Новое сульфированное соединение сахара и его применение в качестве лекарственного средства
Номер патента: 16931
Опубликовано: 30.08.2012
Авторы: Охта Кейсуке, Ямазаки Такаюки, Сугавара Фумио, Такахаси Нобуаки, Миура Масахико, Масаки Казуйоси, Сато Нориюки, Сакагути Кенго, Мори Йоко, Мурата Хироси, Сахара Хироеки





















Формула / Реферат
1. Сульфохиновозилацилпропандиольное соединение, представленное общей формулой (I)

где R1 представляет собой C1-C22 ацильный остаток жирной кислоты и М представляет собой атом водорода или ион натрия либо кальция, или его фармацевтически приемлемые соли.
2. Лекарственное средство, содержащее в качестве активного ингредиента по меньшей мере один активный ингредиент, выбранный из группы, включающей сульфохиновозилацилпропандиольное соединение по п.1, представленное общей формулой (I), и его фармацевтически приемлемые соли.
3. Лекарственное средство по п.2, которое представляет собой радиосенсибилизатор.
4. Лекарственное средство по п.2, которое представляет собой противоопухолевый агент.
Текст
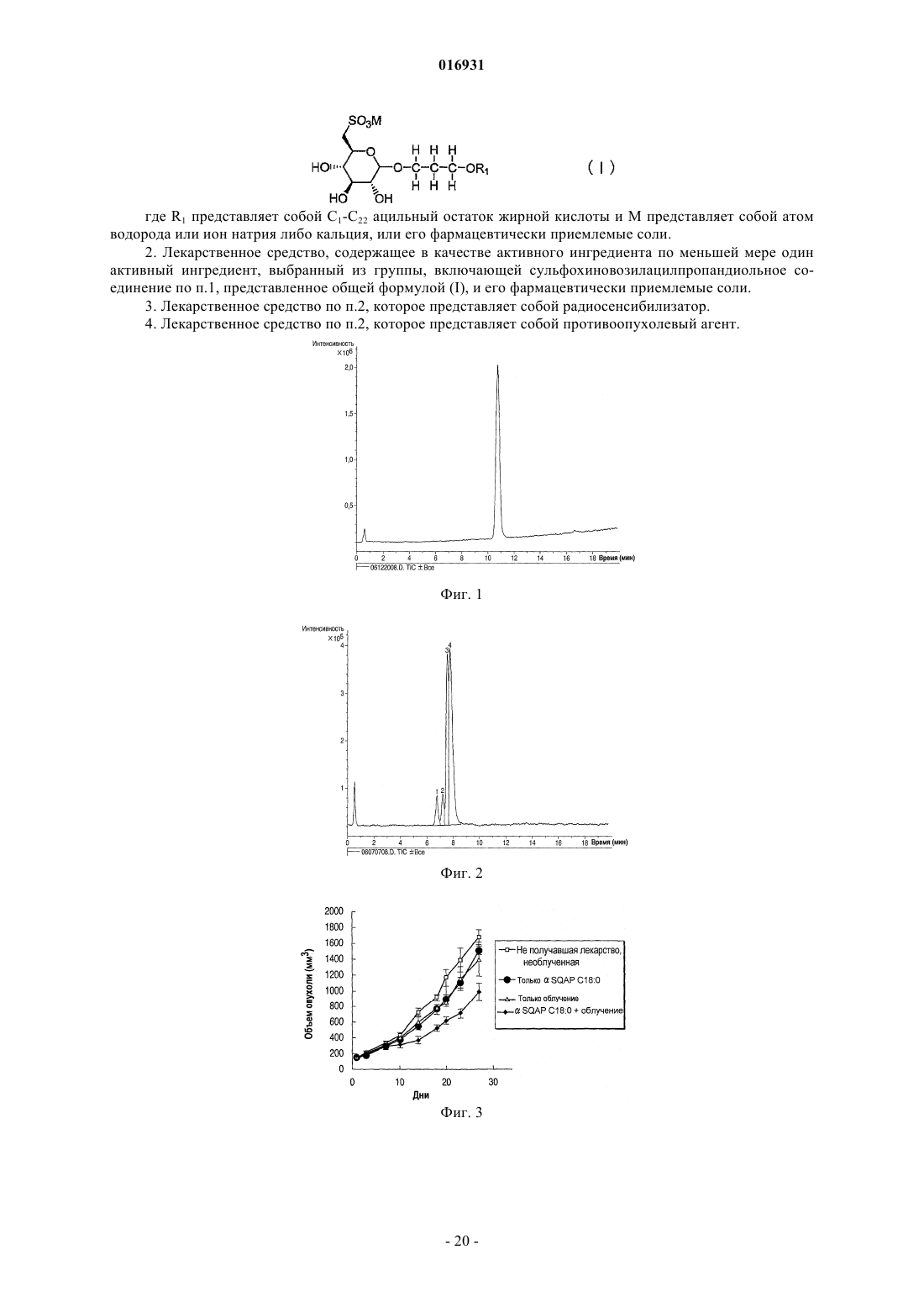
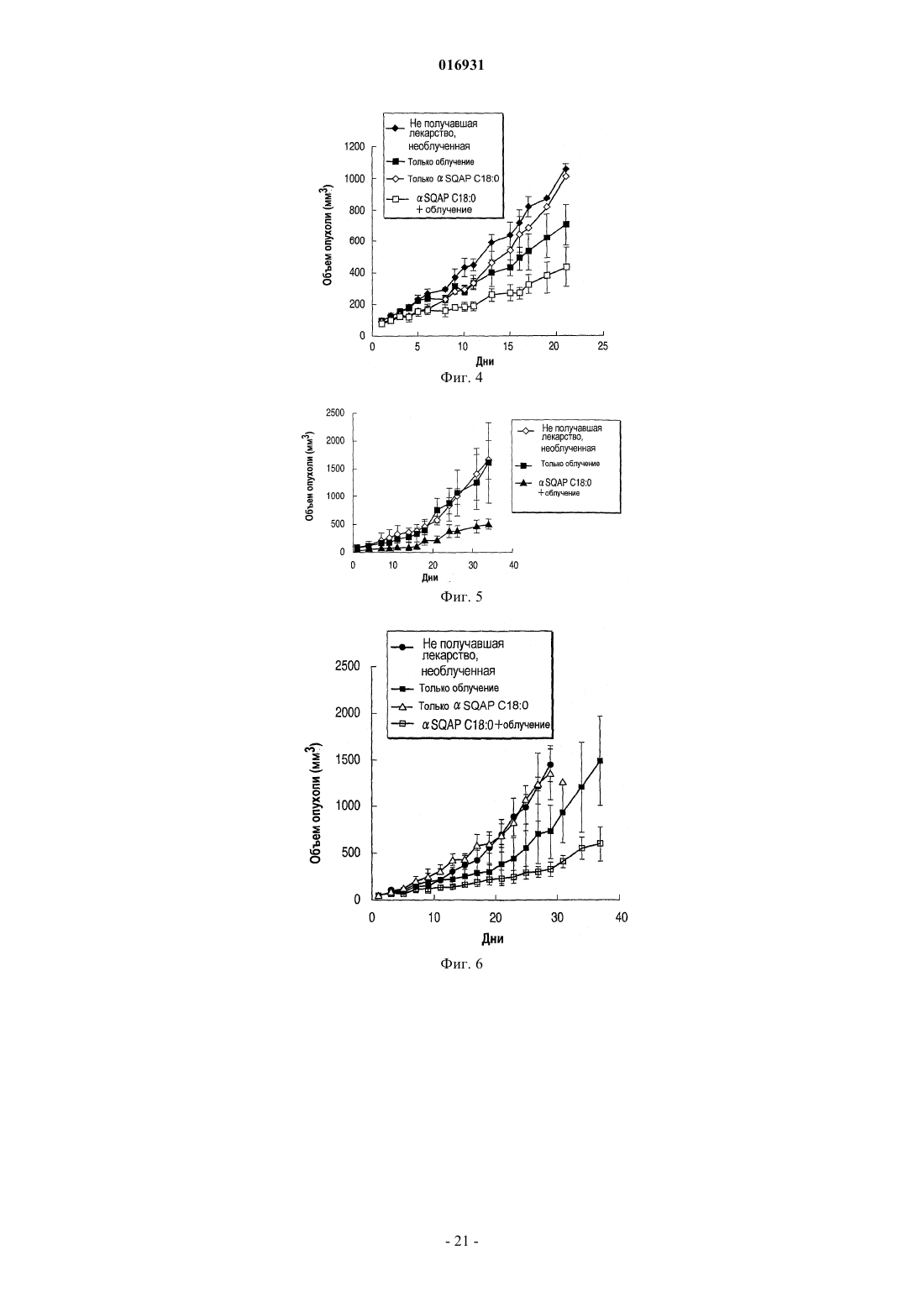
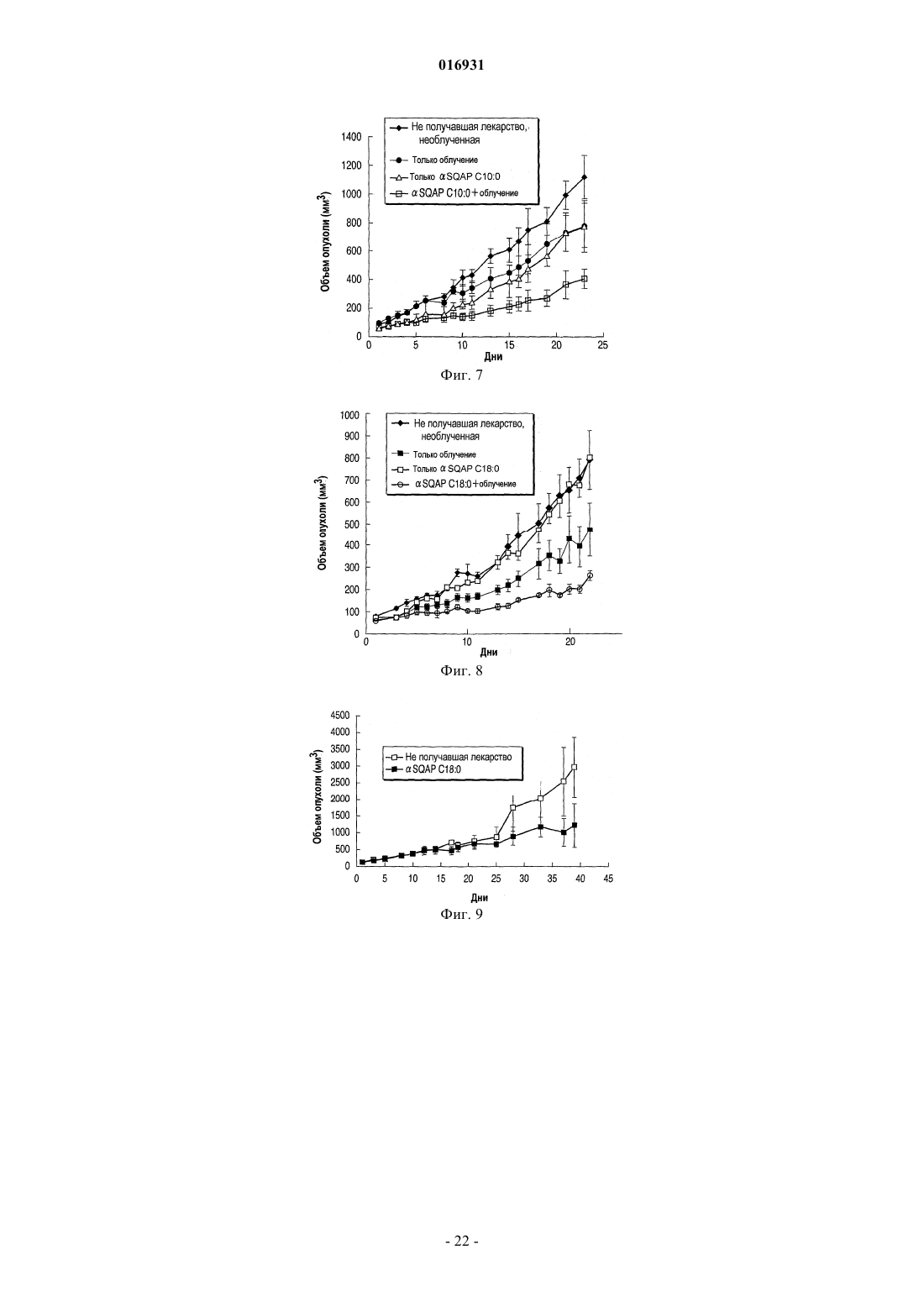
НОВОЕ СУЛЬФИРОВАННОЕ СОЕДИНЕНИЕ САХАРА И ЕГО ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА В изобретении предложено сульфохиновозилацилпропандиольное соединение, представленное общей формулой (I) где R1 представляет собой ацильный остаток жирной кислоты и М представляет собой атом водорода или ион металла либо его фармацевтически приемлемые соли. 016931 Область техники Настоящее изобретение относится к новому сульфированному соединению сахара и к лекарственному средству, содержащему это соединение. Предшествующий уровень техники В настоящее время в Японии злокачественная опухоль, сердечное заболевание и церебральноваскулярное заболевание являются причиной приблизительно 60% случаев смерти. Из них злокачественные опухоли имеют высший показатель из причин смерти, и их число имеет тенденцию увеличиваться. Хирургическое вмешательство, химиотерапия и радиационная терапия известны как три основных способа лечения злокачественных опухолей. В последние годы большое значение придается качеству жизни (КЖ, QOL) пациентов и много внимания уделяется радиационной терапии. При обычной радиационной терапии галогенированный пиримидин и сенсибилизаторы гипоксических клеток известны как химические или фармацевтические вещества, вводимые одновременно с радиационной терапией, в результате чего ее терапевтическое действие усиливается более специфично, как клинически применяемые радиосенсибилизаторы (см., например, Radiobiology for the Radiologist (FourthEdition), Eric J. Hall, et al., J.B. Lippincott Company ("Houshasennkainotameno Hoshasenseibutsugaku", перевод Muneyasu Urano, Shinoharashinsha. Inc.). Примеры известных галогенированных пиримидинов включают 5-йоддеоксиуридин. Примеры известных сенсибилизаторов гипоксических клеток включают мизонидазол. Однако такие известные радиосенсибилизаторы используют лишь при остром применении, так как они продуцируют побочные эффекты, такие как желудочно-кишечные расстройства, периферическая нейротоксичность, и создают другие серьезные проблемы. С другой стороны, для создания новых радиосенсибилизаторов больным дают радиосенсибилизаторы, включающие сульфопиранозилацилглицерин или его соли (Jpn. Pat. Appln. KOKAI Publication2004-374445). Однако в сульфопиранозилацилглицерине атом углерода во 2-м положении глицеринового остатка представляет собой асимметрический атом углерода, поэтому стереоструктуру нельзя контролировать относительно недорогим и простым синтетическим способом, описанным в публикации Jpn. Pat.Appln. KOKAI Publication2004-374445, в котором концевую двойную связь аллильной группы дигидроксилируют с образованием глицеринового скелета. В результате получают R/S диастереомеры с соотношением приблизительно 1:1. Для решения этой проблемы соответствующие диастереомеры могут быть синтезированы отдельно, но такой способ во время синтеза требует связывания глицеринового соединения, имеющего определенную стереоструктуру, с соединением сахара, что приводит к дополнительным проблемам, усложняет процесс синтеза и значительно увеличивает затраты. Кроме того, сульфопиранозилацилглицериновое соединение дает R/S диастереомер в положении 2 глицеринового остатка, а также несколько процентов структурного изомера (2-ацилизомера), где ацильная группа в 1-м положении глицерина перенесена во 2-е положение между молекулами и/или внутри молекул. Такие 2-ацилизомеры образуются во время синтеза и при хранении в растворе. Следовательно,даже если соответствующие диастереомеры получают по отдельности, очень трудно обеспечить высокую чистоту сульфопиранозилацилглицеринового соединения. Хотя сульфопиранозилацилглицериновое соединение проявляет заметный радиосенсибилизирующий эффект, его разработка в качестве лекарственного средства будет создавать очень трудные ситуации из-за проблем, связанных с синтезом и физическими свойствами. Описание изобретения Настоящее изобретение выполнено с учетом названных выше проблем. Цель настоящего изобретения состоит в разработке практичного нового сульфированного соединения сахара и содержащего его лекарственного средства, и, в частности, в разработке практичного нового сульфированного соединения сахара, получаемого с высокой чистотой простым синтетическим способом, и лекарственного средства,содержащего это соединение. В результате специального исследования заявителями найдены средства решения перечисленных выше проблем. Более конкретно:(1) сульфохиновозилацилпропандиольное соединение, представленное общей формулой (I) где R1 представляет собой ацильный остаток жирной кислоты и М представляет собой атом водорода или ион металла, или его фармацевтически приемлемые соли;(2) лекарственное средство, содержащее в качестве активного ингредиента по меньшей мере один активный ингредиент, выбранный из группы, включающей сульфохиновозилацилпропандиольное соединение в соответствии с п.(1), представленное общей формулой (I), и его фармацевтически приемлемые соли; и(3) лекарственное средство в соответствии с п.(2), которое представляет собой радиосенсибилизатор. Настоящее изобретение предлагает практичное новое сульфированное соединение сахара и содержащее его лекарственное средство. В частности, настоящее изобретение предлагает новое сульфированное соединение сахара, получаемое с высокой чистотой простым синтетическим способом и содержащее его лекарственное средство. Эффект настоящего изобретения будет представлен в приведенном ниже описании и будет частично определен описанием или вариантом осуществления настоящего изобретения. Положительный эффект настоящего изобретения будет понятен и реализован с помощью чертежей и описанных ниже комбинаций. Краткое описание чертежей Фиг. 1 представляет собой хроматограмму, показывающую данные анализа SQAP C18:0; фиг. 2 - хроматограмму, показывающую данные анализа SQMG C18:0; фиг. 3 - график, показывающий влияние испытуемого вещества на увеличение объема опухоли; фиг. 4-9 - графики, показывающие влияние испытуемого вещества на увеличение объема опухоли. фиг. 10 - график, показывающий влияние испытуемого вещества на образование трубочек. Наилучший способ осуществления изобретения В соответствии с аспектом настоящего изобретения предложены сульфохиновозилацилпропандиольное соединение, представленное формулой (I), и его фармацевтически приемлемые соли где R1 представляет собой ацильный остаток жирной кислоты и М представляет собой атом водорода или ион металла. В настоящем изобретении, когда "R1" представляет собой ацильный остаток жирной кислоты, число атомов углерода, присутствующих в нем, составляет 26 или меньше и 1 или больше и предпочтительно 22 или меньше. Жирная кислота для образования ацильного остатка жирной кислоты в соответствии с настоящим изобретением может быть линейной или разветвленной, насыщенной или ненасыщенной жирной кислотой. Кольцо хиновозы, присутствующее в сульфохиновозилацилпропандиольном соединении в соответствии с настоящим изобретением, может существовать в форме лодки, в форме кресла или в смешанной форме, но, как правило, существует в форме кресла, так как обычно эта форма является стабильной. Стерическая конфигурация пропандиольного участка в кольце хиновозы может представлять собой аномер, -аномер или их смесь. Сульфохиновозилацилпропандиольное соединение в соответствии с настоящим изобретением может быть далее обозначено как "SQAP" или "соединение SQAP". В обозначении "SQAP Cm:n" означает -аномер, а "Cm:n" показывает, что число атомов углерода, присутствующих в R1 группе SQAP,составляет "m" и число двойных связей равно "n", где "m" принимает целые значения от 1 или выше и"n" принимает целые значения от 0 и выше. Соответственно, например, "SQAP C18:0" означает аномер сульфохиновозилацилпропандиола, где число атомов углерода, присутствующих в ацильном остатке жирной кислоты, равно 18, а число двойных связей равно 0. Способом получения сульфохиновозилацилпропандиольного соединения в соответствии с настоящим изобретением может быть приведенный ниже способ, но без ограничения таким способом. Так как в процессе синтеза сульфохиновозилацилпропандиольное соединение в соответствии с настоящим изобретением не образует новый асимметричный атом углерода, соединение может быть получено легко, просто и с высокой чистотой. Кроме того, соединение можно хранить в структурно стабильном состоянии,так как рядом с R1 группой отсутствует гидроксигруппа, которая легко вызывает перенос."Ph" означает фенильную группу, "Bn" означает бензильную группу, "Ts" означает тозильную группу, "SAc" означает ацетилтиогруппу и "М" означает ион водорода или ион металла.C) литийалюминийгидрид, хлорид алюминия, дихлорметан, диэтиловый эфир, нагревание при кипении с обратным холодильником, 4 ч;I) палладий на активированном угле, водород-газ, этанол, дихлорметан, комнатная температура, 48 ч. Альтернативно, после получения соединения (7) по схеме, включающей стадии A-F, целевое соединение (10) может быть получено с помощью следующих стадий:K) палладий на активированном угле, водород-газ, метанол, дихлорметан, комнатная температура,16 ч; иL) жирная кислота,гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида,4 диметиламинопиридин, N,N-диметилформамид, от 0 С до комнатной температуры, 18 ч. Способ получения сульфохиновозилацетилпропандиольного соединения в соответствии с настоящим изобретением не ограничен приведенным выше конкретным примером и может быть использован следующий дополнительный способ."Ас" означает ацетильную группу, "MP" означает п-метоксифенильную группу, "РМВ" означает пметоксибензильную группу, "Ts" означает тозильную группу, "SAc" означает ацетилтиогруппу и "М" означает ион водорода или ион металла.I') сульфит натрия, этанол, вода, нагревание при кипении с обратным холодильником, 72 ч;J') производное жирной кислоты, 4-диметиламинопиридин, пиридин, дихлорметан, нагревание при кипении с обратным холодильником, 16 ч; иK') 2,3-дихлор-5,6-дициано-1,4-бензохинон, дихлорметан, метанол, вода, комнатная температура, 4 ч. Альтернативно, любые методики, известные специалистам в данной области техники, могут быть объединены с получением сульфохиновозилацилпропандиольного соединения и его фармацевтически приемлемых солей в соответствии с аспектом настоящего изобретения. Такие способы получения также включены в объем настоящего изобретения. Примеры сульфохиновозилацилпропандиольного соединения, представленные общей формулой (I),и его фармацевтически приемлемых солей в соответствии с настоящим изобретением включают, но не ограничиваются ими, соли моновалентных катионов, таких как натрий и калий, и соли двухвалентных катионов, таких как кальций и магний. Соли в соответствии с настоящим изобретением могут быть получены описанными выше способами синтеза или модификациями таких способов синтеза. С другой стороны, продукт, синтезированный описанным выше способом, может быть подвергнут известной ионообменной обработке с получением желаемой соли. Такие способы синтеза солей в соответствии с настоящим изобретением также включены в объем настоящего изобретения. Сульфохиновозилацилпропандиольное соединение и его фармацевтически приемлемые соли в соответствии с настоящим изобретением обладают заметными фармакологическими эффектами, такими как эффект радиационной сенсибилизации и противоопухолевый эффект. Таким образом, в соответствии с другим аспектом настоящего изобретения сульфохиновозилацилпропандиольное соединение и его фармацевтически приемлемые соли могут быть получены в качестве лекарственных средств, использующих их фармакологические эффекты. Таким образом, в соответствии с другим аспектом настоящего изобретения сульфохиновозилацилпропандиольное соединение и его фармацевтически приемлемые соли обладают сенсибилизирующим эффектом в качестве первого фармакологического эффекта. Соответственно сульфохиновозилацилпропандиольное соединение и его фармацевтически приемлемые соли могут быть предложены в качестве радиосенсибилизаторов.-4 016931 Радиосенсибилизатор в соответствии с настоящим изобретением может быть использован для лечения злокачественной неоплазмы. Примерами злокачественной неоплазмы являются, но не ограничиваются ими: нейрогенная опухоль, такая как церебральная опухоль; плоскоклеточный рак и аденокарцинома, такая как рак головы и шеи, рак кожи, рак пищевода, рак щитовидной железы, рак желудка, рак легких, рак желчного пузыря, рак желчных путей, рак поджелудочной железы, рак печени, рак простаты,рак матки, рак яичников, рак молочной железы, рак почек, рак мочевого пузыря и рак толстой кишки; и меланома, остеома, опухоль мягких тканей и лимфома, лейкемия и миелома. Определение "лечение",используемое в данном случае, относится к уменьшению, разрушению и/или ингибированию усиленного роста описанных выше злокачественных неоплазм. Радиосенсибилизатор в соответствии с настоящим изобретением может содержать в качестве активного ингредиента эффективную дозу по меньшей мере одного соединения, выбранного из группы,включающей сульфохиновозилацилпропандиольное соединение, представленное общей формулой (I), и его фармацевтически приемлемые соли. Радиосенсибилизатор может содержать более чем один вид соединений, имеющих различные заместители R1 в общей формуле (I). Кроме того, радиосенсибилизатор может быть смешан с другим(и) радиосенсибилизатором(ами), противоопухолевым(и) агентом(ами) или с другим(и) веществом(ами), имеющим(и) фармакологическую активность и/или фармацевтическую активность без влияния на их активность. Ниже соединения, включающие сульфохиновозилацилпропандиольное соединение, представленное общей формулой (I), и его фармацевтически приемлемые соли в соответствии с настоящим изобретением могут быть названы "радиосенсибилизирующим веществом настоящего изобретения". Радиосенсибилизирующее вещество настоящего изобретения может быть применено, например,путем орального введения или парентерального введения. В соответствии с такими схемами введения радиосенсибилизирующее вещество настоящего изобретения может быть смешано с соответствующей фармацевтически приемлемой лекарственной добавкой, такой как эксципиент или разбавитель, в результате чего получают фармацевтический препарат. Радиосенсибилизатор в соответствии с настоящим изобретением будет содержать эффективную дозу радиосенсибилизирующего вещества настоящего изобретения и может быть представлен в виде фармацевтического препарата, описанного выше. Примерами дозированных форм, приемлемых для орального введения, являются твердые, полутвердые, жидкие и газообразные формы, и их конкретные примеры включают, но не ограничиваются ими, таблетки, капсулы, порошки, гранулы, растворы, суспендирующие агенты, сиропы, эликсиры и аэрозоли. Когда радиосенсибилизирующее вещество настоящего изобретения вводят парентерально, оно может быть применено, например, с помощью инъекции, трансдермального введения, ректального введения или глазного введения. Введение с помощью инъекции может быть проведено, например, путем подкожной, интрадермальной, внутривенной или внутримышечной инъекции. Условия введения радиосенсибилизирующего вещества настоящего изобретения (например, доза,частота введения и интервал введения) могут быть соответствующим образом установлены и подогнаны с учетом дозированной формы, способа введения, заболевания, которое подвергается лечению, например, с учетом статуса злокачественной неоплазмы (например, типа, расположения и стадии); условий,таких как лекарственное средство, которое должно быть комбинировано (например, присутствие или отсутствие комбинируемого лекарства, тип, доза, частота и распределение во времени введения комбинированного лекарства и последовательность введения комбинированного лекарства и радиосенсибилизирующего вещества настоящего изобретения); способа комбинирования с излучением (например, распределения во времени комбинирования и порядка введения радиосенсибилизирующего вещества настоящего изобретения) и состояний субъекта, который подвергается лечению (например, массы тела,пола и возраста). Например, доза радиосенсибилизирующего вещества может составлять, но без ограничения, от 0,001 до 100 мг/кг массы тела в день при оральном введении, от 0,001 до 50 мг/кг массы тела в день через инъекцию, от 0,001 до 100 мг/кг массы тела в день при трансдермальном введении, от 0,001 до 50 мг/кг массы тела в день при ректальном введении; или при капельном введении приблизительно от 0,001 до 3 мас.% раствора несколько раз в день черезглазное введение. При радиотерапевтическом лечении тип, доза и частота облучения могут соответствовать условиям обычного радиотерапевтического лечения. То есть проводят обычное для человека радиотерапевтическое лечение, например, посредством воздействия медицинского облучения, такого как рентгеновское излучение, -излучение, электронный луч, -излучение или пучки других частиц, таких как -мезоны,нейтроны, пучки тяжелых частиц, с дозой облучения приблизительно от 0,1 до 100 Гр за промежуток времени от одной недели до 6 месяцев с получением суммарной дозы облучения приблизительно от 10 до 500 Гр. Типичный пример радиотерапии человека проводят, например, но без ограничения, рентгеновским излучением при дозе 2 Гр за промежуток времени пять раз с получением суммарной дозы 60 Гр за период приблизительно 6 недель. Например, доза и частота облучения могут быть уменьшены. Другие-5 016931 примеры радиотерапевтического метода включают согласованную радиотерапию, стереотаксическое облучение, где очаг злокачественной неоплазмы обстреливают с точечной погрешностью, или модулированную по интенсивности радиотерапию. Кроме того, также можно использовать облучение с помощью капсулированного герметичного радиоактивного источника, -лучевой телетерапии или облучение пучком частиц. Доза облучения за промежуток времени может быть увеличена, а период облучения может быть понижен за счет интервала облучения. Радиационную терапию и введение радиосенсибилизатора настоящего изобретения можно проводить одновременно или последовательно. В этом случае радиосенсибилизатор настоящего изобретения,как ожидается, выполняет функцию противоопухолевого агента, который комбинируется с радиационной терапией. Таким образом, в соответствии с другим аспектом настоящего изобретения новое сульфохиновозилацилпропандиольное соединение или его фармацевтически приемлемые соли настоящего изобретения могут быть предложены в качестве противоопухолевого агента, который комбинируют с радиационной терапией. Как известно специалисту в области радиотерапевтического лечения, условия радиационной терапии и введения радиосенсибилизатора настоящего изобретения могут быть соответствующим образом выбраны специалистами в области здравоохранения или другими специалистами, например, в зависимости от типа источника излучения, способа облучения, участка и периода облучения; типа сенсибилизатора, способа введения и распределения введения во времени; типа и серьезности заболевания, которое подвергается лечению; и возраста, массы тела, состояния здоровья и истории болезни субъекта, который подвергается воздействию облучения. Кроме того, в соответствии еще с одним аспектом настоящего изобретения предложен способ лечения заболевания, в отношении которого радиационная терапия эффективна, включающий введение эффективной дозы радиосенсибилизирующего вещества субъекту, который нуждается в этом веществе. Определение "заболевание, в отношении которого радиационная терапия эффективна" относится к заболеванию, которое эффективно лечится, например, с помощью воздействия радиационной терапии на описанную выше злокачественную неоплазму. Подробности относительно радиосенсибилизирующего вещества и способ и условия его введения могут соответствовать описанным выше. Терапия в соответствии с настоящим изобретением может включать введение эффективной дозы радиосенсибилизирующего вещества субъекту, нуждающемуся в этом веществе, одновременно с радиационной терапией или до или после радиационной терапии. В соответствии с еще одним аспектом настоящего изобретения сульфохиновозилацилпропандиольное соединение и его фармацевтически приемлемые соли обладают противоопухолевым эффектом в качестве второго фармакологического эффекта. Более конкретно, они синергетически усиливают радиационный эффект и могут подавлять злокачественную неоплазму при использовании отдельно. Соответственно сульфохиновозилацилпропандиольное соединение и его фармацевтически приемлемые соли могут быть предложены в качестве противоопухолевого агента. При использовании в качестве противоопухолевого агента сульфохиновозилацилпропандиольное соединение и его фармацевтически приемлемые соли могут быть использованы таким же образом, как и описанный выше радиосенсибилизатор, за исключением того, что они не комбинируются с радиационной терапией. В этом случае условия введения сульфохиновозилацилпропандиольного соединения (например, доза, частота введения и интервал введения) могут быть соответствующим образом установлены и подогнаны в зависимости от дозированной формы; способа введения; заболевания, которое подвергается лечению, например, статуса злокачественной неоплазмы (например, типа, расположения и стадии); условий,таких как лекарство, которое должно быть объединено (например, присутствие или отсутствие комбинируемого лекарства, тип, доза, частота и распределение введения по времени комбинируемого лекарства,и последовательность введения комбинируемого лекарства и радиосенсибилизирующего вещества настоящего изобретения); и состояний субъекта, который подвергается лечению (например, массы тела,пола и возраста). Примеры Примеры настоящего изобретения описаны ниже, но изобретение не ограничивается ими. Примеры синтеза. Пример I. Стадии получения сульфохиновозилацилпропандиольного соединения в соответствии с настоящим изобретением описаны ниже на примере натриевой соли -сульфохиновозилстеароилпропандиола.C) литийалюминийгидрид, хлорид алюминия, дихлорметан, диэтиловый эфир, нагревание при кипении с обратным холодильником, 4 ч, 90,2%;I) палладий на активированном угле, водород-газ, этанол, дихлорметан, комнатная температура, 48 ч, 79,4%. Процесс получения в качестве конечного продукта натриевой соли -сульфохиновозилстеароилпропандиола в соответствии с аспектом настоящего изобретения проводят по направлениям A)-I) приведенной выше схемы. Пример I-1. Стадия А. 1-O-Аллил-4,6-O-бензилиденO-глюкопиранозид (2). Соединение (1) (100 г, 555 ммоль) в качестве исходного материала суспендируют в аллиловом спирте (500 мл), к которому добавляют трифторметансульновую кислоту (1,00 мл) при 0 С, и реакционную жидкость интенсивно перемешивают при 80 С в течение 48 ч. После подтверждения протекания реакции в достаточной степени для остановки реакции добавляют триэтиламин (3 мл) и реакционную жидкость концентрируют при пониженном давлении. Затем остаток суспендируют в безводном ацетонитриле (500 мл), к которому добавляют диметилацеталь бензальдегида (127 г, 1,5 экв.) и моногидрат птолуолсульфоновой кислоты (5,28 г, 0,05 экв.). Реакционную жидкость перемешивают при 40 С в течение 4 ч, затем для остановки реакции добавляют триэтиламин (10 мл) и реакционную жидкость концентрируют при пониженном давлении. Остаток выливают в гексан (2000 мл) и воду (500 мл) и смешанную жидкость интенсивно перемешивают. Образовавшийся осадок отфильтровывают и промывают водой и гексаном. Осадок кристаллизуют из нагретого этанола дважды, получают названное соединение (2) в форме бесцветных игольчатых кристаллов (34,5 г, 112 ммоль, выход 20,2%). Стадия В. 1-О-Аллил-2,3-ди-О-бензил-4,6-О-бензилиденD-глюкопиранозид (3). К раствору соединения (2) (30,0 г, 97,3 ммоль) в безводном N,N-диметилформамиде (ДМФА, 300 мл) добавляют бензилбромид (41,6 г, 2,5 экв.) и гидроксид натрия (11,7 г, 3,0 экв.) и реакционную жидкость интенсивно перемешивают при комнатной температуре 24 ч. После подтверждения протекания реакции в достаточной степени реакционную жидкость выливают в охлажденную воду (900 мл) и экстрагируют этилацетатом (3300 мл). Органические слои объединяют и промывают насыщенным рассолом(2100 мл), сушат над сульфатом натрия, фильтруют, концентрируют при пониженном давлении. Полученный остаток кристаллизуют дважды из нагретого этанола, получают названное соединение в виде бесцветных игольчатых кристаллов (33,5 г). Фильтрат концентрируют, очищают хроматографией на силикагеле (гексан-этилацетат, 15:110:18:1), кристаллизуют из нагретого этанола, получают соединение (3) (6,63 г) (всего 40,1 г, 82,1 ммоль, выход 84,4%). Стадия С. 1-O-Аллил-2,3,4-три-O-бензилD-глюкопиранозид (4). Литийалюминийгидрид (2,02 г, 1,3 экв.) суспендируют в смешанном растворе безводного дихлорметана (100 мл) и безводного диэтилового эфира (100 мл), затем добавляют соединение (3) (20,0 г, 40,9 ммоль). Затем к реакционной жидкости добавляют 200 мл хлорида алюминия (7,09 г, 1,3 экв.) в безводном диэтиловом эфире и смесь перемешивают 4 ч при нагревании и кипячении с обратным холодильником. После подтверждения протекания реакции в достаточной степени медленно по каплям добавляют воду (10 мл), осадок отфильтровывают после выдерживания в течение ночи и затем осадок промывают диэтиловым эфиром. Фильтрат промывают водой (2100 мл), водные слои объединяют и экстрагируют диэтиловым эфиром (2100 мл). Органические слои объединяют и промывают насыщенным рассолом(2200 мл), сушат сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле (гексан-этилацетат, 5:14:13:12:1), получают названное соединение (4) в форме бесцветного маслянистого вещества (18,1 г, 36,9 ммоль, выход 90,2%). Стадия D. 1-O-Аллил-2,3,4-три-O-бензил-6-O-тозилD-глюкопиранозид (5). К раствору соединения (4) (25,1 г, 51,2 ммоль) в безводном пиридине (250 мл) добавляют птолуолсульфонилхлорид (14,6 г, 1,5 экв.) и 4-диметиламинопиридин (62 6 мг, 0,1 экв.) и реакционную жидкость перемешивают при комнатной температуре 16 ч. После подтверждения протекания реакции в достаточной степени добавляют воду (10 мл) для остановки реакции и реакционную жидкость концентрируют при пониженном давлении. Остаток суспендируют в небольшом количестве этилацетата, выливают в 0,5 М соляную кислоту (200 мл) и экстрагируют этилацетатом (3200 мл). Органические слои объединяют, промывают насыщенным раствором бикарбоната натрия (2100 мл) и насыщенным рассолом (2100 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный остаток кристаллизуют дважды из нагретого этанола, получают названное соединение (5) в форме бесцветных игольчатых кристаллов (25,0 г). Фильтрат концентрируют, очищают хроматографией на силикагеле (гексан-этилацетат, 5:14:13:1), получают соединение (5) (4,00 г) (всего 29,0 г, 45,0 ммоль, выход 87,9%). Стадия E. 1-О-(2,3,4-три-O-Бензил-6-O-тозилD-глюкопиранозил)пропан-1,3-диол (6). К раствору соединения (5) (29,0 г, 45,0 ммоль) в безводном тетрагидрофуране (ТГФ, 150 мл) добавляют раствор 0,5 М 9-борабицикло[3,3,1]нонана (9-BBN) в тетрагидрофуране (180 мл, 90,0 ммоль) при 0 С в атмосфере аргона. Через 1 ч реакционную жидкость возвращают к комнатной температуре и непрерывно перемешивают 10 ч. Реакционную жидкость снова охлаждают до 0 С, к жидкости вначале добавляют воду (20 мл) и затем последовательно добавляют 3 М раствор гидроксида натрия (70 мл) и 35% раствор перекиси водорода (70 мл). Через 1 ч реакционную жидкость возвращают к комнатной температуре и перемешивают 12 ч. После подтверждения протекания реакции в достаточной степени раствор экстрагируют этилацетатом (3100 мл), органические слои объединяют и промывают насыщенным рассолом (2100 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле (гексан-этилацетат, 3:21:12:3), получают названное соединение (6) в форме бесцветного маслянистого вещества (28,2 г, 42,5 ммоль, выход 94,4%). Пример 1-6. Стадия F. 1-О-(2,3,4-три-О-Бензил-6-тиоацетилD-хиновопиранозил)пропан-1,3-диол (7). К раствору соединения (6) (28,2 г, 42,5 ммоль) в безводном ДМФА (300 мл) добавляют тиоацетат калия (7,28 г, 1,5 экв.) и смесь перемешивают при 90 С в течение 3 ч. После подтверждения протекания реакции в достаточной степени реакционную жидкость выливают в охлажденную воду (900 мл), экстрагируют этилацетатом (3300 мл). Органические слои объединяют и промывают насыщенным рассолом(2200 мл), сушат сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле (гексан-этилацетат, 2:13:21:12:3), получают названное соединение (7) в форме светло-коричневого маслянистого вещества (21,9 г, 38,6 ммоль, выход 90,8%). Стадия G. 3-O-(2,3,4-три-O-Бензил-6-тиоацетилD-хиновопиранозил)-1-O-стеароилпропан-1,3 диол (8). К раствору соединения (7) (21,9 г, 38,6 ммоль) в безводном дихлорметане (200 мл) добавляют стеароилхлорид (15,2 г, 1,3 экв.) и безводный пиридин (5 мл) и смесь перемешивают при комнатной температуре 2 ч. После подтверждения протекания реакции в достаточной степени добавляют метанол (5 мл) для остановки реакции и смесь концентрируют при пониженном давлении. Остаток суспендируют в небольшом количестве этилацетата, выливают в воду (200 мл) и экстрагируют этилацетатом (3100 мл). Органические слои объединяют и промывают насыщенным рассолом (2100 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле (гексан-этилацетат, 10:18:26:1), получают названное соединение (8) в форме бесцветного маслянистого вещества (31,3 г, 37,6 ммоль, выход 97,4%). Стадия Н. Натриевая соль 3-O-(2,3,4-три-O-бензил-6-сульфоD-хиновопиранозил)-1-Oстеароилпропан-1,3-диола (9). К раствору соединения (8) (31,3 г, 37,6 ммоль) в уксусной кислоте (450 мл) добавляют Oxon (46,2 г) и ацетат калия (11,3 г) и смесь интенсивно перемешивают при комнатной температуре 48 ч. После подтверждения достаточного протекания реакции реакционную жидкость выливают в охлажденный 7,5 М раствор гидроксида натрия (1000 мл) и экстрагируют этилацетатом (4200 мл). Органические слои объединяют, промывают насыщенным раствором гидрокарбоната натрия (2200 мл) и насыщенным рассолом(2200 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле (дихлорметан-метанол, 15:110:18:1), получают названное соединение (9) в форме бесцветного воскообразного вещества (28,7 г, 33,3 ммоль, выход 88,6%). Стадия I. Натриевая соль 3-O-(6-сульфоD-хиновопиранозил)-1-O-стеароилпропан-1,3-диола (10). К раствору соединения (9) (28,7 г, 33,3 ммоль) в этаноле (400 мл) и дихлорметане (150 мл) добавляют 10% палладий на активированном угле (7,00 г) и смесь перемешивают при комнатной температуре 48 ч в атмосфере водорода-газа. После подтверждения протекания реакции в достаточной степени палладий на активированном угле отфильтровывают, фильтрат концентрируют при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле(дихлорметан-метанол,10:15:13:12:11:1) и осаждают из нагретого 98%-ного этанола, получают названное соединение(10) в форме бесцветного порошка (15,6 г, 26,4 ммоль, выход 79,4%). В 60 мл воды растворяют 2,15 г натриевой соли 3-O-(6-сульфоD-хиновопиранозил)-1-Oстеароилпропан-1,3-диола, адсорбируют на колонке WAKOGEL 100C18 (производство Wako PureChemical Industries, Ltd.) и на колонку для замещения выливают 500 мл 1% раствора хлорида кальция и промывают 500 мл дистиллированной воды. Затем проводят элюирование с помощью 200 мл каждого из 50, 80 и 100% метанола и повторно осаждают из нагретого 98%-ного этанола, получают 1,47 г кальцие- 11016931 вой соли 3-O-(6-сульфоD-хиновопиранозил)-1-O-стеароилпропан-1,3-диола. Хотя в примерах не показано, такой же обработкой на колонке можно получить магниевую или калиевую соль за счет замещения раствором хлорида магния или хлорида калия. Пример II. В качестве других примеров -сульфохиновозилацилпропандиольного соединения ниже описан аномер соединений, содержащих 22, 14, 10, 6, 2 и 1 атом углерода в ацильных остатках жирной кислоты. Пример II-1. Натриевая соль 3-O-(6-сульфоD-хиновопиранозил)-1-O-деканоилпропан-1,3-диола. Названное соединение синтезируют таким же образом, что и в примере I, за исключением того, что на стадии G в качестве производного жирной кислоты используют деканоилхлорид.[]22D +57,9 (с 0,76 H2O), LRMS m/z 455 (M-Na)-; HRMS: вычислено для C19H35O10S (M-Na)455,1956, найдено 455,1954. 1 Н ЯМР (400 МГц, ДМСО-d6);5,40 (д, 1 Н, J=3,1 Гц, 4'-ОН), 4,65 (д, 1 Н, J=4,7 Гц, 3'-ОН), 4,56 (д,1 Н, J=3,7 Гц, H1'), 4,52 (д, 1 Н, J=6,48 Гц, 2'-ОН), 4,14-4,08 (м, 2 Н, H1a и H1b), 3,84-3,78 (м, 2 Н, Н 3 а и Н 5'), 3,41-3,33 (м, 2 Н, Н 3b и Н 3'), 3,21-3,16 (м, 1 Н, Н 2'), 2,97-2,92 (м, 2 Н, Н 4' и Н 6'а), 2,61 (дд, 1 Н, J=14,0,6,2 Гц, Н 6'b), 2,29 (т, 2 Н, J=7,4 Гц, СОСН 2), 1,84-1,81 (м, 2 Н, Н 2 а и Н 2b), 1,53-1,50 (м, 2 Н, СОСН 2 СН 2),1,25 (ушир, 12 Н, -СН 2-), 0,86 (т, 3 Н, J=6,8 Гц, Me). 13 С ЯМР (100 МГц, ДМСО- d6);173,1 (С=O), 98,4 (С 1'), 74,8 (С 4'), 73,2 (С 3'), 71,9 (С 2'), 68,4 (С 5'),63,5 (С 3), 61,4 (С 1), 55,2 (С 6'), 33,6 (СОСН 2), 31,4 (-СН 2-), 29,0-28,6 (м, С 2 и -СН 2-), 24,6 (СОСН 2 СН 2),22,2 (-СН 2-), 14,1 (Me). Пример II-2. Натриевая соль 3-O-(6-сульфоD-хиновопиранозил)-1-O-миристоилпропан-1,3 диола. Названное соединение синтезируют таким же образом, что и в примере I, за исключением того, что на стадии G в качестве производного жирной кислоты используют миристоилхлорид.[]23D +49,7 (с 0,67 H2O), LRMS m/z 511 (M-Na)-; HRMS: вычислено для C23H43O10S (M-Na)511,2582, найдено 511,2596. 1 Н ЯМР (400 МГц, ДМСО-d6);5,41 (ушир, 1 Н, 4'-ОН), 4,63-4,61 (м, 1 Н, 3'-ОН), 4,55 (д, 1 Н, J=3,7 Гц, H1'), 4,50-4,48 (м, 1 Н, 2'-ОН), 4,13-4,07 (м, 2 Н, H1a и H1b), 3,83-3,77 (м, 2 Н, Н 3 а и Н 5'), 3,41-3,33 (м,2 Н, Н 3b и Н 3'), 3,20-3,15 (м, 1 Н, Н 2'), 2,98-2,91 (м, 2 Н, Н 4' и Н 6'а), 2,64-2,59 (м, 1 Н, Н 6'b), 2,28 (т, 2 Н,J=7,4 Гц, СОСН 2), 1,86-1,79 (м, 2 Н, Н 2 а и Н 2b), 1,53-1,49 (м, 2 Н, СОСН 2 СН 2), 1,24 (ушир, 20 Н, -СН 2-),0,85 (т, 3 Н, J=6,8 Гц, Me). 13 С ЯМР (100 МГц, ДМСО-d6);173,0 (С=O), 98,4 (С 1'), 74,8 (С 4'), 73,2 (С 3'), 71,9 (С 2'), 68,4 (С 5'),63,5 (С 3), 61,4 (С 1), 55,3 (С 6'), 33,6 (СОСН 2), 31,4 (-СН 2-), 29,1-28,6 (м, С 2 и -СН 2-), 24,6 (СОСН 2 СН 2),22,2 (-СН 2-), 14,0 (Me). Пример II-3. Натриевая соль 3-O-(6-сульфоD-хиновопиранозил)-1-O-бегеноилпропан-1,3-диола. Названное соединение синтезируют таким же образом, что и в примере I, за исключением того, что на стадии G в качестве производного жирной кислоты используют бегеноилхлорид.(м, 2 Н, Н 3b и Н 3'), 3,22-3,17 (м, 1 Н, Н 2'), 2,98-2,90 (м, 2 Н, Н 4' и Н 6'а), 2,60 (дд, 1 Н, J=14,0, 6,7 Гц, Н 6'b),2,28 (т, 2 Н, J=7,22 Гц, СОСН 2), 1,86-1,79 (м, 2 Н, Н 2 а и Н 2b), 1,52-1,49 (м, 2 Н, СОСН 2 СН 2), 1,23 (ушир,36 Н, -СН 2-), 0,85 (т, 3 Н, J=6,1 Гц, Me). 13 С ЯМР (100 МГц, ДМСО-d6);173,4 (С=O), 98,5 (С 1'), 74,7 (С 4'), 73,4 (С 3'), 72,2 (С 2'), 68,6 (С 5'),63,6 (С 3), 61,8 (С 1), 55,0 (С 6'), 33,9 (СОСН 2), 31,7 (-СН 2-), 29,4-28,8 (м, С 2 и -СН 2-), 24,9 (СОСН 2 СН 2),22,5 (-СН 2-), 14,3 (Me). Пример II-4. Кальциевая соль 3-O-(6-сульфоD-хиновопиранозил)-1-O-гексаноилпропан-1,3 диола (10). Натриевую соль синтезируют таким же образом, что и в примере I, за исключением того, что на стадии G в качестве производного жирной кислоты используют гексаноилхлорид.(Me). Натриевую соль затем подвергают ионообменной обработке с получением названного соединения.- 12016931 Пример II-5. Кальциевая соль 3-O-(6-сульфоD-хиновопиранозил)-1-O-ацетилпропан-1,3-диола. Натриевую соль синтезируют таким же образом, что и в примере I, за исключением того, что на стадии G в качестве производного жирной кислоты используют ацетилхлорид.(м, 2 Н, Н 3b и Н 3'), 3,20-3,16 (м, 1 Н, Н 2'), 2,98 (ддд, 1 Н, J=9,0, 9,0, 3,2 Гц, Н 4'), 2,88 (дд, 1 Н, J=13,6, 5,6 Гц,Н 6'а), 2,62 (дд, 1 Н, J=14,0, 5,6 Гц, Н 6'b), 2,00 (с, 3 Н, Me), 1,835 (тт, 1 Н, J=6,4, 6,4 Гц, Н 2). 13 С ЯМР (100 МГц, ДМСО-d6);170,4 (С=O), 98,3 (С 1'), 74,6 (С 4'), 73,2 (С 3'), 71,9 (С 2'), 68,3 (С 5'),63,5 (С 3), 61,5 (С 1), 55,0 (С 6'), 28,5 (С 2), 20,7 (Me). Натриевую соль затем подвергают ионообменной обработке с получением названного соединения. Пример II-6. Натриевая соль 3-О-(6-сульфоD-хиновопиранозил)-1-O-формилоксипропан-1,3 диола (10). Названное соединение получают с помощью стадий от А-1 до F примера I, после которых следуют стадии J-L. Стадия J. Натриевая соль 3-O-(2,3,4-три-O-бензил-6-сульфоD-хиновопиранозил)пропан-1,3 диола (8"). К раствору соединения (7) (542 мг, 956 ммоль) в уксусной кислоте (5,5 г) добавляют Oxon (1,8 г) и ацетат калия (68 мг) и смесь интенсивно перемешивают при комнатной температуре 48 ч. После подтверждения протекания реакции в достаточной степени реакционную жидкость выливают в охлажденный 7,5 М раствор гидроксида натрия (13 мл) и экстрагируют этилацетатом (310 мл). Органические слои объединяют, промывают насыщенным раствором бикарбоната натрия (210 мл) и насыщенным рассолом (210 мл), сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Концентрированный остаток очищают хроматографией на силикагеле (хлорформ-метанол,10:18:16:14:12:11:1), получают названное соединение (8") в форме бесцветного воскообразного вещества (401 мг, 675 ммоль, выход 70,7%). Стадия K. Натриевая соль 3-O-(6-сульфоD-хиновопиранозил)пропан-1,3-диола (9"). К раствору соединения (8") (534 мг, 898 мкмоль) в метаноле (20 мл) и хлороформе (5,0 мл) добавляют 10%-ный палладий на активированном угле (135 мг) и смесь перемешивают при комнатной температуре 16 ч в атмосфере водорода-газа. После подтверждения протекания реакции в достаточной степени палладий на активированном угле отфильтровывают и фильтрат концентрируют при пониженном давлении. К полученному остатку добавляют метанол (20 мл) и толуол (20 мл), смесь интенсивно перемешивают и растворитель удаляют путем упаривания при пониженном давлении, получают смесь в форме бесцветной жидкости (320 мг). Присутствие в смеси названного соединения подтверждают LRMS. Смесь, содержащую названное соединение (9"), затем подвергают последующим реакциям.(10). Смесь, содержащую соединение (9") (70 мг), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (EDClHCl) (93 мг, 487 мкмоль) и 4-диметиламинопиридин (12 мг, 97 мкмоль), растворяют в безводном N,N-диметилформамиде (ДМФА, 10 мл), к раствору по каплям при охлаждении льдом добавляют муравьиную кислоту (14 мг, 259 мкмоль) и дают возможность реагировать при комнат- 13016931 ной температуре 18 ч. После подтверждения протекания реакции в достаточной степени к реакционной жидкости добавляют воду (1,0 мл) для остановки реакции и затем раствор концентрируют при пониженном давлении. Полученный остаток очищают хроматографией на силикагеле (хлорформ-метанол-вода,3:1:0, 12:1:0,11:1:0,1), получают названное соединение (10) в форме бесцветного маслянистого вещества (12 мг, 33 мкмоль, выход 16,7%). Другой пример способа получения -сульфохиновозилацетилпропандиольного соединения в соответствии с настоящим изобретением описан ниже. Пример III-1. Натриевая соль 3-O-(6-сульфоD-хиновопиранозил)-1-O-олеоилпропан-1,3-диола. Названное соединение синтезируют по методике в соответствии с представленной ниже схемой.I') сульфит натрия, этанол, вода, нагревание при кипении с обратным холодильником, 72 ч, 90,2%;J') ангидрид олеиновой кислоты, 4-диметиламинопиридин, пиридин, дихлорметан, нагревание при кипении с обратным холодильником, 16 ч, 67,6%; и(м, С 2 и -СН 2-), 28,1 (-СН 2 СН=СНСН 2-), 26,1 (СОСН 2 СН 2), 23,8 (-СН 2-), 14,5 (Me). Пример III-2. Натриевая соль 3-О-(6-сульфоD-хиновопиранозил)-1-O-стеароилпропан-1,3-диола. Названное соединение синтезируют таким же образом, что и в примере III-1, за исключением того,что вместо ангидрида олеиновой кислоты используют стеароилхлорид.H ЯМР (400 МГц, ДМСО-d6);5,56 (д, 1 Н, J=3,16 Гц, 4'-ОН), 4,81 (д, 1 Н, J=4,92 Гц, 2'-ОН), 4,74 (д,1 Н, J=4,64 Гц, 3'-ОН), 4,09 (д, 1 Н, J=7,76 Гц, H1'), 4,07 (т, 2 Н, J=6, 60 Гц, Н 1 а и H1b), 3,77 (дт, 1 Н, J=10,2,6,27 Гц, Н 3 а), 3,54-3,45 (м, 2 Н, Н 3b и Н 5'), 3,13 (дт, 1 Н, J=8,80, 4,68 Гц, Н 3'), 2,99 (дт, 1 Н, J=9,14, 3,08 Гц,Н 4'), 2,97-2,91 (м, 2 Н, Н 2' и Н 6'а), 2,68 (дд, 1 Н, J=13,9, 5,24 Гц, Н 6'b), 2,27 (т, 2 Н, J=7,40 Гц, СОСН 2), 1,861,78 (м, 2 Н, Н 2 а и Н 2b), 1,54-1,47 (м, 2 Н, СОСН 2 СН 2), 1,24 (ушир, 28 Н, -СН 2-), 0,86 (т, 3 Н, J=6,88 Гц,Me). 13 С ЯМР (100 МГц, ДМСО-d6);172,9 (С=O), 102,8 (С 1'), 76,1 (С 3'), 74,6 (С 4'), 73,4 (С 2'), 72,5 (С 5'),65,2 (С 3), 61,2 (С 1), 55,6 (C6'), 33,6 (СОСН 2), 31,3 (С 2), 29,0-28,5 (м, -СН 2-), 24,5 (СОСН 2 СН 2), 22,1 (-СН 2), 13,9 (Me). Анализ. Пример XV. Пример IV-1. Анализ с помощью высокоэффективной жидкостной хроматографии и массспектрометрии. Натриевую соль 3-О-(6-сульфоD-хиновопиранозил)-1-O-стеароилпропан-1,3-диола и натриевую соль 3-О-(6-сульфоD-хиновопиранозил)-1-О-стеароилглицерина разделяют и идентифицируют с помощью высокоэффективной жидкостной хроматографии и масс-спектроскопии с электрораспылением. Испытуемое соединение растворяют в 5% ацетонитриле в водном растворе 5 ммоль/л ацетата аммония, разбавляют растворителем до желаемой концентрации и затем анализируют с помощью высокоэффективной жидкостной хроматографии, оборудованной колонкой CapCellPak C18MG (размеры колонки: 2,050 мм, производство Shiseido Co., Ltd.). Условия разделения: температура колонки 40 С, скорость потока 0,2 мл в 1 мин, элюирование проводят в течение 20 мин с линейным концентрационным градиентом от 50 до 70% ацетонитрила со ссылкой на описанный выше растворитель. Элюированное испытуемое вещество определяют с помощью ионного масс-спектрометра BrukerEsquire 3000 plus, режим детектирования ионов представляет собой суммарную ионную хроматографию(TIC), а интервал определения массы соответствует m/z=100-1000. Фиг. 1 и 2 представляют собой хроматограммы испытуемого вещества. Результаты анализа показывают, что, как видно на фиг. 1, натриевая соль 3-O-(6-сульфоDхиновопиранозил)-1-O-стеароилпропан-1,3-диола проявляет один пик; тогда как натриевая соль 3-O-(6 сульфоD-хиновопиранозил)-1-O-стеароилглицерина, представленная на фиг. 2, показывает небольшие пики, соответствующие структурному изомеру при 6,8 мин (пик 1) и при 7,3 мин (пик 2), что предполагает ацильный перенос от положения 1 в положение 2 глицеринового остатка, и основные пики, соответствующие диастереомеру SQMG С 18:0 при 7,6 мин (пик 3) и при 7,8 мин (пик 4). Полученные результаты показывают, что натриевая соль 3-O-(6-сульфоD-хиновопиранозил)-1O-стеароилпропан-1,3-диола в соответствии с аспектом настоящего изобретения имеет очень высокую чистоту по сравнению с известным соединением, натриевой солью 3-O-(6-сульфоDхиновопиранозил)-1-O-стеароилглицерина. Пример IV-2. Определение растворимости. По 1 г натриевой соли и кальциевой соли 3-O-(6-сульфоD-хиновопиранозил)-1-Oстеароилпропан-1,3-диола помещают в 5 мл дистиллированной воды для инъекций (производство OtsukaPharmaceutical Co., Ltd.), интенсивно встряхивают при 25 С; соли сразу же растворяются. Этот факт указывает на то, что вещество можно оценить как "легкорастворимое" по критерию, описанному в правилахJapanese Pharmacopeia. Полученные результаты показывают, что SQAP имеет очень высокую растворимость. Кроме того,хотя здесь не показано, серии соединений SQAP в соответствии с настоящим изобретением, отличные от солей 3-O-(6-сульфоD-хиновопиранозил)-1-O-стеароилпропан-1,3-диола, также имеют высокую растворимость. Такая высокая растворимость облегчает растворение необходимого количества вещества в небольшом количестве растворителя. В результате, например, инъекция, которая должна быть введена субъекту, может быть легко получена. Кроме того, такая высокая растворимость в воде дает преимуще- 15016931 ство при получении инъекций, а также других различных препаратов, например оральных средств. Фармакологические тесты. Изучена фармакологическая активность сульфохиновозилацилпропандиольного соединения в соответствии с настоящим изобретением. Пример V. Испытание радиосенсибилизирующего эффекта. Радиосенсибилизирующий эффект оценивают в опытах с мышами-опухоленосителями. Пример V-1. Плоскоклеточный рак пищевода человека ( 1). Клетки плоскоклеточного рака пищевода человека ТЕ-8 трансплантируют в правую бедренную область "голых" мышей KSN в количестве 1106 клеток на отдельное животное. Затем мышей выдерживают приблизительно 14 дней для формирования массы опухоли приблизительно 150 мм 3 у каждого отдельного животного. Затем по четыре мыши приписывают к каждой из следующих групп (1)-(4):(2) не получавшая лекарство, радиотерапевтически обработанная группа (на фиг. 3 показана с помощью пустых треугольников);(4) получавшая SQAP C18:0, радиотерапевтически обработанная группа (на фиг. 3 показана с помощью закрашенных ромбов). Лекарство вводят от 1 до 5 дня, 2 мг/кг один раз в день. Животных подвергают воздействию излучения, испускаемого генератором рентгеновских лучей (HS-225, производство Shimadzu Co., Ltd.) в дозе 2 Гр на 2 и 4 дни. Объем опухоли рассчитывают в соответствии с формулой расчета: [(меньшая ось)2(главная ось)0,5]. Полученные результаты представлены на фиг. 3. Во всех группах объем опухоли стабильно растет от начала до конца испытания. Однако приблизительно с 10 дня прирост объема опухоли в группах (2)-(4) падает ниже, чем в не получавшей лекарство,необлученной группе. Кроме того, подавление роста объема опухоли во (2) не получавшей лекарство,радиотерапевтически обработанной группе и в (3) получавшей SQAP C18:0, необлученной группе находится на одном и том же уровне. По сравнению с другими группами рост объема опухоли подавляется наиболее сильно в (4) получавшей SQAP C18:0, радиотерапевтически обработанной группе. Пример V-2. Плоскоклеточный рак пищевода человека ( 2). Опыт проводят аналогично примеру V-1, за исключением того, что доза лекарства, вводимого от 1 до 5 дня, составляет 1 мг/кг один раз в день, доза облучения составляет 4 Гр, и измеряют объем опухоли. Результаты представлены на фиг. 4. Каждая группа представляет собой:(2) не получавшая лекарство, радиотерапевтически обработанная группа (на фиг. 4 показана с помощью закрашенных квадратов);(4) получавшая SQAP C18:0, радиотерапевтически обработанная группа (на фиг. 4 показана с помощью пустых квадратов). Полученные результаты показывают, что во всех группах объем опухоли растет со временем. Прирост объема опухоли в группах (2)-(4) меньше, чем в (1) не получавшей лекарство, необлученной группе. Кроме того, по сравнению с другими группами рост объема опухоли подавляется наиболее сильно в (4) получавшей SQAP C18:0, радиотерапевтически обработанной группе. Пример V-3. Аденокарцинома ободочной кишки человека. Клетки аденокарциномы ободочной кишки человека SW480 трансплантируют в правую бедренную область "голых" мышей KSN в количестве 1106 клеток на отдельное животное. Затем мышей выдерживают приблизительно 14 дней для формирования массы опухоли приблизительно 150 мм 3 у каждого отдельного животного. Затем по четыре мыши приписывают к каждой из трех групп (1)-(3):(2) не получавшая лекарство, радиотерапевтически обработанная группа (на фиг. 5 показана с помощью закрашенных квадратов);(3) получавшая SQAP C18:0, радиотерапевтически обработанная группа (на фиг. 5 показана с помощью закрашенных треугольников). Лекарство вводят от 1 до 5 дня, 2 мг/кг один раз в день. Животных подвергают воздействию излучения, испускаемого генератором рентгеновских лучей (HS-225, производство Shimadzu Co., Ltd.) в дозе 2 Гр на 1 и 4 дни. Объем опухоли рассчитывают в соответствии с формулой расчета: [(меньшая ось)2(основная ось)0,5]. Полученные результаты представлены на фиг. 5.- 16016931 Результаты показывают, что во всех группах объем опухоли стабильно растет со временем. Однако рост объема опухоли в (1) не получавшей лекарство, необлученной группе и во (2) не получавшей лекарство, радиотерапевтически обработанной группе одинаковый, а рост объема опухоли в (3) получавшейSQAP C18:0, радиотерапевтически обработанной группе подавляется от начальной стадии опыта, и в целом рост объема опухоли подавляется значительно. В следующих примерах V-4, -5, -6 и примерах VI-VIII используют соединение SQAP, которое представляет собой кальциевую соль, полученную ионообменной обработкой. Пример V-4. Аденокарцинома ободочной кишки человека. Клетки аденокарциномы ободочной кишки человека SW480 трансплантируют в правую бедренную область "голых" мышей KSN в количестве 2106 клеток на отдельное животное. После формирования массы опухоли приблизительно 50 мм 3 у каждого отдельного животного по четыре мыши приписывают к каждой из четырех групп (1)-(4):(2) не получавшая лекарство, радиотерапевтически обработанная группа (на фиг. 6 показана с помощью закрашенных квадратов);(4) получавшая SQAP C18:0, радиотерапевтически обработанная группа (на фиг. 6 показана с помощью пустых квадратов). Лекарство вводят через хвостовую вену от 1 до 5 дня, 1 мг/кг один раз в день. Животных подвергают воздействию излучения, испускаемого генератором рентгеновских лучей (HS-225, производство Shimadzu Co., Ltd.) в дозе 2 Гр на 2 и 4 дни. Результаты представлены на фиг. 6, которые показывают, что объем опухоли растет со временем во всех группах. Однако по сравнению с другими группами рост объема опухоли наиболее сильно подавляется в (4) получавшей SQAP C18:0, радиотерапевтически обработанной группе. Пример V-5. Плоскоклеточный рак пищевода человека. Клетки плоскоклеточного рака пищевода человека ТЕ-8 трансплантируют в правую бедренную область "голых" мышей KSN в количестве 1106 клеток на отдельное животное. После формирования массы опухоли приблизительно 50 мм 3 у каждого отдельного животного по четыре мыши приписывают к каждой из четырех групп (1)-(4):(2) не получавшая лекарство, радиотерапевтически обработанная группа (на фиг. 7 показана с помощью закрашенных кругов);(4) получавшая SQAP C10:0, радиотерапевтически обработанная группа (на фиг. 7 показана с помощью пустых квадратов). Лекарство вводят внутрибрюшинно от 1 до 5 дня, 1 мг/кг один раз в день. Животных подвергают воздействию излучения, испускаемого генератором рентгеновских лучей (HS-225, производство Shimadzu Co., Ltd.) в дозе 4 Гр на 1 и 4 дни. Результаты представлены на фиг. 7, которые показывают, что объем опухоли растет со временем во всех группах. Однако по сравнению с другими группами рост объема опухоли наиболее сильно подавляется в (4) получавшей SQAP С 10:0, радиотерапевтически обработанной группе. Пример V-6. Плоскоклеточный рак пищевода человека. Клетки плоскоклеточного рака пищевода человека ТЕ-8 трансплантируют в правую бедренную область "голых" мышей KSN в количестве 1106 клеток на отдельное животное. После формирования массы опухоли приблизительно 50 мм 3 у каждого отдельного животного по четыре мыши приписывают к каждой из четырех групп (1)-(4):(2) не получавшая лекарство, радиотерапевтически обработанная группа (на фиг. 8 показана с помощью закрашенных квадратов);(4) получавшая SQAP C18:0, радиотерапевтически обработанная группа (на фиг. 8 показана с помощью пустых кругов). Лекарство вводят внутрибрюшинно от 1 до 5 дня, 1 мг/кг один раз в день. Животных подвергают воздействию излучения, испускаемого генератором рентгеновских лучей (HS-225, производство Shimadzu Co., Ltd.) в дозе 4 Гр на 1 и 4 дни.- 17016931 Результаты представлены на фиг. 8, которые показывают, что объем опухоли растет со временем во всех группах. Однако по сравнению с другими группами рост объема опухоли наиболее сильно подавляется в (4) получавшей SQAP C18:0, радиотерапевтически обработанной группе. Пример VI. Испытание противоопухолевого эффекта. Клетки аденокарциномы ободочной кишки человека SW480 трансплантируют в правую бедренную область "голых" мышей KSN в количестве 2106 клеток на отдельное животное. После формирования массы опухоли приблизительно 50 мм 3 у каждого отдельного животного по четыре мыши приписывают к каждой из двух групп (1) и (2):(1) не получавшая лекарство группа (на фиг. 9 показана с помощью пустых квадратов) и(2) получавшая SQAP C18:0 группа (на фиг. 9 показана с помощью закрашенных квадратов). Лекарство вводят внутрибрюшинно от 1 до 14 дня, 20 мг/кг один раз в день. Результаты представлены на фиг. 9, которые показывают, что по сравнению с не получавшей лекарство группой рост объема опухоли значительно подавляется в группе, получавшей SQAP С 18:0. Пример VII. Испытание на ингибирование образования трубочек с использованием системы сокультивирования сосудистых эндотелиальных клеток/фиброцитов. С использованием набора для ангиогенеза (KZ-1000), производимого Kurabo Industries Ltd., который представляет собой систему сокультивирования для человеческих сосудистых эндотелиальных клеток и человеческих фиброцитов, проводят оценку влияний SQAP С 10:0, SQAP C14:0, SQAP C18:0,SQAP С 22:0, PSQAP С 18:0 и SQAP C18:1 на образование трубочек. Культивирование для образования трубочек с использованием набора проводят в соответствии с инструкциями производителя. С использованием среды, содержащей конечную концентрацию 10 нг/мл VEGF-A и которая создана специально для ангиогенеза, готовят соответствующие соединения SQAP в необходимых концентрациях. В 1 день культивирования клеток специальные среды, каждая из которых содержит соединенияSQAP в соответствующих концентрациях и ДМСО (отрицательный контроль), добавляют к системам культивирования. Системы культивируют 30 мин и облучают с помощью 2 Гр кобальта 60. Затем на 4,7 и 10 дни культивирования среды заменяют вновь приготовленными специальными средами, содержащимиSQAP или ДМСО. Среды удаляют на 11 день культивирования, фиксируют 70% этанолом и образованные трубочки окрашивают с помощью антитела против CD31 человека. Окрашенные образцы фотографируют в оптическом микроскопе и с помощью анализа изображения рассчитывают количество образовавшихся трубочек. Показатель ангиогенеза рассчитывают в соответствии с инструкциями производителя. Результаты представлены на фиг. 10. По сравнению с контрольной группой, не обработанной соединением SQAP и/или радиационной терапией, группы, подвергнутые только воздействию излучения (2 Гр) и/или соединений SQAP, показывают более низкие показатели ангиогенеза. На фиг. 10 сокращение"РТ" ("RT") означает радиационную терапию. Кроме того, группы, обработанные соединениями SQAP,проявляют более низкие показатели, чем группа, обработанная только излучением 2 Гр. При комбинировании с облучением 2 Гр соединение SQAP C10:0 при конечных концентрациях 5, 10 и 20 мкМ, соединение SQAP C14:0 при конечных концентрациях 5, 10 и 20 мкМ, соединение SQAP C18:0 при конечных концентрациях 5, 10 и 20 мкМ, соединение SQAP С 22:0 при конечных концентрациях 5, 10 и 20 мкМ и соединение PSQAP С 18:0 при конечных концентрациях 5 и 10 мкМ и соединение PSQAP С 18:1 при конечных концентрациях 5 и 10 мкМ ингибируют образование трубочек концентрационнозависимым образом. Пример VIII. Испытание на токсичность.VIII-1. Тест Эймса. Оценку обратимой мутации (тест Эймса) проводят с использованием соединения SQAP C18:0. В качестве индикатора бактериальных штаммов используют пять штаммов, включающих два штамма Salmonella typhimurium, которые представляют собой основную пару мутантов замещения, один штамм Escherichia coli и два штамма Salmonella typhimurium, которые представляют собой мутанты со"сдвигом рамки". Такие штаммы предварительно культивируют в присутствии SQAP C18:0, переносят на агаровые чашки и культивируют на них в течение 48 ч, а затем подсчитывают на чашке число ревертантных колоний. Количество SQAP C18:0, добавленное в соответствующие чашки, составляет 2, 7, 21,62, 185, 556, 1667 и 5000 мкг. Независимо от присутствия или отсутствия смеси S9, добавленной во время предварительного культивирования (где смесь S9 представляет собой раствор, полученный путем добавления кофактора-1 к надосадочной фракции печеночного гомогената, полученного из печени самцов крыс, предварительно обработанных фенобарбиталом и 5,6-бензофлавоном), для всех штаммов число ревертантных колоний не повышается. На основании это факта мутагенность вещества оценивается как отрицательная.VIII-2. Микроядерный тест. Микроядерный тест проводят путем внутривенного введения крысам кальциевой соли SQAPC18:0. По пять самцов SD крыс приписывают к каждой из шести групп (1)-(6):(1) не получавшая лекарство группа;(6) группа положительного контроля (группа, получавшая 2 мг/кг митомицина С). Испытуемые растворы для групп (1)-(5) содержат нормальный физиологический раствор, содержащий 10% CREMOPHOR EL в качестве растворителей, и указанные выше дозы вводят крысам суммарно дважды в течение двух последовательных дней. Для группы положительного контроля указанную выше дозу вводят один раз. Приблизительно через 24 ч после введения готовят мазки костного мозга. Для каждого животного подсчитывают по две тысячи незрелых эритроцитов и рассчитывают долю незрелых эритроцитов,имеющих микроядра. В качестве показателя подавления пролиферации костного мозга рассчитывают незрелые эритроциты, содержащиеся в 1000 эритроцитах. Результаты показывают, что нет значительного увеличения доли микроядер в группе, которой вводилось испытуемое соединение, в сравнении с группой, не получавшей соединение. Кроме того, не обнаружено влияния на подавление пролиферации костного мозга в группе, которой вводилось испытуемое соединение. На основании этих фактов вещество оценивают как невызывающее хромосомную аберрацию в клетках костного мозга.VIII-3. Испытание токсичности единичной дозы. С использованием соединения SQAP C18:0 проводят тест на острую токсичность на крысах. По пять самок и по пять самцов SD крыс возрастом 5-6 недель приписывают к каждой из групп (1)-(7):(1) не получавшая лекарство группа;(7) группа, получавшая 800 мг/кг SQAP C18:0. Испытуемый раствор для (1) и (4)-(7) групп содержит нормальный физиологический раствор, содержащий 10% CREMOPHOR EL, для (2) группы содержит нормальный физиологический раствор, содержащий 2,5% CREMOPHOR EL, и для группы (3) содержит нормальный физиологический раствор,содержащий 5% CREMOPHOR EL, в качестве растворителей. Указанные испытуемые растворы вводят через хвостовую вену крысы и наблюдают клинические признаки в течение двух дней, включая дату введения. Ни одно животное не умерло во время периода испытания, и смертельная доза, по оценкам, составляет больше 800 мг/кг. Другие преимущества и модификации будут легко понятны специалистам в данной области техники. Соответственно, очевидно, что настоящее изобретение в своем наиболее широком аспекте не ограничено конкретными деталями и типичными вариантами осуществления, представленными и описанными выше. Соответственно различные модификации могут быть выполнены без отступления от сути или объема общей изобретательской концепции, которая определена в прилагаемой формуле изобретения и в ее эквивалентах. Соединение настоящего изобретения представляет собой новое вещество и, как описано выше, обладает замечательным радиосенсибилизирующим эффектом и противоопухолевым эффектом. Соединение настоящего изобретения может быть получено с высокой чистотой простым синтетическим способом. Кроме того, соединение настоящего изобретения структурно стабильно и имеет высокую растворимость в воде. Соответственно соединение имеет преимущества при его производстве и при приготовлении препаратов, когда его используют в качестве лекарственного средства, а также имеет преимущества при применении после хранения в виде соединения и лекарства. Кроме того, соединение характеризуется низкой токсичностью. Соответственно соединение имеет большие преимущества в качестве лекарственного средства, которое вводят человеку и другим животным в течение короткого или длительного периода. Настоящее изобретение финансируется специальными координационными фондами по стимулированию науки и технологии, созданными Министерством образования, культуры, спорта, науки и технологии. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Сульфохиновозилацилпропандиольное соединение, представленное общей формулой (I) где R1 представляет собой C1-C22 ацильный остаток жирной кислоты и М представляет собой атом водорода или ион натрия либо кальция, или его фармацевтически приемлемые соли. 2. Лекарственное средство, содержащее в качестве активного ингредиента по меньшей мере один активный ингредиент, выбранный из группы, включающей сульфохиновозилацилпропандиольное соединение по п.1, представленное общей формулой (I), и его фармацевтически приемлемые соли. 3. Лекарственное средство по п.2, которое представляет собой радиосенсибилизатор. 4. Лекарственное средство по п.2, которое представляет собой противоопухолевый агент.
МПК / Метки
МПК: C07H 15/10, A61K 41/00, A61K 31/7028, C07H 15/06, A61P 35/00
Метки: применение, сахара, лекарственного, сульфированное, соединение, средства, качестве, новое
Код ссылки
<a href="https://eas.patents.su/24-16931-novoe-sulfirovannoe-soedinenie-sahara-i-ego-primenenie-v-kachestve-lekarstvennogo-sredstva.html" rel="bookmark" title="База патентов Евразийского Союза">Новое сульфированное соединение сахара и его применение в качестве лекарственного средства</a>
Производные имидазолов, способ их получения и их применение в качестве лекарственного средства

Номер патента: 10392
Опубликовано: 29.08.2008
Авторы: Либератор Анн-Мари, Бигг Денни, Пон Доминик
МПК: C07D 233/64, A61K 31/4164, A61P 35/00...
Метки: качестве, получения, способ, лекарственного, имидазолов, производные, средства, применение
Формула / Реферат:
1. Соединение формулы (I) в форме рацемата, энантиомера или в виде любых сочетаний этих форм, в которой X обозначает один или несколько заместителей, одинаковых или различных, выбранных из Н и галогена; Y обозначает -О- или -S-; А обозначает Н или (C1-C6)алкил; Z обозначает один или несколько заместителей, одинаковых или различных, выбранных из (C1-C6)алкила; арила; арил(C1-C6)алкила; гетероарила; Z1-Z'1; NRN-C(O)-Z'2 или Z2-Z'2; Z1 обозначает...
Стероиды, замещенные в положении 11, способ их получения, их применение в качестве лекарственного средства и содержащие их фармацевтические композиции

Номер патента: 1868
Опубликовано: 22.10.2001
Авторы: Тетш Жан-Жорж, Буали Иамина, Ван Де Вельд Патрик, Ник Франсуа
МПК: A61K 31/565, C07J 41/00, A61P 19/10...
Метки: средства, лекарственного, фармацевтические, положении, содержащие, замещенные, качестве, стероиды, способ, композиции, получения, применение
Формула / Реферат:
1. Соединения общей формулы в которой n - целое число, равно 2 или 3, либо R1 и R2, идентичные или разные, означают атом водорода или алкил, содержащий от 1 до 4 атомов углерода, либо R1 и R2 образуют вместе с атомом азота, с которым они связаны, моно- или полициклический гетероцикл, имеющий от 5 до 15 звеньев, ароматический или не ароматический, содержащий, при необходимости, от 1 до 3 дополнительных гетероатомов, выбираемых из кислорода,...
Сукцинатная и малонатная соли транс-4-((1r,3s )-6 хлор-3-фенилиндан -1-ил)-1,2,2-триметилпиперазина и их применение в качестве лекарственного средства

Номер патента: 14641
Опубликовано: 30.12.2010
Авторы: Ринггар Лоне Мюнк, Лопес Де Диего Хейди, Хауэллз Марк, Люнгсе Ларс Оле, Даль Аллан Карстен, Нильсен Оле, Бан-Андерсен Бенни, Сване Хенрик
МПК: A61K 31/495, C07C 25/22, A61P 25/18...
Метки: средства, соли, применение, малонатная, лекарственного, хлор-3-фенилиндан, качестве, сукцинатная, 1-ил)-1,2,2-триметилпиперазина, транс-4-((1r,3s
Формула / Реферат:
1. Сукцинатная соль или малонатная соль транс-4-((1R,3S)-6-хлор-3-фенилиндан-1-ил)-1,2,2-триметилпиперазина формулы (I)2. Сукцинатная соль по п.1, которая представляет собой гидросукцинатную соль соединения формулы (I).3. Кристаллическая гидросукцинатная соль соединения I, определенного в п.1.4. Соль по п.3, которая представляет собой кристаллическую альфа-форму.5. Соль по п.3 или 4, кристаллическую форму которой характеризуют порошковой...
Калийная соль 5-[4-[2-(n-метил-n-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-диона и ее применение в качестве противодиабетического лекарственного средства

Номер патента: 5110
Опубликовано: 28.10.2004
Автор: Миллан Майкл
МПК: A61P 3/10, A61K 31/4439, C07D 417/12...
Метки: соль, калийная, качестве, средства, применение, противодиабетического, 5-[4-[2-(n-метил-n-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-диона, лекарственного
Формула / Реферат:
1. Калийная соль 5-[4-[2-(N-метил-N-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-диона или ее фармацевтически приемлемый сольват, отличающаяся тем, что характеризуется одним или более из (i) инфракрасным спектром практически в соответствии с фиг.1; (ii) рамановским спектром практически в соответствии с фиг. 2; (iii) порошковой рентгенограммой (XRPD) практически в соответствии с таблицей и фиг. 3; (iv) спектром твердофазного 13C ЯМР практически...
Метансульфонат этилового эфира 3-[(2 -{[4-(гексилоксикарбониламиноиминометил)фениламино]метил}-1- метил-1н-бензимидазол-5-карбонил)пиридин-2-иламино]пропионовой кислоты и его применение в качестве лекарственного средства

Номер патента: 9736
Опубликовано: 28.02.2008
Авторы: Шмид Рольф, Зигер Петер, Зоботта Райнер
МПК: C07D 401/12, A61P 7/02, A61K 31/4439...
Метки: 4-(гексилоксикарбониламиноиминометил)фениламино]метил}-1, 3-[(2, эфира, метил-1н-бензимидазол-5-карбонил)пиридин-2-иламино]пропионовой, кислоты, средства, лекарственного, качестве, применение, этилового, метансульфонат
Формула / Реферат:
1. Метансульфонат этилового эфира 3-[(2-{[4-(гексилоксикарбониламиноиминометил)фениламино]метил}-1-метил-1H-бензимидазол-5-карбонил)пиридин-2-иламино]пропионовой кислоты (BIBR 1048 MS) в кристаллической форме, характеризующийся температурой плавления Тпл, равной 180+3шС (форма I) (определенной с помощью дифференциальной сканирующей калориметрии на основе анализа с использованием максимума пика при скорости нагрева 10шС/мин). 2. Метансульфонат...
Предыдущий патент: Узел клапана, скважинный инструмент и способы управления потоком текучей среды и ее циркуляции
Следующий патент: Преобразователь электромагнитного излучения (варианты)
Случайный патент: Способ производства фосфорной кислоты