Процесс получения этиловых эфиров 4 – (8-хлор-5,6-дигидро-11н-бензо-(5,6)-циклогепта-(1,2в) пиридин-11-илиден)-1-пиперидинкарбоновой кислоты (лоратадина)
Номер патента: 7740
Опубликовано: 29.12.2006
Авторы: Канната Винченцо, Мичиелетто Иван, Поли Стефано, Котарка Ливиус







Формула / Реферат
1. Способ получения лоратадина, включающий:
а) взаимодействие 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина формулы I

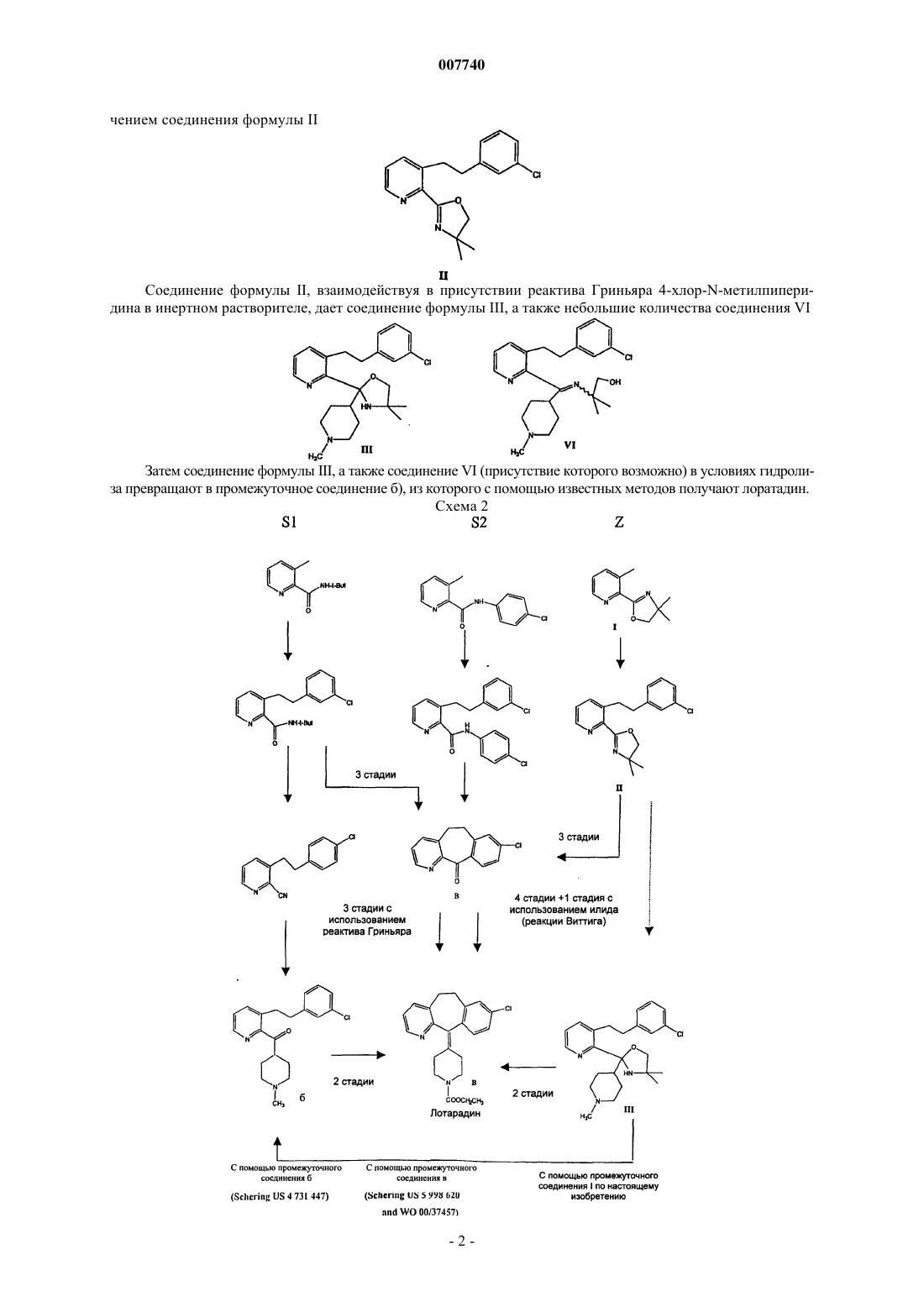
с 3-хлорбензилхлоридом в присутствии сильного основания с получением 3-[2-(3-хлорфенил)этил]-2-(4,4-диметил-4,5-дигидрооксазол-2-ил)пиридина формулы II

б) взаимодействие соединения II с реактивом Гриньяра 4-хлор-N-метилпиперидином в инертном растворителе с получением 3-[2-(3-хлорфенил)этил]-2-[4,4-диметил-2-(1-метилпиперидин-4-ил)оксазолидин-2-ил]пиридина формулы III

в) гидролиз соединения III с получением промежуточного соединения формулы б

которое превращают в лоратадин известными методами.
2. Способ по п.1, отличающийся тем, что в качестве указанного сильного основания используют диизопропиламид лития.
3. Способ по п.1, отличающийся тем, что в качестве указанного инертного растворителя используют тетрагидрофуран.
4. Способ синтеза промежуточного соединения б, включающий:
а) взаимодействие 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина формулы I

с 3-хлорбензилхлоридом в присутствии сильного основания с получением 3-[2-(3-хлорфенил)этил]-2-(4,4-диметил-4,5-дигидрооксазол-2-ил)пиридина формулы II

б) взаимодействие соединения II с реактивом Гриньяра 4-xлop-N-метилпиперидином в инертном растворителе с получением 3-[2-(3-хлорфенил)этил]-2-[4,4-диметил-2-(1-метилпиперидин-4-ил)оксазолидин-2-ил]пиридина формулы III

в) гидролиз соединения III с получением промежуточного соединения формулы б
5. Способ по п.4, отличающийся тем, что в качестве указанного растворителя используют тетрагидрофуран, а в качестве сильного основания используют диизопропиламид лития.
6. Способ по п.4, отличающийся тем, что гидролиз проводят в кислой среде.
7. 2-(4,4-Диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридин формулы I

8. 3-[2-(3-Хлорфенил)этил]-2-(4,4-диметил-4,5-дигидрооксазол-2-ил)пиридин формулы II

9. 3-[2-(3-Хлорфенил)этил]-2-[4,4-диметил-2-(1-метил-4-пиперидил)-2-оксазолидинил]пиридин формулы III

10. Применение 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина формулы I в качестве промежуточного соединения для получения лоратадина.
11. Применение 3-[2-(3-хлорфенил)этил]-2-(4,4-диметил-4,5-дигидрооксазол-2-ил)пиридина формулы II в качестве промежуточного соединения для получения лоратадина.
12. Применение 3-[2-(3-хлорфенил)этил]-2-[4,4-диметил-2-(1-метил-4-пиперидил)оксазолидин-2-ил]пиридина формулы III в качестве промежуточного соединения для получения лоратадина.
Текст
007740 Настоящее изобретение касается процесса получения лоратадина - лекарственного средства, обладающего антигистаминной активностью. Лоратадин представляет собой 4-(8-хлор-5,6-дигидро-11 Нбензо(5,6)циклогепта[1,2-b]пиридин-11-илиден)-1-пиперидин этилкарбоксилат (The Merck Index, 12th ed.,5608, p.953). В частности, настоящее изобретение касается процесса синтеза лоратадина исходя из нового промежуточного соединения: 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина. Лоратадин впервые был описан Schering в патенте США 4.282.233. В указанном патенте при синтезе лоратадина исходят из 8-хлор-11-(1-метилпиперид-4-илиден)-6,11-дигидро-5 Н-бензо[5,6]циклогепта[1,2-b]пиридина (а), который взаимодействует с этилхлорформиатом в бензоле. Эта реакция приведена на схеме 1. Схема 1 Из анализа патентной литературы, касающейся синтеза лоратадина, установлено, что соединение(а) получают из двух основных промежуточных соединений. Первым из них является [3-[2-(3-хлорфенил)этил]пиридин-2-ил]-(1-метил-4-пиперидил)метанон формулы б), а второе представляет собой 8 хлор-5,6-дигидробензо[5,6]циклогепта[1,2-b]пиридин-11-он, имеющий формулу в). В патенте Schering, США 4.731.447 описан синтез соединения а) исходя из соединения б), причем последнее получают четырехстадийным синтезом из 3-метил-2-цианопиридина. Путем циклизации соединения б) с использованием гиперкислоты, кислотная константа которой ниже -12, получают соединение а). В патенте США 4.731.447 описывается одностадийный синтез соединения в) исходя из 3-[2-(3-хлорфенил)этил]-2-пиридинкарбоксамида путем его обработки гиперкислотой, или трехстадийный синтез без применения гиперкислоты. Однако промышленное применение гиперкислот из-за их высокой коррозионной активности проблематично. В патенте Schering, США 4.659.716 описан синтез соединения а) исходя из соединения в). Соединение а) получают взаимодействием соединения в) с реактивом Гриньяра 4-хлор-N-метилпиперидином, с получением 8-хлор-11-(1-метилпиперидин-4-ил)-6,11-дигидро-5 Н-бензо[5,6]циклогепта[1,2-b]пиридин-11-ола, который при последующем дегидрировании дает соединение а). В патенте Schering, заявка WO 00/37457 описан другой процесс, в котором исходным является соединение в), а лоратадин получают без использования соединения а). В этом случае взаимодействие между соединением в) и илидом фосфора протекает по реакции Виттига; в результате реакции получают промежуточное соединение нестабильный -гидроксифосфонат. Из-за своей нестабильности указанный -гидроксифосфонат необходимо стабилизировать, добавив к нему протонирующий агент (воду или уксусную кислоту), и только после этого возможно получение лоратадина с помощью термического разложения. Однако, как отмечается в заявке WO 00/37457, указанный продукт не находится в чистом виде; для того чтобы удалить примеси растворителя, примеси соединения в), а также примеси фосфорсодержащих соединений, необходима его очистка. Такую очистку проводят многократной дистилляцией, а завершают перекристаллизацией. Таким образом, помимо того, что указанный процесс является трудоемким, он вызывает потерю продукта. Следовательно, описанный синтез имеет много недостатков. К ним относятся: большое количество стадий; использование реагентов, с которыми трудно иметь дело в промышленных масштабах; образование нежелательных продуктов побочных реакций, требующих очистки продукта или промежуточного соединения, что снижает общий выход по процессу. Удивительно, что, как было обнаружено в настоящее время, процесс, представляющий собой один из аспектов представленного изобретения, делает возможным синтез лоратадина исходя из 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина формулы I Соединение I взаимодействует с 3-хлорбензилхлоридом в присутствии сильного основания с полу-1 007740 чением соединения формулы II Соединение формулы II, взаимодействуя в присутствии реактива Гриньяра 4-хлор-N-метилпиперидина в инертном растворителе, дает соединение формулы III, а также небольшие количества соединения VI Затем соединение формулы III, а также соединение VI (присутствие которого возможно) в условиях гидролиза превращают в промежуточное соединение б), из которого с помощью известных методов получают лоратадин. Схема 2-2 007740 На схеме 2 процесс, представляющий собой предмет настоящего изобретения, сравнивают с процессами, описанными Schering; эти процессы обозначены аббревиатурой Z, S1 и S2 соответственно. Второй аспект настоящего изобретения представлен новыми соединениями формул I, II и III, а также их применением для получения лоратадина. Третий аспект настоящего изобретения представлен процессом получения промежуточного соединения б) исходя из соединения I, которое после обработки при 0 С диизопропиламидом лития (LDA), а затем 3-хлорбензилхлоридом дает соединение II. Последующая обработка соединения II реактивом Гриньяра 4-хлор-N-метилпиперидином дает соединение III, которое в результате гидролиза приводит к промежуточному соединению б). Четвертый аспект настоящего изобретения представлен способом (альтернативным по отношению к процессам S1 и S2 схемы 2) получения промежуточного соединения в) исходя из соединения II, которое в результате гидролиза дает 3-[2-(3-хлорфенил)этил]пиридин-2-карбоновую кислоту формулы IV Кислотную функция соединения IV превращают в соответствующий хлорид, а затем связывают по реакции Фриделя-Крафта с получением промежуточного соединения в). Пятым аспектом настоящего изобретения является процесс синтеза лоратадина, который осуществляется получением соединения II по описанному выше процессу в целях приготовления соединения в) с последующим превращением последнего соединения в лоратадин по известным методикам. Предпочтительный вариант настоящего изобретения состоит в использовании в качестве исходного соединения 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина формулы I. Очевидно, что использование аналогов оксазолина, таких как 4-метил-, 4,4-диметил- или 4-этилоксазолин, имеющиx такие же заместители в положении 2, входит в границы настоящего изобретения. Выбор 4,4-диметилоксазолина (соединение I) основан исключительно на критерии экономичности процесса. Путь, который, как было обнаружено, является наиболее выгодным путем получения соединения I,описан в работе Fryzuk M.d., Jafarpour L. and Rettig S.J. Tetrahedron: Asymmetry, 1998, 9, 3191. В этой статье приведены экспериментальные условия получения 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина. Последнее соединение получают путем взаимодействия 2-циано-3-метилпиридина и 1,1-диметиламиноэтанола при использовании в качестве катализатора ZnCl2; реакция протекает в течение 15 ч при 140 С в отсутствиe растворителя. Полученное таким образом соединение I взаимодействует с 3-хлорбензилхлоридом или его аналогом (см. схему 3) в присутствии сильного основания, предпочтительно диизопропиламида лития (LDA). Указанная реакция проходит в инертном растворителе (тетрагидрофуран, толуол, диэтиловый эфир или гексан); предпочтителен тетрагидрофуран и диапазон температур от -15 до 25 С, а особенно предпочтителен температурный диапазон от -5 до 5 С. В итоге получают 3-[2-(3-хлорфенил)этил]-2-(4,4-диметил 4,5-дигидрооксазол-2-ил)пиридин формулы II. Схема 3-3 007740 В результате взаимодействия соединения II с реактивом Гриньяра 4-хлор-N-метилпиперидином(приготовленным по стандартной технологии) получают 3-[2-(3-хлорфенил)этил]-2-[4,4-диметил-2-(1 метилпиперидин-4-ил)оксазолидин-2-ил]пиридин формулы III; реакция протекает в тетрагидрофуране при температуре от -40 до 0 С (предпочтительно от -20 до -10 С). Добавление к соединению II реактива Гриньяра происходит избирательно, и поэтому, как видно из схемы 2, исключаются дополнительные стадии, которые, однако, были необходимы для процесса ScheringS1. В завершениe соединение III гидролизом можно превратить в соединение б), из которого по известной технологии получают лоратадин. Следующие далее примеры предназначены для большей иллюстрации настоящего изобретения, не ограничивая его. Пример 1. Синтез 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина.(1,695 мол.) 2-метил-2-аминопропанола и 5,77 г безводного ZnCl2 (0,042 мол.); температуру повысили до 140 С (раствор образуется при температуре, составляющей приблизительно 60 С) и температуру в 140 С поддерживали в течение 15-18 ч. Во время реакции выделяются пары аммиака, и их собирают с помощью ловушки с разбавленной соляной кислотой. Окончание реакции контролируется методом тонкослойной хроматографии. По окончании реакции смесь охлаждают до 60 С и при этой температуре смесь фильтруют через тигель Gooch с получением приблизительно 190,06 г солей белого цвета. Затем охлаждение продолжают при комнатной температуре и добавляют 250 г толуола и 99 г насыщенного раствораNaCl. Водную фазу повторно экстрагируют толуолом, а органические фазы объединяют и вновь промывают водой, удаляя таким образом не прореагировавший диметиламиноэтанол. Раствор толуола упаривают, получив 166,5 г масла блекло-красного цвета; в этом масле по данным титра высокоэффективной жидкостной хроматографии (ВЭЖХ) содержалось 95,3% соединения I, a остальное представляло собой соединение формулы V. В свою очередь, соединение V путем его обработки мезилхлоридом и триэтиламином при -5 С в СН 2 Сl2 можно превратить в оксазолин. Соединение I также можно подвергнуть дистилляции при 105-112 С и давлении в 1,5 мм рт.ст., оксазолиновый титр составляет 97%. Выход 98,5%. Соединение I. 1H-NMR (200 MГц, CDCl3)(ppm): 8,52 (dd, J=4,3 и 1,8 Гц, 1H); 7,57 (dd, J=7,9 и 1,8 Гц,1H); 7,25 (dd, J=7,9 и 4,3 Гц, 1H); 4,14 (s, 2H); 2,59 (s, 3H); 1,42 (s, 6H). Пример 2. Синтез 3-[2-(3-хлорфенил)этил]-2-(4,4-диметил-4,5-дигидроксазол-2-ил)пиридина. В 3 л реактор с рубашкой, снабженный механической мешалкой и термометром, поместили 521,7 мл безводного тетрагидрофурана и 521,57 мл 2 М раствора диизопропиламида лития и, выдерживая ингредиенты в атмосфере азота, температуру внутри реактора довели до -5 С. Затем медленно добавляли раствор, содержащий 158,8 г оксазолина (0,835 мол.) и 834,7 мл тетрагидрофурана, выдерживая при этом температуру, приблизительно равную 0 С. После добавления нескольких капель раствора произошло интенсивное сине-фиолетовое окрашивание. Общее время введения составило приблизительно 1 ч. Затем в течение приблизительно 1,5 ч добавляли 154,6 г (0,96 мол.) 3-хлорбензилхлорида, продолжая при этом поддерживать температуру, равную 0 С. В конце реакции добавили 521,7 г воды, доводя температуру до 20-25 С. Затем две образовавшиеся фазы разделили и органическую фазу выпарили с получением масла. Полученный таким образом сырой продукт перенесли в толуол и отфильтровали. Фильтрат сконцентрировали, получив 268,1 г масла; титр согласно ВЭЖХ составил 79%, конверсия 91,8%, выход 80,2%. 1 Получение реактива Гриньяра. В 400 мл реактор, снабженный механической мешалкой, конденсатором, термометром и капельной воронкой на 100 мл, поместили 10 г опилок магния (0,41 мол.) и 163 г безводного тетрагидрофурана; действия осуществляли в атмосфере азота. Систему довели до 60 С и добавили приблизительно 1 млVitride (70% вес./вес. раствор дигидро-бис(2-метоксиэтокси)алюмината натрия в толуоле), а также приблизительно 5% раствор 4-хлор-N-метилпиперидина (57,7 г; 0,41 мол.) в 163 г безводного тетрагидрофурана. Спустя несколько минут наблюдалось слабое выделение тепла. Затем медленно добавили остаток раствора. По завершении реакции смесь выдерживали в течение ночи при температуре 60 С. Суспензию Гриньяра, легко перемешиваемую, без дальнейших превращений использовали на последующей стадии присоединения. Присоединение. В 1 л реактор, снабженный механической мешалкой, конденсатором, термометром и капельной воронкой на 500 мл, поместили 116 г сырого 3-[2-(3-хлорфенил)этил]-2-(4,4-диметил-4,5-дигидрооксазол-2 ил)пиридина (0,32 мол.) и 368,6 г безводного тетрагидрофурана; действия осуществляли в атмосфере азота. Полученный раствор охладили до -20 С. После этого начали введение реактива Гриньяра, полученного на предыдущей стадии, при этом температуру сохраняли равной приблизительно -20 С. Затем охлаждение прекратили, и система вернулась к комнатной температуре. Протекание реакции контролировали методом ВЭЖХ. После того как смесь пробыла при комнатной температуре в течение ночи, мониторингом методом ВЭЖХ было показано приблизительно 2,9% (по площади) количество непрореагировавшего 3-[2-(3-хлорфенил)этил]-2-(4,4-диметил-4,5-дигидрооксазол-2-ил)пиридина. Смесь разбавили толуолом (200 мл) и медленно добавляли 270 г уксусной кислоты (водный 10% вес./вес. раствор). Получившуюся смесь перемешивали в течение приблизительно 30 мин, а затем отставили еще на 30 мин. Провели разделение фаз. Органическая фаза дала 146 г сырого продукта в виде темного масла, представляющего собой смесь соединения III и небольшого количества соединения VI. В последующих реакциях эта смесь двух продуктов использовалась без дальнейшей очистки. В 250 мл круглодонную колбу, снабженную магнитной мешалкой и конденсатором, поместили 24,1 г сырого продукта (полученного по реакции из примера 3), 51,4 г воды и 10,2 г НСl (31% вес./вес.). Полученный таким образом раствор довели до состояния перегонки. После перегонки с обратным орошением в течение 14 ч контроль методами тонкослойной хроматографии и 1H-ЯМР показали исчезновение субстрата. Полученную смесь охладили до комнатной температуры, разбавили дихлорметаном (СН 2 Сl2)(50 мл) и, добавив гидроксид натрия (NaOH) (30% вес./вес.), рН довели до величины 8,5-9. Органическую фазу высушили над сульфатом натрия (Na2SO4), а растворитель выпарили в вакууме. Сырой продукт в виде темного масла проанализировали методом ВЭЖХ (площадь приблизительно составила 58%). Кетон может быть перекристаллизован из воды в форме гидрохлорида. В 2 л реактор, снабженный механической мешалкой, поместили 268,1 г (0,67 мол.) соединения II и 645,6 г воды; к полученной дисперсии при комнатной температуре добавили 787,3 г 31% НСl. Затем получившуюся в результате смесь довели до состояния перегонки. Контроль за взаимодействием осуществляли методом тонкослойной хроматографии. По окончании реакции смесь охладили до 60 С, добавили 1340,6 г толуола и с помощью 30% NaOH значение рН этой смеси довели до величины 5; образовавшиеся фазы разделили. Водную фазу повторно экстрагировали толуолом, а две органические фазы объединили. После выпаривания толуола было получено 219 г масла темного цвета с 80% титром. Перекристаллизацией из толуола при 0 С из этого темного масла можно получить твердое вещество белого цвета. Пример 6. Синтез 8-хлор-5,6-дигидробензо[5,6]циклогепта[1,2-b]пирид-11-она. 5 г соединения IV с 70% титром поместили в реактор на 250 мл. При комнатной температуре добавляли по каплям 70 г SOCl2, смесь довели до температуры, составляющей 55-60 С, и оставили на 3 ч для дальнейшего взаимодействия. Исчезновение кислоты контролировали методом тонкослойной хроматографии. После этого избыток SOCl2 отогнали, получив при этом 6 г осадка темного цвета. Указанный осадок растворили приблизительно в 10 мл дихлорэтана. Реакционную смесь охладили до 0 С и добавляли порциями 5,3 г АlСl3. Образовавшуюся при этом смесь оставили на ночь при температуре в диапазоне от -5 до 0 С. По окончании реакции смесь подкислили 1N HCl, поддерживая при этом температуру в диапазоне от 10 до 15 С. Образовавшиеся фазы разделили, вторую кислотную экстракцию осуществляли 50 мл воды, водные фазы объединили. Подщелочили с помощью NaOH до рН 12, а затем повторно экстрагировали толуолом. После выпаривания растворителя было получено твердое вещество, которое после перекристаллизации из диизопропилового эфира дало 2 г бледно-желтого твердого вещества, его ЯМР титр был выше 99%, а выход составил 62%. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения лоратадина, включающий: а) взаимодействие 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина формулы I с 3-хлорбензилхлоридом в присутствии сильного основания с получением 3-[2-(3-хлорфенил)этил]2-(4,4-диметил-4,5-дигидрооксазол-2-ил)пиридина формулы II в) гидролиз соединения III с получением промежуточного соединения формулы б которое превращают в лоратадин известными методами. 2. Способ по п.1, отличающийся тем, что в качестве указанного сильного основания используют диизопропиламид лития. 3. Способ по п.1, отличающийся тем, что в качестве указанного инертного растворителя используют тетрагидрофуран. 4. Способ синтеза промежуточного соединения б, включающий: а) взаимодействие 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина формулы I с 3-хлорбензилхлоридом в присутствии сильного основания с получением 3-[2-(3-хлорфенил)этил]2-(4,4-диметил-4,5-дигидрооксазол-2-ил)пиридина формулы II в) гидролиз соединения III с получением промежуточного соединения формулы б-7 007740 5. Способ по п.4, отличающийся тем, что в качестве указанного растворителя используют тетрагидрофуран, а в качестве сильного основания используют диизопропиламид лития. 6. Способ по п.4, отличающийся тем, что гидролиз проводят в кислой среде. 7. 2-(4,4-Диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридин формулы I 10. Применение 2-(4,4-диметил-4,5-дигидрооксазол-2-ил)-3-метилпиридина формулы I в качестве промежуточного соединения для получения лоратадина. 11. Применение 3-[2-(3-хлорфенил)этил]-2-(4,4-диметил-4,5-дигидрооксазол-2-ил)пиридина формулы II в качестве промежуточного соединения для получения лоратадина. 12. Применение 3-[2-(3-хлорфенил)этил]-2-[4,4-диметил-2-(1-метил-4-пиперидил)оксазолидин-2 ил]пиридина формулы III в качестве промежуточного соединения для получения лоратадина.
МПК / Метки
МПК: C07D 221/16, C07D 401/04, C07D 413/04
Метки: пиридин-11-илиден)-1-пиперидинкарбоновой, этиловых, эфиров, кислоты, 8-хлор-5,6-дигидро-11н-бензо-(5,6)-циклогепта-(1,2в, процесс, лоратадина, получения
Код ссылки
<a href="https://eas.patents.su/10-7740-process-polucheniya-etilovyh-efirov-4-8-hlor-56-digidro-11n-benzo-56-ciklogepta-12v-piridin-11-iliden-1-piperidinkarbonovojj-kisloty-loratadina.html" rel="bookmark" title="База патентов Евразийского Союза">Процесс получения этиловых эфиров 4 – (8-хлор-5,6-дигидро-11н-бензо-(5,6)-циклогепта-(1,2в) пиридин-11-илиден)-1-пиперидинкарбоновой кислоты (лоратадина)</a>
Способ получения [is-[1a,2b,3b,4a(s*)]]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3н-имидазо[4,5-b]пиридин-3-ил]-n-этил-2,3-дигидроксициклопентанкарбоксамида.

Номер патента: 1989
Опубликовано: 22.10.2001
Авторы: Рейлли Лоренс В., О'брайен Майкл К, Цуей Чинг Т., Паунер Тори Х., Леон Патрик, Шах Харшавадан К., Гарсиа Эрве, Ванасс Бенуа Дж., Томпсон Майкл Д., Вальтер Фрэнсис Л.
МПК: C07D 409/12
Метки: получения, способ, is-[1a,2b,3b,4a(s*)]]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3н-имидазо[4,5-b]пиридин-3-ил]-n-этил-2,3-дигидроксициклопентанкарбоксамида
Формула / Реферат:
1. Способ получения [1S-[1a,2b,3b,4a(S*)]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамида (соединение (I)), включающий взаимодействие [1S-[1a,2b,3b,4a(S*)]]-4-[[3-амино-4-[[1-[(3-хлор-2-тиенил)метил]пропил]амино]-2-пиридинил]амино]-N-этил-2,3-дигидроксициклопентанкарбоксамида (соединение (IX)) со сложным эфиром ортоформиата, ацетатом формамидина или диметилацеталем...
Способ получения 11-амино-3-хлор-6,11-дигидро-5,5-диоксо-6-метилдибензо[c,f][1,2]тиазепина и его применение для синтеза тианептина

Номер патента: 3770
Опубликовано: 28.08.2003
Авторы: Турбе Юг, Бриго Даньель, Бланшар Жаки
МПК: C07D 281/02
Метки: получения, синтеза, тианептина, применение, 11-амино-3-хлор-6,11-дигидро-5,5-диоксо-6-метилдибензо[c,f][1,2]тиазепина, способ
Формула / Реферат:
1. Способ синтеза 11-амино-3-хлор-6,11-дигидро-5,5-диоксо-6-метилдибензо[c,f][1,2]тиазепина формулы (I) и его аддитивных солей, отличающийся тем, что проводят реакцию кетона формулы (III) с боргидридом натрия в двухфазной среде (хлорированный растворитель, такой как, например, хлороформ, дихлорметан или дихлорэтан/водный раствор гидроксида натрия) в присутствии N-додецил-N-метилдиэтаноламмонийбромида с получением спирта формулы (IV) который в...
Кристаллы, содержащие соль n-[2-(диэтиламино)этил]-5-[(5-фторо-2-оксо- 1,2-дигидро-3h-индол-3-илиден)метил]-2,4 -диметил-1н-пиррол-3- карбоксамида с яблочной кислотой, способы их получения и их композиции

Номер патента: 6445
Опубликовано: 29.12.2005
Авторы: Флек Томас Дж., Прескотт Стивен П., Холи Майкл, Малоуни Марк Т.
МПК: C07D 403/06, A61K 31/404, A61P 35/00...
Метки: 1,2-дигидро-3h-индол-3-илиден)метил]-2,4, получения, композиции, карбоксамида, кислотой, n-[2-(диэтиламино)этил]-5-[(5-фторо-2-оксо, яблочной, способы, кристаллы, соль, диметил-1н-пиррол-3, содержащие
Формула / Реферат:
1. Безводный кристалл, содержащий соль N-[2-(диэтиламино)этил]-5-[(5-фторо-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида, имеющего структуру с яблочной кислотой. 2. Кристалл по п.1, где яблочной кислотой является L-яблочная кислота. 3. Безводный кристалл, содержащий соль N-[2-(диэтиламино)этил]-5-[(5-фторо-1,2-дигидро-2-оксо-3H-индол-3-илиден)метил]-2,4-диметил-1H-пиррол-3-карбоксамида с яблочной кислотой,...
Способ получения эфиров арилиминометилкарбаминовой кислоты

Номер патента: 5006
Опубликовано: 28.10.2004
Авторы: Крёбер Ютта, Бранденбург Йёрг, Зойка Райнер, Андерскевитц Ральф, Бауер Рольф, Шмид Рольф, Хамм Райнер
МПК: C07C 269/00
Метки: получения, способ, арилиминометилкарбаминовой, эфиров, кислоты
Формула / Реферат:
1. Способ получения соединений формулы (I) в которой R1 представляет собой остаток, выбранный из группы, включающей метил, этил, пропил, циклопентил, циклогексил, фенил, бензил и -C(Me2)фенил, каждый из которых может быть одно-, двух- или трехкратно замещен гидроксигруппой, R2 представляет собой остаток, выбранный из группы, включающей метил, этил, пропил и бензил, отличающийся тем, что соединение формулы (II) в которой R1' представляет собой...
Применение эйкозапентаеновой и/или докозагексаеновой жирных кислот и/или их этиловых эфиров для приготовления лекарственного средства для снижения уровня смертности пациентов, перенесших инфаркт миокарда

Номер патента: 4312
Опубликовано: 26.02.2004
Автор: Пампарана Франко
МПК: A61K 31/23, A61P 9/10
Метки: приготовления, этиловых, жирных, перенесших, эйкозапентаеновой, лекарственного, снижения, пациентов, смертности, инфаркт, эфиров, кислот, средства, применение, уровня, докозагексаеновой, миокарда
Формула / Реферат:
1. Применение смеси незаменимых жирных кислот, содержащей эйкозапентаеновую и докозагексаеновую кислоты с высоким содержанием их этиловых эфиров, для приготовлении лекарства, используемого для снижения уровня смертности среди пациентов, перенесших инфаркт миокарда, где суммарное содержание этиловых эфиров в смеси составляет не менее 25 мас.%, причем лекарство предназначено для перорального введения. 2. Применение по п.1, где лекарство...
Предыдущий патент: Гетероциклические трипептиды в качестве ингибиторов вируса гепатита с
Следующий патент: Способ получения 2-(замещенный фенил)-2-гидроксиэтилкарбаматов
Случайный патент: Система дозирования текучих продуктов