Замещенное производное триазолопиридазина, фармацевтические композиции, приготовленные на его основе
Номер патента: 3332
Опубликовано: 24.04.2003
Авторы: Кастро Пинейро Хосе Луис, Мэдин Эндрю, Стрит Лесли Джозеф, Кауден Кэмерон Джон, Дэвис Энтони Джон, Пирс Гарет Эдвард Стефен, Маккабе Джеймс Фрэнсис, Карлинг Уилльям Роберт











Формула / Реферат
1. 7-(1,1-Диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазин.
2. Полиморф A 7-(1,1-диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазина.
3. Фармацевтическая композиция, содержащая 7-(1,1-диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазин в сочетании с фармацевтически приемлемым носителем.
4. Применение 7-(1,1-диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазина для получения лекарственного препарата для лечения и/или профилактики состояния тревоги.
5. Способ получения 7-(1,1-диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазина, включающий взаимодействие соединения формулы III с соединением формулы IV


где L1 представляет подходящую уходящую группу.
6. Способ получения 7-(1,1-диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазина, включающий взаимодействие соединения формулы XI (или его таутомера 1,2,4-триазоло[4,3-b]пиридазин-6-она) с соединением формулы XII


где L3 представляет подходящую уходящую группу.
7. Способ получения 7-(1,1-диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазина, включающий взаимодействие триметилуксусной кислоты с соединением формулы XIII

в присутствии нитрата серебра и персульфата аммония.
8. Способ получения 7-(1,1-диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазина, включающий взаимодействие соединения формулы XIV с соединением формулы XV

где M представляет -B(OH)2 или -Sn(Alk)3, где Alk представляет C1-6 алкильную группу и L4 представляет подходящую уходящую группу, в присутствии катализатора на основе переходного металла.
9. Способ по п.5, где реакцию проводят в 1-метил-2-пирролидиноне в присутствии гидроксида натрия при температуре в области 0шC.
10. Способ лечения и/или профилактики состояния тревоги, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества 7-(1,1-диметилэтил)-6-(2-этил-2H-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло-[4,3-b]пиридазина.
Текст
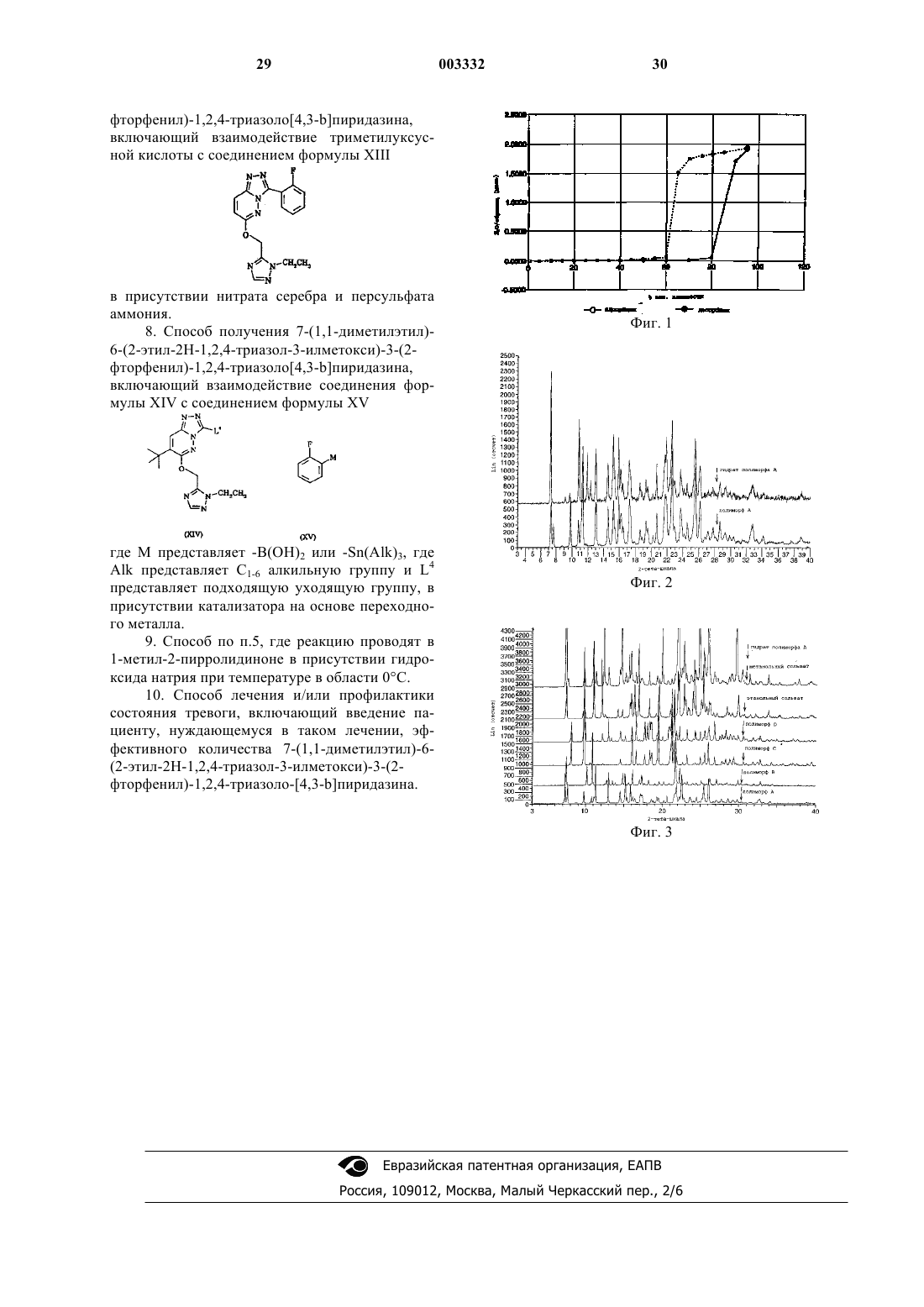
1 Настоящее изобретение относится к замещенному производному триазола-пиридазина и его применению в терапии. Более конкретно,это изобретение касается определенного замещенного производного 1,2,4-триазоло[4,3-b] пиридазина,которое является лигандом ГАМКA-рецепторов и, следовательно, пригодно для терапии патологических психических состояний. Рецепторы основного нейромедиатора ингибирования, гамма-аминомасляной кислоты(ГАМК), разделяются на два основных класса: 1) ГАМКA-рецепторы, которые являются членами суперсемейства ионных каналов, запираемых лигандом; и 2) ГАМКB-рецепторы, которые могут быть членами суперсемейства рецепторов,связанных с G-белком. После того, как были клонированы первые кДНК, кодирующие отдельные субъединицы ГАМКA-рецепторов, число известных членов семейства у млекопитающих возросло с включением, по меньшей мере,шести -субъединиц, четырех -субъединиц,трех -субъединиц, одной -субъединицы, одной -субъединицы и двух -субъединиц. Несмотря на то, что знание о разнообразии гена семейства ГАМКA-рецепторов представляет значительный шаг вперед в нашем понимании этих ионных каналов, запираемых лигандом, понимание степени разнообразия субтипов по-прежнему находится на ранней стадии. Указывалось, что -субъединица, -субъединица и-субъединица составляют минимальные потребности для образования в полной мере функционального ГАМКA-рецептора, экспрессированного при кратковременной трансфекции кДНК в клетки. Как указывалось выше, существуют также ,и -субъединицы, но они находятся на минимальном уровне в популяциях ГАМКA-рецепторов. В результате исследований размера рецептора и получения визуального изображения с помощью электронной микроскопии был сделан вывод, что как другие члены семейства ионных каналов, запираемых лигандом, естественные ГАМКA-рецепторы существуют в пентамерной форме. Выбор, по меньшей мере, одной -, одной - и одной -субъединицы из репертуара семнадцати предоставляет возможность существования более, чем 10000 пентамерным субъединичным комбинациям. Кроме того, этот расчет не учитывает дополнительных перестановок, которые были бы возможны, если расположение субъединиц вокруг ионного канала не имеет сдерживающих факторов (т.е. могло бы быть 120 возможных вариантов для рецептора,состоящего из пяти различных субъединиц). Существующие субтиповые структуры рецепторов включают, среди многих других 122, 22/32, 32/3, 21, 532/3,62, 6 и 4. Субтиповые структуры,содержащие 1-субъединицу, имеются в боль 003332 2 шинстве участков мозга и полагают, что они составляют более 40% ГАМКA-рецепторов у крыс. Полагают, что субтиповые структуры,содержащие 2- и 3-субъединицы соответственно составляют примерно 25% и 17% ГАМКA-рецепторов у крыс. Субтиповые структуры, содержащие 5-субъединицу, экспрессируются преимущественно в гиппокампе и коре,и полагают, что они представляют примерно 4% ГАМКA-рецепторов у крыс. Характерным свойством всех известных ГАМКA-рецепторов является присутствие ряда модуляторных участков, один из которых является участком связывания бензодиазепина (BZ). Участки связывания BZ являются наиболее изученным модуляторным участком ГАМКAрецепторов, и он является участком, через который анксиолитические лекарственные препараты такие, как диазепам и темазепам, проявляют свое действие. До клонирования гена семейства ГАМКA-рецепторов участка связывания бензодиазепина исторически был подразделен на два субтипа BZ1 и BZ2, на основе исследований связывания радиолигандов. Было показано, что субтип BZ1 фармакологически эквивалентен ГАМКA-рецептору, включающему 1-субъединицу в комбинации с -субъединицей и 2. Это наиболее распространенный субтип ГАМКAрецепторов, и считается, что он представляет почти половину всех ГАМКA-рецепторов в мозге. Двумя другими главными популяциями являются субтипы 22 и 32/3. Вместе они составляют примерно еще 35% от общего количества ГАМКA-рецепторов. Оказалось, что фармакологически эта комбинация эквивалентна субтипу BZ2, как было определено ранее с использованием связывания радиолигандов, хотя субтип BZ2 может также включать некоторые 5-содержащие субтиповые структуры. Физиологическая роль этих субтипов до сих пор не ясна, поскольку неизвестны в достаточной мере селективные агонисты или антагонисты. В настоящее время считается, что агенты,действующие, как агонисты BZ в субъединицах 12, 222 или 32, будут обладать желаемыми анксиолитическими свойствами. Соединения, которые являются модуляторами участка связывания бензодиазепина ГАМКAрецепторов, действуя в качестве агонистов BZ,относятся далее к агонистам ГАМКA-рецепторов. 1-селективные агонисты ГАМКABрецепторов алпидем и золпидем клинически назначаются в качестве снотворных средств,предполагая, что, по меньшей мере, некоторые из седативных эффектов, связанных с известными анксиолитическими лекарственными препаратами, которые действуют на участке связывания BZ1, опосредованы через ГАМКAрецепторы, содержащие 1-субъединицу. Следовательно, считается, что агонисты ГАМКA 3 рецепторов, которые более предпочтительно взаимодействуют с 2- и/или 3-субъединицей,чем с 1, будут эффективными при лечении состояния тревоги с пониженной склонностью к проявлению седативного эффекта. Кроме того,агенты, которые являются антагонистами или обратными агонистами для 1, можно использовать для снятия седативного эффекта или гипноза, вызываемых 1-агонистами. Соединения по настоящему изобретению,являясь селективными лигандами для ГАМКAрецепторов, следовательно, полезны для лечения и/или профилактики различных заболеваний центральной нервной системы. Такие заболевания включают состояния тревоги, такие как панические расстройства с или без агорафобии,агорафобия без панических расстройств в анамнезе, страх перед животными и другие виды фобий, включая социальные фобии, обсессивнокомпульсивное нарушение, стрессовые расстройства, включая посттравматическое и острое стрессовое нарушение, и генерализованное или индуцированное химическими веществами состояние тревоги; неврозы; судороги; мигрень; депрессивные или биполярные нарушения, например, единичное эпизодическое или возвратное сильное депрессивное нарушение, дистимическое нарушение, биполярное I и биполярное II маниакальные расстройства и циклотимическое нарушение; психотические заболевания, включая шизофрению; нейродегенерация, возникающая в результате церебральной ишемии; дефицитное гиперактивное нарушение внимания; нарушения циркадных ритмов, например, у субъектов, страдающих от эффектов, связанных с резкой сменой часовых поясов или сдвига рабочего ритма. Дополнительные заболевания, для которых могут быть полезными селективные лиганды ГАМКA-рецепторов, включают боль и передачу боли; рвоту, включая острую, замедленную и антисипатическую рвоту, в частности, рвоту,вызванную химиотерапией или облучением, а также послеоперационную тошноту или рвоту; заболевания, связанные с питанием, включая нервно-психическую анорексию и нервнопсихическую булимию; предменструальный синдром; мышечный спазм или спастичность,например, у пациентов с параплегией, и потерю слуха. Селективные лиганды ГАМКAрецепторов могут быть также эффективными для премедикации перед анестезией или при небольших процедурах, таких как эндоскопия,включая гистроэндоскопию. В WO 98/04559 описывается класс замещенных и с 7,8-конденсированными кольцами производных 1,2,4-триазоло[4,3-b]пиридазина,которые, как указывается, являются селективными лигандами для ГАМКA-рецепторов, полезными для лечения и/или профилактики нев 003332 4 рологических заболеваний, включая состояния тревоги и судороги. Настоящее изобретение раскрывает определенное производное триазолопиридазина и его фармацевтически приемлемые соли, которые обладают требуемыми свойствами связывания с ГАМКA-рецепторами различных субтипов. Соединения по настоящему изобретению обладают хорошей аффинностью в качестве лигандов для 2- и/или 3-субъединицы человеческого ГАМКA-рецептора. Соединения по изобретению взаимодействуют более предпочтительно с 2- и/или 3-субъединицей, чем с 1-субъединицей. Действительно, соединения по изобретению проявляют функциональную избирательность в плане селективной эффективности для 2- и/или 3-субъединицы относительно 1-субъединицы. Соединения по настоящему изобретению являются субтиповыми лигандами ГАМКAрецепторов, имеющими аффинность связывания(Ki) для 2- и/или 3-субъединицы, как определено в тесте, описанном ниже, меньше, чем 1 нМ. Кроме того, соединения по изобретению проявляют функциональную селективность в плане селективной эффективности, для 2 и/или 3-субъединицы относительно 1 субъединицы. Кроме того, соединения по настоящему изобретению обладают интересными фармакокинетическими свойствами, в особенности на основе повышенной биодоступности при пероральном введении. Настоящее изобретение раскрывает 7-(1,1 диметилэтил)-6-(2-этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b] пиридазин формулы I или его фармацевтически приемлемую соль. Соединения по настоящему изобретению входят в общий объем WO 98/04559. Однако в нем отсутствует специфическое раскрытие соединения формулы I, изображенной выше, или его фармацевтически приемлемых солей. Для применения в медицине соли соединения формулы I выше будут фармацевтически приемлемыми солями. Однако могут быть полезными другие соли при получении соединения формулы I или его фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли соединения формулы I включают кислотно-аддитивные соли, которые можно получить, например, при смешивании раствора соединения формулы I с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, метансульфоновая кислота, фумаровая кислота, малеиновая 5 кислота, янтарная кислота, уксусная кислота,бензойная кислота, щавелевая кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Настоящее изобретение также предлагает способ лечения и/или профилактики состояния тревоги, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I, изображенной выше, или его фармацевтически приемлемой соли. Дополнительно настоящее изобретение обеспечивает способ лечения и/или профилактики судорог (например, у пациента, страдающего эпилепсией или родственным заболеванием), включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I, изображенной выше, или фармацевтически приемлемой соли. Аффинность связывания (Ki) соединений по настоящему изобретению с 3-субъединицей человеческого ГАМКA-рецептора соответственно определяют в тесте, описанном ниже. Аффинность связывания с 3-субъединицей (Кi) соединений по изобретению составляет меньше,чем 1 нМ. Соединения по настоящему изобретению вызывают селективное потенцирование ответа ЕС 20 ГАМК в устойчиво трансфектированных рекомбинантных клеточных линиях, экспрессирующих человеческого 3-субъединицу ГАМКA-рецептора, по сравнению с потенцированием ответа EC20 ГАМК, полученного в устойчиво трансфектированных клеточных линиях, экспрессирующих 1-субъединицу человеческого ГАМКA-рецептора. Потенцирование ответа ЕС 20 ГАМК в устойчиво трансфектированных клеточных линиях, экспрессирующих 3- и 4-субъединицы человеческого ГАМA-рецептора, можно соответственно определять способами, аналогичными описанным у Wafford et al., Mol.Pharmacol.,1996, 50, 670-678. Способ соответственно проводят, используя культуры устойчиво трансфектированных эукариотических клеток, обычно устойчиво трансфектированных мышиных Ltkфибробластов. Соединения по настоящему изобретению проявляют анксиолитические свойства, что показано положительной ответной реакцией, выраженной в повышенном плюс сложном и условном подавлении в тестах с питьем (сравниDawson et al., Psychopharmacology, 1995, 121,109-117). Кроме того, соединения по настоящему изобретению являются по существу неседативными, что подтверждено соответствующими данными, полученными в тесте с ответной чувствительностью (оттягивание связи) (сравни 6 Соединения по настоящему изобретению могут также проявлять противосудорожную активность. Это может быть показано способностью блокировать индуцированные пентилентетразолом припадки у крыс и мышей согласно протоколу, аналогичному описанному Bristow etal. in J. Pharmacol. Exp. Ther., 1996, 279, 492-501. Поскольку они вызывают поведенческие эффекты ясно, что соединения по изобретению проникают в мозг; иными словами, соединения способны проникать через так называемый гематоэнцефалический барьер. Преимущественно соединения по изобретению способны проявлять их положительное лечебное действие после перорального введения. Изобретение также раскрывает фармацевтические композиции, содержащие одно или более соединений по изобретению в сочетании с фармацевтически приемлемым носителем. Предпочтительно, эти композиции находятся в единичных дозировочных формах, таких как таблетки, пилюли, капсулы, порошки, гранулы,в стерильных растворах или суспензиях для парентерального введения, дозированных аэрозольных или жидких спреях, каплях, ампулах,устройствах для самоинъекций или суппозиториях; для перорального, парентерального, интраназального, подъязычного или ректального введения, или для введения путем ингаляции или инсуффляции. Для приготовления твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим носителем, например, обычными ингредиентами для таблетирования, такими как кукурузный крахмал, лактоза, сахароза, сорбит,тальк, стеариновая кислота, стеарат магния, дикальций фосфат или камеди, и другими фармацевтическими разбавителями, например, водой,с образованием твердой композиции для последующего формирования, содержащей гомогенную смесь соединения по настоящему изобретению или его фармацевтически приемлемой соли. При упоминании этих композиций для последующего формирования в качестве гомогенных, означает, что активный ингредиент равномерно распределен в композиции так, что композицию можно легко разделить на равноценно эффективные единичные дозировочные формы,такие как таблетки, пилюли и капсулы. Затем эту твердую композицию для последующего формирования разделяют на единичные дозировочные формы вышеописанного типа, содержащие от 0,1 до примерно 500 мг активного ингредиента по настоящему изобретению. Типичные единичные лекарственные формы содержат от 1 до 100 мг, например, 1, 2, 5, 10, 25, 50 или 100 мг активного ингредиента. Таблетки или пилюли новой композиции можно покрывать оболочкой или составлять иначе с обеспечением лекарственной формы, предоставляющей преимущество пролонгированного действия. Например, таблетка или пилюля может содержать 7 компонент внутренней дозы и внешней дозы,последняя находится в форме оболочки над первой. Два компонента могут быть разделены кишечным слоем, который служит для того,чтобы препятствовать распаду в желудке, и позволяет внутреннему компоненту проникать интактным в двенадцатиперстную кишку или замедленно высвобождаться. Для таких кишечных слоев или оболочек можно использовать различные вещества, включающие ряд полимерных кислот и смеси полимерных кислот с такими веществами, как щеллак, цетиловый спирт и ацетилцеллюлоза. Жидкие формы, в которые можно вводить новые композиции по настоящему изобретению для перорального или инъекционного введения,включают водные растворы, соответствующим образом ароматизированные сиропы, водные или масляные суспензии и ароматизированные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические носители. Пригодные диспергирующие или суспендирующие агенты для водных суспензий включают синтетические и натуральные камеди, такие как трагакант, аравийская камедь, альгинат, декстран,натрий, карбоксиметилцелюлоза, метилцеллюлоза, поливинилпирролидон или желатин. При лечении состояния тревоги подходящая доза составляет примерно 0,01-250 мг/кг в день, предпочтительно примерно 0,05-100 мг/кг в день и особенно примерно 0,05-5 мг/кг в день. Соединения можно назначать по схеме 1-4 раза в день. Соединение формулы I, изображенной выше, можно получить способом, который включает взаимодействие соединений формулы где L1 представляет подходящую уходящую группу. Уходящая группа L1 обычно является атомом галогена, особенно хлором. Реакцию между соединениями III и IV соответственно проводят путем перемешивания реагентов в подходящем растворителе в присутствии основания. Обычно растворителем является N,N-диметилформамид, и основание является сильным основанием, таким как гидрид натрия. Предпочтительно растворитель является диметилсульфоксидом и основание является карбонатом цезия. Более предпочтительно растворитель представляет 1-метил-2-пирролидинон, и основание представляет гидроксид натрия, в этом случае реакцию преимущественно проводят при температуре в области 0 С. 8 Промежуточные соединения формулы III выше можно получить взаимодействием соединения формулы V, по существу, с эквимолярным количеством производного гидразина формулы IVL2 представляет подходящую уходящую группу; с последующим, если необходимо, разделением полученной смеси изомеров обычными способами. Уходящая группа L2 обычно является атомом галогена, особенно хлором. В промежуточных соединениях формулы V уходящие группыL1 и L2 могут быть одинаковыми или различными, но лучше одинаковыми, предпочтительно обе являются хлором. Реакцию между соединениями V и VI соответственно проводят нагреванием реагентов в присутствии источника протонов, такого как триэтиламин гидрохлорид, обычно при температуре с образованием флегмы в инертном растворителе, таком как ксилол или 1,4-диоксан. Альтернативно, промежуточные соединения формулы III выше можно получить взаимодействием производного гидразина формулыVII с производным альдегида формулы VIII где L1 имеет значение, определенное выше, с последующей циклизацией промежуточного полученного таким образом основания Шиффа. Реакцию между соединениями VII и VIII соответственно проводят в кислотных условиях,например, в присутствии неорганической кислоты такой, как соляная кислота. Циклизацию полученного промежуточного основания Шиффа затем можно соответственно проводить обработкой хлоридом железа (III) в подходящем растворителе, например, спиртовом растворителе, таком как этанол, при повышенной температуре в интервале 60-70 С. Промежуточные соединения формулы VII выше можно получить взаимодействием соответствующего соединения формулы V, определенного выше, с гидразин гидратом, обычно в изобутиловом спирте при повышенной температуре, например, при температуре в области 90 С, или в 1,4-диоксане при температуре флегмы растворителя с последующим, если необходимо, разделением полученной смеси изомеров обычными способами. 9 При альтернативном подходе, промежуточные соединения формулы III выше можно получить взаимодействием производного гидразина формулы VII, определенного выше, с соединением формулы IX 10 В другом способе, соединение формулы I,изображенной выше, можно получить способом,который включает взаимодействие соединения формулы XI (или его таутомера 1,2,4-триазоло где Q представляет реакционноспособный карбоксилатный фрагмент, с последующей циклизацией полученного таким образом производного гидразида формулы X где L1 является таким, как определено выше. Подходящие значения для реакционноспособного карбоксилатного фрагмента Q включают сложные эфиры, например, C1-4 алкилэфиры; ангидриды кислот, например, смешанные ангидриды C1-4 алифатических кислот; галогенангидриды, например, хлорангидриды; и ацилимидазолы. Подходяще Q представляет хлорангидридную группу. Реакцию между соединениями VII и IX соответственно проводят в щелочных условиях,например, в присутствии триэтиламина, подходяще в инертном растворителе, таком как диэтиловый эфир, и обычно при температуре в области 0 С. Циклизацию полученного соединения формулы Х можно затем соответственно проводить обработкой 1,2-дибром-1,1,2,2-тетрахлорэтаном и трифенилфосфином, в присутствии основания, такого как триэтиламин, подходяще в инертном растворителе, таком как ацетонитрил, и обычно при температуре в области 0 С. В предпочтительном способе, реакцию между соединениями VII и IX можно проводить смешением реагентов в растворителе, таком как 1-метил-2-пирролидинон, при температуре в области 0 С; циклизацию полученного таким образом соединения формулы Х можно затем проводить in situ нагреванием реакционной смеси при температуре в области 130 С. В реакции между соединением V и гидразин гидратом или соединением VI будет, как указано выше, возможно образовываться смесь изомерных продуктов в зависимости от того,которую уходящую группу L1 или L2 замещает атом азота гидразина. Таким образом, в дополнении к необходимому продукту формулы III или VII альтернативный изомер возможно будет получен в некоторой степени. По этой причине может быть необходимым разделять полученную смесь изомеров обычными способами, такими как хроматография. где L3 представляет подходящую уходящую группу. Уходящей группой L3 подходяще является атом галогена, обычно хлора или брома. Реакцию между соединениями XI и XII соответственно проводят перемешиванием реагентов в подходящем растворителе, обычноN,N-диметилформамиде, в присутствии сильного основания, такого как гидрид натрия. Промежуточное соединение формулы XI выше можно соответственно получить взаимодействием соединения формулы III, определенной выше, с гидроксидом щелочного металла,например, гидроксидом натрия. Реакцию соответственно проводят в инертном растворителе,таком как 1,4-диоксан, идеально при температуре флегмы растворителя. В дополнительном способе, соединение формулы I, изображенной выше, можно получить способом, который включает взаимодействие триметилуксусной кислоты с соединением формулы XIII в присутствии нитрата серебра и персульфата аммония. Реакцию соответственно проводят в подходящем растворителе, например, в воде или водном ацетонитриле, необязательно в кислотных условиях, например, используя трифторуксусную кислоту или серную кислоту, обычно при повышенной температуре. Промежуточное соединение формулы XIII соответствует соединению формулы I, изображенной выше, где отсутствует трет-бутиловый заместитель в 7-положении, и промежуточное 11 соединение XIII, следовательно, можно получить способами, аналогичными описанным выше для получения соединения формулы I. В еще дополнительном способе, соединение формулы I, изображенной выше, можно получить способом, который включает взаимодействие соединения формулы XIV с соединением формулы XVAlk представляет C1-6 алкильную группу, обычно н-бутил, и L4 представляет подходящую уходящую группу; в присутствии катализатора на основе переходного металла. Уходящей группой L4 подходяще является атом галогена, например, брома. Подходящий катализатор на основе переходного металла для использования в реакции между соединениями XIV и XV содержит дихлорбис(трифенилфосфин)палладий(II) или тетракис(трифенилфосфин)палладий(0). Реакцию между соединениями XIV и XV соответственно проводят в инертном растворителе, таком как N,N-диметилформамид, обычно при повышенной температуре. Промежуточные соединения формулы XIV можно получить взаимодействием соединения формулы IV, определенной выше, с соединением формулы XVI где L1 и L4 являются такими, как определено выше, в условиях, аналогичных описанным выше для реакции между соединениями III и IV. Промежуточное соединение формулы IV можно получить способами, описанными в ЕРА-0421210 или способами, аналогичными этому. Подходящие способы описываются в сопутствующих примерах. Промежуточные соединения формулы V выше можно получить взаимодействием триметилуксусной кислоты с соединением формулы 12 где L1 и L2 являются такими, как определено выше, в присутствии нитрата серебра или персульфата аммония, в условиях, аналогичных описанным выше для реакции между триметилуксусной кислотой и соединением XIII. Там,где L1 и L2 обе являются хлором в соединенииXVII, реакцию преимущественно проводят в присутствии трифторуксусной кислоты. Когда они коммерчески недоступны, исходные вещества формулы VI, VIII, IX, XII, XV,XVI и XVII можно получить способами, аналогичными описанным в сопутствующих примерах, или обычными способами, хорошо известными в данной области. Во время любой из вышеприведенных последовательностей синтеза может быть необходимым и/или желательным защитить чувствительные или реакционноспособные группы в любой из требуемых молекул. Этого можно достичь с помощью обычных защитных групп,таких как описанные в Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, PlenumSons, 1991. Защитные группы можно удалить на подходящей последующей стадии, используя способы, известные в данной области. Было синтезировано и охарактеризовано четыре безводных полиморфа, два сольвата и дигидрат 7-(1,1-диметилэтил)-6-(2-этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4 триазоло[4,3-b]пиридазина. Все полиморфы и сольваты переходят в наиболее термодинамически стабильную форму, полиморф А (получение и характеристику смотри в примере 3) после перемешивания в виде суспензии в воде. Дигидрат полиморфа А является стабильным, но только при повышенных значениях влажности. Следующие примеры иллюстрируют получение соединений по изобретению. Соединения по данному изобретению эффективно ингибируют связывание[3 Н]флумазенила с участием связывания бензодиазепина человеческих ГАМКA-рецепторов, содержащих 2- или 3-субъединицу, стабильно экспрессируемых в клетках Ltk. Реактивы Забуференный фосфатом физиологический раствор (PBS). Буфер для анализа: 10 мМ KH2PO4, 100 мМ КСl, рН 7,4 при комнатной температуре.[3H]-Флумазенил (18 нМ для 132 клеток; 18 нМ для 232 клеток; 10 нМ для 332 клеток) в буфере для анализа. Флунитразепам 100 мкМ в буфере для анализа. Клетки, ресуспендированные в буфере для анализа (1 поддон до 10 мл). Сбор клеток От клеток отделяют надосадочную жидкость. Добавляют PBS (примерно 20 мл). Клетки 13 соскабливают и помещают в центрифужную пробирку емкостью 50 мл. Процедуру повторяют с дополнительными 10 мл PBS для гарантии того, что большая часть клеток извлечена. Клетки собирают в осадок центрифугированием в течение 20 мин при 3000 об/мин на стендовой центрифуге и затем, если желательно, замораживают. Осадки повторно суспендируют в 10 мл буфера на поддон (25 х 25 см) с клетками. Анализ Можно проводить в глубоких 96-луночных планшетах или пробирках. Каждая пробирка содержит: 300 мкл буфера для анализа; 50 мкл [3 Н] -флумазенила (конечная концентрация для 132: 1,8 нМ; для 232: 1,8 нМ; для 332: 1,0 нМ); 50 мкл буфера или растворяющего носителя (например, 10% ДМСО), если соединения растворяются в 10% ДМСО (полностью); испытуемое соединение или флунитразепам (для определения неспецифического связывания), конечная концентрация 10 мкМ; 100 мкл клеток. Пробы для анализа инкубируют в течение 1 ч при 40 С, затем фильтруют, используя коллектор клеток Tomtec или Brandel, на фильтрыGF/B с последующим промыванием (3 х 3 мл) охлажденным на льду буфером для анализа. Фильтры высушивают и радиоактивность определяют с использованием жидкой сцинтилляции. Предполагаемые значения общего связывания составляют 3000-4000 распадов в минуту при полном обсчете и менее 200 распадов в минуту для неспецифического связывания при использовании жидкой сцинтилляции, или 15002000 распадов в минуту при полном обсчете и менее 200 распадов в минуту для неспецифического связывания, если радиоактивность определяют с плавким твердым сцинтиллянтом. Параметры связывания определяют нелинейным регрессионным анализом наименьших площадей, из которого можно рассчитать константу ингибирования Ki для каждого испытуемого соединения. Соединение из сопутствующих примеров тестировали вышеописанным методом, и было найдено, что оно обладает значением Ki вытеснения [3H]-флумазенила из 2- и/или 3 субъединиц человеческого ГАМКA-рецептора меньше, чем 1 нМ. Пример 1. 7-(1,1-Диметилэтил)-6-(2-этил 2 Н-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазин.a) 3,6-Дихлор-4-(1,1-диметилэтил)пиридазин. Концентрированную серную кислоту (53,6 мл, 1,0 моль) осторожно добавляли к перемешиваемой суспензии 3,6-дихлорпиридазина (50,0 г,0,34 моль) в воде (1,25 л). Затем смесь нагревали до 70 С (внутренняя температура) перед добав 003332 14 лением триметилуксусной кислоты (47,5 мл,0,41 моль). Затем в течение примерно одной минуты добавляли раствор нитрата серебра(11,4 г, 0,07 моль) в воде (20 мл). Это привело к тому, что реакционная смесь по внешнему виду стала молочной. Затем в течение 20-30 мин добавляли раствор персульфата аммония (230 г,1,0 моль) в воде (0,63 л). Внутренняя температура поднималась до примерно 85 С. Во время добавления образовывался продукт в виде липкого осадка. После полного добавления реакционную смесь перемешивали еще 10 мин, затем давали охладиться до комнатной температуры. Смесь затем выливали на лед и подщелачивали концентрированным водным аммиаком с добавлением еще льда, сколько потребуется для поддержания температуры ниже 10 С. Водный слой экстрагировали дихлорметаном (3 х 300 мл). Объединенные экстракты высушивали (MgSO4),фильтровали и выпаривали с получением 55,8 г сырого продукта в виде масла. Его очищали хроматографией на силикагеле, используя 0-15% этилацетат в гексане в качестве элюента с получением 37,31 г (53%) требуемого соединения. Данные для титульного соединения: 1H ЯМР (360 МГц, d6-ДМСО)1,50 (9 Н, с), 7,48b) 6-Хлор-7-(1,1-диметилэтил)-3-(2-фторфенил)-1,2,4-триазоло [4,3-b]пиридазин. Смесь 3,6-дихлор-4-(1,1-диметилэтил)пиридазина (20 г, 0,097 моль), 2-фторбензгидразида (22,6 г, 0,145 моль) и триэтиламина гидрохлорида (20 г, 0,0145 моль) в диоксане (1,2 л) перемешивали и нагревали при температуре образования флегмы в потоке азота в течение 4 дней. После охлаждения летучие вещества удаляли в вакууме и остаток растирали с дихлорметаном (200 мл), фильтровали и концентрировали в вакууме. Остаток очищали хроматографиеи на силикагеле,элюируя 0%-25% этилацетат/дихлорметан с получением титульного соединения (12,95 г, 44%) в виде белого твердого вещества. Данные для титульного соединения: 1H ЯМР (360 МГц, СDCl3)1,57 (9 Н, с), 7,267,35 (2 Н, м), 7,53-7,60 (1 Н, м) 7,89-7,93 (1 Н, м),8,17 (1 Н, с), MC(ES+) m/e 305[МН]+, 307[MH]+. с) (2-Этил-2 Н-1,2,4-триазол-3-ил)метанол. К раствору 1,2,4-триазола (10 г, 0,145 моль) в N,N-диметилформамиде (DMF) (150 мл) при комнатной температуре добавляли гидрид натрия (6,4 г 60% дисперсии в масле, 0,16 моль) порциями в течение 15 мин. Когда добавление заканчивалось, реакционной смеси давали охладиться до комнатной температуры, затем охлаждали на ледяной бане и добавляли по каплям йодэтан (14 мл, 0,174 моль) в течение 10 мин. Реакционной смеси давали нагреться до комнатной температуры и после перемешивания в течение 3 ч растворители удаляли под высоким вакуумом с получением остатка, который распределяли между водой (300 мл) и этилацета 15 том (3 х 300 мл). Объединенные органические слои промывали насыщенным рассолом и высушивали (MgSO4), фильтровали и концентрировали под вакуумом с получением маслянистого остатка, который очищали перегонкой (120 С 20 мм рт.ст.) с получением 1-этил-1,2,4 триазола, загрязненного 15% DMF (2,4 г). Сырой продукт (2,4 г, 0,25 моль) растворяли в тетрагидрофуране сухом (ТГФ) (35 мл), охлаждали до -40 С и медленно в течение 20 мин добавляли н-бутиллитий (16,2 мл 1,6 М раствора в гексане, 0,026 моль), поддерживая температуру постоянной. Затем добавляли DMF (2,03 мл,0,026 моль) и через 15 мин реакционной смеси давали медленно нагреться до комнатной температуры в течение 2 ч. К реакционной смеси добавляли метанол (20 мл) с последующим добавлением борогидрида натрия (1 г, 0,026 моль) и раствор перемешивали в течение 14 ч. Растворители удаляли под вакуумом и остаток распределяли между рассолом (50 мл) и дихлорметаном (6 х 50 мл). Объединенные органические слои высушивали (MgSO4) фильтровали и концентрировали под вакуумом с получением остатка, который очистили хроматографией на силикагеле, используя 0-5% метанол в дихлорметане в качестве элюента с получением титульного соединения в виде не совсем белого твердого вещества (0,5 г, 3%). Данные для титульного соединения: 1H ЯМР (250 МГц, СDСl3)1,48 (3H, т, J=7,3 Гц), 4,25 (2 Н, кв, J=7,3 Гц),4,75(2 Н, с), 5,14 (1 Н, шир.с), 7,78 (1 Н, с).(10 мл) добавляли гидрид натрия (0,024 г 60% дисперсии в масле, 1,1 моль-экв) и реакционную смесь перемешивали при комнатной температуре в течение 30 мин. После этого времени реакционную смесь разбавляли водой (80 мл) и твердое вещество, которое выпало в осадок,собирали фильтрованием и несколько раз промывали водой на воронке из металлокерамики. Твердое вещество перекристаллизовывали из этилацетата/гексана с получением чистого титульного соединения (0,085 г, 44%). Данные для титульного соединения: 1H ЯМР (250 МГц,CDCl3)1,40-1,47 (12 Н, м), 4,14 (2 Н, т, J=7,3 Гц), 5,26 (2 Н, с), 7,26-7,38 (2 Н, м), 7,53-7,58 (1 Н,м), 7,86-7,90 (1 Н, м), 7,93 (1 Н, с), 7,99 (1 Н, с); 16 а) 3,6-Дихлор-4-(1,1-диметилэтил)пиридазин. Смесь дихлорпиридазина (100 г, 0,67 моль), триметилуксусной кислоты (96 г, 0,94 моль) и воды (800 мл) в колбе емкостью 10 л с подвесной мешалкой нагревали до 55 С, получая двухфазный раствор. Одной порцией добавляли раствор АgNO3 (11,4 г, 0,134 моль в воде(125 мл), что приводило к образованию непрозрачного раствора. Одной порцией добавляли трифторуксусную кислоту (10,3 мл, 0,134 моль). Персульфат аммония (245 г, 1,07 моль) растворяли в воде (500 мл) и добавляли по каплям к суспензии в течение 45-60 мин, что приводило к выделению тепла (обычно температура поднимается до 75-80 С и ее можно контролировать скоростью добавления персульфата). Температуру поддерживали при 75 С еще 1 ч и затем охлаждали до комнатной температуры. Реакционную смесь экстрагировали изобутиловым спиртом (1 л) и водный слой отбрасывали. Органический слой промывали водой (250 мл) и водную фракцию отбрасывали. По данным анализа ВЭЖХ выход составлял 134 г (97%). Изобутиловый спиртовый раствор использовали как таковой на следующей стадии.b) 6-Хлор-5-(1,1-диметилэтил)пиридазин 3-илгидразин. Гидразин гидрат (95 мл, 1,95 моль) добавляли к изобутиловому спиртовому раствору со стадии а) в колбе емкостью 3 л и нагревали при 90 С в течение 20 ч. Реакционную смесь охлаждали до комнатной температуры и нижний водный слой отбрасывали. Реакционную смесь промывали водой (450 мл) и водную фракцию отбрасывали. Реакционную смесь перегоняли при пониженном давлении до тех пор, пока продукт не начал кристаллизоваться, и затем добавляли 1-метил-2-пирролидинон (NMP) (550 мл). Перегонку продолжали до полного удаления изобутилового спирта. Раствор использовали как таковой на следующей стадии.c) 6-Хлор-7-(1,1-диметилэтил)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазин. 2-Фторбензоилхлорид (103 г, 0,65 моль) добавляли по каплям к охлажденному (0 С) раствору NMP со стадии b), поддерживая внутреннюю температуру 5 С. После добавления реакционную смесь нагревали при 130 С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, что приводило к кристаллизации продукта. В течение 30 мин по каплям добавляли воду (1,3 л). Суспензию охлаждали до 10 С и твердое вещество отделяли фильтрованием, затем высушивали при пониженном давлении с получением продукта (145 г, выход 71% из 3,6-дихлорпиридазина).d) 1-Этил-1,2,4-триазол. 1,2,4-Триазол (100,0 г, 1,45 моль) в безводном ТГФ (950 мл) охлаждали до 0 С и одной порцией добавляли 1,8-диазабицикло [5.4.0] ундец-7-ен (DBU) (220 г, 1,45 моль). Реакцион 17 ную смесь перемешивали в течение 30 мин, до тех пор, пока не наблюдали полного растворения. Поддерживая на охлаждающей бане лед/ вода добавляли по каплям в течение 15 мин иодэтан (317 г, 2,03 моль), что приводило к повышению внутренней температуры до 30 С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, после чего DBU гидроиодид удаляли фильтрованием. Фильтратный раствор использовали как таковой на следующей стадии. е) (2-Этил-2 Н-1,2,4-триазол-3-ил)метанол. Перемешиваемый раствор с предыдущей стадии охлаждали до внутренней температуры-75 С на бане с суспензией твердый СО 2 ацетон. В течение 25 мин добавляли по каплям гексиллитий (458 мл 33% раствора в гексане), поддерживая внутреннюю температуру ниже -55 С. Реакционную смесь выдерживали в течение 30 мин (обратно до -75 С) и затем добавляли по каплям чистый DMF (108 мл, 1,39 моль) в течение 10 мин, поддерживая внутреннюю температуру ниже -60 С. Реакционную смесь выдерживали при -70 С в течение 90 мин перед удалением охлаждающей бани и реакционной смеси давали нагреться до 0 С в течение 30 мин. Добавляли в течение 10 мин технический этиловый спирт, денатурированный метиловым спиртом (340 мл). Затем порциями добавляли борогидрид натрия (26,3 г, 0,695 моль), поддерживая внутреннюю температуру ниже 6 С. После добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 1 ч при этой температуре. Реакцию гасили осторожным добавлением 2 М H2SO4 (200 мл) и затем перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь концентрировали до 675 мл и одной порцией добавляли сульфат натрия (135 г). Реакционную смесь нагревали до 35 С и перемешивали в течение 15 мин. Раствор экстрагировали теплым(45 С) изобутиловым спиртом (2 х 675 мл). Объединенные органические фракции концентрировали при пониженном давлении до 450 мл,когда продукт кристаллизуется. Добавляли гептан (1,125 л) и суспензию концентрировали при пониженном давлении для удаления большей части изобутилового спирта. Добавляли гептан с получением конечного объема суспензии 680 мл. После охлаждения до 0 С фильтрование давало титульное соединение (137 г, 74% из 1,2,4-триазола). 18 порцией добавляли диметилсульфоксид (2,5 л) и реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Продукт кристаллизовался. Реакционную смесь поддерживали при 25 С, пока к перемешиваемой суспензии добавляли по каплям воду (5 л) в течение 45 мин. После охлаждения до 10 С продукт отделяли фильтрованием и осадок на фильтре промывали водой (1,75 л). Высушивание при 50 С в вакууме давало титульное соединение(317 г, 98%) в виде белого твердого вещества. Способ В. 6-Хлор-7-(1,1-диметилэтил)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазин (10 г, 32,12 ммоль) и (2-этил-2 Н-1,2,4-триазол-3-ил)метанол(5,01 г, 38,55 ммоль) загружали в колбу емкостью 500 мл, снабженную подвесной мешалкой. Одной порцией добавляли NMP (1-метил-2 пирролидинон) (100 мл) и реакционную смесь перемешивали до достижения полного растворения. Реакционную смесь охлаждали до 0 С и одной порцией добавляли 48% (маc./маc.) раствор гидроксида натрия (4,02 г, 46 ммоль). После перемешивания в течение 1 ч при 0 С продукт кристаллизовался. В течение 15 мин по каплям добавляли воду (100 мл) и суспензию выдерживали в течение 30 мин. Продукт отделяли фильтрованием и осадок на фильтре промывали водой (100 мл). Высушивание при 50 С в вакууме давало титульное соединение (12,50 г,98%) в виде белого твердого вещества. Пример 3. Получение и характеристика полиморфов и сольватов 7-(1,1-диметилэтил)-6(2-этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2 фторфенил)-1,2,4-триазоло[4,3-b]пиридазина. 7-(1,1-Диметилэтил)-6-(2-этил-2 Н-1,2,4 триазол-3-илметокси)-3-(2-фторфенил)-1,2,4 триазоло[4,3-b]пиридазин (далее называемое как соединение I) перекристаллизовывали из выбранных органических растворителей и полученные твердые вещества высушивали в вакууме в течение ночи при 60 С, если не указано иначе. Каждую партию затем охарактеризовывали, используя оптическую микроскопию,дифференциальную сканирующую калориметрию (ДСК), термогравиметрический анализ(ТГА) и порошковую рентгенографию (ПРГ). Была дана характеристика четырем различным безводным полиморфам, гидрату и двум сольватам, как обобщено в табл. 1. Полиморф А Таблица 1. Полиморфы соединения I Растворитель при Форма перекристаллизакристаллов ции Удлиненные Метанол прямоугольные Этилацетат или неправильАцетонитрил ной формы Ацетон Уксусная кислота/ Н 2 О Изобутанол Удлиненные прямоугольные или неправильной формы 003332 Игольчатые Игольчатые Неправильной формы Неправильной формы Полиморф А Полиморф А состоит из прямоугольных двоякопреломляющих кристаллов неправильной или удлиненной формы. Они показывают одну основную эндотерму при ДСК при примерно 186 С за счет плавления. В случаях некоторых из полученных образцов эта кривая плавления показывает несколько превращений, имеющих место внутри нее, выраженных плечами на эндотерме плавления. Полиморф А является безводным и не показывает потерь при ТГА. Он имеет уникальную дифрактограмму в ПРГ, выраженную двумя пиками при 27,3. Полиморф В Полиморф В состоит из прямоугольных двоякопреломляющих кристаллов неправильной или удлиненной формы. Полиморф В показывает одну основную эндотерму при ДСК при примерно 181 С за счет плавления. Он является безводным и не показывает потерю в массе при ТГА. Он имеет уникальную дифрактограмму вXRPD. Полиморф С Полиморф С состоит из игольчатых двоякопреломляющих кристаллов. Термограммы ДСК полиморфа С показывают эндотерму при примерно 170 С, экзотерму при примерно 173 С, небольшую эндотерму при примерно 181 С и эндотерму за счет плавления при примерно 186 С. Не наблюдают потерь при термогравиметрическом анализе. Полиморф D Полиморф D состоит из игольчатых двоякопреломляющих кристаллов. Дифрактограмма ПРГ полиморфа D является аналогичной для полиморфа С. Однако наблюдали значительные различия, особенно дополнительные пики при 2 = 9,861, 15,113, 18,015 и 22,224. В линии ДСК полиморфа D показывает очень широкую экзотерму при примерно 108 С с последующей эндотермой и экзотермой при 170 С и 173 С,как у полиморфа С. Основное плавление наблюдается при примерно 181 С, и видно несколько превращений, имеющих место внутри эндотермы плавления с минимальным плавлением при 186 С. Не наблюдали потерь при ТГА. Метанольный сольват Термограмма ДСК метанольного сольвата показывает одну эндотерму при примерно 157 С за счет потери метанола и одну эндотерму при 186 С за счет плавления. Наблюдается постепенная потеря при ТГА вплоть до при 20 мерно 150 С, в этой точке наблюдается ступенчатая потеря, совпадающая с эндотермой, наблюдаемой при ДСК. Эта ступенчатая потеря в некоторых случаях состоит из более, чем одного превращения, и количество варьировало среди перекристаллизованных образцов, и оказалось,что не соответствует стехиометрическому сольвату. Однако исследования сорбции паров метанола на полиморфе А показывают наличие отдельного гемисольвата. Дифрактограмма ПРГ сольвата является уникальной. Этанольный сольват Этанольный сольват выражается его термограммой ДСК, которая показывает одну эндотерму при примерно 111 С за счет потери этанола, и эндотерму при примерно 186 С за счет плавления. ТГА показал, что потеря различается для различных перекристаллизованных образцов и составляет между примерно 4 и 6,5% и не соответствует стехиометрическому сольвату. Дифрактограмма ПРГ является уникальной. Гидрат полиморфа А На фиг. 1 показана изотерма адсорбции/ десорбции полиморфа А соединения I при 25 С. Эти исследования сорбции водяных паров показывают образование дигидрата при относительной влажности выше 80% (RH) при 25 С. Виден гистерезис, указывающий на образование гидрата с десорбцией, имеющей место при RH ниже примерно 60%. На фиг. 2 рядом показаны дифрактограммы ПРГ безводного полиморфа А соединения I и дигидрата полиморфа А. Дифрактограмма ПРГ дигидрата, полученного на влажном образце полиморфа А, показывает отдельные различия, наблюдаемые между двумя формами. Основные видимые изменения в дифракции заключаются в потере пика при 2 = 11,2 и появлении двух основных пиков при 2 = 11,9 и 12,3. Превращение полиморфов соединения I в полиморф А При суспендировании в воде все описанные выше полиморфы и сольваты превращаются в полиморф А в течение 1-4 дней, указывая,что он является наиболее стабильной формой при комнатной температуре. Это превращение является медленным за счет низкой растворимости соединения I в воде по отношению к большому избытку суспендированного твердого вещества. Данные порошковой рентгенографии лучей На фиг. 3 рядом показаны дифрактограммы ПРГ безводных полиморфов А, В, С и D соединения I, метанольных и этанольных сольватов и дигидрата полиморфа А. Цифровые данные, связанные с ними, представлены ниже. Полиморф А соединения I: данные порошковой рентгенографии Интенсивность, ИнтенсивИнтенсивность, ИнтенсивУгол 2 Значение d Угол 2 Значение d отсчет ность, % отсчет ность, % 7,243 12,19523 2214 88,5 22,885 3,88289 195 7,8 7,559 11,68660 613 24,5 23,621 3,76351 307 12,3 8,601 10,27227 50 2 23,67 3,75587 256 10,2 9,694 9,11647 211 8,4 23,767 3,74068 253 10,1 10,703 8,25928 270 10,8 24,12 3,68684 102 4,1 11,027 8,01719 213 8,5 24,518 3,62778 281 11,2 11,239 7,86640 1464 58,5 25,223 3,52803 162 6,5 12,943 6,83420 2501 100 25,275 3,52082 185 7,4 13,441 6,58218 41 1.6 25,478 3,49324 808 32,3 14,497 6,10492 740 29,6 26,088 3,41292 2178 87,1 15,178 5,83251 1211 48,4 26,686 3,33787 86 3,4 15,376 5,75802 187 7,5 27,093 3,28857 138 5,5 15,42 5,74169 144 5,8 27,74 3,21330 186 7,4 15,854 5,58536 391 15,6 28,056 3,17789 87 3,5 16,081 5,50706 236 9,4 28,607 3,11788 248 9,9 16,134 5,48931 250 10 28,703 3,10770 155 6,2 16,419 5,39437 135 5,4 29,135 3,06255 131 5,2 16,76 5,28552 36 1,4 29,267 3,04911 284 11,4 17,165 5,16167 304 12,1 29,353 3,04034 193 7,7 17,199 5,15149 235 9,4 29,787 2,99703 71 2,8 17,24 5,13933 329 13,2 30,357 2,94205 106 4,2 17,355 5,10550 281 11,2 30,954 2,88666 38 1,5 18,556 4,77774 213 8,5 31,436 2,84343 52 2,1 18,829 4,70917 85 3,4 32,618 2,74309 163 6,5 19,27 4,60224 372 14,9 32,773 2,73047 161 6,5 19,499 4,54892 233 9,3 33,254 2,69202 64 2,6 20,203 4,39192 118 4,7 33,897 2,64245 66,4 2,7 20,635 4,30098 238 9,5 34, 14 2,62417 162 6,5 21,584 4,11385 250 10 36,522 2,45827 53 2,1 21,689 4,09416 704 28,2 38,532 2,33459 40 1,6 21,867 4,06119 1621 64,8 38,859 2,31564 78 3,1 22.28 3,98692 154 6,2 39,594 2,27434 47 1,9 Полиморф В соединения I: данные порошковой рентгенографии Интенсивность, ИнтенсивИнтенсивность, ИнтенсивУгол 2 Значение d Угол 2 Значение d отсчет ность, % отсчет ность, % 7,379 11,97023 651 49,2 20,179 4,39705 119 9 10,096 8,75424 481 36,4 20,9 4,24699 54 4,1 10,821 8,16974 1323 100 21,718 43,08884 1300 98,3 12,482 7,08575 61 4,6 22,285 3,98602 446 33,7 12,739 6,94328 149 11,3 22,96 3,87033 87 6,6 13,036 6,78588 194 14,7 23,304 3,81394 111 8,4 13,475 6,56586 123 9,3 24,018 3,70224 73 5,5 13,828 6,39890 69 5,2 24,459 3,63648 75 5,7 14,815 5,97480 322 24,3 25,081 3,54765 141 10,7 15,078 5,87109 307 23,2 25,837 3,44555 47 3,6 15,584 5,68147 86 6,5 26,204 3,39810 193 14,6 16,082 5,50675 391 29,6 27,078 3,29031 176 13,3 16,979 5,21789 147 11,1 28,449 3,13490 91 6,9 17,02 5,20548 163 12,3 28,889 3,08812 51 3,9 17,287 5,12570 257 19,4 29,84 2,99177 166 12,6 17,616 5,03052 72 5,4 29,922 2,98380 121 9,1 18,125 4,89033 83,1 6,3 30,227 2,95440 58 4,4 18,161 4,88081 129 9,7 31,94 2,79975 70,5 5,3 18,192 4,87269 143 10,8 32,837 2,72528 117 8,8 19,022 4,66174 69 5,2 33,295 2,68885 40 3 Полиморф С соединения I: данные порошковой рентгенографии Интенсивность, ИнтенсивИнтенсивность, ИнтенсивУгол 2 Значение d Угол 2 Значение d отсчет ность, % отсчет ность, % 7,254 12,17653 25 0,6 24,647 3,60908 75 1,8 8,023 11,01169 798 19.3 24,961 3,56448 398 9,6 9,686 9,12421 4130 100 25,341 3,51181 182 4,4 10,489 8,42726 64 1,5 25,549 3,48378 617 14,9 10,813 8,17559 107 2,6 25,981 3,42671 832 20,1 11,277 7,83989 31 0,8 26,794 3,32466 328 7,9 12,193 7,25327 48 1,2 27,782 3,20863 166 4 12,961 6,82506 40 1 27,985 3,18576 158 3,8 14,357 6,16430 146 3,5 28,257 3,15573 241 5,8 14,497 6,10512 213 5,2 28,788 3,09868 257 6,2 15,225 5,81495 35 0,8 29,242 3,05164 322 7,8 16,007 5,53248 917 22,2 29,301 3,04560 322 7,8 16.545 5,35385 681 16,5 29,56 3,01950 111 2,7 16,951 5,22646 61 1,5 30,544 2,92444 158 3,8 17,672 5,01479 402 9,7 30,942 2,88768 70 1,7 18,337 4,83428 536 13 31,839 2,80834 156 3,8 18,578 4,77228 533 12,9 32,457 2,75628 157 3,8 19,396 4,57272 3486 84,4 32,745 2,73271 369 8,9 20,034 4,42851 57 1,4 34,246 2,61626 150 3,6 20,913 4,24430 193 4,7 34,979 2,56310 76 1,8 21,189 4,18959 1917 46,4 35,42 2,53225 74 1,8 21,523 4,12543 476 11,5 35,753 2,50941 92 2,2 21,876 4,05969 329 8 36,71 2,44615 91 2,2 22,654 3,92185 151 3,7 37,036 2,42537 145 3,5 23,207 3,82968 463 11,2 37,549 2,39339 137 3,3 23,238 3,82472 462 11,2 38,915 2,31247 74 1,8 24,316 3,65757 87 2,1 39,41 2,28456 166 4 Полиморф D соединения I: данные порошковой рентгенографии ИнтенсивИнтенсивИнтенсивУгол 2 Значение d Угол 2 Значение d ность, отсчет ность, % ность, отсчет 8,024 11,00971 501 33,4 22,224 3,99690 414 9,664 9,14458 1501 100 22,654 3,92192 112 9,861 8,96259 502 33.5 23,22 3,82757 322 10,497 8,42115 38 2,5 24,052 3,69702 89 10,8 8,18489 229 15,3 24,313 3,65791 295 11,462 7,71362 40 2,7 24,948 3,56619 272 12,105 7,30563 216 14,4 25,3 3,51744 120 12,951 6,83035 573 38,2 25,543 3,48455 304 13,312 6,64558 107 7,1 25,977 3,42728 362 13,817 6,40392 39 2,6 26,036 3,41965 416 14,478 6,11305 282 18,8 26,789 3,32518 539 14,823 5,97145 76 5,1 27,479 3,24329 173 15,113 5,85746 162 10,8 27,754 3,21176 198 16 5,53485 396 26,4 28,019 3,18199 170 16,533 5,35742 774 51,6 28,261 3,15525 314 16,952 5,22613 72 4,8 28,478 3,13170 144 17,661 5,01784 509 33,9 28,809 3,09646 205 18,015 4,92003 479 31,9 29,257 3,05012 176 18,316 4,83994 545 36,3 29,83 2,99276 178 18,46 4,80255 512 34,1 30,533 2,92545 157 18,543 4,78110 498 33,2 30,904 2,89114 96 19,239 4,60966 196 13,1 31,818 2,81015 131 Метанольный сольват соединения I: данные порошковой рентгенографии Интенсивность, ИнтенсивИнтенсивность, ИнтенсивУгол 2 Значение d Угол 2 Значение d отсчет ность, % отсчет ность, % 7,357 12,00677 9531 90,4 21,153 4,19677 224 2,1 7,608 11,61023 260 2,5 22,081 4,02236 10545 100 9,616 9,19052 260 2,5 22,588 3,93327 526 5 9,755 9,05958 433 4,1 22,825 3,89290 1150 10,9 10,943 8,07881 1145 10,9 23,14 3,84065 362 3.4 11,274 7,84189 406 3,9 23,742 3,74460 291 2,8 12,36 7,15546 3517 33,4 24,295 3,66064 440 4,2 12,942 6,83511 472 4,5 24,818 3,58463 3357 31,8 12,981 6,81447 530 5 25,434 3,49921 933 8,8 14,42 6,13754 440 4,2 25,967 3,42863 2020 19,2 14,69 6,02552 3179 30,1 26 3,42428 2000 19 15,196 5,82583 371 3,5 26,761 3,32860 402 3,8 15,651 5,65758 767 7,3 27,151 3,28169 230 2,2 15,913 5,56476 954 9 28,642 3,11415 273 2,6 16,507 5,36597 379 3,6 28,951 3,08162 298 2,8 16,539 5,35556 426 4 29,56 3,01950 1793 17 16,871 5,25102 1970 10,1 30,365 2,94130 455 4,3 17,226 5,14355 237 2,2 31,319 2,85382 313 3 18,226 4,86354 372 3,5 32,074 2,78834 173 1,6 18,613 4,76331 112 1,1 32,739 2,73323 134 1,3 19,283 4,59933 291 2,8 33,77 2,65210 344 3,3 19,545 4,53827 195 1,8 35,185 2,54858 129 1,2 19,961 4,44452 1703 16,2 37,166 2,41715 145 1,4 20,675 4,29264 161 1,5 37,694 2,38454 218 2,1 Этанольный сольват соединения I: данные порошковой рентгенографии Интенсивность, ИнтенсивИнтенсивность, ИнтенсивУгол 2 Значение d Угол 2 Значение d отсчет ность, % отсчет ность, % 7,357 12,00679 943 52,5 25,061 3,55045 462 25,7 9,541 9,26191 201 11,2 25,341 3,51184 1795 100 9,809 9,00980 208 11,6 25,712 3,46196 290 16,2 10,951 8,07238 1365 76,1 26,055 3,41713 463 25,8 12,031 7,35019 1500 83,6 26,235 3,39415 401 22,4 12,499 7,07637 115 6,4 26,599 3,34857 178 9,9 14,158 6,25067 245 13,7 27,054 3,29329 159 8,9 14,707 6,01841 236 13,2 28,355 3,14498 311 17,3 15,719 5,63302 428 23,8 28,491 3,13034 190 10,6 15,899 5,56980 657 36,6 28,667 3,11154 150 8,4 16,304 5,43218 869 48,4 29,029 3,07353 142 7,9 16,895 5,24359 1076 60 29,658 3,00975 207 11,5 18,227 4,86334 500 27,9 29,819 2,99388 604 33,7 19,109 4,64073 148 8,2 30,421 2,93596 132 7,4 19,49 4,55085 1045 58,2 30,609 2,91837 133 7,4 20,998 4,22731 211 11,8 31,383 2,84817 131 7,3 21,177 4,19195 168 9,3 32,048 2,79050 154 8,6 21,982 4,04035 1230 68,6 32,116 2,78480 146 8,1 22,124 4,01471 1146 63,9 33,78 2,65134 233 13 Гидрат полиморфа А соединения I: данные порошковой рентгенографии Интенсивность, ИнтенсивИнтенсивность, ИнтенсивУгол 2 Значение d Угол 2 Значение d отсчет ность, % отсчет ность, % 7.261 12,16414 447 100 18,657 4,75222 48,4 10,8 9,1 9,70988 25,6 5,7 18,779 4,72155 35,7 8 9,626 9,18058 31 6,9 18,859 4,70171 43,7 9,8 10,796 8,18860 283 63,2 19,276 4,60089 82 18,3 11,858 7,45707 170 38 19,504 4,54766 46 10,3 12,218 7,23806 51,8 11,6 20,22 4,38821 28 6,3 12,261 7,21317 79,4 17,7 20,655 4,29677 133 29,7 12,302 7,18922 78 17,4 21,68 4,09588 163 36,4 12,97 6,82039 173 38,7 21,86 4,06263 223 49,8 14,462 6,11993 132 29,5 22,316 3,98062 88,8 19,8 14,518 6,09637 138 30,8 22,537 3,94207 254 56,7 15,19 5,82798 229 51,2 22,579 3,93478 281 62, 7 15,402 5,74824 114 25,6 22,901 3,88013 78 17,4 15,866 5,58141 222 49,6 23,673 3,75541 84 18,8 15,906 5,56750 196 43,9 24,507 3,62946 46 10,3 16,08 5,50763 102 22,7 25,469 3,49445 203 45,4 16,127 5,49137 117 26,1 26,087 3,41311 128 28,6 16,178 5,47423 112 25,1 27,754 3,21175 56 12,5 16,221 5,46004 78 17,4 28,613 3,11725 65 14,5 16,357 5,41482 65,9 14,7 29,291 3,04663 57 12,7 16,418 5,39480 71,3 15,9 29,935 2,98248 43 9,6 17,189 5,15467 146 32,6 32,747 2,73253 63 14,1 17,345 5,10866 105 23,5 34,176 2,62146 32 7,2 18,57 4,77428 71 15,9 38,848 2,31630 31 6,9 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. 7-(1,1-Диметилэтил)-6-(2-этил-2 Н-1,2,4 триазол-3-илметокси)-3-(2-фторфенил)-1,2,4 триазоло[4,3-b]пиридазин. 2. Полиморф А 7-(1,1-диметилэтил)-6-(2 этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазина. 3. Фармацевтическая композиция, содержащая 7-(1,1-диметилэтил)-6-(2-этил-2 Н-1,2,4 триазол-3-илметокси)-3-(2-фторфенил)-1,2,4 триазоло[4,3-b]пиридазин в сочетании с фармацевтически приемлемым носителем. 4. Применение 7-(1,1-диметилэтил)-6-(2 этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2-фторфенил)-1,2,4-триазоло[4,3-b]пиридазина для получения лекарственного препарата для лечения и/или профилактики состояния тревоги. 5. Способ получения 7-(1,1-диметилэтил)6-(2-этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2 фторфенил)-1,2,4-триазоло[4,3-b]пиридазина,включающий взаимодействие соединения формулы III с соединением формулы IV где L1 представляет подходящую уходящую группу. 6. Способ получения 7-(1,1-диметилэтил)6-(2-этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2 фторфенил)-1,2,4-триазоло[4,3-b]пиридазина,включающий взаимодействие соединения формулы XI (или его таутомера 1,2,4-триазоло[4,3b]пиридазин-6-она) с соединением формулы XII где L3 представляет подходящую уходящую группу. 7. Способ получения 7-(1,1-диметилэтил)6-(2-этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2 29 фторфенил)-1,2,4-триазоло[4,3-b]пиридазина,включающий взаимодействие триметилуксусной кислоты с соединением формулы XIII в присутствии нитрата серебра и персульфата аммония. 8. Способ получения 7-(1,1-диметилэтил)6-(2-этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2 фторфенил)-1,2,4-триазоло[4,3-b]пиридазина,включающий взаимодействие соединения формулы XIV с соединением формулы XVAlk представляет C1-6 алкильную группу и L4 представляет подходящую уходящую группу, в присутствии катализатора на основе переходного металла. 9. Способ по п.5, где реакцию проводят в 1-метил-2-пирролидиноне в присутствии гидроксида натрия при температуре в области 0 С. 10. Способ лечения и/или профилактики состояния тревоги, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества 7-(1,1-диметилэтил)-6(2-этил-2 Н-1,2,4-триазол-3-илметокси)-3-(2 фторфенил)-1,2,4-триазоло-[4,3-b]пиридазина.
МПК / Метки
МПК: C07D 487/04, A61K 31/5025
Метки: композиции, замещенное, приготовленные, производное, триазолопиридазина, фармацевтические, основе
Код ссылки
<a href="https://eas.patents.su/16-3332-zameshhennoe-proizvodnoe-triazolopiridazina-farmacevticheskie-kompozicii-prigotovlennye-na-ego-osnove.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенное производное триазолопиридазина, фармацевтические композиции, приготовленные на его основе</a>
Замещенное производное тиазолидиндиона, способ его получения и его фармацевтическое применение

Номер патента: 2833
Опубликовано: 31.10.2002
Авторы: Линч Ян Роберт, Сэсс Майкл Джон, Чаудари Бернадетт Мари
МПК: C07D 417/12, A61K 31/427
Метки: тиазолидиндиона, замещенное, способ, производное, получения, применение, фармацевтическое
Формула / Реферат:
1. Гидрат соли малеиновой кислоты 5-[4-[2-(N-метил-N-(2-пиридил)амино)этокси]бензил]тиазолидин-2,4-диона, отличающийся тем, что он (i) включает воду в интервале от 0,3 до 0,6 мольных эквивалента; и (ii) имеет инфракрасный спектр, содержащий пики при 1757, 1331, 1290, 1211 и 767 см-1; и/или (iii) имеет спектр Рамана, содержащий пики при 1758, 1610, 1394, 1316 и 1289 см-1; и/или (iv) в твердом состоянии имеет спектр ядерно-магнитного резонанса,...
Фармацевтические композиции на основе тизоксанида и нитазоксанида

Номер патента: 2908
Опубликовано: 31.10.2002
Автор: Россиньоль Жан Франсуа
МПК: A61K 31/426, A61P 33/00, C07D 277/58...
Метки: тизоксанида, нитазоксанида, фармацевтические, основе, композиции
Формула / Реферат:
1. Фармацевтическая композиция для орального применения, содержащая в качестве активного агента, по крайней мере, одно соединение, выбранное из группы, состоящей из соединения формулы I и соединения формулы II причем упомянутый активный агент находится в форме активных частиц, имеющих размеры менее 200 мкм и средний размер более 5 мкм. 2. Композиция по п.1, в которой средний размер твердых активных частиц составляет от 10 до 100 мкм. 3....
Фармацевтические композиции на основе тизоксанида и нитазоксанида

Номер патента: 2920
Опубликовано: 31.10.2002
Автор: Россиньоль Жан Франсуа
МПК: C07D 277/58, A61K 31/426, A61P 33/00...
Метки: основе, тизоксанида, нитазоксанида, фармацевтические, композиции
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного агента, по крайней мере, одно соединение, выбранное из группы, состоящей из соединения формулы I и соединения формулы II и фармацевтически приемлемую кислоту в количестве, улучшающем ее стабильность. 2. Композиция по п.1, в которой фармацевтически приемлемая кислота выбрана из группы, состоящей из лимонной, глутаминовой, янтарной, этансульфокислоты, уксусной, винной, аскорбиновой,...
Е – 2 – [4 - (4 - хлор - 1, 2 - дифенил - бут - 1 - енил) фенокси] этанол и фармацевтические композиции на его основе

Номер патента: 2919
Опубликовано: 31.10.2002
Авторы: Мянтюля Эро, Седервалл Марья-Лииса, Калапудас Арья, Виитанен Антти
МПК: C07C 43/23, A61K 31/085, A61P 9/10...
Метки: хлор, основе, этанол, енил, фенокси, дифенил, фармацевтические, бут, композиции
Формула / Реферат:
1. Е-2-[4-(4-хлор-1,2-дифенил-бут-1-енил)-фенокси]этанол или его фармацевтически приемлемый сложный эфир. 2. Фармацевтическая композиция, которая включает Е-2-[4-(4-хлор-1,2-дифенил-бут-1-енил)-фенокси]этанол или его фармацевтически приемлемый эфир в качестве активного ингредиента, вместе с фармацевтически приемлемым носителем. 3. Способ снижения уровня холестерина в сыворотке, который включает введение пациенту, нуждающемуся в таком лечении,...
Пептид, способный связываться с интерлейкин-1-beta-конвертирующим ферментом (ice), и фармацевтические композиции на его основе

Номер патента: 1866
Опубликовано: 22.10.2001
Авторы: Муцио Марта, Интрона Мартино, Мантовани Альберто
МПК: A61P 31/04, C07K 14/54, A61K 38/20...
Метки: ice, пептид, связываться, композиции, основе, ферментом, фармацевтические, способный, интерлейкин-1-beta-конвертирующим
Формула / Реферат:
1. Пептид, способный связываться с интерлейкин-1-b-конвертирующим ферментом (ICE), имеющий, согласно Списку последовательностей, аминокислотную последовательность SEQ ID NO: 1, в которой Хаа представляет собой Asp или Аlа. 2. Пептид по п.1, имеющий, согласно Списку последовательностей, аминокислотную последовательность SEQ ID NO: 2. 3. Пептид по п.1, имеющий, согласно Списку последовательностей, аминокислотную последовательность SEQ ID NO: 3....
Предыдущий патент: Способ образования гранул путем влажного гранулирования
Следующий патент: Производные аминоантрациклинона и их применение в лечении амилоидоза
Случайный патент: Нефтяное топливо с наночастицами и способ его приготовления