Способ получения 5-(2-{[6-(2,2-дифтор-2-фенилэтокси)гексил]амино}-1-гидроксиэтил)-8-гидроксихинолин-2(1h)-oha







Формула / Реферат
1. Способ получения соединения 5-(2-{[6-(2,2-дифтор-2-фенилэтокси)гексил]амино}-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она формулы (I) или его фармацевтически приемлемой соли

который включает:
а) реакцию в ксилольном растворителе соединения формулы (V)

где P1 и Р2 представляют собой защитные группы для гидроксигрупп и L представляет собой уходящую группу,
с 6-(2,2-дифтор-2-фенилэтокси)гексан-1-амином формулы (IV)

с получением соединения формулы (III)

b) осуществление стадии снятия защитной группы P1 и стадии снятия защитной группы Р2 для удаления защитных групп P1 и Р2 и получения соединения формулы (I).
2. Способ по п.1, в котором стадия (b) включает осуществление указанной стадии снятия защитной группы Р2 в соединении формулы (III) с получением соединения формулы (II)

в котором Р1 является таким, как определено выше; и осуществление указанной стадии снятия защитной группы Р1 в соединении формулы (II) с получением соединения формулы (I)

3. Способ по п.1 или 2, в котором:
(i) стадию снятия защитной группы Р2 осуществляют при температуре в диапазоне около 30-60°С в течение до 8 ч и/или
(ii) стадию снятия защитной группы Р1 осуществляют в присутствии растворителя, который представляет собой уксусную кислоту или смесь уксусной кислоты со спиртом или сложным эфиром.
4. Способ по любому из предшествующих пунктов, в котором:
(а) Р1 представляет собой бензильную группу, и стадию снятия защитной группы Р1 осуществляют гидрированием и/или
(b) Р2 представляет собой трет-бутилдиметилсилильную группу, и стадию снятия защитной группы P2 осуществляют реакцией с тригидратом фторида тетра-н-бутиламмония или с хлоридом водорода.
5. Способ по любому из предшествующих пунктов, в котором L представляет собой бром.
6. Способ по любому из предшествующих пунктов, в котором стадию снятия защитной группы P2 осуществляют с помощью тригидрата фторида тетра-н-бутиламмония в тетрагидрофуране.
7. Способ по любому из предшествующих пунктов, в котором стадию снятия защитной группы Р2 осуществляют при температуре 40-50°С в течение периода времени, не превышающего 6 ч.
8. Способ по п.7, в котором период времени не превышает 4 ч.
9. Способ по любому из предшествующих пунктов для получения геминападисилатной или мезилатной соли соединения формулы (I).
10. Способ по п.9 для получения геминападисилатной соли соединения формулы (I), в котором после стадии (b) добавляют нафтален-1,5-дисульфоновую кислоту без выделения 5-[2-{[6-(2,2-дифтор-2-фенилэтокси)гексил]амино}-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она формулы (I).
11. Способ по п.3, в котором промежуточное соединение формулы (II), полученное со стадии снятия защитной группы Р2, очищают кристаллизацией с тетрагидратом 1,5-нафталендисульфоновой кислоты в этаноле.
12. Способ по любому из предшествующих пунктов, в котором P1 представляет собой бензил, и стадию снятия защитной группы P1 осуществляют гидрированием в присутствии катализатора палладия на угле в количестве менее 10 вес.% относительно количества соединения формулы (II).
13. Способ по п.12, в котором используемое количество катализатора составляет менее 5%.
14. Способ по любому из предшествующих пунктов, в котором стадию снятия защитной группы P1 осуществляют в присутствии растворителя, которым является уксусная кислота или смесь уксусной кислоты со спиртом или сложным эфиром.
15. Способ по п.14, в котором растворителем является уксусная кислота или смесь метанол/уксусная кислота (1:1).
16. Способ по п.14, в котором растворителем является смесь метанол/уксусная кислота (1:1).
17. Способ по любому из предшествующих пунктов, в котором:
(i) стадию снятия защитной группы P2 осуществляют при температуре в диапазоне 30-60°С в течение периода до 8 ч и
(ii) стадию снятия защитной группы P1 осуществляют в присутствии растворителя, которым является уксусная кислота или смесь уксусной кислоты со спиртом или сложным эфиром.
Текст
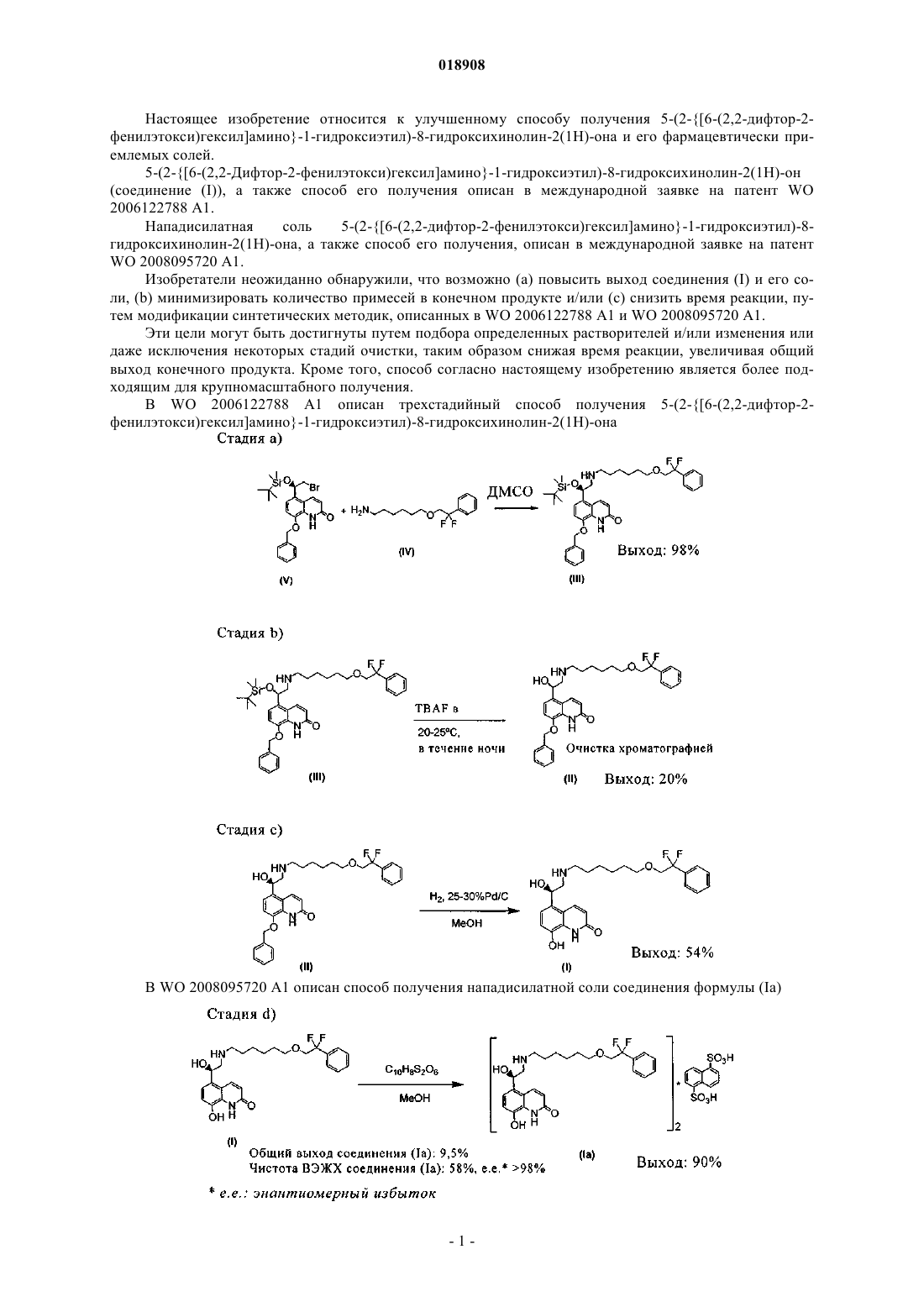
Марчуэта Эреу Иоланда, Моьес Вальс Энрике (ES) Веселицкая И.А., Кузенкова Н.В.,Веселицкий М.Б., Каксис Р.А.,Белоусов Ю.В., Куликов А.В.,Кузнецова Е.В. (RU) Способ получения соединения 5-(2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1 гидроксиэтил)-8-гидроксихинолин-2(1H)-она формулы (I) или его фармацевтически приемлемой соли, который включает взаимодействие соединения формулы (V) с соединением формулы (IV) в ксилольном растворителе с получением соединения формулы (III) и последующее осуществление стадии снятия защитной группы Р 1 и стадии снятия защитной группы P2. Настоящее изобретение относится к улучшенному способу получения 5-(2-[6-(2,2-дифтор-2 фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она и его фармацевтически приемлемых солей. 5-(2-[6-(2,2-Дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-он(соединение (I, а также способ его получения описан в международной заявке на патент WO 2006122788 A1. Нападисилатная соль 5-(2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8 гидроксихинолин-2(1H)-она, а также способ его получения, описан в международной заявке на патентWO 2008095720 A1. Изобретатели неожиданно обнаружили, что возможно (а) повысить выход соединения (I) и его соли, (b) минимизировать количество примесей в конечном продукте и/или (с) снизить время реакции, путем модификации синтетических методик, описанных в WO 2006122788 A1 и WO 2008095720 A1. Эти цели могут быть достигнуты путем подбора определенных растворителей и/или изменения или даже исключения некоторых стадий очистки, таким образом снижая время реакции, увеличивая общий выход конечного продукта. Кроме того, способ согласно настоящему изобретению является более подходящим для крупномасштабного получения. В WO 2006122788 A1 описан трехстадийный способ получения 5-(2-[6-(2,2-дифтор-2 фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она В WO 2008095720 A1 описан способ получения нападисилатной соли соединения формулы (Ia) Как следствие, для получения нападисилатной соли соединения формулы (Ia) из промежуточных соединений (V) и (IV) согласно методам, описанным в предшествующем уровне техники, должны быть выполнены четыре стадии реакции, на которой каждое промежуточное соединение при его получении было выделено и очищено перед его использованием в качестве исходного материала на следующей стадии. Стадии очистки в методиках предшествующего уровня техники осуществляли, используя обычные методы очистки, известные из уровня техники, такие как, например, экстракция растворителем или хроматографические методики. Рассчитанный общий выход получения нападисилатного соединения (Ia) составлял около 9,5%, в то время как уровень примесей, определенных анализом ВЭЖХ, составлял около 5-6%. Неожиданно было обнаружено, что способ получения 5-(2-[6-(2,2-дифтор-2-фенилэтокси)гексил] амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она и его солей может быть значительно улучшен путем модификации условий реакции, особенно модификацией или даже исключением процессов очистки на некоторых стадиях, таким образом упрощая многочисленные стадии реакции, в то же время повышая общий выход реакций. Кроме того, было обнаружено, что в зависимости от выбора растворителей,необходимый продукт может быть получен с более высоким выходом и в более чистой форме по сравнению с известным раннее способом. Соответственно, настоящее изобретение относится к способу получения соединения 5-(2-[6-(2,2 дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она формулы (I) или его фармацевтически приемлемой соли который включает: а) реакцию в ксилольном растворителе соединения формулы (V) где P1 и Р 2 представляют собой защитные группы для гидроксигруппы и L представляет собой уходящую группу,с 6-(2,2-дифтор-2-фенилэтокси)гексан-1-амином формулы (IV) с получением соединения формулы (III)b) осуществление стадии снятия защитной группы P1 и стадии снятия защитной группы P2 для удаления защитных групп P1 и P2 и получения соединения формулы (I). Обычно(i) стадию снятия защитной группы P2 осуществляют при температуре в диапазоне 30-60 С в течение периода до 8 ч и/или(ii) стадию снятия защитной группы P1 осуществляют в присутствии растворителя, которым является уксусная кислота или смесь уксусной кислоты со спиртом или сложным эфиром. Стадию (а) проводят в ксилольном растворителе. Наоборот, соответствующую стадию реакции,описанную в WO 2006/122788, проводят в ДМСО. Неожиданным открытием настоящего изобретения является использование конкретного ксилольного растворителя, способного существенно улучшить чистоту соединения формулы (III). В предпочтительном варианте осуществления описанная выше стадия (b) включает осуществление описанной выше стадии снятия защитной группы P2 в соединении формулы (III) с получением соединения формулы (II) где P1 является таким, как определено выше; и осуществление описанной выше стадии снятия защитной группы Р 1 в соединении формулы (II) с получением соединения формулы (I) Таким образом, в этом варианте осуществления настоящее изобретение относится к способу получения соединения 5-(2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она формулы (I) или его фармацевтически приемлемой соли который включает: а) реакцию в ксилольном растворителе соединения формулы (V) где P1 и Р 2 представляют собой защитные группы для гидроксигруппы и L представляет собой уходящую группу,с 6-(2,2-дифтор-2-фенилэтокси)гексан-1-амином формулы (IV) с получением соединения формулы (III)b) снятие защитной группы с соединения формулы (III) с получением соединения формулы (II) где Р 1 является таким, как определено выше; и с) снятие защитной группы с соединения формулы (II) с получением соединения формулы (I)(i) стадию b) осуществляют при температуре в диапазоне 30-60 С в течение периода до 8 ч и/или(ii) стадию с) осуществляют в присутствии растворителя, которым является уксусная кислота или смесь уксусной кислоты со спиртом или сложным эфиром. В другом варианте осуществления способ согласно настоящему изобретению включает: а) реакцию в ксилольном растворителе соединения формулы (V) где P1 и Р 2 представляют собой защитные группы для гидроксигруппы и L представляет собой уходящую группу,с 6-(2,2-дифтор-2-фенилэтокси)гексан-1-амином формулы (IV) с получением соединения формулы (III)b) снятие защитной группы с соединения формулы (III) с получением соединения формулы (II) где P2 является таким, как определено выше; и с) снятие защитной группы с соединения формулы (II) с получением соединения формулы (I)(i) стадию b) осуществляют в присутствии растворителя, которым является уксусная кислота или смесь уксусной кислоты со спиртом или сложным эфиром, и/или(ii) стадию с) осуществляют при температуре в диапазоне 30-60 С в течение периода до 8 ч.P1 и Р 2 представляют собой защитные группы для гидроксигруппы. P1 и P2 могут быть одинаковыми или различными. Предпочтительно они являются различными. Специалист в данной области техники легко подберет подходящие защитные группы для гидроксигрупп для положений Р 1 и Р 2. Например,подходящие защитные группы описаны в книге Т.W. Greene и G.M. Wuts, Protecting Groups in OrganicSynthesis, 3-е издание, Wiley, New York, 1999 и приведенных в ней ссылках. Примеры подходящих защитных групп для гидроксигрупп включают алкильные группы, такие как метил, этил и трет-бутил; ацильные группы, например алканоильные группы, такие как ацетил; арилметильные группы, такие как бензил (Bn), п-метоксибензил (РМВ), 9-флуоренилметил (Fm) и дифенилметил (бензгидрил, DPM); силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил(TBS) и им подобные. Обычно Р 1 представляет собой бензильную группу. В данном варианте осуществления стадию снятия защитной группы Р 1 обычно осуществляют гидрированием предпочтительно в присутствии катализатора, такого как гидроксид палладия(II) (Pd(OH)2) или палладий(0) (Pd(0. Предпочтительно катализатором является палладий(0) на угле. Обычно в данном варианте осуществления реакцию гидрирования стадии снятия защитной группыP1 осуществляют в присутствии катализатора в количестве менее 10%, предпочтительно менее 5%, наиболее предпочтительно около 4 вес.% от количества используемого реагента. Использование катализатора в этих количествах обычно позволяет снизить уровень образующихся примесей. В частности, можно снизить образование дефторированных примесей, т.е. 5-(2-[6-(2-фенилэтокси)гексил]амино-1 гидроксиэтил)-8-гидроксихинолин-2(1 Н)-она. Также можно снизить образование дигидрохинолиновых примесей. Обычно Р 2 представляет собой трет-бутилдиметилсилильную группу. В этом варианте осуществления стадию снятия защитной группы Р 2, как правило, осуществляют реакцией с тригидратом тетра-нбутилфторида аммония (TBAF), предпочтительно в растворителе, таком как тетрагидрофуран (ТГФ), или с хлоридом водорода в растворителе, выбранном из простых эфиров, сложных эфиров и спиртов. Предпочтительно в этом варианте осуществления настоящего изобретения стадию снятия защитной группы Р 2 осуществляют с хлоридом водорода в растворителе, выбранном из диэтилового эфира, третбутилметилового эфира (ТВМЕ), этанола и изопропилацетата. Альтернативно, в этом варианте осуществления настоящего изобретения стадию снятия защитной группы Р 2 предпочтительно осуществляют с помощью TBAF в тетрагидрофуране (ТГФ) или 2 метилтетрагидрофуране, предпочтительно в ТГФ. Альтернативно, в этом варианте осуществления настоящего изобретения стадию снятия защитной группы P2 предпочтительно осуществляют с помощью нафтален-1,5-дисульфоновой кислоты в тетрагидрофуране (ТГФ).L представляет собой уходящую группу. Специалист-химик легко способен подобрать подходящие уходящие группы для L-положения. Примеры подходящих уходящих групп включают атомы галогена,мезилатные группы (-O-S(О)2-СН 3) и трифлатные (-OS(O)2-CF3) группы. Предпочтительно L представляет собой атом галогена. Более предпочтительно L представляет собой атом брома. Обычно растворитель, используемый на стадии (а), по существу, не содержит ДМСО. Более предпочтительно он, по существу, не содержит ДМСО и диоксан. Использование ксилольного растворителя, подробно описанного выше на стадии (а), способно в целом улучшить чистоту и/или выход по сравнению с аналогичными процессами, в которых стадию (а) проводят в растворителях, таких как ДМСО. В другом варианте осуществления настоящего изобретения стадию снятия защитной группы Р 2 осуществляют при температуре в пределах 40-50 С в течение периода времени, не превышающего 6 ч,предпочтительно не дольше 4 ч, более предпочтительно не дольше 2 ч, наиболее предпочтительно до 1 ч. Сокращение времени реакции для стадии снятия защитной группы Р 2 позволяет неожиданно снизить образование нежеланных побочных продуктов. В другом варианте осуществления настоящего изобретения гидрирование на стадии снятия защитной группы Р 2 необязательно осуществляют в присутствии фторида тетрабутиламмония в количестве около 0,3-0,9 г TBAF на 1 г реагента. Обычно реагентом является соединение формулы (II). В другом варианте осуществления настоящего изобретения соединение, полученное на стадии снятия защитной группы P2, очищают кристаллизацией. Обычно кристаллизацию осуществляют с помощью 1,5-нафталендисульфоновой кислоты в спирте, предпочтительно этаноле. Очистка соединения, полученного на стадии снятия защитной группы Р 2, перекристаллизацией, в отличие от хроматографии, позволяет повысить чистоту и/или выход. Предпочтительно в этом варианте осуществления настоящего изобретения стадию снятия защитной группы Р 2 осуществляют перед стадией снятия защитной группы Р 1, и соединение, полученное на стадии снятия защитной группы Р 2, поэтому представляет собой соединение формулы (II). В предпочтительном варианте осуществления настоящего изобретения стадию снятия защитной группы Р 1 осуществляют в присутствии растворителя, которым является уксусная кислота или смесь уксусной кислоты со спиртом или сложным эфиром. Предпочтительно в этом варианте осуществления растворитель представляет собой уксусную кислоту отдельно или смесь уксусной кислоты/метанола (1:1),более предпочтительно смесь уксусной кислоты/метанола (1:1). Обычно указанный растворитель содержит менее 5% (об./об.), предпочтительно менее 3%, более предпочтительно менее 1% любых жидкостей, отличных от уксусной кислоты, спирта и сложного эфира,предпочтительно любых жидкостей, отличных от уксусной кислоты и метанола. В предпочтительном варианте осуществления изобретения получают фармацевтически приемлемую соль соединения формулы (I). Предпочтительно указанная соль представляет собой нападисилатную соль или мезилатную соль. Нападисилатные соли обычно являются теми, которые описаны в WO 2008/095720. Предпочтительно нападисилатная соль представляет собой геминападисилатную соль или мононападисилатную соль. Мононападисилатная соль обычно содержит от около 0,8 и 1,2 мол.экв. нафтален-1,5 дисульфоновой кислоты на 1 мол.экв. свободного основания, обычно около 1,0 мол.экв. нафтален-1,5 дисульфоновой кислоты на 1 мол.экв. свободного основания. Геминападисилатная соль обычно содержит от около 0,35 до 0,65 мол.экв. нафтален-1,5-дисульфтновой кислоты на 1 мол.экв. свободного основания, обычно около 0,5 мол.экв. нафтален-1,5-дисульфтновой кислоты на 1 мол.экв. свободного основания. Настоящее изобретение также относится к 5-(2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1 гидроксиэтил)-8-гидроксихинолин-2(1H)-ону или его фармацевтически приемлемой соли, полученным способом по настоящему изобретению. Предпочтительно настоящее изобретение относится к нападисилатной соли или мезилатной соли 5-(2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8 гидроксихинолин-2(1H)-она, полученной способом по настоящему изобретению. Более предпочтительно соль представляет собой нападисилатную соль. Описанные выше молярные соотношения могут определяться стандартными методиками, например 1 Н ЯМР, элементным анализом и методами ВЭЖХ. Когда получают нападисилатную соль соединения формулы (I), обычно после стадии (b) добавляют нафтален-1,5-дисульфоновую кислоту без выделения 5-(2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она формулы (I). Получая конечный продукт одностадийной реакции этим способом, без выделения свободного основания, можно повысить чистоту и/или выход. Кроме того, такая одностадийная реакция также предпочтительна, поскольку способна повысить эффективность способа. В предпочтительном варианте осуществления настоящего изобретения стадию (b) и, при необходимости, последующую стадию солеобразования, каждую, проводят без очистки промежуточного соединения, полученного на предыдущей стадии реакции. Соединения формулы (V) могут быть получены известными способами или аналогично известным способам. Например, соединение, в котором Р 1 представляет собой бензил и Р 2 представляет собой TBS,может быть получено методиками синтеза, описанными в документах US 2004059116 (пример 9 С), WO 2004/011416 (пример 2) и WO 2004/016578 (пример lii). 6-(2,2-Дифтор-2-фенилэтокси)гексан-1-амин (IV) получают по методикам синтеза, описанным вWO 2006122788 А 1 (промежуточное соединение 9). Реагенты и растворители, используемые в настоящем изобретении, являются коммерчески доступными, например от Aldrich Chemical Company, Inc. или Fluka Chemie GmbH. Предпочтительными условиями способа стадии (а) являются следующие. К раствору 10,30-11,30 г (40-44 ммоль) 6-(2,2-дифтор-2-фенилэтокси)гексан-1-амина (IV) в 15-25 мл ксилольного растворителя добавляли 19,9 г (40 ммоль) (R)-8-(бензилокси)-5-(2-бром-1-(третбутилдиметилсилилокси)этил)хинолин-2(1H)-она (V) и 9-12 г бикарбоната натрия или 15-20 г карбоната калия. Реакционную смесь нагревали при кипении с обратным холодильником в течение 4-6 ч. После охлаждения до комнатной температуры осажденные неорганические соли отфильтровывали и промывали 80-120 мл ксилола. Растворитель удаляли, получая маслянистый остаток, который использовали на следующей стадии без дополнительной очистки. Предпочтительными условиями стадии снятия защитной группы Р 2 являются следующие. Маслянистый остаток, полученный на предыдущей стадии, растворяли в 300-350 мл ТГФ. Затем к реакционной смеси добавляли 20-25 г TBAF. Реакционную смесь перемешивали в течение 1-2 ч при 4050 С. После охлаждения до комнатной температуры растворитель удаляли в вакууме. Всего 250-300 мл смеси вода/органический растворитель (1:1) добавляли к остатку. Органический слой отделяли и водный слой экстрагировали дважды органическим растворителем (220-30 мл). Органические слои объединяли и концентрировали в вакууме для удаления растворителя. Предпочтительными органическими растворителями, используемыми для экстракции, являются толуол, дихлорметан, изопропилацетат или метилизобутилкетон (MIK), более предпочтительно толуол, изопропилацетат или дихлорметан, наиболее предпочтительно изопропилацетат или дихлорметан. В альтернативном способе полученный остаток, когда удаляют растворитель реакции (ТГФ), может использоваться непосредственно на последующей кристаллизационной очистке без водной экстракции. Остаток очищали кристаллизацией с 8-9 г тетрагидрата 1,5-нафталендисульфоновой кислоты в 300400 мл этанола. Полученный продукт отфильтровывали и промывали 50-70 мл этанола. Полученный сырой остаток обрабатывали 250-260 мл смеси метанол/дихлорметан (1:2), метанол/изопропилацетат (1:2) или метанол/толуол (1:2). К этой суспензии добавляли раствор 3,5-4 г NaOH в 170-190 мл воды. Реакционную смесь перемешивали при 20-30 С в течение 40-50 мин. Органическую фазу отделяли и растворитель удаляли в вакууме. Предпочтительными условиями способа стадии снятия защитной группы Р 1 являются следующие. Промежуточное соединение (II) растворяли в общем объеме 160-170 мл смеси уксусная кислота/спирт (1:1), предпочтительно уксусная кислота/метанол. К раствору добавляли 1-1,5 г 10% Pd/C, 50% воды. Затем к раствору необязательно добавляли около 5-15 г TBAF. После нескольких насыщений азотом реакционную смесь гидрировали при температуре 20-30 С при давлении менее 4 бар, предпочтительно при давлении 1-2 бар в течение 6-8 ч. Катализатор затем отфильтровывали и промывали 190-200 мл метанола. К фильтрату добавляли около 200-250 мл уксусной кислоты и к этому фильтрату добавляли раствор 6-6,5 г тетрагидрата 1,5-нафталендисульфоновой кислоты в 50-70 мл смеси метанол/уксусная кислота (1:1). Смесь нагревали при кипении с обратным холодильником в течение 30 мин. После охлаждения до комнатной температуры продукт отфильтровывали и промывали 25-30 мл метанола. Полученный продукт необязательно может быть очищен суспендированием в метаноле в горячих условиях, таких как при температуре кипения метанола. Конечный продукт (Ia) сушили в вакууме при 50 С. Способ синтеза, описанный в настоящем изобретении, далее иллюстрируется следующими примерами. Примеры приведены только для иллюстрации и не должны рассматриваться как ограничивающие. Структуры полученных соединений подтверждали данными 1 Н-ЯМР и MS. ЯМР записывали с помощью ЯМР-спектрометра Varian Gemini-200 при частоте 200 или 300 МГц. Тетраметилсилан использовали в качестве стандарта и образцы растворяли в дейтерированном диметилсульфоксиде (ДМСО-d6) или дейтерированном хлороформе (CDCl3). Их чистоту определяли по данным ВЭЖХ с помощью оборудования Alliance 2795 Waters, оснащенного детектором с диодной матрицей (DAD) и ZMD или ZQ масс-детектором (ионизация электроспреем). Метод ВЭЖХ с помощью колонки Symmetry C18 (3,5 мкм, 21100 мм) и подвижной фазы, состоящей из двух фаз: фаза А: забуференный (муравьиная кислота/аммиак) водный раствор при рН 3; фаза В: 50:50 смесь ацетонитрил/метанол с формиатом аммония. Градиент составлял от 0 до 95% фазы В в течение 10 мин. Эксперименты препаративной ВЭЖХ-MS осуществляли с помощью оборудования Gilson, оснащенного двойным насосом (поршневой насос Gilson 321); вакуумным дегазатором (Gilson 864); инжектором-сборником фракций (жидкостной манипулятор Gilson 215); двумя инъекционными модулями,аналитическим и препаративным (Gilson 819); клапаном (Gilson Valvemate 7000); 1/1000 разветвителем(Acurate by LC Packings); приставным насосом (Gilson 307); детектором с диодной матрицей (Gilson 170) и MS-детектором (Thermoquest Finnigan aQa, квадрупольный масс-спектрометр с ES и APCI видами ионизации). Оборудование ВЭЖХ-MS контролировали с помощью IBM PC. Экспериментальный раздел Сравнительный пример I (в соответствии с WO 2006122788 и WO 2008095720). Промежуточное соединение III. 8-(Бензилокси)-5-1R)-1-[трет-бутил(диметил)силил]окси-2-([6(2,2-дифтор-2-фенилэтокси)гексил]аминоэтил)хинолин-2(1H)-он. К раствору (8-(бензилокси)-5-1R)-2-бром-1-[трет-бутил(диметил)силил]оксиэтил)хинолин 2(1H)-она (V) (4,80 г, 9,83 ммоль) и 6-(2,2-дифтор-2-фенилэтокси)гексил]амина (IV) (3,04 г, 11,8 ммоль) в диметилсульфоксиде (13,5 мл) добавляли бикарбонат натрия (2,49 г, 29,4 ммоль) и йодид натрия (2,22 г,14,8 ммоль). Смесь нагревали при 140 С в течение 2 ч. После охлаждения реакционную смесь разбавляли водой (40 мл) и экстрагировали диэтиловым эфиром (220 мл). Объединенные органические экстракты промывали водой (210 мл) и насыщенным раствором хлорида натрия (20 мл), сушили (Na2SO4) и растворитель удаляли при пониженном давлении. Указанное в заголовке соединение получали (6,40 г, 98%) в виде масла. Промежуточное соединение (II). 8-(Бензилокси)-5-1R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил] амино-1-гидроксиэтил)хинолин-2(1 Н)-он. К раствору промежуточного соединения (III) (6,4 г, 9,63 ммоль) в тетрагидрофуране (60 мл) добавляли TBAF (5,02 г, 19,26 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Очистка колоночной хроматографией с помощью метиленхлорида/метанола (от 95:5 до 85:15) в качестве элюента приводила к получению 8-(бензилокси)-51R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)хинолин-2(1H)-она (II) (1,1 г,20%) в виде масла. 5-1R)-2-[6-(2,2-Дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин 2(1H)-он (I). К промежуточному соединению (II) (1,10, 2,0 ммоль) в метаноле (50 мл) добавляли 20% палладия на угле (300 мг). Смесь гидрировали при 2 бар в течение 3 ч. Катализатор отфильтровывали через целит и растворитель концентрировали. Полученное масло очищали колоночной хроматографией на силикагеле, элюируя смесью метиленхлорид/метанол (95:5) с получением указанного в заголовке соединения(0,50 г, 54%) в виде масла. Нападисилат 5-1R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она (Ia). 5-1R)-2-[6-(2,2-Дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин 2(1H)-он (6,63 г; 14,4 ммоль) растворяли в 134 мл метанола с получением 1,075 М раствора, который нагревали до температуры около 50 С. Затем к нагретому раствору добавляли 7,74 ммоль тетрагидрата нафтален-1,5-дисульфоновой кислоты. Смесь затем перемешивали в течение 30 мин при температуре кипения и затем охлаждали до 20/25 С и перемешивали при этой температуре в течение 1 ч. Полученный осадок выделяли фильтрацией, промывали метанолом и сушили в вакууме при температуре 50 С (15,67 г, 90%). Пример II (в соответствии с настоящим изобретением). Промежуточное соединение III. 8-(Бензилокси)-5-1R)-1-[трет-бутил(диметил)силил]окси-2-[6(2,2-дифтор-2-фенилэтокси)гексил]аминоэтил)хинолин-2(1H)-он. К раствору [6-(2,2-дифтор-2-фенилэтокси)гексил]амина (IV) (11,0 г, 42,8 ммоль) в ксилоле (20 мл) добавляли (8-(бензилокси)-5-1R)-2-бром-1-[трет-бутил(диметил)силил]оксиэтил)хинолин-2(1H)-он(V) (19,9 г, 40,7 ммоль) и бикарбонат натрия (10,4 г, 123 ммоль). Смесь нагревали при кипении с обратным холодильником в течение 6 ч. После охлаждения до комнатной температуры к реакционной смеси добавляли дополнительно ксилол (176 мл) и осажденные неорганические соли отфильтровывали и промывали ксилолом (100 мл). Полученный фильтрат концентрировали в вакууме для удаления растворителя, получая маслянистый остаток (промежуточное соединение (III, который использовали на следующей стадии без очистки. Промежуточное соединение (II). 8-(Бензилокси)-5-1R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил] амино-1-гидроксиэтил)хинолин-2(1H)-он. Промежуточное соединение (III) растворяли в тетрагидрофуране (330 мл). Затем к этому раствору добавляли TBAF (23,3 г, 73 ммоль). Смесь перемешивали при 45 С в течение 1 ч. После охлаждения до комнатной температуры растворитель удаляли в вакууме и полученный остаток необязательно экстрагировали 266 мл смеси вода/дихлорметан (1:1). Органические слои извлекали и затем упаривали в вакууме. Затем добавляли 352 мл 96% этанола и смесь нагревали до 50-60 С. При этой температуре добавляли раствор 8,5 г тетрагидрата 1,5-нафталендисульфоновой кислоты в 35 мл 96% этанола в течение 1 ч. Систему добавления промывали 29 мл 96% этанола, который добавляли к реакционной смеси. Реакционную смесь перемешивали при кипении с обратным холодильником в течение 30 мин и затем охлаждали до комнатной температуры. Продукт отфильтровывали и промывали 60 мл этанола. Сырой продукт на фильтре обрабатывали 252 мл смеси метанол/дихлорметан (1:2). Затем добавляли раствор 3,6 г NaOH в 116 мл воды и реакционную смесь перемешивали при 20-25 С в течение 45 мин. Водный слой отделяли и экстрагировали дихлорметаном (342 мл). Органические фазы извлекали и перемешивали вместе с раствором 4,2 г NaCl в 168 мл воды. Органическую фазу отделяли и растворитель удаляли в вакууме, получая 8-(бензилокси)-5-1R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)хинолин 2(1H)-он (II) (16,8 г, 75%) в виде масла. Нападисилат 5-1R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино)-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она (Ia). К раствору промежуточного соединения (II) (16,8 г, 30,5 ммоль) в смеси метанола (69 мл) и уксусной кислоты (77 мл) добавляли суспензию 10% палладия на угле, 50% воды (1,33 г) в смеси метанола (15 мл) и уксусной кислоты (7 мл). Смесь гидрировали при 1-2 бар в течение 8 ч. Катализатор отфильтровывали через целит и промывали метанолом (193 мл). К этому фильтрату добавляли уксусную кислоту (220 мл). Затем к фильтрату медленно добавляли раствор тетрагидрата 1,5-нафталендисульфоновой кислоты(6,33 г) в смеси метанола (54 мл) и уксусной кислоты (27 мл). Реакционную смесь нагревали при кипении с обратным холодильником в течение 30 мин и затем охлаждали до комнатной температуры. Осадок отфильтровывали и промывали метанолом (27 мл). Сырой продукт на фильтре растворяли в метаноле (800 мл) и нагревали при кипении с обратным холодильником в течение 30 мин. Продукт отфильтровывали и промывали дополнительно метанолом (34 мл). Полученное твердое вещество сушили в вакууме при 50 С, получая нападисилат 5-1R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8 гидроксихинолин-2(1H)-она (14,9 г, 81%). Общий выход нападисилата 5-1R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1 гидроксиэтил)-8-гидроксихинолин-2(1H)-она (Ia) составлял около 60,7% (7581%), и его чистота составляла ВЭЖХ примесей = 1,5%, е.е. 98%. Таблица 1 Сравнительные результаты стадия снятия защитной группы Р 2 стадия снятия защитной группы P1 Как видно из табл. 1, применение ксилольного растворителя на стадии (а) существенно снижает количество примесей в промежуточном соединении формулы (III). Кроме того, общий выход нападисилата 5-1R)-2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она существенно повышается при снижении примесей до низкого уровня по сравнению со сравнительным примером. Это достигается модификацией некоторых методик очистки, таким образом упрощая реакционные стадии и снижая количество различных реагентов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения 5-(2-[6-(2,2-дифтор-2-фенилэтокси)гексил]амино-1 гидроксиэтил)-8-гидроксихинолин-2(1H)-она формулы (I) или его фармацевтически приемлемой соли который включает: а) реакцию в ксилольном растворителе соединения формулы (V) где P1 и Р 2 представляют собой защитные группы для гидроксигрупп и L представляет собой уходящую группу,с 6-(2,2-дифтор-2-фенилэтокси)гексан-1-амином формулы (IV) с получением соединения формулы (III)b) осуществление стадии снятия защитной группы P1 и стадии снятия защитной группы Р 2 для удаления защитных групп P1 и Р 2 и получения соединения формулы (I). 2. Способ по п.1, в котором стадия (b) включает осуществление указанной стадии снятия защитной группы Р 2 в соединении формулы (III) с получением соединения формулы (II) в котором Р 1 является таким, как определено выше; и осуществление указанной стадии снятия защитной группы Р 1 в соединении формулы (II) с получением соединения формулы (I)(i) стадию снятия защитной группы Р 2 осуществляют при температуре в диапазоне около 30-60 С в течение до 8 ч и/или(ii) стадию снятия защитной группы Р 1 осуществляют в присутствии растворителя, который представляет собой уксусную кислоту или смесь уксусной кислоты со спиртом или сложным эфиром. 4. Способ по любому из предшествующих пунктов, в котором:(а) Р 1 представляет собой бензильную группу, и стадию снятия защитной группы Р 1 осуществляют гидрированием и/или(b) Р 2 представляет собой трет-бутилдиметилсилильную группу, и стадию снятия защитной группыP2 осуществляют реакцией с тригидратом фторида тетра-н-бутиламмония или с хлоридом водорода. 5. Способ по любому из предшествующих пунктов, в котором L представляет собой бром. 6. Способ по любому из предшествующих пунктов, в котором стадию снятия защитной группы P2 осуществляют с помощью тригидрата фторида тетра-н-бутиламмония в тетрагидрофуране. 7. Способ по любому из предшествующих пунктов, в котором стадию снятия защитной группы Р 2 осуществляют при температуре 40-50 С в течение периода времени, не превышающего 6 ч. 8. Способ по п.7, в котором период времени не превышает 4 ч. 9. Способ по любому из предшествующих пунктов для получения геминападисилатной или мезилатной соли соединения формулы (I). 10. Способ по п.9 для получения геминападисилатной соли соединения формулы (I), в котором после стадии (b) добавляют нафтален-1,5-дисульфоновую кислоту без выделения 5-[2-[6-(2,2-дифтор-2 фенилэтокси)гексил]амино-1-гидроксиэтил)-8-гидроксихинолин-2(1H)-она формулы (I). 11. Способ по п.3, в котором промежуточное соединение формулы (II), полученное со стадии снятия защитной группы Р 2, очищают кристаллизацией с тетрагидратом 1,5-нафталендисульфоновой кислоты в этаноле. 12. Способ по любому из предшествующих пунктов, в котором P1 представляет собой бензил, и стадию снятия защитной группы P1 осуществляют гидрированием в присутствии катализатора палладия на угле в количестве менее 10 вес.% относительно количества соединения формулы (II). 13. Способ по п.12, в котором используемое количество катализатора составляет менее 5%. 14. Способ по любому из предшествующих пунктов, в котором стадию снятия защитной группы P1 осуществляют в присутствии растворителя, которым является уксусная кислота или смесь уксусной кислоты со спиртом или сложным эфиром. 15. Способ по п.14, в котором растворителем является уксусная кислота или смесь метанол/уксусная кислота (1:1). 16. Способ по п.14, в котором растворителем является смесь метанол/уксусная кислота (1:1). 17. Способ по любому из предшествующих пунктов, в котором:(i) стадию снятия защитной группы P2 осуществляют при температуре в диапазоне 30-60 С в течение периода до 8 ч и(ii) стадию снятия защитной группы P1 осуществляют в присутствии растворителя, которым является уксусная кислота или смесь уксусной кислоты со спиртом или сложным эфиром.
МПК / Метки
МПК: A61K 31/4704, C07D 215/26, A61P 11/00
Метки: 5-(2-{[6-(2,2-дифтор-2-фенилэтокси)гексил]амино}-1-гидроксиэтил)-8-гидроксихинолин-2(1h)-oha, получения, способ
Код ссылки
<a href="https://eas.patents.su/12-18908-sposob-polucheniya-5-2-6-22-diftor-2-feniletoksigeksilamino-1-gidroksietil-8-gidroksihinolin-21h-oha.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения 5-(2-{[6-(2,2-дифтор-2-фенилэтокси)гексил]амино}-1-гидроксиэтил)-8-гидроксихинолин-2(1h)-oha</a>
1,1-дифтор-1,4-дихлорбутан и способ его получения.

Номер патента: 624
Опубликовано: 29.12.1999
Авторы: Браун Стивен Мартин, Боуден Мартин Чарльз, Вилльямс Альфред Глин
МПК: C07C 19/10
Метки: способ, получения, 1,1-дифтор-1,4-дихлорбутан
Формула / Реферат:
1. 1,1-дифтор-1,4-дихлорбутан. 2. Способ получения 1,1-дифтор-1,4-дихлорбутана, включающий взаимодействие 1,1,1,4-тетрахлорбутана с фтороводородом в жидкой фазе при автогенном давлении. 3. Способ по п.2, отличающийся тем, что реакцию проводят в присутствии катализатора, выбранного из галогенидов поливалентных металлов и оксидов алюминия. 4. Способ по п.3, отличающийся тем, что галогенид металла выбирают из хлорида железа, фторида алюминия,...
Способ получения эфиров 4,4-дифтор-3-оксобутановой кислоты

Номер патента: 11019
Опубликовано: 30.12.2008
Авторы: Эренфройнд Йозеф, Корси Камилла, Тоблер Ханс, Ламберт Клеменс, Вальтер Харальд
МПК: C07C 67/343, C07C 69/716
Метки: эфиров, получения, 4,4-дифтор-3-оксобутановой, кислоты, способ
Формула / Реферат:
1. Способ получения соединения формулы (I) в которой R обозначает С1-С12алкил, который включает взаимодействие соединения общей формулы (II) в которой R1 и R2 все независимо обозначают С1-С12алкил или R1 и R2 совместно с атомом азота, к которому они присоединены, образуют алициклический кольцевой амин, содержащий от 4 до 7 атомов углерода, или морфолиновое кольцо, с эфиром уксусной кислоты общей формулы (III) в которой R является таким, как...
Промежуточное соединение и способ получения обогащенных β-аномером 21-дезокси-21, 21-дифтор-d-рибофуранозилнуклеозидов

Номер патента: 11558
Опубликовано: 28.04.2009
Авторы: Панда Биджан Кумар, Бхатт Дипендра, Маикап Голак Чандра
МПК: C07H 13/08, C07H 19/073
Метки: beta;-аномером, 21-дифтор-d-рибофуранозилнуклеозидов, способ, обогащенных, 21-дезокси-21, промежуточное, соединение, получения
Формула / Реферат:
1. Соединение формулы (I) где Р представляет собой водород или гидроксизащитную группу. 2. Соединение формулы (I) по п.1, где защитную группу Р выбирают из формила, 2-хлорацетила, бензила, дифенилметила, трифенилметила, 4-нитробензила, феноксикарбонила, третичного бутила, метоксиметила, тетрагидропиранила, аллила, тетрагидротиенила, 2-метоксиэтоксиметила, метоксиацетила, феноксиацетила, изобутирила, этоксикарбонила, бензилоксикарбонила, мезила,...
Производные оптически активных изомеров 2-амино-4-(бициклил) амино-6-(алкилзамещенного)-1,3,5-триазина, способы их получения, их применение в качестве гербицидов и регуляторов роста растений, гербицидная или регулирующая рост растений композиция, способ борьбы с сорными растениями и промежуточные соединения

Номер патента: 12406
Опубликовано: 30.10.2009
Авторы: Менне Хуберт, Минн Клеменс, Бирингер Херманн, Хилльс Мартин, Аулер Томас, Аренс Хартмут, Кене Хайнц, Дитрих Хансёрг
МПК: A01N 43/68, C07D 311/68, C07D 251/18...
Метки: растений, регуляторов, композиция, растениями, применение, соединения, получения, 2-амино-4-(бициклил, амино-6-(алкилзамещенного)-1,3,5-триазина, сорными, способы, качестве, рост, изомеров, оптически, борьбы, производные, регулирующая, способ, промежуточные, роста, гербицидная, гербицидов, активных
Формула / Реферат:
1. Производные оптически активных изомеров 2-амино-4-(бициклил)амино-6-(алкилзамещенного)-1,3,5-триазина формулы (I) где R1 означает H, галоид, (С1-С6)алкил, (С1-С6)галоидалкил, [(C1-C4)алкокси](С1-С6)алкил, (С3-С6)циклоалкил, который не замещен или замещен одним или несколькими радикалами, выбираемыми из группы, которая включает галоид, (С1-С4)алкил и (С1-С4)галоидалкил, или означает (С2-С6)алкенил, (С2-С6)алкинил, (С2-С6)галоидалкенил,...
Способы получения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила

Номер патента: 7953
Опубликовано: 27.02.2007
Авторы: Лендерс Рубен Герардус Жорж, Паскье Элизабет Тереза Жанна, Виллемс Йоаннес Йозефус Мария, Хэрес Ян, Схилс Дидье Филипп Робер, Гийемон Жером Эмиль Жорж, Жанссен Поль Адриан Ян, Медар Барт Петрус Анна Мария Йозеф
МПК: C07C 253/30, C07C 253/20, A61K 31/505...
Метки: 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила, способы, получения
Формула / Реферат:
1. Способ получения 4-[[4-[[4-(2-цианоэтенил)-2,6-диметилфенил]амино]-2-пиримидинил]амино]бензонитрила формулы (I) его N-оксида, фармацевтически приемлемой кислотно-аддитивной соли, четвертичного амина или стереохимически изомерной формы, который включает осуществление взаимодействия промежуточного соединения формулы (II) его подходящей кислотно-аддитивной соли или стереохимически изомерной формы с промежуточным соединением формулы (III) его...
Предыдущий патент: Кристаллический гидрохлорид n-{(1s)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1н-пиразол-5-ил)-2-тиофенкарбоксамида
Следующий патент: Способ выделения никельсодержащих сульфидов из руд
Случайный патент: Штепсель с гильзой