Замещенные пиримидиноновые соединения, композиция и способ лечения, ингибирования или предупреждения тромботических состояний у млекопитающих
Номер патента: 4867
Опубликовано: 26.08.2004
Авторы: Хамме Эштон Т.II, Джоунс Дэрин Е., Руппел Мелвин Л., Ньюманн Уилльям, Саут Майкл С.






















Формула / Реферат
1. Соединение, имеющее формулу

или его фармацевтически приемлемая соль, где B выбран из группы, состоящей из фенила, C3-C6-циклоалкила, C1-C6-алкила;
A выбран из группы, состоящей из -CH2SO2-, -CH2-, -CH2-CH2- или простой ковалентной связи;
y представляет NH;
M представляет CH;
R2 выбран из группы, состоящей из фенила, -S-фенила, аминофенила, необязательно замещенного амино, карбокси, алкоксикарбонилом или алкиламинокарбонилом, пиридила;
-K-E0- представляет -CH2-C(O)NH-;
Y0 представляет бензил, замещенный амидино.
2. Соединение по п.1, имеющее формулу

или его фармацевтически приемлемая соль, где B представляет фенил;
A выбран из группы, состоящей из -CH2SO2-, -CH2-CH2-;
R2 представляет фенил.
3. Соединение по п.2 или его фармацевтически приемлемая соль, где A представляет -CH2SO2-.
4. Соединение по п.2 или его фармацевтически приемлемая соль, где A представляет -CH2-CH2-.
5. Соединение по п.1, имеющее формулу

или его фармацевтически приемлемая соль, где B выбран из группы, состоящей из фенила, C1-C6-алкила;
A выбран из группы, состоящей из -CH2-CH2- или простой ковалентной связи;
R2 представляет пиридил.
6. Соединение по п.1, имеющее формулу

или его фармацевтически приемлемая соль, где B выбран из группы, состоящей из C1-C6-алкила, C3-C6-циклоалкила;
A представляет простую ковалентную связь;
R2 представляет аминофенил, необязательно замещенный амино, карбокси, алкоксикарбонилом или алкиламинокарбонилом, пиридил.
7. Композиция для ингибирования тромботических состояний в крови, включающая соединение по любому из пп.1-6 и фармацевтически приемлемый носитель.
8. Способ ингибирования тромботических состояний в крови, предусматривающий введение в кровь терапевтически эффективного количества композиции по любому из пп.1-6.
9. Способ ингибирования образования агрегации тромбоцитов в крови, предусматривающий введение в кровь терапевтически эффективного количества композиции по любому из пп.1-6.
10. Способ ингибирования образования тромбов в крови, предусматривающий введение в кровь терапевтически эффективного количества композиции по любому из пп.1-6.
11. Способ лечения или предупреждения тромбоэмболии вены и тромбоэмболии легких у млекопитающих, предусматривающий введение указанному млекопитающему терапевтически эффективного количества композиции по любому из пп.1-6.
12. Способ лечения или предупреждения тромбоза глубокой вены у млекопитающих, предусматривающий введение указанному млекопитающему терапевтически эффективного количества композиции по любому из пп.1-6.
13. Способ лечения или предупреждения кардиогенной тромбоэмболии у млекопитающих, предусматривающий введение указанному млекопитающему терапевтически эффективного количества композиции по любому из пп.1-6.
14. Способ лечения или предупреждения тромбоэмболического шока у человека и других млекопитающих, предусматривающий введение указанным млекопитающим терапевтически эффективного количества композиции по любому из пп.1-6.
15. Способ лечения или предупреждения тромбоза, ассоциированного с раком и противораковой химиотерапией у человека и других млекопитающих, предусматривающий введение указанным млекопитающим терапевтически эффективного количества композиции по любому из пп.1-6.
16. Способ лечения или предупреждения нестабильной стенокардии у человека и других млекопитающих, предусматривающий введение указанным млекопитающим терапевтически эффективного количества композиции по любому из пп.1-6.
17. Способ ингибирования образования тромбов в крови, предусматривающий введение в кровь терапевтически эффективного количества соединения по любому из пп.1-6 в сочетании с терапевтически эффективным количеством антагониста рецептора фибриногена.
18. Применение соединения по любому из пп.1-6 или их фармацевтически приемлемой соли в целях изготовления лекарственного препарата для ингибирования образования тромбов, лечения тромботических состояний или предупреждения образования тромбов у млекопитающих.
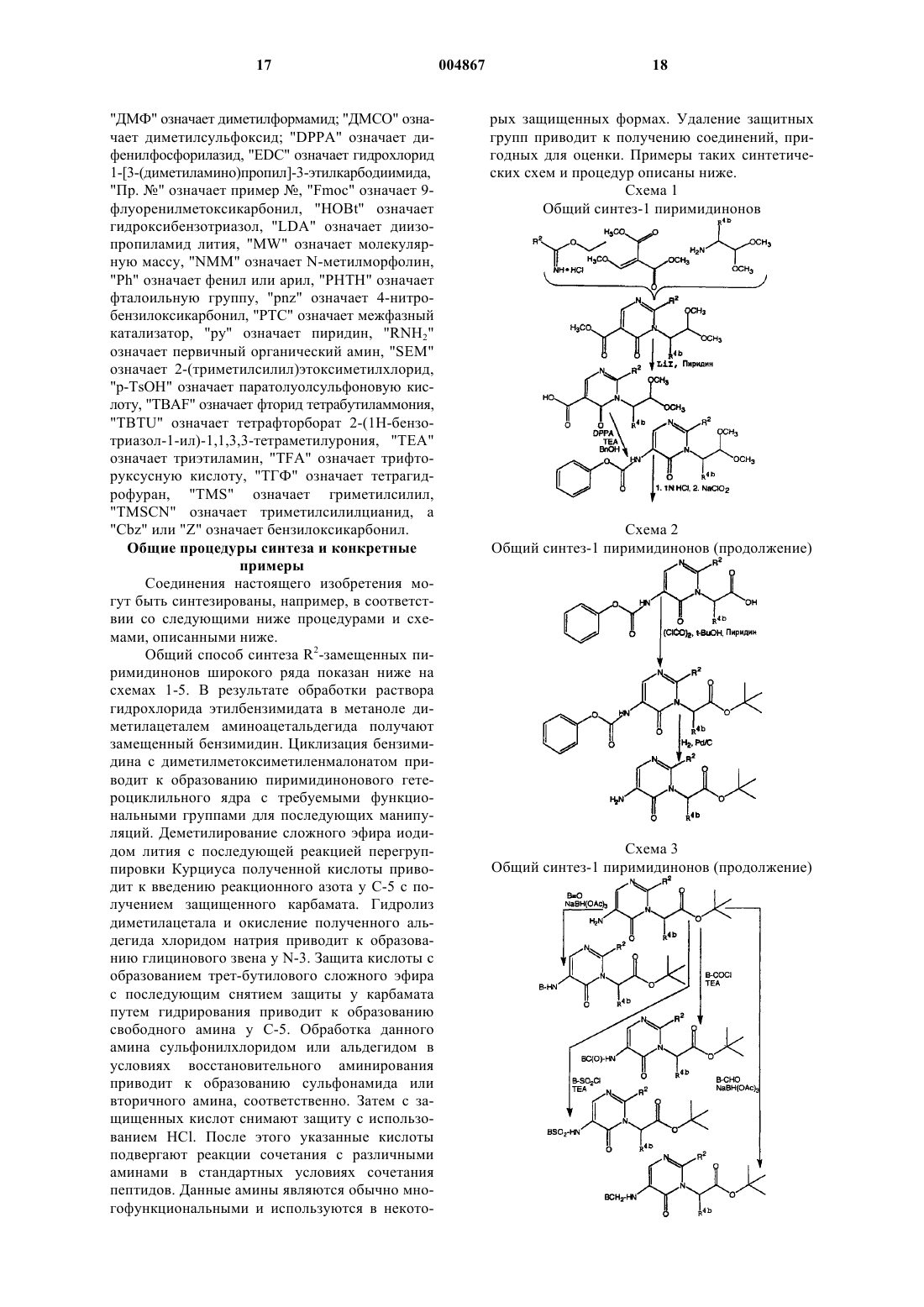
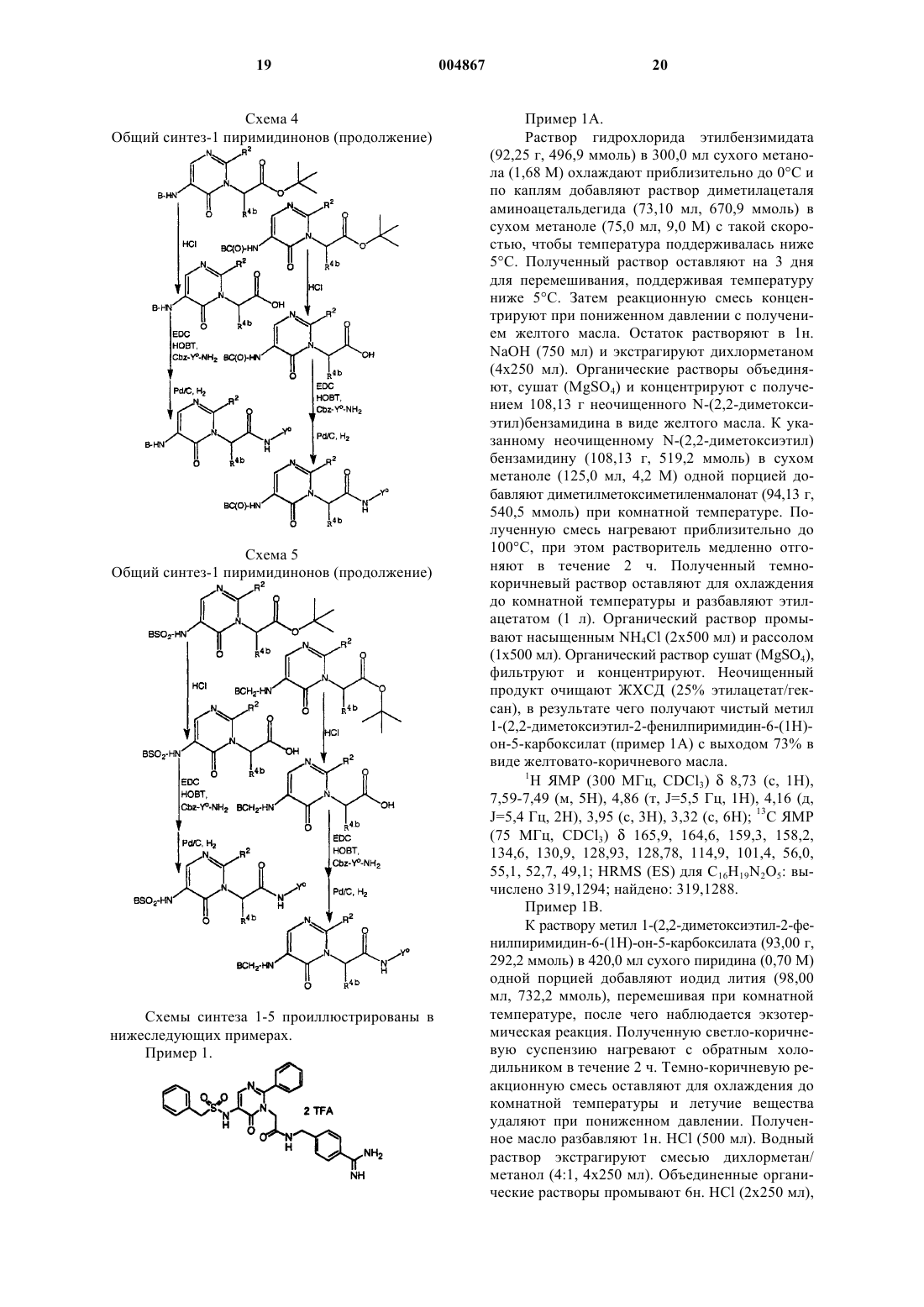
Текст
1 Область, к которой относится изобретение Настоящее изобретение относится к области противокоагулянтной терапии, а в частности к соединениям, композициям и к способам предупреждения и лечения тромботических состояний, таких как заболевание коронарной артерии и цереброваскулярные заболевания. Более конкретно, настоящее изобретение относится к замещенным полициклическим арил- и гетероарилпиримидиноновым соединениям, которые ингибируют сериновые протеазы, участвующие в каскаде коагуляции. Предпосылки создания изобретения Физиологические системы регулируют текучесть крови у млекопитающих [Majerus, P.W.Basis of Therapeutics. 9th edition. New York,McGraw-Hill Book Co., 1996, pp.1341-1343]. Кровь должна оставаться жидкой в сосудистых системах, и при этом она должна быть способна подвергаться быстрому гемостазу, прекращению потери крови из поврежденного сосуда. Гемостаз, или свертывание крови, начинается с того, что сначала тромбоциты прилипают к макромолекулам в субэндотелиальных областях пораженных и/или поврежденных сосудов. Эти тромбоциты агрегируются с образованием первичной гемостатической пробки и стимулируют локальную активацию факторов коагуляции плазмы, что приводит к образованию фибринового сгустка, который укрепляет агрегированные тромбоциты. Факторами коагулирования (свертывания) плазмы являются факторы II, V, VII, VIII, IX, X,XI и XII, которые также называются проферментами (зимогенами) протеазы. Эти факторы свертывания, или зимогены протеазы, активируются сериновыми протеазами, что приводит к коагуляции в так называемом "каскаде реакций свертывания крови", или цепной реакцииBook Co., 1991, p.350]. Коагуляция, или свертывание крови, происходит двумя путями в соответствии с различными механизмами. Внутренний или контактный путь начинается с превращений XIIXIIaXIaIXa и заканчивается превращением Х в Ха. Фактор Ха вместе с фактором Va превращает протромбин (II) в тромбин(IIа) с последующим превращением фибриногена в фибрин. Полимеризация фибрина приводит к образованию фибринового сгустка. Внешний путь начинается с превращения фактора свертывания VII в VIIa под действием фактора Ха. Присутствие тканевого фактора и фактора VIIa ускоряет образование Ха в присутствии ионов кальция и фосфолипидов. Продуцирование Ха приводит к образованию тромбина, фибрина и фибринового сгустка, как описано выше. При 004867 2 сутствие одного или нескольких из многих различных факторов свертывания крови и существование двух различных путей образования сгустков позволяет осуществлять эффективный селективный контроль за некоторыми стадиями процесса коагуляции, или свертывания крови,или лучше понять определенные стадии этого процесса. Хотя свертывание крови в результате повреждения кровеносных сосудов является важным физиологическим процессом у млекопитающих, таких как человек, однако, образование сгустков может также приводить к патологическим состояниям. Патологический процесс, называемый тромбозом, происходит в результате агрегации тромбоцитов и/или блокирования(т.е. закупорки) кровеносного сосуда фибриновым сгустком. Артериальный тромбоз может приводить к ишемическому некрозу ткани,снабжаемой артериальной кровью. При тромбозе, происходящем в коронарной артерии, может возникнуть инфаркт миокарда или сердечный приступ. Тромбоз, возникающий в вене, может вызывать дренирование ткани через вену, что приводит к образованию отека и воспаления. Тромбоз в глубокой вене может быть осложнен эмболией легких. Ингибирование образования агрегатов тромбоцитов, ингибирование образования фибрина, ингибирование образования тромбов и ингибирование возникновения эмболии может оказаться терапевтически эффективным для предупреждения образования или удаления сгустков в кровеносном сосуде и для лечения или предупреждения нестабильной стенокардии, застойной стенокардии, инфаркта миокарда, преходящих приступов ишемии,фибрилляции предсердий, тромботического шока, эмболического шока, тромбоза глубокой вены, диссеминированного внутрисосудистого свертывания, глазного отложения фибрина и повторной окклюзии или рестеноза реканализированных сосудов. Существует несколько работ о непептидных и пептидных пиримидиноновых соединениях, которые действуют как ингибиторы фактора свертывания крови, участвующего в каскаде реакций коагуляции или процесса свертывания крови. В патентной заявке РСТ WO 98/47876, Van Boekel и др. были описаны пептидные 6-алкилпиридоны и 2-алкилпиримидиноны в качестве антитромботических соединений. В патентной заявке РСТ WO 98/16547 Zhu и Scarborough описывают 3-(N-гетероциклиламино)-4,5,6-замещенные пиридонилацетамиды и 2,4-замещенные 5-(N-гетероциклиламино)пиримидинонилацетамиды, содержащие амидные заместители и обладающие активностью против фактора Ха млекопитающего. В патенте США 5656645 Tamura и др. описывают 4,5,6 замещенные 3-аминопиридонилацетамиды, 1,6 замещенные 5-аминоурацинилацетамиды и 2,4 замещенные 5-аминопиримидинонилацетамиды, 3 содержащие амидные заместители, имеющие формильную функциональную группу, и обладающие активностью против тромбина. В патенте США 5658930 Tamura и др. также описывают 4,5,6-замещенные 3-аминопиридонилацетамиды, 1,6-замещенные 5-аминоурацинилацетамиды и 2,4-замещенные 5-аминопиримидинонилацетамиды, содержащие амидные заместители, имеющие формильную функциональную группу и обладающие активностью против тромбина. В патентных заявках РСТ 96/18644 и 97/46207 Tamura и др. описывают 4,5,6 замещенныe 3-аминопиридонилацетамиды, 1,6 замещенные 5-аминоурацинилацетамиды и 2,4 замещенные 5-аминопиримидинонилацетамиды,содержащие амидные заместители, имеющие формильную функциональную группу и обладающие активностью против тромбина. В патентной заявке РСТ WO 98/09949 Suzuki и др. описывают 2-гетероциклилацетамидо-производные 1,2-дикетонов и сообщают, что они являются ингибиторами протеаз, а в частности ингибиторами химазы. Краткое описание изобретения Целью настоящего изобретения являются соединения или их фармацевтически приемлемые соли, которые могут быть использованы в антикоагулянтной терапии и имеют общую структуру Формула I где В выбран из группы, состоящей из фенила, С 3-С 6-циклоалкила, C1-С 6-алкила; А выбран из группы, состоящей из -СН 2SO2-,-СН 2-, -СН 2-СН 2- или простой ковалентной связи;представляет NH; М представляет СН;Y0 представляет бензил, замещенный амидино. Другой целью настоящего изобретения является разработка способов предупреждения и лечения тромботических состояний, таких как поражения коронарной артерии, цереброваскулярные заболевания и другие расстройства, связанные со свертыванием крови. Предупреждение и лечение таких тромботических состояний осуществляют путем введения пациенту, нуждающемуся в этом, эффективного количества соединений формулы (I). Другие цели и преимущества настоящего изобретения будут очевидны из нижеследующего описания изобретения. 4 Описание изобретения Настоящее изобретение относится к классу соединений, включающему замещенные пиримидиноны или их фармацевтически приемлемые соли, которые при антикоагулянтной терапии являются эффективными для лечения и предупреждения ряда тромботических состояний, таких как поражения коронарной артерии и цереброваскулярные заболевания, и которые представлены формулой (I) Формула I где В выбран из группы, состоящей из фенила,С 3-С 6-циклоалкила, C1-С 6-алкила; А выбран из группы, состоящей из -CH2SO2-,-СН 2-, -СН 2-СН 2- или простой ковалентной связи;представляет NH; М представляет СН;Y0 представляет бензил, замещенный амидино. В предпочтительном варианте соединений формулы I или его фармацевтически приемлемой соли В представляет фенил; А выбран из группы, состоящей изR2 представляет фенил. В других вариантах соединений формулы I или их фармацевтически приемлемой соли А представляет -CH2SO2- или -СН 2-СН 2-. В предпочтительном варианте соединений формулы I или его фармацевтически приемлемой соли В выбран из группы, состоящей из фенила,C1-С 6-алкила; А выбран из группы, состоящей из -СН 2 СН 2- или простой ковалентной связи;R2 представляет пиридил. Еще в одном предпочтительном варианте соединений формулы I или его фармацевтически приемлемой соли В выбран из группы, состоящей из C1-С 6 алкила, C3-С 6-циклоалкила; А представляет простую ковалентную связь;R2 представляет аминофенил, необязательно замещенный амино, карбокси, алкоксикарбонилом или алкиламинокарбонилом, пиридил. Соединения настоящего изобретения могут быть использованы в антикоагулянтной терапии для лечения и предупреждения и тромботических состояний, таких как поражения коронарной артерии и цереброваскулярные заболе 5 вания. Соединения настоящего изобретения могут быть использованы для ингибирования сериновых протеаз, ассоциированных с каскадом реакций коагуляции, и факторов II, V, VII,VIII, IX, X, XI или XII. Соединения настоящего изобретения могут ингибировать агрегацию тромбоцитов крови, образование фибрина, образование тромбов, возникновения эмболии у млекопитающих в крови, в продуктах крови и в органах млекопитающих. Указанные соединения могут быть также использованы для лечения или предупреждения нестабильной стенокардии, застойной стенокардии, инфаркта миокарда, преходящих приступов ишемии, фибрилляции предсердий, тромботического шока, эмболического шока, тромбоза глубокой вены,диссеминированного внутрисосудистого свертывания, глазного отложения фибрина и повторной окклюзии или рестеноза реканализированных сосудов. Указанные соединения могут быть также использованы для профилактического лечения индивидуумов с риском развития подобных расстройств. Указанные соединения могут быть также использованы для снижения риска возникновения атеросклероза. Соединения формулы (I) могут быть также использованы для предупреждения нарушения мозгового кровообращения (CVA) или инсульта. Помимо использования для лечения человека указанные соединения могут быть также использованы в ветеринарии для лечения домашних животных, экзотических животных и сельскохозяйственных животных, включая млекопитающих, грызунов и т.п. Более предпочтительными животными являются лошади, собаки и кошки. В другом варианте осуществления настоящего изобретения новые соединения выбирают из соединений, представленных в примерах 1-19 и табл. 1. Для ясности, ниже приводится определение общеупотребимых терминов, используемых в настоящем описании. Стандартные однобуквенные символы элементов, если это не оговорено особо, используются для обозначения конкретных типов атомов. Символ "С" представляет атом углерода. Символ "О" представляет атом кислорода. Символ "N" представляет атом азота. Символ "Р" представляет атом фосфора. Символ "S" представляет атом серы. Символ "Н" представляет атом водорода. Двухбуквенные символы элементов используются для обозначения элементов Периодической таблицы элементов (то есть Сl представляет хлор, Se представляет селен и т.п.). Используемый здесь термин "алкил", отдельно или в других терминах, таких как "алкоксикарбонил" и "алкиламинокарбонил", означает ациклический алкильный радикал, содержащий от 1 до около 10, предпочтительно от 3 до около 8 атомов углерода, а более предпочти 004867 6 тельно от 3 до около 6 атомов углерода. Указанные алкильные радикалы могут быть, но не обязательно, замещены. Примерами таких радикалов являются метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил,пентил, изоамил, гексил и т.п. Термин "гидридо" означает простой атом водорода (Н). Указанный гидридорадикал может быть связан, например, с атомом кислорода с образованием "гидроксильного" радикала,один гидридорадикал может быть связан с атомом углерода с образованием "метинового" радикала -СН=, либо два гидридных радикала могут быть связаны с атомом углерода с образованием "метиленового" радикала (-СН 2-). Термин "углеродный" радикал означает атом углерода, не имеющий каких-либо ковалентных связей и способный образовывать четыре ковалентные связи. Термин "алкиленовый" радикал означает линейный или разветвленный радикалы, имеющие от 1 до около 10 атомов углерода и имеющие положения присоединения для двух или более ковалентных связей. Примерами указанных радикалов являются метилен, этилен, метилэтилен и иэопропилиден. Термин "арил", отдельно или в комбинации, означает карбоциклическую ароматическую систему, содержащую одно, два или три кольца, где указанные кольца могут быть соединены вместе с боковой группой, либо они могут быть конденсированы. Термин "конденсированный" означает, что присутствует второе кольцо, т.е. присоединенное или образованное посредством двух смежных атомов, которые являются общими (т.е. общими атомами) с первым кольцом. Термин "конденсированный" эквивалентен термину "присоединенный с кондесацией". Термин "арил" означает ароматические радикалы, такие как фенил, нафтил, тетрагидронафтил, индан и бифенил. Термин "сульфонил", независимо от того,используется ли он отдельно или в связи с другими терминами, такими как алкилсульфонил,означает, соответственно, двухвалентные радикалы -SO2-. Термин "алкилсульфонил" охватывает алкильные радикалы, присоединенные к сульфонильному радикалу, где "алкил" определен выше. Термин "циклоалкил" охватывает радикалы, имеющие от 3 до 15 атомов углерода. Более предпочтительными циклоалкильными радикалами являются "низшие циклоалкильные" радикалы, имеющие от 3 до 7 атомов углерода. Примерами таких радикалов являются циклопропил,циклобутил, циклопентил, циклогексил и циклопентил. Термин "циклоалкил" охватывает радикалы, имеющие от 7 до 15 атомов углерода и имеющие от двух до четырех колец. Примерами таких радикалов являются норборнил (то есть бицикло[2,2,1]гептил) и адамантил. 7 Термин "арилтио" охватывает арильные радикалы, определенные выше, присоединенные к атому серы. Примером таких радикалов является фенилтио. Термин "карбонил" означает углеродный радикал, имеющий две из четырех ковалентных связей с общим атомом кислорода. Термин"карбокси" охватывает гидроксильный радикал,определенный выше и присоединенный к одной из двух не общих связей в карбонильной группе. Термин "ацил", употребляемый отдельно или в комбинации, означает карбонильную или тионокарбонильную группу, связанную с радикалом, выбранным, например, из гидридо, алкила, алкенила, алкинила, галогеналкила, алкокси,алкоксиалкила, галогеналкокси, арила, гетероциклила, гетероарила, алкилсульфинилалкила,алкилсульфонилалкила, аралкила, циклоалкила,циклоалкилалкила, циклоалкенила, алкилтио,арилтио, амино, алкиламино, диалкиламино,аралкокси, арилтио и алкилтиоалкила. Примерами "ацила" являются формил, ацетил, бензоил, трифторацетил, фталоил, малонил, никотинил и т.п. Термин "амино" означает атом азота, содержащий два заместителя, такие как гидридо,гидрокси или алкил, и имеющий одну ковалентную связь, служащую для связывания с одним атомом, таким как углерод. Примерами таких аминорадикалов являются, например, -NH2-,-NНСН 3, -NHOH и -NHOCH3. Термин "амидино" охватывает замещенную или незамещенную аминогруппу, связанную с одной из двух имеющихся связей иминокарбонильного радикала. Примерами таких амидинорадикалов являются, например, NH2-C=NH,NН 2-С=NСН 3, NH2-С=NOСН 3 и CH3NH-C=NOH. Термин "гуанидино" означает амидиногруппу,связанную с аминогруппой, определенной выше, где указанная аминогруппа может быть связана с третьей группой. Примерами таких гуанидинорадикалов являются, например, NH2C(NH)-NH-, NH2-C(NCH3)-NH-, NH2-C(NOCH3)NH- и СН 2NН-С(NOH)-NH-. Термин "спейсер" может включать ковалентную связь и линейную группу, имеющую основу из 1-7 смежных атомов. В прямой цепи может быть 1 или 2, или 3 атома. Эта цепь может быть образована одним или более радикалами,выбранными из-CH2CH2-,-S(O)2CH2-, -C(O)NH- и других радикалов, определенных выше, или, в основном, известных или установленных любым специалистом. Соединения настоящего изобретения могут существовать в форме таутомера, геометрического изомера или стереоизомера. Настоящее изобретение относится ко всем указанным соединениям, включая цис- и транс-геометрические изомеры, Е- и Z-геометрические изомеры,R- и S-энантиомеры, диастереоизомеры, Dизомеры, L-изомеры, их рацемические смеси и 8 другие их смеси, входящие в объем настоящего изобретения. В объем настоящего изобретения также входят фармацевтически приемлемые соли указанных таутомерных, геометрических или стереоизомерных форм. Термины "цис" и "транс" означают форму геометирической изомерии, в которой каждый из двух атомов углерода, соединенных двойной связью, имеют атом водорода на одной и той же стороне двойной связи ("цис") или на противоположной стороне двойной связи ("транс"). Некоторые из описанных соединений содержат алкенильные группы, что означает, что они включают как "цис", так и "транс", либо "Е" и "Z"-геометрические формы. Некоторые из описанных соединений содержат один или несколько стереоцентров, что означает, что они включают R-, S-формы, и смеси R и S-форм для каждого присутствующего стереоцентра. Некоторые из описанных здесь соединений могут содержать одну или несколько кетоновых или альдегидных карбонильных групп или их комбинаций, взятых отдельно или как часть гетероциклической кольцевой системы. Указанные карбонильные группы могут присутствовать частично или, в основном, в "кето" форме и частично или, в основном, в одной или нескольких "енольных" формах каждой присутствующей альдегидной и кетоновой группы. Считается, что соединения настоящего изобретения,имеющие альдегидные или кетоновые карбонильные группы, присутствуют как в "кето", так и в "енольной" таутомерных формах. Некоторые из описываемых соединений могут содержать одну или несколько амидных карбонильных групп или их комбинаций, взятых отдельно или как часть гетероциклической кольцевой системы. Указанные карбонильные группы могут присутствовать частично или, в основном, в "кето" форме и частично или, в основном, в одной или нескольких "енольных" формах каждой присутствующей амидной группы. Считается, что соединения настоящего изобретения, имеющие амидные карбонильные группы, присутствуют как в "кето", так и в"енольной" таутомерных формах. Указанными амидными карбонильными группами могут быть группы как типа оксо (С=O), так и типа тионо (C=S). Некоторые из описываемых соединений могут содержать одну или несколько иминовых или енаминовых групп или их комбинаций. Указанные карбонильные группы могут присутствовать частично или, в основном, в "иминовой" форме и частично или, в основном, в одной или нескольких "енаминовых" формах каждой присутствующей группы. Считается, что соединения настоящего изобретения, имеющие иминовые или енаминовые группы, присутствуют как в "иминовой", так и в "енаминовой" таутомерных формах. 9 Настоящее изобретение также относится к лечению и профилактике путем антикоагулянтной терапии, используемой для лечения и предупреждения ряда тромботических состояний,включая поражения коронарной артерии и цереброваскулярные заболевания у индивидуума,и предусматривающей введение указанному индивидууму, страдающему такими заболеваниями, терапевтически эффективного количества соединения (I) или его фармацевтически приемлемой соли. В другом варианте осуществления изобретения соединения настоящего изобретения формулы (I) или его фармацевтически приемлемые соли, определенные выше, используются для лечения и профилактики поражения коронарной артерии, цереброваскулярных заболеваний и других расстройств, ассоциированных с каскадом реакций свертывания крови у индивидуума,предусматривающих введение указанному индивидууму, страдающему такими заболеваниями, терапевтически эффективного количества соединения формулы (I) настоящего изобретения или его фармацевтически приемлемой соли. Соединения настоящего изобретения формулы (I) или их фармацевтически приемлемые соли могут быть также использованы во всех случаях, когда требуется ингибирование свертывания крови, для предупреждения свертывания консервированной цельной крови и для предупреждения свертывания в других биологических образцах, предназначенных для анализа или консервирования. Таким образом, ингибиторы свертывания крови настоящего изобретения могут быть добавлены к консервированной цельной крови или они могут быть подвергнуты контактированию с цельной консервированной кровью и с любой средой, которая содержит или предположительно содержит факторы свертывания плазмы и в которой желательно ингибирование свертывания крови, например, в случае контактирования крови млекопитающего с материалом, выбранным из группы, состоящей из сосудистых трансплантатов,стентов, ортопедических протезов, сердечных протезов и систем экстракорпорального кровообращения. Соединения формулы (I) способны ингибировать активность сериновых протеаз, связанных с каскадом реакций свертывания крови,а поэтому они могут быть использованы для изготовления лекарственных препаратов, в способе профилактики или терапевтического лечения заболеваний, опосредованных сериновыми протеазами, участвующими в каскаде реакций свертывания крови, например, для ингибирова 004867 10 ния образования агрегатов тромбоцитов, ингибирования образования фибрина, ингибирования образования тромбов и ингибирования возникновения эмболии у млекопитающих, в крови, продуктах крови и в органах млекопитающих. Соединения настоящего изобретения могут быть также использованы для лечения или предупреждения нестабильной стенокардии,застойной стенокардии, инфаркта миокарда,преходящих приступов ишемии, фибрилляции предсердий, тромботического шока, эмболического шока, тромбоза глубокой вены, диссеминированного внутрисосудистого свертывания,глазного отложения фибрина и повторной окклюзии или рестеноза реканализированных сосудов. Указанные соединения могут быть также использованы для изучения механизма действия сериновых протеаз, участвующих в каскаде реакций коагуляции, в целях получения лучших ингибиторов и разработки лучших аналитических методов. Соединения формулы (I) могут быть также использованы для предупреждения нарушения мозгового кровообращения (CVA) или инсульта. В семейство соединений формулы (I) также входят их фармацевтически приемлемые соли. Термин "фармацевтически приемлемая соль" охватывает соли, обычно используемые для образования солей щелочных металлов и для образования аддитивных солей свободных кислот или свободных оснований. Природа данной соли не имеет решающего значения, при условии, что она является фармацевтически приемлемой. Подходящие фармацевтически приемлемые кислотно-аддитивные соли соединений формулы (I) могут быть получены из неорганической кислоты или из органической кислоты. Примерами таких неорганических кислот являются соляная, бромисто-водородная, иодисто-водородная, азотная, угольная, серная и фосфорная кислоты. Подходящие органические кислоты могут быть выбраны из органических кислот класса алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоциклических и сульфоновых кислот, примерами которых являются муравьиная, уксусная, пропионовая, янтарная, гликолевая, глюконовая, молочная, яблочная, винная,лимонная, аскорбиновая, глюкороновая, малеиновая, фумаровая, пирувиновая, аспартановая,глутаминовая, бензойная, антраниловая, мезиловая, салициловая, п-гидроксибензойная, фенилуксусная, миндальная, эмбоновая (памовая),метансульфоновая, этилсульфоновая, бензолсульфоновая, сульфаниловая, стеариновая, циклогексиламиносульфоновая, альгеновая, галактуроновая кислота. Подходящими фармацевтически приемлемыми основно-аддитивными солями соединений формулы (I) являются соли металлов, образованные алюминием, кальцием,литием, магнием, калием, натрием и цинком,или органические соли, образованные N,N' 11 дибензилэтилендиамином, холином, хлорпрокаином, диэтаноламином, этилендиамином, меглумином (N-метилглукамином) и прокаином. Все указанные соли могут быть получены стандартными методами из соответствующего соединения формулы (I) путем реакции, например,соответствующей кислоты или основания с соединением формулы (I). Настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) в сочетании по крайней мере с одним фармацевтически приемлемым носителем, адъювантом или разбавителем. Фармацевтические композиции настоящего изобретения могут содержать активные соединения формулы(I) в сочетании с нетоксичными фармацевтически приемлемыми носителями, и/или разбавителями, и/или адъювантами (называемыми в настоящем описании общим термином "материалы-носители") и, если это необходимо, другие активные ингредиенты. Активные соединения настоящего изобретения могут быть введены подходящим способом, предпочтительно,в форме фармацевтической композиции, пригодной для такого способа введения, и в дозе,эффективной для предусматриваемого лечения. Активные соединения и композиции могут быть, например, введены перорально, интраваскулярно, внутрибрюшинно, подкожно, внутримышечно, офтальмически или местно. Для лечения глазного отложения фибрина данные соединения могут быть введены офтальмически или местно, а также перорально или парентерально. Данные соединения могут быть введены в форме депо-инъекции или в форме препаратаимплантата, который может быть изготовлен так, чтобы обеспечивалось пролонгированное высвобождение активного ингредиента. Этот активный ингредиент может быть спрессован в таблетки или небольшие цилиндры и подкожно или внутримышечно имплантирован в виде депо-инъекций или имплантатов. Для имплантатов могут быть использованы инертные материалы, такие как биологически разлагаемые материалы или синтетические силиконы, например силастик, силоксановый каучук или другие кремнийсодержащие полимеры. Указанные соединения могут быть также введены в форме липосомных систем доставки,таких как небольшие однослойные везикулы,крупные однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Указанные соединения могут быть также доставлены с использованием моноклональных антител в качестве отдельных носителей, с которыми связывается молекула данного соединения. Указанные соединения могут быть также связаны с растворимыми полимерами, служащими в качестве носителей для доставляемого 12 лекарственного средства. Такими полимерами могут быть поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный пальмитоильными остатками. Кроме того, указанные соединения могут быть присоединены к биологически разлагаемым полимерам, используемым для осуществления регулируемого высвобождения лекарственного средства, например к полимолочной кислоте, полигликолевой кислоте,сополимерам полимолочной и полигликолевой кислоты, полиэпсилонкапролата, полигидроксимасляной кислоте, сложным полиортоэфирам,полиацеталям, полидигидропиранам, полицианоакрилатам и перекрестно-сшитым или амфипатическим блоксополимерам гидрогелей. Для перорального введения фармацевтические композиции могут быть получены, например, в форме таблеток, капсул (каждая из которых включает композиции пролонгированного или замедленного высвобождения), драже, порошков, гранул, эликсиров, настоек, суспензий,жидкостей, включая сиропы, и эмульсий. Фармацевтические композиции предпочтительно изготавливают в виде разовой лекарственной формы, содержащей конкретное количество активного ингредиента. Примерами таких унифицированных лекарственных форм являются таблетки или капсулы. Активный ингредиент может быть также введен путем инъекции в виде композиции, где в качестве подходящего носителя могут быть использованы, например,физиологический раствор, декстроза или вода. Количество терапевтически активных соединений, предназначенных для введения, и схема введения доз для лечения патологических состояний соединениями и/или композициями настоящего изобретения зависят от ряда факторов, включая возраст, вес, пол и состояние индивидуума, от тяжести заболевания, от способа и частоты введения и от конкретно используемого соединения, и этот ряд факторов может быть очень широким. Фармацевтические композиции могут содержать активные ингредиенты в количестве от около 0,1 до 2000 мг, а предпочтительно от около 0,5 до 500 мг. Подходящей ежедневной дозой может быть доза от около 0,01 до 100 мг/кг массы тела, а предпочтительно от около 0,5 до около 20 мг/кг массы тела. Такая ежедневная доза может быть введена 1-4 раз в день. Соединения могут быть включены в мазь или крем для наружного применения или в суппозитории, содержащие активные ингредиенты в расчете на полное количество композиции, в количестве, например, 0,075-30 мас.%, предпочтительно 0,2-20 мас.%, а наиболее предпочтительно 0,4-15 мас.%. При приготовлении мази активные ингредиенты могут быть использованы в сочетании либо с парафиновой основой,либо со смешиваемой с водой основой для мази. 13 Альтернативно, активные ингредиенты могут быть включены в крем, приготовленный на основе для крема типа "масло в воде". Если это необходимо, водная фаза основы для крема может включать, например по крайней мере 30 мас.% многоатомного спирта, такого как пропиленгликоль, бутан-1,3-диол, маннит, сорбит,глицерин, полиэтиленгликоль и их смеси. Композиция для местного применения может, что желательно, включать соединение, повышающее степень абсорбции или проникновения активного ингредиента через кожу или другие пораженные участки. Примерами таких усилителей проникновения через кожу являются диметилсульфоксид и родственные аналоги. Соединения настоящего изобретения также могут быть введены с использованием приспособления для чрескожной доставки. Местное введение может быть предпочтительно осуществлено с использованием пластыря либо типа резервуара и пористой мембраны, либо типа твердой матрицы. В любом случае, непрерывная доставка активного агента осуществляется из указанного резервуара или микрокапсул через мембрану в адгезив, проницаемый для активного агента, который контактирует с кожей или слизистой реципиента. Если активный агент абсорбируется через кожу, то реципиенту вводят регулируемый и предварительно определенный поток активного агента. В случае использования микрокапсул, инкапсулирующий агент может также функционировать как мембрана. Масляная фаза эмульсий настоящего изобретения может быть приготовлена из известных ингредиентов известным способом. Хотя эта фаза может включать лишь эмульгатор, однако, она может включать смесь по крайней мере одного эмульгатора с жиром или с маслом либо и с жиром, и с маслом. Предпочтительно,если гидрофильный эмульгатор включен вместе с липофильным эмульгатором, который действует как стабилизатор. Предпочтительно также,чтобы он также включал как масло, так и жир. Помимо этого, данный эмульгатор(ы) со стабилизатором(ами) или без него образует(ют) так называемый эмульгирующийся воск, и этот воск вместе с указанным маслом и жиром составляет так называемую эмульгирующую основу для мази, которая образует диспергированную масляную фазу данного крема. Эмульгаторами и стабилизаторами эмульсий, которые могут быть использованы в композиции настоящего изобретения, являются Твин 60, Спан 80, цетостеариловый спирт, миристиловый спирт, глицерилмоностеарат, лаурилсульфат натрия и т.п. Поскольку растворимость активного соединения в большинстве масел, которые обычно используются в фармацевтических эмульсиях,является очень низкой, то для достижении нужных косметических свойств необходимо выбрать подходящие масла или жиры для данной композиции. Таким образом, данный крем дол 004867 14 жен быть предпочтительно нежирным, неокрашивающим и смываемым продуктом с подходящей консистенцией во избежание вытекания из тюбиков или других контейнеров. Могут быть использованы прямые или разветвленные моно- или диосновные алкиловые сложные эфиры, такие как диизоадипат, изоцетилстеарат,сложный диэфир пропиленгликоля и жирных кислот кокосового ореха, изопропилмиристат,децилолеат, изопропилпальмитат, бутилстеарат,2-этилгексилпальмитат или смесь разветвленных сложных эфиров. Эти эфиры могут быть использованы отдельно или в комбинации, в зависимости от требуемых свойств. Альтернативно, могут быть использованы липиды с высокой температурой плавления, такие как белый полутвердый парафин и/или вазелиновое масло,или другие минеральные масла. Для терапевтических целей активные соединения настоящего изобретения обычно объединяют с одним или несколькими адъювантами, подходящими для указанного способа введения. При пероральном введении данные соединения могут быть смешаны с лактозой, сахарозой, порошком крахмала, сложными эфирами целлюлозы и алкановых кислот, алкиловыми сложными эфирами целлюлозы, тальком, стеариновой кислотой, стеаратом магния, окисью магния, натриевыми и кальциевыми солями фосфорной и серной кислот, желатином, аравийской камедью, альгинатом натрия, поливинилпирролидоном и/или с поливиниловым спиртом, а затем из них могут быть изготовлены таблетки или капсулы для соответствующего введения. Такие капсулы или таблетки могут содержать композицию с регулируемым высвобождением, поскольку они могут быть изготовлены в виде дисперсии активного соединения в гидроксипропилметилцеллюлозе. Композиции для парентерального введения могут быть изготовлены в форме водных или безводных изотонических стерильных растворов или суспензий для инъекций. Эти растворы и суспензии могут быть получены из стерильных порошков или гранул, имеющих один или несколько носителей или разбавителей, используемых в композициях для перорального введения. Указанные соединения могут быть растворены в воде, полиэтиленгликоле, пропиленгликоле, этаноле,кукурузном масле, масле семян хлопчатника,арахисовом масле, кунжутном масле, бензиловом спирте, хлориде натрия и/или в различных буферах. Хорошо известны и широко применяются в фармацевтической практике и другие адъюванты и способы введения. При осуществлении способов настоящего изобретения для лечения и предупреждения ряда тромботических состояний, включая поражения коронарной артерии и цереброваскулярные заболевания, соединения и фармацевтические композиции настоящего изобретения вводят отдельно, или в комбинации друг с другом, 15 либо в комбинации с другими терапевтическими или in vivo-диагностическими агентами. Ингибиторы каскада реакций коагуляции настоящего изобретения могут быть также введены вместе с подходящими агентами, ингибирующими агрегацию тромбоцитов, включая, но не ограничиваясь ими, тиклопидин или клопидрогель; с антагонистами рецептора фибриногена(т.е. для лечения или предупреждения нестабильной стенокардии или для предупреждения повторной окклюзии после ангиопластики и рестеноза); с антикоагулянтами, такими как аспирин, варфарин или гепарины; с тромболитическими агентами, такими как активаторы плазминогена или стрептокиназа для достижения синергических эффектов при лечении различных патологий; с агентами, снижающими уровень липидов в крови, включая средства,снижающие уровень холестерина в крови (например, ингибиторы СоА-редуктазы HMG, такие как мевастатин, ловастатин, симвастатин,правастатин и флувастатин, ингибиторы СоАсинтетазы HMG и т.п.); с противодиабетическими лекарственными средствами или с другими сердечно-сосудистыми средствами (диуретиками, действующими в петле Генле, диуретиками типа тиазида, нитратами, антагонистами альдостерона (т.е. спиронолактоном и эпоксимекслереноном), ингибиторами фермента превращения ангиотензина (например, АСЕ), антагонистами рецептора ангиотензина II, бетаблокаторами, средствами против аритмии, гипотоническими агентами и блокаторами кальциевых каналов) для лечения или предупреждения атеросклероза. Так, например, у пациентов,страдающих поражением коронарной артерии, и у пациентов, подвергнутых операции ангиопластики, должно наблюдаться улучшение состояния после совместного введения антагонистов рецептора фибриногена и ингибиторов каскада реакции свертывания крови настоящего изобретения. Кроме того, ингибиторы каскада реакции свертывания крови могут повышать эффективность тромболитической реперфузии, опосредованной тканевым активатором плазминогена. Обычные дозы ингибиторов каскада реакций свертывания крови настоящего изобретения с другими подходящими антитромбоцитарными агентами, антикоагулирующими агентами, сердечнососудистыми терапевтическими средствами или тромболитическими агентами могут быть такими же, как и дозы ингибиторов каскада реакций свертывания крови, вводимых без совместного введения дополнительных антитромбоцитарных агентов, антикоагулирующих агентов, сердечно-сосудистых терапевтических средств или тромболитических агентов, либо они могут быть, в основном, меньше, чем дозы ингибиторов каскада реакций свертывания крови, вводимых без совместного введения дополнительных антитромбоцитарных агентов, антикоагулирующих агентов, сердечнососудистых 16 терапевтических средств или тромболитических агентов, в зависимости от терапевтических доз,требующихся для пациента. Все указанные выше цитируемые работы введены в настоящее описание в качестве ссылки. Хотя настоящее изобретение описано на конкретных вариантах его осуществления, детали этих вариантов не должны рассматриваться как ограничивающие объем изобретения. Следующие ниже примеры приводятся для иллюстрации настоящего изобретения и не должны рассматриваться как ограничивающие его объем. Без излишнего экспериментирования очевидно, что любой специалист в данной области,используя приведенное выше описание, может реализовать настоящее изобретение в полной мере. Поэтому следующие ниже предпочтительные конкретные варианты осуществления изобретения приводятся лишь в иллюстративных целях и никоим образом не должны рассматриваться как ограничение изобретения. Рассматриваются также соединения, содержащие множество вариантов структурных модификаций, проиллюстрированных в схемах или в следующих ниже примерах. Каждому специалисту в данной области будет ясно, что для получения данных соединений могут быть использованы различные условия и способы осуществления следующих ниже процедур приготовления. Каждый специалист в данной области может применить общие методы для получения соединений нижеследующих конкретных примеров, которые были или могут быть соответствующим образом охарактеризованы с помощью 1H-ЯМР, масс-спектрометрии, элементного анализа и аналогичных процедур. Данные соединения могут быть также образованы in vivo. Следующие ниже примеры включают подробное описание способов получения соединений формулы (I). Подробное описание, входящее в объем изобретения, приводится лишь в иллюстративных целях, а поэтому не должно рассматриваться как ограничение объема изобретения. Все части даны по массе, а температура приводится в градусах Цельсия, если это не оговорено особо. Для осуществления настоящего изобретения могут быть использованы общепринятые последовательности общего синтеза. В схемах и таблицах были использованы следующие сокращения: "АА" означает аминокислоты; "AcCN" означает ацетонитрил, "АсОН" означает уксусную кислоту; "BINAP" означает 2,2'-бис(дифенилфосфино)-1,1'-бинафтил; "ВnОН" означает бензиловый спирт, "ВnСНО" означает 2-фенилэтаналь; "BnSO2Cl" означает бензилсульфонилхлорид; "Воc" означает трет-бутилоксикарбонил;"ВОР" означает бензотриазол-1-ил-окситрис(диметиламино), "bu" означает бутил; "dba" означает дибензилиденацетон; "DCC" означает 1,3 дициклогексилкарбодиимид; "DCM" означает дихлорметан или метиленхлорид; "DIBAH" или"ДМФ" означает диметилформамид; "ДМСО" означает диметилсульфоксид; "DPPA" означает дифенилфосфорилазид, "EDC" означает гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида,"Пр. " означает пример , "Fmoc" означает 9 флуоренилметоксикарбонил, "HOBt" означает гидроксибензотриазол, "LDA" означает диизопропиламид лития, "MW" означает молекулярную массу, "NMM" означает N-метилморфолин,"Ph" означает фенил или арил, "РНТН" означает фталоильную группу, "рnz" означает 4-нитробензилоксикарбонил, "РТС" означает межфазный катализатор, "ру" означает пиридин, "RNH2" означает первичный органический амин, "SEM" означает 2-(триметилсилил)этоксиметилхлорид,"p-TsOH" означает паратолуолсульфоновую кислоту, "TBAF" означает фторид тетрабутиламмония,"TBTU" означает тетрафторборат 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония, "TEA" означает триэтиламин, "TFA" означает трифторуксусную кислоту, "ТГФ" означает тетрагидрофуран, "TMS" означает гриметилсилил,"TMSCN" означает триметилсилилцианид, a"Cbz" или "Z" означает бензилоксикарбонил. Общие процедуры синтеза и конкретные примеры Соединения настоящего изобретения могут быть синтезированы, например, в соответствии со следующими ниже процедурами и схемами, описанными ниже. Общий способ синтеза R2-замещенных пиримидинонов широкого ряда показан ниже на схемах 1-5. В результате обработки раствора гидрохлорида этилбензимидата в метаноле диметилацеталем аминоацетальдегида получают замещенный бензимидин. Циклизация бензимидина с диметилметоксиметиленмалонатом приводит к образованию пиримидинонового гетероциклильного ядра с требуемыми функциональными группами для последующих манипуляций. Деметилирование сложного эфира иодидом лития с последующей реакцией перегруппировки Курциуса полученной кислоты приводит к введению реакционного азота у С-5 с получением защищенного карбамата. Гидролиз диметилацетала и окисление полученного альдегида хлоридом натрия приводит к образованию глицинового звена у N-3. Защита кислоты с образованием трет-бутилового сложного эфира с последующим снятием защиты у карбамата путем гидрирования приводит к образованию свободного амина у С-5. Обработка данного амина сульфонилхлоридом или альдегидом в условиях восстановительного аминирования приводит к образованию сульфонамида или вторичного амина, соответственно. Затем с защищенных кислот снимают защиту с использованием НСl. После этого указанные кислоты подвергают реакции сочетания с различными аминами в стандартных условиях сочетания пептидов. Данные амины являются обычно многофункциональными и используются в некото 004867 18 рых защищенных формах. Удаление защитных групп приводит к получению соединений, пригодных для оценки. Примеры таких синтетических схем и процедур описаны ниже. Схема 1 Общий синтез-1 пиримидинонов(92,25 г, 496,9 ммоль) в 300,0 мл сухого метанола (1,68 М) охлаждают приблизительно до 0 С и по каплям добавляют раствор диметилацеталя аминоацетальдегида (73,10 мл, 670,9 ммоль) в сухом метаноле (75,0 мл, 9,0 М) с такой скоростью, чтобы температура поддерживалась ниже 5 С. Полученный раствор оставляют на 3 дня для перемешивания, поддерживая температуру ниже 5 С. Затем реакционную смесь концентрируют при пониженном давлении с получением желтого масла. Остаток растворяют в 1 н.(4 х 250 мл). Органические растворы объединяют, сушат (МgSO4) и концентрируют с получением 108,13 г неочищенного N-(2,2-диметоксиэтил)бензамидина в виде желтого масла. К указанному неочищенному N-(2,2-диметоксиэтил) бензамидину (108,13 г, 519,2 ммоль) в сухом метаноле (125,0 мл, 4,2 М) одной порцией добавляют диметилметоксиметиленмалонат (94,13 г,540,5 ммоль) при комнатной температуре. Полученную смесь нагревают приблизительно до 100 С, при этом растворитель медленно отгоняют в течение 2 ч. Полученный темнокоричневый раствор оставляют для охлаждения до комнатной температуры и разбавляют этилацетатом (1 л). Органический раствор промывают насыщенным NH4Cl (2 х 500 мл) и рассолом(1 х 500 мл). Органический раствор сушат (MgSO4),фильтруют и концентрируют. Неочищенный продукт очищают ЖХСД (25% этилацетат/гексан), в результате чего получают чистый метил 1-(2,2-диметоксиэтил-2-фенилпиримидин-6-(1 Н)он-5-карбоксилат (пример 1 А) с выходом 73% в виде желтовато-коричневого масла. 1(75 МГц, CDCl3)165,9, 164,6, 159,3, 158,2,134,6, 130,9, 128,93, 128,78, 114,9, 101,4, 56,0,55,1, 52,7, 49,1; HRMS (ES) для C16H19N2O5: вычислено 319,1294; найдено: 319,1288. Пример 1 В. К раствору метил 1-(2,2-диметоксиэтил-2-фенилпиримидин-6-(1 Н)-он-5-карбоксилата (93,00 г,292,2 ммоль) в 420,0 мл сухого пиридина (0,70 М) одной порцией добавляют иодид лития (98,00 мл, 732,2 ммоль), перемешивая при комнатной температуре, после чего наблюдается экзотермическая реакция. Полученную светло-коричневую суспензию нагревают с обратным холодильником в течение 2 ч. Темно-коричневую реакционную смесь оставляют для охлаждения до комнатной температуры и летучие вещества удаляют при пониженном давлении. Полученное масло разбавляют 1 н. НСl (500 мл). Водный раствор экстрагируют смесью дихлорметан/ метанол (4:1, 4 х 250 мл). Объединенные органические растворы промывают 6 н. НСl (2 х 250 мл), 21 сушат (МgSO4), фильтруют и концентрируют. Неочищенный продукт очищают кристаллизацией (этилацетат/гексан) с получением чистой 1-(2,2-диметоксиэтил-2-фенилпиримидин-6-(1 Н)он-5-карбоновой кислоты (пример 1 В) с выходом 63% в виде белого твердого вещества. 1(0,27 М) добавляют триэтиламин (50,0 мл, 358,7 ммоль), а затем одной порцией добавляют дифенилфосфорилазид (51,40 мл, 238,5 ммоль) при комнатной температуре. Полученный раствор медленно нагревают с обратным холодильником в течение 2 ч. Затем к реакционной смеси добавляют бензиловый спирт (45,00 мл,434,8 ммоль) и нагревание с обратным холодильником продолжают приблизительно в течение 14 ч. Черный раствор оставляют для охлаждения до комнатной температуры и летучие вещества удаляют в вакууме. Полученный остаток разбавляют этилацетатом (1,5 л). Органический раствор промывают насыщенным NH4Cl (2 х 500 мл), 1 н. NaOH (1 х 500 мл) и рассолом (1 х 500 мл). Органический раствор сушат (MgSO4),фильтруют и концентрируют с получением неочищенного продукта. После очистки с помощью ЖХСД (15-30% этилацетат/гексан) получают чистый диметилацеталь [5-[(бензилоксикарбонил)амино]-2-фенил-6-оксо-1,6-дигидро-1-пиримидинил]ацетальдегида (пример 1 С) в виде светло-коричневого твердого вещества с выходом 46%. 1HRMS (EI) для C22H24N3O5: вычислено 410,1716; найдено: 410,1741. Пример 1D. К раствору диметилацеталя [5-[(бензилоксикарбонил)амино]-2-фенил-6-оксо-1,6-дигидро-1 пиримидинил]ацетальдегида (17,24 г, 42,11 ммоль) в 103,0 мл тетрагидрофурана добавляют 35,0 мл 1 н HCl. Полученную двухфазную смесь нагревают с обратным холодильником в течение 12 ч. Затем реакционную смесь охлаждают до комнатной температуры и летучие вещества удаляют в вакууме. Полученный остаток разбавляют водой (200 мл) и рН доводят до 7 добавлением твердого NаНСО 3. Полученную эмульсию экст 004867 22 рагируют дихлорметаном (4 х 150 мл). Объединенные органические растворы промывают водой (1 х 200 мл), сушат (MgSO4), фильтруют и концентрируют, в результате чего получают 15,74 г неочищенного [5-[(бензилоксикарбонил) амино]-2-фенил-6-оксо-1,6-дигидро-1-пиримидинил]ацетальдегида. Раствор неочищенного [5-[(бензилоксикарбонил)амино]-2-фенил-6-оксо-1,6-дигидро-1 пиримидинил]ацетальдегида (15,30 г, 42,11 ммоль) в 198,0 мл тетрагидрофурана, трет-бутилового спирта и 2-метил-2-бутена (1:1:1,3, 0,21 М) охлаждают до 0 С. Затем в раствор медленно добавляют раствор хлорита натрия (29,94 г, 331,1 ммоль) и моногидрата дигидрофосфата натрия(35,42 г, 256,7 ммоль) в 102,0 мл воды (3,2 М,исходя из концентрации хлорита натрия). Полученный золотистый двухфазный раствор перемешивают в течение 10 мин и охлаждающую баню удаляют. Реакционную смесь перемешивают в течение 1 ч при комнатной температуре. Летучие вещества удаляют при пониженном давлении. Полученный раствор разбавляют водой (200 мл) и рН доводят до 3 добавлением насыщенного NаНСО 3 и 1 н. НСl. Водный раствор экстрагируют смесью тетрагидрофурана и дихлорметана (1:2, 4 х 180 мл). Объединенные органические растворы сушат (MgSO4), фильтруют и концентрируют с получением неочищенного продукта. После очистки растиранием с этиловым эфиром получают [5-[(бензилоксикарбонил)амино]-2-фенил-6-оксо-1,6-дигидро-1 пиримидинил]уксусную кислоту с выходом 88% в виде белого твердого вещества. 1(с, 2 Н), 4,56 (с, 2 Н); 13 С ЯМР (75 МГц, d-ДМСО)169,4, 158,0, 154,6, 154,3, 137,1, 134,8, 130,9,129,4, 129,1, 128,78, 128,72, 128,50, 125,5, 128,50,125,5, 67,7, 48,8; HRMS (EI) для С 20 Н 18N3 О 5: вычислено 380,1246; найдено: 380,1246. Пример 1E. Суспензию [5-[(бензилоксикарбонил)амино]-2-фенил-6-оксо-1,6-дигидро-1-пиримидинил] уксусной кислоты (5,2503 г, 13,84 ммоль) в 70,0 мл хлороформа (0,2 М) охлаждают на бане со льдом до приблизительно 0 С. Затем к охлажденной суспензии по каплям шприцем добавляют оксалилхлорид (6,00 мл, 68,78 ммоль). После интенсивного выделения газа образуется золотистый гомогенный раствор. После перемешивания в течение 5 мин охлаждающую баню удаляют и раствор перемешивают еще 2 ч при комнатной температуре. Растворитель удаляют при пониженном давлении и помещают на 10 мин в систему высокого вакуума для удаления остаточных растворителей. Полученное желтое твердое вещество разбавляют 70,0 мл хлороформа (0,2 М) и добавляют пиридин (1,70 мл,21,02 ммоль) и 2-метил-2-пропанол (3,50 мл,36,60 ммоль). Полученный желтовато-коричне 23 вый раствор перемешивают в течение 1 ч при комнатной температуре, а затем нагревают с обратным холодильником в течение 12 ч. Реакционную смесь охлаждают до комнатной температуры и разбавляют хлороформом (300 мл). Органический раствор промывают водой (1 х 100 мл), насыщенным NаНСО 3 (1 х 100 мл) и рассолом (1 х 100 мл). Органический раствор сушат(MgSO4), фильтруют и концентрируют. Неочищенную реакционную смесь очищают с помощью ЖХСД (25% этилацетат/гексан), в результате чего получают продукт с выходом 49%. 1H ЯМР (300 МГц, CDCl3)8,81 (шир.с,1 Н), 7,57-7,38 (м, 11 Н), 5,27 (с, 2 Н), 4,57 (с, 2 Н),1,47 (с, 9 Н); 13 С ЯМР (75 МГц, СDСl3)166,4,158,0, 153,2, 135,9, 135,0, 134,4, 130,6, 129,1,128,9, 128,7, 128,5, 128,4, 125,4, 83,4, 67,7, 49,1,28,2; HRMS (ES) для С 24 Н 26N3O5: вычислено 436,1872; найдено: 436,1876. Пример 1F. К раствору трет-бутилового эфира [5 амино-2-фенил-6-оксо-1,6-дигидро-1-пиримидинил]уксусной кислоты (1,8647 г, 4,282 ммоль) в 21,0 мл метанола (0,2 М) одной порцией добавляют 211,3 мг 10% Pd/C. Полученную смесь перемешивают в атмосфере водорода (баллонного) при комнатной температуре в течение приблизительно 16 ч. Неочищенную реакционную смесь фильтруют через слой целита 545 и растворитель удаляют при пониженном давлении. Неочищенный продукт растирают с этиловым эфиром и получают чистый продукт третбутиловый эфир [5-амино-2-фенил-6-оксо-1,6 дигидро-1-пиримидинил]уксусной кислоты (пример 1F) с выходом 99%. 1N-метилморфолин (1,20 мл, 10,91 ммоль). Раствор охлаждают до 0 С на ледяной бане. После перемешивания в течение 10 мин по каплям в течение 5 мин добавляют раствор 718,2 мг толуолсульфонилхлорида (3,767 ммоль) в 5,5 мл тетрагидрофурана (0,68 М). Реакционную смесь перемешивают в течение 2 ч при 0 С. Реакционную смесь разбавляют этилацетатом (150 мл). Органический раствор промывают 1 н. НСl (2 х 25 мл), насыщенным NaHCO3 (2 х 25 мл) и рассолом(МgSO4), фильтруют и концентрируют при пониженном давлении. Полученное желтое твердое вещество растирают с этиловым эфиром, 004867 24 фильтруют и сушат в вакууме с получением чистого продукта (пример 1G) в виде белого твердого вещества с выходом 74%. 1(с, 2 Н), 1,32 (с, 9 Н); 13 С ЯМР (100 МГц, d-ДМСО)167,0, 158,8, 156,5, 142,1, 134,5, 131,7, 131,0 130,1, 129,4, 129,0, 128,94, 128,58, 124,8, 83,0,59,6, 49,2, 28,1; HRMS (EI) для С 23 Н 26N3O5S: вычислено 456,1593; найдено: 456,1597. Пример 1H. Раствор (пример 1G) (1,0643 г, 2,336 ммоль) в 9,0 мл 4 М НСl в диоксане (0,1 М) перемешивают в течение 12 ч при комнатной температуре. Неочищенную реакционную смесь концентрируют при пониженном давлении. Полученный остаток растирают с этиловым эфиром и получают чистый продукт (пример 1H) с выходом 87% в виде белого твердого вещества. 1(с, 2 Н); 13 С ЯМР (100 МГц, d-ДМСО)169,2,158,7, 156,6, 141,9, 134,5, 131,7 131,0 130,1, 129,4,129,0, 128,90, 128,56, 128,8, 59,6, 48,9; HRMS (EI) для C19H18N3O5S: вычислено 400,0967; найдено: 400,0959. Пример 1I. Раствор кислоты (пример 1H) (406,8 мг,1,018 ммоль) в 10,0 мл диметилформамида (0,10 М) добавляют N,N-диизопропилэтиламин (0,900 мл,5,167 ммоль), N-гидроксибензотриазол (167,7 мг,1,241 ммоль) и гидрохлорид 1-[3-(диметиламино)пропил]-3-этилкарбодиимида (236,1 мг,1,232 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 30 мин,после чего смесь становится гомогенной. Затем к реакционной смеси одной порцией добавляют 4-(Cbz-амидин)бензиламин (324,6 мг, 1,126 ммоль) при комнатной температуре. После этого полученную смесь перемешивают в течение 18 ч. Реакционную смесь разбавляют этилацетатом (50 мл). Органический раствор промывают 5% лимонной кислотой (1 х 25 мл), насыщеннымNaHCO3 (1 х 25 мл) и рассолом (1 х 25 мл). Органический раствор сушат (МgSO4), фильтруют и концентрируют. Неочищенную реакционную смесь очищают растиранием с этиловым эфиром и получают чистый продукт (пример 1I) в виде белого твердого вещества. 1HRMS (EI) для C35H33N6O6S2: вычислено 665,2182; найдено: 665,2177. Раствор Cbz-амидина (пример 1I) (237,7 мг, 357,6 ммоль) в 3,5 мл метанола и 4 М НСl в диоксане (4:1, 0,1 М) одной порцией добавляют 42,1 мг 10% Pd/C. Полученную смесь перемешивают в атмосфере водорода (баллонного) при комнатной температуре в течение приблизи 25 тельно 16 ч. Неочищенную реакционную смесь фильтруют через слой целита 545 и растворитель удаляют при пониженном давлении. Неочищенный продукт очищают с помощью ВЭЖХ (градиент, 5-95% ацетонитрил/вода с 0,1% трифторуксусной кислотой) и получают чистый продукт в виде белого твердого вещества. 1 Н ЯМР (300 МГц, d-ДМСО)9,31-9,28 (м,4 Н), 8,88 (шир.с, 1 Н), 7,81-7,77 (м, 3 Н), 7,60-7,54 Пример 3 А. К раствору трет-бутилового эфира [5 амино-2-фенил-6-оксо-1,6-дигидро-1-пиримидинил]уксусной кислоты (пример 1F) (613,7 мг,2,037 ммоль) в 7,0 мл тетрагидрофурана и дихлорметана (1:1, 0,3 М) добавляют 25,0 мл уксусной кислоты и фенилацетальдегид (0,475 мл,4,060 ммоль). Раствор охлаждают до 0 С на ледяной бане и одной порцией добавляют триацетоксиборогидрид натрия (1,9131 г, 9,027 ммоль). После перемешивания в течение 5 мин, ледяную баню удаляют и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Затем реакцию гасят добавлением 1 н. NaOH (5 мл) и смесь перемешивают в течение 5 мин. Реакционную смесь разбавляют 0,5 н. NaOH (100 мл). Водный раствор экстрагируют этилацетатом (3 х 25 мл). Объединенные органические растворы промывают 0,5 н. NaOH (1 х 25 мл) и рассолом (1 х 25 мл). Раствор сушат (MgSO4),фильтруют и концентрируют при пониженном давлении. После очистки с помощью ЖХСД(25% этилацетат/гексан) получают соединение примера 3 А в виде желтого масла с выходом 74%. 1 26 2 Н), 1,40 (с, 9 Н); HRMS (EI) для C24H28N3O3: вычислено 406,2131; найдено: 406,2125. Раствор соединения примера 3 А (521,3 мг,1,286 ммоль) в 13,0 мл 4 М НСl в диоксане (0,1 М) перемешивают в течение 12 ч при комнатной температуре. Неочищенную реакционную смесь концентрируют при пониженном давлении. Полученный остаток растирают с этиловым эфиром и получают чистый продукт (пример 3 В) с количественным выходом в виде желтого твердого вещества. 1HRMS (EI) для C20H20N3O3: вычислено 350,1505; найдено: 350,1520. К раствору соединения примера 3 В (497,8 мг, 1,290 ммоль) в 13,0 мл диметилформамида(302,5 мг, 1,578 ммоль). Полученную смесь перемешивают при комнатной температуре 30 мин,после чего смесь становится гомогенной. Затем к реакционной смеси одной порцией добавляют 4-(Сbz-амидин)бензиламин (410,9 мг, 1,425 ммоль) при комнатной температуре. После этого полученную смесь перемешивают 18 ч. Реакционную смесь разбавляют этилацетатом (50 мл). Органический раствор промывают 5% лимонной кислотой (1 х 25 мл), насыщенным NаНСО 3(1 х 25 мл) и рассолом (1 х 25 мл). Органический раствор сушат (MgSO4), фильтруют и концентрируют. Неочищенную реакционную смесь очищают растиранием с этиловым эфиром и получают чистый продукт (пример 3 С) в виде белого твердого вещества. 1H ЯМР (400 МГц, d-ДМСО)9,08 (шир.с,3 Н), 8,62 (т, J=5,8 Гц, 1 Н), 7,90 (д, J=8,3 Гц, 2 Н),7,44-7,15 (м, 17 Н), 5,35 (т, J=5,9 Гц, 1 Н), 5,07 (с,2 Н), 4,44 (с, 2 Н), 4,29 (д, J=5,4 Гц, 2 Н), 3,31-3,26 (м,2 Н), 2,88-2,85 (м, 2 Н); HRMS (EI) для C36H35N6O4: вычислено 615,2720; найдено: 615,2688. К раствору соединения примера 3 С (222,6 мг, 362,1 ммоль) в 4,0 мл метанола и 4 М НСl в диоксане (3:1, 0,1 М) одной порцией добавляют 53,0 мг 10% Pd/C. Полученную смесь перемешивают в атмосфере водорода (баллонного) при комнатной температуре в течение приблизительно 16 ч. Неочищенную реакционную смесь фильтруют через слой целита 545 и растворитель удаляют при пониженном давлении. Неочищенный продукт очищают растиранием с этиловым эфиром и получают чистый продукт в виде желтого твердого вещества. 1(шир.м, 5 Н), 7,88 (шир.м, 2 Н), 7,58-6,86 (шир.м,15 Н), 4,59 (шир.с, 2 Н), 4,36 (шир.с, 2 Н), 3,38 (шир.с, 27 2 Н), 2,91 (шир.с, 2 Н); HRMS (EI) для C28H29N6O2: вычислено 481,2352; найдено: 481,2348. Пиримидиноны, имеющие, например, непосредственно связанный 2-арил, 2-гетероарил или 2-гетероатом, связывающий органическую группу через линкерную группу или непосредственно через гетероатом с пиримидиноновым кольцом, могут быть получены в соответствии со схемой 6 и схемой 7, представленными ниже. Гетероатомом обычно является сера, азот, кислород или другой подходящий гетероатом. Использование процедуры, проиллюстрированной на схеме 6 для получения конкретных замещенных гетероатомом пиримидинонов, описано в примерах 4 и 5. Схема 6 Получение 2-замещенных пиримидинонов Пример 4 А. К раствору 5-нитро-2,4(1 Н,3 Н)пиримидиндиона в диметилсульфоксиде (0,2 М) одной порцией добавляют, размешивая, 1,1 эквивалента карбоната калия. Приблизительно через 10 мин по каплям добавляют раствор 1 эквивалента 2-(триметилсилил)этоксиметилхлорида в диметилсульфоксиде. Затем реакционную смесь нагревают до 40 С и перемешивают в течение 18 ч. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 4 А. 28 Пример 4 В. К раствору 5-нитро-1-SЕМ-2,4(1 Н,3 Н)пиримидиндиона (пример 4 А) в диметилсульфоксиде (0,2 М) одной порцией добавляют, размешивая, 1,1 эквивалента карбоната калия. Приблизительно через 10 мин по каплям добавляют раствор 1 эквивалента метилбромацетата в диметилсульфоксиде. Затем реакционную смесь нагревают до 40 С и перемешивают в течение 18 ч. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 4 В. Пример 4 С. Раствор 5-нитро-1-SЕМ-3-метоксикарбонилметил-2,4(1 Н,3 Н)пиримидиндиона (пример 4 В) в метаноле дегазируют газообразным водородом. Затем к раствору добавляют 5% Pd/C и перемешивают в атмосфере водорода в течение 24 ч при комнатной температуре. Неочищенную реакционную смесь фильтруют через слой целита 545 и растворитель удаляют при пониженном давлении. После очистки с помощью колоночной хроматографии получают чистый 5-амино-1SЕМ-3-метоксикарбонилметил-2,4(1 Н,3 Н)пиримидиндиона примера 4 С. Пример 4D. К раствору 5-амино-1-SЕМ-3-метоксикарбонилметил-2,4(1 Н,3 Н)пиримидиндиона (пример 4 С) в тетрагидрофуране и дихлорметане(1:1, 0,3 М) добавляют каталитическое количество уксусной кислоты и 1 эквивалент фенилацетальдегида. Раствор охлаждают до 0 С на ледяной бане и одной порцией добавляют 1 эквивалент триацетоксиборогидрид натрия. После перемешивания в течение 5 мин ледяную баню удаляют и реакционную смесь перемешивают 2 ч при комнатной температуре. Затем реакцию гасят добавлением 1 н. NaOH и смесь перемешивают в течение 5 мин. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 4D. Пример 4 Е. К раствору 1-SЕМ-3-метоксикарбонилметил-5-(2-фенилэтил)амино-2,4(1 Н,3 Н)пиримидиндиона (пример 4D) в тетрагидрофуране и метаноле (1:1, 0,2 М) добавляют 1 эквивалент гидроксида лития в воде. После завершения реакции летучие вещества удаляют при пониженном давлении. Оставшийся водный раствор охлаждают на ледяной бане и подкисляют до рН 1 добавлением 1,0 н. НСl. После экстракции органическим растворителем и удаления растворителя при пониженном давлении получают чистый продукт примера 4 Е. Пример 4F. К раствору диметилформамида 1-SEM-3 метиленкарбокси-5-(2-фенилэтил)амино-2,4(1 Н,3 Н)пиримидиндиона (0,1 М) добавляют 5 эквивалентов N,N-диизопропилэтиламина, 1 эквивалент N-гидроксибензотриазола и 1 эквивалент гидрохлорида 1-[3-(диметиламино)пропил]-3 этилкарбодиимида. Полученную смесь переме 29 шивают в течение 30 мин. Затем реакционную смесь обрабатывают 1 эквивалентом соответствующего амина и перемешивают в течение ночи. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 4F. Пример 4G. К раствору 1-SЕМ-3-метиленкарбамид-5(2-фенилэтил)амино-2,4(1 Н,3 Н)пиримидиндиона в тетрагидрофуране (0,3 М) добавляют 2 эквивалента фторида тетрабутиламмония в тетрагидрофуране. Полученный раствор нагревают с обратным холодильником в течение нескольких часов. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 4G. Пример 4 Н. К раствору 3-метиленкарбоксамид-5-(2 фенилэтил)амино-2,4(1 Н,3 Н)пиримидиндиона(пример 4G) в N,N-диметиланилине (0,3 М) добавляют 1 эквивалент оксихлорида фосфора. Полученный раствор нагревают с обратным холодильником в течение нескольких часов. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 4 Н. Пример 4I. К раствору 2-хлор-3-метиленкарбоксамид 5-(2-фенилэтил)амино-2,4(1 Н,3 Н)пиримидиндиона (пример 4 Н) в диоксане (0,3 М) добавляют 2 эквивалента фенилтиола. Полученный раствор нагревают с обратным холодильником в течение нескольких часов. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 4I. Раствор 2-тиофенил-3-метиленкарбоксамид 5-(2-фенилэтил)амино-2,4(1 Н,3 Н)пиримидиндиона (пример 4I) в метаноле и 4 М НСl-диоксане(3:1, 0,1 М) дегазируют газообразным водородом. Затем к раствору добавляют 5% Pd/C и перемешивают в атмосфере водорода в течение 24 ч при комнатной температуре. Неочищенную реакционную смесь фильтруют через слой целита 545 и концентрируют при пониженном давлении. После очистки с помощью колоночной хроматографии получают чистый продукт. Пример 5.(1 Н,3 Н)пиримидиндиона и 1 эквивалента 3-пиридинбороновой кислоты в 1-пропаноле (0,5 М) добавляют 2 эквивалента 2,0 М карбоната натрия, а затем 1 мол.% тетракис(трифенилфосфин)палладия. Полученную смесь нагревают с 30 обратным холодильником в течение нескольких часов. После охлаждения до комнатной температуры и после обычной водной обработки с последующей хроматографией получают чистый продукт. Пример 5 А. Раствор 2-(3-пиридинил)-3-метиленкарбоксамид-5-(2-фенилэтил)амино-2,4(1 Н,3 Н)пиримидиндиона (пример 5 А) в метаноле и смеси 4 М HCl-диоксан (3:1, 0,1 М) дегазируют газообразным водородом. Затем к раствору добавляют 5% Pd/C и перемешивают в атмосфере водорода в течение 24 ч при комнатной температуре. Неочищенную реакционную смесь фильтруют через слой целита 545 и концентрируют при пониженном давлении. После очистки с помощью колоночной хроматографии получают чистый продукт. Пиримидиноны, имеющие, например, непосредственно связанный 2-арил, 2-гетероарил или 2-гетероатом, связывающий органическую группу через линкерную группу или непосредственно через гетероатом с пиримидиноновым кольцом, могут быть получены в соответствии со схемой 7, представленной ниже. В этой реакционной схеме арил вводят путем образования углерод-углеродной связи. Гетероатомом, подходящим для образования связанных через гетероатом арилпиримидинонов, является сера,азот, кислород или другой подходящий гетероатом. Общая процедура, представленная на схеме 7 для получения конкретных замещенных гетероатомом пиримидинонов, описана в примерах 6 и 7. Схема 7 Альтернативный синтез пиримидинонов Пример 6 А. 2-Тиоурацил (66,7 г, 520,5 ммоль) растворяют в растворе гидроксида натрия (41,6 г твердого NaOH в 365 мл воды). Затем смесь обрабатывают метилиодидом (37 мл, 583 ммоль) и полученную реакционную смесь оставляют на 16 ч при комнатной температуре для перемешивания. После этого раствор подкисляют ледяной уксусной кислотой (30 мл). Белый осадок собирают вакуумным фильтрованием и твердое вещество несколько раз промывают холодной водой и сушат, в результате чего получают 74 г примера 6 А в виде белого кристаллического твердого вещества с количественным выходом. Пример 6 В. Раствор примера 6 А (74,0 г, 520,5 ммоль) в ледяной уксусной кислоте (2275 мл) охлаждают до 0 С на бане со льдом и обрабатывают Br2. Реакционную смесь оставляют для нагревания до комнатной температуры и перемешивают в течение 16 ч. Образовавшийся желтый осадок фильтруют и промывают 3 раза эфиром. В результате выделяют 97,2 г соединения примера 6 В с выходом 62%. Пример 6 С. Смесь гидрида кальция в ТГФ охлаждают до 0 С и обрабатывают чистым продуктом примера 6 В, а затем чистым трет-бутилбромацетатом. Реакционную смесь перемешивают 1 ч при 0 С. Затем, после нагревания смеси до комнатной температуры, реакционную смесь перемешивают в течение 2 ч. После этого реакционную смесь нагревают с обратным холодильником в течение 16 ч. После охлаждения смеси до комнатной температуры смесь медленно выливают в 1 л водной суспензии со льдом. Погашенную смесь экстрагируют дихлорметаном (3 х 500 мл). Органические слои объединяют и промывают водой и рассолом. После сушки органического слоя МgSO4 и фильтрования летучие компоненты удаляют в вакууме с получением 28,81 г(90%) соединения примера 6 С в виде беловатого твердого вещества, представляющего собой смесь N-алкилированного и О-алкилированного изомеров (9:1). N-алкилированный изомер: 1 32 Пример 6D. В заполненном аргоном боксе с перчатками,в колбу Фишера-Портера емкостью 12 унций,содержащую магнитную мешалку, загружают соединение (пример 6 С) (5,00 г, 15,0 ммоль),Pd(OAc)2 (168 мг, 0,75 ммоль, 5 мол.%), racBINAP (654 мг, 1,05 ммоль, 7 мол.%), Сs2 СО 3(6,84 г, 21,0 ммоль) и безводный дегазированный толуол (65,0 мл). К этой смеси добавляют изопропиламин (3,00 мл, 35,2 ммоль). Колбу закрывают колпаком с гидростатическим напором, снабженным манометром, и удаляют из бокса. Закрытую систему нагревают на масляной бане при 118-120 С с последующим размешиванием магнитной мешалкой в течение 16 ч. Во время реакции гидростатический напор в пространстве составил 10 фунт/кв.дюйм. Колбу Фишера-Портера, содержащую реакционную смесь, удаляют из масляной бани, оставляют на 30 мин для охлаждения, вентилируют системой потока аргона и берут шприцем образцы. ЖХМС анализ показывает наличие 35% продукта и 65% остаточного исходного продукта. Снимают гидростатический напор в токе аргона и реакционную смесь обрабатывают Pd(OAc)2 (337 мг,1,5 ммоль, 10 мол.%), rac-BINAP (1,0 г, 1,6 ммоль, 11 мол.%), Сs2 СO3 (10,0 г, 30,7 ммоль) и изопропиламином (6,00 мл, 70,4 ммоль). Колбу закрывают колпаком с гидростатическим напором и снова нагревают до 118-120 С, размешивая магнитной мешалкой, в течение 16 ч. Затем повторяют процедуру взятия образцов и завершение реакции определяют по ЖХ-МС. Реакционную смесь оставляют охлаждаться до комнатной температуры и фильтруют через воронку со стеклянным фильтром со средним размером пор. Твердые вещества тщательно промывают и очищают с помощью флеш-хроматографии(SiO2, 230-400 меш, Merck, 10% этилацетат в гексане), в результате чего получают 3,80 г (выход 81%) соединения (пример 6D) в виде желтовато-коричневого твердого вещества. 1(75 МГц, СDСl3)165,7 (с), 158,7 (с), 147,4 (с),130,5 (с), 123,6 (д), 82,9 (с), 45,9 (т), 44,1 (д),28,0 (кв.), 22,1 (кв.), 14,8 (кв.); HRMS (ESI) для С 14H24N3SO3 [М+Н]+: вычислено 314,1538; найдено: 314,1539. Пример 6 Е. В заполненном аргоном боксе с перчатками,в колбу Фишера-Портера емкостью в 3 унции,содержащую магнитную мешалку, загружают соединение примера 6D (1,00 г, 3,20 ммоль), 3-нитрофенилбороновую кислоту (634 мг, 3,80 ммоль),Cu(I)-2-тиофенкарбоксилат (1,21 г, 6,37 ммоль) и Pd(PPh3)4 (100 мг, 0,86 ммоль, 2,7 мол.%). Затем добавляют ТГФ (25 мл) и колбу закрывают колпаком с гидростатическим напором, снабженным манометром, и удаляют из бокса. За 33 крытую систему нагревают на масляной бане при 70 С с последующим размешиванием магнитной мешалкой в течение 16 ч. Реакционную смесь оставляют охлаждаться до комнатной температуры, вентилируют и разбавляют эфиром (200 мл). Смесь фильтруют через воронку со стеклянным фильтром со средним размером пор. Зеленое твердое вещество промывают эфиром (100 мл) и отбрасывают. Фильтрат концентрируют и очищают с помощью флеш-хроматографии (SiO2, 230-400 меш, Merck, 10% этилацетат в гексане до 30%), в результате чего получают 961 мг (выход 78%) соединения примера 6 Е в виде желтой пены. 1[M+H]+: вычислено 389,1815; найдено: 389,1825. Пример 6F. В 250-мл круглодонную колбу, снабженную магнитным стержнем для перемешивания,загружают соединение примера 6 Е (755 мг, 1,9 ммоль) и трифторуксусную кислоту (30 мл). Эту смесь перемешивают при КТ в токе аргона в течение 30 мин и концентрируют на роторном испарителе. Остаток растирают с эфиром (50 мл) и выпаривают вместе с гептаном (2 х 50 мл),в результате чего получают 801 мг (выход 95%) соединения примера 6F в виде светло-желтого стеклообразного вещества: 1C15H17N4O5 [M+H]+ свободного oснования: вычислено 333,1223; найдено: 333,1199. Из соединения примера 6F, полученного в соответствии с процедурой, описанной для CBZ-защищенного предшественника, получают продукт: 1[М+Н]+: вычислено 598,2463; найдено: 598,2414. Из соединения примера 6G, полученного в соответствии с процедурой, описанной для SC81703, получают продукт: 1 Пример 7 А. Нужный продукт получают из соединения примера 6D в соответствии с процедурой, описанной для примера 6 Е, за исключением того,что используют основание Сs2 СО 3. В основном,использование соединения примера 6D (213 мг,0,68 ммоль), циклического сложного эфира 1,3 пропандиола и пиридин-3-бороновой кислоты(166 мг, 1,02 ммоль) и Сs2 СО 3 (771 мг, 2,37 ммоль) после флеш-хроматографии (SiO2 230400 меш, Merck, 2% МеОН в СНСl3) дает 143 мг соединения примера 7 А (выход 61%) в виде светло-желтого стеклообразного вещества. 1H ЯМР (300 МГц, СDСl3)8,79-8,66 (м,2 Н), 7,90-7,26 (комплексный м, 3 Н), 7,16 (с, 1 Н),4,53 (с, 2 Н), 3,57 (септет, J=6,3 Гц, 1 Н), 1,44 (с,9 Н), 1,27 (д, J=6,3 Гц, 6 Н); HRMS (ESI) для С 18 Н 25N4O3 [M+H]+: вычислено 319,1927; найдено: 345,1928. Пример 7 В. Из соединения примера 7 А (143 мг, 0,41 ммоль) в соответствии с процедурой, описанной для соединения примера 6F, получают 212 мгH ЯМР (300 МГц, CDCl3)8,72-8,64 (м,2 Н), 7,95-7,89 (м, 1 Н), 7,62-7,52 (комплексный м, 2 Н), 7,42-7,37 (м, 1 Н), 7,20 (с, 1 Н), 4,52 (с,2 Н), 3,57 (септет, J=6,3 Гц, 1 Н), 1,18 (д, J=6,3 Гц, 6 Н); HRMS (ESI) для C14H17N4O3 [M+H]+: вычислено 289,1301; найдено: 289,1296. Пример 7 С. Из соединения примера 7 В в соответствии с процедурой, описанной для CBZ-защищенного соединения, получают соединение примера 7 С.HRMS (ESI) для C30H32N7 О 4: [M+H]+: вычислено 554,2516; найдено: 554,2523. Из соединения примера 7 С в соответствии с процедурой, описанной для соединения примера 6, получают нужный продукт. Пример 8.HRMS (ESI) для C24H27N7 О 2: [M+H]+: вычислено 446,2304; найдено: 446,2309. Метиленовые аналоги пиримидинонов, где в положении N-3 пиримидинона вместо карбонила ацетиламида присутствует метилен, могут быть получены в соответствии со схемой 8"Общий метод получения метиленпиримидинонов", подробно описанной ниже вместе с конкретным примером 9. Схема 8 Общий синтез метиленпиримидинонов(1:1, 0,3 М) добавляют каталитическое количество уксусной кислоты и 1 эквивалент соответствующего амина. Раствор охлаждают до 0 С на ледяной бане и одной порцией добавляют 1 эквивалент триацетоксиборогидрида натрия. После перемешивания в течение 5 мин ледяную баню удаляют и реакционную смесь перемешивают в течение 2 ч для перемешивания при комнатной температуре. Затем реакцию гасят добавлением 1 н. NaOH и смесь перемешивают в течение 5 мин. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 9 А. 36 Пример 9 В. К раствору [5-[(бензилоксикарбонил)амино]2-фенил-6-оксо-1,6-дигидро-1-пиримидинил]ацетамида (пример 9 А) в метаноле (0,2 М) добавляют одной порцией 10% Pd/C. Полученную смесь перемешивают в атмосфере газообразного водорода (баллонного) в течение 16 ч при комнатной температуре. Неочищенную реакционную смесь фильтруют через слой целита 545 и концентрируют при пониженном давлении. После растирания остатка с этиловым эфиром получают чистый продукт примера 9 В. Пример 9 С. К раствору [5-амино-2-фенил-6-оксо-1,6 дигидро-1-пиримидинил]ацетамида (пример 9 В) в тетрагидрофуране и дихлорметане (1:1, 0,3 М) добавляют каталитическое количество уксусной кислоты и 1 эквивалент фенилацетальдегида. Раствор охлаждают до 0 С на ледяной бане и одной порцией добавляют 1 эквивалент триацетоксиборогидрид натрия. После перемешивания в течение 5 мин ледяную баню удаляют и реакционную смесь перемешивают при комнатной температуре 2 ч. Затем реакцию гасят добавлением 1 н NaOH и смесь перемешивают в течение 5 мин. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 9 С. Раствор [5-(2-фенилэтил)амино-2-фенил-6 оксо-1,6-дигидро-1-пиримидинил]ацетамида в 4 М HCl-диоксане (0,2 М) перемешивают в течение нескольких часов. После завершения реакции летучие вещества удаляют при пониженном давлении. Остаток очищают растиранием с этиловым эфиром и получают чистый продукт 5. Сульфониловые аналоги пиримидинонов,где в положении N-3 пиримидинона вместо карбонила ацетиламида присутствует метилен, могут быть получены в соответствии с общими методами, в основном, описанными в схемах 15, с использованием вместо 1,1-диметоксиэтиламина соответствующего аминометансульфонамида в N-Вос-амидинозащищенном амине. Так,например, может быть использован N-(4-амидинобензил)-2-аминометансульфонамид. Этим методом могут быть получены сульфониловые аналоги в примерах 10 и 11. Пример 10. 37 Триазиноновые аналоги пиримидинонов,где вместо углерода в 4-положении пиримидинона присутствует азот, могут быть получены в соответствии со схемой 9 "Общий метод получения 4-азапиримидинона" описанной ниже вместе с конкретным примером 12. Схема 9 Общий метод синтеза 4-азапиримидинона Пример 12 А. Раствор гидрохлорида трет-бутилового сложного эфира глицина (1 ммоль) в дихлорметане обрабатывают бензоилхлоридом (1 ммоль) и триэтиламином (2 ммоль). Реакционную смесь перемешивают в течение на 16 ч при комнатной температуре. Затем смесь промывают водой и экстрагируют дихлорметаном. Объединенные органические слои сушат над MgSО 4. После удаления летучих веществ в вакууме выделяют чистый продукт примера 12 А. Пример 12 В. Смесь трет-бутилового сложного эфира Nбензоилглицина (1 ммоль; пример 12 А), реактив Лавессона (0,5 ммоль) и толуол нагревают до 80 С в течение 16 ч. Реакционную смесь концентрируют при пониженном давлении. После очистки колоночной хроматографией на силикагеле получают чистый продукт примера 12 В. 38 Пример 12 С. Смесь трет-бутилового сложного эфира Nтиобензоилглицина (1 ммоль; пример 12 В) в дихлорметане обрабатывают тетрафторборатом триметилоксония (1,1 ммоль) при -78 С. После перемешивания реакционной смеси в течение 2 ч. смесь промывают NaHCO3 (вод.) и экстрагируют дихлорметаном. Объединенные органические слои сушат MgSO4 и фильтруют. После концентрирования летучих органических компонентов выделяют нужный продукт примера 12 С. Пример 12D. Раствор трет-бутилового эфираNтиометилбензилглицина (1 ммоль; пример 12 С) в метаноле обрабатывают гидразином (1 ммоль). Летучие вещества удаляют в вакууме и выделяют нужное соединение примера 12D без дополнительной очистки. Пример 12 Е. Смесь соединения примера 12D (1 ммоль) и этилтиооксамата (1 ммоль) в метаноле кипятят с обратным холодильником в течение 4 ч. Затем выпадает осадок в виде бесцветных кристаллов и кристаллы нужного продукта примера 12 Е выделяют вакуумным фильтрованием. Раствор соединения примера 12 Е (1 ммоль) и пиридина (5 ммоль) в ацетонитриле обрабатывают раствором а-толуолсульфонилхлорида (3 ммоль) в ацетонитриле. Реакционную смесь перемешивают в течение 3 ч при температуре от-10 до 0 С. После завершения реакции образуется белый осадок. Кристаллы нужного продукта примера 12F выделяют вакуумным фильтрованием. Пример 12G. Трифторуксусную кислоту добавляют к смеси соединения примера 12F (1 ммоль) в анизоле при 0 С. Реакционную смесь оставляют на 1 ч для перемешивания при 0 С. Реакционную смесь экстрагируют органическим растворителем. После удаления органического растворителя при пониженном давлении получают чистый продукт пpимера 12G. Пример 12 Н. Соединение пpимера 12G (1 ммоль), EDC(1,3 ммоль) и HOBt (1,3 ммоль) смешивают в ДМФ и смесь перемешивают при 20 С в течение 15 мин. К полученной смеси добавляют раствор гидрохлоридной соли бензил-(4 аминоме-тилфенил)иминометил]аминометил] амино]карбамата (1,3 ммоль) и DIEA (1,3 ммоль) в ДМФ. После обычной водной обработки с последующей хроматографией получают чистый продукт примера 12 Н. Соединение примера 12 Н (1 ммоль), моногидрат п-толуолсульфоновой кислоты (1 ммоль) и 10% Pd на активированном угле (0,1 ммоль) смешивают с метанолом. Полученную смесь перемешивают в течение 2 ч в атмосфере водорода, который вводят и поддерживают с помощью резинового баллона. После отфильтровы 39 вания катализатора и удаления метанола выделяют нужный продукт. Соединения формулы (I) настоящего изобретения, содержащие гидроксильную, тиоловую и аминовую функциональные группы, могут быть превращены в производные широкого ряда. Альтернативно, производные соединения формулы (I) могут быть получены сначала получением производных одного или нескольких промежуточных соединений в данных способах получения, с последующим превращением данного производного промежуточного соединения в соединения формулы (I). Гидроксильная группа в форме спирта или фенола может быть легко преобразована в сложные эфиры карбоновой,сульфоновой, карбаминовой, фосфоновой и фосфорной кислот. Ацилирование с образованием сложного эфира карбоновой кислоты может быть легко осуществлено с использованием подходящего ацилирующего реагента, такого как ангидрид алифатической кислоты или хлорангидрид. Могут быть также использованы соответствующие ангидриды ариловых и гетероариловых кислот и хлорангидриды. Указанные реакции обычно осуществляют с использованием аминового катализатора, такого как пиридин,в инертном растворителе. Аналогично, сложные эфиры карбаминовых кислот (уретаны) могут быть получены путем реакции гидроксильной группы с изоцианатами и карбамоилхлоридами. Сложные эфиры сульфоновой, фосфоновой и фосфорной кислот могут быть получены с использованием соответствующего хлорангидрида и аналогичных реагентов. Соединения формулы(I), которые содержат по крайней мере одну тиольную группу, могут быть преобразованы в соответствующие производные сложных тиоэфиров, аналогично производным спиртов и фенолов, с использованием аналогичных реагентов и подходящих реакционных условий. Соединения формулы (I), которые содержат по крайней мере одну первичную или вторичную аминную группу, могут быть преобразованы в соответствующие производные амида. Амиды карбоновых кислот могут быть получены с использованием соответствующего хлорангидрида или ангидридов кислот в реакционных условиях, аналогичных условиям с использованием спиртов и фенолов. Карбамиды соответствующего первичного или вторичного амина могут быть получены непосредственно с использованием изоцианатов и карбамоилхлоридов в присутствии акцептора кислоты, такого как триэтиламин или пиридин. Сульфонамиды могут быть получены из соответствующего сульфонилхлорида в присутствии водного гидроксида натрия или третичного амина. Подходящие процедуры и методы получения указанных производных можно найти в House's Modern Synthetic Reactions, W.A. Benjamin, Inc., Shriner, Fuson,FieserFieser in Reagents for Organic Synthesis,Volume 1, John WileySons. Реагенты широкого ряда, которые могут быть использованы для получения производных из гидроксилов, тиолов и аминов соединений формулы (I), являются коммерчески доступными, или их получение описано в указанных выше работах, которые введены в настоящее описание в качестве ссылки. Соединения формулы (I) настоящего изобретения, содержащие гидроксильную, тиольную и аминную функциональные группы, могут быть алкилированы с образованием производных широкого ряда. Альтернативно, алкилированные соединения формулы (I) могут быть получены сначала путем алкилирования одного или нескольких промежуточных соединений с последующим превращением алкилированных промежуточных соединений в соединения формулы (I). Гидроксильная группа соединений формулы (I) может быть легко превращена в простые эфиры. Алкилирование с образованием простого эфира может быть легко осуществлено с использованием подходящего алкилирующего реагента, такого как алкилбромид, алкилиодид или алкилсульфонат. Могут быть также использованы соответствующие аралкил-, гетероаралкил-, алкоксиалкил-, аралкилоксиалкил- и гетероаралкилоксиалкилбромиды, -иодиды и -сульфонаты. Указанные реакции обычно проводят с использованием алкоксидобразующего реагента, такого как гидрид натрия, трет-бутоксид калия, амид натрия, амид лития и н-бутиллитий в инертном полярном растворителе, таком как ДМФ, ДМСО, ТГФ, и аналогичных подходящих растворителях, и аминового катализатора, такого как пиридин в инертном растворителе. Соединения формулы (I), которые имеют по крайней мере одну тиольную группу, могут быть превращены в соответствующие тиоэфирные производные, аналогичные спиртам и фенолам,с использованием тех же самых реагентов и подобных реакционных условий. Соединения формулы (I), которые имеют по крайней мере одну первичную, вторичную или третичную аминогруппу, могут быть превращены в соответствующие вторичные, третичные или четвертичные аммониевые производные. Производные четвертичного аммония могут быть получены с использованием соответствующих бромидов,иодидов и сульфонатов, аналогичных тем, которые получают с использованием спиртов и фенолов. Условия включают реакцию взаимодействия амина при его нагревании с алкилирующим агентом со стехиометирическим количеством амина (то есть один эквивалент с третичным амином, два эквивалента с вторичным амином и три эквивалента с первичным амином). В случае первичного и вторичного аминов, одновременно используются два эквивалента и один эквивалент акцептора кислоты соответственно. Вторичные или третичные амины могут быть 41 получены из соответствующего первичного и вторичного амина. Первичный амин может быть диалкилирован путем восстановительного аминирования с использованием альдегида, такого как формальдегид, и цианоборогидрида натрия в присутствии ледяной уксусной кислоты. Первичный амин может быть сначала моноалкилирован путем монозащиты амина легко отщепляемой защитной группой, такой как трифторацетил. Алкилирующий агент, такой как диметилсульфат, в присутствии ненуклеофильного основания, такого как основание Бартона (2 трет-бутил-1,1,3,3-тетраметилгуанидин),дает монометилированный защищенный амин. Удаление защитной группы с использованием водного раствора гидроксида калия дает нужный моноалкилированный амин. Дополнительные подходящие процедуры и методы получения указанных производных можно найти в House'sOrganic Synthesis, Volume 1, John WileySons. Перфторалкиловые производные могут быть получены, как описано DesMarteau в J. Chem.Soc. Chem. Commun. 2241 (1998). Реагенты широкого ряда, которые могут быть использованы для получения производных из гидроксилов,тиолов и аминов соединений формулы (I), являются коммерчески доступными, или они могут быть получены, как описано в указанных выше работах, которые введены в настоящее описание в качестве ссылки. Результаты указанных выше методов синтеза, используемых для получения пиримидинонов через производные по нуклеофильному заместителю, такому, который может присутствовать в В, R1, R2 и Y0, представлены в табл. 1 для конкретных примеров 13-19. Конкретные примеры, представленные ниже, приводятся в иллюстративных целях и не должны рассматриваться как ограничение объема изобретения. Таблица 1 Структуры пиримидинонов, полученных общими методами дериватизации Анализы на биологическую активность Анализ на TF-VIIa В данном анализе 100 нМ рекомбинантный растворимый тканевый фактор и 2 нМ рекомби 004867 42 нантный фактор VIIa человека добавляют в 96 луночный аналитический планшет, содержащий 0,4 мМ субстрат, N-метилсульфонил-D-Рhе-GlyАrg-п-нитроанилин, и либо ингибитор, либо буфер (5 мМ CaC2, 50 мМ Трис-HCl, рН 8,0, 100 мМ NaCl, 0,1% BSA). Данную реакцию, в конечном объеме 100 мкл, сразу измеряют при 405 нМ для определения фоновой оптической плотности. Указанный планшет инкубируют при комнатной температуре в течение 60 мин и на данной стадии измеряют степень гидролиза субстрата путем мониторинга реакции при 405 нм на высвобождение п-нитроанилина. Процент ингибирования активности TF-VIIa вычисляют,исходя из величины ОП 405 нм для экспериментальных и контрольных образцов. Анализ на Ха 0,3 нМ фактора Ха человека и 0,15 мМ N-бензилоксикарбонил-D-аргинил-L-глицил-Lаргинин-п-нитроанилиндигидрохлорид (S-2765) добавляют в 96-луночный аналитический планшет, содержащий либо ингибитор, либо буферBSA). Данную реакцию, в конечном объеме 100 мкл, сразу измеряют при 405 нМ для определения фоновой оптической плотности. Указанный планшет инкубируют при комнатной температуре в течение 60 мин и на данной стадии измеряют степень гидролиза субстрата путем мониторинга реакции при 405 нм на высвобождение п-нитроанилина. Процент ингибирования активности фактора Ха вычисляют, исходя из величины ОП 405 нм для экспериментальных и контрольных образцов. Анализ на тромбин 0,28 нМ человеческого тромбина и 0,06 мМ дигидрохлорида Н-D-фенилаланил-L-пипеколил-L-аргинин-п-нитроанилина добавляют в 96-луночный аналитический планшет, содержащий либо ингибитор, либо буфер (50 мМ Трис-HCl, рН 8,0, 100 мМ NaCl, 0,1% BSA). Данную реакцию, в конечном объеме 100 мкл,сразу измеряют при 405 нМ для определения фоновой оптической плотности. Указанный планшет инкубируют при комнатной температуре в течение 60 мин и на данной стадии измеряют степень гидролиза субстрата путем мониторинга реакции при 405 нм на высвобождение п-нитроанилина. Процент ингибирования активности тромбина вычисляют, исходя из величины ОП 405 нм для экспериментальных и контрольных образцов. Анализ на трипсин 5 мкг/мл трипсина типа IX, выделенного из поджелудочной железы свиньи, и 0,375 мМBSA). Данные реакции, в конечном объеме 100 мкл, сразу измеряют при 405 нМ для определе 43 ния фоновой оптической плотности. Указанный планшет инкубируют при комнатной температуре в течение 60 мин и на данной стадии измеряют степень гидролиза субстрата путем мониторинга реакции при 405 нм на высвобождение п-нитроанилина. Процент ингибирования активности тромбина вычисляют, исходя из величины ОП 405 нм для экспериментальных и контрольных образцов. Рекомбинантный растворимый TF, состоящий из аминокислот 1-219 последовательности зрелого белка, экспрессируют в E.coli и очищают с использованием ЖЭХБ (жидкостной экспресс-хроматографии белков) на сефарозеMono Q. Рекомбинантный фактор человека был закуплен у American Diagnostica, Greenwich CT,a хромогенный субстрат N-метилсульфонил-DРhе-Gly-Аrg-п-нитроанилин был получен отAmerican Peptide Company, Inc., Sunnyvale, CA. Фактор Ха был получен из лаборатории EnzymeResearch Laboratories, South Bend IN, тромбин был получен от Calbiochem, La Jolla, CA, а трипсин и L-BAPNA от Sigma, St. Louis, МО. Хромогенные субстраты S-2765 и S-2238 были закуплены у Chromogenix, Sweden. Биологические активности соединений примеров 1-19, определенные в биологических анализах, систематизированы в табл. 2. Таблица 2 Ингибирующая активность пиримидинонов по отношению к фактору Ха, TF-VIIA, тромбину II и трипсину II или его фармацевтически приемлемая соль, где В выбран из группы, состоящей из фенила,С 3-С 6-циклоалкила, C1-C6-алкила; А выбран из группы, состоящей из-CH2SO2-, -СН 2-, -СН 2-СН 2- или простой ковалентной связи;представляет NH; М представляет СН; или его фармацевтически приемлемая соль,где В представляет фенил; А выбран из группы, состоящей изR2 представляет фенил. 3. Соединение по п.2 или его фармацевтически приемлемая соль, где А представляет-СН 2SО 2-. 4. Соединение по п.2 или его фармацевтически приемлемая соль, где А представляет или его фармацевтически приемлемая соль,где В выбран из группы, состоящей из фенила, C1-C6-алкила; А выбран из группы, состоящей из -СН 2 СН 2- или простой ковалентной связи; или его фармацевтически приемлемая соль,где В выбран из группы, состоящей из C1 С 6-алкила, С 3-С 6-циклоалкила; А представляет простую ковалентную связь;R2 представляет аминофенил, необязательно замещенный амино, карбокси, алкоксикарбонилом или алкиламинокарбонилом, пиридил. 7. Композиция для ингибирования тромботических состояний в крови, включающая соединение по любому из пп.1-6 и фармацевтически приемлемый носитель. 8. Способ ингибирования тромботических состояний в крови, предусматривающий введение в кровь терапевтически эффективного количества композиции по любому из пп.1-6. 9. Способ ингибирования образования агрегации тромбоцитов в крови, предусматривающий введение в кровь терапевтически эффективного количества композиции по любому из пп.1-6. 10. Способ ингибирования образования тромбов в крови, предусматривающий введение в кровь терапевтически эффективного количества композиции по любому из пп.1-6. 11. Способ лечения или предупреждения тромбоэмболии вены и тромбоэмболии легких у млекопитающих, предусматривающий введение указанному млекопитающему терапевтически эффективного количества композиции по любому из пп.1-6. 12. Способ лечения или предупреждения тромбоза глубокой вены у млекопитающих,предусматривающий введение указанному млекопитающему терапевтически эффективного количества композиции по любому из пп.1-6. 13. Способ лечения или предупреждения кардиогенной тромбоэмболии у млекопитающих, предусматривающий введение указанному млекопитающему терапевтически эффективного количества композиции по любому из пп.1-6. 14. Способ лечения или предупреждения тромбоэмболического шока у человека и других млекопитающих, предусматривающий введение указанным млекопитающим терапевтически эффективного количества композиции по любому из пп.1-6. 46 15. Способ лечения или предупреждения тромбоза, ассоциированного с раком и противораковой химиотерапией у человека и других млекопитающих, предусматривающий введение указанным млекопитающим терапевтически эффективного количества композиции по любому из пп.1-6. 16. Способ лечения или предупреждения нестабильной стенокардии у человека и других млекопитающих, предусматривающий введение указанным млекопитающим терапевтически эффективного количества композиции по любому из пп.1-6. 17. Способ ингибирования образования тромбов в крови, предусматривающий введение в кровь терапевтически эффективного количества соединения по любому из пп.1-6 в сочетании с терапевтически эффективным количеством антагониста рецептора фибриногена. 18. Применение соединения по любому из пп.1-6 или их фармацевтически приемлемой соли в целях изготовления лекарственного препарата для ингибирования образования тромбов,лечения тромботических состояний или предупреждения образования тромбов у млекопитающих.
МПК / Метки
МПК: A61K 31/505, C07D 239/46, A61P 9/00
Метки: композиция, способ, тромботических, лечения, пиримидиноновые, млекопитающих, соединения, замещенные, предупреждения, состояний, ингибирования
Код ссылки
<a href="https://eas.patents.su/24-4867-zameshhennye-pirimidinonovye-soedineniya-kompoziciya-i-sposob-lecheniya-ingibirovaniya-ili-preduprezhdeniya-tromboticheskih-sostoyanijj-u-mlekopitayushhih.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные пиримидиноновые соединения, композиция и способ лечения, ингибирования или предупреждения тромботических состояний у млекопитающих</a>
Композиция и способ приготовления фармацевтического состава, лиофилизированная композиция и ее применение, способ ингибирования spla2-опосредованного высвобождения жирной кислоты, способ лечения млекопитающих, способ лечения или профилактики сепсиса, закрытая емкость.

Номер патента: 3864
Опубликовано: 30.10.2003
Авторы: Конфер Уильям Лестер, Тай Хидеаки
МПК: A61P 29/00, A61K 31/4045
Метки: сепсиса, профилактики, емкость, spla2-опосредованного, высвобождения, фармацевтического, жирной, композиция, закрытая, ингибирования, млекопитающих, применение, лиофилизированная, кислоты, лечения, способ, приготовления, состава
Формула / Реферат:
1. Композиция, содержащая [[3-(2-амино-1,2-диоксоэтил)-2-этил-1-(фенилметил)-1H-индол-4-ил]окси]ацетат натрия в качестве активного компонента, солюбилизатор, выбранный по крайней мере из одного соединения, выбранного из группы, включающей лимонную кислоту, эдетовую кислоту, полифосфорную кислоту и их соли, и стабилизатор, выбранный по крайней мере из одного соединения, выбранного из группы, включающей маннит, ксилит, сорбит, глюкозу, фруктозу,...
Способ лечения заболеваний млекопитающих и цвиттерионная композиция

Номер патента: 1929
Опубликовано: 22.10.2001
Авторы: Теодоре Рональд Т., Ван Зандт Роское Л.
МПК: A61K 31/205, A61P 35/00
Метки: способ, заболеваний, лечения, цвиттерионная, композиция, млекопитающих
Формула / Реферат:
1. Применение цвиттерионной композиции, содержащей по существу в качестве активного ингредиента амфотерное цвиттерионное соединение или его фармацевтически приемлемую соль, при этом цвиттерионное соединение является алифатическим или гетероциклическим соединением, имеющим, по меньшей мере, одну связь углерод-азот, и кислотную группу выбирают из SО3 и СОО, и щелочную группу выбирают из ОН, N+, N+H и NH2 для производства лекарственного средства...
Производные циклической аминокислоты или их фармацевтически приемлемые соли, фармацевтическая композиция и способ лечения депрессии, состояний паники, страха и болевых ощущений

Номер патента: 1534
Опубликовано: 23.04.2001
Авторы: Брайэнс Джастин С., Ниин Клэр О., Хорвелл Дейвид Си, Ретклифф Джильс С.
МПК: C07D 309/04, A61P 25/00, A61K 31/351...
Метки: паники, приемлемые, ощущений, композиция, соли, депрессии, фармацевтически, болевых, аминокислоты, способ, циклической, страха, фармацевтическая, лечения, производные, состояний
Формула / Реферат:
1. Производные циклической аминокислоты общей формулы (I) где X означает О, S, S(O), S(O)2 или NR1, где R1 означает водород, линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, бензил, -C(O)R2, где R2 означает линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, бензил или фенил или -CO2R3, где R3 означает линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода, или бензил, и где бензильная...
Производные никотинамида, их применение, фармацевтическая композиция, способ лечения и способ ингибирования изоферментов фдэ4 d

Номер патента: 3528
Опубликовано: 26.06.2003
Авторы: Клайнмен Эдуард Фокс, Даплэнтьер Аллен Джейкоб, Ченг Джон Бин, Марфэт Энтони, Чеймберс Роберт Джеймс, Уотсон Джон Уэсли
МПК: A61K 31/455, C07D 213/82, A61P 1/04...
Метки: способ, фармацевтическая, композиция, изоферментов, фдэ4, лечения, никотинамида, производные, применение, ингибирования
Формула / Реферат:
1. Производные никотинамида формулы (I) или их фармацевтически приемлемые соли, где t равно 0 или 1; A представляет собой кислород или NH; R1 представляет собой (C3-C7)циклоалкил, (C6-C10)арил или насыщенную или ненасыщенную циклическую или бициклическую (C3-C7)гетероциклическую группу, содержащую в качестве гетероатома от одного до четырех гетероатомов, независимо выбранных из группы, состоящей из кислорода, серы, азота и NR9, где R9 является...
Производные аминогуанидина и алкоксигуанидина (варианты) и способ их получения (варианты), фармацевтическая композиция и способ ингибирования протеолиза у млекопитающего, способ лечения различных заболеваний млекопитающего, способы ингибирования индуцированной тромбином агрегации тромбоцитов и свертывания фибриногена в плазме, тромбина, агрегации тромбоцитов и образования тромбов в крови, устройство для сбора крови, искусственного кровообращения и хранения крови

Номер патента: 3461
Опубликовано: 26.06.2003
Авторы: Сайедем Коллин, Маркоутэн Томас П., Солл Ричард М., Лу Тианбао, Томкзук Брюс Э.
МПК: A61P 7/02, A61K 31/445, C07C 279/00...
Метки: устройство, способ, агрегации, получения, алкоксигуанидина, тромбоцитов, способы, млекопитающего, хранения, индуцированной, кровообращения, искусственного, тромбов, плазме, варианты, образования, аминогуанидина, сбора, заболеваний, лечения, протеолиза, ингибирования, различных, производные, крови, фармацевтическая, тромбина, композиция, фибриногена, тромбином, свертывания
Формула / Реферат:
1. Соединение формулы I или его сольват, гидрат или фармацевтически приемлемая соль, где L - означает -C(O)- или -SO2-; R1 означает группу R2 означает группу или R1 и R2 могут быть объединены с атомом азота, к которому они присоединены, с образованием цикла, содержащего от трех до семи атомов, который необязательно содержит дополнительный атом азота или кислорода, и который по выбору является бензо- или пиридоконденсированным циклом, причем...
Предыдущий патент: Метаболиты агониста/антагониста эстрогена
Следующий патент: Производные сульфонамида
Случайный патент: Буровая железобетонная колонна и способ ее возведения