Промышленный способ синтеза 17-ацетокси-11b-[4-(диметиламино) фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона и ключевые промежуточные соединения данного способа
Номер патента: 15478
Опубликовано: 31.08.2011
Авторы: Араньи Антал, Санта Чаба, Терди Ласло, Молнар Чаба, Хорват Золтан, Боди Йожеф, Махо Шандор, Туба Золтан, Селеш Янош, Виски Дьёрдь, Чёргей Янош, Балог Габор







Формула / Реферат
1. Промышленный способ синтеза 17-ацетокси-11b-[4-(диметиламино)фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона формулы (I)

из 3,3-[1,2-этандиил-бис-(окси)]оэстр-5(10),9(11)-диен-17-она формулы (II)

отличающийся тем, что включает стадии:
i) образование эпоксида по двойной связи в положении 5(10) взаимодействием 3,3-[1,2-этандиил-бис-(окси)]оэстр-5(10),9(11)-диен-17-она формулы (II)

с перекисью водорода;
ii) присоединение полученного in situ циановодорода в положение 17 полученного 5,10a-эпокси-3,3-[1,2-этандиил-бис-(окси)]-5a-оэстр-9(11)-ен-17-она формулы (III)

iii) силилирование гидроксильной группы в положении 17 образовавшегося 5,10a-эпокси-3,3-[1,2-этандиил-бис-(окси)]-17a-гидрокси-5a-оэстр-9(11)-ен-17b-карбонитрила формулы (IV)

триметилхлорсиланом;
iv) взаимодействие полученного 5,10a-эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-[триметилсилилокси]-5a-оэстр-9(11)-ен-17b-карбонитрила формулы (V)

с реактивом Гриньяра 4-(диметиламино)фенилмагнийбромидом в присутствии CuCl ("Teutsch reaction");
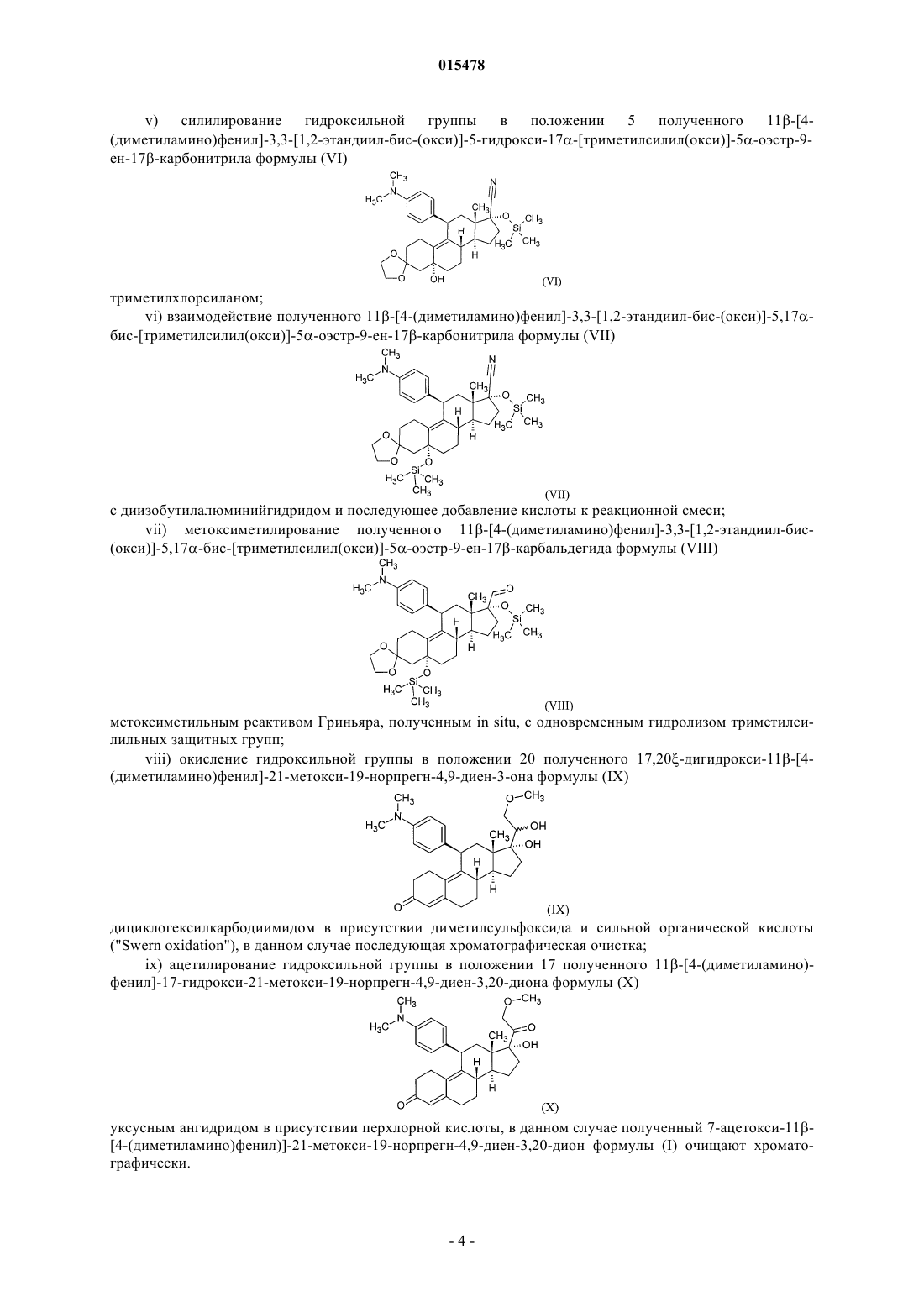
v) силилирование гидроксильной группы в положении 5 полученного 11b-[4-(диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5-гидрокси-17a-[триметилсилил(окси)]-5a-оэстр-9-ен-17b-карбонитрила формулы (VI)

триметилхлорсиланом;
vi) взаимодействие полученного 11b-[4-(диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5,17a-бис-[триметилсилил(окси)]-5a-оэстр-9-ен-17b-карбонитрила формулы (VII)

с диизобутилалюминийгидридом и последующее добавление кислоты к реакционной смеси;
vii) метоксиметилирование полученного 11b-[4-(диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5,17a-бис-[триметилсилил(окси)]-5a-оэстр-9-ен-17b-карбальдегида формулы (VIII)

метоксиметильным реактивом Гриньяра, полученным in situ, с одновременным гидролизом триметилсилильных защитных групп;
viii) окисление гидроксильной группы в положении 20 полученного 17,20x-дигидрокси-11b-[4-(диметиламино)фенил]-21-метокси-19-норпрегн-4,9-диен-3-она формулы (IX)

дициклогексилкарбодиимидом в присутствии диметилсульфоксида и сильной органической кислоты ("Swern oxidation") и в данном случае последующая хроматографическая очистка;
ix) ацетилирование гидроксильной группы в положении 17 полученного 11b-[4-(диметиламино)фенил]-17-гидрокси-21-метокси-19-норпрегн-4,9-диен-3,20-диона формулы (X)

уксусным ангидридом в присутствии перхлорной кислоты и в данном случае последующая хроматографическая очистка полученного 7-ацетокси-11b-[4-(диметиламино)фенил)]-21-метокси-19-норпрегн-4,9-диен-3,20-диона формулы (I).
2. Способ по п.1, отличающийся тем, что на стадии iv) используют 0,25±0,025 экв. избытка реактива Гриньяра 4-(диметиламино)фенилмагнийбромида относительно 5,10a-эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-[триметилсилилокси]-5a-оэстр-9(11)-ен-17b-карбонитрила формулы (V).
3. Способ по п.1, отличающийся тем, что на стадии viii) используют трифторуксусную кислоту в качестве сильной органической кислоты.
4. 11b-[4-(Диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5,17a-бис-[триметилсилил(окси)]-5a-оэстр-9-ен-17b-карбонитрил формулы (VII).
5. 11b-[4-(Диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5,17a-бис-[триметилсилил(окси)]-5a-оэстр-9-ен-17b-карбальдегид формулы (VIII).
Текст
ПРОМЫШЛЕННЫЙ СПОСОБ СИНТЕЗА 17-АЦЕТОКСИ-11-[4(ДИМЕТИЛАМИНО)ФЕНИЛ]-21-МЕТОКСИ-19-НОРПРЕГН-4,9-ДИЕН-3,20-ДИОНА И КЛЮЧЕВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДАННОГО СПОСОБА Настоящее изобретение относится к способу синтеза известного 17-ацетокси-11-[4-(диметиламино)фенил]-21-метокси 19-норпрегн-4,9-диен-3,20-диона (в дальнейшем обозначается CDB-4124) формулы (I) из 3,3-[1,2-этандиил-бис(окси)]оэстр-5(10),9(11)-диен-17-она формулы (II). Соединение CDB-4124 принадлежит к группе антигормонов. Способ по настоящему изобретению состоит в следующем: i) образование эпоксида по двойной связи в положении 5(10) взаимодействием 3,3-[1,2-этандиил-бис-(окси)]оэстр-5(10),9(11)-диен-17-она формулы (II) с перекисью водорода; ii) присоединение полученного in situ циановодорода в положение 17 полученного 5,10-эпокси-3,3-[1,2-этандиил-бис-(окси)]-5 оэстр-9(11)-ен-17-она формулы (III); iii) силилирование гидроксильной группы в положении 17 образовавшегося 5,10 эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-гидрокси-5-оэстр-9(11)-ен-17-карбонитрила формулы (IV) триметилхлорсиланом; iv) взаимодействие полученного 5,10-эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-[триметилсилилокси]-5-оэстр 9(11)-ен-17-карбонитрила формулы (V) с реактивом Гриньяра 4-(диметиламино)фенилмагнийбромидом в присутствииCuCl ("Teutsch reaction"); v) силилирование гидроксильной группы в положении 5 полученного 11-[4(диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5-гидрокси-17-[триметилсилил(окси)]-5-оэстр-9-ен-17 карбонитрила формулы (VI) триметилхлорсиланом; vi) взаимодействие полученного 11-[4-(диметиламино)фенил]-3,3[1,2-этандиил-бис-(окси)]-5,17-бис-[триметилсилил(окси)]-5-оэстр-9-ен-17-карбонитрила формулы (VII) с диизобутилалюминийгидридом и последующее добавление кислоты к реакционной смеси; vii) метоксиметилирование полученного 11-[4-(диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5,17-бис-[триметилсилил(окси)]-5-оэстр-9-ен-17 карбальдегида формулы (VIII) метоксиметильным реактивом Гриньяра, полученным in situ, с одновременным гидролизом триметилсилильных защитных групп; viii) окисление гидроксильной группы в положении 20 полученного 17,20 дигидрокси-11-[4-(диметиламино)фенил]-21-метокси-19-норпрегн-4,9-диен-3-она формулы (IX) дициклогексилкарбодиимидом в присутствии диметилсульфоксида и сильной органической кислоты ("Swern oxidation"), в данном случае последующая хроматографическая очистка; ix) ацетилирование гидроксильной группы в положении 17 полученного 11[4-(диметиламино)фенил]-17-гидрокси-21-метокси-19-норпрегн-4,9-диен-3,20-диона формулы (X) уксусным ангидридом в присутствии перхлорной кислоты, в данном случае полученный 7-ацетокси-11-[4-(диметиламино)фенил)]-21-метокси 19-нопрегн-4,9-диен-3,20-дион формулы (I) очищают хроматографически. Настоящее изобретение также относится к новым промежуточным соединениям формул (VII) и (VIII). 015478 Область техники, к которой относится изобретение Настоящее изобретение относится к способу синтеза известного 17-ацетокси-11-[4(диметиламино)фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона (в дальнейшем именуемый CDB4124) формулы (I) Предшествующий уровень техники Соединение CDB-4124 принадлежит к группе антигормонов. Антигормоны могут дезактивировать действие гормонов в организме за счет ингибирования связывания гормонов, например мужских и женских половых гормонов или гормонов, продуцируемых надпочечником, с местом приложения в органемишени; вследствие этого индуцируемые гормонами функции могут быть блокированы введением антигормонов. Соединения, ингибирующие синтез прогестерона или его связывание с рецептором, можно потенциально использовать в контрацепции и в случаях патологий, где прогестерон играет заметную роль. Идеальное антипрогестогенное соединение специфично присоединяется только к тому рецептору, который необходимо блокировать; обладает высоким сродством к рецептору и медленно диссоциирует; не оказывает другого биологического или фармакологического эффекта. Первый антипрогестин, использованный в клинической практике, был описан в 1981 г. [ЕР 57115] и назывался мифепристон. После этого были синтезированы некоторые аналоги и проверена связь структура-активность для полученных соединений, особенно селективность, главным образом соотношение антипрогестинной и антиглюкокортикоидной активности. В настоящее время ни одно из известных антипрогестогенных соединений не удовлетворяет полностью требованиям к селективности. Соединение CDB-4124, которое можно синтезировать способом по настоящему изобретению, согласно проведенным к настоящему моменту клиническим испытаниям представляет собой такое перспективное соединение, промышленный синтез которого является экономически выгодным. В литературе описаны несколько способов лабораторного синтеза CDB-4124 формулы (I), которые различаются по исходным материалам или порядку стадий реакции. Синтез различных функциональных групп осуществляют похожими способами. Упомянутые способы синтеза характеризуются тем, что в них обычно не учитывают аспекты безопасности при масштабировании, особенно воспламеняемость растворителей (реакционных сред), а также то, что данные растворители могут быть вредны для здоровья и что некоторые реактивы могут быть дорогими. Целью первых синтезов было получение такого количества соединения/соединений, которое было бы достаточно для проведения фармакологических тестов. Для удовлетворения всем требования чистоты для терапевтического использования соединения необходима дальнейшая разработка. Промышленное осуществление экономичного синтеза обычно представляет собой модификацию оригинального способа или способов или может представлять собой улучшенный синтез. Первый синтез соединения CDB-4124 был описан в патенте WO 97/41145, объектом которого был синтез 11- и 21-замещенных 19-норпрогестероновых производных и их аналогов. Данные соединения обладают значительной антипрогестогенной активностью. На схеме 3 проиллюстрирован синтез соединения CDB-4124. Исходное вещество для данного синтеза представляло собой 17-[(бромметил)диметилсилилокси]3,3-[1,2-этандиил-бис-(окси)]-5(10),9(11)-диен-17-карбонитрил, который можно получать из коммерчески доступного 3,3-[1,2-этандиил-бис-(окси)]-17-гидроксиоэстр-5(10),9(11)-диен-17-карбонитрила(Davos Chemical Inc. в Нью-Джерси) с выходом 69,5% силилированием гидроксильной группы в положении 17 (бромметил)диметилсилилхлоридом. Очистку осуществляли флэш-хроматографией.-1 015478 Исходное вещество вводили в реакцию с диизопропиламидом лития в тетрагидрофурановом растворе при -78 С, а выделение продукта осуществляли экстрагированием этилацетатом и очисткой эфиром. 21-Бром соединение, полученное с выходом 60,4%, вводили в реакцию с ацетатом калия (99%), затем гидролизовали гидрокарбонатом калия, получая 21-гидрокси производное с выходом 57,6%. Для защиты кетогрупп в положении 3 и 20 применяли образование бис-кеталя (с выходом 62,5%). Ключевую стадию синтеза - монометилирование по положению 21 17,21-дигидрокси производного, защищенного по положениям 3 и 20, - осуществляли в присутствии 1:1 смеси тетрафторборатной соли триметилоксония и "протонной губки" [1,8-бис-(диметиламино)нафталина]. Полученный продукт 3,3;20:20-бис-[1,2-этандиил-бис-(окси)]-17-гидрокси-21-метокси-19-норпрегн-5(10),9(11)-диен - выделяли из дихлорметана с выходом 79% и использовали в следующей стадии. Образование эпоксида по двойной связи в положении 5(10) полученного сырого 21-метокси производного осуществляли взаимодействием с перекисью водорода в присутствии тригидрата гексафторацетона. Согласно данным ЯМР-спектроскопии полученный продукт содержал четыре типа эпоксидов (основной продукт представлял собой 5,10-эпоксид с выходом 66%). Полученную сырую смесь эпоксидов использовали в реакции Гриньяра, катализируемой ионом меди(I). После выделения из эфирного раствора продукт очищали колоночной флэш-хроматографией. Гидролиз дикетальной защитной группы в 11-[4-(диметиламино)фенил] производном осуществляли смесью трифторуксусная кислота/вода (3:1) в тетрагидрофуране. Продукт получали с выходом 96,3% после экстракции дихлорметаном, упаривания и обработки маслообразного остатка водой. Последняя стадия синтеза представляла собой ацетилирование гидроксильной группы в положении 17, которое осуществляли смесью трифторуксусного ангидрида и уксусной кислоты в дихлорметане в присутствии п-толуолсульфокислоты (катализатор) при 0 С. По окончании реакции смесь разбавляли водой, нейтрализовали раствором гидроксида аммония, экстрагировали дихлорметаном и промывали рассолом. Объединенные органические слои упаривали и очищали остаток колоночной флэшхроматографией, получая соединение CDB-4124 с выходом 75,8%. Исходное вещество для синтеза, описанного в вышеуказанном патенте, представляло собой 17-[(бромметил)диметилсилилокси]-3,3-[1,2-этандиил-бис-(окси)]-5(10),9(11)-диен-17-карбонитрил,который синтезировали из кетокеталя добавлением цианидного иона с последующим силилированием гидроксильной группы. 17-Силилоксибром соединение превращали в 21-бром производное диизопропиламидом лития при -78 С. Введение метоксигруппы в положение 21 осуществляли не напрямую в несколько стадий - 21-бром соединение, 21-ацетокси производное - через 21-гидроксисоединение при совместном использовании 6 экв. (относительно исходного вещества) тетрафторборатной соли триметилоксония и "протонной губки" (SNAP реакция). Указанный способ долгий и дорогостоящий, удаление избытка "протонной губки" затруднительно, во многих случаях необходима многократная очистка продукта. Образование эпоксида по двойной связи в положении 5(10) дало согласно данным ЯМР-спектроскопии четыре типа эпоксидов, из которых только 66% представляли собой желаемый 5,10-эпоксид. Несмотря на тот факт, что сырой продукт содержал около 34% нежелательного продукта 3-эпоксид), его использовали в реакции Гриньяра. В реакции Гриньяра использовали пятикратный избыток 4-бром-диметиланилина, что способствовало образованию монометильного производного и димеризации этого реактива, поэтому выделения и очистка продукта были затруднены, выход выделенного продукта снижался. Со стратегической точки зрения эпоксидирование, приводящее к получению сырой смеси, на седьмой стадии синтетической цепочки не является экономически выгодным. Флэшхроматографию использовали на 4 стадиях 11-стадийного синтеза. В некоторых случаях при выделении и очистке промежуточных соединений использовался эфир, что является опасным в условиях промышленного осуществления синтеза. Выход конечного продукта снижался вследствие стадий очистки, поэтому общий выход синтеза составлял только 3,22%. На схемах 1 и 2 патента WO 01/47945 показаны две другие последовательности реакций синтеза соединения CDB-4124. Исходное вещество в обоих случаях синтеза представляло собой 3,3-[1,2-этандиил-бис-(окси)]-17 гидроксиоэстр-5(10),9(11)-диен-17-карбонитрил, который силилировали описанным выше способом, но 21-галогеновые производные - хлор и бром - тоже образовывались в той же реакционной смеси. Следующие стадии, замещение атома брома ацетоксигруппой и гидролиз, были идентичны известным способам. Последующие стадии синтеза, показанного на схеме 1, идентичны стадиям, описанным в предыдущем патенте. Согласно схеме 2 синтезировали 3-монокетальное производное 21-гидрокси производного, после чего проводили SNAP реакцию для введения 21-метоксифункции и кетогруппы в положение 20 (временная защита) восстановлением литийалюминийгидридом. Образование эпоксида осуществляли в положении 5(10) полученного 20-гидрокси производного. Раскрытие эпоксидного кольца и удаление кетальной группы были идентичны описанным ранее способам. Йодоксибензойную кислоту использовали для повторного окисления гидроксильной группы в положении 20. Конечную стадию, синтез желаемого-2 015478 17-ацетоксипродукта, осуществляли согласно способу, представленному на схеме 1. Данный синтез проводили в 12 стадий с общим выходом 3,89%. Хотя объединение первых двух стадий синтеза было хорошим решением, но, например, образование эпоксидного производного из-за стадии очистки, которая приводила к пониженному выходу, на более поздней фазе синтетической цепочки было не современным, дорогостоящим. Другое невыгодное решение заключалось в том, что для очистки промежуточных соединений и конечного продукта использовали флэш-хроматографию. В нескольких случаях выделение продуктов осуществляли обработкой эфиром, который нельзя использовать в крупномасштабном синтезе. Введение метоксигруппы осуществляли в несколько стадий, как описано ранее. Удаление "протонной губки", используемой в SNAP реакции,достигалось только многократной очисткой. Восстановление литийалюминийгидридом, которое применяли для временной защиты оксофункциональной группы в положении 20, является особенно опасным в промышленном масштабе. Последующая регенерация оксогруппы в положении 20 путем окисления йод-оксибензойной кислотой является дорогостоящей и поэтому непригодна для промышленного получения. Сущность изобретения Согласно вышеперечисленным фактам не существует такого известного способа, который является пригодным для осуществления синтеза CDB-4124 в промышленном масштабе в простых реакционных условиях. Цель данного изобретения заключалась в разработке легко масштабируемого способа, промышленное осуществление которого безопасно, экономично и при котором чистота действующего вещества удовлетворяет требованиям фармакопеи. Неожиданно было обнаружено, что следующий способ удовлетворяет вышеуказанным требованиям:i) образование эпоксида по двойной связи в положении 5(10) взаимодействием 3,3-[1,2-этандиилбис-(окси)]оэстр-5(10),9(11)-диен-17-она формулы (II)iii) силилирование гидроксильной группы в положении 17 образовавшегося 5,10-эпокси-3,3-[1,2 этандиил-бис-(окси)]-17-гидрокси-5-оэстр-9(11)-ен-17-карбонитрила формулы (IV)v) силилирование гидроксильной группы в положении 5 полученного 11-[4(диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5-гидрокси-17-[триметилсилил(окси)]-5-оэстр-9 ен-17-карбонитрила формулы (VI) с диизобутилалюминийгидридом и последующее добавление кислоты к реакционной смеси; метоксиметильным реактивом Гриньяра, полученным in situ, с одновременным гидролизом триметилсилильных защитных групп;viii) окисление гидроксильной группы в положении 20 полученного 17,20-дигидрокси-11-[4(диметиламино)фенил]-21-метокси-19-норпрегн-4,9-диен-3-она формулы (IX)("Swern oxidation"), в данном случае последующая хроматографическая очистка;ix) ацетилирование гидроксильной группы в положении 17 полученного 11-[4-(диметиламино)фенил]-17-гидрокси-21-метокси-19-норпрегн-4,9-диен-3,20-диона формулы (X) уксусным ангидридом в присутствии перхлорной кислоты, в данном случае полученный 7-ацетокси-11[4-(диметиламино)фенил)]-21-метокси-19-норпрегн-4,9-диен-3,20-дион формулы (I) очищают хроматографически.-4 015478 Кетокеталь формулы (II) предпочтительно вводят в реакцию с 50%-ным раствором перекиси водорода в сухом дихлорметане в присутствии пиридина и гексахлорацетона при 0-1 С в течение 20-24 ч. После завершения реакции смесь разбавляют дихлорметаном, избыток перекиси водорода разлагают,органический слой отделяют, водную фазу дважды экстрагируют дихлорметаном, объединенные органические слои промывают водой, высушивают и упаривают. Маслообразный остаток обрабатывают смесью 1:3 этилацетат/диизопропиловый эфир. Полученный кетокеталь-эпоксид формулы (III), чистота которого составляет 98,8% и который содержит 95,3% 5,10-эпоксида согласно данным ВЭЖХ, используют в стадии ii) без дополнительной очистки. Стадию ii) предпочтительно осуществляют суспендированием кетокеталь-эпоксида формулы (III) в метаноле, при 20-25 С добавляют порошкообразный цианид калия, затем после осторожного добавления уксусной кислоты реакционную смесь нагревают до 50-55 С. Реакционную смесь охлаждают до 20-25 С в течение 1 ч, затем перемешивают при этой же температуре в течение 5 ч. После завершения реакции смесь разбавляют водой, перемешивают 1 ч и отфильтровывают выпавший в осадок кристаллический эпоксикарбонитрил формулы (IV). Полученный продукт можно использовать в стадии iii) без дополнительной очистки. На стадии iii) эпоксикарбонитрил формулы (IV) предпочтительно растворяют в дихлорметане при интенсивном перемешивании, затем после высушивания проверяют содержание воды. К сухому раствору добавляют имидазол, затем в течение 1 ч при 20-25 С добавляют триметилхлорсилан. После завершения реакции раствор разбавляют дихлорметаном и разлагают избыток триметилхлорсилана добавлением воды. Органический слой отделяют, промывают водой, высушивают и упаривают. Остаток кристаллизуют из метанола, фильтруют и сушат. Полученный таким образом TMSO-карбонитрил формулы (V)(73,3%) можно использовать в следующей стадии без дополнительной очистки.TMSO-карбонитрил формулы (V), полученный на стадии iv), предпочтительно вводят в реакцию следующим образом. Сначала к сухому тетрагидрофурану добавляют магний и 1,2-дибромэтан. Температура реакционной смеси начинает повышаться, указывая на успешное осуществление активации. Затем 4-бром-диметиланилин и небольшое количество раствора 1,2-дибромэтана в сухом тетрагидрофуране и толуол добавляют при перемешивании в реакционную смесь, содержащую магний (реактив Гриньяра). Закипание реакционной смеси указывает на успешное осуществление активации. Затем к раствору реактива Гриньяра добавляют CuCl и после 5 мин перемешивания смесь охлаждают до 8-13 С. Добавляют раствор TMSO-карбонитрила в дихлорметане, поддерживая температуру между 10-15 С. После завершения реакции смесь добавляют к перемешиваемому и охлажденному 10%-ному раствору хлорида аммония, содержащему пиросульфит щелочного металла. Теплый раствор охлаждают до комнатной температуры, разбавляют дихлорметаном, отделяют органический слой и экстрагируют водную фазу дихлорметаном. Объединенные органические слои промывают водой, обрабатывают силикагелем, сушат над безводным сульфатом натрия, фильтруют и упаривают в вакууме. Остаток кристаллизуют из метанола. Кристаллический продукт отделяют и сушат. Полученный A-TMSO-карбонитрил формулы (VI) (80,79%) можно использовать в следующей стадии без дополнительной очистки. Стадию v) предпочтительно осуществляют растворением полученного A-TMSO-карбонитрила формулы (VI) в дихлорметане при 20-25 С, затем после добавления имидазола гидроксильную группу в положении 5 силилируют триметилхлорсиланом. Время реакции около 2 ч, затем смесь разбавляют дихлорметаном и водой, органический слой отделяют, промывают водой, сушат над безводным сульфатом натрия, фильтруют и упаривают. Остаток обрабатывают метанолом, полученный кристаллический А-бис-TMSO-карбонитрил формулы (VII) (89,09%) отфильтровывают и высушивают. Его можно использовать в следующей стадии без дополнительной очистки. На стадии vi) полученный А-бис-TMSO-карбонитрил формулы (VII) растворяют в смеси метилтрет-бутилового эфира и тетрагидрофурана, охлаждают до -(15-20)С и 1 М раствор DIBAL-H (диизобутилалюминийгидрид) в циклогексане добавляют в течение около 30 мин, поддерживая температуру, затем реакционную смесь перемешивают еще 1 ч при той же температуре. После завершения реакции смесь воды и уксусной кислоты (2:1) добавляют при -(5-10)С, затем смесь перемешивают в течение 20 мин. Органический слой отделяют, промывают водой, 0,3 М раствором гидрокарбоната натрия и водой. Органический слой упаривают без высушивания при 40-45 С, остаток растворяют в метаноле и упаривают до заданного объема (см. примеры). Кристаллическую суспензию охлаждают до 5-10 С, фильтруют после 1 ч стояния, промывают метанолом с температурой 0-(-5)С и сушат. Полученный А-бис-TMSO-карбальдегид формулы (VIII) (83,6%) можно использовать в следующей стадии без дополнительной очистки. Полученный А-бис-TMSO-карбальдегид формулы (VIII) на стадии vii) превращают в 21-метокси производное с удлинением цепи. Это превращение осуществляют активацией магниевых стружек, как описано выше, в сухом тетрагидрофуране 1,2-дибромэтаном, затем добавляют хлорид ртути(II) с образованием амальгамы. Полученную смесь разбавляют толуолом, затем проверяют активность амальгамы, как описано в примерах 7, 8 и 19. После проверки активности реактива добавляют раствор метоксиметилхлорида в толуоле. Одновременно с этим А-бис-TMSO-карбальдегид растворяют в толуоле-5 015478 и добавляют этот раствор к раствору амальгамы в течение 30 мин при 0-5 С. После завершения реакции смесь добавляют к 1 М водному раствору гидросульфата калия, поддерживая температуру ниже 30 С. После перемешивания в течение 2 ч слои разделяют, водную фазу добавляют к смеси 1 М раствора гидрокарбоната натрия и дихлорметана и перемешивают 10-15 мин. Органический слой отделяют, водную фазу экстрагируют дихлорметаном, объединенные органические слои сушат и обрабатывают углем,фильтруют и упаривают. Остаток представляет собой твердый диол формулы (IX) (84,1%), который можно использовать в следующей стадии без дополнительной очистки. Полученный диол формулы (IX) далее вводят в реакцию согласно стадии viii) настоящего изобретения. Предпочтительно его растворяют в сухом толуоле и добавляют в атмосфере азота диметилсульфоксид, пиридин и трифторуксусную кислоту при 20-25 С. Затем к смеси добавляют раствор дициклогексилкарбодиимида в толуоле (окисление по Шверну). Реакционную смесь перемешивают при 40 С в течение 2 ч, затем охлаждают до 20-25 С и добавляют 1 М водный раствор гидросульфата калия. После перемешивания в течение 30 мин выпавшее в осадок кристаллическое соединение отфильтровывают и промывают 1 М водным раствором гидросульфата калия. Две фазы фильтрата разделяют, водную фазу добавляют к 1 М раствору гидроксида натрия, выпавший в осадок сырой продукт отфильтровывают,промывают водой и сушат. Полученный кетон формулы (X) (79,5%) используют в следующей стадии после очистки. Синтез чистого CDB-4124 формулы (I), удовлетворяющего требованиям чистоты для терапевтического применения, включает две стадии очистки методом ВЭЖХ. Первая представляет собой очистку кетона формулы (X). Ацетилирование очищенного кетона формулы (X) в результате дает сыройCDB-4124, очистка которого методом ВЭЖХ дает действующее вещество с чистотой 99%. Хроматографию кетона формулы (X) и сырого CDB-4124 предпочтительно осуществляют с использованием силикагеля в качестве неподвижной фазы и 53:35:12 смеси циклогексан/метил-трет-бутиловый эфир/ацетон в качестве элюента как в лабораторном, так и в промышленном способе. Н-гексан и н-гептан также можно использовать вместо циклогексана. Соотношение растворителей в элюенте может варьироваться в определенных пределах (циклогексан, н-гексан, н-гептан: 40-60%; метил-третбутиловый эфир: 25-45%; ацетон: 10-20%). В конце стадии viii) кетон формулы (X) предпочтительно очищают следующим образом: силикагелевым адсорбентом (ZEOPREP C-GEL C-490L, производство ZEOCHEM; размер частиц 15-35 мкм; высота слоя около 60 см) заполняют ВЭЖХ колонку методом суспензионной набивки и колонку уравновешивают элюентом (53:35:12 смесь циклогексан/метил-трет-бутиловый эфир/ацетон). Сырой кетон формулы (X) растворяют в смеси ацетона и метил-трет-бутилового эфира и к раствору добавляют циклогексан. Полученный таким образом раствор фильтруют и наносят на колонку путем вкола. Используют УФ-детектирование. Отделяют первую фракцию, содержащие чистое соединение фракции собирают и упаривают. Согласно другому способу после упаривания фракций отгоняют от остатка дихлорметан и растворяют продукт в дихлорметане. Содержание примесей в обоих случаях: менее 4%. Полученный дихлорметановый раствор можно использовать в следующей стадии.CDB-4124 формулы (I) синтезируют из очищенного кетона формулы (X) согласно стадии ix) настоящего изобретения с использованием уксусного ангидрида в присутствии перхлорной кислоты: 70%-ную перхлорную кислоту добавляют к перемешиваемому и охлажденному до -(20-25)С уксусному ангидриду с такой скоростью, чтобы температура не поднималась выше -15 С. Затем добавляют раствор очищенного 11-[4-(диметиламино)фенил)]-17-гидрокси-21-метокси-19-норпрегн-4,9-диен-3,20-диона формулы (X) в дихлорметане. После завершения реакции смесь разбавляют дихлорметаном, охлаждают до -10 С и добавляют воду для разложения уксусного ангидрида. Уровень рН смеси доводят до 7-8 добавлением раствора гидроксида аммония. Затем водную фазу отделяют, экстрагируют дихлорметаном,объединенные органические слои промывают водой, сушат и упаривают. Полученный сырой конечный продукт CDB-4124 формулы (I) очищают методом ВЭЖХ согласно описанному выше способу. Преимущества способа по настоящему изобретению по сравнению с известными способами можно суммировать следующим образом.a) Исходное вещество для синтеза [кетокеталь формулы (II)] можно легко синтезировать из оэстр-4-ен-3,17-диона известными способами.b) Согласно заявленному авторами способу образование эпоксида по двойной связи в положении 5(10) представляет собой первую стадию синтеза. Из получаемой изомерной смеси эпоксидов только 5,10-эпоксид приводит к образованию требуемого соединения, соответственно другие изомеры являются побочными продуктами. Согласно описанному авторами способу данную стадию, приводящую к большим потерям вещества, проводят в начале синтетической цепочки; поэтому происходит потеря исходного вещества, а не потеря промежуточного соединения с более поздней стадии, являющегося более ценным. Способ по настоящему изобретению является более экономичным. Другой недостаток эпоксидирования, проводимого на более поздней стадии синтетической цепочки в известных методиках, заключается в более сложной очистке полученного соединения.c) Согласно описанному авторами способу введение стратегически важной 21-метоксигруппы осуществляют в две стадии через новые промежуточные соединения формулы (VII) и (VIII). Применение нового промежуточного соединения формулы (VII) сделало возможным образование альдегидной группы в положении 21, и полученное новое соединение формулы (VIII) обеспечивало простое и промышленно применимое введение метоксигруппы в положение 21. Силилированный циангидрин формулы(VII) взаимодействует с раствором DIBAL-H в циклогексане с получением А-бис-TMSO-карбальдегида формулы (VIII), который реагирует с метоксиметилхлоридом или бромидом по реакции Гриньяра с получением диола формулы (IX). Окисление 20-гидрокси производного, полученного в реакции Гриньяра,до 20-кетопроизводного осуществляют окислением по Шверну - вместо йод-оксибензойной кислоты,которую используют в литературных примерах - дициклогексилкарбодиимидом в присутствии диметилсульфоксида и сильной органической кислоты. Преимущество данного способа по настоящему изобретению заключается в том, что используемый реактив устойчив и его применение экономически выгодно.d) Исходным веществом в методиках, описанных в литературе, является силилированный циангидрин, из которого синтезировали 21-метокси производное в четыре стадии последовательно окружным путем. Циангидрин превращали в 21-хлор или 21-бром производное литийдиизопропиламидом при-78 С, затем галогеновые заместители заменяли на ацетоксигруппу и гидролизовали последнюю с получением 21-гидрокси производного, которое вводили в реакцию с тетрафторборатом триметилоксонияe) В способе по настоящему изобретению отсутствуют опасные реактивы, такие как алюмогидрид лития, и крайне огнеопасные растворители, такие как эфир.f) Другое преимущество способа по настоящему изобретению заключается в том, что получаемые промежуточные соединения в большинстве случаев достаточно чистые для использования в следующих стадиях без очистки.g) Хотя хроматографическую очистку используют только в двух последних стадиях способа по настоящему изобретению, чистота полученного действующего вещества CDB-4124 составляет 99%, что удовлетворяет требованиям для терапевтического применения.h) Другое преимущество способа по настоящему изобретению заключается в высоких выходах в индивидуальных синтетических стадиях (48,65; 73,36; 80,79; 89,09; 83,6; 84,1; 99,56; 83,78%). Общий выход 11-стадийного синтеза составляет 8,23% в отличие от общих выходов по известным методикамi) Условия реакций, используемые в некоторых стадиях способа по настоящему изобретению, отличаются от условий, используемых в известных методиках, поэтому выходы и чистота полученных продуктов выше. Например, в реакции Гриньяра - замещение в положении 11 - соотношение стероидного исходного вещества и 4-бромдиметиланилина составляет 1:1.25 в отличие от соотношения 1:5, которое применяется в известных методиках. Такое проведение реакции менее затратно, а выделение продукта легче, так как образуется меньше примесей. Способ по настоящему изобретению проиллюстрирован следующими неограничивающими примерами. Сведения, подтверждающие возможность осуществления изобретения Пример 1. 5,10-Эпокси-3,3-[1.2-этандиил-бис-(окси)]-5-оэстр-9(11)-ен-17-он [соединение формулы (III)]. В атмосфере азота растворяли 3,3-[1,2-этандиил-бис-(окси)]оэстр-5(10),9(11)-диен-17-он (46,7 г,149 ммоль) при интенсивном перемешивании в смеси пиридина (2,46 мл, 0,2 моль-эквивалент) и дихлорметана (234 мл) и охлаждали полученный раствор до -(6-8)С. После добавления гексахлорацетона(5,46 мл, 35,95 ммоль) к перемешиваемому раствору добавляли 50%-ную перекись водорода, имеющую температуру от 0 до -2 С (60 мл, 1058 ммоль) с такой скоростью, чтобы температура не поднималась выше 0 С. Реакционную смесь перемешивали при температуре от 1 до -1 С в течение 20-24 ч, затем разбавляли дихлорметаном, имеющим температуру 0-5 С (390 мл), избыток перекиси водорода разлагали добавлением раствора пентагидрата тиосульфата натрия (327 г, 1318 ммоль, 8,87 моль-эквивалент) в ледяной воде (1500 мл). Реакционную смесь перемешивали в течение 1,5 ч, затем отделяли органическую фазу. Водную фазу экстрагировали дихлорметаном, объединенные органические слои промывали водой,сушили над безводным сульфатом натрия, фильтровали и упаривали. Маслообразный остаток кристаллизовали из смеси 1:3 этилацетат/диизопропиловый эфир (435 мл), содержащей 0,1% пиридина Полученный таким образом продукт сушили, получая 23,87 г (48,66%) целевого соединения. Чистота целевого соединения составляла 98,5-98,8% (определено методом ВЭЖХ); содержание -эпоксида составляло 95,3%. Температура плавления: 153-155 С.(O-CH2-CH2-O); 107,0 (С-3); 125,7 (С-11); 136,7 (С-9); 221,1 (С-17). Пример 2. 5,10-Эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-гидрокси-5-оэстр-9(11)-ен-17 карбонитрил [соединение формулы (IV)]. 5,10-Эпокси-3,3-[1,2-этандиил-бис-(окси)]-5-оэстр-9(11)-ен-17-он (33 г, 0,1 моль), полученный в примере 1, суспендировали в метаноле (132 мл), затем добавляли порошкообразный цианид калия(19,5 г, 0,3 моль) при 20-25 С. После осторожного добавления уксусной кислоты (11,5 мл, 0,2 моль) гетерогенную реакционную смесь нагревали до 55 С в течение 15 мин, затем охлаждали до 25 С в течение 1 ч и перемешивали при этой температуре еще 5 ч. После завершения реакции добавляли воду (132 мл) в течение 30 мин, полученный кристаллический продукт отфильтровывали, промывали водой и использовали в следующей стадии без высушивания. Температура плавления высушенного образца: 143-144 С.(O-СН 2-СН 2-O); 77,3 (С-17); 106,9 (С-3); 120,7 (С-20); 125,9 (С-11); 135,7 (С-9). Пример 3. 5,10-Эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-[(триметилсилил)окси]-5-оэстр-9(11)ен-17-карбонитрил [соединение формулы (V)]. 5,10-Эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-гидрокси-5-оэстр-9(11)-ен-17-карбонитрил, полученный в примере 2, растворяли при интенсивном перемешивании в дихлорметане (300 мл), полученный раствор сушили над безводным сульфатом натрия, затем отгоняли из раствора 200 мл дихлорметана. К полученному таким образом раствору добавляли имидазол (10,1 г, 0,148 моль), затем добавляли по каплям триметилхлорсилан (15,5 мл, 0,121 моль) при 20-25 С в течение 20 мин. После перемешивания в течение 1 ч раствор разбавляли дихлорметаном (66 мл) и водой (66 мл). Органический слой отделяли,промывали водой, сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток обрабатывали метанолом (60 мл), охлаждали до 0 С, выпавший в осадок кристаллический продукт отфильтровывали, промывали метанолом, имеющим температуру 0 С, и сушили при 40 С в вакууме, получая 31,5 г(73,36%) целевого соединения. Полученный продукт использовали в следующей стадии. Температура плавления: 167-170 С.[]25D=12,5 (с=1%, хлороформ). 1 Н ЯМР (300 МГц, CDCl3 (ТМС),(м.д.: 0,15 (9 Н, с, 17-O-Si(CH3)3); 0,83 (3 Н, д, 18-СН 3); 1,83 (1H,дд, Нх-4); 2,08 (1H, д, Hx-4); 3,76-3.94 (4 Н, м, O-СН 2-СН 2-O); 6,01 (1H, м, Н-11); 13 С ЯМР (75 МГц, CDCl3 (ТМС),(м.д.: 0,9 (17-O-Si(CH3)3); 16,3 (С-18); 40,1 (С-4); 59,9 (С-10); 61,5 (С-5); 63,9 и 64,1 (O-СН 2-СН 2-О); 78,2 (С-17); 106,8 (С-3); 120,5 (С-20); 126,3 (С-11); 135,4 (С-9). Пример 4. 11-[4-(Диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5-гидрокси-17-[(триметилсилил)окси]-5-оэстр-9-ен-17-карбонитрил [соединение формулы (VII)]. В атмосфере азота магниевые стружки (3,3 г, 0,136 моль), сухой тетрагидрофуран (24 мл) и 1,2-дибромэтан (0,12 мл, 0,00131 моль) помещали в колбу, оснащенную мешалкой, термометром, капельной воронкой, подводом и отводом газа, при 20-25 С. После перемешивания в течение 5-10 мин температура начинала повышаться, указывая на успешное осуществление активации. Одновременно при 25 С в атмосфере азота готовили следующий раствор: сухой тетрагидрофуран(15 мл), сухой толуол (84 мл), 4-бром-N,N-диметиланилин (25 г, 0,125 моль) и 1,2-дибромэтан (0,16 мл,0,00186 моль); 2 мл данного раствора добавляли к раствору, содержащему магниевые стружки, и полученную таким образом перемешиваемую реакционную смесь нагревали до 60 С. Если интенсивное кипение реакционной смеси указывало на успешное осуществление активации, тогда добавляли по каплям остаток раствора 4-бром-N,N-диметиланилина после охлаждения и поддерживали охлаждением температуру 14-16 С еще в течение 2 ч. Хлорид меди(I) (0,4 г, 4,04 ммоль) добавляли к полученному раствору реактива Гриньяра, затем реакционную смесь перемешивали при 20-25 С в течение 5 мин. После охлаждения до 8-13 С 5,10-эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-[(триметилсилил)окси]-5-оэстр-9(11)-ен-17-карбонитрил (42,96 г, 0,1 моль) в дихлорметане (180 мл) прикапывали к перемешиваемому и охлаждаемому раствору с такой скоростью, чтобы температура не превышала 10-15 С. Затем охлаждение прекращали и реакционную смесь перемешивали еще 4 ч. После завершения реакции реакционную смесь добавляли к интенсивно перемешиваемому раствору хлорида аммония (100 мл, 10%-ный водный раствор), который содержал пиросульфит натрия (0,4 г,2,1 ммоль), разбавляли дихлорметаном (100 мл), перемешивали и оставляли для разделения. После отделения органического слоя водную фазу экстрагировали дихлорметаном, объединенные органические слои промывали водой, сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток перекристаллизовали из метанола, получая 44.5 г (80,79%) целевого соединения. Температура плавления: 243-256 С.(1H, д, ОН); 6,64 (2 Н, м, Н-3' и Н-5'); 7,05 (2 Н, м, Н-2' и Н-6'); 13 С ЯМР (75 МГц, CDCl3 (TMC),(м.д.: 1,1 (17-O-Si(CH3)3); 16,9 (С-18); 38,8 (С-11); 40,7 (N-СН 3); 47,5 (С-4); 64,1 и 64,5 (O-СН 2-СН 2-O); 70,1 (С-5); 78,9 (С-17); 108,8 (С-3); 112,6 (С-3' и С-5'); 121,0 (С-20); 127,6 (С-2' и С-6'); 133,9, 134,0, 134,1 (С-9, С-10, С-1'); 148,4 (С-4'). Полученный таким образом продукт использовали в следующей стадии без дополнительной очистки. Пример 5. 11-[4-(Диметиламино)фенил)]-3.3-[1,2-этандиил-бис-(окси)]-5,17-бис-[(триметилсилил)окси]-5-оэстр-9-ен-17-карбонитрил [соединение формулы (VII)]. 11-[4-(Диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5-гидрокси-17-[(триметилсилил)окси]-5-оэстр-9-ен-17-карбонитрил (55 г, 0,1 моль) и имидазол (10,2 г, 0,15 моль) растворяли при перемешивании в дихлорметане (225 мл) при 20-25 С. К раствору прикапывали триметилхлорсилан(15,75 мл, 0,123 моль) в течение 20 мин. Во время добавления реагента начинал осаждаться гидрохлорид имидазола, указывая на протекание реакции. После перемешивания в течение 2 ч реакционную смесь разбавляли дихлорметаном (100 мл) и водой (100 мл), перемешивали в течение нескольких минут, оставляли для разделения слоев, затем органический слой отделяли, промывали водой, сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток кристаллизовали из метанола, отфильтрованный продукт сушили в вакууме, получая 55,5 г (89,09%) целевого соединения. Температура плавления: 164-166 С.(С-3' и С-5'); 120,7 (С-20); 127,4 (С-2' и С-6'); 132,3, 133,2, 134,9 (С-9, С-10, С-1'); 148,1 (С-4'). Пример 6. 11-[4-Диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5,17-бис-[(триметилсилил)окси]-5-оэстр-9-ен-17-карбальдегид [соединение формулы (VIII)]. В атмосфере азота 11-[4-(диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5,17-бис[(триметилсилил)окси]-5-оэстр-9-ен-17-карбонитрил (40 г, 62,4 ммоль) растворяли в смеси метилтрет-бутилового эфира (220 мл) и тетрагидрофурана (17 мл). Полученный раствор охлаждали до(160 мл) в течение 30 мин при -(15-20)С. Реакционную смесь перемешивали 1 ч, затем в атмосфере азота добавляли при интенсивном перемешивании смесь воды (160 мл) и уксусной кислоты (80 мл), имеющую температуру -(5-10)С в течение 15-20 мин. Полученную таким образом реакционную смесь перемешивали при 20-25 С в течение 30 мин, затем органический слой отделяли, промывали водой (200 мл), 0,3 М раствором гидрокарбоната натрия (2200 мл) и водой (200 мл). Органический слой без высушивания упаривали в вакууме при 40-45 С. Остаток растворяли в метаноле (140 мл) и упаривали в вакууме до объема 30 мл. Полученную кристаллическую суспензию охлаждали до 5-10 С, фильтровали после 1 ч стояния, промывали и сушили при температуре ниже 60 С в вакууме, получая 33,6 г (83,6%) целевого соединения, которое использовали в следующей стадии. Температура плавления: 154-158 С.(С-11); 40,7 (N-CH3); 63,4 и 64,4 (O-СН 2-СН 2-O); 73,7 (С-5); 91,1 (С-17); 108,5 (С-3); 112,8 (С-3' и С-5'); 127,6 (С-2' и С-6'); 133,6 (С-10); 134,3 (С-1'); 135,9 (С-9); 148,3 (С-4'); 203,3 (С-20). Пример 7. 11-[4-(Диметиламино)фенил)]-17,20-дигидрокси-21-метокси-19-норпрегн-4,9-диен-3 он [соединение формулы (IX)]. В атмосфере азота магниевые стружки (4,2 г, 173 ммоль), сухой тетрагидрофуран (60 мл) и 1,2-дибромэтан (2,4 мл, 28 ммоль) помещали в 4-горлую колбу объемом 500 мл, оснащенную мешалкой,термометром, капельной воронкой, обратным холодильником, подводом и отводом газа, при 20-25 С. После нескольких минут перемешивания смесь достигала температуры кипения. Затем реакционную смесь охлаждали до 35-40 С и добавляли хлорид ртути(II) (0,23 г, 0,85 ммоль), после перемешивания в течение 15 мин смесь охлаждали до 20-25 С и добавляли сухой толуол (20 мл). Метоксиметилхлорид(12,8 мл, 168 ммоль) растворяли в сухом толуоле (50 мл) и 6 мл полученного таким образом раствора добавляли к смеси в 4-горлую колбу. Через несколько минут температура реакционной смеси поднималась до 35 С. Реакционную смесь охлаждали до 0-(-5)С и добавляли остаток раствора метоксиметил-9 015478 хлорида в толуоле в течение 2-2,5 ч, поддерживая температуру 0-(-5)С. По окончании добавления в течение 1 ч добавляли раствор 11-[(4-диметиламино)фенил]-3,3-[(1,2-этандиил)-бис-(окси)]-5,17-бис[(триметилсилил(окси)]-5-оэстр-9-ен-17-карбальдегида (20,0 г, 32 ммоль) в сухом толуоле (80 мл),поддерживая температуру 0-(-5)С. После завершения реакции реакционную смесь добавляли к 1 М водному раствору гидросульфата калия (200 мл) с такой скоростью, чтобы температура не поднималась выше 30 С. Смесь перемешивали при 20-25 С в течение 2 ч, затем органический слой отделяли и промывали 1 М раствором гидросульфата калия (110 мл). Объединенные водные фазы добавляли к перемешиваемой смеси 1 М раствора гидрокарбоната натрия (225 мл) и дихлорметана (75 мл). После перемешивания в течение 10-15 мин отделяли органический слой. Водную фазу экстрагировали дихлорметаном(550 мл), объединенные органические слои сушили над безводным сульфатом натрия (2 г), фильтровали, промывали дихлорметаном (220 мл) и фильтрат перемешивали с углем (2,5 г) в течение 10 мин. Уголь отфильтровывали, промывали дихлорметаном (220 мл) и фильтрат упаривали, получая 12,51 г(N-СН 3); 59,2 (O-СН 3); 71,9 (С-20); 74,9 (С-21); 84,4 (С-17); 112,8 (С-3' и С-5'); 122,5 (С-4); 127,6 (С-2' и С 6'); 128,6 (С-10); 132,0 (С-1'); 147,2 (С-9); 148,5 (С-4'); 157,1 (С-5); 199,7 (С-3). Пример 8. 11-[4-(Диметиламино)фенил)]-17,20-дигидрокси-21-метокси-19-норпрегн-4,9-диен-3 он [соединение формулы (IX)]. В атмосфере азота магниевые стружки (4,2 г, 173 ммоль), сухой тетрагидрофуран (60 мл) и 1,2-дибромэтан (2,4 мл, 28 ммоль) помещали в 4-горлую колбу объемом 500 мл, оснащенную мешалкой,термометром, капельной воронкой, обратным холодильником, подводом и отводом газа, при 20-25 С. После нескольких минут перемешивания смесь достигала температуры кипения. Затем реакционную смесь охлаждали до 35-40 С и добавляли хлорид ртути(II) (0,23 г, 0,85 ммоль), после перемешивания в течение 15 мин смесь охлаждали до 20-25 С и добавляли сухой толуол (20 мл). Метоксиметилбромид(13,7 мл, 168 ммоль) растворяли в сухом толуоле (50 мл) и 6 мл полученного таким образом раствора добавляли к смеси в 4-горлую колбу. Через несколько минут температура реакционной смеси поднималась до 30-35 С. Реакционную смесь охлаждали до 10-15 С и добавляли остаток раствора метоксиметилбромида в толуоле в течение 2-2,5 ч, поддерживая температуру 10-15 С. По окончании добавления в течение 1 ч добавляли раствор 11-[(4-диметиламино)фенил]-3,3-[(1,2-этандиил)-бис-(окси)]-5,17-бис[(триметилсилил(окси)]-5-оэстр-9-ен-17-карбальдегида (20,0 г, 32 ммоль) в сухом толуоле (80 мл),поддерживая температуру 10-15 С. После завершения реакции реакционную смесь добавляли к 1 М водному раствору гидросульфата калия (200 мл) с такой скоростью, чтобы температура не поднималась выше 30 С. Смесь перемешивали при 20-25 С в течение 2 ч, затем органический слой отделяли и промывали 1 М раствором гидросульфата калия (110 мл). Объединенные водные фазы добавляли к перемешиваемой смеси 1 М раствора гидрокарбоната натрия (225 мл) и дихлорметана (75 мл). После 10-15 мин перемешивания отделяли органический слой. Водную фазу экстрагировали дихлорметаном (550 мл),объединенные органические слои сушили над безводным сульфатом натрия (2 г), фильтровали, промывали дихлорметаном (220 мл) и фильтрат перемешивали с углем (2,5 г) в течение 10 мин. Уголь отфильтровывали, промывали дихлорметаном (220 мл) и фильтрат упаривали, получая 10,76 г (72,3%) целевого соединения. Небольшой образец продукта перемешивали с н-пентаном. Температура плавления: 105 С (стеклообразная структура, размягчение).[соединение формулы (Х)]. В атмосфере азота 17,20-дигидрокси-11-[(4-диметиламино)фенил]-21-метокси-19-норпрегн-4,9 диен-3-он (11,94 г, 25,7 ммоль) и сухой толуол (68 мл) помещали в 4-горлую колбу объемом 500 мл, оснащенную мешалкой, термометром, капельной воронкой, обратным холодильником, подводом и отводом газа. К полученному таким образом раствору добавляли сухой диметилсульфоксид (9,9 мл,139,5 ммоль), пиридин (2,9 мл, 36,0 ммоль) и трифторуксусную кислоту (0,99 мл, 12,85 ммоль) при 20-25 С. Затем к реакционной смеси добавляли раствор дициклогексилкарбодиимида (10,6 г, 51,4 ммоль) в толуоле (54 мл) и полученную таким образом смесь перемешивали при 40 С. Время реакции составляло 3 ч. По окончании реакции реакционную смесь охлаждали до 20-25 С и добавляли 1 М раствор гидросульфата калия (78 мл). После 30 мин перемешивания выпавшие в осадок кристаллы отфильтровывали и промывали 1 М раствором гидросульфата калия (419 мл). Две фазы фильтрата разделяли, водную фазу добавляли к 1 M раствору гидроксида натрия (254 мл) при 10-20 С. После 30 мин перемешивания вы- 10015478 павший в осадок сырой продукт отфильтровывали, промывали водой и сушили, получая 9,45 г (79,5%) целевого соединения. Температура плавления: 105-110 С. Сырой продукт очищали методом ВЭЖХ согласно способу, описанному в следующем примере. 1 Н ЯМР (500 МГц, ДМСО-d6 (ТМС),(м.д.: 0,20 (3 Н, с, 18-СН 3); 2,82 (6 Н, с, N-СН 3); 3,26 (3 Н, с,O-СН 3); 4,20 (1 Н, д, Нх-21); 4,35 (1H, м, Н-11); 4,49 (1 Н, д, Ну-21); 5,37 (1H, с, ОН); 5,67 (1H, с, Н-4); 6,62(С-9); 148,2 (С-4'); 156,5 (С-5); 197,9 (С-3); 208,9 (С-20). Пример 10. Очистка сырого 11-[4-(диметиламино)фенил)]-17-гидрокси-21-метокси-19-норпрегн 4,9-диен-3,20-диона [соединение формулы (X)] методом ВЭЖХ (лабораторный масштаб). Силикагелем (510 г, ZEOPREP C-GEL C-490L, размер частиц 15-35 мкм; высота слоя около 60 см) заполняли ВЭЖХ колонку с аксиальным сжатием диаметром 5 см методом суспензионной набивки и колонку уравновешивали элюентом - смесью 45:40:15 циклогексан/метил-трет-бутиловый эфир/ацетон. 10,0 г сырого целевого соединения (содержание действующего вещества: 80%) растворяли в смеси ацетона (30 мл) и метил-трет-бутилового эфира (80 мл), к перемешиваемому раствору добавляли циклогексан (90 мл). Полученный таким образом раствор фильтровали и наносили на колонку путем вкола. Продукт элюировали со скоростью потока 85 мл/мин, используя УФ-детектирование. Первая фракция составляла около 50 мл, основная фракция, содержащая чистое целевое соединение, составляла около 600 мл. Твердое целевое соединение получали упариванием элюированной основной фракции. Выход: 7,4 г (74%) чистого целевого соединения. Содержание примесей: менее 4%. Температура плавления: 108-110 С.(-(20-25)С) уксусному ангидриду (45 мл) с такой скоростью, чтобы температура не поднималась выше-15 С. Затем добавляли раствор 11-[4-(диметиламино)фенил)]-17-гидрокси-21-метокси-19-норпрегн 4,9-диен-3,20-диона (15,5 г) в дихлорметане (60 мл) при -(20-25)С. По окончании реакции, которую контролировали методом тонкослойной хроматографии, реакционную смесь разбавляли дихлорметаном(50 мл), охлаждали до -10 С и добавляли пропущенную через ионообменник воду (52 мл) для разложения уксусного ангидрида. После 10 мин перемешивания добавляли 25%-ный раствор гидроксида аммония (77 мл) с такой скоростью, чтобы температура не поднималась выше 25 С (рН 7-8). Затем выпавший в осадок карбамидный побочный продукт отфильтровывали, водную фазу отделяли, экстрагировали дихлорметаном (230 мл) и объединенные органические слои упаривали, получая 16,2 г (95,8%) целевого соединения, которое очищали методом ВЭЖХ согласно способу, описанному в следующем примере. 1 Н ЯМР (500 МГц, CDCl3 (ТМС),(м.д.: 0,40 (3 Н, с, 18-СН 3); 2,10 (3 Н, с, О-СО-СН 3); 2,90 (6 Н, с,N-CH3); 3,41 (3 Н, с, O-СН 3); 4,09 (1 Н, д, Нх-21); 4,38 (1 Н, м, Н-11); 4,29 (1H, д, Ну-21); 5,77 (1H, ушир.,Н-4); 6,62 (2 Н, м, Н-3' и Н-5'); 6,96 (2 Н, м, Н-2' и Н-6'); 13 С ЯМР (125 МГц, CDCl3 (ТМС),(м.д.: 15,6 (С-18); 21,1 (O-СО-СН 3); 39,3 (С-11); 40,6 (N-СН 3); 59,4 (O-СН 3); 76,0 (С-21); 93,9 (С-17); 112,8 (С-3' и С-5'); 123,0 (С-4); 127,3 (С-2' и С-6'); 129,4 (С-10); 131,3 (С-1'); 145,5 (С-9); 148,7 (С-4'); 156,4 (С-5); 170,7 (O-СО-СН 3); 199,4 (С-3); 202,7 (С-20). Пример 12. Очистка сырого CDB-4124 методом ВЭЖХ (элюент: циклогексан:метил/третбутиловый эфир:ацетон=60:30:10) (лабораторный масштаб) [соединение формулы (I)]. Силикагелем (510 г, ZEOPREP C-GEL C-490L, размер частиц 15-35 мкм; высота слоя около 60 см) заполняли ВЭЖХ колонку с аксиальным сжатием диаметром 5 см методом суспензионной набивки и колонку уравновешивали элюентом - смесью 60:30:10 циклогексан/метил-трет-бутиловый эфир/ацетон. 5,1 г сырого соединения формулы (I) (CDB-4124), полученного в предыдущем примере (содержание примесей: менее 4%), растворяли в элюенте (100 мл), фильтровали и наносили на колонку путем вкола. Продукт элюировали со скоростью потока 85 мл/мин, применяя УФ-детектирование. Первая фракция составляла около 40 мл, основная фракция, содержащая чистый CDB-4124, составляла около 560 мл. Твердое целевое соединение получали упариванием элюированной основной фракции. Выход: 4,25 г (83,33%).[]25D=127,2 (с=1%, хлороформ). 1 Н ЯМР (500 МГц, CDCl3 (TMC),(м.д.: 0,40 (3 Н, с, 18-СН 3); 2,10 (3 Н, с, О-СО-СН 3); 2,90 (6 Н, с,N-CH3); 3,41 (3 Н, с, O-СН 3); 4,09 (1H, д, Нх-21); 4,38 (1H, м, Н-11); 4,29 (1H, д, Ну-21); 5,77 (1H, ушир., Н 4); 6,62 (2 Н, м, Н-3' и Н-5'); 6,96 (2 Н, м, Н-2' и Н-6'); 13 С ЯМР (125 МГц, CDCl3 (ТМС),(м.д.: 15,6 (С-18); 21,1 (O-СО-СН 3); 39,3 (С-11); 40,6 (N-CH3); 59,4 (O-СН 3); 76,0 (С-21); 93,9 (С-17); 112,8 (С-3' и С-5'); 123,0 (С-4); 127,3 (С-2' и С-6'); 129,4 (С-10); 131,3 (С-1'); 145,5 (С-9); 148,7 (С-4'); 156,4 (С-5); 170,7 (O-СО-СН 3); 199,4 (С-3); 202,7 (С-20). Пример 13. 5,10-Эпокси-3,3-[1,2-этандиил-бис-(окси)]-5-оэстр-9(11)-ен-17-он [соединение формулы (III)]. Оборудование: кислотоупорный стальной реактор емкостью 500 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. В атмосфере азота растворяли 3,3-[1,2-этандиил-бис-(окси)]оэстр-5(10),9(11)-диен-17-он (21,0 кг) при интенсивном перемешивании в смеси пиридина (1,106 л, 0,2 моль-эквивалент) и сухого дихлорметана (105,2 л), полученный раствор охлаждали до -(6-8)С. После добавления гексахлорацетона (2,455 л) к перемешиваемому раствору добавляли 50%-ную перекись водорода, имеющую температуру 0-(-2)С(26,97 л) с такой скоростью, чтобы температура не поднималась выше 0 С. Реакционную смесь перемешивали при 1-(-1)С в течение 20-24 ч, затем разбавляли дихлорметаном, имеющим температуру 0-5 С(175 л), разлагали избыток перекиси водорода добавлением раствора пентагидрата тиосульфата натрия(8,87 моль-эквивалент) в ледяной воде (150 л). Реакционную смесь перемешивали в течение 1,5 ч, затем отделяли органическую фазу. Водную фазу экстрагировали дихлорметаном, объединенные органические слои промывали водой, сушили над безводным сульфатом натрия, фильтровали и упаривали. Маслообразный остаток кристаллизовали из смеси 1:3 этилацетат/диизопропиловый эфир (195,6 л), содержащей 0,1% пиридина. Полученный таким образом продукт сушили, получая 11,500 кг (52,13%) целевого соединения. Чистота целевого соединения составляла 98,5-98,9% (определено методом ВЭЖХ); содержание[]25D=125,0 (с=1%, хлороформ). Пример 14. 5,10-Эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-гидрокси-5-оэстр-9(11)-ен-17-карбонитрил [соединение формулы (IV)]. Оборудование: эмалированный реактор емкостью 250 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. 5,10-Эпокси-3,3-[1,2-этандиил-бис-(окси)]-5-оэстр-9(11)-ен-17-он (9,9 кг), полученный в примере 13, суспендировали в метаноле (39,6 л), затем добавляли порошкообразный цианид калия (5,85 кг,0,3 моль) при 20-25 С. После осторожного добавления уксусной кислоты (3,48 л) гетерогенную реакционную смесь нагревали до 55 С в течение 15 мин, затем охлаждали до 25 С в течение 1 ч и перемешивали при этой температуре еще 5 ч. По окончании реакции добавляли воду (39,6 л) в течение 30 мин, полученный кристаллический продукт отфильтровывали, промывали водой и использовали в следующей стадии без высушивания. Температура кипения высушенного образца: 140-143 С.[]25D=13,0 (с=1%, хлороформ). Пример 15. 5,10-Эпокси-3,3-[1.2-этандиил-бис-(окси)]-17-[(триметилсилил)окси]-5-оэстр-9(11)ен-17-карбонитрил [соединение формулы (V)]. Оборудование: эмалированный реактор емкостью 250 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. 5,10-Эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-гидрокси-5-оэстр-9(11)-ен-17-карбонитрил, полученный в примере 14, растворяли при интенсивном перемешивании в дихлорметане (90 л), полученный раствор сушили над безводным сульфатом натрия, затем 60 л дихлорметана отгоняли из раствора. Имидазол (0,303 кг) добавляли к полученному таким образом раствору, затем прикапывали триметилхлорсилан (7,2 л) при 20-25 С в течение 20 мин. После перемешивания в течение 1 ч раствор разбавляли дихлорметаном (19,8 л) и водой (19,8 л). Органический слой отделяли, промывали водой, сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток обрабатывали метанолом (18 л), охлаждали до 0 С, выпавший в осадок кристаллический продукт отфильтровывали, промывали метанолом,имеющим температуру 0 С, и сушили при 40 С в вакууме, получая 10,1 кг (78,4%) целевого соединения. Полученный продукт использовали в следующей стадии без дополнительной очистки. Температура плавления: 167-170 С.- 12015478 Пример 16. 11-[4-(Диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5-гидрокси-17-[(триметилсилил)окси]-5-оэстр-9-ен-17-карбонитрил [соединение формулы (VI)]. Оборудование: эмалированный реактор емкостью 250 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. В атмосфере азота магниевые стружки (0,768 кг), сухой тетрагидрафуран (5,59 л) и 1,2-дибромэтан(27,94 мл) помещали в реактор при 20-25 С. После перемешивания в течение 5-10 мин температура начинала повышаться, указывая на успешное осуществление активации. Одновременно при 25 С в атмосфере азота готовили следующий раствор: сухой тетрагидрофуран(3,5 л), сухой толуол (19,6 л), 4-бром-N,N-диметиланилин (5,8 кг) и 1,2-дибромэтан (34,25 мл). 400 мл этого раствора добавляли к раствору, содержащему магниевые стружки, и полученную таким образом перемешиваемую реакционную смесь нагревали до 60 С. Интенсивное кипение реакционной смеси указывало на успешное осуществление активации, остаток раствора 4-бром-N,N-диметиланилина добавляли по каплям после охлаждения и охлаждением поддерживали температуру равной 14-16 С еще в течение 2 ч. Хлорид меди(I) (93,11 г) добавляли к полученному раствору реактива Гриньяра, затем реакционную смесь перемешивали при 20-25 С в течение 5 мин. После охлаждения до 8-13 С прикапывали к перемешиваемому и охлаждаемому раствору 5,10-эпокси-3,3-[1,2-этандиил-бис-(окси)]-17-[(триметилсилил)окси]-5-оэстр-9(11)-ен-17-карбонитрил (10,0 кг) в дихлорметане (42 л) с такой скоростью, чтобы поддерживать температуру 10-15 С. Затем прекращали охлаждение и перемешивали реакционную смесь еще 4 ч. По окончании реакции смесь добавляли к интенсивно перемешиваемому раствору хлорида аммония(23,3 л, 10%-ный водный раствор), содержащему пиросульфит натрия (93,1 г), разбавляли дихлорметаном (23,3 л), перемешивали и оставляли расслаиваться. После отделения органического слоя водную фазу экстрагировали дихлорметаном, объединенные органические слои промывали водой, сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток перекристаллизовали из метанола, получая 10,5 кг (85,71%) целевого соединения. Температура плавления: 243-256 С.[]25D=-12,4 (с=1%, хлороформ). Полученный таким образом продукт использовали в следующей стадии без дополнительной очистки. Пример 17. 11-[4-(Диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5,17-бис-[(триметилсилил)окси]-5-оэстр-9-ен-17-карбонитрил [соединение формулы (VII)]. Оборудование: эмалированный реактор емкостью 250 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. 11-[4-(Диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5-гидрокси-17-[(триметилсилил)окси]-5-оэстр-9-ен-17-карбонитрил (10,45 кг) и имидазол (1,93 кг) растворяли при перемешивании в дихлорметане (42,75 л) при 20-25 С. К раствору прикапывали триметилхлорсилан (3,0 л) в течение 20 мин. Во время добавления реактива начинал выпадать в осадок гидрохлорид имидазола, указывая на протекание реакции. После перемешивания в течение 2 ч реакционную смесь разбавляли дихлорметаном(19 л) и водой (19 л), перемешивали несколько минут, оставляли расслаиваться, затем органический слой отделяли, промывали водой, сушили над безводным сульфатом натрия, фильтровали и упаривали. Остаток кристаллизовали из метанола, отфильтрованный продукт сушили в вакууме, получая 10,25 кг (87,0%) целевого соединения. Температура плавления: 164-166 С.[]25D=14,7 (с=1%, хлороформ). Пример 18. 11-[4-(Диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5,17-бис-[(триметилсилил)окси]-5-оэстр-9-ен-17-карбальдегид [соединение формулы (VIII)]. Оборудование: эмалированный реактор емкостью 250 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. В атмосфере азота 11-[4-(диметиламино)фенил)]-3,3-[1,2-этандиил-бис-(окси)]-5,17-бис[(триметилсилил)окси]-5-оэстр-9-ен-17-карбонитрил (8 кг) растворяли в смеси метил-трет-бутилового эфира (44 л) и тетрагидрофурана (3,4 л). Полученный раствор охлаждали до -(15-20)С, затем добавляли 1 M раствор DIBAL-H в циклогексане (32 л) в течение 30 мин при -(15-20)С. Реакционную смесь перемешивали в течение 1 ч, затем при интенсивном перемешивании в атмосфере азота добавляли смесь воды (32 л) и уксусной кислоты (16 л), имеющую температуру -(5-10)С, в течение 15-20 мин. Полученную таким образом реакционную смесь перемешивали при 20-25 С в течение 30 мин, затем органический слой отделяли, промывали водой (40 л), 0,3 М раствором гидрокарбоната натрия (240 л) и водой (40 л). Органический слой без высушивания упаривали в вакууме при 40-45 С. Остаток растворяли в метаноле(28 л) и упаривали в вакууме до объема 6 л Полученную кристаллическую суспензию охлаждали до 5-10 С, фильтровали после 1 ч стояния, промывали и сушили при температуре ниже 60 С в вакууме, получая 6,95 кг (86,46%) целевого соединения, которое использовали в следующей стадии без дополнительной очистки.[]25D=7,7 (с=1%, хлороформ). Пример 19. 11-[4-(Диметиламино)фенил)]-17,20-дигидрокси-21-метокси-19-норпрегн-4.9-диен-3 он [соединение формулы (IX)]. Оборудование: эмалированный реактор емкостью 250 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. В атмосфере азота магниевые стружки (1,05 кг), сухой тетрагидрофуран (15 л) и 1,2-дибромэтан(600 мл) добавляли в реакционный аппарат при 20-25 С. После нескольких минут перемешивания смесь достигала температуры кипения. Затем реакционную смесь охлаждали до 35-40 С и добавляли хлорид ртути(II) (57,5 г), после перемешивания в течение 15 мин смесь охлаждали до 20-25 С и добавляли сухой толуол (5 л). Метоксиметилхлорид (3,2 л) растворяли в сухом толуоле (12,5 л) и 1,5 л полученного таким образом раствора добавляли к реакционной смеси. Через несколько минут температура реакционной смеси поднималась до 35 С. Реакционную смесь охлаждали до 0-(-5)С и остаток раствора метоксиметилхлорида в толуоле добавляли в течение 2-2,5 ч, поддерживая температуру 0-(-5)С. По окончании добавления в течение 1 ч добавляли раствор 11-[(4-диметиламино)фенил]-3,3-[(1,2-этандиил)-бис-(окси)]5,17-бис-[(триметилсилил(окси)]-5-оэстр-9-ен-17-карбальдегида (5,0 кг) в сухом толуоле (20 л), поддерживая температуру 0-(-5)С. По окончании реакции реакционную смесь добавляли к 1 М водному раствору гидросульфата калия (50 л) с такой скоростью, чтобы температура не поднималась выше 30 С. Смесь перемешивали при 20-25 С в течение 2 ч, затем органический слой отделяли и промывали 1 М раствором гидросульфата калия (12,5 л). Объединенные водные фазы добавляли к перемешиваемой смеси 1 М раствора гидрокарбоната натрия (56 л) и дихлорметана (19 л). После 10-15 мин перемешивания отделяли органический слой. Водную фазу экстрагировали дихлорметаном (512,5 л), объединенные органические слои сушили над безводным сульфатом натрия (500 г), фильтровали, промывали дихлорметаном (22 л) и фильтрат перемешивали с углем (625 г) в течение 10 мин. Уголь отфильтровывали,промывали дихлорметаном (25 л) и фильтрат упаривали, получая 3,3 кг (88,73%) целевого соединения. Температура плавления: 105 С (размягчение).[]25D=156,2 (с=1%, дихлорметан). Пример 20. 11-[4-(Диметиламино)фенил)]-17-гидрокси-21-метокси-19-норпрегн-4.9-диен-3.20 дион [соединение формулы (X)]. Оборудование: эмалированный реактор емкостью 250 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. В атмосфере азота 17,20-дигидрокси-11-[(4-диметиламино)фенил]-21-метокси-19-норпрегн-4,9 диен-3-он (3,2 кг) и сухой толуол (18 л) помещали в реактор. К полученному таким образом раствору добавляли сухой диметилсульфоксид (2,7 л), пиридин (0,78 л) и трифторуксусную кислоту (0,265 л) при 20-25 С. Затем к реакционной смеси добавляли раствор дициклогексилкарбодиимида (2,84 кг) в толуоле(14,5 л) и полученную таким образом смесь перемешивали при 40 С. Время реакции составляло 3 ч. По окончании реакции реакционную смесь охлаждали до 20-25 С и добавляли 1 М раствор гидросульфата калия (21 л). После 30 мин перемешивания выпавшие в осадок кристаллы отфильтровывали и промывали 1 М раствором гидросульфата калия (45 л). Две фазы фильтрата разделяли, водную фазу добавляли к 1 М раствору гидроксида натрия (68 л) при 10-20 С. После 30 мин перемешивания выпавший в осадок сырой продукт отфильтровывали, промывали водой и сушили, получая 2,556 кг (80,0%) целевого соединения. Сырой продукт очищали методом ВЭЖХ согласно способу, описанному в следующем примере. Пример 21. Очистка сырого 11-[4-(диметиламино)фенил)]-17-гидрокси-21-метокси-19-норпрегн 4,9-диен-3,20-диона [соединение формулы (Х)] методом ВЭЖХ (промышленный масштаб). Силикагелем (8 кг, ZEOPREP C-GEL C-490L, размер частиц 15-35 мкм; высота слоя около 60 см) заполняли ВЭЖХ колонку с аксиальным сжатием диаметром 20 см методом суспензионной набивки и колонку уравновешивали элюентом - смесью 53:35:12 циклогексан/метил-трет-бутиловый эфир/ацетон. 160 г сырого целевого соединения (содержание действующего вещества: 80%) растворяли в смеси ацетона (0,48 л) и метил-трет-бутилового эфира (1,28 л), к перемешиваемому раствору добавляли циклогексан(1,44 л). Полученный таким образом раствор фильтровали и наносили на колонку путем вкола. Продукт элюировали при скорости потока 80 л/ч, применяя УФ-детектирование. Первая фракция составляла около 1 л, основная фракция, содержащая чистое целевое соединение, составляла около 14 л. Твердое целевое соединение получали упариванием элюированной основной фракции, но предпочтительно после упаривания основной фракции дихлорметан отгоняли из остатка и продукт растворяли в дихлорметане. Полученный дихлорметановый раствор использовали в следующей стадии. Выход: 120 г (75%) чистого, твердого целевого соединения или содержащего действующее вещество дихлорметанового раствора. Содержание примесей: менее 4%. Температура плавления: 105-110 С.- 14015478 Пример 22. Сырой 17-ацетокси-11-[4-(диметиламино)фенил)]-21-метокси-19-норпрегн-4,9-диен 3,20-дион [соединение формулы (I)]. Оборудование: эмалированный реактор емкостью 250 л, оснащенный пропеллерной мешалкой с разными скоростями вращения, обратным холодильником и термометром. 70%-ную перхлорную кислоту (1,8 л) добавляли к перемешиваемому и охлаждаемому (-(20-25)С) уксусному ангидриду (13,5 л) с такой скоростью, чтобы температура не поднималась выше -15 С. Затем добавляли раствор 11-[4-(диметиламино)фенил)]-17-гидрокси-21-метокси-19-норпрегн-4,9-диен-3,20 диона (4,65 кг) в дихлорметане (18 л) при -(20-25)С. По окончании реакции, которую контролировали методом тонкослойной хроматографии, реакционную смесь разбавляли дихлорметаном (15 л), охлаждали до -10 С и добавляли пропущенную через ионообменник воду (15,5 л) для разложения уксусного ангидрида. После 10 мин перемешивания добавляли 25%-ный раствор гидроксида аммония (23 л) с такой скоростью, чтобы температура не поднималась выше 25 С (рН 7-8). Затем выпавший в осадок побочный карбамидный продукт отфильтровывали, отделяли водную фазу, экстрагировали дихлорметаном (29 л) и объединенные органические слои упаривали, получая 4,73 кг (93,79%) целевого соединения(CDB-4124), которое очищали методом ВЭЖХ согласно способу, описанному в следующем примере. Пример 23. Очистка сырого CDB-4124 методом ВЭЖХ (промышленный масштаб) [соединение формулы (I)]. Силикагелем (8 кг, ZEOPREP C-GEL C-490L, размер частиц 15-35 мкм; высота слоя около 60 см) заполняли ВЭЖХ колонку с аксиальным сжатием диаметром 20 см методом суспензионной набивки и уравновешивали колонку элюентом - смесью 53:35:12 циклогексан/метил-трет-бутиловый эфир/ацетон. 80 г сырого соединения формулы (I) (CDB-4124), полученного в предыдущем примере (содержание примесей: менее 4%), растворяли в элюенте (1,6 л), фильтровали и наносили на колонку путем вкола. Продукт элюировали при скорости потока 80 л/ч, применяя УФ-детектирование. Первая фракция составляла около 0,7 л, основная фракция, содержащая чистый CDB-4124, составляла около 10 л. Твердое целевое соединение получали упариванием элюированной основной фракции или его можно получать в виде метанольного раствора после упаривания основной фракции и растворения продукта в метаноле. Выход: 70 г твердого целевого соединения или содержащего действующее вещество метанольного раствора. Содержание примесей: менее 0,5%. Температура плавления: 118 С.[]25D=127,2 (с=1%, хлороформ). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Промышленный способ синтеза 17-ацетокси-11-[4-(диметиламино)фенил]-21-метокси-19 норпрегн-4,9-диен-3,20-диона формулы (I)i) образование эпоксида по двойной связи в положении 5(10) взаимодействием 3,3-[1,2-этандиилбис-(окси)]оэстр-5(10),9(11)-диен-17-она формулы (II)iii) силилирование гидроксильной группы в положении 17 образовавшегося 5,10-эпокси-3,3-[1,2 этандиил-бис-(окси)]-17-гидрокси-5-оэстр-9(11)-ен-17-карбонитрила формулы (IV)v) силилирование гидроксильной группы в положении 5 полученного 11-[4-(диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5-гидрокси-17-[триметилсилил(окси)]-5-оэстр-9-ен-17-карбонитрила формулы (VI) с диизобутилалюминийгидридом и последующее добавление кислоты к реакционной смеси; метоксиметильным реактивом Гриньяра, полученным in situ, с одновременным гидролизом триметилсилильных защитных групп;viii) окисление гидроксильной группы в положении 20 полученного 17,20-дигидрокси-11-[4(диметиламино)фенил]-21-метокси-19-норпрегн-4,9-диен-3-она формулы (IX)("Swern oxidation") и в данном случае последующая хроматографическая очистка;ix) ацетилирование гидроксильной группы в положении 17 полученного 11-[4(диметиламино)фенил]-17-гидрокси-21-метокси-19-норпрегн-4,9-диен-3,20-диона формулы (X) уксусным ангидридом в присутствии перхлорной кислоты и в данном случае последующая хроматографическая очистка полученного 7-ацетокси-11-[4-(диметиламино)фенил)]-21-метокси-19-норпрегн-4,9 диен-3,20-диона формулы (I). 2. Способ по п.1, отличающийся тем, что на стадии iv) используют 0,250,025 экв. избытка реактива Гриньяра 4-(диметиламино)фенилмагнийбромида относительно 5,10-эпокси-3,3-[1,2-этандиил-бис(окси)]-17-[триметилсилилокси]-5-оэстр-9(11)-ен-17-карбонитрила формулы (V). 3. Способ по п.1, отличающийся тем, что на стадии viii) используют трифторуксусную кислоту в качестве сильной органической кислоты. 4. 11-[4-(Диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5,17-бис-[триметилсилил(окси)]5-оэстр-9-ен-17-карбонитрил формулы (VII). 5. 11-[4-(Диметиламино)фенил]-3,3-[1,2-этандиил-бис-(окси)]-5,17-бис-[триметилсилил(окси)]5-оэстр-9-ен-17-карбальдегид формулы (VIII).
МПК / Метки
МПК: C07J 21/00, C07J 51/00, C07J 41/00
Метки: способ, фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона, синтеза, промышленный, соединения, способа, данного, ключевые, 17-ацетокси-11b-[4-(диметиламино, промежуточные
Код ссылки
<a href="https://eas.patents.su/18-15478-promyshlennyjj-sposob-sinteza-17-acetoksi-11b-4-dimetilamino-fenil-21-metoksi-19-norpregn-49-dien-320-diona-i-klyuchevye-promezhutochnye-soedineniya-dannogo-sposoba.html" rel="bookmark" title="База патентов Евразийского Союза">Промышленный способ синтеза 17-ацетокси-11b-[4-(диметиламино) фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона и ключевые промежуточные соединения данного способа</a>
Промышленный способ синтеза 17a-ацетокси-11b-[4-(n,n-диметиламино)фенил]-19-норпрегна-4,9-диен-3,20-диона и новые промежуточные соединения, полученные в данном способе

Номер патента: 15302
Опубликовано: 30.06.2011
Авторы: Молнар Чаба, Данчи Лайошне, Туба Золтан, Мадьяри Эндрене, Виски Дьёрдь, Чёргей Янош
МПК: C07J 21/00, C07J 31/00, C07J 41/00...
Метки: способ, 17a-ацетокси-11b-[4-(n,n-диметиламино)фенил]-19-норпрегна-4,9-диен-3,20-диона, полученные, способе, данном, соединения, промежуточные, новые, промышленный, синтеза
Формула / Реферат:
1. Способ синтеза несольватированного 17a-ацетокси-11b-[4-(N,N-диметиламино)фенил]-19-норпрегна-4,9-диен-3,20-диона формулы (I), включающий следующие стадии:i) взаимодействие 3-(этилендиокси)эстра-5(10),9(11)диен-17-она формулы (X) с ацетилидом калия, образующимся in situ в сухом тетрагидрофуране,ii) взаимодействие полученного 3-(этилендиокси)-17a-этинил-17b-гидроксиэстра-5(10),9(11)диена формулы (IX) с фенилсульфенилхлоридом в дихлорметане в...
Способ получения 17бета-гидрокси-11бета-{4-(диметиламино)фенил} 17альфа-(проп-1- инил)эстра-4,9-диен-3-она

Номер патента: 14
Опубликовано: 30.12.1997
Авторы: Климова Людмила Игоревна, Морозова Людмила Сергеевна, Густова Ольга Валериевна, Гриненко Галина Семеновна, Долгинова Елена Максовна, Кочев Дмитрий Михайлович, Ряховская Маргарита Игоревна, Турчин Константин Федорович
МПК: A61K 31/565, C07J 1/00
Метки: способ, 17бета-гидрокси-11бета-{4-(диметиламино)фенил, получения, инил)эстра-4,9-диен-3-она, 17альфа-(проп-1
Формула / Реферат:
Способ получения 17b -гидрокси-11b -[4-(диметиламино) фенил]-17a -(проп-1-инил)эстра-4,9-диен-3-она формулы I взаимодействием производного стероида с 4-диметиламинофенилмагнийбромидом в присутствии катализатора в среде тетрагидрофурана, выделением соответствующего арилкеталя с использованием насыщенного водного раствора хлористого аммония, дегидратацией и гидролизом в присутствии кислотного агента в среде растворителя при комнатной...
Способ разложения n2o, катализатор для данного способа и получение данного катализатора

Номер патента: 11258
Опубликовано: 27.02.2009
Авторы: Питерсе Йоханнис Алауисиус Захариас, Ван Ден Бринк Рудольф Виллем
МПК: B01J 29/10, B01J 29/06, B01D 53/86...
Метки: катализатора, способа, катализатор, разложения, получение, данного, способ
Формула / Реферат:
1. Способ каталитического разложения N2O в газе, содержащем N2O, в присутствии катализатора, при этом катализатор содержит цеолит, который наполнен первым металлом, выбранным из группы благородных металлов, состоящей из рутения, родия, серебра, рения, осмия, иридия, платины и золота, и вторым металлом, выбранным из группы переходных металлов, состоящей из ванадия, хрома, марганца, железа, кобальта, никеля и меди, и при этом наполнение цеолита...
Способ получения (альфа s, бета r)-6-бром-альфа-[2-(диметиламино)этил]-2-метокси-альфа-1-нафталенил-бета-фенил-3-хинолинэтанола

Номер патента: 11770
Опубликовано: 30.06.2009
Авторы: Порстманн Франк Ральф, Хорнс Штефан, Бадер Томас
МПК: C07F 9/09, C07D 215/22
Метки: альфа, получения, r)-6-бром-альфа-[2-(диметиламино)этил]-2-метокси-альфа-1-нафталенил-бета-фенил-3-хинолинэтанола, способ, beta
Формула / Реферат:
1. Способ выделения (aS,bR)-6-бром-a-[2-(диметиламино)этил]-2-метокси-a-1-нафталенил-b-фенил-3-хинолинэтанола из смеси стереоизомерных форм 6-бром-a-[2-(диметиламино)этил]-2-метокси-a-1-нафталенил-b-фенил-3-хинолинэтанола оптическим разделением с хиральным 4-гидроксидинафто[2,1-d:1',2'-f][1,3,2]диоксафосфепин 4-оксидом или его производным в качестве разделяющего агента. 2. Способ по п.1, в котором хиральный...
Новый способ получения производных n-(1-бензгидрилазетидин-3-ил)-n-фенилметилсульфонамида и промежуточные соединения для осуществления этого способа

Номер патента: 12523
Опубликовано: 30.10.2009
Авторы: Наит Буда Лалу, Лампила Максим, Грондар Люк, Мальпар Жоэль, Боффелли Филипп, Рике-Цапп Йорг, Дельтиль Мишель, Мютти Стефан
МПК: A61K 31/397, C07D 205/04, A61P 3/00...
Метки: промежуточные, производных, новый, n-(1-бензгидрилазетидин-3-ил)-n-фенилметилсульфонамида, способа, соединения, осуществления, способ, этого, получения
Формула / Реферат:
1. Способ получения N-(1-бензгидрилазетидин-3-ил)-N-фенилметилсульфонамида общей формулы (I), в которой R, R' и R'' обозначают, независимо друг от друга, один или несколько радикалов, представляющих собой атом водорода, атом галогена (Cl, F, Br, I), цианогруппу, нитрогруппу, алкильную группу, линейную или разветвленную, содержащую 1-6 атомов углерода, алкоксильную группу, линейную или разветвленную, содержащую 1-6 атомов углерода,...
Предыдущий патент: Способ получения поликристаллического кремния из раствора кремнефтористо-водородной кислоты и установка для получения поликристаллического кремния
Следующий патент: Способ оценки поперечного сцепления пневматической шины с заснеженной дорогой
Случайный патент: Способ и устройство для изготовления коксового пирога для коксования