Фармацевтическая композиция glp-1
Номер патента: 12287
Опубликовано: 28.08.2009
Авторы: Лакомб Фредерик, Кордеро Риголь Хосе-Антонио, Тобалина Маэстре Мария Долорес, Шериф-Шейк Ролан, Аллоса Миравете Ресуррексион, Донг Чжен Ксин



















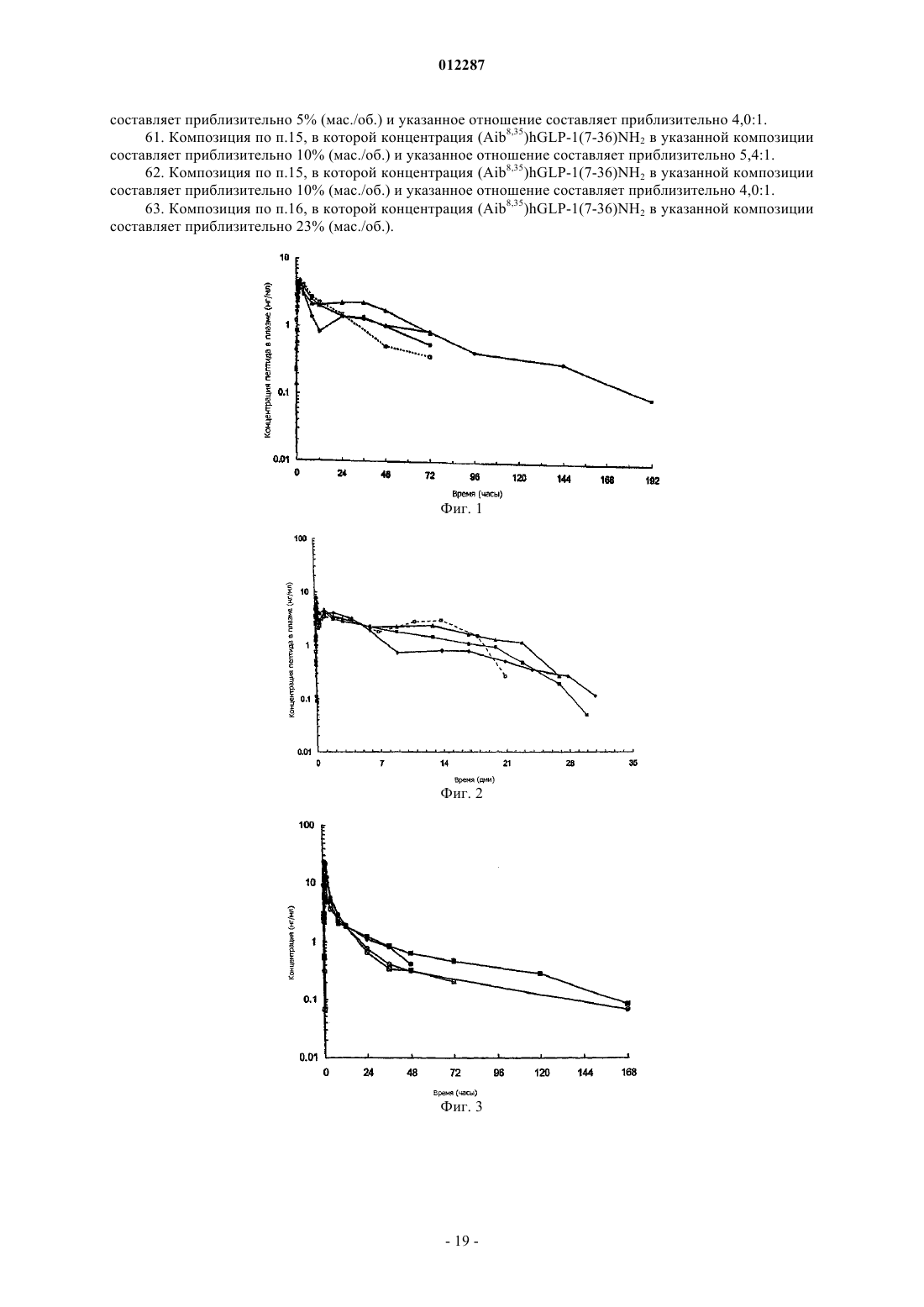
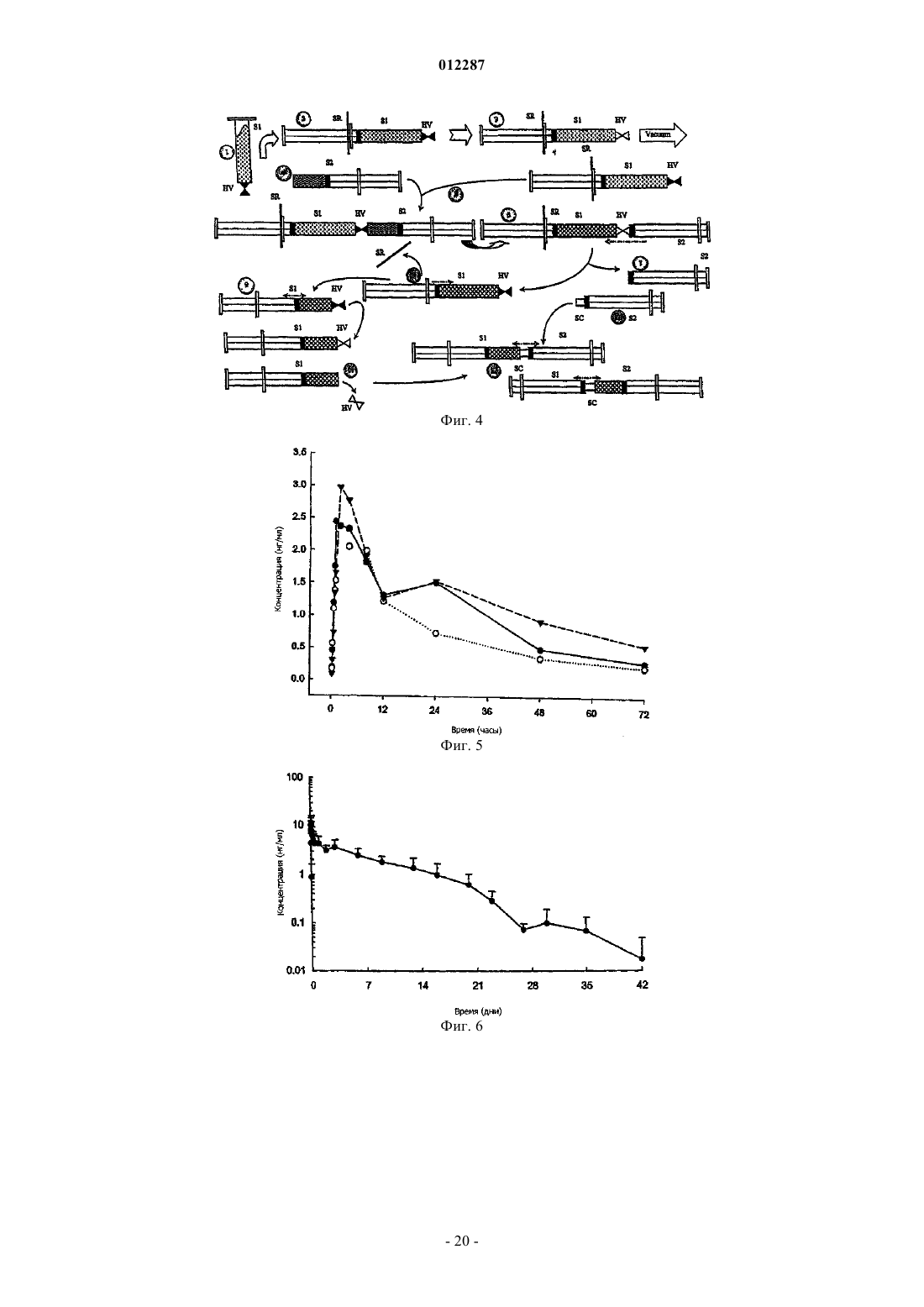
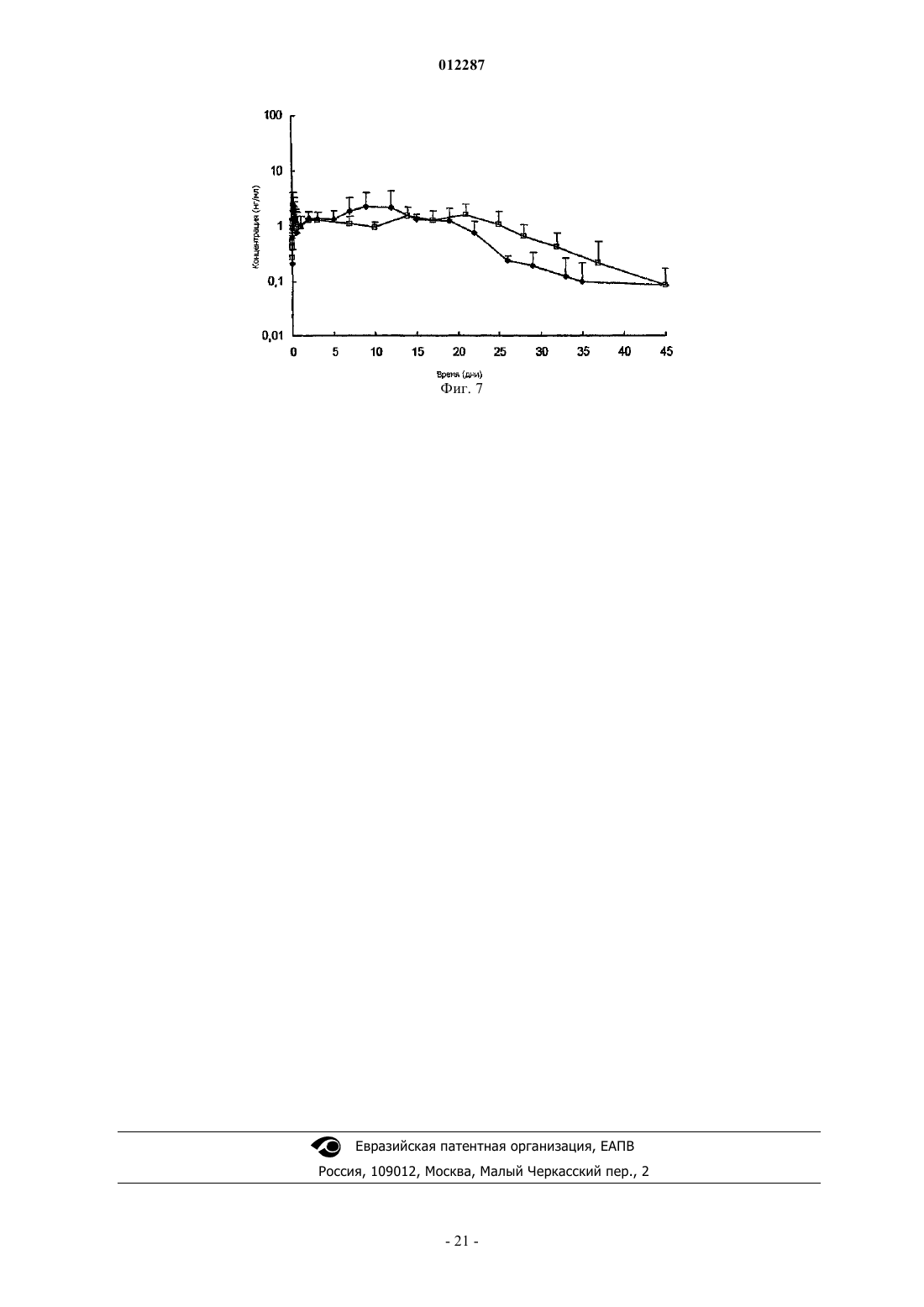
Формула / Реферат
1. Фармацевтическая композиция, включающая аналог, соответствующий формуле
![]()
вместе с цинком и фармацевтически приемлемым носителем или разбавителем, при условии, что указанная композиция не состоит из чистого водного раствора ZnCl2, имеющего рН 4, в котором указанное (Aib8,35)hGLP-1(7-36)NH2 присутствует в концентрации 4 мг/мл, и указанный ZnCl2 присутствует в концентрации 0,5 мг/мл.
2. Фармацевтическая композиция по п.1, в которой указанный цинк присутствует в концентрации от 0,0005 до 50 мг/мл.
3. Фармацевтическая композиция по п.2, в которой указанный цинк присутствует в концентрации от 0,01 до 0,50 мг/мл.
4. Фармацевтическая композиция по п.1, в которой указанный разбавитель включает фармацевтически приемлемый водный раствор.
5. Фармацевтическая композиция по п.4, в которой указанный разбавитель включает стерильную воду.
6. Фармацевтическая композиция по п.1, в которой указанная фармацевтическая композиция включает водную смесь, суспензию или раствор и в которой указанное соединение формулы (I) присутствует в концентрации приблизительно от 0,5 до 30% (мас./мас.).
7. Фармацевтическая композиция по п.6, в которой концентрация указанного соединения формулы (I) в указанной водной смеси, суспензии или растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30% (мас./мас.).
8. Фармацевтическая композиция по п.7, в которой концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 14, 15, 16, 19, 20, 21, 22, 23, 24, 25, 26, 29 или 30% (мас./мас.).
9. Фармацевтическая композиция по п.8, в которой концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 9, 10, 11, 22, 23, 24, 25 или 26% (мас./мас.).
10. Фармацевтическая композиция по п.9, в которой концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 10, 22, 23, 24, 25 или 26% (мас./мас.).
11. Фармацевтическая композиция по п.10, в которой концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1, 2, 5, 10, 23 или 25% (мас./мас.).
12. Фармацевтическая композиция по п.6, в которой молярное отношение указанного соединения формулы (I) к цинку в указанной фармацевтической композиции составляет от приблизительно 6:1 до приблизительно 1:1.
13. Фармацевтическая композиция по п.12, в которой указанное отношение варьирует от приблизительно 5,5:1 до приблизительно 1:1.
14. Фармацевтическая композиция по п.13, в которой указанное отношение варьирует от приблизительно 5,4:1 до приблизительно 1,5:1.
15. Фармацевтическая композиция по п.14, в которой указанное отношение составляет приблизительно 5,4:1, 4,0:1 или 1,5:1.
16. Фармацевтическая композиция по п.15, в которой указанное отношение составляет приблизительно 1,5:1.
17. Фармацевтическая композиция по п.6, в которой указанный цинк присутствует в виде хлорида цинка или ацетата цинка.
18. Фармацевтическая композиция по п.6, в которой указанный ацетат цинка присутствует в виде ZnAc2Ч2H2O.
19. Фармацевтическая композиция по п.1, в которой рН указанной фармацевтической композиции регулируют с помощью основания.
20. Фармацевтическая композиция по п.19, в которой указанное регулирование рН осуществляют с помощью NaOH.
21. Фармацевтическая композиция по п.20, в которой рН указанной фармацевтической композиции регулируют с помощью NaOH так, что при разбавлении до приблизительно 1/2 от начальной концентрации с помощью 0,9% NaCl, получают значение рН приблизительно от 5,0 до 5,5.
22. Фармацевтическая композиция по п.6, в которой рН указанной фармацевтической композиции регулируют с помощью основания.
23. Фармацевтическая композиция по п.22, в которой указанное регулирование рН осуществляют с помощью NaOH.
24. Фармацевтическая композиция по п.23, в которой рН указанной фармацевтической композиции регулируют с помощью NaOH так, что при разбавлении до приблизительно 1/2 от начальной концентрации с помощью 0,9% NaCl получают значение рН приблизительно от 5,0 до 5,5.
25. Фармацевтическая композиция по п.1, в которой соединение формулы (I) высвобождается в организме нуждающегося в этом субъекта в течение пролонгированного периода времени.
26. Фармацевтическая композиция по п.25, в которой указанное высвобождение указанного соединения продолжается по крайней мере от приблизительно 1 до приблизительно 12 ч.
27. Фармацевтическая композиция по п.26, в которой указанное высвобождение указанного соединения продолжается в течение не менее приблизительно 24 ч.
28. Фармацевтическая композиция по п.27, в которой соединение согласно формуле (I) высвобождается в течение не менее приблизительно 48 ч, более предпочтительно не менее приблизительно 72 ч и еще более предпочтительно не менее приблизительно 96 ч.
29. Фармацевтическая композиция по п.28, в которой соединение согласно формуле (I) высвобождается в организме субъекта в течение по меньшей мере от приблизительно 5 до приблизительно 7 дней, более предпочтительно по меньшей мере приблизительно 14 дней, более предпочтительно по меньшей мере приблизительно 2 недель и еще более предпочтительно по меньшей мере приблизительно 4 недель.
30. Фармацевтическая композиция по п.6, в которой соединение согласно формуле (I) высвобождается в организме субъекта, нуждающегося в этом, в течение пролонгированного периода времени.
31. Фармацевтическая композиция по п.30, в которой указанное высвобождение указанного соединения продолжается по меньшей мере от приблизительно 1 до приблизительно 12 ч.
32. Фармацевтическая композиция по п.31, в которой указанное высвобождение указанного соединения продолжается по меньшей мере приблизительно 24 ч.
33. Фармацевтическая композиция по п.32, в которой соединение согласно формуле (I) высвобождается в течение по меньшей мере приблизительно 48 ч, более предпочтительно по меньшей мере приблизительно 72 ч, еще более предпочтительно по меньшей мере приблизительно 96 ч.
34. Фармацевтическая композиция по п.33, в которой соединение согласно формуле (I) высвобождается в организме субъекта в течение по меньшей мере от приблизительно 5 до приблизительно 7 дней, более предпочтительно по меньшей мере приблизительно 14 дней, более предпочтительно по меньшей мере приблизительно 2 недель и еще более предпочтительно по меньшей мере приблизительно 4 недель.
35. Фармацевтическая композиция по любому из пп.25-29, в которой указанным субъектом является млекопитающее, предпочтительно человек.
36. Фармацевтическая композиция по любому из пп.30-34, в которой указанным субъектом является млекопитающее, предпочтительно человек.
37. Способ выявления эффекта агониста GLP-1, указанный способ включает контактирование рецептора лиганда GLP-1(7-36)NH2 с соединением согласно формуле (I), указанное соединение согласно формуле (I) доставляется к указанному рецептору непосредственно или опосредованно с помощью композиции по п.1.
38. Способ выявления эффекта агониста рецептора GLP-1 у нуждающегося в этом субъекта, указанный способ включает введение указанному субъекту фармацевтической композиции по п.1.
39. Способ по п.38, в котором указанный рецептор лиганда GLP-1(7-36)NH2 присутствует у субъекта, являющегося животным.
40. Способ по п.39, в котором указанный субъект является человеком.
41. Способ по п.40, в котором указанный субъект, являющийся человеком, страдает или подвержен риску развития заболевания или состояния, выбранного из группы, включающей диабет I типа, диабет II типа, гестационный диабет, ожирение, булимию, недостаточное чувство насыщения и метаболическое расстройство.
42. Способ по п.41, в котором указанэюх заболевание является диабетом I типа или диабетом II типа.
43. Способ по п.40, в котором указанный субъект, являющийся человеком, страдает или подвержен риску развития заболевания или состояния, выбранного из группы, состоящей из глюкагоном, секреторных расстройств дыхательных путей, артрита, остеопороза, заболевания центральной нервной системы, рестеноза, нейродегенеративного заболевания, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза печени, отека легких, гипертензии и расстройств, при которых желательно уменьшение приема пищи, заболеваний или расстройств центральной нервной системы, болезни Паркинсона, болезни Альцгеймера, болезни Хантингтона, бокового амиотрофического склероза, удара, синдрома дефицита внимания и психоневрологических синдромов, синдрома раздраженной кишки, инфаркта миокарда, удара, острого коронарного синдрома, послеоперационных катаболических изменений, гибернации миокарда или диабетической кардиомиопатии, недостаточного выделения натрия с мочой, повышенной концентрации калия в моче, состояний или расстройств, связанных с токсической гиперволемией, (например, почечной недостаточности, сердечной недостаточности, нефротического синдрома, цирроза печени, отека легких и гипертензии), синдрома поликистоза яичников, респираторного дистресс-синдрома, нефропатии, систолической дисфункции левого желудочка, гастроинтестинальных расстройств, таких как диарея, послеоперационный демпинг-синдром и синдром раздраженной кишки, критической полинейропатии (CIPN), синдрома системной воспалительной реакции (SIRS), дислипидемии, поражения тканей органов, вызванного реперфузией кровотока после ишемии и синдрома фактора риска ишемической болезни сердца (CHDRF).
44. Способ преобразования стволовых клеток/клеток-предшественников печени в функциональные панкреатические клетки, предотвращения деградации b-клеток и стимулирования пролиферации b-клеток, снижения уровней норепинефрина в плазме крови, стимулирования инотропной реакции и увеличения сократимости миокарда, улучшения питания через неалиментарный путь, предварительной подготовки субъекта к проведению эндоскопических процедур и модулирования уровней триглицеридов у нуждающегося в этом субъекта, указанный способ включает введение указанному субъекту фармацевтической композиции по п.1.
45. Способ по п.44, в котором указанный субъект является млекопитающим, более предпочтительно приматом, еще более предпочтительно человеком.
46. Способ выявления эффекта агониста GLP-1, указанный способ включает контакт рецептора лиганда GLP-1(7-36)NH2 с соединением согласно формуле (I), указанное соединение согласно формуле (I) доставляется к указанному рецептору непосредственно или опосредованно с помощью композиции по п.6.
47. Способ выявления эффекта агониста рецептора GLP-1 у нуждающегося субъекта, указанный способ включает введение указанному субъекту фармацевтической композиции по п.6.
48. Способ по п.47, в котором указанный рецептор лиганда GLP-1(7-36)NH2 присутствует у субъекта, являющегося животным.
49. Способ по п.48, в котором указанный субъект является человеком.
50. Способ по п.49, в котором субъект, являющийся человеком, страдает или подвержен риску развития заболевания или состояний, выбранных из группы, состоящей из диабета I типа, диабет II типа, гестационного диабета, ожирения, повышенного аппетита, недостаточного чувства насыщения и метаболического расстройства.
51. Способ по п.50, в котором указанное заболевание является диабетом I типа или диабетом II типа.
52. Способ по п.49, в котором субъект, являющийся человеком, страдает или подвержен риску развития заболевания или состояния, выбранного из группы, состоящей из глюкагона, секреторных расстройств дыхательных путей, артрита, остеопороза, заболевания центральной нервной системы, рестеноза, нейродегенеративного заболевания, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза печени, отека легких, гипертензии и расстройств, при которых желательно уменьшение приема пищи, заболевания или расстройства центральной нервной системы, болезни Паркинсона, болезни Альцгеймера, болезни Хантингтона, бокового амиотрофического склероза, удара, синдрома дефицита внимания и нейропсихиатрических синдромов, синдрома раздраженной кишки, инфаркта миокарда, удара, острого коронарного синдрома, постхирургических катаболических изменений, гибернации миокарда или диабетической кардиомиопатии, недостаточного выделения натрия с мочой, повышенной концентрации калия в моче, состояний или расстройств, связанных с токсической гиперволемией, (например, почечная недостаточность, сердечная недостаточность, нефротический синдром, цирроз печени, отек легких и гипертензия), синдрома поликистоза яичников, респираторного дистресс-синдрома, нефропатии, систолической дисфункции левого желудочка, гастроинтестинальных расстройств, таких как диарея, послеоперационный демпинг-синдром и синдром раздраженной кишки, критической полиневропатии (CIPN), синдрома системной воспалительной реакции (SIRS), дислипидемии, поражения тканей органов, вызванного восстановлением кровотока после ишемии, и синдрома фактора риска ишемической болезни сердца (CHDRF).
53. Способ преобразования стволовых клеток/клеток-предшественников печени в функциональные панкреатические клетки, предотвращения деградации b-клеток и стимулирования пролиферации b-клеток, снижения уровней норепинефрина в плазме крови, стимулирования инотропной реакции и увеличения сократимости миокарда, улучшения питания через не-алиментарный путь, предварительной подготовки субъекта к проведению эндоскопических процедур и модулирования уровней триглицеридов у нуждающегося субъекта, указанный способ включает введение указанному субъекту фармацевтической композиции по п.6.
54. Способ по п.53, в котором указанный субъект является млекопитающим, более предпочтительно приматом, еще более предпочтительно человеком.
55. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 1% (мас./об.).
56. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 2% (мас./об.).
57. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 10% (мас./об.).
58. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 25% (мас./об.).
59. Композиция по п.15, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 5% (мас./об.) и указанное отношение составляет приблизительно 5,4:1.
60. Композиция по п.15, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 5% (мас./об.) и указанное отношение составляет приблизительно 4,0:1.
61. Композиция по п.15, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 10% (мас./об.) и указанное отношение составляет приблизительно 5,4:1.
62. Композиция по п.15, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 10% (мас./об.) и указанное отношение составляет приблизительно 4,0:1.
63. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 23% (мас./об.).
Текст
012287 Родственные заявки Эта заявка испрашивает приоритет предварительной заявки США 60/696142, поданной 30 июня 2006 г. Уровень техники Настоящее изобретение относится к пептидным аналогам глюкагоноподобного пептида-1, к их фармацевтически приемлемым солям, к способам применения таких аналогов для лечения млекопитающих и к фармацевтически эффективным композициям, включающим указанные аналоги. Глюкагоноподобный пептид-1 (7-36)амид (GLP-1) синтезируется в L-клетках кишечника с помощью тканеспецифичного посттрансляционного процессинга предшественника глюкагона препроглюкагона (Vamdell, J.M., et al., J. Histochem Cytochem, 1985:33:1080-6) и высвобождается в кровоток в ответ на прием пищи. Концентрация GLP-1 в плазме повышается с уровня натощак, составляющего приблизительно 15 пмоль/л, до максимального постпрандиального уровня, составляющего 40 пмоль/л. Показано,что для определенного повышения концентрации глюкозы в плазме, увеличение содержания инсулина в плазме приблизительно в три раза больше при оральном введении глюкозы, чем при внутривенном(Kreymann, В., et al., Lancet 1987:2, 1300-4). Это алиментарное усиление высвобождения инсулина, известное как инкретиновый эффект, является первично гуморальным, и GLP-1 считается наиболее активным физиологическим инкретином у человека. В дополнение к инсулинотропному эффекту GLP-1 подавляет секрецию глюкагона, замедляет опорожнение желудка (Wettergren A., et al., Dig Dis Sci 1993:38:665-73) и может усиливать периферическое использование глюкозы (D'Alessio, D.A. et al., J. ClinInvest 1994:93:2293-6). В 1994 г. было высказано предположение о терапевтическом потенциале GLP-1, исходя из наблюдения, что единичная подкожная (s/c) доза GLP-1 может полностью нормализовать постпрандиальный уровень глюкозы у пациентов с инсулиннезависимым диабетом (NIDDM) (Gutniak, M.K., et al., DiabetesCare 1994:17:1039-44). Полагалось, что этот эффект опосредован как увеличением высвобождения инсулина, так и уменьшением секреции глюкагона. Более того, внутривенное введение GLP-1 замедляло постпрандиальное освобождение желудка у пациентов с NIDDM (Williams, В., et al., J. Clin Endo Metab 1996:81:327-32). В отличие от сульфонилуреаз, инсулинотропное действие GLP-1 зависит от концентрации глюкозы в плазме (Holz, G.G. 4th, et al., Nature 1993:361:362-5). Таким образом, отсутствие GLP-1 опосредованного высвобождения инсулина при низкой концентрации глюкозы в плазме защищает от тяжелой гипогликемии. Эта комбинация действий придает GLP-1 уникальный набор терапевтических преимуществ перед другими агентами, использовавшимися ранее для лечения NIDDM. Во многих исследованиях показано, что в случае назначения здоровым субъектам GLP-1 сильно влияет на гликемические уровни, а также на концентрации инсулина и глюкагона (Orskov, С., Diabetologia 35:701-711, 1992; Hoist, J.J., et al., Potential of GLP-1 in diabetes management in Glucagon III, Handbookof Experimental Pharmacology, Lefevbre PJ, Ed. Berlin, Springer Verlag, 1996, p. 311-326), эти эффекты являются глюкозозависимыми (Kreymann, В., et al., Lancet ii: 1300-1304, 1987; Weir, G.C., et al., Diabetes 38:338-342, 1989). Более того, он также эффективен у больных диабетом (Gutniak, M., N. Engl J Med 226:1316-1322, 1992; Nathan, D.M., et al., Diabetes Care 15:270-276, 1992), нормализует уровень глюкозы в крови у пациентов с диабетом 2 типа (Nauck, M.A., et al., Diabetologia 36:741-744, 1993) и улучшает гликемический контроль у пациентов с диабетом 1 типа (Creutzfeldt, W.O., et al., Diabetes Care 19:580-586,1996), помимо этого, демонстрирует способность повышать чувствительность к инсулину/уменьшать резистентность к инсулину. GLP-1 и его агонисты были предложены для применения субъектам, имеющим риск развития инсулиннезависимого диабета (см. WO 00/07617), а также для лечения гестационного диабета (публикация патента США 20040266670). В дополнение к вышеизложенному, существует множество применений в терапии млекопитающих,например людей, для которых предложены GLP-1 и его агонисты, включая, но не ограничиваясь ими,улучшение обучаемости, усиление нейропротекции и/или облегчение симптомов заболеваний или расстройств центральной нервной системы, например, через модуляцию нейрогенеза, и, например, болезнь Паркинсона, болезнь Альцгеймера, болезнь Гентингтона, боковой амиотрофический склероз, инсульт,синдром нарушения внимания и нейропсихиатрические синдромы (патенты США 20050009742 и 20020115605); конверсию стволовых клеток/клеток-предшественников печени в функциональные клетки поджелудочной железы (WO 03/033697); предотвращение дегенерации -клеток (патенты США 20040053819 и 20030220251) и стимулирование пролиферации -клеток (публикация патента США 20030224983); лечение ожирения (публикация патента США 20040018975; WO 98/19698); подавление аппетита и стимулирование чувства насыщения (публикация патента США 20030232754); лечение синдрома раздраженного кишечника (WO 99/64060); сокращение заболеваемости и/или смертности, связанных с инфарктом миокарда (публикация патента США 20040162241, WO 98/08531) и ударом (см.WO 00/16797); лечение острого коронарного синдрома, характеризующегося отсутствием инфаркта миокарда с Q-зубцом (публикация патента США 20040002454); уменьшение послеоперационных катаболических изменений (публикация патента США 6006753); лечение гибернированного миокарда или диабетической кардиомиопатии (публикация патента США 20050096276); снижение уровня норэпи-1 012287 нефрина в плазме крови (публикация патента США 20050096276); увеличение выделения натрия с мочой, уменьшение концентрации калия в моче (публикация патента США 20050037958); лечение состояний или расстройств, связанных с токсической гиперволемией, например почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза печени, отека легких и гипертензии (публикация патента США 20050037958); стимулирование инотропной реакции и увеличение сократимости миокарда (публикация патента США 20050037958); лечение синдрома поликистоза яичников (патенты США 20040266678 и 20040029784); лечение дыхательной недостаточности(публикация патента США 20040235726); улучшение питания посредством не алиментарного пути, то есть посредством внутривенных, подкожных, внутримышечных, перитонеальных или других инъекций или инфузий (публикация патента США 20040209814); лечение нефропатии (публикация патента США 20040209803); лечение систолической дисфункции левого желудочка, например, с аномальной фракцией выброса левого желудочка (публикация патента США 20040097411); подавление антродуоденальной перистальтики, например, для лечения или предотвращения гастроинтестинальных расстройств, таких как диарея, послеоперационный демпинг-синдром и спастический колит, и в качестве премедикации при эндоскопических процедурах (публикация патента США 20030216292); лечение критической полиневропатии (CIPN) и синдрома системной воспалительной реакции (SIRS) (публикация патента США 20030199445); модулирование уровней триглицеридов и лечение дислипидемии (патенты США 20030036504 и 20030143183); лечение повреждений тканей органов, вызванных реперфузией кровотока после ишемии (публикация патента США 20020147131); лечение синдрома риска ишемической болезни сердца (CHDRF) (публикация патента США 20020045636); и другие. Однако GLP-1 является метаболически нестабильным, имеет период полувыведения in vivo (t1/2) только 1-2 мин. При наружном применении GLP-1 также быстро распадается (Deacon, С.F., et al., Diabetes 44:1126-1131, 1995). Эта метаболическая нестабильность ограничивает терапевтический потенциал природного GLP-1. Было предпринято несколько попыток улучшения терапевтического потенциалаGLP-1 и его аналогов посредством усовершенствования композиции. Например, в международной патентной публикацииWO 01/57084 описан процесс получения кристаллов аналогов GLP-1, которые считаются эффективными при получении фармацевтических композиций, таких как инъекционные лекарственные средства, включающих кристаллы и фармацевтически приемлемый носитель. Гетерогенные микрокристаллические кластеры GLP-1(7-37)ОН образуются из солевого раствора и исследуются после обработки кристаллов пропиткой цинком и/или m-крезолом (Kim and Haren, Pharma. Res. Vol. 12 No. 11(1995. Необработанную кристаллическую суспензию GLP(7-36)NH2, состоящую из игольчатых кристаллов и бесформенного преципитата, получают из фосфатных растворов, содержащих цинк или протамин (Pridal, et. al., International Journal of Pharmaceutics Vol. 136, pp. 53-59 (1996. В публикации европейского патента ЕР 0619322 А 2 описано получение микрокристаллических форм GLP-1(7-37)ОН с помощью смешивания растворов белков в буфере с рН 7-8,5 с определенными комбинациями солей и полиэтиленгликолей с низкой молекулярной массой (PEG). В патенте США 6566490 описано образование микрокристаллов, помимо других, GLP-1, которые, как считают, помогают при изготовлении очищенных пептидных продуктов. В патенте США 6555521 (US '521) раскрыты кристаллы GLP-1, имеющие четырехугольный плоский стержень или форму пластины, которые, как полагают, улучшают чистоту и проявляют пролонгированную активность in vivo. В US '521 сообщается, что такие кристаллы относительно однородны и сохраняются в суспензии в течение большего периода времени, чем предшествующие кристаллические кластеры и аморфные кристаллические суспензии, которые, как полагают, быстро образуют осадок, агрегируют и группируются вместе, закупоривают иглы шприцов и обычно ухудшают непрогнозируемое дозированное введение. Биодеградируемый триблоксополимер сополимер[(dl-лактид-о-гликолид)этиленгликоль(лактид-о-гликолид)] был предложен для применения в композиции с контролируемым высвобождениемGLP-1. Однако подобно другим полимерным системам, производство триблоксополимера включает сложные протоколы и образование неустойчивых частиц. Подобным образом, биодеградируемые полимеры, например сополимер молочной и гликолевой кислот (PLGA), были предложены для применения в композициях пептидов с замедленной доставкой. Однако от применения в области техники таких биодеградируемых полимеров отказались, поскольку эти полимеры обычно имеют плохую растворимость в воде и требуют органических не смешивающихся с водой растворителей, например метиленхлорида, и/или жестких условий изготовления в процессе производства. Считается, что такие органические растворители и/или жесткие условия изготовления увеличивают риск индукции конформационного изменения интересующего пептида или протеина, в результате чего уменьшается структурная прочность и нарушается биологическая активность (Choi et al., Pharm.Research, Vol. 21, No. 5, (2004. У полокапсомеров обнаружили подобные недостатки (Id.). Композиции GLP-1, описанные в предыдущих ссылках, менее чем идеальны для получения фармацевтических композиций GLP, поскольку они имеют склонность к захвату примесей и/или иным образом сложны для воспроизводства и применения. Кроме того, известно, что аналоги GLP вызывают тошноту при высоких концентрациях, поэтому существует необходимость в обеспечении замедленного эффекта лекарственного средства при уменьшении начальных концентраций в плазме. Следовательно, существу-2 012287 ет потребность в композициях GLP-1, которые можно более легко и безопасно производить, которые можно более легко и воспроизводимо вводить пациенту и которые обеспечивают пониженные начальные концентрации в плазме для уменьшения или устранения нежелательных побочных эффектов. Сущность изобретения Сущность изобретения может быть изложена ниже, а также в формуле изобретения. Соответственно, первой целью изобретения является предоставление фармацевтической композиции, включающей аналог GLP-1 в соответствии с формулой (I) или его фармацевтически приемлемой соли, получение указанной композиции обеспечивает лучшее производство, применение, фармакокинетические и фармакодинамические свойства, а также уменьшение отрицательных побочных эффектов. Предпочтительно фармацевтическая композиция изобретения не состоит из чистого водного раствора ZnCl2, имеющего рН 4, в котором указанный [Aib8,35]hGLP-1(736)NH2 присутствует в концентрации 4 мг/мл, а указанный ZnCl2 присутствует в концентрации 0,5 мг/мл. В первом аспекте указанной первой цели изобретение предоставляет фармацевтическую композицию, имеющую улучшенный профиль высвобождения лекарственного средства, предпочтительно с уменьшенным начальным импульсом. Во втором аспекте указанной первой цели изобретение предоставляет фармацевтическую композицию, включающую соединение формулы (I), имеющее пролонгированную продолжительность действия. В третьем аспекте указанной первой цели изобретение предоставляет фармацевтическую композицию, включающую соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель или разбавитель. Предпочтительно указанный носитель или разбавитель включает воду. В первом предпочтительном варианте осуществления указанного третьего аспекта указанной первой цели указанная фармацевтическая композиция далее включает цинк. Наиболее предпочтительно,цинк в упомянутой фармацевтической композиции присутствует в концентрации от приблизительно 0,0005 до 50 мг/мл. Еще более предпочтительно цинк в указанной фармацевтической композиции присутствует в концентрации от приблизительно 0,01 до 0,50 мг/мг. Более предпочтительно указанная фармацевтическая композиция включает разбавитель, при этом указанный разбавитель включает фармацевтически приемлемый водный раствор. Разбавитель может включать стерильную воду. Более предпочтительно упомянутая фармацевтическая композиция включает водную смесь, суспензию или раствор, в которой указанное соединение формулы (I) присутствует в концентрации приблизительно от 0,5 до 30% (мас./мас.). Более предпочтительно концентрация указанного соединения формулы (I) в водной смеси, суспензии или растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30% (мас./мас.). Более предпочтительно концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 14, 15, 16, 19, 20, 21, 22, 23, 24, 25, 26, 29 или 30% (мас./мас.). Еще более предпочтительно концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 9, 10, 11, 22, 23, 24, 25 или 26% (мас./мас.). Еще более предпочтительно концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 10, 22, 23, 24, 25 или 26% (мас./мас.). Еще более предпочтительно концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1,2, 5, 10, 23 или 25% (мас./мас). Под "приблизительно" понимается следующее: для концентраций от приблизительно 0,5 до приблизительно 4% желаемый интервал составляет 0,5% от заданного значения (например, от 0,5 до 1,5% соответствует приблизительно 1%); для заданных концентраций от приблизительно 5% и выше желаемый интервал составляет 20% от заданного значения (например, от 8 до 12% соответствует приблизительно 10%). Во втором предпочтительном варианте осуществления указанного третьего аспекта первой цели указанная фармацевтическая композиция далее включает цинк, при этом молярное отношение указанного соединения формулы (I) к цинку в указанной фармацевтической композиции варьирует от приблизительно 6:1 до приблизительно 1:1. Более предпочтительно указанное отношение варьирует от приблизительно 5,5:1 до приблизительно 1:1. Еще более предпочтительно указанное отношение варьирует от приблизительно 5,4:1 до приблизительно 1,5:1. Еще более предпочтительно указанное отношение составляет приблизительно 5,4:1, 4,0:1 или 1,5:1. Наиболее предпочтительно указанное отношение составляет приблизительно 1,5:1. В этом аспекте изобретения под термином приблизительно имеется в виду отношение 1,5:110% от каждого заданного значения, соответственно ожидаемые отношения включают отношения, входящие в этот интервал, например, 1,35-1,65:0,85-1,15. Желательно, чтобы в указанном втором предпочтительном варианте осуществления указанного третьего аспекта первой цели изобретения концентрация [Aib8,35]hGLP-1(7-36)NH2 в указанной фармацевтической композиции составляла приблизительно 1% (вес./об.), и молярное отношение [Aib8,35]hGLP1(7-36)NH2 к цинку составляло приблизительно 1,5:1. Также желательно, чтобы в указанном втором-3 012287 предпочтительном варианте осуществления указанного третьего аспекта первой цели изобретения концентрация [Aib8,35]hGLP-1(7-36)NH2 в указанной фармацевтической композиции составляла приблизительно 2% (вес./об.) и молярное отношение [Aib8,35]hGLP-1(7-36)NH2 к цинку составляло приблизительно 1,5:1. Более того, желательно, чтобы в указанном втором предпочтительном варианте осуществления указанного третьего аспекта первой цели изобретения концентрация [Aib8,35]hGLP-1(7-36)NH2 в указанной фармацевтической композиции составляла приблизительно 10% (вес./об.) и молярное отношение[Aib8,35]hGLP-1(7-36)NH2 к цинку составляло приблизительно 1,5:1. Еще более предпочтительно, чтобы в указанном втором предпочтительном варианте осуществления указанного третьего аспекта первой цели изобретения концентрация [Aib8,35]hGLP-1(7-36)NH2 в указанной фармацевтической композиции составляла приблизительно 23 или 25% (вес./об.), а молярное отношение [Aib8,35]hGLP-1(7-36)NH2 к цинку составляло приблизительно 1,5:1. Желательно, чтобы в указанном втором предпочтительном варианте осуществления указанного третьего аспекта первой цели изобретения концентрация [Aib8,35]hGLP-1(7-36)NH2 в указанной фармацевтической композиции составляла приблизительно 5% (вес./об.), а указанное соотношение составляло приблизительно 5,4:1. Также предпочтительно, чтобы в указанном втором предпочтительном варианте осуществления указанного третьего аспекта первой цели изобретения концентрация [Aib8,35]hGLP-1(736)NH2 в указанной фармацевтической композиции составляла приблизительно 5% (вес./об.), а указанное соотношение составляло приблизительно 4,0:1. Также желательно, чтобы в указанном втором предпочтительном варианте осуществления указанного третьего аспекта первой цели изобретения концентрация[Aib8,35]hGLP-1(7-36)NH2 в указанной фармацевтической композиции составляла приблизительно 10%(вес./об.) и указанное соотношение составляло приблизительно 5,4:1. Еще более желательно, чтобы в указанном втором предпочтительном варианте осуществления указанного третьего аспекта первой цели изобретения концентрация [Aib8,35]hGLP-1(7-36)NH2 в указанной фармацевтической композиции составляла приблизительно 10% (вес./об.) и указанное соотношение составляло приблизительно 4,0:1. Желательно, чтобы в указанном втором предпочтительном варианте осуществления указанного третьего аспекта указанной первой цели цинк был представлен в виде хлорида цинка или ацетата цинка. Более предпочтительно, чтобы указанный ацетат цинка был представлен в виде ZnAc22H2O. Предпочтительно, чтобы в указанных первом и втором предпочтительных вариантах осуществления третьего аспекта первой цели, рН указанной фармацевтической композиции регулировали на повышение с помощью основания. Более предпочтительно, чтобы упомянутое регулирование рН осуществляли с помощью NaOH. Еще более предпочтительно, чтобы рН в данной фармацевтической композиции регулировали с помощью NaOH так, чтобы при разбавлении приблизительно до 1/2 от начальной концентрации с помощью 0,9% NaCl, было получено значение рН приблизительно от 5,0 до 5,5 с помощью прямого потенциометрического определения. Специалисту в области фармацевтических композиций ясно, что рН композиции изобретения можно регулировать даже в более широких пределах, чем описано выше, с помощью соответствующих фармацевтически приемлемых кислот и оснований. Подобное дальнейшее регулирование рН конечной композиции предусматривает модулирующие параметры, такие как, например, концентрация пептидов, концентрация цинка и профиль высвобождения in vivo. В первом предпочтительном варианте осуществления указанного второго аспекта указанной первой цели в изобретении описана фармацевтическая композиция согласно третьему аспекту, включающая,независимо от наличия каждого, каждый из указанных предпочтительных вариантов осуществления третьего аспекта, в котором композицию составляют так, что соединение согласно формуле (I) высвобождается в организме нуждающегося субъекта, например млекопитающего, предпочтительно человека, в течение пролонгированного периода времени. Предпочтительно, чтобы высвобождение указанного соединения продолжалось по крайней мере в течение 1 ч, более предпочтительно в течение не менее 4, 6,12 или 24 ч. Еще более предпочтительно, чтобы указанная композиция была составлена так, чтобы соединение согласно формуле (I) высвобождалось в организме субъекта в течение не менее 36, 48, 60, 72, 84 или 96 ч. Еще более предпочтительно, чтобы указанная композиция была составлена так, чтобы соединение согласно формуле (I) высвобождалось в организме субъекта в течение не менее приблизительно 5, 6, 7, 8, 9,10, 11, 12, 13 или 14 дней. Еще более предпочтительно, чтобы указанная композиция была составлена так, чтобы соединение согласно формуле (I) высвобождалось в организме субъекта в течение приблизительно 2, 3 или 4 недель. Еще более желательно, чтобы указанная композиция была составлена так, чтобы соединение согласно формуле (I) высвобождалось в организме субъекта в течение не менее приблизительно 1, 1,5, 2 или 3 месяцев или дольше. Второй целью настоящего изобретения является предоставление способа достижения эффекта агониста GLP-1, указанный способ включает контактирование рецептора лиганда GLP-1(7-36)NH2 с соединением согласно формуле (I), указанное соединение согласно формуле (I) доставляется к указанному рецептору непосредственно или опосредованно с помощью композиции, согласно указанному третьему аспекту, включающему, независимо от наличия каждого, каждый из указанных предпочтительных вариантов осуществления указанного третьего аспекта.-4 012287 В первом варианте осуществления указанной второй цели изобретения рецептор лиганда GLP-1(736)NH2 присутствует в организме субъекта, являющегося животным, предпочтительно в организме примата, более предпочтительно в организме человека. Таким образом, в этом варианте осуществления настоящее изобретение предоставляет способ достижения эффекта агониста рецептора GLP-1 у нуждающегося субъекта, который включает в себя введение указанному субъекту композиции настоящего изобретения, в которой указанная композиция включает эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли. В наиболее предпочтительном варианте осуществления указанной второй цели изобретения, указанный субъект является человеком, страдающим или имеющим риск развития заболевания или состояния, выбранного из группы, включающей диабет I типа, диабет II типа, гестационный диабет, ожирение,повышенный аппетит, недостаточное чувство насыщения и метаболическое расстройство. Предпочтительно указанное заболевание является диабетом I типа или диабетом II типа. В другом наиболее предпочтительном варианте осуществления указанной второй цели изобретения,указанный субъект является человеком, страдающим или имеющим риск развития заболевания или состояния, выбранного из группы, включающей диабет I типа, диабет II типа, ожирение, глюкагоному, секреторные нарушения дыхательных путей, артрит, остеопороз, заболевание центральной нервной системы, рестеноз, нейродегенеративное заболевание, почечную недостаточность, застойную сердечную недостаточность, нефротический синдром, цирроз печени, отек легких, гипертензию и расстройства, при которых желательно уменьшение потребления пищи, заболевание или расстройство центральной нервной системы (например, посредством модуляции нейрогенеза, и, например, болезнь Паркинсона, болезнь Альцгеймера, болезнь Гентингтона, боковой амиотрофический склероз, инсульт, синдром нарушения внимания и нейропсихиатрические синдромы), синдром раздраженной кишки, инфаркт миокарда (например, уменьшение заболеваемости и/или смертности, связанных с этим заболеванием), инсульт, острый коронарный синдром (например, характеризуемый отсутствием Q-зубца) при инфаркте миокарда,послеоперационные катаболические изменения, гибернированный миокард или диабетическая кардиомиопатия, недостаточное выделение натрия с мочой, повышенную концентрацию калия в моче, состояния или расстройства, связанные с токсической гиперволемией (например, почечная недостаточность,застойная сердечная недостаточность, нефротический синдром, цирроз печени, отек легких и гипертензия), синдром поликистоза яичников, дыхательную недостаточность, нефропатию, систолическую дисфункцию левого желудочка (например, с патологической фракцией выброса левого желудочка), гастроинтестинальные расстройства, такие как диарея, послеоперационный демпинг-синдром и синдром раздраженной кишки (то есть с помощью угнетения антродуоденальной перистальтики), критическую болезненную полиневропатию (CIPN), синдром системной воспалительной реакции (SIRS), дислипидемию,повреждение тканей внутренних органов, вызванное реперфузией кровотока после ишемии, и синдром фактора риска ишемической болезни сердца (CHDRF). В другом аспекте упомянутой второй цели в изобретении описан способ превращения стволовых клеток/клеток-предшественников печени в функциональные клетки поджелудочной железы, предотвращения дегенерации -клеток и стимуляции пролиферации -клеток, снижение уровня норэпинефрина в плазме крови, стимуляции инотропной реакции и усиления сердечной сократимости, улучшения питания через не алиментарный путь (то есть посредством внутривенных, подкожных, внутримышечных, перитонеальных или других инъекций или инфузий), предварительного лечения субъекта перед эндоскопическими процедурами и модулирования уровней триглицеридов у нуждающегося субъекта, вышеуказанный способ включает введение субъекту композиции настоящего изобретения, включающей эффективный объем соединения формулы (I) или его фармацевтически приемлемой соли. Желательно, чтобы указанный субъект был млекопитающим, более предпочтительно приматом, еще более предпочтительно человеком. Краткое описание чертежей На фиг. 1 представлены профили плазмы (средние значения), полученные после единичного подкожного (s.c.) введения собаке приблизительно 1 мг [Aib8,35]hGLP-1(7-36)NH2. В каждом случае пептид вводят в виде водной цинковой композиции, включающей приблизительно 1% (мас./об.) пептида и имеющей молярное отношение пептид:цинк приблизительно 1,5:1. Закрашенные квадраты и прозрачные квадраты изображают композиции, в которых рН регулируется с помощью NaOH, как описано здесь; закрашенные треугольники изображают композицию, в которой рН не регулируется с помощью NaOH; закрашенные круги изображают композицию, буферизованную с помощью АсОН/АсО-. На фиг. 2 изображены профили плазмы (средние значения), полученные после единичного подкожного (s.c.) введения собаке приблизительно 15 мг [Aib8,35]hGLP-1(7-36)NH2. В каждом случае пептид вводят в виде водной цинковой композиции, включающей приблизительно 10% (мас./об.) пептида и имеющей молярное отношение пептид:цинк приблизительно 1,5. Закрашенные квадраты и прозрачные квадраты изображают композиции, в которых рН регулируется с помощью NaOH, как описано здесь; закрашенные треугольники изображают композицию, в которой рН не регулируется с помощью NaOH; закрашенные круги изображают композицию, буферизованную с помощью АсОН/АсО-. На фиг. 3 изображены профили плазмы (средние значения), полученные после единичного подкож-5 012287 ного (s.c.) введения собаке приблизительно 1 мг [Aib8,35]hGLP-1(7-36)NH2. В каждом случае пептид вводят в виде полутвердой водной цинковой композиции, как указано далее, закрашенный круг: приблизительно 5% (мас./об.) пептида, молярное отношение пептид:цинк приблизительно 5,4:1, без регулирования рН; прозрачный круг: приблизительно 10% (мас./об.) пептида, молярное отношение пептид:цинк приблизительно 5,4:1, без регулирования рН; прозрачный квадрат: приблизительно 10% (мас./об.) пептида, молярное отношение пептид:цинк приблизительно 5,4:1, без регулирования рН с помощью NaOH; закрашенный квадрат: приблизительно 10% (мас./об.) пептида, молярное отношение пептид:цинк приблизительно 4:1, рН регулируют с помощью NaOH. На фиг. 4 схематически представлены различные устройства, необходимые при изготовлении определенных композиций настоящего изобретения. На фиг. 5 изображены профили плазмы (средние значения), полученные после единичного подкожного (s.c.) введения собаке приблизительно 1 мг [Aib8,35]hGLP-1(7-36)NH2. В каждом случае пептид вводят в виде водной цинковой композиции, включающей приблизительно 2% пептида и имеющей молярное отношение пептид:цинк приблизительно 1,5:1. На фиг. 6 изображены профили плазмы (средние значения), полученные после единичного подкожного (s.c.) введения собаке приблизительно 15 мг [Aib8,35]hGLP-1(7-36)NH2. Пептид вводят в виде полутвердой цинковой композиции, имеющей концентрацию пептида приблизительно 25% и имеющей молярное отношение пептид:цинк приблизительно 4:1. На фиг. 7 изображены профили плазмы (средние значения), полученные после единичного подкожного (s.c.) введения собаке приблизительно 15 мг [Aib8,35]hGLP-1(7-36)NH2. Пептид вводят в виде полутвердой цинковой композиции, имеющей концентрацию пептида приблизительно 23% и имеющей молярное отношение пептид:цинк приблизительно 1,5:1. Подробное описание Пептид данного изобретения описан следующим образом, например, [Aib8,35]hGLP-1(7-36)NH2, с природной последовательностью с замещенными аминокислотами, расположенными между первой парой круглых скобок (например, Aib8,35 означает, что Aib замещена Ala8 и Gly35 в hGLP-1). Aib - это аббревиатура -аминоизомасляной кислоты. Аббревиатура GLP-1 означает глюкагоноподобный пептид-1;hGLP-1 означает человеческий глюкагоноподобный пептид-1. Числа между второй парой круглых скобок относятся к числу аминокислот, присутствующих в пептиде (например, hGLP-1(7-36) относится к аминокислотам с 7 по 36 из пептидной последовательности человеческого GLP-1). ПоследовательностьhGLP-1(7-37) перечислена в Mojsov, S., Int. J. Peptide Protein Res., 40, 1992, pp. 333-342. Обозначение NH2 в hGLP-1(7-36)NH2 указывает, что С-окончание пептида амидировано. hGLP-1(7-36) означает, что Сокончание является свободной кислотой. В hGLP-1(7-38) остатки в положениях 37 и 38 представлены,соответственно, Gly и Arg, если не обозначено иначе. Пептиды, применяемые в данном изобретении,можно предпочтительно применять в форме фармацевтически приемлемых солей. Примеры подобных солей включают, но не ограничиваясь ими, соли, образованные из органических кислот (например, уксусной, молочной, малеиновой, лимонной, яблочной, аскорбиновой, янтарной, бензойной, метансульфоновой, толуолсульфоновой или памовой кислоты), неорганических кислот (например, соляной кислоты,серной кислоты или ортофосфорной кислоты) и полимерных кислот (например, дубильной кислоты, карбоксиметилцеллюлозы, полимолочной, полигликолевой или сополимеров полимолочной-гликолевой кислот). Обычный способ получения соли пептида настоящего изобретения хорошо известен в области техники и может быть осуществлен с помощью стандартных способов солевого обмена. Следовательно,TFA соль пептида в настоящем изобретении (TFA соль получают в результате очистки пептида с помощью препаративной HPLC, элюирования с TFA, содержащим буферные растворы) может быть преобразована в другую соль, такую как соль уксусной кислоты, с помощью растворения пептида в небольшом количестве 0,25 N водного раствора уксусной кислоты. Полученный раствор наносят на колонку для полупрепаративной HPLC (Zorbax, 300 SB, С-8). Колонку элюируют (1) 0,1N водным раствором ацетата аммония в течение 0,5 ч, (2) 0,25N водным раствором уксусной кислоты в течение 0,5 ч и (3) линейным градиентом (от 20 до 100% раствора в течение более 30 мин) со скоростью потока 4 мл/мин (раствор А является 0,25N водным раствором уксусной кислоты; раствор В является 0,25N уксусной кислотой в ацетонитриле/воде, 80:20). Фракции, содержащие пептид, собирают и лиофилизуют до высушивания. Специалистам в области техники известно о множестве и разнообразии известных и потенциальных областей применения GLP-1 (см. Todd, J.F., et al., Clinical Science, 1998, 95, pp. 325-329; и Todd, J.F. et al.,European Journal of Clinical Investigation, 1997, 27, pp.533-536). Соответственно, применение соединений данного изобретения с целью достижения эффекта агониста может иметь такие же эффекты и области применения, как и собственно GLP-1. Эти многообразные области применения можно обобщить, как указано далее, как лечение диабета I типа, диабета II типа, ожирения, глюкагономы, секреторных расстройств дыхательных путей, метаболического расстройства, артрита, остеопороза, заболеваний центральной нервной системы, рестеноза, нейродегенеративных заболеваний, почечной недостаточности,застойной сердечной недостаточности, нефротического синдрома, цирроза печени, отека легких, гипертензии, расстройств, при которых желательно уменьшение потребления пищи, а также других описанных здесь различных состояний и расстройств. Соответственно, в объем настоящего изобретения входят-6 012287 фармацевтические композиции, как определено здесь, включающие в качестве активного компонента соединение формулы (I). Доза активного компонента в композициях данного изобретения может варьировать; тем не менее,необходимо, чтобы количество активного компонента было таким, чтобы была достигнута подходящая доза. Выбранная доза зависит от желаемого терапевтического эффекта, пути введения и от продолжительности лечения и обычно определяется лечащим врачом. В целом, эффективная доза для активности данного изобретения находится в диапазоне от 110-7 до 200 мг/кг/день, желательно от 110-4 до 100 мг/кг/день, которую можно вводить в виде одной дозы либо разделив на несколько доз. Композиции данного изобретения предпочтительно вводить парентерально, например внутримышечно, внутрибрюшинно, внутривенно, подкожно и так далее. К препаратам согласно данному изобретению для парентерального введения относятся стерильные водные или неводные растворы, суспензии, гели или эмульсии, необходимые для достижения желаемого профиля высвобождения in vivo. К примерам неводных растворителей или наполнителей относятся пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло,желатин, и инъекционные органические эфиры, такие как этилолеат. Такие лекарственные формы могут также включать вспомогательные вещества, такие как консервирующие, смачивающие, эмульгирующие и диспергирующие агенты. Их можно простерилизовать с помощью, например, фильтрации через фильтр, задерживающий бактерии, включения в композиции стерилизующих агентов, облучения композиций или нагревания композиций. Их также можно производить в форме стерильных твердых композиций, которые можно растворять в стерильной воде или в какой-либо другой стерильной инъекционной среде непосредственно перед применением. Синтез пептидов. Пептиды, подходящие для практического осуществления настоящего изобретения, могут быть и были получены с помощью стандартного твердофазного синтеза пептидов. См., например, Stewart, J.M.,et al., Solid Phase Synthesis (Pierce Chemical Co., 2d ed. 1984). Следующие примеры описывают способы синтеза, которые могут быть и были использованы для получения пептидов, с которыми настоящее изобретение может быть предпочтительно практически осуществлено, указанные способы синтеза хорошо известны специалистам в области техники. Другие способы также хорошо известны специалистам в области техники. Примеры предоставлены с целью иллюстрации и никоим образом не служат для ограничения объема данного изобретения. Вос-Ala-OH, Boc-D-Arg(Tos)-ОН и Boc-D-Asp(OcHex) приобрели у Nova Biochem, San Diego, California. Boc-Aun-OH приобрели у Bachem, King of Prussia, PA. Boc-Ava-OH и Boc-Ado-OH приобрели уChem-lmpex International, Wood Dale, IL. Boc-2Nal-OH приобрели у Synthetech, Inc. Albany, OR. Полные названия к другим применяемым здесь аббревиатурам следующие: Boc для tбутилоксикарбонила, HF для фтороводорода, Fm для формила, Xan для ксантила, BzI для бензила, Tos для тозила, DNP для 2,4-динитрофенила, DMF для диметилформамида, DCM для дихлорметана, HBTU для 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметил-уран-гексафторфосфата, DIEA для диизопропилэтиламина, НОАс для уксусной кислоты, TFA для трифторуксусной кислоты, 2ClZ для 2 хлорбензилоксикарбонила, 2BrZ для 2-бромбензилоксикарбонила, ОсНех для О-циклогексила, Fmoc для 9-фторенилметоксикарбонила, HOBt для N-гидроксибензотриазола; РАМ камедь для камеди 4 гидроксиметилфенилацетамидометила; Tris для трис(гидроксиметил)аминометана; и Bis-Tris для бис(2 гидроксиэтил)амино-трис(гидроксиметил)метана (то есть 2-бис(2-гидроксиэтил)амино-2-(гидроксиметил)-1,3-пропандиол). Термин "галоген" включает фтор, хлор, бром и йод. Если не определено иначе, все применяемые здесь технические и научные термины имеют те же значения, под которыми они обычно понимаются специалистами в области техники, к которой относится данное изобретение. Также все публикации, заявки на патенты, патенты и другие ссылки, упомянутые здесь, приведены в качестве ссылки. Пример 1. [Aib8,35]hGLP-1(7-36)NH2. Подробное описание синтеза [Aib8,35]hGLP-1(7-36)NH2 представлено в международной публикации патентаWO 00/34331 (РСТ/ЕР 99/09660), содержание которого приведено здесь в полном объеме. Вкратце, соединение синтезировали на синтезаторе пептидов модель 430 А Applied Biosystems (FosterCity, CA), который был модифицирован для осуществления ускоренного Вос-химического твердофазного синтеза пептидов. (См. Schnolzer, et al., Int. J. Peptide Protein Res., 90:180 (1992. Применяли камедь 4 метилбензгидриламина (MBHA) (Peninsula, Belmont, CA) с замещением 0,91 ммоль/г. Вос-аминокислоты(Bachem, CA, Torrance, CA; Nova Biochem., LaJoIIa, CA) применяли со следующей защитой боковой цепи: Вос-Ala-OH, Boc-Arg(Tos)-ОН, Boc-Asp(OcHex)-OH, Boc-Tyr(2BrZ)-OH, Boc-His(DNP)-OH, Boc-ValOH, Boc-Leu-OH, Boc-Gly-OH, Boc-Gln-OH, Boc-lle-OH, Boc-Lys(2ClZ)-OH, Boc-Thr(Bzl)-OH, BocSer(Bzl)-OH, Boc-Phe-OH, Boc-Aib-OH, Boc-Glu(ОсНех)-ОН и Boc-Trp(Fm)-ОН. Вос-группы удаляли с помощью 100% TFA в течение 21 мин. Вос-аминокислоты (2,5 моль) были предварительно активированы с помощью HBTU (2,0 моль) и DIEA (1,0 мл) в 4 мл DMF и были связаны без предварительной нейтрализации TFA соли пептида-смолы. Время связывания составило 5 мин, исключая время для Boc-Aib-7 012287OH остатков и следующих остатков, Boc-Lys(2ClZ)-ОН и Boc-His(DNP)-ОН, при которых время связывания составило 2 ч. В конце сборки пептидной цепи, камедь обработали раствором 20% меркаптоэтанола/10% DIEA вDMF в течение 230 мин, для удаления DNP-группы на His стороне цепочки. Затем N-концевую Восгруппу удаляли с помощью обработки 100% TFA в течение 22 мин. После нейтрализации пептидасмолы с помощью 10% DIEA в DMF (11 мин), формильные группы на боковой цепи Trp удалили с помощью обработки раствором 15% этаноламина/15% воды/70% DMF в течение 230 мин. Пептид-камедь промыли с помощью DMF и DCM и высушивали при пониженном давлении. Финальное расщепление производили с помощью взбалтывания пептида-камеди в 10 мл HF, содержащего 1 мл анизола и дитиотреитола (24 мг) при 0 С в течение 75 мин. HF удалили с помощью потока азота. Остаток промыли эфиром (610 мл) и извлекли с помощью 4N НОАс (610 мл). Пептидную смесь в водном экстракте очистили с помощью препаративной жидкостной хроматографии высокого давления с обращенной фазой (HPLC) с помощью обращено-фазной VYDAC(R) C18 колонки (Nest Group, Southborough, MA). Колонку элюировали с помощью линейного градиента (от 20 до 50% раствора В более 105 мин) со скоростью потока 10 мл/мин (раствор А=вода, содержащая 0,1% TFA; раствор В=ацетонитрил, содержащий 0,1% TFA). Фракции собрали и проверили с помощью аналитической HPLC. Эти фракции, содержащие чистый продукт, объединили и подвергли лиофилизации для высушивания. В одном примере синтеза данного соединения получили 135 мг белого твердого вещества. Чистота составила 98,6%, что подтвердили с помощью аналитического HPLC анализа. В результате массспектрометрического (MS(ESS анализа с электро-спрей ионизацией получили молекулярную массу 3339,7 (совпадает с вычисленной молекулярной массой 3339,7). Пример 2. Способ получения композиции I. 2.1. Материалы, исходные растворы, вычисления.(i) ZnCl2, pH=3. 1. При перемешивании добавляют 35% HCl к WFI до достижения рН=3. 2. В мерную колбу помещают навеску ZnCl2. При перемешивании добавляют рН=3 HCl до получения конечной концентрации приблизительно 1-4 мг ZnCl2/мл.(ii) ZnCl2, pH=2. 1. При перемешивании добавляют 35% HCl к WFI до достижения рН=2. 2. В мерную колбу помещают навеску ZnCl2. При перемешивании добавляют рН=2 HCl до получения конечной концентрации приблизительно 4-12 мг ZnCl2/мл.(iii) NaOH, от 0,1 до 10 мг/мл. 1. В мерную колбу помещают навеску NaOH. При перемешивании добавляют WFII до достижения конечной концентрации приблизительно 0,1-10 мг NaOH/мл.(iv) аликвота 20 мг высушенного сублимацией (Aib8,35)HGLP-1(7-36)NH2/пробирка. 1. Получают 0,04% (об./об.) слабого раствора уксусной кислоты и WFI. 2. В мерную колбу переливают навеску (Aib8,35)HGLP-1(7-36)NH2 (соль уксусной кислоты). При перемешивании добавляют 0,04% уксусную кислоту в количестве, достаточном, чтобы довести конечную концентрацию до 20 мг (Aib8,35)HGLP-1(7-36)NH2/мл. После стерилизации с помощью фильтрации, с применением фильтров 0,45 мкм, 1 мл аликвоты раствора переносят в пробирки для лиофилизации, высушивают сублимацией, и высушенный продукт хранят при температуре -22 С.(v) Высушенная сублимацией аликвота 50 мг (Aib8,35)HGLP-1(7-36)NH2/пробирка. 1. Получают 0,1% (об./об.) слабого раствора уксусной кислоты и WFI. 2. В мерную колбу помещают навеску [Aib8,35]hGLP-1(7-36)NH2 (соль уксусной кислоты). При перемешивании добавляют 0,1% уксусную кислоту в количестве, достаточном, чтобы довести конечную концентрацию до 50 мг [Aib8,35]hGLP-1(7-36)NH2/мл. После стерилизации с помощью фильтрации, с применением фильтров 0,45 мкм, 1 мл аликвоты раствора переносят в пробирки для лиофилизации, высушивают сублимацией, и высушенный продукт хранят при температуре -22 С. С) Вычисления.(i) Для определения общей массы/объема наполнителя (Е) для композиции где Е - наполнитель (мг); А - содержание чистого пептида (мг); Т - заданная концентрация композиции; например, 2, если заданная концентрация 2%; и Р - концентрация чистого пептида (мг пептида/100 мг композиции). Относительно общего объема наполнителя применяют условие, что 1 мл=1 г.(ii) Для определения объема/массы (W) ZnCl2, который следует добавить к каждому мл или г рас-8 012287 твора композиции:a) W=100% E для композиций, в которых не осуществляют регулирование рН;b) W=80% E для жидких композиций, в которых доля пептида составляет приблизительно 1%, или приблизительно 2%, или до приблизительно 10%, и рН регулируют с помощью основания;c) W=50% E для полутвердых или гелевых композиций, в которых доля пептида составляет приблизительно 1%, или приблизительно 2%, или до приблизительно 10%, и рН регулируют с помощью основания;d) W=66,66% E для полутвердых или гелевых композиций, в которых доля пептида составляет приблизительно 25%, и рН регулируют с помощью основания;e) W=90% E для композиций, в которых пептид восстанавливают из высушенного сублимацией препарата, и рН регулируют с помощью основания.(iii) Для определения объема/массы (W) NaOH, который необходимо добавить к каждому мл или г раствора композиции:a) W=20% E для композиций, в которых доля пептида составляет приблизительно 1%, или приблизительно 2%, или до приблизительно 10%, и рН регулируют с помощью основания;b) W=50% E для полутвердых или гелевых композиций, в которых доля пептида составляет приблизительно 1%, или приблизительно 2%, или до приблизительно 10%, и рН регулируют с помощью основания;c) W=33,33% E для полутвердых или гелевых композиций, в которых доля пептида составляет приблизительно 25%, и рН регулируют с помощью основания;d) W=10% E для композиций, в которых пептид восстанавливают из высушенного сублимацией препарата, и рН регулируют с помощью основания.(iv) Для определения концентрации ZnCl2 (мг/мл или мг/г), которую необходимо применять в каждой композиции: где А - содержание чистого пептида (мг);R - 1,5 для композиций, в которых доля пептида составляет приблизительно 1%, или приблизительно 2%, или приблизительно 10%, или до приблизительно 23%;R - 4,0 для композиций, в которых доля пептида составляет приблизительно 25%; иW - вес (г) или объем (мл) раствора ZnCl2, который необходимо добавлять к каждому г или мл раствора композиции. 2.2. Получение композиций 1-10% высушенного сублимацией пептида и ZnCl2, без регулирования рН. Применяемый здесь термин композиция, включающая определенный процент пептидов, характеризует композицию, включающую определенный вес пептида на общий вес композиции, например, 1% пептида, характеризует композицию, включающую 1 г пептида на 100 г общего веса композиции. Композиции, включающие приблизительно 1%, или приблизительно 2%, до приблизительно 10% пептида получают следующим образом. Высушенные сублимацией образцы [Aib8,35]hGLP-1(7-36)NH2,полученные, как описано выше, тщательно смешивают с исходным раствором ZnCl2 с рН 3, со 100% общего объема наполнителя и соотношением [пептид:Zn] = 1,5:1.[Aib8,35]hGLP-1(7-36)NH2 (см. 2.1 В (v) выше) с 0,45 мл раствора ZnCl2 (3,023 мг/мл; см. 2.1 В (i) выше). Лиофилизированным пептидам и растворам позволяют достичь комнатной температуры. Определенный объем раствора ZnCl2 вводят в пробирку, содержащую лиофилизированный пептид, и позволяют гидратироваться в течение от приблизительно 2 мин для 1 или 2% пептидных композиций, до приблизительно 60 мин для 10% пептидной композиции, или до полной гидратации всех лиофилизированных пептидов, и освобождения раствора от пептидных групп. После гидратации восстановленный пептид встряхивают в течение приблизительно 1 мин. Подходящее количество растворенного пептида может быть удалено для дозирования, например,100 мкл 1% раствора пептида, полученного согласно пункту А, приравнивают к дозе 1 мг, 50 мкл 2% раствора пептида, полученного согласно пункту В, приравнивают к дозе 1 мг, 150 мкл 10% раствора пептида, составленного согласно пункту С, приравнивают к дозе 15 мг и т.д. Используя положения настоящей заявки, специалист в области техники может изменять количество пептида и ZnCl2 для получения композиций, отличных от 1, 2 и 10% композиций, описанных ниже, а также желательных доз. 2.3. Получение композиций с 1-10% лиофилизированного пептида и ZnCl2 с регулированием рН. Композиции, включающие приблизительно 1%, или приблизительно от 2 до приблизительно 10% пептида, получают следующим образом. Лиофилизированные образцы (Aib8,35)hGLP-1(7-36)NH2, полу-9 012287 ченные, как описано, тщательно смешивают с концентрированным раствором ZnCl2 с рН 3 при общем объеме наполнителя 90%. Необходимое значение рН раствора достигают с помощью добавления разбавленного раствора NaOH.A) 1% композиции получают с помощью смешивания 20 мг лиофилизированного (Aib8,35)hGLP-1(736)NH2 (см. 2.1 В (i) выше) с 1,8 мл раствора ZnCl2 (см. 2.1 В (i) выше),B) 2% композиции получают с помощью смешивания 20 мг лиофилизированного (Aib8,35)hGLP-1(736)NH2 (см. 2.1 В (i) выше) с 0,9 мл раствора ZnCl2 (см. 2.1 В (i) выше),C) 10% композиции получают с помощью смешивания 50 мг лиофилизированного (Aib8,35)hGLP1(7-36)NH2 (см. 2.1 В (v) выше) с 0,40 мл раствора ZnCl2 (см. 2.1 В (i) выше). Для достижения заданной концентрации и рН к вышеуказанным растворам добавляют необходимый объем (10% от общего объема наполнителя) разбавленного раствора NaOH. Например: 1% композиция: добавляют 0,2 мл раствора NaOH подходящей концентрации; 2% композиция: добавляют 0,1 мл раствора NaOH подходящей концентрации; 10% композиция: добавляют 0,05 мл раствора NaOH подходящей концентрации. Используя положения настоящей заявки, специалист в области техники может изменять количества пептида и ZnCl2 для получения композиций, отличных от 1, 2 и 10%, описанных ниже. 2.4. Получение жидких композиций с 1-10% пептида и ZnCl2 без регулирования рН. Жидкие композиции, включающие приблизительно 1%, или приблизительно 2% до приблизительно 10% пептида, получают следующим образом. Образцы (Aib8,35)hGLP-1(7-36)NH2 взвешивают и перемешивают с исходным раствором ZnCl2 с рН 3 для достижения заданной концентрации пептида 1, 2 до 10%. После смешивания композицию стерилизуют с помощью фильтрации и сохраняют до последующего применения. 2.5. Получение жидких композиций с 1-10% пептида и ZnCl2 с регулированием рН. Жидкие композиции, включающие приблизительно 1%, или приблизительно 2% до приблизительно 10% пептида, получают следующим образом. Образцы (Aib8,35)hGLP-1(7-36)NH2 взвешивают и перемешивают с исходным раствором ZnCl2 с рН 3 при общем объеме наполнителя 80%. Раствор цинка может содержать либо ZnCl2, либо ZnAc22H2O. Желаемый рН раствора достигают с помощью добавления разбавленного раствора NaOH. Препараты от С 5 до С 13 получают с помощью этого способа. Используя положения настоящей заявки, специалист в области техники может изменять количества пептида и ZnCl2 для получения композиций, отличных от описанных здесь 1, 2 и 10%. 2.6. Получение полутвердых/гелевых композиций с 25% пептида и ZnCl2 без регулирования рН. Полутвердые или гелевые композиции, включающие приблизительно 25% пептида, получают следующим образом. Образцы (Aib8,35)hGLP-1(7-36)NH2 взвешивают и полностью перемешивают с концентрированным раствором ZnCl2 с рН 2 при общем объеме наполнителя 66,66%. Раствор цинка может содержать либо ZnCl2, либо ZnAc22H2O. Препараты С 1 и С 2 получают с помощью этого способа. Конкретнее, полутвердую или гелевую композицию получают с помощью способа смешивания к себе-от себя: а) необходимое количество пептида набирают в цилиндр одноразового шприца S1, предварительно оснащенного специальным двухходовым ручным клапаном HV (внутренний диаметр 0,5 мм), и трубкой,установленной в отверстие Luer шприца;b) поршень шприца скрепляют со стержнем из нержавеющей стали SR;c) HV в S1 соединяют с источником создания вакуума и HV открывают. Через 10 мин HV закрывают;d) раствор цинка аккуратно набирают в цилиндр второго одноразового шприца S2;e) затем S2 соединяют со свободной частью HV;g) HV закрывают и шприц S2 с растворителем отсоединяют, таким образом, вызывая гидратацию порошка пептида в S1;h) SR удаляют и поршень шприца медленно выводят;i) поршень шприца перемещают (к себе-от себя), не открывая HV, так, чтобы порошковая масса полностью пропиталась растворителем;j) двухходовой нержавеющий коннектор SC (внутренний диаметр 1,0 мм) помещают в шприц S2 с трубкой, установленной в отверстие Luer шприца, и его поршень выталкивают до конца;k) HV в S1 открывают для удаления вакуума и затем HV удаляют. Поршень шприца перемещают так, чтобы содержание воздуха в цилиндре шприца было минимальным; иl) S1 и S2 соединяют с помощью SC и композицию перемешивают от S1 до S2 через SC. Используя положения настоящей заявки, специалист в области техники может изменять количества пептида и ZnCl2 для получения композиций, отличных от описанных здесь 25%. 2.7. Получение полутвердых/гелевых композиций с 25% пептида и ZnCl2 с регулированием рН.- 10012287 Полутвердые или гелевые композиции, включающие приблизительно 25% пептида, получают следующим образом. Образцы (Aib8,35)hGLP-1(7-36)NH2 взвешивают и полностью перемешивают с концентрированным раствором ZnCl2 с рН 2 при общем объеме наполнителя 66,66%. Раствор цинка может содержать либо ZnCl2, либо ZnAc22H2O. Желаемый рН раствора достигают с помощью добавления разбавленного раствора NaOH. В этом примере общий объем жидкости, добавляемой к порошку, должен быть разделен между растворами цинка и NaOH. Поэтому концентрацию раствора цинка регулируют так, чтобы общий объем необходимого раствора цинка был уменьшен до 50% от общего объема жидкости, добавляемой к порошку пептида (шаг d). Оставшиеся 50% общего объема жидкости, добавляемой к порошку пептида, добавляют в виде раствора NaOH, как описано ниже. Образцы C3 и С 4 получают с помощью этого способа. Полутвердые или гелевые композиции с отрегулированным рН получают с помощью способа перемешивания "к себе-от себя":a) желаемое количество пептида завешивают в цилиндр одноразового шприца S1, предварительно оснащенного специальным двухходовым ручным клапаном HV (диаметр сечения=0,5 мм), и трубку помещают в отверстие Luer шприца;b) поршень шприца снабжен стержнем из нержавеющей стали SR;d) раствор цинка аккуратно набирают в цилиндр второго одноразового шприца S2;e) затем S2 соединяют со свободной частью HV;g) HV закрывают и шприц S2 с растворителем отсоединяют, таким образом, вызывая гидратацию порошка пептида в S1;h) SR удаляют и поршень шприца медленно выводят;i) поршень шприца перемещают (к себе-от себя), не открывая HV, так, чтобы порошковая масса полностью пропиталась растворителем;j) двухходовой нержавеющий коннектор SC (внутренний диаметр 1,0 мм), помещают в шприц S2 с трубкой, установленной в отверстие Luer шприца, и его поршень выталкивают до конца;k) HV в S1 открывают для удаления вакуума и затем HV удаляют. Поршень шприца перемещают так, чтобы содержание воздуха в цилиндре шприца было минимальным; иl) S1 и S2 соединяют с помощью SC и композицию перемешивают от S1 до S2 через SC;m) после гомогенизации аликвоту перемешанного продукта удаляют для определения концентрации пептида;n) оставшийся промежуточный основной объем продукта точно взвешивают и вычисляют количество раствора NaOH, необходимое для достижения желаемого рН; о) раствор NaOH аккуратно набирают в третий одноразовый шприц S3; и р) поршни шприца подвергают медленному сжатию, чтобы минимизировать содержание воздуха в камерах шприца. Оба шприца соединяют с помощью SC и композицию перемешивают через SC. Используя положения настоящей заявки, специалист в области техники может изменить количества пептида и ZnCl2 для получения композиции с концентрацией, отличной от 25%, описанной здесь. Таблица 1 Показано заданное значение. Фактическое значение было в пределах 5% от заданного во всех случаях. Показано заданное значение. Фактическое значение было в пределах 10% от заданного во всех случаях.- 11012287 3.0. Определение сродства GLP-1 рецептора. Соединение, применяемое для практического осуществления настоящего изобретения, может быть проверено на его способность связываться с рецептором GLP-1 с помощью следующей процедуры. Клеточная культура. Клетки крысиной инсулиномы RIN 5F (АТСС- CRL-2058, American Type Culture Collection, Manassas, VA), экспрессирующие рецептор GLP-1, культивируют на среде Eagle's в модификации Dulbecco(DMEM), содержащей 10% эмбриональную сыворотку теленка, и выдерживают при температуре приблизительно 37 С во влажной атмосфере, состоящей из 5% CO2/95% воздуха. Радиолигандное связывание. Для исследований радиолигандного связывания получают мембраны с помощью гомогенизацииRIN клеток в 20 мл ледяном 50 мкМ Трис-HCl в Brinkman Polytron (Westbury, NY) (настройка 6, 15 с). Гомогенаты дважды промывают с помощью центрифугирования (39000 об./10 мин) и полученные гранулы ресуспендируют в 50 мкМ Трис-HCI, содержащем 2,5 мкМ MgCl2, 0,1 мг/мл бацитрацина (SigmaChemical, St. Louis, МО) и 0,1% BSA. Для анализа аликвоты (0,4 мл) инкубируют с 0,05 нМ (125I)GLP-1(736) (2200 Ci/mmol, New England Nuclear, Boston, MA), с и без 0,05 мл немеченных пептидов конкурирующего теста. После 100 мин выдержки (25 С), связанный (125I)GLP-1(7-36) отделяют от несвязанного с помощью быстрой фильтрации через фильтры GF/C (Brandel, Gaithersburg, MD), которые предварительно пропитывают 0,5% полиэтиленимином. Затем фильтры трижды промывают в 5 мл аликвот ледяного 50 мкМ Трис-HCl, и связанную радиоактивность, задержанную на фильтрах, подсчитывают с помощью гамма-спектрометрии (Wallac LKB, Gaithersburg, MD). Специфическое связывание определяют как общее количество связанного (125I)GLP-1(7-36) минус связанное в присутствии 1000 нМ GLP-l(7-36)(Bachem, Torrence, CA). 4. Определение растворимости в зависимости от рН. 4.1. Определение растворимости соединения в зависимости от рН в буферном солевом растворе. Соединение, которое можно эффективно применять при практическом осуществлении изобретения,исследуют для определения его растворимости в PBS при различных рН и температурах, применяя следующую процедуру. Исходный буферный раствор PBS получают с помощью растворения одной упаковки предварительно перемешанного порошка (SIGMA, Product No.: Р-3813) в 1 л деионизированной воды для получения 10 мкМ фосфатно-буферного солевого раствора с 138 мкМ NaCl, 2,7 мМ KCl и рН 7,4. Буферы PBS с различными значениями рН можно получить с помощью регулирования рН этого исходного раствора с помощью фосфорной кислоты и/или гидроксида натрия. Образцы соединения по 2 мг, которое необходимо исследовать, например 2 мг соединения примера 1, можно завесить в стеклянных пробирках. В каждую пробирку добавляют аликвоты по 50 мкл буфераPBS с известным рН. Раствор интенсивно перемешивают, а в случае необходимости обрабатывают ультразвуком до прозрачного состояния. Для каждого тестируемого значения рН регистрируют общий объем буфера, необходимый для растворения 2 мг соединения, и вычисляют растворимость. Растворы пептида, прозрачные при комнатной температуре (20-25 С), помещают в рефрижератор(4 С) на ночь и затем исследуют растворимость пептида при 4 С. 4.2. Определение растворимости соединения в зависимости от рН в солевом растворе. Соединение, которое можно эффективно применять для практического осуществления изобретения,можно исследовать для определения его растворимости в солевом растворе при различных значениях рН и температурах, применяя следующую процедуру. С помощью растворения 9 г NaCl в одном литре деионизированной воды получают исходный солевой раствор. Солевые растворы с различными значениями рН получают с помощью регулирования рН этого исходного раствора с помощью HCl и/или NaOH. Образцы по 2 мг исследуемого соединения, например 2 мг соединения примера 1, завешивают в стеклянные пробирки. В каждую пробирку добавляют аликвоты по 50 мкл солевого раствора с известным рН. Пробирку интенсивно перемешивают, а в случае необходимости обрабатывают ультразвуком до прозрачного состояния. Для каждого исследуемого рН регистрируют общий объем солевого раствора,необходимый для растворения 2 мг соединения, и вычисляют растворимость. Растворы, прозрачные при комнатной температуре (20-25 С), помещают в рефрижератор (4 С) на ночь и затем исследуют растворимость при 4 С. 4.3. Определение растворимости соединения в солевом растворе при рН 7,0. Соединения, которые можно эффективно применять в практическом осуществлении изобретения,исследуют для определения их растворимости при комнатной температуре в солевом растворе, имеющем рН 7, с помощью следующей процедуры. Солевой раствор получают с помощью растворения 9 г NaCl в одном литре деионизированной воды. Образец 2 мг исследуемого соединения, например соединения примера 1, завешивают в стеклянную пробирку и добавляют аликвоты по 1 мл солевого раствора, при интенсивном перемешивании и воздействии ультразвука, до прозрачного состояния. Затем регистрируют общий объем солевого раствора, необходимый для растворения 2 мг пептида и вычисляют растворимость при комнатной температуре. 4.4. Определение растворимости соединения в солевом растворе при различных рН.- 12012287 Соединения, которые можно эффективно применять в практическом осуществлении изобретения,исследуют для определения их растворимости при комнатной температуре в солевых растворах, имеющих различные значения рН, с помощью следующей процедуры. Исходный солевой раствор получают с помощью растворения 9 г NaCl в одном литре деионизированной воды. Солевые растворы с различными значениями рН получают с помощью обработки аликвот этого исходного солевого раствора с помощью HCl и NaOH. Образец 2 мг исследуемого соединения, например соединения примера 1, завешивают в стеклянную пробирку. Добавляют аликвоты по 50 мкл солевого буфера с известным рН. Раствор интенсивно перемешивают и подвергают воздействию ультразвука, до прозрачного состояния. Регистрируют общий объем буфера, использованного для растворения 2 мг пептида и вычисляют растворимость. 5. Определение растворимости соединения в воде в зависимости от концентрации цинка. Соединение, которое можно эффективно применять в практическом осуществлении изобретения,исследуют для определения его растворимости в воде при рН 7 при различных концентрациях цинка, с помощью следующей процедуры. Исходный раствор цинка получают с помощью растворения ZnCl2 в деионизированной воде в концентрации 100 мг/мл и регулирования рН до 2,7 с помощью HCl. Растворы с различными концентрациями ZnCl2 ("исследуемые растворы цинка") получают с помощью соответствующего разбавления исходного раствора. 1 мг исследуемого соединения, например 1 мг соединения примера 1, растворяют в 250 мкл каждого исследуемого раствора цинка для получения раствора с концентрацией соединения 4 мг/мл. Затем регулируют рН раствора, используя 0,2N NaOH, до формирования видимых белых преципитатов. Осажденный раствор центрифугируют и маточный раствор анализируют с помощью HPLC. Определяют область пика поглощения UV для исследуемого соединения и определяют концентрацию исследуемого соединения в маточном растворе в сравнении с калибровочной кривой. В качестве показательного примера соединения, которое можно применять для практического осуществления изобретения, исследовали соединение примера 1 в непосредственно нижеприведенном анализе и получили следующие результаты (водный солевой раствор, рН 7,0, комнатная температура). Таблица 2 6. Определение pl с помощью гелей IEF. Гели Invitrogen's Novex IEF pH 3-10 можно применять для определения pl пептидов GLP-1. Пептидильные соединения, которые необходимо исследовать, растворяют в воде в концентрации 0,5 мг/мл. 5 мкл полученного раствора каждого соединения смешивают с 5 мкл Novex Sample Buffer 2X (включающим 20 мМ аргинина в виде свободного основания, 20 мМ лизина в виде свободного основания и 15% глицерина) и полученные 10 мкл образца раствора загружают на гель вместе со стандартным образцом белка. Подвижные буферы также получают от Invitrogen, и гель двигается согласно инструкциям изготовителя, в основном, следующим образом: 100 V постоянно в течение 1 ч, затем 200 V постоянно в течение 1 ч, затем 500 V постоянно в течение 30 мин. После этого гель фиксируют в 12% ТСА, содержащем 3,5% сульфосалициловой кислоты в течение 30 мин, и затем окрашивают в течение 2 ч Colloidal Coomassie Blue согласно инструкциям от Novex Colloidal Blue Kit, затем обесцвечивают в воде в течение ночи. Гель сканируют и анализируют с помощью программы Fragment Analysis 1.2. pl неизвестных пептидов вычисляют относительно pl стандартных соединений, имеющих значения pl: 10,7, 9,5, 8,3, 8,0, 7,8,7,4, 6,9, 6,0, 5,3, 5,2, 4,5, 4,2 и 3,5. 7. Исследования на крысах in vivo. Композиции настоящего изобретения можно исследовать для определения их способности активировать и усиливать эффект in vivo, применяя следующие анализы. 7.1. Процедура эксперимента. В день, предшествующий эксперименту, взрослым самцам крыс Sprague-Dawley (Taconic, Germantown, NY), имеющим вес приблизительно 300-350 г, под воздействием хлоргидратного анестетика через- 13012287 яремную вену имплантировали канюлю в правое предсердие. Затем крыс подвергали голоданию в течение 18 ч до инъекции соответствующей исследуемой композиции или контрольного носителя в момент времени 0. Крыс продолжали подвергать голоданию в течение всего эксперимента. 0,5 мг/мл раствора ZnCl2 получают с помощью разбавления 100 мг/мл раствора ZnCl2 в водном растворе HCl, имеющем рН 2,7. 1 мг соединения формулы (I) (Aib8,35)hGLP-1(7-36)NH2 растворяют в 250 мкл этого раствора для получения чистого раствора, содержащего 4 мг/мл соединения и 0,5 мг/мл цинка при рН 4. В нулевой момент времени крысам вводят подкожно (sc) либо (а) непосредственно полученный раствор (Aib8,35)hGLP-1(7-36)NH2, либо контрольный носитель. В обоих случаях объем инъекции очень мал (4-6 мкл), а доза соединения GLP-1, вводимого субъекту, составляет 75 мкг/кг. Через подходящее время после sc инъекций производят забор образцов крови 500 мкл через внутривенную (iv) канюлю, и крысам проводят iv нагрузку глюкозой для теста на наличие усиленной секреции инсулина. Нагрузку глюкозой проводят через 0,25, 1, 6, 12 и 24 ч после инъекции соединения. После забора начального образца крови, iv вводят глюкозу (1 г/кг), и канюлю промывают 500 мкл гепаринизированного солевого раствора (10 U/мл). Затем через 2,5, 5, 10 и 20 мин после инъекции глюкозы производят забор образцов крови по 500 мкл. После каждого забора незамедлительно производят iv инъекцию 500 мкл гепаринизированного солевого раствора (10 U/мл) через канюлю. Образцы крови центрифугируют, из каждого образца удаляют плазму, и образцы хранят при -20 С, пока их не проанализируют на содержание инсулина. Количество инсулина в каждом образце определяют с помощью набора для твердофазного иммуноферментного анализа крысиного инсулина (ELISA) (American Laboratory Products Co., Windham, NH). 7.1.1. Результаты. В течение всех 24 ч эксперимента наблюдалась продолжительная активность, усиливающая действие инсулина, индуцированная с помощью инъекции глюкозы. 8. Анализы in vivo на собаках. В области техники известно множество анализов in vivo, которые позволяют специалисту определить способность композиции вызвать пролонгированное высвобождение активного соединения in vivo. 8.1. 1% Пептидная композиция. В качестве примера получают исследуемую водную композицию, включающую 1% (мас.) соединения с формулой (I) в буферном растворе ZnCl2 (соотношение пептид:Zn составляет 1,5:1,0). Мужских особей собак породы бигль общим числом 6 в возрасте от 42 до 78 месяцев и массой тела от 14 до 21 кг содержат, обеспечивая свободный доступ к воде и однократное ежедневное кормление(приблизительно 400 г сухой стандартной диеты (SAFE 125. За 18 ч до назначения исследуемой композиции собак перестают кормить. Исследуемую композицию вводят с помощью подкожной инъекции в межлопаточную область. Назначенный объем (приблизительно 20 мкл каждому животному) вводили с помощью шприцев Terumo объемом 0,3 мл с 0,33-12 мм (BS=30M2913). Таким образом, достигается теоретическая доза приблизительно 0,2 мг пептида. Периодически производят забор образцов крови, приблизительно через 0, 8, 15,30, 45 мин, 1, 2, 4, 8 и 12 ч и через 1, 2, 3, 4, 5 и 6 дней после введения. После забора образцов кровь быстро охлаждают до центрифугирования, плазму декантируют и быстро замораживают до последующего анализа. Определение концентрации пептида в плазме производят после твердофазной экстракции в режиме офф-лайн, с последующей фазной экстракцией в режиме он-лайн, связанной с LC-MS/MS, и полученные данные обрабатывают с помощью программного обеспечения Analyst v1.2. Композиция демонстрирует пролонгированное высвобождение активного пептида в течение по крайней мере 2 дней. 8.2. 1% Раствор (Aib8,35)hGLP-l(7-36)NH2. Применяя, в основном, тот же способ исследования in vivo, который описан выше, в разделе 8.1,следующие композиции исследуют на их способность высвобождать исследуемый пептид в течение длительного периода времени. Для каждой из следующих четырех композиций концентрация пептида составляет приблизительно 1% (вес/вес.), соотношение пептида к цинку составляет приблизительно 1,5:1, и доза вводимого пептида составляет приблизительно 1 мг. Раствор 8.2. A: (Aib8,35)hGLP-1(7-36)NH2 в растворе, содержащем (i) 90% ZnCl2 (0,298 мг/мл) и (ii) 10% NaOH (0,975 мг/мл). Раствор 8.2. В: (Aib8,35)hGLP-1(7-36)NH2 в растворе ZnCl2 (0,286 мг/мл). Раствор 8.2. С: в основном, подобный раствору 8.2. В, но забуференный с помощью АсОН/АсО-. Раствор 8.2. D: в основном, подобный раствору 8.2. А. Композиции обеспечивают пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как показано на фиг. 1. 8.3. 1% Раствор (Aib8,35)hGLP-1(7-36)NH2. Применяя, в основном, тот же способ исследования in vivo, который описан выше, в разделе 8.1,следующие композиции исследуют на их способность высвобождать исследуемый пептид в течение длительного периода времени. Для следующей композиции концентрация пептида составляет приблизительно 2% (вес./вес.), соотношение пептида к цинку составляет приблизительно 1,5:1, и доза вводимого- 14012287 пептида составляет приблизительно 1 мг. Раствор 8.3. (Aib8,35)hGLP-1(7-36)NH2 в растворе, содержащем (i) 80% ZnCl2 (0,695 мг/мл) и (ii) 20%NaOH (1,75 мг/мл). Композиция обеспечивает пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как показано на фиг. 5. 8.4. 10% Растворы пептида. Применяя, в основном, тот же способ исследования in vivo, который описан выше, в разделе 8.1,следующие композиции исследуют на их способность высвобождать исследуемый пептид в течение длительного периода времени. Для каждой из следующих четырех композиций концентрация пептида составляет приблизительно 10% (вес./вес.), соотношение пептида к цинку составляет приблизительно 1,5:1,и доза вводимого пептида составляет приблизительно 15 мг. Раствор 8.4. A: (Aib8,35)hGLP-1(7-36)NH2 в растворе, содержащем (i) 90% ZnCl2 (3,367 мг/мл) и (ii) 10% NaOH (5,01 мг/мл). Раствор 8.4. В: (Aib8,35)hGLP-1(7-36)NH2 в растворе ZnCl2 (2,993 мг/мл). Раствор 8.4. С: в основном, подобный раствору 8.4. В, но забуференный с помощью АсОН/AcO-. Раствор 8.4. D: в основном, подобный раствору 8.4. А. Композиции обеспечивают пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как показано на фиг. 2. 8.5. Полутвердые композиции. Применяя, в основном, тот же способ исследования in vivo, который описан выше, в разделе 8.1,следующие полутвердые композиции исследуют на их способность высвобождать исследуемый пептид в течение длительного периода времени. Для композиции 8.5. А, концентрация пептида составляет приблизительно 5%, тогда как для композиций 8.5. В, 8.4. С и 8.5. D, концентрация пептида составляет приблизительно 10% (вес./вес.). Соотношение пептида к цинку для композиций 8.5. А, 8.5. В и 8.5. С составляет приблизительно 5,4:1, тогда как для композиции 8.5. D это отношение составляет приблизительно 4,0:1. Для всех четырех композиций доза вводимого пептида составляет приблизительно 1 мг. Композиция 8.5. A: (Aib8,35)hGLP-1(7-36)NH2 в полутвердой композиции, содержащей ZnCl2 (0,40 мг/мл) в WFI. Композиция 8.5. В: в основном подобна композиции 8.5. А, в которой концентрация ZnCl2 отрегулирована на повышение для достижения соотношения пептид:Zn, составляющего приблизительно 5,4:1. Композиция 8.5. С: (Aib8,35)hGLP-1(7-36)NH2 в полутвердом состоянии, содержащая (i) 50% ZnCl2(2,28 мг/мл) и (ii) 50% NaOH (1 мг/мл). Композиции обеспечивают пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как показано на фиг. 3. 8.6. Полутвердые композиции. Применяя, в основном, тот же способ исследования in vivo, который описан выше, в разделе 8.1,следующую полутвердую композицию исследуют на ее способность высвобождать исследуемый пептид в течение длительного периода времени. Эту композицию получают с применением раствора ZnCl2 5,22 мг/мл, при рН 2,0. Предоставляют достаточное количество пептида для получения 25% полутвердой пептидной композиции, имеющей соотношение пептида к цинку приблизительно 4:1. рН композиции регулируют, как представлено здесь, применяя NaOH в концентрации 10 мг/мл. Доза вводимого пептида составляет приблизительно 15 мг. Композиция 8.6 обеспечивает пролонгированное высвобождение (Aib8,35)hGLP-1(7-36)NH2, как показано на фиг. 6. 8.7. Полутвердые композиции. Применяя, в основном, тот же способ исследования in vivo, который описан выше, в разделе 8.1,следующую полутвердую композицию исследуют на ее способность высвобождать исследуемый пептид в течение длительного периода времени. Эту композицию получают с применением раствора ZnCl2 8,5 мг/мл, при рН 2,0. Предоставляют достаточное количество пептида для получения 23% полутвердой пептидной композиции, имеющей соотношение пептида к цинку приблизительно 1,5:1. Композицию получают согласно процессу, описанному выше, в разделе 2.6. Доза вводимого пептида составляет приблизительно 15 мг (соответствует приблизительно 65 мкл композиции). Композиция 8.6 обеспечивает пролонгированное высвобождение (Aib8,35)hGLP-l(7-36)NH2, как показано на фиг. 7. Дальнейшие анализы с различными изменениями раскрытой композиции подобным образом являются объектом анализа in vivo и подтверждают, что композиции настоящего изобретения обеспечивают эффективную платформу для доставки лекарственного средства для соединения формулы (I). Применяя положения настоящей заявки, специалист в области техники может изменять количества пептида, ZnCl2 и рН для получения композиций настоящего изобретения, как здесь описано.- 15012287 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Фармацевтическая композиция, включающая аналог, соответствующий формуле вместе с цинком и фармацевтически приемлемым носителем или разбавителем, при условии, что указанная композиция не состоит из чистого водного раствора ZnCl2, имеющего рН 4, в котором указанное(Aib8,35)hGLP-1(7-36)NH2 присутствует в концентрации 4 мг/мл, и указанный ZnCl2 присутствует в концентрации 0,5 мг/мл. 2. Фармацевтическая композиция по п.1, в которой указанный цинк присутствует в концентрации от 0,0005 до 50 мг/мл. 3. Фармацевтическая композиция по п.2, в которой указанный цинк присутствует в концентрации от 0,01 до 0,50 мг/мл. 4. Фармацевтическая композиция по п.1, в которой указанный разбавитель включает фармацевтически приемлемый водный раствор. 5. Фармацевтическая композиция по п.4, в которой указанный разбавитель включает стерильную воду. 6. Фармацевтическая композиция по п.1, в которой указанная фармацевтическая композиция включает водную смесь, суспензию или раствор и в которой указанное соединение формулы (I) присутствует в концентрации приблизительно от 0,5 до 30% (мас./мас.). 7. Фармацевтическая композиция по п.6, в которой концентрация указанного соединения формулы(I) в указанной водной смеси, суспензии или растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9,10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29 или 30% (мас./мас.). 8. Фармацевтическая композиция по п.7, в которой концентрация указанного соединения формулы(I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 14, 15, 16, 19, 20,21, 22, 23, 24, 25, 26, 29 или 30% (мас./мас.). 9. Фармацевтическая композиция по п.8, в которой концентрация указанного соединения формулы(I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 9, 10, 11, 22, 23, 24, 25 или 26% (мас./мас.). 10. Фармацевтическая композиция по п.9, в которой концентрация указанного соединения формулы(I) в указанном водном растворе составляет приблизительно 1, 2, 3, 4, 5, 6, 10, 22, 23, 24, 25 или 26%(мас./мас.). 11. Фармацевтическая композиция по п.10, в которой концентрация указанного соединения формулы (I) в указанном водном растворе составляет приблизительно 1, 2, 5, 10, 23 или 25% (мас./мас.). 12. Фармацевтическая композиция по п.6, в которой молярное отношение указанного соединения формулы (I) к цинку в указанной фармацевтической композиции составляет от приблизительно 6:1 до приблизительно 1:1. 13. Фармацевтическая композиция по п.12, в которой указанное отношение варьирует от приблизительно 5,5:1 до приблизительно 1:1. 14. Фармацевтическая композиция по п.13, в которой указанное отношение варьирует от приблизительно 5,4:1 до приблизительно 1,5:1. 15. Фармацевтическая композиция по п.14, в которой указанное отношение составляет приблизительно 5,4:1, 4,0:1 или 1,5:1. 16. Фармацевтическая композиция по п.15, в которой указанное отношение составляет приблизительно 1,5:1. 17. Фармацевтическая композиция по п.6, в которой указанный цинк присутствует в виде хлорида цинка или ацетата цинка. 18. Фармацевтическая композиция по п.6, в которой указанный ацетат цинка присутствует в видеZnAc22H2O. 19. Фармацевтическая композиция по п.1, в которой рН указанной фармацевтической композиции регулируют с помощью основания. 20. Фармацевтическая композиция по п.19, в которой указанное регулирование рН осуществляют с помощью NaOH. 21. Фармацевтическая композиция по п.20, в которой рН указанной фармацевтической композиции регулируют с помощью NaOH так, что при разбавлении до приблизительно 1/2 от начальной концентрации с помощью 0,9% NaCl, получают значение рН приблизительно от 5,0 до 5,5. 22. Фармацевтическая композиция по п.6, в которой рН указанной фармацевтической композиции регулируют с помощью основания. 23. Фармацевтическая композиция по п.22, в которой указанное регулирование рН осуществляют с помощью NaOH. 24. Фармацевтическая композиция по п.23, в которой рН указанной фармацевтической композиции регулируют с помощью NaOH так, что при разбавлении до приблизительно 1/2 от начальной концентрации с помощью 0,9% NaCl получают значение рН приблизительно от 5,0 до 5,5.- 16012287 25. Фармацевтическая композиция по п.1, в которой соединение формулы (I) высвобождается в организме нуждающегося в этом субъекта в течение пролонгированного периода времени. 26. Фармацевтическая композиция по п.25, в которой указанное высвобождение указанного соединения продолжается по крайней мере от приблизительно 1 до приблизительно 12 ч. 27. Фармацевтическая композиция по п.26, в которой указанное высвобождение указанного соединения продолжается в течение не менее приблизительно 24 ч. 28. Фармацевтическая композиция по п.27, в которой соединение согласно формуле (I) высвобождается в течение не менее приблизительно 48 ч, более предпочтительно не менее приблизительно 72 ч и еще более предпочтительно не менее приблизительно 96 ч. 29. Фармацевтическая композиция по п.28, в которой соединение согласно формуле (I) высвобождается в организме субъекта в течение по меньшей мере от приблизительно 5 до приблизительно 7 дней,более предпочтительно по меньшей мере приблизительно 14 дней, более предпочтительно по меньшей мере приблизительно 2 недель и еще более предпочтительно по меньшей мере приблизительно 4 недель. 30. Фармацевтическая композиция по п.6, в которой соединение согласно формуле (I) высвобождается в организме субъекта, нуждающегося в этом, в течение пролонгированного периода времени. 31. Фармацевтическая композиция по п.30, в которой указанное высвобождение указанного соединения продолжается по меньшей мере от приблизительно 1 до приблизительно 12 ч. 32. Фармацевтическая композиция по п.31, в которой указанное высвобождение указанного соединения продолжается по меньшей мере приблизительно 24 ч. 33. Фармацевтическая композиция по п.32, в которой соединение согласно формуле (I) высвобождается в течение по меньшей мере приблизительно 48 ч, более предпочтительно по меньшей мере приблизительно 72 ч, еще более предпочтительно по меньшей мере приблизительно 96 ч. 34. Фармацевтическая композиция по п.33, в которой соединение согласно формуле (I) высвобождается в организме субъекта в течение по меньшей мере от приблизительно 5 до приблизительно 7 дней,более предпочтительно по меньшей мере приблизительно 14 дней, более предпочтительно по меньшей мере приблизительно 2 недель и еще более предпочтительно по меньшей мере приблизительно 4 недель. 35. Фармацевтическая композиция по любому из пп.25-29, в которой указанным субъектом является млекопитающее, предпочтительно человек. 36. Фармацевтическая композиция по любому из пп.30-34, в которой указанным субъектом является млекопитающее, предпочтительно человек. 37. Способ выявления эффекта агониста GLP-1, указанный способ включает контактирование рецептора лиганда GLP-1(7-36)NH2 с соединением согласно формуле (I), указанное соединение согласно формуле (I) доставляется к указанному рецептору непосредственно или опосредованно с помощью композиции по п.1. 38. Способ выявления эффекта агониста рецептора GLP-1 у нуждающегося в этом субъекта, указанный способ включает введение указанному субъекту фармацевтической композиции по п.1. 39. Способ по п.38, в котором указанный рецептор лиганда GLP-1(7-36)NH2 присутствует у субъекта, являющегося животным. 40. Способ по п.39, в котором указанный субъект является человеком. 41. Способ по п.40, в котором указанный субъект, являющийся человеком, страдает или подвержен риску развития заболевания или состояния, выбранного из группы, включающей диабет I типа, диабет II типа, гестационный диабет, ожирение, булимию, недостаточное чувство насыщения и метаболическое расстройство. 42. Способ по п.41, в котором указанное заболевание является диабетом I типа или диабетом II типа. 43. Способ по п.40, в котором указанный субъект, являющийся человеком, страдает или подвержен риску развития заболевания или состояния, выбранного из группы, состоящей из глюкагоном, секреторных расстройств дыхательных путей, артрита, остеопороза, заболевания центральной нервной системы,рестеноза, нейродегенеративного заболевания, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза печени, отека легких, гипертензии и расстройств, при которых желательно уменьшение приема пищи, заболеваний или расстройств центральной нервной системы, болезни Паркинсона, болезни Альцгеймера, болезни Хантингтона, бокового амиотрофического склероза, удара, синдрома дефицита внимания и психоневрологических синдромов, синдрома раздраженной кишки, инфаркта миокарда, удара, острого коронарного синдрома, послеоперационных катаболических изменений, гибернации миокарда или диабетической кардиомиопатии, недостаточного выделения натрия с мочой, повышенной концентрации калия в моче, состояний или расстройств, связанных с токсической гиперволемией, (например, почечной недостаточности, сердечной недостаточности, нефротического синдрома, цирроза печени, отека легких и гипертензии), синдрома поликистоза яичников, респираторного дистресс-синдрома, нефропатии, систолической дисфункции левого желудочка, гастроинтестинальных расстройств, таких как диарея, послеоперационный демпинг-синдром и синдром раздраженной кишки, критической полинейропатии (CIPN), синдрома системной воспалительной реакции (SIRS),дислипидемии, поражения тканей органов, вызванного реперфузией кровотока после ишемии и синдрома фактора риска ишемической болезни сердца (CHDRF).- 17012287 44. Способ преобразования стволовых клеток/клеток-предшественников печени в функциональные панкреатические клетки, предотвращения деградации -клеток и стимулирования пролиферации клеток, снижения уровней норепинефрина в плазме крови, стимулирования инотропной реакции и увеличения сократимости миокарда, улучшения питания через неалиментарный путь, предварительной подготовки субъекта к проведению эндоскопических процедур и модулирования уровней триглицеридов у нуждающегося в этом субъекта, указанный способ включает введение указанному субъекту фармацевтической композиции по п.1. 45. Способ по п.44, в котором указанный субъект является млекопитающим, более предпочтительно приматом, еще более предпочтительно человеком. 46. Способ выявления эффекта агониста GLP-1, указанный способ включает контакт рецептора лиганда GLP-1(7-36)NH2 с соединением согласно формуле (I), указанное соединение согласно формуле (I) доставляется к указанному рецептору непосредственно или опосредованно с помощью композиции по п.6. 47. Способ выявления эффекта агониста рецептора GLP-1 у нуждающегося субъекта, указанный способ включает введение указанному субъекту фармацевтической композиции по п.6. 48. Способ по п.47, в котором указанный рецептор лиганда GLP-1(7-36)NH2 присутствует у субъекта, являющегося животным. 49. Способ по п.48, в котором указанный субъект является человеком. 50. Способ по п.49, в котором субъект, являющийся человеком, страдает или подвержен риску развития заболевания или состояний, выбранных из группы, состоящей из диабета I типа, диабет II типа,гестационного диабета, ожирения, повышенного аппетита, недостаточного чувства насыщения и метаболического расстройства. 51. Способ по п.50, в котором указанное заболевание является диабетом I типа или диабетом II типа. 52. Способ по п.49, в котором субъект, являющийся человеком, страдает или подвержен риску развития заболевания или состояния, выбранного из группы, состоящей из глюкагона, секреторных расстройств дыхательных путей, артрита, остеопороза, заболевания центральной нервной системы, рестеноза, нейродегенеративного заболевания, почечной недостаточности, застойной сердечной недостаточности, нефротического синдрома, цирроза печени, отека легких, гипертензии и расстройств, при которых желательно уменьшение приема пищи, заболевания или расстройства центральной нервной системы,болезни Паркинсона, болезни Альцгеймера, болезни Хантингтона, бокового амиотрофического склероза,удара, синдрома дефицита внимания и нейропсихиатрических синдромов, синдрома раздраженной кишки, инфаркта миокарда, удара, острого коронарного синдрома, постхирургических катаболических изменений, гибернации миокарда или диабетической кардиомиопатии, недостаточного выделения натрия с мочой, повышенной концентрации калия в моче, состояний или расстройств, связанных с токсической гиперволемией, (например, почечная недостаточность, сердечная недостаточность, нефротический синдром, цирроз печени, отек легких и гипертензия), синдрома поликистоза яичников, респираторного дистресс-синдрома, нефропатии, систолической дисфункции левого желудочка, гастроинтестинальных расстройств, таких как диарея, послеоперационный демпинг-синдром и синдром раздраженной кишки, критической полиневропатии (CIPN), синдрома системной воспалительной реакции (SIRS), дислипидемии,поражения тканей органов, вызванного восстановлением кровотока после ишемии, и синдрома фактора риска ишемической болезни сердца (CHDRF). 53. Способ преобразования стволовых клеток/клеток-предшественников печени в функциональные панкреатические клетки, предотвращения деградации -клеток и стимулирования пролиферации клеток, снижения уровней норепинефрина в плазме крови, стимулирования инотропной реакции и увеличения сократимости миокарда, улучшения питания через не-алиментарный путь, предварительной подготовки субъекта к проведению эндоскопических процедур и модулирования уровней триглицеридов у нуждающегося субъекта, указанный способ включает введение указанному субъекту фармацевтической композиции по п.6. 54. Способ по п.53, в котором указанный субъект является млекопитающим, более предпочтительно приматом, еще более предпочтительно человеком. 55. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 1% (мас./об.). 56. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 2% (мас./об.). 57. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 вуказанной композиции составляет приблизительно 10% (мас./об.). 58. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 25% (мас./об.). 59. Композиция по п.15, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 5% (мас./об.) и указанное отношение составляет приблизительно 5,4:1. 60. Композиция по п.15, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции- 18012287 составляет приблизительно 5% (мас./об.) и указанное отношение составляет приблизительно 4,0:1. 61. Композиция по п.15, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 10% (мас./об.) и указанное отношение составляет приблизительно 5,4:1. 62. Композиция по п.15, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 10% (мас./об.) и указанное отношение составляет приблизительно 4,0:1. 63. Композиция по п.16, в которой концентрация (Aib8,35)hGLP-1(7-36)NH2 в указанной композиции составляет приблизительно 23% (мас./об.).
МПК / Метки
МПК: C07K 14/605
Метки: фармацевтическая, glp-1, композиция
Код ссылки
<a href="https://eas.patents.su/22-12287-farmacevticheskaya-kompoziciya-glp-1.html" rel="bookmark" title="База патентов Евразийского Союза">Фармацевтическая композиция glp-1</a>
Композиция, обладающая антивирусной активностью, способ её получения и фармацевтическая композиция на её основе

Номер патента: 112
Опубликовано: 27.08.1998
Автор: Аликс Ролан Коммэн
МПК: A61K 35/78
Метки: фармацевтическая, активностью, способ, композиция, антивирусной, основе, получения, обладающая
Формула / Реферат:
1. Композиция на основе продуктов природного происхождения, обладающая антивирусной активностью, отличающаяся тем, что она содержит настой порошка кокосового ореха в уксусной кислоте, водный раствор минеральных солей и фармацевтически приемлемые экстракты кактусовых, лилейных, анакардиевых и молочайных культур. 2. Композиция по п.1, отличающаяся тем, что она содержит настой порошка кокосового ореха в уксусной кислоте, водный раствор минеральных...
Фармацевтическая композиция, содержащая фозиноприл

Номер патента: 8171
Опубликовано: 27.04.2007
Автор: Эйёлфссон Рейнар
МПК: A61K 31/675, A61K 47/14, A61K 9/20...
Метки: фозиноприл, фармацевтическая, композиция, содержащая
Формула / Реферат:
1. Фармацевтическая композиция, содержащая от приблизительно 1 до 25% фозиноприла или его соли, отличающаяся тем, что в качестве смазочного материала содержит дибегенат глицерина. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что она содержит натриевую соль фозиноприла. 3. Фармацевтическая композиция по п.1, отличающаяся тем, что она содержит от приблизительно 0,25 до приблизительно 5% дибегената глицерина. 4. Фармацевтическая...
Диспергируемая во рту фармацевтическая композиция ивабрадина

Номер патента: 7681
Опубликовано: 29.12.2006
Авторы: Жюльен Марк, Роллан Эрве, Вутрих Патрик
МПК: A61P 9/00, A61K 31/55, A61K 9/20...
Метки: рту, ивабрадина, фармацевтическая, композиция, диспергируемая
Формула / Реферат:
1. Твердая диспергируемая в ротовой полости фармацевтическая композиция ивабрадина или его фармацевтически приемлемых солей, применяемая для лечения устойчивой стенокардии, сердечной недостаточности и острой ишемии, отличающаяся тем, что она включает ивабрадин или его фармацевтически приемлемую соль, гранулы, состоящие из совместно высушенных лактозы и крахмала. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что она включает по...
Фармацевтическая композиция для интраназального введения пирибедила

Номер патента: 11041
Опубликовано: 30.12.2008
Авторы: Вутрих Патрик, Роллан Эрве
МПК: A61K 31/506, A61K 47/40, A61K 9/08...
Метки: введения, пирибедила, композиция, фармацевтическая, интраназального
Формула / Реферат:
1. Фармацевтическая композиция в виде водного раствора или порошка для интраназального введения пирибедила, которая содержит пирибедил или его фармацевтически приемлемую соль, необязательно циклодекстрин, один или несколько фармацевтически приемлемых наполнителей. 2. Фармацевтическая композиция в соответствии с п.1, отличающаяся тем, что пирибедил находится в виде основания. 3. Фармацевтическая композиция в соответствии с п.1 или 2, отличающаяся...
Диспергируемая во рту фармацевтическая композиция периндоприла

Номер патента: 7181
Опубликовано: 25.08.2006
Авторы: Роллан Эрве, Вутрих Патрик, Жюльен Марк
МПК: A61K 38/55, A61P 9/00, A61K 9/20...
Метки: фармацевтическая, диспергируемая, рту, композиция, периндоприла
Формула / Реферат:
1. Твердая диспергируемая в ротовой полости фармацевтическая композиция периндоприла или его фармацевтически приемлемых солей, отличающаяся тем, что она включает периндоприл или его фармацевтически приемлемую соль, гранулы, состоящие из совместно высушенных лактозы и крахмала. 2. Фармацевтическая композиция в соответствии с п.1, отличающаяся тем, что она включает по отношению к общему весу композиции от 0,1 до 10 вес.% периндоприла или его...