Композиции, содержащие антигрибковый агент и ацетатный буфер.
Номер патента: 1386
Опубликовано: 26.02.2001
Авторы: Ханке Вильям А., Неруркар Маниш Дж., Кауфман Майкл Дж.







Формула / Реферат
1. Фармацевтическая композиция для внутривенного введения пациенту, содержащая
а) фармацевтически эффективное количество соединения, имеющего формулу

и его фармацевтически приемлемых солей,
б) фармацевтически приемлемое количество эксципиента, такого как сахар-наполнитель или комбинация сахаров-наполнителей, эффективных для образования лиофилизованной лепёшки; и
в) фармацевтически приемлемое количество ацетатного буфера, эффективное для обеспечения фармацевтически приемлемого рН.
2. Фармацевтическая композиция по п.1, содержащая
а) фармацевтически эффективное количество соединения I;
б) между около 10-200 мг/мл эксципиента, такого как сахар-наполнитель или комбинация сахаров-наполнителей, эффективных для образования лиофилизованной лепёшки; и
в) фармацевтически приемлемое количество ацетатного буфера, эффективного для обеспечения рН между около 5 и 7.
3. Композиция по п.2, содержащая около 5-200 мг/мл соединения I или его фармацевтически приемлемой соли, от около 12,5 до около 200 мМ ацетатного буфера, около 10-200 мг/мл наполняющего агента и воду.
4. Композиция по п.3, содержащая около 30-50 мг/мл соединения I или его фармацевтически приемлемой соли, около 20-60 мМ ацетатного буфера, около 30-70 мг/мл сахара-наполнителя или комбинации сахаров-наполнителей, эффективных для образования лиофилизованной лепёшки, и воду.
5. Композиция по п.4, содержащая 42 мг/мл соединения I или его фармацевтически приемлемой соли, 25 мМ ацетатного буфера, 30 мг/мл сахарозы, 20 мг/мл маннита и воду.
6. Композиция по п.4, содержащая 42 мг/мл соединения 1 или его фармацевтически приемлемой соли, 50 мМ ацетатного буфера, 30 мг/мл сахарозы, 20 мг/мл маннита и воду.
7. Способ лечения и/или профилактики грибковых инфекций млекопитающего, включающий внутривенное введение млекопитающему фармацевтически эффективного количества композиции по п.1.
8. Способ по п.7, где млекопитающим является человек.
9. Способ лечения инфекции, вызванной Candida sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.1.
10. Способ лечения инфекции, вызванной Candida sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.5.
11. Способ лечения инфекции, вызванной Candida sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.6.
12. Способ лечения инфекции, вызванной Aspergillus sp, у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.1.
13. Способ лечения инфекции, вызванной Aspergillus sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.5.
14. Способ лечения инфекции, вызванной Aspergillus sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.6.
15. Способ лечения или профилактики инфекции или состояния, вызванного Pneumocystis carinii у пациента, нуждающегося в таком лечении или профилактике, который предусматривает введение этому пациенту профилактического или терапевтического количества композиции по п. 1.
16. Способ лечения или профилактики инфекции или состояния, вызванного Pneumocystis carinii, у пациента, нуждающегося в таком лечении или профилактике, который предусматривает введение этому пациенту профилактического или терапевтического количества композиции по п.5.
17. Способ лечения или профилактики инфекции или состояния, вызванного Pneumocystis carinii, у пациента, нуждающегося в таком лечении или профилактике, который предусматривает введение этому пациенту профилактического или терапевтического количества композиции по п.6.
18. Способ получения фармацевтической композиции, содержащей соединение, имеющее формулу

и его фармацевтически приемлемые соли, который содержит стадии
а) растворения наполнителя или комбинации наполнителей в воде,
б) добавления уксусной кислоты и установления рН до около 3,7;
в) добавления соединения I и установление рН до от около 5 до около 7 основанием;
г) фильтрования таким образом полученного раствора;
д) замораживания такого раствора и
е) лиофилизации замороженного раствора.
Текст
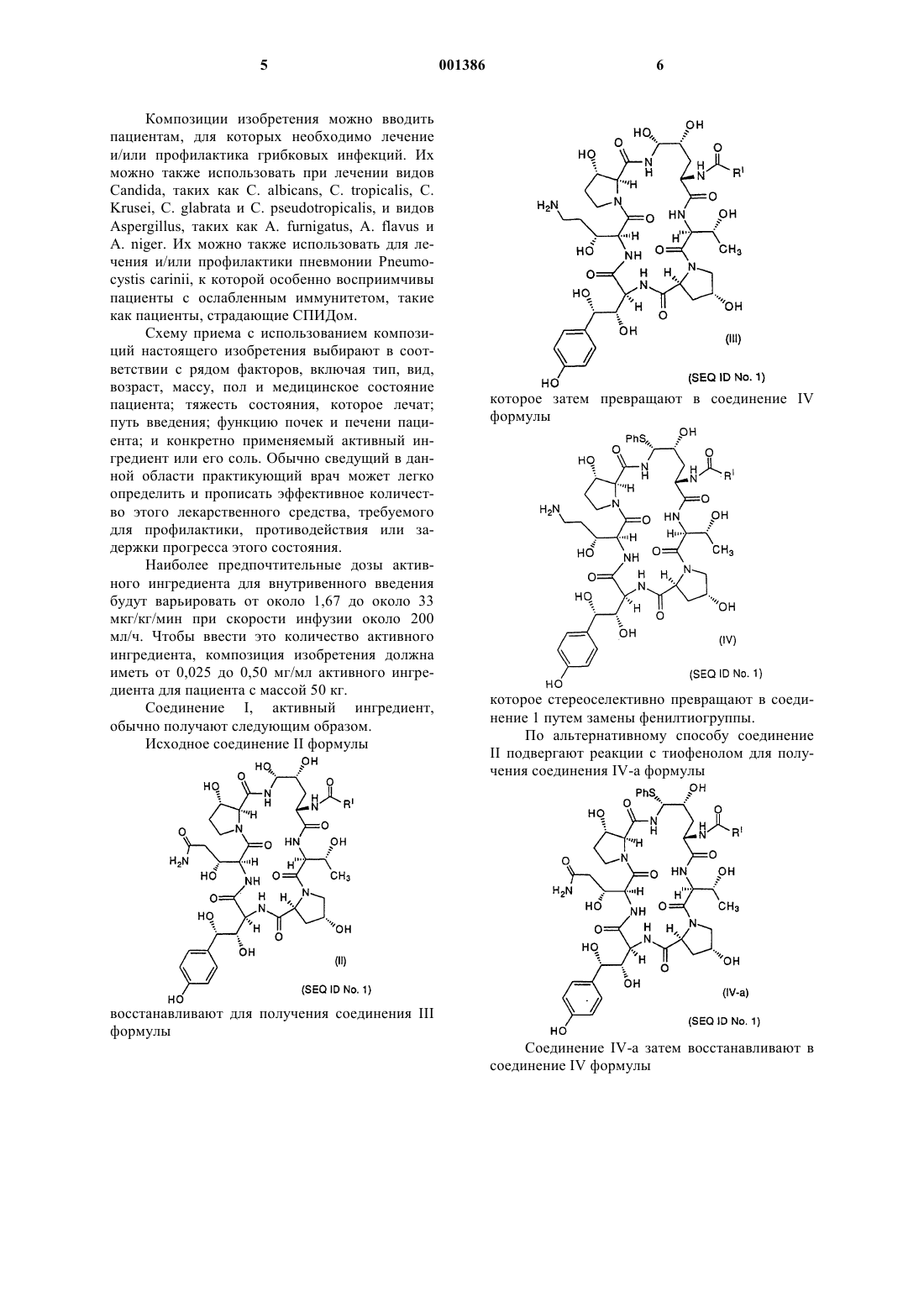
1 Предпосылки создания изобретения Это изобретение относится к композициям для лечения и/или профилактики грибковых инфекций. Существует возрастающая потребность в новых антигрибковых средствах, которые эффективны против условно-патогенных микотических инфекций, вызванных такими агентами,как Candida, Aspergillus, Cryptococcus и Pneumocystis carinii. Лечение их в настоящее время, т.е. лечение амфотерицином В и флюконазолом,неадекватно, вследствие их токсичности и отбора по устойчивости. Композиции по настоящему изобретению рассматриваются как безопасные и фунгицидные. Композиции по настоящему изобретению содержат соединение, которое можно использовать в качестве антибиотика, особенно в качестве антигрибкового средства или в качестве средства против простейших. В качестве антигрибкового средства его можно использовать для борьбы с нитеобразными грибами и дрожжами. Особенно его легко можно приспособить для применения при лечении микотических инфекций у млекопитающих, в особенности инфекций, вызванных видами Candida, такими какpseudotropicalis, и видами Aspergillus, такими как A. fumigatus, A. flavus и A. niger. В частности, композиции содержат соединение, которое,как было обнаружено, эффективно против устойчивых к амфотерицину В и флюконазолу изолятов Candida. Композиции можно также использовать для лечения и/или профилактики пневмонии Pneumocystis carinii, к которой особенно восприимчивы пациенты с ослабленным иммунитетом, такие как пациенты, страдающие СПИДом. Композиции настоящего изобретения являются безопасными, стабильными, лиофилизованными лекарственными формами для пересоздания, которые особенно полезны для доставки антигрибковых средств пациентам, нуждающимся в таких средствах. Краткое изложение сущности изобретения Это изобретение относится к фармацевтической композиции, содержащей а) фармацевтически эффективное количество соединения (именуется здесь также активным ингредиентом), имеющего формулу 2 б) фармацевтически приемлемое количество ацетатного буфера, эффективного для обеспечения рН около от 4 до 7; и в) фармацевтически приемлемое количество эксципиента, такого как смесь сахароза/ маннит, для образования лиофилизованной лепешки. В специфическом воплощении этого изобретения готовую препаративную форму получают в виде раствора 42 мг/мл соединения I, 30 мг/мл сахарозы, 20 мг/мл маннита, 1,5 мг/мл (25 мМ) уксусной кислоты, рН которого устанавливают около 6 гидроксидом натрия. Раствором затем заполняют пузырек на 1,25 мл и лиофилизуют. Полученная таким образом лиофилизованная лепешка содержит 52,5 мг соединения I,62,5 мг cахаров и 31,25 микромолей ацетатного буфера на пузырек. Лиофилизованную лепешку затем пересоздают для использования путем разбавления 21 мл разбавителя. 20 мл продукта разбавления отбирают и воссоздают в мешке для инфузии на 200 мл. Пациенту затем вводят инфузией раствор, содержащий около 0,25 мг/мл соединения I, около 0,03 миллимолей или 0,15 миллимолярного ацетатного буфера, около 0,3 мг/мл наполняющих агентов, причем получаемая композиция имеет рН около от 5 до 7. Подробное описание изобретения Готовые препаративные формы изобретения обеспечивают повышенную химическую стабильность фармацевтических композиций. Одним преимуществом такой стабильности является длительный срок годности фармацевтического продукта. Предыдущие готовые препаративные формы, с тартратным буфером, содержали фармацевтически значительные количества нежелательных продуктов деградации. Использование забуференной ацетатом готовой препаративной формы приводит к генерации меньшего количества продуктов деградации и более стабильной готовой препаративной форме. Длительный срок фармацевтической годности обеспечивает значительные экономические преимущества. Было обнаружено, что соединение формулы и его фармацевтически приемлемые соли значительно более стабильны при хранении, когда их готовят в присутствии ацетатного буфера. и его фармацевтически приемлемых солей; 3 Соединение формулы I заявляется и описывается в патенте US A 5378804. Способы его получения раскрываются в этом патенте, а также в патенте US A 5552521, который выдан 3 сентября 1996. Это соединение само очень нестабильно и разрушается различными путями, включая, но не ограничиваясь ими, гидролиз, димеризацию и окисление. Однако с этой нестабильностью ранее боролись путем лиофилизации этого соединения в забуференной тартратом готовой препаративной форме. Эта готовая препаративная форма, однако, хотя она относительно стабильна, приводила к генерации продуктов деградации с относительно высокой скоростью. При использовании ацетатного буфера лиофилизованный продукт более стабилен, содержит меньше нежелательных продуктов деградации при повышенном сроке годности композиции. Это делает его привлекательным в качестве коммерческого продукта. Настоящее изобретение относится также к способу лечения грибковых инфекций, вызванных Candida, Aspergillus и Pneumocystis carinii,который предусматривает введение композиции, содержащей соединение формулы I, пациенту, нуждающемуся в таком лечении, в количестве, эффективном для лечения грибковой инфекции. Изобретение дополнительно относится к способу профилактики инфекций Pneumocystis carinii у пациента, который предусматривает введение профилактического количества соединения формулы I. Забуференная ацетатом готовая препаративная форма изобретения включает количество ацетата, эффективное для обеспечения фармацевтически приемлемого значения рН, например, для обеспечении рН окружающей среды в диапазоне от 5 до 8, предпочтительно около от 6 до 7. Для обеспечения фармацевтически приемлемого количества ацетатного буфера, эффективного для достижения желаемого значения рН, можно использовать подходящие количества ацетата натрия и уксусной кислоты или подходящие количества уксусной кислоты и гидроксида натрия. Буфер обычно присутствует в диапазоне от около 12,5 до около 200 мМ, предпочтительно в диапазоне от около 25 до около 50 мМ. Эксципиенты, такие как наполнители, т.е. сахара-наполнители, используют для обеспечения образования эстетически подходящей лиофилизованной лепешки, твердого разбавления активного ингредиента и сорбции имеющейся влаги. Сахара, которые можно использовать в изобретении, включают сахарозу, лактозу, маннит или их комбинации. Было обнаружено, что сахароза и маннит обеспечивают получение более стабильной готовой препаративной формы и образуют фармацевтически изящную лепешку для композиции. Эксципиенты обычно присутствуют в количествах около 10 - 200 мг/мл, 001386 4 предпочтительное количество их составляет около 4,0 - 60 мг/мл. Композиции не ограничиваются активным ингредиентом, ацетатным буфером и наполнителем, и могут также включать другие фармацевтически приемлемые разбавители, эксципиенты или носители. Такие готовые препаративные формы пригодны для долговременного хранения в стеклянных контейнерах, обычно используемых в фармацевтической промышленности, например, в концентрированной форме в стандартных боросиликатных стеклянных контейнерах типа I USP. Композиции изобретения обычно получают следующим образом 1) наполнитель или комбинацию таких наполнителей растворяют в воде; 2) добавляют уксусную кислоту и, если нужно, рН устанавливают около 3,7; 3) добавляют соединение I и растворяют с последующим установлением рН основанием от около 5 до около 6; 4) раствор фильтруют и наполняют им пузырек для лиофилизации и замораживают при-50 С; 5) замороженную готовую препаративную форму лиофилизуют при -20 С со второй сушкой при 15 С (полный цикл требует свыше двух дней); и 6) лиофилизованные пузырьки закрывают пробкой и хранят приблизительно при 5 С. Лиофилизованные готовые препаративные формы можно разбавить во время введения подходящим разбавителем для получения конечной концентрации, например, около 5,0 мг/мл, которая пригодна для переноса в мешок для инфузии для использования пациентом, нуждающимся в целевом активном ингредиенте. Термин фармацевтически приемлемые соли означает нетоксичные соли активного ингредиента, включая моно-, ди- и трикислотные формы, которые обычно получают реакцией свободного основания с подходящей органической или неорганической кислотой. Фармацевтически приемлемые соли, пригодные в виде кислотно-аддитивных солей, а также соли, обеспечивающие анион четвертичной соли,являются солями, полученными из кислот, таких как хлористо-водородная, бромистоводородная, фосфорная, серная, малеиновая,лимонная, уксусная, винная, янтарная, щавелевая, яблочная, глутаминовая, памоевая и тому подобные кислоты, и включают другие кислоты,связанные с фармацевтически приемлемыми солями, перечисленными в Journal of Pharmaceutical Science, 66, 2 (1977). Термин фармацевтически эффективное количество будет означать такое количество активного ингредиента, которое будет вызывать биологическую или медицинскую реакцию ткани/системы или животного, которую добивается исследователь или практикующий врач. 5 Композиции изобретения можно вводить пациентам, для которых необходимо лечение и/или профилактика грибковых инфекций. Их можно также использовать при лечении видовA. niger. Их можно также использовать для лечения и/или профилактики пневмонии Pneumocystis carinii, к которой особенно восприимчивы пациенты с ослабленным иммунитетом, такие как пациенты, страдающие СПИДом. Схему приема с использованием композиций настоящего изобретения выбирают в соответствии с рядом факторов, включая тип, вид,возраст, массу, пол и медицинское состояние пациента; тяжесть состояния, которое лечат; путь введения; функцию почек и печени пациента; и конкретно применяемый активный ингредиент или его соль. Обычно сведущий в данной области практикующий врач может легко определить и прописать эффективное количество этого лекарственного средства, требуемого для профилактики, противодействия или задержки прогресса этого состояния. Наиболее предпочтительные дозы активного ингредиента для внутривенного введения будут варьировать от около 1,67 до около 33 мкг/кг/мин при скорости инфузии около 200 мл/ч. Чтобы ввести это количество активного ингредиента, композиция изобретения должна иметь от 0,025 до 0,50 мг/мл активного ингредиента для пациента с массой 50 кг. Соединение I, активный ингредиент,обычно получают следующим образом. Исходное соединение II формулы восстанавливают для получения соединения III формулы которое затем превращают в соединение IV формулы которое стереоселективно превращают в соединение 1 путем замены фенилтиогруппы. По альтернативному способу соединениеII подвергают реакции с тиофенолом для получения соединения IV-a формулы которое стереоселективно превращают в соединение I путем замены фенилтиогруппы. Получение соединения I. 1. Синтез и выделение соединения III. Соединение II (15,9 г, чистота 89% по площади пика, 3,4% воды, 0,0128 моль) добавляли к сухому ТГФ (0,64 л) и суспензию сушили до содержания 10 мол.% воды кипячением с обратным холодильником через слой молекулярных сит 3 А. Добавляли дополнительный сухой ТГФ для воссоздания смеси до первоначального объема и суспензию охлаждали до 4 С на бане лед/вода/метанол. В течение десяти минут добавляли неразбавленный ВН 3SМе 2 (10,91 г, 0,144 моль) и реакционную смесь сохраняли при 0-4 С. Протекание реакции контролировали по ВЭЖХ до тех пор, пока отношение исходного материала к продукту не было 1:1, что указывало на окончание реакции (3,5 ч). После 4 ч смесь охлаждали до -12 С и медленно гасили 2 Н HCl (0,036 л). Раствор разбавляли до 1,14 л водой. Анализ показал, что выход соединения III был 6,60 г(47%). Раствор после гашения разбавляли до 4 л и загружали в колонку под средним давлением с адсорбентом LiChroprep RP-C18 (158 г). После загрузки колонку промывали 1,2 л воды и амин элюировали 1,9 л смеси 1:4 (об./об.) ацетонитрил/вода и затем 0,38 л смеси 1:3 (об./об.) ацетонитрил-вода. Обогащенные фракции (80% по площади) объединяли и разбавляли водой до получения раствора 1:7,3 (об./об.) ацетонитрил-вода (всего 1,70 л). Эту смесь загружали в ту же самую колонку, описанную выше, и колонку промывали 0,57 л воды. Целевое соединение элюировали 0,57 л метанола. Обогащенные фракции (85% по площади) объединяли и концентрировали роторным испарением при статическом высоком вакууме, получая 6,81 г (чистота 87 мас.%,6,8 мас.% воды) содержащей 5,92 г соединения 8 охлаждали до -5 С, в этой точке добавляли тиофенол (3,10 г, 0,028 моль). Для того чтобы поддерживать температуру реакционной смеси ниже 0 С TFA (трифторуксусную кислоту) (36 г,24,5 мл, 0,318 моль.) добавляли на протяжении 20 мин. Реакционную смесь выдерживали при температуре от -10 до 0 С до тех пор, пока анализ ВЭЖХ не показал 3% (по площади) исходного материала (3,75 ч). В это время медленно(1 ч) добавляли охлажденную воду (0,56 л) при охлаждении реакционной смеси для поддержания температуры ниже 5 С. Анализ показал, что выход - и -фенилсульфидного аддукта в виде трифторацетатной соли был 4,82 г (71%). Этот раствор загружали в ту же самую колонку, описанную в стадии 1, и колонку промывали водой (0,57 л), затем адсорбированные органические соединения элюировали метанолом(0,50 л). Обогащенные фракции концентрировали роторным испарением в статическом высоком вакууме. Это давало 7,20 г (чистота 57 мас.%, 5,1 мас.% воды) сырой трифторацетатной соли фенилсульфида в виде аморфного пенистого твердого продукта. Исправленный выход в стадии выделения фенилсульфида в виде смеси 93:7 диастереомеров - и -аминаля был 4,10 г (61%). 3. Превращение соединения IV в соединение I-1. Сырую трифторметансульфонатную соль фенилсульфида (8,4 г сырой соли, чистота 57 мас.%, 0,00377 моль) добавляли к этилендиамину (24 мл) при перемешивании при комнатной температуре. Получаемый раствор перемешивали 1,5 ч до завершения замещения, затем добавляли метанол (40 мл), затем уксусную кислоту(45 мл), поддерживая температуру ниже 25 С охлаждением на ледяной бане. Получали густую суспензию. Для растворения суспензии добавляли воду (160 мл) и водный слой экстрагировали, осторожно встряхивая с гексаном (75 мл). Слой гексана обратно экстрагировали водой (40 мл) и объединенный водный слой фильтровали через сплавленную с фильтром стеклянную воронку со средней пористостью, затем очищали препаративной ВЭЖХ с использованием колонки С 18 с диаметром 50 мм и с использованием в качество элюента смеси 22% ацетонитрила/78% 0,15% водной уксусной кислоты. Обогащенную фракцию лиофилизовали, получая 4,2 г соединения I-1 в виде диацетатной соли с чистотой 85% и с выходом на стадии выделения 78%. 4. Кристаллизация соединения I-1. Твердый продукт (2,3 г) растворяли в этаноле (25 мл) и затем добавляли воду (2,7 мл). Раствор пропускали через сплавленную с фильтром стеклянную воронку для удаления постороннего материала. К этому фильтрату добавляли уксусную кислоту (0,14 мл), затем медленно добавляли (1,75 ч) этилацетат (14 мл). В раствор вносили затравку и слой затравки выдерживали в течение 1 ч. Остальную часть этилацетата (32 мл) добавляли в течение 5 ч и выдерживали дополнительно 1 ч. Кристаллический твердый продукт собирали на сплавленной с фильтром стеклянной воронке и промывали раствором этанол/этилацетат/вода (6 мл/9 мл/0,5 мл, соответственно). Сырой осадок сушили потоком азота, получая 1,91 г (1,75 г по данным анализа, 88% выделение) диацетатной соли соединения I-1. Пример I. Получение готовой препаративной формы I. Ингредиент Соединение I Сахароза Маннит Уксусная кислота Гидроксид натрия по необходимости до рН Объем наполнения Типично к мерной колбе на 25 мл добавляли 0,75 г сахарозы и 0,5 г маннита, около 17,5 мл воды, 0,5 мл раствора 75 мг/мл уксусной кислоты и 42 мг/мл эквивалента соединения I. Раствор смешивали и рН устанавливали до 6 с использованием 1 М NaOH. Объем устанавливали добавлением воды и подтверждали рН. Раствор фильтровали через фильтр-шприцMillex-GV и вносили в стеклянные трубчатые склянки на 10 мл по 1,75 мл в каждую. Склянки частично закрывали пробками для лиофилизации и лиофилизовали, получая твердую лиофилизованную лепешку на дне склянки. Лиофилизованную готовую препаративную форму разбавляли 10,5 мл и 10 мл отбирали и разбавляли в 200 мл, получая конечную концентрацию 0,25 мг/мл, перед введением пациенту. Были получены, как описано выше, дополнительные готовые препаративные формы, содержащие следующие ингредиенты (каждую из этих готовых препаративных форм получали при концентрации раствора 40-42 мг/мл активного ингредиента). Пример Буфер 2 50 мМ тартрата (7,5 мг/мл) 3 50 мМ тартрата (7,5 мг/мл) Готовые препаративные формы хранили в лиофилизованном состоянии при 5 С и испытывали с интервалами около 4 недель на стабильность. Стабильность и образование продуктов деградации определяли градиентной ВЭЖХ с использованием стандартных способов, известных специалистам в данной области. Неожиданно было обнаружено, что готовые препаративные формы 1, 5, 7 и 8 были значительно более стабильны и обнаруживали значительно меньше нежелательных продуктов деградации, чем другие готовые препаративные формы. СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙNO:1: Хаа Thr Xaa Xaa Хаа Хаа 1 5 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Фармацевтическая композиция для внутривенного введения пациенту, содержащая а) фармацевтически эффективное количество соединения, имеющего формулу и его фармацевтически приемлемых солей,б) фармацевтически приемлемое количество эксципиента, такого как сахар-наполнитель или комбинация сахаров-наполнителей, эффективных для образования лиофилизованной лепшки; и в) фармацевтически приемлемое количество ацетатного буфера, эффективное для обеспечения фармацевтически приемлемого рН. 2. Фармацевтическая композиция по п.1,содержащая а) фармацевтически эффективное количество соединения I; б) между около 10-200 мг/мл эксципиента,такого как сахар-наполнитель или комбинация сахаров-наполнителей, эффективных для образования лиофилизованной лепшки; и в) фармацевтически приемлемое количество ацетатного буфера, эффективного для обеспечения рН между около 5 и 7. 3. Композиция по п.2, содержащая около 5-200 мг/мл соединения I или его фармацевтически приемлемой соли, от около 12,5 до около 200 мМ ацетатного буфера, около 10-200 мг/мл наполняющего агента и воду. 4. Композиция по п.3, содержащая около 30-50 мг/мл соединения I или его фармацевтически приемлемой соли, около 20-60 мМ ацетатного буфера, около 30-70 мг/мл сахаранаполнителя или комбинации сахаровнаполнителей, эффективных для образования лиофилизованной лепшки, и воду. 5. Композиция по п.4, содержащая 42 мг/мл соединения I или его фармацевтически приемлемой соли, 25 мМ ацетатного буфера, 30 мг/мл сахарозы, 20 мг/мл маннита и воду. 12 6. Композиция по п.4, содержащая 42 мг/мл соединения 1 или его фармацевтически приемлемой соли, 50 мМ ацетатного буфера, 30 мг/мл сахарозы, 20 мг/мл маннита и воду. 7. Способ лечения и/или профилактики грибковых инфекций млекопитающего, включающий внутривенное введение млекопитающему фармацевтически эффективного количества композиции по п.1. 8. Способ по п.7, где млекопитающим является человек. 9. Способ лечения инфекции, вызваннойCandida sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.1. 10. Способ лечения инфекции, вызваннойCandida sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.5. 11. Способ лечения инфекции, вызваннойCandida sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.6. 12. Способ лечения инфекции, вызваннойAspergillus sp, у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.1. 13. Способ лечения инфекции, вызваннойAspergillus sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.5. 14. Способ лечения инфекции, вызваннойAspergillus sp., у пациента, который предусматривает введение этому пациенту эффективного количества композиции по п.6. 15. Способ лечения или профилактики инфекции или состояния, вызванного Pneumocystiscarinii у пациента, нуждающегося в таком лечении или профилактике, который предусматривает введение этому пациенту профилактического или терапевтического количества композиции по п. 1. 16. Способ лечения или профилактики инфекции или состояния, вызванного Pneumocystiscarinii, у пациента, нуждающегося в таком лечении или профилактике, который предусматривает введение этому пациенту профилактического или терапевтического количества композиции по п.5. 17. Способ лечения или профилактики инфекции или состояния, вызванного Pneumocystiscarinii, у пациента, нуждающегося в таком лечении или профилактике, который предусматривает введение этому пациенту профилактического или терапевтического количества композиции по п.6. 18. Способ получения фармацевтической композиции, содержащей соединение, имеющее формулу 14 а) растворения наполнителя или комбинации наполнителей в воде,б) добавления уксусной кислоты и установления рН до около 3,7; в) добавления соединения I и установление рН до от около 5 до около 7 основанием; г) фильтрования таким образом полученного раствора; д) замораживания такого раствора и е) лиофилизации замороженного раствора. и его фармацевтически приемлемые соли, который содержит стадии
МПК / Метки
МПК: A61K 38/12
Метки: буфер, содержащие, агент, антигрибковый, ацетатный, композиции
Код ссылки
<a href="https://eas.patents.su/8-1386-kompozicii-soderzhashhie-antigribkovyjj-agent-i-acetatnyjj-bufer.html" rel="bookmark" title="База патентов Евразийского Союза">Композиции, содержащие антигрибковый агент и ацетатный буфер.</a>
Мутантный il-6 человека и его внутренний фрагмент, кодирующие их последовательности днк, способы их получения, содержащие их фармацевтические композиции, содержащие их векторы, линии клеток- хозяев испособ лечения il-6 опосредованных заболеваний

Номер патента: 852
Опубликовано: 26.06.2000
Авторы: Розе-Йон Штефан, Гротзингер Йоахим, Элерс Марк
МПК: A61K 38/20, A61P 19/10, A61P 35/00...
Метки: клеток, фрагмент, внутренний, композиции, испособ, линии, заболеваний, способы, человека, последовательности, фармацевтические, кодирующие, получения, лечения, хозяев, опосредованных, днк, векторы, мутантный, содержащие
Формула / Реферат:
1. Мутантный интерлейкин-6 (IL-6) человека, имеющий аминокислотную последовательность содержащую следующие точечные мутации по сравнению с природным IL-6 человека: Pro 54, Glu 159, Pro 162, Leu 170 и Аrg 176. 2. Внутренний фрагмент мутантного IL-6 человека по п.1 формулы, обладающий аналогичной биологической активностью. 3. Последовательность ДНК, кодирующая мутантный IL-6 человека по п.1 формулы. 4. Последовательность ДНК, кодирующая...
Фармацевтические композиции, содержащие ингибиторы агрегации тромбоцитов

Номер патента: 628
Опубликовано: 29.12.1999
Автор: Джелотт Карл М.
МПК: A61K 47/02
Метки: содержащие, композиции, агрегации, ингибиторы, тромбоцитов, фармацевтические
Формула / Реферат:
1. Фармацевтическая композиция для внутривенного введения пациенту, содержащая а) фармацевтически эффективное количество соединения формулы где R4 представляет арил, C1-10-алкил или C1-10-арилалкил; R5 представляет где R8 является гидрокси или C1-10-алкилокси; р равен нулю или единице; и m равен целому числу от двух до шести; и его фармацевтически приемлемых солей; б) фармацевтически приемлемое количество цитратного буфера,...
Композиции, содержащие фосфатидные кислоты.

Номер патента: 1159
Опубликовано: 30.10.2000
Авторы: Шиницкий Мейр, Шенфельд Авнер
МПК: A61K 31/683
Метки: композиции, кислоты, фосфатидные, содержащие
Формула / Реферат:
1. Способ лечения симптомов синдрома отмены у человека, включающий введение больному эффективного количества липидного препарата, содержащего, по крайней мере, около 10% (мас./мас.) фосфатидной кислоты. 2. Способ по п.1 лечения симптомов синдрома отмены, связанных с прекращением курения. 3. Способ по п.1, где состоянием субъекта или заболеванием являются симптомы синдрома отмены в процессе реабилитации от привыкания к чрезмерному употреблению...
Бензотиофены, содержащие их фармацевтические композиции и способы лечения патологических состояний с их использованием

Номер патента: 1257
Опубликовано: 25.12.2000
Авторы: Хартауэр Керри Дж., Дэлдер Брайан В., Эрбутнот Гордон Н., Стратфорд Роберт Е., Льюк Вейн Д.
МПК: A61K 31/381, C07D 333/56
Метки: фармацевтические, способы, содержащие, лечения, использованием, патологических, состояний, бензотиофены, композиции
Формула / Реферат:
1. Соединение формулы I и его фармацевтически приемлемые соли и сольваты, характеризующееся тем, что оно находится в форме частиц, причем эти частицы имеют средний размер менее 25 микрон. 2. Соединение по п.1, где указанные частицы имеют средний размер в диапазоне примерно от 5 до 20 микрон. 3. Соединение по п.1 или 2, где, по меньшей мере, 90% указанных частиц имеют размер менее чем 50 микрон. 4. Соединение по п.3, где, по меньшей мере, 90%...
Фармацевтические композиции для парентерального применения, содержащие индолкарбоновые кислоты

Номер патента: 211
Опубликовано: 24.12.1998
Авторы: Лоди Андреа, Россато Мария Тереза
МПК: A61K 47/10
Метки: парентерального, фармацевтические, композиции, кислоты, индолкарбоновые, применения, содержащие
Формула / Реферат:
1. Фармацевтическая композиция в приемлемой для парентерального введения форме, содержащая раствор (Е)-3-[2-(фенилкарбамоил)этенил]-4,6-дихлориндол-2-карбоновой кислоты или ее физиологически приемлемой соли в изотоническом растворе сахара, содержащем смешивающийся с водой органический растворитель для указанного соединения, причем рН указанного препарата находится в пределах от 7 до 9. 2. Фармацевтическая композиция по п.1, отличающаяся тем,...
Предыдущий патент: Промежуточное соединение для использования в синтезе доцетаксела и способ его получения
Следующий патент: Хелатные соединения металлов с макроциклическими полиаминокарбоновыми соединениями и их использование для диагностических исследований
Случайный патент: Непрерывный способ получения хлоргидринов