Способ получения морфологически чистой модификации “н” натеглинида
Номер патента: 11409
Опубликовано: 27.02.2009
Авторы: Газдаг Мария, Тарканьи Габор, Семзё Аттила, Гизур Тибор, Хегедуш Бела, Бабьяк Моника, Тёрли Йожеф






Формула / Реферат
1. Способ кристаллической модификации "H" N-(транс-4-изопропилциклогексилкарбонил)-D-фенилаланина (натеглинида) формулы (I)

включающий кипячение в алкане, предпочтительно в н-гексане или н-гептане, исходного материала, которым является иная кристаллическая модификация натеглинида, имеющая более низкую температуру плавления, или кипячение в алкане смеси таких модификаций до получения продукта в стабильной кристаллической форме "Н".
2. Способ по п.1, отличающийся тем, что в качестве исходного материала используется натеглинид в кристаллической модификации "G".
Текст
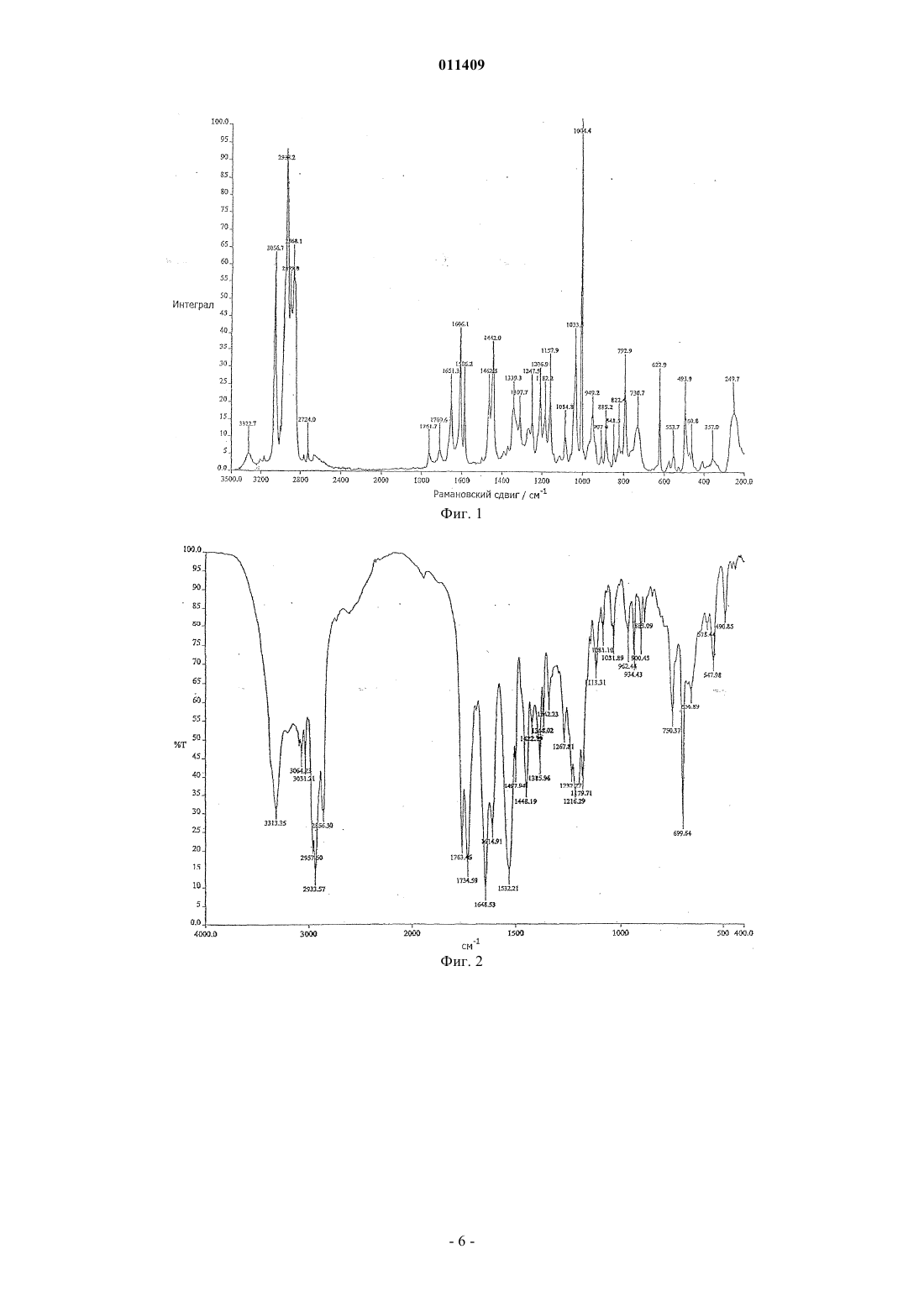
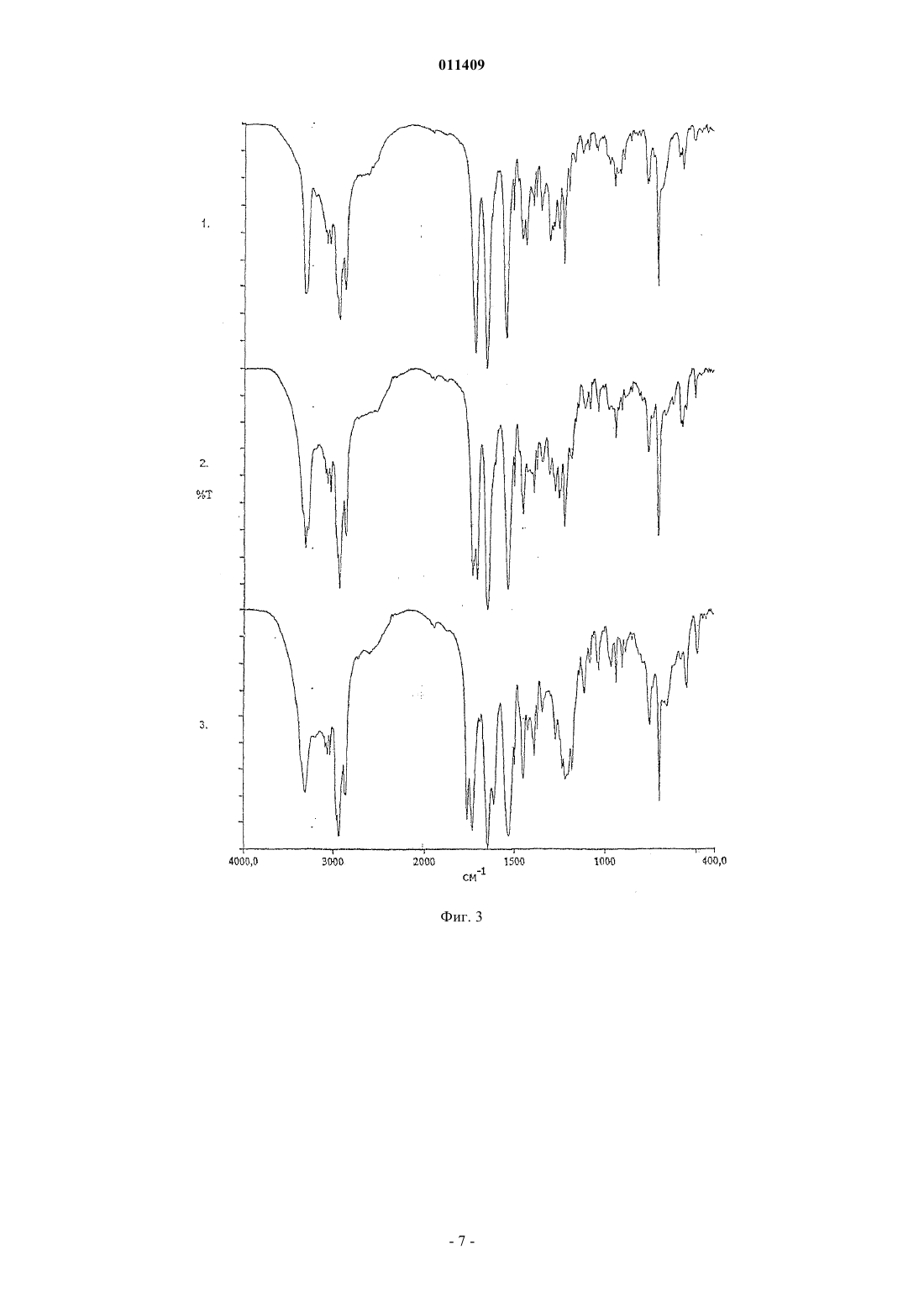
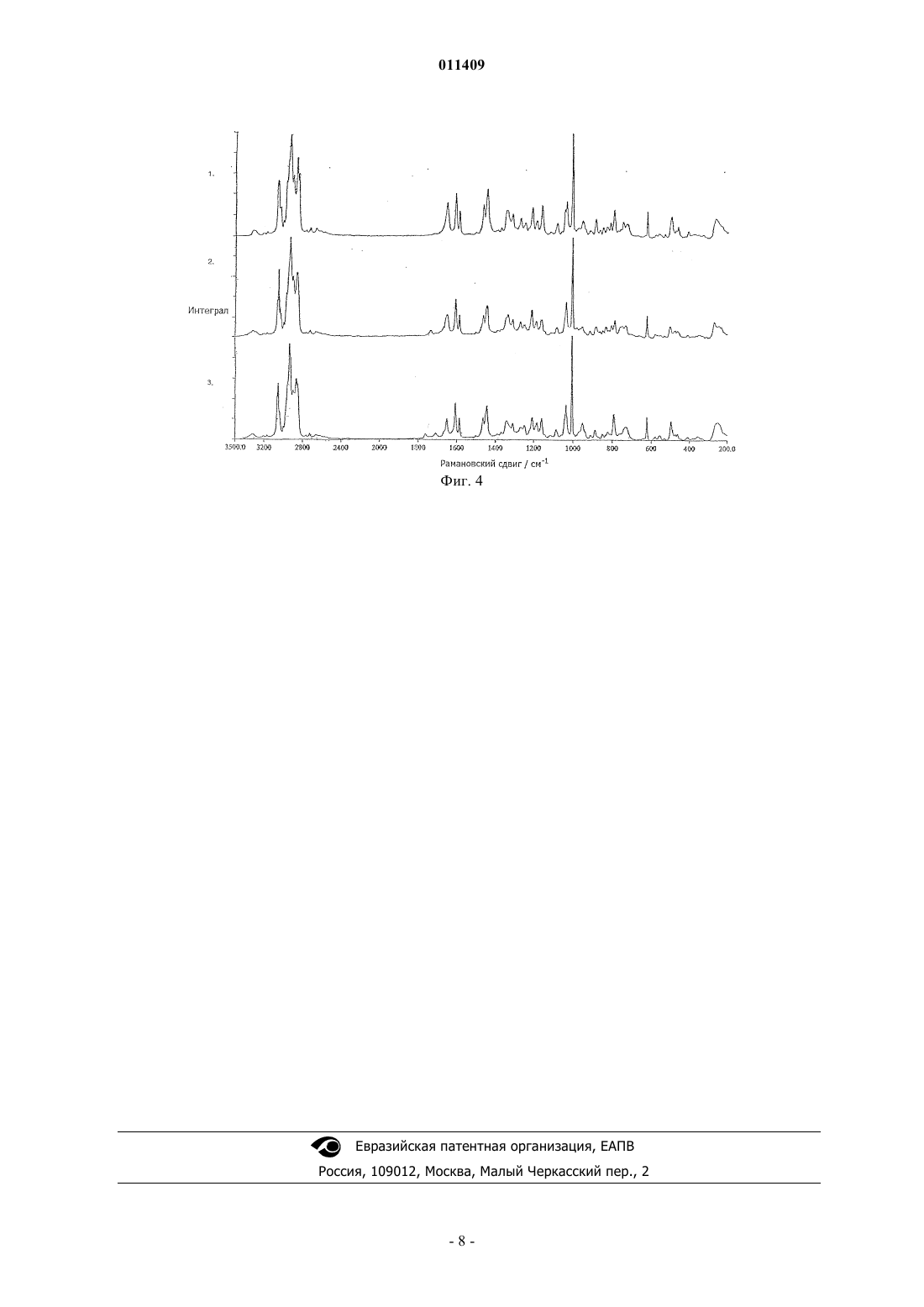
011409 Область техники, к которой относится изобретение Изобретение касается способа получения модификации "Н" натеглинида в морфологически чистой кристаллической форме из других модификаций или форм натеглинида в качестве исходных морфологически не чистых форм натеглинида "Н" формулы (I) из соединения общей формулы (II) где R означает низшую алкильную группу (С 1-С 4) или водород, который включает обработку соединения общей формулы (II) основанием с образованием соли щелочного металла, а затем добавление кислоты надлежащим образом к данной соли щелочного металла с высвобождением модифицированного продукта. Уровень техники Натеглинид известен как активный ингредиент композиций для лечения диабета 2-го типа (J. Med.Chem. 32, 1436 (1989. Также известны способы получения этого препарата и его двух кристаллических форм (неустойчивая форма "В" с Т.пл. 127-129 С и устойчивая форма "H" с Т.пл. 139 С). Получение кристаллической формы "В" описано в J. Med. Chem. (там же); при реакции этот препарат образуется в каждом случае. В патенте US 5488150 раскрыто получение кристаллической формы "H" из неустойчивой кристаллической модификации "В". Такая перестройка происходит при хранении модификации "В" в водном органическом растворителе (ацетоне, ацетонитриле или спиртах) с перемешиванием в течение 24 ч. Однако этот процесс имеет тот недостаток, что для получения устойчивой модификации "H", содержащейся в композиции, нужна дополнительная трудоемкая стадия. Другим недостатком является то, что модификация "В" с трудом поддается фильтрованию, что представляет серьезную проблему при промышленном производстве. Еще одним недостатком является то, что применение водной системы затрудняет извлечение органического растворителя. В соответствии с J. Med. Chem. (там же) и описанием патента US 4816484 препарат получают при щелочном гидролизе метилового эфира натеглинида с образованием соответствующей соли щелочного металла, которую, в свою очередь, обрабатывают неорганической кислотой с получением продукта. Ни в одной из вышеприведенных публикаций не указана оптическая чистота продукта, хотя это может иметь существенное значение вследствие различий в биологической активности энантиомеров. Следовательно,нужно предпринять все меры для сведения к минимуму содержания энантиомерной примеси. Из химической литературы известно, что хиральный атом углерода в -аминокислотах и дипептидах более или менее подвержен рацемизации. Такая подверженность столь сильно выражена, что рацемизация протекает даже в присутствии слабого основания типа гидроокиси бария (Hoppe-Seyler's Z.Physiol. Chem. 33, 173 (1901 или гидроокиси кальция, что ведет к энантиомерному загрязнению конечного продукта - натеглинида. При воспроизведении описанного в US 4816484 способа мы обнаружили 0,2-0,3% энантиомерной примеси в продукте; такая величина не удовлетворяет строгим требованиям, налагаемым как фармакопеей, так и органами здравоохранения и директивами по качеству, поскольку допустимый максимум хиральных примесей равен 0,1%. Вследствие этого, продукт, полученный указанным выше способом, нуждается в дополнительной очистке, что можно сделать посредством нескольких стадий перекристаллизации из очень разбавленных растворов с весьма низким выходом (10-20%). Другой возможный способ очистки заключается в использовании хирального реагента в количестве, рассчитанном, исходя из энантиомерной примеси. Этот последний способ, однако, нерентабелен в промышленных масштабах, так как он ведет к существенному возрастанию затрат и продолжительности обработки. Целью настоящего изобретения является обеспечение способа, пригодного для заводского получе-1 011409 ния хирально чистого натеглинида с высоким выходом и коротким временем реакции в кристаллической форме "H", требуемой для фармацевтических композиций, либо получения кристаллической формы "H" из других кристаллических модификаций. Сущность изобретения Во время наших опытов неожиданно оказалось, что при выделении натеглинида из его соли в присутствии водорастворимого органического растворителя при температуре ниже 20C образуется неизвестная в данной области кристаллическая модификация с Т.пл. 100-109C и обладающая такими свойствами фильтрования, которые превосходят известные кристаллические модификации. Мы именуем эту новую форму кристаллической модификацией "G". Из вышеизложенной модификации "G" получают кристаллическую форму "H" при нагревании в алкане или циклоалкане типа н-гексана или н-гептана, не используя каких-либо водных органических растворителей. Кроме того, весьма неожиданно оказалось, что если выделение продукта либо из соли щелочного металла, полученной по окончании щелочного гидролиза алкильного эфира натеглинида, либо из соли натеглинида и щелочного металла, содержащей энантиомерную примесь, проводить не в одну стадию добавлением эквивалентного количества минеральной кислоты, а добавлять кислоту двумя порциями таким образом, что сначала добавить кислоту в количестве меньше эквимолярного, получая смесь натеглинида и его соли щелочного металла, затем выделить эту смесь и добавить в нее остальную часть минеральной кислоты, то получится хирально чистый натеглинид, т.е. не содержащий энантиомерной примеси. При получении натеглинида из его соли можно получать различные кристаллические модификации в зависимости от температуры реакции. Просто поразительно, что при добавлении минеральной кислоты в раствор соли вещества и щелочного металла образуется не кислый продукт в количестве, эквивалентном реагенту, а смесь кислоты и соли. Также поразительно то, что при реакции соли натеглинида, содержащей энантиомерную примесь, с нехиральной кислотой образуется чистый продукт, не содержащий энантиомерной примеси, без добавления какого-либо хирального реагента/вспомогательного вещества. Такой способ позволяет избежать многократной кристаллизации продукта из очень разбавленных растворов - такая операция требует затрат и труда, но она даже не обеспечивает хиральной чистоты. В наших опытах также осуществляли очистку натеглинида, содержащего энантиомерную примесь,таким образом, что в продукт, содержащий энантиомерную примесь, добавляли основание с образованием соответствующей соли, а выделение продукта из данной соли проводили не в одну стадию добавлением эквивалентного количества минеральной кислоты, но добавляли кислоту двумя порциями таким образом, что сначала кислоту добавляли в количестве меньше эквимолярного, получая смесь натеглинида и его соли щелочного металла, затем выделяли эту смесь и добавляли в нее остальную часть минеральной кислоты, получая хирально чистый натеглинид. Соответственно, изобретение предусматривает способ получения кристаллических модификаций натеглинида формулы (I) путем обработки соединения общей формулы (II) основанием с получением соли продукта и щелочного металла и выделения продукта из этой соли таким образом, что высвобождение продукта кислотой проводится при температуре ниже комнатной, предпочтительно в интервале температур от 0 до 20C, получая натеглинид в кристаллической модификации "G"; либо при температуре выше комнатной, предпочтительно в интервале температур от 65 до 70C, получая натеглинид в кристаллической модификации "H". Натеглинид в кристаллической форме "G" - модификация, неизвестная в этой области, - также входит в объем притязаний изобретения. Изобретение также предусматривает способ получения натеглинида формулы (I) путем обработки соединения общей формулы (II) основанием с получением соли продукта и щелочного металла и выделения продукта из этой соли таким образом, что высвобождение продукта осуществляется добавлением эквивалентного количества минеральной кислоты порциями, предпочтительно двумя порциями (избирательное осаждение), т.е. сначала кислоту добавляют в количестве меньше эквимолярного, получая смесь натеглинида и его соли щелочного металла, затем выделяют эту смесь и добавляют в нее остальную часть минеральной кислоты, получая хирально чистый натеглинид. Кроме того, изобретение предусматривает способ получения устойчивой кристаллической формы"H" из других кристаллических модификаций, обладающих более низкими температурами плавления. В соответствии с одним воплощением изобретения метиловый эфир натеглинида подвергают гидролизу в водном алканоле при 15-30C в присутствии 1-1,5 экв., предпочтительно 1,2 экв. гидроокиси натрия. Раствор, содержащий полученную соль щелочного металла, обрабатывают минеральной кислотой, сначала в количестве из расчета 0,4-0,6 экв. эфира плюс избыток основания. Полученную при этом смесь натеглинида и его соли щелочного металла выделяют фильтрованием, осадок на фильтре растворяют и раствор нагревают до температуры - в случае кристаллической модификации "H" - 65-70C, подходящей для дальнейшего высвобождения продукта водным раствором минеральной кислоты. Выпавший осадок продукта выделяют фильтрованием и сушат при 50-60C. При получении кристаллической модификации "G", неизвестной в данной области, высвобождение продукта осуществляется при температуре ниже 20C и продукт сушат при 30-35C. В случае кристалли-2 011409 ческой модификации "В", описанной в литературе, подкисление проводят при 30-35C и продукт сушат при 40-45C. Перестройка кристаллических модификаций с более низкими температурами плавления в устойчивую кристаллическую модификацию "H" проводится без применения водных растворителей; она осуществляется в алканах или циклоалканах типа н-гексана или н-гептана с кратковременным кипячением. Основание, используемое в способе, может представлять собой гидроокись щелочного металла,предпочтительно гидроокись натрия, гидроокись калия или гидроокись лития; наиболее предпочтительно оно представляет собой гидроокись натрия. Минеральная кислота, используемая в способе, может представлять собой соляную кислоту, серную кислоту; предпочтительно она представляет собой соляную кислоту. Продукт, содержащий хиральную примесь, подвергают очистке добавлением эквивалентного количества гидроокиси щелочного металла в метанольном растворе, а затем избирательным осаждением, как описано выше. Хиральную чистоту продукта, полученного в соответствии с изобретением, можно легко и точно определить методами HPLC и ЯМР-спектроскопии. При растворении конечного продукта, полученного в соответствии с нашим способом, в соответствующей смеси растворителей (CCl4:CD2Cl2 = 5:7, об./об.) и получении его ЯМР-спектра в условиях, изложенных ниже, соотношение энантиомеров в конечном продукте можно определить без применения какого-нибудь внешнего хирального вспомогательного вещества; определение просто основывается на отчетливых сигналах 1 Н-ЯМР, вызванных автоидентификацией энантиомеров. Способ по изобретению обладает тем преимуществом, что хирально чистый продукт может быть получен простым способом с хорошим выходом без выполнения нескольких стадий очистки, причем можно получить любую из кристаллических модификаций; более того, проводя высвобождение продукта при соответствующей температуре, можно получить и кристаллическую модификацию "G", которая легко поддается фильтрованию. Поскольку для получения кристаллической модификации "H" из других кристаллических форм, обладающих более низкими температурами плавления, применяется растворитель, отличный от смеси водного и органического растворителя, то извлечение органического растворителя легко осуществляется, что, опять же, является преимуществом. Краткое описание фигур Прилагаются четыре фигуры, представляющие определенные спектры различных кристаллических модификаций, а именно на фиг. 1 представлен рамановский спектр натеглинида в кристаллической модификации "G" по изобретению; на фиг. 2 - инфракрасный спектр натеглинида в кристаллической модификации "G" по изобретению; на фиг. 3 - инфракрасные спектры натеглинида в кристаллических модификациях "H", "В" и "G" по изобретению, обозначенных 1, 2 и 3 соответственно; на фиг. 4 - рамановские спектры натеглинида в кристаллических модификациях "H", "В" и "G" по изобретению, обозначенных 1, 2 и 3 соответственно. Ниже приведены спектроскопические данные по индивидуальным модификациям (см-1), причем интенсивные полосы подчеркнуты. Натеглинид в модификации "H": ИК спектр: 3315, 3065, 3031, 2926, 2861, 1714, 1650, 1541, 1446, 1425, 1292, 1214, 1187, 934, 756,742, 700, 558 Рамановский спектр: 3059, 2935, 2902, 2862, 2844, 1652, 1606, 1587, 1463, 1443, 1337, 1310, 1208,1158, 1080, 1004, 950, 884, 828, 811, 794, 748, 623, 494, 408, 263 Натеглинид в модификации "В": ИК спектр: 3313, 3064, 3028, 2934, 2858, 1732, 1706, 1648, 1536, 1446, 1386, 1298, 1217, 1178, 1078,934, 755, 702, 569, 498 Рамановский спектр: 3055, 3040, 2936, 2903, 2866, 1735, 1650, 1606, 1586, 1462, 1442, 1333, 1209,1158, 1081, 1004, 911, 880, 832, 805, 750, 732, 623, 577, 499, 474, 268 Натеглинид в модификации "G": ИК спектр: 3313, 3064, 3031, 2934, 2856, 1763, 1735, 1648, 1614, 1533, 1448, 1386, 1368, 1216, 1180,1113, 1081, 934, 750, 700, 574, 491 Рамановский спектр: 3057, 2938, 2868, 1762, 1710, 1651, 1606, 1586, 1462, 1442, 1339, 1207, 1182,1158, 1085, 1004, 949, 885, 822, 793 Условия HPLC при определении хиральной чистоты: колонка: CHIRALCEL OD-RH, 1504,6 мм, 5 мкл элюент: 0,1 M К-гексафторфосфатный буфер: метанол = 30:70 скорость протока: 0,3 мл/мин температура: 40C детектирование: 214 нм-3 011409 вводимый объем: 20 мкл подготовка образцов: 1 мг тестируемого продукта растворяли в 5 мл элюента. Соотношение энантиомеров измеряли методом спектроскопии 1 Н-ЯМР в условиях, приведенных ниже: рабочая частота: 500 МГц растворитель: CCl4:CD2Cl2 = 7:5 об./об. опорный сигнал: CD2Cl2 = 5,32 ppm температура: 21,5C. Растворяли 22 мг конечного продукта в указанном выше растворителе. Для анализа использовали порции раствора по 0,7 мл без фильтрования. Использовали сопряжение на несущей в 4,8 ppm при 10 децибелах. Соотношение энантиомеров определяли из группы сигналов в интервале 3,08-3,17 ppm методом деконволюции. Далее изобретение раскрывается на следующих неограничивающих примерах. Пример 1. Получение хирально чистого натеглинида в кристаллической модификации "G". В 4-горлую колбу на 1 л, снабженную поворотной лопастной мешалкой, холодильником, термометром и загрузочной воронкой, вносили 200 г (250 мл) метанола и 33,1 г (0,1 моль) метилового эфира натеглинида. В образовавшуюся суспензию по каплям при 20-25C добавляли 4,8 г (0,12 моль) гидроокиси натрия, растворенной в 110 мл воды, охлаждая смесь холодной водой. Реакционную смесь держали при 20-25C с перемешиванием в течение 4 ч, что вызывало превращение суспензии в раствор. После того как эфир был израсходован в реакции, из раствора фильтрованием удаляли небольшое количество твердых веществ. К фильтрату по каплям добавляли 6,9 г (5,85 мл, 0,07 моль) концентрированной соляной кислоты в 5,5 мл воды при 10-15C. Полученную густую суспензию перемешивали при 13-18 С в течение 30 мин, а затем фильтровали. Осадок на фильтре промывали сначала 43 г (50 мл) смеси метанол/вода(2:1 об./об., 26,3 г метанола + 16,7 г воды), а затем 200 мл воды. Влажное вещество растворяли в 514 г(650 мл) метанола при 25-30C, а затем раствор охлаждали до 15-20C и добавляли 5,5 г (4,7 мл, 0,056 моль) концентрированной соляной кислоты в 5 мл воды с тем, чтобы после добавления величина рН раствора была между 2 и 3. После дальнейшего перемешивания в течение 10 мин добавляли воду с температурой 5C (750 мл) и полученный осадок перемешивали в течение 20 мин. Продукт отфильтровывали,промывали водой (200 мл) и высушивали под инфракрасной лампой при 30-35C, получая 26,3 г (94,4%) натеглинида в кристаллической модификации "G". Т.пл.: 100-109C. Энантиомерная примесь была ниже предела обнаружения методом HPLC; общее содержание других примесей составляло менее 0,1%. Пример 2. Получение хирально чистого натеглинида в кристаллической модификации "H". В 4-горлую колбу на 1 л, снабженную поворотной лопастной мешалкой, холодильником, термометром и загрузочной воронкой, вносили 200 г (250 мл) метанола и 33,1 г (0,1 моль) метилового эфира натеглинида. В образовавшуюся суспензию по каплям при 20-25C добавляли 4,8 г (0,12 моль) гидроокиси натрия, растворенной в 110 мл воды, охлаждая смесь холодной водой. Реакционную смесь держали при 20-25C с перемешиванием в течение 4 ч, что вызывало превращение суспензии в раствор. После того как эфир был израсходован в реакции, из раствора фильтрованием удаляли небольшое количество твердых веществ. К фильтрату по каплям добавляли 6,9 г (5,85 мл, 0,07 моль) концентрированной соляной кислоты в 5,5 мл воды при 10-15C. Полученную густую суспензию перемешивали при 13-18 С в течение 30 мин, а затем фильтровали. Осадок на фильтре промывали сначала 43 г (50 мл) смеси метанол/вода(2:1 об./об., 26,3 г метанола + 16,7 г воды), а затем 200 мл воды. Влажное вещество растворяли в 514 г(650 мл) метанола при 50-60C и при той же температуре добавляли 5,5 г (4,7 мл, 0,056 моль) концентрированной соляной кислоты в 5 мл воды с тем, чтобы после добавления величина рН раствора была между 2 и 3. После дальнейшего перемешивания в течение 10 мин добавляли воду (750 мл) при указанной выше температуре и полученный осадок перемешивали в течение 20 мин при той же температуре. Продукт отфильтровывали, промывали водой (200 мл) и высушивали под инфракрасной лампой при 60-70C, получая 26,3 г (94,4%) натеглинида в кристаллической форме "H". Т.пл.: 138-139 С. Энантиомерная примесь была ниже предела обнаружения методом HPLC; общее содержание других примесей составляло менее 0,1%. Пример 3. Получение хирально чистого натеглинида в кристаллической модификации "H" через кристаллическую форму "G". В 4-горлую колбу на 1 л, снабженную поворотной лопастной мешалкой, холодильником, термометром и загрузочной воронкой, вносили 200 г (250 мл) метанола и 33,1 г (0,1 моль) метилового эфира натеглинида. В образовавшуюся суспензию по каплям при 20-25C добавляли 4,8 г (0,12 моль) гидроокиси натрия, растворенной в 110 мл воды, охлаждая смесь холодной водой. Реакционную смесь держали при 20-25C с перемешиванием в течение 4 ч, что вызывало превращение суспензии в раствор. После того как эфир был израсходован в реакции, из раствора фильтрованием удаляли небольшое количество твердых веществ. К фильтрату по каплям добавляли 6,9 г (5,85 мл, 0,07 моль) концентрированной соляной кислоты в 5,5 мл воды при 10-15C. Полученную густую суспензию перемешивали при 13-18 С в течение 30 мин, а затем фильтровали. Осадок на фильтре промывали сначала 43 г (50 мл) смеси метанол/вода(2:1 об./об., 26,3 г метанола + 16,7 г воды), а затем 200 мл воды. Влажное вещество растворяли в 514 г(650 мл) метанола при 25-30C, а затем раствор охлаждали до 15-20C и добавляли 5,5 г (4,7 мл, 0,056 моль) концентрированной соляной кислоты в 5 мл воды с тем, чтобы после добавления величина рН раствора была между 2 и 3. После дальнейшего перемешивания в течение 10 мин добавляли воду (750 мл) при указанной выше температуре и полученный осадок перемешивали в течение 20 мин. Кристаллы натеглинида в модификации "G" отфильтровывали и промывали 200 мл воды. Влажное вещество переносили в круглодонную колбу и кипятили в 513 г (750 мл) н-гептана с перемешиванием в течение 1,5 ч. Суспензию охлаждали до 20-25C и перемешивали в течение 20 мин при этой температуре. Продукт отфильтровывали, промывали 2100 мл (268 г) н-гептана и высушивали под инфракрасной лампой при 50C, получая 25,68 г (80,9%) натеглинида в кристаллической форме "H". Т.пл.: 139-140C. Энантиомерная примесь была ниже предела обнаружения методом HPLC; общее содержание других примесей составляло менее 0,1%. Пример 4. Очистка натеглинида, содержащего энантиомерную примесь. Растворяли 6,34 г (0,02 моль) натеглинида (хиральная чистота: 98%) в 50 мл метанола. В раствор добавляли 0,8 г (0,02 моль) гидроокиси натрия в 22 мл воды. В этот раствор по каплям добавляли смесь из 0,83 мл концентрированной соляной кислоты и 1 мл воды при температуре 10-15C. После 20 мин перемешивания осадок отфильтровывали, промывали смесью метанол/вода 2:1 об./об. (25 мл), а затем 50 мл воды. Влажное вещество растворяли в метаноле (130 мл) и добавляли смесь из 0,83 мл концентрированной соляной кислоты и 1 мл воды. Смесь перемешивали в течение 10 мин, добавляли 150 мл воды и продолжали перемешивать в течение 20 мин. Продукт фильтровали, промывали, суспендировали в нгептане (100 мл) и кипятили в течение 1,5 ч. После охлаждения продукт отфильтровывали и высушивали при 50C, получая 4,2 г (66,4%) натеглинида. Т.пл.: 138-139C. Энантиомерная примесь была ниже предела обнаружения методом HPLC; общее содержание других примесей составляло менее 0,1%. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ кристаллической модификации "H" N-(транс-4-изопропилциклогексилкарбонил)-Dфенилаланина (натеглинида) формулы (I) включающий кипячение в алкане, предпочтительно в н-гексане или н-гептане, исходного материала, которым является иная кристаллическая модификация натеглинида, имеющая более низкую температуру плавления, или кипячение в алкане смеси таких модификаций до получения продукта в стабильной кристаллической форме "Н". 2. Способ по п.1, отличающийся тем, что в качестве исходного материала используется натеглинид в кристаллической модификации "G".
МПК / Метки
МПК: C07C 231/24, C07C 231/12, C07C 233/63
Метки: натеглинида, получения, способ, морфологически, модификации, чистой
Код ссылки
<a href="https://eas.patents.su/9-11409-sposob-polucheniya-morfologicheski-chistojj-modifikacii-n-nateglinida.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения морфологически чистой модификации “н” натеглинида</a>
Способ получения хирально чистой кристаллической модификации натеглинида

Номер патента: 10569
Опубликовано: 30.10.2008
Авторы: Гизур Тибор, Газдаг Мария, Тёрли Йожеф, Тарканьи Габор, Бабьяк Моника, Семзё Аттила, Хегедуш Бела
МПК: C07C 231/24, C07C 233/63, C07C 231/12...
Метки: хирально, натеглинида, способ, чистой, получения, модификации, кристаллической
Формула / Реферат:
1. Способ получения хирально чистого N-(транс-4-изопропилциклогексилкарбонил)-D-фенилаланина (натеглинида) формулы (I) путем обработки соединения общей формулы (II) где R означает низшую алкильную группу (С1-С4) или водород, основанием с образованием соли щелочного металла, и высвобождения продукта из данной соли неорганической кислотой, включающий кислотное высвобождение продукта, выполняемое путем добавления этой кислоты двумя порциями таким...
Полиморфные модификации телмисартана, способ их получения и их применение для изготовления лекарственного средства

Номер патента: 3065
Опубликовано: 26.12.2002
Автор: Шнайдер Генрих
МПК: A61P 9/12, C07D 235/18, A61K 31/4184...
Метки: лекарственного, полиморфные, средства, получения, применение, модификации, изготовления, способ, телмисартана
Формула / Реферат:
1. Полиморфная кристаллическая модификация В (форма В) телмисартана формулы I отличающаяся тем, что эндотермический максимум, проявляющийся при термическом анализе методом дифференциальной сканирующей калориметрии (ДСК), составляет 183+2шС. 2. Телмисартан, отличающийся тем, что он содержит форму В по п.1. 3. Способ получения полимерной формы В телмисартана, отличающийся тем, что а) телмисартан растворяют в смеси растворителей, состоящей из...
5-хлор-3(4-метансульфонилфенил)-6′-метил-[2,3']бипиридинил в чистой кристаллической форме и способ синтеза

Номер патента: 4809
Опубликовано: 26.08.2004
Авторы: Котляр Эндрю, Осифчин Ричард Дж., Крокер Луис С., Дэвис Ян В.
МПК: C07D 213/61, A61P 29/00, A61K 31/444...
Метки: 5-хлор-3(4-метансульфонилфенил)-6'-метил-[2,3']бипиридинил, способ, кристаллической, чистой, синтеза, форме
Формула / Реферат:
1. Полиморфная форма соединения формулы A обозначенная как форма V. 2. Полиморфная форма по п.1, отличающаяся наличием межплоскостного расстояния d, определенного посредством порошковой рентгенограммы, Cu K альфа, приблизительно 13,7 ангстрем. 3. Полиморфная форма по п.2, отличающаяся, кроме того, наличием по меньшей мере одного дополнительного межплоскостного расстояния d, определенного посредством порошковой рентгенограммы, Cu K альфа, как...
Способ получения полимера, обладающего бимодальным молекулярно-массовым распределением, способ модификации молекулярно-массового распределения статистического сополимера изобутилена, полимерный продукт и полимерная композиция на его основе

Номер патента: 1222
Опубликовано: 25.12.2000
Автор: Уайт Дональд Эндрю
МПК: C08F 8/00
Метки: получения, распределением, полимерный, продукт, способ, бимодальным, полимера, модификации, молекулярно-массового, обладающего, полимерная, сополимера, распределения, молекулярно-массовым, статистического, композиция, изобутилена, основе
Формула / Реферат:
1. Способ получения полимера, обладающего бимодальным молекулярно-массовым распределением, из полимера, обладающего мономодальным молекулярно-массовым распределением, молекулярная масса которого уменьшается при перемешивании с высокой сдвиговой деформацией необязательно в присутствии инициатора свободнорадикальной полимеризации, характеризующийся тем, что исходный полимер выбирают из группы, включающей полипропилен, сополимеры пропилена и...
Способ поверхностной модификации резиновой крошки

Номер патента: 1545
Опубликовано: 23.04.2001
Автор: Гиззатуллина Яна Леонидовна
МПК: C08J 7/12
Метки: крошки, способ, модификации, поверхностной, резиновой
Формула / Реферат:
Способ поверхностной модификации резиновой крошки озонированием, отличающийся тем, что озонирование крошки размером 0,5-0,7 мм осуществляют озоно-кислородной смесью с объемной концентрацией в ней озона от 1 до 5% в течение 10-40 мин при постоянном перемешивании, при этом удаление озоносодержащей смеси осуществляют через слой резиновой крошки подлежащей озонированию.
Предыдущий патент: Новый продукт, способ и промежуточные продукты получения производных азетидина
Следующий патент: Способ одновременного получения бензола и этилена конверсией ацетилена
Случайный патент: Способ получения 6-[3-(1-адамантил)-4-метоксифенил]-2-нафтойной кислоты