Фармацевтическая композиция эффективного ингибитора всг для перорального введения
Номер патента: 22272
Опубликовано: 30.12.2015
Авторы: Виллагра Мария-Фернанда, Чэнь Фэнцзин, Хель Хуан-Франсиско








Формула / Реферат
1. Жидкая фармацевтическая композиция, содержащая:
а) 4,6 мас.% или менее соединения (1) или его фармацевтически приемлемой соли

б) по крайней мере от 10 до 30 мас.% ПАВа и
в) по крайней мере 60 мас.% фармацевтически приемлемого растворителя;
где массовое соотношение ПАВ и соединения (1) или его фармацевтически приемлемой соли равно 2,7 или более;
композиция содержит количество липида, равное 5 мас.% или менее, и
композиция образует прозрачную дисперсию со средним размером частиц менее 1 мкм после разбавления в модельном желудочном соке.
2. Фармацевтическая композиция по п.1, где гидрофильно-липофильный баланс ПАВ составляет более 10.
3. Фармацевтическая композиция по п.1, где ПАВ представляет собой витамин E-TPGS, полиэтоксилированное касторовое масло, полиоксилгидрированное касторовое масло, эфир полиоксиэтиленсорбита и жирной кислоты, каприлокапроилмакроголглицерид или их смесь.
4. Фармацевтическая композиция по п.1, где фармацевтически приемлемый растворитель представляет собой пропиленгликоль, полипропиленгликоль, полиэтиленгликоль, глицерин, этанол, триацетин, диметилизосорбид, гликофурол, пропиленкарбонат, вода, диметилацетамид или их смесь.
5. Фармацевтическая композиция по п.1, где растворитель представляет собой смесь воды, полиэтиленгликоля со средней молекулярной массой более 300, но менее 600, и пропиленгликоля.
6. Фармацевтическая композиция по п.1, которая не содержит липид.
7. Фармацевтическая композиция по п.1, которая содержит количество пропиленгликоля, равное 8 мас.% или менее.
8. Фармацевтическая композиция по п.1, которая содержит количество амина, равное 2 мас.% или менее.
9. Жидкая фармацевтическая композиция, содержащая:
а) 6,3 мас.% или менее соединения (1) или его фармацевтически приемлемой соли

б) по крайней мере от 10 до 30 мас.% ПАВа и
в) по крайней мере 60 мас.% фармацевтически приемлемого растворителя;
где массовое соотношение ПАВ и соединения (1) или его фармацевтически приемлемой соли равно 4,3 или более;
композиция содержит количество липида, равное 5 мас.% или менее, и
композиция образует прозрачную дисперсию со средним размером частиц менее 1 мкм после разбавления в модельном желудочном соке.
10. Фармацевтическая композиция по п.9, где гидрофильно-липофильный баланс ПАВ составляет более 10.
11. Фармацевтическая композиция по п.9, где ПАВ представляет собой витамин E-TPGS, полиэтоксилированное касторовое масло, полиоксилгидрированное касторовое масло, эфир полиоксиэтиленсорбита и жирной кислоты, каприлокапроилмакроголглицерид или их смесь.
12. Фармацевтическая композиция по п.9, где фармацевтически приемлемый растворитель представляет собой пропиленгликоль, полипропиленгликоль, полиэтиленгликоль, глицерин, этанол, триацетин, диметилизосорбид, гликофурол, пропиленкарбонат, вода, диметилацетамид или их смесь.
13. Фармацевтическая композиция по п.9, где растворитель представляет собой смесь воды, полиэтиленгликоля со средней молекулярной массой более 300, но менее 600, и пропиленгликоля.
14. Фармацевтическая композиция по п.9, которая не содержит липид.
15. Фармацевтическая композиция по п.9, которая содержит количество пропиленгликоля, равное 8 мас.% или менее.
16. Фармацевтическая композиция по п.9, которая содержит количество амина, равное 2 мас.% или менее.
Текст
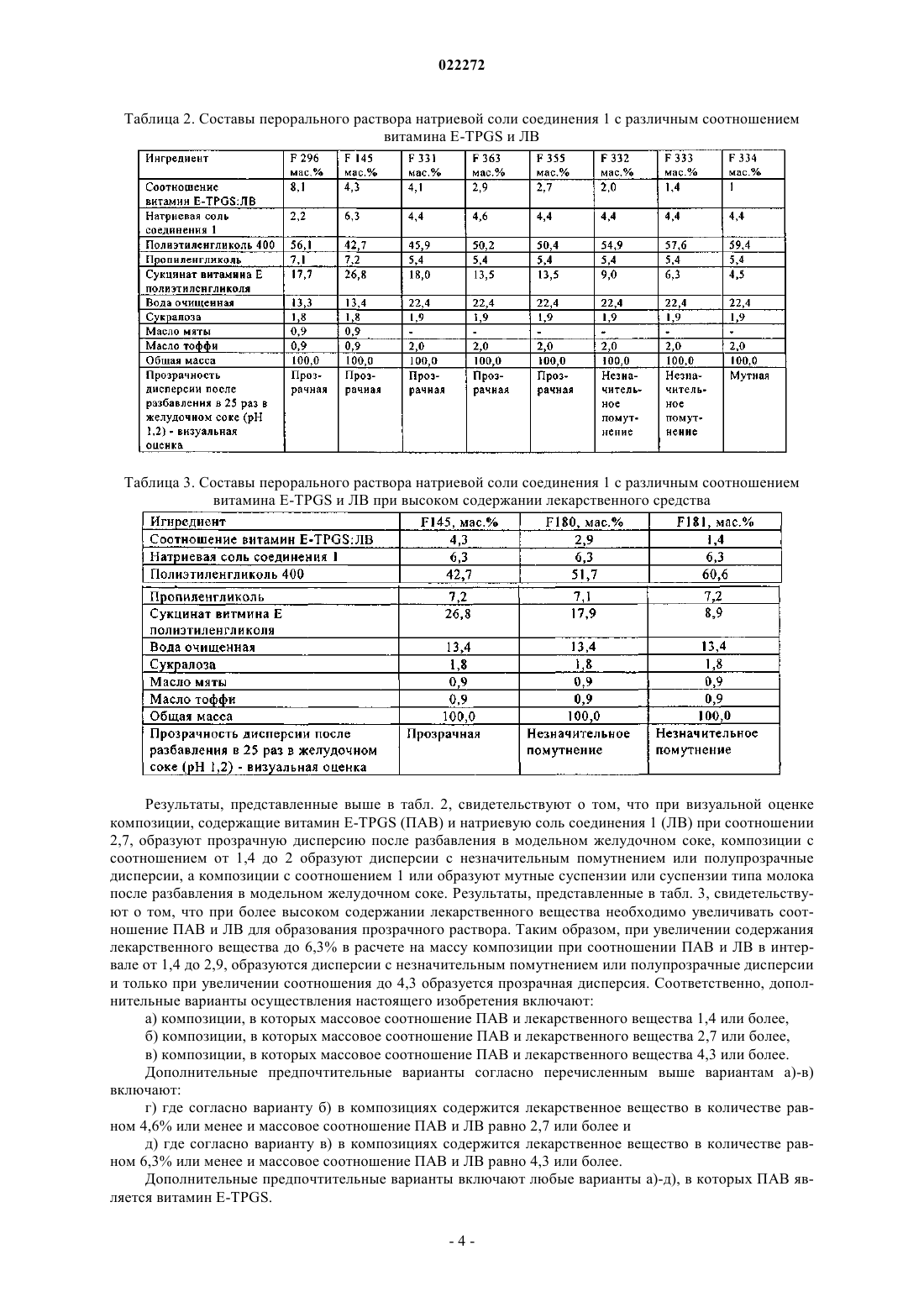
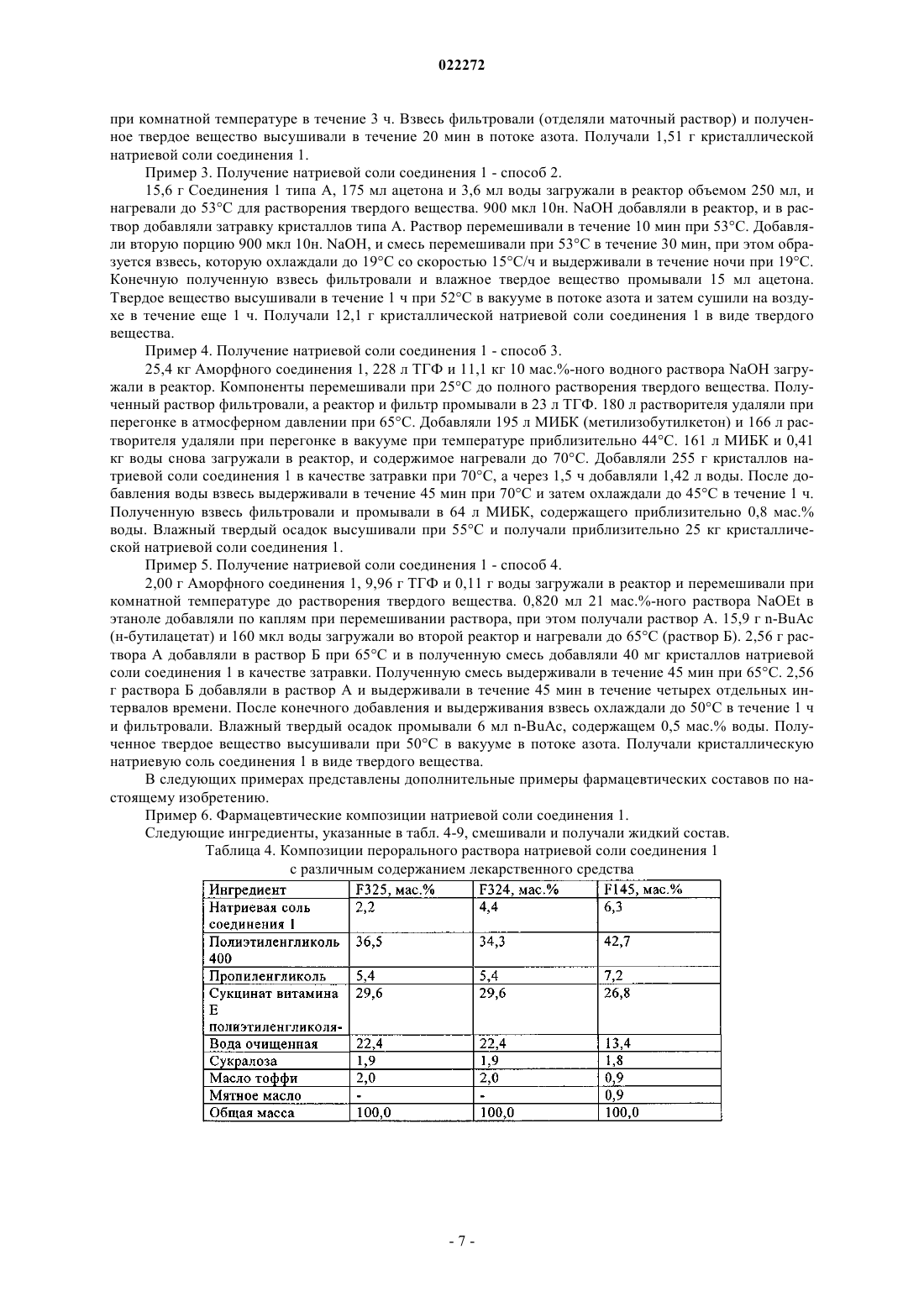
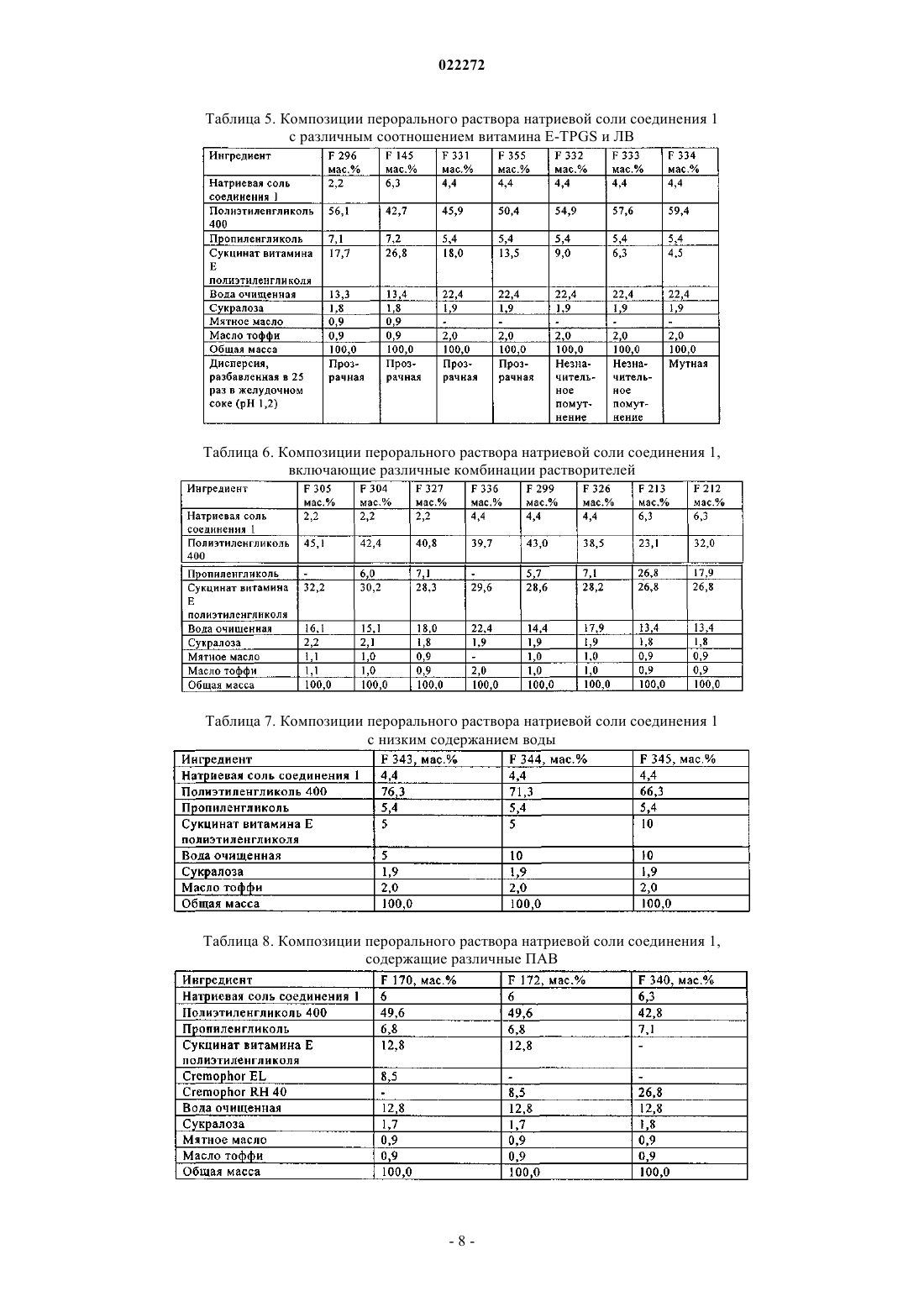
ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ЭФФЕКТИВНОГО ИНГИБИТОРА ВСГ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ Изобретение относится к жидким фармацевтическим композициям эффективного ингибитора вируса гепатита С (ВГС) для перорального введения, содержащим соединение (1), указанное ниже, или его фармацевтически приемлемую соль, поверхностно-активное вещество (ПАВ),фармацевтически приемлемый растворитель и липид, где ПАВ и указанное соединение(1) находятся при определенных массовых соотношениях. Указанные композиции образуют прозрачную дисперсию со средним размером частиц менее 1 мкм после разбавления в модельном желудочном соке.(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) В настоящей заявке испрашивается приоритет в связи с временной заявкой США 61/116789, поданной 21 ноября 2008 г. Предпосылки создания настоящего изобретения 1. Область техники. В настоящем изобретении предлагается фармацевтическая композиция эффективного ингибитора вируса гепатита С (ВГС) для перорального введения. 2. Предпосылки создания настоящего изобретения. Следующее соединение 1: известно в качестве селективного и эффективного ингибитора сериновой протеазы NS3 ВГС. Соединение 1 является цвиттерионом и относится к серии ациклических пептидов-ингибиторов ВГС, описанных в патентах США 7323180, 7514557 и 7585845. Соединение 1 подробно описано как соединение 1055 в патенте США 7585845 и как соединение 1008 в патенте США 7514557. Соединение 1 можно получить по общим методикам, описанным в указанных выше патентах, которые включены в настоящее описание в качестве ссылок. Предпочтительные формы соединения 1 включают кристаллические формы, прежде всего кристаллическую форму натриевой соли, которую получают, как описано в разделе "Примеры" в данном описании. Соединение 1 можно также характеризовать следующей химической формулой, которая эквивалентна описанной выше структуре:, L0 обозначает MeO-, L1 обозначает Br, a R2 обозначает В патенте США 6531139 и соответствующей международной заявке WO 9906024 описана фармацевтическая композиция, которая содержит липофильный фармацевтически приемлемый агент, липид,который представляет собой смесь моно- и диглицеридов, растворитель и ПАВ. Перечислены несколько фармацевтически приемлемых растворителей, включая полиэтиленгликоль, однако пропиленгликоль является более предпочтительным растворителем. Перечислены несколько фармацевтически приемлемых ПАВ, причем предпочтительны Cremophor RH 40 или Cremophor EL. Витамин E-TPGS не включен в список фармацевтически приемлемых ПАВ. В этих цитированных документах указано, что композицию,описанную в данном контексте, которая является жидкостью, можно использовать для заполнения капсул для перорального введения и что ее можно также перерабатывать в жидкий раствор для перорального, парентерального, ректального или местного введения. В патенте США 6121313 (опубликованная заявка WO 9906043), содержание которого аналогично патенту 6531139 (опубликованная заявка WO 9906024), описанному выше, но фармацевтически активный агент ограничен определенными пиранонами. В патенте США 6231887 (опубликованная заявка WO 9906044) описана фармацевтическая композиция, которая содержит пиранон в качестве фармацевтически активного агента, основный амин, растворитель, ПАВ и необязательно липид, который представлен моно- и диглицеридами. Перечислены несколько фармацевтически приемлемых растворителей, включая полиэтиленгликоль, хотя пропиленгликоль является предпочтительным растворителем. Перечислены несколько фармацевтически приемлемых ПАВ, причем предпочтительны Cremophor RH 40 или Cremophor EL. Витамин E-TPGS не включен в список фармацевтически приемлемых ПАВ. Указано, что описанную в данной заявке композицию, которая является жидкостью, можно использовать для заполнения капсул для перорального введения и что ее можно также переработать в жидкий раствор для перорального, парентерального, ректального или местного введения. В патенте США 6555558 (опубликованная заявка WO 0236110) описана фармацевтическая композиция, которая содержит пираноновый ингибитор протеазы (кроме того включая, но не ограничиваясь только им, типранавир), ПАВ, растворитель полиэтиленгликоль, липид, который представляет собой смесь моно- и диглицеридов, и необязательно основный амин. Композиция практически не содержит этанол и пропиленгликоль. Перечислены несколько фармацевтически приемлемых ПАВ, причем предпочтителен Cremophor EL. Витамин E-TPGS не включен в список фармацевтически приемлемых ПАВ. Указано, что описанная выше композиция, которая является жидкостью, прежде всего пригодна для заполнения мягких желатиновых капсул, предназначенных для перорального введения. Витамин E-TPGS (сукцинат D-альфа токоферилполиэтиленгликоля) является водорастворимой формой витамина Е и эксципиентом для ускорения эмульгирования липофильных веществ, и по своему действию является неиногенным ПАВ, а также улучшает биодоступность определенных лекарственных веществ. Например, в статье Sokol R.J. и др., Lancet, 338, 212-214, (1991) установлено, что совместное введение витамина E-TPGS и циклоспорина улучшает биодоступность циклоспорина. В патенте США 6193985 (опубликованная заявка WO 09531217) описано использование токоферолов в качестве растворителей и/или ПАВ для лекарственных препаратов, которые практически нерастворимы в воде, прежде всего для получения лекарственной формы для местного введения. Использование витамина E-TPGS упоминается на страницах 7-8 и 12 в качестве ПАВ для получения составов с высоким содержанием альфа-токоферола в качестве липидного слоя. Примеры описанных составов для местного введения, содержащих витамин E-TPGS, такие как примеры 1-5, обычно включают липидный слой (альфа-токоферол), лекарственный агент и витамин E-TPGS в качестве ПАВ, в количестве менее 25 мас.% в расчете на массу состава. В заявке WO 9636316 указано, что витамин E-TPGS можно использовать для повышения эффективности доставки липофильных соединений в виде самоэмульгирующегося преконцентрата состава, включающего а) липофильное лекарственное средство (прежде всего циклоспорин), б) витамин E-TPGS и в) липофильную фазу. В типичных примерах описанных составов, таких как примеры 2 и 4, содержится менее 14 мас.% витамина E-TPGS в качестве ПАВ, липидный слой и лекарственное средство. Ссылка на состав ингибитора протеазы ВИЧ отсутствует. Наконец, в патенте США 6730679 (опубликованная заявка WO 09735587) и статье Yu и др., PharmRes. 16(12), 1812-1817 (1999) описаны фармацевтические композиции, содержащие ампренавир, ингибитор протеазы ВИЧ и витамин Е-TPGS. Таким образом, в настоящее время отсутствуют какие-либо составы соединения 1, прежде всего предназначенные для перорального введения в виде неинкапсулированной жидкости. Такую лекарственную форму можно было бы использовать прежде всего для детей, а также взрослых, для которых существуют проблемы при проглатывании твердых веществ. Таким образом, объектом настоящего изобретения является получение такого жидкого состава соединения 1. Краткое изложение сущности настоящего изобретения В настоящем изобретении предлагается фармацевтически приемлемый состав соединения 1, или его фармацевтически приемлемой соли, в виде раствора для перорального введения. На основании физико-химических свойств лекарственного вещества в объем настоящего изобретения включено получение раствора, способного образовывать эмульсию, микроэмульсию или мицеллярный раствор при контактировании с водной средой. Состав включает по крайней мере один растворитель для повышения растворимости лекарственного средства и по крайней мере одно ПАВ, гидрофильнолипофильный баланс (ГЛБ) которого составляет 10 и которое добавляют для поддержания лекарственного вещества в растворенном состоянии после разбавления в модельных жидкостях желудочнокишечного тракта. Состав по настоящему изобретению может дополнительно содержать воду в качестве сорастворителя и маскирующие вкус компоненты, такие как подсластители и ароматизаторы. Можно добавлять также антиоксидант для предотвращения окисления лекарственного вещества. В табл. 1 приведены два примера композиции такого состава в двух различных дозировках. Таблица 1. Состав перорального раствора натриевой соли соединения 1 (F330 и F335). Иногда из-за физико-химических характеристик лекарственное вещество в комбинации с ПАВ затвердевает или может стать недостаточно жидким для перорального введения. В одном объекте настоящего изобретения предлагается получение раствора, пригодного для перорального введения, в неинкапсулированной форме. Предпочтительно такой состав является текучим при комнатной температуре, таким образом его можно использовать для перорального введения в жидком виде и для упрощения дозировки. Было установлено, что добавление по крайней мере одного сорастворителя в комбинации с ПАВ обеспечивает поддержание указанного состава в виде жидкого текучего раствора при комнатной температуре, таким образом достигаются преимущества, упомянутые выше. Концентрация лекарственного вещества, присутствующего в композиции, составляет такой уровень, который обеспечивает возможность дозировки в широком интервале, т.е. можно изменять вводимую дозу, изменяя объем. Такая возможность позволяет изменять дозировку в широком интервале: от низкой дозы для введения маленьким детям до высокой дозы для введения взрослым, неспособным проглатывать твердые формы. Подробное описание изобретения Жидкая композиция по настоящему изобретению включает: а) соединение 1 или его фармацевтически приемлемую соль: б) по крайней мере одно ПАВ,в) по крайней мере один фармацевтически приемлемый растворитель, причем композиция практически не содержит липид. Активный ингредиент, соединение 1, или его фармацевтически приемлемая соль, присутствует в количестве от 1 до 40 мас.% в расчете на общую массу композиции, предпочтительно от 2 до 10 мас.% в расчете на общую массу композиции и даже более предпочтительно от 2 до 8 мас.% в расчете на общую массу композиции. Предпочтительные формы соединения 1, которые можно использовать в составе,включают его кристаллические формы, прежде всего кристаллическую форму натриевой соли соединения 1. ПАВ, пригодные для применения в композиции по настоящему изобретению, включают ПАВ, ГЛБ которых составляет более 10. Примеры пригодных ПАВ включают витамин E-TPGS, полиэтоксилированное касторовое масло (например, CREMOPHOR EL), полиоксилгидрированное касторовое масло(например, CREMOPHOR RH), эфир полиоксиэтиленсорбита и жирной кислоты (например, полисорбат 80), каприлокапроилмакроголглицерид (например, LABRASOL) или их смесь. Предпочтительным ПАВ является витамин E-TPGS. Содержание ПАВ составляет от 2 до 50 мас.% в расчете на общую массу композиции, предпочтительно от 10 до 30 мас.% в расчете на общую массу композиции. Различное содержание ПАВ и лекарственного вещества (ЛВ) позволяют получить различные водные дисперсии. Примеры этих композиций представлены в табл. 2 ниже. Таблица 2. Составы перорального раствора натриевой соли соединения 1 с различным соотношением витамина E-TPGS и ЛВ Таблица 3. Составы перорального раствора натриевой соли соединения 1 с различным соотношением витамина E-TPGS и ЛВ при высоком содержании лекарственного средства Результаты, представленные выше в табл. 2, свидетельствуют о том, что при визуальной оценке композиции, содержащие витамин E-TPGS (ПАВ) и натриевую соль соединения 1 (ЛВ) при соотношении 2,7, образуют прозрачную дисперсию после разбавления в модельном желудочном соке, композиции с соотношением от 1,4 до 2 образуют дисперсии с незначительным помутнением или полупрозрачные дисперсии, а композиции с соотношением 1 или образуют мутные суспензии или суспензии типа молока после разбавления в модельном желудочном соке. Результаты, представленные в табл. 3, свидетельствуют о том, что при более высоком содержании лекарственного вещества необходимо увеличивать соотношение ПАВ и ЛВ для образования прозрачного раствора. Таким образом, при увеличении содержания лекарственного вещества до 6,3% в расчете на массу композиции при соотношении ПАВ и ЛВ в интервале от 1,4 до 2,9, образуются дисперсии с незначительным помутнением или полупрозрачные дисперсии и только при увеличении соотношения до 4,3 образуется прозрачная дисперсия. Соответственно, дополнительные варианты осуществления настоящего изобретения включают: а) композиции, в которых массовое соотношение ПАВ и лекарственного вещества 1,4 или более,б) композиции, в которых массовое соотношение ПАВ и лекарственного вещества 2,7 или более,в) композиции, в которых массовое соотношение ПАВ и лекарственного вещества 4,3 или более. Дополнительные предпочтительные варианты согласно перечисленным выше вариантам а)-в) включают: г) где согласно варианту б) в композициях содержится лекарственное вещество в количестве равном 4,6% или менее и массовое соотношение ПАВ и ЛВ равно 2,7 или более и д) где согласно варианту в) в композициях содержится лекарственное вещество в количестве равном 6,3% или менее и массовое соотношение ПАВ и ЛВ равно 4,3 или более. Дополнительные предпочтительные варианты включают любые варианты а)-д), в которых ПАВ является витамин E-TPGS. В предпочтительном варианте осуществления настоящего изобретения композиции образуют прозрачную, с незначительным помутнением или полупрозрачную дисперсию после разбавления в модельном желудочном соке. В другом предпочтительном варианте композиции образуют прозрачные дисперсии после разбавления в модельном желудочном соке. Если композиции образуют прозрачные, полупрозрачные дисперсии или дисперсии с незначительным помутнением при разбавлении, то это означает, что в дисперсиях отсутствует или содержится только небольшое количество осадка соединения 1 и что активный ингредиент остается практически в солюбилизированном состоянии. Такие системы являются предпочтительными, т.к. в этом случае ожидается повышение биодоступности активного ингредиента после проглатывания, по сравнению с мутными дисперсиями, в которых активный ингредиент практически выпадает в осадок. Прозрачность конечной дисперсии можно оценивать известными методами. Прозрачность можно оценивать по размеру капель и частиц, измеренному методом лазерного светорассеяния (например, динамическое светорассеяние или статическое светорассеяние), которые известны в данной области. При различных соотношениях ПАВ и лекарственного вещества образуются частицы/капли различного размера с различной степенью прозрачности. Чем меньше размер капли эмульсии, микроэмульсии или мицеллярных частиц, тем больше прозрачность образующегося раствора. Типичное значение среднего размера частиц для прозрачной конечной дисперсии составляет менее 1 мкм, а для дисперсии с незначительным помутнением или мутной дисперсии размер частиц составляет более 1 мкм. Примеры композиций такого состава с различными прозрачностью и размерами капель или частиц описаны в примере 7. Таким образом, в дополнительном варианте средний размер частиц композиции составляет менее 1 мкм после разбавления в модельном желудочном соке. Фармацевтически приемлемыми растворителями, пригодными для применения в контексте настоящего изобретения, являются пропиленгликоль, полипропиленгликоль, полиэтиленгликоль (например,низкомолекулярный полиэтиленгликоль, но не ограничиваясь только ими, ПЭГ 300, 400, 600 и т.п.), глицерин, этанол, триацетин, диметилизосорбид, гликофурол, пропиленкарбонат, вода, диметилацетамид или их смесь. В одном варианте по крайней мере одним растворителем является низкомолекулярный полиэтиленгликоль, например, полиэтиленгликоль 300, полиэтиленгликоль 400, полиэтиленгликоль 600 или их смеси. Предпочтительным растворителем является смесь воды, полиэтиленгликоля со средней молекулярной массой более 300, но менее 600, и пропиленгликоля. Еще более предпочтительным растворителем является смесь воды, пропиленгликоля и полиэтиленгликоля 400. В другом предпочтительном варианте растворителем является смесь воды и полиэтиленгликоля 400. Растворитель или смесь растворителей составляет от 10 до 90 мас.% в расчете на общую массу композиции, предпочтительно от 60 до 90 мас.% в расчете на общую массу композиции. В предпочтительном варианте вода в качестве сорастворителя присутствует в количестве от 0 до 50 мас.% в расчете на общую массу композиции, более предпочтительно от 0 до 30 мас.% в расчете на общую массу композиции, даже более предпочтительно от 5 до 20 мас.% в расчете на общую массу композиции. В композициях по настоящему изобретению предпочтительно практически отсутствует пропиленгликоль. В данном контексте практически отсутствует обозначает количество равное 8 мас.% или менее, более предпочтительно 2 мас.% или менее пропиленгликоля в композиции. В предпочтительном варианте в композиции по настоящему изобретению пропиленгликоль не содержится. В композициях по настоящему изобретению также предпочтительно практически отсутствуют амины. В данном контексте "практически отсутствует" обозначает количество равное 2 мас.% или менее,более предпочтительно 1 мас.% или менее, даже более предпочтительно 0,5 мас.% или менее амина в композиции. В предпочтительном варианте в композиции по настоящему изобретению амин не содержится. В композициях по настоящему изобретению практически отсутствует липид, поскольку такие соединения существенно влияют на вкус. Таким образом, если исключить добавление или значительно снизить содержание таких веществ, то можно улучшить вкус, прежде всего для использования в педиатрии. В данном контексте "практически отсутствует" обозначает количество равное 5 мас.% или менее,более предпочтительно 2 мас.% или менее липида в композиции. В предпочтительном варианте в композиции настоящего изобретения липид не содержится. Композиция по настоящему изобретению необязательно включает дополнительные эксципиенты,такие как антиоксиданты (например, -токоферол, пропилгаллат, пальмитат аскорбиновой кислоты, БГТ(бутилгидрокситолулол), БГА (бутилгидроксианизол) или их смеси) и/или подсластители (например,сукралоза, ацесульфам калия, натриевая соль сахарина или их смеси) и ароматизаторы (например, масло тоффи, мятное масло, жевательная резинка, ароматизаторы виноград, вишня, клубника или их смеси). Таким образом, например, предпочтительно, чтобы подсластить или придать аромат лекарственной форме, следует добавлять указанные агенты. Специалисты в области фармацевтики обычно могут выбрать приемлемые подсластители или ароматизаторы. В одном предпочтительном варианте фармацевтическая композиция по настоящему изобретению включает: а) от 1 до 40 мас.% соединения 1 или его фармацевтически приемлемой соли,б) от 2 до 50 мас.% ПАВ,в) от 10 до 90 мас.% растворителя или смеси растворителей,причем в композиции практически отсутствует липид или более предпочтительно липид не содержится. В другом предпочтительном варианте фармацевтическая композиция по настоящему изобретению содержит: а) от 2 до 10 мас.% соединения 1 или его фармацевтически приемлемой соли,б) от 10 до 30 мас.% ПАВ,в) от 60 до 90 мас.% растворителя или смеси растворителей,причем в композиции практически отсутствует липид или более предпочтительно липид не содержится. В еще одном предпочтительном варианте фармацевтическая композиция по настоящему изобретению содержит: а) от 2 до 10 мас.% соединения 1 или его фармацевтически приемлемой соли,б) от 10 до 30 мас.% витамина E-TPGS,в) от 60 до 90 мас.% смеси воды, пропиленгликоля и полиэтиенгликоля 400,причем в композиции практически отсутствует липид или более предпочтительно липид не содержится. В другом предпочтительном варианте фармацевтическая композиция по настоящему изобретению содержит: а) от 2 до 10 мас.% соединения 1 или его фармацевтически приемлемой соли,б) от 10 до 30 мас.% витамина E-TPGS,в) от 60 до 90 мас.% смеси воды и полиэтиенгликоля 400,причем в композиции практически отсутствует липид или более предпочтительно липид не содержится. Дополнительные варианты включают любой из четырех вариантов, перечисленных выше, где в композиции: (1) практически отсутствует пропиленгликоль, или не содержится пропиленгликоль, и/или(2) практически отсутствует амин, или не содержится амин. Пример методики получения составов по изобретению заключается в следующем: смешивают растворители при температуре 40-50C, добавляют ПАВ и перемешивают. Затем добавляют лекарственное вещество и перемешивают до полного растворения. Добавляют растворенный в воде подсластитель и перемешивают. Понижают температуру вплоть до 35-37C, добавляют ароматизаторы и перемешивают. Самодиспергирующиеся составы по настоящему изобретению образуют мицеллярные растворы при смешивании с водной средой. Состав перед введением можно смешивать с водной средой, такой как вода, фруктовый сок и т.п. Состав можно проглатывать в жидком виде, при этом он смешивается с желудочным соком, образуя мицеллярный раствор in situ. В определенных условиях соединение 1 выпадает в осадок из раствора при смешивании состава с желудочным соком, что приводит к образованию мутной суспензии или суспензии типа молока. Композиции по настоящему изобретению можно использовать для лечения инфекции вируса гепатита С (ВГС) и вводить их по известной методике, как описано в патенте США 7585845. Квалифицированный терапевт может выбрать соответствующую дозировку для каждого конкретного пациента с использованием общих рекомендаций, изложенных в указанном патенте, и мнения медицинских работников с учетом возраста, размера, общего состояния здоровья, тяжести состояния и других характеристик конкретного пациента, нуждающегося в лечении. В примерах 1-5 описано получение различных кристаллических форм соединения 1. Пример 1. Получение кристаллической формы соединения 1 типа А. Аморфное соединение 1 (партия 7, 13,80 г) загружали в трехгорлую колбу, объемом 1000 мл. В колбу добавляли чистый этанол (248,9 г). При перемешивании содержимое колбы нагревали со скоростью 60C/ч до приблизительно 74C. (Твердые вещества не растворяются при температуре 74C). Затем добавляли воду (257,4 г) в течение 4 ч, при этом получали взвесь при перемешивании при температуре 74C. После завершения добавления воды, температуру постепенно снижали до температуры окружающей среды со скоростью 8C/ч и затем выдерживали при указанной температуре в течение 6 ч при постоянном перемешивании. Полученное твердое вещество отделяли фильтрованием и промывали 50 мл раствора EtOH/вода (1:1). Влажное твердое вещество высушивали на воронке в течение 30 мин, продувая через осадок азот. (Результаты порошковой рентгеновской дифракции этого образца свидетельствуют о том, что образец представляет собой сольват этанола). Затем твердое вещество высушивали при 65-70C в вакууме (P= 25 мм рт.ст.) в потоке азота в течение 1,5 ч. Структуру полученного твердого вещества(12,6 г, корректированный выход 95,5% ) подтверждали методом рентгеновской порошковой дифракции,т.е. подтверждали соответствие соединения 1 типу А. Пример 2. Получение натриевой соли соединения 1 - способ 1. 2,1 г Аморфной натриевой соли соединения 1 и 8,9 г ацетона загружали в сосуд и перемешивали при комнатной температуре в течение 3 ч. Взвесь фильтровали (отделяли маточный раствор) и полученное твердое вещество высушивали в течение 20 мин в потоке азота. Получали 1,51 г кристаллической натриевой соли соединения 1. Пример 3. Получение натриевой соли соединения 1 - способ 2. 15,6 г Соединения 1 типа А, 175 мл ацетона и 3,6 мл воды загружали в реактор объемом 250 мл, и нагревали до 53C для растворения твердого вещества. 900 мкл 10 н. NaOH добавляли в реактор, и в раствор добавляли затравку кристаллов типа А. Раствор перемешивали в течение 10 мин при 53C. Добавляли вторую порцию 900 мкл 10 н. NaOH, и смесь перемешивали при 53C в течение 30 мин, при этом образуется взвесь, которую охлаждали до 19C со скоростью 15C/ч и выдерживали в течение ночи при 19C. Конечную полученную взвесь фильтровали и влажное твердое вещество промывали 15 мл ацетона. Твердое вещество высушивали в течение 1 ч при 52C в вакууме в потоке азота и затем сушили на воздухе в течение еще 1 ч. Получали 12,1 г кристаллической натриевой соли соединения 1 в виде твердого вещества. Пример 4. Получение натриевой соли соединения 1 - способ 3. 25,4 кг Аморфного соединения 1, 228 л ТГФ и 11,1 кг 10 мас.%-ного водного раствора NaOH загружали в реактор. Компоненты перемешивали при 25C до полного растворения твердого вещества. Полученный раствор фильтровали, а реактор и фильтр промывали в 23 л ТГФ. 180 л растворителя удаляли при перегонке в атмосферном давлении при 65C. Добавляли 195 л МИБК (метилизобутилкетон) и 166 л растворителя удаляли при перегонке в вакууме при температуре приблизительно 44C. 161 л МИБК и 0,41 кг воды снова загружали в реактор, и содержимое нагревали до 70C. Добавляли 255 г кристаллов натриевой соли соединения 1 в качестве затравки при 70C, а через 1,5 ч добавляли 1,42 л воды. После добавления воды взвесь выдерживали в течение 45 мин при 70C и затем охлаждали до 45C в течение 1 ч. Полученную взвесь фильтровали и промывали в 64 л МИБК, содержащего приблизительно 0,8 мас.% воды. Влажный твердый осадок высушивали при 55C и получали приблизительно 25 кг кристаллической натриевой соли соединения 1. Пример 5. Получение натриевой соли соединения 1 - способ 4. 2,00 г Аморфного соединения 1, 9,96 г ТГФ и 0,11 г воды загружали в реактор и перемешивали при комнатной температуре до растворения твердого вещества. 0,820 мл 21 мас.%-ного раствора NaOEt в этаноле добавляли по каплям при перемешивании раствора, при этом получали раствор А. 15,9 г n-BuAc(н-бутилацетат) и 160 мкл воды загружали во второй реактор и нагревали до 65C (раствор Б). 2,56 г раствора А добавляли в раствор Б при 65C и в полученную смесь добавляли 40 мг кристаллов натриевой соли соединения 1 в качестве затравки. Полученную смесь выдерживали в течение 45 мин при 65C. 2,56 г раствора Б добавляли в раствор А и выдерживали в течение 45 мин в течение четырех отдельных интервалов времени. После конечного добавления и выдерживания взвесь охлаждали до 50C в течение 1 ч и фильтровали. Влажный твердый осадок промывали 6 мл n-BuAc, содержащем 0,5 мас.% воды. Полученное твердое вещество высушивали при 50C в вакууме в потоке азота. Получали кристаллическую натриевую соль соединения 1 в виде твердого вещества. В следующих примерах представлены дополнительные примеры фармацевтических составов по настоящему изобретению. Пример 6. Фармацевтические композиции натриевой соли соединения 1. Следующие ингредиенты, указанные в табл. 4-9, смешивали и получали жидкий состав. Таблица 4. Композиции перорального раствора натриевой соли соединения 1 с различным содержанием лекарственного средства Таблица 5. Композиции перорального раствора натриевой соли соединения 1 с различным соотношением витамина E-TPGS и ЛВ Таблица 6. Композиции перорального раствора натриевой соли соединения 1,включающие различные комбинации растворителей Таблица 7. Композиции перорального раствора натриевой соли соединения 1 с низким содержанием воды Таблица 8. Композиции перорального раствора натриевой соли соединения 1,содержащие различные ПАВ Таблица 9. Композиции перорального раствора натриевой соли соединения 1, содержащие амины Пример 7. Фармацевтические композиции натриевой соли соединения 1. Следующие ингредиенты, перечисленные в табл. 10, смешивали и получали жидкий состав. Отбирали образец (10 мл) такой композиции и диспергировали его при перемешивании в 250 мл желудочного сока (pH 1,2) в течение 1 ч. Образец полученной дисперсии анализировали методом статического светорассеяния или методом динамического светорассеяния (другое название метода: фотон-корреляционная спектроскопия или ФКС). Результаты визуальной оценки и размеры частиц указаны в табл. 10. Таблица 10. Композиции перорального раствора натриевой соли соединения 1 с различным соотношением витамина E-TPGS и ЛВ ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Жидкая фармацевтическая композиция, содержащая: а) 4,6 мас.% или менее соединения (1) или его фармацевтически приемлемой соли б) по крайней мере от 10 до 30 мас.% ПАВа и в) по крайней мере 60 мас.% фармацевтически приемлемого растворителя; где массовое соотношение ПАВ и соединения (1) или его фармацевтически приемлемой соли равно 2,7 или более; композиция содержит количество липида, равное 5 мас.% или менее, и композиция образует прозрачную дисперсию со средним размером частиц менее 1 мкм после разбавления в модельном желудочном соке. 2. Фармацевтическая композиция по п.1, где гидрофильно-липофильный баланс ПАВ составляет более 10. 3. Фармацевтическая композиция по п.1, где ПАВ представляет собой витамин E-TPGS, полиэтоксилированное касторовое масло, полиоксилгидрированное касторовое масло, эфир полиоксиэтиленсорбита и жирной кислоты, каприлокапроилмакроголглицерид или их смесь. 4. Фармацевтическая композиция по п.1, где фармацевтически приемлемый растворитель представляет собой пропиленгликоль, полипропиленгликоль, полиэтиленгликоль, глицерин, этанол, триацетин,диметилизосорбид, гликофурол, пропиленкарбонат, вода, диметилацетамид или их смесь. 5. Фармацевтическая композиция по п.1, где растворитель представляет собой смесь воды, полиэтиленгликоля со средней молекулярной массой более 300, но менее 600, и пропиленгликоля. 6. Фармацевтическая композиция по п.1, которая не содержит липид. 7. Фармацевтическая композиция по п.1, которая содержит количество пропиленгликоля, равное 8 мас.% или менее. 8. Фармацевтическая композиция по п.1, которая содержит количество амина, равное 2 мас.% или менее. 9. Жидкая фармацевтическая композиция, содержащая: а) 6,3 мас.% или менее соединения (1) или его фармацевтически приемлемой соли б) по крайней мере от 10 до 30 мас.% ПАВа и в) по крайней мере 60 мас.% фармацевтически приемлемого растворителя; где массовое соотношение ПАВ и соединения (1) или его фармацевтически приемлемой соли равно 4,3 или более; композиция содержит количество липида, равное 5 мас.% или менее, и композиция образует прозрачную дисперсию со средним размером частиц менее 1 мкм после разбавления в модельном желудочном соке. 10. Фармацевтическая композиция по п.9, где гидрофильно-липофильный баланс ПАВсоставляет более 10. 11. Фармацевтическая композиция по п.9, где ПАВ представляет собой витамин E-TPGS, полиэтоксилированное касторовое масло, полиоксилгидрированное касторовое масло, эфир полиоксиэтиленсорбита и жирной кислоты, каприлокапроилмакроголглицерид или их смесь. 12. Фармацевтическая композиция по п.9, где фармацевтически приемлемый растворитель представляет собой пропиленгликоль, полипропиленгликоль, полиэтиленгликоль, глицерин, этанол, триацетин, диметилизосорбид, гликофурол, пропиленкарбонат, вода, диметилацетамид или их смесь. 13. Фармацевтическая композиция по п.9, где растворитель представляет собой смесь воды, полиэтиленгликоля со средней молекулярной массой более 300, но менее 600 и пропиленгликоля. 14. Фармацевтическая композиция по п.9, которая не содержит липид. 15. Фармацевтическая композиция по п.9, которая содержит количество пропиленгликоля, равное 8 мас.% или менее. 16. Фармацевтическая композиция по п.9, которая содержит количество амина, равное 2 мас.% или менее.
МПК / Метки
МПК: A61K 31/44, A61K 9/10, A61K 47/10, A61K 9/08, A61K 47/14
Метки: ингибитора, фармацевтическая, композиция, всг, введения, эффективного, перорального
Код ссылки
<a href="https://eas.patents.su/11-22272-farmacevticheskaya-kompoziciya-effektivnogo-ingibitora-vsg-dlya-peroralnogo-vvedeniya.html" rel="bookmark" title="База патентов Евразийского Союза">Фармацевтическая композиция эффективного ингибитора всг для перорального введения</a>
Фармацевтическая полимерная композиция для перорального введения тербуталина сульфата с контролируемым высвобождением

Номер патента: 13969
Опубликовано: 30.08.2010
Авторы: Бэдван Эднан, Ол-Римеви Мейас
МПК: A61K 31/137, A61P 43/00, A61K 9/22...
Метки: перорального, сульфата, контролируемым, полимерная, композиция, высвобождением, тербуталина, фармацевтическая, введения
Формула / Реферат:
1. Фармацевтическая композиция с контролируемым высвобождением, содержащая, по меньшей мере, тербуталин сульфат или его производное в качестве активного агента и дополнительно содержащая неактивную матрицу, причем упомянутая матрица содержит смесь гидрофильных полисахаридных полимеров, упомянутая смесь содержит хитозан или его производное и дополнительно содержит ксантановую смолу или ее производное, где соотношение ксантановой смолы и хитозана...
Антимикробная композиция для перорального введения

Номер патента: 10199
Опубликовано: 30.06.2008
Авторы: Чичерин Дмитрий Сергеевич, Киселёв Николай Александрович
МПК: A61K 45/08, A61K 47/26
Метки: антимикробная, введения, композиция, перорального
Формула / Реферат:
1. Антимикробная композиция для перорального введения, отличающаяся тем, что она содержит антибиотик, выбранный из группы, включающей пенициллины широкого спектра действия, цефалоспорины, тетрациклины, линкозамиды, макролиды, и лактулозу при соотношении активных компонентов 1:1-1:100, причем средний размер частиц лактулозы составляет 100 нм-200 мкм. 2. Антимикробная композиция по п.1, отличающаяся тем, что она содержит дополнительно...
Стабильная композиция фенофибрата для перорального введения и ее применение

Номер патента: 12842
Опубликовано: 30.12.2009
Авторы: Радди Стивен Б., Пэйтел Рэйкеш, Гастоу Ивэн И., Уилкинс Майкл Джон, Райд Туула, Джейн Рэйджив
МПК: A61K 9/20, A61K 31/216, A61K 9/14...
Метки: фенофибрата, стабильная, введения, применение, композиция, перорального
Формула / Реферат:
1. Стабильная композиция фенофибрата для перорального введения, содержащая: (а) частицы фенофибрата, имеющие средний эффективный размер частиц менее примерно 2000 нм; и (б) по меньшей мере один поверхностный стабилизатор, причем эта композиция демонстрирует биоэквивалентность при введении человеку в сытом состоянии по сравнению с введением человеку в состоянии натощак; причем биоэквивалентность устанавливается по: (1) 90% доверительному...
Фармацевтическая композиция для интраназального введения пирибедила

Номер патента: 11041
Опубликовано: 30.12.2008
Авторы: Роллан Эрве, Вутрих Патрик
МПК: A61K 31/506, A61K 47/40, A61K 9/08...
Метки: интраназального, пирибедила, введения, композиция, фармацевтическая
Формула / Реферат:
1. Фармацевтическая композиция в виде водного раствора или порошка для интраназального введения пирибедила, которая содержит пирибедил или его фармацевтически приемлемую соль, необязательно циклодекстрин, один или несколько фармацевтически приемлемых наполнителей. 2. Фармацевтическая композиция в соответствии с п.1, отличающаяся тем, что пирибедил находится в виде основания. 3. Фармацевтическая композиция в соответствии с п.1 или 2, отличающаяся...
Фармацевтическая композиция тгп и ингибитора апф

Номер патента: 3470
Опубликовано: 26.06.2003
Авторы: Бундулис Юрис, Веверис Марис, Калвиньш Иварс, Скарда Илзе
МПК: A61K 31/205, A61P 9/00
Метки: тгп, ингибитора, апф, композиция, фармацевтическая
Формула / Реферат:
1. Фармацевтическая композиция, содержащая дигидрат 3-(2,2,2-триметилгидразиний)пропионата (ТГП) и ингибитор ангиотензинпревращающего фермента (АПФ). 2. Фармацевтическая композиция по п.1, в которой указанный ингибитор АПФ является (S)-1-{N-[1-(этоксикарбонил)-3-фенилпропил]-L-аланил}-L-пролином, т.е. эналаприлом. 3. Фармацевтическая композиция по п.2, в которой соотношение указанных активных соединений дигидрата...
Предыдущий патент: Гетероциклические замещенные соединения арила в качестве ингибиторов hif
Следующий патент: Втулка для уплотнения шиберного клапана
Случайный патент: Щетинное изделие типа щетки, кисти и т.п.