Твёрдый фармацевтический дозированный состав
Номер патента: 14446
Опубликовано: 30.12.2010
Авторы: Липольд Бернд, Берндль Гунтер, Брайтенбах Йорг, Розенберг Йорг, Райнхольд Ульрих, Алани Ламан, Гхош Соумоджит


















Формула / Реферат
1. Способ лечения ВИЧ/СПИДа, включающий введение субъекту, нуждающемуся в таком лечении, твердой фармацевтической дозированной формы, содержащей нерастворенную форму лопинавира в терапевтически эффективном количестве, причем указанная дозированная форма принимается указанным пациентом без еды или в условиях голодной выдержки.
2. Способ по п.1, в котором указанная дозированная форма содержит твердый раствор или твердую дисперсию лопинавира в матрице, причем указанная матрица содержит по меньшей мере один фармацевтически приемлемый водорастворимый полимер и по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество, причем указанное поверхностно-активное вещество имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10.
3. Способ по п.2, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера имеет температуру стеклования Тс по меньшей мере 50°С.
4. Способ по п.3, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер включает сополимер N-винил пирролидона и винил ацетата и указанное фармацевтически приемлемое поверхностно-активное вещество представляет собой моносложные эфиры жирной кислоты сорбитана.
5. Способ по п.3, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер включает коповидон и указанное поверхностно-активное вещество представляет собой монолаурат сорбитана.
6. Способ по п.5, в котором указанная дозированная форма дополнительно содержит ритонавир.
7. Способ по п.1, в котором указанная дозированная форма содержит твердый раствор лопинавира в матрице, где указанная матрица содержит по меньшей мере один фармацевтически приемлемый водорастворимый полимер и по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество, причем указанное фармацевтически приемлемое поверхностно-активное вещество имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10.
8. Способ по п.7, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера имеет температуру стеклования Тс по меньшей мере 50°С и каждое из указанного по меньшей мере одного фармацевтически приемлемого поверхностно-активного вещества имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10.
9. Способ по п.8, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера выбран из группы, состоящей из гомополимера N-виниллактама, сополимера N-виниллактама, сложного эфира целлюлозы, простого эфира целлюлозы, оксида полиалкилена, полиакрилата, полиметакрилата, полиакриламида, поливинилового спирта, полимера винил ацетата, олигосахарида и полисахарида, и где каждое из указанного фармацевтически приемлемого поверхностно-активного вещества выбрано из группы, состоящей из простого эфира полиоксиэтиленалкила, полиоксиэтилен алкиларилового простого эфира, сложного эфира полиэтиленгликолевой жирной кислоты, алкиленгликолевого моносложного эфира жирной кислоты, сложного эфира жирной кислоты сахарозы и моносложного эфира жирной кислоты сорбитана.
10. Способ по п.8, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера выбран из группы, состоящей из гомополимера N-винилпирролидона, сополимера N-винилпирролидона, сополимера N-винилпирролидона и винилацетата, сополимера N-винилпирролидона или винилпропионата, поливинилпирролидона, метилцеллюлозы, этилцеллюлозы, гидроксиалкилцеллюлозы, гидроксипропилцеллюлозы, гидроксиалкилалкилцеллюлозы, гидроксипропилметилцеллюлозы, фталата целлюлозы, сукцината целлюлозы, фталата ацетилцеллюлозы, фталата гидроксипропилметилцеллюлозы, сукцината гидроксипропилметилцеллюлозы, ацетат сукцината гидроксипропилметилцеллюлозы, полиэтиленоксида, полипропиленоксида, сополимера оксида этилена и оксида пропилена, метакриловой кислоты/сополимеры этилакрилата, метакриловой кислоты/сополимера метилметакрилата, бутилметакрилата/сополимера 2-диметиламиноэтил метакрилата, поли(гидроксиалкил акрилата), поли(гидроксиалкил метакрилата), сополимера винилацетата и кротоновой кислоты, частично гидролизованного поливинилацетата, каррагинана, галактоманнана и ксантановой смолы, и где каждое из указанного фармацевтически приемлемого поверхностно-активного вещества выбрано из группы, состоящей из полиоксиэтилен (3) лаурилового эфира, полиоксиэтилен (5) цетилового эфира, полиоксиэтилен (2) стеарилового эфира, полиоксиэтилен (5) стеарилового эфира, полиоксиэтилен (2) нонилфенилового эфира, полиоксиэтилен (3) нонилфенилового эфира, полиоксиэтилен (4) нонилфенилового эфира, полиоксиэтилен (3) октилфенилового эфира, ПЭГ-200 монолаурата, ПЭГ-200 дилаурата, ПЭГ-300 дилаурата, ПЭГ-400 дилаурата, ПЭГ-300 дистеарата, ПЭГ-300 диолеата, монолаурата пропиленгликоля, моностеарата сахарозы, дистеарата сахарозы, монолаурата сахарозы, дилаурата сахарозы, монолаурата сорбитана, моноолеата сорбитана, монопальмитата сорбитана, стеарата сорбитана.
11. Способ по п.8, в котором указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество составляет по меньшей мере 50 мас.% от общего количества поверхностно-активных веществ, содержащихся в дозированной форме.
12. Способ по п.8, в котором указанная дозированная форма содержит 50-85 мас.% от дозированной формы указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера и 2-20 мас.% от дозированной формы указанного по меньшей мере одного фармацевтически приемлемого поверхностно-активного вещества.
13. Способ по п.12, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер включает сополимер N-винилпирролидона и винилацетата и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество включает моносложный эфир жирной кислоты сорбитана.
14. Способ по п.12, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер представляет собой коповидон и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество представляет собой монолаурат сорбитана.
15. Способ по п.14, в котором указанная дозированная форма дополнительно содержит ритонавир.
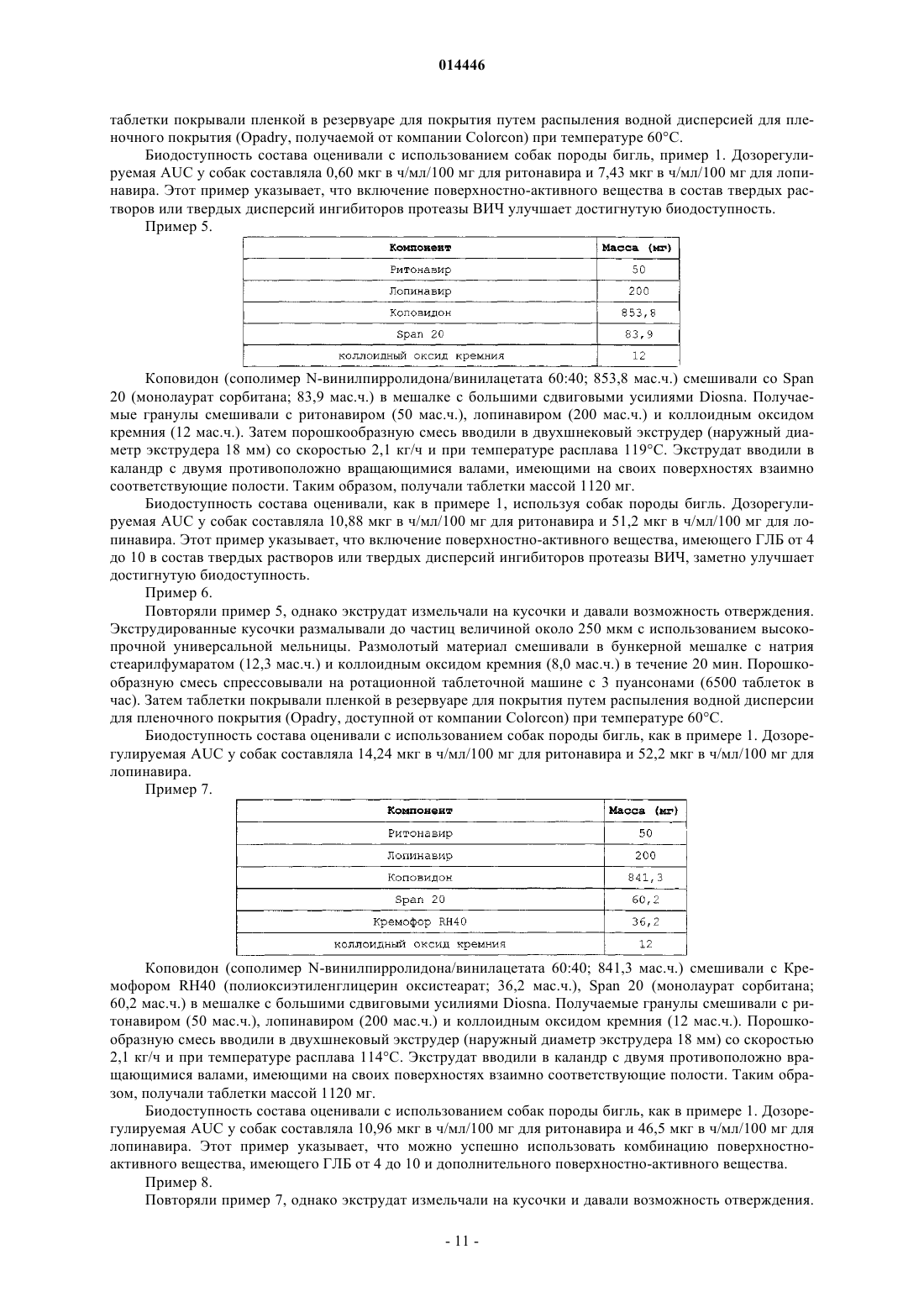
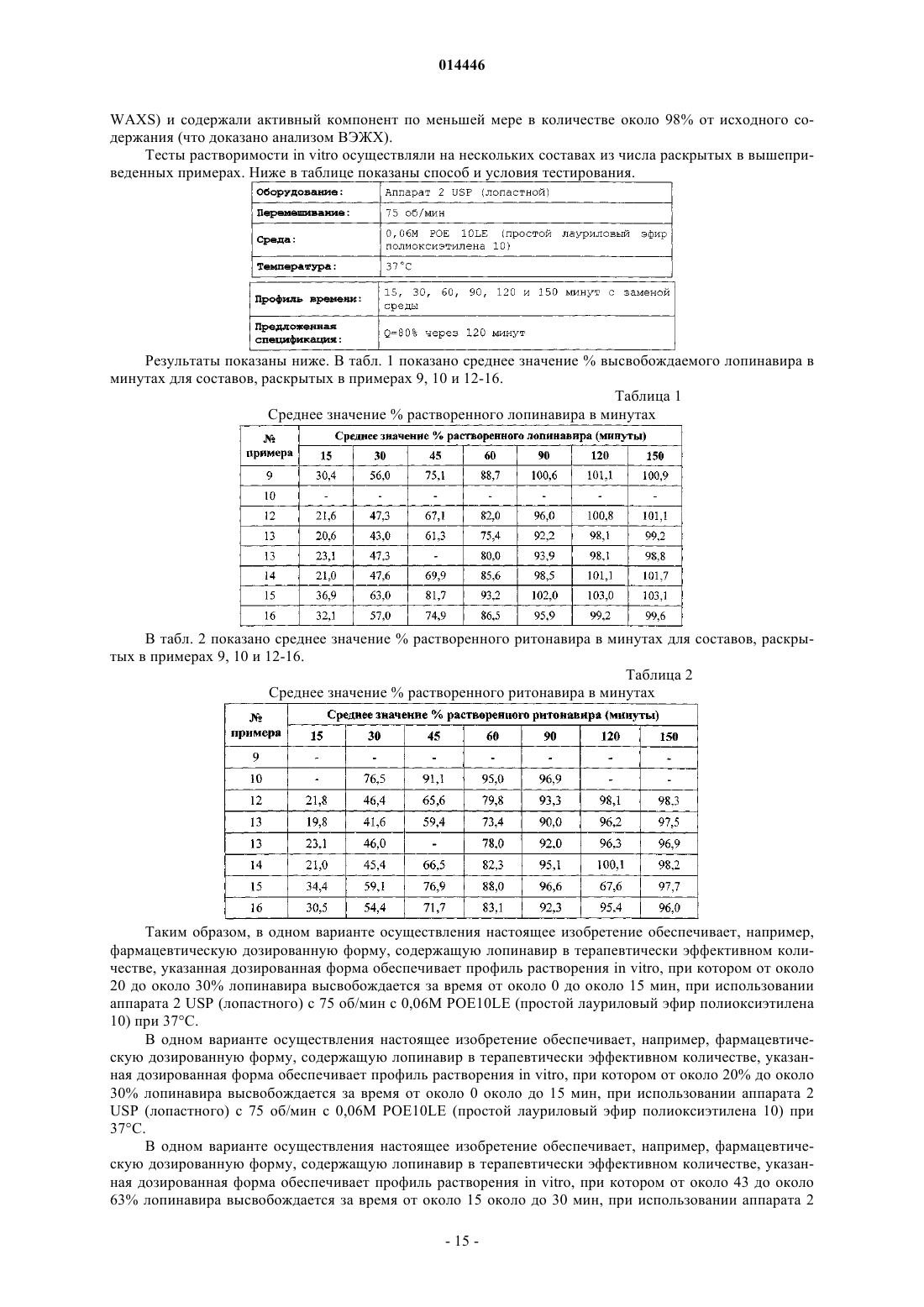
16. Способ по п.1, в котором при растворении указанной дозированной формы in vitro при использовании аппарата 2 USP (лопастного) с 75 об/мин с 0,06М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37°С, от около 20 до около 30% лопинавира высвобождается за время от около 0 до около 15 мин, от около 43 до около 63% лопинавира высвобождается за время от около 15 около до 30 мин, от около 61,3 до около 81,7% лопинавира высвобождается за время от около 30 около до 45 мин, от около 75,4 до около 93,2% лопинавира высвобождается за время от около 45 до около 60 мин.
17. Способ по п.1, в котором указанная дозированная форма дополнительно содержит ритонавир, причем при растворении указанной дозированной формы in vitro при использовании аппарата 2 USP (лопастного) с 75 об/мин с 0,06М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37°С, от около 19,8 до около 34,4% ритонавира высвобождается за время от около 0 до около 15 мин, от около 41,6 до около 76,5% ритонавира высвобождается за время от около 15 до около 30 мин, от около 59,4 до около 91,1% ритонавира высвобождается за время от около 30 до около 45 мин, от около 73,4 до около 95% ритонавира высвобождается за время от около 45 до около 60 мин.
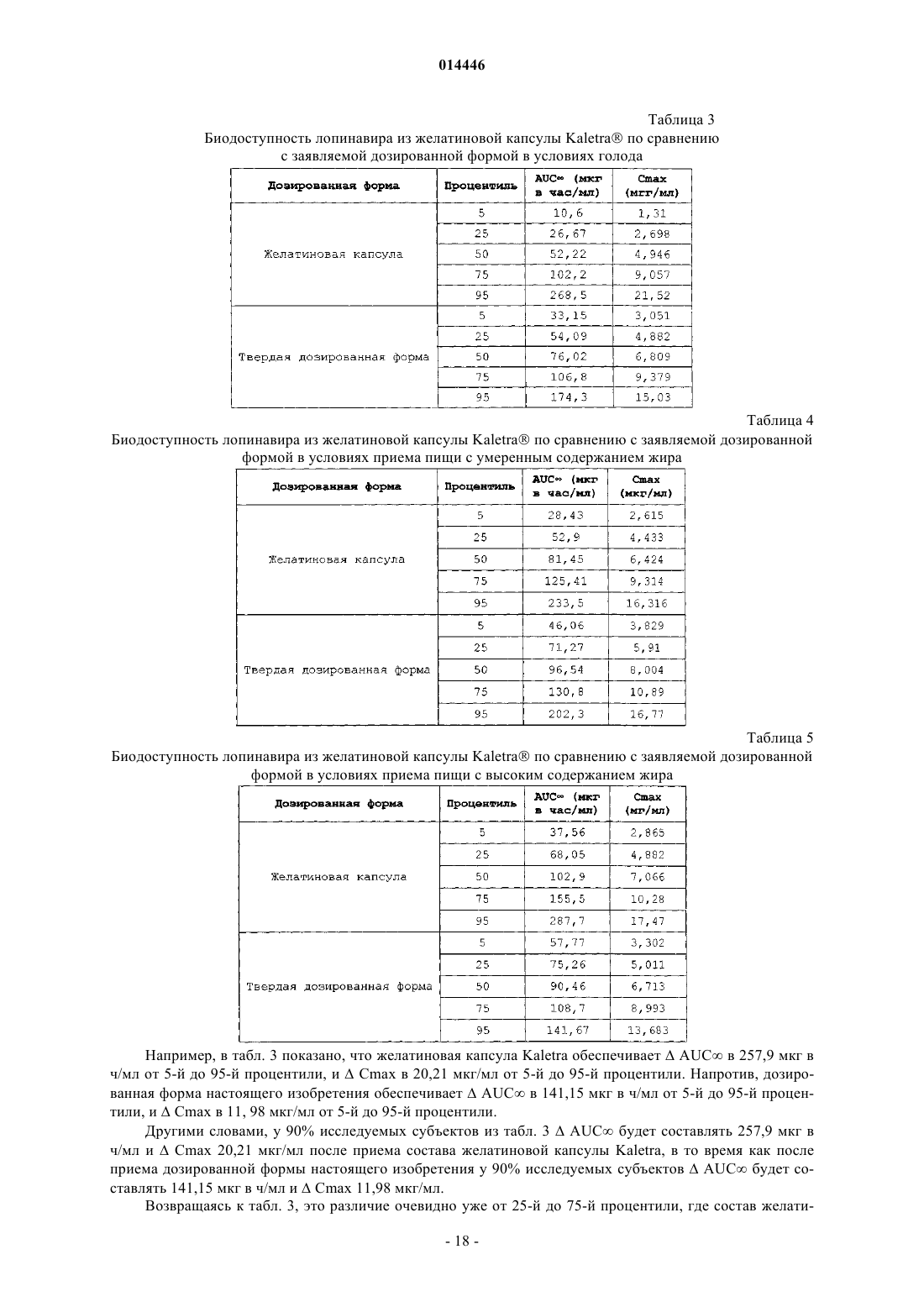
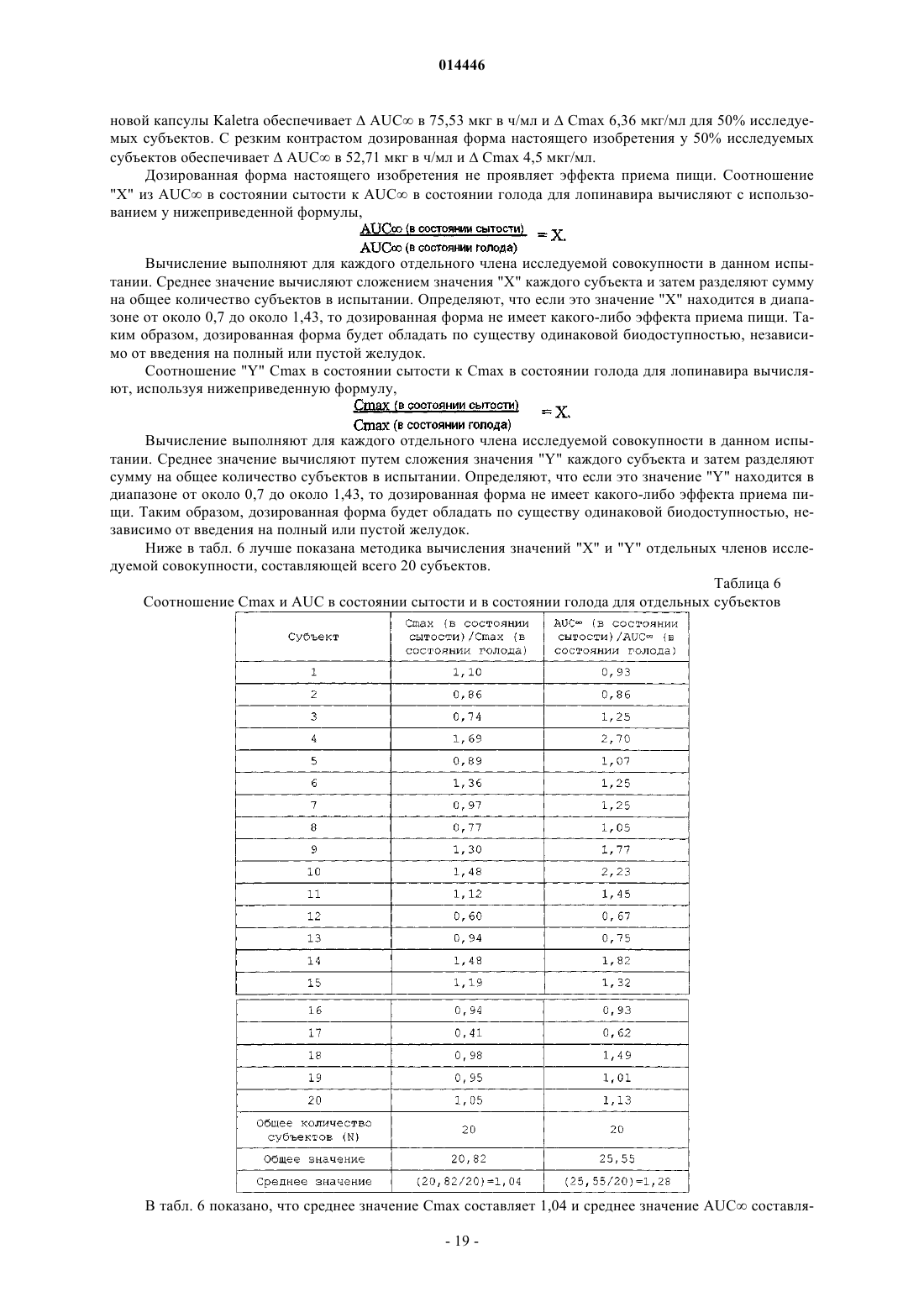
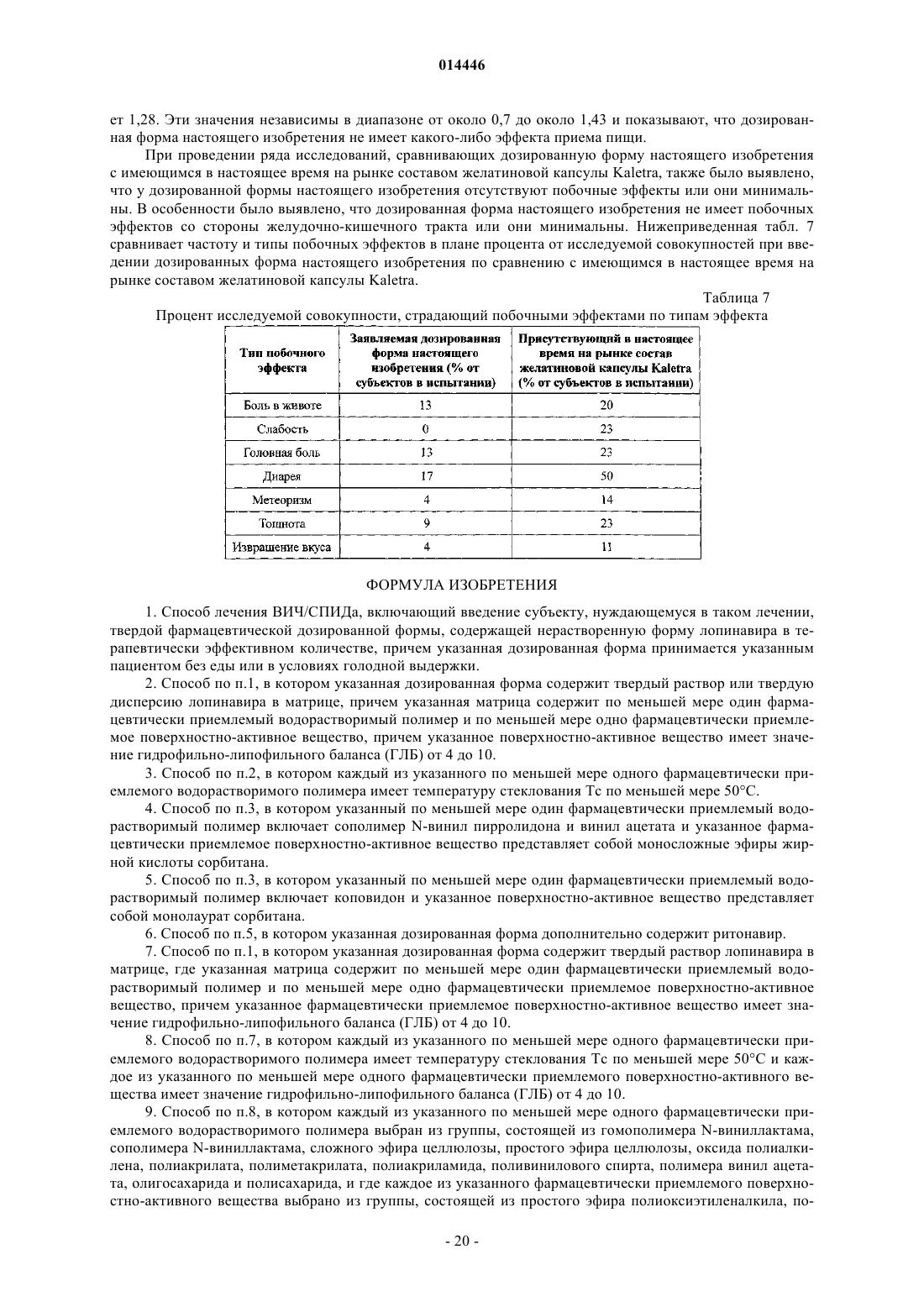
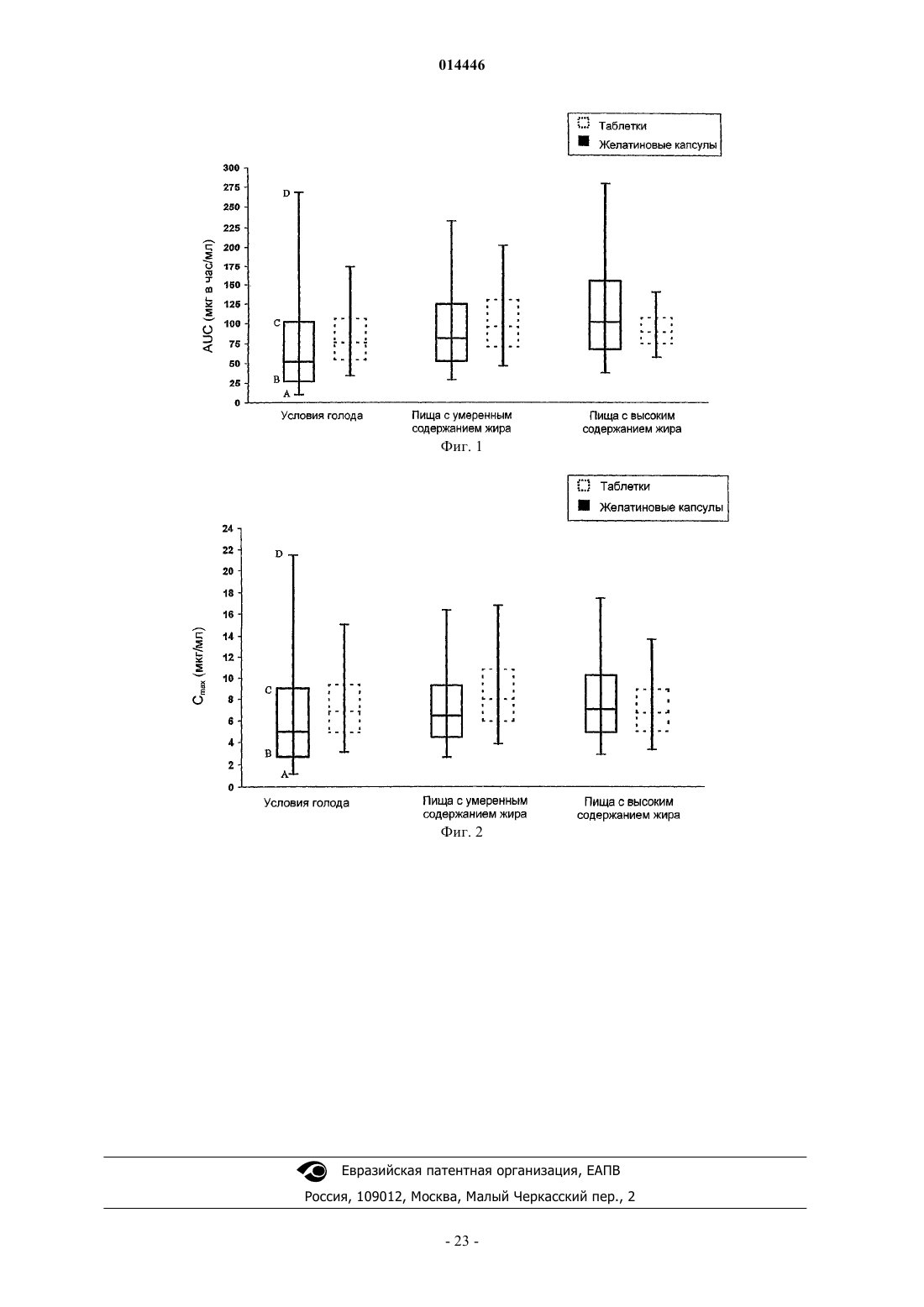
18. Способ по п.1, в котором после введения однократной дозы ингибитора протеазы каждому члену исследуемой совокупности отношение среднего значения AUC¥ (в состоянии сытости) к AUC¥ (в состоянии голода) для членов исследуемой совокупности находится в диапазоне от около 0,7 до около 1,43.
19. Способ по п.1, в котором после введения однократной дозы ингибитора протеазы каждому члену исследуемой совокупности отношение среднего значения Cmax (в состоянии сытости) к Cmax (в состоянии голода) для членов исследуемой совокупности находится в диапазоне от около 0,7 до около 1,43.
20. Способ по п.1, в котором после введения указанной дозированной формы каждому члену исследуемой совокупности в условиях голода, различие в показателе AUC¥ лопинавира между 95-й процентилью и 5-й процентилью исследуемой совокупности составляет меньше чем 170.
21. Способ по п.1, в котором после введения указанной дозированной формы каждому члену исследуемой совокупности в условиях голода различие в показателе Cmax лопинавира между 95-й процентилью и 5-й процентилью исследуемой совокупности составляет меньше чем 15.
22. Способ по п.1, в котором после введения указанной дозированной формы каждому члену исследуемой совокупности в условиях голода, от 5-й процентили до 95-й процентили значения AUC¥ исследуемой совокупности находятся в диапазоне от около 33 до около 175 мкг в ч/мл.
23. Способ по п.1, в котором указанная нерастворимая форма представляет собой аморфную форму.
24. Способ по п.7, в котором указанный водорастворимый полимер имеет кажущуюся вязкость при растворении в 2% (мас./об.) водном растворе при 20°С, составляющую от около 1 до около 5000 мПа×с.
25. Способ по п.7, в котором после хранения в течение 6 недель при 40°С и влажности 75% указанная дозированная форма содержит лопинавир в количестве по меньшей мере 98% от исходного содержания.
26. Способ лечения пациента с ВИЧ, включающий введение такому пациенту независимо от приема пищи твердой фармацевтической дозированной формы, причем указанная дозированная форма содержит по меньшей мере один ингибитор ВИЧ протеазы, по меньшей мере один фармацевтически приемлемый водорастворимый полимер и по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество, в котором указанный ингибитор ВИЧ протеазы представляет собой лопинавир и указанное поверхностно-активное вещество имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10.
27. Способ по п.26, в котором указанная дозированная форма содержит твердый раствор или твердую дисперсию указанного по меньшей мере одного ингибитора ВИЧ протеазы в матрице, где указанная матрица содержит указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество.
28. Способ по п.27, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера имеет температуру стеклования Тс по меньшей мере 50°С и каждое из указанного по меньшей мере одного фармацевтически приемлемого поверхностно-активного вещества имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10.
29. Способ по п.28, в котором указанная дозированная форма содержит 50-85 мас.% от дозированной формы указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера и 2-20 мас.% от дозированной формы указанного по меньшей мере одного фармацевтически приемлемого поверхностно-активного вещества.
30. Способ по п.29, в котором указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество составляет по меньшей мере 50 мас.% от общего количества поверхностно-активных веществ, содержащихся в дозированной форме.
31. Способ по п.29, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер включает сополимер N-винилпирролидона и винилацетата и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество включает моносложный эфир жирной кислоты сорбитана.
32. Способ по п.29, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер представляет собой коповидон и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество представляет собой монолаурат сорбитана.
33. Способ по п.32, в котором указанный твердый раствор или твердая дисперсия представляет собой твердый раствор.
34. Способ по п.33, в котором указанная дозированная форма дополнительно содержит ритонавир.
Текст
ТВРДЫЙ ФАРМАЦЕВТИЧЕСКИЙ ДОЗИРОВАННЫЙ СОСТАВ Настоящее изобретение обеспечивает фармацевтическую дозированную форму, более конкретно фармацевтическую дозированную форму, содержащую ингибитор протеазы ВИЧ. 014446 Область изобретения Настоящее изобретение относится к фармацевтическому дозированному составу, более конкретно относится к фармацевтическому дозированному составу, содержащему ингибитор протеазы ВИЧ. Уровень техники изобретения Миллионы людей во всем мире больны ВИЧ/СПИДом, и каждый год вероятность заражения получают миллионы других людей. В настоящее время для лечения ВИЧ/СПИДа доступны многочисленные лекарства, включающие в себя ингибиторы протеазы ВИЧ (ИП), нуклеозидные/нуклеотидные ингибиторы обратной транскриптазы (НИОТ) и ненуклеозидные ингибиторы обратной транскриптазы (ННИОТ). Наиболее современные схемы лечения требуют комбинации по меньшей мере трех лекарств, чаще всего двух НИОТ и/или ИП либо ННИОТ. ИП являются плохо растворимыми и представляют большую трудность при создании составов. Первоначально ИП обеспечивались в виде жидких составов, в которых был растворен компонент ИП. В настоящее время наиболее широко используемыми дозированными формами ИП являются желатиновые капсулы, содержащие раствор-заполнитель, в котором растворен активный компонент. Растворызаполнители, необходимые для растворения ИП, часто содержат наполнители, вызывающие дискомфорт или раздражающие желудочно-кишечный тракт. Кроме того, в этих дозированных формах можно растворять только ограниченное количество ИП, что таким образом ограничивает количество ИП, помещаемого в каждую желатиновую капсулу. Для получения необходимой дозы отдельного ИП больной должен принимать несколько желатиновых капсул за любое заданное время приема, который повторяют несколько раз в день. Как упомянуто выше, лечение больных ВИЧ включает в себя множество лекарств, в число которых обычно входит ИП. Кроме того, для этих больных часто необходимы дополнительные лекарства, такие как антибиотики и гиполипидемические средства для контроля оппортунистических инфекций и других заболеваний или состояний, которыми они могут быть подвержены. Следовательно, эти больные в течение одного дня могут принимать чрезвычайно большое количество лекарств различных дозированных форм. Такие схемы лечения дополнительно осложняются тем фактом, что для некоторых из дозированных форм (включающие в себя некоторые ИП) необходимо хранение в прохладных условиях для предотвращения распада активных компонентов. Для субъектов, проживающих в экономически неразвитых или развивающихся странах, где холодильники в домашнем хозяйстве не распространены, такие условия хранения представляют особенно сложную дилемму. Также наблюдалась вариабельность уровней в крови активного компонента у различных субъектов и даже у одного и того же субъекта после введения ИП из желатиновых капсул. Таким образом, у некоторых больных, получающих лечение, уровни ИП в крови могут быть очень высокими или очень низкими. В свою очередь, у больных с высоким уровнем лекарственного средства в крови это может приводить к нежелательным побочным эффектам, или у больных, имеющих низкие уровни в крови лекарственного средства, лечение может стать менее эффективным или неэффективным. Для повышения биодоступности ИП больным с целью повышения общей биодоступности активного компонента рекомендовано принимать состав в желатиновой капсуле после еды. Биодоступность может дополнительно варьировать в зависимости от содержания жира в каждом приеме пищи. К сожалению, многие больные не всегда придерживаются этого порядка из-за сложности схем своего лечения или по другим причинам. Часто больные осуществляют прием лекарства натощак, что приводит к низкой биодоступности лекарственного средства и, возможно, к неэффективности лечения. В этой связи желательно получить дозированную форму ИП, в которой снижены или отсутствуют побочные эффекты со стороны желудочно-кишечного тракта. Также желательно получить такую дозированную форму, которая для уменьшения лекарственной нагрузки на больного может вмещать большее количество активного компонента. Кроме того, желательно получить дозированную форму, которая обеспечивает малую вариабельность уровня в крови ИП у субъекта и в пределах популяции больных. Другим желательным признаком будет дозированная форма, которая обеспечивает сходные уровни ИП в крови независимо от времени приема лекарства по отношению к еде. Еще одним желательным признаком будет дозированная форма, которая не требует охлаждения для предотвращения распада ИП. Сущность изобретения Было неожиданно выявлено, что при создании состава нерастворенной формы ИП (в частности, лопинавира и комбинации лопинавира/ритонавира) в фармацевтической дозированной форме можно преодолеть все вышеупомянутые недостатки, связанные с дозированными формами, содержащими растворенный ИП. В частности, фармацевтические дозированные формы, содержащие нерастворенный ИП,снижают лекарственную нагрузку у больных ВИЧ, в основном в силу возможного увеличения содержания лекарственного средства в этих составах. Дополнительно, такие составы способны храниться при комнатной температуре и не требуют охлаждения. Кроме того, эти составы обеспечивают более постоянный уровень ИП в крови у больных, получающих такое лечение, что способствует обеспечению эффективной терапевтической пользы и снижает побочные эффекты. Дополнительно, с составом согласно настоящему изобретению можно достигнуть таких постоянных уровней в крови независимо от приема пищи больным и от типа пищи. Считается, что в настоящем изобретении впервые была создана нерас-1 014446 творенная форма лопинавира в твердой дозированной форме. Учитывая преимущества, присущие такому составу, это представляет дальнейший прорыв в лечении ВИЧ, который будет способствовать облегчению сложных схем лечения, назначаемых в настоящее время больным ВИЧ. Описание чертежей Фиг. 1 показывает графики с прямоугольниками (нижние и верхние части) и штрихами (5 и 95 процентилей) для области под кривой AUC лопинавира в условиях различных приемов пищи; фиг. 2 показывает графики с прямоугольниками (нижние и верхние части) и штрихами (5 и 95 процентилей) для максимальной концентрации Cmax лопинавира в условиях различных приемов пищи. Подробное описание изобретения Определения. Термин "AUC" означает область под кривой концентрации от времени (AUC), экстраполируемую к бесконечности, или область под кривой AUC последней отмеченной точки времени + (последняя измеренная концентрация/постоянную скорости выведения). Термин "Cmax" означает наблюдаемую максимальную концентрацию активного компонента в плазме. Используемый в настоящем изобретении термин "фармацевтически приемлемый" определяют как функциональные группы или соединения, которые в рамках обоснованного медицинского заключения подходят для использования при контакте с тканями человека и низших млекопитающих без нежелательной токсичности, раздражения, аллергической реакции и т.п. и имеют соизмеримое приемлемое соотношение пользы/риска. Термин "процент по массе", или "массовый процент", или "мас.%" определяют как массу отдельного компонента в соединении, разделенную на общую массу всех компонентов в составе и затем умноженную на 100. В некоторых случаях, когда состав имеет внешнее покрытие, массу покрытия можно или включать в общую массу, или исключать из нее. Выражение "голодная выдержка/состояние или условие голода" в общем определяют как 10 ч воздержания от приема пищи до приема лекарства и 4 ч после приема лекарства, вместе с тем специалисты в данной области техники признают другие различные режимы, которые также можно считать голоданием или состоянием голода. Выражение "условие приема умеренно жирной пищи" определяют как получение пищи, калорийность которой составляет около 500-600 ккал, в которой 20-30% калорий получают из жиров, принимаемых примерно за 30 мин до приема лекарства. Выражение "условие приема пищи с высоким содержанием жиров" определяют как получение пищи, калорийность которой составляет около 1000 килокалорий, в которой 50-55% калорий получают из жиров, принимаемых примерно за 30 мин до приема лекарства, и в настоящем изобретении используется по отношению к "состоянию сытости", вместе с тем специалисты в данной области техники признают различные условия приема пищи, которые также считаются состоянием сытости. Термин "твердый раствор" определяют как систему в твердом состоянии, в которой лекарственное средство диспергировано на молекулярном уровне во всей матрице таким образом, что вся система является химически и физически однородной или гомогенной. Термин "твердая дисперсия" определяют как систему, имеющую частицы малого размера, обычно менее 400 мкм, более обычный размер которых составляет менее 100 мкм и наиболее обычный размер составляет менее 10 мкм, одной фазы, диспергированные в другой фазе (в фазе носителя). Подходящие ИП для использования согласно настоящему изобретению включаютв себя (2S,3S,5S)2-(2,6-диметилфеноксиацетил)амино-3-гидрокси-5-[2S-(1-тетрагидропиримид-2-онил)-3 метилбутаноил]амино-1,6-дифенилгексан (АВТ-378; лопинавир) или комбинацию из (2S,3S,5S)-2-(2,6 диметилфеноксиацетил)амино-3-гидрокси-5-[2S-(1-тетрагидропиримид-2-онил)-3-метилбутаноил]амино 1,6-дифенилгексана (АВТ-378; лопинавира) и (2S,3S,5S)-5-(N-(N-(N-метил-N-2-изопропил-4 тиазолил)метил)амино)карбонил)-L-валинил)амино-2-(N-5-тиазолил)метоксикарбонил)амино)амино 1,6-дифенил-3-гидроксигексана (ритонавира), используемые или для действия ИП, или другим образом,таким как в случае с ритонавиром, который иногда можно применять в качестве ингибитора цитохромР 450 монооксигеназы (иначе называемого "рК-бустером"). Предпочтительными ИП являются лопинавир или комбинация из лопинавира и ритонавира. В общем, дозированные формы настоящего изобретения будут содержать терапевтически эффективное количество по меньшей мере одного ИП. Конкретный терапевтически эффективный уровень дозы для любого конкретного больного будет зависеть от множества факторов, включающих в себя тяжесть заболевания; от действия конкретного используемого соединения; конкретной используемой композиции; возраста, массы тела, общего состояния здоровья, пола и питания больного; от времени введения и скорости выведения используемого конкретного соединения; от продолжительности лечения; лекарственных средств, используемых в комбинации, или симптоматических средств с конкретным используемым соединением; и от других факторов, известных рядовому специалисту в области медицины. Например, в данной области техники известно, что для достижения желательного терапевтического эффекта начинают с уровня дозы соединения ниже требуемого и постепенно увеличивают дозировку до достиже-2 014446 ния желательного эффекта. Вместе с тем, обычно фармацевтическая дозированная форма настоящего изобретения будет содержать ингибитор протеазы ВИЧ или комбинацию ингибиторов протеазы ВИЧ в количестве от около 5 до около 30% от общей массы дозированной формы, предпочтительно от около 10 до около 25% от общей массы дозированной формы. Предпочтительно, дозированная форма будет содержать ИП в количестве между около 10 до около 1500 мг. Наиболее предпочтительно дозированная форма будет содержать лопинавир и ритонавир в примерном соотношении соответственно 4:1. Предпочтительная доза лопинавира и ритонавира составляет соответственно 400 и 100 мг, которые можно равномерно разделять между многократными дозами дозированной формы, предпочтительно между двумя. Подразумевается, что многократные дозы, обычно две, можно давать в заданный день. Обычно фармацевтические дозированные формы, обеспечиваемые настоящим изобретением, будут содержать "нерастворенное" ИП. В отличие от существующих желатиновых капсул, наполненных растворенным в растворителе ИП, нерастворенные ИП, согласно использованию в настоящем изобретении,означают, что ИП находятся в твердой форме и не растворены в жидком носителе в его конечной дозированной форме. Например, твердые формы ИП могут включать в себя кристаллические, тонкодисперсные кристаллические формы, кристаллические наночастицы, аморфные, тонкодисперсные аморфные формы, аморфные наночастицы или предпочтительно аморфные твердые формы ИП. Для использования согласно настоящему изобретению подходят многие фармацевтические дозированные формы; выбор этих форм, основанный на свойствах дозированных форм, обеспечиваемых настоящим изобретением, известен рядовому специалисту в этой области техники. Например, вводимые перорально твердые дозированные формы включают в себя капсулы, драже, гранулы, пилюли, порошки и таблетки, но не ограничены вышеперечисленным. Наполнители, обычно используемые для создания состава таких дозированных форм, включают в себя обволакивающие материалы или составные добавки,такие как усилители всасывания, антиоксиданты, связующие вещества, буферные вещества, вещества для покрытия, красители, разбавители, разрыхляющие вещества, эмульгаторы, заменители, наполнители,ароматические вещества, увлажняющие вещества, смазывающие вещества, консерванты, пропелленты,разделительные вещества, стерилизующие вещества, подсластители, солюбилизаторы и их смеси. Наполнители для соединений, вводимых перорально в твердых дозированных формах, включают в себя агар, альгиновую кислоту, гидроксид алюминия, бензилбензоат, 1,3-бутиленгликоль, касторовое масло,целлюлозу, ацетилцеллюлозу, масло какао, кукурузный крахмал, кукурузное масло, хлопковое масло,этиловый спирт, этилацетат, этилкарбонат, этилцеллюлозу, этиллаурат, этилолеат, желатин, масло пшеничных зародышей, глюкозу, глицерин, арахисовое масло, изопропиловый спирт, изотонический раствор, лактозу, гидроксид магния, стеарат магния, солод, оливковое масло, ореховое масло, соли фосфата калия, картофельный крахмал, пропиленгликоль, тальк, трагакант, воду, сафлоровое масло, сезамовое масло, натриевую соль карбоксиметилцеллюлозы, лаурилсульфат натрия, соли фосфата натрия, соевое масло, сахарозу, тетрагидрофурфуриловый спирт и их смеси. Предпочтительная дозированная форма обычно будет содержать по меньшей мере один ингибитор протеазы ВИЧ в терапевтически эффективном количестве, по меньшей мере один фармацевтически приемлемый растворимый в воде полимер и по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество. Более предпочтительно в составе одной из вышеупомянутых фармацевтических дозированных форм можно вводить твердый раствор или твердую дисперсию. Такие растворы или дисперсии можно производить с подходящими фармацевтически приемлемыми растворимыми в воде полимерами, включающими в себя, без ограничения перечисленными, растворимые в воде полимеры, имеющие температуру стеклования Тс по меньшей мере около 50 С, предпочтительно по меньшей мере около 60 С, наиболее предпочтительно от около 80 до около 180 С. Способы определения значения Тс органических полимеров описаны во втором выпуске "Introduction to Physical Polymer Science", 2nd Edition, L.H. Sperling,опубликованном John WileySons, Inc, 1992. Значение Тс можно рассчитывать как взвешенную сумму значений Тс для гомополимеров, полученную из значений Тс каждого из отдельных мономеров, т.е. мономеров, которые составляют полимер: Tc=WiXi, где W является процентом массы мономера i в органическом полимере и X является значением Тс для гомополимера, получаемого из мономера i. Значения Тс для гомополимеров можно взять из второго выпуска руководства "Polymer Handbook", 2nd Edition,авторов J. Brandrup и Е.Н. Immergut, Editors, опубликованном John WileySons, Inc., 1975. Растворимые в воде полимеры, имеющие Тс согласно приведенному выше определению, позволяют изготавливать твердые растворы или твердые дисперсии, являющиеся механически устойчивыми и имеющими достаточную температурную устойчивость в пределах обычных температурных диапазонов,таким образом, чтобы можно было использовать твердые растворы или твердые дисперсии в качестве дозированных форм без дальнейшей обработки, или их можно было прессовать в таблетки, задействуя только небольшое количество вспомогательных веществ для таблетирования. Растворимый в воде полимер, содержащийся в предпочтительной дозированной форме, является полимером, который при растворении в 2% (мас./об.) водном растворе при 20 С предпочтительно имеет кажущуюся вязкость, составляющую от около 1 до около 5000 мПас, более предпочтительно от около 1-3 014446 до около 700 мПас и наиболее предпочтительно от около 5 до около 100 мПас. Растворимые в воде полимеры, подходящие для использования в предпочтительной дозированной форме настоящего изобретения, включают в себя, без ограничения перечисленными, гомополимеры и сополимеры N-виниллактамов, в особенности гомополимеры и сополимеры N-винилпирролидона, например поливинилпирролидон (ПВП), сополимеры N-винилпирролидона и винилацетата или винилпропионата, сложные эфиры целлюлозы и простые эфиры целлюлозы, в частности метилцеллюлозу и этилцеллюлозу, гидроксиалкилцеллюлозы, в частности гидроксипропилцеллюлозу, гидроксиалкилалкилцеллюлозы, в частности гидроксипропилметилцеллюлозу, фталаты или сукцинаты целлюлозы, в частности фталат ацетилцеллюлозы и фталат гидроксипропилметилцеллюлозы, сукцинат гидроксипропилметилцеллюлозы или ацетат сукцинат гидроксипропилметилцеллюлозы; высокомолекулярные оксиды полиалкилена, такие как полиэтиленоксид и полипропиленоксид и сополимеры оксида этилена и оксида пропилена, полиакрилаты и полиметакрилаты, такие как метакриловая кислота/сополимеры этилакрилата, метакриловая кислота/сополимеры метилметакрилата, бутилметакрилат/сополимеры 2-диметиламиноэтил метакрилата, поли(гидроксиалкил акрилаты), поли(гидроксиалкил метакрилаты), полиакриламиды, полимеры винилацетата, такие как сополимеры виниладетата и кротоновой кислоты, частично гидролизованного поливинилацетата (также называемого частично омыленным "поливиниловым спиртом"), поливиниловый спирт, олиго- и полисахариды, такие как каррагинаны, галактоманнаны и ксантановая смола,или смеси одного или более этих компонентов. Из них предпочтительными являются гомополимеры или сополимеры N-винилпирролидона, в особенности сополимер N-винилпирролидона и винилацетата. Особенно предпочтительным полимером является сополимер, состоящий из сополимера N-винилпирролидона в количестве около 60 мас.% и сополимера винилацетата в количестве около 40 мас.%. Согласно предпочтительной дозированной форме настоящего изобретения фармацевтическая дозированная форма содержит водорастворимый полимер или любую комбинацию таких полимеров в количестве от около 50 до около 85 мас.% от общей массы дозированной формы, предпочтительно от около 60 около до 80% от общей массы дозированной формы. Используемый в настоящем изобретении термин "фармацевтически приемлемое поверхностноактивное вещество" относится к фармацевтически приемлемому неионному поверхностно-активному веществу. В одном варианте осуществления настоящее изобретение обеспечивает дозированную форму,содержащую по меньшей мере одно поверхностно-активное вещество, имеющее значение гидрофильнолипофильного баланса (ГЛБ) от около 4 до около 10, предпочтительно от около 7 до около 9. Система ГЛБ (Fiedler, H.B., Fiedler, H.B., Encylopedia of Excipients, 5th ed., Aulendorf: ECV-Editio-Cantor-Verlag(2002, классифицирует поверхностно-активные вещества по числовым значениям, при этом липофильные вещества получают низкие значения ГЛБ и гидрофильные вещества получают высокие значения ГЛБ. Поверхностно-активные вещества, имеющие значение ГЛБ от около 4 до около 10, подходящие для использования в настоящем изобретении, включают в себя, без ограничения перечисленными, простые эфиры полиоксиэтиленалкила, например полиоксиэтилен (3) лауриловый эфир, полиоксиэтилен (5) цетиловый эфир, полиоксиэтилен (2) стеариловый эфир, полиоксиэтилен (5) стеариловый эфир; полиоксиэтилен простые алкилариловые эфиры, например полиоксиэтилен (2) нонилфениловый эфир, полиоксиэтилен (3) нонилфениловый эфир, полиоксиэтилен (4) нонилфениловый эфир, полиоксиэтилен (3) октилфениловый эфир; сложные эфиры полиэтиленгликолевой жирной кислоты, например ПЭГ-200 монолаурат,ПЭГ-200 дилаурат, ПЭГ-300 дилаурат, ПЭГ-400 дилаурат, ПЭГ-300 дистеарат, ПЭГ-300 диолеат; алкиленгликолевые моносложные эфиры жирной кислоты, например монолаурат пропиленгликоля (Lauroglycol); сложные эфиры жирной кислоты сахарозы, например моностеарат сахарозы, дистеарат сахарозы,монолаурат сахарозы, дилаурат сахарозы; или моносложные эфиры жирной кислоты сорбитана, такие как монолаурат сорбитана (Span 20), сорбитана моноолеат, монопальмитат сорбитана (Span 40), или сорбитана стеарат, или смеси одного или более этих компонентов. Предпочтительными являются сложные моноэфиры жирной кислоты сорбитана и особенно предпочтительными - монолаурат сорбитана и монопальмитат сорбитана. Предпочтительная дозированная форма настоящего изобретения содержит поверхностно-активное вещество или комбинацию поверхностно-активных веществ в количестве от около 2 до около 20% от общей массы дозированной формы, предпочтительно от около 3 до около 15% от общей массы дозированной формы. Кроме поверхностно-активного вещества, имеющего значение ГЛБ от около 4 до около 10, предпочтительная дозированная форма может содержать дополнительные фармацевтически приемлемые поверхностно-активные вещества, такие как полиоксиэтиленовые производные касторового масла, например полиоксиэтиленглицерина тририцинолеат или полиоксил 35 касторовое масло (Cremophor EL;BASF Corp.) или полиоксиэтиленглицерина оксистеарат, такой как полиэтиленгликоль 40 гидрированное касторовое масло (Cremophor RH 40) или полиэтиленгликоль 60 гидрированное касторовое масло-4 014446 полиоксиэтиленовые полиоксипропиленовые блок-сополимеры или полиоксиэтиленполипропиленгликоли, такие как Полоксамер 124, Полоксамер 188, Полоксамер 237, Полоксамер 388, Полоксамер 407 (BASF Wyandotte Corp.); или моносложный эфир жирной кислоты полиоксиэтилена (20) сорбитана, например полиоксиэтилена (20) сорбитана моноолеат (Tween 80), полиоксиэтилена (20) сорбитана моностеарат (Tween 60), полиоксиэтилена (20) монопальмитат сорбитана (Tween 40), полиоксиэтилена (20) монолаурат сорбитана (Tween 20). При использовании таких дополнительных поверхностно-активных веществ обычно по меньшей мере около 50 мас.%, предпочтительно по меньшей мере около 60 мас.% от общего количества используемого поверхностно-активного вещества составляет поверхностно-активное вещество, имеющее значение ГЛБ от около 4 до около 10. Дозированная форма настоящего изобретения может включать в себя дополнительные наполнители или добавки, такие как, например, регуляторы текучести, смазывающие вещества, вещества для придания объема (наполнители) и разрыхляющие вещества. Такие дополнительные наполнители могут составлять от около 0 до около 15% от общей массы дозированной формы. Предпочтительную дозированную форму настоящего изобретения на основе твердой дисперсии или твердого раствора можно изготовлять путем приготовления твердого раствора или твердой дисперсии ингибитора протеазы ВИЧ или комбинации ингибиторов протеазы ВИЧ, в матрице водорастворимого полимера и поверхностно-активного вещества, и затем придавая ей требуемую форму таблетки. Альтернативно, твердый раствор или твердый диспергированный продукт можно измельчать в гранулы, например, путем размалывания или дробления, после чего гранулы можно прессовать в таблетки. Для изготовления твердых растворов или твердых дисперсий существуют различные способы,включающие в себя экструзию из расплава, распылительную сушку и выпаривание из раствора с предпочтительной экструзией из расплава. Способ экструзии из расплава содержит этапы приготовления гомогенного расплава ингибитора протеазы ВИЧ или комбинации ингибиторов протеазы ВИЧ, водорастворимого полимера и поверхностно-активного вещества, и охлаждения расплава до его отвердения."Плавление" означает переход в жидкое или эластичное состояние, в котором один компонент может становиться гомогенно введенным в другой компонент. Обычно один продукт будет расплавляться,и другие продукты будут растворяться в расплаве, образуя таким образом раствор. Плавление обычно охватывает нагревание водорастворимого полимера выше температуры размягчения. Изготовление расплава можно осуществлять множеством путей. Смешивание компонентов может происходить перед образованием расплава, во время или после него. Например, компоненты сначала можно смешивать и затем расплавлять или одновременно смешивать и расплавлять. Обычно расплав гомогенизируют для эффективного диспергирования активных компонентов. Также, может быть удобным сначала расплавлять водорастворимый полимер и затем смешивать и гомогенизировать активные компоненты. Обычно температура плавления находится в диапазоне от около 70 до около 250 С, предпочтительно от около 80 около до 180 С, наиболее предпочтительно от около 100 до около 140 С. Активные компоненты можно использовать как есть или в виде раствора или дисперсии в подходящем растворителе, таком как спирты, алифатические углеводороды или сложные эфиры. Другим возможным для использования растворителем является жидкая двуокись углерода. После приготовления расплава растворитель удаляют, например выпариванием. В расплав можно включать различные добавки, например регуляторы текучести, такие как коллоидный оксид кремния; смазывающие вещества, наполнители, разрыхляющие вещества, пластификаторы,стабилизаторы, такие как антиоксиданты, вещества для светоустойчивости, акцепторы радикалов, стабилизаторы против микробного заражения. Плавление и/или смешивание осуществляют в общепринятом для этой цели устройстве. Особенно подходящими устройствами являются экструдеры или смесители. Подходящие экструдеры включают в себя одношнековые экструдеры, экструдеры со шнеками, находящимися в зацеплении или другие многошнековые экструдеры, предпочтительно двухшнековые экструдеры, шнеки которых могут вращаться в одном направлении или в противоположных направлениях и, необязательно, экструдеры могут оснащаться месильными дисками. Следует понимать, что рабочие температуры также будут определяться видом экструдера или видом конфигурации используемого экструдера. Часть энергии, необходимой для расплавления, смешивания и растворения компонентов в экструдере, может обеспечиваться нагревательными элементами. Вместе с тем, трение и сдвиг материала в экструдере также могут обеспечить существенное количество энергии для смеси и способствовать образованию гомогенного расплава компонентов. Расплав находится в диапазоне от пастообразного до вязкого состояния. Придание формы экструдату удобно осуществлять каландром с двумя встречно вращающимися валами с взаимно соответствующими выемками на их поверхности. При использовании валов с различными формами выемок можно получать широкий диапазон форм таблеток. Альтернативно, экструдат разрезают на кусочки или перед отверждением (горячая нарезка), или после него (холодная нарезка). Получаемый продукт твердого раствора или твердой дисперсии необязательно размалывают или-5 014446 измельчают в гранулы. Затем гранулы можно прессовать. Прессование означает способ, посредством которого порошковая масса, содержащая гранулы, уплотняется под высоким давлением для получения прессованного продукта с низкой пористостью, например таблетки. Сжатие порошковой массы обычно осуществляют в таблетирующем прессе, более конкретно в стальной пресс-форме между двумя движущимися пуансонами. Безусловно, если твердая дозированная форма настоящего изобретения содержит комбинацию более чем одного ингибитора протеазы ВИЧ (или комбинацию ингибитора протеазы ВИЧ с другими одним или более активными компонентами), возможно раздельное приготовление продуктов отдельных активных компонентов из твердого раствора или твердой дисперсии и смешивание размолотых или измельченных продуктов перед прессованием. Предпочтительно для прессования гранул используют по меньшей мере одну добавку, выбираемую из регуляторов текучести, разрыхляющих веществ, веществ для придания объема (наполнителей) и смазывающих веществ. Разрыхляющие вещества содействуют быстрому распаду прессованного продукта в желудке и сохраняют высвобождаемые гранулы раздельно друг от друга. Подходящие разрыхляющие вещества представляют собой сшитые полимеры, такие как сшитый поливинилпирролидон и сшитая карбоксиметилцеллюлоза натрия. Подходящие вещества для придания объема (также называемые "наполнителями") выбирают из лактозы, вторичного кислого фосфата кальция, микрокристаллической целлюлозы (Avicell), силикатов, в особенности из диоксида кремния, оксида магния, талька, картофельного или кукурузного крахмала, изомальтозы, поливинилового спирта. Подходящие регуляторы текучести выбирают из тонкодисперсного кремнезема (Aerosil) и животных или растительных жиров или восков. Смазывающее вещество предпочтительно используют при прессовании гранул. Подходящие смазывающие вещества выбирают из полиэтиленгликоля (например, имеющего молярную массу от 1000 до 6000), стеаратов магния и кальция, натрия стеарилфумарата и т.п. веществ. Можно использовать различные другие добавки, например красители, такие как азокрасители, органические или неорганические пигменты, такие как оксид алюминия или диоксид титана или красители природного происхождения; стабилизаторы, такие как антиоксиданты, вещества для придания светоустойчивости, акцепторы радикалов, стабилизаторы против микробного заражения. Дозированные формы согласно настоящему изобретению могут обеспечиваться в виде дозированных форм, состоящих из нескольких слоев, например пластинчатых или многослойных таблеток. Они могут находиться в открытой или закрытой форме. "Закрытые дозированные формы" представляют собой формы, в которых один слой полностью окружен по меньшей мере одним другим слоем. Преимущество многослойных форм состоит в том, что можно обрабатывать два несовместимых друг с другом активных компонента или что можно регулировать параметры высвобождения активного компонента(компонентов). Например, возможно обеспечивать начальную дозу путем включения в один из наружных слоев активного компонента и поддерживающую дозу путем включения активного компонента(компонентов) во внутренний слой. Можно производить таблетки многослойного типа путем прессования гранул с двумя или больше слоями. Альтернативно, можно производить многослойные дозированные формы способом, известным, как "соэкструзия". В сущности, способ содержит изготовление по меньшей мере двух различных композиций расплава, как объясняется выше, и пропускание этих расплавленных композиций через объединенный пуансон соэкструзии. Форма пуансона соэкструзии зависит от требуемой формы лекарственного средства. Например, подходящими являются пуансоны с плоским просветом, называемые щелевыми матрицами и матрицы с кольцевой щелью. Для облегчения приема млекопитающим такой дозированной формы является предпочтительным придание соответствующей формы лекарственному средству. Поэтому большие таблетки, которые можно удобно глотать, предпочтительно имеют не округлую, а удлиненную форму. Пленочное покрытие на таблетке дополнительно способствует легкости ее проглатывания. Пленочное покрытие также улучшает вкус и обеспечивает приятный внешний вид. Если желательно, пленочное покрытие может быть энтеросолюбильным покрытием. Пленочное покрытие обычно включает в себя полимерный пленкообразующий материал, такой как гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза и сополимеры метакрилата или акрилата. Помимо пленкообразующего полимера пленочное покрытие может дополнительно содержать пластификатор, например полиэтиленгликоль, поверхностноактивное вещество, например типа Tween, и необязательно пигмент, например диоксид титана или оксиды железа. Пленочное покрытие может также содержать тальк в качестве противоадгезивного вещества. Пленочное покрытие обычно составляет менее чем около 5 мас.% дозированной формы. Сейчас считается, что выгоды, обеспечиваемые согласно настоящему изобретению, относятся к фармакокинетическим (фК) свойствам дозированной формы. Обычно под фармакокинетическими свойствами понимают способ и степень всасывания лекарственного средства. Общепринятые фК параметры включают в себя AUC (или "область под кривой"), которая обычно относится к количеству средства, определяемому в крови или производных крови человека, принимающего средство на протяжении времени. Иначе AUC указывает на контакт больного с лекарственным средством. Cmax является другим фК термином, который относится к максимальному уровню в крови (или в производных крови) лекарственного-6 014446 средства в течение данной схемы его приема. Схемы приема средства, для которых измеряют фК параметры, включают в себя "клинические испытания". Некоторые клинические испытания проводят в конечной совокупности здоровых пациентов-добровольцев, и они разработаны для определения фК параметров лекарственного средства (такого, как упомянутого выше), а не для лечения больных. Таким образом, каждого пациента называют членом исследуемой совокупности. Осуществляют тщательный контроль и мониторинг таких клинических испытаний, и вместе с тем, между клиническими испытаниями фК параметры могут варьировать в высокой степени, поскольку разные клинические испытания проводят на разных совокупностях пациентов. Хотя между клиническими испытаниями существуют варианты,для специалистов в данной области техники будет легко понять, что создание состава лекарственного средства для достижения сходного набора фК параметров является стандартной практикой, поскольку конкретный набор фК параметров является общеизвестным. Как упомянуто ранее, настоящее изобретение обеспечивает дозированную форму, которую можно принимать, не учитывая факт приема пищи больным, иногда называемой "безотносительной к приемам пищи", "возможной для приема с едой или без еды", "не имеющей эффекта приема пищи" или сходными выражениями. В частности, у больных натощак ("состояние голода") Cmax лекарственного средства иAUC лекарственного средства являются сходными по сравнению с этими параметрами у больных после еды ("состояние сытости"). Следовательно, обеспечиваемая настоящим изобретением дозированная форма имеет преимущество приема в любое время независимо от того, ели или не ели больные за последнее время. Несмотря на предыдущее определение, не существует каких-либо полностью стандартных определений для состояний сытости и голода. Вместе с тем, обычно состояние голода относится к факту, что больной не ел определенное количество времени перед приемом дозы лекарства, а также не ел определенное количество времени после приема дозированной формы. Эти периоды времени перед приемом и после приема лекарства являются вопросом выбора и могут располагаться, например, в интервале от 2 до 24 ч. Состояние сытости обычно относится к факту, что больной ел в пределах определенного периода времени приема конкретного лекарства. Этот период времени является вариабельным, но может составлять, например, прием пищи непосредственно перед приемом лекарства, во время, или сразу после приема лекарства, обычно пища принимается в течение около часа после приема лекарства. Количество съеденной пищи, которое будет считаться состоянием сытости, также является вариабельным, но обычно может содержать от около 500 до около 1500 килокалорий пищи. Обеспечиваемые в настоящем изобретении дозированные формы будут иметь, по существу, сходные значения Cmax и AUC у больных в состоянии голода, а также в состоянии сытости, независимо от принимаемой дозы. В частности, значения отдельных отношений пациентов в совокупности пациентов как для Cmax, так и для AUC в состоянии сытости к состоянию голода будут находиться в диапазоне от около 0,7 до около 1,43; более предпочтительно в диапазоне от около 0,75 до около 1,35 и наиболее предпочтительно от около 0,8 до около 1,25. Таким образом, например, в исследуемой совокупности 30 человек каждому пациенту дают дозу лекарственного средства в состоянии сытости и, через соответствующий период времени, дозу средства в состоянии голода. AUC и Cmax для обоих условий приема пищи вычисляют для каждого пациента. Затем значение AUC для состояния сытости делят на значениеAUC для состояния голода для каждого пациента. Затем значения отдельных пациентов суммируют,после чего делят на число пациентов, завершивших испытание, чтобы прийти к среднему значениюAUC для всех пациентов, завершивших испытание. Среднее значение Cmax вычисляют подобным образом. Если среднее значение отношений состояний сытости к состоянию голода для значений Cmax илиAUC всех пациентов в данном испытании находится в диапазоне, например, от 0,7 до 1,43, то дозированную форму, предоставляемую пациентам, можно считать возможной для введения безотносительно к состоянию сытости или голода у пациента. Как также ранее упомянуто, дозированные формы, обеспечиваемые настоящим изобретением,имеют меньшую вариабельность, чем другие составы на основе желатиновых капсул, содержащие растворенную форму лекарственного средства или средств. Это отсутствие вариабельности доказано на фиг. 1 и 2, на которых приведено сравнение данных AUC и Cmax варианта осуществления настоящего изобретения и данных имеющейся на рынке желатиновой капсулы, содержащей растворенный ИП. Как показано на фигурах, данные AUC и Cmax, связанные с вариантом осуществления настоящего изобретения, показывают меньшую вариабельность. В частности, схемы представляют собой графики данных"прямоугольников и штрихов", сравнивающих два указанных состава, в которых любой нижний приведенный "штрих" (обозначенный А в первом прямоугольнике и штриховой график на фиг. 1) называют"5-й процентилью", что означает, что значения 5% пациентов в испытании находятся ниже определяемых значений AUC или Cmax для конкретного штриха. Верхний штрих (обозначенный D в первом прямоугольнике и штриховом графике на фиг. 1) отражает "95-ю процентиль", что означает, что значенияAUC или Cmax у 5% пациентов в испытании были выше верхних значений, определяемых любым конкретным штрихом. Точно так же основание любого конкретного прямоугольника (обозначенного В в первом прямоугольнике и штриховом графике на фиг. 1) представляет 25-ю процентиль, и верхняя гра-7 014446 ница любого конкретного прямоугольника (обозначенного С в первом прямоугольнике и штриховом графике на фиг. 1) представляет 75-ю процентиль. Прямая, проложенная через любой конкретный прямоугольник, является 50-й процентилью или медианой любой конкретной исследуемой совокупности. Как показано на фигурах, обычно данные демонстрируют, что вариабельность, связанная с вариантом осуществления настоящего изобретения, является меньшей, чем связанная с существующим составом желатиновых капсул. При анализе фиг. 1, показывающей дозированные формы, даваемые в условиях голода (например), различия между 95-й процентилью и 5-й процентилью желатиновой капсулы является больше, чем различия между 95-й процентилью и 5-й процентилью варианта осуществления настоящего изобретения. Это трансформируется в факт, что терапевтический эффект в большей части исследуемой совокупности достигается с ИП без проявления побочных эффектов в силу передозировки средства. Например, в целях снижения побочных эффектов и достижения терапевтических уровней, обычно предпочтительно, что различия между 95-й процентилью AUC и 5-й процентилью AUC любой заданной исследуемой совокупности, которая принимает дозированную форму, обеспечиваемую настоящим изобретением (независимо от приема пищи или голода в совокупности), составляют меньше чем около 180, более предпочтительно меньше чем около 175, еще более предпочтительно меньше чем около 165 и наиболее предпочтительно меньше чем около 160. В условиях голода предпочтительно, что различия между 95-й процентилью AUC и 5-й процентилью AUC в любой заданной исследуемой совокупности, которая принимает дозированную форму, обеспечиваемую настоящим изобретением, составляют меньше чем около 170, более предпочтительно меньше чем около 160 и наиболее предпочтительно меньше чем около 150. В условиях сытости является предпочтительным, что различия между 95-й процентилью AUC и 5-й процентилью AUC в любой данной исследуемой совокупности, которая принимает дозированную форму, обеспечиваемую настоящим изобретением, составляют меньше чем около 130, более предпочтительно меньше чем около 120 и наиболее предпочтительно меньше чем около 110. Аналогично различиям между 95-й и 5-й процентилями, представленными выше, различия между 75-й процентилью и 25-й процентилью данных AUC на фиг. 1 также являются очень важными для демонстрации отсутствия вариабельности в дозированных формах настоящего изобретения. Обычно предпочтительно, что различия между 75-й процентилью AUC и 25-й процентилью AUC любой заданной исследуемой совокупности, которая принимает дозированную форму, обеспечиваемую настоящим изобретением (независимо от приема пищи или голода в совокупности), составляют меньше чем около 60,более предпочтительно меньше чем около 55, еще более предпочтительно меньше чем около 50. В условиях голода предпочтительно, что различия между 75-й процентилью AUC и 25-й процентилью AUC любой заданной исследуемой совокупности, которая принимает дозированную форму, обеспечиваемую настоящим изобретением, составляют меньше чем около 65, более предпочтительно меньше чем около 60 и наиболее предпочтительно меньше чем около 55. В условиях сытости предпочтительно, что отличие между 75-й процентилью AUC и 25-й процентилью AUC в любой заданной исследуемой совокупности, которая принимает дозированную форму, обеспечиваемую настоящим изобретением, составляет меньше чем около 60, более предпочтительно меньше чем около 50 и наиболее предпочтительно меньше чем около 40. В плане диапазонов значений AUC является предпочтительным, что в условиях голода от 5-й процентили до 95-й процентили значения AUC в любой заданной исследуемой совокупности, принимающей дозированную форму, обеспечиваемую настоящим изобретением, находятся в диапазоне от около 33 до около 175 мкг в ч/мл; и от 25-й процентили до 75-й процентили значения AUC в любой заданной исследуемой совокупности, принимающей дозированную форму, обеспечиваемую настоящим изобретением, варьируют от около 54 до около 107 мкг в ч/мл. В условиях приема пищи является предпочтительным, что от 5-й процентили до 95-й процентили значения AUC в любой заданной исследуемой совокупности, которая принимает дозированную форму, обеспечиваемую настоящим изобретением,находятся в диапазоне от около 57 до около 142 мкг в ч/мл; и от 25-й процентили до 75-й процентили значения AUC в любой заданной исследуемой совокупности, принимающей дозированную форму,обеспечиваемую настоящим изобретением, находятся в диапазоне от около 75 до около 109 мкг в ч/мл. Также предпочтительно, что 5-я процентиль AUC любой заданной исследуемой совокупности, которая принимает дозированную форму, обеспечиваемую настоящим изобретением, составляет больше чем около 30 мкг в ч/мл в условиях голода и больше чем около 50 мкг в ч/мл в условиях сытости. Наконец, в отношении AUC предпочтительно, что в условиях голода среднее значение AUC составляет от около 60 до около 95 мкг в ч/мл для любой заданной исследуемой совокупности, которая принимает обеспечиваемую дозированную форму. Аналогично параметрам AUC, показанным на фиг. 1, параметры Cmax, показанные на фиг. 2, также демонстрируют отсутствие вариабельности, связанной с дозированными формами, обеспечиваемыми настоящим изобретением. Например, исходя из графической схемы прямоугольников и штрихов фиг. 2 для пациентов в условиях голода, принимающих дозу ИП, созданную согласно настоящему изобретению, является предпочтительным, что различия между 95-й процентилью и 5-й процентилью составляют меньше чем около 15, более предпочтительно меньше чем около 13 и наиболее предпочтительно меньше-8 014446 чем около 11. В условиях голода также предпочтительно, что 5-я процентиль Cmax заданной исследуемой совокупности, принимающая дозу активного компонента, созданного согласно настоящему изобретению, составляет больше чем около 2,5 мкг/мл. Обращаясь к графику прямоугольников и штрихов фиг. 2 для условий сытости при приеме дозированной формы настоящего изобретения, предпочтительно, что различие между 95-й процентилью и 5-й процентилью составляет меньше чем около 12, более предпочтительно меньше чем около 11. В отношении описания представленных выше чисел необходимо подчеркнуть, что при упоминании больного, принимающего дозированную форму настоящего изобретения, такие больные получают дозу ИП в многократных дозированных формах. Более конкретно, так называемая дозированная форма содержала 400 мг лопинавира и 100 мг ритонавира, равномерно разделенных между двумя дозированными формами. Лопинавир был единственным лекарственным средством, для которого проводили измерения в этих испытаниях, вследствие того, что ритонавир добавляли не в силу его действия в качестве ИП, но в качестве фармакокинетического усилителя или бустера (ритонавир замедляет метаболизм лопинавира). Кроме того, подразумевается, что данные значения могут меняться, например, в силу вариабельности в режиме приема пищи и в ее количестве, а также телосложения в исследуемой совокупности. Известно,что в исследуемых совокупностях различных национальностей можно наблюдать различную скорость метаболизма лекарственного средства. Соответственно, в случаях, где данные испытаний взяты из таких совокупностей, данные, скорее всего, придется нормировать согласно хорошо известным в данной области техники способам. Кроме того, в случаях, когда в исследуемой совокупности обеспечивается, например, увеличение дозы или уменьшение дозы лопинавира, для данных, получаемых от такого приема средства, может потребоваться нормирование с использованием соответствующего моделирования, известного в данной области техники. Наконец, в отношении вышеупомянутого обсуждения фигур, считается, что "пища с высоким содержанием жира", как описано на фигурах, является состоянием сытости. В дополнение к обеспечению способов лечения больных людей, зараженных ВИЧ/СПИДом, настоящее изобретение обеспечивает способы снижения побочных эффектов, связанных с лечением ВИЧ,способы повышения биодоступности ИП, способы снижения лекарственной нагрузки у больных ВИЧ/СПИДом, способы уменьшения вариабельности уровней в крови ИП у больных, получающих ИП терапию и способы обеспечения больных ИП, получающих ИП терапию. Все эти способы содержат этап обеспечения фармацевтические дозированные формы, содержащего терапевтически эффективное количество нерастворенной формы ИП для больного. Предпочтительно ИП представляет собой (2S,3S,5S)-2(2,6-диметилфеноксиацетил)амино-3-гидрокси-5-[2S-(1-тетрагидропиримид-2-онил)-3-метилбутаноил]амино-1,6-дифенилгексан (АВТ-378; лопинавир). Более предпочтительно дозированная форма будет содержать комбинацию (2S,3S,5S)-2-(2,6-диметилфеноксиацетил)амино-3-гидрокси-5-[2S-(1-тетрагидропиримид-2-онил)-3-метилбутаноил]амино-1,6-дифенилгексана (АВТ-378; лопинавир) и (2S,3S,5S)-5-(N(N-(N-метил-N-2-изопропил-4-тиазолил)метил)амино)карбонил)-L-валинил)амино-2-(N-5 тиазолил)метоксикарбонил)амино)амино-1,6-дифенил-3-гидроксигексана (ритонавир). Примеры Следующие примеры представлены для дополнительного понимания и иллюстрации настоящего изобретения и не ограничивают сущность и объем настоящего изобретения согласно определению в прилагаемой формуле изобретения. Пример 1.(4,17 мас.ч.), лопинавиром (16,67 мас.ч.) и коллоидным оксидом кремния (1,0 мас.ч.). Затем порошкообразную смесь вводили в двухшнековый экструдер (наружный диаметр экструдера 18 мм) со скоростью 2,0 кг/ч и температуре расплава 133 С. Чистый, полностью прозрачный расплав вводили в каландр с двумя противоположно вращающимися валами, имеющими на своих поверхностях взаимно соответствующие полости. Таким образом, получали таблетки 1080 мг. Анализы дифференциальной сканирующей калориметрии ДСК и широкоугловое рассеяние рентгеновских лучей WAXS не выявляли доказательства присутствия кристаллического лекарственного материала в составе. Биодоступность состава оценивали с использованием собак породы бигль (обоего пола, массой около 10 кг), которые получали сбалансированную диету с 27% содержанием жира и свободным доступом к воде. Примерно за 30 мин до приема лекарства каждая собака получала гистамин в дозе 100 мкг/кг-9 014446 подкожно. Каждой собаке вводили единственную дозу, соответствующую около 200 мг лопинавира, около 50 мг ритонавира или около 200 мг лопинавира и около 50 мг ритонавира соответственно. К дозе добавляли около 10 мл воды. Образцы крови получали от каждого животного перед введением и через 0,25,0,5, 1,0, 1,5, 2, 3, 4, 6, 8, 10, 12 и 24 ч после введения лекарственного средства. Плазму отделяли от эритроцитов посредством центрифугирования и замораживали (-30 С) до проведения анализа. Концентрации ингибиторов протеазы ВИЧ определяли обращенно-фазовой высокоэффективной жидкостной хроматографией ВЭЖХ с низковолновым УФ-детектированием с последующим экстрагированием жидкости жидкостью образцов плазмы. Область под кривой (AUC) вычисляли трапецоидальным способом по ходу времени исследования. Каждую дозированную форму оценивали в группе, состоявшей из 8 собак; представленные данные являются средними значениями для каждой группы собак. Дозорегулируемая AUC у собак составляла 0,52 мкг в ч/мл/100 мг для ритонавира и 4,54 мг в ч/мл/100 мг для лопинавира. Этот пример указывает, что твердые растворы или твердые дисперсии ингибиторов протеазы ВИЧ без добавленного поверхностно-активного вещества в результате приводят к очень малой биодоступности. Пример 3. Коповидон (сополимер N-винилпирролидона/винилацетата 60:40; 78,17 мас.ч.) смешивали с ритонавиром (4,16 мас.ч.), лопинавиром (16,67 мас.ч.) и коллоидным оксидом кремния (1,0 мас.ч.). Затем порошкообразную смесь вводили в двухшнековый экструдер (наружный диаметр экструдера 18 мм) со скоростью 2,0 кг/ч и температуре расплава 133 С. Чистый, полностью прозрачный расплав вводили в каландр с двумя противоположно вращающимися валами, имеющими на своих поверхностях взаимно соответствующие полости. Таким образом, получали таблетки массой 1080 мг. Анализы ДСК и WAXS не выявляли доказательства присутствия кристаллического лекарственного материала в составе. Пример 4. Коповидон (сополимер N-винилпирролидона/винилацетата 60:40; 68,17 мас.ч.) смешивали с Кремофором RH40 (полиоксиэтиленглицерин оксистеарат; 10,00 мас.ч.) в мешалке с большими сдвиговыми усилиями Diosna. Получаемые гранулы смешивали с ритонавиром (4,17 мас.ч.), лопинавиром(16,67 мас.ч.) и коллоидным оксидом кремния (1,00 мас.ч.). Затем порошкообразную смесь вводили в двухшнековый экструдер Leistritz Micro 18 со скоростью 2,3 кг/ч и с температурой расплава 126 С. Экструдат размельчали на кусочки и давали возможность отверждения. Экструдированные кусочки размалывали с использованием высокопрочной универсальной мельницы. Размолотый материал (86,49 мас.ч.) смешивали в бункерной мешалке с моногидратом лактозы (6,00 мас.ч.), сшитым ПВП (6,00 мас.ч.), коллоидным оксидом кремния (1,00 мас.ч.) и стеаратом магния (0,51 мас.ч.). Порошкообразную смесь спрессовывали в таблетки массой 1378,0 мг на таблетирующем однопуансонном прессе Fette E 1. Затем- 10014446 таблетки покрывали пленкой в резервуаре для покрытия путем распыления водной дисперсией для пленочного покрытия (Opadry, получаемой от компании Colorcon) при температуре 60 С. Биодоступность состава оценивали с использованием собак породы бигль, пример 1. Дозорегулируемая AUC у собак составляла 0,60 мкг в ч/мл/100 мг для ритонавира и 7,43 мкг в ч/мл/100 мг для лопинавира. Этот пример указывает, что включение поверхностно-активного вещества в состав твердых растворов или твердых дисперсий ингибиторов протеазы ВИЧ улучшает достигнутую биодоступность. Пример 5. Коповидон (сополимер N-винилпирролидона/винилацетата 60:40; 853,8 мас.ч.) смешивали со Span 20 (монолаурат сорбитана; 83,9 мас.ч.) в мешалке с большими сдвиговыми усилиями Diosna. Получаемые гранулы смешивали с ритонавиром (50 мас.ч.), лопинавиром (200 мас.ч.) и коллоидным оксидом кремния (12 мас.ч.). Затем порошкообразную смесь вводили в двухшнековый экструдер (наружный диаметр экструдера 18 мм) со скоростью 2,1 кг/ч и при температуре расплава 119 С. Экструдат вводили в каландр с двумя противоположно вращающимися валами, имеющими на своих поверхностях взаимно соответствующие полости. Таким образом, получали таблетки массой 1120 мг. Биодоступность состава оценивали, как в примере 1, используя собак породы бигль. Дозорегулируемая AUC у собак составляла 10,88 мкг в ч/мл/100 мг для ритонавира и 51,2 мкг в ч/мл/100 мг для лопинавира. Этот пример указывает, что включение поверхностно-активного вещества, имеющего ГЛБ от 4 до 10 в состав твердых растворов или твердых дисперсий ингибиторов протеазы ВИЧ, заметно улучшает достигнутую биодоступность. Пример 6. Повторяли пример 5, однако экструдат измельчали на кусочки и давали возможность отверждения. Экструдированные кусочки размалывали до частиц величиной около 250 мкм с использованием высокопрочной универсальной мельницы. Размолотый материал смешивали в бункерной мешалке с натрия стеарилфумаратом (12,3 мас.ч.) и коллоидным оксидом кремния (8,0 мас.ч.) в течение 20 мин. Порошкообразную смесь спрессовывали на ротационной таблеточной машине с 3 пуансонами (6500 таблеток в час). Затем таблетки покрывали пленкой в резервуаре для покрытия путем распыления водной дисперсии для пленочного покрытия (Opadry, доступной от компании Colorcon) при температуре 60 С. Биодоступность состава оценивали с использованием собак породы бигль, как в примере 1. Дозорегулируемая AUC у собак составляла 14,24 мкг в ч/мл/100 мг для ритонавира и 52,2 мкг в ч/мл/100 мг для лопинавира. Пример 7. Коповидон (сополимер N-винилпирролидона/винилацетата 60:40; 841,3 мас.ч.) смешивали с Кремофором RH40 (полиоксиэтиленглицерин оксистеарат; 36,2 мас.ч.), Span 20 (монолаурат сорбитана; 60,2 мас.ч.) в мешалке с большими сдвиговыми усилиями Diosna. Получаемые гранулы смешивали с ритонавиром (50 мас.ч.), лопинавиром (200 мас.ч.) и коллоидным оксидом кремния (12 мас.ч.). Порошкообразную смесь вводили в двухшнековый экструдер (наружный диаметр экструдера 18 мм) со скоростью 2,1 кг/ч и при температуре расплава 114 С. Экструдат вводили в каландр с двумя противоположно вращающимися валами, имеющими на своих поверхностях взаимно соответствующие полости. Таким образом, получали таблетки массой 1120 мг. Биодоступность состава оценивали с использованием собак породы бигль, как в примере 1. Дозорегулируемая AUC у собак составляла 10,96 мкг в ч/мл/100 мг для ритонавира и 46,5 мкг в ч/мл/100 мг для лопинавира. Этот пример указывает, что можно успешно использовать комбинацию поверхностноактивного вещества, имеющего ГЛБ от 4 до 10 и дополнительного поверхностно-активного вещества. Пример 8. Повторяли пример 7, однако экструдат измельчали на кусочки и давали возможность отверждения.- 11014446 Экструдированные кусочки размалывали до частиц величиной около 250 мкм с использованием высокопрочной универсальной мельницы. Размолотый материал смешивали в бункерной мешалке с натрия стеарилфумаратом (13,9 мас.ч.), коллоидным оксидом кремния (7,0 мас.ч.), изомальтозы DC100(159,4 мас.ч.) и силиката кальция (7,0 мас.ч.) в течение 20 мин. Смесь спрессовывали и затем покрывали пленкой в резервуаре для покрытия путем распыления водной дисперсии для пленочного покрытия (Opadry, доступной от компании Colorcon) при температуре 60 С. Биодоступность состава оценивали, используя собак породы бигль, как в примере 1. Дозорегулируемая AUC у собак составляла 10,38 мкг в ч/мл/100 мг для ритонавира и 42,7 мкг в ч/мл/100 мг для лопинавира. Пример 9. Коповидон (сополимер N-винилпирролидона/винилацетата 60:40; 683,3 мас.ч.) смешивали со Span 40 (монопальмитат сорбитана; 67,2 мас.ч.) в мешалке с большими сдвиговыми усилиями Diosna. Получаемые гранулы смешивали с лопинавиром (200 мас.ч.) и коллоидным оксидом кремния (9,6 мас.ч.). Затем порошкообразную смесь вводили в двухшнековый экструдер (наружный диаметр экструдера 18 мм) со скоростью 2,1 кг/ч и при температуре расплава 119 С. Экструдат измельчали на кусочки и давали возможность отверждения. Экструдированные кусочки размалывали с использованием высокопрочной универсальной мельницы. Размолотый материал смешивали в бункерной мешалке с натрия стеарилфумаратом (7,9 мас.ч.), коллоидным оксидом кремния (11,3 мас.ч.), изомальтозой DC100 (129,1 мас.ч.) и додецилсульфатом натрия (15,6 мас.ч.). Смесь спрессовывали и затем покрывали пленкой в резервуаре для покрытия путем распыления водной дисперсии для пленочного покрытия (Opadry, доступной от компании Colorcon) при температуре 60 С. Биодоступность состава оценивали, как в примере 1, используя собак породы бигль. Таблетки, соответствующие 200 мг лопинавира, совместно вводили собакам вместе с 50 мг ритонавира. Дозорегулируемая AUC лопинавира составляла 38,8 мкг в ч/мл/100 мг. Пример 11.- 12014446 Экструдированный материал размалывали, спрессовывали с наполнителями для таблетирования и наносили покрытие. Состав состоял из лопинавира (200 мг на таблетку), ритонавира (50 мг на таблетку),коповидона в качестве полимерного носителя и полиоксил 40 гидрированного касторового масла в качестве поверхностно-активного вещества. К размолотому экструдату для спрессовывания добавляли наполнители внешней фазы. Поверхностно-активное вещество вводили перед экструзией путем грануляции с частью полимера. Пример 12. Таблетированный состав спрессовывали из раздельно экструдированного лопинавира и смесей порошкового ритонавира. Поверхностно-активное вещество вводили перед экструзией путем грануляции с частью полимера. Пример 13. Состав приготавливали путем размалывания экструдата, смешивания с наполнителями для таблетирования и спрессовывания в таблетки. На спрессованные таблетки наносили пленочное покрытие на водной, гидроксипропилметилцеллюлозной основе для усиления фармацевтической ясности. Поверхностно-активное вещество вводили перед экструзией путем грануляции с частью полимера. Состав приготавливали путем размалывания экструдата, смешивания с наполнителями для таблетирования и спрессовывания в таблетки. На спрессованные таблетки наносили пленочное покрытие на водной, гидроксипропилметилцеллюлозной основе для усиления фармацевтической ясности. Поверхностно-активное вещество вводили перед экструзией путем грануляции с частью полимера. Пример 15. Состав экструдировали в форме таблеток без дополнительных этапов обработки в виде размалывания, прессования и нанесения покрытия. Состав композиции включал в себя ритонавир, лопинавир, коповидон, поверхностно-активное вещество и коллоидный диоксид кремния, и эти два состава отличались по типу используемого поверхностно-активного вещества. Экструдированный таблетированный состав содержал монолаурат сорбитана в качестве поверхностно-активного вещества, которое вводили перед экструзией путем грануляции с частью полимера. Пример 16. Состав экструдировали в форме таблеток без дополнительных этапов обработки в виде размалывания, прессования и нанесения покрытия. Состав композиции включал в себя ритонавир, лопинавир, коповидон, поверхностно-активное вещество и коллоидный диоксид кремния, и эти два состава отличались по типу используемого поверхностно-активного вещества. Экструдированный таблетированный состав содержал в качестве поверхностно-активных веществ и полиоксил 40 гидрированное касторовое масло, и монолаурат сорбитана. Поверхностно-активные вещества вводили перед экструзией путем грануляции с частью полимера. Эта дозированная форма отличалась превосходной стабильностью и, в частности, проявляла высокую устойчивость против рекристаллизации или разложения активного компонента (компонентов). Таким образом, после хранения в течение 6 недель при 40 С и влажности 75% (например, при хранении во флаконах из полиэтилена высокой плотности (ПЭВП) без десиканта), дозированные формы согласно настоящему изобретению не показывали признаков кристалличности (что доказано анализами ДСК илиWAXS) и содержали активный компонент по меньшей мере в количестве около 98% от исходного содержания (что доказано анализом ВЭЖХ). Тесты растворимости in vitro осуществляли на нескольких составах из числа раскрытых в вышеприведенных примерах. Ниже в таблице показаны способ и условия тестирования. Результаты показаны ниже. В табл. 1 показано среднее значение % высвобождаемого лопинавира в минутах для составов, раскрытых в примерах 9, 10 и 12-16. Таблица 1 Среднее значение % растворенного лопинавира в минутах В табл. 2 показано среднее значение % растворенного ритонавира в минутах для составов, раскрытых в примерах 9, 10 и 12-16. Таблица 2 Среднее значение % растворенного ритонавира в минутах Таким образом, в одном варианте осуществления настоящее изобретение обеспечивает, например,фармацевтическую дозированную форму, содержащую лопинавир в терапевтически эффективном количестве, указанная дозированная форма обеспечивает профиль растворения in vitro, при котором от около 20 до около 30% лопинавира высвобождается за время от около 0 до около 15 мин, при использовании аппарата 2 USP (лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С. В одном варианте осуществления настоящее изобретение обеспечивает, например, фармацевтическую дозированную форму, содержащую лопинавир в терапевтически эффективном количестве, указанная дозированная форма обеспечивает профиль растворения in vitro, при котором от около 20% до около 30% лопинавира высвобождается за время от около 0 около до 15 мин, при использовании аппарата 2USP (лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С. В одном варианте осуществления настоящее изобретение обеспечивает, например, фармацевтическую дозированную форму, содержащую лопинавир в терапевтически эффективном количестве, указанная дозированная форма обеспечивает профиль растворения in vitro, при котором от около 43 до около 63% лопинавира высвобождается за время от около 15 около до 30 мин, при использовании аппарата 2USP (лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С. В одном варианте осуществления настоящее изобретение обеспечивает, например, фармацевтическую дозированную форму, содержащую лопинавир в терапевтически эффективном количестве, указанная дозированная форма обеспечивает профиль растворения in vitro, при котором от около 61,3% до около 81,7% лопинавира высвобождается за время от около 30 около до 45 мин, при использовании аппарата 2 USP (лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С. В одном варианте осуществления настоящее изобретение обеспечивает, например, фармацевтическую дозированную форму, содержащую лопинавир в терапевтически эффективном количестве, указанная дозированная форма обеспечивает профиль растворения in vitro, при котором от около 75,4% до около 93,2% лопинавира высвобождается за время от около 45 до около 60 мин, при использовании аппарата 2 USP (лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С. В одном варианте осуществления настоящее изобретение обеспечивает, например, фармацевтическую дозированную форму, содержащую лопинавир в терапевтически эффективном количестве, указанная дозированная форма обеспечивает профиль растворения in vitro с использованием аппарата 2 USP(лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С, в которой от около 20 до около 30% лопинавира высвобождается за время от около 0 до около 15 мин; от около 43 до около 63% лопинавира высвобождается за время от около 15 до около 30 мин; от около 61,3 до около 81,7% лопинавира высвобождается за время от около 30 до около 45 мин и от около 75,4 до около 93,2% лопинавира высвобождается за время от около 45 до около 60 мин. В одном варианте осуществления настоящее изобретение обеспечивает, например, фармацевтическую дозированную форму, содержащую ритонавир и лопинавир в терапевтически эффективном количестве, указанная дозированная форма обеспечивает in vitro профиль растворения, при использовании аппарата 2 USP (лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С, в которой от около 19,8 до около 34,4% ритонавира и от около 20 до около к 30% лопинавира высвобождается за время от около 0 до около до 15 мин; от около 41,6 до около 76,5% ритонавира и от около 43 до около 63% лопинавира высвобождается за время от около 15 до около до 30 мин; от около 59,4 до около 91,1% ритонавира и от около 61,3 до около 81,7% лопинавира высвобождается за время от около 30 до около 45 мин и от около 73,4 до около 95% ритонавира и от около 75,4 до около 93,2% лопинавира высвобождается за время от около 45 до около 60 мин. Для понимания воздействие лопинавира на людей, получающих дозированную форму настоящего изобретения и в настоящее время имеющуюся на рынке в виде желатиновых капсул Kaletra, исходя из нижеописанных исследований, были построены нормальные распределения. Предполагалось, что натуральные логарифмы Cmax и AUC лопинавира для каждого состава исходили из нормальных распределений со средним значениеми вариансой (2). Эти значения были получены от единственной дозы 400/100 мг лопинавира/ритонавира, в 4- или 5-этапных, рандомизированных, открытых перекрестных испытаниях у здоровых людей-добровольцев в условиях регулируемого приема пищи (или голодания,или приема пищи с умеренным содержанием жиров, или с высоким содержанием жиров). В каждом испытании находилось от 48 до 63 субъектов с периодами выведения по меньшей мере 7 дней. Средние значения Cmax и AUC лопинавира в условиях приема пищи с умеренным содержанием жиров были получены из центральных значений мета-анализа перекрестного испытания биоэквивалентности, что в общем является общеизвестным рядовым специалистам в данной области техники. Значения вариансы для распределения были получены из вариабельности у субъектов, оцениваемой для дозированной формы настоящего изобретения и имеющейся в настоящее время на рынке желатиновой капсулы Kaletra,используя смешанную методику системы статистического анализа SAS, которая в общем известна специалистам в данной области техники. Нормальные распределения Cmax и AUC лопинавира в условиях голода и приема пищи с высоким содержанием жиров подбирали с использованием точечной оценки из нижеописанных испытаний С и А для дозированной формы настоящего изобретения и имеющейся в настоящее время на рынке желатиновой капсулы Kaletra. Вариансу для обоих условий голода и приема пищи еды с высоким содержанием жиров составляли согласно величине вариабельности относительно условий приема умеренно жирной пищи, используя данные из испытаний А, В и С, описанные ниже более подробно. Плотность вероятности в отношении AUC для каждого состава вычисляли на основе среднего значения и вариансы, с использованием следующей формулы: Нормальное распределение Cmax лопинавира составляли тем же образом. Испытание А представляло собой пятиэтапное, рандомизированное открытое базовое исследование биодоступности после однократного введения дозы (лопинавир/ритонавир 400/100 мг), у 63 здоровых субъектов. Первые четыре этапа проводили согласно полностью перекрестной схеме испытания. Субъекты были эквивалентно рандомизированы к четырем последовательным схемам А, В, С и D в течение от 1 до 4 этапа. Осуществляли рандомизированный отбор пяти субъектов из каждой последовательной группы, завершившей от 1 до 4 этапа, для участия в 5 этапе и получения схемы Е. Пять этапов приема лекарства в испытании были разделены периодами выведения по меньшей мере 7 дней. Эти пять этапов представляли собой следующее. Схема А. Три имеющихся в настоящее время на рынке желатиновых капсулы Kaletra лопинавира/ритонавира 133,3/33,3 мг после завтрака с умеренным содержанием жиров. Схема В. Три имеющиеся в настоящее время на рынке желатиновые капсулы Kaletra лопинавира/ритонавира 133,3/33,3 мг в условиях голода. Схема С. Две дозированные формы настоящего изобретения лопинавира/ритонавира 200/50 мг после завтрака с умеренным содержанием жиров. Схема D. Две дозированные формы настоящего изобретения лопинавира/ритонавира 200/50 мг в условиях голода. Схема Е. Две дозированные формы настоящего изобретения лопинавира/ритонавира 200/50 мг настоящего изобретения после завтрака с высоким содержанием жиров. Испытание В представляло собой открытое четырехэтапное, рандомизированное, полностью перекрестное базовое исследование биодоступности в условиях не голода, приема пищи с умеренным содержанием жиров, после однократного введения дозы (лопинавир/ритонавир 400/100 мг), у 48 здоровых субъектов. Субъектов распределяли рандомизированным образом в равном количестве для достижения одной из четырех последовательностей схем А, В, С и D, определяемых следующим образом. Схема А. Две дозированные формы настоящего изобретения лопинавира/ритонавира 200/50 мг(партия 1). Схема В. Две дозированные формы настоящего изобретения лопинавира/ритонавира 200/50 мг (партия 2). Схема С. Две дозированные формы настоящего изобретения лопинавира/ритонавира 200/50 мг (партия 3). Схема D. Три имеющиеся в настоящее время на рынке желатиновые капсулы Kaletra лопинавира/ритонавира 133,3/33,3 мг. Указанные дозы вводили однократно утром в 1 день каждого этапа испытания после завтрака с умеренным содержанием жиров. Период выведения в 7 дней разделял дозы четырех этапов испытания. Испытание С было 1 фазой открытого пятиэтапного, рандомизированного, частично перекрестного одноцентрового исследования в условиях голода и приема пищи, у 56 здоровых субъектов. Вводили присутствующие в настоящее время на рынке составы жидкости и желатиновых капсул Kaletra для обеспечения однократной дозы лопинавира/ритонавира 400/100 мг. Оба состава давали в условиях голода и последующих приемов пищи с умеренным и высоким содержанием жиров. Было выявлено, что дозированная форма настоящего изобретения обеспечивает существенно более низкую вариабельность Cmax и AUC от 5-й до 95-й процентили в случае введения субъекту лопинавира вне зависимости от состояния голода или сытости, чем состав желатиновой капсулы. Таким образом,дозированная форма настоящего изобретения обеспечивает меньшуюCmax иAUC от 5-й до 95-й процентили для лопинавира, чем состав желатиновой капсулы Kaletra. Это показано как графически на фиг. 1 и 2, а также в цифровой форме в табл. 3-5. Дозированная форма настоящего изобретения также обеспечивает существенно более низкую вариабельность Cmax и AUC от 25-й до 75-й процентили в случае введения субъекту лопинавира вне зависимости от состояния голода или сытости, чем состав желатиновой капсулы. Таким образом, дозированная форма настоящего изобретения обеспечивает меньшуюCmax иAUC от 25-й до 75-й процентили для лопинавира, чем состав желатиновой капсулы Kaletra. Это показано как графически на фиг. 1 и 2, а также в цифровой форме в табл. 3-5.- 17014446 Таблица 3 Биодоступность лопинавира из желатиновой капсулы Kaletra по сравнению с заявляемой дозированной формой в условиях голода Таблица 4 Биодоступность лопинавира из желатиновой капсулы Kaletra по сравнению с заявляемой дозированной формой в условиях приема пищи с умеренным содержанием жира Таблица 5 Биодоступность лопинавира из желатиновой капсулы Kaletra по сравнению с заявляемой дозированной формой в условиях приема пищи с высоким содержанием жира Например, в табл. 3 показано, что желатиновая капсула Kaletra обеспечиваетAUC в 257,9 мкг в ч/мл от 5-й до 95-й процентили, иCmax в 20,21 мкг/мл от 5-й до 95-й процентили. Напротив, дозированная форма настоящего изобретения обеспечиваетAUC в 141,15 мкг в ч/мл от 5-й до 95-й процентили, иCmax в 11, 98 мкг/мл от 5-й до 95-й процентили. Другими словами, у 90% исследуемых субъектов из табл. 3AUC будет составлять 257,9 мкг в ч/мл иCmax 20,21 мкг/мл после приема состава желатиновой капсулы Kaletra, в то время как после приема дозированной формы настоящего изобретения у 90% исследуемых субъектовAUC будет составлять 141,15 мкг в ч/мл иCmax 11,98 мкг/мл. Возвращаясь к табл. 3, это различие очевидно уже от 25-й до 75-й процентили, где состав желати- 18014446 новой капсулы Kaletra обеспечиваетAUC в 75,53 мкг в ч/мл иCmax 6,36 мкг/мл для 50% исследуемых субъектов. С резким контрастом дозированная форма настоящего изобретения у 50% исследуемых субъектов обеспечиваетAUC в 52,71 мкг в ч/мл иCmax 4,5 мкг/мл. Дозированная форма настоящего изобретения не проявляет эффекта приема пищи. Соотношение"X" из AUC в состоянии сытости к AUC в состоянии голода для лопинавира вычисляют с использованием у нижеприведенной формулы,Вычисление выполняют для каждого отдельного члена исследуемой совокупности в данном испытании. Среднее значение вычисляют сложением значения "X" каждого субъекта и затем разделяют сумму на общее количество субъектов в испытании. Определяют, что если это значение "X" находится в диапазоне от около 0,7 до около 1,43, то дозированная форма не имеет какого-либо эффекта приема пищи. Таким образом, дозированная форма будет обладать по существу одинаковой биодоступностью, независимо от введения на полный или пустой желудок. Соотношение "Y" Cmax в состоянии сытости к Cmax в состоянии голода для лопинавира вычисляют, используя нижеприведенную формулу,Вычисление выполняют для каждого отдельного члена исследуемой совокупности в данном испытании. Среднее значение вычисляют путем сложения значения "Y" каждого субъекта и затем разделяют сумму на общее количество субъектов в испытании. Определяют, что если это значение "Y" находится в диапазоне от около 0,7 до около 1,43, то дозированная форма не имеет какого-либо эффекта приема пищи. Таким образом, дозированная форма будет обладать по существу одинаковой биодоступностью, независимо от введения на полный или пустой желудок. Ниже в табл. 6 лучше показана методика вычисления значений "X" и "Y" отдельных членов исследуемой совокупности, составляющей всего 20 субъектов. Таблица 6 Соотношение Cmax и AUC в состоянии сытости и в состоянии голода для отдельных субъектов В табл. 6 показано, что среднее значение Cmax составляет 1,04 и среднее значение AUC составля- 19014446 ет 1,28. Эти значения независимы в диапазоне от около 0,7 до около 1,43 и показывают, что дозированная форма настоящего изобретения не имеет какого-либо эффекта приема пищи. При проведении ряда исследований, сравнивающих дозированную форму настоящего изобретения с имеющимся в настоящее время на рынке составом желатиновой капсулы Kaletra, также было выявлено,что у дозированной формы настоящего изобретения отсутствуют побочные эффекты или они минимальны. В особенности было выявлено, что дозированная форма настоящего изобретения не имеет побочных эффектов со стороны желудочно-кишечного тракта или они минимальны. Нижеприведенная табл. 7 сравнивает частоту и типы побочных эффектов в плане процента от исследуемой совокупностей при введении дозированных форма настоящего изобретения по сравнению с имеющимся в настоящее время на рынке составом желатиновой капсулы Kaletra. Таблица 7 Процент исследуемой совокупности, страдающий побочными эффектами по типам эффекта ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ лечения ВИЧ/СПИДа, включающий введение субъекту, нуждающемуся в таком лечении,твердой фармацевтической дозированной формы, содержащей нерастворенную форму лопинавира в терапевтически эффективном количестве, причем указанная дозированная форма принимается указанным пациентом без еды или в условиях голодной выдержки. 2. Способ по п.1, в котором указанная дозированная форма содержит твердый раствор или твердую дисперсию лопинавира в матрице, причем указанная матрица содержит по меньшей мере один фармацевтически приемлемый водорастворимый полимер и по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество, причем указанное поверхностно-активное вещество имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10. 3. Способ по п.2, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера имеет температуру стеклования Тс по меньшей мере 50 С. 4. Способ по п.3, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер включает сополимер N-винил пирролидона и винил ацетата и указанное фармацевтически приемлемое поверхностно-активное вещество представляет собой моносложные эфиры жирной кислоты сорбитана. 5. Способ по п.3, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер включает коповидон и указанное поверхностно-активное вещество представляет собой монолаурат сорбитана. 6. Способ по п.5, в котором указанная дозированная форма дополнительно содержит ритонавир. 7. Способ по п.1, в котором указанная дозированная форма содержит твердый раствор лопинавира в матрице, где указанная матрица содержит по меньшей мере один фармацевтически приемлемый водорастворимый полимер и по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество, причем указанное фармацевтически приемлемое поверхностно-активное вещество имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10. 8. Способ по п.7, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера имеет температуру стеклования Тс по меньшей мере 50 С и каждое из указанного по меньшей мере одного фармацевтически приемлемого поверхностно-активного вещества имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10. 9. Способ по п.8, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера выбран из группы, состоящей из гомополимера N-виниллактама,сополимера N-виниллактама, сложного эфира целлюлозы, простого эфира целлюлозы, оксида полиалкилена, полиакрилата, полиметакрилата, полиакриламида, поливинилового спирта, полимера винил ацетата, олигосахарида и полисахарида, и где каждое из указанного фармацевтически приемлемого поверхностно-активного вещества выбрано из группы, состоящей из простого эфира полиоксиэтиленалкила, по- 20014446 лиоксиэтилен алкиларилового простого эфира, сложного эфира полиэтиленгликолевой жирной кислоты,алкиленгликолевого моносложного эфира жирной кислоты, сложного эфира жирной кислоты сахарозы и моносложного эфира жирной кислоты сорбитана. 10. Способ по п.8, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера выбран из группы, состоящей из гомополимера Nвинилпирролидона, сополимера N-винилпирролидона, сополимера N-винилпирролидона и винилацетата,сополимера N-винилпирролидона или винилпропионата, поливинилпирролидона, метилцеллюлозы,этилцеллюлозы, гидроксиалкилцеллюлозы, гидроксипропилцеллюлозы, гидроксиалкилалкилцеллюлозы,гидроксипропилметилцеллюлозы, фталата целлюлозы, сукцината целлюлозы, фталата ацетилцеллюлозы,фталата гидроксипропилметилцеллюлозы, сукцината гидроксипропилметилцеллюлозы, ацетат сукцината гидроксипропилметилцеллюлозы, полиэтиленоксида, полипропиленоксида, сополимера оксида этилена и оксида пропилена, метакриловой кислоты/сополимеры этилакрилата, метакриловой кислоты/сополимера метилметакрилата,бутилметакрилата/сополимера 2-диметиламиноэтил метакрилата,поли(гидроксиалкил акрилата), поли(гидроксиалкил метакрилата), сополимера винилацетата и кротоновой кислоты, частично гидролизованного поливинилацетата, каррагинана, галактоманнана и ксантановой смолы, и где каждое из указанного фармацевтически приемлемого поверхностно-активного вещества выбрано из группы, состоящей из полиоксиэтилен (3) лаурилового эфира, полиоксиэтилен (5) цетилового эфира, полиоксиэтилен (2) стеарилового эфира, полиоксиэтилен (5) стеарилового эфира, полиоксиэтилен(2) нонилфенилового эфира, полиоксиэтилен (3) нонилфенилового эфира, полиоксиэтилен (4) нонилфенилового эфира, полиоксиэтилен (3) октилфенилового эфира, ПЭГ-200 монолаурата, ПЭГ-200 дилаурата,ПЭГ-300 дилаурата, ПЭГ-400 дилаурата, ПЭГ-300 дистеарата, ПЭГ-300 диолеата, монолаурата пропиленгликоля, моностеарата сахарозы, дистеарата сахарозы, монолаурата сахарозы, дилаурата сахарозы,монолаурата сорбитана, моноолеата сорбитана, монопальмитата сорбитана, стеарата сорбитана. 11. Способ по п.8, в котором указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество составляет по меньшей мере 50 мас.% от общего количества поверхностно-активных веществ, содержащихся в дозированной форме. 12. Способ по п.8, в котором указанная дозированная форма содержит 50-85 мас.% от дозированной формы указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера и 2-20 мас.% от дозированной формы указанного по меньшей мере одного фармацевтически приемлемого поверхностно-активного вещества. 13. Способ по п.12, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер включает сополимер N-винилпирролидона и винилацетата и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество включает моносложный эфир жирной кислоты сорбитана. 14. Способ по п.12, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер представляет собой коповидон и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество представляет собой монолаурат сорбитана. 15. Способ по п.14, в котором указанная дозированная форма дополнительно содержит ритонавир. 16. Способ по п.1, в котором при растворении указанной дозированной формы in vitro при использовании аппарата 2 USP (лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С, от около 20 до около 30% лопинавира высвобождается за время от около 0 до около 15 мин, от около 43 до около 63% лопинавира высвобождается за время от около 15 около до 30 мин, от около 61,3 до около 81,7% лопинавира высвобождается за время от около 30 около до 45 мин, от около 75,4 до около 93,2% лопинавира высвобождается за время от около 45 до около 60 мин. 17. Способ по п.1, в котором указанная дозированная форма дополнительно содержит ритонавир,причем при растворении указанной дозированной формы in vitro при использовании аппарата 2 USP (лопастного) с 75 об/мин с 0,06 М POE10LE (простой лауриловый эфир полиоксиэтилена 10) при 37 С, от около 19,8 до около 34,4% ритонавира высвобождается за время от около 0 до около 15 мин, от около 41,6 до около 76,5% ритонавира высвобождается за время от около 15 до около 30 мин, от около 59,4 до около 91,1% ритонавира высвобождается за время от около 30 до около 45 мин, от около 73,4 до около 95% ритонавира высвобождается за время от около 45 до около 60 мин. 18. Способ по п.1, в котором после введения однократной дозы ингибитора протеазы каждому члену исследуемой совокупности отношение среднего значения AUC (в состоянии сытости) к AUC (в состоянии голода) для членов исследуемой совокупности находится в диапазоне от около 0,7 до около 1,43. 19. Способ по п.1, в котором после введения однократной дозы ингибитора протеазы каждому члену исследуемой совокупности отношение среднего значения Cmax (в состоянии сытости) к Cmax (в состоянии голода) для членов исследуемой совокупности находится в диапазоне от около 0,7 до около 1,43. 20. Способ по п.1, в котором после введения указанной дозированной формы каждому члену исследуемой совокупности в условиях голода, различие в показателе AUC лопинавира между 95-й процентилью и 5-й процентилью исследуемой совокупности составляет меньше чем 170.- 21014446 21. Способ по п.1, в котором после введения указанной дозированной формы каждому члену исследуемой совокупности в условиях голода различие в показателе Cmax лопинавира между 95-й процентилью и 5-й процентилью исследуемой совокупности составляет меньше чем 15. 22. Способ по п.1, в котором после введения указанной дозированной формы каждому члену исследуемой совокупности в условиях голода, от 5-й процентили до 95-й процентили значения AUC исследуемой совокупности находятся в диапазоне от около 33 до около 175 мкг в ч/мл. 23. Способ по п.1, в котором указанная нерастворимая форма представляет собой аморфную форму. 24. Способ по п.7, в котором указанный водорастворимый полимер имеет кажущуюся вязкость при растворении в 2% (мас./об.) водном растворе при 20 С, составляющую от около 1 до около 5000 мПас. 25. Способ по п.7, в котором после хранения в течение 6 недель при 40 С и влажности 75% указанная дозированная форма содержит лопинавир в количестве по меньшей мере 98% от исходного содержания. 26. Способ лечения пациента с ВИЧ, включающий введение такому пациенту независимо от приема пищи твердой фармацевтической дозированной формы, причем указанная дозированная форма содержит по меньшей мере один ингибитор ВИЧ протеазы, по меньшей мере один фармацевтически приемлемый водорастворимый полимер и по меньшей мере одно фармацевтически приемлемое поверхностноактивное вещество, в котором указанный ингибитор ВИЧ протеазы представляет собой лопинавир и указанное поверхностно-активное вещество имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10. 27. Способ по п.26, в котором указанная дозированная форма содержит твердый раствор или твердую дисперсию указанного по меньшей мере одного ингибитора ВИЧ протеазы в матрице, где указанная матрица содержит указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество. 28. Способ по п.27, в котором каждый из указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера имеет температуру стеклования Тс по меньшей мере 50 С и каждое из указанного по меньшей мере одного фармацевтически приемлемого поверхностно-активного вещества имеет значение гидрофильно-липофильного баланса (ГЛБ) от 4 до 10. 29. Способ по п.28, в котором указанная дозированная форма содержит 50-85 мас.% от дозированной формы указанного по меньшей мере одного фармацевтически приемлемого водорастворимого полимера и 2-20 мас.% от дозированной формы указанного по меньшей мере одного фармацевтически приемлемого поверхностно-активного вещества. 30. Способ по п.29, в котором указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество составляет по меньшей мере 50 мас.% от общего количества поверхностно-активных веществ, содержащихся в дозированной форме. 31. Способ по п.29, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер включает сополимер N-винилпирролидона и винилацетата и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество включает моносложный эфир жирной кислоты сорбитана. 32. Способ по п.29, в котором указанный по меньшей мере один фармацевтически приемлемый водорастворимый полимер представляет собой коповидон и указанное по меньшей мере одно фармацевтически приемлемое поверхностно-активное вещество представляет собой монолаурат сорбитана. 33. Способ по п.32, в котором указанный твердый раствор или твердая дисперсия представляет собой твердый раствор. 34. Способ по п.33, в котором указанная дозированная форма дополнительно содержит ритонавир.
МПК / Метки
МПК: A61K 31/513, A61K 9/28, A61K 31/426, A61K 9/20
Метки: состав, твёрдый, фармацевтический, дозированный
Код ссылки
<a href="https://eas.patents.su/24-14446-tvyordyjj-farmacevticheskijj-dozirovannyjj-sostav.html" rel="bookmark" title="База патентов Евразийского Союза">Твёрдый фармацевтический дозированный состав</a>
Состав аэрозоля, дозированный ингалятор под давлением и способ его заполнения

Номер патента: 10311
Опубликовано: 29.08.2008
Авторы: Гандертон Дэвид, Льюис Дэвид, Миакин Брайан, Дельканале Маурицио, Пиветти Фаусто
МПК: A61M 15/00, A61K 31/4704, A61K 9/12...
Метки: ингалятор, состав, способ, давлением, дозированный, заполнения, аэрозоля
Формула / Реферат:
1. Состав аэрозоля, который содержит 8-гидрокси-5-[(1R)-1-гидрокси-2-[[(1R)-2-(4-метоксифенил)-1-метилэтил]амино]этил]-2(1Н)-хинолинон или его соль, в частности гидрохлоридную соль, сжиженный пропеллент, представляющий собой гидрофторалкан, сорастворитель, выбранный из фармацевтически приемлемых спиртов, и высококонцентрированную фосфорную кислоту, причём состав находится в виде раствора, в котором...
Фармацевтический состав оланзапина

Номер патента: 8516
Опубликовано: 29.06.2007
Авторы: Банко Иванка, Перц Станка, Коленц Иванка
МПК: A61P 25/18, A61K 31/551, A61K 9/20...
Метки: состав, оланзапина, фармацевтический
Формула / Реферат:
1. Фармацевтический состав, содержащий в качестве активного ингредиента оланзапин или его фармацевтически приемлемую соль, получаемый гомогенным смешиванием (а) оланзапина или его фармацевтически приемлемой соли с (b) моносахаридом, и/или олигосахаридом, и/или их восстановленной или окисленной формой, (с) полисахаридом с последующим прямым прессованием смеси в таблетки в отсутствие какого-либо растворителя. 2. Фармацевтический состав по п.1,...
Твердый или полутвёрдый состав для снижения разрушения зубов

Номер патента: 2972
Опубликовано: 26.12.2002
Автор: Паркер Дейвид Майатт
МПК: A23L 1/304, A61K 7/16
Метки: зубов, снижения, полутвёрдый, разрушения, твердый, состав
Формула / Реферат:
1. Твердый или полутвердый состав для орального применения, содержащий соединение кальция и подкислитель, отличающийся тем, что кальций присутствует в диапазоне от 0,3 до 0,65 молей на моль кислоты, а соотношение кальция и подкислителя в составе выбрано таким, чтобы эффективное значение рН состава было от 3,5 до 4,5. 2. Состав по п.1, в котором кальций присутствует в диапазоне 0,3-0,55 моля на моль кислоты. 3. Состав по любому из пп.1-2, в...
Фармацевтический состав, содержащий хинаприл или фармацевтически приемлемую его соль

Номер патента: 7981
Опубликовано: 27.02.2007
Автор: Эйолфссон Рейнир
МПК: A61K 31/47, A61K 31/40, A61K 38/05...
Метки: фармацевтически, фармацевтический, приемлемую, соль, содержащий, состав, хинаприл
Формула / Реферат:
1. Фармацевтический состав, содержащий: (a) 0,5-50 вес.% хинаприла или любой фармацевтически приемлемой его соли; (b) 5-90 вес.% карбоната щелочного или щелочно-земельного металла; (c) 5-90 вес.% нерастворимой соли гидрофосфата щелочно-земельного металла, при этом состав включает меньше, чем 5 вес.% сахаридного соединения. 2. Состав по п.1, отличающийся тем, что карбонат щелочного или щелочно-земельного металла выбирают из карбоната магния,...
Фармацевтический состав с замаскированным вкусом и способ его получения

Номер патента: 6016
Опубликовано: 25.08.2005
Авторы: Шовен Жозиан, Бекур Филипп, Швабе Детлев
МПК: A61K 9/00
Метки: получения, вкусом, способ, фармацевтический, замаскированным, состав
Формула / Реферат:
1. Фармацевтический состав с замаскированным вкусом, остающимся замаскированным во время приема состава, в частности, в форме суспензии в водном носителе, отличающийся тем, что он содержит, по меньшей мере, следующие элементы: а) смесь, состоящую из целлюлозного полимера, растворимого в органических растворителях, но практически не растворимого в воде при любом значении pH; метакрилового полимера, растворимого в кислой среде и практически не...
Предыдущий патент: Низкомолекулярные ингибиторы mdm2 и их применения
Следующий патент: Способ извлечения металлов из отработанных катализаторов
Случайный патент: Заготовка для пластикового контейнера, разработанного специально для упаковки продуктов питания