Циклопентаноиндолы, фармацевтические композиции и способы лечения
Номер патента: 6765
Опубликовано: 28.04.2006
Авторы: Стурино Клаудио, Рой Бруно, Лябелль Марк, Бертелетт Карл, Бойд Майкл, Шайгетц Джон, Ляшанс Николя








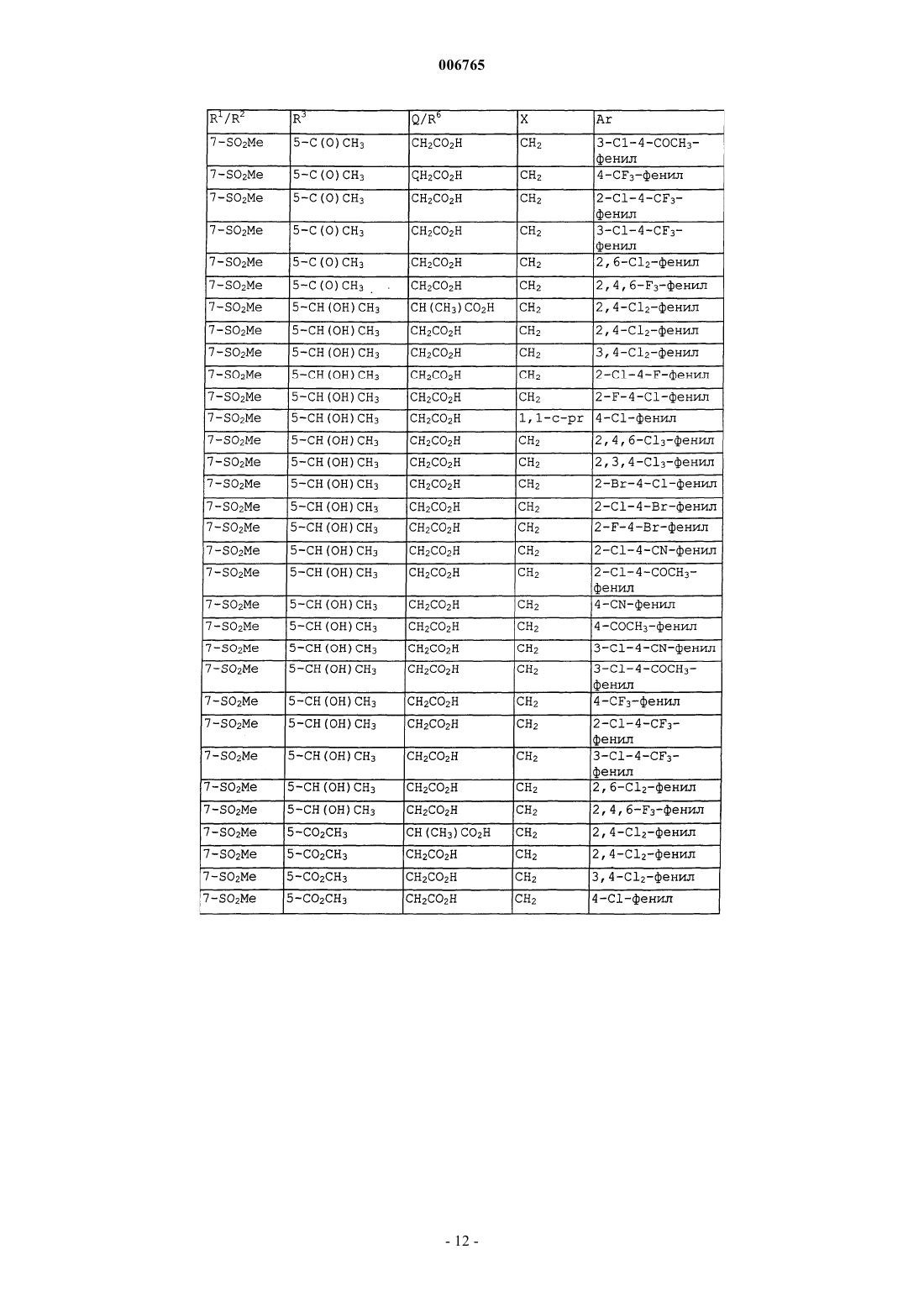
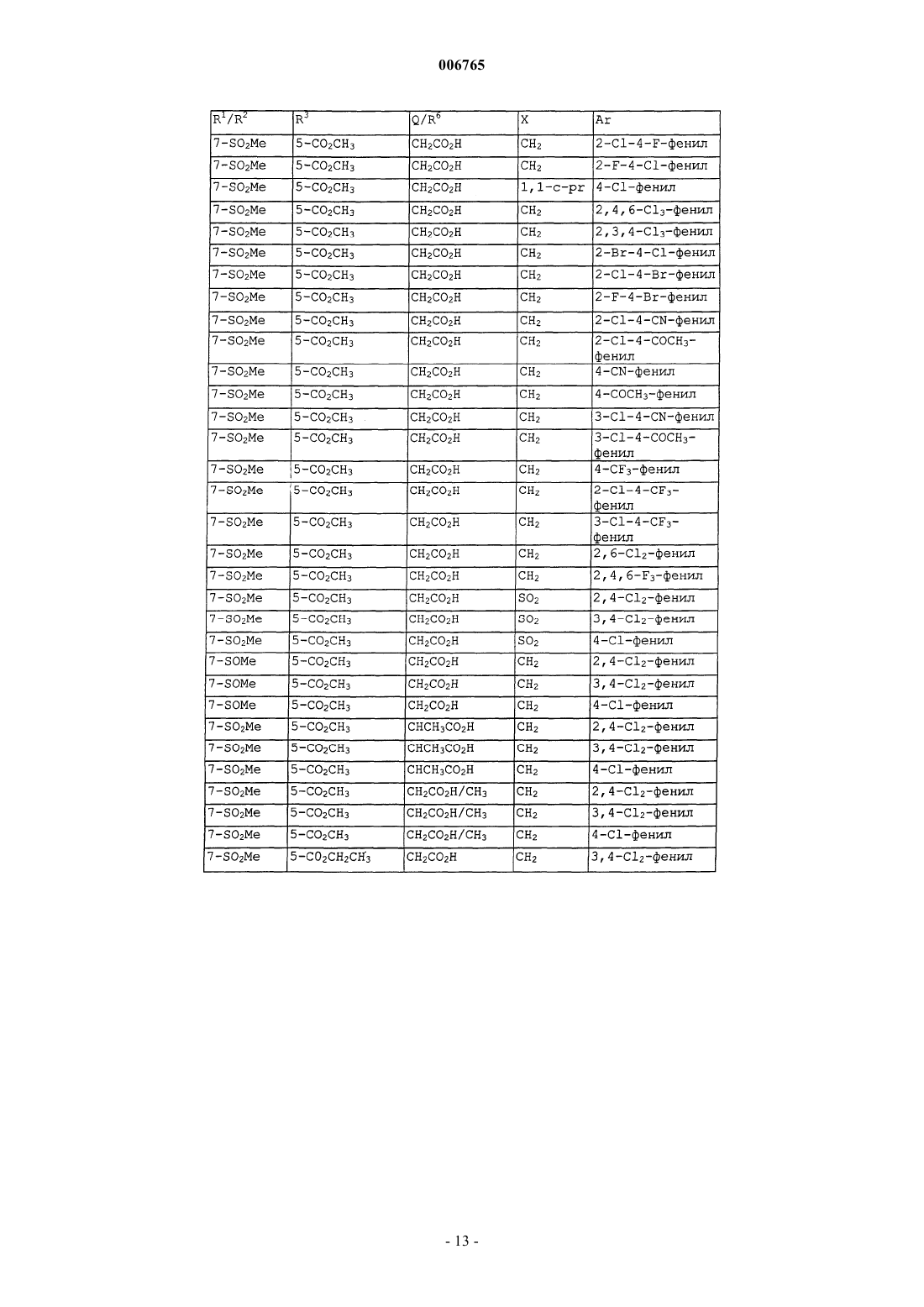
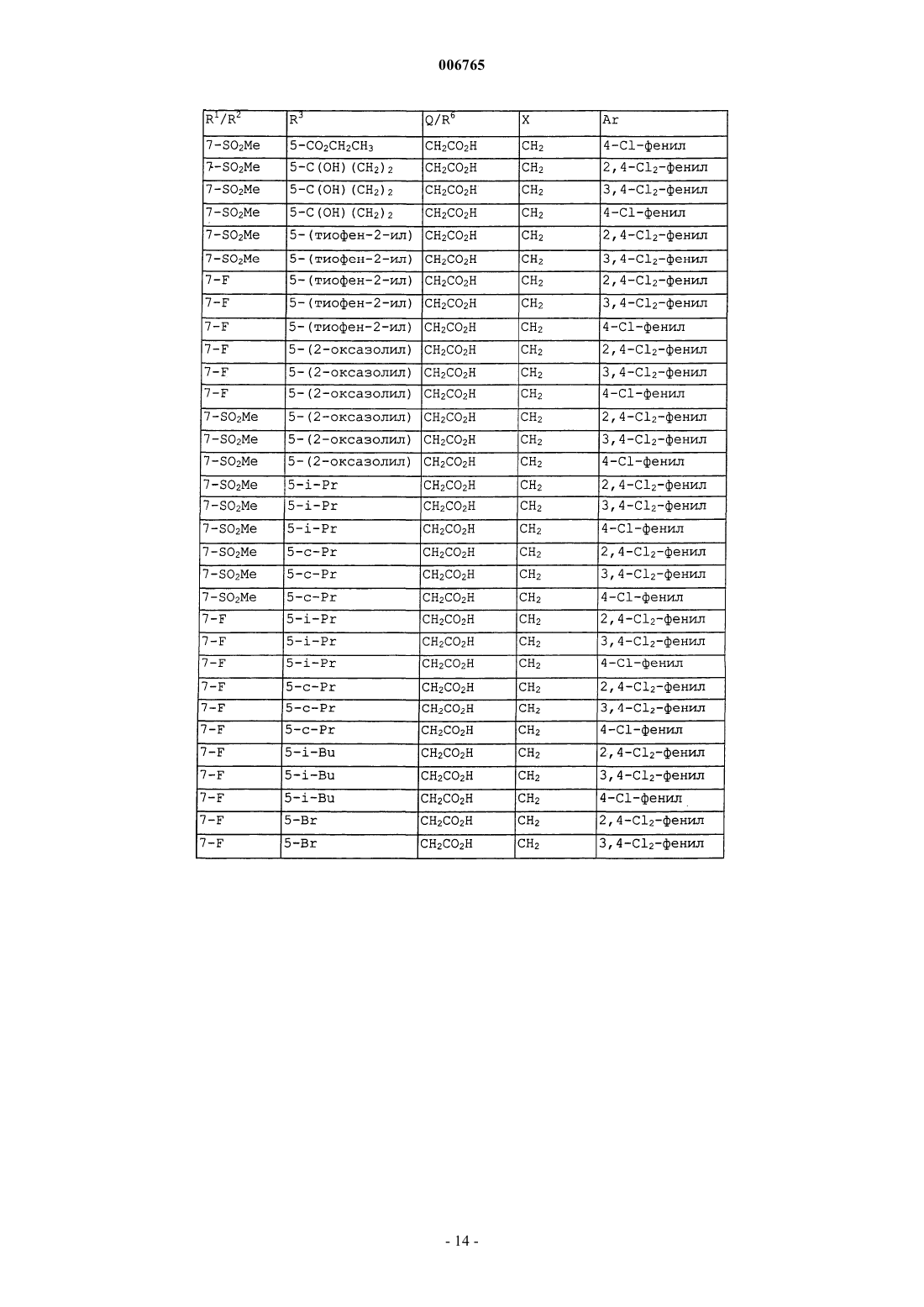
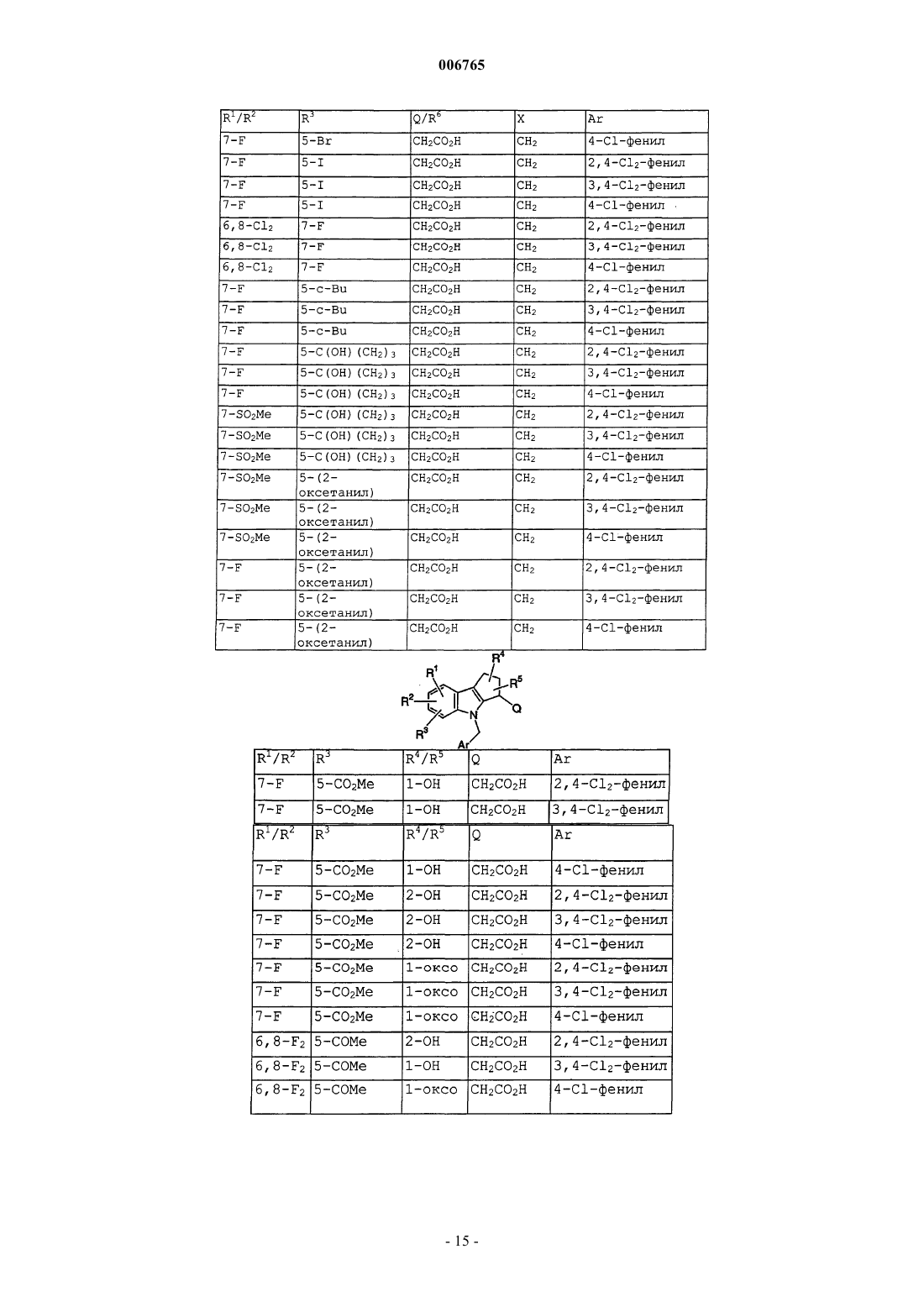
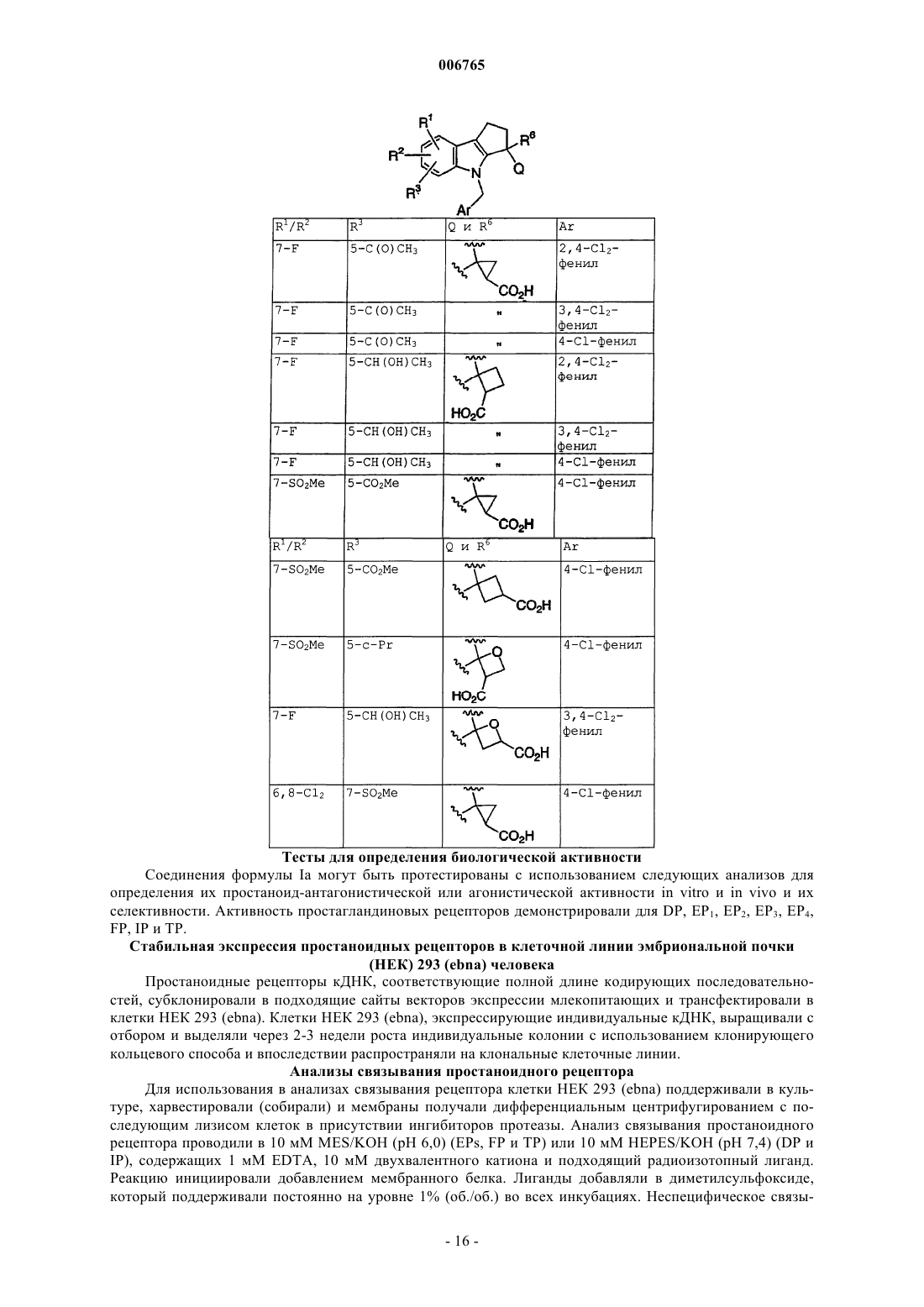











Формула / Реферат
1. Соединение, имеющее формулу Iа

где Ar представляет хлорфенил или дихлорфенил;
R1 и R2 независимо выбирают из галогена, -SO2CH3, -SO2N(CH3)2, C(O)CH3, -СН(ОН)СН3, -СН=СН2, циклопропила и тиофенила;
или его фармацевтически приемлемые соли, гидраты и эфиры.
2. Соединение по п.1, где R1 находится в 7 положении.
3. Фармацевтическая композиция, включающая соединение формулы Iа по любому из пп.1, 2 или его фармацевтически приемлемую соль, гидрат или сложный эфир и фармацевтически приемлемый носитель.
4. Способ лечения простагландин-опосредованных заболеваний, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1.
5. Способ лечения простагландин-D2 опосредованных заболеваний, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1.
6. Способ лечения закупорки носового канала, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1.
7. Способ лечения аллергической астмы, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1.
8. Способ лечения аллергического ринита, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1.
9. Применение соединения формулы Ia по любому из пп.1, 2 или его фармацевтически приемлемой соли, гидрата или сложного эфира для получения лекарственного средства для лечения простагландин-опосредованных заболеваний.
10. Применение соединения формулы Iа по любому из пп.1, 2 или его фармацевтически приемлемой соли, гидрата или сложного эфира для получения лекарственного средства для лечения закупорки носового канала, аллергической астмы или аллергического ринита.
11. Антагонистическая фармацевтическая композиция в отношении закупоривающего действия носовых каналов и легких под действием простагландинов D-типа, включающая приемлемое антагонистическое количество соединения формулы Iа по любому из пп.1, 2 или его фармацевтически приемлемой соли, гидрата или сложного эфира в сочетании с фармацевтически приемлемым носителем.
Текст
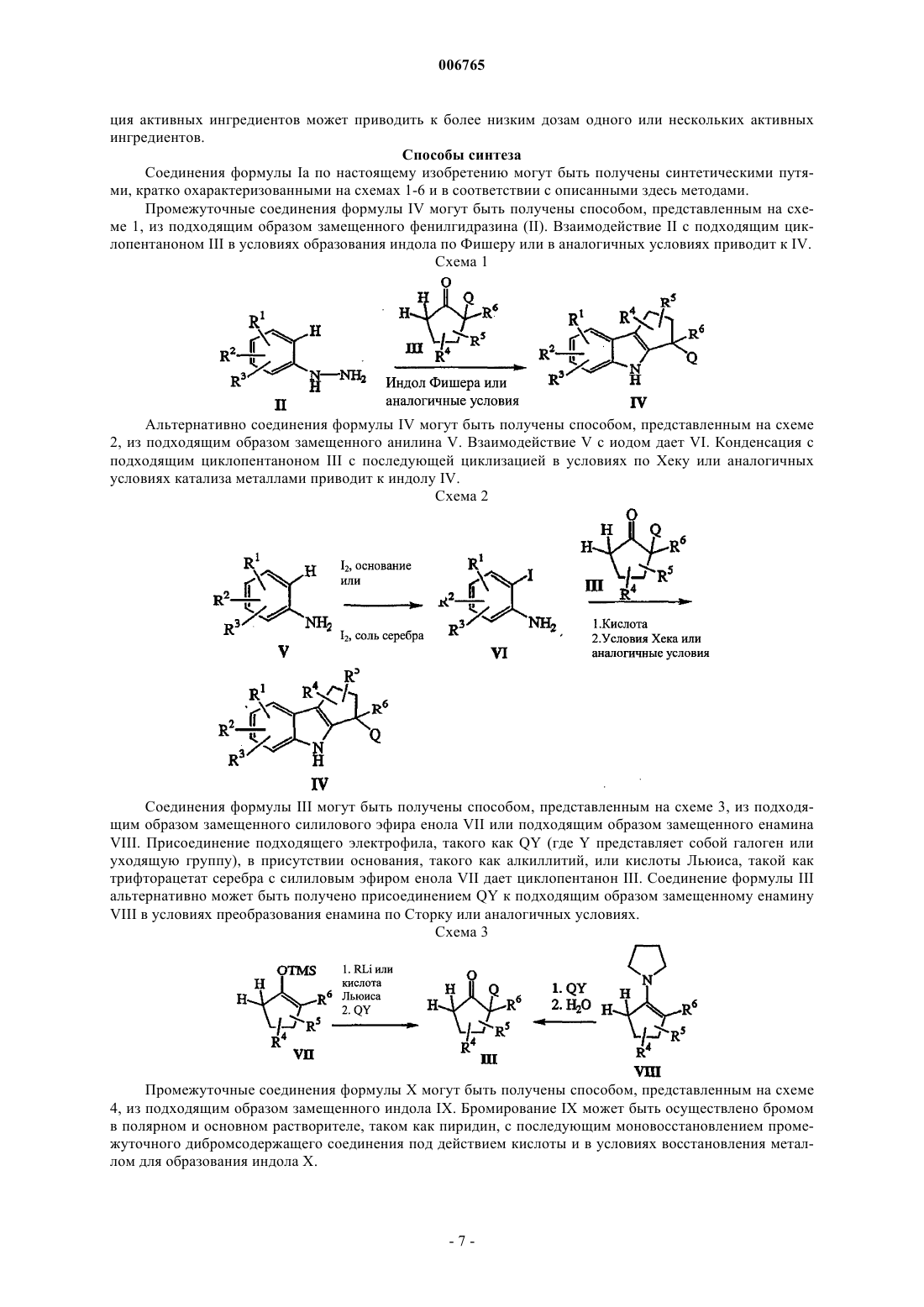
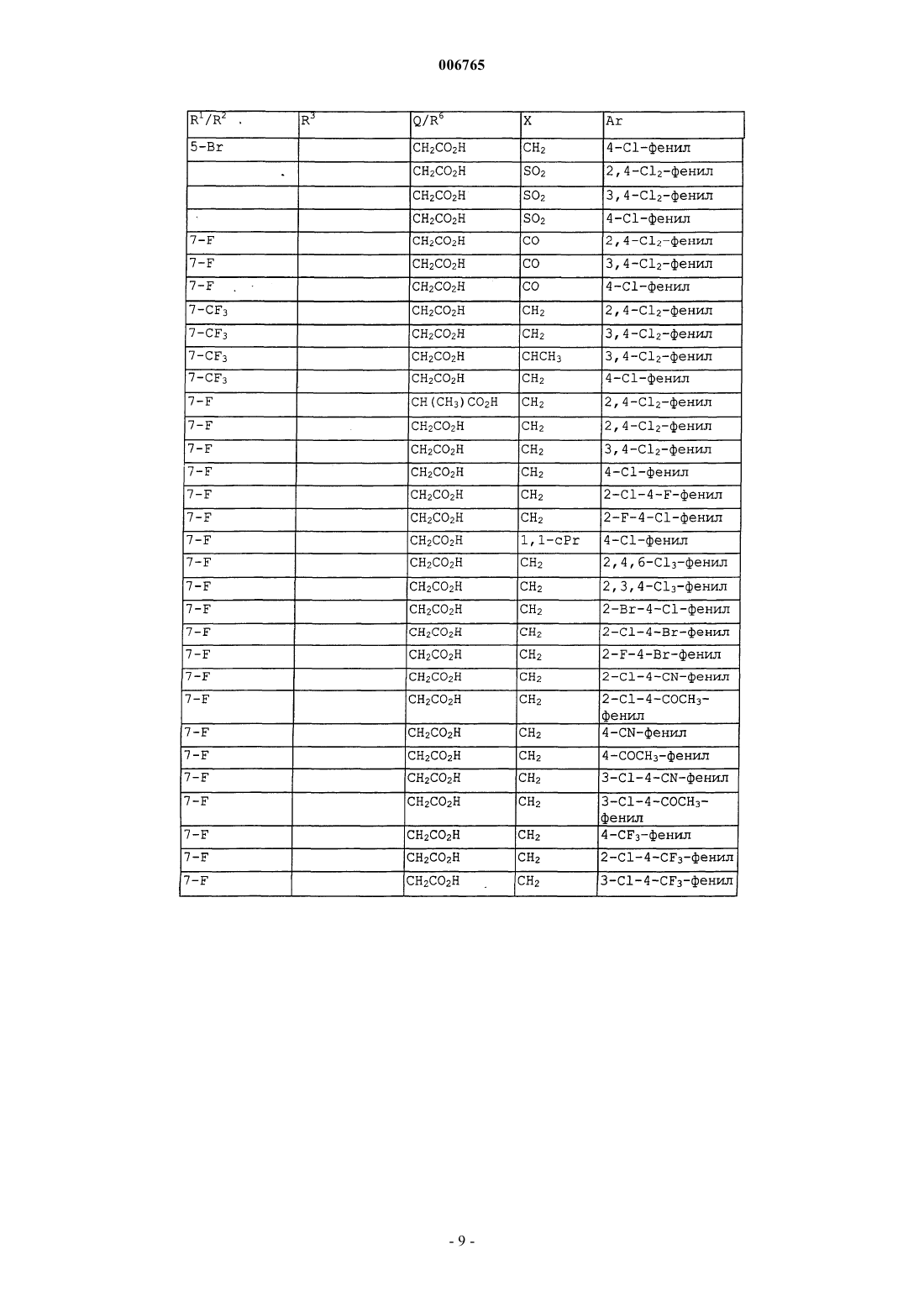
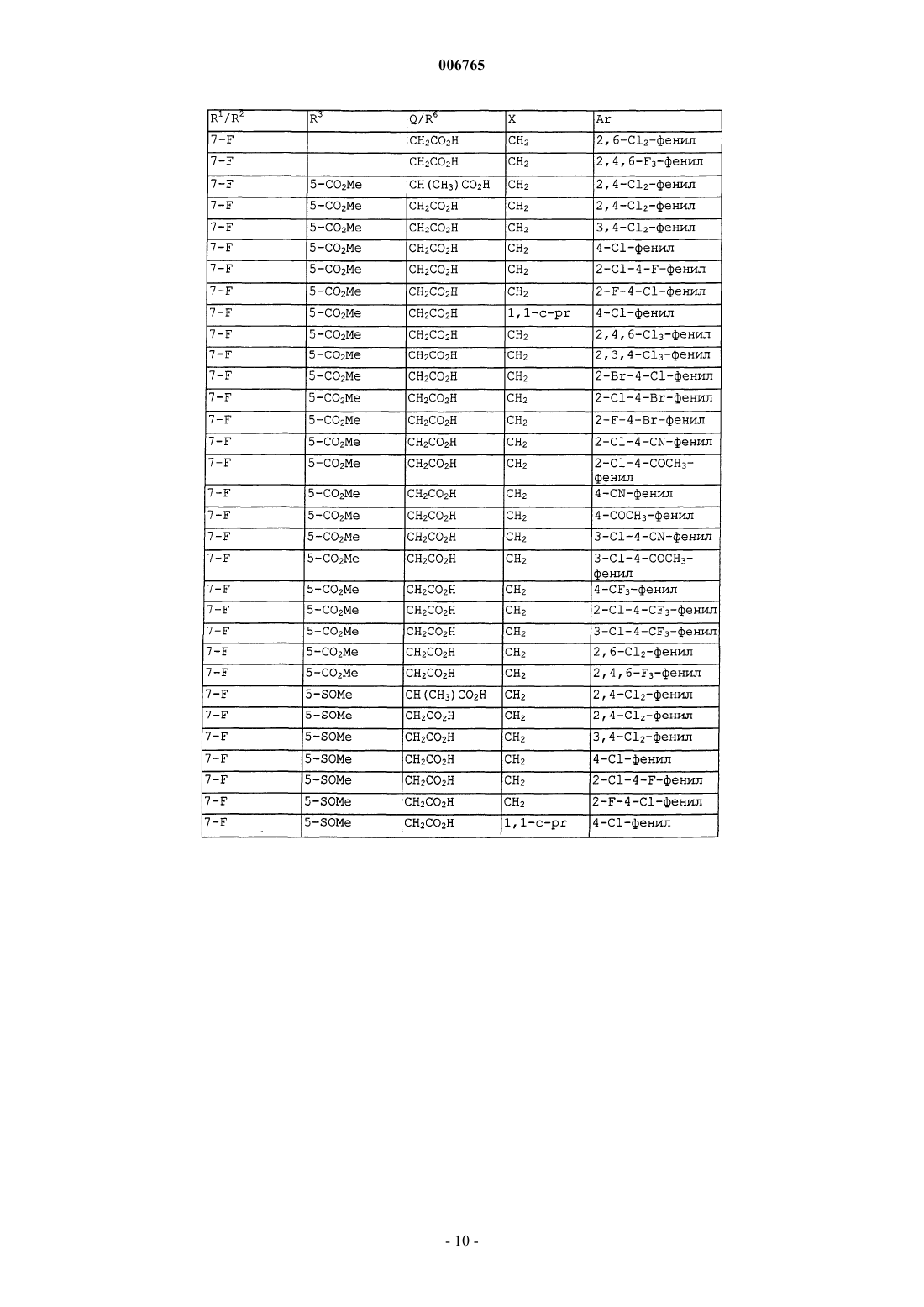
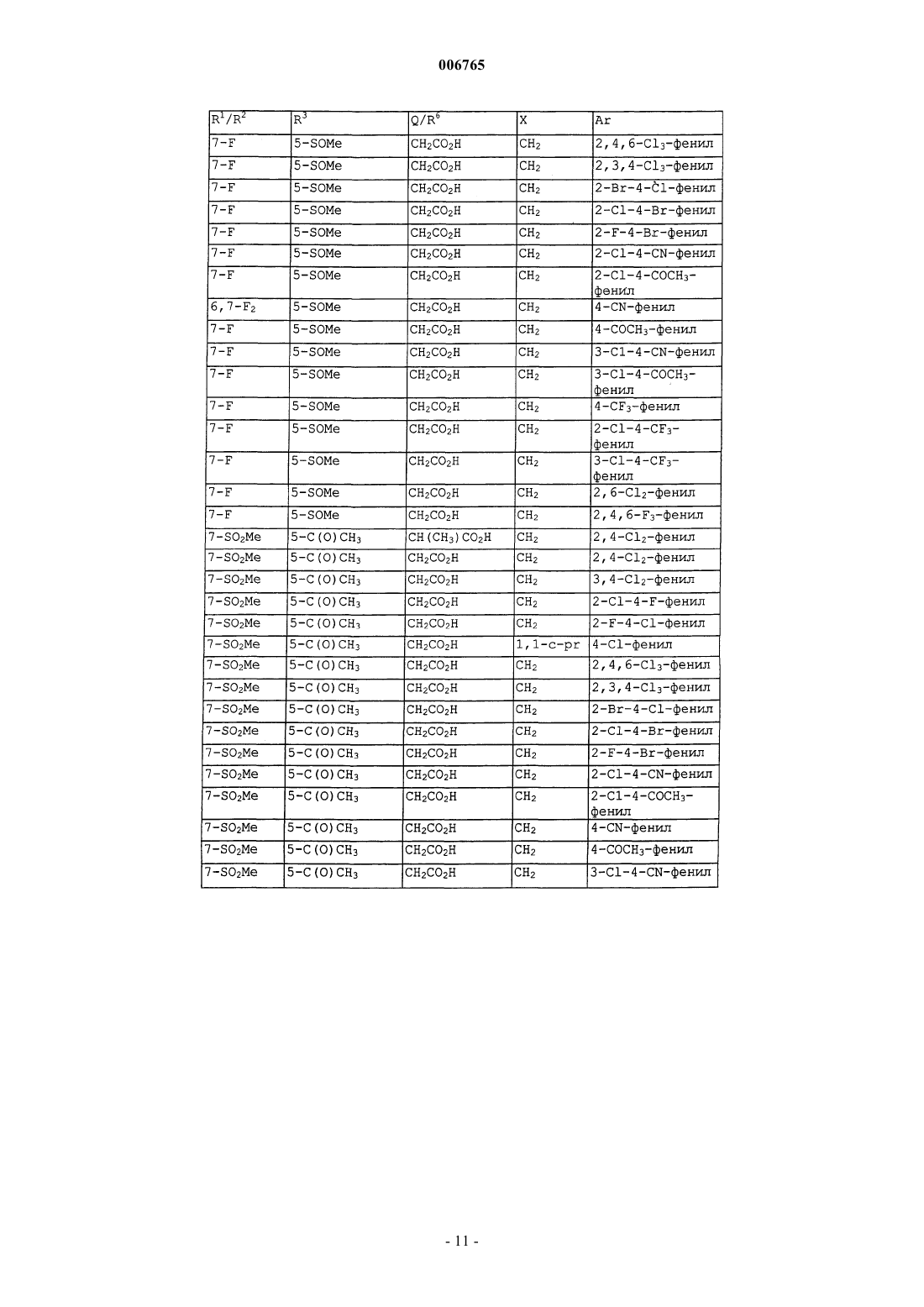
006765 Настоящее изобретение относится к соединениям и способам лечения простагландинопосредованных заболеваний и их некоторым фармацевтическим композициям. Более конкретно, соединения по изобретению структурно отличаются от стероидов, антигистаминов или адренергических агонистов и являются антагонистами действия простагландинов D-типа, закупоривающего носовые каналы и легкие. В двух обзорных статьях описаны характеристики и терапевтическая значимость простаноидных рецепторов, а также большинства обычно используемых селективных агонистов и антагонистов: Eicosanoids: From Biotechnology to Therapeutic Applications (Эйкозаноиды: от биотехнологии к терапевтическим применениям), ред. Folco, Samuelsson, Maclouf and Velo, Plenum Press, New York, 1996, часть 14,137-154, и Journal of Lipid Mediators and Cell Signalling, 1996, 14,83-87. В статье T.Tsuri et al., опубликованной в 1997 г. в Journal of Medicinal Chemistry, vol.40, pp.3504-3507, указано, что PGD2 рассматривается как важный медиатор при различных аллергических заболеваниях, таких как аллергический ринит,неинфекционно-аллергическая (атопическая) бронхиальная астма, аллергический конъюнктивит и атопический дерматит. Совсем недавно, в статье Matsuoka et al., в Science (2000), 287:2013-7, PGD2 описан как ключевой медиатор при аллергической астме. Кроме того, в патентах, таких как патент США 4808608, упоминается, что антагонисты простагландина могут использоваться при лечении аллергических заболеваний и подробно при лечении аллергической астмы. Антагонисты PGD2 описаны, например,в заявке на Европейский патент 837052 и заявке РСТ WO98/25919, а также WO99/62555. В патенте США 4808608 описаны производные тетрагидрокарбазол-1-алкановой кислоты в качестве антагонистов простагландина. В заявке на Европейский патент 468785 описано соединение 4-[(4-хлорфенил)метил]-1,2,3,4 тетрагидро-7-(2-(хинолинилметокси)циклопент[b]индол)-3-уксусная кислота, которое является разновидностью рода, считающегося ингибиторами биосинтеза лейкотриена. В патенте США 3535326 описаны соединения, обладающие противовоспалительным действием,формулы Краткое изложение сущности изобретения Настоящее изобретение относится к новым соединениям, которые представляют собой антагонисты простагландиновых рецепторов; более конкретно, они являются антагонистами простагландин D2 рецептора (DP рецептор). Соединения настоящего изобретения полезны для лечения различных простагландин-опосредованных заболеваний и болезненных состояний; соответственно, настоящее изобретение относится к способу лечения простагландин-опосредованных заболеваний с использованием описанных в нем новых соединений, а также содержащих их фармацевтических композиций. Подробное описание изобретения Настоящее изобретение относится к соединениям формулы Iа и их фармацевтически приемлемым солям, гидратам и сложным эфирам,где Ar представляет хлорфенил или дихлорфенил;R1 и R2 независимо выбирают из галогена, -SO2CH3, -SO2N(CH3)2, С(O)СН 3, -СН(OН)СН 3, -СН=СН 2,циклопропила и тиофенила; В объем изобретения также включены фармацевтические композиции, содержащие соединение формулы Iа и способы лечения или профилактики простагландин-опосредованных заболеваний с использованием соединений формулы Iа. Нумерация ядра трициклической кольцевой системы показана ниже-1 006765 Изобретение описано с использованием следующих определений, если не указано другого. Термин галоген включает F, Cl, Br и I. Для данного описания следующие сокращения имеют указанные значения: ВОС = трет-бутилоксикарбонилLDA = диизопропиламид лития МСРВА = метахлорпербензойная кислотаTfO= трифторметансульфонат =трифлат ТСХ= тонкослойная хроматография Галоген предпочтительно представляет собой фтор. Предпочтительными соединениями формулы Iа являются соединения, где R1 не является водородом в 7-положении. Оптические изомеры - Диастереомеры - Геометрические изомеры -Таутомеры Соединения формулы Iа содержат один или несколько асимметрических центров и следовательно могут существовать в виде рацематов и рацемических смесей, отдельных энантиомеров, диастереомерных смесей и отдельных диастереомеров. Подразумевается, что настоящее изобретение включает в себя все такие изомерные формы соединений формулы Iа. Некоторые из описанных здесь соединений содержат олефиновые двойные связи, и подразумевается, что, если не указано другого, включают оба геометрических изомера Е и Z. Некоторые из описанных здесь соединений, которые могут существовать при различных местах присоединения водорода, упоминаются как таутомеры. Таким примером может быть кетон и его енольная форма, известные как кето-енольные таутомеры. Соединения формулы Iа охватывают индивидуальные таутомеры, а также их смеси. Соединения формулы Iа могут быть разделены на диастереоизомерные пары энантиомеров, например, дробной кристаллизацией из подходящего растворителя, например метанола или этилацетата или из их смеси. Полученная таким образом пара энантиомеров может быть разделена на индивидуальные стереоизомеры обычными методами, например, при использовании в качестве разделяющего агента оптически активной кислоты. Альтернативно, любой энантиомер соединения общей формулы Iа может быть получен стереоспецифическим синтезом с использованием оптически чистых исходных веществ или реагентов известной конфигурации. Соли Термин фармацевтически приемлемые соли относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди,железа (II), железа (III), лития, магния, марганца (III), марганца (II), калия, натрия, цинка и тому подоб-2 006765 ные. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины,циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кафеин, холин, N,N'дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и тому подобные. Когда соединение настоящего изобретения является основным, соли могут быть получены из фармацевтически приемлемых нетоксичных кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромисто-водородную, хлористо-водородную,изетионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную,памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуолсульфоновую кислоту и тому подобные. Особенно предпочтительными являются лимонная, бромисто-водородная, хлористоводородная, малеиновая, фосфорная, серная и винная кислоты. Следует понимать, что если не указано другого, подразумевается, что ссылки на соединения формулы Iа также включают фармацевтически приемлемые соли. Применение Способность соединений формулы Iа взаимодействовать с простагландиновыми рецепторами делает их полезными для профилактики или исчезновения нежелательных симптомов, вызванных простагландинами у млекопитающих, в особенности у человека. Такая имитация или антагонизм действия простагландинов указывают на то, что соединения и их фармацевтические композиции полезны для лечения, профилактики или улучшения самочувствия у млекопитающих и особенно у людей при заболевании дыхательных путей, аллергических состояниях, боли,воспалительных заболеваниях, нарушениях выделений слизистой, костных заболеваниях, нарушений сна, нарушений способности к воспроизведению потомства, нарушений свертываемости крови, проблем зрения, а также иммунных и аутоиммунных заболеваний. Кроме того, такое соединение может ингибировать клеточные трансформации, относящиеся к новообразованиям, и рост метастазов опухолей и, следовательно, может использоваться при лечении рака. Соединения формулы Iа также могут использоваться для лечения и/или профилактики простагландин-опосредованных нарушений пролиферации, таких,которые могут существовать при диабетической ретинопатии и ангиогенезе опухолей. Соединения формулы Iа также могут ингибировать простаноид-индуцированное сокращение гладкой мускулатуры за счет антагонизирования сокращающихся простаноидов или имитации расслабляющих простаноидов и вследствие этого могут использоваться при лечении дисменореи (расстройства менструального цикла),преждевременных родов и относящихся к эозинофилам заболеваний. Более конкретно, соединения формулы Iа представляют собой антагонисты простагландина D2. Соответственно, другой аспект изобретения относится к способу лечения или профилактики простагландин-опосредованного заболевания, включающему введение нуждающемуся в таком лечении пациенту-млекопитающему соединения формулы Iа в количестве, которое является эффективным для лечения или профилактики указанного простагландин-опосредованного заболевания. Простагландин-опосредованные заболевания включают, но не ограничиваются ими, аллергический ринит, закупорку носового канала, ринорею, продолжительный ринит, воспаление носового канала, астму, включая аллергическую астму, хронические обструктивные заболевания легких и другие формы воспаления легких; нарушения сна и нарушения цикла засыпание-просыпание; простаноид-индуцированное сокращение гладкой мускулатуры, связанное с дисменореей (расстройством менструального цикла) и преждевременными родами; связанные с эозинофилами заболевания; тромбоз; глаукому и нарушения зрения; заболевания закупорки сосудов; застойную сердечную недостаточность; заболевания или болезненные состояния, требующие антикоагулирующего лечения, такие как посттравматическое или постхирургическое лечение; воспаление; гангрену; болезнь Рейнауда (Raynaud); нарушения секреции слизистой, включая цитозащиту; боль и мигрень; заболевания, требующие борьбы с образованием и рассасыванием костной ткани, такие, например, как остеопороз; шок; терморегуляцию, включая высокую температуру; и иммунные нарушения или болезненные состояния, при которых желательно иммунорегулирование. Более конкретно, предназначенное для лечения заболевание является заболеванием, опосредованным простагландином D2, таким как закупорка носового канала, закупорка легких и астма, включая аллергическую астму. Одним вариантом осуществления изобретения является способ лечения или профилактики простагландин-опосредованного заболевания, включающий введение нуждающемуся в таком лечении пациентумлекопитающему соединения формулы Iа в количестве, которое является эффективным для лечения или профилактики простагландин-опосредованного заболевания, где простагландин-опосредованное заболевание представляет собой закупорку носового канала, ринит, включая аллергический и продолжительный ринит, и астму, включая аллергическую астму.-3 006765 Другим вариантом осуществления настоящего изобретения является способ лечения или профилактики простагландин D2-опосредованного заболевания, включающий введение нуждающемуся в таком лечении пациенту-млекопитающему соединения формулы Iа в количестве, которое является эффективным для лечения или профилактики простагландин D2-опосредованного заболевания, где указанное простагландин D2-опосредованное заболевание представляет собой закупорку носового канала или астму. Другим вариантом осуществления настоящего изобретения является способ лечения закупорки носового канала у нуждающегося в таком лечении пациента, который включает введение указанному пациенту терапевтически эффективного количества соединения формулы Iа. Еще одним вариантом осуществления настоящего изобретения является способ лечения астмы, в частности аллергической астмы, у нуждающегося в таком лечении пациента, который включает введение указанному пациенту терапевтически эффективного количества соединения формулы Iа. Диапазон дозировок Величина профилактической или терапевтической дозы соединения формулы Iа, конечно, будет изменяться в зависимости от природы и серьезности подвергаемого лечению болезненного состояния,конкретного соединения формулы Iа и пути введения. Она также будет изменяться в соответствии со множеством факторов, включая возраст, вес, общее состояние здоровья, пол, режим питания, время введения, скорость выделения, комбинацию лекарственных средств и ответную реакцию индивидуального пациента. Обычно дневная доза составляет примерно от 0,001 до примерно 100 мг на кг веса тела млекопитающего, предпочтительно от 0,01 до примерно 10 мг на кг. С другой стороны, в некоторых случаях может оказаться необходимым использовать дозировки, находящиеся за данными границами. Количество активного ингредиента, которое можно объединять с веществами носителя для получения отдельной препаративной лекарственной формы, будет изменяться в зависимости от организмахозяина, подвергаемого лечению и конкретного способа введения. Например, состав, предназначаемый для перорального введения людям, может содержать от 0,05 мг до 5 г активного агента, компаундированного с соответствующим и подходящим количеством материала носителя, которое будет изменяться примерно от 5 до 99,95% от общей композиции. Единичные препаративные лекарственные формы обычно будут содержать примерно от 0,1 мг до примерно 0,4 г активного ингредиента, обычно 0,5, 1, 2, 5, 10,25, 50, 100, 200 или 400 мг. Фармацевтические композиции Другой аспект настоящего изобретения относится к фармацевтическим композициям, включающим соединение формулы Iа с фармацевтически приемлемым носителем. Термин композиция, как он употреблен для фармацевтической композиции, предназначен для охвата продукта, включающего активный(е) ингредиент(ы) и инертный ингредиент(ы) (фармацевтически приемлемые эксципиенты), который составляет носитель, а также любого продукта, который возникает, непосредственно или косвенно, при комбинации, комплексообразовании или агрегации любых двух или более ингредиентов, или при диссоциации одного или нескольких ингредиентов, или в результате других типов реакций или взаимодействий одного или нескольких ингредиентов. Соответственно фармацевтические композиции настоящего изобретения охватывают любую композицию, полученную смешиванием соединения формулы Iа, дополнительного активного ингредиента(ов) и фармацевтически приемлемых эксципиентов. Для лечения любого из простаноид-опосредованных заболеваний соединения формулы Iа можно вводить орально, путем ингаляционного распыления, местно, парентерально или ректально в виде единичных препаративных лекарственных составов, содержащих обычные нетоксичные фармацевтически приемлемые носители, адьюванты и наполнители. Термин парентеральный, как он использован в данном описании, включает подкожные инъекции, внутривенные, внутримышечные, интрастенальные инъекции или методики вливания. В дополнение к лечению теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, кошки и т.д., соединение по изобретению эффективно для лечения людей. Фармацевтические композиции, содержащие активный ингредиент, могут находиться в форме,подходящей для перорального применения, например, в виде таблеток, пилюль, лепешек для рассасывания, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких желатиновых капсул, или сиропов, или эликсиров. Композиции, предназначенные для перорального применения, могут быть получены в соответствии с любым способом, известным в данной области для производства фармацевтических композиций, и такие композиции могут содержать один или несколько агентов, выбранных из группы, состоящей из подсластителей, вкусовых агентов, красителей и консервантов для получения фармацевтически превосходных и приятных препаратов. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые являются подходящими для производства таблеток. Такие эксципиенты могут представлять собой, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал или альгиновую кислоту; связующие агенты, например крахмал, желатин или гуммиарабик, и смазочные агенты, например стеарат магния, стеариновую кислоту или тальк. Таблетки могут быть не покрыты-4 006765 оболочкой, или они могут быть покрыты оболочкой известными способами для замедления разложения и поглощения в желудочно-кишечном тракте и обеспечения таким образом продолжительного действия в течение длительного периода времени. Например, можно использовать вещество для отсрочки времени, такое как глицерилмоностеарат или глицерилдистеарат. Они также могут быть покрыты оболочкой способом, описанным в патентах США 4256108; 4166452 и 4265874, для образования осмотических лечебных таблеток для контролируемого высвобождения. Составы для перорального применения также могут быть представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активные ингредиенты смешивают со смешивающимися с водой растворителями, такими как пропиленгликоль, PEG (полиэтиленгликоли) и этанол, или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Водные суспензии содержат активный ингредиент в смеси с эксципиентами, подходящими для получения водных суспензий. Такие эксципиенты представляют собой суспендирующие агенты, например карбоксиметилцеллюлозу натрия, метилцеллюлозу, гидроксипропилметилцеллюлозу, альгинат натрия,поливинилпирролидон, трагакантовую камедь и гуммиарабик; диспергаторы или смачивающие агенты могут представлять собой природный фосфатид, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гекситола, такие как моноолеат полиоксиэтиленсорбита, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и ангидридов гекситола, например полиэтиленсорбитан моноолеат. Водные суспензии также могут содержать один или несколько консервантов,например, этил- или н-пропил, п-гидроксибензоат, один или несколько красителей, один или несколько ароматизаторов, один или несколько подсластителей, таких как сахароза, сахарин или аспартам. Масляные суспензии могут быть получены суспендированием активного ингредиента в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле, или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Для улучшения вкуса перорального препарата можно добавлять подсластители, такие как указанные выше, и вкусовые агенты. Данные композиции можно сохранять при добавлении антиоксиданта, такого как аскорбиновая кислота. Диспергируемые порошки и гранулы, подходящие для получения водной суспензии при добавлении воды, обеспечивают активный ингредиент в смеси с диспергирующим или смачивающим агентом,суспендирующим агентом и одним или несколькими консервантами. Подходящие диспергирующие или смачивающие агенты и суспендирующие агенты, которые могут быть использованы, упоминались выше. Также могут присутствовать дополнительные эксципиенты, например подсластители, вкусовые агенты и красители. Фармацевтические композиции по изобретению также могут быть в форме эмульсии масло-в-воде. Масляная фаза может представлять собой растительное масло, например оливковое масло, или арахисовое масло, или минеральное масло, например жидкий парафин, или их смеси. Подходящие эмульгирующие агенты могут представлять собой природные фосфатиды, например соевое масло, лецитин и сложные эфиры или частичные сложные эфиры, полученные из жирных кислот и ангидридов гекситола, например сорбитанмоноолеат, и продукты конденсации указанных частичных сложных эфиров с этиленоксидом, например полиоксиэтиленсорбитанмоноолеат. Эмульсии также могут содержать подсластители и вкусовые агенты. Сиропы и эликсиры могут быть получены с использованием подсластителей, например глицерина,пропиленгликоля, сорбита или сахарозы. Такие составы также могут содержать средство, уменьшающее раздражение, консервант и вкусовые или красящие агенты. Фармацевтические композиции могут быть в форме стерильной инъецируемой водной или масляной суспензии. Такая суспензия может быть получена в соответствии с известным уровнем техники с использованием таких подходящихдиспергирующих или смачивающих агентов и суспендирующих агентов, которые были отмечены выше. Стерильные инъецируемые препараты также могут представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3 бутандиоле. Приемлемыми носителями и растворителями, которые можно использовать, являются вода,раствор Рингера и изотонический раствор хлорида натрия. Также можно использовать сорастворители,такие как этанол, пропиленгликоль или полиэтиленгликоли. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные жирные (нелетучие) масла. Для данной цели можно использовать любое мягкое жирное (нелетучее) масло, включая синтетические моно- или диглицериды. Кроме того, при получении препаратов для инъекций находят применение жирные кислоты. Соединения формулы Iа также можно вводить в форме суппозиториев для ректального введения лекарственного средства. Такие композиции могут быть получены смешиванием лекарственного средства с подходящим нераздражающим эксципиентом, который является твердым при температурах окру-5 006765 жающей среды, но жидким при ректальной температуре, и следовательно, будет расплавляться в прямой кишке, высвобождая лекарственное средство. Такими веществами являются кокосовое масло и полиэтиленгликоли. Для местного применения используют кремы, мази, гели, растворы или суспензии и т.д., содержащие соединение формулы Iа. (Для такого применения местное применение будет включать жидкости для полоскания рта и полоскания для горла). Местные составы обычно могут включать фармацевтический носитель, сорастворитель, эмульгатор, усилитель проницаемости, систему консервантов и мягчительное средство. Комбинации с другими лекарственными средствами. Для лечения и профилактики простагландин-опосредованных заболеваний соединение формулы Iа можно вводить совместно с другими терапевтическими агентами. Таким образом, в другом аспекте настоящее изобретение относится к фармацевтическим композициям для лечения простагландинопосредованных заболеваний, включающим терапевтически эффективное количество соединения формулы Iа и один или несколько других терапевтических агентов. Подходящие терапевтические агенты для комбинационной терапии с соединением формулы Iа включают (1) антагонист простагландина D2, такой как S-5751; (2) кортикостероид, такой как триамцинолон ацетонид; (3) -агонист, такой как салметерол,форметерол, тербуталин, метапротеренол, албутерол и тому подобные; (4) лейкотриеновый модификатор, включая антагонист лейкотриена или ингибитор липооксигеназы, такой как монтелукаст, зафирлукаст, пранлукаст или цилейтон; (5) антигистамин, такой как бромфенирамин, хлорфенирамин, дексхлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин,метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамин, пириламин, астемизол, норастемизол, терфенадин, лоратадин, цетиризин, левоцетиризин, фексофенадин, дезкарбоэтоксилоратадин и тому подобные; (6) противоотечное лекарственное средство, включая фенилеприн, фенилпропаноламин, псевдофедрин, оксиметазолин, эфинеприн, нафазолин, ксилометазолин, пропилгекседрин или леводезоксиэфедрин; (7) наркотические анальгетики, включая кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; (8) другие простагландиновые лиганды, включая агонист простагландина F, такой как латанопрост; мизопростол, энпростил, риопростил, орнопростол или розапростол; (9) диуретик; (10) нестероидные противовоспалительные средства (НСПВС), такие как производные пропионовой кислоты (аминопрофен, беноксапрофен, буклоксановая кислота, карпрофен, фенбуфен,фенпрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен,оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (индометацин, ацеметацин, альклофенак, клиданак, диклофенак, фенклофенак, фенклозиновая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенаминовой кислоты (флуфенаминовая кислота, меклофенаминовая кислота, мефенаминовая кислота, нифлуминовая кислота и толфенаминовая кислота), производные бифенилкарбоновой кислоты (дифлузиналь и флуфенизаль), оксикамы (изоксикам, пироксикам, судоксикам и теноксикам), салицилаты (ацетилсалициловая кислота, сульфазалазин) и пиразолоны(апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (11) ингибиторы циклооксигеназы-2 (СОХ-2), такие как целекоксиб и рофекоксиб, эторикоксиб и валдекоксиб; (12) ингибиторы фосфодиэстеразы типа IV (PDE-IV), например арифло, рофлумиласт; (13) антагонисты хемокиновых рецепторов, особенно CCR-1, CCR-2 и CCR-3; (14) агенты, снижающие уровень холестерина, такие как ингибиторы HMG-СоА редуктазы (ловастатин, симвастатин и правастатин, флувастатин, аторвастатин и другие статины), секвестранты (холестирамин и колестипол), никотиновая кислота, производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (15) антидиабетические агенты, такие как инсулин, сульфонилмочевины, бигуанидины (метформин), ингибиторы аглюкозидазы (акарбоза) и глитазоны (троглитазон, пиоглитазон, энглитазон, розиглитазон и тому подобные); (16) препараты бета-интерферона (интерферон бета-la, интерферон бета-lb); (17) антихолинергические агенты, такие как мускариновые антагонисты (ипратропийбромид и тиотропийбромид), а также селективные мускариновые М 3 антагонисты; (18) стероиды, такие как беклометазон, метилпреднизолон,бетаметазон, преднизон, дексаметазон и гидрокортизон; (19) триптаны, обычно используемые для лечения мигрени, такие как сумитриптан и ризатриптан; (20) алендронат и другие лекарственные средства для остеопороза; (21) другие соединения, такие как 5-аминосалициловая кислота и ее пролекарства, антиметаболиты, такие как азатиоприн и 6-меркаптопурин, цитотоксические противораковые химиотерапевтические агенты, антагонисты брадикинина (ВК 2), антагонисты ТР рецептора, такие как сератродаст,антагонисты нейрокинина (NK1/NK2), VLA-4 антагонисты, такие как описанные в патенте США 5510332, WO97/03094, WO97/02289, WO96/40781, WO96/22966, WO96/20216, WO96/01644,WO96/06108, WO95/15973 и WO96/31206. Кроме того, изобретение охватывает способ лечения простагландин D2 опосредованных заболеваний, включающий введение нуждающемуся в таком лечении пациенту нетоксичного терапевтически эффективного количества соединения формулы Iа, необязательно совместно с одним или несколькими такими ингредиентами, которые только что были перечислены выше. Количества активных ингредиентов могут представлять собой количества, обычно используемые для каждого из активных ингредиентов, при их введении по отдельности, или в некоторых случаях комбина-6 006765 ция активных ингредиентов может приводить к более низким дозам одного или нескольких активных ингредиентов. Способы синтеза Соединения формулы Iа по настоящему изобретению могут быть получены синтетическими путями, кратко охарактеризованными на схемах 1-6 и в соответствии с описанными здесь методами. Промежуточные соединения формулы IV могут быть получены способом, представленным на схеме 1, из подходящим образом замещенного фенилгидразина (II). Взаимодействие II с подходящим циклопентаноном III в условиях образования индола по Фишеру или в аналогичных условиях приводит к IV. Схема 1 Альтернативно соединения формулы IV могут быть получены способом, представленным на схеме 2, из подходящим образом замещенного анилина V. Взаимодействие V с иодом дает VI. Конденсация с подходящим циклопентаноном III с последующей циклизацией в условиях по Хеку или аналогичных условиях катализа металлами приводит к индолу IV. Схема 2 Соединения формулы III могут быть получены способом, представленным на схеме 3, из подходящим образом замещенного силилового эфира енола VII или подходящим образом замещенного енаминаVIII. Присоединение подходящего электрофила, такого как QY (где Y представляет собой галоген или уходящую группу), в присутствии основания, такого как алкиллитий, или кислоты Льюиса, такой как трифторацетат серебра с силиловым эфиром енола VII дает циклопентанон III. Соединение формулы III альтернативно может быть получено присоединением QY к подходящим образом замещенному енаминуVIII в условиях преобразования енамина по Сторку или аналогичных условиях. Схема 3 Промежуточные соединения формулы X могут быть получены способом, представленным на схеме 4, из подходящим образом замещенного индола IX. Бромирование IX может быть осуществлено бромом в полярном и основном растворителе, таком как пиридин, с последующим моновосстановлением промежуточного дибромсодержащего соединения под действием кислоты и в условиях восстановления металлом для образования индола X. Соединения формулы Iа могут быть получены способом, представленным на схеме 5, из подходящим образом замещенного индола IV. Алкилирование IV подходящим электрофилом, таким как ArXY(где Y представляет собой галоген или уходящую группу) в присутствии основания и в подходящем растворителе, таком как ДМФ, дает Iа. Схема 5 Альтернативно, соединения формулы Iа могут быть получены способом, представленным на схеме 6, из подходящим образом замещенного броминдола XI из соединения формулы X после реакции связывания, описанной на схеме 5. Связывание в присутствие палладия или аналогичные реакции броминдолаXI с подходящим металлоорганическим соединением R3M (где М представляет собой металл, такой как В, Mg, Zn или Sn) приводят к соединению Iа. Тот же самый броминдол XI альтернативно первоначально может взаимодействовать с подходящим металлирующим агентом, таким как n-BuLi, с последующим захватом электрофилом, таким как R3Y, давая соединение Iа. Схема 6 Иллюстративные соединения Иллюстративные соединения формулы Iа представлены в следующих таблицах; заместители являются такими, как указано, и Н предназначен для случаев, где не имеется значения конкретной переменной. Подразумевается, что каждое соединение включает рацемическую или диастереомерную смесь и индивидуальные энантиомеры и/или диастереомеры. Способы разделения энантиомеров и разделения диастереомеров хорошо известны специалистам в данной области; избранные иллюстрации таких методов также описаны далее в примерах. Тесты для определения биологической активности Соединения формулы Iа могут быть протестированы с использованием следующих анализов для определения их простаноид-антагонистической или агонистической активности in vitro и in vivo и их селективности. Активность простагландиновых рецепторов демонстрировали для DP, EP1, EP2, EP3, ЕР 4,FP, IP и ТР. Стабильная экспрессия простаноидных рецепторов в клеточной линии эмбриональной почки(НЕК) 293 (ebna) человека Простаноидные рецепторы кДНК, соответствующие полной длине кодирующих последовательностей, субклонировали в подходящие сайты векторов экспрессии млекопитающих и трансфектировали в клетки НЕК 293 (ebna). Клетки НЕК 293 (ebna), экспрессирующие индивидуальные кДНК, выращивали с отбором и выделяли через 2-3 недели роста индивидуальные колонии с использованием клонирующего кольцевого способа и впоследствии распространяли на клональные клеточные линии. Анализы связывания простаноидного рецептора Для использования в анализах связывания рецептора клетки НЕК 293 (ebna) поддерживали в культуре, харвестировали (собирали) и мембраны получали дифференциальным центрифугированием с последующим лизисом клеток в присутствии ингибиторов протеазы. Анализ связывания простаноидного рецептора проводили в 10 мМ MES/KOH (рН 6,0) (EPs, FP и ТР) или 10 мМ HEPES/KOH (рН 7,4) (DP иIP), содержащих 1 мМ EDTA, 10 мМ двухвалентного катиона и подходящий радиоизотопный лиганд. Реакцию инициировали добавлением мембранного белка. Лиганды добавляли в диметилсульфоксиде,который поддерживали постоянно на уровне 1% (об./об.) во всех инкубациях. Неспецифическое связы- 16006765 вание определяли в присутствии 1 мкМ соответствующего нерадиоактивного простаноида. Инкубации проводили в течение 60 мин при комнатной температуре или при 30 С и завершали быстрым фильтрованием. Специфическое связывание рассчитывали путем вычитания неспецифического связывания из общего связывания. Рассчитывали остаточное специфическое связывание при каждой концентрации лиганда и выражали как функцию концентрации лиганда для создания сигмоидальных кривых концентрацияответ для определения аффинности лиганда. Анализы агониста и антагониста простаноидного рецептора Для определения того, являются рецепторные лиганды агонистами или антагонистами, проводили анализы измерительной стимуляции (ЕР 2, ЕР 4, DP и IP в НЕК 293 (ebna) клетках) или ингибирования(ЕР 3 в клетках эритролейкемии человека (HEL межклеточной цАМФ аккумуляции или мобилизации внутриклеточного кальция (EP1, FP и ТР в НЕК 293 (ebna) клетках, стабильно трасфектированных апоаэкворином) для вторичных мессенджеров целой клетки. Для цАМФ анализов клетки харвестировали и повторно суспендировали в HBSS, содержащем 25 мМ HEPES, рН 7,4. Инкубаты содержат 100 мкМ RO20174 (ингибитор фосфодиэстеразы типа IV, доступный от Biomol) и, только в случае анализа ЕР 3 ингибирования, 15 мкМ форсколина для стимуляции продуцирования цАМФ. Образцы инкубировали при 37 С в течение 10 мин, реакцию завершали и затем измеряли уровни цАМФ. Для анализов мобилизации кальция клетки заряжали кофакторами, снижающими глютатион и коэлентеразин, собирали и повторно суспендировали в Ham F12 среде. Мобилизацию кальция измеряли путем контроля люминесценции, вызываемой связыванием кальция с внутриклеточным фотобелком аэкворином. Лиганды добавляли в диметилсульфоксиде, уровень которого поддерживали постоянным 1% (об./об.) во всех инкубациях. Для агонистов ответные реакции вторичных мессенджеров выражали как функцию концентрации лиганда и рассчитывали как значения EC50, так и максимальную ответную реакцию в сравнении с простаноидным стандартом. Для антагонистов способность лиганда ингибировать ответную реакцию агониста определяли анализом Шилда (Schild) и рассчитывали как КВ, так и значения наклона. Предотвращение PGD2- или аллерген-индуцированной закупорки носового канала у овцы, склонной к аллергии Подготовка животного: Использовали здоровых взрослых овец (18-50 кг). Данных животных отбирали на основе естественной положительной реакции шкуры на подкожное введение экстракта Ascarissuum. Измерения закупорки носового канала: Эксперимент проводили на находящихся в сознании животных. Их двигательную активность ограничивали в тележке в лежачем положении с фиксированной в неподвижном состоянии головой. Устойчивость носовых дыхательных путей (NAR) измеряли с использованием модифицированной техники ринометрической маски. Для введения носо-трахейной трубки проводили местную анестезию (2% лидокаин) носоглотки. Максимальный конец трубки связывают с пневмотахографом и регистрируют сигнал тока и давления на осциллографе, связанном с компьютером для расчета NAR в оперативном режиме. Вызывают раздражение носа (назальной области) путем введения аэрозольного раствора (10 выпусков/ноздря). Изменения закупорки NAR регистрируют до и через 60-120 мин после введения в организм вещества, провоцирующего выделение антител. Предотвращение PGD2- или аллерген-индуцированной закупорки носового канала у обезьяныcynomolgus Подготовка животного: Использовали здоровых взрослых обезьян cynomolgus (4-10 кг). Данных животных отбирали на основе естественной положительной реакции шкуры на подкожное введение экстракта Ascaris suum. Перед каждым экспериментом обезьяну, отобранную для исследования, не кормили в течение ночи, обеспечивая водой по желанию. На следующее утро животное успокаивали кетамином(10-15 мг/кг, внутримышечно) перед удалением его из своей клетки. Его помещали на подогретый стол(36 С) и инъецировали болюсной дозой (5-12 мг/кг внутривенно) пропофола. Животному делали интубацию с помощью эндотрахеальной трубки с манжетами (внутренний диаметр 4-6 мм) и поддерживали анестезию путем постоянного внутривенного вливания пропофола (25-30 мг/кг/ч). Во время эксперимента контролировали признаки жизни (сокращения сердца, давление крови, частота дыхания, температура тела). Измерения закупорки носового канала: Устойчивость дыхательных путей животного измеряли с использованием пневмотахографа, связанного с эндотрахеальной трубкой для того, чтобы гарантировать,что измерения являются нормальными. Для оценки закупорки носового канала использовали акустический ринометр Ecovision. Такой метод дает неинвазивную 2D эхограмму внутри носа. Объем носа и минимальную поперечно-рассеченную область вдоль длины носовой полости вычисляли в течение 10 с с использованием небольшого портативного компьютера, оборудованного обычным программным обеспечением (Hood Laboratories, Mass, США). Вещество для носового канала, провоцирующее выделение антител, вводили непосредственно в носовую полость животного (объем 50 мкл). Регистрировали изменения закупорки носового канала до и через 60-120 мин после введения в организм вещества, провоцирующего выделение антител. Если происходит закупорка носового канала, ее переводят в снижение объема носового канала.- 17006765 Легочная механика у дрессированных находящихся в сознании беличьих обезьян (саймири) Процедура исследования включает помещение дрессированных беличьих обезьян в кресла в камеры аэрозольного действия. В целях контроля измерения легочной механики параметров дыхания регистрировали в течение примерно 30 мин для установления нормальных контрольных значений для каждой обезьяны в этот день. Для перорального введения соединения растворяли или суспендировали в 1%-ном растворе метоцеля (метилцеллюлоза, 65HG, 400 сП) и давали в объеме 1 мг/кг веса тела. Для аэрозольного введения соединений использовали ультразвуковой распылитель De Vilbiss. Периоды перед обработкой изменялись от 5 мин до 4 ч перед тем, как обезьянам вводили аэрозольные дозы либо PGD2 или антигена Ascaris suum. После введения каждую минуту данные рассчитывали с помощью компьютера как процент изменения от контрольных значений для каждого параметра дыхания, включая устойчивость дыхательных путей (RL) и динамическое соответствие (Cdyn). Результаты для каждого тестируемого соединения впоследствии получали для минимального периода в 60 мин после введения вещества, которые затем сравнивали с предварительно полученными историческими контрольными значениями базисной линии для данной обезьяны. Кроме того, общие значения в течение 60 мин после введения вещества для каждой обезьяны(значения исторической базисной линии и значения в испытании) усредняли отдельно и использовали для расчета суммарного процента ингибирования тестируемым соединением ответной реакции медиатора или Ascaris антигена. Для статистического анализа использовали спаренный t-тест (Ссылки: McFarlaneC.S. et al., Prostaglandins, 28, 173-182 (1984) и McFarlane C.S. et al., Agents Actions, 22, 63-68 (1987. Предотвращение индуцированного бронхостеноза у овец, склонных к аллергии Подготовка животного: Использовали взрослую овцу со средним весом 35 кг (диапазон 18-50 кг). Все тестируемые животные отвечали двум критериям: а) они имели естественную кожную реакцию к 1:1000 или 1:10000 разведению экстракта Ascaris suum (Greer Diagnostics, Lenois, NC); и b) они ранее давали ответную реакцию на введение ингаляцией Ascaris suum в виде острого бронхостеноза и позднего бронихиального затруднения дыхания (W.M.Abraham et al., Am.Rev.Resp.Dis., 128, 839-44 (1983. Измерения механики дыхательных путей: Двигательную активность овцы без введения успокоительного средства ограничивали в тележке в лежачем положении с фиксированной в неподвижном состоянии головой. После местной анестезии носоглотки 2%-ным раствором лидокаина баллонный катетер вводили в нижний эзофагус через одну ноздрю. Животных затем интубировали с помощью эндотрахеальной трубки с манжетами через другую ноздрю, используя гибкий оптико-волоконный бронхоскоп в качестве направляющего. Плевральное давление оценивали с помощью балонного катетера для пищевода (наполненного 1 мл воздуха), который устанавливали таким образом, что вдыхание давало отрицательное отклонение давления с четко различимыми кардиогенными колебаниями. Латеральное давление в трахее измеряли с использованием бокового ствола катетера (внутренний размер 2,5 мм) продвинутого вперед и расположенного на периферии к вершине носотрахеальной трубки. Транслегочное давление,разницу между трахеальным давлением и плевральным давлением измеряли с дифференциальным датчиком давления (DP45, Validyne Corp., Northridge, CA). Для измерения легочной устойчивости (RL) максимальный конец носотрахеальной трубки связывали с пневмотахографом (Fleisch, Dyna Sciences, BlueBell, PA). Сигналы движения и транслегочное давление регистрировали на осциллоскопе (модель DR-12;Equipment Corp., Maynard, MA) для расчета в оперативном режиме RL от транслегочного давления, дыхательного объема, полученного интегрированием и движения. Для определения RL использовали анализ 10-15 дыханий. Измеряли грудной объем газа (Vtg) в плетизмографе для тела для получения специфической легочной устойчивости (SRL=RLVtg). Далее изобретение будет проиллюстрировано следующими неограничивающими примерами в которых, если не указано другого: 1. Все конечные продукты формулы Ia были проанализированы ЯМР, ТСХ и элементным анализом или масс-спектрометрией. 2. Промежуточные соединения анализировали с использованием ЯМР и ТСХ. 3. Большинство соединений очищали флэш-хроматографией на силикагеле, перекристаллизацией и/или энергичным растиранием (суспензия в растворителе с последующим фильтрованием твердого вещества). 4. Ход реакций контролировали с помощью тонкослойной хроматографии (ТСХ) и время реакций приведено только для иллюстрации. 5. Энантиомерный избыток измеряли-ВЭЖХ с нормальной фазой с использованием хиральной колонки: ChiralPak AD; 250x4,6 мм. Следующие промежуточные соединения получали в соответствии с литературными методиками,или они были закуплены от следующего поставщика: 1. Этил 2-(2-оксоциклопентил)'ацетат: Acros/Fisher Scientific. 2. 4-фтор-2-иоданилин: Beugelmans, R.; Chbani, M. Bull. Soc. Chim. Fr. 1995, 132, 306-313. 3. Этил 2-(1-метил-2-оксоциклопентил)ацетат: Hudlicky, Т.; Short, R. P.; Revol, J.-M.; Ranu, В. С. J.(+/-)-2-5-ацетил-4-[(4-хлорфенил)метилЛ]-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3 В]индол-3-илуксусная кислота Стадия 1: 2-йод-4-(метилсульфонил)фениламин К интенсивно перемешиваемому раствору 100 г 4-(метилсульфонил)-фениламина, растворенного в 5,5 л EtOH при 50 С добавляли смесь 49,3 г иода и 110 г сульфата серебра в 1 л EtOH. Данную процедуру повторяли после перемешивания в течение 1 ч. Еще через час добавляли смесь 49,3 г иода и 43,8 г сульфата серебра в 1 л EtOH и смесь перемешивали в течение ночи. Горячий раствор затем фильтровали через целит и растворитель удаляли. Остаток растирали с 1 л EtOH при 50 С в течение 45 мин и охлаждали до 0 С. Продукт отфильтровывали и собирали, получая 140 г указанного в заголовке соединения в виде коричневого твердого вещества. 1 Н ЯМР (ацетон-d6)7,95 (1 Н, д), 7,54 (1 Н, дд), 6,79 (1 Н, д), 6,19. (2 Н, ушир. с), 3,08 (3 Н, с). MS К раствору 150 г 2-иод-4-(метилсульфонил)фениламина и 4,8 г PTSA в 30 мл дегазированного и хранящегося в N2-атмосфере ДМФ добавляли последовательно 135 г тетраэтоксисилана и 129 г этил 2-(2 оксоциклопентил)ацетата. Полученную смесь нагревали до 130-140 С и перемешивали в течение 6 ч. Затем добавляли 30 мл ДМФ и раствор дегазировали перед последующим последовательным добавлением 270 мл основания Ханига (Hunig) и 3,4 г Pd(OAc)2. Раствор нагревали при 120 С в течение 2 ч, затем охлаждали до комнатной температуры. Для гашения реакции добавляли 300 мл 1 н HCl и 200 мл изопропилацетата и смесь фильтровали через целит. Фазы разделяли и кислую фазу экстрагировали дважды 200 мл изопропилацетата. Органические слои объединяли, промывали насыщенным раствором соли, сушили над безводным Na2SO4, фильтровали через целит и концентрировали. Неочищенное вещество дополнительно очищали флэш-хроматографией, элюируя 50% EtOAc в гексанах, получая 63 г указанного в заголовке соединения в виде желтого твердого вещества. 1 Н ЯМР (ацетон-d6)10,23 (1 Н, ушир. с), 7,98 (1 Н, с), 7,58 (2 Н, м), 4,14 (2 Н, кв), 3,63 (1 Н, с), 3,04 К раствору 10,4 г сложного эфира со стадии 2 в 80 мл ТГФ при комнатной температуре добавляли 40 мл МеОН, после чего 40 мл 2 н NaOH. Через 1,5 ч реакционную смесь выливали в делительную воронку, содержащую EtOAc/1 н HCl. Фазы разделяли и кислую фазу экстрагировали дважды EtOAc. Органические слои объединяли, промывали насыщенным раствором соли, сушили над безводным Na2SO4 и упаривали досуха. Неочищенное твердое вещество энергично растирали в смеси EtOAc/гексаны, получая 9,1 г указанной в заголовке кислоты в виде тускло-коричневого твердого вещества. 1 Н ЯМР (ацетон-d6)10,86 (1 Н, ушир. с), 10,25 (1 Н, ушир. с), 7,98 (1 Н, с), 7,58 (2 Н, м), 3,62 (1 Н, м),3,04 (3 Н, с), 2,89-2,68 (5 Н, м), 2,21 (1 Н, м). MS (+APCI) m/z 294,0 (М+Н)+. Стадия 4: (+/-)-2-[5-бром-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]индол-3-ил]уксусная кислота Трибромид пиридиния (154 г) добавляли к раствору 2-[7-(метилсульфонил)-1,2,3 тригидроциклопента[2,3-b]индол-3-ил] уксусной кислоты (50,8 г) в пиридине при -25 - -30 С. Раствор нагревали до 0 С в течение 15 мин и затем до комнатной температуры в течение 30 мин. Добавляли 1250- 19006765 мл смеси 1:1 ТГФ/простой эфир и 2500 мл смеси 1:1 насыщенный раствор соли/6 н HCl, фазы отделяли,водный слой промывали смесью 1:1 ТГФ/простой эфир и объединенные органические слои сушилиNa2SO4. Органическую фазу охлаждали до 10 С, добавляли уксусную кислоту (50,5 мл) с последующим медленным прибавлением цинка (70,2 г) (поддерживая температуру ниже 15 С). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Добавляли 3000 мл 1 н HCl и 1250 мл EtOAc, фазы отделяли и водный слой промывали 2000 млEtOAc. Объединенные органические слои сушили Na2SO4 и растворитель удаляли. Полученный коричневатый порошок энергично растирали с 1000 мл смеси 20% EtOAc/гексаны. Выделяли 52 г (81%) указанного в заголовке соединения. 1 Н ЯМР (ацетон-d6)10,38 (1 Н, ушир. с), 8,00 (1 Н, д), 7,76 (1 Н, д), 3,66 (1 Н, м), 3,13 (3 Н, с), 3,002,75 (4 Н, м), 2,62 (1 Н, дд), 2,26 (1 Н, м). MS (-APCI) m/z 372,2, 370,2 (М-Н)-. Стадия 5: (+/-)-метил 2-[5-бром-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]индол-3 ил]ацетат Кислоту, полученную на стадии 4, этерифицировали в ТГФ с использованием эфирного раствораCH2N2. После удаления растворителей, указанный в заголовке сложный эфир получали с количественным выходом в виде бледно-коричневого твердого вещества. 1 Н ЯМР (ацетон-d6)10,41 (1 Н, ушир. с), 8,00 (1 Н, д), 7,76 (1 Н, д), 3,68 (4 Н, м), 3,13 (3 Н, с), 3,002,75 (4 Н, м), 2,62 (1 Н, дд), 2,23 (1 Н, м). Стадия 6: (+/-)-метил 2-5-бром-4-[(4-хлорфенил)метил]-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]индол-3-илацетат К раствору 3,70 г индола со стадии 5 в 30 мл ДМФ при -78 С добавляли 790 мг суспензии NaH (60% в масле). Полученную суспензию перемешивали в течение 10 мин при 0 С, опять охлаждали до -78 С и обрабатывали 2,36 г 4-хлорбензилбромида. Через 5 мин температуру повышали до 0 С и перемешивали 20 мин. В этот момент реакционную смесь гасили добавлением 1 мл АсОН и данную смесь выливали в делительную воронку, содержащую 1 н HCl/EtOAc. Слои разделяли и органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали. Неочищенное вещество дополнительно очищали флэш-хроматографией, элюируя 10%-ным EtOAc в толуоле и энергично растирали в смеси EtOAc/гексаны, получая 4,33 г указанного в заголовке соединения в виде белого твердого вещества. 1 К раствору 3,0 г броминдола со стадии 6 в 15 мл ДМФ добавляли 3,97 мл 1 этоксивинилтрибутилолова. Полученную смесь дегазировали, барботируя через раствор N2 в течение нескольких минут. В отдельную колбу помещали 538 мг Pd2 (dba)3, 720 мг Ph3As вместе с 9,0 мл ДМФ и данную смесь подвергали действию ультразвука в течение 1 мин. Затем в реакционную колбу вводили каталитическую смесь и нагревали при 90 С в течение 30 мин, позволяя реакционной смеси охладиться до комнатной температуры, в реакционную колбу добавляли 4 мл 1 н раствора HCl и оставляли перемешиваться до тех пор, пока ТСХ анализ не показывал расходование аддукта винилового простого эфира. Реакционную смесь разбавляли водой, экстрагировали изопропилацетатом, сушили над безводнымNa2SO4 и концентрировали. Полученное вещество дополнительно очищали флэш-хроматографией,- 20006765 элюируя 20%-ным ацетоном в толуоле, получая 2,7 г указанного в заголовке кетона в виде белого твердого вещества. 1 Н ЯМР (CDCl3)8,19 (1 Н, с), 7,74 (1 Н, с), 7,16 (2 Н, д), 6,55 (2 Н, д), 5,35 (1 Н, д), 5,29 (1 Н, д), 3,71(1 Н, м), 3,65 (3 Н, с), 3,06 (3 Н, с), 3,05-2,80 (3 Н, м), 2,66 (1 Н, дд), 2,50 (1 Н, дд), 2,32 (1 Н, м), 2,12 (3 Н, с). Стадия 8: (+/-)-2-5-Ацетил-4-[(4-хлорфенил)метил]-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]индол-3-илуксусная кислота Указанное в заголовке соединение получали из 2,08 г сложного эфира со стадии 7 в соответствии с методикой стадии 3, получая 1,99 г коричневого твердого вещества. 1 Н ЯМР (ацетон-d6)10,76 (1 Н, ушир. с), 8,19 (1 Н, д), 7,87 (1 Н, д), 7,27 (2 Н, д), 6,73 (2 Н, д), 5,48(+)-2-5-Ацетил-4-[(4-хлорфенил)метил]-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-В]индол-3-илуксусная кислота [(+)-изомер соединения примера 1] 150-200 мг (+/-)-2-5-Ацетил-4-[(4-хлорфенил)метил]-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]индол-3-ил уксусной кислоты (пример 1, стадия 8), растворенной в 10 мл горячего EtOH,разделяли препаративной хиральной ВЭЖХ с нормальной фазой [ChiralPak AD колонка: 50x5 см, 20 мкм; подвижная фаза: гексан/2-пропанол/уксусная кислота (70:30:0,4); поток: 70-75 мл/мин; давление: 280-300 фт/кв.дюйм; УФ: 265 нм]. Времена удерживания двух энантиомеров составляли 38 и 58 мин. Указанное в заголовке соединение получали в качестве менее полярного энантиомера с 98% ее. ее=98%; Время удерживания =12,1 мин [ChiralPak AD колонка: 250x4,6 мм, гексан/2-пропанол/уксусная кислота(-)-2-5-Ацетил-4-[(4-хлорфенил)метил]-7-(метилсульфонил)-1,2,3-тригидоциклопента[2,3-В]индол 3-илуксусная кислота [(-)-изомер соединения примера 1] 150-200 мг (+/-)-2-5-Ацетил-4-[(4-хлорфенил)метил]-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]индол-3-ил уксусной кислоты (пример 1, стадия 8) , растворенной в 10 мл горячего EtOH,разделяли препаративной хиральной ВЭЖХ с нормальной фазой [ChiralPak AD колонка: 50x5 см, 20 мкм; подвижная фаза: гексан/2-пропанол/уксусная кислота (70:30:0,4); поток: 70-75 мл/мин; давление: 280-300 фт/кв.дюйм (20,636-21,093 кгс/см 2; УФ:265 м). Времена удерживания двух энантиомеров составляли 38 и 58 мин. Указанное в заголовке соединение получали в качестве более полярного энантиомера с 96,7% ее. Данный энантиомер перекристаллизовывали из смеси 80% 2-пропанол/гексаны, для увеличения ее. ее=96,7%; Время удерживания = 15,3 мин [ChiralPak AD колонка: 250x4,6 мм, гексан/2 пропанол/уксусная кислота (75:25:0,1)];[]D21=-10,9 (с 0,45, МеОН). Элементный анализ. Вычислено для C23H22ClNO5S: С, 60,06; Н, 4,82; N, 3,05; S, 6,97. Найдено: С,59,96; Н, 4,81; N, 3,01; S, 7,22. Т.пл. 219,5 С. Пример 3 А. Альтернативный способ получения соединения примера 3. А. Разделение (+)-2-[5-бром-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]индол-3-ил]уксусной кислоты Суспензию 300 мг (+/-)-2-[5-бром-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]индол-3 ил]уксусной кислоты (пример 1, стадия 4) и 138 мг (R) - (+)-1-(нафтил) этиламина в 15 мл 2-пропанола и 5 мл ацетона растворяли путем нагревания при кипении с обратным холодильником. Растворители затем удаляли при пониженном давлении и остаток перекристаллизовывали из смеси 1:1 2-пропанол/ацетон (7 мл). После фильтрования полученную белую твердую соль суспендировали в 5 мл метанола и обрабатывали 3 н HCl до рН 1. Осадок отфильтровывали и сушили на воздухе, получая 78 мг указанного в заголовке энантиомера. Времена удерживания двух энантиомеров составляли 6,5 и 8,2 мин [ChiralPak AD колонка, гексан/2-пропанол/уксусная кислота (75:25:0,2)]. Указанное в заголовке соединение получали в качестве более полярного -энантиомера с 90% ее. Данную процедуру повторяли для получения вышеуказанного соединения с 99% ее. ее=99%; Время удерживания = 8,2 мин [ChiralPak AD колонка: 250x4,6 мм,гексан/2-пропанол/уксусная кислота (75:25:0,2)]; []D21=+11,0 (с 0,5, МеОН). В. Для получения соединения примера 3 следовали методике, описанной в примере 1, стадии 5-8,используя вышеуказанный (+)энантиомер вместо рацемата.(+)-2-4-[(4-Хлорфенил)метил]-5-(гидроксиэтил)-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-В]индол-3-илуксусная кислота (диастереомер А) Стадия 1. Восстановление кетона В сухую колбу помещали 350 мг (-)-2-5-ацетил-4-[(4-хлор-фенил)метил]-7-(метилсульфонил)1,2,3-тригидроциклопента[2,3-b]индол-3-илуксусной кислоты (пример 3; ее=99%) вместе с 30 мл МеОН. К данному перемешиваемому раствору порциями добавляли NaBH4 (примерно 50 мг/порция) с 10-15 минутными интервалами до тех пор, пока израсходование кетона не отмечалось по ТСХ анализу. В данный момент реакционную смесь выливали в делительную воронку, содержащую 100 мл насыщенного водного раствора NH4C1/10 мл 1 н раствора HCl и 100 мл EtOAc. Слои разделяли и водный слой экстрагировали EtOAc, объединенные органические слои сушили над безводным Na2SO4 и концентрировали. Полученное вещество очищали хиральным ВЭЖХ следующим образом: Стадия 2. Хиральная очистка 150-200 мг предшествующей смеси спиртов, растворенной в 10 мл горячего EtOH разделяли на препаративной хиральной ВЭЖХ с нормальной фазой [ChiralPak AD колонка: 50x5 см, 20 мкм; подвижная фаза: гексан/2-пропанол/уксусная кислота (80:20:0,4); поток: 70-75 мл/мин; давление: 280-300 фт/кв.дюйм, УФ: 245 нм]. Времена удерживания двух диастереоизомеров составляли 33 и 51 мин. Указанное в заголовке соединение получали в качестве менее полярного диастереоизомера с de99%. ее=99%; de99%. Время удерживания = 6,0 мин [ChiralPak AD колонка: 250x4,6 мм, гексан/2 пропанол/уксусная кислота (75:25:0,2)]; []D21=+7,6 (с 1,0, МеОН). 1 Н ЯМР (ацетон-d6)7,95 (1 Н, д), 7,83 (1 Н, д), 7,32 (2 Н, д), 6,90 (2 Н, д), 5,92 (1 Н, д), 5,65 (1 Н, д),5,19 (1 Н, кв), 3,60 (1 Н, м), 3,07 (3 Н, с), 2,99 (1 Н, м), 2,83 (2 Н, м), 2,65 (1 Н, дд), 2,39 (1 Н, м), 2,31 (1 Н, м),1,45 (3 Н, д). 13 С ЯМР (ацетонов)173,3, 152,0, 141,1, 139,4, 133,3, 133,2, 132,6, 129,7, 127,8, 126,4, 121,7, 118,8,117,3, 64,6, 50,3, 45,0, 39,1, 36,5, 36,2, 24,6, 23,5.MS (-APCI) m/z 4 62,8,4 60,5 (М-Н)-. Элементный анализ. Вычислено для C23H24ClNO5S: С, 59,80; Н,5,24; N, 3,03; S, 6,94. Найдено: С, 59,47; Н, 5,22; N, 2,96; S, 7,14. Т.пл. 212,4 С. Пример 5. 2-4-[(4-Хлорфенил)метил]-5-(гидроксиэтил)-7-(метилсульфонил)-1,2,3-тригидроциклопента-[2,3 В]индол-3-илуксусная кислотаА (диастереомер В) Вещество примера 4, стадия 1 (150-200 мг, ее=99%), растворенное в 10 мл горячего EtOH, разделяли с помощью препаративной хиральной ВЭЖХ с нормальной фазой [ChiralPak AD колонка: 50x5 см, 20 мкм; подвижная фаза: гексан/2-пропанол/уксусная кислота (80:20:0,4); потока: 70-75 мл/мин; давление: 280-300 фт/кв.дюйм; УФ: 245 нм]. Времена удерживания двух диастереоизомеров составляли 33 и 51 мин. Указанное в заголовке соединение получали в виде более полярного диастереоизомера с 95% de. ее=99%; de95%; Время удерживания = 7,9 мин [ChiralPak AD колонка: 250x4,6 мм, гексан/2 пропанол/уксусная кислота (75:25:0,2)]. 1(+/-)-2-4-[(2,4-дихлорфенил)метил]-5-бром-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3 В]индол-3-илуксусная кислота Следуя методике конденсации, описанной в примере 1, стадия 6, используя 104 мг метил 2-[5-бром 7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-b]-индол-3-ил]ацетата (пример 1, стадия 5) и 50 мкл 2,4-дихлорбензилхлорида получали 50 мг метилового сложного эфира указанного в заголовке соединения в виде белого твердого вещества (чистота 95%). Н ЯМР (ацетон-d6)8,05 (1 Н, д), 7,73 (1 Н, д), 7,58 (1 Н, д), 7,24 (1 Н, дд), 6,28 (1 Н, д), 5,91 (1 Н, д),5,85 (1 Н, д), 3,73 (1 Н, м), 3,55 (3H, с), 3,15 (3H, с), 3,01 (1 Н, м), 2,95-2,75 (2 Н, м), 2,68 (1 Н, дд), 2,49 (1 Н,дд), 2,30 (1 Н, м). Указанное в заголовке соединение получали из 50 мг вышеуказанного метилового сложного эфира в соответствии с методикой, описанной в примере 1, стадия 3, получая 34 мг белого твердого вещества(+/-)-2-4-[(4-Хлорфенил)метил]-7-(метилсульфонил)-5-винил-1,2,3-тригидроциклопента[2,3 В]индол-3-илуксусная кислота С использованием способа, описанного в примере 1, стадия 7, из 112 мг метил 2-5-бром-4-[(4 хлорфенил)метил]-7-(метилсульфонил)-1,2,3-тригидроциклопента-[2,3-b]индол-3-ил ацетата (пример 1,стадия 6) и 128 мкл винилтрибутилолова получали 94 мг метилового сложного эфира указанного в заголовке соединения в виде желтого твердого вещества. Данное вещество использовали без дополнительной очистки на следующей стадии. Указанное в заголовке соединение получали из 17 мг вышеуказанного метилового сложного эфира в соответствии с методикой, описанной в примере 1, стадия 3, получая 16,5 мг бесцветного масла (чистота 95%). 1(+/-)-2-4-[(4-Хлорфенил)метил]-5-циклопропил-7-(метилсульфонил)-1,2,3-тригидроциклопента[2,3-В]индол-3-илуксусная кислота В круглодонную колбу, содержащую 27,6 мг метил 2-4-[(4-хлорфенил)метил]-7-(метилсульфонил)-5-винил-1,2,3-тригидроциклопента[2,3-b]-индол-3-илацетата (полученного в примере 7) и 2 мл ТГФ, охлажденных до 0 С, добавляли несколько мг Pd(OAc)2 и 4 мл эфирного раствора CH2N2 и оставляли перемешиваться при данной температуре. К реакционной смеси добавляли дополнительные порции Pd(OAc)2 и CH2N2 до тех пор, пока 1 Н ЯМР анализ аликвоты реакционной смеси не выявил отсутствие винильных протонов. Реакционную смесь фильтровали через слой силикагеля и концентрировали для получения метилового сложного эфира указанного в заголовке соединения. 1 Н ЯМР (ацетон-d6)8,00 (1 Н, с), 7,34 (3H, м), 6,92 (2 Н, д), 6,04 (1 Н, д), 5,93 (1 Н, д), 3,54 (4 Н, м),3,05 (3H, с), 2,96 (1 Н, м), 2,81 (2 Н, м), 2,65 (1 Н, дд), 2,43 (1 Н, дд), 2,25 (1 Н, м), 2,06 (1 Н, м), 0,95-0,70 (4 Н,м). MS (+APCI) m/z 495,8, 493,8 (M+Na)+. Указанное в заголовке соединение получали из 42 мг вышеуказанного метилового сложного эфира в соответствии с методикой, описанной в примере 1, стадия 3, получая 34 мг бесцветного масла. 1(+/-)-2-4-[(4-Хлорфенил)метил]-7-(метилсульфонил)-5-(2-тиенил)-1,2,3-тригидроциклопента[2,3 В]индол-3-илуксусная кислота С использованием способа, описанного в примере 1, стадия 7, из 500 мг метил 2-5-бром-4-[(4 хлорфенил)метил]-7-(метилсульфонил)-1,2,3-тригидроциклопента-[2,3-b]индол-3-илацетата (пример 1,стадия 6) и 600 мкл 2-тиофенилтрибутилолова получали 480 мг метилового сложного эфира указанного в заголовке соединения в виде тускло-желтого масла. Данное вещество использовали без дополнительной очистки на следующей стадии. 1(2 Н, д), 5,03 (1 Н, д), 4,96 (1 Н, д), 3,61 (3H, с), 3,49 (1 Н, м), 3,07 (3H, с), 3,05-2,70 (3H, м), 2,51 (1 Н, дд),2,39 (1 Н, дд), 2,25 (1 Н, м). Указанное в заголовке соединение получали из 480 мг вышеуказанного метилового сложного эфира в соответствии с методикой, описанной в примере 1, стадия 3, получая 450 мг белого вспененного вещества. 1 Н ЯМР натриевой соли (ДМСО-d6)8,00 (1 Н, д), 7,59 (1 Н, дд), 7,31 (1 Н, д), 7,18 (2 Н, д), 7,04 (1 Н,дд), 6,94 (1 Н, м), 6,37 (2 Н, д), 5,26 (1 Н, д), 5,04 (1 Н, д), 3,42 (1 Н, м), 3,20 (3H, с), 2,88 (1 Н, м), 2,78 (1 Н, м),2,62 (1 Н, м), 2,25-2,05 (2 Н, м), 1,92 (1 Н, дд). MS (-APCI) m/z 500,3, 498,2 (М-Н)-. Пример 10.(+/-)-2-7-[(Диметиламино)сульфонил]-4-[(4-хлорфенил)метилЛ]-1,2,3-тригидроциклопента[2,3 В]индол-3-илуксусная кислота Стадия 1: [(4-Аминофенил)сульфонил]диметиламин К раствору 40,2 г сульфаниламида в 1,5 л МеОН, охлажденному до 0 С, добавляли одновременно в течение 3 ч с помощью двух насосов со шприцами 220 мл диметилсульфата и 464 мл 5 н NaOH. После расходования исходного вещества органический растворитель удаляли в вакууме, добавляли водныйNH4Cl и продукт экстрагировали EtOAc. Органический слой промывали водой, насыщенным раствором соли и сушили над безводным Na2SO4. Органическую фазу концентрировали досуха и неочищенное твердое вещество перекристаллизовывали из 90% EtOAc в гексанах, получая 26,2 г указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (CDCl3)7,53 (2 Н, д), 6,69 (2 Н, д), 4,12 (2 Н, ушир. с), 2,64 (6 Н, с). Стадия 2: [(4-Амино-3-иодфенил)сульфонил]диметиламин С использованием способа, описанного в примере 1, стадия 1, исходя из 6,1 г [(4 аминофенил)сульфонил]диметиламина, получали 4,2 г указанного в заголовке соединения в виде коричневого твердого вещества. 1 Указанное в заголовке соединение получали из 3,46 г- 24006765 С использованием способа, описанного в примере 1, стадия 6, из 497 мг сложного эфира стадии 3 и 354 мг 4-хлорбензилбромида получали этиловый сложный эфир указанного в заголовке соединения, который гидролизовали в соответствии с примером 1, стадия 3, получая 500 мг указанного в заголовке соединения в виде белого твердого вещества. 1 Этиловый сложный эфир указанного в заголовке соединения получали из 10,00 г 4-фтор-2 иодфениламина и 6,57 г этил 2-(2-оксоциклопентил)ацетата в соответствии с примером 1, стадия 2, получения 5,36 г желтого твердого вещества. 1 Н ЯМР (ацетон-d6)9,76 (1 Н, ушир. с), 7,34 (1 Н, дд), 7,03 (1 Н, д), 6,78 (1 Н, тд), 4,14 (2 Н, кв), 3,57(1 Н, м), 2,85-2,55 (5 Н, м), 2,15 (1 Н, м), 1,22 (3H, т). Указанное в заголовке соединение получали из 1,24 г вышеуказанного этилового сложного эфира в соответствии с примером 1, стадия 3, получая 1,08 г неочищенного и нестабильного воскообразного коричневого масла, которое использовали как таковое на следующей стадии (чистота 90 %). 1 С использованием способа, описанного в примере 1, стадия 4, исходя из 2,2 г кислоты со стадии 2(чистота 90%), получали 2,13 г указанного в заголовке соединения в виде неочищенного и нестабильного коричневого твердого вещества. Данное вещество использовали без дополнительной очистки на следующей стадии. 1H ЯМР (ацетон-d6)10,77 (1 Н, ушир. с), 9,84 (1 Н, ушир. с), 7,09 (2 Н, м), 3,60 (1 Н, м), 2,95-2,65 (4 Н,м), 2,56 (1 Н, дд), ,19 (1 Н, м). Стадия 4: (+/-)-2-5-Бром-4-[(4-хлорфенил)метил]-7-фтор-1,2,3-тригидроциклопента[2,3-b]индол-3 илуксусная кислота Этерификация 2,13 г предшествующей кислоты диазометаном с последующим алкилированием 1,7 г 4-хлорбензилбромида в соответствии со способами, описанными в примере 1, стадии 5 и 6 давали метиловый сложный эфир указанного в заголовке соединения, которое гидролизовали с использованием методики примера 1, стадия 3. Получали 2,35 г указанного в заголовке соединения в виде коричневого твердого вещества. 1 Н ЯМР (ацетон-d6)10,70 (1 Н, ушир. с), 7,31 (2 Н, д), 7,18 (1 Н, д), 7,06 (1 Н, д), 6,92 (2 Н, д), 5,90(+)-2-5-Бром-4-[(4-хлорфенил)метил]-7-фтор-1,2,3-тригидроциклопента[2,3-В]индол-3 илуксусная кислота [(+)-изомер соединения примера 11] К раствору 2,35 г (+/-)-2-5-бром-4-[(4-хлорфенил)метил]-7-фтор-1,2,3-тригидроциклопента[2,3b]индол-3-илуксусной кислоты (пример 11, стадия 4) в 130 мл EtOH при 80 С добавляли 780 мкл (S)-(-)1-(нафтил)этиламина. Раствор охлаждали до комнатной температуры и перемешивали в течение ночи. 1,7 г выделенной соли повторно перекристаллизовывали из 200 мл EtOH. После фильтрования полученное белое твердое вещество нейтрализовали 1 н HCl и продукт экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали. Ве- 25006765 щество фильтровали через слой SiO2, элюируя EtOAc, получая 500 мг указанного в заголовке энантиомера в виде белого твердого вещества. Времена удерживания обоих энантиомеров составляли, соответственно, 7,5 мин и 9,4 мин [ChiralPak AD колонка, гексан/2-пропанол/уксусная кислота (95:5:0,1)]. Болееполярный энантиомер имел 98% ее. ее=98%; Время удерживания =9,4 мин [ChiralPak AD колонка: 250x4,6 мм, гексан/2-пропанол/уксусная кислота (75:25:0,1)]; []D21 = +39,2 (с 1,0, МеОН). Пример 13.(-)-2-5-Бром-4-[(4-хлорфенил)метил]-7-фтор-1,2,3-тригидроциклопента[2,3-В]индол-3-илуксусная кислота [(-)-изомер соединения примера 11] К раствору 1,58 г (+/-)-2-5-бром-4-[(4-хлорфенил)-метил]-7-фтор-1,2,3-тригидроциклопента[2,3b]индол-3-илуксусной кислоты (выделенной из надосадочных жидкостей, полученных при разделении в примере 12) в 180 мл EtOH при 80 С добавляли 530 мл (R)-(+)-l-(нафтил)этиламина. Раствор охлаждали до комнатной температуры и перемешивали в течение ночи. 1,07 г выделенной соли опять перекристаллизовывали из 120 мл EtOH. После фильтрования, полученную белую твердую соль нейтрализовали 1 нHCl и продукт экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили над безводным Na2SO4 и концентрировали. Вещество фильтровали через слой SiO2, элюируяEtOAc, получая 640 мг указанного в заголовке энантиомера в виде белого твердого вещества. Времена удерживания двух энантиомеров составляли 7,5 и 9,4 мин [ChiralPak AD колонка, гексан/2 пропанол/уксусная кислота (95:5:0,1)]. Менее полярный энантиомер получали с 99% ее. ее 99%; Время удерживания =7,4 мин [ChiralPak AD колонка: 250x4,6 мм, гексан/2-пропанол/уксусная кислота(+/-)-2-5-Бром-4-[(4-хлорфенил)метил]-3-метил-1,2,3-тригидроциклопента[2,3-В]индол-3 илуксусная кислота Стадия 1: (+/-)-Этил 2-(5-бром-3-метил-1,2,3-тригидроциклопента[2,3-b]индол-3-ил)ацетат Суспензию 3,52 г 2-бромфенилгидразина и 2,90 г этил 2-(1-метил-2-оксоциклопентил)ацетата в 40 мл АсОН нагревали при 100 С в течение 1 ч. Затем по прошествии данного времени добавляли 20 мл толуола и растворители удаляли при пониженном давлении. Неочищенное вещество очищали флэшхроматографией, получая 1,40 г указанного в заголовке соединения в виде желтого масла. 1 С использованием способа, описанного в примере 1, стадия 6, 390 мг предшествующего сложного эфира со стадии 1 и 222 мг 4-хлорбензилхлорида давали 120 мг этилового сложного эфира указанного в заголовке соединения в виде не совсем белого твердого вещества. 1 Н ЯМР (ацетон-d6)7,44 (1 Н, д), 7,30 (2 Н, д), 7,22 (1 Н, д), 6,92 (1 Н, т), 6,90 (2 Н, д), 6,05 (1 Н, д),5,85 (1 Н, д), 3,90 (2 Н, кв), 2,80 (3H, м), 2,65 (2 Н, д), 2,36 (1 Н, м), 1,35 (3H, с), 1,00 (3H, т). MS (-APCI) m/z 458,3 (М-Н)-. Указанное в заголовке соединение получали из 105 мг вышеуказанного этилового сложного эфира указанного в заголовке соединения в соответствии с примером 1, стадия 3, получая 90 мг белого твердого вещества. 1 где Ar представляет хлорфенил или дихлорфенил;R1 и R2 независимо выбирают из галогена, -SO2CH3, -SO2N(CH3)2, C(O)CH3, -СН(ОН)СН 3, -СН=СН 2,циклопропила и тиофенила; или его фармацевтически приемлемые соли, гидраты и эфиры. 2. Соединение по п.1, где R1 находится в 7 положении. 3. Фармацевтическая композиция, включающая соединение формулы Iа по любому из пп.1, 2 или его фармацевтически приемлемую соль, гидрат или сложный эфир и фармацевтически приемлемый носитель. 4. Способ лечения простагландин-опосредованных заболеваний, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1. 5. Способ лечения простагландин-D2 опосредованных заболеваний, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1. 6. Способ лечения закупорки носового канала, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1. 7. Способ лечения аллергической астмы, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1. 8. Способ лечения аллергического ринита, включающий введение нуждающемуся в таком лечении пациенту терапевтически эффективного количества соединения по п.1. 9. Применение соединения формулы Ia по любому из пп.1, 2 или его фармацевтически приемлемой соли, гидрата или сложного эфира для получения лекарственного средства для лечения простагландинопосредованных заболеваний. 10. Применение соединения формулы Iа по любому из пп.1, 2 или его фармацевтически приемлемой соли, гидрата или сложного эфира для получения лекарственного средства для лечения закупорки носового канала, аллергической астмы или аллергического ринита. 11. Антагонистическая фармацевтическая композиция в отношении закупоривающего действия носовых каналов и легких под действием простагландинов D-типа, включающая приемлемое антагонистическое количество соединения формулы Iа по любому из пп.1, 2 или его фармацевтически приемлемой соли, гидрата или сложного эфира в сочетании с фармацевтически приемлемым носителем.
МПК / Метки
МПК: A61P 11/02, A61P 11/06, C07D 209/80, A61K 31/403
Метки: циклопентаноиндолы, фармацевтические, лечения, способы, композиции
Код ссылки
<a href="https://eas.patents.su/28-6765-ciklopentanoindoly-farmacevticheskie-kompozicii-i-sposoby-lecheniya.html" rel="bookmark" title="База патентов Евразийского Союза">Циклопентаноиндолы, фармацевтические композиции и способы лечения</a>
Мостиковые инденопирролокарбазолы, фармацевтические композиции и способы лечения

Номер патента: 4066
Опубликовано: 25.12.2003
Авторы: Трипатхи Рабиндранатх, Малламо Джон П., Сингх Джасбир, Андерайнер Теодор Л., Хадкинз Роберт Л.
МПК: A61P 35/00, C07D 498/22, A61K 31/553...
Метки: способы, композиции, инденопирролокарбазолы, фармацевтические, лечения, мостиковые
Формула / Реферат:
1. Инденопирролокарбазолы формулы II где R1, R4, R6, R7 представляют собой водород; Y представляют собой O; n представляет собой 1; m представляет собой 1 или 2; R2 представляет собой водород, OH или алкил, имеющий от 1 до 4 атомов углерода; R3 представляет собой водород, галоген, C1-C4алкокси, 3-(3'-NH2-Ph); R5 представляет собой водород, C1-C4алкокси; R8 представляет собой водород, C1-C4алкил, CO2Et, CH2OH; Z представляет собой связь или O....
Конъюгаты эритропоэтина, их применение, фармацевтические композиции и способы лечения

Номер патента: 3777
Опубликовано: 28.08.2003
Автор: Бейлон Паскаль Себастиан
МПК: A61K 38/18, A61P 7/06, C07K 14/505...
Метки: эритропоэтина, конъюгаты, композиции, способы, применение, фармацевтические, лечения
Формула / Реферат:
1. Конъюгат, который включает гликопротеин эритропоэтина, имеющий, по меньшей мере, одну свободную аминогруппу и обладающий биологической активностью in vivo, обусловливающей увеличение продуцирования ретикулоцитов и эритроцитов клетками костного мозга, и выбранный из группы, включающей человеческий эритропоэтин и его аналоги, которые имеют последовательность человеческого эритропоэтина, модифицированную добавлением от 1 до 6 сайтов...
Антибактериальные соединения карбапенема, содержащие их фармацевтические композиции и способы лечения

Номер патента: 1296
Опубликовано: 25.12.2000
Авторы: Рэтклифф Рональд В., Близзард Тимоти А., Вилкенинг Роберт Р.
МПК: A61K 31/428, C07D 477/14
Метки: антибактериальные, карбапенема, лечения, композиции, фармацевтические, способы, содержащие, соединения
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R1 обозначает Н или метил; СО2М обозначает карбоновую кислоту, карбоксилатный анион, фармацевтически приемлемую сложноэфирную группу или карбоновую кислоту, защищенную защитной группой; Р обозначает водород, гидроксил, F или гидроксил, защищенный гидроксилзащитной группой; каждый R независимо выбирают из -R*; -Q; водорода; галогена; -CN; -NO2; -NRaRb; -ORc; -SRc; -C(O)NRaRb;...
Производные пиразолотриазинов, фармацевтические композиции, содержащие их, способы лечения

Номер патента: 4403
Опубликовано: 29.04.2004
Авторы: Хорват Роберт Джон, Арванитис Арджириос Георгиос
МПК: A61P 1/04, A61K 31/4162, A61K 31/53...
Метки: композиции, способы, лечения, пиразолотриазинов, содержащие, их, производные, фармацевтические
Формула / Реферат:
1. Соединение формулы (1) и его изомеры, его стереоизомерные формы или смеси его стереоизомерных форм и его фармацевтически приемлемые соли или пролекарства, где A обозначает N; Z обозначает CR2; Ar выбран из группы фенил, пиридил, где каждый Ar необязательно замещен 1-5 группами R4; R1 независимо выбран в каждом случае из группы H, C1-C4-алкил; R2 независимо выбран из группы H, C1-C4-алкил; R3 выбран из группы -H, OR7, NR6aR7a, или фенил,...
Кальцийдикарбоксилатные эфиры, способы их получения, фармацевтические композиции и способ лечения

Номер патента: 4862
Опубликовано: 26.08.2004
Авторы: Эндо Хауард Йошихиса, Батлер Доналд Юджин, Доузмэн Гэри Джей
МПК: A61P 3/10, C07C 59/125, A61K 31/19...
Метки: эфиры, кальцийдикарбоксилатные, фармацевтические, лечения, композиции, получения, способ, способы
Формула / Реферат:
1. Кристаллическая Форма 1 соединения монокальциевой соли 6-(5-карбокси-5-метилгексилокси)-2,2-диметилгексановой кислоты, имеющая картину дифракции рентгеновских лучей на порошке, содержащую характеристический пик, выраженный в градусах 2q , при 6,760. 2. Кристаллическая Форма 1 соединения монокальциевой соли 6-(5-карбокси-5-метилгексилокси)-2,2-диметилгексановой кислоты по п.1, имеющая картину дифракции рентгеновских лучей на порошке,...
Предыдущий патент: Частица, содержащая органические вещества, имеющая повышенную температуру самовозгорания
Следующий патент: Фармацевтические композиции, включающие метформин и глибенкламид, применяемые для лечения сахарного диабета типа ii
Случайный патент: Усовершенствованное интермодальное железнодорожное транспортное средство для формирования поезда