Соединение альфа-(n-сульфонамидо) ацетамида в качестве ингибитора продуцирования бета-амилоидного пептида


























Формула / Реферат
1. Соединение (2R)-2-[[(4-хлорфенил)сульфонил][[2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил] амино]-5,5,5-трифторпентанамид.
2. Фармацевтическая композиция, включающая терапевтически эффективное количество (2R)-2-[[(4хлорфенил)сульфонил][[2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида совместно с фармацевтически приемлемым адъювантом, носителем или разбавителем.
3. Способ лечения или отсрочки начала болезни Альцгеймера, церебральной амилоидной ангиопатии, умеренного когнитивного нарушения, синдрома Дауна, который включает введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества (2R)-2-[[(4-хлорфенил)сульфонил][[2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида.
4. Способ по п.3 для лечения болезни Альцгеймера.
5. Соединение формулы

или его фармацевтически приемлемая соль.
6. Соединение формулы

или его фармацевтически приемлемая соль.
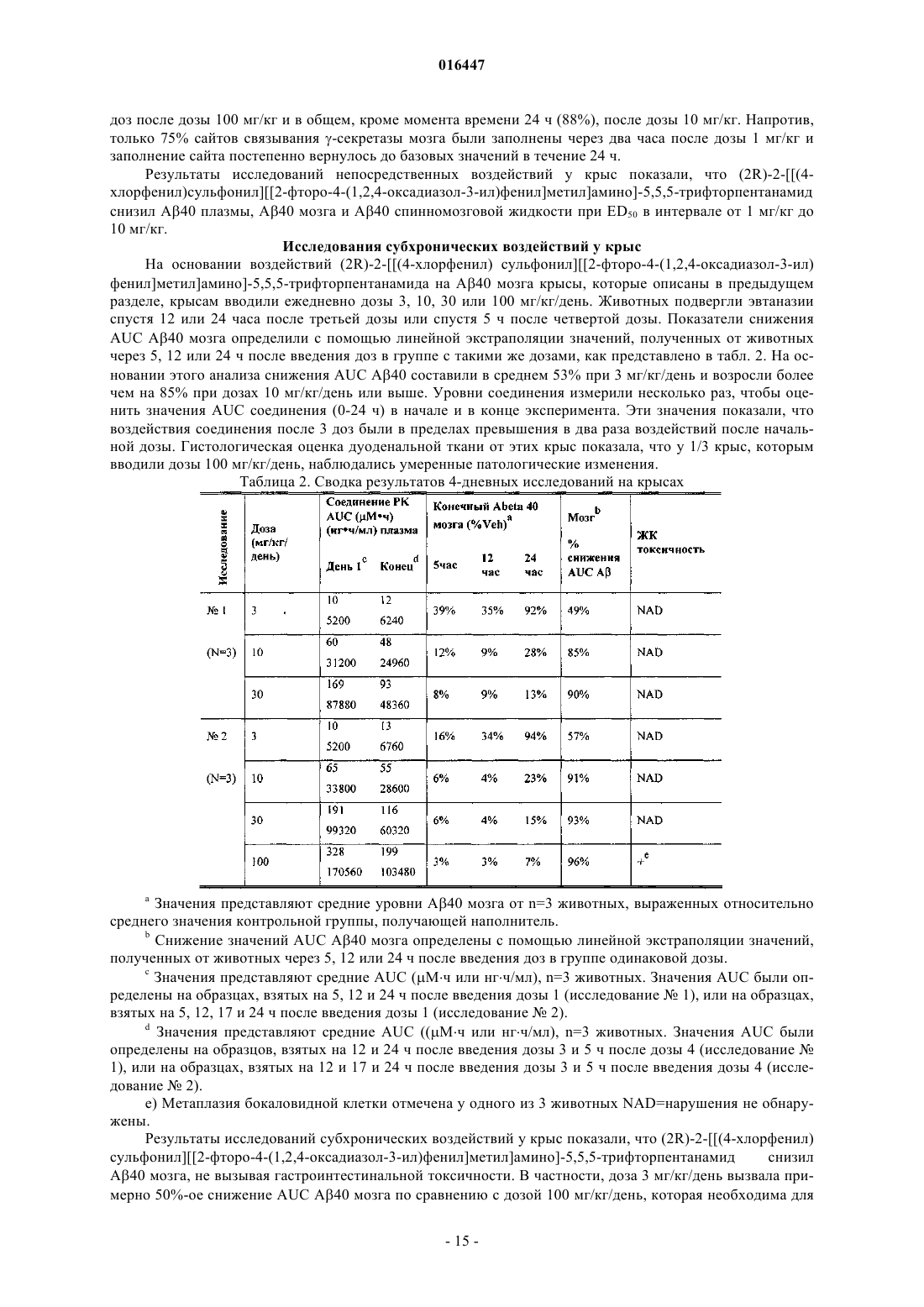
Текст
СОЕДИНЕНИЕ АЛЬФА-(N-СУЛЬФОНАМИДО) АЦЕТАМИДА В КАЧЕСТВЕ ИНГИБИТОРА ПРОДУЦИРОВАНИЯ БЕТА АМИЛОИДНОГО ПЕПТИДА В изобретении предлагается новое соединение (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид, его фармацевтический состав, технологические процессы и способ лечения болезни Альцгеймера и других состояний,ассоциированных с -амилоидным пептидом. 016447 Область техники, к которой относится изобретение Изобретение относится к (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамиду, обладающему лекарственными и биовоздействующими свойствами, его фармацевтическим составам, технологическим процессам и способам применения. Новое соединение обладает специфическим ингибированием выработки А пептида, тем самым способствуя предотвращению накопления А пептидов и/или отложений амилоидного белка в мозгу и пригоден при лечении или отсрочке начала болезни Альцгеймера (AD), синдрома Дауна, умеренных когнитивных нарушений и других состояний, ассоциированных с -амилоидным пептидом. Предпосылки Болезнь Альцгеймера (AD) - прогрессирующая нейродегенеративная болезнь, которая начинается с потери памяти и развивается с присоединением тяжелых когнитивных нарушений, измененного поведения и снижения двигательной функции (Grundman, M. с соавт., Arch Neurol. (2004) 61: 59-66; Walsh, D.M. с соавт., Neuron (2004) 44: 181-193). Эта болезнь является самой общей формой деменции и находится на третьем месте по смертности после сердечно-сосудистых заболеваний и рака. Плата за AD огромна и включает страдание пациентов и семей и потерю производительности пациентов и ухаживающих. В настоящее время не существует лечения, которое эффективно предупреждает AD или полностью исключает клинические симптомы и лежащую в основе патофизиологию. Окончательный диагноз AD у дементного пациента требует гистопатологической оценки количества и локализации нейритных гемолитических бляшек и нейрофибриллярных клубков после аутопсии(Consensus recommendations for the postmortem diagnosis of Alzheimer's disease - Усредненные рекомендации о согласии на посмертный диагноз болезни Альцгеймера. Neurobiol Aging (1997) 18: S1-2). Сходные изменения наблюдаются у больных Трисомией 21 (синдром Дауна). Бляшки сначала состоят из амилоидных (А) пептидов, которые образуются постадийным протеолитическим расщеплением амилоидного белка-предшественника (АРР) с помощью АРР-отщепляющего фермента (ВАСЕ) с получениемN-конца и -секретазы с получением С-конца (Selkoe, D.J., Physiol Rev. (2001) 81: 741-766). -Секретаза это трансмембранный белковый комплекс, который включает Никастрин, Aph-1, PEN-2 и любой Презенилин-1 (PS-1) или Презенилин-2 (PS-2) (Wolfe, M.S. с соавт., Science (2004) 305: 1119-1123). Предполагают, что PS-1 и PS-2 содержат каталитические сайты -секретазы. А 40 - это самая богатая форма синтезированного А (80-90%), тогда как А 42 наиболее близко связан с патогенезом AD. В частности, мутации в АРР, PS-1 и PS-2 генах, которые приводят к редким,семейным формам AD, вовлекают А 42 скопления как первичные токсические виды (Selkoe, D.J., PhysiolRev., (2001) 81: 741-766). Имеющийся опыт показывает, что олигомерные, протофибриллярные и внутриклеточные АР 42 играют значительную роль в процессе заболевания (Cleary, J.P. с соавт., Nat Neurosci.(2005) 8: 79-84). Ингибиторы ферментов, образующие A42, как, например, -секретазу, представляют потенциальную изменяющую болезнь терапию для лечения болезни Альцгеймера.-Секретаза расщепляет многие трансмембранные белки типа 1 помимо АРР (Pollack, S.J. с соавт.,Curr Opin Investig Drugs (2005) 6: 35-47). Несмотря на то, что физиологическое значение большинства этих случаев расщепления неизвестно, генетический опыт показывает, что расщепление у-секретазойNotch необходимо для передачи сигналов Notch (Artavanis-Tsakonas, S. с соавт., Science (1999) 284(5415): 770-6; Kadesch, Т.; Exp Cell Res. (2000) 260(1): 1-8). У грызунов, которым дозами давали ингибиторы секретазы, в желудочно-кишечном (GI) тракте, тимусе и селезенке была определена связанная с лекарственным средством токсичность (Searfoss, G.H.; Jordan с соавт., J Biol Chem. (2003) 278: 46107-46116;Wong, G.T. с соавт., J Biol Chem. (2004) 279: 12876-12882; Milano, J. с соавт., Toxicol Sci. (2004) 82: 341358). Эти виды токсичности вероятно связаны с ингибированием передачи сигналов Notch (Jensen, J. с соавт., Nat Genet. (2000) 24: 36-44). Идентификация основанной на механизме действия токсичности ставит вопрос, можно ли получить приемлемый терапевтический показатель с помощью ингибиторов -секретазы. Селективное ингибирование образования А при Notch обработке, фармакокинетика, распределение лекарственного средства и/или тканеспецифичная фармакодинамика могут повлиять на терапевтический интервал. Опыт показывает, что снижение в мозгу уровней А за счет ингибирования -секретазы может предупредить начало и развитие AD (Selkoe, D. Physiol. Rev. (2001) 81: 741-766; Wolfe, M., J. Med. Chem.(2001) 44: 2039-2060). Появляются данные о роли А в других заболеваниях, включая умеренные когнитивные нарушения (MCI), синдром Дауна, церебральную амилоидную ангиопатию (САА), деменцию с тельцами Леви (DLB), боковой амиотрофический склероз (ALS-D), миозит с включенными тельцами(IBM) и возрастную дегенерацию желтого пятна. Предпочтительно соединения, которые ингибируют секретазу и снижают выработку A, можно было бы использовать при лечении этих или других Aзависимых заболеваний. Избыточная выработка и/или сниженный клиренс А вызывает САА Thal, D. с соавт., J. Neuropath. Exp. Neuro, (2002) 61: 282-293). У этих пациентов сосудистые амилоидные отложения приводят к разрушению стенок сосудов и аневризмам, которые могут быть причиной 10-15% геморрагических уда-1 016447 ров у пожилых пациентов. Как при AD, мутации в гене, кодирующем А, приводят к форме САА с ранним началом, именуемой внутримозговым кровоизлиянием с амилоидозом голландского типа, и у мышей, экспрессирующих этот мутантный белок, развивается САА, сходная с имеющейся у пациентов. Соединения, которые специфически нацелены на -секретазу, могли бы ослабить или предупредить САА.DLB проявляется зрительными галлюцинациями, бредом и паркинсонизмом. Интересно, что мутации семейной болезни Альцгеймера, которые вызывают отложения АР, могут также вызвать тельца Леви и симптомы DLB (Yokota, О. с соавт., Acta Neuropathol (Berl) (2002) 104: 637-648). Кроме того, у пациентов со спорадическим DLB отложения А подобны отложениям при AD (Deramecourt, V. с соавт., J Neuropathol Exp Neurol (2006) 65: 278-288). На основании этих данных А, вероятно, побуждают патологию телец Леви при DLB и поэтому ингибиторы у-секретазы могут ослабить или предупредить DLB. Приблизительно 25% пациентов с ALS имеют значительную деменцию или афазию (Hamilton, R.L. с соавт., Acta Neuropathol (Berl) (2004) 107: 515-522). Большинство (60%) этих пациентов, у которых установлено ALS-D, содержит убиквитин-положительные включения, содержащие первоначально TDP43 белок (Neumann, M. с соавт., Science (2006) 314: 130-133). Примерно 30% пациентов с ALS-D имеют амилоидные бляшки, совместимые с A, вызывающие у них деменцию (Hamilton, R.L. с соавт., Acta Neuropathol (Berl) (2004) 107: 515-522). Эти пациенты должны идентифицироваться амилоидным радиоактивным препаратом и потенциально поддаваться лечению ингибиторами -секретазы.IBM - это редкое возрастное дегенеративное заболевание скелетных мышц. Появление отложений А в мышце, пораженной IBM, и рекапитуляция нескольких картин заболевания путем направления сверхэкспрессии АРР на мышцу у трансгенных мышей подтверждает роль А в IBM (рассмотрено в работе Murphy, M.P. с соавт., Neurology (2006) 66: S65-68). Соединения, которые специфически нацелены на -секретазу, могут ослабить или предупредить IBM. При возрастной дегенерации желтого пятна А идентифицировали как один из нескольких компонентов друз, внеклеточных отложений под ретинальным пигментным эпителием (RPE) (Anderson, D.H. с соавт., ExpEye Res (2004) 78: 243-256). Последние исследования показали потенциальные связи между А и дегенерацией желтого пятна у мышей (Yoshida, Т. с соавт., J Clin Invest (2005) 115: 2793-2800) 115: 2793-2800). У пациентов с болезнью Альцгеймера были обнаружены увеличения отложений А и супрануклеарные катаракты(Goldstein, L.E. с соавт., Lancet (2003) 361: 1258-1265). Соединения, которые специфически нацелены на секретазу, могли бы уменьшить или предупредить возрастную дегенерацию желтого пятна. Основанные на роли Notch передачи сигналов при онкогенезе, ингибирующие -секретазу соединения могут быть также пригодны в качестве терапевтических агентов для лечения рака (Shih, I.-M., с соавт., Cancer Research (2007) 67: 1879-1882). Смит с соавт. [Smith, с соавт.] в международной заявке WO 00/50391, опубликованной 31 августа 2000 года, предлагает ряд сульфамидных соединений, которые могут способствовать модулированию выработки амилоидногобелка в качестве средства лечения ряда заболеваний, особенно болезни Альцгеймера и других заболеваний, относящихся к отложению амилоида. В японском патенте 11343279, опубликованном 14 декабря 1999 года, предложен ряд производных соединений сульфамида, которые являются TNF-альфа ингибиторами, пригодными для лечения аутоиммунных заболеваний. Паркер с соавт. [Parker, с соавт.] в международной заявке WO 03/053912, опубликованной 3 июля 2003 года, предлагают ряд -(N-сульфонамидо) ацетамидных производных соединений в качестве амилоидных ингибиторов, которые пригодны для лечения болезни Альцгеймера и других состояний,ассоциированных с -амилоидным пептидом. Новое соединение в соответствии с настоящим изобретением, которое попадает в рамки определения формулы в WO 03/053912, не раскрыто или описано в работе Parker, с соавт. К удивлению, было обнаружено, что (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид обладает уникальными свойствами, которые делают его пригодным для лечения болезни Альцгеймера и других состояний, ассоциированных с -амилоидным пептидом. Описание изобретения Изобретение относится к(2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3 ил)фенил]метил]амино]-5,5,5-трифторпентанамиду, имеющему Формулу I, его фармацевтическим составам и его использованию в ингибировании выработки А у больных, страдающих болезнью Альцгеймера или восприимчивых к болезни Альцгеймера, или другим нарушениям, ассоциированным с -амилоидным пептидом.-2 016447 В другом примере осуществления настоящее изобретение предусматривает фармацевтический состав, включающий терапевтически эффективное количество (2R)-2-(4-хлорфенил)сульфонил]2-фторо 4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида совместно с фармацевтически приемлемым адъювантом, носителем или разбавителем. В еще одном примере осуществления настоящее изобретение предусматривает способ лечения,смягчения или отсрочки начала нарушений, ассоциированных с -амилоидным пептидом, особенно болезни Альцгеймера, церебральной амилоидной ангиопатии, умеренных когнитивных нарушений и синдрома Дауна, который включает введение вместе с обычным адъювантом, носителем или разбавителем терапевтически эффективного количества (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида или его сольвата или гидрата. В другом аспекте настоящее изобретение предусматривает способ получения (2R)-2-(4 хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида, включающий стадию вступления в реакцию (R)-2-(4-хлорфенилсульфонамидо)-5,5,5-трифторпентанамида с 3-(4-(бромметил)-3-фторфенил)-1,2,4-оксадиазол в инертном органическом растворителе при наличии основания и предпочтительно неорганического основания, как например карбонат цезия. В следующем аспекте настоящее изобретение предусматривает способ получения (2R)-2-(4 хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида, включающий следующие стадии:(b) обработку получившегося (R)-2-(4-хлоро-N-(2-фторо-4-(N'-гидроксикарбамимидоил)бензил)фенилсульфонамидо)-5,5,5-трифторпентанамида триэтоксиметаном в инертном органическом растворителе при наличии кислотного катализатора. Поскольку соединение в соответствии с настоящим изобретением имеет асимметрический атом углерода, настоящее изобретение включает рацемат, а также отдельные энантиомерные формы соединения формулы I и хиральные и рацемические интермедиаты согласно приведенному здесь описанию. Использование одиночного обозначения, например, (R) или (S), предназначено для включения главным образом одного стереоизомера. Смеси изомеров можно разделить на отдельные изомеры в соответствии с известными методами, например, фракционная кристаллизация, адсорбционная хроматография или другие подходящие процессы разделения. Полученные рацематы можно разделить на антиподы обычном способом после введения пригодных солеобразующих группировок, например, путем образования смеси диастереоизомерных солей с оптически активными солеобразующими агентами, разделения смеси на диастереомерные соли и конверсии разделенных солей в свободные соединения. Энантиомерные формы можно также разделить фракционированием, пропустив через колонки хиральной жидкостной хроматографии высокого давления. В способе в соответствии с настоящим изобретением термин "терапевтически эффективное количество" означает общее количество каждого активного компонента способа, достаточное для демонстрации существенной пользы пациенту, то есть исцеление состояний, ассоциированных с -амилоидным пептидом. Когда термин применяется к отдельному активному ингредиенту, который вводится отдельно, он относится только к этому ингредиенту. Когда термин применяется к комбинации, он относится к объединенным количествам активных ингредиентов, вызывающих терапевтический эффект, вводятся ли они в комбинации, последовательно или одновременно. Термины "лечить, лечение" в соответствии с использованием в настоящем описании и в формуле изобретения означает предупреждение, отсрочку, ослабление или улучшение заболеваний, ассоциированных с -амилоидным пептидом. В следующем примере осуществления изобретения соединение формулы I можно использовать в комбинации с другими лекарственными средствами, которые используются для лечения/профилактики/ ослабления или улучшения болезней или состояний, для которых пригодно соединение формулы I. Эти другие препараты можно вводить способом и в количестве, которые обычно для этого используются,одновременно или последовательно с соединением в соответствии с настоящим изобретением. Когда соединение формулы I используется одновременно с одним или несколькими другими лекарственными средствами, предпочтителен фармацевтический состав, содержащий такие лекарственные средства дополнительно к соединению формулы I. Следовательно, фармацевтические составы настоящего изобретения включают составы, которые также содержат один или несколько других активных ингредиентов дополнительно к соединению формулы I. Примеры других активных ингредиентов, которые могут объединяться с (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]5,5,5-трифторпентанамидом, вводимым либо отдельно, либо в тех же фармацевтических составах, для лечения болезни Альцгеймера включают, но не ограничиваются этим: класс лекарственных средств, которые являются ингибиторами холинэстеразы, например донепезил (Aricept), ривастигмин (Exelon),галантамин (Reminyl, сейчас Razadyne); другие лекарственные средства, являющиеся антагонистамиNMDA, как например, мемантин (Namenda) и ингибиторами PDE4, например, циломиласт (Ariflo); класс нестероидных противовоспалительных средств (NSAID), например, R-флурбипрофен (Flurizan);-3 016447 понижающие холестерин статиновые лекарства, например, правастатин, симвастатин и аторвастатин; антиамилоидная и анти-А иммунная терапия; соединения, которые ингибируют агрегацию А, например, сциллоинозитол и склиоквинол; другие соединения, которые ингибируют или изменяют выработку или обработку А, например, ингибиторы -секретазы, ингибиторы -секретазы, модуляторы секретазы, модуляторы А и ингибиторы GSK-3; соединения, регулирующие метаболизм А, например,ингибиторы PAI-1; соединения, регулирующие тау фосфорилирование, например, ингибиторы GSK-3 иCDK-5; PPAR агонисты, например, росиглитазон; соединения, регулирующие тау или фосфор-тау метаболизм, или олигомеризацию, например, HSP90 ингибиторы, HDAC ингибиторы и анти-тау иммунная терапия; и соединения, которые стабилизируют микротрубки или связываются с ними, например, производные соединения таксана и производные соединения эпотилона; и соединения, регулирующие функцию митохондрии, например, Димебон. При лечении рака соединение в соответствии с настоящим изобретении можно использовать с известными противоопухолевыми агентами или препаратами. Такие агенты и препараты включают цитотоксические/цитостатические агенты, модуляторы андрогенового рецептора, модуляторы эстрогенного рецептора, модуляторы ретиноидного рецептора, ингибиторы пренилпротеин трансферазы, ингибиторы ангиогенеза, агенты, которые сталкиваются с контрольными точками клеточного цикла, и лучевую терапию. Кроме того, соединения в соответствии с настоящим изобретением могут быть пригодны при лечении иммунологических нарушений, как например, волчанка. Описанные выше терапевтические агенты при использовании в комбинации с соединением в соответствии с настоящим изобретением можно применять, например, в количествах, указанных в Настольном справочнике врача (PDR). Однако ясно, что количество фактически вводимого соединения будет определять врач в свете сложившихся обстоятельств, включая состояние, которое необходимо лечить, выбор вводимого соединения,выбранный способ введения, возраст, вес и реакцию конкретного пациента, а также серьезность симптомов у пациента. Общие схемы реакций Соединение в соответствии с настоящим изобретением можно приготовить самыми разными способами, известными специалистам в области органического синтеза. Соединение формулы I можно приготовить с помощью методов, описанных ниже в схемах реакций 1-5. Приемлемые варианты описанных процедур вместе с синтетическими методами, которые были бы очевидны специалистам в данной области, вписываются в объем настоящего изобретения. Схема реакций 1 В одном методе изготовления, который иллюстрируется в схеме реакций 1, начальный (-амино) ацетамид формулы II, используемый в основном в энантиомерно чистой форме, можно приготовить по известным в литературе процедурам, как например, с помощью метода асимметричного синтеза Штрекера, описанного в схеме реакций 3 для конверсии трифторобутиральдегида в (-амино)ацетамид формулыII, или альтернативно из (R)-5,5,5-трифторонорвалина (см.; I. Ojima, J. Org. Chem. (1989) 54: 4511-4522),и метода, описанного в схеме реакций 4, с последующими общими процедурами для приготовления амида: R.C. Larock "Comprehensive Organic Transformations", VCH Publishers, New York, 1989, pp. 972 - 976.(-амино)ацетамид формулы II обрабатывается соответствующим основанием и сульфонилируется рхлорсульфонил хлоридом в подходящем апротонном растворителе, например, CH2Cl2 примерно при комнатной температуре, чтобы получить (-сульфонамидо) ацетамид формулы III. Подходящие основания включают триэтиламин, диизопропиламин, пиридин и т.п. Конверсия соединения формулы III в сульфамид формулы I выполняется при наличии основания при помощи реакции (-сульфонамидо)ацетамида формулы III с оксадиазол фторбензил алкилирующим агентом формулы IV в подходящем апротонном растворителе с нагреванием или без него. Фторбензил оксадиазол формулы IV можно без труда приготовить известными методами, в которых X - уходящая группа, и методом, описанным в схеме реакций 6. Подходящие основания для этого алкилирования включают неорганические основания, как например, карбонат калия и карбонат цезия. Предпочтительные растворители включают DMF и ацетонитрил. Обычный температурный диапазон для реакции от 20 до 100 С. В другом методе приготовления, который иллюстрируется в схеме реакций 2,1,2,4-океадиазол формулы I приготовлен алкилированием соединения формулы III производным 2-циано-4-фторбензила формулы VI, в котором X - это уходящая группа при наличии основания в подходящем растворителе для получения нитрила формулы VII. Требуемое соединение формулы I затем получают из нитрильного соединения формулы VII с помощью известных специалистам методов (ссылка: Joule, J.A, с соавт., Heterocyclic Chemistry, 3rd ed., ChapmanHall, London (1995) 452-456 и указанная в настоящем описании библиография). Например, реакция нитрила формулы VII с гидроксиламином в спиртовом растворителе,например, метаноле или этаноле, при температурах вплоть до обратного потока обеспечивает промежуточный амид оксим, который впоследствии обрабатывается ортоформиатом (например, триэтил или триметил ортоформиатом) при наличии кислотного источника, например, трифторуксусной кислоты или эфирата трехфтористого бора в инертном органическом растворителе, например, CH2Cl2, ацетонитриле,тетрагидрофуране и т.п., чтобы получить 1,2,4-оксадиазол формулы I. Схема реакций 3 В схеме реакций 3 приводится описание приготовления (-амино)ацетамида из формулы II, начиная с коммерчески доступного трифторобутиральдегида и (R)метил бензил амина в условиях по Штрекеру с уксусной кислотой и источником цианида, например, цианистый натрий, цианистый калий или триметилсилилцианид в подходящем растворителе, например, метанол, для получения аминонитрила из формулы VIII в виде смеси диастереомеров. Исходный трифторобутиральдегид можно также приготовить окислением трифторбутанола. Гидролиз нитрила из формулы VIII до соответствующего амида из формулы IX выполняется при помощи серной кислоты и нейтрализацией реакции с последующим окислением и рекристаллизацией из пригодного растворителя, например, метанола, изопропанола, этилацетата, метил-трет-бутилового эфира или их смесей, для получения амида из формулы X при 99% диастереоизомерном избытке. Бензильную группу можно затем удалить гидрированием при наличии подходящего катализатора, например, гидроокиси палладия или палладия на углероде, чтобы получить амино амид из формулы II, который может быть сульфонилирован р-хлорсульфонил хлоридом для получения сульфамида из формулы III. В другом методе подготовки (-амино) ацетамид формулы II можно создать стереоизбирательно,используя ферментативный процесс, начиная с 5,5,5-трифторо-2-левулиновой кислоты, как проиллюстрировано в схеме реакций 4. (R)-5,5,5-трифторонорвалин формулы XIV можно приготовить в основном в энантиомерно чистой форме из соединения формулы XIII с помощью коммерчески доступного фермента(R)-аминотрансферазы известными специалистам методами. В альтернативном методе ферментативный процесс может быть проведен с использованием коммерчески доступного (R)-фермента дегидразы аминокислоты. Ферментные процессы проводятся с использованием описанных ниже методов и известных специалистам методов. Конверсию (R)-5,5,5- трифторонорвалина формулы XIV в соединение формулы II можно выполнить при помощи общих процедур приготовления амида, известных специалистам. Схема реакций 5 Бромистый бензил формулы VIa можно приготовить бромированием коммерчески доступного 2 фторо-4-цианотолуола N-бромсукцинимидом в подходящем растворителе, например, дихлорметане, дихлорэтане или тетрахлорметане с использованием инициатора, например, AIBN, как проиллюстрировано в схеме реакций 5. Бромирование протекает с высоким выходом и, при желании, соединение ФормулыVIa можно без труда преобразовать в соединение формулы VI, в котором X - уходящая группа, с помощью методов, известных специалистам в данной области. Схема реакций 6 В качестве альтернативы применения соединения формулы VI в линейной последовательности сульфамид оксадиазолу из формулы I, описанной выше в схеме реакций 2, приготовление соединения формулы IV для использования в конвергентном пути, изображенном в схеме реакций 1, показан в схеме реакций 6. Обработка коммерчески доступного 2-фторо-4-цианотолуола гидроксиламином при комнатной температуре в спиртовом растворителе дает сырой амид оксим из формулы XI, который можно непосредственно использовать в последующей реакции. Циклизация амид оксима из формулы XI обработкой эфиратом трехфтористого бора и триэтоксиметаном дает оксадиазол из формулы XII с выходом более 90% в две стадии. В качестве альтернативы применения трехфтористого бора циклизацию можно также чисто выполнить, используя трифторуксусную кислоту в виде кислотного источника. Бромирование N-бромсукцинимидом в подходящем растворителе, например, дихлорметане, дихлорэтане или тетрахлорметане, с использованием инициатора, например, AIBN, дает соединение монобромооксадиазола формулы IVa. При необходимости избежать возможных смесей моно- и дибромидов функцию толуола в соединении формулы XI можно специально сверхбромировать N-бромсукцинимидом и AIBN, чтобы получить соответствующий дибромид, который затем можно восстановить диэтиловым фосфитом до получения монобромида из формулы VIa с выходом более 90%. Дибромирование и редуцирование можно выполнять в одном сосуде без выделения дибромида с общим выходом более 90%. Альтернативно,соединение формулы IVa можно также приготовить из соединения формулы XII избыточным броматом натрия и бисульфитом натрия в подходящей двухфазной смеси растворителей, например, этилацетат/вода, дихлорметан/вода, бутилацетат/вода, трифторотолуол/вода и т.п., чтобы получить смесь монои дибромидных интермедиатов, которая редуцируется на месте диэтилфосфатом/ диизопропиламином до получения монобромида формулы IVa. По желанию соединение формулы IVa можно без труда преобразовать в соединение формулы IV, в котором X -уходящая группа, с помощью методов, известных спе-6 016447 циалистам в данной области. В другом примере осуществления настоящее изобретение включает фармацевтические составы,включающие соединение формулы I в комбинации с фармацевтическим адъювантом, носителем или разбавителем. В следующем примере осуществления настоящее изобретение касается способа лечения нарушений, реагирующих на ингибирование -амилоидного пептида, у млекопитающих, нуждающихся в этом,который включает введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или его сольвата или гидрата. В следующем примере осуществления настоящее изобретение касается способа лечения, облегчения или отсрочки начала болезни Альцгеймера, церебральной амилоидной ангиопатии, системного амилоидоза, наследственного внутримозгового кровоизлияния с амилоидозом голландского типа, мультиинфарктной деменции, умеренных когнитивных нарушений и синдрома Дауна у нуждающегося в этом пациента, который включает введение указанному пациенту терапевтически эффективного количества соединения формулы I или его сольвата или гидрата. Для терапевтического применения фармакологически активное соединение формулы I будет, как правило, вводиться как фармацевтический состав, включающий в качестве определенного (или какоголибо) основного активного ингредиента, как минимум, одно такое соединение совместно с твердым или жидким фармацевтически приемлемым носителем и, функционально, с фармацевтически приемлемыми адъювантами и эксципиентами при использовании стандартных и традиционных методик. Фармацевтические составы включают пригодные лекарственные формы для перорального, парэнтерального (включая подкожное, внутримышечное, внутрикожное и внутривенное), трансдермального,подъязычного, бронхиального или назального введения. Таким образом, при использовании твердого носителя состав может быть таблетированным, помещен в твердую желатиновую капсулу в виде порошка или гранул, или может быть в форме пастилки или лепешки. Твердый носитель может содержать обычные эксцепиенты, например, связующие вещества, наполнители, таблетирующие смазывающие вещества, разрыхлители, увлажняющие средства и т.п. Таблетка, по желанию, может быть покрыта пленкой в соответствии с традиционными методиками. Составы для орального введения включают твердые[push-fit] капсулы из желатина, а также мягкие, со шкалой, капсулы из желатина и покрытие, как например, глицерин или сорбит. Твердые капсулы могут содержать активные ингредиенты, смешанные с наполнителем или связующими компонентами, например, лактоза или крахмалы, смазывающие вещества,например, тальк или стеарат магния, и, функционально, стабилизаторы. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, например, нелетучих жидких маслах, жидкости или жидком полиэтиленгликоле со стабилизаторами или без них. При использовании жидкого носителя состав может быть в виде сиропа, эмульсии, мягкой желатиновой капсулы,стерильного наполнителя для инъекции, водной или неводной жидкой суспензии, или состав может быть сухим продуктом, который восстанавливается в воде или другом подходящем наполнителе перед использованием. Жидкие составы могут содержать обычные добавки, например, суспендирующие агенты,эмульгирующие агенты, увлажняющие средства, неводный носитель (включая пищевые масла), консерванты, а также ароматизирующие и/или красящие вещества. Для парентерального введения носитель обычно включает стерилизованную воду, по меньшей мере в основном, хотя можно использовать физиологические растворы, растворы глюкозы и подобные средства. Инъецируемые суспензии также можно использовать, в этом случае можно применять обычные суспендирующие агенты. Обычные консерванты, буферные агенты и т.п. также можно добавлять к парэнтеральным лекарственным формам. Для локального или назального введения при составлении рецептуры используют пенетранты или проникающие агенты, которые соответствуют специфическому барьеру, через который проникают вещества. Такие пенетранты хорошо известны в данной области. Фармацевтические составы приготовлены по обычным технологиям, соответствующим нужному составу, содержащему адекватные количества активного ингредиента, то есть соединение формулы I в соответствии с настоящим изобретением. См., например,Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, 17th edition, 1985. Дозировка соединения формулы I для достижения терапевтического эффекта будет зависеть не только от таких факторов, как возраст, вес и пол пациента и способа введения, но также и от необходимой степени ингибирования А и активности соединения формулы I для конкретного рассматриваемого нарушения или болезни. Также предусматривается, что лечение и дозировка соединения формулы I можно проводить в дозированной лекарственной форме и что дозированная лекарственная форма может регулироваться соответственно специалистом для отображения относительного уровня активности. Решение в отношении конкретной применяемой дозировки (и число раз введения в сутки) принимает врач,и может изменяться титрованием дозировки в соответствии с конкретными обстоятельствами в соответствии с настоящим изобретением для достижения нужного терапевтического эффекта. Пригодная доза соединения формулы I или его фармацевтического состава для млекопитающего,включая человека, у которого наблюдается или может наблюдаться любое состояние, связанное с выработкой А пептида, как описано здесь, обычно суточная доза, составит примерно от 0,01 мг/кг до при-7 016447 мерно 10 мг/кг и предпочтительно, примерно 0,1-2 мг/кг при введении парэнтерально. Для перорального введения доза может быть в диапазоне от примерно 0,01 до примерно 20 мг/кг и предпочтительно от 0,1 до 10 мг/кг веса тела. Активный ингредиент будет предпочтительно вводиться в равных дозах от одного до четырех раз в день. Однако обычно вводится малая дозировка, и постепенно дозировка увеличивается,пока не будет определена оптимальная дозировка для объекта лечения. В соответствии с приемлемой клинической практикой предпочтительно вводить не требующее приготовления соединение при уровне концентрации, создающей эффективный антиамилоидный эффект, не вызывая никаких вредных или неблагоприятных побочных действий. Однако ясно, что количество фактически вводимого соединения будет определять врач, в свете сложившихся обстоятельств, включая состояние, лечение которого проводится, выбор вводимого соединения, выбранный способ введения, возраст, вес и реакцию конкретного пациента, а также серьезность симптомов у пациента. Биологические данные Трансактивация PXR Прегнан-Х-рецептор (PXR) - это ядерный гормональный рецептор, преимущественно ответственный за индукцию цитохрома Р 450 (CYP) 3A4, который играет главную роль в метаболизировании многих клинически предписанных препаратов. Известно, что индукция CYP3A4 может вызвать либо межлекарственное взаимодействие увеличением метаболического клиренса совместно вводимых CYP3A4 субстратов (Bertilsson, G. с соавт., Proc. Nat. Acad. Sci. USA (1998) 95: 12208-12213; Lehmann, J.M. с соавт., J,Clin. Invest. (1998) 102: 1016-1023) или может вызвать потерю экспозиции лекарственного средства вследствие автоиндукции. Характеризация индукционного потенциала открытия или разработки кандидатов лекарственных средств стала важным фильтром во всей фармацевтической промышленности. Анализ трансактивации PXR используется для оценки индукционного потенциала CYP3A4, а анализ цитотоксичности HepG2 клеток используется для контроля интерференции анализа вследствие цитотоксичности. Используемая клеточная культуральная среда - DMEM. Липофектамин 2000, PBS [фосфатносолевой буферный раствор], трипсин-EDTA (0,25%) и пенициллинстрептомицин приобретены уGIBCO/Invitrogen (Карлсбад, шт. Калифорния). Термоинактивированная эмбриональная бычья сыворотка (FBS) приобретена у фирмы Sigma (Сент Луис, шт. Миссури). Эмбриональная бычья сыворотка, обработанная активированным углем/декстраном (FBS), приобретена у фирмы Hyclone (Логан, шт. Юта).HepG2 клетки получены в АТСС (Манассас, шт. Вирджиния). Человеческий PXR-pcDNA3 и репортер люциферазы, содержащий CYP3A4 промотор, CYP3A-Luc, созданы компанией Bristol-Myers Squibb. Черные стандартные 384-луночные планшеты приобретены у BD Biosciences (Лексингтон, шт. Кентукки). Субстрат люциферазы (Steady-Glo) приобретен у фирмы Promega (Мадисон, шт. Висконсин). Контрольное соединение рифампицин приобретено у фирмы Sigma (Сент Луис, шт. Миссури). Культура HepG2 клеток производится в Т 175 склянках с использованием DMEM, содержащего 10%FBS. Трансфекционная смесь содержит 1 /мл PXR-pcDNA3 плазмидной ДНК, 20 /мл Сур 3 А-Luc плазмидной ДНК, 90 /мл липофектамина 2000 и бессывороточную среду. После инкубирования при комнатной температуре в течение 20 мин трансфекционная смесь (1 мл на склянку) добавляется к клеткам в свежей питательной среде (20 мл на склянку), и склянки инкубируются при 37 С (5% CO2) в течение ночи. Клетки в каждой склянке промывают с помощью PBS, добавляют 4 мл Трипсина-EDTA (0,25%) и инкубируют в течение одной минуты при комнатной температуре. Трипсин затем аспирируют и склянки инкубируют еще в течение пяти минут при комнатной температуре. По склянкам после этого энергично стучат, чтобы разбить скопления клеток. После добавления 10 мл DMEM, содержащего 5% обработанного активированным углем/декстраном FBS, вся смесь переносится в конические пробирки. Пул клеток в суспензии далее разъединяют пипетированием через наконечник пипетки 1 мл. После этого клетки считают и разбавляют в среде до 3,0105 клеток/мл. Пятьдесят л клеточной смеси добавляют в каждую лунку черного стандартного 384-луночного планшета, содержащего 0,25 л тест-соединения, растворенного в 100% DMSO. Планшеты инкубируют при 37 С (5% CO2) в течение ночи перед добавлением в каждую лунку 25 л субстрата люциферазы (Steady-Glo, Promega). Через пятнадцать минут планшеты считывают на Viewlux (Perkin-Elmer) планшет-ридере. В каждый планшет включен рифампицин (10 цМ), известный агонист PXR, в качестве внутреннего стандарта и положительного контроля. Данные затем были представлены как процент активации (%Act),где общий сигнал - это сигнал от М рифампицина, а сигнал гашения - это сигнал от DMSO наполнителя. Соединения проверяют при десяти концентрациях (50 М - 2,5 пМ, 1:3 серийное разведение), и XLFit (IDBS, Inc), используемом для подбора кривой. Известно о концентрациях, при которых происходит 20 и 60%-ая активация (ЕС 20 А и ЕС 60 А соответственно). Человеческий прегнан-Х-рецептор в основном отвечает за индукцию CYP3A4, а также CYP2B6,-8 016447CYP2C8/9, энзимы фазы 2, например, UGT, и несколько транспортеров, например, P-gp, MRP2 и ОАТР 2. Вышеприведенный тест продемонстрировал, что (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4 оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид имеет EC20A (20% реакции на рифампицин при 10 цМ) 6,9 М и EC60A (60% реакции на рифампицин при 10 М) более 16,7 М при анализеhPXR трансактивации. Эти результаты указывают на то, что (2R)-2-(4-хлорфенил)сульфонил]2-фторо 4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид может иметь потенциал, чтобы быть индуктором человеческого CYP3A4 путем активации hPXR. Индукция цитохрома Р 450 в Fa2N-4 клетках Способность(2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил] амино]-5,5,5-трифторпентанамида индуцировать CYP3A4 мРНК была оценена in vitro при помощи Fa2N4 клеточной линии. Fa2N-4 клетка - это иммортализованная линия человеческого гепатоцита, которая была создана с помощью Т антигена обезьяньего вируса 40 (SV40), как иммортализующего гена, при сохранении базовой экспрессии и индуцибильности нескольких CYP изоформ, включающих CYP3A4.Fa2N-4 клетки нанесли на 12-луночные, покрытые коллагеном планшеты с плотностью посева 0,67106 клеток на лунку. После приема клеток питательную среду заменили и клетки выдержали в течение ночи во влажном, заполненном CO2 инкубаторе. После этого адаптационного периода клетки проверили под оптическим микроскопом и установили морфологическое нормальное состояние клеток и их пригодность к использованию. В течение периода обработки клетки подвергали ежедневному визуальному осмотру; клеточная морфология, конфлуентность и симптомы токсичности, обусловленные тестируемым препаратом, регистрировали по мере необходимости. Клеточные культуры были обработаны 4 концентрациями (0,5, 2, 8, и 20 М при окончательной инкубации) (2R)-2-(4-хлорфенил)сульфонил]2 фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида и контрольным растворителем-наполнителем (0,1% DMSO) в трех повторностях. Рифампицин (10 М), прототипичныйCYP3A4 индуктор, использовали в качестве положительного контроля. Fa2N-4 клетки подвергли воздействию тестируемого препарата всего в течение 3 дней. Культуральную среду заменяли ежедневно свежей средой, содержавшей тестируемый препарат. Через 72 ч воздействия питательную среду отсосали, а клетки промыли один раз теплым забуференным фосфатом физиологическим раствором перед добавлением буфера для лизиса клеток (Qiagen, Валенсия, шт. Калифорния). Способность(2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил] амино]-5,5,5-трифторпентанамида индуцировать CYP3A4 мРНК экспрессию исследовали на Fa2N-4 иммортализованных человеческих гепатоцитах. Обработка Fa2N-4 гепатоцитов рифампицином (10 М) в течение 3 дней вызвала значительное (12-кратное) увеличение клеточной CYP3A4 мРНК экспрессии, что указывает на надлежащее функционирование клеток. Инкубация гепатоцитов при помощи (2R)-2-(4 хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида в течение 3 дней дала в результате зависящее от концентрации увеличение CYP3A4 мРНК экспрессии,с достижением 2,6-кратного значения по сравнению с контрольным наполнителем при самой высокой проверяемой концентрации (20 цМ). Эта величина индукции была эквивалентна 22% индукционной реакции, вызванной рифампицином, при предположении, что (2R)-2-(4-хлорфенил)сульфонил]2-фторо 4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид смог бы незначительно индуцировать CYP3A4 при концентрациях свыше 20 цМ. Относительно минимальная индукция CYP3A4 (2,0 кратной) наблюдалась при более низких концентрациях (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4(1,2,4-оксадиазол-3-ил)фенил]метил] амино]-5,5,5-трифторпентанамида (8,0 М). Метаболическая устойчивость в микросомах печени Микросомы печени мыши, крысы, собаки, человека и обезьяны циномолгус были получены от BDGentest (Вобурн, шт. Массачусетс). Количество в партии составило 13 (мышь), 8 (крыса), 8 (собака), 19 и 26 (человек), и 3 (обезьяна циномолгус). Окислительный метаболизм (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида изучили в микросомах печени при трех комплексах условий. Инкубационные смеси для человека и собаки (полный объем 3 мл, содержание органического растворителя 0,3%) содержали (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид (1 М), микросомальный белок (1 мг/мл), NADPH (1 mM), Трис хлорид буфер (100 mM, рН 7,4) и хлорид магния (3,3mM). Реакция, проведенная в трех повторностях, была инициирована добавлением NADPH с последующей инкубацией при 37 С в течение 50 минут. Аликвоты образцов (0,25 мл) брали на 0, 5, 10, 20, 30, 40 и 50 минуте, а реакцию погасили добавлением 3 объемов ацетонитрила. Инкубационные смеси для обезьяны циномолгус и крысы (полный объем 3 мл, содержание органического растворителя 0,3%) содержали(2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид (1 М), микросомальный белок (0,1 мг/мл), NADPH (1 mM), Трис хлорид буфер (100mM, рН 7.4) и хлорид магния (3,3 мм). Реакция, проведенная в трех повторностях, была инициирована добавлением NADPH с последующей инкубацией при 37 С в течение 50 минут. Аликвоты образцов (0,25-9 016447 мл) брали на 0, 5, 10, 20, 30, 40 и 50 минуте, а реакцию погасили добавлением 3 объемов ацетонитрила. Инкубационные смеси для мыши и обезьяны циномолгус (полный объем 3 мл, содержание органического растворителя 0,3%) содержали (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3 ил)фенил]метил]амино]-5,5,5-трифторпентанамид (1 М), микросомальный белок (0,1 мг/мл), NADPH (1mM), Трис хлорид буфер (100 mM, рН 7,4) и хлорид магния (3,3 mM). Реакция, проведенная в двух повторностях, была инициирована добавлением NADPH с последующей инкубацией при 37 С в течение 40 мин. Аликвоты образцов (0,25 мл) брали на 0, 5, 10, 15, 20, 30 и 40 минуте, а реакцию погасили добавлением 3 объемов ацетонитрила. Во всех исследованиях образцы были проанализированы сразу же методом LC/MS. Скорость исчезновения родителя рассчитали из соотношений площади пика (2R)-2-(4 хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида в каждый момент времени. Собственный клиренс печени (CLh, int, мл/мин/кг) (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида в разных видах вычислили на основании данных о микросоме печени с помощью метода, описанного в работе Houston JB., BiochemPharmacol Exp Ther (1997) 283:46-58. Скорость метаболизма in vitro определили для (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4 оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида при наличии NADPH-обогащнных микросом печени разных видов. Соединение в соответствии с настоящим изобретением (2R)-2-(4 хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид(1 М) было метаболизировано при расходе 690, 630, 40, 495 и 32 пкмоль/мин/мг белка при наличии микросом мыши, крысы, собаки, обезьяны циномолгус и человека соответственно. Когда скорости потребления(2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]5,5,5-трифторпентанамида были приведены в соответствие с клиренсом in vivo, предсказанные значения клиренса in vivo (серологического) (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил) фенил]метил]амино]-5,5,5-трифторпентанамида составили приблизительно 87, 52, 14, 39 и 8 мл/мин/кг для мыши, крысы, собаки, обезьяны циномолгус и человека соответственно. Эти данные указывают на то, что (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил] амино]-5,5,5 трифторпентанамид, как ожидается, - это соединение с низким клиренсом у человека. Фармакология in vitro Анализ связывания презенилина Анализы вытеснения радиолигандов при помощи [3 Н](R)-4-N-(1-амино-4-метил-1-оксопентан-2 ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил)бензамида были проведены с помощью метода,описанного ранее для [3 Н] (2R,3S)-2-изобутил-N1-S)-2-оксо-1-(3-феноксибензил)азепан-3-ил)-3 пропилсукцинамида (RE987, соединение A [Seiffert D, с соавт. Presenilin-1 and -2 are molecular targets for-secretase inhibitors -Презенилин-1 и -2 представляет молекулярные цели для ингибиторов -секретазы. J.Biol. Chem. (2000) 275 (44): 34086-34091]). Было проведено несколько экспериментов для подтверждения, что [3 Н] (R)-4-N-(1-амино-4-метил-1-оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2 метоксиэтил)бензамид связывается с -секретазой. Во-первых, специфическое связывание [3 Н] (R)-4-N(1-амино-4-метил-1-оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил) бензамида с мембранами от фибробластов дикого типа и PS-1/PS-2 нокаутных фибробластов показали, что [3 Н] (R)-4N-(1-амино-4-метил-1-оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил)бензамид специфически связывается с мембранами от фибробластов дикого типа, но не нокаутных фибробластов,и что со специфическим сигналом конкурируют структурно отличающиеся ингибиторы у-секретазы. Вовторых, фармакологию связывания [3 Н] (R)-4-N-(1-амино-4-метил-1-оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил)бензамида исследовали несколькими ингибиторами секретазы. Эти результаты показали, что вытеснение связывания [3 Н] (R)-4-N(1-амино-4-метил-1 оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил)бензамида с ТНР-1 мембранами хорошо согласуется с потенцией -секретазы, как при измерении с помощью прежних анализов связывания радиолигандов, так и ингибирования образования А образования в культивируемых клетках. ТНР-1 клетки были выращены в спинкультурах в RPMI 1640, содержавшем L-глутамин (Life Technologies Inc) и М -монотиогликоля до плотности клеток 5105/мл. Клетки собрали центрифугированием, и гранулы, содержащие 210 клеток, быстро заморозили сухим льдом/этанолом и хранили при -70 С до использования. В день проведения анализа клетки разморозили при 2108 клеток на 10 мл гомогенизационного буфера, состоящего из 50 mM HEPES рН 7,0, содержащего коктейль ингибитора протеазы из 104 М 4-(2-аминоэтил) бензилсульфонил фторид гидрохлорида, 80 nM апротина, 2 M лейпептина, 4M бестатина, 1,5 M пепстатина А и 1,4 M E-64 (0,1% коктейля ингибитора протеазы Р 8340, SigmaAldrich, Сент Луис, шт. Миссури) при 4 С. Клетки гомогенизировали с помощью Polytron (Brinkman,Вестбери, шт. Нью-Йорк) при 18000 об./мин в течение 15 с, затем центрифугировали при 18000 об.мин,4 С, 15 мин в центрифуге Sorval RC5B. Полученную гранулу ресуспендировали в буфере при 4 С до получения общей концентрации белка 5 мг/мл. Для использования в анализе связывания клеточный гомо- 10016447 генат разбавили до концентрации 300 г/мл в аналитическом буфере, состоявшем из 50 mM HEPES рН 7.0, 0,1% CHAPSO. Анализы были инициированы на полипропиленовых 96-луночных планшетах (Costar,Кембридж, шт. Массачусетс) добавлением 200 л клеточного гомогената к 50 л аналитического буфера,содержавшего 0,75 пМ радиолиганда ([3 Н] (R)-4-N-(1-амино-4-метил-1-оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил)бензамид, 110 Ки/ммоль) и различные концентрации немаркированных соединений, и инкубированы в течение 1 ч при 37 С. Конечная концентрация DMSO составила 1%. Выделение связи из свободного радиолиганда было выполнено фильтрацией на GFF стекловолоконных фильтрах (Innotech Biosystems International, Лансинг, шт. Мичиган) с помощью коллектора клеток отInnotech. Фильтры промыли три раза забуференным фосфатом физиологического раствора, 0,3 мл на лунку, рН 7,0 при 4 С, радиоактивность измерили жидкостным сцинтилляционным счтчиком Wallac 1450 Microbeta (PerkinElmer, Бостон, шт. Массачусетс). Ki значения конкурирующих соединений были получены при помощи коррекции Ченг-Пруссова [Cheng-Prussoff] значений IC50, рассчитанных с помощью XLfit (IDBS, Гилдфорд, Великобритания). Описанный выше анализ связывания радиолигандов использовали для измерения аффинности соединений к целевому сайту презенилина комплекса -секретазы. (2R)-2-(4-хлорфенил)сульфонил]2 фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид вызвал дозозависимое ингибирование связывания [3 Н] (R)-4-N-(1-амино-4-метил-1-оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил) бензамида с презенилином в ТНР-1 мембранах. Анализ кратных экспериментов определил Ki=0,48 nM0,24 nM (среднее значениеSD, n=8). Ингибирование образования A в культивируемых клетках Соединения подвергли анализу на ингибирование А 40 или А 42 в клетках с использованием Н 4 АРР 751 SWE клона 8.20, разработанного компанией Bristol-Myers Squibb, H4 нейроглиомной клеточной линии, устойчиво экспрессирующей шведский мутант АРР 751. Клетки выдержали в лог-фазе посредством пассирования дважды в неделю при разбавлении 1:20. Для определений IC50, 30 л клеток (1,5104 клеток/лунку) в среде DMEM, содержавшей 0,0125% BSA (Sigma A8412), высеяли непосредственно на 384-луночные планшеты (Costar 3709), содержавшие 0,1 л последовательно разбавленного соединения вDMSO. После инкубации в течение 19 ч в 5% CO2 при 37 С планшеты в течение короткого периода центрифугировали (1000 об/мин, 5 мин). Аликвоту 10 л из каждой лунки перенесли на второй аналитический планшет (Costar 3709) для измерений АР 40. Коктейли из антител, свежеприготовленные разбавлением 40 mM Трис-HCl (рН 7,4) 0,2% бычьим сывороточным альбумином (BSA), добавили в аналитические планшеты. Для измерений A42 антитела, специфические для А 42 неоэпитопа (565, разработкаA пептида (26D6, разработка SIBIA/Bristol-Myers Squibb; конъюгирован с АРС (Perkin Elmer, смешали и 20 л смеси добавили в каждую лунку планшета с инкубированными клетками до получения конечной концентрации 0,8 нг/лунку 565 и 75 нг/лунку 26D6. Для измерений А 40 антитела, специфические для А 40 неоэпитопа (TSD, разработка Bristol-Myers Squibb; конъюгирован с реагентом Wallac (PerkinElmer и 26D6 согласно вышеприведенному описанию смешали и 20 л смеси добавили к 10-л аликвотам, которые были ранее удалены из клеточного планшета, до получения конечной концентрации 1,6 нг/лунку TSD и 17,5 нг/лунку 26D6. Аналитические планшеты, содержавшие антитела, герметизировали алюминиевой фольгой и инкубировали в течение ночи при 4 С. Сигнал был определен с помощью счетчика Viewlux (Perkin Elmer), а значения IC50 определили путем подбора кривой в CurveMaster (на основе(2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5 трифторпентанамид эффективно ингибировал образование А 40 и А 42 в H4-8Sw клетках. Анализ кратных экспериментов показал IC50=0,30 0,15 nM (среднее значение SD, n=50) для ингибирования A40 иIC50=0,27 0,12 nM (среднее значениеSD, n=45) для ингибирования А 42. Ингибирование передачи сигналов Notch в культивируемых клетках Для передачи сигналов семейством Notch трансмембранных рецепторов требуется активность секретазы. Поскольку ингибирование передачи сигналов Notch вызывает нежелательные, основанные на механизме побочные эффекты, использовали клеточный анализ для передачи сигналов Notch1-Е, чтобы контр-скринировать ингибиторы у-секретазы. С помощью PCR [полимеразная цепная реакция] была создана структура экспрессии Notch1 мыши с использованием стандартных методик молекулярной биологии и проверена секвенированием. Эта структура была создана в pCDNA3.1+Hyg векторе (Invitrogen), модифицированном таким образом, чтобы содержал N-концевую 20 аминокислотную сигнальную последовательность и С-концевую 7 Х myc метку. Сигнальную последовательность получили от Notch1 мыши; 7 Х myc-метка была создана с помощью накладывающихся праймеров и субклонированием на HindIII/XhoI сайты pCDNA3.1 + Hyg вектора. Структура Notch1-Е мыши содержит сигнальную последовательность Notch1 мыши и M1727V мутацию в пределах трансмембранного домена для подавления инициирования внутреннего перемещения. Кодирующая последовательность Notch1-Е мыши от аминокислоты 1704 до 2193 была выбрана из биб- 11016447 лиотеки кДНК быстрого клона селезенки мыши (Clontech) и субклонирована в модифицированный вектор, содержащий 7 Х myc-метку и сигнальную последовательность в виде XbaI/HindII фрагмента.Hela клетки выдержали в DMEM (GibcoBRL), содержавшем 10% FBS (GibcoBRL), Пенициллин/Стрептомицин (GibcoBRL) и 2 mM L-глутамина (GibcoBRL). Клетки были временно трансфицированы с помощью TransIT-HelaMONSTER (Mirus) согласно указаниям изготовителя. Hela клетки (АТСС) высеяли за 16 ч до трансфекции с плотностью 4106 клеток на Т 175 склянку в среде для выращиванияHela (DMEM (высокая глюкоза с HEPES) с глутамином, пенициллином, стрептомицином и 10% зародышевой бычьей сывороткой). Клетки были трансфицированы в среде для выращивания: плазмида, содержащая 6 г Notch1-E мыши, плазмида, содержащая 15,6 г носителя (pCDNA3.1+hyg), плазмида, содержащая 14,4 г CBF1 (репортер люциферазы) при помощи HelaMonster реагента трансфекции (Mirus). Структура репортера CBF1-люциферазы состоит из 4 Х реплик CBF1 связывающего элемента до SV40 промотора (pGL3-промотор, Promega). Репортер CBF1-люциферазы был создан с помощью перекрывающихся праймеров для получения 4 Х CBF1 связывающей области. Этот фрагмент был субклонирован на NheI/BgIII сайты структур pGL3-Промотора. Целостность этой структуры была подтверждена секвенированием. Запасы ДНК разбавили в ТЕ буфере (10 mM Трис, 1 mM EDTA, рН 8,0) для трансфекции. Через пять -шесть часов после добавления ДНК клетки удалили из склянки с помощью Трипсин-EDTA и ресуспендировали в заданной среде (DMEM (высокая глюкоза с HEPES) с глутамином, пенициллином,стрептомицином, 0,0125% бычьим сывороточным альбумином и заменимыми аминокислотами) при концентрации 5104 клеток/мл. Клетки высеяли на 96-луночные черные планшеты Clearview (Packard) при объеме 200 л/лунку (1104 клеток) и инкубировали при 37 С в течение 1,5 ч, чтобы клетки прилипли к планшетам. Испытательные соединения сначала разбавили на 96-луночных полипропиленовых планшетах в 100% DMSO. Запасы соединения DMSO затем разбавили 47,6-кратно переносом на планшет, содержавший заданную среду с получением 2,1% DMSO концентрации. Разбавленные растворы соединения (10 л) добавили к клеточным планшетам с получением конечной 0,1% DMSO концентрации. Клеточные планшеты, содержавшие соединение, инкубировали в течение ночи при 37 С. После ночной инкубации питательную среду осторожно удалили и в каждую лунку добавили 25 л забуференного фосфатом физиологического раствора (с кальцием и магнием). Luc-Screen (Applied Biosystems) реагенты А и В смешали в равных пропорциях и в каждую лунку добавили 50 л смеси. Планшет инкубировали при комнатной температуре в течение 10 минут перед тем, как ко дну планшета прикрепили черную подложку, и сигнал считали на TopCount (Packard). Затем для определения значений IC50 подобрали концентрационные кривые при помощи нелинейной регрессии. Для передачи сигналов семейством Notch трансмембранных рецепторов требуется активность усекретазы. Поскольку ингибирование передачи сигналов Notch вызывает нежелательные, основанные на механизме побочные эффекты, использовали клеточный анализ для передачи сигналов Notch1-Е, чтобы контр-скринировать ингибиторы -секретазы. Анализ кратных экспериментов для ингибирования передачи сигналов Notch1-Е с помощью (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3 ил)фенил] метил] амино]-5,5,5-трифторпентанамида дал IC50=58 23 nM (среднее значениеSD, n=58). Основанная на клеточной активности для ингибирования A40 (0,3 nM) и Notch (58 nM), селективность Notch/APP для (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил] метил] амино]-5,5,5-трифторпентанамида составляет 193 Х (95% CI=163-232). Фармакология in vivo Виды A от животных измеряли с помощью сэндвич-анализов ELISA. Краткое описание этих анализов включено в настоящий документ, так как особенности эпитопов для отдельных антител определяют выявляемые виды A. A мыши и крысы имеют общую A последовательность, которая отличается от человеческого A. В результате этих отличий последовательностей антитела, распознающие Nконцевую область человеческого A, как например, 26D6, слабо связываются с A грызуна. Аналогично,антитела, которые сильно связываются с A грызуна, как например, 252, слабо связываются с человеческим A. Были разработаны два анализа для измерения A40 грызунов: 252-TSD и 4G8-TSD. С помощью анализа TSD-4G8 можно измерять не только A40, но и другие продукты расщепления ВАСЕ-секретазы (A11-40) и продукты расщепления -секретазысекретазы (Р 3). В табл. 1 представлена сводка анализов, представленных в настоящей заявке, и их использование. Таблица 1. Сводка пар антител, используемых для анализа образцов in vivo- 12016447 Точная локализация х неизвестна. Несмотря на то, что 252 распознает N-концевую область A,неизвестно, влияет ли аминоконцевое усечение A на связывание 252. Эта неопределенность вряд ли будет проблемой в случае с крысами, так как усечение N-конца встречается редко. Каждый из этих анализов был утвержден применением нескольких методов. Во-первых, к биологической матрице добавили изменяющиеся количества синтетического A, и увеличение сигнала сравнили с полученным с помощью синтетического A в буферном растворе. Во-вторых, A убавили в биологическом образце с помощью анти-A антител. В-третьих, проанализировали образцы от животных, которых лечили высокими дозами ингибитора -секретазы. Утвержденный анализ, эффективно определяющий экзогенно добавленный A ( 80% выздоровлений), имел значительно ослабленный сигнал после иммунного убавления A ( 80% выздоровлений по сравнению с неспецифическими контрольными группами), и имел сигнал, ослабленный до значений, приближающихся или перекрывающихся с минимальным уровнем анализа при использовании образцов от животных, которых лечили высокими дозами ингибитора -секретазы. Оптимизированные и утвержденные A анализы все еще содержали сигнал небольшой величины (5-20% контрольной группы, получающей носитель), который нельзя было убавить анти-A антителами или лечением ингибиторами -секретазы. Этот сигнал вряд ли является A и упоминается, следовательно, как минимальный уровень анализа. Минимальный уровень анализа не использовался для коррекции какого-либо из измерений A и, следовательно, значения, о которых сообщают, это, видимо, недооценки фактического объема ингибирования A.A40 использовали в качестве заменителя A42 in vivo. A40 приблизительно в 10 раз более богат биологическими образцами, чем A42, A40 - хороший заменитель для A42 на основании экспериментов в культивируемых клетках, где A40 и A42 одинаково ингибировались (2R)-2-(4-хлорфенил) сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамидом и другими ингибиторами -секретазы. Исследования на крысах На живом организме Самкам крыс Harlan Sprague-Dawley (200-250 г) ежедневно утром вводили определенными дозами с помощью ротового зондового питания носитель 99% PEG-400, 1% Tween-80 при 4 мл/кг. Дозирующие растворы приготовили один раз в начале исследования. Для растворения соединения в дозирующем растворе применили нагревание при 56 С и обработку ультразвуком. Все процедуры выполнялись в соответствии с руководящими указаниями ACUC. Окончательные пробы крови получили при помощи сердечной пункции после эвтаназии CO2 и собрали в EDTA пробирки. Плазму получили после центрифугирования. Мозговая ткань была иссечена, взвешена и заморожена сухим льдом до проведения анализа. Образцы CSF центрифугировали для удаления клеток или дебриса перед разбавлением 1:2 в 4% BSA и заморозили для последующего анализа. Гистопатологические образцы поместили в нейтральный буферный формалин до обработки. Образцы, собранные для заполнения, были заделаны в матрицу и заморожены при (-25 С) - (-30 С) в 2-метилбутановой ванне, а затем сухим льдом. Образцы плазмы на живом организме получили при помощи ретроорбитального кровопускания. Анализ Abeta40 мозга Мозг крысы (половина полушария) гомогенизировали с помощью политрона при 4 мл/г в PBS, рН 7,8, 2% CHAPS, полные ингибиторы протеазы (Roche). Крупный дебрис удалили центрифугированием в течение 30 минут при 20800 г и получившийся супернатант разбавили 1:2 в PBS, 2,5% BSA. Белые планшеты Microlite II ELISA (Thermo Electron) инкубировали с помощью 50 г/мл TSD9S3.2 антитела вPBS в течение 1 часа при 37 С. Планшеты блокировали с помощью 200 л 5%-ого бычьего сывороточного альбумина (BSA; вес/объем приготовлен в PBS) в течение 2 ч при комнатной температуре на планшетном шейкере, а затем промыли 5 раз с помощью 500 л/лунку PBS, 0,05% Tween-20. Осветленные мозговые гомогенаты загрузили 6 репликами, 50 л на лунку, и инкубировали в течение 1 ч при комнатной температуре. Планшеты промыли, как описано ранее, и затем инкубировали конъюгированным пероксидазой хрена (HRP) 252 антителом (Biosource), разбавленным 1:2000 в PBS, 0,05% Tween, 0,1% BSA в течение 1 ч. Три реплики содержали только 252-HRP антитело, а три реплики содержали 252-HRP антитело с 1 г/мл крысиного A1-14 (Anaspec), который конкурировал со специфически связанным антителом; этот фоновый сигнал вычли из общего сигнала и получили специфический сигнал. Связанное 252HRP антитело определили с помощью Pierce Supersignal Pico хемилюминесцентного субстрата в течение 10 минут и выразили количество с помощью счетчика Packard TopCount. Образцы были нормализованы до эталона мозгового гомогената, помещенного на каждый планшет. На основании A40 стандартных кривых LLQ составил 10 пг/мл, a LLD составил 20 пг/г ткани. Анализ A40 плазмы Планшеты покрыли TSD антителом и промыли как для A40 анализа мозга. Образцы плазмы разбавили 1:3 в PBS буфере, рН 7,8, 0,25% nonidet P40, 2,5% BSA. Образцы загрузили 6 репликами, 50 л на лунку, инкубировали в течение 1-2 ч при комнатной температуре. Образцы детектировали с помощью 4G8-биотина (Signet), разбавленного 1:8000 в PBS, 0,05% Tween, 0,1% BSA в течение 1 ч. Три реплики- 13016447 имели только антитело 4G8-биотина, а три реплики имели антитело 4G8-биотина с 1 г/мл A17-24, который конкурировал со специфическим сигналом и, таким образом создавал фоновое значение для каждого образца. После промывки, как описано выше, планшеты инкубировали стрептавидином-HRP(Zymed), разбавленным 1:50000 в PBS, 0,05% Tween, 0,1% BSA в течение 10 мин. Детекция и количественный анализ были выполнены, как и в A40 анализах мозга. Образцы были нормализованы до эталона плазмы, помещенного на каждый планшет. На основании A40 стандартных кривых LLQ составил 7,5 пг/мл, а LLD составил 23 пг/мл плазмы. Анализ Abeta40 CSF [цереброспинальная жидкость] Планшеты покрыли TSD антителом и промыли, как для анализа A40 мозга. Образцы CSF разбавили 1:10 в PBS, рН 7,8, 0,1% Tween-20. Во время сбора цереброспинальную жидкость предварительно разбавили 1:2 в 4% BSA в воде. Образцы загрузили в виде 3 реплик, 50 л на лунку, и инкубировали в течение 1-2 ч при комнатной температуре. Образцы детектировали с помощью 4G8-биотина (Signet),разбавленного 1:8000 в PBS, 0.05% Tween, 0,1% BSA в течение 1 ч. Поскольку фон был низкий, не было необходимости прогонять повторные образцы с конкурирующим пептидом для этого анализа. Связанный 4G8-HRP детектировали и количественно определили, как для анализа A40 плазмы, образцы были нормализованы до эталона CSF, помещенного на каждый планшет. На основании А 40 стандартных кривых LLQ составил 20 пг/мл, a LLD составил 400 пг/мл CSF. Заполнение сайта гамма-секретазы Мозговые секции были рассечены коронально на глубине 20 м на криостате и помещены после оттаивания на предметные стекла Superfrost Plus. Секции сохраняли на уровне рострального гиппокампа,по 3 секции на предметное стекло с интервалами примерно 120 м, и заморозили при -20 С до использования. Для исследования заполнения мозговые секции нагрели при комнатной температуре, высушили,инкубировали в течение 10 мин в 50 мм HEPES буфере, рН 7,4, затем перенесли в такой же буфер, содержащий 1,5 nM [3H]IN973 (Goldstein, М.Е. с соавт, J Pharmacol Exp Ther (2007) 323:102-108) или [3 Н](R)-4-(N-(1-амино-4-метил-1-оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил) бензамид и инкубировали при комнатной температуре в течение 10 мин. Чтобы определить неспецифическое связывание, смежные секции инкубировали в буфере, содержащем [3H]IN973 или [3 Н] (R)-4-N(1-амино-4-метил-1-оксопентан-2-ил)-4-хлорфенилсульфонамидо)метил)-N-(2-метоксиэтил) бензамид,но включающий немеченый ингибитор -секретазы при 0,5 М. После инкубации предметные стекла промыли три раза в течение 2 мин каждое в ледяном PBS, рН 7,2, погрузили в ледяную дистиллированную воду и высушили феном холодным воздухом. Предметные стекла поместили под чувствительные к тритию люминесцентные экраны с большим временем послесвечения (Amersham Biosciences, Арлингтон Хайтс, шт. Иллинойс) и выдержали в темноте в течение 7 дней. Изображения получили от экранов с большим временем послесвечения фосфора с помощью циклонного люминесцентного сканера (Packard,Мериден, шт. Коннектикут) и программного обеспечения сбора данных и анализа OptiQuant (Packard). Оптическую плотность (выраженная в виде цифровых световых единиц на квадратный миллиметр) на рассматриваемых площадях и измерили, и представили в виде процента от контрольной группы, получающей наполнитель. Методы патологической гистологии После эвтаназии при помощи CO2 из брюшной полости удалили секции проксимальной двенадцатиперстной кишки длиной примерно 3 см, промыли ледяным забуференным фосфатом физиологическим раствором и поместили в 10% буферный формалин перед изготовлением срезов. Секции ткани двенадцатиперстной кишки залили медицинским парафиновым маслом, закрепленным на блоках, и рассекли на три сигиттальные (перекрестно-люминальные) кольцевые секции и окрасили с помощью метода Шиффиодной кислоты перед микроскопической оценкой. Все животные были живы до выполнения запланированной аутопсии. Исследования непосредственных воздействий у крыс Однократная доза (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида при 30 мг/кг значительно снизила и A40 плазмы, и A40 мозга до уровней 2% и 16% контрольной группы, получающей наполнитель, соответственно. Для дальнейшего исследования влияния (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида на крыс однократную дозу 2R)-2-(4-хлорфенил)сульфонил]2 фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида ввели в расчете 1 мг/кг,10 мг/кг и 100 мг/кг, а ткани собрали через 2, 5, 8, 12 и 24 ч для измерений A40 плазмы, A40 мозга и заполнения сайта -секретазы. Эти измерения показали, что 10 мг/кг и 100 мг/кг снизили A40 плазмы иA40 мозга до менее 25% базовых значений A40 от 2 до 24 ч после введения доз. Напротив, 1 мг/кг не вызвал статистически значимого изменения A40 мозга, но вместо этого вызвал переходное повышениеA40 плазмы. В частности, A40 плазмы повысился до 250% от исходных значений в течение 8 ч перед возвратом к базовым значениям в течение 24 ч. Заполнение сайта -секретазы показало, что более 94% сайтов связывания ингибитора -секретазы в мозге были заполнены в продолжение интервала введения- 14016447 доз после дозы 100 мг/кг и в общем, кроме момента времени 24 ч (88%), после дозы 10 мг/кг. Напротив,только 75% сайтов связывания -секретазы мозга были заполнены через два часа после дозы 1 мг/кг и заполнение сайта постепенно вернулось до базовых значений в течение 24 ч. Результаты исследований непосредственных воздействий у крыс показали, что (2R)-2-(4 хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид снизил A40 плазмы, A40 мозга и A40 спинномозговой жидкости при ED50 в интервале от 1 мг/кг до 10 мг/кг. Исследования субхронических воздействий у крыс На основании воздействий (2R)-2-(4-хлорфенил) сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил) фенил]метил]амино]-5,5,5-трифторпентанамида на А 40 мозга крысы, которые описаны в предыдущем разделе, крысам вводили ежедневно дозы 3, 10, 30 или 100 мг/кг/день. Животных подвергли эвтаназии спустя 12 или 24 часа после третьей дозы или спустя 5 ч после четвертой дозы. Показатели сниженияAUC A40 мозга определили с помощью линейной экстраполяции значений, полученных от животных через 5, 12 или 24 ч после введения доз в группе с такими же дозами, как представлено в табл. 2. На основании этого анализа снижения AUC A40 составили в среднем 53% при 3 мг/кг/день и возросли более чем на 85% при дозах 10 мг/кг/день или выше. Уровни соединения измерили несколько раз, чтобы оценить значения AUC соединения (0-24 ч) в начале и в конце эксперимента. Эти значения показали, что воздействия соединения после 3 доз были в пределах превышения в два раза воздействий после начальной дозы. Гистологическая оценка дуоденальной ткани от этих крыс показала, что у 1/3 крыс, которым вводили дозы 100 мг/кг/день, наблюдались умеренные патологические изменения. Таблица 2. Сводка результатов 4-дневных исследований на крысах Значения представляют средние уровни А 40 мозга от n=3 животных, выраженных относительно среднего значения контрольной группы, получающей наполнитель.b Снижение значений AUC A40 мозга определены с помощью линейной экстраполяции значений,полученных от животных через 5, 12 или 24 ч после введения доз в группе одинаковой дозы. с Значения представляют средние AUC (Мч или нгч/мл), n=3 животных. Значения AUC были определены на образцах, взятых на 5, 12 и 24 ч после введения дозы 1 (исследование 1), или на образцах,взятых на 5, 12, 17 и 24 ч после введения дозы 1 (исследование 2).d Значения представляют средние AUC Мч или нгч/мл), n=3 животных. Значения AUC были определены на образцов, взятых на 12 и 24 ч после введения дозы 3 и 5 ч после дозы 4 (исследование 1), или на образцах, взятых на 12 и 17 и 24 ч после введения дозы 3 и 5 ч после введения дозы 4 (исследование 2). е) Метаплазия бокаловидной клетки отмечена у одного из 3 животных NAD=нарушения не обнаружены. Результаты исследований субхронических воздействий у крыс показали, что (2R)-2-(4-хлорфенил) сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид снизилA40 мозга, не вызывая гастроинтестинальной токсичности. В частности, доза 3 мг/кг/день вызвала примерно 50%-ое снижение AUC A40 мозга по сравнению с дозой 100 мг/кг/день, которая необходима для- 15016447 индуцирования гастроинтестинальной токсичности. Микроскопия проксимальной двенадцатиперстной кишки у отдельного животного в группе высокой дозы (1 из 3 животных оценивали при этом уровне дозы), которым давали 100 мг/кг/день (2R)-2-(4 хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида один раз ежедневно в течение 4 дней выявила небольшую метаплазию бокаловидной клетки у этого животного. Никаких изменений, связанных с лекарством изменений со стороны двенадцатиперстной кишки у крыс, которым давали (2R)-2-(4-хлорфенил) сульфонил]2-фторо-4-(1,2,4-оксадиазол-3 ил)фенил] метил] амино]-5,5,5-трифторпентанамид при дозе 30 мг/кг, не было отмечено. В другом эксперименте крысам дозированно ежедневно вводили в течение либо 4, либо 7 дней 30 мг/кг/день или 300 мг/кг/день (табл. 3). Уровни A40 мозга в конце (5 ч после введения дозы) составили 13% от контрольной группы, получающей наполнитель, и таким образом, сходны с предыдущими исследованиями у крыс с применением (2R)-2-(4-хлорфенил) сульфонил]2-фторо-4-(1,2,4-оксадиазол-3 ил)фенил]метил]амино]-5,5,5-трифторпентанамида. Уровни соединения в плазме были одинаковыми через 5 часов после введения первой и четвертой дозы. Некоторые крысы в группе дозы 300 мг/кг/день (4 из 5) показали гастроинтестинальную токсичность после 4 дней дозированного введения, но ни у одной из крыс в других лечебных группах не была выявлена гастроинтестинальная токсичность, включая животных, которым дозы вводили в течение 7 дней. Таблица 3. Сводка результатов исследования на крысах 3N=протестированы 5 животных/группа дозирования/момент окончания (день 4 или день 7). Метаплазия бокаловидной клетки отмечена у 4 из 5 животных. NAD=нарушения не обнаружены. Описанные выше результаты подтверждают, что (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид является мощным и селективным ингибитором -секретазы. Эти результаты поддерживают использование (2R)-2-(4-хлорфенил) сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида в качестве терапевтического лечения болезни Альцгеймера и других нарушений, ассоциированных с -амилоидным пептидом. Следующие примеры приведены в целях иллюстрации и не должны рассматриваться как ограничивающие изобретение каким-либо образом, поскольку в пределах объема изобретения возможны варианты изобретения. Соединения в соответствии с настоящей заявкой можно приготовить многими способами, известными специалистам в области органического синтеза. Соединения в соответствии с настоящей заявкой можно синтезировать при помощи методов, описанных ниже, вместе с синтетическими методами, известными специалистам в области в синтетической органической химии, или их вариантов по выбору специалистов. Предпочтительные методы включают, но не ограничиваются этим, описанные ниже методы. Вся библиография, приведенная в настоящем описании, включена во всей полноте путем ссылки. Соединения можно приготовить при помощи реакций и технологий, описанных в этом разделе. Реакции выполняются в растворителях, подходящих для используемых реагентов и материалов и пригодных для производимых трансформаций. Кроме того, из описания синтетических методов, приведенных ниже, ясно, что все предложенные условия реакции, включая выбор растворителя, реакционные атмосферы, температуру реакции, продолжительность эксперимента и процедуры обработки, выбраны как стандартные условия для реакции, которые должны быть знакома специалисту в данной области. Специалисту в области органического синтеза ясно, что функциональные свойства, присутствующие на различных частях молекулы, должны быть совместимы с предлагаемыми реагентами и реакциями. Такие ограничения в отношении заменителей, которые совместимы с условиями реакции, очевидны специалисту и, следовательно, должны применяться альтернативные методы. Описание конкретных примеров осуществления изобретения В следующих примерах все значения температуры приведены в градусах Цельсия. Значения температуры плавления зарегистрированы на капиллярном приборе для определения точки плавления ThomasScientific Unimelt и нескорректированы. Спектры протонного магнитного резонанса (1 Н NMR [ЯМРядерный магнитный резонанс]) регистрировали на спектрометре Broker Avance 300, Broker Avance 400 или Broker Avance 500. Все спектры определены в указанных растворителях, химические сдвиги указаны- 16016447 вединицах в сторону слабого поля от тетраметилсилана внутреннего стандарта (TMS), а межпротонные постоянные взаимодействия в герцах (Hz). Мультиплетные структуры определяются следующим образом: s, синглет; d, дуплет; t, триплет; q, квартет; m, мультиплет; br, размытый максимум; dd, дублет дублетов; br d, широкий дублет; dt, дублет триплетов; br s, широкий синглет; dq, дублет квартетов. Инфракрасные (IR) спектры с помощью пленки бромистого калия (KBr) или хлористого натрия определены на спектрометре Perkin Elmer 2000 FT-IR от 4000 см-1 до 400 см-1, калиброваны на 1601 см-1 поглощение полистирольной пленки и указаны в обратных сантиметрах (см-1). Оптические вращения []D определены на поляриметре Rudolph Scientific Autopol IV в указанных растворителях; концентрации приводятся в мг/мл. Масс-спектры с низкой разрешающей способностью (MS) и видимый молекулярный (МН+) или(М-Н)+ определены на Finnegan SSQ7000. Масс-спектры с высокой разрешающей способностью определены на Finnegan MAT900. Жидкостная хроматография (LC)/масс-спектры выполнены на Shimadzu LC,соединенным с Water Micromass ZQ. Использована следующая аббревиатура: DMF (диметилформамид); THF (тетрагидрофуран); DMSO диметилсульфоксид, Leu (лейцин); TFA (трифторуксусная кислота); МТВЕ (метилтретбутилэфир); DAST Дихлорметан (4.2 L) загрузили в четыре 20-литровые колбы с горлышком и круглым дном, снабженные ванной с механическим перемешиванием и охлаждением. Начали перемешивание и реакционную смесь охладили до 0 - минус 2 С. Загрузили 4,4-трифторобутанол (750,0 г) и реакционную смесь охладили далее до минус 5 - минус 8 С. TEMPO; добавили (2,2,6,6-тетраметил-1-пиперидинилокси, свободнорадикальный) (9,15 г) при поддержании температуры между минус 5 - минус 8 С. Водный раствор бромистого калия (60 г в 1,17 л воды) добавили к описанному выше раствору, температуру поддерживали при минус 5 - минус 8 С. Водный раствор NaOCl (8,8 л, 6-7 вес.%, забуференный до рН=8,5 с помощью бикарбонат натрия), добавили к реакционной смеси (предостережение: экзотермическая реакция) при поддержании температуры реакционной смеси при -5 С. Аналогично этому, йоднокислый натрий(NaIO4) может заменить NaOCl в качестве окисляющего агента. После полного введения слой дихлорметана отделили, а водный слой промыли дихлорметаном (1750 мл). Слои дихлорметана объединили и высушили с помощью безводного сульфата натрия. Осушитель профильтровали и концентрацию раствора 4,4,4-трифторобутанала определили с помощью ЯМР. Раствор, содержащий титульное соединение,использовали непосредственно на следующей стадии без дополнительной обработки. 1 Н NMR (CDCl3)Rметил бензил амин (528,5 г) загрузили в соответствующий сосуд, оснащенный механическим перемешиванием, ванной для охлаждения и содержащийся под слоем азота. Загрузили раствор 4,4,4 трифторбутиральдегида (со стадии А, 550 г), а затем метанол (3,3 л). Реакционную смесь затем охладили приблизительно до 0 - минус 3 С. Уксусную кислоту (ледяную, 260 мл) добавили по каплям при сохранении температуры около 0 С, а затем добавили триметилсилильный цианид (581 мл) в течение 15 мин. Аналогично этому в качестве источника цианида можно использовать цианистый натрий (NaCN) или цианистый калий. Реакционную смесь нагрели до 25-27 С и перемешивали в течение ночи. Завершение реакции определили с помощью TLC [тонкослойная хроматография]. Охлажденную воду (10,0 л) загрузили в реакционную смесь, а реакционную смесь экстрагировали с помощью дихлорметана (110.0 л). Слой дихлорметана промыли водой (210,0 л), а затем соляным раствором (15,0 л). Слой дихлорметана высушили на безводном сульфате натрия и концентрировали при пониженном давлении с получением титульного аминонитрила (смесь диастереомеров в виде вязкой жидкости, средний выход продукта 90%. 1 Н NMR (CDCl3) (400 MHz)1.42 (dm, 5 Н), 2.152.35 (два m, 1 Н каждый), 3.10 - 3.20 (m, 1H), 4.104.15 (m, 1 Н), 7.10 -7.35 (m, 6 Н). Стадия С. 5,5,5-Трифторо-2-(1-фенилэтиламино)пентанамид (смесь диастереомеров)- 17016447 5,5,5-Трифторо-2-(1-фенилэтиламино)пентанамид (сырая смесь диастереомеров стадии В, 1,10 кг) растворили в дихлорметане (5,5 л) в соответствующем сосуде, оснащенном механическим перемешиванием, ледяной ванной для охлаждением и содержащимся под слоем азота. Начали перемешивание и реакционную смесь охладили до 0 - минус 5 С. Концентрированную серную кислоту (1,75 л) добавили по каплям в течение 1 ч в описанную выше смесь, поддерживая температуру ниже 0 С; после завершения процесса добавления получился прозрачный раствор. Температуру реакционной смеси повысили до 2527 С и перемешивали в течение ночи (12-14 ч). Завершение реакции определили с помощью HPLC. Реакционную смесь медленно вылили на измельченный лед (15,0 кг) и нейтрализовали нашатырным спиртом (25 об.%). Водный слой отделили и экстрагировали с помощью дихлорметана (23,0 л). Объединенный слой дихлорметана промыли водой (112,0 л), а затем соляным раствором (13,0 л). Богатый продуктом органический слой высушили на сульфате натрия и концентрировали при пониженном давлении с получением 0,85 кг (72,0%) сырого титульного соединения 1 Н NMR (CDCl3) (400 MHz) (смесь диастереомеров)1.36 (dm, 4 Н, СН 3 (J=8.0 Hz 1 Н из СН 2), 1.90 (m, 1H из СН 2), 2.152.35 (два m, 1 Н каждый из CH2-CF3), 2.80 - 2.90 (m, 1H, CH-Ph), 3.60 - 3.70 (m, 1H, -(CONH2)CH(NH), 5.906.45 (1 Н каждый из CONH2 с минорными пиками для других диастереомеров), 7.20 - 7.40 (m, 6H, Ar + NH). Стадия D. (R)-5,5,5-трифторо-2-R)-1-фенилэтиламино)пентанамид гидрохлорид 5,5,5-трифторо-2-R)-1-фенилэтиламино)пентанамид (смесь диастереомеров) (850 г) загрузили в соответствующий сосуд, оснащенный механическим перемешиванием и ванной для охлаждения. Загрузили метанол (2,55 л), этилацетат (1,7 л) и воду (1,06 л), реакционную смесь охладили до 0 - 5 С. РастворHCl в диоксане (4,5 М, 1,72 л) добавили по каплям в течение 30-45 мин. Аналогично этому в качестве растворителя можно использовать смеси изопропанола и метил трет-бутил эфира, а водный или концентрированный HCl можно использовать в качестве источника HCl, температуру реакционной смеси повысили до 25-27 С и перемешивали в течение 2 ч. Завершение реакции определили с помощью TLC. Осажденные твердые вещества профильтровали и осадок промыли соответствующим растворителем, например, этилацетатом (1,8 л), а затем петролейным эфиром (2,5 л), или смесью изопропанола и метил трет-бутил эфира. Твердое вещество оставили для высушивания при температуре окружающей среды на открытом поддоне и получили титульный R-амино амид (480 г, 50% выход, диастереоизомерный избыток=99,9 %). 1 Н NMR (CDCl3) (400 MHz)1.73 (d, 3H,CH3, J=8.0 Hz), 2.08 - 2.09 (m, 2H из СН 2), 2.20 - 2.40 (m, 2H, CH2-CF3), 3.50 - 3.55 ( m, 1 Н, CH-Ph), 4.40 4.41 (m, 1H, -(CONH2)CH(NH), 7.48 - 7.53 (br s, 5H, Ar). Стадия Е. (R)-2-амино-5,5,5- трифторпентанамид гидрохлорид В соответствующий сосуд, работающий под давлением, загрузили вместе с метанолом (15,0 л) (R)5,5,5-трифторо-2-R)-1-фенилэтиламино)-пентанамид гидрохлорид (1,50 кг). После этого добавили воду(701,0 мл), а затем 20% гидроокись палладия на углероде (225 г). Аналогично в качестве гидрогенизирующего катализатора можно использовать палладий на углероде (Pd/C). Сосуд промыли азотом три раза, а затем в сосуд наддули газообразный водород (3-4 кг/см 2) при 60 С. Контроль окончания реакции проводился с помощью HPLC. После завершения реакционную смесь оставили для охлаждения до 3035 С и профильтровали через целитную подложку, а затем промыли метанолом. Фильтрат после этого концентрировали при пониженном давлении. После полной концентрации оставшуюся реакционную смесь обработали дихлорметаном (2,5 л на промывку), отфильтровали и высушили при 45 С в течение 12 часов, в результате получили титульное соединение (915 г, 91,0%; чистота=97%). 1 Н NMR (DMSO-d6)- 18016447 Выходы продукта были увеличены за счет добавления вспомогательных ферментов для превращения пирувата в лактат. Для лактатдегидрогеназы в качестве кофермента требуется НАДН [NADH]. НАДН был восстановлен с помощью формиатдегидрогеназы. Раствор, содержащий 5,5,5-трифторо-2 левулиновую кислоту (100 мг, 0,588 ммоль), D, L-аланин (200 мг, 2,244 ммоль), 0,02 mM пиридоксаль фосфата, формиат натрия (68 мг, 1 ммоль), NAD (3,31 мг, 5 моль) L-лактатдегидрогеназу, клонированную из мышцы кролика (Biocatalytics LDH-103, 0,107 мг, 15 единиц), и формиатдегидрогеназу (0,5 мл, 15 единиц, клонированных из Pichia pastoris и экспрессированных в Escherichia coli) в 0,1 М буфере фосфорнокислого калия, рН 7,5, инкубировали R-трансаминазой АТ-103 производства Biocatalytics (5 мг, 44 единицы) или клонированной R-трансаминазой от Bacillus thuringiensis SC16569 (0,5 мл, 10 единиц) при 30 С в полном объеме 5 мл в 15-мл пробирках. Выходы реакции (R)-5,5,5-трифторо-2-аминопентановой кислоты 94% и 91% были получены с помощью АТ-103 и BMS трансаминаз соответственно. Э.и. составил 100% в обоих случаях. Метод В. Дегидрогеназа (R)-аминокислотная (производства Biocatalytics и BMS). Процедура. 1. 5,5,5-трифторо-2-левулиновую кислоту (60,00 г, 0,353 моль), NH4Cl (64,19 г, 1,2 моль), глюкозу (86,4 г, 0,479 моль) и воду (975 мл) загрузили в 2-L реактор с рубашкой. Добавили NaOH(36 мл из 10 N) и смесь перемешали с помощью магнита при 30 С для растворения твердых веществ. Значение рН было равно примерно 7. Добавили Na2CO3 (12,72 г, 0,12 моль), после чего значение рН стало примерно 8,5. Затем добавили НАДФ [NADP] (458 мг, 0,60 ммоль), глюкозодегидразу (33,7 мг, 5277 единиц производства Amano Enzyme Company) и R-аминокислотную дегидразу (600 мг D-AADH-102,производства Biocatalytics) в указанном порядке. Реакционную смесь довели до рН 9 добавлением по каплям 10 N NaOH. Реакционную смесь перемешали при 30 С и выдержали при рН 9,00 добавлением 5(36 мл из 10 N) и смесь перемешали с помощью магнита при 30 С для растворения твердых веществ. Значение рН было равно примерно 7. Добавили Na2CO3 (12,72 г, 0,12 моль), после чего значение рН стало примерно 8,5. Затем добавили НАДФ [NADP] (458 мг, 0,60 ммоль), глюкозодегидразу (33,7 мг, 5277 единиц производства Amano Enzyme Company) и D-аминокислотную дегидразу (50 мл экстракта, содержащего 1500 единиц, BMS энзим) в указанном порядке. Реакционная смесь была приведена до рН 9 добавлением по каплям 10N NaOH. Реакционную смесь перемешали при 30 С и выдержали при рН 9,00 добавлением 5N NaOH из рН-стат. Через 15 часов выход раствора (R)-5,5,5- трифторо-2- аминопентановой кислоты составил 51,04 г, 84,7% выход, 99,1% э.и. Приготовление С. 4-(Бромметил)-3-фторбензонитоил Метод A. NBS/AIBN бромирование. В соответствующий сосуд загрузили 1,2 дихлорэтан (151 кг) вместе с 4-циано-2-фтортолуолом (24 кг) и AIBN (2 кг). Смесь нагрели до 7074 С. Как только температура загрузки достигла 70 С, частями добавили N-бромсукцинимид (47,4 кг) при расходе 12 кг/ч при поддержании температуры 7074 С (важно контролировать расход во время добавления во избежание экзотермической реакции). Пробу смеси отобрали путем GC детекции после добавления 24 кг N-бромсукцинимида, температуру реакции поднимали до 70-74 С, пока не была определена завершенная реакция. Смесь охладили до 0-5 С и выдержали еще в течение 2 ч. Смесь профильтровали и кек промыли с помощью МТВЕ (24 кг). Фильтрат промыли водой (365 кг). Органический слой высушили сульфатом натрия (10,3 кг) в течение 6 ч, профильтровали и кек промыли с помощью МТВЕ (24 кг). Раствор испарили под пониженным давлением, добавили этанол (12 кг) и смесь нагрели до 40-45 С, затем охладили медленно до 0-5 С при перемешивании до кристаллизации. Смесь профильтровали и кек промыли холодным этанолом (5 кг). Сырое твердое вещество перекристаллизовали из петролейного эфира, профильтровали и промыли петролейным эфиром (10 кг), в результате получили титульное соединение 4-(бромметил)-3- флорбензонитрил в виде желтоватого твердого вещества (21 кг, выход 55%). 1 Н NMR (300 MHz, CDCl3)ppm 4.46 - 4.50 (m, 2 H) 7.36 (dd, J=8.85,1.32 Hz, 1H) 7.44 (dd, J=7.91, 1.32 Hz, 1H) 7.52 (dd, J=7.91, 7.16 Hz, 1 H). 13C NMR (75 MHz, CDCl3)ppm 23.65 (d, J=4.60 Hz, 1C) 113.76 (d, J=9.77 Hz, 1C) 117.09 (d, J=2.87 Hz, 1C) 119.44 (d, J=24.71 Hz, 1C) 128.44 (d, J=4.02 Hz, 1C) 130.66 - 130.81 (s, 1C) 130.81 - 131.06 (s, 1C) 132.18 (d, J=3.45 Hz, 1C) 159.86 (d,J=254.03 Hz, 1 С). IR: (KBr) 3088, 3077, 3040, 2982, 2250, 1571, 1508, 1439, 1248 cm-1. Рассчитано аналитическим методом для C8H5BrFN: расчет C 44.89; Н 2.35; N 6.54; F 8.88; определено: С 44.94; Н 2.73; N 6.56; F 8.73. Метод В. Бромирование броматом натрия.- 19016447 В соответствующий реактор добавили дихлорметан (40 л) и 3-фторо-4-метилбензонитрил (4 кг, 18,7 моль), а затем раствор бромата натрия в воде (13,45 кг, 89,1 моль, растворенных в 53,6 л воды). Реакционную смесь охладили до 0-5 С. Раствор гидросульфита натрия (9,25 кг, растворенного в 42 л воды) добавили в течение 2-3 часов при поддержании температуры загрузки 10-20 С (реакция экзотермическая). После завершения процесса добавления на реакторе загорелась лампа на 200 Вт и температура загрузки повысилась до 25-30 С. Свет и температура сохранялись до тех пор, пока продукт не составил 70-75% с помощью HPLC. Свет убрали, перемешивание прекратили и реакцию оставили для оседания на 15 минут. Органический слой удалили, а оставшийся водный слой экстрагировали дихлорметаном дважды. Органические слои объединили и промыли четыре раза 10% раствором тиосульфата натрия. Затем органический слой промыли соляным раствором (10 л) и высушили сульфатом натрия. Органический слой концентрировали, а затем добавили петролейный эфир и дистиллировали до сухого состояния дважды для удаления всего дихлорметана. Добавили петролейный эфир (3 л) и суспензию охладили до 5-10 С в течение 1 ч. Суспензию профильтровали и промыли холодным петролейным эфиром. Продукт высушили в вакуумной печи при 40-45 С и получили титульное соединение (3,2 кг, 50,4% выход) в виде желтоватого твердого вещества. Типовая процедура для восстановления титульного соединения из маточного раствора; сырую массу (36% 4-(бромметил)-3-флорбензонитрил и 59% гем-дибромид), полученную после концентрации маточного раствора (300 г) и 2 эквивалентов диизопропил -этиламина (на основании гем-дибромида) растворили в ацетонитриле (3 л) и воде (50 мл). Реакцию охладили до 0-5 С и добавили диэтилфосфит(169 г, 1.22 моль) в течение 30 мин (добавление было экзотермическим). Реакцию перемешали в течение 60-90 минут при 0-5 С, мониторинг осуществлялся с помощью TLC, Когда по показаниям TLC наличие дибромида больше не отмечалось, добавили воду (3,3 л) и получившуюся суспензию профильтровали. Фильтрационный кек промыли водой и высушили в вакуумной печи (до достижения влажности 1%) с получением 202 г (98 АР с помощью HPLC - высокоэффективная жидкостная хроматография) дополнительного титульного соединения. Приготовлеиие D. Приготовление 3-фторо-N'-гидрокси-4-метилбензимидамида. В соответствующий сосуд, оснащенный механической мешалкой в атмосфере N2, загрузили 202,0 г 3-фторо-4-метилбензонитрила, а затем 1,0 л этанола. Гидроксиламин (144 мл 50% раствора в воде) добавили через дополнительную воронку в течение 20 мин. Смесь перемешали при температуре окружающей среды, пока HPLC анализ не показал, что реакция завершилась (исходного материала не осталось). Воду(3,0 л) добавили по каплям к светло-желтому раствору в течение 1 ч до получения густой суспензий. Суспензию охладили в ледяной водяной бане до 2 С в течение 1,5 ч, профильтровали и высушили в вакууме при 35 С в течение 22 ч и получили тигульное соединение (230,3 г. 91,6%) в виде белого твердого вещества. 1H-NMR (CDCl3, 500 MHz) 7.30 (m. 2H), 7.19 (m, 1H), 4.87 (s, 2H), 2.28 (s, 3H); 13C-NMR(CDCl3, 500 MHz) 162.15, 160.19, 151.58, 131.82, 131.76, 131.66, 131.62, 127.01, 126.87. 121.06. 112.69,112.51, 14.48; 19F-NMR (CDCl3, 500 MHz) -116.35,-116.37,-116.39. LC-MS М-Н 169.19. Приготовление E. Приготовление 3-(3-фторо-4-метилфенил)-1,2,4-оксадиазола. Метод А. Трифторидбора. Сырой амид оксим 3-фторо-N'-гидрокси-4-метилбензимидамид (118.6 г) и триэтоксиметан (292 мл,260 г, 1,76 моль) суспендировали в дихлорметане (800 мл) и добавили диэтилэфират трифторидабора(14,8 мл, 16,6 г, 0,12 моль) при комнатной температуре. Получившийся желтый раствор нагрели до 45 С в течение 1 ч и выдержали при комнатной температуре в течение 16 ч, что обеспечило 60% конверсию с помощью HPLC. Раствор нагрели до 45 С в течение 2,5 ч, что обеспечило конверсию до 1% остаточного исходного материала. После охлаждения до комнатной температуры к смеси добавили 1N HCl (600 мл). Фазы разделили, а водный слой экстрагировали дихлорметаном (200 мл). Объединенный органический слой высушили на сульфате натрия и концентрировали при 25 С при пониженном давлении до получения белого твердого вещества. После высушивания под высоким вакуумом в течение 16 ч получили титульное соединение (102,3 г, 98 % выход в течение двух стадий). Метод В. Трифторуксусная кислота. В соответствующий сосуд, оснащенный механической мешалкой в атмосфере N2, загрузили 3 фторо-N'-гидрокси-4-метилбензимидамид (302,52 г, 1,79 моль), триэтоксиметан (382,5 мл, 341,1 г, 2,3 моль) и ацетонитрил (1512 мл). Реакционную смесь нагрели до 45 С и добавили трифторуксусную кислоту (6,72 мл, 10,08 г, 87,6 ммоль) при этой температуре. Реакционную смесь нагревали до 60 С в течение еще 90 мин, затем охладили до комнатной температуры. Добавили по каплям воду (3 L) в течение 60 мин. Суспензию охладили до 4 С и перемешали при этой температуре в течение 30 мин. Твердое вещество профильтровали и высушили в вакууме при 50 С в течение 12 ч и получили 3-(3-фторо-4 метилфенил)-1,2,4-оксадиазол в виде белого твердого вещества (306 г, 95,6%). HPLC анализ показал химическую чистоту 99,8%. 1H-NMR (CDCl3, 400 MHz) 8.67 (s, 1H), 7.71 (m, 2H), 7.23 (t, J=8 HZ, 1H), 2.28 (s,3H); 13C-NMR (CDCl3, 400 MHz) 166.9447, 164.7258, 162.5473, 160.1066, 132.0480, 132.0077, 128.5987,122.9910, 122.9507, 114.2568, 114.0147,14.6902.19F-NMR (CDCl3, 400 MHz) -115.94, -115.96. Рассчитано аналитическим методом для C9H7N2O: С 60.67; Н 3.96; N 15.72. Обнаружено: С 60.54; Н 3.78; N 15.69.- 20016447 Приготовление F. Приготовление 3-(4-(бромметил)-3-фторфенил)-1,2,4-оксадиазола. Метод А. Постадийное бромирование. К смеси перемешивания 3-(3-фторо-4-метилфенил)-1,2,4-оксадиазола (5,34 г, 30 ммоль), CCl4 (50 мл) и NBS (11,7g, 66 ммоль) добавили AIBN (246 мг, 1,5 ммоль). Реакционную смесь нагрели до 80 С в течение 3 ч в атмосфере азота, охладили до комнатной температуры и добавили 50 мл насыщенного раствора бикарбоната натрия, Органический слой отделили, а водный слой экстрагировали дихлорметаном(50 мл). Органический слой высушили на сульфате натрия, профильтровали и концентрировали на роторном испарителе и получили 3-(4-(дибромметил)-3-фторфенил)-1,2,4-оксадиазол (9,34 г, 93 %) в виде белого твердого вещества, которое использовали на следующей стадии без дальнейшей очистки. В колбу с круглым дном объемом 200 мл загрузили 3-(4-(дибромметил)-3-фторфенил)-1,2,4-оксадиазол (8,37 г,25 ммоль) и THF [тетрагидрофуран] (60 мл). Смесь охладили до 0 С и добавили по каплям диизопропил этиламин (3,48 г, 27 ммоль) в течение 15 мин, а затем диэтилфосфит (3,7 г, 26,8 ммоль). Смесь перемешали при комнатной температуре в течение 60 мин и быстро охладили 40 мл воды. Водный слой экстрагировали эфиром (280 мл). Объединенный органический слой промыли 20 мл насыщенного водногоNH4Cl и 20 мл насыщенного раствора натрий хлорида. Органический слой высушили на сульфате натрия, профильтровали, концентрировали на роторном испарителе и получили сырое твердое вещество,которое очистили с помощью низкой подушки из двуокиси кремния и получили 3-(4-(бромметил)-3 фторфенил)-1,2,4-оксадиазол (6,03 г, 94 %). mp [температура плавления] 87,3 С. 1 Н-NMR (CDCl3, 400MHz)8.80 (s, 1H), 7.94 (dd, 1H), 7.87 (dd, 1H), 7.58 (t, 1H), 4.57 (s, 2H); 13C-NMR (CDCl3, 400 MHz)166.97, 166.95, 165.45, 162.29, 159.73, 132.34, 132.30, 128.99, 128.90, 128.81, 124.04, 124.01, 115.56, 115.32,25.22, 25.18; 19F-NMR (CDCl3, 400 MHz)-115.81, -115.84, -115.86. Рассчитано аналитическим методом для C9H6BrFN2O: С 42.05; Н 2.35; N 10.90. Определено: С 42.17; Н 2.17; N 10.63. Метод В. Альтернативное однореакторное бромирование. 3-(3-фторо-4-метилфенил)-1,2,4-оксадиазол (101,8 г, 0,57 моль) и N-бромсукцинимид (206 г, 1,16 моль) растворили в ацетонитриле (1 л) и добавили азобисизобутиронитрил (4,2 г, 26 ммоль) при комнатной температуре. Смесь нагрели до 70 С в течение 2 ч, в этой точке HPLC показала полную конверсию исходного материала в смесь монобромида и дибромида. Смесь охладили до 0 С и добавили диизопропил этиламин (73 мл, 54,2 г, 0,42 моль) при поддержании температуры реакции ниже 5 С. Медленно добавили диэтилфосфит (54,7 мл, 58,6 г, 0,42 моль) и реакционную смесь нагрели до комнатной температуры. Через 2 ч HPLC анализ показал полную конверсию дибромида в монобромид. Добавили воду(1,2 л) и получившийся осадок профильтровали. После промывки водой (2200 мл) и высушивания получили 3-(4-(бромметил)-3-фторфенил)-1,2,4-оксадиазол (135 г, 92% выход). Метод С. Бромирование броматом натрия (прямое выделение). Бромат натрия (2,54 г) растворили в воде (8,4 мл). К этому раствору добавили раствор 3-(3-фторо-4 метилфенил)-1,2,4-оксадиазола (1,0 г) в EtOAc (12 мл) при комнатной температуре. Раствор NaHSO3(1,75 г) в воде (17 мл) добавили по каплям (предостережение: реакция экзотермическая). Смесь перемешали при комнатной температуре в течение 2 ч, и затем хранили в холодном помещении в течение ночи. Органический слой отделили, промыли 10%-ым Na2S2O3 и водой, после чего концентрировали. Получившееся твердое вещество растворили в EtOAc (6 мл). Медленно добавили гептан (30 мл). Суспензию перемешали при комнатной температуре в течение 3 ч, затем профильтровали. Твердое вещество промыли гептаном (15 мл), после этого высушили и получили 0,74 г (51%) титульного соединения:HPLC: 99,42 АР. Второе восстановление посева: фильтрат концентрировали до объема 30 мл и получившуюся суспензию профильтровали и получили титульное соединение в виде белого твердого вещества 0,23 (16%) с помощью HPLC 97,09 АР. Метод D. Бромирование броматом натрия (двустадийная процедура с использованием восстановления дибромида). Раствор 3-(3-фторо-4-метилфенил)-1,2,4-оксадиазола (20,0 г, 112,2 мммоль) в дихлорметане(200 мл) добавили к раствору NaBrO3 (50,8 г, 336,7 ммоль) в воде (200 мл) при комнатной температуре. Получившуюся двухфазную смесь охладили до 0 С. Раствор NaHSO3 (35,7 г, 336,7 ммоль) в воде (160 мл) добавили по каплям для поддержания температуры закладки ниже 20 С (1 ч). Получившийся красный раствор перемешивали при комнатной температуре, пока 3-(3-фторо-4-метилфенил)-1,2,4 оксадиазол не стал меньше 1,0 АР с помощью HPLC (2 ч). Органический слой (нижний слой) отделили,а водный слой экстрагировали дихлорметаном (200 мл). Объединенный раствор дихлорметана промыли 10% водным раствором Na2S2O3 (200 мл), водой (200 мл) и 15% соляным раствором (200 мл). После концентрации под вакуумом получили белое твердое вещество (смесь монобромида и дибромида). Это твердое вещество растворили во влажном MeCN (200 мл, KF: 1,5-4%), а раствор охладили до минус 5 - 0 С. Добавили N,N-диизо-пропилэтиламин (6,0 г, 8,1 мл, 46,4 ммоль), а затем по каплям диэтилфосфит (6,0 г,46,1 ммоль) в течение 15 мин. Смесь перемешали при минус 5 - 0 С, пока 3-(4-(дибромметил)-3 фторфенил)-1,2,4-оксадиазол не стал 0,5 АР (1,5-2 часа). Добавили воду (500 мл) в течение 30 мин, в результате получили белую суспензию. Эту суспензию перемешали при комнатной температуре в течение 1-3 ч, а затем профильтровали. Кек промыли водой (2200 мл), затем высушили под вакуумом при- 21016447 45 С в течение 20 ч. Рассчитано аналитическим методом для C9H6BrFN2O: Расчет С, 42.05; Н, 2.35; N, 10.89; Вг, 31.08; F,7.39. Определено: С, 42.10; Н, 2.24; N, 10.90; Br, 31.18; F, 7.00. Пример 1. (2R)-2-(4-Хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид. Стадия А. 5,5,5-Трифторо-2-(1-фенилэтиламино)пентаннитрил. К раствору (R)-фенетиламина (9,60 г, 79,4 ммоль) и уксусной кислоты (5,08 г, 79,6 ммоль) в МеОН(150 мл) добавили NaCN (3,88 г, 79,6 ммоль). Реакцию охладили до 0 С и добавили раствор 4,4,4 трифторбутиральдегида (10,0 г, 79,6 ммоль) в МеОН (50 мл). Реакцию нагрели до комнатной температуры и перемешали в течение 20 часов. Реакцию разбавили водой (400 мл) и экстрагировали при помощиCH2Cl2 (3300 мл). Объединенный органический слой высушили на Na2SO4 и концентрировали под вакуумом до получения аминитрилового титульного соединения (18,1 г, 89%, как 4:1 смесь диастереомеров) в виде светло-желтого масла: 1 Н NMR (300 MHz, CD3OD)7.38-7.27 (m, 5 Н), 4.15-4.02 (m, 1 Н), 3.69(d, J=6.5 Hz, 2.34H), 1.36 (d, J=6.5 Hz, 0.66H); ESI MS 257 [C13H15F3N2+H]. Стадия В. (R)-5,5,5-Трифторо-2-R)-1- фенилэтиламино)пентанамид гидрохлорид. К раствору 5,5,5-трифторо-2-(1-фенилэтиламино) пентаннитрила (18,0 г, 70,31 ммоль, 4:1 смесь диастереомеров) в CH2Cl2 (100 мл) добавили H2SO4 (100 мл). Реакцию перемешали при комнатной температуре в течение 22 ч, вылили на измельченный лед и нейтрализовали с помощью NH4OH. Смесь экстрагировали с помощью EtOAc (3500 мл). Объединенный органический слой высушили на Na2SO4 и концентрировали под вакуумом до получения свободного основания титульного соединения как смесь диастереомеров (18,94 г, 98%) в виде апельсинового масла: 1 Н NMR (300 MHz, CDCl3)7.40-7.18 (m,5 Н), 6.78 (br s, 0.23 Н), 6.50 (br s, 0.77H), 6.00 (br s, 0.77H), 5.81 (br s, 0.23H), 3.82 (q, J=6.5 Hz, 0.23H), 3.70(q, J=6.5 Hz, 0.77H), 3.14 (t, J=6.0 Hz, 0.23H), 2.86 (t, J=7.0 Hz, 0.77H), 2.35-1.86 (m, 2H), 1.84-1.64 (m, 2H),1.39 (d, J=6.5 Hz, 0.69H), 1.35 (d, J= 6.5 Hz, 2.31H); ESI MS m/z 275 [C13H17N2O+H]. Соль гидрохлорида К раствору свободного основания титульного соединения в виде смеси диастереомеров (11,9 г, 43,4 ммоль) в Et2O/МеОН (7:1, 40 мл) добавили раствор 1 N HCl в Et2O (70 мл). Образовавшийся белый осадок повторно растворили путем нагрева смеси и добавления МеОН (до конечного соотношения 4:1Et2O/MeOH). Раствор охладили при комнатной температуре и оставили на ночь. Выделили соль аминоамид гидрохлорида титульного соединения как отдельный диастереомер (3,11 г, 23%) в виде белого твердого вещества: 1 Н NMR (300 MHz, CD3OD)7.93 (br s, 1H), 7.69 (br s, 1H), 7.54-7.44 (m, 5H), 4.39 (q,J=7.0 Hz, 1H), 3.50 (t, J=6.5 Hz, 1H), 2.29-2.20 (m, 2H), 2.10-2.01 (m, 2H), 2.07 (d, J=7.0 Hz, 3H); ESI MS(40 psi=фунтов на квадратный дюйм) в течение 4 ч при 50 С. Реакцию профильтровали через целит, а фильтрат концентрировали под вакуумом до получения промежуточного амингидрохлорида в виде белого твердого вещества. К суспензии амина в CH2Cl2 (100 мл) добавили N, N-диизо-пропилэтиламин (5,25 мл, 30,0 ммоль) и 4-хлорбензолсульфонил хлорид (2,53 г, 12,0 ммоль). Реакцию перемешали при комнатной температуре в течение 18 ч и разбавили EtOAc (200 мл), промыли NaHCO3 (250 мл) и соляным раствором (250 мл), высушили на Na2SO4 и концентрировали под вакуумом. Титульное соединение (2,91 г,84%) получили в виде белого твердого вещества растиранием в порошок остатка с CH2Cl2/гексаны (2:1): 1 Н NMR (300 MHz, CD3OD)7.84 (dt, J=8.5, 2.0 Hz, 2H), 7.55 (dt, J=8.5, 2.0 Hz, 2H), 3.85 (dd, J=8.5, 5.0Hz, 1H), 2.34-2.05 (m, 2H), 1.97-1.68 (m, 2H); ESI MS m/z 345 [C11H12ClF3N2O3S+H]. Стадия D. (R)-2-(4-хлоро-N-(2-фторо-4-(1,2,4-оксадиазол-3-ил) бензил) фенилсульфонамидо)-5,5,5 трифторпентанамид. К раствору сульфонамид (R)-2-(4-хлорфенилсульфонамидо)-5,5,5-трифторпентанамида (130 мг,0,37 ммоль) в DMF (2 мл) добавили Cs2CO3 (241 мг, 0,74 ммоль) и 3-(4-бромметил-3-фторбензил)-1,2,4 оксадиазол (257 мг, 0,48 ммоль). Реакцию перемешали при комнатной температуре в течение 2 ч, разбавили водой (50 мл) и экстрагировали с помощью EtOAc (250 мл). Объединенный органический экстракт промыли водой (250 мл) и соляным раствором (50 мл) и концентрировали под вакуумом. Остаток очистили колоночной хроматографией (силикагель, 0-55% EtOAc/гексаны) и получили титульное соединение оксадиазола (92 мг, 45%) в виде белого твердого вещества: mp 66-68 С; 1 Н NMR (500 MHz, CDCl3)8.77 (s, 1H), 7.90 (dd, J= 80, 1.5 Hz, 1H), 7.77-7.71 (m, 3H), 7.64 (dd, J=7.5, 7.5 Hz, 1H), 7.51 (d, J= 8.5 Hz,2H), 6.34 (s, 1H), 5.28 (s, 1H), 4.66 (d, J=15.6 Hz, 1H), 4.51 (d, J=15.6 Hz, 1H), 4.39 (dd, J=8.9, 6.3 Hz, 1H),2.25-1.82 (m, 3H), 1.54-1.47 (m, 1H); ESI MS m/z 521 [C20H17ClF4N4O4S+H]+; HPLC 98.9% (AUC), tR=19,4 мин. Пример 2. (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5- трифторпентанамид.- 22016447 Стадия А. 4-(Бромметил)-3-фторбензонитрил. К раствору 3-фторо-4-метилбензонитрила (5,0 г, 0,23 моль) в 100 мл тетрахлорида углерода добавили N-бромсукцинимид (4,97 г, 0,28 моль) и AIBN (100 мг, 0,61 ммоль) и смесь дефлегмировали за шесть часов. Реакцию охладили и профильтровали. Фильтрат промыли водой, осушили на сульфате магния,профильтровали и растворители удалили под вакуумом до получения 5,44 г титульного соединения в виде желтоватого твердого вещества. 1 Н NMR показал присутствие 20% исходного материала. 1 Н NMR(400 MHz, CDCl3) для титульного соединения :7. 54-7.30 (m, 3H), 4.83 (s, 2H). Стадия В.(R)-2-(4-Хлоро-N-(4-циано-2-фторбензил)фенилсульфонамидо)-5,5,5-трифторпентанамид. К раствору (R)-2-(4-хлорфенилсульфонамидо)-5,5,5-трифторпентанамида (6,88 г, 20,0 ммоль) и 4(бромметил)-3-фторбензонитрила (6,43 г, 30 ммоль) в DMF (35 мл) добавили безводный Cs2CO3 (19,56 г,60 ммоль). Получившуюся смесь перемешали при комнатной температуре в течение 45 минут, а затем разбавили EtOAc (200 мл), промыли водой (100 мл X 4) и высушили на Na2SO4. Продукт очистили при помощи Biotage (40+M колонка, 3-80% EtOAc в гексанах, 651 мл). Получили титульное соединение в виде белого твердого вещества (6,50 г, 68,1% выход). 1 Н NMR (DMSO-d6, 400 MHz)7.80-7.88 (m, 3H),7,70-7.75 (m, 2H), 7.67 (d, 2H, J=8), 7.60 (s, 1H), 7.26 (s, 1H), 4.99 (d, 1H, J=16), 4.68 (d, 1H, J=16), 4.14 (t,1H, J=8), 1.99-2.17 (m, 2H), 1.80-1.94 (m, 1H), 1.40-1.56 (m, 1H). LC/MS M+H 478.14, 94%. Стадия С. (2R)-2-(4-Хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5- трифторпентанамид. К раствору (R)-2-(4-хлоро-N-(4-циано-2-фторбензил)фенилсульфонамидо)-5,5,5-трифторпентанамида (6,5 г, 13,6 ммоль) в EtOH (70 мл) добавили NH2OH (50% в Н 2 О, 2,6 мл 40,8 ммоль). Получившуюся смесь перемешали при 80 С в атмосфере азота в течение одного часа, а затем охладили при комнатной температуре. Растворители испарили под пониженным давлением. Остаток растворили в EtOAc и промыли водой и высушили на Na2SO4. После испарения растворителя получили белое твердое вещество,которое перекристаллизовали из EtOAc и гексанов до получения промежуточного амид оксима в виде белого твердого вещества (6,93 г, количественный выход). К смеси промежуточного (R)-2-(4-хлоро-N-(2 фторо-4-(N'-гидроксикарбамимидоил)бензил)фенилсульфонамидо)-5,5,5-трифторпентанамида (6,93 г,13,6 ммоль) и триэтоксиметана (6,77 мл, 40,8 ммоль) в дихлорэтане (30 мл) добавили BF3OEt2 (0,17 мл,1,36 ммоль). Получившуюся смесь перемешали при 70 С в течение одного часа, а затем охладили до комнатной температуры. Хроматография (силикагель, biotage, 40+M колонка, 3-80% EtOAc в гексанах,651 мл) обеспечила титульное соединение в виде белого твердого вещества (4,9 г, 69,3% выход). Указанные выше 4,9 г продукта объединили с второй закладкой 9,8 г (2R)-2-(4-хлорфенил) сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида (приготовленного в соответствии с процедурой, описанной в примере 1). К объединенным закладкам (14,7 г) добавили изопропиловый спирт (75 мл). Смесь дефлегмировали до почти полного растворения, а затем профильтровали. Фильтрат перемешали при комнатной температуре 16 ч и профильтровали. После высушивания до постоянной массы образовалось белое мелкокристаллическое твердое вещество и получили (2R)-2(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид (13,7 г). 1 Н NMR (CDCl3, 500 MHz)8.77 (s, 1 Н), 7.90 (dd, J=8.0, 1.5 Hz, 1H), 7.77-7.71 (m, 3H),7.64 (dd, J=7.5, 7.5 Hz, 1H), 7.51 (d, .1=8.5 Hz, 2H), 6.34 (s, 1H), 5.28 (s, 1H), 4.66 (d, J=15.6 Hz, 1H), 4.51 (d,J=15.6 Hz, 1H), 4.39 (dd, J=8.9, 6.3 Hz, 1H), 2.25-1.82 (m, 3H), 1.54-1.47 (m, 1H); ESI MS m/z 521 В соответствующий сухой сосуд добавили (R)-2-амино-5,5,5-трифторпентанамид гидрохлорид(199,52 г 0,966 моль, 1,0 эквив.), а затем 4-хлорбензолсульфонил хлорид (215,22 г 0,989 моль, 1,02 эквив.,97% вес/вес%) и 1,6 л THF при комнатной температуре. Триэтиламин (206,5 г, 2,04 моль, 2,1 эквив.) добавили в течение 20 мин, поддерживая температуру в сосуде 15-25 С, и получившуюся белую суспензию перемешали при 15-25 С в течение 30 мин. В реакционную смесь добавили (1,4 л, 7 об.) воду при 2025 С, а затем дистилляцией под вакуумом удалили THF (1,4 л, 7 об.) (в течение процесса дистилляции температура в сосуде поддерживалась 40-60 С при 250-400 мм рт.ст.). После завершения процесса дистилляции 1,4 л (7 об.) воды добавили в течение 30 минут при поддержании температуры в сосуде 5060 С, и получившуюся белую суспензию перемешали при 50-60 С в течение 30 минут, а затем охладили при 10 С. Суспензию взбалтывали не менее одного часа, продукт профильтровали. Фильтрационный кек промыли водой (600 мл, каждая промывка), пока значение рН промывки кека после измерения не стало 5. Кек высушили под вакуумом при температуре не выше 70 С (темп. рубашки), пока потери при вы- 23016447 сушивании не стали 0,5 вес/вес %, и получили титульное соединение в виде белого твердого вещества(300 г, 91% выход). 1 Н NMR (DMSO-d6) (400 MHz)160 - 1.90 (два m, 1H каждый из СН 2), 2.10 - 2.35 (m,2H из СН 2-CF3), 3.85 - 3.88 (m,1 Н, -(CONH2)CH(NH), 7.137.37 (br s, 1H, каждый из CONH2), 7.61 (m,2 Н, Ar-На), 7.64 (m, 2 Н, Ar-Hb), 8.18 (d, 1H, J=8.0 Hz, NH-SO2). 13C NMR (DMSO-d6) (100.0 MHz)171.75,140.27, 137.77, 131.71, 129.56, 128.95, 126.22, 55.12, 30.1, 29.82, 29.53,29.25,25.82,25.79. Стадия В. (2R)-2-(4-Хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5- трифторпентанамид Стадия В. Процедура 1. В соответствующий сосуд загрузили 3-(4-(бромметил)-3-фторфенил)-1,2,4-оксадиазол (492,14 г,1,10 эквив., 1,914 моль), (R)-2-(4-хлорфенилсульфонамидо)-5,5,5-трифторпентанамид (600 г, 1,00 эквив.; 1,74 моль) карбонат цезия (312,22 г, 0,55 эквив.; 0,98 моль), тетра-n-бутил аммоний йодид (64,29 г, 0,10 эквив.; 0,17 ммоль) и ацетонитрил (9 мл/г; 5,4 л). Рубашку нагрели до 40 С (внутренняя температура 38 С). Контроль реакции осуществляли с помощью HPLC, пока (R)-2-(4-хлорфенилсульфонамидо)-5,5,5 трифторпентанамид не стал 5 АР. Сосуд охладили до внутренней температуры 20 С и добавили воду(5,4 л). Фазы разделили, обогащенный продуктом слой оказался сверху Нижний слой отбросили. Воду(3,7 л) загрузили в течение 18 мин, реакцию выдержали в течение 20 ч при 20 С, а затем профильтровали. В сосуд добавили воду (6 л) и перемешали, чтобы убрать твердые частицы, прилипшие к мешалке и стенкам сосуда. Сырой кек промыли один раз водой, используемой для промывки реактора. Сырой кек высушили на поддонах под вакуумом при 50 С. Сухой сырой кек весил 699 г. Кек перенесли в 10 литровый реактор и загрузили THF (2,025 л), а затем раствор гидроксиламина (50% в воде) (42,97 мл, 0,40 эквив., 0,696 моль). Рубашку реактора нагрели до 40 С. Контроль реакции осуществляли с помощью HPLC,пока (R)-2-(4-хлоро-N-(4-циано-2-фторбензил) фенилсульфонамидо)-5,5,5-трифторпентанамид не стал 0,4 АР. Реакцию охладили до 20 С, добавили воду (2 л) и реакцию перемешали при 20 С в течение 30 мин. Фазы разделили и органическую фазу обработали гептаном (8 л) при перемешивании, и реакция стала масляной, а затем выпала в осадок. Суспензии дали отстояться при 20 С в течение 2 ч, и реакцию профильтровали, а кек промыли гептаном (2 л). Сырой кек высушили на поддоне под вакуумом при 4050 С, его вес составил 613 г после высушивания до 1% потерь при высушивании. Сырой продукт перенесли обратно в сосуд вместе с МеОН (3,678 л) и MeCN (1,226 л). Рубашку нагрели до 60 С (внутренняя температура 52 С) до полного растворения, и при этой температуре медленно добавили воду (2,023 л),поддерживая внутреннюю температуру 50 С. После завершения процесса добавления воды раствор охладили до внутренней температуры 15 С в течение 4 ч по мере того, как происходила кристаллизация. В реактор загрузили дополнительно воду (400 мл), и реакцию профильтровали и маточный раствор возвратили в сосуд. Маточный раствор взболтали в течение 2 мин, чтобы отделить весь налипший продукт в сосуде. Сырой кек промыли маточным раствором, а затем гептанами (1,500 л). Продукт высушили на поддоне под вакуумом при 40-50 С, пока потери при высушивании не стали 0.5%, и получили (2R)-2(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамид в виде блестящего желтоватого твердого вещества (577,6 г, 63,76% выход). Стадия В. Процедура 2. В соответствующий сосуд добавили (R)-2-(4- хлорфенилсульфонамидо)-5,5,5-трифторпентанамид(2,68 кг, 7,77 моль, 1 экв.), 3-(4-(бромметил)-3-фторфенил)-1,2,4-оксадиазол (2,00 кг, 7,78 моль, 1 экв.),карбонат цезия (1,65 кг, 5,06 моль, 0,65 экв.), тетрабутиламмоний йодид (0,29 кг, 0,78 моль, 0,1 экв.) и ацетонитрил (12,0 л 4,5 л/кг). Реакцию нагрели до 35 С до завершения с помощью HPLC (3-(4(бромметил)-3-фторфенил)-1,2,4-оксадиазол 0,5 относительный АР с помощью HPLC). Реакцию охладили до 15 С и добавили воду (10,72 л, 4 л/кг) при перемешивании, а затем ледяную уксусную кислоту(0,22 кг), чтобы довести значение рН реакции до 6,5. Перемешивание прекратили и фазы разделили(верхний слой содержал продукт). К обогащенному продуктом слою добавили толуол (26,8 кг, 31 л, 10 кг/кг), а затем концентрированный соляной раствор (20% вес/вес, 6,9 кг, 2 л/кг), слои разделили (верхний слой содержал продукт). Смесь дистиллировали при 50 С под вакуумом (200 мбар) до удаления ацетонитрила. Концентрацию отрегулировали с помощью дополнительного толуола, при необходимости, по- 24016447 сле дистилляции для обеспечения полного объема в реакторе 10 л/кг. Загрузили изопропиловый спирт(0,48 кг, 0,2 л/кг), и закладку охладили до 15 С для инициирования кристаллизации. Получившуюся суспензию профильтровали и промыли холодным толуолом (18,65 кг, 21,56 л, 8 л/кг). Сырой кек высушили на поддоне под вакуумом при 50 С, пока потери при высушивании не стали 1,0%. Сухой кек добавили в 100-литровый реактор вместе с изопропиловым спиртом (27,34 кг, 34,8 л, 13 л/кг) и гидроксиламином(50% водный раствор, 0,05 кг, 1,51 моль, 0,2 экв.). Смесь нагрели до 65 С, контроль смеси осуществляли с помощью HPLC, пока (R)-2-(4-хлоро-N-(4-циано-2-фторбензил)фенилсульфонамидо)-5,5,5-трифторпентанамид не стал 0,4 АР. Затем реакцию дистиллировали (температура в сосуде 50 С, вакуум 300 мбар), пока объем реакции не стал 60% от исходного. Ацетонитрил (5,36 кг, 2 л/кг) загрузили и температуру реакции увеличили до 70 С для достижения полного растворения. Медленно добавили воду(11,26 л, 4,2 л/кг) при поддержании температуры реакции 65 С. Реакцию охладили до 15 С в течение 2 ч, произошла кристаллизация. Суспензию профильтровали и промывали холодным водным изопропиловым спиртом (2:1 IPA: вода по объему), Кек высушили в вакуумной печи, пока потери при высушивании не стали 1%. Продукт затем перекристаллизовали растворением в ацетонитриле (2 л/кг на основании веса загрузки высушенного кека) и метаноле (6 л/кг) и после этого нагрели до 50 С. Медленно добавили воду (4 л/кг) при поддержании температуры реакции 50 С. Реакцию охладили до 15 С в течение 2 ч. Получившуюся суспензию профильтровали и промыли раствором метанол:ацетонитрил:вода (6:2:4, 5 л/кг), получили (2R)-2-(4-хлорфенил) сульфонил]2-флоро-4-(1,2,4-оксадиазол-3-ил)фенил]метил] амино]-5,5,5-трифторпентанамид (2,02 кг, 50% выход) в виде белого твердого вещества. Пример 4. (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5- трифторпентанамид. Стадия А. (2R)-2-(4-Хлорфенил)сульфонил][(4-циано-2-фторфенил)метил]амино]-5,5,5-трифторпентанамид(R)-2-(4-хлорфенилсульфонамидо)-5,5,5-трифторпентанамид (3,444 кг), карбонат калия (2,774 кг),тетрабутиламмоний бромид (0,484 кг) и 4-(бромметил)-3-фторбензонитрил (2,092 кг) загрузили в реактор. Затем загрузили этилацетат (17,2 л) и воду (3,44 л) и загрузку нагрели до 50 С до завершения с помощью HPLC (1 относительный АР исходного материала). Реакция обычно завершается приблизительно через 15 ч. Загрузку охладили до 15-20 С, загрузили воду (6,88 л) и отделили нижнюю водную фазу. Загрузили раствор фосфорнокислого натрия одноосновного (0,2 М в воде, 20,66 л) и отделили нижнюю водную фазу, проверили рН, чтобы значение было 6,5. (Примечание: Если рН 6,5, можно загрузить дополнительно 20,66 л 0,2 М раствора фосфорнокислого натрия одноосновного и повторить экстракцию и измерение рН). Затем растворитель сменили вакуумной дистилляцией постоянного объема. Реактор поместили в вакуум (270 мм рт.ст.), рубашку нагрели до 75-80 С. Как только началась дистилляция этилацетата, добавили изопропанол (41,34 л) при такой же скорости сбора дистиллята, и весь объем загрузки поддерживали на постоянном уровне. Когда добавили весь изопропанол, вакуум сбросили и загрузили воду (13,76 л). Во время добавления воды поддерживали температуру загрузки приблизительно 50 С. После этого загрузку охладили до 15-20 С и профильтровали. Влажный кек промыли 50%-ым (объем/объем) водным изопропанолом (421,6 кг), а затем высушили под вакуумом при 50 С и получили титульное соединение в виде желтоватого твердого вещества (3,648 кг, 78% выход). 1 Н NMR (300 MHz, DMSO- d6)ppm 1.42 1.55 (m, 1 H) 1.80-1.93 (m, 1 H) 2.00-2.15 (m, 2 H) 4.44 (dd, J=7.91, 1.13 Hz, 1 H) 4.68 (d, J=17.71 Hz, 1 H) 4.99 (d, J=17.71 Hz, 1 H) 7.26 (s, 1 H) 7.50 (s, 1 H) 7.63 -7.73 (m, 4 H) 7.78 - 7.87 (m, 3 H). 13C NMR (75(KBr): 3443, 3342, 3210, 2955, 2245, 1699, 1577, 1476, 1163 см-1. Рассчитано аналитическим методом для C19H16ClF4N3O3S Расчет С 47.75; Н 3.37; N 8.79; S 6.71; F 15.90; Cl 7.41. Определено: С 47.95; Н 3.31; N 8.67; S 6.72; F 15.59; Cl 7.49. Стадия В. (R)-2-(4-Хлоро-N'-(2-фторо-4-(N'-гидроксикарбамимидоил)бензил)фенилсульфонамидо)5,5,5- трифторпентанамид(399 г) и метанол (1,6 л) загрузили в реактор, а затем гидроксиламин (50% раствор в воде, 93 мл, 1,8 экв.). Смесь нагрели до 45-50 С до завершения реакции с помощью HPLC (0,15 относительного АР исходного материала). Медленно загрузили воду (0,5 л), поддерживая температуру загрузки в интервале 30-50 С. Загрузку оставили, пока не началась кристаллизация, а затем загрузили воду (2,7 л). Загрузку охладили до 15-20 С и профильтровали. Кек промыли раствором 2:1 МеОН:вода (2 л), а затем высушили под вакуумом при 50 С и получили титульное соединение в виде белого твердого вещества (415 г, 96% выход). 1 Н NMR (300 MHz, DMSO-d6)ppm 1.43 - 1.64 (m, 1 H) 1.77 - 1.93 (m, 1 H) 1.93 - 2.17 (m, 2 H) 4.41 (dd,J=8.48, 6.03 Hz, 1 H) 4.60 (d, J=17.14 Hz, 1H) 4.94 (d, J=16.77 Hz, 1 H) 5.81 - 5.98 (m, 2 H) 7.19 - 7.27 (m, 1(m, 1 H). IR (KBr): 3491, 3379, 1680, 1651, 1592, 1433, 1343. Рассчитано аналитическим методом для C19H19ClF4N4O4S Расчет С, 44.66; Н, 3.74; N, 10.96; S, 6.27;R)-2-(4-хлоро-N-(2-фторо-4-(N'-гидроксикарбамимидоил)бензил)фенилсульфонамидо)-5,5,5 трифторпентанамид (246 г) загрузили в реактор, а затем сухой ацетонитрил (509 мл), триэтоксиметан(120 мл) и трифторуксусную кислоту (7 мл). Раствор нагрели до 40-50 С, пока реакция не была завершена с помощью HPLC (0,15 относительный АР исходного материала). Метанол (1,48 л) загрузили одной порцией, а затем воду (1,034 л), поддерживая температуру загрузки 45-50 С. Затем загрузку охладили до 15-20 С и профильтровали. Кек промыли с помощью раствора 2:6:5 MeCN:МеОН:вода и высушили на поддоне под вакуумом при 50-60 С, получили титульное соединение в виде белого твердого вещества(d, J=30.0 Hz, 1C), 22.90. 19F NMR, (CDCl3, 282 MHz) : -116.3, -66.5. IR (KBr): 3454, 334, 3286, 2952,1705, 1432, 1325, 1260, 1167, 1084, 828 см-1. Рассчитано аналитическим методом для C20H17ClF4N4O4S Расчет С 46.11; Н, 3.29; N 10.71; S 6.15; F 14.58; Cl 6.80. Определено С 46.06; Н 3.24; N 10.71; S 6.25; F 14.60; Cl 6.88. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил] метил]амино]-5,5,5-трифторпентанамид. 2. Фармацевтическая композиция, включающая терапевтически эффективное количество (2R)-2(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида совместно с фармацевтически приемлемым адъювантом, носителем или разбавителем. 3. Способ лечения или отсрочки начала болезни Альцгеймера, церебральной амилоидной ангиопатии, умеренного когнитивного нарушения, синдрома Дауна, который включает введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества (2R)-2-(4-хлорфенил)сульфонил]2-фторо-4-(1,2,4-оксадиазол-3-ил)фенил]метил]амино]-5,5,5-трифторпентанамида. 4. Способ по п.3 для лечения болезни Альцгеймера. 5. Соединение формулы или его фармацевтически приемлемая соль. 6. Соединение формулы или его фармацевтически приемлемая соль.
МПК / Метки
МПК: C07C 317/32, C07C 235/30, A61K 31/4245, A61P 25/28, C07C 237/06, C07C 317/36, C07C 255/50, C07D 271/06
Метки: ингибитора, ацетамида, качестве, пептида, продуцирования, альфа-(n-сульфонамидо, соединение, beta;-амилоидного
Код ссылки
<a href="https://eas.patents.su/28-16447-soedinenie-alfa-n-sulfonamido-acetamida-v-kachestve-ingibitora-producirovaniya-beta-amiloidnogo-peptida.html" rel="bookmark" title="База патентов Евразийского Союза">Соединение альфа-(n-сульфонамидо) ацетамида в качестве ингибитора продуцирования бета-амилоидного пептида</a>
Антитела против бета-амилоидного пептида

Номер патента: 15654
Опубликовано: 31.10.2011
Авторы: Кумар Умеш, Бербидж Стивен Энтони, Соден Питер Эрнест, Гермашевски Фолькер, Филпотт Карен Луиз, Эллис Джонатан Генри, Форд Сюзанна К.
МПК: A61P 25/28, A61K 39/395, C07K 16/18...
Метки: против, beta;-амилоидного, антитела, пептида
Формула / Реферат:
1. Терапевтическое антитело, представляющее собой антитело или его антигенсвязывающий фрагмент и/или производное, которые связываются с b-амилоидным пептидом и которые содержат следующие CDR (области, определяющие комплементарность):CDRH1: DNGMA (SEQ ID NO:1);CDRH2: FISNLAYSIDYADTVTG (SEQ ID NO:2);CDRH3: GTWFAY (SEQ ID NO:3)в вариабельной области человеческой тяжелой цепи, происходящей из семейства генов VH3, иCDRL1: RVSQSLLHSNGYTYLH (SEQ ID...
Антитела, направленные против бета-амилоидного пептида, и способы их использования

Номер патента: 16193
Опубликовано: 30.03.2012
Авторы: Розенталь Арнон, Хо Вэй-Сень, Понз Хауме
МПК: A61P 25/00, C07K 16/18
Метки: использования, направленные, пептида, против, beta;-амилоидного, способы, антитела
Формула / Реферат:
1. Моноклональное антитело, специфически связывающееся с амилоидным пептидом Аβ, где указанное антитело связывается с эпитопом на Aβ1-40, включающим аминокислоты 25-34 и 40, причем указанное антитело связывается с Aβ1-40 с более высокой аффинностью, чем с Aβ1-42 и Aβ1-43, и связывается с Аβ22-37 с KD меньше 1 мкМ, и где указанное антитело содержит вариабельную область тяжелой цепи, содержащую три CDR, которые по...
Антитела, направленные против бета-амилоидного пептида, и способы их применения

Номер патента: 16357
Опубликовано: 30.04.2012
Авторы: Гримм Ян Маркус, Понс Хауме, Хо Вей-Хсиен, Розенталь Арнон
МПК: C07K 16/18, A61P 25/00
Метки: beta;-амилоидного, антитела, направленные, способы, применения, пептида, против
Формула / Реферат:
1. Способ лечения заболевания, характеризующегося аномальным отложением β-амилоида у субъекта, включающий введение данному субъекту эффективного количества антитела, которое специфически связывается с Aβ-пептидом или агрегированной формой Aβ-пептида, где это антитело включает Fc-область с нарушенной эффекторной функцией и где указанное антитело выбрано из группы, состоящей из:1) антитела, включающего вариабельную область тяжелой...
Иммуногенные композиции, содержащие циклические пептиды, полученные из бета-амилоидного пептида

Номер патента: 10586
Опубликовано: 30.10.2008
Автор: Цурбригген Ринальдо
МПК: C07K 14/47, C07K 7/64, G01N 33/68...
Метки: пептида, пептиды, содержащие, полученные, композиции, beta;-амилоидного, иммуногенные, циклические
Формула / Реферат:
1. Антигенный пептид формулы где Ab представляет собой пептид из 12-20 аминокислотных остатков, производный от b-амилоидного пептида (Ab1-26; SEQ ID NO:2), предпочтительно Ab1-16 (SEQ ID NO:3) или Ab11-26 (SEQ ID NO:4), или его аналога, содержащего консервативную аминокислотную замену; m равно от 1 до 6; А и B ковалентно связаны с образованием циклического пептида; A и B представляют собой прямую связь, линкерный фрагмент или обозначают...
Способ получения (альфа s, бета r)-6-бром-альфа-[2-(диметиламино)этил]-2-метокси-альфа-1-нафталенил-бета-фенил-3-хинолинэтанола

Номер патента: 11770
Опубликовано: 30.06.2009
Авторы: Хорнс Штефан, Бадер Томас, Порстманн Франк Ральф
МПК: C07F 9/09, C07D 215/22
Метки: beta, способ, получения, r)-6-бром-альфа-[2-(диметиламино)этил]-2-метокси-альфа-1-нафталенил-бета-фенил-3-хинолинэтанола, альфа
Формула / Реферат:
1. Способ выделения (aS,bR)-6-бром-a-[2-(диметиламино)этил]-2-метокси-a-1-нафталенил-b-фенил-3-хинолинэтанола из смеси стереоизомерных форм 6-бром-a-[2-(диметиламино)этил]-2-метокси-a-1-нафталенил-b-фенил-3-хинолинэтанола оптическим разделением с хиральным 4-гидроксидинафто[2,1-d:1',2'-f][1,3,2]диоксафосфепин 4-оксидом или его производным в качестве разделяющего агента. 2. Способ по п.1, в котором хиральный...
Предыдущий патент: Производные n5-(2-этоксиэтил)-n3-(2-пиридинил)-3,5-пиперидиндикарбоксамида, предназначенные для применения в качестве ингибиторов ренина
Следующий патент: Способ синтеза фенофибрата
Случайный патент: Способ производства стерильного обезжиренного молока