Способ получения триазолонов
Номер патента: 16913
Опубликовано: 30.08.2012
Авторы: Сарма Кешаб, Арзено Умберто Бартоломе, Чжу Цзян, Ли Гэри М., Мартин Майкл















Формула / Реферат
1. Способ получения соединения формулы 2

в которой Ar обозначает фенил, замещенный с помощью 2 или 3 групп, независимо выбранных из группы, включающей галоген, цианогруппу и C1-С6-галогеналкил;
Rf обозначает водород;
R1 и R3 обозначают С6-С10-алкил,
способ включает следующие стадии:

(a) взаимодействие 4 с сильным основанием в инертном растворителе с образованием сопряженного основания 4 и взаимодействие указанного сопряженного основания с 6 с получением 8;
(b) обработку 8 при условиях, которые приводят к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты с получением 2.
2. Способ по п.1, в котором Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил, R1 обозначает метил и R3 обозначает трет-Bu.
3. Способ по п.2, в котором указанное сильное основание представляет собой трет-бутоксид калия, указанный инертный растворитель представляет собой тетрагидрофуран (ТГФ) и указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником.
4. Способ по п.3, в котором Ar обозначает 3-циано-5-дифторметилфенил.
5. Способ получения соединения формулы 12 и 14, который включает стадии по п.1 и дополнительно включает следующие стадии:

(a) взаимодействие 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12;
(b) взаимодействие 12 с диазотирующим реагентом и Cu(I)Br/LiBr или Cu(I)Cl/LiCl с получением 14, в котором R2 обозначает бром и хлор соответственно.
6. Способ по п.5, в котором Ar обозначает 3-циано-5-дифторметилфенил, R1 обозначает метил, R2 обозначает бром, R3 обозначает трет-бутил, указанное сильное основание представляет собой трет-бутоксид калия, указанный инертный растворитель представляет собой тетрагидрофуран (ТГФ), указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником, указанный восстановительный реагент представляет собой водород, Pd/C и ванадилацетилацетонат (VO(acac)2), указанный диазотирующий реагент представляет собой трет-бутилнитрит и указанная соль меди(I) представляет собой Cu(I)Br.
7. Способ, который включает стадии по п.6 и дополнительно включает стадию превращения 14, где R2 обозначает Br, в тозилат.
8. Способ получения соединения формулы 20

который включает стадии по п.6 и дополнительно включает следующие стадии:
(a) обработку 14 окислительным реагентом, способным окислять сульфид в сульфон 18;
(b) взаимодействие 18 с уксусной кислотой/уксусным ангидридом при условиях, которые приводят к расщеплению связи S-алкил и гидролизу образовавшегося сульфона, с получением 20.
9. Способ по п.8, в котором Ar обозначает 3-циано-5-дифторметилфенил, R1 обозначает метил, R2 обозначает бром, R3 обозначает трет-бутил, указанное сильное основание представляет собой трет-бутоксид калия, указанный инертный растворитель представляет собой тетрагидрофуран (ТГФ), указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником, указанный восстановительный реагент представляет собой водород, Pd/C и ванадилацетилацетонат (VO(acac)2), указанный диазотирующий реагент представляет собой трет-бутилнитрит, указанная соль меди(I) представляет собой Cu(I)Br и указанный окислительный реагент представляет собой метахлорпербензойную кислоту (МХПБК).
10. Соединение формулы 4

в которой R1 и R3 независимо обозначают С6-С10-алкил.
11. Соединение по п.10, в котором R1 обозначает метил и R3 обозначает трет-бутил.
12. Способ получения соединения по п.10, который включает следующие стадии:

(a) взаимодействие моноэфира малоновой кислоты 19 с карбонилдиимидазолом в инертном растворителе с образованием эфира 3-имидазол-1-ил-3-оксопропионовой кислоты (21);
(b) взаимодействие ацилимидазола, полученного на стадии (а), с тиосемикарбазидом 22;
(c) обработку полученного 5-тиооксо-4,5-дигидро-1Н-[1,2,4]триазол-3-карбоксилата 24 алкилирующим реагентом с получением 4, в котором R1 и R3 обозначают С6-С10-алкил.
13. Способ по п.12, в котором указанный моноэфир малоновой кислоты представляет собой трет-бутилгидромалонат и указанный алкилирующий реагент представляет собой метилйодид.
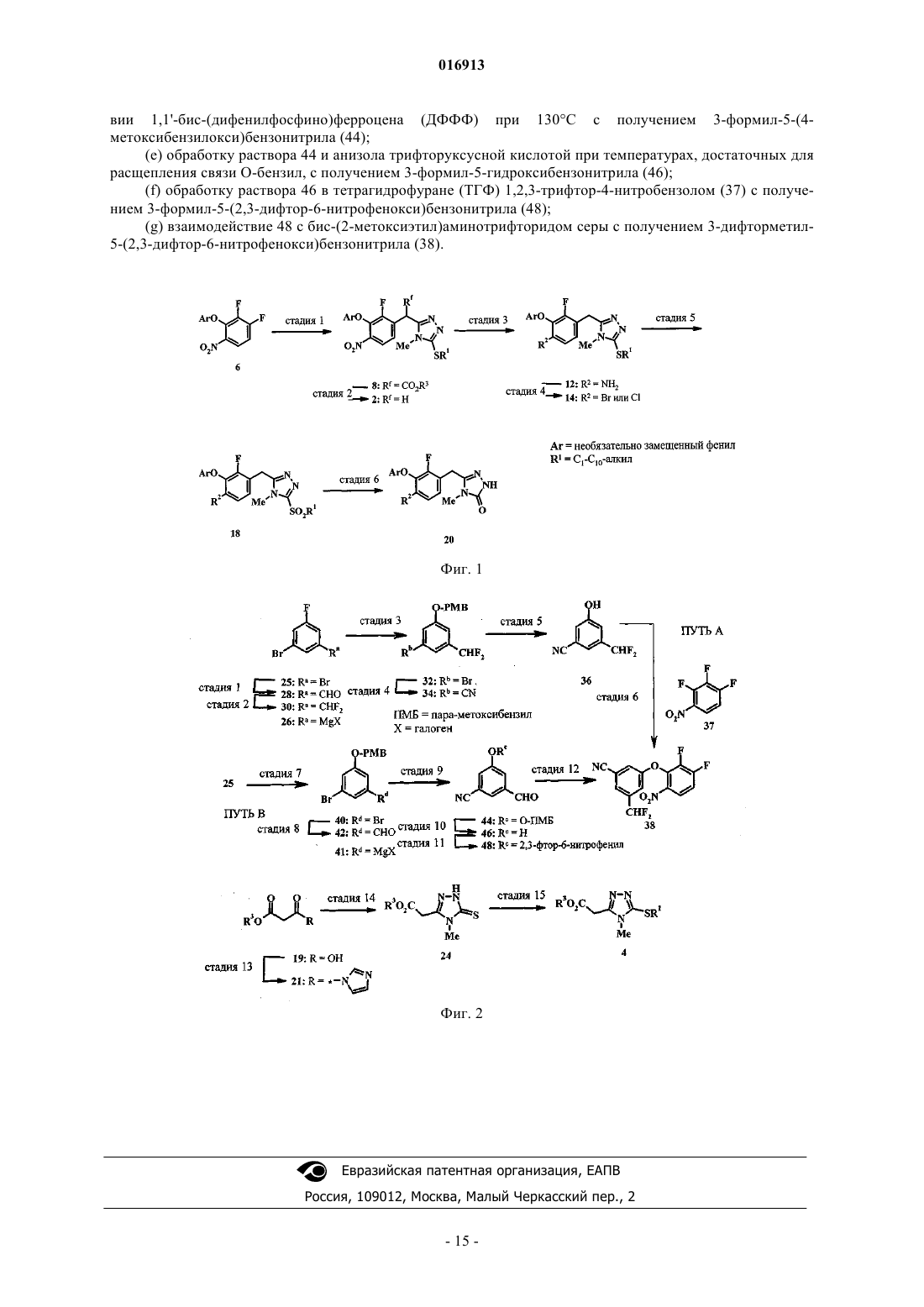
14. Способ получения 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (38), который включает стадии по п.8 и дополнительно включает следующие стадии:
(a) взаимодействие 3,5-дибромфторбензола (25) с изопропилмагнийхлоридом с получением 3-бром-5-фторфенилмагнийгалогенида (26);
(b) взаимодействие 26 с N,N-диметилформамидом (ДМФ) в метил-трет-бутиловом эфире (МТБЭ) с получением 3-бром-5-фторбензальдегида (28);
(c) взаимодействие 28 с бис-(2-метоксиэтил)аминотрифторидом серы с получением 3-фтор-5-дифторметил-1-бромбензола (30);
(d) взаимодействие 30 с п-метоксибензиловым спиртом в тетрагидрофуране (ТГФ) с получением 1-бром-3-дифторметил-5-(4-метоксибензилокси)бензола (32);
(e) взаимодействие раствора 32 и N-метилпирролидона (NMP) с ферроцианидом калия в присутствии 1,1'-бис-(дифенилфосфино)ферроцена (ДФФФ) при 130°С с получением 3-дифторметил-5-(4-метоксибензилокси)бензонитрила (34);
(f) обработку раствора 34 и анизола трифторуксусной кислотой при температурах, достаточных для расщепления связи О-бензил, и с получением 3-дифторметил-5-гидроксибензонитрила (36);
(g) обработку раствора 36 в тетрагидрофуране (ТГФ) 1,2,3-трифтор-4-нитробензолом (37) с получением 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (38).
15. Способ получения 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (38), который включает стадии по п.8 и дополнительно включает следующие стадии:
(a) взаимодействие 25 с п-метоксибензиловым спиртом в тетрагидрофуране (ТГФ) с получением 1,3-дибром-5-(4-метоксибензилокси)бензола (40);
(b) взаимодействие 40 с изопропилмагнийхлоридом с получением 3-бром-5-(4-метоксибензилокси)бензолмагнийгалогенида (41);
(c) взаимодействие 41 с N,N-диметилформамидом (ДМФ) в метил-трет-бутиловом эфире (МТБЭ) с получением 3-бром-5-(4-метоксибензилокси)бензальдегида (42);
(d) взаимодействие раствора 42 и N-метилпирролидона (NMP) с ферроцианидом калия в присутствии 1,1'-бис-(дифенилфосфино)ферроцена (ДФФФ) при 130°С с получением 3-формил-5-(4-метоксибензилокси)бензонитрила (44);
(e) обработку раствора 44 и анизола трифторуксусной кислотой при температурах, достаточных для расщепления связи О-бензил, с получением 3-формил-5-гидроксибензонитрила (46);
(f) обработку раствора 46 в тетрагидрофуране (ТГФ) 1,2,3-трифтор-4-нитробензолом (37) с получением 3-формил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (48);
(g) взаимодействие 48 с бис-(2-метоксиэтил)аминотрифторидом серы с получением 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (38).
Текст
В изобретении описан улучшенный способ получения алкилсульфанилзамещенных триазолов 2, которые являются полезными промежуточными продуктами в новом способе получения триазолонов 20 016913 Настоящее изобретение относится к способу получения производных 3-[3-(4-метил-5 метилсульфанил-4 Н-[1,2,4]триазол-3-илметил)фенокси]-5-дифторметилбензонитрила формулы 2. Соединения формулы 2 применимы для получения триазолонов формулы 20 с помощью дополнительных стадий, раскрытых в настоящем изобретении. Триазолоны формулы 20 являются полезными ингибиторами обратной транскриптазы ВИЧ-1 и применимы для лечения синдромов, опосредуемых с помощью СПИД и ВИЧ-1. Настоящее изобретение также относится к соединениям формулы 4, которые являются полезными реагентами в способе, раскрытом в настоящем изобретении. Вирус иммунодефицита человека, ВИЧ, является возбудителем синдрома приобретенного иммунодефицита (СПИД), т.е. заболевания, характеризующегося поражением иммунной системы, в частностиCD4+ Т-клеток, при сопутствующей восприимчивости к инфекциям, вызываемым условно-патогенными организмами. Инфекция ВИЧ также связана с предшественником, СПИД-ассоциированным комплексом(ПСП), т.е. синдромом, характеризующимся такими симптомами, как хроническая генерализованная лимфаденопатия, лихорадка и потеря массы. Существующая в настоящее время химиотерапия направлена на два критически важных вирусных фермента: протеазу ВИЧ и обратную транскриптазу ВИЧ (J.S.G. Montaner et al., Biomed.Pharmacother. 1999, 53:63-72; R.W. Shafer and D.A. Vuitton, Biomed.Pharmacother, 1999, 53:73-86; E. De Clercq, Curr.Med. Chem. 2001, 8:1543-1572). Выявлены два общих класса ингибиторов ОТ: нуклеозидные ингибиторы обратной транскриптазы (НИОТ) и ненуклеозидные ингибиторы обратной транскриптазы (ННИОТ). В настоящее время корецептор CCR5 стал потенциальной мишенью для химиотерапии ВИЧ (D. Chantry,Expert Opin. Emerg. Drugs 2004 9(1): 1-7; С.G. Barber, Curr. Opin. Invest. Drugs 2004 5(8):851-861; D.Dev. 2003 6(4):451-461). На рынок поступили лекарственные средства, воздействующие на новые ферменты-мишени, включая ингибиторы интегразы, примерами которых являются ралтегравир (Merck), утвержденные к применению FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) и элвитегравир (Gilead Sciences и Japan Tobacco), находящийся в фазе II клинических исследований. Антагонист CCR5 маравирок (SELZENTRY, Pfizer) также утвержден FDA к применению в качестве средства лечения ВИЧ-1. ННИОТ впервые были открыты в 1989 г. ННИОТ являются аллостерическими ингибиторами, которые обратимо связываются с несубстратным центром связывания обратной транскриптазы ВИЧ и тем самым меняют форму активного центра или активность блокирующей полимеразы (R.W. Buckheit, Jr.,Expert Opin. Investig. Drugs 2001, 10(8):1423-1442; E. De Clercq, Antiviral Res. 1998, 38:153-179; E. DeClercq, Current medicinal Chem. 2001, 8(13):1543-1572; G. Moyle, Drugs 2001, 61(1):19-26). Хотя ННИОТ сначала рассматривались как перспективный класс соединений, проведенные in vitro и in vivo исследования быстро показали, что ННИОТ создают низкий барьер для появления резистентных по отношению к лекарственным препаратам штаммов ВИЧ и обладают низкой классоспецифической токсичностью. Хотя в лаборатории идентифицированы более 30 структурных классов ННИОТ, для лечения ВИЧ утверждены к применению только 3 соединения: эфавиренц, невирапин и делавирдин. Охраняется необходимость в более безопасных лекарственных препаратах, обладающих активностью по отношению к штаммам дикого типа и широко распространенным резистентным штаммам ВИЧ. 5-Арилалкил-2,4-дигидро-[1,2,4]триазол-3-оны являются ненуклеозидными ингибиторами обратной транскриптазы и их раскрыли J.P. Dunn et al. в патенте US7208509, выданном 24 апреля 2007 г., и J.P.Dunn et al. в публикации патента US20060025462, поданного 27 июня 2005 г. Пиридазиноновые ненуклеозидные ингибиторы обратной транскриптазы раскрыли J.P. Dunn et al. в патенте US7208509,выданном 13 марта 2007 г., и в публикации патента US20050215554, опубликованной 28 сентября 2005 г. Способ получения пиридазинононовых ненуклеозидных ингибиторов обратной транскриптазы раскрыл D.J. Kertesz в публикации патента US20050234236, опубликованной 20 октября 2005 г. Настоящее изобретение относится к улучшенному способу синтеза производных 3-[3-(1,4-диметил 5-оксо-4,5-дигидро-1 Н-[1,2,4]триазол-3-илметил)-2-фтор-фенокси]-5-дифторметилбензонитрила, которые являются ингибиторами обратной транскриптазы ВИЧ-1 и применимы для лечения опосредуемого ВИЧ-1 заболевания. Настоящее изобретение относится к способу получения триазолов формулы 2, которые можно превратить в искомые триазолоны способом, описанным в настоящем изобретении. Способ включает конденсацию 6 и сопряженного основания 4, где Ar обозначает фенил, замещенный с помощью 2 или 3 групп, независимо выбранных из группы, включающей галоген, цианогруппу и C1-С 6 галогеналкил; R1 и R3 обозначают С 6-С 10-алкил; способ включает следующие стадии:(a) взаимодействие 4 с сильным основанием в инертном растворителе с образованием сопряженного основания 4 и взаимодействие указанного сопряженного основания с 6, где Ar обозначает фенил, замещенный с помощью 2 или 3 групп, независимо выбранных из группы, включающей галоген, цианогруппу и C1-С 6-галогеналкил, с получением 8;(b) обработка 8 при условиях, которые приводят к гидролизу сложного эфира с декарбоксилированием полученной кислоты с получением 2. Настоящее изобретение также относится к способу замещения нитрогруппы 2 на хлор или бром и последующее превращение триазола 14 в триазолон 20, этот способ включает следующие стадии:(c) взаимодействие 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12;(e) обработка 14 окислительным реагентом, способным селективно окислять сульфид в сульфон 18;(f) взаимодействие 18 с уксусной кислотой/уксусным ангидридом при условиях, которые приводят к расщеплению связи S-алкил и гидролизу образовавшегося триазола с получением 20. Настоящее изобретение также относится к новым соединениям формулы 4, в которых R1 и R3 независимо обозначают С 6-С 10-алкил, которые применимы для получения триазолов формулы 2 и триазолонов формулы 20. На фиг. 1 представлен способ получения производных 3-арилокси-2-фтор-1-(4-метил-5 метилсульфанил-4 Н-[1,2,4]триазол-3-илметил)фенила 2 и производных 5-(4-галоген-2-фтор-3 арилоксибензил)-4-метил-2,4-дигидро-[1,2,4]триазол-3-она 20. На фиг. 2 представлен способ получения 3-дифторметил-5-(2,3-дифтор-6 нитрофенокси)бензонитрила (38) и трет-бутилового эфира (4-метил-5-метилсульфанил-4 Н[1,2,4]триазол-3-ил)уксусной кислоты 4 (R1 и R3 - метил). При использовании в настоящем изобретении объект в форме единственного числа относится к одному или большему количеству объектов; например, "соединение" означает одно или большее количество соединений или не менее одного соединения. Форма единственного числа и выражения "один или большее количество" и "не менее чем один" в настоящем изобретении можно использовать, как взаимозаменяемые. Выражение "определенная выше в настоящем изобретении" относится к самому широкому определению каждой группы, приведенному в "Кратком изложении сущности изобретения". Для всех остальных вариантов осуществления, приведенных ниже, заместители, которые могут использоваться в каждом варианте осуществления и которые не определены явно, соответствуют самому широкому определению,приведенному в "Кратком изложении сущности изобретения". Термин "необязательно" или "необязательный" при использовании в настоящем изобретении означает, что последующее описанное событие или обстоятельство может, но не должно осуществиться и что описание включает случаи, когда событие или обстоятельство осуществляется, и случаи, когда оно не осуществляется. Например, "необязательно замещенный" означает, что необязательно замещенный фрагмент может включать водород или заместитель. При использовании в настоящем описании в промежуточной фразе или в содержании пункта формулы изобретения термины "включает" и "включающий" следует интерпретировать, как допускающее изменения значения. Таким образом, термины следует интерпретировать, как синонимичные с выражениями "содержащий не менее" или "включающий не менее". При использовании в контексте способа термин "включающий" означает, что способ включает, по меньшей мере, указанные стадии, но может включать дополнительные стадии. При использовании в контексте соединения или композиции термин"включающий" означает, что соединение или композиция включает, по меньшей мере, указанные характеристики или компоненты, но также может включать дополнительные характеристики или компоненты. Термин "примерно" при использовании в настоящем изобретении означает приблизительно, в области, ориентировочно или около. Если термин "примерно" используется в связи с числовым диапазоном, то он изменяет этот диапазон путем расширения границ за пределы верхней и нижней границы указанного числового диапазона. Обычно термин "примерно" при использовании в настоящем изобретении изменяет числовое значение выше и ниже указанного значения с отклонением на 20%. При использовании в настоящем изобретении указания числового диапазона переменной означает,что настоящее изобретение можно осуществить при значении переменной, равном любым значениям,-2 016913 находящимся в этом диапазоне. Так, переменная, которая по определению является дискретной, может быть равна любому целому числу в этом числовом диапазоне, включая конечные значения диапазона. Аналогичным образом, переменная, которая по определению является непрерывной, может быть равна любому вещественному числу в этом числовом диапазоне, включая конечные значения диапазона. Например, переменная, которая описана, как обладающая значениями, равными от 0 до 2, может быть равна 0, 1 или 2 в случае, если она по определению является дискретной, и может быть равна 0,0, 0,1, 0,01,0,001 или любому другому вещественному числу, если она по определению является непрерывной."Стабильное" соединение означает соединение, которое можно получить и выделить и структура и свойства которого остаются в основном неизменными в течение периода времени, достаточного для того, чтобы обеспечить применение соединения для задач, описанных в настоящем изобретении (например,для терапевтического или профилактического введения субъекту). Если явно не указано иное, то все диапазоны, указанные в настоящем изобретении, включают границы. Например, описание гетероциклического кольца, как содержащего "от 1 до 4 гетероатомов", означает, что кольцо может содержать 1, 2, 3 или 4 гетероатома. Также следует понимать, что любой диапазон, указанный в настоящем изобретении, включает все поддиапазоны, входящие в этот диапазон. Так,например, арил или гетероарил, описанный как необязательно содержащий "от 1 до 5 заместителей",включает в качестве вариантов любой арил, необязательно содержащий от 1 до 4 заместителей, от 1 до 3 заместителей, от 1 до 2 заместителей, от 2 до 5 заместителей, от 2 до 4 заместителей, от 2 до 3 заместителей, от 3 до 5 заместителей, от 3 до 4 заместителей, от 4 до 5 заместителей, 1 заместитель, 2 заместителя,3 заместителя, 4 заместителя и 5 заместителей. Значок в конце связи или ", проведенный через связь, означают положение присоединения функциональной группы или другого химического фрагмента к остальной части молекулы, частью которой она является. Так, например Термин "инертный органический растворитель" или "инертный растворитель" означает, что растворитель является инертным в условиях описываемой с его участием реакции, включая, например, бензол,толуол, MeCN, ТГФ, N,N-диметилформамид, хлороформ, ДХМ, дихлорэтан, диэтиловый эфир, EtOAc,ацетон, метилэтилкетон, МеОН, EtOH, пропанол, ИПС, трет-бутанол, диоксан, пиридин и т.п. Если не указано иное, то растворители, применяющиеся в реакциях в настоящем изобретении, являются инертными растворителями. Инертные растворители, совместимые с сильными основаниями, не должны содержать кислых протонов, которые могут отщепляться, и обычно включают алифатические и арильные углеводороды, простые эфиры, такие как ТГФ, ДМЭ (диметиловый эфир), диэтиловый эфир и диоксан,или полярные апротонные растворители, такие как ДМФ, NMP и ДМСО. Термин "сильное основание" при использовании в настоящем изобретении означает основное соединение, обладающее достаточной основностью, чтобы отщепить протон от метиленового атома углерода, находящегося между сложноэфирным фрагментом и триазольным кольцом формулы 4. Типичные основания, которые можно использовать, включают, но не ограничиваются только ими, диалкиламиды лития, такие как диизопропиламид лития, дициклогексиламид лития, трет-бутоксид калия или натрия,гексаметилдисилазан лития или натрия и гидрид натрия или калия. Селективный гидролиз трет-бутиловых эфиров можно провести в кислой среде, такой как ТФК илиHCl в эфирных растворителях. Термин "диазотирующий реагент" означает реагент, способный превратить ариламин в арилдиазониевую соль (например, PhN+X-). Обычные реагенты, использующиеся для превращения ароматического амина в диазониевую соль, включают азотистую кислоту (нитрит натрия в кислом растворе) или алкилнитрит, такой как трет-бутилнитрит. Окисление триазола в сульфоксид или сульфон обычно является легким и известны многочисленные реагенты, способные проводить такое превращение. Окисление серы обычно проводят водными растворами пероксида водорода, NaIO4, трет-бутилгипохлоритом, ацилнитритами, перборатом натрия, гидроперсульфатом калия и надкислотами, такими как надуксусная кислота и мета-хлорпербензойная кислота (МХПБК). Обычно при использовании примерно 1 экв. окислителя можно выделить сульфоксид. Использование двух или большего количества эквивалентов приводит к окислению в сульфон. Без отклонения от сущности настоящего изобретения в способе, предлагаемом в настоящем изобретении, можно использовать любой окислитель. Восстановление нитрогруппы можно провести с помощью множества хорошо известных восстановительных реагентов, например активированный металл, такой как активированное железо, цинк или олово (полученный например, промывкой порошкообразного железа разбавленным раствором кислоты,такой как разбавленная хлористо-водородная кислота). Восстановление также можно провести в атмосфере водорода в присутствии инертного растворителя и в присутствии металла, эффективного для катализа реакций гидрирования, такого как платина или палладий. Другие реагенты, которые можно исполь-3 016913 зовать для восстановления нитросоединений в амины, включают AlH3-AlCl3, гидразин и катализатор,TiCl3, Al-NiCl2-ТГФ, муравьиную кислоту и Pd/C и сульфиды, такие как NaHS, (NH4)2S или полисульфиды (т.е. реакция Цинна). Ароматические нитрогруппы восстанавливали с помощью NaBH4 или ВН 3 в присутствии катализаторов, таких как NiCl2 и CoCl2. Так, например, восстановление можно провести путем нагревания нитросоединения в присутствии достаточно активированного металла, такого как Fe, и растворителя или разбавителя, такого как Н 2 О и спирт, например МеОН или EtOH, при температуре в диапазоне от 50 до 150 С, обычно примерно при 70 С (J. March, Advanced Organic Chemistry, John WileySons: New York, NY, 1992, p. 1216). Все условия восстановления, применимые для селективного восстановления нитрогруппы в промежуточных продуктах, описанных в настоящем изобретении, входят в объем настоящего изобретения. Термин "алкил" при использовании в настоящем изобретении означает обладающий разветвленной или неразветвленной цепью насыщенный одновалентный углеводородный остаток, содержащий от 1 до 10 атомов углерода. Термин "низш. алкил" означает обладающий разветвленной или неразветвленной цепью углеводородный остаток, содержащий от 1 до 6 атомов углерода. "С 6-С 10-Алкил" при использовании в настоящем изобретении означает алкил, содержащий от 1 до 10 атомов углерода. Примеры алкильных групп включают, но не ограничиваются только ими, низшие алкильные группы, включая метил,этил, пропил, изопропил, н-бутил, изобутил, трет-бутил или пентил, изопентил, неопентил, гексил, гептил или октил. Термин "галоген" при использовании в настоящем изобретении означает фтор, хлор, бром или йод. Термин "галогеналкил" при использовании в настоящем изобретении означает обладающую разветвленной или неразветвленной цепью алкильную группу, определенную выше, в которой 1, 2, 3 или большее количество атомов водорода замещены на галоген. Примерами являются 1-фторметил, 1 хлорметил, 1-бромметил, 1-йодметил, дифторметил, трифторметил, трихлорметил, трибромметил,трийодметил, 1-фторэтил, 1-хлорэтил, 1-бромэтил, 1-йодэтил, 2-фторэтил, 2-хлорэтил, 2-бромэтил, 2 йодэтил, 2,2-дихлорэтил, 3-бромпропил или 2,2,2-трифторэтил. В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; и (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, где Ar обозначает фенил, замещенный с помощью 2 или 3 групп, независимо выбранных из группы, включающей галоген, цианогруппу и C1-С 6-галогеналкил, и R1 и R3 обозначают С 6 С 10-алкил. Во втором варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; и (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, где Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5 дифторметилфенил, R1 обозначает метил и R3 обозначает трет-Bu. В третьем варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; и (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, где Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5 дифторметилфенил, R1 обозначает метил и R3 обозначает трет-Bu, указанное сильное основание представляет собой трет-бутоксид калия, указанный инертный растворитель представляет собой ТГФ и указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником. В четвертом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном-4 016913 растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; и (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, где Ar обозначает 3-циано-5-дифторметилфенил, R1 обозначает метил и R3 обозначает трет-Bu, указанное сильное основание представляет собой трет-бутоксид калия, указанный инертный растворитель представляет собой ТГФ и указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником. В пятом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а; (с) взаимодействия 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12; и (d) взаимодействия 12 с диазотирующим реагентом иCu(I)Br/LiBr или Cu(I)Cl/LiCl с получением 14, в котором R2 обозначает бром и хлор соответственно, Ar обозначает фенил, замещенный с помощью 2 или 3 групп, независимо выбранных из группы, включающей галоген, цианогруппу и C1-С 6-галогеналкил и R1 и R3 обозначают С 6-С 10-алкил. Специалист в данной области техники должен понимать, что без отклонения от сущности настоящего изобретения вместо солей лития, указанных в настоящем изобретении, можно использовать другие хлориды и бромиды. В шестом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, (с) взаимодействия 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12; и (d) взаимодействия 12 с диазотирующим реагентом иCu(I)Br/LiBr с получением 14, в котором R2 обозначает бром, Ar обозначает 3-циано-5 дифторметилфенил, R1 обозначает метил, R3 обозначает трет-бутил, указанное сильное основание представляет собой трет-бутоксид калия, указанный инертный растворитель представляет собой ТГФ, указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником, указанный восстановительный реагент представляет собой водород, Pd/C и VO(acac)2 и указанный диазотирующий реагент представляет собой трет-бутилнитрит. В седьмом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, (с) взаимодействия 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12; (d) взаимодействия 12 с диазотирующим реагентом иCu(I)Br/LiBr с получением 14, в котором R2 обозначает бром, Ar обозначает 3-циано-5 дифторметилфенил, R1 обозначает метил, R3 обозначает трет-бутил, указанное сильное основание представляет собой трет-бутоксид калия, указанный инертный растворитель представляет собой ТГФ, указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником, указанный восстановительный реагент представляет собой водород, Pd/C и VO(acac)2, указанный диазотирующий реагент представляет собой трет-бутилнитрит; и (е) превращение 14 в тозилат и перекристаллизацию указанной соли. В восьмом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а; (с) взаимодействия 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12; (d) взаимодействия 12 с диазотирующим реагентом иCu(I)Br/LiBr с получением 14; (е) обработки 14 окислительным реагентом, способным окислять сульфид в сульфон 18; и (f) взаимодействия 18 с уксусной кислотой/уксусным ангидридом при условиях, которые приводят к расщеплению связи S-гетероарил и гидролизу образовавшегося ацетата, с получением 20, в котором R2 обозначает бром, Ar обозначает 3-циано-5-дифторметилфенил, R1 обозначает метил и R3 обозначает трет-бутил. В девятом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8, (b) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, (с) взаимодействия 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12; (d) взаимодействия 12 с диазотирующим реагентом иCu(I)Br/LiBr с получением 14, (е) обработки 14 окислительным реагентом, способным окислять сульфид в сульфон 18; и (f) взаимодействия 18 с уксусной кислотой/уксусным ангидридом при условиях, которые приводят к расщеплению связи S-гетероарил и гидролизу образовавшегося ацетата с получением 20, в котором R2 обозначает бром; где Ar обозначает 3-циано-5-дифторметилфенил, R1 обозначает метил, R3 обозначает трет-бутил, указанное сильное основание представляет собой трет-бутоксид калия, указанный инертный растворитель представляет собой ТГФ, указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником, указанный восстановительный реагент представляет собой водород, Pd/C и VO(acac)2, указанный диазотирующий реагент представляет собой трет-бутилнитрит, и указанный окислительный реагент представляет собой МХПБК. В десятом варианте осуществления настоящее изобретение относится к соединению формулы 4, в котором R1 и R3 независимо обозначают С 6-С 10-алкил. В одиннадцатом варианте осуществления настоящее изобретение относится к соединению формулы 4, в котором R1 обозначает метил и R3 обозначает трет-бутил. В двенадцатом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 4, указанный способ включает стадии (а) взаимодействия моноэфира малоновой кислоты с КДИ в инертном растворителе с образованием эфира 3-имидазол-1-ил-3-оксопропионовой кислоты (21), (b) взаимодействия ацилимидазола, полученного на стадии (а), с тиосемикарбазидом 22; и (с) обработки полученного 5-тиооксо-4,5-дигидро-1 Н-[1,2,4]триазол-3-карбоксилата 24 алкилирующим реагентом с получением 4, в котором R1 и R3 обозначают C1-C10. В тринадцатом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 4, указанный способ включает стадии (а) взаимодействия моноэфира малоновой кислоты с КДИ в инертном растворителе с образованием эфира 3-имидазол-1-ил-3-оксопропионовой кислоты (21), (b) взаимодействия ацилимидазола, полученного на стадии (а), с тиосемикарбазидом 22; и (с) обработки полученного 5-тиооксо-4,5-дигидро-1 Н-[1,2,4]триазол-3-карбоксилата 24 алкилирующим реагентом с получением 4, в котором R1 обозначает Me и R3 обозначает трет-Bu, указанный моноэфир малоновой кислоты представляет собой трет-бутилгидромалонат и указанный алкилирующий реагент представляет собой метилйодид. В четырнадцатом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 3,5-дибромфторбензола (25) с изопропилмагнийхлоридом с получением 3-бром-5-фторфенилмагнийгалогенида (26); (b) взаимодействия 26 с ДМФ и затем с водным раствором кислоты и МТБЭ с получением 3-бром-5-фторбензальдегида (28); (с) взаимодействия 28 с DEOXO-FLUOR и ДХМ с получением 3-фтор-5-дифторметил-1-бромбензола (30);(d) взаимодействия 30 с п-метоксибензиловым спиртом и трет-бутоксидом калия в ТГФ с получением 1 бром-3-дифторметил-5-(4-метоксибензилокси)бензола (32); (е) взаимодействия раствора 32 и NMP с ферроцианидом калия, Na2CO3, Pd(OAc)2 и ДФФФ примерно при 130 С с получением 34; (f) обработки раствора 34 и анизола с помощью ТФК при температурах, достаточных для расщепления связи О-бензил,с получением 36; (g) обработки раствора 36 и ТГФ 1,2,3-трифтор-4-нитробензолом (37) и K2CO3 с получением 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (38); (h) взаимодействия 4 с сильным основанием в инертном растворителе, указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; (i) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, (j) взаимодействия 2 с восстановительным реагентом,способным селективно восстанавливать нитрогруппу, с получением 12; (k) взаимодействия 12 с диазотирующим реагентом и Cu(I)Br/LiBr или Cu(I)Cl/LiCl, с получением 14, (l) обработки 14 окислительным реагентом, способным окислять сульфид в сульфон 18; и (m) взаимодействия 18 с уксусной кислотой/уксусным ангидридом при условиях, которые приводят к расщеплению связи S-гетероарил и гидролизу образовавшегося ацетата с получением 20, в котором R2 обозначает бром и хлор соответственно, Ar обозначает фенил, замещенный с помощью 2 или 3 групп, независимо выбранных из группы, включающей галоген, цианогруппу и C1-С 6-галогеналкил, и R1 и R3 обозначают С 6-С 10-алкил.-6 016913 В пятнадцатом варианте осуществления настоящее изобретение относится к способу получения соединения формулы 2, способ включает стадии (а) взаимодействия 25 с п-метоксибензиловым спиртом и трет-бутоксидом калия в ТГФ с получением 40; (b) взаимодействия 40 с изопропилмагнийхлоридом с получением 41; (b) взаимодействия 41 с ДМФ и затем с водным раствором кислоты и МТБЭ с получением 42; (d) взаимодействия раствора 42 и NMP с ферроцианидом калия Na2CO3, Pd(OAc)2 и ДФФФ при 130 С с получением 44; (е) обработки раствора 44 и анизола с помощью ТФК при температурах, достаточных для расщепления связи О-бензил, с получением 46; (g) обработки раствора 46 и ТГФ 1,2,3 трифтор-4-нитробензолом (37) и карбонатом калия с получением 48; (h) взаимодействия 48 с DEOXOFLUOR и ДХМ с получением 38; (g) взаимодействия 4 с сильным основанием в инертном растворителе,указанное сильное основание способно образовать сопряженное основание 4 и взаимодействия указанного сопряженного основания с 6 с получением 8; (h) обработки 8 при условиях проведения реакции, которые могут привести к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты 8 а, (i) взаимодействия 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12; (j) взаимодействия 12 с диазотирующим реагентом и Cu(I)Br/LiBr илиCu(I)Cl/LiCl с получением 14, (k) обработки 14 окислительным реагентом, способным окислять сульфид в сульфон 18; и (l) взаимодействия 18 с уксусной кислотой/уксусным ангидридом при условиях, которые приводят к расщеплению связи S-гетероарил и гидролизу образовавшегося ацетата с получением 20, в котором R2 обозначает бром и хлор соответственно, Ar обозначает фенил, замещенный с помощью 2 или 3 групп, независимо выбранных из группы, включающей галоген, цианогруппу и C1-С 6-галогеналкил, и(Et2O), уксусная кислота (НОАс), высокоэффективная жидкостная хроматография (ВЭЖХ), изопропанол(ТСХ), тетрагидрофуран (ТГФ), триметилсилил или Me3Si (TMC), моногидрат пара-толуолсульфоновой кислоты (TsOH или pTsOH), 4-Ме-C6H4SO2- или тозил (Ts). При использовании для алкильного фрагмента общепринятая номенклатура, включая приставки нормальный (н), изо, вторичный (втор-), третичный(трет-) и нео, обладает обычными значениями (J. Rigaudy and D.P. Klesney, Nomenclature in OrganicChemistry, IUPAC 1979, Pergamon Press, Oxford.). Способ. 5-Арилалкилтриазолоны А-2 получали конденсацией ацилгидразида A-1b с метилизоцианатом с образованием N-ацил-N-карбамоилгидразида А-1 с, который циклизовали в А-2 путем обработки метанольным раствором гидроксида калия. Схема А Эта последовательность дает возможность получить триазолоновые ННИОТ, эксперименты показывают, что реакция может быть нестабильной и непригодна для крупномасштабного синтеза. Согласно изобретению было установлен новый путь, который оказался общим, удобным и применимым для крупномасштабного синтеза. Способ, предлагаемый в настоящем изобретении, включает SNAr замещение ароматического фторида енолятом, образованным из алкил(4-алкил-5-алкилсульфанил-4 Н-[1,2,4]триазол-3-ил)ацетата.-7 016913 Полученный арилалкиловый эфир гидролизуют, декарбоксилируют и алкилтиотриазол превращают в искомый триазолон при мягких условиях проведения реакции. В публикации патента US 2005/0234236, опубликованном 20 октября 2005 г., D.J. Kertesz et al., раскрыли арилирование алкил(5-алкил-6-оксо-1,6-дигидропиридазин-3-ил)ацетатов и диалкилмалонатов 2 арилокси-3,4-дифторнитробензолами с получением производных 6-бензил-4-метил-2 Н-пиридазин-3-она и производных 3-арилоксифенилуксусной кислоты. Искомые 2-арилокси-3,4-дифторнитробензолы получали путем обработки 2,3,4-трифторнитробензола подходящим замещенным фенолом, приводящей к замещению 2-фторидных заместителей с хорошей региоселективностью. Аналогичную последовательность, приводящую к 3-арилоксифенилуксусным кислотам, представили J.P. Dunn et al. в патенте US7166730, опубликованном 23 января 2007 г. Два пути получения 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила представлены на фиг. 2. Оба пути начинаются с 3,5-дибромфторбензола (25) с использованием сходных реакций, но последовательности реакций различаются. Путь А начинается с селективного монометаллирования 25 и формилирования полученного арильного реагента Гриньяра. Фторирование альдегида приводит к введению искомого дифторметильного заместителя с образованием 30. Альдегиды и кетоны превращают и превращаются в дифторпроизводные фторирующими реагентами, такими как SF4/кислоты Льюиса, ДАТС (диэтиламинотрифторид серы), бис-(2 метоксиэтил)аминотрифторид серы в неполярном и неосновном растворителе. Арилфториды обычно значительно более лабильны, чем содержащие другие галогенидные заместители. В то время как сильные нуклеофильные реагенты, такие как вода и гидроксид, не замещают фтор,слабые нуклеофильные реагенты, такие как фенолы, имидазолы, амины, тиолы и некоторые амиды, легко замещают фтор при комнатной температуре (D. Boger et al., Biorg. Med. Chem. Lett. 2000, 10:1471-75; F.Terrier Nucleophilic Aromatic Displacement: The Influence of the Nitro Group VCH Publishers, New York, NY 1991). Замена фторида калиевой солью п-метоксибензилового спирта дает защищенный фенол. Катализируемое палладием замещение брома с использованием ферроцианидом калия и Pd(OAc)2 в присутствии ДФФФ дает искомый бензонитрил 34, у которого можно удалить защитную группу путем обработки кислотой с удалением п-метоксибензильного карбониевого иона, который захватывается анизолом, с получением 36. Сообщали, что реакция метоксида натрия с 2,3,4-трифторнитробензолом в метаноле дает неразделяющуюся смесь соответствующих 2- и 4-монометокси- и 2,4-диметоксипроизводных (P.M. O'Neill et al.,J. Med. Chem. 1994, 37:1362-70). Также описана замена орто-атома фтора в 2,4-дифторнитробензоле с помощью аминосодержащих нуклеофильных реагентов. (W.С. Lumma, Jr. et al., J. Med. Chem. 1981,24:93-101). Реакция 2,3,4-трифторнитробензола (Aldrich catalog No. 33, 836-2) с 3-дифторметил-5 гидроксибензонитрилом приводит к региоспецифическому замещению 2-фторидного фрагмента с получением 38. Специалист в данной области техники должен легко понять, что, хотя реакция описана на примере 36, легко получить большое количество замещенных фенолов или гидроксизамещенных гетероароматических соединений и ее можно использовать для получения многих других соединений для борьбы с ВИЧ. Реакцию замещения можно проводить в различных органических растворителях, включая, но не ограничиваясь только ими, простые эфиры (например, диэтиловый эфир, ТГФ, ДМЭ и диоксан) и спирты(например, изопропанол и втор-бутанол). Ясно, что исключены растворители, способные взаимодействовать с фторнитробензолом, поскольку они являются растворителями, которые могут привести к потере региохимического контроля. Так, вторичные и третичные спирты являются приемлемыми растворителями, но первичные спирты могут привести к замещению фтора. Опытный химик должен без чрезмерного количества экспериментов быть способен определить приемлемые растворители. Фенол обрабатывают основанием с получением фенолята. В способе, предлагаемом в настоящем изобретении, можно использовать соль любого щелочного металла, но реакцию обычно проводят с использованием солей лития,натрия или калия. Феноляты натрия легко получить путем обработки фенола трет-бутоксидом натрия или трет-амилатом натрия в трет-бутаноле или трет-амиловом спирте соответственно. Алкоголят натрия можно получить путем обработки спирта металлическим натрием или гидридом натрия. Феноляты калия можно получить аналогично. Альтернативно, фенол можно объединить с алкоголятом натрия в ТГФ с получением соли. Реакцию можно провести при температуре от примерно -30 до примерно 40 С без значительного нарушения региоселективности. Обычно реагенты объединяют при низкой температуре и после начального перемешивания им дают нагреться до КТ. При этих условиях протекает ароматическое нуклеофильное замещение с высокой региоселективностью по положению 2 субстрата. Альтернативный путь (фиг. 2, путь В) проводят путем проводимого сначала введения фрагмента РМВ, который затем формилируют и обрабатывают ферроцианидом калия и Pd(OAc)2 в присутствии ДФФФ с получением 44. Катализируемое кислотой дебензилирование и конденсация с 2,3,4 трифторнитробензолом дает 48. Заключительное фторирование формильного фрагмента с помощьюDEOXOFLUOR дает 36. трет-Бутил-(4-метил-5-метилсульфанил-4 Н-[1,2,4]триазол-3-ил)ацетат (4, R1 - метил, R3 - трет-8 016913 бутил) получали путем взаимодействия трет-бутилгидромалоната с карбонилдиимидазолом с образованием ацилимидазола, который ацилируют 4-метил-3-тиосемикарбазидом, который затем подвергается внутримолекулярной циклизации с образованием 24 (фиг. 2). S-Алкилирование протекает быстро при обработке 24 алкилирующим реагентом. Если, например, используют метилйодид, то специалист в данной области техники должен понимать, что другие тиоалкильные группы реагируют аналогичным образом и пример схемы реакции с участием тиометильной группы не следует рассматривать, как ограничивающий. Аналогичным образом, пример реакции приведен с использованием трет-бутилового эфира и эту группу обычно удаляют путем обработки кислотой в мягких условиях. Без затруднений можно использовать другие сложные эфиры, которые могут эффективнее гидролизоваться в щелочной среде. Взаимодействие трет-бутил (4-метил-5-метилсульфанил-4 Н-[1,2,4]триазол-3-ил)ацетата с третбутоксидом калия и 36 приводит к замещению 4-фторидного заместителя с образованием 8, который гидролизуют метансульфоновой кислотой (фиг. 1). Если реакцию проводить при повышенных температурах, то кислота декарбоксилируется с образованием 2. Каталитическое гидрирование нитрогруппы проводили в присутствии Pd на угле и ацетилацетоната ванадия, что легко дает соответствующий амин,который можно превратить в соответствующий бромидный или хлоридный заместитель путем диазотирования амина трет-бутилнитритом в присутствии бромида меди (I) и LiBr (или CuCl/LiCl с получением соответствующего хлорида), что дает 14 (R1 - Me, R2 - Br и Ar - 3-циано-5-дифторметилфенил). Заключительное образование триазолонового кольца завершали путем окисления тиометильного производного в сульфоксид. Реакции S-окисления можно провести с использованием 30% водного раствора пероксида водорода или с помощью других окислительных реагентов, таких как, NaIO4, третбутоксихлорид, ацилнитриты, перборат натрия и надкислоты. Сульфиды можно окислить в сульфоксиды, которые затем можно окислить в сульфоны путем прибавления еще одного эквивалента пероксида водорода, KMnO4, пербората натрия, гидроперсульфата калия, надкислот или аналогичных реагентов. Если содержится достаточное количество окислительного реагента, то сульфиды можно превратить непосредственно в сульфоны без выделения сульфоксидов. Обработка сульфона уксусным ангидридом и уксусной кислотой приводит к превращению метилсульфона в ацетат и гидролизу промежуточного ацетата с получением 20. Пример 1. 3-Дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрил (фиг. 2; путь А). Стадия 1. К раствору изопропилмагнийхлорида в ТГФ (500 мл 2 М раствора в ТГФ, 1,0 моль) и ТГФ(200 мл) добавляли раствор 3,5-дибромфторбензола (25; 200 г, 0,79 моль) в ТГФ (100 мл), поддерживая температуру равной примерно 0 С. После промывания с помощью ТГФ (320 мл) смесь выдерживали примерно при 0 С в течение 2 ч и затем нагревали примерно до 20 С и выдерживали в течение 0,5 ч. Отбирали пробу реакционной смеси и анализировали с помощью ВЭЖХ и затем смесь охлаждали примерно до 0 С. В течение 0,5 ч добавляли ДМФ, поддерживая температуру равной примерно 0 С. Смесь выдерживали примерно при 0 С в течение 1,5 ч и затем в течение ночи ее медленно нагревали примерно до 20 С. После отбора пробы реакционной смеси и анализа с помощью ВЭЖХ смесь разбавляли гептаном(200 мл) и затем концентрированной HCl (120 мл), разбавленной водой до объема 360 мл. Добавляли концентрированную HCl (50 мл) для обеспечения рН 7. Органическую фазу отделяли, промывали водой(400 мл) и выпаривали досуха и получали 160,8 г (100,5%) соединения 28 в виде желтого масла, которое при выдерживании затвердевало. Стадия 2. К раствору соединения 28 (144,1 г, 0,71 моль) в ТГФ, охлажденному примерно до 0 С,одной порцией добавляли DEOXO-FLUOR (бис-(2-метоксиэтил)аминтрифторид серы; 218 мл, 261,6 г,1,18 моль). Смесь нагревали до КТ, выдерживали в течение 3 ч и за протеканием реакции следили с помощью ВЭЖХ. Избыток реагента нейтрализовали путем переноса реакционной смеси в насыщенный раствор NaHCO3 (1200 мл). Органическую фазу отделяли, промывали 1,5 н. раствором HCl (1000 мл),смесью воды (250 мл) и насыщенного раствора NaHCO3 (250 мл) и в заключение водой (500 мл). Органическую фазу концентрировали и получали масло, которое подвергали фракционной перегонке в вакууме,и получали 98,1 г (61,3%) соединения 30. Стадии 3-5. К смеси трет-бутоксида калия (28,7 г, 255,5 моль) и ТГФ (250 мл) медленно добавляли пара-метоксибензиловый спирт (36,8 г, 266,7 ммоль). После перемешивания в течение примерно 15 мин добавляли соединение 30 (50,0 г, 222,2 ммоль) и реакционную смесь нагревали примерно до 65 С. После перемешивания при 65 С в течение 2 ч реакционную смесь анализировали с помощью ВЭЖХ. После охлаждения до КТ добавляли смесь насыщенного раствора NaHCO3 (150 мл) и воды (150 мл). Добавляли толуол (300 мл), органическую фазу отделяли и промывали смесью насыщенного раствора NaHCO3 (75 мл) и воды (75 мл). Фильтрование через тонкий фильтр и концентрирование в вакууме давали 83,9 г неочищенного соединения 32 в виде масла, которое использовали без дополнительной очистки. К раствору неочищенного соединения 32 в NMP (180 мл) добавляли ферроцианид калия (31,1 г,84,44 ммоль) и Na2CO3 (23,55 г, 222,2 ммоль). Полученную взвесь тщательно дегазировали путем обработки с помощью нескольких циклов вакуумирование/продувание азотом. Взвесь нагревали примерно до 100 С и добавляли раствор Pd(OAc)2 (150 мг, 0,67 ммоль) и ДФФФ (505 мг, 0,91 ммоль) в дегазирован-9 016913 ном NMP (20 мл). Смесь нагревали примерно при 130 С в течение примерно 3 ч. Анализ с помощью ВЭЖХ показывал, что в смеси осталось примерно 5% исходного вещества. Повторно добавлялиPd(OAc)2 (50 мг, 0,22 ммоль) и нагревание продолжали при 130 С в течение 1,5 ч, после чего анализ с помощью ВЭЖХ указывал на полное превращение исходных веществ. После охлаждения смеси добавляли толуол (400 мл) и насыщенный раствор сульфита натрия(10 мл) и смесь нагревали примерно при 40 С в течение примерно 1 ч. Добавляли Solka-floc (функционирующая целлюлоза) (10 г) и смесь фильтровали через слой Solka-floc и отфильтрованный осадок промывали толуолом (всего примерно 100 мл). Фильтрат последовательно промывали разбавленным раствором сульфита натрия (1400 мл) и водой (2200 мл). Объединенные водные фазы экстрагировали толуолом(1100 мл) и фазу, содержащую толуол, подвергали обратной экстракции водой (250 мл). Объединенные органические фазы фильтровали через тонкий фильтр и концентрировали в вакууме и получали 70,4 г соединения 34 в виде темного масла (70,4 г), которое использовали на следующей стадии без дополнительной очистки. К раствору неочищенного соединения 36 в толуоле (190 мл) и анизоле (65 мл) добавляли ТФК(25,3 г, 222,2 ммоль). Реакционную смесь нагревали примерно до 65 С и перемешивали в течение примерно 2 ч до завершения реакции по данным ВЭЖХ. Смесь перегоняли в вакууме для удаления большей части ТФК. После охлаждения смесь дважды экстрагировали примерно 10% раствором Na2CO3 (300 мл,затем 150 мл). Объединенные водные фазы подкисляли до рН 5,5 концентрированной HCl и экстрагировали с помощью EtOAc (2200 мл). Объединенные органические фазы промывали водой (1150 мл),фильтровали через тонкий фильтр и растворитель отгоняли вакууме и заменяли на толуол. Раствор концентрировали до объема примерно равного 200 мл, затем медленно добавляли гептан (200 мл) и смесь нагревали до 80 С. Смесь охлаждали до КТ, выдерживали в течение ночи, фильтровали и промывали 50% раствором гептана в толуоле (примерно 30 мл). Выделенный продукт сушили в вакууме примерно при 60 С и получали 29,0 г (выход 77,2% за 3 стадии) соединения 36. Стадия 6. К раствору соединения 36 (0,80 г, 4,73 ммоль) в ТГФ (4,0 мл) при 0 С шприцевым насосом медленно (в течение примерно 4,5 ч) добавляли смесь соединения 37 (0,57 мл, 0,88 г, 4,97 ммоль) иK2CO3 (1,96 г, 14,2 ммоль) в ТГФ (2,4 мл). Реакционную смесь выдерживали при 0 С до завершения реакции. Добавляли уксусную кислоту (0,82 мл, 0,85 г, 14,2 ммоль), поддерживая температуру равной 5 С,затем воду (4,0 мл) и смесь нагревали до КТ. Фазы разделяли. Затем органический слой промывали насыщенным раствором NaCl (5 мл), концентрировали и продукт очищали с помощью хроматографии наSiO2, элюируя смесью 20% EtOAc/гексан, и получали 1,24 г (80%) соединения 38 в виде масла, которое кристаллизовалось при выдерживании. Аналитическую пробу получали путем перекристаллизации из смеси ИПС/гексан. Пример 2. 3-Дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрил (фиг. 2; путь В). Стадия 7. К смеси трет-бутоксида калия (10,0 кг, 89,4 моль) и ТГФ (78 л) медленно добавляли параметоксибензиловый спирт (12,4 кг, 89,8 моль), давая температуре реакционной смеси повыситься примерно до 35 С. После перемешивания при 35-40 С в течение 0,5 ч медленно добавляли соединение 25(21,4 кг, 84,3 моль), давая температуре реакционной смеси повыситься примерно до 60 С. После перемешивания при 60 С в течение 2 ч за протеканием реакции следили с помощью ВЭЖХ. После охлаждения смеси до КТ добавляли НОАс (примерно 600 г) и затем воду (30 л) и фазы разделяли. Водную фазу экстрагировали с помощью EtOAc (20 л) и объединенные органические фазы промывали смесью насыщенного рассола (10 кг) и воды (10 л). Органическую фазу концентрировали в вакууме (примерно 27 дюймов рт.ст., температура кожуха примерно 65 С) и получали масло. Добавляли МеОН (примерно 43 кг) и получали двухфазную смесь, которую выдерживали при 45-50 С. Продукт осаждался и взвесь перемешивали до получения однородной консистенции. После охлаждения до КТ и выдерживания в течение ночи продукт отфильтровывали, промывали с помощью МеОН (9,8 кг) и сушили в вакууме при 50 С и получали 26,06 кг соединения 40. Вещество, оставшееся в реакторе, и осадок на фильтре растворяли в ТГФ (примерно 10 л) и раствор выпаривали досуха в роторном испарителе и получали еще 3,44 кг(общий выход 94%). Стадия 8. К раствору соединения 40 (387 г, 1,04 моль) в ТГФ (1,2 л) при КТ в течение примерно 15 мин добавляли изопропилмагнийхлорид (0,7 л 2 М раствора в ТГФ, 1,4 моль), поддерживая температуру равной от 20 до 25 С (незначительное выделение тепла). После выдерживания в течение 3-4 ч отбирали пробу реакционной смеси для установления степени протекания реакции (ВЭЖХ). Смесь охлаждали в бане со смесью соль/лед ( -5 С) и в течение нескольких минут добавляли ДМФ (250 мл, 3,2 моль)(при добавлении происходит выделение тепла и следует поддерживать температуру 30 С). После выдерживания смеси в течение 30 мин реакцию останавливали путем добавления смеси третбутилметилового эфира (1 л) и 1 М раствора H2SO4 (2 л). Органическую фазу отделяли и промывали насыщенным раствором NaHCO3 (1 л), водой (1 л), сушили (MgSO4), фильтровали и выпаривали досуха. Продукт растворяли в EtOAc (0,4 л) и гептане (0,8 л), добавляли SiO2 (340 г, 230-400 меш) и перемешивали в течение 2 ч. SiO2 отфильтровывали, промывали 33% раствором EtOAc в гептане (0,6 л) и выпари- 10016913 вали досуха и получали 345 г (выход 107%) соединения 42. Стадия 9. К раствору соединения 42 (333 г, 1,037 моль) в NMP (1,7 л) добавляли безводный порошкообразный ферроцианид калия (115 г, 0,312 моля, высушенный в вакууме при 100 С), безводныйNa2CO3 (110 г, 1,037 моль), Pd(OAc)2 (0,23 г, 0,001 моль) и ДФФФ (1,15 г, 0,002 моль). Реакционную смесь обрабатывали с помощью по меньшей мере 3 циклов вакуумирование/продувание азотом и затем нагревали при 130 С до завершения реакции по данным ВЭЖХ (в течение от 3 до 6 ч). Охлажденную реакционную смесь фильтровали через слой целита, добавляли ТВМЕ (трет-бутилметиловый эфир) (4 л) и затем смесь промывали водой (31 л). Органическую фазу обесцвечивали активированным древесным углем (25 г). После замены растворителя путем обмена с EtOAc (0,4 л) и гексанами (0,4 л) смесь охлаждали примерно до 0 С. Продукт отфильтровывали, промывали смесью 20% EtOAc/гексаны (20,2 л) и сушили в вакууме при 60 С в течение ночи и получали 223 г (81%) соединения 44. Стадия 10. Смесь соединения 44 (201 г, 752 ммоль), толуола (603 мл) и анизола (201 мл) нагревали примерно до 50 С. Одной порцией добавляли ТФК (90,0 г, 790 ммоль) и полученную смесь нагревали примерно до 65 С и выдерживали в течение примерно 1 ч. В ходе реакции возможна кристаллизация продукта, что связано с повышением температуры примерно на 10 С. За протеканием реакции следили с помощью ВЭЖХ и после завершения реакции смесь охлаждали до КТ. Продукт отфильтровывали, последовательно промывали толуолом (2x50 мл) и гептаном (1x100 мл), сушили в вакууме при 70 С и получали 106,1 г (95,9%) соединения 46. Стадия 11. Раствор соединения 46 (95,0 г, 646 ммоль) в ТГФ (665 мл) охлаждали до -10 С и в течение 15 мин добавляли раствор трет-бутоксида калия в ТГФ (646 мл 1 М раствора, 646 ммоль). Полученную взвесь выдерживали при 0 С в течение 45 мин, охлаждали до -10 С и затем быстро добавляли соединение 37 (182,9 г, 1,03 моль). Взвесь в течение 3 ч нагревали до 10 С, при этом смесь становилась гомогенной. Смесь концентрировали в вакууме до 1/3 ее объема и затем при интенсивном перемешивании выливали в холодную воду (2,4 л). После перемешивания в течение 30 мин твердое вещество отфильтровывали, промывали водой (примерно 150 мл) и частично сушили в вакууме при 45 С. Затем твердое вещество при 0 С растирали с Et2O в количестве, достаточном для получения поддающейся перемешиванию взвеси (примерно 150 мл). Взвесь фильтровали, промывали холодным Et2O (всего примерно 150 мл) и затем сушили в вакууме при 45 С и получали 141,4 г (72,0%) соединения 48. Стадия 12. К раствору соединения 48 (140,0 г, 460 ммоль) в ДХМ (1,4 л) добавляли DEOXOFLUOR (203,6 г, 920 ммоль), поддерживая температуру равной от 20 до 30 С. После выдерживания смеси в течение ночи реакцию останавливали, по каплям добавляя воду (380 мл), при охлаждении с помощью бани с температурой равной -15 С. Фазы разделяли и органическую фазу промывали водой(380 мл), затем насыщенным раствором NaHCO3 (2380 мл). ДХМ выпаривали при пониженном давлении и остаток переносили в ИПС (700 мл) и затем добавляли 25% раствор бисульфита натрия (115 мл). Мутную смесь выдерживали при 45 С в течение 30 мин и затем примерно 70% объема ИПС отгоняли при пониженном давлении и заменяли водой. После перемешивания в течение ночи смесь кристаллов и затвердевших комков отделяли фильтрованием, измельчали пестиком в ступке и на фильтре промывали водой (примерно 250 мл). После частичной сушки в вакууме при 50 С твердое вещество растирали с минимальным количеством холодного Et2O (примерно 80 мл; 0 С), фильтровали и промывали холоднымEt2O (примерно 50 мл). Продукт сушили в вакууме при 50 С и получали 116,5 г (77,4%) соединения 38. Пример 3. трет-Бутиловый эфир (4-метил-5-метилсульфанил-4 Н-[1,2,4]триазол-3-ил)уксусной кислоты. Стадии 1 и 2. К раствору трет-бутилгидромалоната (93,7 г, 585 ммоль) в MeCN (1,6 л) при КТ в течение 20 мин добавляли 1,1'-карбонилдиимидазол (93,9 г, 579 ммоль). Через 1 ч в течение примерно 20 мин добавляли 4-метилтиосемикарбазид (92,3 г, 878 ммоль). После перемешивания в течение 1 ч взвесь кипятили с обратным холодильником в течение 30 ч и затем охлаждали до КТ. Концентрирование смеси в вакууме и замена растворителя на воду давали взвесь. После выдерживания при 0 С продукт отфильтровывали, промывали водой и сушили в вакууме при 50 С и получали 98,86 г (73,7%) соединения 24, которое перекристаллизовывали из EtOAc. Стадия 3. Взвесь соединения 24 (125,0 г, 550 ммоль) в MeCN (600 мл) обрабатывали метилйодидом(93,7 г, 660 ммоль). После перемешивания в течение ночи раствор выпаривали и получали темнокоричневое масло. Остаток растворяли в ДХМ (250 мл) и последовательно промывали насыщенным раствором NaHCO3 (75 мл), 25% раствором бисульфита натрия (75 мл), водой (75 мл) и насыщенным раствором NaCl (75 мл). Органическую фазу сушили (Na2SO4), фильтровали и выпаривали и получали 128,8 г (96,3%) соединения 4 (R1 - Me и R3 - трет-Bu) в виде масла, которое затвердевало при выдерживании при КТ. Пример 4. Стадии 1-3. К раствору соединения 6 (Ar - 3-циано-5-дифторметилфенил, 18,5 г, 56,7 ммоль) и соединения 4 (16,55 г, 68,0 ммоль) в ТГФ (93 мл) медленно добавляли трет-бутоксид калия (113,5 мл 1 М раствора в ТГФ, 113,4 ммоль), поддерживая температуру равной от -20 до -10 С. Смесь нагревали до 0 С и добавляли НОАс (6,5 мл, 113,4 ммоль), затем воду (110 мл). После нагревания смеси до КТ органиче- 11016913 скую фазу отделяли. Большую часть ТГФ выпаривали в вакууме, добавляли MeCN (65 мл) и раствор(примерно 70 мл) фильтровали через слой целита. Добавляли метансульфоновую кислоту (11 мл,170 ммоль) и раствор кипятили с обратным холодильником до завершения реакции (в течение примерно 4 ч). После охлаждения смесь последовательно разбавляли с помощью EtOAc (60 мл), водой (60 мл) и количеством насыщенного раствора K2CO3, достаточным для доведения рН примерно до 7. Водную фазу отделяли и экстрагировали с помощью EtOAc (20 мл). Объединенные органические слои фильтровали через слой целита и добавляли катализатор Pd/C (тип Johnson Matthey A503023-5, 3,0 г) и ванадилацетилацетонат (0,77 г, 2,8 ммоль). Смесь перемешивали в атмосфере водорода до завершения восстановления нитрогруппы. Добавляли целит (5 г) и затем смесь фильтровали через слой целита (10 г) и осадок на фильтре промывали с помощью MeCN (520 мл). Фильтрат промывали смесью насыщенного раствораNaCl (40 мл) и насыщенного раствора NaHCO3 (40 мл), затем насыщенным раствором NaCl (30 мл). Органическую фазу концентрировали и продукт кристаллизовали из EtOAc (40 мл). К взвеси добавляли гексан (10 мл), охлаждали до 0 С и выдерживали в течение по меньшей мере 2 ч. Продукт отфильтровывали, промывали 17% раствором гексана в EtOAc (310 мл) и сушили в вакууме при 55 С и получали 16,86 г (выход 71%) соединения 12. Стадия 4. В колбе, закрытой алюминиевой фольгой, смесь, содержащую соединение 12 (Ar - 3 циано-5-дифторметилфенил, 41,45 г, 98,8 ммоль), Cu(I)Br (57,86 г, 395 ммоль), LiBr (26,54 г, 296 ммоль) и MeCN (620 мл), нагревали до 58 С. Через 15 мин в течение 30 мин добавляли трет-бутилнитрит(20,04 мл, 198 ммоль), поддерживая температуру равной примерно 58 С. После завершения реакции смесь концентрировали до минимального объема (собирали примерно 600 мл растворителя). Добавляли ДХМ (400 мл), затем 3 М раствор HCl (200 мл). Органическую фазу отделяли и промывали 3 М раствором HCl (5100 мл). После нейтрализации смеси водным раствором K2CO3 до рН, равного примерно 7,органический слой промывали 6% раствором тиосульфата натрия (690 г), насыщенным растворомNaHCO3 (250 мл), насыщенным раствором NaCl (250 мл) и затем фильтровали через слой целита. Добавляли пара-толуолсульфоновую кислоту (21 г, 108,7 ммоль) и растворитель выпаривали при пониженном давлении и заменяли на EtOH (250 мл). Объем взвеси уменьшали примерно до 125 мл путем выпаривания при пониженном давлении. Взвесь охлаждали до КТ и выдерживали в течение по крайней мере 2 ч. Продукт отфильтровывали и твердое вещество промывали с помощью EtOH (250 мл) и сушили при 65 С в вакууме и получали 43,0 г (66,4%) тозилата соединения 14 (R2 - Br). Стадия 5. К смеси соединения 14 (Ar - 3-циано-5-дифторметилфенил, R2 - Br; 25,0 г, 38 ммоль) и НСО 2 Н (4,49 г, 114,4 ммоль) в ДХМ (250 мл) в течение 5 мин добавляли 30% раствор Н 2 О 2 (25,95 г,228,8 ммоль) и смесь кипятили с обратным холодильником до завершения реакции. Реакцию останавливали раствором сульфита натрия (12,5 г, 99,1 ммоль) в воде (75 мл) и 60% раствором K2CO3 (примерно 25 мл) значение рН доводили примерно до 10. Водную фазу отделяли и экстрагировали с помощью ДХМ(2100 мл). Объединенные органические экстракты промывали насыщенным раствором NaCl (200 мл) и фильтровали через слой целита. Растворитель заменяли на ИПС и смесь концентрировали до объема равного примерно 200 мл. Добавляли гексан (50 мл) и после образования кристаллов смесь выдерживали при 60 С в течение 2 ч. Взвесь охлаждали до 25 С и выдерживали в течение 2 ч. Продукт отфильтровывали, промывали 25% раствором гексана в ИПС (325 мл) и сушили в вакууме при 65 С и получали 17,7 г (90%) соединения 18. Стадия 6. Смесь соединения 18 (Ar - 3-циано-5-дифторметилфенил, R2 - Br; 9,29 кг, 18,1 моль),Ас 2 О (3,25 кг, 31,8 моль) и НОАс (36,0 кг) нагревали при температуре, равной от 105 до 110 С. Смесь выдерживали в течение примерно 5 ч и за протеканием реакции следили с помощью ВЭЖХ. После завершения реакции смесь охлаждали до температуры равной примерно от 35 до 45 С и добавляли воду(7,5 л). После выдерживания смеси примерно при 45 С в течение 8 ч смесь анализировали с помощью ВЭЖХ. Смесь охлаждали до температуры равной от 15 до 25 С, разбавляли водой (168 л) и затем экстрагировали с помощью EtOAc (102 кг). Органическую фазу последовательно промывали водой (47 л), 10% раствором NaHCO3 (277 л) и водой (19 л). Органическую фазу концентрировали при атмосферном давлении до объема, равного примерно 33 л, и смесь охлаждали до температуры, равной от 18 до 25 С. После начала кристаллизации медленно добавляли гептан (3,9 кг). После охлаждения до 2 С продукт отфильтровывали, промывали смесью EtOAc и гептана состава 1:1 и затем сушили в вакууме при температуре, равной от 50 до 60 С, и получали 6,12 кг (75%) соединения 20. Отличительные признаки, раскрытые в приведенном выше описании или в приведенной ниже формуле изобретения, или в прилагаемых чертежах, выраженные в конкретных формах или с помощью средств для осуществления раскрытого назначения, или методика или способ для достижения раскрытого результата, если это целесообразно, могут по отдельности или в виде любой комбинации таких отличительных признаков быть использованы для осуществления настоящего изобретения в его различных формах. Приведенное выше изобретение описано довольно подробно для иллюстрации и примера с целью обеспечения ясности и понимания. Для специалиста в данной области техники должно быть очевидно,что в объеме прилагаемой формулы изобретения возможны изменения и модификации. Поэтому следует- 12016913 понимать, что приведенное выше описание является иллюстративным, а не ограничивающим. Поэтому объем настоящего изобретения следует определять не с учетом приведенного выше описания, а по прилагаемой формуле изобретения вместе со всем объемом эквивалентов, на которые распространяется такая формула изобретения. Все патенты, заявки на патенты и публикации, цитированные в настоящем изобретении, включены в него в качестве ссылки во всей своей полноте и для всех объектов в такой степени, как если бы по отдельности был отмечен каждый отдельный патент, заявка на патент или публикация. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы 2 в которой Ar обозначает фенил, замещенный с помощью 2 или 3 групп, независимо выбранных из группы, включающей галоген, цианогруппу и C1-С 6-галогеналкил;(a) взаимодействие 4 с сильным основанием в инертном растворителе с образованием сопряженного основания 4 и взаимодействие указанного сопряженного основания с 6 с получением 8;(b) обработку 8 при условиях, которые приводят к гидролизу сложного эфира с декарбоксилированием полученной карбоновой кислоты с получением 2. 2. Способ по п.1, в котором Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5 дифторметилфенил, R1 обозначает метил и R3 обозначает трет-Bu. 3. Способ по п.2, в котором указанное сильное основание представляет собой трет-бутоксид калия,указанный инертный растворитель представляет собой тетрагидрофуран (ТГФ) и указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником. 4. Способ по п.3, в котором Ar обозначает 3-циано-5-дифторметилфенил. 5. Способ получения соединения формулы 12 и 14, который включает стадии по п.1 и дополнительно включает следующие стадии:(a) взаимодействие 2 с восстановительным реагентом, способным селективно восстанавливать нитрогруппу, с получением 12;(b) взаимодействие 12 с диазотирующим реагентом и Cu(I)Br/LiBr или Cu(I)Cl/LiCl с получением 14, в котором R2 обозначает бром и хлор соответственно. 6. Способ по п.5, в котором Ar обозначает 3-циано-5-дифторметилфенил, R1 обозначает метил, R2 обозначает бром, R3 обозначает трет-бутил, указанное сильное основание представляет собой третбутоксид калия, указанный инертный растворитель представляет собой тетрагидрофуран (ТГФ), указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником, указанный восстановительный реагент представляет собой водород,Pd/C и ванадилацетилацетонат (VO(acac)2), указанный диазотирующий реагент представляет собой третбутилнитрит и указанная соль меди(I) представляет собой Cu(I)Br. 7. Способ, который включает стадии по п.6 и дополнительно включает стадию превращения 14, гдеR2 обозначает Br, в тозилат. 8. Способ получения соединения формулы 20,который включает стадии по п.6 и дополнительно включает следующие стадии:(a) обработку 14 окислительным реагентом, способным окислять сульфид в сульфон 18;(b) взаимодействие 18 с уксусной кислотой/уксусным ангидридом при условиях, которые приводят к расщеплению связи S-алкил и гидролизу образовавшегося сульфона, с получением 20. 9. Способ по п.8, в котором Ar обозначает 3-циано-5-дифторметилфенил, R1 обозначает метил, R2 обозначает бром, R3 обозначает трет-бутил, указанное сильное основание представляет собой третбутоксид калия, указанный инертный растворитель представляет собой тетрагидрофуран (ТГФ), указанные условия гидролиза включают использование метансульфоновой кислоты в ацетонитриле при кипячении с обратным холодильником, указанный восстановительный реагент представляет собой водород,Pd/C и ванадилацетилацетонат (VO(acac)2), указанный диазотирующий реагент представляет собой третбутилнитрит, указанная соль меди(I) представляет собой Cu(I)Br и указанный окислительный реагент представляет собой метахлорпербензойную кислоту (МХПБК). 10. Соединение формулы 4 в которой R1 и R3 независимо обозначают С 6-С 10-алкил. 11. Соединение по п.10, в котором R1 обозначает метил и R3 обозначает трет-бутил. 12. Способ получения соединения по п.10, который включает следующие стадии:(a) взаимодействие моноэфира малоновой кислоты 19 с карбонилдиимидазолом в инертном растворителе с образованием эфира 3-имидазол-1-ил-3-оксопропионовой кислоты (21);(b) взаимодействие ацилимидазола, полученного на стадии (а), с тиосемикарбазидом 22;(c) обработку полученного 5-тиооксо-4,5-дигидро-1 Н-[1,2,4]триазол-3-карбоксилата 24 алкилирующим реагентом с получением 4, в котором R1 и R3 обозначают С 6-С 10-алкил. 13. Способ по п.12, в котором указанный моноэфир малоновой кислоты представляет собой третбутилгидромалонат и указанный алкилирующий реагент представляет собой метилйодид. 14. Способ получения 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (38), который включает стадии по п.8 и дополнительно включает следующие стадии:(f) обработку раствора 34 и анизола трифторуксусной кислотой при температурах, достаточных для расщепления связи О-бензил, и с получением 3-дифторметил-5-гидроксибензонитрила (36);(g) обработку раствора 36 в тетрагидрофуране (ТГФ) 1,2,3-трифтор-4-нитробензолом (37) с получением 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (38). 15. Способ получения 3-дифторметил-5-(2,3-дифтор-6-нитрофенокси)бензонитрила (38), который включает стадии по п.8 и дополнительно включает следующие стадии:(e) обработку раствора 44 и анизола трифторуксусной кислотой при температурах, достаточных для расщепления связи О-бензил, с получением 3-формил-5-гидроксибензонитрила (46);
МПК / Метки
МПК: C07D 249/10, C07C 205/11
Метки: способ, получения, триазолонов
Код ссылки
<a href="https://eas.patents.su/16-16913-sposob-polucheniya-triazolonov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения триазолонов</a>
Способ получения защищенного 4-аминометилпирролидин-3-она, промежуточные соединения и способ получения 3-аминометил-4-алкоксииминопирролидина

Номер патента: 2499
Опубликовано: 27.06.2002
Авторы: Ким Вон Суп, Моон Кванг Юл, Чанг Джей Хиок, Ли Тае Хи
МПК: C07D 207/24
Метки: защищенного, 4-аминометилпирролидин-3-она, соединения, промежуточные, 3-аминометил-4-алкоксииминопирролидина, получения, способ
Формула / Реферат:
1. Способ получения соединения формулы (1) в которой Р1 и Р2 обозначают защитные группы; включающий а) восстановление соединения формулы (5) где Р1 такой, как определено для формулы (1); в присутствии катализатора никель Ренея в растворителе в атмосфере водорода в присутствии одной или более добавок с получением соединения формулы (6) где Р1 такой, как определено для формулы (1); b) защиту аминогруппы в присутствии одного или более оснований...
Способ получения дихлорпропанола, способ получения эпихлоргидрина, способ получения эпоксидных смол и применение оборудования, обладающего коррозионной стойкостью, в способе получения дихлорпропанола

Номер патента: 14241
Опубликовано: 29.10.2010
Авторы: Краффт Филипп, Франк Кристиан, Жильбо Патрик, Бальтазар Доминик, Сметс Валентин
МПК: C07C 31/36, B01J 19/02, C07C 29/62...
Метки: эпоксидных, способе, стойкостью, обладающего, коррозионной, дихлорпропанола, эпихлоргидрина, применение, способ, смол, получения, оборудования
Формула / Реферат:
1.Способ получения дихлорпропанола, содержащий:(a) стадию, на которой глицерин или сложный эфир глицерина или их смесь вводят во взаимодействие с агентом хлорирования, содержащим хлороводород,(b) по меньшей мере одну другую стадию, осуществляемую на оборудовании, выполненном или имеющем покрытие из материалов, обладающих стойкостью по отношению к агенту хлорирования, в условиях осуществления этой стадии,причем другая стадия является стадией...
Катализатор для получения сложных эфиров,способ получения сложного эфира и способ получения сложного полиэфира с участием такого катализатора

Номер патента: 11171
Опубликовано: 27.02.2009
Авторы: Партридж Мартин Грэхэм, Хэнратти Алан Джозеф, Макинтош Кэлам Гарри
МПК: C08G 63/85, B01J 31/04
Метки: участием, такого, катализатор, способ, сложного, катализатора, эфира, эфиров,способ, получения, полиэфира, сложных
Формула / Реферат:
1. Катализатор для получения сложного эфира в реакции этерификации, состоящий из продукта взаимодействия: a) соединения титана, циркония или гафния; b) 2-оксикарбоновой кислоты; c) четвертичного аммониевого соединения, выбранного из группы, состоящей из гидроксида тетраэтиламмония и гидроксида тетраметиламмония, и d) соединения цинка. 2. Катализатор по п. 1, в котором соединением титана, циркония или гафния является алкоголят, имеющий формулу...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Крок Вероник, Ларкин Джон Патрик, Колладан Колетт, Руссель Патрик
МПК: C07D 487/04
Метки: производные, терапевтически, получения, 10-диоксо-6н-пиридазино, кислоты, применение, способ, 1,2-а, активных, соединений, октагидро-6, диазепин-1-карбоновой, 1,2
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Стабильная полиморфная модификация флибансерина, способ её промышленного получения и её применение для получения лекарственных средств

Номер патента: 6400
Опубликовано: 29.12.2005
Авторы: Эцайя Антоине, Дубини Энрика, Шнайдер Генрих, Бомбарда Карло
МПК: A61K 31/495, A61P 25/00, C07D 403/06...
Метки: применение, стабильная, полиморфная, способ, модификация, лекарственных, получения, средств, флибансерина, промышленного
Формула / Реферат:
1. Кристаллическая полиморфная модификация A (форма A) флибансерина формулы 1 у которой эндотермический максимум при ее термическом анализе дифференциальной сканирующей калориметрией приходится на температуру 161шC. 2. Флибансерин формулы 1, включающий форму A по п.1. 3. Способ получения флибансерина формулы 1 по п.1 или 2 отличающийся тем, что на первой стадии бензимидазолон формулы 2 в которой R обозначает аминозащитную группу, подвергают...
Предыдущий патент: Производные арилоазол-2-илцианоэтиламина, способ их получения и способ их применения
Следующий патент: Способ получения 2,6-диэтил-4-метилфенилуксусной кислоты
Случайный патент: Способ и устройство для обработки трехкомпонентных сейсмических данных