Способ получения некоторых азациклогексапептидов








Формула / Реферат
1. Способ получения азациклогексапептидных соединений формулы

где R1 обозначает CH2CH2NH2;
RI обозначает С9-С21-алкил, С9-С21-алкенил, C1-С10-алкоксифенил, C1-C10-алкоксинафтил или C1-С10-алкокситерфенил;
RII обозначает Н, C1-4-алкил, С3-С4-алкенил, (СН2)2-4OН или (СН2)2-4NRIVRV; или
RIII обозначает Н, C1-4-алкил, С3-С4-алкенил, (СН2)2-4OН или (CH2)2-4NRIVRV или
RII и RIII, взятые вместе, обозначают (СН2)4, (СН2)5, (СН2)2O(СН2)2 или (CH2)2NH(CH2)2;
RIV обозначает Н или С1-4-алкил;
RV обозначает Н или C1-4-алкил;
Q обозначает N или О;
или их фармацевтически приемлемых кислотно-аддитивных солей, который предусматривает стадии
а) взаимодействия соединения II формулы

с фенилбороновой кислотой, п-метоксифенилбороновой кислотой или метанбороновой кислотой с получением соединения III формулы

b) которое затем восстанавливают до амина и гидролизуют с получением соединения IV формулы

с) которое взаимодействует с тиофенолом в подходящем растворителе с образованием соединения V формулы

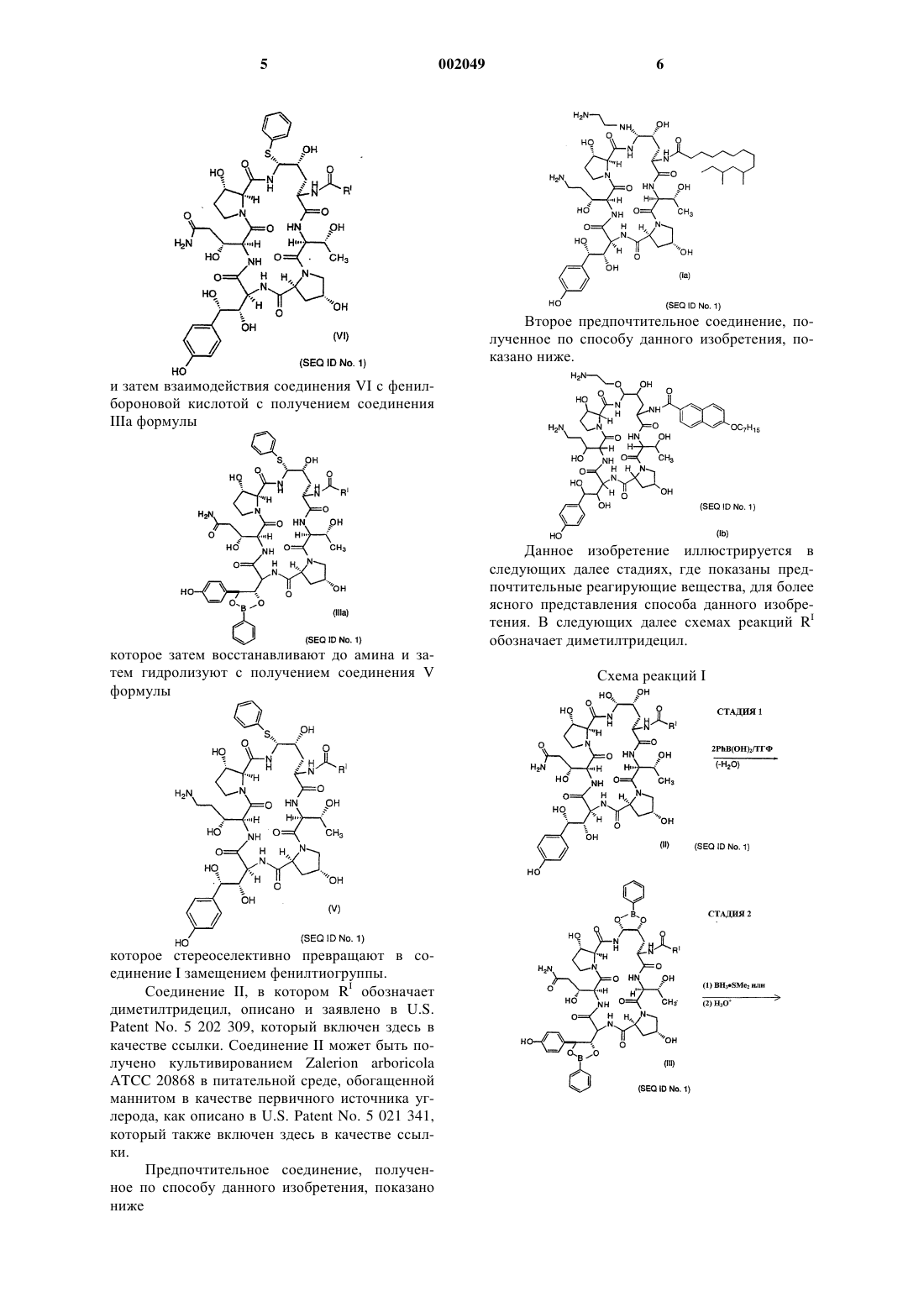
которое стереоселективно превращают в соединение I замещением фенилтиогруппы.
2. Способ по п.1, где восстановление на стадии (b) выполняют с использованием боранового комплекса или борида металла.
3. Способ по п.2, где борид металла представляет собой ZrCl4/NaBH4 или TiCl4/NaBH4, а борановый комплекс представляет собой боран, комплексированный с диметилсульфидом, дибензилсульфидом, дифенилсульфидом, ТГФ или 1,4-оксатианом или BH2Cl с диметилсульфидом.
4. Способ по п.1, где подходящим растворителем в стадии (с) является ацетонитрил.
5. Способ по п.1, где замещение фенилтиогруппы имеет место в неразбавленном этилендиамине или с этилендиамином, растворенным в подходящем растворителе, при температуре примерно от 10 до примерно 40шС.
6. Способ по п.5, где подходящий растворитель выбран из воды, метанола, этанола, тетрагидрофурана, изопропанола, трифторэтанола, ацетонитрила или дихлорметана.
7. Способ получения азациклогексапептидных соединений формулы

где R1 обозначает CH2CH2NH2;
RI обозначает С9-С21-алкил, С9-С21-алкенил, C1-С10-алкоксифенил, C1-С10-алкоксинафтил или C1-С10-алкокситерфенил;
RII обозначает Н, C1-4-алкил, С3-С4-алкенил, (СН2)2-4OН или (CH2)2-4NRIVRV; или
RIII обозначает Н, C1-4-алкил, С3-С4-алкенил, (СН2)2-4OН или (CH2)2-4NRIVRV или RII и RIII, взятые вместе, обозначают (СН2)4, (CH2)5, (СН2)2O(СН2)2 или (CH2)2NH(CH2)2;
RIV обозначает Н или C1-4-алкил;
RV обозначает Н или C1-4-алкил;
или их фармацевтически приемлемых кислотно-аддитивных солей, который предусматривает стадии
а) взаимодействия соединения II формулы

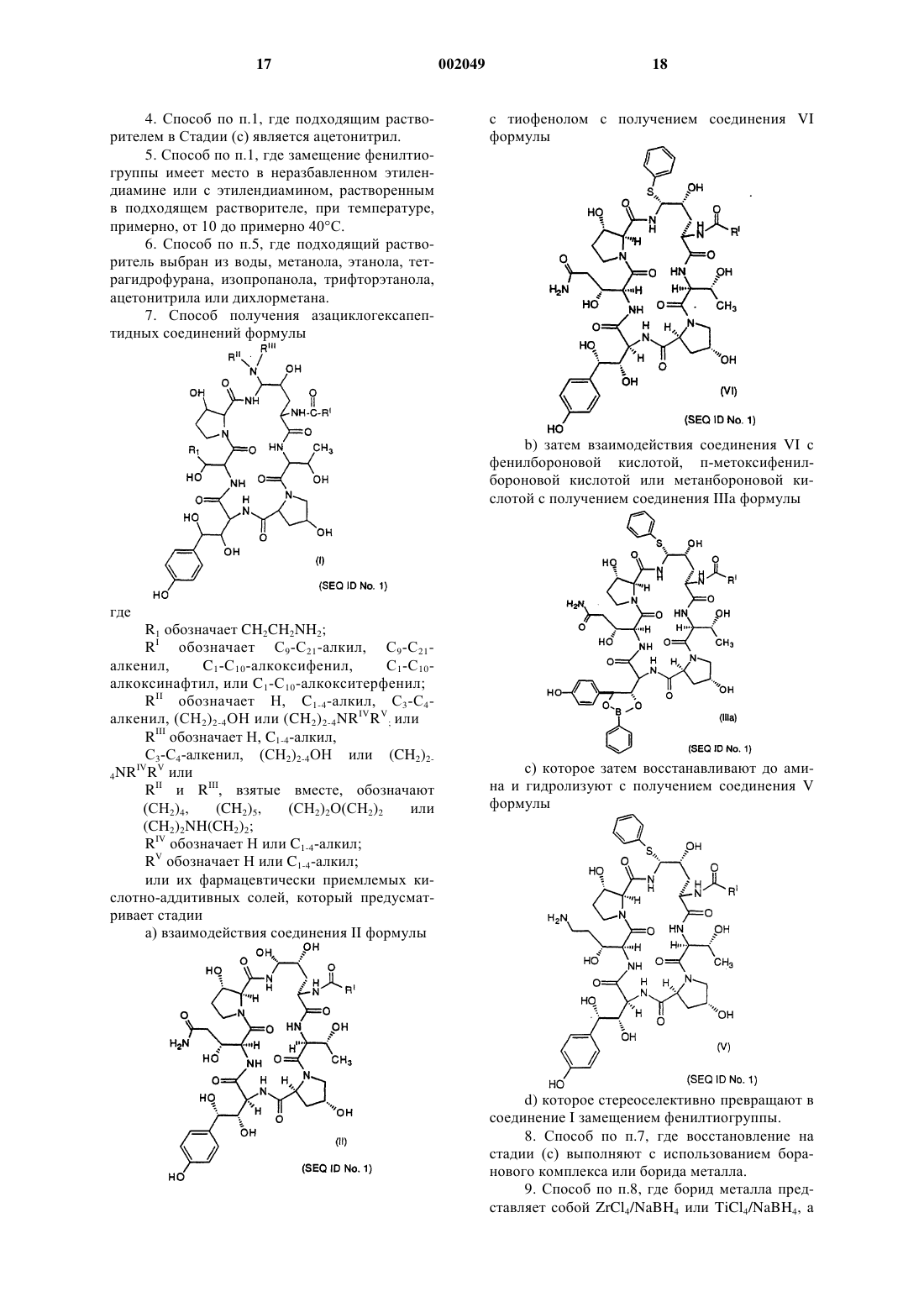
с тиофенолом с получением соединения VI формулы

b) затем взаимодействия соединения VI с фенилбороновой кислотой, п-метоксифенилбороновой кислотой или метанбороновой кислотой с получением соединения IIIa формулы

с) которое затем восстанавливают до амина и гидролизуют с получением соединения V формулы

d) которое стереоселективно превращают в соединение I замещением фенилтиогруппы.
8. Способ по п.7, где восстановление на стадии (с) выполняют с использованием боранового комплекса или борида металла.
9. Способ по п.8, где борид металла представляет собой ZrCl4/NaBH4 илиTiCl4/NaBH4, а борановый комплекс представляет собой боран, комплексированный с диметилсульфидом, дибензилсульфидом, дифенилсульфидом, ТГФ или 1,4-оксатианом или ВН2Сl с диметилсульфидом.
10. Способ по п.7, где соединение II превращают в фенилсульфид реакцией с тиофенолом в подходящем растворителе.
11. Способ по п.10, где подходящим растворителем является ацетонитрил.
12. Способ по п.7, где замещение фенилтиогруппы имеет место в неразбавленном этилендиамине или с этилендиамином, растворенном в подходящем растворителе, при температуре от примерно 10 до примерно 40шС.
13. Способ по п.12, где подходящий растворитель выбран из воды, метанола, этанола, тетрагидрофурана, изопропанола, трифторэтанола, ацетонитрила или дихлорметана.
14. Способ по п.1, где получают соединение формулы

или его фармацевтически приемлемую соль.
15. Способ по п.7, где получают соединение формулы

или его фармацевтически приемлемую соль.
16. Способ по п.1, где получают соединение формулы

или его фармацевтически приемлемую соль.
17. Соединение формулы

где RI обозначает С9-С21-алкил, С9-С21-алкенил, C1-С10-алкоксифенил, C1-С10-алкоксинафтил или С1-С10-алкокситерфенил.
18. Соединение формулы

где RI обозначает С9-С21-алкил, С9-С21-алкенил, C1-C10-алкоксифенил, C1-С10-алкоксинафтил или C1-С10-алкокситерфенил.
Текст
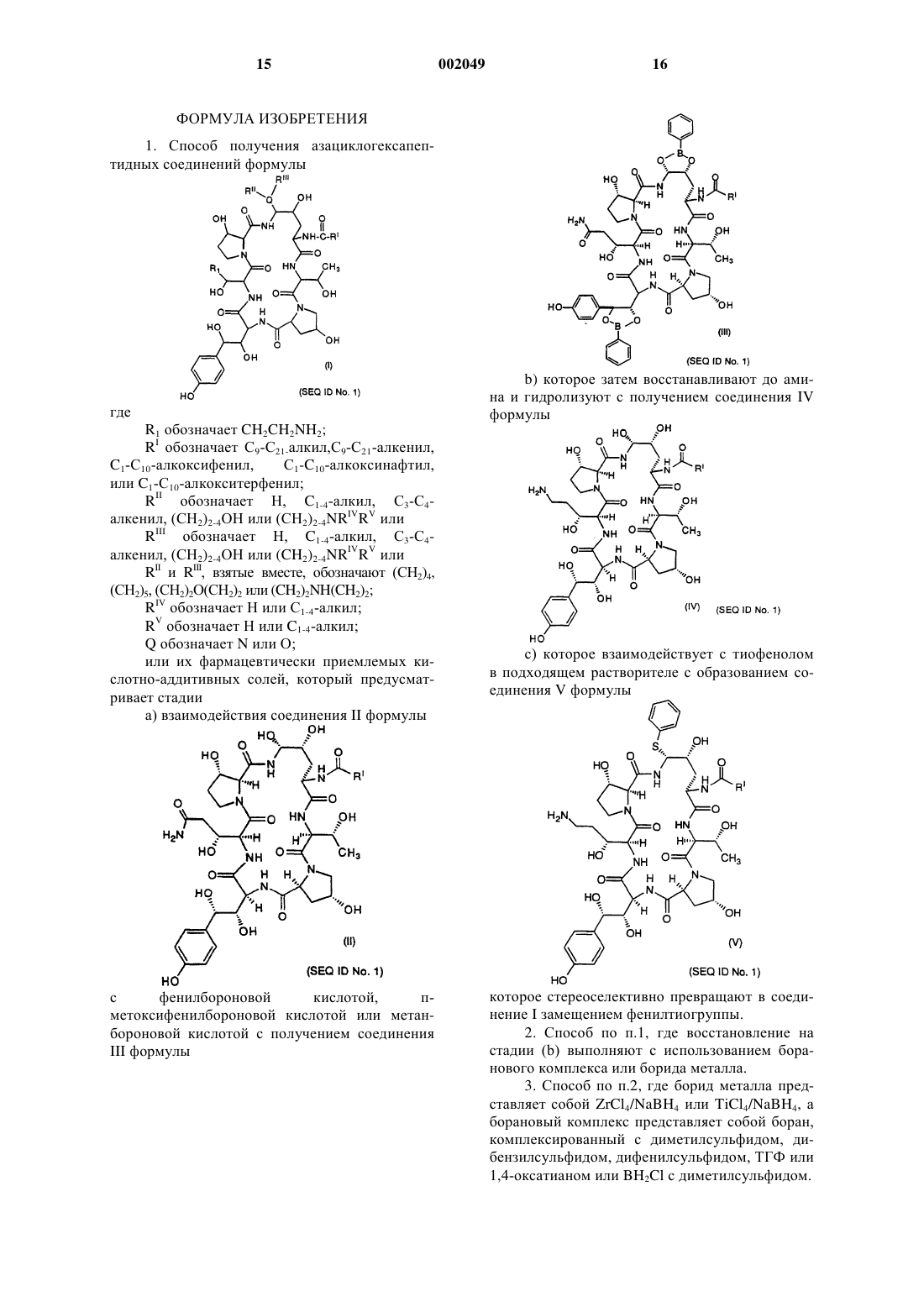
1 Предпосылки изобретения Данное изобретение относится к усовершенствованному способу получения некоторых азациклогексапептидов типа, описанного в U.S.Patent5 378 804, выданном 3 января 1995 года. Первоначальный описанный способ для синтеза этих соединений требовал пяти стадий и не был очень стереоселективным или дающим высокие выходы. Известные способы восстановления первичных амидов, такие как гидрирование, восстановление гидридом металла и электрохимическое восстановление, требовали форсированных (сильнодействующих) условий,не совместимых с другими амидами и функциональными группами в ряду пневмокандина. Эти способы восстановления страдают отсутствием хемоселективности среди различным образом замещенных амидов. Усовершенствованный трехстадийный способ был описан в находящейся в процессе одновременного рассмотрения заявке (Application serial no. 08/386 618), однако этот способ имеет максимальный химический выход в диапазоне приблизительно 23-25%. Новый способ, описанный здесь, приводит к более высоким выходам и более легкому синтезу аналогов этих соединений. Сущность изобретения Данное изобретение относится к способу получения азациклогексапептидов формулы;Q обозначает N или О; или их фармацевтически приемлемых кислотно-аддитивных солей. Было обнаружено, что соединения, получаемые по способу данного изобретения, применимы в лечении грибковых инфекций, в частности, вызываемых Candida, Aspergillus, Histo 002049plasma, Coccidioides и Blastomyces. Было также обнаружено, что они применимы для лечения и предупреждения инфекций, вызываемых Pneumocystis carinii, которые часто обнаруживаются у пациентов с ослабленным иммунитетом, таких как пациенты, имеющие СПИД. Описаны также новые промежуточные соединения, применимые в способе данного изобретения. Сведения, подтверждающи возможность осуществления изобретения Данное изобретение относится к способу получения соединений формулы (I) при помощи стереоскопического способа с высоким выходом. Во всем описании и прилагаемой формуле изобретения представленная химическая формула или название будут включать все оптические изомеры и стереоизомеры, а также рацемические смеси в случаях, когда такие изомеры и смеси существуют. Термин алкил относится к углеводородным группам с прямой, разветвленной или циклической цепью, таким как, например, метил,этил, н-пропил, изопропил, н-бутил, пентил,гексил, гептил, циклопентил, циклогексил, циклогексилметил и т.п. Термин циклоалкил относится к виду алкила, содержащему 3-15 атомов углерода, без перемежающихся или резонирующих двойных связей между атомами углерода. Термин алкенил относится к группам, таким как, например, винил, 1-пропен-2-ил, 1 бутен-4-ил, 2-бутен-4-ил, 1-пентен-5-ил и т.п. Термин алкокси относится к оксиалькильным группам с прямой или разветвленной цепью, таким как, например, метокси, этокси, бутокси, гептокси, додецилокси и т.п. Соединения данного изобретения обычно получают в виде смесей стереоизмерных форм,в которых обычно преобладает одна форма. Условия могут быть скорректированы при помощи средств, находящихся в пределах квалификации специалиста в данной области, для преимущественного получения желательного изомера. Соединениями с предпочтительной стереоизомерной формой, обозначенной здесь как нормальная форма, являются соединения, в которых группа при положении "С-5-орн" находится ниже плоскости при этом положении. Обозначение эпи применялось для тех соединений, в которых группа при положении "С-5-орн" находится выше этой плоскости. Положение "С-5 орн" определено как углерод-5 на 4 гидроксиорнитиновом компоненте. Соединения данного изобретения могут быть введены в форме фармацевтически приемлемых солей. Термин фармацевтически приемлемая соль включает все приемлемые соли. Примерами кислотно-аддитивных солей являются соли хлористо-водородной, азотной, серной, фосфорной, муравьиной, уксусной, триф 3 торуксусной, пропионовой, малеиновой, янтарной, малоновой, метансульфоновой и др. кислот, которые могут быть использованы в виде дозированной формы для модификации характеристик растворимости или гидролиза или могут быть использованы в виде композиций устойчивого длительного высвобождения или композиций пролекарств. В зависимости от конкретной функциональности соединения данного изобретения фармацевтически приемлемые соли данного изобретения включают соли, образованные из катионов, таких как натрий, калий,алюминий, кальций, литий, магний, цинк, и из оснований, таких как аммиак, этилендиамин, Nметилглутамин, лизин, аргинин, орнитин, холин, N,N'-дибензилэтилендиамин, хлорпрокаин,диэтаноламин, прокаин, N-бензилфенетиламин,диэтиламин,пиперазин,трис(гидроксиметил)аминометан и гидроксид тетраметиламмония. Эти соли могут быть получены стандартными процедурами, например, реакцией свободной кислоты с подходящим органическим или неорганическим основанием, или альтернативно реакцией свободного основания с подходящей органической или неорганической кислотой. Также, в случае присутствия кислотной (СООН) или спиртовой группы могут применяться фармацевтически приемлемые эфиры, например,метиловый, этиловый, бутиловый, ацетатный,малеатный, пивалоилоксиметиловый и т.п., и сложные эфиры, о которых в данной области известно, что они модифицируют характеристики растворимости или гидролиза, для использования в качестве композиций устойчивого продолжительного высвобождения или композиций пролекарств. В предпочтительном варианте, способ данного изобретения предусматривает стадии взаимодействия соединения II формулы которое затем восстанавливают до амина и затем гидролизуют с получением соединения IV формулы которое стереоселективно превращают в соединение I через соединение V формулы путем замещения фенилтиогруппы. В альтернативном варианте способ предусматривает стадии взаимодействия соединения Второе предпочтительное соединение, полученное по способу данного изобретения, показано ниже. и затем взаимодействия соединения VI с фенилбороновой кислотой с получением соединения которое затем восстанавливают до амина и затем гидролизуют с получением cоединения V формулы которое стереоселективно превращают в cоединение I замещением фенилтиогруппы. Соединение II, в котором RI обозначает диметилтридецил, описано и заявлено в U.S.Patent No. 5 202 309, который включен здесь в качестве ссылки. Соединение II может быть получено культивированием Zalerion arboricolaATCC 20868 в питательной среде, обогащенной маннитом в качестве первичного источника углерода, как описано в U.S. Patent No. 5 021 341,который также включен здесь в качестве ссылки. Предпочтительное соединение, полученное по способу данного изобретения, показано ниже Данное изобретение иллюстрируется в следующих далее стадиях, где показаны предпочтительные реагирующие вещества, для более ясного представления способа данного изобретения. В следующих далее схемах реакций RI обозначает диметилтридецил. Схема реакций I Схема I. Как показано выше, стадия 1 включает образование соединения бис(фенилбороната) (Соединения III) при взаимодействии соединения II и сухого ТГФ с фенилбороновой кислотой, пметоксифенилбороновой кислотой или метанбороновой кислотой. Можно использовать 1-10 мол. экв. кислоты, предпочтительно 1-3 мол. экв. кислоты. Стадия 2 включает восстановление соединения III до амина (соединения IV) с использованием боранового комплекса, такого как боран с тетрагидрофураном (ТГФ), диметилсульфидом, дифенилсульфидом, дибензилсульфидом,1,4-оксатианом или ВН 2 Сl с диметилсульфидом или боридом металла, таким как ZrCl4/NaBH4 или TiCl4/NaBH4 в ТГФ или другом подходящем растворителе. Восстановление можно также проводить с использованием борановых комплексов с аммиаком, диметиламином, пиридином или пиперазином. Предпочтительные восстановители включают борановые комплексы с тетрагидрофураном (ТГФ), диметилсульфидом,дифенилсульфидом, дибензилсульфидом, 1,4 оксатианом или ВН 2 Сl с диметилсульфидом или боридом металла, такие как ZrCl4/NaBH4 илиTiCl4/NaBH4 в ТГФ или другом подходящем растворителе. Любой амид, не превращенный этим восстановлением, отделяют при помощи обратнофазовой хроматографии. После восстановления стадия 2 включает также удаление фенилборонатных групп во время обработки водной кислотой. Стадия 3 включает две части. Прежде всего, взаимодействие соединения IV с тиофенолом в ацетонитриле и трифторуксусной кислоте(TFA) для получения содержащего фенил сульфид промежуточного соединения. Ожидается,что любая кислота умеренной силы будет давать это промежуточное соединение с хорошим вы 002049 8 ходом. Примеры таких кислот умеренной силы включают (но не ограничиваются ими) трифторуксусную кислоту, фосфорную кислоту и трихлоруксусную кислоту. Могут быть использованы другие сульфиды, такие как 4 метокситиофенол, 2-меркапто-1-метилимидазол и 2-меркаптобензимидазол. Соединение III экстрагируют нанесением разбавленного реакционного раствора на колонку С-18 с обращенной фазой с последующей элюцией метанолом. Количество TFA является решающим для скорости замещения, а также для последующего образования нежелательного сульфида при сегменте гомотирозина циклического пептида. Было найдено, что от примерно 5 до примерно 25%TFA в ацетонитриле давала наилучшие выход и время проведения процесса. Было обнаружено,что предпочтительный диапазон концентрацийTFA был от примерно 7 до примерно 15%. Количество тиофенола, используемого в этой стадии, также является критическим для выхода конечного продукта. Наилучший выход обеспечивали 3-5 экв тиофенола. Предпочтительными условиями для образования сульфида были 5 экв тиофенола в смеси 10% TFA/ацетонитрил при 0 С. Эти условия давали выход 65-70% после твердофазной экстракции. Вторая часть стадии 3 включает замещение фенилтиогруппы. Фенилсульфид реагирует в неразбавленном этилендиамине (1:3) при температуре окружающей среды, давая соединение 1 а с выходом 95%. Эта реакция может происходить при температуре от примерно 10 до примерно 40 С в течение приблизительно 0,5-6,0 ч. Предпочтительно реакция имеет место при комнатной температуре в течение приблизительно 1,5 ч. Реакцию можно также проводить с использованием этилендиамина, растворенного в подходящем растворителе, таком как вода, метанол, этанол, изопропанол, тетрагидрофуран,трифторэтанол, дихлорэтан или ацетонитрил. Схема реакций II Схема II. Как показано выше, стадия I включает взаимодействие соединения II с тиофенолом в ацетонитриле и трифторуксусной кислоте (TFA) с образованием содержащего фенилсульфид промежуточного соединения. Ожидается, что любая кислота умеренной силы будет давать это промежуточное соединение с хорошим выходом. Примеры таких кислот умеренной силы включают (но не ограничиваются ими) трифторуксусную кислоту, фосфорную кислоту и трихлоруксусную кислоту. Могут быть использованы другие сульфиды, такие как 4 метокситиофенол, 2-меркапто-1-метилимидазол и 2-меркаптобензимидазол. Соединение VI осаждают добавлением воды, выделяют фильтрованием. Количество TFA является решающим для скорости замещения, а также для последующего образования нежелательного сульфида при сегменте гомотирозина циклического пептида. Было найдено, что примерно 5-25% TFA в ацетонитриле давала наилучшие выход и время про 002049 10 ведения процесса с предпочтительным диапазоном концентраций TFA от 7 до примерно 15%. Количество тиофенола, используемого в этой стадии, также является критическим для выхода конечного продукта. Наилучший выход обеспечивали 3-5 экв тиофенола. Предпочтительными условиями для образования сульфида были 5 экв тиофенола в смеси 10% TFA/ацетонитрил при 0 С. Эти условия давали выход 65-70% после твердофазной экстракции. Стадия 2 включает дериватизацию (получение производного) содержащего фенил сульфид промежуточного соединения реакцией его с фенилбороновой кислотой, п-метоксифенилбороновой кислотой или метанбороновой кислотой в ТГФ. Можно использовать 1-10 мол. экв. кислоты, предпочтительно 1-3 мол. экв. кислоты. Стадия 3 включает восстановление соединения IIIa до амина (соединения V) с использованием боранового комплекса, такого как боран с тетрагидрофураном (ТГФ), диметилсульфидом, дифенилсульфидом, дибензилсульфидом,1,4-оксатианом или BH2Cl с диметилсульфидом или боридом металла, таким как ZrCl4/NaBH4 или TiCl4/NaBH4 в ТГФ или другом подходящем растворителе. Восстановление можно также проводить с использованием борановых комплексов с аммиаком, диметиламином, пиридином или пиперазином. Предпочтительные восстановители включают борановые комплексы с тетрагидрофураном (ТГФ), диметилсульфидом,дифенилсульфидом, дибензилсульфидом, 1,4 оксатианом или ВН 4 Сl с диметилсульфидом или боридом металла, таким как ZrCl4/NaBH4 илиTiCl4/NaBH4 в ТГФ или другом подходящем растворителе. Любой амид, не превращенный этим восстановлением, отделяют при помощи обратнофазовой хроматографии. Стадия 3 включает также удаление фенилборонатных групп во время обработки водной кислотой. Наконец, стадия 4 включает замещение фенилтиогруппы. Фенилсульфид реагирует в неразбавленном этилендиамине (1:3) при температуре окружающей среды, давая соединениеIа с выходом 95%. Эта реакция может происходить при температуре, примерно, от 10-40C в течение приблизительно 0,5-6,0 ч. Предпочтительно реакция имеет место при комнатной температуре в течение приблизительно 1,5 ч. Реакцию можно также проводить с использованием этилендиамина, растворенного в подходящем растворителе, таком как вода, метанол,этанол, изопропанол, тетрагидрофуран, трифторэтанол, дихлорэтан или ацетонитрил. Соединения III, IIIa, V и VI являются новыми промежуточными соединениями, которые применимы в способе данного изобретения. Данное изобретение описано более подробно в следующих далее примерах, в которых 11 все части, препараты, соотношения и проценты даны в расчете на вес, если нет иных указаний. В этом примере RI был диметилтридецилом. Пример 1. а) Синтез соединения IV из соединения II(через соединение III). Соединение II (60 г навеска (грубо), 52,6 г согласно ВЖХ-анализу, 49,4 ммоль) ) добавляли к сухому ТГФ (1480 мл). К этой суспезнии добавляли PhB(OH)2 (14,56 г, 119 ммоль). Суспензию выдерживали при комнатной температуре,затем нагревали с обратным холодильником. Во время выдерживания при комнатной температуре и нагревания с обратным холодильником реакционный раствор становился гомогенным. Конденсат дефлегмации пропускали через молекулярные сита 3 А в приборе для экстракции твердого тела жидкостью таким образом, чтобы высушить раствор до 25 мол.% воды относительно соединения II. Реакционную смесь охлаждали до температуры окружающей среды и разбавляли 490 мл сухого ТГФ. Полученный,как описано выше, раствор бис(фенилбороната) охлаждали до примерно -7 С и добавляли ВН 3S(СН 3)2 (10 М, 33,3 мл, 6,7 мол. экв.). Реакцию поддерживали при -12-0 С и выдерживали раствор в течение 6,5 ч, после чего медленно добавляли водный раствор НСl (2 М, 140 мл, 280 ммоль). ВЖХ-анализ показал 61% выход соединения IV. Часть погашенного раствора разбавляли водой до получения раствора 1:5,7 об/об ТГФ/вода. Этот раствор наносили на колонку ВЖХ среднего давления с адсорбентом С-18 с обращенной фазой. После загрузки соединение IV элюировали растворами 1:4 об/об ацетонитрил/вода и затем 1:3 об/об ацетонитрал/вода. Обогащенные фракции (80% по площади ВЖХ) объединяли и разбавляли водой до раствора 1:7,3 об/об ацетонитрил/вода. Эту смесь наносили на описанную выше колонку и колонку элюировали метанолом. Обогащенные фракции (85% по площади ВЖХ) объединяли и концентрировали с получением типичного извлечения 88-92% соединения IV для хроматографии и выделения.V). Соединение IV (5,80 г согласно анализу,0,00533 моль) загружали в 0,23 л сухого ацетонитрила и охлаждали до -5 С, после чего добавляли тиофенол (3,10 г, 0,028 моль). На протяжении 20 мин добавляли TFA (36 г, 24,5 мл, 0,318 моль) для поддержания температуры реакционной смеси ниже 0 С. Реакцию выдерживали при-10 С-0 С, пока ВЖХ-анализ не показывал 3% по площади исходного материала (3,75 ч), в этот момент медленно добавляли охлажденную воду(0,56 л) (1 ч) с охлаждением реакционной смеси для поддержания температуры ниже 5. Соглас 002049 12 но анализу выход - и -фенилсульфидного аддукта в виде соли трифторуксусной кислоты был 4,82 г (71%). Этот раствор нагружали на ту же колонку,описанную в стадии а), и колонку промывали водой(0,57 л), затем адсорбированные органические соединения элюировали метанолом (0,50 л). Обогащенные фракции концентрировали роторным испарением и статистическим высоким вакуумом. Это давало 7,20 г (57 мас.% чистоты, 5,1 мас.% воды) неочищенной трифторацетатной соли фенилсульфида в виде аморфного пенообразного твердого вещества. Корректированный выход в стадии выделения для фенилсульфида был 4,10 г (61%) в виде смеси 93:7 - и -диастереомеров аминаля.c) Превращение фенилсульфида в соединение Iа. Неочищенную трифторметансульфонатную соль фенилсульфида (8,4 г неочищенной соли, 57 мас.% чистоты, 0,00377 моль) добавляли к этилендиамину (24 мл) при перемешивании при температуре окружающей среды. Полученный раствор перемешивали в течение 1,5 ч до полного замещения, затем добавляли метанол(40 мл) и затем уксусную кислоту (45 мл) с поддерживанием температуры ниже 25 С с охлаждением на бане со льдом. Получали густую суспензию. Для растворения этой суспензии добавляли воду (160 мл) и водный слой экстрагировали осторожным встряхиванием с гексаном (75 мл). Гексановый слой обратно-экстрагировали водой (40 мл) и объединенный водный слой фильтровали через стеклянный фильтр средней пористости, затем очищали препаративной ВЖХ с использованием колонки С 18 с диаметром 50 мм с элюцией смесью 22% ацетонитрил/78% 0,15% водной уксусной кислоты. Обогащенную фракцию лиофилизировали с получение 4,2 г 85 мас.% чистого соединения I-1 в виде диацетатной соли с выходом в стадии выделения 78%.d) Кристаллизация соединения Iа. Твердое вещество (2,3 г) растворяли в этаноле (25 мл) и затем добавляли воду (2,7 мл). Раствор пропускали через стеклянный фильтр для удаления постороннего материала. К этому фильтрату добавляли уксусную кислоту (0,14 мл) с последующим медленным добавлением(1,75 ч) этилацетата (14 мл). В раствор вносили затравку кристаллизации и содержащий затравку слой выдерживали в течение 1 ч. Оставшийся этилацетат (32 мл) добавляли на протяжении 5 ч и выдерживали еще в течение 1 ч. Кристаллическое твердое вещество собирали на стеклянном фильтре и промывали раствором этанол/этилацетат/вода (6 мл/9 мл/0,5 мл, соответственно). Сырой фильтровальный осадок сушили током азота с получением 1,91 г (1,75 г согласно анализу, 88% извлечение) диацетатной соли соединения 1 а.VI). Соединение II (2,48 кг согласно анализу,2,33 моль) загружали в 78 л сухого ацетонитрила и охлаждали до -8 С, после чего добавляли тиофенол (1,08 кг, 9,8 моль). Для поддержания температуры реакционной смеси ниже 0 С добавляли TFA (12,8 кг, 8,65 л, 112 моль) на протяжении 30 мин. Реакцию выдерживали при -13-0 С,пока ВЖХ-анализ не показал 3% по площади исходного материала (5 ч). В этот момент медленно добавляли охлажденную воду (35 л) при охлаждении реакционной смеси для поддержания температуры ниже 5 С. Продукт VI осаждается во время добавления воды. Добавляли дополнительное количество воды для доведения смеси до 1:3 об/об ацетонитрил/вода, пока рН фильтрата не становился рН 5. Твердое вещество сушили под током азота. Выход (согласно анализу) с 7 оединения VI в виде трифторацетатной соли был 2,03 кг (76%).(через соединение IIIa). Соединение VI (922 г, согласно анализу,0,94 моль) добавляли к сухому ТГФ (44 л). К этой суспензии добавляли PhB(OH)2 (119 г, 0,98 моль). Суспензию выдерживали при комнатной температуре в течение 12 ч, затем нагревали с обратным холодильником. Конденсат дефлегмации пропускали через молекулярные сита 3 А в приборе для экстракции твердого тела жидкостью таким образом, чтобы высушить раствор до 25 мол.% воды относительно cоединенияVI. Реакционную смесь охлаждали и добавляли дополнительное количество сухого ТГФ для восстановления исходного объема смеси. Смесь охлаждали до-4 С. Добавляли неразбавленный ВН 3SМе 2 (494 г, 6,51 моль) на протяжении 15 мин и температуру реакционной смеси поддерживали при -4-0 С. Развитие реакции наблюдали при помощи ВЖХ, пока не оставалось 30% исходного материала, что свидетельствовало о конце выдерживания реакции (9 ч). Смесь охлаждали до -10 С и медленно гасили 2 Н НСl (2,98 л). Выход согласно анализу Соединения V в виде гидрохлоридной соли был 537 г (61%). Погашенный раствор разбавляли до смеси 1:3,76 об/об ТГФ/вода и наносили на колонку адсорбента RP-C18 (ОФ-С 18) среднего давления(16,8 кг). После загрузки колонку элюировали смесью 1:2,64 об/об ацетонитрил/вода и затем смесью 1:2,45 об/об ацетонитрил/вода. Обогащение фракции (80% по ВЖХ-площади) объединяли с получением 90% выхода соединенияV. Объединенные обогащенные фракции разбавляли водой до раствора 1:2 об/об ацетонитрил/вода. Эту смесь объединяли с разбавленными обогащенными фракциями серии восстановления такого же размера и нагружали на ту жеV элюировали метанолом. Обогащенные фракции (85% по площади) объединяли и концентрировали роторным испарением с получением 98% извлечения соединения V. Соединение V превращали в cоединениеIа, как описано выше в примере 1 с и 1d. СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ(ii) НАЗВАНИЕ ИЗОБРЕТЕНИЯ: Способ получения некоторых азациклогексапептидов ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения азациклогексапептидных соединений формулыQ обозначает N или О; или их фармацевтически приемлемых кислотно-аддитивных солей, который предусматривает стадии а) взаимодействия соединения II формулы с фенилбороновой кислотой,пметоксифенилбороновой кислотой или метанбороновой кислотой с получением соединенияb) которое затем восстанавливают до амина и гидролизуют с получением соединения IV формулы которое стереоселективно превращают в соединение I замещением фенилтиогруппы. 2. Способ по п.1, где восстановление на стадии (b) выполняют с использованием боранового комплекса или борида металла. 3. Способ по п.2, где борид металла представляет собой ZrCl4/NaBH4 или TiCl4/NaBH4, а борановый комплекс представляет собой боран,комплексированный с диметилсульфидом, дибензилсульфидом, дифенилсульфидом, ТГФ или 1,4-оксатианом или BH2Cl с диметилсульфидом. 17 4. Способ по п.1, где подходящим растворителем в Стадии (с) является ацетонитрил. 5. Способ по п.1, где замещение фенилтиогруппы имеет место в неразбавленном этилендиамине или с этилендиамином, растворенным в подходящем растворителе, при температуре,примерно, от 10 до примерно 40 С. 6. Способ по п.5, где подходящий растворитель выбран из воды, метанола, этанола, тетрагидрофурана, изопропанола, трифторэтанола,ацетонитрила или дихлорметана. 7. Способ получения азациклогексапептидных соединений формулыb) затем взаимодействия соединения VI с фенилбороновой кислотой, п-метоксифенилбороновой кислотой или метанбороновой кислотой с получением соединения IIIa формулыRV обозначает Н или C1-4-алкил; или их фармацевтически приемлемых кислотно-аддитивных солей, который предусматривает стадии а) взаимодействия соединения II формулы с) которое затем восстанавливают до амина и гидролизуют с получением соединения V формулыd) которое стереоселективно превращают в соединение I замещением фенилтиогруппы. 8. Способ по п.7, где восстановление на стадии (с) выполняют с использованием боранового комплекса или борида металла. 9. Способ по п.8, где борид металла представляет собой ZrCl4/NaBH4 или TiCl4/NaBH4, а борановый комплекс представляет собой боран,комплексированный с диметилсульфидом, дибензилсульфидом, дифенилсульфидом, ТГФ или 1,4-оксатианом или ВН 2 Сl с диметилсульфидом. 10. Способ по п.7, где соединение II превращают в фенилсульфид реакцией с тиофенолом в подходящем растворителе. 11. Способ по п.10, где подходящим растворителем является ацетонитрил. 12. Способ по п.7, где замещение фенилтиогруппы имеет место в неразбавленном этилендиамине или с этилендиамином, растворенном в подходящем растворителе, при температуре от примерно 10 до примерно 40 С. 13. Способ по п.12, где подходящий растворитель выбран из воды, метанола, этанола,тетрагидрофурана, изопропанола, трифторэтанола, ацетонитрила или дехлорметана. 14. Способ по п.1, где получают соединение формулы или его фармацевтически приемлемую соль. 17. Соединение формулы или его фармацевтически приемлемую соль. 15. Способ по п.7, где получают соединение формулы или его фармацевтически приемлемую соль. 16. Способ по п.1, где получают соединение формулы
МПК / Метки
МПК: C07K 7/56
Метки: способ, некоторых, получения, азациклогексапептидов
Код ссылки
<a href="https://eas.patents.su/11-2049-sposob-polucheniya-nekotoryh-azaciklogeksapeptidov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения некоторых азациклогексапептидов</a>
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Мейллян Пьер, Бьенейм Юг, Ансель Жан-Эрик
МПК: C07C 39/19, A61K 31/355, B01J 31/24...
Метки: cпособ, фенолов, способ, замещенных, использованием, витамина, получения
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Свинделл Чарльз С., Чандер Мадхави С., Систи Николас Дж.
МПК: C07D 305/14
Метки: соединение, соединения, промежуточное, способ, получения, паклитаксела, промежуточного
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Производные индолилпирролиденметилпиррола, способ их получения, фармацевтическая композиция и комбинированный препарат на их основе, способ лечения с их использованием и промежуточный продукт для их получения

Номер патента: 1055
Опубликовано: 28.08.2000
Авторы: Феррари Марио, Тиболла Марчеллино, Д`алессио Роберто, Колотта Франческо, Изетта Анна Мария, Барджотти Альберто
МПК: C07F 5/02, C07D 403/14
Метки: композиция, препарат, фармацевтическая, индолилпирролиденметилпиррола, лечения, продукт, использованием, основе, производные, получения, способ, промежуточный, комбинированный
Формула / Реферат:
1. Производные (1Н-индол-2-ил)-5-[(2Н-пиррол-2-илиден)метил]-1H-пиррола формулы (I) где каждый из R1, R2, R3 и R4, которые являются одинаковыми или разными, представляет независимо водород, (C1-C6)-алкил, галоген, циано, нитро, гидрокси, (C1-C6)-алкокси, незамещенный или замещенный фенилом, (C1-C6)-алкилкарбонилокси, -NRaRb, в котором каждый из Ra и Rb независимо представляет водород или (C1-C6)-алкил, (C1-С6)-алкилкарбониламино, карбокси,...
Циклическое соединение, способ его получения и способ получения смеси диастереомеров

Номер патента: 867
Опубликовано: 26.06.2000
Авторы: Акиба Тосифуми, Симизу Садахиро, Охта Наоки, Тодзо Тосиаки, Саито Татсуру, Ебата Тутому, Хираи Кеиити
МПК: C07C 253/30, C07D 209/54
Метки: соединение, получения, циклическое, смеси, диастереомеров, способ
Формула / Реферат:
1. Циклическое соединение, представленное формулой (I) где n является целым числом от 2 до 5; R1 представляет атом водорода или заместитель, изображаемый формулой где каждый из Ra, Rb и Rc отличается один от другого и представляет фенильную, фенилметильную или нафтильную группу, которая может быть замещена, по меньшей мере, одним заместителем, выбираемым из группы, состоящей из алкильной группы с 1-4 атомами углерода, алкоксильной...
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Бонне Алан, Мазюри Алан, Дельтиль Мишель
МПК: C07H 17/08
Метки: активных, производные, применение, способ, получения, биологически, 5-0-дезозаминил-6-0-метилэритронолида, продуктов
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Предыдущий патент: Фрезерное устройство, размещаемое в скважине
Следующий патент: Электронный многотарифный счетчик электрической энергии
Случайный патент: Способ и устройство для обработки жидкости