактамный эфир анализировали посредством ВЭЖХ (CHIRALPAKAD-H, 4,6 мм 250 мм), используя подвижную фазу из гексана и EtOH (95:5), скорость потока 1,0 мл/мин,объем инъекции 10 мкл, температуру колонки 35C и детекцию при 200 нм. Пример 9. Определение оптической чистоты (э.и.) прегабалина (формула 9) Оптическую чистоту прегабалина анализировали способом дериватизации. Образец прегабалина дериватизировали, используя реагент Marfey (1-фтор-2,4-динитрофенил-5-L-аланинамид) и затем анализировали посредством ВЭЖХ (LUNA, колонка C18(2) 0,46 мм 150 мм, 3 мкм), используя подвижную фазу из водного NaPO4 (20 нМ, pH 2,0) и ACN (90:10 в течение 10 мин, 10:90 в течение 3 мин, 90:10 в течение 5 мин), скорость потока 1,2 мл/мин, объем инъекции 10 мкл, температуру колонки 35C и детекцию при 200 нм. Пример 10. Ферментативное разделение этилового эфира (R/S)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 20) с получением натриевой соли (3S)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 23) и этилового эфира (R)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 22) В реактор (16000 л), оборудованный верхней мешалкой, загружали кальций-ацетатный буфер (254 кг), деионизованную воду (1892,7 кг) и LIPOZYME TL 100L (LIPOLASE пищевого качества, 983,7 кг). После завершения перемешивания загружали этиловый эфир (R/S)-3-циано-2-этоксикарбонил-5 метилгексановой кислоты (формула 20, 9000 кг, 85%-ная чистота) и эту смесь перемешивали в течение 24 ч. Добавляли NaOH (2068 кг 30%-ного раствора) в процессе протекания реакции для поддержания pH 7,0. Степень реакции контролировали посредством ВЭЖХ (колонка C18, 4,6 мм 150 мм, детекция при 200 нм). После достижения приблизительно 42-45%-ного превращения (например через приблизительно 20-25 ч) титратор и мешалку останавливали. Органическую фазу немедленно отделяли, а водную фазу промывали дважды толуолом (780 кг). Водный слой, содержащий натриевую соль (3S)-3-циано-2 этоксикарбонил-5-метилгексановой кислоты (формула 23), использовали в следующих превращениях(пример 11) без отделения. Органические слои, содержащие этиловый эфир (R)-3-циано-2 этоксикарбонил-5-метилгексановой кислоты (формула 22), объединяли и концентрировали. Полученный в результате диэтиловый эфир затем рацемизировали в соответствии с примером 6. Пример 11. Получение этилового эфира (S)-3-циано-5-метилгексановой кислоты (формула 24) из натриевой соли (3S)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты (формула 23) В реактор (16000 л), оборудованный верхней мешалкой, загружали конечный водный раствор из примера 10 (9698,6 л, содержащий натриевую соль (3S)-3-циано-2-этоксикарбонил-5-метилгексановой кислоты, формула 23), NaCl (630 кг) и толуол (900 л). Смесь перемешивали в течение 2 ч в условиях дефлегмации (75-85C). Перемешивание прекращали; органическую фазу немедленно отделяли, а водную фазу дважды промывали толуолом (900 л). Органические слои, которые содержат этиловый эфир (S)-3 циано-5-метилгексановой кислоты (формула 24), объединяли и концентрировали. Этиловый эфир (формула 24) затем подвергали гидролизу в соответствии с примером 12. Пример 12. Получение калиевой соли (S)-3-циано-5-метилгексановой кислоты (формула 26) из этилового эфира (S)-3-циано-5-метилгексановой кислоты (формула 24) В реактор (12000 л), оборудованный верхней мешалкой, загружали этиловый эфир (S)-3-циано-5 метилгексановой кислоты (формула 24, 2196 л, из примера 11). К этой реакционной смеси добавлялиKOH (1795,2 кг, 45%-ный раствор, об./об.) и H2O (693,9 кг) при энергичном перемешивании. Температуру поддерживали при 25C. Через 4 ч эту реакционную смесь загружали в сосуд для гидрирования (пример 13) без какой-либо дополнительной обработки. Пример 13. Получение прегабалина (формула 9) из калиевой соли (S)-3-циано-5-метилгексановой кислоты (формула 26) В гидрогенизатор (12000 л) загружали воду (942,1 л) и реакционную смесь из примера 12, которая содержит калиевую соль (S)-3-циано-5-метилгексановой кислоты (формула 26, 4122,9 л). Добавляли суспензию никеля Ренея (219,6 кг, 50 мас./мас.%, в H2O). Гидрирование проводили при избыточном давлении 50 фунт/дюйм 2 (344,74 кПа) при 35C. Через 6 ч никель Ренея отфильтровывали и полученный в результате фильтрат переносили в реактор (16000 л) для кристаллизации. После добавления H2O (1098 л)pH раствора доводили до 7,0-7,5, используя HOAc (864,7 кг). Полученный в результате осадок отфильтровывали и промывали один раз H2O (549 л) и два раза IPA (каждый раз по 2,586 л). Твердое вещество перекристаллизовывали в IPA (12296 л) и H2O (6148 л). Смесь нагревали до 70C и затем охлаждали до 4C. Через 5-10 ч кристаллическое твердое вещество отфильтровывали, промывали IPA (5724 л) и сушили в вакуумной печи при 45C в течение 24 ч с получением прегабалина в виде белого кристаллического твердого вещества (1431 кг, общий выход 30,0%, чистота 99,5% и 99,75% э.и.). Следует отметить, что упоминание в данном описании и прилагаемой формуле изобретения какоголибо объекта в единственном числе может относиться к одному объекту или к множеству объектов, если из контекста явно не следует иное. Так, например, ссылка на композицию, содержащую "соединение",может включать одно соединение или два или более соединений. Следует понимать, что представленное выше описание предназначено для иллюстрации, но не для ограничения. Многие воплощения будут очевидны специалистам в данной области техники при прочтении данного описания. Таким образом, объем данного изобретения следует определять со ссылкой на прилагаемую формулу изобретения, и он включает полный объем эквивалентов, которые такие пункты защищают. Описания всех статей и ссылок,включая патенты, заявки на патенты и публикации, включены в данное описание ссылкой во всей полноте и для любых целей. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы 1 или его фармацевтически приемлемых комплекса, соли, сольвата или гидрата, где R1 и R2 являются разными, и каждый независимо выбран из атома водорода, C1-12 алкила, C3-12 циклоалкила и замещенного(а) взаимодействие соединения формулы 2 или его соли с кислотой и водой с получением соединения формулы 1 или его соли; и(б) возможно превращение соединения формулы 1 или его соли в фармацевтически приемлемые комплекс, соль, сольват или гидрат, где R1 и R2 в формуле 2 являются такими, как определено в формуле 1. 2. Способ по п.1, дополнительно включающий восстановление цианогруппировки соединения формулы 3 или его соли с получением соединения формулы 2 или его соли, гдеR3 в формуле 3 представляет собой C1-12 алкил, C3-12 циклоалкил или арил-C1-6 алкил. 3. Способ получения соединения формулы 1 или его фармацевтически приемлемых комплекса, соли, сольвата или гидрата, где R1 и R2 являются разными, и каждый независимо выбран из атома водорода, C1-12 алкила, C3-12 циклоалкила и замещенного(а) восстановление цианогруппировки соединения формулы 6 или его соли с получением соединения формулы 7(б) декарбоксилирование соединения формулы 7 или его соли с получением соединения формулы 1 или его соли; и(в) возможно превращение соединения формулы 1 или его соли в фармацевтически приемлемые комплекс, соль, сольват или гидрат, где R1 и R2 в формуле 6 и в формуле 7 являются такими, как определено в формуле 1.
МПК / Метки
МПК: C07C 229/08, C07C 253/30, C07C 227/22, C07C 227/18, C07C 255/19
Метки: получение, родственных, соединений, прегабалина
Код ссылки
<a href="https://eas.patents.su/29-15418-poluchenie-pregabalina-i-rodstvennyh-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Получение прегабалина и родственных соединений</a>
Получение прегабалина и родственных соединений

Номер патента: 11765
Опубликовано: 30.06.2009
Авторы: Мартинес Карлос Альберто, Келлехер Патрик Жерар Томас, Талли Уилльям Юджин, Дюмон Ив Рене, Ху Шангхуи, Тао Йунхуа
МПК: C07C 227/18, C07C 255/03, C07C 253/30...
Метки: соединений, прегабалина, получение, родственных
Формула / Реферат:
1. Способ получения соединения формулы 3 или его соли, где R1 и R2 являются разными, и каждый независимо выбран из атома водорода, С1-12алкила, С3-12циклоалкила и замещенного С3-12циклоалкила, и R3 представляет собой С1-12алкил, С3-12циклоалкил или арил-С1-6алкил, включающий: (а) приведение соединения формулы 4 в контакт с ферментом с получением соединения формулы 3 и соединения формулы 5 где данный фермент адаптирован к энантиоселективному...
Синтез гидроксисульфона и родственных соединений.

Номер патента: 1246
Опубликовано: 25.12.2000
Авторы: Матр Дэвид Дж., Муди Дэвид, Сохар Пол, Блэкер Эндрю Дж.
МПК: C07D 495/04
Метки: синтез, гидроксисульфона, соединений, родственных
Формула / Реферат:
1. Способ получения соединения формулы IV, имеющего структурную формулу где R представляет водород, С1-4 алкил или С1-4 алкокси-С1-4 алкил, включающий добавление ангидрида к первому раствору, содержащему растворитель и соединение формулы I где R определен выше, при поддержании температуры от примерно -5 до примерно 50шС, с получением второго раствора, содержащего соединение структурной формулы II где R определен выше, добавление ко...
Способы получения производных 4-(фенокси-5-метилпиримидин-4-илокси)пиперидин-1-карбоновой кислоты и родственных соединений

Номер патента: 14156
Опубликовано: 29.10.2010
Авторы: Гхарбауи Тавфик, Труп Беверли Уолгаст, Като Наоми С., Фрич Джон Р., Кришнан Ашвин М.
МПК: C07D 401/12
Метки: получения, 4-(фенокси-5-метилпиримидин-4-илокси)пиперидин-1-карбоновой, соединений, производных, родственных, способы, кислоты
Формула / Реферат:
1. Способ получения соединения формулы IгдеX представляет собой N;Y представляет собой N;Z представляет собой C1-8алкил;R1 представляет собой Н;R2 представляет собой -C(O)O-R22;R4 представляет собой Н;R11 представляет собой C1-4алкилсульфинил, C1-4алкилсульфонил;R12, R13, R14 каждый независимо представляет собой Н;R15 представляет собой C2-6алкенил, C1-4алкокси, C1-8алкил, C2-6алкинил, циано, галоген, C1-4галогеналкокси, C1-4галогеналкил,...
Получение промежуточных соединений эндотелина реакцией ассиметрического сопряженного присоединения с использованием хиральной добавки.

Номер патента: 2056
Опубликовано: 24.12.2001
Авторы: Щаен Дэвид М., Ксу Фенг, Тилльер Ричард Д.
МПК: A61K 31/44, C07D 213/55, C07B 37/02...
Метки: ассиметрического, получение, хиральной, соединений, сопряженного, эндотелина, реакцией, использованием, добавки, промежуточных, присоединения
Формула / Реферат:
1. Способ получения соединения формулы I где представляет а) 5- или 6-членный гетероциклил, содержащий одну, две или три двойные связи, по крайней мере одну двойную связь, и 1, 2 или 3 гетероатома, выбранных из О, N и S, незамещенный или замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из ОН, CO2R4, Br, Cl, F, I, СF3, N(R5)2, C1-C8 алкокси, C1-C8 алкила, C2-C8 алкенила, C2-C8 алкинила или C3-C8 циклоалкила,...
Получение олефинов посредством конверсии кислородсодержащих соединений

Номер патента: 14002
Опубликовано: 30.08.2010
Авторы: Миллер Лоренс В., Паджадо Питер Р., Вора Бипин В., Сенетар Джон Дж.
МПК: C07C 5/48
Метки: посредством, олефинов, конверсии, кислородсодержащих, получение, соединений
Формула / Реферат:
1. Интегрированный способ синтеза кислородсодержащих соединений и их конверсии в легкие олефины, где указанный способ включаетконтактирование исходного сырья (16), содержащего синтез-газ, в зоне (22) реактора конверсии синтез-газа с каталитическим материалом конверсии синтез-газа при реакционных условиях, эффективных для образования исходящего потока (24) зоны реактора конверсии синтез-газа, содержащего, по меньшей мере, метанол;контактирование...
Предыдущий патент: Пестицидные композиции с риском кристаллизации и способ их получения
Следующий патент: Новые производные фенантридина в качестве антагонистов брадикинина