Способ получения бензилэпоксидов и промежуточные соединения
















Формула / Реферат
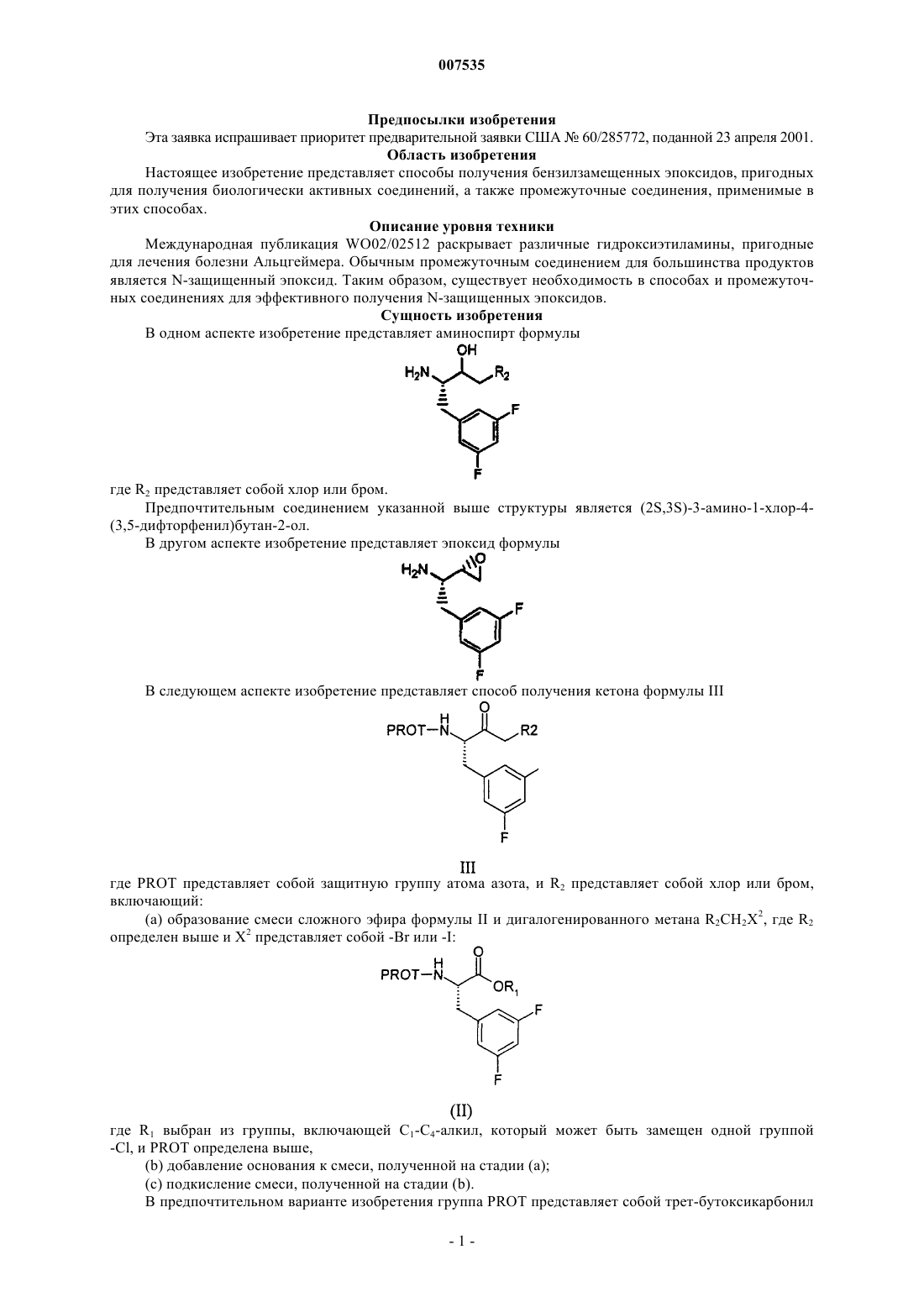
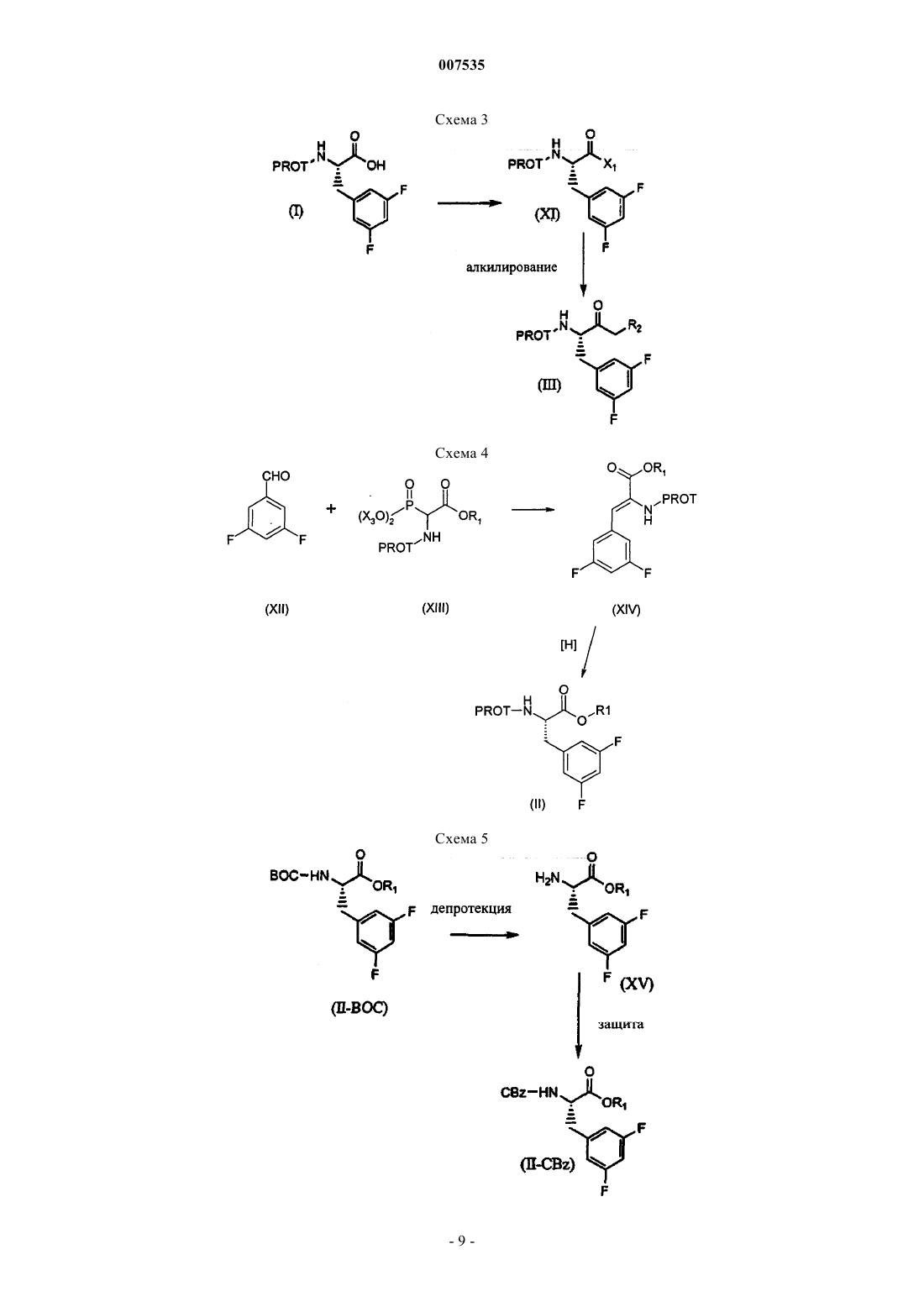
1. Соединение формулы

где R2 представляет собой хлор или бром.
2. Соединение по п.1, где R2 представляет собой -Cl.
3. Соединение по п.1, где R2 представляет собой -Вr.
4. Соединение по п.1, которое представляет собой (2S,3S)-3-амино-1-хлор-4-(3,5-дифторфенил)бутан-2-ол.
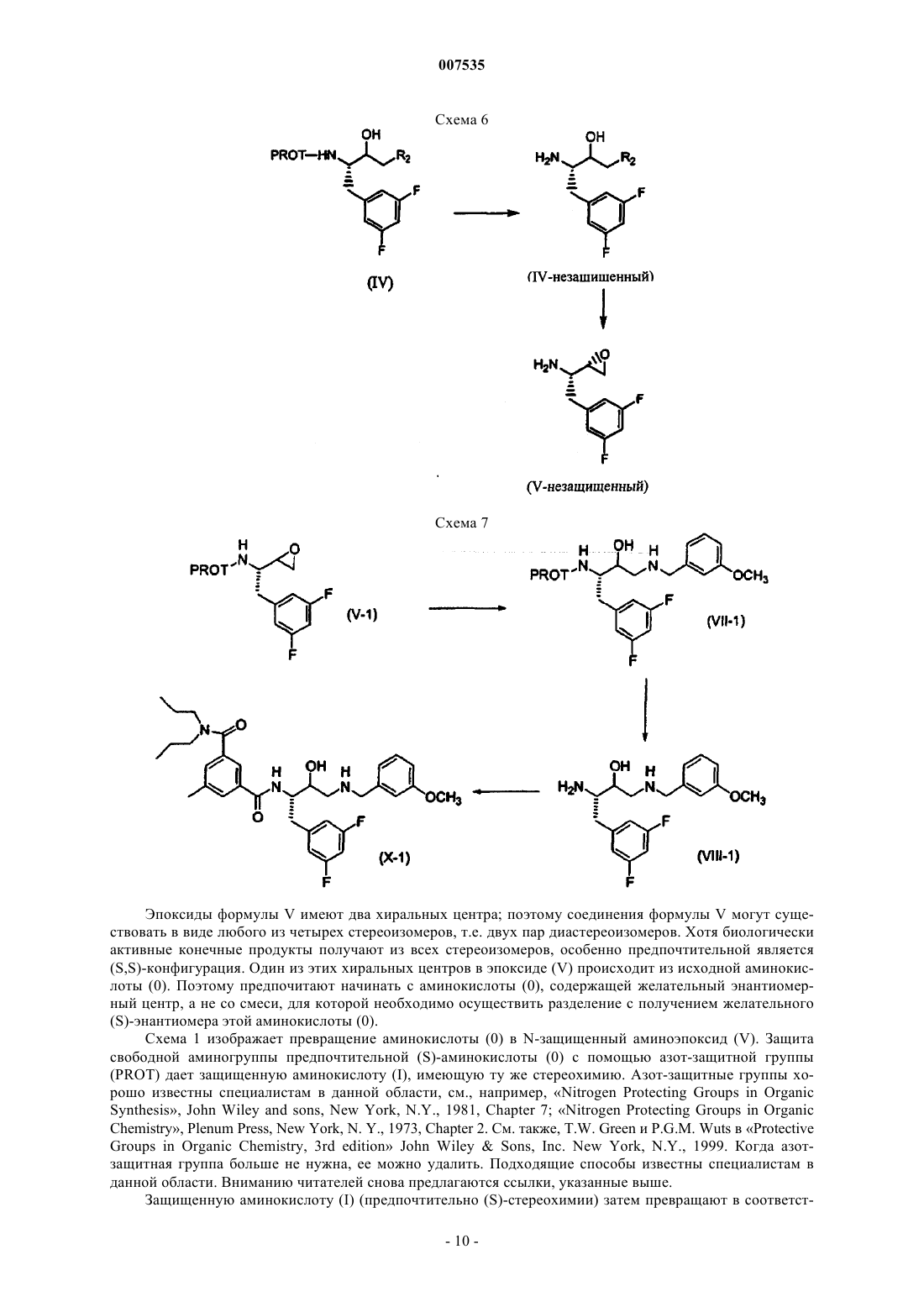
5. Соединение формулы

6. Способ получения кетона формулы III

где PROT представляет собой азотзащитную группу и R2 представляет собой хлор или бром, включающий:
(а) образование смеси сложного эфира формулы II и дигалогенированного метана R2CH2X2, где R2 определен выше и X2 представляет собой -Вr или -I:

где R1 выбран из группы, включающей С1-С4-алкил, который может быть замещен одной группой -Сl, и PROT определена выше,
(b) добавление основания к смеси, полученной на стадии (а);
(c) подкисление смеси, полученной на стадии (b).
7. Способ по п.6, где PROT представляет собой трет-бутоксикарбонил или бензилоксикарбонил.
8. Способ по п.6, где R1 представляет собой С1-С2-алкил.
9. Способ по п.6, где R1 представляет собой С1-алкил.
10. Способ по п.6, где R2 представляет собой хлор.
11. Способ по п.6, где CH2R2X2 присутствует в количестве от примерно 1 до примерно 1,5 эквивалентов, исходя из количества сложного эфира II.
12. Способ по п.6, где X2 представляет собой йод.
13. Способ по п.6, где сильное основание представляет собой диизопропиламид лития, (С1-С8-алкил)литий.
14. Способ по п.13, где сильное основание представляет собой диизопропиламид лития.
15. Способ по п.6, где сильное основание используют в количестве примерно от 2 до 2,5 эквивалентов, исходя из количества сложного эфира II.
16. Способ по п.6, где добавляют вторую порцию основания, представляющего собой (С1-С4)алкиллитий.
17. Способ по п.16, где второе основание представляет собой н-бутиллитий, втop-бутиллитий, трет-бутиллитий, метиллитий.
18. Способ по п.17, где второе основание представляет собой н-бутиллитий.
19. Способ по п.6, где количество второго основания составляет от примерно 1 до примерно 1,5 эквивалентов, исходя из количества сложного эфира II.
20. Способ по п.6, где подкисление осуществляют с помощью кислоты, имеющей рКа менее примерно 10.
21. Способ по п.20, где кислота выбрана из группы, включающей уксусную и соляную кислоту и их смеси.
22. Способ по п.6, где кетон (III) представляет собой трет-бутил-(1S)-3-хлор-1-(3,5-дифторбензил)-2-оксопропилкарбамат.
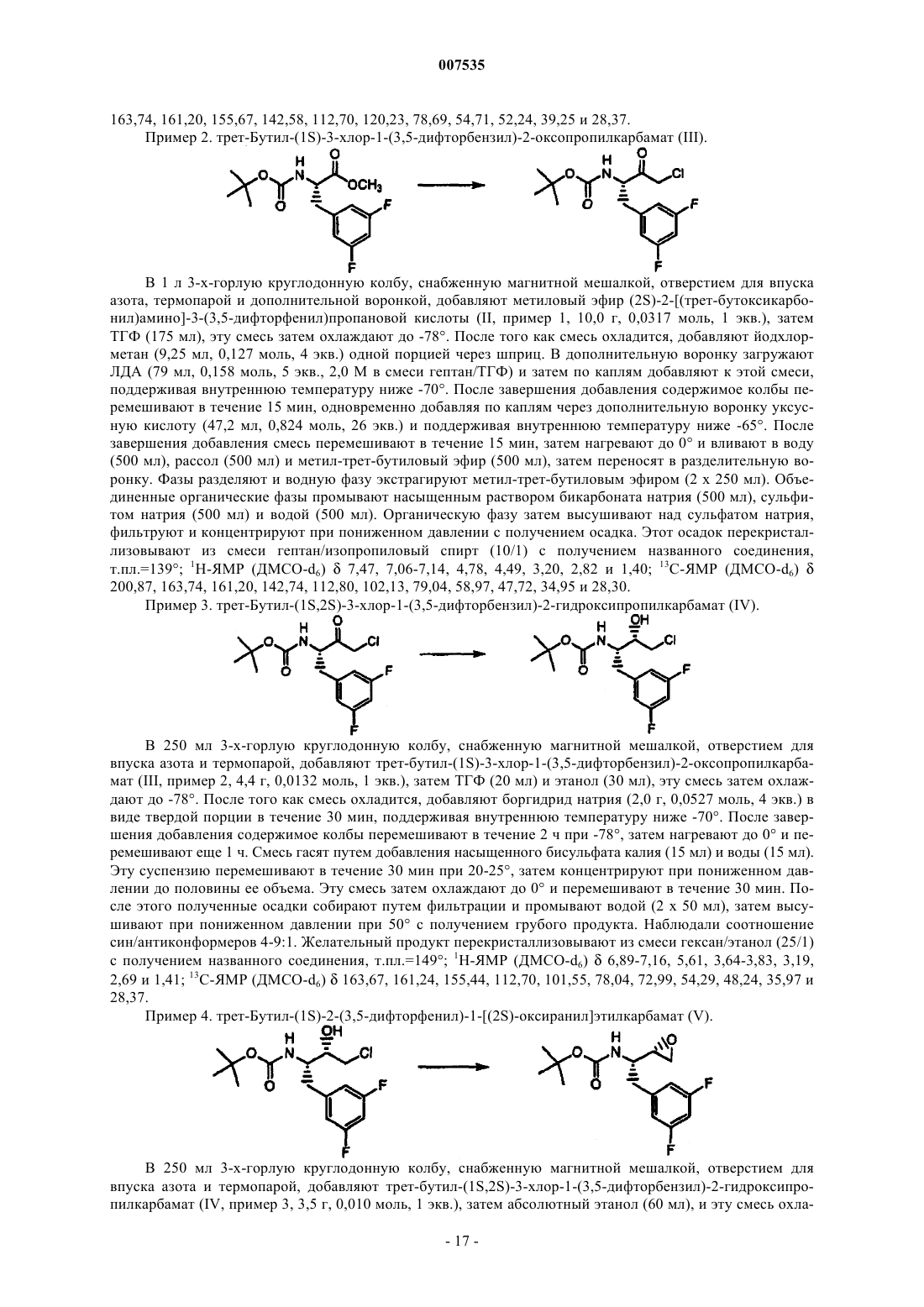
23. Способ получения эпоксида формулы V-R

где R представляет собой фенил, который может быть замещен 1, 2, 3 или 4 группами, независимо выбранными из галогенов, включающий:
(а) превращение сложного эфира формулы II

в кетон формулы III

(b) восстановление кетона до соответствующего спирта формулы IV

и
(с) обработку спирта основанием с получением эпоксида.
24. Способ по п.23, дополнительно включающий этерификацию кислоты формулы (0)

с образованием сложного эфира формулы II.
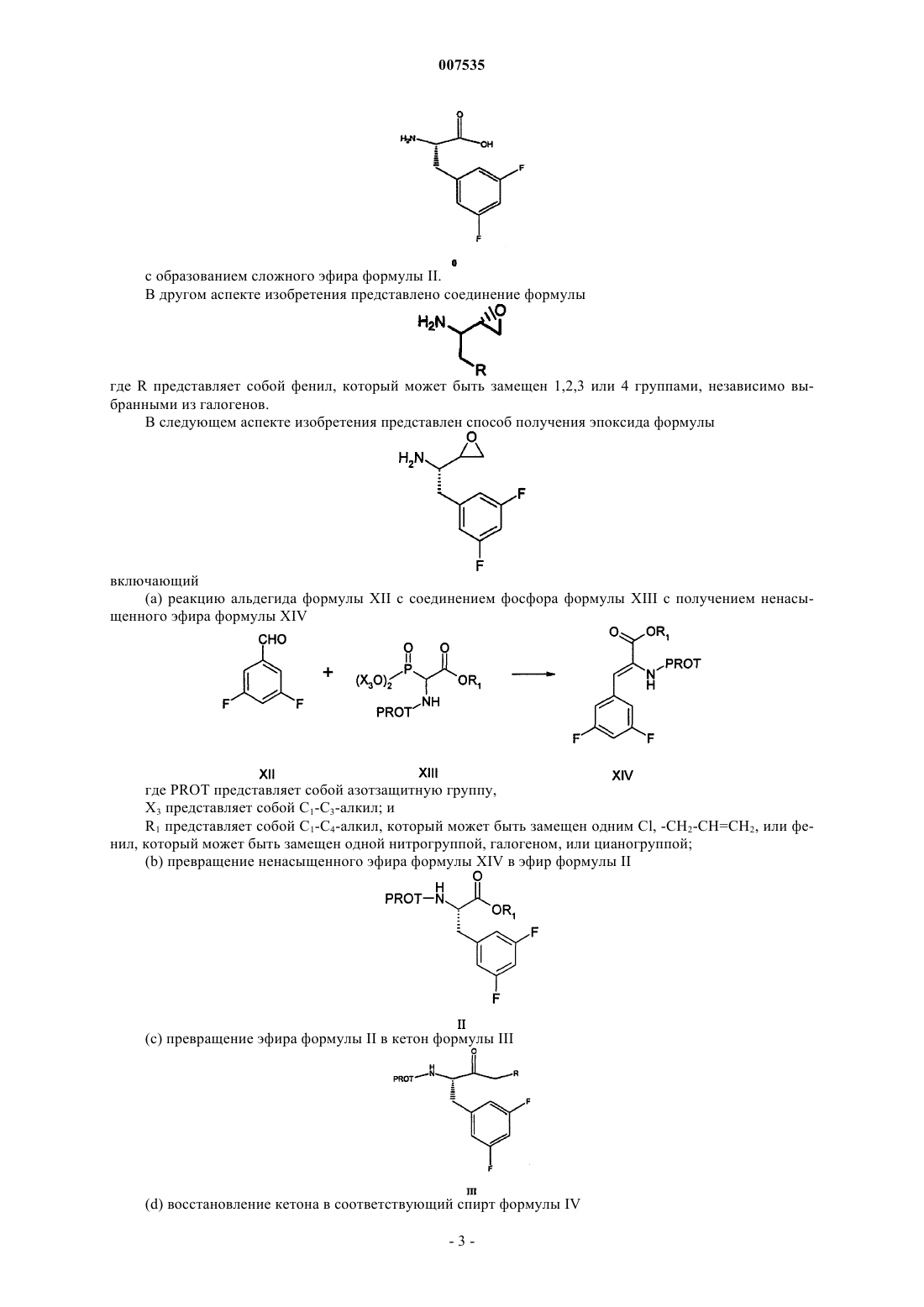
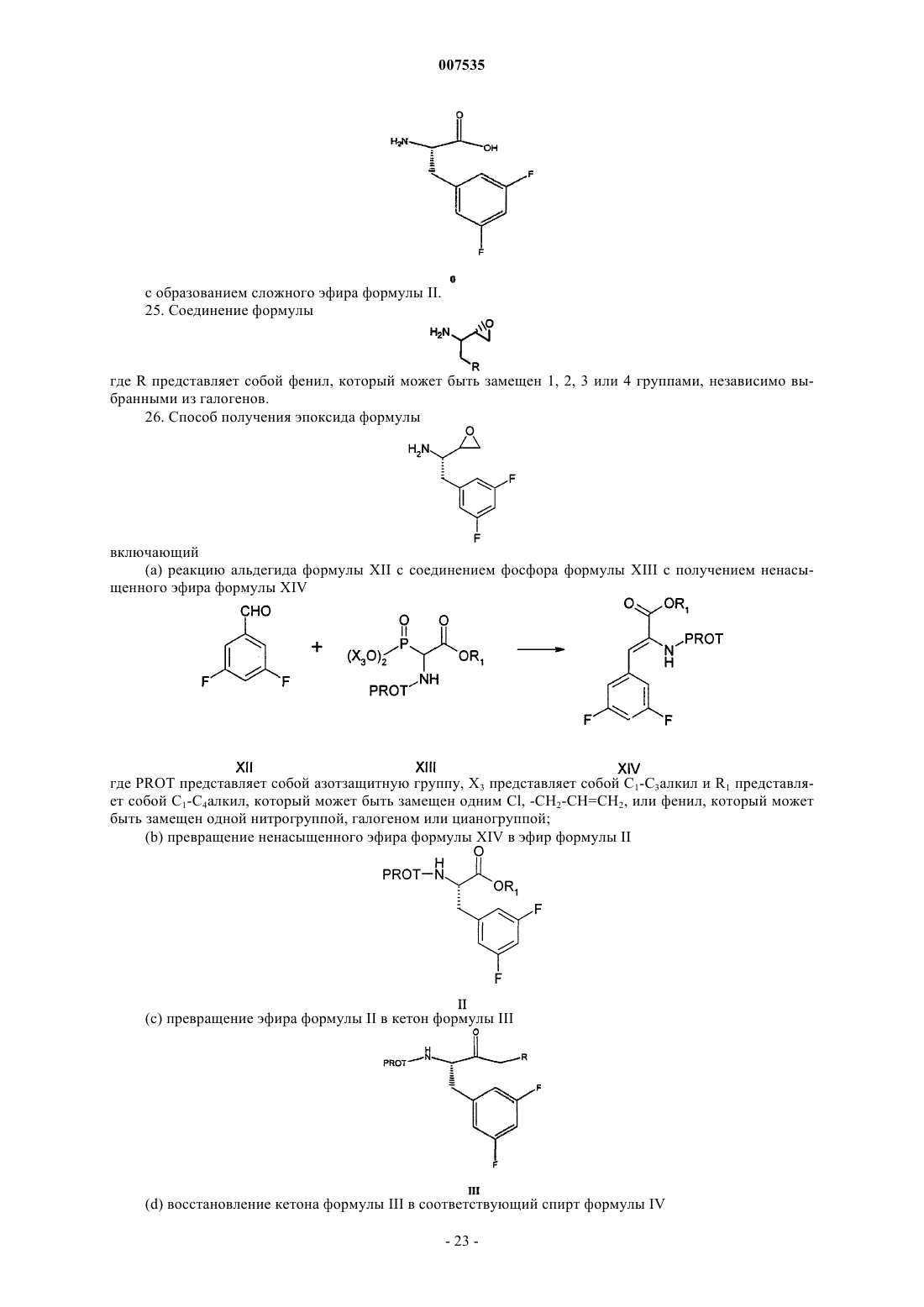
25. Соединение формулы

где R представляет собой фенил, который может быть замещен 1, 2, 3 или 4 группами, независимо выбранными из галогенов.
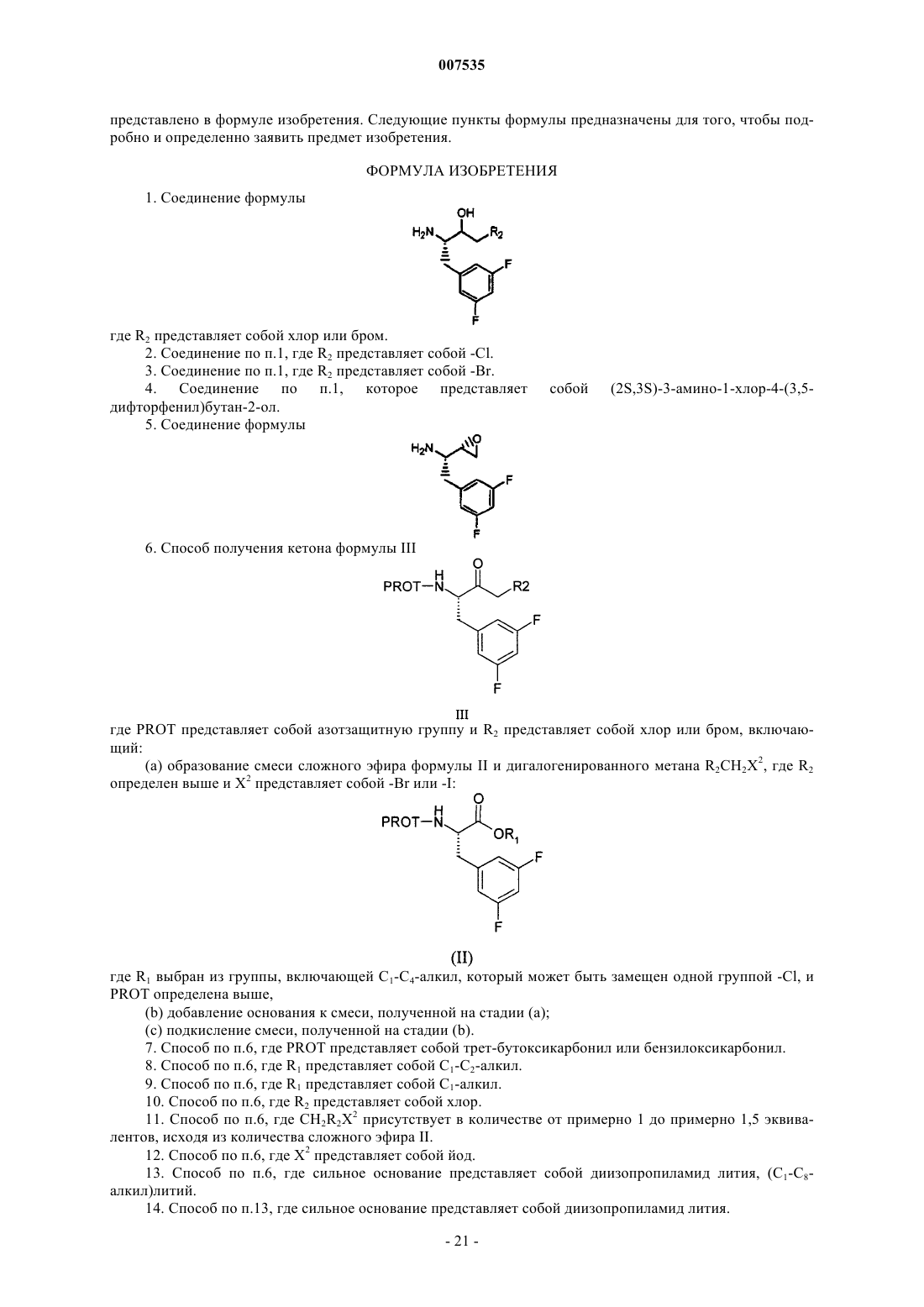
26. Способ получения эпоксида формулы

включающий
(а) реакцию альдегида формулы XII с соединением фосфора формулы XIII с получением ненасыщенного эфира формулы XIV

где PROT представляет собой азотзащитную группу, Х3 представляет собой C1-С3алкил и R1 представляет собой С1-С4алкил, который может быть замещен одним Cl, -СН2-СН=СН2, или фенил, который может быть замещен одной нитрогруппой, галогеном или цианогруппой;
(b) превращение ненасыщенного эфира формулы XIV в эфир формулы II

(с) превращение эфира формулы II в кетон формулы III

(d) восстановление кетона формулы III в соответствующий спирт формулы IV

и (е) обработку спирта основанием с получением эпоксида.
Текст
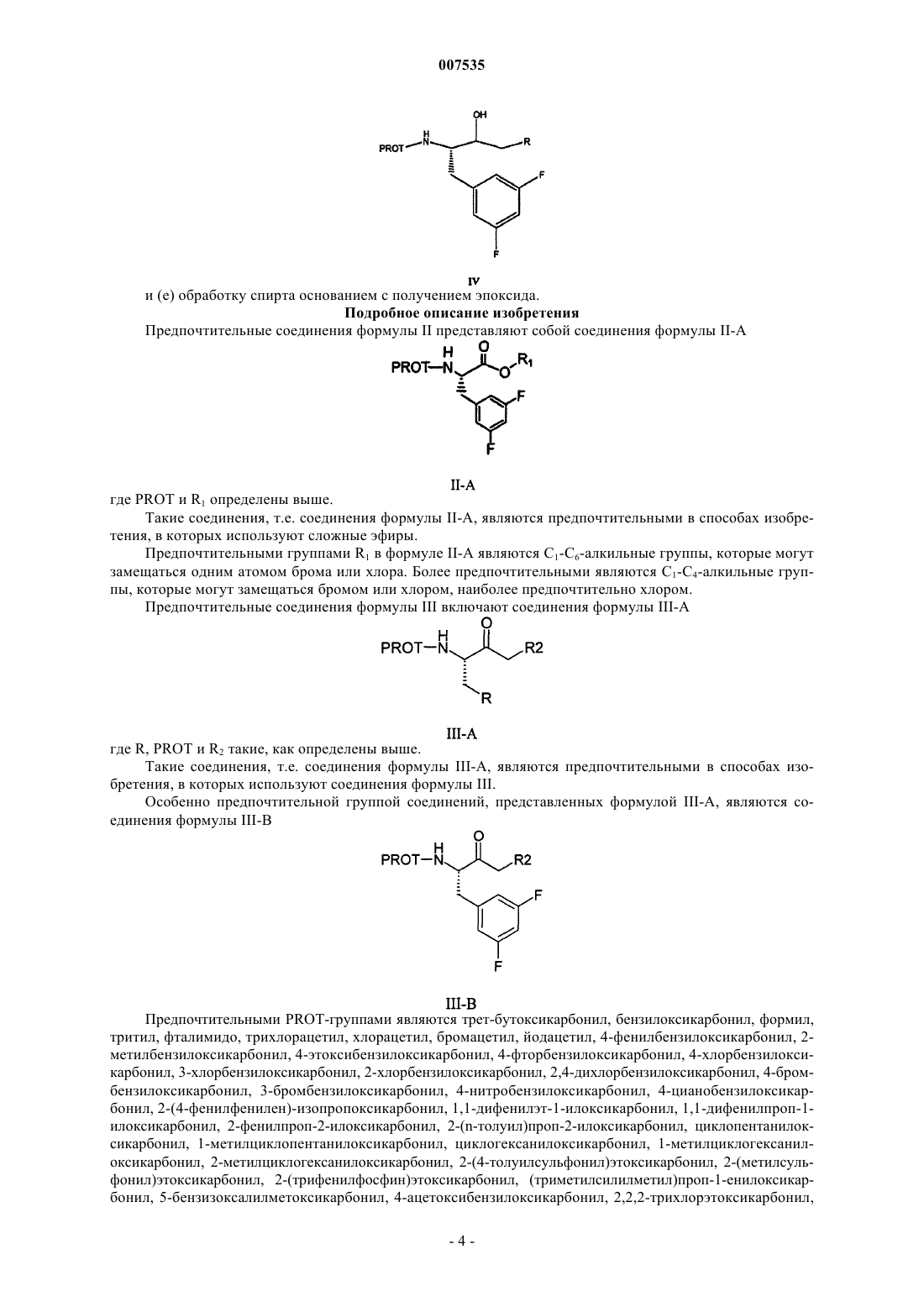
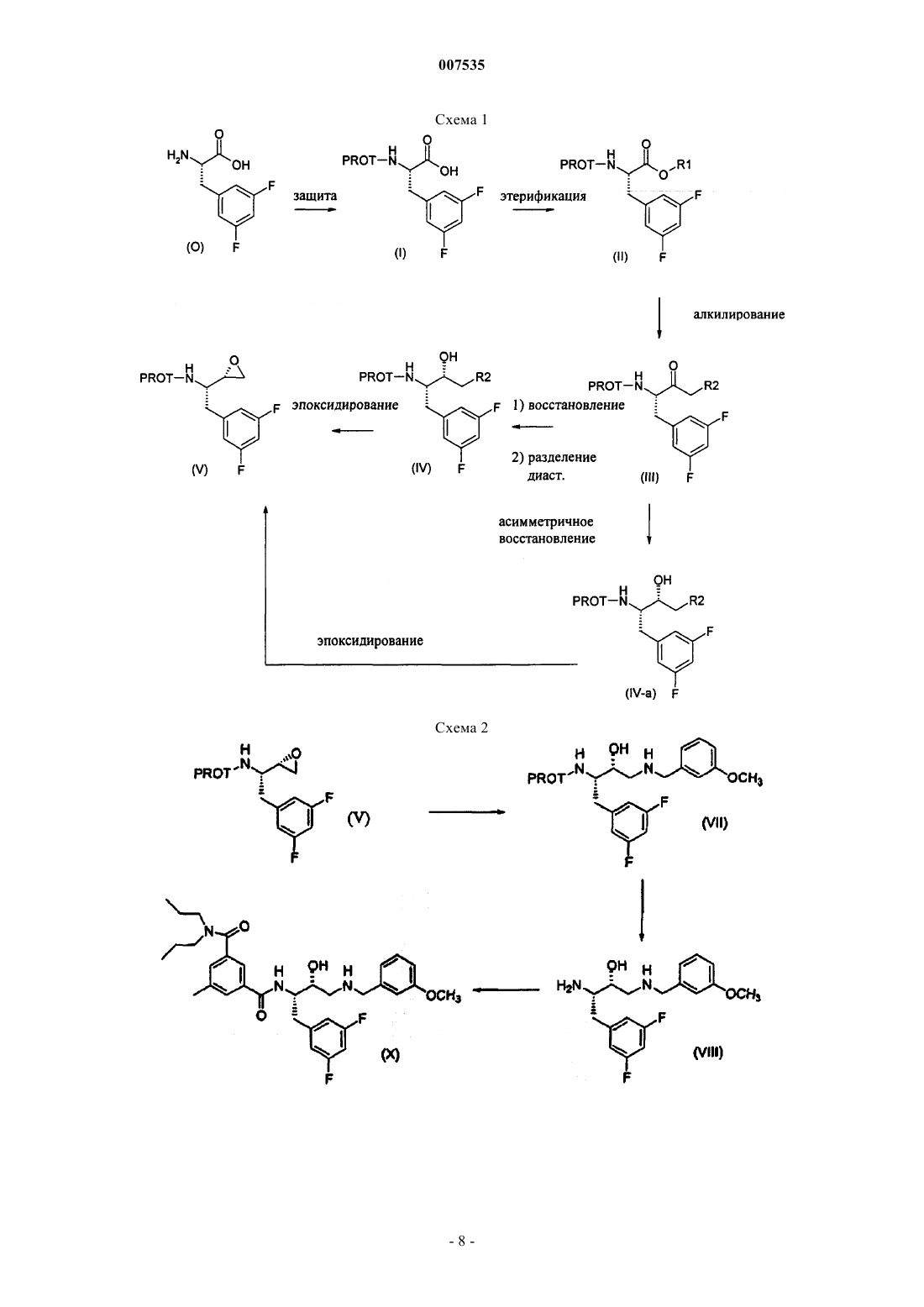
007535 Предпосылки изобретения Эта заявка испрашивает приоритет предварительной заявки США 60/285772, поданной 23 апреля 2001. Область изобретения Настоящее изобретение представляет способы получения бензилзамещенных эпоксидов, пригодных для получения биологически активных соединений, а также промежуточные соединения, применимые в этих способах. Описание уровня техники Международная публикация WO02/02512 раскрывает различные гидроксиэтиламины, пригодные для лечения болезни Альцгеймера. Обычным промежуточным соединением для большинства продуктов является N-защищенный эпоксид. Таким образом, существует необходимость в способах и промежуточных соединениях для эффективного получения N-защищенных эпоксидов. Сущность изобретения В одном аспекте изобретение представляет аминоспирт формулы где R2 представляет собой хлор или бром. Предпочтительным соединением указанной выше структуры является (2S,3S)-3-амино-1-хлор-4(3,5-дифторфенил)бутан-2-ол. В другом аспекте изобретение представляет эпоксид формулы В следующем аспекте изобретение представляет способ получения кетона формулы III где PROT представляет собой защитную группу атома азота, и R2 представляет собой хлор или бром,включающий:(а) образование смеси сложного эфира формулы II и дигалогенированного метана R2CH2X2, где R2 определен выше и X2 представляет собой -Вr или -I: где R1 выбран из группы, включающей С 1-С 4-алкил, который может быть замещен одной группой-Сl, и PROT определена выше,(b) добавление основания к смеси, полученной на стадии (а);(c) подкисление смеси, полученной на стадии (b). В предпочтительном варианте изобретения группа PROT представляет собой трет-бутоксикарбонил-1 007535 или бензилоксикарбонил, R1 представляет собой С 1-С 2-алкил, более предпочтительно C1-алкил, R2 представляет собой хлор, X2 представляет собой йод. В предпочтительном варианте способа получения кетона формулы III количество используемогоCH2R2X2 составляет от примерно 1 до примерно 1,5 эквивалентов, исходя из количества сложного эфираII. В предпочтительном варианте способа сильное основание представляет собой диизопропиламид лития, (С 1-С 8-алкил)литий, более предпочтительно диизопропиламид лития. В предпочтительном варианте способа сильное основание используют в количестве примерно от 2 до 2,5 эквивалентов, исходя из количества сложного эфира II. В предпочтительном варианте способа на стадии b) добавляют вторую порцию основания, представляющего собой (С 1-С 4)алкиллитий. Наиболее предпочтителен способ, где второе основание представляет собой н-бутиллитий, вторбутиллитий, трет-бутиллитий, метиллитий, более предпочтительно н-бутиллитий. В предпочтительном варианте способа количество второго основания составляет от примерно 1 до примерно 1,5 эквивалентов, исходя из количества сложного эфира II. В предпочтительном варианте способа на стадии с) подкисление осуществляют с помощью кислоты, имеющей рКа менее примерно 10, более предпочтительно с помощью уксусной или соляной кислоты или их смесью. В предпочтительном варианте способа кетон (III) представляет собой трет-бутил-(1S)-3-хлор-1-(3,5 дифторбензил)-2-оксопропилкарбамат. В другом аспекте изобретение представляет способ получения эпоксида формулы V-R где R представляет собой фенил, который может быть замещен 1,2,3 или 4 группами, независимо выбранными из галогенов, включающий:(а) превращение сложного эфира формулы II(b) восстановление кетона до соответствующего спирта формулы IV и (с) обработку спирта основанием с получением эпоксида. В предпочтительном осуществлении способа дополнительно осуществляют этерификацию кислоты формулы (0) с образованием сложного эфира формулы II. В другом аспекте изобретения представлено соединение формулы где R представляет собой фенил, который может быть замещен 1,2,3 или 4 группами, независимо выбранными из галогенов. В следующем аспекте изобретения представлен способ получения эпоксида формулы(а) реакцию альдегида формулы XII с соединением фосфора формулы XIII с получением ненасыщенного эфира формулы XIV где PROT представляет собой азотзащитную группу,Х 3 представляет собой С 1-С 3-алкил; иR1 представляет собой С 1-С 4-алкил, который может быть замещен одним Сl, -СН 2-СН=СН 2, или фенил, который может быть замещен одной нитрогруппой, галогеном, или цианогруппой;(b) превращение ненасыщенного эфира формулы XIV в эфир формулы II(с) превращение эфира формулы II в кетон формулы III(d) восстановление кетона в соответствующий спирт формулы IV и (е) обработку спирта основанием с получением эпоксида. Подробное описание изобретения Предпочтительные соединения формулы II представляют собой соединения формулы II-А где PROT и R1 определены выше. Такие соединения, т.е. соединения формулы II-А, являются предпочтительными в способах изобретения, в которых используют сложные эфиры. Предпочтительными группами R1 в формуле II-А являются С 1-С 6-алкильные группы, которые могут замещаться одним атомом брома или хлора. Более предпочтительными являются С 1-С 4-алкильные группы, которые могут замещаться бромом или хлором, наиболее предпочтительно хлором. Предпочтительные соединения формулы III включают соединения формулы III-А где R, PROT и R2 такие, как определены выше. Такие соединения, т.е. соединения формулы III-А, являются предпочтительными в способах изобретения, в которых используют соединения формулы III. Особенно предпочтительной группой соединений, представленных формулой III-А, являются соединения формулы III-В(ВОС) или бензилоксикарбонил (CBZ), более предпочтительней, чтобы PROT представляла собой третбутоксикарбонил. Специалистам в данной области будут понятны предпочтительные способы введения третбутоксикарбонильной или бензилоксикарбонильной групп, и они могут дополнительно проконсультироваться в справочнике Т.W. Green и P.G.M. Wuts в Protective Groups in Organic Chemistry, 3-е изданиеJohn WileySons, Inc. New York, N.Y., 1999. Предпочтительные соединения формулы IV-незащищенный включают соединения формулы IV-Aнезащищенный где R и R2 такие, как определены выше. Такие соединения, т.е. соединения формулы IV-A-незащищенный, являются предпочтительными для применения в способах изобретения, в которых используют соединения формулы IV-незащищенный. Предпочтительные соединения формулы IV-A-незащищенный включают соединения формулы IVB-незащищенный Предпочтительные группы Rp и Rq в формуле IV-B-незащищенный представляют собой независимо выбранные галогены. Более предпочтительно, чтобы Rp и Rq представляли собой атомы фтора. Особенно предпочтительными соединениями формулы IV-B-незащищенный являются соединения, где Rp и Rq представляют собой атомы фтора в 3- и 5-положениях относительно точки присоединения фенильного кольца к исходному метилену. Предпочтительные соединения формулы V-незащищенный включают соединения формулы V-Aнезащищенный где R такой, как определен выше. Такие соединения, т.е. соединения формулы V-A-незащищенный, являются предпочтительными в способах изобретения, в которых используют соединения формулы V-незащищенный. Предпочтительные соединения формулы V-A-незащищенный включают соединения, где R представляет собой фенил, замещенный вплоть до двух групп Rp и Rq. Предпочтительные группы Rp и Rq в формуле V-A-незащищенный представляют собой независимо выбранные галогены. Более предпочтительно, чтобы Rp и Rq представляли собой атомы фтора. Особенно предпочтительными соединениями формулы V-A-незащищенный являются соединения, где Rp и Rq представляют собой атомы фтора в 3- и 5-положениях относительно точки присоединения фенильного кольца к исходному метилену. Предпочтительные соединения формулы XI включают соединения формулы XI-А где R такой, как определен выше,X1 представляет собой хлор, бром или имидазолил. Такие соединения, т.е. соединения формулы XI-A, являются предпочтительными в способах изобретения, в которых используют соединения формулы XI. Предпочтительные соединения формулы XI-A включают соединения, где R представляет собой фенил, замещенный вплоть до двух групп Rp и Rq. Предпочтительные группы Rp и Rq в формуле XI-A представляют собой независимо выбранные галогены. Более предпочтительно, чтобы Rp и Rq представляли собой атомы фтора. Особенно предпочтительными соединениями формулы XI-A являются соединения, где Rp и Rq представляют собой атомы фтора в 3- и 5-положениях относительно места присоединения фенильного кольца к исходному метилену. Другими предпочтительными соединениями формулы II являются соединения формулы XV В соединениях формулы XV R1 такой, как определен выше для формулы II. Предпочтительные алкилирующие агенты для этерификации соединения формулы I в соединение формулы II включают:(а) Х 4-С 1-С 4-алкил, который может быть замещен одним из атомов йода, брома или хлора, предпочтительно хлором;(b) Х 4-СН 2-СН=СН 2,(c) Х 4-СН 2-фенил, где фенильное кольцо может быть замещено нитрогруппой, галогеном, цианогруппой; и где Х 4 представляет собой йод, бром, хлор, -О-тозилат, -О-мезилат или -О-трифлат. Предпочтительными соединениями формулы I являются соединения, имеющие (S)-стереохимию, и где R представляет собой фенил, замещенный двумя атомами галогена, предпочтительно атомами фтора. Предпочтительно, чтобы фенил был замещен в 3- и 5-положениях, более предпочтительно атомами фтора в 3- и 5-положениях. Следующие типичные соединения перечислены для того, чтобы читатель мог представить себе соединения формулы X, которые могут быть получены при использовании данного изобретения. Если не указано иначе, то все названия здесь произведены с помощью программы, разработанной AdvancedN ,N -дипропилизофталамид. Схемы 1-7 в общих чертах представляют способы изобретения; несмотря на то, что в этих схемах в качестве промежуточных и исходных соединений используются различные предпочтительные соединения данного изобретения, следует понимать, что эти способы применимы также к соединениям, не имеющим специфической стереохимии или примеров заместителей, изображенных на схемах. Схема 1 в общих чертах представляет способ получения N-защищенного эпоксида V из известной аминокислоты (0). Эпоксиды формулы V используют в качестве промежуточных соединений для получения биологически активных соединений, например фармацевтических препаратов для лечения болезни Альцгеймера. Схема 2 раскрывает способ превращения эпоксида V в желательные соединения формулы X. Схема 3 раскрывает альтернативный способ превращения защищенной аминокислоты (I) в соответствующий кетон (III). Схема 4 раскрывает альтернативный способ получения сложного эфира (II). Схема 5 раскрывает способ замены защитной группы в сложном эфире (II). Схема 6 раскрывает способ получения незащищенного спирта (IV-незащищенный) и незащищенного эпоксида (V-незащищенный). Схема 7 раскрывает способ превращения эпоксида V-1 в желательные соединения формулы Х-1. Типичные примеры способов получения соединений настоящего изобретения представлены ниже. Эпоксиды формулы V имеют два хиральных центра; поэтому соединения формулы V могут существовать в виде любого из четырех стереоизомеров, т.е. двух пар диастереоизомеров. Хотя биологически активные конечные продукты получают из всех стереоизомеров, особенно предпочтительной является(S,S)-конфигурация. Один из этих хиральных центров в эпоксиде (V) происходит из исходной аминокислоты (0). Поэтому предпочитают начинать с аминокислоты (0), содержащей желательный энантиомерный центр, а не со смеси, для которой необходимо осуществить разделение с получением желательного(S)-энантиомера этой аминокислоты (0). Схема 1 изображает превращение аминокислоты (0) в N-защищенный аминоэпоксид (V). Защита свободной аминогруппы предпочтительной (S)-аминокислоты (0) с помощью азот-защитной группы(PROT) дает защищенную аминокислоту (I), имеющую ту же стереохимию. Азот-защитные группы хорошо известны специалистам в данной области, см., например, Nitrogen Protecting Groups in OrganicGroups in Organic Chemistry, 3rd edition John WileySons, Inc. New York, N.Y., 1999. Когда азотзащитная группа больше не нужна, ее можно удалить. Подходящие способы известны специалистам в данной области. Вниманию читателей снова предлагаются ссылки, указанные выше. Защищенную аминокислоту (I) (предпочтительно (S)-стереохимии) затем превращают в соответст- 10007535 вующий защищенный сложный эфир (II) (сохраняющий предпочтительную (S)-стереохимию). Это превращение может осуществляться множеством способов. Когда R1 представляет собой (а) С 1-С 4-алкил, который может быть замещен одной группой -О, (b) СН 2-СН=СН 2, или (с) фенил, который может быть замещен нитрогруппой, галогеном или цианогруппой,превращение соединения I в соединение II включает:(1) этерификацию защищенной аминокислоты формулы I алкилирующим агентом в присутствии основания. Подходящие алкилирующие агенты включают:(a) агенты, представленные формулой Х 4-С 1-С 4-алкил, который может быть замещен одной группой(d) бензил, замещенный на метальной группе группой Х 4, где значение Х 4 определено выше и где фенильное кольцо может быть замещено нитрогруппой, галогеном или цианогруппой. Хотя для этой этерификации подходит множество оснований, предпочтительным основанием является гидроксид, карбонат, бикарбонат, ЛДА, н-(С 1-С 8-алкил)литий, LiHMDS, NaHMDS или KHMDS. Более предпочтительным основанием является гидроксид, карбонат или бикарбонат. Еще более предпочтительным основанием является карбонат. Предпочтительными алкилирующими агентами являются диметилсульфат, метилйодид и метилтрифлат. Более предпочтительным алкилирующим агентом является диметилсульфат. Когда основание представляет собой ЛДА, н-(С 1-С 8-алкил)литий, LiHMDS, NaHMDS или KHMDS, раствор сложного эфира перед добавлением основания предпочтительно охлаждают в интервале температур примерно от-78 С и более предпочтительно примерно от -20 до 25 С. После добавления алкилирующиего агента эту смесь предпочтительно нагревают примерно до 20 - 50 С. Нагревание особенно полезно, когда алкилирующим агентом является диметилсульфат. Альтернативно, когда R1 является возможно замещенной бензильной группой, этерификацию можно осуществлять путем(a) взаимодействия защищенной аминокислоты формулы (I) с активирующим агентом, т.е. активации аминокислоты или образования активированной аминокислоты; и(b) добавления к смеси, полученной на стадии (а), фенола, который может быть замещен в фенильном кольце нитрогруппой, галогеном или цианогруппой. Применение активирующих агентов, например алкилхлорформиатов, таких как изобутилхлорформиат, КДИ и ДЦК, в процессе этерификации кислот спиртами хорошо известно специалистам в данной области. Предпочтительными активирующими агентами здесь являются КДИ и ДЦК. Предпочтительными сложными эфирами в этом способе являются эфиры, где R1 представляет собой метил или этил, более предпочтительно метил. Особенно предпочтительным сложным эфиром является метиловый эфир (2S)2-[(трет-бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановой кислоты. Схема 4 демонстрирует альтернативный путь получения сложного эфира (II). См. также примеры 9 и 10. В способе, представленном на схеме 4, предпочтительный 3,5-дифторбензальдегид (XII), который коммерчески доступен, например, от фирмы Aldrich (Milwaukee, Wisconsin, США), подвергают реакции с соединением фосфора (XIII), где Х 3 является подходящей уходящей группой, с получением олефина(XIV). Подходящие уходящие группы известны специалистам в данной области. Особенно предпочтительным олефином (XIV) является метил-(2Z)-2-[(бензилокси)карбонил]-3-(3,5-дифторфенил)-2 пропенонат. Соединения фосфора (XIII) известны специалистам в данной области. Х 3 является предпочтительно С 1-С 3-алкильной группой, более предпочтительно -метилом. Альдегид (XII) и соединение фосфора (XIII) обычно объединяют в полярном апротонном органическом растворителе, например, ТГФ, МТБЭ, диоксане, эфире или ДМЭ, и полученную смесь, предпочтительней раствор, затем охлаждают до примерно 0. Прибавляют основание, например ДБУ или ТМГ, и содержимое смеси нагревают примерно до 20-25 С и перемешивают до окончания реакции, т.е. до достижения превращения, предпочтительно превышающего примерно 90%, более предпочтительно примерно 95% и наиболее предпочтительно примерно 99%. Как только реакция завершится, Е- и Z-олефиновые изомеры (XIV) предпочтительно разделяют, посколькуZ-изомер имеет предпочтительную, а в некоторых случаях необходимую стереохимию олефина для получения желательного продукта. Разделение выполняют способами, известными специалистам в данной области, например, с помощью хроматографии на силикагеле. Затем олефин (XIV) гидрируют подходящим гидрирующим катализатором для получения желательного сложного эфира (II). Реакцию можно проводить при давлении от примерно 1 до примерно 100psi. Специалистам в данной области известно множество подходящих катализаторов. Пример класса подходящих катализаторов представлен формулой [Rh(диен)L]+Х-, гдеRh представляет собой родий; диен представляет собой циклооктадиен и норборнадиен;X- представляет собой СlO4-, BF4-, CF3-SO3-, Сl-, Br-, PF6- и SbF6-. Этот класс является предпочтительным для применения в этом аспекте способа данного изобретения, особенно в тех случаях, когда L представляет собой DIPMAP или EtDuPhos. Специалисты в данной области смогут ознакомиться с подходящими специфическими методами восстановления, т.е. гидрирования. Обычно олефин XIV сначала растворяют в растворителе, либо в реакционном сосуде, либо раствор позже переносят в сосуд. Затем в сосуд вводят водород и желательный катализатор. Водород обычно добавляют под давлением, например, от примерно 25 до 75 psi водорода. Катализатор можно добавлять в чистом виде или в виде раствора катализатора, например, в метаноле. Некоторые реакции гидрирования будут давать рацемическую смесь сложных эфиров (II). Поскольку предпочтительной стереохимией сложного эфира (II) является (S)-стереохимия, предпочтительней использовать Z-олефин (XIV) с соответствующим гидрирующим катализатором. Подходящие растворители для гидрирования включают полярные растворители, такие как ТГФ и различные спирты, предпочтительно С 1-С 5-спирты и наиболее предпочтительно метанол, этанол, изопропанол. Еще одним предпочтительным растворителем является ТГФ. Растворитель предпочтительно дегазируют. Кроме того, предпочтительно продувать реакционный сосуд после растворения олефина (XIV) в растворителе и до введения катализатора. Предпочтительно, чтобы гидрирование представляло собой хиральное гидрирование, которое осуществляют в интервале температур от примерно 0 до примерно температуры кипения; предпочтительно,чтобы эта реакция проводилась в интервале температур от примерно 0 до примерно комнатной температуры (20-25). Хиральное гидрирование осуществляют при давлении от примерно одной атмосферы до примерно 100 psig. Предпочтительно проводить хиральное гидрирование при давлении от примерно 1 атмосферы до примерно 70 psi; еще более предпочтительно проводить хиральное гидрирование при давлении от примерно 10 psi до примерно 40 psi. Сложный эфир (II) получают с энантиомерной чистотой,превышающей 90%, предпочтительней с энантиомерной чистотой, превышающей 95%. Гидрирование можно выполнять многими способами, например, в периодическом режиме или в непрерывном режиме. Схема 5 и примеры 11 и 12 раскрывают еще один альтернативный способ получения сложного эфира II. Способ, представленный на схеме 5, позволяет заменить одну азот-защитную группу на другую и,кроме того, дает свободный амин XV. Например, если имеется ВОС-защищенный сложный эфир (II), а необходим СВZ-защищенный сложный эфир (II), то ВОС-защищенный сложный эфир (II) обычно подвергают реакции с кислотой, например соляной кислотой, в подходящем растворителе, например метаноле, при температуре примерно от -20 до температуры кипения с получением свободного амина(XV). Предпочтительным амином XV является метил-(2S)-2-амино-3-(3,5-дифторфенил)пропионат. Свободный амин (XV) затем защищают с помощью другой азот-защитной группы, например CBZ, с получением соответствующего и желательного СВZ-защищенного сложного эфира (И). Защищенный сложный эфир (II), предпочтительно (S)-стереохимии, затем превращают в соответствующий предпочтительно (S)-защищенный кетон (III) с помощью любого из множества способов.R2 предпочтительно представляет собой -Сl или -Вr, более предпочтительно R2 представляет собой-Сl. Один из способов превращения (S)-защищенного сложного эфира (II) в соответствующий (S)защищенный кетон (III) поясняется в примере 16. Обычно защищенный сложный эфир (II), предпочтительно (S)-стереохимии, объединяют с дигалогенированным метановым реагентом, и к этой смеси затем добавляют подходящее основание. Предпочтительней добавлять основание к смеси сложного эфира и дигалогенированного метана, а не наоборот. Затем к полученной смеси основание/сложный эфир/дигалогенированный метан прибавляют вторую порцию основания. Предпочтительней добавлять вторую порцию основания к уже имеющейся смеси. Наконец, основание/сложный эфир/дигалогенированный метан обрабатывают кислотой. Предпочтительно, чтобы X2 представлял собой -I. Предпочтительней использовать примерно от 1 до 1,5 эквивалентовR2CH2X2. Сильное основание должно иметь рКb более примерно 30. Предпочтительно, чтобы сильное основание было выбрано из группы, включающей ЛДА, (С 1-С 8-алкил)литий, LiHMDS, NaHMDS и KHMDS; более предпочтительно, чтобы сильное основание представляло собой ЛДА. Предпочтительно, чтобы сильное основание присутствовало в количестве от примерно 2 до примерно 2,5 эквивалентов. Примеры второго основания включают соединения, выбранные из группы, включающей (С 1-С 4)алкиллитий, фениллитий, (С 1-С 4)алкил-Гриньяр и фенил-Гриньяр. Предпочтительно, чтобы второе основание было выбрано из группы, включающей фениллитий, н-бутиллитий, бромметилмагний, хлорметилмагний, бромфенилмагний или хлорфенилмагний; более предпочтительно, чтобы второе основание представляло собой н-бутиллитий. Предпочтительно, чтобы второе основание присутствовало в количестве от примерно 1 до примерно 1,5 эквивалентов. Подходящими кислотами являются кислоты, которые имеют рКа менее примерно 10. Предпочтительно, чтобы кислота была выбрана из группы, включающей уксусную, серную, соляную, лимонную,фосфорную, бензойную кислоты и их смеси; более предпочтительно, чтобы кислота представляла собой- 12007535 соляную или уксусную кислоту. Для этого способа используют множество растворителей; предпочтительным растворителем для этого способа является ТГФ. Реакцию можно проводить в интервале температур примерно от -80 до -50; предпочтительней проводить реакцию в интервале температур примерно от -75 до -65. Предпочтительно,чтобы кетон (III) представлял собой трет-бутил-(1S)-3-хлор-1-(3,5-дифторбензил)-2-оксопропилкарбамат. Способ превращения (S)-защищенного сложного эфира (II) в соответствующий (S)-защищенный кетон (III) можно осуществлять также без добавления второго основания, см. пример 2. Этот способ требует присутствия избытка СН 2(R2)Х 2 и трех или более эквивалентов сильного основания, которое имеет рКb более примерно 30, и последующего добавления кислоты. Кроме того, (S)-защищенный сложный эфир (II) также можно превращать в соответствующий кетон(2) взаимодействие смеси, полученной на стадии (1), со сложным эфиром формулы (II); и(3) взаимодействие смеси, полученной на стадии (2), с кислотой. В этом способе предпочтительно, чтобы сильное основание было выбрано из группы, включающей ЛДА, (С 1-С 8-алкил)литий, LiHMDS, NaHMDS и KHMDS; более предпочтительно, чтобы основание представляло собой ЛДА. Предпочтительней использовать примерно от 2 до 2,5 эквивалентов сильного основания. Те же кислоты, которые обсуждались выше, используются также и здесь. Схема 3 и пример 15 представляют альтернативный путь получения кетона (III) из аминокислоты(I). В этом способе аминокислоту (I) сначала превращают в промежуточное соединение (XI), а затем промежуточное соединение (XI) превращают в желательный кетон (III). Превращение аминокислоты (I) в промежуточное соединение (XI) включает:(1) взаимодействие защищенной аминокислоты формулы (I) с реагентом, выбранным из группы,включающей тионилхлорид, SO2Cl2, треххлористый фосфор, оксалилхлорид, трехбромистый фосфор,трифенилдибромофосфоран, оксалилбромид, 1,2-фенилентрихлорфосфат и 2,4,6-трихлор-1,3,5-триазин. Предпочтительно, чтобы реагент представлял собой тионилхлорид или оксалилхлорид. Промежуточное соединение (XI) не выделяют. Предпочтительно, чтобы промежуточное соединение (XI) представляло собой трет-бутил-(1S)-2-хлор-1-[3,5-дифторбензил]-2-оксоэтилкарбамат. Промежуточное соединение(1) взаимодействие карбонильного соединения формулы (XI), где X1 представляет собой -Cl, -Вr и имидазолил, с LiСН 2 Сl или LiСН 2 Вr. Это соединение затем подвергают реакции с анионом, полученным из реагента CH2R2X2. Используют различные растворители, которые известны специалистам в данной области; предпочтительным растворителем является ТГФ. Реакцию следует проводить на холоду в интервале температур примерно от -78 до -50. Затем (S)-защищенный кетон (III) восстанавливают до соответствующего (S)-спирта (IV) или (IV-a) способами, известными специалистам в данной области, например, путем восстановления кетона до соответствующего вторичного спирта, см. ПРИМЕР 3. Кроме этого, Европейская патентная заявка ЕР 0963972 А 2 и международная заявка PCT/US01/21012 (W002/02512) раскрывают альтернативные реагенты, которые используются и хорошо работают в восстановлении. Реакции восстановления проводят в течение периода примерно от 1 часа до 3 дней при температурах, варьирующих примерно от -78 до повышенных температур, вплоть до точки кипения использующегося растворителя. Предпочтительно проводить восстановление в интервале примерно от -78 до 0. В случае использования борана, его можно применять в виде комплекса, например боран-метилсульфидного комплекса, боран-пиперидинового комплекса или боран-тетрагидрофуранового комплекса. Предпочтительная комбинация восстанавливающих агентов и необходимые условия реакции известны специалистам в данной области, см., например, Larock R.C. в Comprehensive Organic Transformations, VCH Publishers, 1989. Восстановление (S)-защищенного соединения (III) до соответствующего спирта (IV) дает второй хиральный центр и смесь диастереоизомеров по второму центру, (S,R/S)-спирт (IV). Эту диастереоизомерную смесь затем разделяют способами, известными специалистам в данной области, такими как селективная перекристаллизация при низких температурах или хроматографическое разделение, наиболее предпочтительными способами являются перекристаллизация, колоночная хроматография или применение коммерчески доступных хиральных колонок. В другой реализации диастереоизомерную смесь, полученную путем неселективного восстановления (S)-защищенного соединения (III), не разделяют, а сразу превращают в эпоксид. Эпоксидные диастереоизомеры могут быть затем разделены способами, хорошо известными в данной области. Или эпоксидные диастереоизомеры могут быть подвергнуты реакции с амином RCNH(R57) с образованием соединений, структура которых аналогична структуре (VII-1, где RC представляет собой 3-метоксибензил и R57 представляет собой Н.) Диастереоизомеры могут быть разделены на этой стадии, либо могут быть осуществлены дополнительные превращения до того, как будут разделяться диастереоизомеры. Например,разделение диастереоизомеров может быть осуществлено после депротекции (снятия защитных групп) спирта (VII) с образованием свободного амина (VIII), либо разделение может быть осуществлено после превращения амина (VIII) в структуру (X).- 13007535 Альтернативно, (S)-защищенное соединение (III) может быть селективно восстановлено в S- или Rформу спирта, как показано на схеме 1, где селективно образуется S-спирт. Селективное восстановление уменьшит необходимость разделения диастереоизомеров, которое обсуждалось выше, и увеличит количество образующегося желательного изомера. В идеале, во время восстановления кетона в спирт образуется единственный диастереоизомер, и разделения не требуется. Спирт (IV) превращают в соответствующий эпоксид (V) способами, известными специалистам в данной области, см. схему 6 (выше) и пример 4. При образовании эпоксида (V) поддерживают стереохимию (S)-(IV) центра. Предпочтительный способ подразумевает реакцию с основанием, например, но не только, с гидроксид-ионом, полученным из гидроксида натрия, гидроксида калия, гидроксида лития и тому подобного. Условия реакции включают применение С 1-С 6-спиртовых растворителей, предпочтительно - этанола. Реакции проводят при температурах, варьирующих примерно от -45 до температуры кипения использующегося спирта; предпочтительные интервалы температур составляют примерно от -20 до 40. Защищенные эпоксиды аминокислот (V) известны специалистам в данной области в качестве промежуточных соединений для получения фармацевтических агентов, пригодных в качестве ингибиторов ренина и HIV, см., например, патенты US 5482947, 5508294, 5510349, 5510388, 5521219, 5583238,5610190, 5639769, 5760064 и 5965588. Кроме того, защищенные эпоксиды (V) являются промежуточными соединениями, использующимися для изготовления фармацевтических агентов для лечения болезни Альцгеймера. Эпоксиды (V) превращают в полезные соединения с помощью способа, представленного на схемах 2 и 3 и в примерах 5-8. Предпочтительным соединением является соединение из примера 8. Незащищенный эпоксид (V-незащищенный) используют аналогичным образом. Его можно легко защитить с образованием эпоксида (V) или можно подвергнуть реакции в незащищенной форме. В некоторых случаях свободная аминогруппа может мешать последующим реакциям, но в иных случаях она будет работать достаточно хорошо. В некоторых случаях можно сначала провести реакцию по N-концу и затем раскрыть эпоксид с получением желательного соединения (X). Соединения (X) представляют собой амины и способны образовывать соли при взаимодействии с кислотой. Фармацевтически приемлемые соли предпочтительнее, чем соответствующие соединения (X),поскольку они часто дают соединения, которые являются более водорастворимыми, стабильными и/или более кристаллическими. Фармацевтически приемлемые соли представляют собой любую соль, которая сохраняет активность исходного соединения и не оказывает какого-либо вредного или нежелательного воздействия на субъект, которому ее вводят, и в контексте, в котором ее вводят. Фармацевтически приемлемые соли включают соли как неорганических, так и органических кислот. Предпочтительные фармацевтически приемлемые соли включают соли следующих кислот: соляной, бромисто-водородной, йодисто-водородной, азотной, серной, фосфорной, лимонной, метансульфокислоты, СН 3-(СН 2)n1-СООН,где n1 означает 0 - 4, НООС-(СН 2)n1-СООН, где n1 определено выше, НООС-СН=СН-СООН, -СООН. Информацию о других приемлемых солях см. в Int. J. Pharm., 33, 201-217 (1986). Соединения (X) и их фармацевтически приемлемые соли пригодны для лечения людей, страдающих от болезни Альцгеймера, для предупреждения или замедления наступления болезни Альцгеймера,для лечения пациентов с синдромом слабого когнитивного снижения (МКС) и предупреждения или замедления наступления болезни Альцгеймера у тех, у кого, возможно, будет наблюдаться прогрессия от МКС к болезни Альцгеймера (БА), для лечения синдрома Дауна, для лечения людей с наследственной церебральной геморрагией, ассоциированной с амилоидозом голландского типа, для лечения церебральной амилоидной ангиопатии и предупреждения ее возможных последствий, например однократных и рецидивирующих лобарных геморрагий, для лечения других дегенеративных деменций, включая деменции смешанного сосудистого и дегенеративного происхождения, деменцию, ассоциированную с болезнью Паркинсона, деменцию, ассоциированную с прогрессирующим супрануклеарным параличом, деменцию, ассоциированную с кортикальной базальной дегенерацией, болезнь диффузных телец Леви альцгеймеровского типа. Соединения предпочтительней применяют для лечения, предупреждения и/или облегчения болезни Альцгеймера. Определения и условные обозначения Ниже даны определения и объяснения терминов, которые используются во всем документе, включая описание и формулу изобретения. Термином алкил и С 1-С 6-алкил в настоящем изобретении обозначают алкильные группы с прямой или разветвленной цепью, имеющие от 1 до 6 атомов углерода, например метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, 2-пентил, изопентил, неопентил, гексил, 2-гексил, 3 гексил и 3-метилпентил. Очевидно, что в тех случаях, когда алкильная цепь заместителя (например, алкильной, алкокси- или алкенильной группы) короче или длиннее 6 атомов углерода, то это будет указано у второго атома С, как, например, С 1-С 10 указывает максимальное количество атомов углерода - 10. Термином галоген в настоящем изобретении обозначают фтор, бром, хлор и йод. Все температуры указаны в градусах Цельсия. ТСХ означает тонкослойную хроматографию. ВЭЖХ означает высокоэффективную жидкостную хроматографию.psi означает давление в фунтах на квадратный дюйм. КДИ означает 1,1'-карбонилдиимидазол. ДЦК означает дициклогексилкарбодиимид. ТМГ означает 1,1,3,3-тетраметилгуанидин. ДМФА означает диметилформамид. ДБУ означает 1,8-диазабицикло[5,4,0]ундец-7-ен. ДБН означает 1,5-диазабицикло[4,3,0]нон-5-ен. ЛДА означает лития диизопропиламид.KHMDS означает бис(триметилсилил)амид калия. ВОС означает трет-бутоксикарбонил; 1,1-диметилэтоксикарбонил; (СН 3)3 С-О-СО-. Основание Хюнига означает ДИПЭА, диизопропилэтиламин, [(СН 3)2 СН]2-N-СН 2 СН 3. ДМАП означает диметиламинопиридин, (СН 3)2N-пиридин-1-ил. Рассол означает водный насыщенный раствор хлорида натрия. Хроматография (колоночная и флэш-хроматография) означает очистку/разделение соединений, определяется как (носитель; элюент). Очевидно, что соответствующие фракции объединяют и концентрируют с получением желательного соединения (соединений). 13 С-ЯМР означает 13 С магнитно-резонансную спектроскопию, химические сдвиги описываются в-1 млнотносительно ТМС. 1 Н-ЯМР означает ядерную (протонную) магнитно-резонансную спектроскопию, химические сдвиги описываются в млн-1 (d) относительно ТМС. ТМС означает триметилсилил.означает фенил (C6H5). МС означает масс-спектрометрию, выраженную в единицах т/е, m/z или масса/заряд. [М+Н]+ относится к положительному иону исходного атома плюс атом водорода. ЭУ означает электронный удар. ХИ означает химическую ионизацию. ББА означает бомбардировку быстрыми атомами (от англ. FAB - fastatom bombardment). ЭСМС означает электроспрей-масс-спектрометрию. МСВР означает масс-спектрометрию высокого разрешения. Фармацевтически приемлемый означает такие свойства и/или вещества, которые приемлемы для пациента с фармакологической/токсикологической точки зрения, а для изготовителя-фармацевта с физической/химической точки зрения, что касается композиции, препарата, стабильности, переносимости пациентом и биодоступности. Фармацевтически приемлемые анионные соли включают соли следующих кислот: метансульфокислоты, соляной, бромисто-водородной, серной, фосфорной, азотной, бензойной, лимонной, винной,фумаровой, малеиновой, СН 3-(СН 2)n-СООН, где n означает 0-4, HOOC-(CH2)n-COOH, где n определено выше.-О-трифлат означает -О-трифторуксусную кислоту. Когда используют пары растворителей, то соотношение растворителей представляют в виде отношения их объемов (об./об.). Когда используют растворимость осадка в растворителе, то соотношение осадка к растворителю представляют как вес/объем (вес/об.).PNNP означает N,N'-бис(дифенилфосфин)-N,N'-бис[(R)-1-фенил]этилендиамин. Тионилхлорид означает SOCl2. Треххлористый фосфор означает РСl3. Оксалилхлорид означает (СОСl)2. Трехбромистый фосфор означает РВr3. Трифенилдибромофосфоран означает 3 РВr2. Оксалилбромид означает (СОВr)2. Эфир означает диэтиловый эфир. 1,2-Фенилентрихлорфосфат означает МТБЭ означает метил-трет-бутиловый эфир. ДМЭ означает диметоксиэтан. Все статьи и ссылки, раскрытые в этой заявке, включая патенты, включены здесь посредством ссылки. Кроме того, изобретение проиллюстрировано последующими примерами, которые не следует рассматривать в качестве примеров, ограничивающих данное изобретение рамками или сущностью специфических методов, описанных в них. Исходные соединения и различные промежуточные соединения могут быть получены из коммерческих источников, приготовлены из коммерчески доступных органических соединений или получены с использованием хорошо известных способов синтеза. Примеры Следующие подробные примеры описывают, как получить различные соединения и/или осуществить различные способы изобретения, и должны рассматриваться лишь в качестве иллюстративных и не ограничивающих предшествующее раскрытие примеров. Специалисты в данной области познакомятся с соответствующими модификациями методов, которые касаются как реагентов, так и условий реакций. Пример 1. Метиловый эфир (2S)-2-[(трет-бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановой кислоты (II). В 1 л 3-х-горлую круглодонную колбу, снабженную магнитной мешалкой, отверстием для впуска азота и термопарой, добавляют (2S)-2-[(тpeт-бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановую кислоту (I, 40 г, 0,133 моль, 1 экв.), затем ТГФ (240 мл). Гидроксид лития моногидрат (5,6 г, 0,133 моль, 1 экв.) прибавляют одной порцией и перемешивают в течение 30 мин, одновременно охлаждая содержимое колбы до 0. После того как реакционная смесь охладится, через шприц по каплям добавляют диметилсульфат (12,6 мл, 0,133 моль, 1 экв.) и перемешивают в течение 30 мин. Затем эту смесь нагревают до примерно 50 и осуществляют контроль реакции (с помощью ВЭЖХ) до тех пор, пока не будет достигнута 90%-ная конверсия. В то же время смесь охлаждают до температуры ниже 20 (образуется осадок). Затем смесь вливают в бикарбонат натрия (200 мл), перемешивают в течение 15 мин и затем экстрагируют метил-тpeт-бутиловым эфиром (200 мл). Фазы разделяют и водный слой экстрагируют метил-третбутиловым эфиром (2 х 200 мл). Объединенные органические фазы промывают водой (400 мл), высушивают над сульфатом натрия, фильтруют и концентрируют при пониженном давлении с получением осадка. Этот продукт затем перекристаллизовывают из гексана с получением названного соединения,т.пл.=81; 1 Н-ЯМР (ДМСО-d6)7,51, 7,15-7,25, 4,43, 3,81, 3,00-3,26 и 1,49; 13 С-ЯМР (ДМСО-d6)172,43,- 16007535 163,74, 161,20, 155,67, 142,58, 112,70, 120,23, 78,69, 54,71, 52,24, 39,25 и 28,37. Пример 2. трет-Бутил-(1S)-3-хлор-1-(3,5-дифторбензил)-2-оксопропилкарбамат (III). В 1 л 3-х-горлую круглодонную колбу, снабженную магнитной мешалкой, отверстием для впуска азота, термопарой и дополнительной воронкой, добавляют метиловый эфир (2S)-2-[(трет-бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановой кислоты (II, пример 1, 10,0 г, 0,0317 моль, 1 экв.), затем ТГФ (175 мл), эту смесь затем охлаждают до -78. После того как смесь охладится, добавляют йодхлорметан (9,25 мл, 0,127 моль, 4 экв.) одной порцией через шприц. В дополнительную воронку загружают ЛДА (79 мл, 0,158 моль, 5 экв., 2,0 М в смеси гептан/ТГФ) и затем по каплям добавляют к этой смеси,поддерживая внутреннюю температуру ниже -70. После завершения добавления содержимое колбы перемешивают в течение 15 мин, одновременно добавляя по каплям через дополнительную воронку уксусную кислоту (47,2 мл, 0,824 моль, 26 экв.) и поддерживая внутреннюю температуру ниже -65. После завершения добавления смесь перемешивают в течение 15 мин, затем нагревают до 0 и вливают в воду(500 мл), рассол (500 мл) и метил-трет-бутиловый эфир (500 мл), затем переносят в разделительную воронку. Фазы разделяют и водную фазу экстрагируют метил-трет-бутиловым эфиром (2 х 250 мл). Объединенные органические фазы промывают насыщенным раствором бикарбоната натрия (500 мл), сульфитом натрия (500 мл) и водой (500 мл). Органическую фазу затем высушивают над сульфатом натрия,фильтруют и концентрируют при пониженном давлении с получением осадка. Этот осадок перекристаллизовывают из смеси гептан/изопропиловый спирт (10/1) с получением названного соединения,т.пл.=139; 1 Н-ЯМР (ДМСО-d6)7,47, 7,06-7,14, 4,78, 4,49, 3,20, 2,82 и 1,40; 13 С-ЯМР (ДМСО-d6)200,87, 163,74, 161,20, 142,74, 112,80, 102,13, 79,04, 58,97, 47,72, 34,95 и 28,30. Пример 3. трет-Бутил-(1S,2S)-3-хлор-1-(3,5-дифторбензил)-2-гидроксипропилкарбамат (IV). В 250 мл 3-х-горлую круглодонную колбу, снабженную магнитной мешалкой, отверстием для впуска азота и термопарой, добавляют трет-бутил-(1S)-3-хлор-1-(3,5-дифторбензил)-2-оксопропилкарбамат (III, пример 2, 4,4 г, 0,0132 моль, 1 экв.), затем ТГФ (20 мл) и этанол (30 мл), эту смесь затем охлаждают до -78. После того как смесь охладится, добавляют боргидрид натрия (2,0 г, 0,0527 моль, 4 экв.) в виде твердой порции в течение 30 мин, поддерживая внутреннюю температуру ниже -70. После завершения добавления содержимое колбы перемешивают в течение 2 ч при -78, затем нагревают до 0 и перемешивают еще 1 ч. Смесь гасят путем добавления насыщенного бисульфата калия (15 мл) и воды (15 мл). Эту суспензию перемешивают в течение 30 мин при 20-25, затем концентрируют при пониженном давлении до половины ее объема. Эту смесь затем охлаждают до 0 и перемешивают в течение 30 мин. После этого полученные осадки собирают путем фильтрации и промывают водой (2 х 50 мл), затем высушивают при пониженном давлении при 50 с получением грубого продукта. Наблюдали соотношение син/антиконформеров 4-9:1. Желательный продукт перекристаллизовывают из смеси гексан/этанол (25/1) с получением названного соединения, т.пл.=149; 1 Н-ЯМР (ДМСО-d6)6,89-7,16, 5,61, 3,64-3,83, 3,19,2,69 и 1,41; 13 С-ЯМР (ДМСО-d6)163,67, 161,24, 155,44, 112,70, 101,55, 78,04, 72,99, 54,29, 48,24, 35,97 и 28,37. Пример 4. трет-Бутил-(1S)-2-(3,5-дифторфенил)-1-[(2S)-оксиранил]этилкарбамат (V). В 250 мл 3-х-горлую круглодонную колбу, снабженную магнитной мешалкой, отверстием для впуска азота и термопарой, добавляют тpeт-бутил-(1S,2S)-3-хлop-1-(3,5-дифторбензил)-2-гидроксипропилкарбамат (IV, пример 3, 3,5 г, 0,010 моль, 1 экв.), затем абсолютный этанол (60 мл), и эту смесь охла- 17007535 ждают до 0. К этой смеси в течение 1 ч добавляют гидроксид калия (0,73 г, 0,013 моль, 1,25 экв.), растворенный в абсолютном этаноле (10 мл), и полученную суспензию нагревают до 15-20 и перемешивают в течение 1 ч. За это же время добавляют воду (100 мл), и содержимое реакции охлаждают до -5 и перемешивают в течение 30 мин. Осадки собирают путем фильтрации и промывают холодной водой (2 х 25 мл), затем высушивают при пониженном давлении при 45 с получением названного соединения,т.пл.=133; 1 Н-ЯМР (ДМСО-d6)7,03, 3,61, 2,68-2,98 и 1,33; 13 С-ЯМР (ДМСО-d6)163,72, 161,29,155,55, 143,35, 112,65, 101,80, 78,17, 53,42, 52,71, 44,90, 36,98 и 28,36. Анти-диастереомер, т.пл.=101. Пример 5. трет-Бутил-(1S,2R)-1-(3,5-дифторбензил)-2-гидрокси-3-[(3-метоксибензил)амино]пропилкарбамат(VII). трет-Бутил-(1S)-2-(3,5-дифторфенил)-1-[(2S)-оксиранил]этилкарбамат (V, пример 4, 245 мг, 0,82 ммоль) суспендируют в изопропиловом спирте (6 мл) и добавляют при перемешивании 3-метоксибензиламин(160 мкл, 1,22 ммоль) при 20-25. Эту смесь нагревают до слабого кипения (температура бани 85) в токе азота в течение 2 ч, после чего полученную смесь концентрируют при пониженном давлении с получением названного соединения. Названное соединение очищают посредством флэш-хроматографии (2-5% метанол/хлористый метилен, градиентная элюция) с получением очищенного названного соединения. Пример 6. (2R,3S)-3-Амино-4-(3,5-дифторфенил)-1-[(3-метоксибензил)амино]-2-бутанол (VIII). трет-Бутил-(1S,2R)-1-(3,5-дифторбензил)-2-гидрокси-3-[(3-метоксибензил)амино]пропилкарбамат(VII, пример 5, 258 мг, 0,59 ммоль) растворяют в хлористом метилене (1 мл) при 20-25 и при перемешивании в токе азота добавляют трифторуксусную кислоту (1 мл). Эту смесь перемешивают в течение 1 ч при 20-25, после чего концентрируют при пониженном давлении с получением названного соединения. Названное соединение используют в следующей реакции без дополнительной очистки. Пример 7. N1-(1S,2R)-1-(3,5-Дифторбензил)-2-гидрокси-3-[(3-метоксибензил)амино]пропил-5-метил-N3,N3-дипропилизофталамид (X).(2R,3S)-3-Амино-4-(3,5-дифторфенил)-1-[(3-метоксибензил)амино]-2-бутанол (VIII, пример 6) растворяют в безводном ДМФА (3 мл) и охлаждают до 0. Добавляют триэтиламин (500 мкл, 3,6 ммоль) и 5 метил-N,N-дипропилизофталамид (IX, 156 мг, 0,59 ммоль) при перемешивании. Эту смесь кратковременно нагревают до 20-25 для полного растворения карбоновой кислоты, предварительно охлажденной до 0. Прибавляют при перемешивании 1-гидроксибензотриазол (157 мг, 1,2 ммоль), затем 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (229 мг, 1,2 ммоль). Полученную смесь перемешивают при 0 в течение 5 мин, затем нагревают до 20-25 в течение 15 ч. Эту смесь затем гасят водным раствором лимонной кислоты (10%), и полученную смесь трижды экстрагируют этилацетатом. Объединенные органические экстракты промывают насыщенным раствором бикарбоната натрия, рассолом, высушивают над сульфатом натрия, фильтруют и концентрируют при пониженном давлении с получением названного соединения в сыром виде. Этот продукт очищают посредством флэш-хроматографии (2-10% метанол/хлористый метилен, градиентная элюция) с получением очищенного названного соединения, ЭСМС МН+=582,3. Пример 8. N1-(1S,2R)-1-(3,5-Дифторбензил)-2-гидрокси-3-[(3-йодбензил)амино]пропил-5-метил 3 3 3,5-Дифторбензальдегид (XII, 2,87 г, 0,02 моль, 1 экв.) и ТГФ (100 мл) перемешивают и охлаждают до примерно 0. N-(Бензилоксикарбонил)фосфонилглицинтриметиловый эфир (XIII, 8,7 г, 0,026 моль, 1,3 экв.) прибавляют к смеси 3,5-дифторбензальдегид (ХII)/ТГФ. Затем по каплям прибавляют 1,1,3,3-тетраметилгуанидин (4,0 мл, 0,032 моль, 1,56 экв.). Реакционную смесь перемешивают в течение 5 мин при 0, затем нагревают до 20-25. Через 2 ч, после того как реакция завершится (анализ с помощью ТСХ), добавляют воду (100 мл) и этилацетат (100 мл). Фазы разделяют и водную фазу экстрагируют этилацетатом (100 мл),объединенные органические фазы промывают рассолом (100 мл), высушивают над сульфатом натрия,фильтруют и концентрируют при пониженном давлении с получением сырого продукта. Этот продукт очищают с помощью хроматографии на силикагеле (этилацетат/гексан; 15/85) с получением названного соединения, т.пл.=112; 1 Н-ЯМР (CDCl3)7,19, 7,06, 6,86, 6,15, 6,43, 4,97 и 3,69; 13 С-ЯМР (CDCl3)165,56, 164,54, 164,41, 162,07, 137,39, 136,02, 128,97, 128,80, 128,62, 128,57, 128,47, 126,25, 112,57, 112,38,105,22, 104,97, 104,72, 68,17 и 53,33. Получают дополнительный продукт, который представляет собой смесь Е- и Z-олефинов. Пример 10. Метил-(2S)-2-[(бензилокси)карбонил]амино-3-(3,5-дифторфенил)пропаноат (II). Метил-(2Z)-2-[(бензилокси)карбонил]-3-(3,5-дифторфенил)-2-пропенонат (XIV, XIV, пример 9, 0,100 г,0,228 ммоль) и дегазированный метанол (10 мл) перемешивают в 100 мл бомбе Хастеллой (Hastelloy bomb). Смесь трижды продувают водородом (60 psig) и затем перемешивают при давлении водорода 60 psig в течение 60 мин при 20-25. Затем добавляют R,R)-DIPAP)Rh (5,2 мг, 3 моль%), растворенный в метаноле (1 мл, дегазированный), и систему продувают водородом (3 х 60 psig). Содержимое сосуда затем перемешивают при давлении водорода 20 psig в течение ночи при 25, за это время реакция завершается,что определяют с помощью ВЭЖХ. Затем систему продувают и фильтруют для удаления катализатора, а растворитель удаляют при пониженном давлении с получением названного соединения. Пример 11. Метил-(2S)-2-амино-3-(3,5-дифторфенил)пропаноат (XV). Перемешивают метиловый эфир (2S)-2-[(трет-бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановой кислоты (II, пример 1, 0,60 г, 0,002 моль, 1 экв.), метанол (20 мл) и соляную кислоту (3N, 20 мл). Эту смесь затем нагревают до 50 и перемешивают до тех пор, пока реакция не завершится, что определяют с помощью ВЭЖХ. После завершения реакции содержимое охлаждают до 20-25, подводят рН смеси до 8 насыщенным раствором бикарбоната натрия и затем концентрируют при пониженном давлении. Эту смесь экстрагируют этилацетатом (2 х 20 мл) и объединенные органические фазы высушивают над сульфатом натрия, фильтруют и концентрируют, ВЭЖХ (Время удержания = 2,89 мин; Zorbax RX-C8 ацетонитрил/0,05 М дигидрофосфата калия, 60/40; 1,0 мл/мин, =210 нм). Этот продукт используют без дополнительной очистки на следующей стадии. Пример 12. Метил-(2S)-2-(бензилокси)карбонил]амино]-3-(3,5-дифторфенил)пропаноат (II). Метил-(2S)-2-амино-3-(3,5-дифторфенил)пропаноат (XV, пример 11, 0,300 г, 1,40 ммоль, 1 экв.) и воду (10 мл) перемешивают. Добавляют карбонат натрия (0,15 г, 1,40 ммоль, 1 экв.), затем бензилхлорформиат (0,2 мл, 0,24 г, 1,4 ммоль, 1 экв.) и смесь перемешивают при 20-25 до тех пор, пока реакция не завершится, что определяют с помощью ВЭЖХ. После завершения реакци добавляют этилацетат (20 мл) и разделяют фазы. Водную фазу экстрагируют этилацетатом (2 х 20 мл) и объединенные органические фазы высушивают над сульфатом натрия, фильтруют и концентрируют. Концентрированный остаток кристаллизуют из смеси гексан/этилацетат с получением названного соединения, т.пл.=54; 1 Н-ЯМР(2S)-2-[(трет-Бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановую кислоту (I, 5,0 г, 0,017 моль,1,0 экв.) и карбонат калия (2,5 г, 0,018 моль, 1,1 экв.) перемешивают в ТГФ (100 мл). Затем к этой гетерогенной смеси прибавляют диметилсульфат (1,6 мл, 2,1 г, 0,017 моль, 1,0 экв.) и содержимое перемешивают в течение ночи при 20-25. После того как реакция завершится, что определяют с помощью ВЭЖХ,прибавляют гидроксид аммония (10%, 20 мл) и перемешивают еще 1 ч, после чего содержимое экстрагируют этилацетатом (3 х 50 мл). Объединенные органические фазы промывают водой (50 мл) и рассолом(50 мл), высушивают над сульфатом натрия, фильтруют и концентрируют при пониженном давлении с получением названного соединения. Пример 14. Метиловый эфир (2S)-2-[(трет-бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановой кислоты (II).(2S)-2-[(трет-Бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановую кислоту (I, 5,0 г, 0,017 моль,1,0 экв.), карбонат калия (2,5 г, 0,018 моль, 1,1 экв.) и ДМФА (100 мл) перемешивают. Затем к этой гете- 19007535 рогенной смеси прибавляют диметилсульфат (1,6 мл, 2,1 г, 0,017 моль, 1,0 экв.) и содержимое перемешивают в течение ночи при 20-25. После того как реакция завершится, что определяют с помощью ВЭЖХ,прибавляют гидроксид аммония (10%, 20 мл) и реакционную смесь перемешивают еще 1 ч. Затем содержимое перемешивают еще в течение 30 мин, затем охлаждают до 0 и фильтруют. Осадки промывают холодной водой (20 мл) и высушивают при пониженном давлении с получением названного соединения. Пример 15. трет-Бутил-(1S)-3-хлор-1-(3,5-дифторбензил)-2-оксопропилкарбамат (III).(2S)-2-[(трет-Бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановую кислоту (I) растворяют в ТГФ и перемешивают при 20-25. Прибавляют оксалилхлорид (1 экв.) и смесь перемешивают в течение примерно 15 мин с получением трет-бутил-(1S)-2-хлор-1-[3,5-дифторбензил]-2-оксоэтилкарбамата (XI). Смесь охлаждают до 0 и прибавляют LiСН 2 Сl (более 2 экв.). Эту смесь перемешивают до тех пор, пока реакция не завершится. Реакцию гасят водой, и продукт экстрагируют в этилацетате. Объединенные органические фазы промывают рассолом, высушивают над сульфатом натрия и концентрируют при пониженном давлении с получением названного соединения. Пример 16. трет-Бутил-(1S)-3-хлор-1-(3,5-дифторбензил)-2-оксопропилкарбамат (III).IСН 2 Сl (3,54 г, 1,46 мл, 19,82 ммоль, 1,25 экв.) и ТГФ (5 мл) прибавляют к метиловому эфиру (2S)2-[(трет-бутоксикарбонил)амино]-3-(3,5-дифторфенил)пропановой кислоты (II, пример 1, 5 г, 15,86 ммоль,1 экв.). Эту смесь охлаждают до -78 и по каплям добавляют ЛДА (22,3 мл, 44,60 ммоль, 2,25 экв., 2,0 М),поддерживая внутреннюю температуру ниже -60. После завершения добавления, содержимое перемешивают в течение 30 мин при -78, одновременно добавляя по каплям н-бутиллитий (15,3 мл, 19,82 ммоль,1,25 экв.; 1,3 М в гексане) и поддерживая внутреннюю температуру ниже примерно -60. Реакционную смесь перемешивают в течение 30 мин, затем гасят в охлажденной до 0 соляной кислоте (1N). Прибавляют этилацетат, фазы разделяют и водную фазу экстрагируют этилацетатом. Объединенные органические фазы промывают насыщенным раствором бикарбоната натрия, высушивают над сульфатом натрия,фильтруют и концентрируют при пониженном давлении с получением названного соединения, 1 Н-ЯМР трет-Бутил-(1S,2S)-3-хлор-1-(3,5-дифторбензил)-2-гидроксипропилкарбамат (IV, пример 3, 1,0 г,2,98 ммоль), смолу Dowex50WX2-400 (4,6 г, 23,8 ммоль) и метанол (25 мл) перемешивают. Затем эту смесь помещают на качалку J-Kim с нагреванием при 50 на 2 ч. Анализ ЭСМС не обнаружил присутствия исходного продукта в смеси. Содержимое реакции фильтруют через керамическую воронку и смолу промывают метанолом (25 мл) и смесью метанол/хлористый метилен (1/1, 25 мл). Полученную смесь элюируют аммиаком в метаноле (2N, 2 х 25 мл). Элюат концентрируют при пониженном давлении с получением названного соединения, ЭСМС=236,1. Пример 18. (1S)-2-(3,5-Дифторфенил)-1-[(2S)-оксиран-2-ил]этиламин.(1S,2S)-3-Хлор-1-(3,5-дифторбензил)-2-гидроксипропиламин (пример 17, 33 мг, 0,14 ммоль) и абсолютный этанол (1,5 мл) перемешивают. К этой смеси прибавляют гидроксид калия (9,8 мг, 0,175 ммоль) в абсолютном этаноле (0,5 мл) и полученную смесь перемешивают при 20-25 в течение 30 мин. В то же время ЭСМС указывает на образование продукта (МН+=200,1). Добавляют воду (2 мл) и смесь концентрируют при пониженном давлении до половины объема, затем разбавляют этилацетатом (15 мл). Органическую фазу отделяют и водную фазу экстрагируют этилацетатом (2 х 10 мл). Органические фазы объединяют, промывают рассолом и высушивают над безводным сульфатом магния. Растворитель удаляют при пониженном давлении с получением названного соединения, МН+=200,1. Изобретение и способы его осуществления и применения описаны теперь настолько полно, ясно и точно, что позволяют любому специалисту в данной области осуществить и использовать то же самое. Следует понимать, что выше описаны предпочтительные реализации настоящего изобретения и что допускаются модификации без отклонения от сущности или объема настоящего изобретения, которое- 20007535 представлено в формуле изобретения. Следующие пункты формулы предназначены для того, чтобы подробно и определенно заявить предмет изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы где R2 представляет собой хлор или бром. 2. Соединение по п.1, где R2 представляет собой -Cl. 3. Соединение по п.1, где R2 представляет собой -Вr. 4. Соединение по п.1, которое представляет дифторфенил)бутан-2-ол. 5. Соединение формулы(2S,3S)-3-амино-1-хлор-4-(3,5 6. Способ получения кетона формулы III где PROT представляет собой азотзащитную группу и R2 представляет собой хлор или бром, включающий:(а) образование смеси сложного эфира формулы II и дигалогенированного метана R2CH2X2, где R2 определен выше и X2 представляет собой -Вr или -I: где R1 выбран из группы, включающей С 1-С 4-алкил, который может быть замещен одной группой -Сl, иPROT определена выше,(b) добавление основания к смеси, полученной на стадии (а);(c) подкисление смеси, полученной на стадии (b). 7. Способ по п.6, где PROT представляет собой трет-бутоксикарбонил или бензилоксикарбонил. 8. Способ по п.6, где R1 представляет собой С 1-С 2-алкил. 9. Способ по п.6, где R1 представляет собой С 1-алкил. 10. Способ по п.6, где R2 представляет собой хлор. 11. Способ по п.6, где CH2R2X2 присутствует в количестве от примерно 1 до примерно 1,5 эквивалентов, исходя из количества сложного эфира II. 12. Способ по п.6, где X2 представляет собой йод. 13. Способ по п.6, где сильное основание представляет собой диизопропиламид лития, (С 1-С 8 алкил)литий. 14. Способ по п.13, где сильное основание представляет собой диизопропиламид лития.- 21007535 15. Способ по п.6, где сильное основание используют в количестве примерно от 2 до 2,5 эквивалентов, исходя из количества сложного эфира II. 16. Способ по п.6, где добавляют вторую порцию основания, представляющего собой (С 1 С 4)алкиллитий. 17. Способ по п.16, где второе основание представляет собой н-бутиллитий, втop-бутиллитий, третбутиллитий, метиллитий. 18. Способ по п.17, где второе основание представляет собой н-бутиллитий. 19. Способ по п.6, где количество второго основания составляет от примерно 1 до примерно 1,5 эквивалентов, исходя из количества сложного эфира II. 20. Способ по п.6, где подкисление осуществляют с помощью кислоты, имеющей рКа менее примерно 10. 21. Способ по п.20, где кислота выбрана из группы, включающей уксусную и соляную кислоту и их смеси. 22. Способ по п.6, где кетон (III) представляет собой трет-бутил-(1S)-3-хлор-1-(3,5-дифторбензил)2-оксопропилкарбамат. 23. Способ получения эпоксида формулы V-R где R представляет собой фенил, который может быть замещен 1, 2, 3 или 4 группами, независимо выбранными из галогенов, включающий:(а) превращение сложного эфира формулы II(b) восстановление кетона до соответствующего спирта формулы IV(с) обработку спирта основанием с получением эпоксида. 24. Способ по п.23, дополнительно включающий этерификацию кислоты формулы (0) с образованием сложного эфира формулы II. 25. Соединение формулы где R представляет собой фенил, который может быть замещен 1, 2, 3 или 4 группами, независимо выбранными из галогенов. 26. Способ получения эпоксида формулы(а) реакцию альдегида формулы XII с соединением фосфора формулы XIII с получением ненасыщенного эфира формулы XIV где PROT представляет собой азотзащитную группу, Х 3 представляет собой C1-С 3 алкил и R1 представляет собой С 1-С 4 алкил, который может быть замещен одним Cl, -СН 2-СН=СН 2, или фенил, который может быть замещен одной нитрогруппой, галогеном или цианогруппой;(b) превращение ненасыщенного эфира формулы XIV в эфир формулы II(с) превращение эфира формулы II в кетон формулы III(d) восстановление кетона формулы III в соответствующий спирт формулы IV и (е) обработку спирта основанием с получением эпоксида.
МПК / Метки
МПК: C07D 301/26, C07D 303/36
Метки: способ, соединения, бензилэпоксидов, промежуточные, получения
Код ссылки
<a href="https://eas.patents.su/25-7535-sposob-polucheniya-benzilepoksidov-i-promezhutochnye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения бензилэпоксидов и промежуточные соединения</a>
Нафтильные соединения, промежуточные соединения для их получения, применение нафтильных соединений, способ снижения холестерина

Номер патента: 1600
Опубликовано: 25.06.2001
Авторы: Палковиц Алан Д., Брайант Генри У., Кроуелл Томас А., Джонс Чарльз Д.
МПК: A61K 31/33, C07C 47/546, A61P 19/10...
Метки: нафтильные, холестерина, соединения, соединений, промежуточные, нафтильных, способ, получения, снижения, применение
Формула / Реферат:
1. Соединение формулы I где R1 является -Н, -ОН, -O(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr; где Аr является фенилом или замещенным фенилом, OCO(С1-С6-алкилом), -O(СО)O(С1-С6-алкилом) или -ОSО2(С4-С6-алкилом); R2 является -Н, -F, -Cl, -ОН, -О(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr, где Аr является фенилом или замещенным фенилом, -OCO(С1-С6-алкилом),...
Конъюгаты соединения, содержащего сульфгидрильную группу, и производного жирной кислоты, способ получения конюгатов, промежуточные соединения для их получения, способы повышения абсорбции и пролонгированного сохранения в крови и тканях млекопитающего соединения, содержащего сульфгидрильную группу

Номер патента: 584
Опубликовано: 29.12.1999
Авторы: Шен Вей Чанг, Икрами Хуссейн М.
МПК: C07D 213/70, A61K 31/44, C07H 19/048...
Метки: получения, повышения, содержащего, производного, млекопитающего, группу, сохранения, способы, промежуточные, конъюгаты, абсорбции, пролонгированного, кислоты, конюгатов, соединения, крови, способ, сульфгидрильную, тканях, жирной
Формула / Реферат:
1. Соединение общей формулы VI где Р является фрагментом соединения, содержащего сульфгидрильную группу, выбранного из группы, включающей пептиды, белки или олигонуклеотиды; R1 представляет собой водород, низший алкил или арил; R2 представляет собой фрагмент, содержащий липидную группу; а R3 представляет собой гидроксил, фрагмент, содержащий липидную группу или аминокислотную последовательность, включающую 1 или 2 аминокислоты и...
Промежуточные соединения и способ получения оланзапина

Номер патента: 1642
Опубликовано: 25.06.2001
Авторы: Баннелл Чарлз А., Стефенсон Грегори А., Рётзель Сюзн М., Николс Джон Р., Ларсен Сэмюель Д.
МПК: C07D 495/04
Метки: способ, оланзапина, соединения, получения, промежуточные
Формула / Реферат:
1. Соединение, которое представляет собой дигидрат оланзапина. 2. Соединение по п.1, где дигидрат является промежуточным соединением для получения оланзапина формы II. 3. Соединение по п.1, где дигидратом является кристаллический полиморф дигидрата В оланзапина, имеющий типичную порошковую рентгенограмму, как представлено следующими межплоскостными расстояниями (d), как показано ниже d 9.9045 ...
Способ получения иопамидола и новые промежуточные соединения, получаемые в этом способе

Номер патента: 5922
Опубликовано: 25.08.2005
Авторы: Каппеллетти Энрико, Анелли Пьер Лучио, Лукс Джованна, Броккетта Марино
МПК: C07C 237/46
Метки: иопамидола, способе, этом, способ, новые, получаемые, получения, соединения, промежуточные
Формула / Реферат:
1. Способ получения (S)-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-[(2-гидрокси-1-оксопропил)амино]-2,4,6-трииодо-1,3-бензолдикарбоксамида формулы (I) исходя из соединения формулы (II) причем указанный способ включает а) введение соединения формулы (II) в реакцию с подходящим защитным агентом с образованием соединения формулы (III) где -R - группа формулы A или B где R1 - атом водорода, C1-C4 линейная или разветвленная алкильная группа или...
4-галогенированные стероиды, способ их получения промежуточные соединения, лекарственные средства и фармацевтические композиции

Номер патента: 2116
Опубликовано: 24.12.2001
Авторы: Ван Де Вельд Патрик, Ник Франсуа, Буали Иамина, Тетш Жан-Жорж
МПК: A61K 31/565, C07J 41/00, A61P 9/00...
Метки: фармацевтические, стероиды, способ, промежуточные, композиции, лекарственные, соединения, 4-галогенированные, средства, получения
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает атом водорода или радикал -(СH2)m-СН2-СО2Н, в котором m равно 0, 1, 2 или 3, R2 обозначает алкил, содержащий 1-6 атомов углерода, D обозначает остаток пятичленного цикла, при необходимости замещенный, Х обозначает атом галогена, Y выбирается из О, S, SO, SO2, n обозначает целое число от 2 до 5, R3 и R4, одинаковые или разные, означают атом водорода, (C1-C6)-алкил, линейный,...
Предыдущий патент: Стентовое устройство на основе хинолиновых и хиноксалиновых соединений в качестве ингибиторов pdgf-рецептора и/или тирозинкиназы lck
Следующий патент: Способ извлечения капролактама
Случайный патент: Фунгицидные смеси