Трициклические диазепины в качестве антагонистов вазопрессина, способ их получения и способ лечения с использованием трициклических диазепинов
Номер патента: 56
Опубликовано: 30.04.1998
Авторы: Венкатесан Аранапакан Мудумбай, Олбрайт Джей Дональд, Гросу Джордж Теодор



















Формула / Реферат
1. Соединение, выбранное среди соединений с общей формулой I:

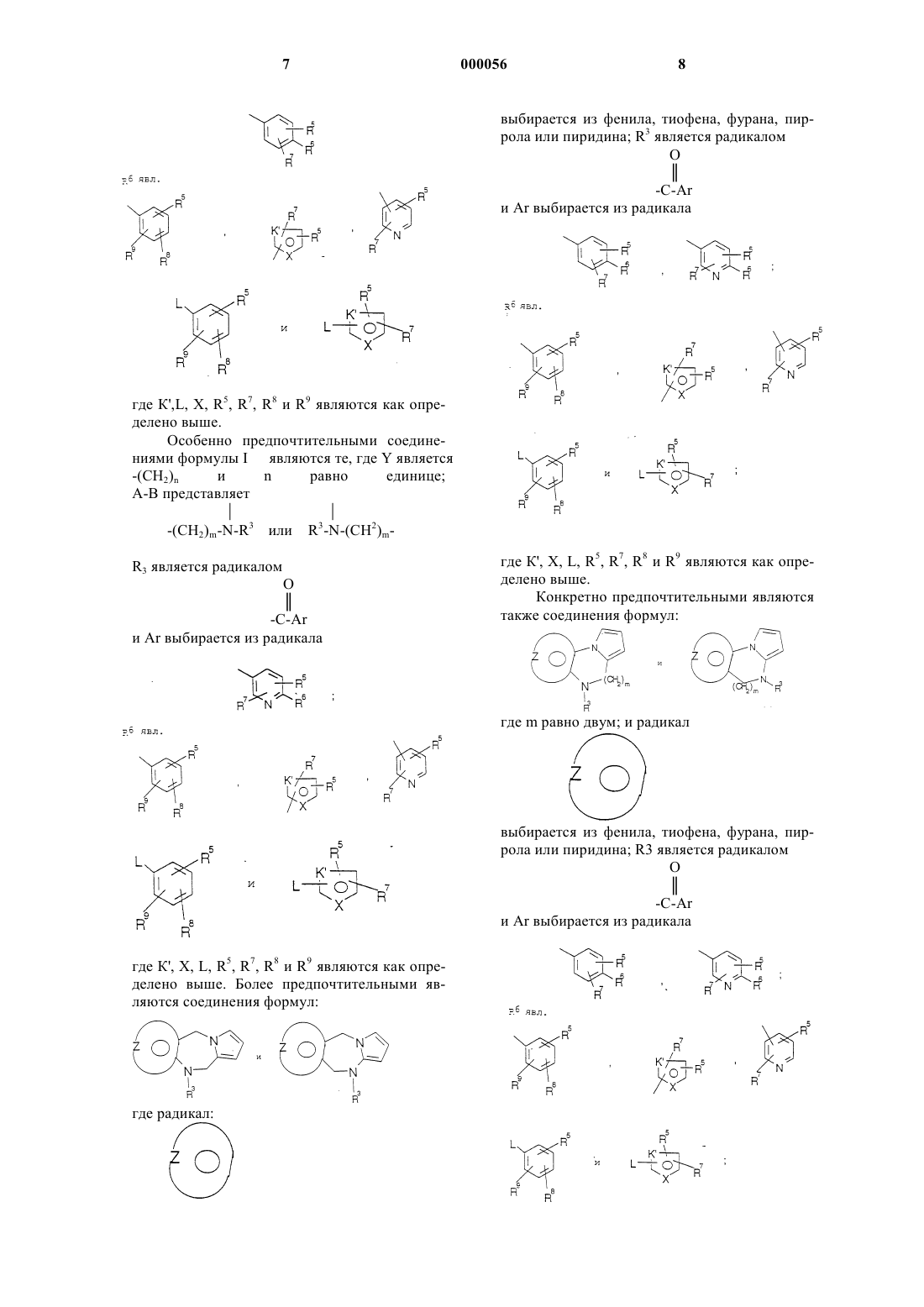
где Y является радикалом, выбранным из -(СН2)n-, где n является целым числом, 0 или 1, и А-В является радикалом, выбранным из
![]()
где m является целым числом от 1 до 2; и радикал:

представляет: (1) фенил или замещенный фенил, необязательно замещенный одним или двумя заместителями, выбранными из (C1-С3) низшего алкила, галогена, амина, (C1-С3) низшего алкокси или (C1-С3) низшего алкиламина; (2) 5-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один гетеро-атом, выбранный из О, N, или S; (3) 6-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один азот; (4) 5- или 6-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее два атома азота; (5) 5-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один атом азота, вместе с или одним атомом кислорода, или одним атомом серы; где 5- или 6-членные гетероциклические кольца необязательно замещены (C1-С3) низшим алкилом, галогеном или (C1-С3) низшим алкокси; радикал:

является пятичленным ароматическим (ненасыщенным), содержащим азот кольцом, где D, E и F являются выбранными из углерода или азота и где атомы углерода могут быть необязательно замещенными галогеном, (C1-С3) низшим алкилом, гидрокси, -СОСl3, -СОСF3,


-СНО, амино, (C1-С3) низшим алкокси и (C1-С3) низшим алкиламином, -CONH-(C1-С3) низшим алкилом, -СON [низшим алкилом (C1-С3)]2; q равен 1 или 2; Rb является независимо выбранным из водорода, -СН3 и -C2H5; R3 является радикалом формулы:

где Аr является

где R6 выбирается из

где L является О, S, SO, SO2, -CO-, -CH2-,
-CєC-;
К' является CH или N;
X является O, S, N-низшим алкилом (C1-С3); и
W' выбирается из О, S, NH, N-низшего алкила(C1-С3) и N-бензила;
R4 выбирается из водорода, низшего алкила (C1-С3), -СО-низшего алкила (C1-С3);
R5 выбирается из водорода, низшего алкила (C1-С3), низшего алкокси (C1-С3),
-O-СН2-СН=СН2 и галогена;
R7 выбирается из водорода, низшего алкила (C1-С3), галогена, О-низшего алкила (C1-С3) и СF3;
R8 и R9 являются независимо выбранными из водорода, низшего алкила (C1-С3), -S-низшего алкила (C1-С3), галогена, -NH-низшего алкила (C1-С3), -ОСF3, -ОН, -CN,
-S-СF3, -NO2, -NH2, О-низшего алкила (C1-С3), СО-низшего алкила (C1-С3) и СF3; и их фармацевтически приемлемых солей.
2. Соединение по п. 1, где А-В является

3. Соединение по п.1, где радикал
является фенилом или замещенным фенилом, необязательно замещенным одним или двумя заместителями, выбранными из (C1-С3) низшего алкила, галогена, амина, (C1-С3) низшего алкокси или (C1-С3) низшего алкиламина.
4. Соединение по п.1, где радикал
является 5-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим один гетероатом, выбранный из О, N или S, необязательно замещенным одним или двумя заместителями, выбранными из необязательно замещенного (C1-С3) низшего алкила, галогена или (C1-С3) низшего алкокси.
5. Соединение по п.1, где радикал
является 6-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим один атом азота, где кольцо необязательно замещено (C1-С3) низшим алкилом, галогеном или (C1-С3) низшим алкокси.
6. Соединение по п.1, где радикал
является 5- или 6-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим два атома азота, где кольцо необязательно замещено (C1-С3) низшим алкилом, галогеном или (C1-С3) низшим алкокси.
7. Соединение по п.1, где радикал
является 5-членным ароматическим (ненасыщенным) гетероциклическим кольцом, обладающим одним атомом азота вместе, или с одним атомом кислорода, или одним атомом серы, где 5-членное гетероциклическое кольцо необязательно замещено (C1-С3) низшим алкилом, галогеном или (C1-С3) низшим алкокси.
8. Соединение по п.1, где радикал
является пятичленным ароматическим (ненасыщенным), содержащим азот кольцом, где D, Е и F являются атомами углерода, где один из атомов углерода необязательно замещен заместителем, выбранным из галогена, (C1-С3) низшего алкила, гидрокси, -СОСl3, -СОСF3,

-СНО, амино, (C1-С3) низшего алкокси и (C1-С3) низшего алкил-амина, -CONH-(C1-С3) низшего алкила, -СON[низшего алкила (C1-С3)]2; q равен 1 или 2; Rb является независимо выбранным из водорода, -СН3 и -С2Н5.
9. Соединение по п.1, где радикал
является пятичленным ароматическим (ненасыщенным) гетероциклическим кольцом, в котором D является азотом и Е и F являются углеродом: где один из атомов углерода необязательно замещен заместителем, выбранным из галогена, (C1-С3) низшего алкила и (C1-С3) низшего алкокси.
10. Соединение по п.1, где Y является -(СН2)n-, n равен 0 или 1, где радикал
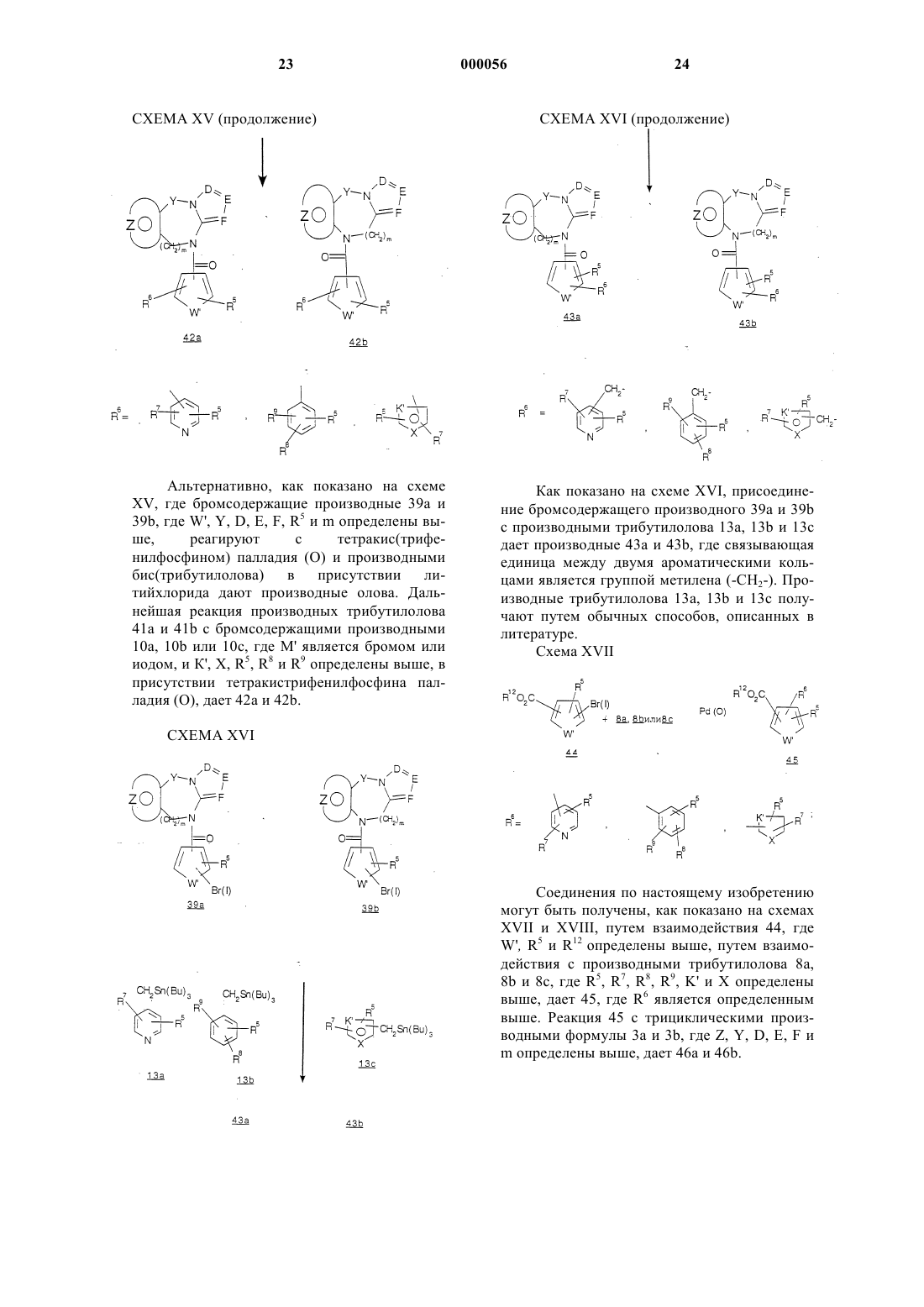
является фенилом или замещенным фенилом, необязательно замещенным одним или двумя заместителями, выбранными из (C1-С3) низшего алкила, галогена, амина,(C1-С3) низшего алкокси или (C1-С3) низшего алкиламина; радикал
является пятичленным ароматическим (ненасыщенным) гетероциклическим кольцом, в котором D, E и F являются атомами углерода, где один из атомов углерода необязательно замещен заместителем, выбранным из галогена, (C1-С3) низшего алкила, гидрокси, -СОСl3, -СОСF3,
-СНО, амино, (C1-С3) низшего алкокси и (C1-С3) низшего алкиламина, -CONH-(C1-С3) низшего алкила, -CON [низшего алкила (C1-С3)]2; q равен 1 или 2; Rb является независимо выбранным из водорода, -СН3 и -C2H5.
11. Соединение по п.1, где Y является -(СН2)n-, n равен 0 или 1, где радикал
является фенилом или замещенным фенилом, необязательно замещенным одним или двумя заместителями, выбранными из (C1-С3) низшего алкила, галогена, амина, (C1-С3) низшего алкокси или (C1-С3) низшего алкиламина; радикал
является пятичленным ароматическим (ненасыщенным) гетероциклическим кольцом, где D является азотом, а Е и F являются атомами углерода, где один из атомов углерода необязательно замещен заместителем, выбранным из галогена, (C1-С3) низшего алкила и (C1-С3) низшего алкокси.
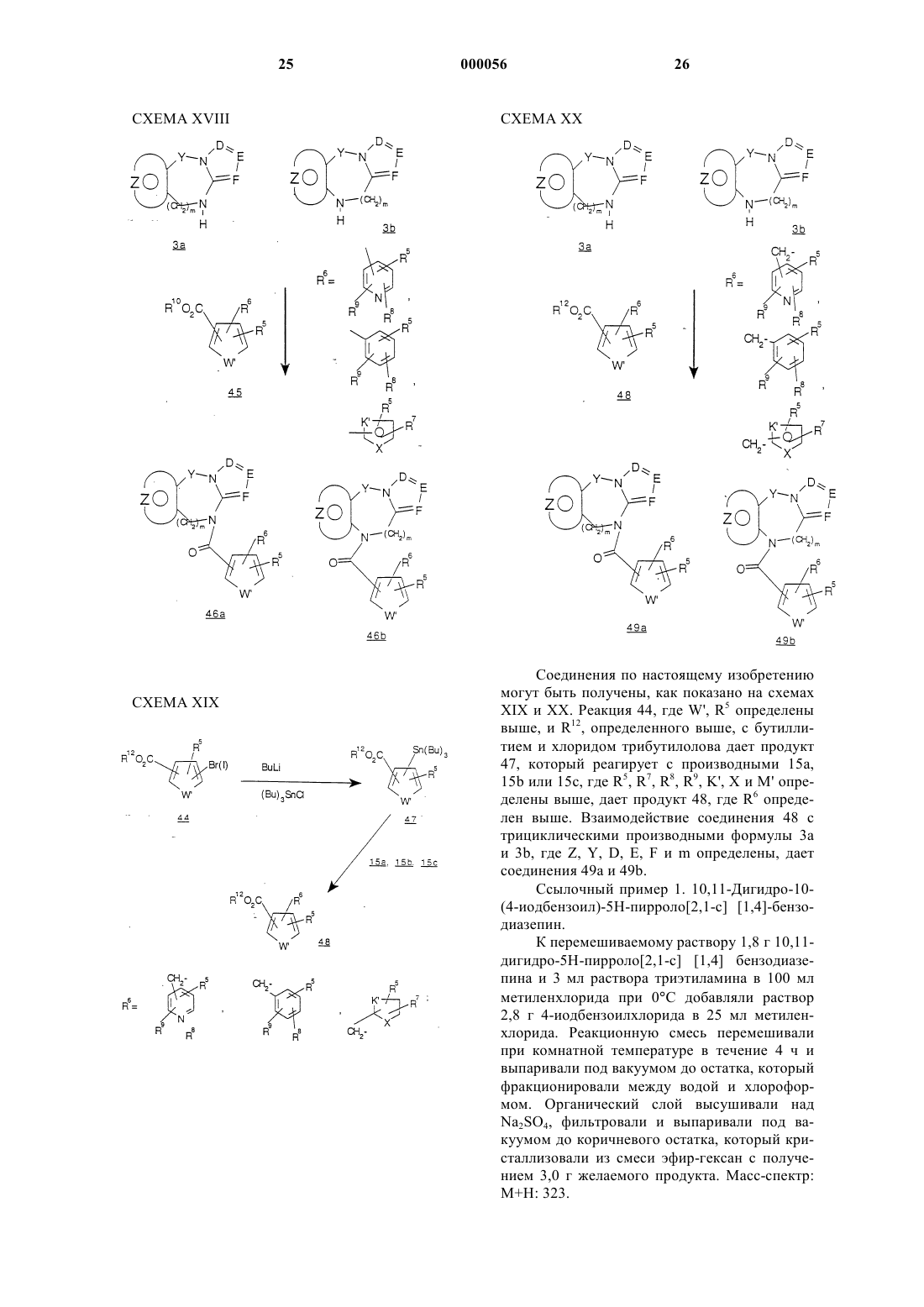
12. Соединение по п.1, где Y является
-(CH2)n-, n равен 0 или 1, где радикал
является 5-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим один гетероатом, выбранный из О или S, где кольцо необязательно замещено (C1-С3) низшим алкилом, галогеном или (C1-С3) низшим алкокси; радикал
является пятичленным ароматическим (ненасыщенным) гетероциклическим кольцом, где D, Е и F являются атомами углерода, где один из атомов углерода необязательно замещен заместителем, выбранным из галогена, (C1-С3) низшего алкила, гидрокси, -СОСl3, -СОСF3,
-CHO, амино, (C1-С3) низшего алкокси и (C1-С3) низшего алкиламина, -CONH- (C1-С3) низшего алкила, -СОN[низшего алкила (C1-С3)]2; q равен 1 или 2; Rb является независимо выбранным из водорода, -СН3 и -C2H5.
13. Соединение по п.1, где Y является -(СН2)n-, n равен 0 или 1, где радикал
является 5-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим один гетероатом, выбранный из O или S, где кольцо необязательно замещено (C1-С3) низшим алкилом, галогеном или (C1-С3) низшим алкокси; радикал
является пятичленным ароматическим (ненасыщенным) гетероциклическим кольцом, где D является азотом, а Е и F являются атомами углерода, где один из атомов углерода необязательно замещен заместителем, выбранным из галогена, (C1-С3) низшего алкила и (C1-С3) низшего алкокси.
14. Соединение по п.1, выбранное среди соединений с общей формулой I:
где Y является - (CH2)n-, где n=0 или 1; А-В является радикалом, выбранным из
![]()
где m является 1 или 2; и радикал:
является 6-членным ароматическим (ненасыщенным) кольцом, имеющим один азот; радикал:
является пятичленным ароматическим (ненасыщенным), содержащим азот гетероциклическим кольцом, где D, E и F являются атомами углерода, где атомы углерода могут быть необязательно замещены заместителем, выбранным среди галогена, (C1-С3) низшего алкила, гидрокси, -СОСl3, -СОСF3.
-СНО, амино, (C1-С3) низшего алкокси и (C1-С3) низшего алкиламина, -CONH-(C1-С3) низшего алкила, -СON[низшего алкила (C1-С3)]2; q равен 1 или 2; Rb является независимо выбранным из водорода, -СН3 и -C2H5; R3 является радикалом формулы:
где Ar является
где R6 выбирается из
где L является O, S, SO, SO2, -CO-, -CH2-,
-CєC-;
К' является СН или N;
Х является О, S, N-низшим алкилом (C1-С3); и
W' выбирается из О, S, NH, N-низшего алкила(C1-С3) и N-бензила;
R4 выбирается из водорода, низшего алкила (C1-С3), -СО-низшего алкила (C1-С3);
R5 выбирается из водорода, низшего алкила (C1-С3), низшего алкокси (C1-С3),
-O-СН2-СН=СН2 и галогена; R7 выбирается из водорода, низшего алкила (C1-С3), галогена, О-низшего алкила (C1-С3) и СF3;
R8 и R9 являются независимо выбранными из водорода, низшего алкила (C1-С3), -S-низшего алкила (C1-С3), галогена, -NH-низшего алкила (C1-С3), -ОСF3, -ОН, -СN,
-S-CF3, -NO2, -NH2, О-низшего алкила (C1-С3), СО-низшего алкила (C1-С3) и СF3; и их фармацевтически приемлемых солей.
15. Соединение по п.1, выбранное среди соединений с общей формулой I:
где Y является -(СН2)n-, где n=0 или 1; А-В является радикалом, выбранным из
![]()
где m является 1 или 2; и радикал:
является 6-членным ароматическим (ненасыщенным) гетероциклическим кольцом, имеющим один азот; радикал:
является пятичленным ароматическим (ненасыщенным), содержащим азот гетероциклическим кольцом, где D является азотом, а Е и F являются атомами углерода, где атомы углерода могут быть необязательно замещены заместителем, выбранным среди галогена, (C1-С3) низшего алкила, (C1-С3) низшего алкокси; R3 является радикалом формулы:
где Аr является
где R6 выбирается из
где L является О, S, SO, SO2, -CO-, -CH2-,
-CєC-;
К' является СН или N;
Х является О, S, N-низшим алкилом (C1-С3); и
W' выбирается из О, S, NН, N-низшего алкила(C1-С3) и N-бензила;
R4 выбирается из водорода, низшего алкила (C1-С3), -СО-низшего алкила (C1-С3);
R5 выбирается из водорода, низшего алкила (C1-С3), низшего алкокси (C1-С3),
-O-СН2-СН=СН2 и галогена;
R7 выбирается из водорода, низшего алкила (C1-С3), галогена, О-низшего алкила (C1-С3) и СF3;
R8 и R9 являются независимо выбранными из водорода, низшего алкила (C1-С3), -S-низшего алкила (C1-С3), галогена, -NН-низшего алкила (C1-С3), -ОСF3, -ОН, -СN,
-S-СF3, -NO2, -NH2, О-низшего алкила (C1-С3), СО-низшего алкила (C1-С3) и СF3; и их фармацевтически приемлемых солей.
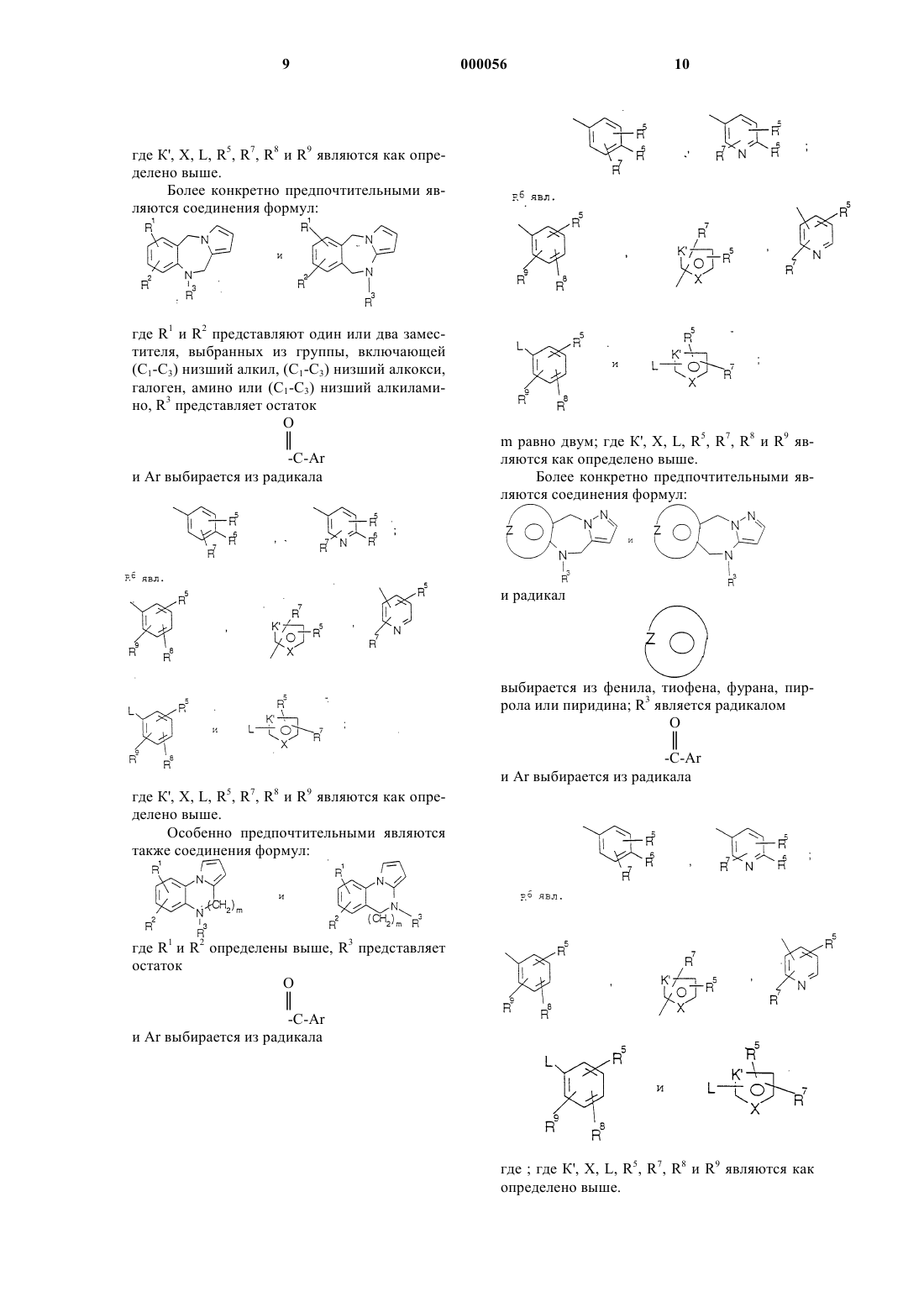
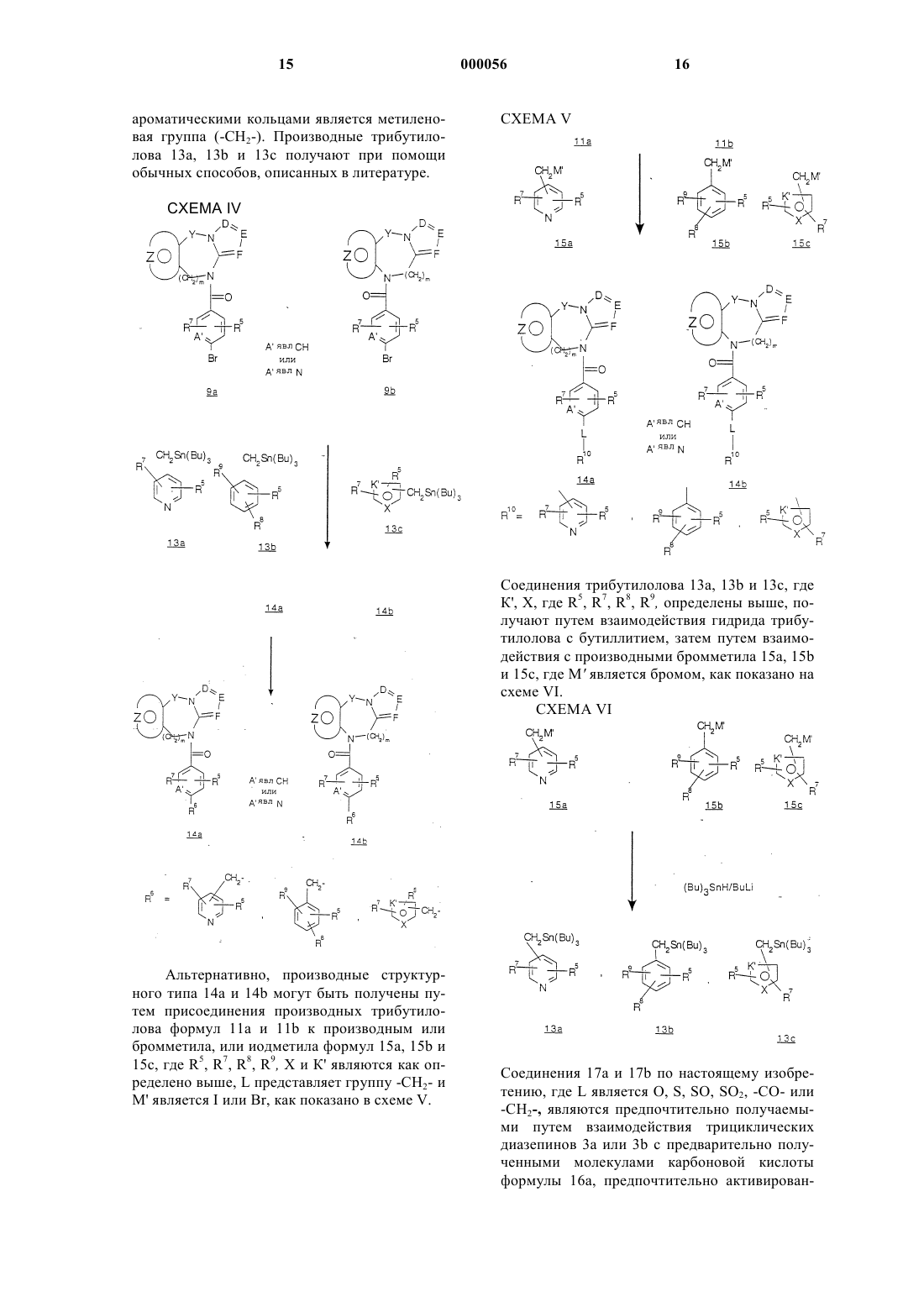
16. Соединение по п.11, выбранное из группы, включающей:
10,11-дигидро-10-[4-(2-тиенил)бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[4-(2-нитрофенил)бензоил]-5Н-пирроло[2,1-c] [1,4]-бензодиазепин;
10,11-дигидро-10-[4-(3,5-дифторфенил)бензоил]-5Н-пирроло[2,1-c]-[1,4]-бензодиазепин;
10,11-дигидро-10-[4-(фенилэтинил)-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[4-(2-метилфенил)бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-3-[(диметиламино)метил]-10-[4-(2-тиенил)-бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
5-([1,1'-бифенил]-4-илкарбонил)-5,10-дигидро-4Н-пиразоло[5,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[2'-(трифторметил)[1,1'-бифенил]-4-ил]карбонил-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[4-(2-пиридинил)бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[4-(2-тиазолил)бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[4-[(4-метилфенил)тио]бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[[4-фенилсульфонил]бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[4-[(4-метилфенил)сульфонил]бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10,11-дигидро-10-[[4'-(2-пропенилокси)[1,1'-бифенил]-4-ил]карбонил]-5Н-пирроло[2,1-c] [1,4]-бензодиазепин;
10,11-дигидро-10-[4-(фенилтио)бензоил]-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10-(4-бензоилбензоил)-10,11-дигидро-5Н-пирроло[2,1-c] [1,4]-бензодиазепин;
5-([1,1'-бифенил]4-илкарбонил)-4,5-дигидро-пирроло[1,2-а] хиноксалин;
10,11-дигидро-10-[4-(4-мeтилфeнoкcи) бензоил]-5Н-пирроло-[2,1-с] [1,4]-бензодиазепин;
10-([1,1'-бифенил]-4-илкарбонил)-10,11-дигидро-5Н-пирроло[2,1-с] [1,4]-бензодиазепин;
10-([1,1'-бифенил]-4-илкарбонил)-10,11-дигидpo-N,N-диметил-5Н-пирроло[2,1-с] [1,4]-бензодиазепин-3-метанамин;
10,11-дигидро-10-[(4'-пропил[1,1'-бифенил]-4-ил)карбонил]-5Н-пирроло[2,1-c] [1,4]-бензодиазепин;
10,11-дигидpo-10-[5[(2-тиенил)пиридинил] карбонил]-5Н-пирроло[2,1-c] [1,4]-бензодиазепин.
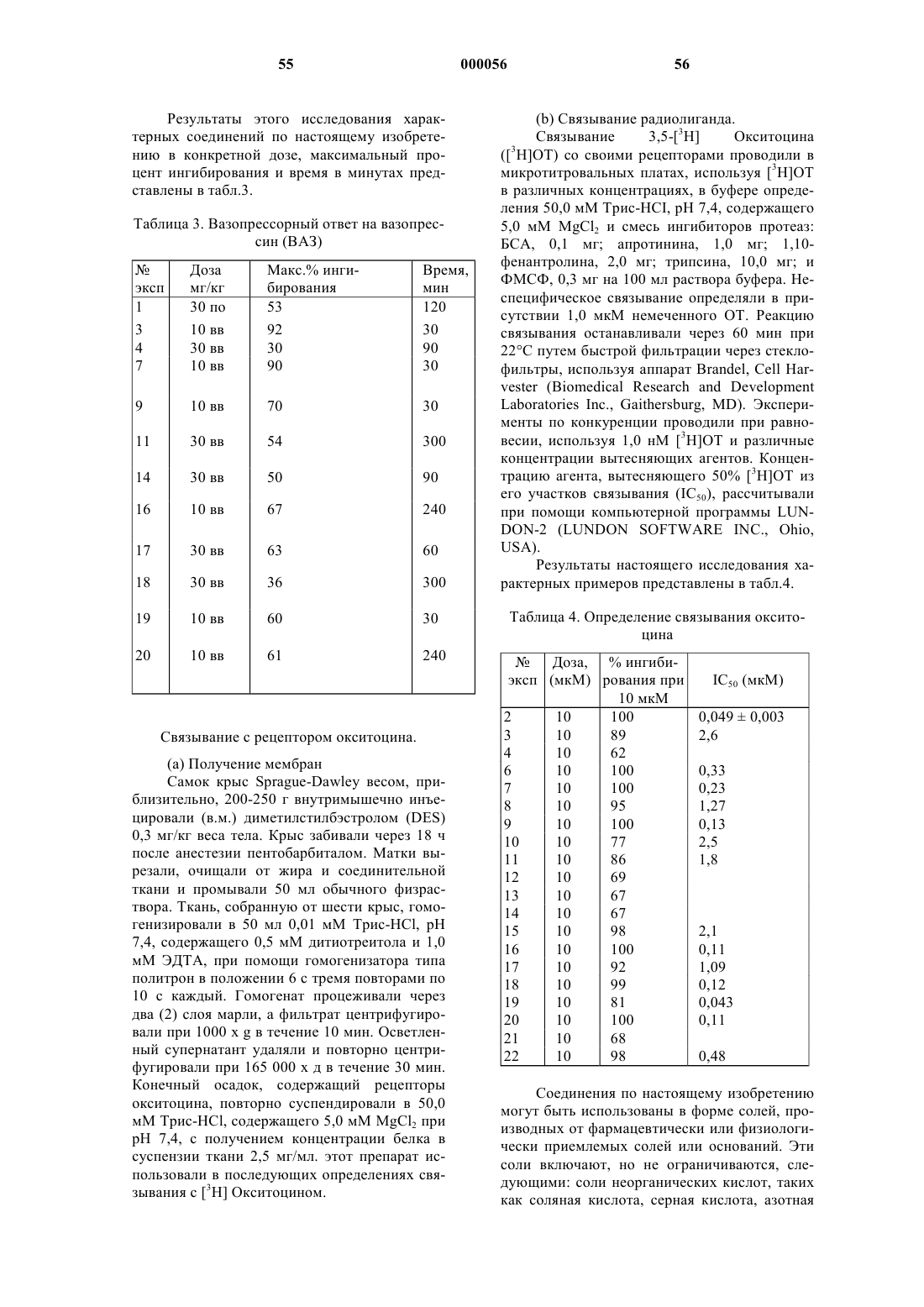
17. Способ лечения заболеваний, характеризуемых избыточной почечной реабсорбцией воды, а также застойной сердечной недостаточностью, циррозом печени, почечным синдромом, поражением центральной нервной системы, заболеванием легких и гипонатрийемией у млекопитающего, отличающийся тем, что вводят указанному млекопитающему соединение по п.1 для облегчения состояния в количестве 0,5-500 мг/кг веса тела.
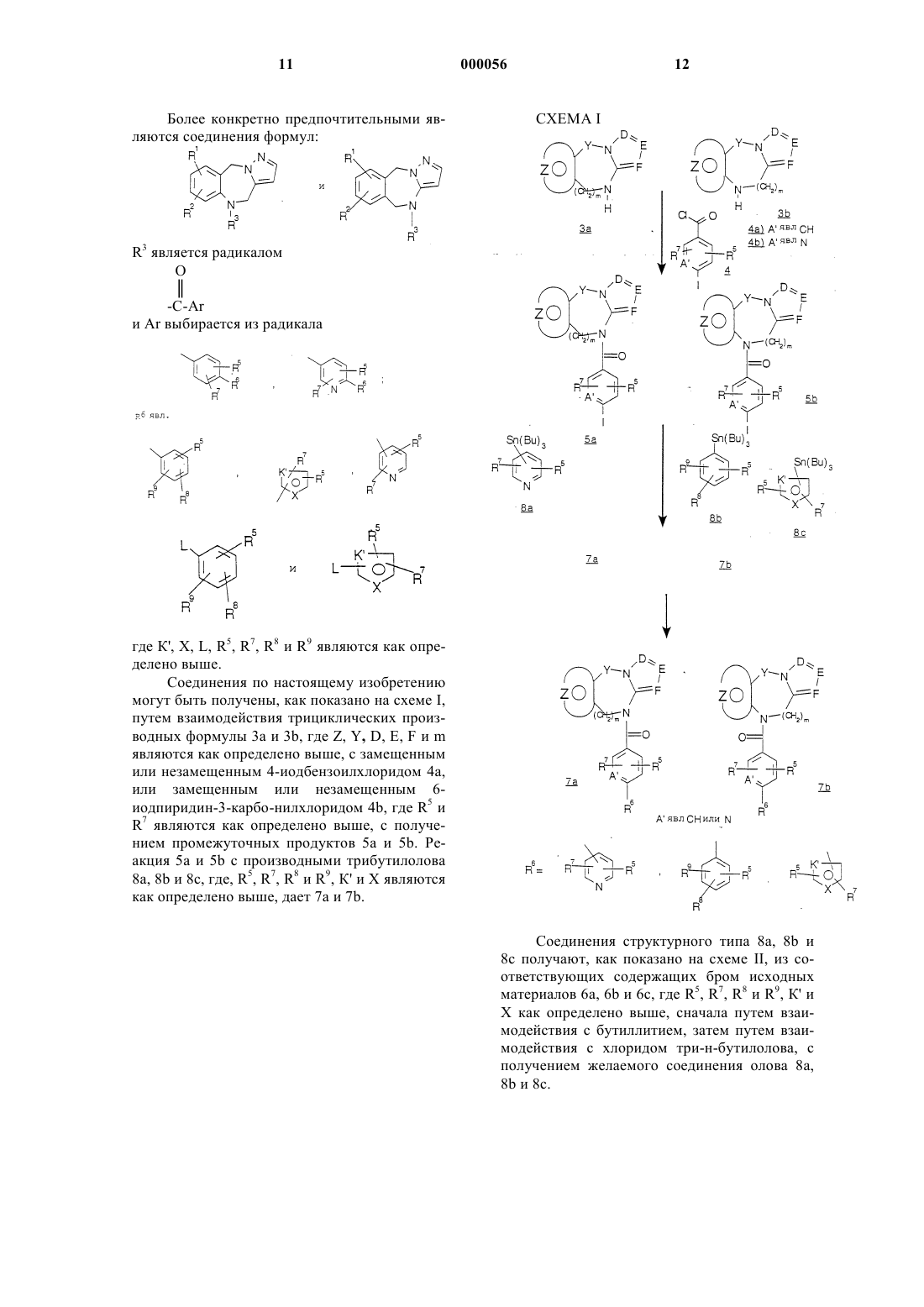
18. Способ получения соединения, выбранного среди соединений с общей формулой I:
где Y является радикалом, выбранным из -(СН2)n-, где n является целым числом, 0 или 1 и А-В является радикалом, выбранным из
![]()
где m является целым числом от 1 до 2; и радикал:
представляет: (1) фенил или замещенный фенил, необязательно замещенный одним или двумя заместителями, выбранными из группы (C1-С3) низшего алкила, галогена, амина, (C1-С3) низшего алкокси или (C1-С3) низшего алкиламина; (2) 5-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один гетероатом, выбранный из О, N, или S: (3) 6-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один азот; (4) 5- или 6-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее два атома азота; (5) 5-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один атом азота, вместе с или одним атомом кислорода, или одним атомом серы; где 5- или 6-членное гетероциклические кольца являются необязательно замещенными группой (C1-С3) низшим алкилом, галогеном или (C1-С3) низшим алкокси; радикал:
является пятичленным ароматическим (ненасыщенным), содержащим азот кольцом, где D, E и F являются выбранными из углерода или азота и где атомы углерода могут быть необязательно замещены галогеном, (C1-С3) низшим алкилом, гидрокси, -СОСl3, -СОСF3,
-СНО, амино, (C1-С3) низшим алкокси и (C1-С3) низшим алкиламином, -CONH-(C1-С3) низшим алкилом, -СОN [низшим алкилом (C1-С3)]2; q равен 1 или 2; Rb является независимо выбранным из водорода,
-СН3 и -C2H5; R3 является радикалом формулы:
где Аr является
где R6 выбирается из
где L является O, S, SO, SO2, -CO-, -CH2-,
-CєC-;
K' является ch или N;
X является O, S, N-низшим алкилом (C1-С3); и
W' выбирается из О, S, NH, N-низшего алкила(C1-С3) и N-бензила;
R4 выбирается из водорода, низшего алкила (C1-С3), -СО-низшего алкила (C1-С3);
R5 выбирается из водорода, низшего алкила (C1-С3), низшего алкокси (C1-С3),
-O-СН2-СН=СН2 и галогена;
R7 выбирается из водорода, низшего алкила (C1-С3), галогена, О-низшего алкила (C1-С3) и СF3;
R8 и R9 являются независимо выбранными из водорода, низшего алкила (C1-С3), -S-низшего алкила (C1-С3), галогена, -NH-низшего алкила (C1-С3), -ОСF3, -ОН, -CN,
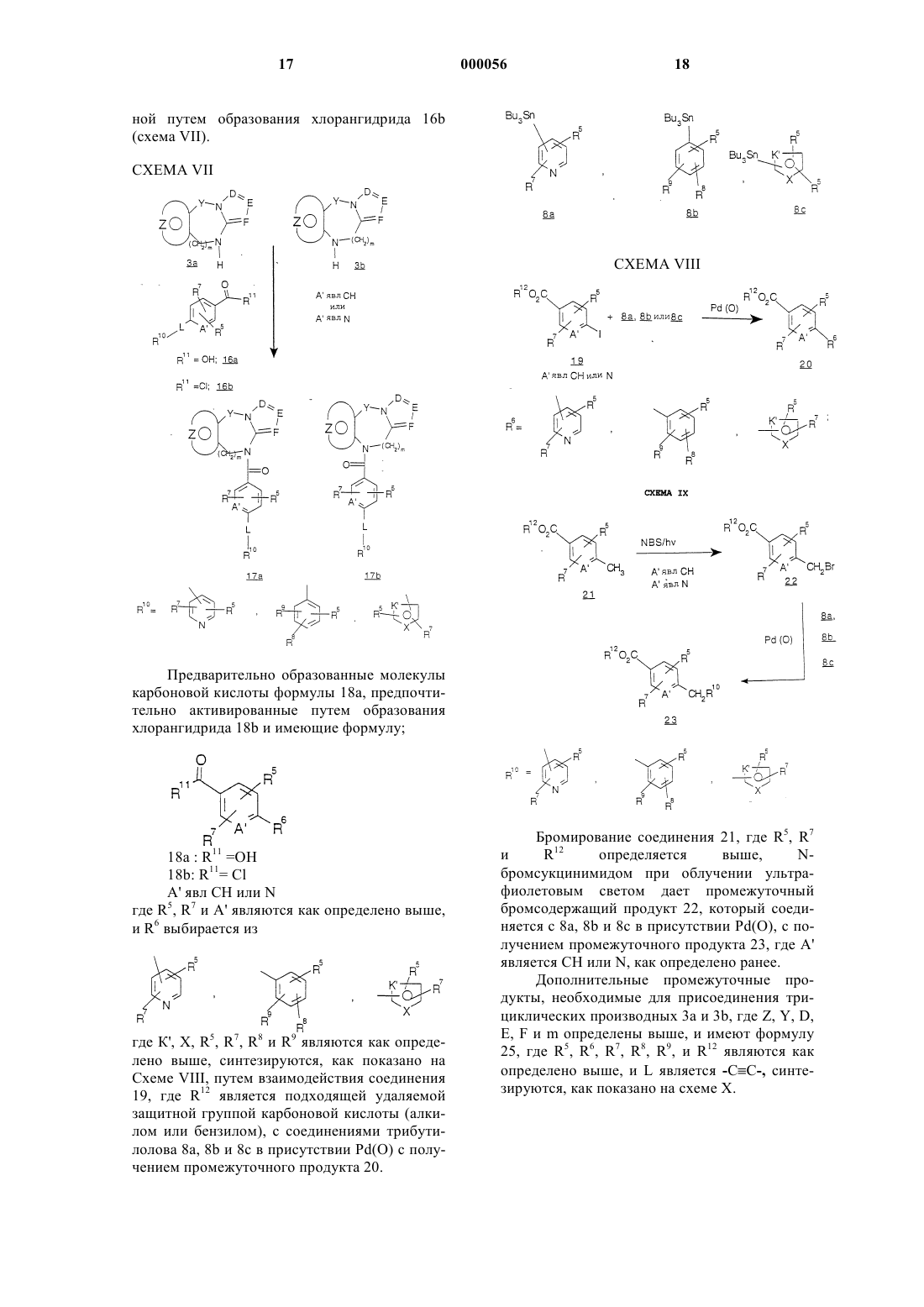
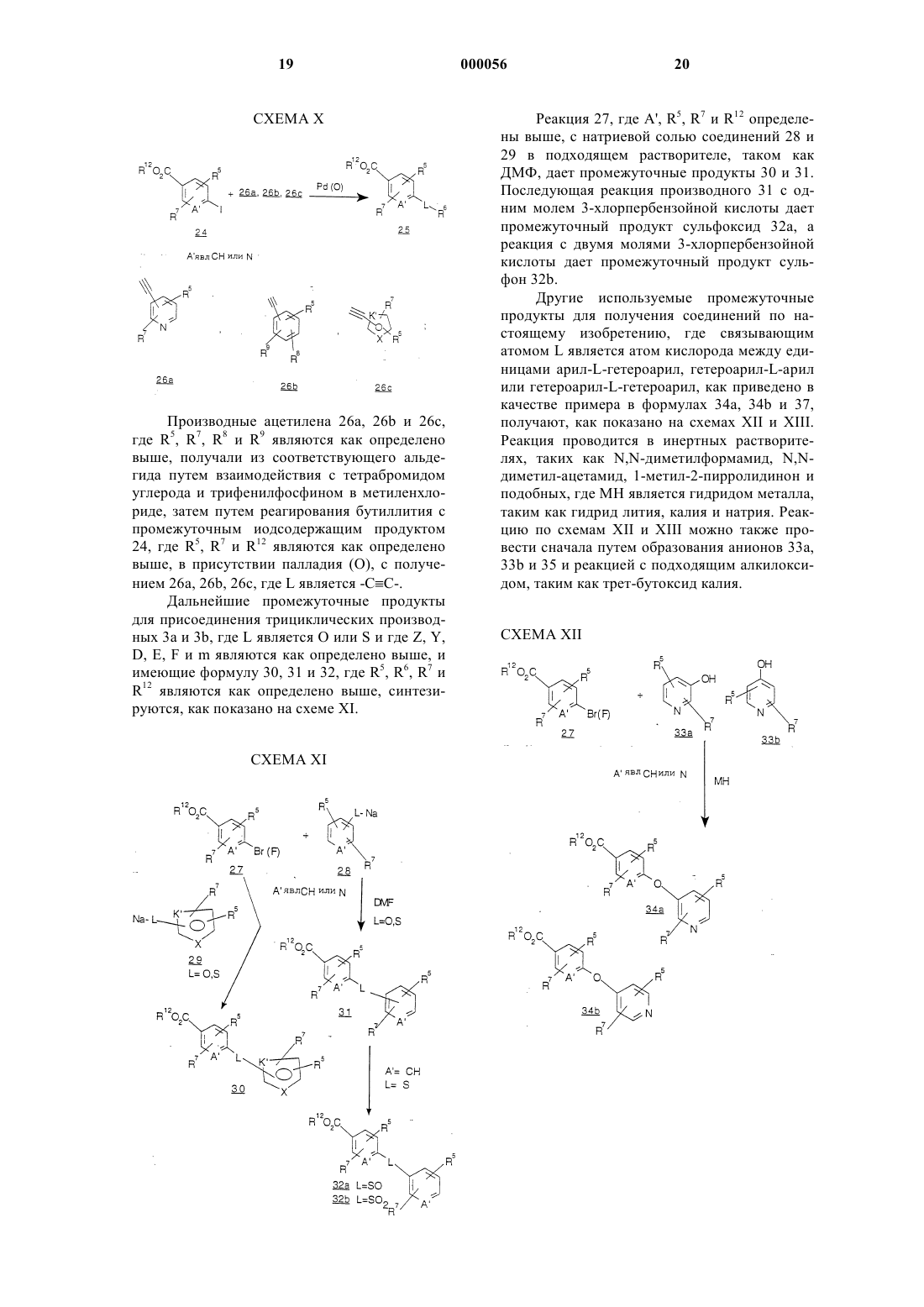
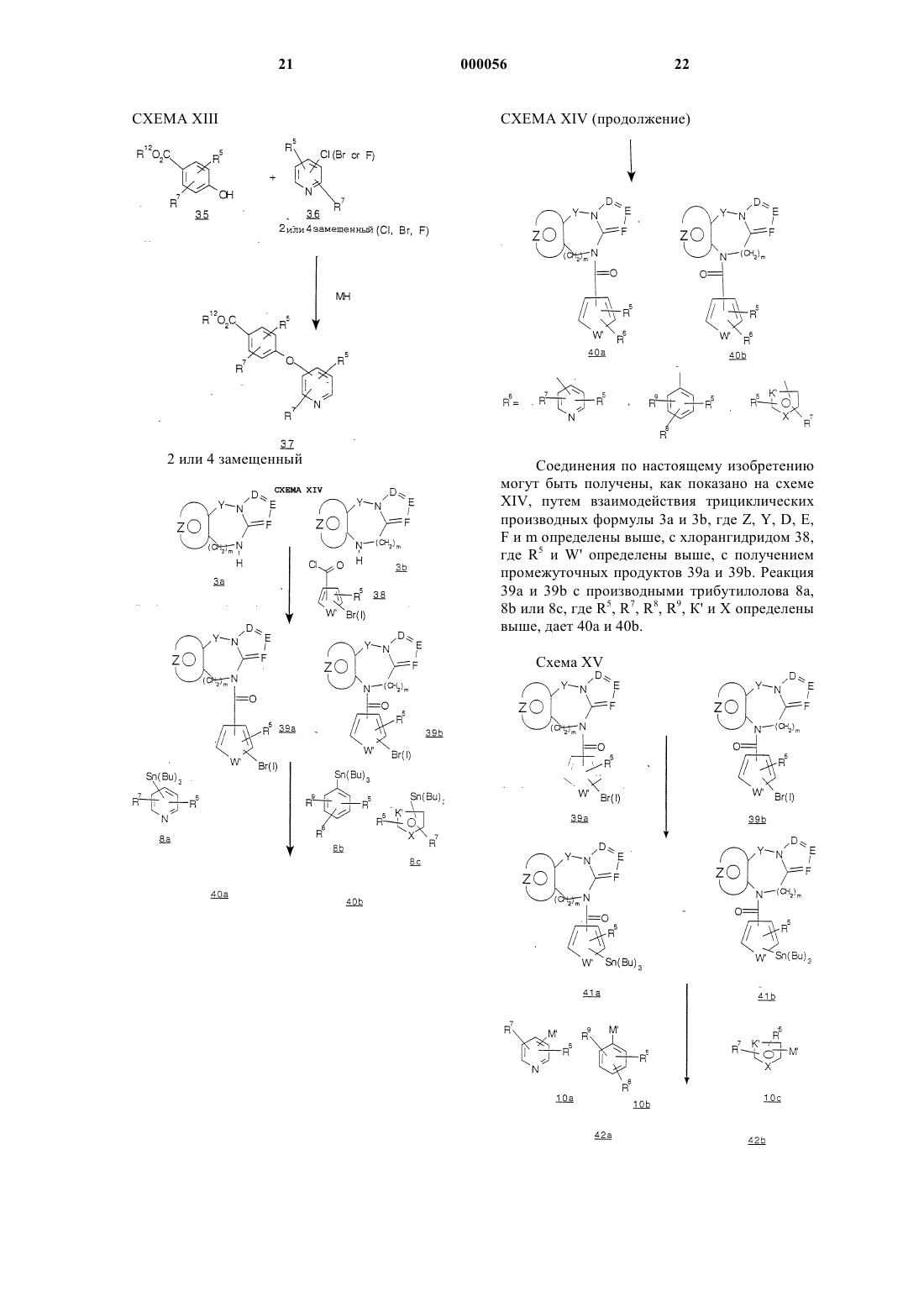
-S-СF3, -NO2, -NH2, О-низшего алкила (C1-С3), СО-низшего алкила (C1-С3) и СF3; включающий осуществление взаимодействия соединения формулы:

с соединением формулы:

где Q является галогеном или активирующей группой, которая получается при превращении арилкарбоновой кислоты в смешанный ангидрид, или при активации реагента, сшивающего пептид, с получением соединений формулы I.
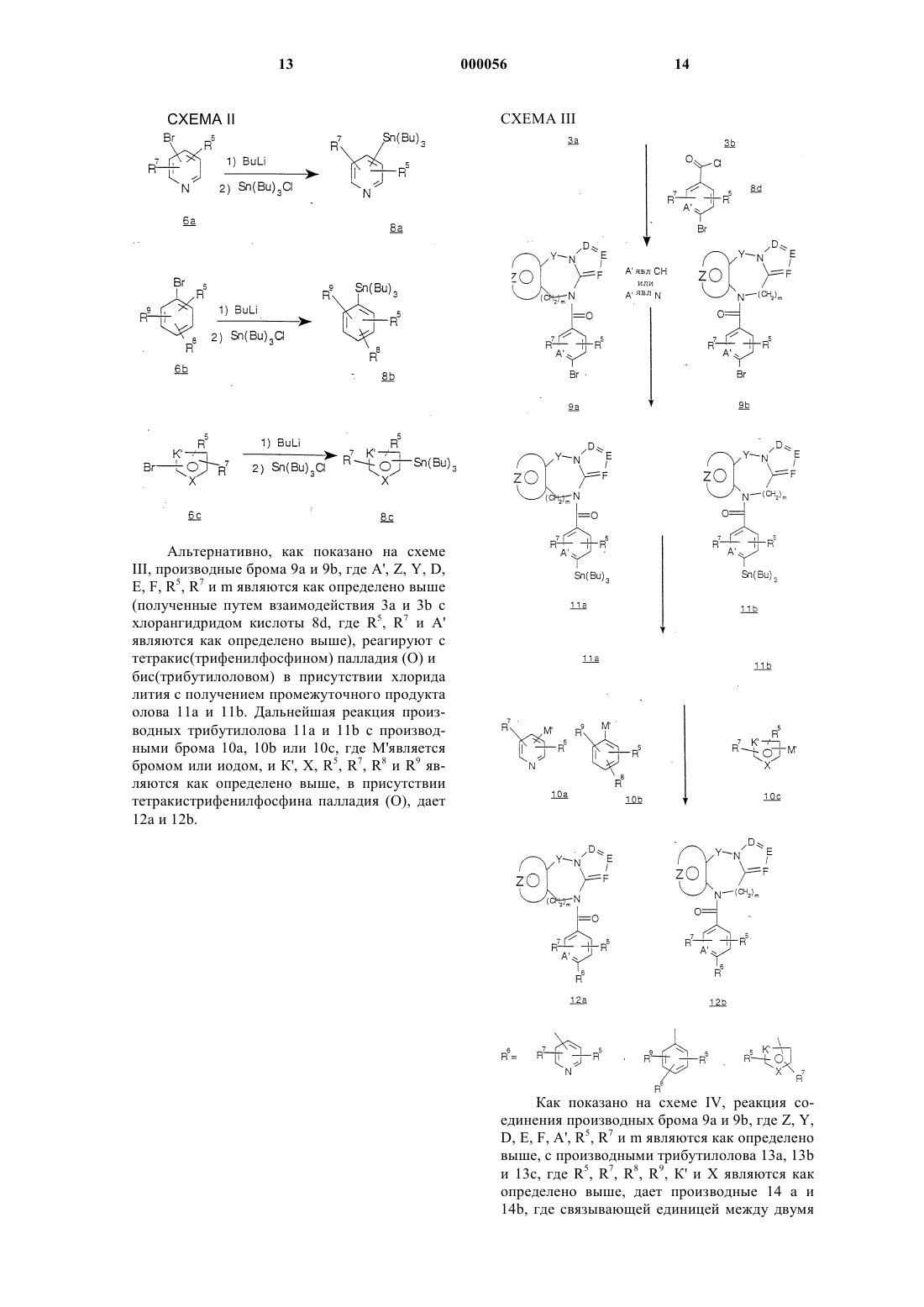
Текст
1 Настоящее изобретение относится к новым непептидным трициклическим антагонистам вазопрессина, которые используются для лечения состояний, когда желателен пониженный уровень вазопрессина, таких как застойная сердечная недостаточность, для болезненных состояний с повышенной реабсорбцией воды в почке и состояний с повышенной сосудистой сопротивляемостью и коронарным вазоспазмом. Вазопрессин высвобождается из заднего гипофиза либо в ответ на повышенную осмомолярность плазмы, регистрируемую осморецепторами мозга, либо на пониженный объем крови и давление крови, определяемые рецепторами объема низкого давления и артериальными барорецепторами. Гормон осуществляет свое действие посредством двух хорошо известных подтипов рецептора: рецептор V1 в сосудах и рецептор V2 в эпителии почки. Задержка экскреции мочи, опосредуемая V2 рецепторами эпителия почки, помогает поддерживать нормальную осмомолярность плазмы,объем крови и давление крови. Вазопрессин вовлечен в некоторые случаи застойной сердечной недостаточности,когда повышена периферическая сопротивляемость. Антагонисты V1 рецепторов могут снижать системное сосудистое сопротивление,увеличивать сердечный выброс и предотвращать индуцированный вазопреccином спазм коронарных сосудов. Таким образом, при индуцированных вазопрессином состояниях с повышенным общим периферическим сопротивлением и нарушенном местном кровотоке антагонисты вазопрессина могут быть лекарственными препаратами. Антагонисты V1 рецепторов могут снижать давление крови, вызывать гипотензивное действие и быть, таким образом, терапевтически полезными при лечении некоторых типов гипертонии. Блокада V2 рецепторов является полезной при лечении заболеваний, характеризующихся повышенной почечной реабсорбцией свободной воды. Антидиурез регулируется посредством выброса вазопрессина (антидиуретического гормона) из гипоталамуса, который связывается со специфическими рецепторами тубулярных клеток почки, осуществляющих абсорбцию воды. Это связывание стимулирует аденилатциклазу и приводит к повышению опосредованного цАМФ встраивания пор для воды в люминальную поверхность данных клеток. V2 антагонисты могут регулировать задержку жидкости при застойной сердечной недостаточности, циррозе печени, почечном синдроме, поражениях центральной нервной системы, заболеваниях легких и гипонатрийемии. Повышенный уровень вазопрессина наблюдается при застойной сердечной недостаточности, которая более часто встречается у пожилых больных с сердечной недостаточно 000056 стью. Антагонисты V2 вазопрессина могут быть полезными у больных с гипонатрийемической застойной сердечной недостаточностью и повышенным уровнем вазопрессина,способствуя экскреции свободной воды путем блокирования действия антидиуретического гормона. На основании биохимического и фармакологического действий гормона ожидается, что антагонисты вазопрессина будут терапевтически полезными при лечении и/или профилактике гипертонии, сердечной недостаточности, коронарного вазоспазма, ишемии сердца, почечного вазоспазма, цирроза печени,застойной сердечной недостаточности, почечного синдрома, отека мозга, ишемии мозга,геморрагического инсульта мозга, тромболитического кровотечения и состояния нарушения задержки воды. Следующие ссылки предшествующего уровня техники описывают пептидные антагонисты вазопрессина: М. Manning et al., J. Med.P.D. Williams et al., сообщали об эффективном гексапептиде антагонисте окситоцина[J. Med. Chem., 35, 3905 (1992)], который также проявлял слабую активность антагониста вазопрессина по связыванию с V1 и V2 рецепторами. Пептидные антагонисты вазопрессина имеют недостаток из-за отсутствия активности при приеме через рот, и многие из этих пептидов не являются селективными антагонистами,поскольку они проявляют также активность частичного агониста. Недавно были описаны непептидные антагонисты вазопрессина Y. Yamamura et al.,Science, 252, 579 (1991); Y. Yamamura et al., Br.Fujisava Co., Ltd., EP 620216-A1 Ogawa et al.,(Otsuka Pharm. Co.) EP 470514A описывают производные карбостирила и фармацевтические композиции, содержащие их. Непептидные антагонисты окситоцина и вазопрессина описаны Merck and Co., M.G. Bock and P.D.Williams, EP 0533242A; M.G. Bock et al., EP 0533244A; J. M. Erb, D.F. Verber, P.D. Villiams,EP 0533240A; K. Gilbert et al., EP 0533243A. Рождение недоношенного ребенка может быть причиной проблем со здоровьем и смерти, и ключевым медиатором в механизме родов является пептидный гормон окситоцин. На основании фармакологического действия окситоцина антагонисты этого гормона исполь 3 зуются для предотвращения преждевременных родов (В.Е. Evans et al., J. Med. Chem. 35, 3919(1992), J. Med. Chem., 36, 3993 (1993) и включенные в них ссылки). Соединения по настоящему изобретению являются антагонистом пептидного гормона окситоцина и используются для регуляции преждевременных родов. Настоящее изобретение относится к новым трициклическим производным, которые проявляют активность антагониста по отношению к V1 и/или V2 рецепторам и проявляют активность антагониста вазопрессина in vivo. Соединения проявляют также активность антагониста по отношению к рецепторам окситоцина. Настоящее изобретение относится к новым соединениям, выбранным из группы с общей формулой I:E и F выбираются из углерода или азота и где атомы углерода могут быть необязательно замещенными заместителем, выбранным из галогена, (C1-С 3) низшего алкила, гидрокси, СОСl3, -СОСF3, где Y является радикалом, выбранным из-(СН 2)n-, где n является целым числом 0 или 1,и А-В является радикалом, выбранным из-СНО, амино, (C1-С 3) низшего алкокси и (C1 С 3) низшего алкиламина, -CONH-( C1-С 3) низшего алкила, -CON[низший алкил (C1-С 3]2; q равен 1 или 2, Rb является независимо выбранным из водорода, -СН 3 и -C2H5; R3 представляет радикал формулы:R3 где m является целым числом от 1 до 2; и радикал: представляет: (1) фенил или замещенный фенил, необязательно замещенный одним или двумя заместителями, выбранными из группы(C1-С 3) низшего алкила, галогена, амина, (C1 С 3) низшего алкокси или (C1-С 3) низшего алкиламина; (2) 5-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один гетероатом, выбранный из О, N, или(ненасыщенное) гетероциклическое кольцо,имеющее два атома азота; (5) 5-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один атом азота, вместе с или кислородом, или атомом серы; где 5- или 6-членные гетероциклические кольца являются необязательно замещенными группой (C1 С 3) низшим алкилом, галогеном или (C1-С 3) низшим алкокси; радикал: является пятичленным ароматическим (ненасыщенным), содержащим азот кольцом, где D, где R6 выбирается из где L является O, S, SO, SO2, -CO-, -СН 2-, СС-; К' является СН или N; Х является О, S, N-низшим алкилом (C1-С 3) и W' выбирается из О, S, NH, N-низшего алкила (C1-С 3) и N-бензила;R8 и R9 являются независимо выбранными из водорода, низшего алкила (C1-С 3), -Sнизшего алкила (C1-С 3), галогена, -NHнизшего алкила (C1-С 3), N-[низшего алкила(C1-С 3)]2,-ОСF3,-ОН, -CN, -S-СF3, -NO2, - NH2, О-низшего алкила (C1-С 3), СО-низшего алкила (C1-С 3) и СF3; и их фармацевтически приемлемые соли. Внутри группы соединений, определенных формулой I, определенные подгруппы соединений являются особенно предпочтительными. Особенно применимыми являются те соединения, где R3 является радикалом: где R5, R6 и R7 являются как определено выше. Особенно предпочтительными являются соединения, где R3 является радикалом-C-Ar и Аг выбирается из радикала Наиболее предпочтительными соединениями формулы I являются те, где Y является (CH2)n и n равно единице; А-В представляет где K', Х, L, R5, R7, R8 и R9 являются как определено выше. Наиболее предпочтительными соединениями формулы I являются те, где Y является (CH2)n и n равно нулю или единице; где радикал-(CH2)m-N-R3 или R3-N-(CH2)m где K', X, L, R5, R7, R8 и R9 являются как определено выше. Особенно предпочтительными являются также соединения, где Y в формуле I является-(CH2)n и n равно нулю или единице;-(CH2)m-N-R3 или R3-N-(CH2)mи К', Х, L, R3, R5, R6, R7, R8 и R9 являются как определено выше, и m является целым числом от 1 до 2.m равно единице, когда n равно единице, и m равно двум, когда n равно нулю; D, E, F, К', L,X, R3, R5, R6, R7, R8 и R9 являются как определено ранее. Особенно предпочтительными являются соединения, где R3 является радикалом где К',L, Х, R5, R7, R8 и R9 являются как определено выше. Особенно предпочтительными соединениями формулы I являются те, где Y является где К', X, L, R5, R7, R8 и R9 являются как определено выше. Конкретно предпочтительными являются также соединения формул:-C-Ar и Аr выбирается из радикала где К', Х, L, R5, R7, R8 и R9 являются как определено выше. Более предпочтительными являются соединения формул: где К', X, L, R5, R7, R8 и R9 являются как определено выше. Более конкретно предпочтительными являются соединения формул: где R1 и R2 представляют один или два заместителя, выбранных из группы, включающейm равно двум; где К', Х, L, R5, R7, R8 и R9 являются как определено выше. Более конкретно предпочтительными являются соединения формул:-С-Аr и Аr выбирается из радикала где К', X, L, R5, R7, R8 и R9 являются как определено выше. Особенно предпочтительными являются также соединения формул: где ; где К', Х, L, R5, R7, R8 и R9 являются как определено выше. 11 Более конкретно предпочтительными являются соединения формул: где К', Х, L, R5, R7, R8 и R9 являются как определено выше. Соединения по настоящему изобретению могут быть получены, как показано на cхеме I,путем взаимодействия трициклических производных формулы 3 а и 3b, где Z, Y, D, Е, F и m являются как определено выше, с замещенным или незамещенным 4-иодбензоилхлоридом 4 а,или замещенным или незамещенным 6 иодпиридин-3-карбо-нилхлоридом 4b, где R5 иR7 являются как определено выше, с получением промежуточных продуктов 5 а и 5b. Реакция 5 а и 5b с производными трибутилолова 8 а, 8b и 8 с, где, R5, R7, R8 и R9, К' и X являются как определено выше, дает 7 а и 7b. Соединения структурного типа 8 а, 8b и 8 с получают, как показано на схеме II, из соответствующих содержащих бром исходных материалов 6 а, 6b и 6c, где R5, R7, R8 и R9, К' иX как определено выше, сначала путем взаимодействия с бутиллитием, затем путем взаимодействия с хлоридом три-н-бутилолова, с получением желаемого соединения олова 8 а,8b и 8 с. Альтернативно, как показано на схемеIII, производные брома 9 а и 9b, где A', Z, Y, D,Е, F, R5, R7 и m являются как определено выше(полученные путем взаимодействия 3 а и 3b с хлорангидридом кислоты 8d, где R5, R7 и А' являются как определено выше), реагируют с тетракис(трифенилфосфином) палладия (О) и бис(трибутилоловом) в присутствии хлорида лития с получением промежуточного продукта олова 11 а и 11b. Дальнейшая реакция производных трибутилолова 11 а и 11b с производными брома 10 а, 10b или 10 с, где M'является бромом или иодом, и К', X, R5, R7, R8 и R9 являются как определено выше, в присутствии тетракистрифенилфосфина палладия (О), дает 12 а и 12b. Как показано на схеме IV, реакция соединения производных брома 9 а и 9b, где Z, Y,D, Е, F, A', R5, R7 и m являются как определено выше, с производными трибутилолова 13 а, 13b и 13 с, где R5, R7, R8, R9, К' и X являются как определено выше, дает производные 14 а и 14b, где связывающей единицей между двумя 15 ароматическими кольцами является метиленовая группа (-СН 2-). Производные трибутилолова 13 а, 13b и 13 с получают при помощи обычных способов, описанных в литературе. Соединения трибутилолова 13 а, 13b и 13 с, где К', X, где R5, R7, R8, R9, определены выше, получают путем взаимодействия гидрида трибутилолова с бутиллитием, затем путем взаимодействия с производными бромметила 15 а, 15b и 15 с, где M' является бромом, как показано на схеме VI. СХЕМА VI Альтернативно, производные структурного типа 14 а и 14b могут быть получены путем присоединения производных трибутилолова формул 11 а и 11b к производным или бромметила, или иодметила формул 15 а, 15b и 15 с, где R5, R7, R8, R9, X и К' являются как определено выше, L представляет группу -СH2- и М' является I или Вr, как показано в схеме V.-CH2-, являются предпочтительно получаемыми путем взаимодействия трициклических диазепинов 3 а или 3b с предварительно полученными молекулами карбоновой кислоты формулы 16 а, предпочтительно активирован 17 ной путем образования хлорангидрида 16b Предварительно образованные молекулы карбоновой кислоты формулы 18 а, предпочтительно активированные путем образования хлорангидрида 18b и имеющие формулу; 18 а : R =ОН 18b: R11= Сl А' явл СН или N где R5, R7 и А' являются как определено выше,и R6 выбирается из где К', X, R5, R7, R8 и R9 являются как определено выше, синтезируются, как показано на Схеме VIII, путем взаимодействия соединения 19, где R12 является подходящей удаляемой защитной группой карбоновой кислоты (алкилом или бензилом), с соединениями трибутилолова 8 а, 8b и 8 с в присутствии Pd(O) с получением промежуточного продукта 20.R12 определяется выше,Nбромсукцинимидом при облучении ультрафиолетовым светом дает промежуточный бромсодержащий продукт 22, который соединяется с 8 а, 8b и 8 с в присутствии Pd(O), с получением промежуточного продукта 23, где А' является СН или N, как определено ранее. Дополнительные промежуточные продукты, необходимые для присоединения трициклических производных 3 а и 3b, где Z, Y, D,Е, F и m определены выше, и имеют формулу 25, где R5, R6, R7, R8, R9, и R12 являются как определено выше, и L является -CC-, синтезируются, как показано на схеме X. Производные ацетилена 26 а, 26b и 26 с,где R5, R7, R8 и R9 являются как определено выше, получали из соответствующего альдегида путем взаимодействия с тетрабромидом углерода и трифенилфосфином в метиленхлориде, затем путем реагирования бутиллития с промежуточным иодсодержащим продуктом 24, где R5, R7 и R12 являются как определено выше, в присутствии палладия (О), с получением 26 а, 26b, 26 с, где L является -CC-. Дальнейшие промежуточные продукты для присоединения трициклических производных 3 а и 3b, где L является О или S и где Z, Y,D, Е, F и m являются как определено выше, и имеющие формулу 30, 31 и 32, где R5, R6, R7 иR12 являются как определено выше, синтезируются, как показано на cхеме XI. СХЕМА XI Реакция 27, где A', R5, R7 и R12 определены выше, с натриевой солью соединений 28 и 29 в подходящем растворителе, таком как ДМФ, дает промежуточные продукты 30 и 31. Последующая реакция производного 31 с одним молем 3-хлорпербензойной кислоты дает промежуточный продукт сульфоксид 32 а, а реакция с двумя молями 3-хлорпербензойной кислоты дает промежуточный продукт сульфон 32b. Другие используемые промежуточные продукты для получения соединений по настоящему изобретению, где связывающим атомом L является атом кислорода между единицами арил-L-гетероарил, гетероарил-L-арил или гетероарил-L-гетероарил, как приведено в качестве примера в формулах 34 а, 34b и 37,получают, как показано на схемах XII и XIII. Реакция проводится в инертных растворителях, таких как N,N-диметилформамид, N,Nдиметил-ацетамид, 1-метил-2-пирролидинон и подобных, где МН является гидридом металла,таким как гидрид лития, калия и натрия. Реакцию по схемам XII и XIII можно также провести сначала путем образования анионов 33 а,33b и 35 и реакцией с подходящим алкилоксидом, таким как трет-бутоксид калия. СХЕМА XII Соединения по настоящему изобретению могут быть получены, как показано на схемеXIV, путем взаимодействия трициклических производных формулы 3 а и 3b, где Z, Y, D, Е,F и m определены выше, с хлорангидридом 38,где R5 и W' определены выше, с получением промежуточных продуктов 39 а и 39b. Реакция 39 а и 39b с производными трибутилолова 8 а,8b или 8 с, где R5, R7, R8, R9, К' и X определены выше, дает 40 а и 40b. Альтернативно, как показано на схемеXV, где бромсодержащие производные 39 а и 39b, где W', Y, D, Е, F, R5 и m определены выше,реагируют с тетракис(трифенилфосфином) палладия (О) и производными бис(трибутилолова) в присутствии литийхлорида дают производные олова. Дальнейшая реакция производных трибутилолова 41 а и 41b с бромсодержащими производными 10 а, 10b или 10 с, где M' является бромом или иодом, и К', X, R5, R8 и R9 определены выше, в присутствии тетракистрифенилфосфина палладия (О), дает 42 а и 42b. Как показано на схеме XVI, присоединение бромсодержащего производного 39 а и 39b с производными трибутилолова 13 а, 13b и 13 с дает производные 43 а и 43b, где связывающая единица между двумя ароматическими кольцами является группой метилена (-CH2-). Производные трибутилолова 13 а, 13b и 13 с получают путем обычных способов, описанных в литературе. Схема XVII Соединения по настоящему изобретению могут быть получены, как показано на схемахW', R5 и R12 определены выше, путем взаимодействия с производными трибутилолова 8 а,8b и 8 с, где R5, R7, R8, R9, K' и X определены выше, дает 45, где R6 является определенным выше. Реакция 45 с трициклическими производными формулы 3 а и 3b, где Z, Y, D, Е, F и Соединения по настоящему изобретению могут быть получены, как показано на схемахXIX и XX. Реакция 44, где W', R5 определены выше, и R12, определенного выше, с бутиллитием и хлоридом трибутилолова дает продукт 47, который реагирует с производными 15 а,15b или 15 с, где R5, R7, R8, R9, K', X и М' определены выше, дает продукт 48, где R6 определен выше. Взаимодействие соединения 48 с трициклическими производными формулы 3 а и 3b, где Z, Y, D, Е, F и m определены, дает соединения 49 а и 49b. Ссылочный пример 1. 10,11-Дигидро-10(4-иодбензоил)-5 Н-пирроло[2,1-с] [1,4]-бензодиазепин. К перемешиваемому раствору 1,8 г 10,11 дигидро-5 Н-пирроло[2,1-с] [1,4] бензодиазепина и 3 мл раствора триэтиламина в 100 мл метиленхлорида при 0 С добавляли раствор 2,8 г 4-иодбензоилхлорида в 25 мл метиленхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч и выпаривали под вакуумом до остатка, который фракционировали между водой и хлороформом. Органический слой высушивали надNa2SO4, фильтровали и выпаривали под вакуумом до коричневого остатка, который кристаллизовали из смеси эфир-гексан с получением 3,0 г желаемого продукта. Масс-спектр: М+Н: 323. 27 Ссылочный пример 2. 10,11-Дигидро-10[4-(трибутилстаннил)бензоил-5 Н-пирроло[2,1-с][1,4]бензодиазепин. Смесь 4,1 г 10,11-дигидро-10-(4 иодбензоил)-5 Н-пирроло[2,1-с][1,4]бензодиазепина, 200 мг тетракис(трифенилфосфина) палладия (О), 11,6 г бис (трибутил)олова и 4,0 г хлорида лития в 100 мл безводного диоксана нагревали с обратным холодильником в течение 24 ч, реакционную смесь фильтровали и остаток промывали диоксаном. Объединенные фильтраты выпаривали под вакуумом до остатка, который очищали путем хроматографии на колонке с силикагелем, элюируя 30%-ной смесью этилацетат-гексан с получением 5,0 г желаемого продукта в виде твердого вещества. Масс-спектр: М+Н: 583. Ссылочный пример 3. 2-(Трибутилстаннил) толуол. К перемешиваемому раствору 3,4 г 2 бромтолуола в 100 мл сухого тетрагидрофурана медленно добавляли 8 мл 2,5 М бутиллития в гексане при -78 С. Реакционную смесь перемешивали в течение 30 мин и добавляли 6,5 г хлорида три-н-бутил олова в 25 мл тетрагидрофурана. Реакционную смесь перемешивали дополнительно 1 ч, гасили водой и экстрагировали эфиром. Эфирные экстракты высушивали над Na2SO4, фильтровали и фильтраты выпаривали под вакуумом с получением 7,0 г остатка. Масс-спектр: М+Н: 381. Ссылочный пример 4. 1-(2-Нитрофенил)1 Н-пиррол-2-карбоксальдегид. К раствору 3,76 г 1-(2-нитрофенил) пиррола в 20 мл N,N-диметилформамида при 0 С по каплям при перемешивании добавляли 3 мл оксихлорида фосфора. Перемешивание продолжали в течение 30 мин и реакционную смесь нагревали при 90 С в течение 1 ч. После охлаждения до комнатной температуры смесь обрабатывали размельченным льдом и доводили рН до 12 путем добавления 2 н гидроксида натрия. Полученную суспензию фильтровали, промывали водой и высушивали с получением 5,81 г желаемого продукта в виде светложелтого твердого вещества с т. пл. 119-122 С. Ссылочный пример 5. 4,5-Дигидропирроло[1,2-а]хиноксалин. К раствору 1,0 г 1-(2-нитрофенил)-1 Нпиррол-2-карбоксальдегида в 40 мл этилового спирта и 40 мл этилацетата в атмосфере аргона добавляли 40 мг 10% Pd/C. Смесь гидрогенизировали при давлении 40 ф/дюйм 2 в течение 2 ч и фильтровали через диатомовую землю. Фильтрат концентрировали под вакуумом до остатка, который растворяли в эфире и обрабатывали гексанами с получением 0,35 г желаемого продукта в виде твердого бежевого вещества с т. пл. 108-110 С. Ссылочный пример 6. N-(2-Нитробензоил)пиррол-2-карбоксальдегид. К раствору 5,6 г 2-пирролкарбоксальдегида в 40 мл тетрагидрофурана при охлаждении в ледяной бане добавляли 2,4 г 60%ного раствора гидрида натрия в минеральном масле. Температура повышается до 40 С. После перемешивания в течение 20 мин добавляли раствор 11,0 г 2-нитробензоилхлорида в 20 мл тетрагидрофурана по каплям в течение 20 мин. После перемешивания на холоду в течение 45 мин реакционную смесь вливали в воду со льдом и эфиром, затем фильтровали. Лепешку дополнительно промывали эфиром. Фильтрат второй фазы отделяли и слой эфира высушивали и концентрировали под вакуумом с получением 10 г остатка в виде темного сиропа, который соскабливали в этаноле с получением кристаллов, которые собирали путем фильтрования, промывали эфиром и затем высушивали с получением 3,2 г твердого продукта, т. пл. 95-99 С. Ссылочный пример 7. 10,11-Дигидро-5 Нпирроло[2,1-с][1,4]бензодиазепин-5-он. Смесь 1,5 г N-(2-нитpoбeнзoил)пиppoл-2 кapбoкcaльдerидa в 50 мл этилацетата, 2-х капель концентрированной НСl и 0,3 г 10%-ногоPd/C встряхивали в аппарате Парра под давлением водорода в течение 2 ч. Реакционную смесь фильтровали через диатомовую землю и фильтрат концентрировали под вакуумом с получением 1,0 г желтого масла. Остаток очищали на хроматографических пластинах с толстым слоем, элюируя смесью этилацетат:гексан (4:1), с получением 107 мг желаемого продукта в виде маслянистого твердого вещества. Ссылочный пример 8. 1-(2-Нитробензил)-2-пирролкарбоксальдегид. К 5,56 г 60%-ного гидрида натрия в минеральном масле, промытом три раза гексаном, добавляли 300 мл N,N-диметилформамида в атмосфере аргона. Реакционную смесь охлаждали в ледяной бане и медленно добавляли 13,2 г пиррол-2-карбоксальдегида. Реакционная смесь полностью растворялась и перемешивалась дополнительно в течение 10 мин. Во время перемешивания медленно добавляли 30,0 г 2-нитробензил бромида. После завершения добавления реакционную смесь перемешивали в течение 30 мин, ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение 24 ч. N,N-диметилформамид концентрировали под вакуумом с получением остатка, который перемешивали с ледяной водой в течение 1 ч. Полученный твердый продукт собирали,высушивали на воздухе, затем высушивали под вакуумом с получением 30,64 г желаемого продукта в виде рыжевато-коричневого твердого вещества, т. пл. 128-132 С. Ссылочный пример 9. 10,11-Дигидро-5 Нпирроло[2,1-с][1,4]бензодиазепин.Pd/C в 400 мл этилацетата и 400 мл этилового спирта гидрогенизировали более 18 ч. Реакционную смесь фильтровали через диатомовую землю и фильтрат обрабатывали активированным углем и фильтровали через диатомовую землю. Фильтрат концентрировали под вакуумом с получением остатка, который растворяли в метиленхлориде, содержащем этиловый спирт. Раствор пропускали через прокладку из силикагеля и прокладку промывали смесью гексан-этилацетат (7:1) с получением 16,31 г желаемого продукта в виде твердого вещества,т. пл. 145-148 С. Ссылочный пример 10. 1-(о-Нитробензил)-имидазол-2-карбоксальдегид. Порцию 2,0 г гидрида натрия (60% в масле) промывали два раза пентаном. К остатку добавляли 110 мл N,N-диметилформамида в аргоне. При перемешивании и наружном охлаждении добавляли 4,80 г 2 имидазолкарбоксальдегида и охлаждение удаляли. Легкое нагревание снаружи приводит к образованию желтого раствора. Реакционную смесь охлаждали во льду и добавляли 10,8 г 2 нитробензил бромида, реакционную смесь перемешивали при 0 С в течение 18 ч. Летучую часть удаляли под вакуумом до остатка,который перемешивали с ледяной водой,фильтровали, лепешку хорошо промывали водой и высушивали при отсасывании с получением 10,9 г желаемого продукта в виде твердого вещества, т. пл. 141-144 С. МН+ 232. Ссылочный пример 11. 10,11-Дигидро 5 Н-имидазо[2,1-с] [1,4] бензодиазепин. Образец 5,0 г 1-(о-нитробензил)-имидазол-2-карбоксальдегида растворяли в 150 мл горячего этилового спирта, охлаждали до комнатной температуры и фильтровали. К фильтрату добавляли 0,5 г 10%-ного Pd/C и смесь гидрогенизировали при давлении 48 ф/дюйм 2 в течение 4 ч. Добавляли дополнительные 0,5 г 10%-ного Pd/C и продолжали гидрогенизацию в течение 25 мин при давлении 65 ф/дюйм 2. Смесь фильтровали через диатомовую землю и лепешку промывали этилацетатом. Фильтрат выпаривали под вакуумом до остатка, который растворяли в метиленхлориде, обрабатывали активированным углем, фильтровали через диатомовую землю и к фильтрату при кипении добавляли гексаны с получением 1,86 г желаемого продукта в виде твердого кристаллического вещества, т. пл. 164-170 С. Ссылочный пример 12. 10,11-Дигидро 5 Н-имидазо[2,1-с] [1,4] бензодиазепин. К суспензии 4 ммоль литийалюмогидрида в 20 мл безводного тетрагидрофурана добавляли 1 ммоль раствор 10,11-дигидро-11 оксо-5 Н-имидазо[2,1-с] [1,4] бензодиазепина и смесь нагревали с образным холодильником в течение 24 ч и охлаждали при 0 С. К смеси добавляли по каплям 0,12 мл воды и 6 мл 1 н гидроксида натрия. Смесь экстрагировали этилацетатом и растворитель удаляли с получением желаемого продукта в виде твердого вещества. Перекристаллизация из смеси метиленхлоридгексан дает кристаллы, т. пл. 164170 С. Ссылочный пример 13. 10-[(6-Бромо-3 пиридинил)карбонил]-10,11-дигидро-5 Нпирроло[2,1-с] [1,4]бензодиазепин. К раствору 1,0 г 10,11-дигидро-5 Нпирроло[2,1-с] [1,4]-бензодиазепина и 10 мл триэтиламина в 50 мл дихлорметана в атмосфере аргона добавляли 3 г 6-бромпиридин-3 карбонилбромида. Смесь перемешивали при комнатной температуре в течение 16 ч и затем вливали в 100 мл воды. Органический слой отделяли и промывали 2%-ной НСl, водой,насыщенным раствором NaHCO3 и высушивали (Na2SО 4), растворитель отгоняли под вакуумом, и остаток хроматографировали на силикагеле этиловым эфиром в качестве растворителя с получением твердого вещества. Ссылочный пример 14. 9,10-Дигидро-4 Нфуро[2,3-о]пирроло[1,2-а][1,4]диазепин. К суспензии 4 ммоль литийалюмогидрида в 25 мл безводного тетрагидрофурана добавляли 1 ммоль 9,10-дигидро-4 Н-фуро[2,3-е] пирроло[1,2-а][1,4]диазепин-9-она. Смесь нагревали с обратным холодильником в течение 12 ч и затем отстаивали в течение ночи. К смеси добавляли по каплям 0,12 мл воды и затем 6 мл 1 н гидроксида натрия. Смесь экстрагировали этилацетатом и экстракты высушивали(Na2SO4). Летучую часть удаляли под вакуумом с получением желаемого твердого вещества. Ссылочный пример 15. 9,10-Дигидро-4 Нфуро[2,3-е]пирроло[1,2-а][1,4]диазепин. Раствор 1 ммоль 4 Н-фуро[2,3-е] пирроло[1,2-а][1,4]диазепина и 2,0 г 10% Pd/C-ного в 10 мл этанола гидрогенизировали в течение 18 ч. Реакционную смесь фильтровали через диатомовую землю и фильтрат выпаривали под вакуумом с получением желаемого твердого вещества. Ссылочный пример 16. 10-[(6-Иодо-3 пиридинил)карбонил]-10,11-дигидро-5 Нпирроло[2,1-с][1,4]бензодиазепин. К раствору 1,0 г 10,11-дигидро-5 Нпирроло[2,1-с] [1,4]-бензодиазепина и 10 мл триэтиламина в 50 мл дихлорметана в атмосфере аргона добавляли 3,2 г 6-иодопиридин 3-карбонилхлорида. Смесь перемешивали при комнатной температуре в течение 16 ч и затем вливали в 100 мл воды. Органический слой отделяли и промывали 2%-ной НСl, водой,насыщенным раствором NaHCO3 и высушивали (Na2SO4). Растворитель отгоняли под вакуумом и остаток хроматографировали на силикагеле смесью этилацетат-гексан в качестве растворителя с получением твердого вещества. 31 Ссылочный пример 17. 9,10-Дигидро-4 Нпирроло[1,2-а]тиено[2,3-е] [1,4] диазепин. К смеси 7,0 г 9-оксо-9,10-дигидро-4 Нпирроло[1,2-а]тиено[2,3-е][1,4]диазепина в 25 мл безводного тетрагидрофурана добавляли 9 мл 10 молярного диметилсульфида бора в тетрагидрофуране. Смесь перемешивали в течение 6 ч. Раствор охлаждали до комнатной температуры и по каплям добавляли 25 мл метанола. Летучую часть удаляли под вакуумом. К остатку добавляли 100 мл 2 н NaOH. Смесь нагревали с обратным холодильником в течение 5 ч и фильтровали. Твердый продукт экстрагировали дихлорметаном и экстракт промывали 2 н лимонной кислотой, водой и высушивали (Na2SO4). Растворитель удаляли под вакуумом с получением желаемого твердого вещества. Ссылочный пример 18. 4,10-Дигидро-5 Нпирроло[1,2-а]тиено[3,2-е] [1,4] диазепин. К суспензии 7,0 г 5-оксо-4,5-дигидропирроло[1,2-а]тиено[3,2-е][1,4]диазепина в 25 мл безводного тетрагидрофурана добавляли 9 мл 10 молярного диметилсульфида бора в тетрагидрофуране. Смесь нагревали с обратным холодильником в течение 6 ч. Раствор охлаждали до комнатной температуры и по каплям добавляли 25 мл метанола. Летучую часть удаляли под вакуумом. К остатку добавляли 100 мл 2 н NaOH. Смесь нагревали с обратным холодильником в течение 5 ч и фильтровали. Твердый продукт экстрагировали дихлорметаном и экстракт промывали 2 н лимонной кислотой, водой и высушивали(Na2SO4). Растворитель удаляли под вакуумом с получением желаемого твердого вещества. Ссылочный пример 19. 5,6-Дигидро-4 Н[1,2,4] триазоло [4,3-а] [1,5] бензодиазепин. Смесь 7,0 г 5,6-дигидро-4 Н-[1,2,4]триазоло-[4,3-а]-[1,5] бензодиазепин-5-она в 25 мл тетрагидрофурана добавляли к 9 мл 10 молярного диметилсульфида бора в тетрагидрофуране. Смесь нагревали с обратным холодильником в течение 6 ч, охлаждали до комнатной температуры и по каплям добавляли 25 мл метанола. Летучую часть удаляли под вакуумом, а к остатку добавляли 100 мл 2 н гидроксида натрия. Смесь нагревали с обратным холодильником в течение 5 ч, охлаждали и экстрагировали дихлорметаном. Экстракт промывали 2 н лимонной кислотой, водой и высушивали (Na2SO4). Растворитель удаляли под вакуумом, получая твердое вещество, которое очищали путем хроматографии на силикагеле с получением желаемого продукта. Ссылочный пример 20. 1-(2-Нитрофенил)-1 Н-пиррол-2-карбоксальдегид. Образец 4,7 гидрида натрия (60% в масле) промывали гексаном (под аргоном), к гидриду натрия добавляли 200 мл сухого N,Nдиметилформамида и смесь охлаждали до 0 С. К смеси добавляли 10,11 г пиррол-2 000056 карбоксальдегида по маленьким порциям. Смесь перемешивали 10 мин и по каплям добавляли 15,0 г 1-фтор-2-нитробензола. После добавления смесь перемешивали при комнатной температуре 16 ч и смесь концентрировали (65 С) при высоком вакууме. К остатку добавляли 400 мл дихлорметана и смесь промывали по 150 мл каждого H2 О, рассолом и высушивали (Na2SO4). Растворитель удаляли под вакуумом с получением желтого твердого продукта. Кристаллизация из смеси этилацетат-гексан (9:1) дает 17,0 г светло-желтых кристаллов, т. пл. 119-122 С. Ссылочный пример 21. 4,10-Дигидро-5 Нпирроло[1,2-а]тиено[3,2-е] [1,4] диазепин. К охлажденной на льду смеси 2,1 г пиррол-2-карбоновой кислоты и 3,2 г метил 3 амино-тиофен-2-карбоксилата в 40 мл сухого дихлорметана добавляли 4 г N,N-дициклогексилкарбодиимида. Смесь перемешивали при комнатной температуре в течение 3 ч и фильтровали. Лепешку фильтрата промывали дихлорметаном и затем экстрагировали дважды 60 мл ацетона. Ацетоновый экстракт концентрировали досуха с получением 0,8 г твердого продукта, т. пл. 214-218 С. К суспензии предыдущего соединения (1,19 г) в 20 мл сухого тетрагидрофурана добавляли 0,2 г гидрида натрия (60% в масле). После выхода водорода смесь перемешивали и нагревали с обратным холодильником в течение 4,5 ч, охлаждали и вливали в воду со льдом. Осадившийся твердый продукт фильтровали и твердый продукт растирали с петролейным эфиром (т. кип. 30-60 С) с получением 0,75 г 4,10 дигидро-4,10-диоксо-5 Н-пирроло[1,2-а]тиено(0,362 г) добавляли к охлажденному водой со льдом раствору 1 М диборана в тетрагидрофуране. Смесь перемешивали при комнатной температуре в течение 65 ч. Раствор концентрировали досуха и к остатку добавляли воду со льдом. Смесь подкисляли разбавленной НСl, перемешивали и подщелачивали твердымNaHCO3. Смесь фильтровали с получением 0,223 г твердого продукта (пена), т. пл. 8085 С. Ссылочный пример 22. 10,11-Дигидро 5 Н-1,2,4-триазоло[3,4-е][1,4]бензодиазепин. Смесь 2,2 г 2-цианоанилина, 2,0 г метилбромацетила и 1,3 г карбоната калия в 12 мл сухого N,N-диметилформамида нагревали при 150-155 С в течение 40 мин. Охлажденную смесь вливали в воду со льдом и смесь фильтровали с получением 2 г метил[N-(2 циaнoфeнил)aминo] ацетата в виде желтого твердого продукта, т. пл. 70-78 С. Предыдущее соединение (2,0 г) добавляли к раствору 0,5 г натрия метоксида в 50 мл метанола. Смесь встряхивали в атмосфере водорода с катализатором Raney-Ni в течение 19 ч. Смесь 33 фильтровали через диатомовую землю и фильтрат выпаривали. К остатку добавляли воду и смесь фильтровали с получением 2,3,4,5-тетрагидро-1 Н-1,4-бензодиазепин-3 она в виде желтого твердого продукта, т. пл. 167-170 С. Смесь предыдущего соединения (1,6 г) и 0,84 г пентасульфида фосфора в 10 мл сухого(высушенного над КОН) пиридина перемешивали и нагревали при 80-85 С в течение 15 мин. Смесь выливали в Н 2O и перемешивали 30 мин. Фильтрация дает 1,0 г 1,2,4,5 тетрагидро-3 Н-1,4-бензодиазепин-3-тиона в виде твердого желтого продукта, т. пл. 150153 С. Предыдущее соединение (0,5 г) и 0,5 г Nформилгидразина в 6 мл сухого н-бутанола нагревали с обратным холодильником в течение 16 ч и растворитель удаляли. Смолянистый остаток растирали в холодной воде и смесь фильтровали. Твердый продукт растирали в ацетоне с получением 0,19 г желтого твердого вещества, т. пл. 232-237 С. Ссылочный пример 23. 4,5-Дигидро-6 Н[1,2,4]триазоло[4,3-а] [1,5] бензодиазепин. Смесь 2,3,4,5-тетрагидро-1 Н-1,5 бензодиазепин-2-тиона (0,8 г) и 0,80 г Nформилгидразина в 8 мл н-бутанола перемешивали и нагревали с обратным холодильником в течение 18 ч, растворитель удаляли под вакуумом. К остатку твердого продукта добавляли ледяную воду и смесь фильтровали с получением 0,312 г серого твердого вещества, т. пл. 162-165 С. Ссылочный пример 24. 4,5-Дигидро-6 Нимидазо[1,2-а][1,5]бензодиазепин. Смесь 30 г акриловой кислоты, 33 г о-фенилендиамина нагревали на паровой бане в течение 1,5 ч,охлажденную черную смесь растирали в ледяной воде. Водную фазу декантировали и к остатку добавляли лед и водный раствор гидроксида аммония. Смесь экстрагировали дихлорметаном и экстракт концентрировали досуха. Остаток растирали в четыреххлористом углероде и фильтровали. Маслянистый твердый продукт растирали в небольшом количестве этанола с получением 9,7 г твердого вещества. Растирание твердого вещества в этилацетате дает 2,3,4,5-тетрагидро-1 Н-1,5 бензодиазепин-2-он в виде твердого продукта с примесями, т. пл. 75-107 С. Смесь предшествующего соединения(11,3 г) и 5,9 г пентасульфида фосфора в 70 мл сухого пиридина перемешивали и нагревали при, приблизительно, 80 С в течение 20 мин. Смесь вливали в воду и смесь перемешивали в течение 30 мин. Фильтрация дает 8,6 г 2,3,4,5 тетрагидро-1 Н-1,5-бензодиазепин-2-тиона в виде твердого продукта, т. пл. 154-157 С. Смесь предыдущего соединения (0,70 г),1,0 г аминоацетальдегиддиметилацеталя и 15 мг моногидрата 4-метилбензолсульфоновой кислоты в 6 мл сухого н-бутанола нагревали с обратным холодильником в течение 4 ч и растворитель удаляли под вакуумом. Остаток нагревали с обратным холодильником с 10 мл 3 н соляной кислоты в течение 55 мин. К охлажденной смеси добавляли лед и смесь подщелачивали твердым NaHCO3. Смесь экстрагировали дихлорметаном и экстракт высушивали(Na2SO4). Растворитель удаляли с получением оранжевого сиропа, который становился твердым при стоянии. Маслянистое твердое вещество растирали в ацетоне с получением светло-желтого твердого продукта (0,185 г), т. пл. 119-122 С. Ссылочный пример 25. Этиловый эфир 1(2-нитрофенил)-2-пирролуксусной кислоты. К перемешиваемой смеси 1,88 г 1-(2 нитрофенил) пиррола, 4,80 г этилиодацетата и 2,22 г FeSO47H2O в 40 мл диметилсульфоксида по каплям добавляли 10 мл 30% перекиси водорода, поддерживая реакционную смесь при комнатной температуре при помощи бани с холодной водой, смесь перемешивали при комнатной температуре в течение одного дня. Добавляли дополнительно 2,4 г этилиодацетата, 1,1 г FeS047H2O и 5 мл 30%ной перекиси водорода и смесь перемешивали при комнатной температуре в течение 1 дня. Смесь разбавляли водой и экстрагировали диэтиловым эфиром. Органический экстракт промывали водой, рассолом и высушивали(2,12 г) хроматографировали на силикагеле смесью этилацетат-гексан (1:4) в качестве растворителя с получением 0,30 г продукта в виде коричневой смолы. Ссылочный пример 26. 6,7-Дигидро-5 Нпирроло[1,2-а] [1,5] бензодиазепин-6-он. К раствору 0,8 ммоль 1-(2-нитрофенил)2-пирролуксусной кислоты этилового эфира в 3 мл этанола добавляли дигидрат хлорида олова (SnCl27H2O) в 2 мл концентрированной соляной кислоты (при охлаждении в водяной бане). Смесь перемешивали при комнатной температуре в течение 5 ч и охлаждали на ледяной бане. К смеси медленно добавляли насыщенный раствор карбоната натрия. Осадившийся твердый продукт фильтровали и твердое вещество промывали водой и затем экстрагировали этилацетатом. Этилацетатный экстракт высушивали (Na2SO4) и растворитель удаляли с получением 0,16 г твердого вещества, которое растирали в эфире с получением 0,11 г продукта в виде не совсем белого твердого вещества. Ссылочный пример 27. 6,7-Дигидро-5 Нпиррол[1,2-а] [1,5] бензодиазепин. К раствору 0,070 г 6,7-дигидро-5 Нпиррол[1,2-а][1,5]бензодиазепин-6-она в 2 мл тетрагидрофурана добавляли 0,45 мл 2,0 М раствора диметилсульфида бора в тетрагидрофуране. Смесь нагревали с обратным холо 35 дильником в течение 3 ч, вливали в воду и подщелачивали 2 н NaOH. Тетрагидрофуран удаляли под вакуумом и оставшуюся водную смесь экстрагировали диэтиловым эфиром. Экстракт промывали рассолом, высушивали(Na2SO4) и растворитель удаляли с получением 0,065 г бесцветного масла; одно пятно при тонкослойной хроматографии (силикагель) в смеси этилацетат-гексан (1:2) в качестве растворителя (Rf 0,81). Ссылочный пример 28. 1-[2-Нитро 5(этоксикарбонил)бензил]-пиррол-2-карбоксальдегид. К перемешанной взвеси 2,2 г гидрида натрия (60% в масле, промытого гексаном) в тетрагидрофуране добавляли при 0 С раствор 4,5 г пиррол-2-карбоксальдегида в 25 мл тетрагидрофурана. После завершения добавления медленно добавляли раствор 15 г этил 4 нитро-3-бромметилбензоата в 30 мл сухого тетрагидрофурана под азотом. Реакционную смесь перемешивали при 20 С в течение 8 ч и осторожно гасили водой. Реакционную смесь экстрагировали хлороформом, промывали водой, высушивали Na2SO4 и концентрировали под вакуумом с получением 12 г желаемого твердого продукта; масс-спектр (М+Н) 349. Ссылочный пример 29. 1[2-Нитро-4(этоксикарбонил)бензил]-пиррол-2-карбоксальдегид. Для получения 13,0 г желаемого продукта в виде твердого вещества использовали условия ссылочного примера 28 и этил 3-нитро 4-бромметилбензоат; масс-спектр (М+Н) 349. Ссылочный пример 30. Этил 10,11 дигидро-5 Н-пирроло[2,1-с][1,4]бензодиазепин-7-карбоксилат. Раствор 10,0 г 1-[2-нитро-5(этоксикарбонил)бензил]-пиррол-2-карбоксальдегида в 150 мл абсолютного этанола, содержащего 1,0 г 10% Pd/C, гидрогенизировали в аппарате Парра в течение 16 ч при давлении водорода 40 ф/дюйм 2. Реакционную смесь фильтровали через диатомовую землю и фильтрат концентрировали под вакуумом до остатка 5,5 г желаемого продукта в виде твердого вещества; масс-спектр (М+Н) 255. Ссылочный пример 31. Этил 10,11 дигидро-5 Н-пирроло[2,1-с] [1,4] бензодиазепин-8-карбоксилат. Использовали условия гидрогенизации ссылочного примера 30 и 1-[2-нитро-4(этоксикарбонил)бензил]-пиррол-2-карбоксальдегид для получения 5,0 г желаемого продукта в виде твердого вещества; масс-спектр(М+Н) 255. Ссылочный пример 32. 4-[(4-Метилфенил)тио]бензойная кислота. Смесь 6,0 г меркаптотолуола и 9,2 г третбутоксида калия перемешивали при комнатной температуре при атмосфере азота в 50 мл диметилсульфоксида в течение 10 мин, затем добавляли 11,5 г 4-бромбензойной кислоты и 0,2 г металлической меди. Реакционную смесь перемешивали при 210 С в течение 16 ч. Реакционную смесь вливали в размельченный лед и фильтровали через диатомовую землю. Фильтрат подкисляли НСl до рН 2. Полученное твердое вещество собирали, промывали водой и 60 мл петролейного эфира с получением 8,5 г желаемого продукта, т. пл. 105107 С, М+Н=245. Ссылочный пример 33. 4-(Фенилтио)бензойная кислота. Использовали условия получения соединения по ссылочному примеру 32 с 1384847 с 6,0 г меркаптобензола, 12,22 г третбутоксида калия и 13,15 г 4-бромбензойной кислоты для получения 12,0 г желаемого продукта в виде твердого вещества, т. пл. 101-103 С; М+Н=231. Ссылочный пример 34. 4-(Фенилтио)бензоил хлорид. Раствор 2,0 г соединения по ссылочному примеру 33 в 30 мл тионилхлорида нагревали с обратным холодильником в атмосфере азота в течение 40 мин. Летучую часть выпаривали под вакуумом, а осадок выпаривали два раза с 30 мл четыреххлористого углерода с получением желаемого продукта в виде остатка, который растворяли в 30 мл метиленхлорида и использовали на следующем этапе. Ссылочный пример 35. 4-[(4-Метилфенил)тио]бензоил хлорид. Применяли условия для получения соединения по ссылочному примеру 34 и 2,0 г соединения по ссылочному примеру 32 и 30 мл тионилхлорида для получения 2,16 г желаемого продукта в растворе из 30 мл метиленхлорида. Ссылочный пример 36. 4-(Бензоил) бензоил хлорид. Применяли условия для получения соединения по ссылочному примеру 34 и 2,0 г 4 бензоилбезойной кислоты и 30 мл тионилхлорида для получения желаемого продукта в растворе 30 мл метиленхлорида. Ссылочный пример 37. 4-[(4 Метилфенил)сульфонил]бензоил хлорид. Применяли условия для получения соединения по ссылочному примеру 34 и 2,0 г соединения по ссылочному примеру 38 и 30 мл тионилхлорида для получения желаемого продукта в растворе 30 мл метиленхлорида. Ссылочный пример 38. 4[(4-Метилфенил)сульфонил]бензойная кислота. Смесь 4,0 г соединения по ссылочному примеру 32, 11,3 г м-хлорпербензойной кислоты в 100 мл хлороформа перемешивали при нагревании с обратным холодильником под азотом в течение 16 ч. Реакционную смесь вливали в воду и отделяли органический слой,промывали 2 н НСl и водой. Органический слой высушивали (Na2SO4) и упаривали досу 37 ха под вакуумом с получением 6,5 г желаемого продукта в виде желтоватого масла. Ссылочный пример 39. 4-(Фенилсульфонил)бензойная кислота. Смесь 7,0 г соединения по ссылочному примеру 33 и 11,5 г м-хлорпербензойной кислоты в 80 мл хлороформа перемешивали при нагревании с обратным холодильником под азотом в течение 16 ч. Реакционную смесь выпаривали под вакуумом и остаток суспендировали в 200 мл ледяной воды. Полученный нерастворимый твердый продукт собирали и высушивали в вакуумной печи при 60 С с получением 7,2 г желаемого продукта, т. пл. 128132 С. М+Н=263. Ссылочный пример 40. Метиловый эфир 4-[(4-метилфенил)сульфонил]бензойной кислоты. Раствор 6,5 г соединения по ссылочному примеру 38 в 200 мл метилового спирта, содержащего несколько капель серной кислоты,нагревали с обратным холодильником в течение 16 ч. Летучую часть удаляли под вакуумом и к остатку добавляли 150 мл ледяной воды. Полученное твердое вещество собирали фильтрацией и промывали 200 мл воды. Твердое вещество высушивали в вакуумной печи при 60 С в течение 16 ч с получением 3,5 г белого пушистого продукта, М+Н=291,1;M+Na=313,1. Ссылочный пример 41. 4-[(4-Метилфенил)сульфонил]бензойная кислота. Смесь 2,5 г соединения по ссылочному примеру 40 растворяли в 60 мл смеси 1:1 вода: метанол и 20 мл 5%-ного NaOH и перемешивали при комнатной температуре в течение 8 ч. Летучую часть выпаривали под вакуумом до остатка, который перемешивали в 50 мл ледяной воды и подкисляли приблизительно 20 мл 10 н раствора НСl. Полученное твердое вещество собирали, промывали 200 мл воды и высушивали в вакуумной печи при 60 С с получением 2,0 г желаемого продукта, М+Н=277. Ссылочный пример 42. 4-[(4-Метилфенил)сульфонил]бензоил хлорид. Смесь 2,0 г соединения по ссылочному примеру 41 и 30 мл тионилхлорида нагревали при 80 С под азотом в течение 45 мин. Летучую часть выпаривали под вакуумом до остатка, который выпаривали с толуолом и четыреххлористым углеродом. Конечный остаток растворяли в 30 мл метиленхлорида для использования на последующей стадии. Ссылочный пример 43. Этиловый эфир 4'-(2-пропенилокси)-[1,1'-бифенил]-4-карбоновой кислоты. Смесь 10,0 г этилового эфира 4'гидрокси-[1,1'-бифенил]-4-карбоновой кислоты растворяли в 100 мл ацетона и добавляли 8,02 г аллилбромида в атмосфере азота. Во время перемешивания добавляли 15,0 г карбоната калия и реакционную смесь нагревали с обратным холодильником в течение 16 ч. Реакционную смесь охлаждали до 0 С, вливали в 200 мл ледяной воды и экстрагировали хлороформом (3 х 150 мл). Объединенные экстракты промывали 1 н НСl и водой, высушивали(Na2SO4) и выпаривали под вакуумом с получением 12,0 г желаемого продукта, т. пл. 8992 С. М+Н=283. Ссылочный пример 44. 4'-(2-Пропенилокси)-[1,1'-бифенил]-4-карбоновая кислота. Смесь 6,0 г соединения по ссылочному примеру 43 в 60 мл этанола и 30 мл 5 н NaOH перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь вливали в 200 мл ледяной воды, подкисляли НСl, экстрагировали хлороформом (3 х 150 мл), промывали насыщенным раствором NаНСО 3 и воды. Органический слой высушивали (Na2SO4) и выпаривали под вакуумом с получением 4,4 г желаемого продукта в виде твердого вещества,т. пл. 360 С, М+Н=255. Ссылочный пример 45. 4'-(2-Пропенилокси)-[1,1'-бифенил]-4-карбонил хлорид. Смесь 9,0 г соединения по ссылочному примеру 44 в 45 мл тионилхлорида нагревали с обратным холодильником под азотом в течение 45 мин. Летучую часть выпаривали под вакуумом и из четыреххлористого углерода (2 х 30 мл) выпаривали остаток под вакуумом до остатка, который растворяли в 30 мл метиленхлорида для использования на последующей стадии. Ссылочный пример 46. Трибутил[2(трифторметил) фенил] олово. К раствору 10,0 г 2-бромтрифторметилбензола в 100 мл тетрагидрофурана добавляли по каплям через шприц под азотом при охлаждении в бане из сухого льда и ацетона 30,6 мл 1,6 М раствора бутиллития. Перемешивание продолжали в течение 1 ч. К реакционной смеси добавляли по каплям 15,9 г хлорида трибутилолова в 30 мл тетрагидрофурана. Перемешивали 7 дополнительных часов. Реакционную смесь гасили 20 мл воды и после перемешивания в течение 20 мин экстрагировали хлороформом (3 х 100 мл). Органический слой фильтровали через диатомовую землю, фильтрат промывали насыщенным раствором NaHCO3 и водой. Органический слой высушивали (Na2SO4) и выпаривали под вакуумом с получением 16,8 г желаемого продукта. МС 379,1. Ссылочный пример 47. 2-(Трибутилстаннил) пиридин. К раствору 10,0 г 2-бромпиридина в 100 мл тетрагидрофурана под азотом при охлаждении на бане из сухого льда и ацетона по каплям через шприц добавляли 47,0 мл 1,6 М раствора бутиллития. Перемешивание продолжали 1 ч. К реакционной смеси по каплям добавляли 24,7 г хлорида трибутилолова в 30 мл тетрагидрофурана и дополнительно пере 39 мешивали в течение часа. Реакционную смесь гасили 20 мл воды и добавляли дополнительно 100 мл воды. После перемешивания в течение 20 мин реакционную смесь экстрагировали хлороформом (3 х 100 мл). Органический слой фильтровали через диатомовую землю и фильтрат промывали насыщенным растворомNaHCO3 и воды. Органический слой высушивали (Na2SO4) и выпаривали под вакуумом с получением 21,2 г желаемого продукта. МС 379,1. Ссылочный пример 48. 2-(Трибутилстаннил)тиазол. К раствору 10,0 г 2-бромтиазола в 50 мл тетрагидрофурана под азотом при охлаждении на бане из сухого льда и ацетона по каплям через шприц добавляли 36,6 мл 1,6 М раствора бутиллития. Перемешивание продолжали 1 ч. К реакционной смеси по каплям добавляли 29,8 г хлорида трибутилолова в 20 мл тетрагидрофурана. Дополнительно перемешивали в течение 1 ч. Реакционную смесь гасили 20 мл воды и добавляли дополнительно 50 мл воды. После перемешивания в течение 20 мин реакционную смесь экстрагировали хлороформом(Na2SO4) и выпаривали под вакуумом с получением 11,7 г желаемого продукта. Ссылочный пример 49. 10-[(6-Хлор-3 пиридинил)карбонил]-10,11-дигидро-5 Н-пирроло [2,1-с][1,4]бензодиазепин. К раствору 1,0 г 10,11-дигидро-5 Нпирроло [2,1-с][1,4]бензодиазепина в 50 мл сухого метиленхлорида в атмосфере азота и 7,0 мл триэтиламина добавляли 1,43 г 6 хлорпиридин-3-карбонилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч и вливали в 100 мл воды. Органический слой отделяли, промывали 2%-ной НСl, водой, насыщенным раствором NaHCO3 и высушивали (Na2SO4). Растворитель выпаривали под вакуумом с получением остатка, который хроматографировали на колонке с силикагелем, элюируя смесью этилацетат-гексан (от 7:1 до 1:1), с получением 1,76 г желаемого продукта в виде желтоватого кристаллического твердого вещества. М+Н=324. Ссылочный пример 50. 4,5-Дигидро-4,4 диметил-2-(2-тиенил)-оксазол. Раствор 30,4 г 2-амино-2-метил-1 пропанола в 125 мл метиленхлорида добавляли по каплям к раствору 25,0 г 2 тиофенкарбоксилхлорида в 125 мл метиленхлорида при поддерживании температуры ниже 20 С. Смесь перемешивали при комнатной температуре в течение 2 ч и промывали водой. Органический слой высушивали над MgSO4 и выпаривали под вакуумом до остатка. Остаток суспендировали в метиленхлориде и по каплям добавляли 67,7 г тионилхлорида при поддерживании температуры ниже 30 С. Реакци 000056 онную смесь перемешивали при комнатной температуре в течение 18 ч и летучую часть выпаривали под вакуумом до остатка, который растворяли в воде. Водный раствор переводили в щелочной 1 М NaOH и экстрагировали эфиром. Органический слой высушивалиMgS04 и концентрировали под вакуумом с получением 31,1 г желаемого продукта в виде желтоватого масла. Ссылочный пример 51. 4,5-Дигидро-4,4 диметил-2-[3-(трибутилстаннил)-2-тиенил]-оксазол. К раствору 5,0 г 4,5-дигидро-4,4 диметил-2-(2-тиенил)-оксазола в 20 мл эфира добавляли по каплям 18,75 мл бутиллития,поддерживая температуру -70 С при перемешивании в атмосфере азота. Реакционную смесь перемешивали при -70 С в течение 15 мин и 30 мин при 0 С. Реакционную смесь охлаждали до -70 С и добавляли 10,0 г хлорида трибутилолова. Реакционную смесь гасили водой и экстрагировали эфиром. Органический слой промывали водой (2 х 100 мл), высушивали и выпаривали под вакуумом с получением 17,4 г остатка, который очищали путем хроматографии на силикагеле, элюируя смесью гексан : этилацетат 7:1, с получением 12,4 г желтоватого масла. МС (М+Н=472), следующая стадия примера 19. Ссылочный пример 52. 4-(4-Метилфенокси)бензойная кислота. К раствору 4,36 г 4-метилфенола в 70 мл сухого диметилсульфоксида под азотом при перемешивании добавляли 9,0 г третбутоксида калия, после перемешивания в течение 15 мин добавляли 0,1 г металлической меди, а затем 10,0 г 4-иодбензойной кислоты. Реагенты перемешивали в атмосфере азота при нагревании до 210 С в течение 18 ч. Охлажденную реакционную смесь экстрагировали хлороформом (2 х 900 мл) и оставшуюся воду подкисляли 2 н НС 1 (рН=3) при охлаждении в ледяной бане с получением 8,1 г желаемого продукта. МС (М+Н=229,0). Ссылочный пример 53. 4-(4-Метилфенокси)бензоил хлорид. Раствор 2,0 г 4-(4-метилфенокси)бензойной кислоты в 30 мл метиленхлорида добавляли к 1,67 г оксалилхлорида и смесь нагревали с обратным холодильником в течение 30 мин при перемешивании в атмосфере азота. Реакционную смесь выпаривали под вакуумом до остатка, который выпаривали с четыреххлористым углеродом (2 х 30 мл). Ссылочный пример 54. 4-[4-Пропилфенил]бензоил хлорид. Смесь 2,0 г 4-[4-пропилфенил]бензойной кислоты и 30 мл тионилхлорида перемешивали при нагревании с обратным холодильником в течение 45 мин. Реакционную смесь выпаривали до остатка, который выпаривали из четыреххлористого углерода (2 41 х 50 мл) с получением остатка, который растворяли в метиленхлориде. Ссылочный пример 55. Метиловый эфир 6-гидроксиникотиновой кислоты. К 500 мл метилового спирта, охлажденного до 0-5 С добавляли газообразный НС 1 в течение более 30 мин. Раствор оставляли нагреваться до комнатной температуры и добавляли 50 г 6-гидроксиникотиновой кислоты. Реакционную смесь перемешивали при нагревании с обратным холодильником в течение 2 дней. Метиловый спирт удаляли под вакуумом и остаток суспендировали в 200 мл воды и вливали в 300 мл насыщенного водного раствора NaHCO3. Нерастворяющиеся кристаллы собирали путем фильтрации, промывали 500 мл воды и высушивали под вакуумом при 60 С с получением желаемого продукта(53,9 г). Ссылочный пример 56. о-Трифталат метил 6-гидроксипиридин-3-карбоксилата. Перемешиваемый раствор 8,0 г пиридина и 5,0 г метилового эфира 6 гидроксиникотиновой кислоты в 50 мл метиленхлорида охлаждали до 0 С в атмосфере азота при добавлении 22,0 г ангидрида трифторметансульфоновой кислоты. Реакционную смесь нагревали с обратным холодильником в течение 16 ч, выпаривали под вакуумом до остатка, который перемешивали с 200 мл ледяной воды, и собирали твердый продукт. Твердый продукт высушивали в вакуумной печи при 30 С с получением 7,0 г желаемого продукта. Ссылочный пример 57. 2-(Трибутилстаннил)тиофен. Раствор 8,4 г тиофена в 200 мл эфира перемешивали, охлаждали до 0 С в атмосфере азота при добавлении по каплям через шприц 48,0 г бутиллития. После перемешивания в течение 1 ч реакционную смесь охлаждали до-78 С и по каплям через шприц добавляли 35,0 мл хлорида трибутилолова. После перемешивания при комнатной температуре в течение 30 мин реакционную смесь гасили 60 мл воды,вливали на размолотый лед и экстрагировали эфиром (3 х 200 мл). Органический слой высушивали и выпаривали под вакуумом с получением 46,2 г желаемого продукта в виде остатка. Ссылочный пример 58. Метил 6-(2 тиенил)пиридин-3-карбоксилат. Раствор 2,0 г (о-трифталата метил 6 гидроксипиридин-3-карбоксилата) и 5,23 г 2(трибутилстаннил)тиофена в 50 мл сухого толуола перемешивали под азотом при нагревании с обратным холодильником в течение 16 ч в присутствии тетракис(трифенилфосфин) палладия (О). Реакционную смесь разбавляли 50 мл хлороформа и фильтровали через прокладку из диатомовой земли. Фильтрат выпаривали под вакуумом до остатка, который экс 000056 трагировали и декантировали (2 х 100 мл) смесью эфир : петролейный эфир 1:1. Объединенные экстракты выпаривали под вакуумом с получением 1,6 г желаемого продукта в виде остатка. Ссылочный пример 59. 6-(2-Тиенил) пиридин-3-карбоновая кислота. Раствор 2,0 г метил 6-(2-тиенил)пиридин 3-карбоксилата в 100 мл метилового спирта и 50 мл 5 н гидроксида натрия перемешивали при комнатной температуре в течение 16 ч в атмосфере азота. Реакционную смесь выпаривали под вакуумом до около одной четверти объема, затем разбавляли 150 мл холодной воды. Уксусной кислотой доводили рН до 4 и собирали желаемый белый продукт, промывали 400 мл холодной воды до нейтрального состояния. Твердое вещество промывали 50 мл петролейного эфира и высушивали под вакуумом при 40 С с получением желаемого продукта. Ссылочный пример 60. 6-(2-Тиенил) пиридин-3-карбонил хлорид. Смесь 1,8 г 6-(тиенил)пиридин-3 карбоновой кислоты в 150 мл тиенилхлорида нагревали с обратным холодильником в течение 1 ч. Реакционную смесь выпаривали досуха и снова выпаривали из 50 мл четыреххлористого углерода до остатка. Остаток растворяли в 60 мл метиленхлорида и использовали в дальнейших реакциях. Пример 1. 10,11-Дигидро-10-[4-(2 тиенил)бензоил]-5 н-пирроло[2,1-с] [1,4] бензодиазепин. Смесь 1,64 г 10,11-дигидро-10-(4 иодбензоил)-5 Н-пирроло [2,1-с][1,4] бензодиазепина, 1,9 г 2-три-н-бутилстаннил тиофена и 200 мг тетракис(трифенилфосфин) палладия (О) в 200 мл толуола нагревали с обратным холодильником в атмосфере азота в течение 16 ч. Реакционную смесь выпаривали под вакуумом до остатка, который очищали путем хроматографии на колонке с силикагелем,элюируя смесью гексан : этилацетат 5:1, с получением 1,2 г желаемого продукта в виде твердого вещества. Масс-спектр: М+Н:371. Пример 2. 10,11-Дигидро-10-[4-(2 нитрофенил)бензоил]-5 Н-пирроло [2,1-с][1,4] бензодиазепин. Смесь 3,0 г 10,11-дигидро-10-[4-(трибутилстаннил)бензоил]-5 Н-пирроло[2,1-с][1,4] бензодиазепина, 200 мг тетракис (трифенилфосфин) палладия (О) и 2,0 г 1-бром-2 нитробензола в 200 мл толуола нагревали с обратным холодильником в течение 16 ч. Реакционную смесь фильтровали и фильтрат выпаривали под вакуумом до остатка, который очищали путем хроматографии на колонке с силикагелем, элюируя 30%-ной смесью этилацетат-гексан, с получением 1,2 г желаемого продукта в виде твердого вещества. Массспектр: М+Н:410. 43 Пример 3. 10,11-Дигидро-10-[4-(3,5 дифторфенил)бензоил]-5 Н-пирроло[2,1-с] [1,4] бензодиазепин. Смесь 1,5 г 10,11-дигидро-10-[4-(трибутилстаннил)бензоил]-5 Н-пирроло[2,1-с][1,4] бензодиазепина, 1 мл 3,5-дифтор-1-бромбензола и 200 мг тетракис(трифенилфосфин) палладия (О) в 200 мл толуола нагревали с обратным холодильником в течение 16 ч. Реакционную смесь фильтровали и фильтрат выпаривали под вакуумом до остатка, который очищали путем хроматографии на колонке с силикагелем, элюируя 30%-ной смесью этилацетатгексан, с получением 700 мг желаемого продукта в виде твердого вещества. Масс-спектр: М+Н:401. Пример 4. 10,11-дигидро-10-[4-(фенилэтинил)бензоил]-5 Н- пирроло [2,1-с][1,4] бензодиазепин. Смесь 2,0 г 10,11-дигидро-10-[4 иодбензоил)-5 Н-пирроло [2,1-с][1,4] бензодиазепина, 1,0 г фенилацетилена и 200 мг хлоридаPd(II) нагревали с обратным холодильником в 250 мл ацетонитрила в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и собирали полученное желтое кристаллическое вещество, высушивали и кристаллизовали из метилового спирта с получением 1,2 г желаемого продукта. Масс-спектр: М+Н:389. Пример 5. 10,11-Дигидро-10-[4-(2 метилфенил)бензоил]-5 Н-пирроло[2,1-с][1,4] бензодиазепин. Смесь 3,0 г 10,11-дигидро-10-(4 иодбензоил)-5 Н-пирроло(О) в 200 мл толуола нагревали с обратным холодильником в течение 16 ч. Реакционную смесь фильтровали и фильтрат выпаривали под вакуумом до остатка, который очищали путем хроматографии на колонке с силикагелем, элюируя 30%-ной смесью этилацетатгексан, с получением 1,0 г желаемого продукта в виде твердого вещества. Масс-спектр: М+Н:379. Пример 6. 10,11-Дигидро-3-[(диметиламино)метил]-10-[4-(2-тиенил)бензоил]-5 нпирроло[2,1-с] [1,4] бензодиазепин. Смесь 400 мг 10,11-дигидро-10-[4-(2-тиенил)бензоил]-5 Н-пирроло[2,1-с][1,4]бензодиазепина, 10 мл 40%-ного формалина и 10 мл 40%-ного N,N-диметиламина в 50 мл смеси 1:1 тетрагидрофуран : метиловый спирт нагревали с обратным холодильником в течение 3 ч. Реакционную смесь выпаривали под вакуумом до остатка, который экстрагировали хлороформом, промывали водой, высушивали надNa2SO4, фильтровали и фильтрат выпаривали под вакуумом с получением остатка, который очищали путем хроматографии на колонке с силикагелем, элюируя 30%-ной смесью этилацетат-гексан, с получением 300 мг желаемого продукта в виде твердого вещества. Массспектр: М+Н:428. Пример 7. 5-([1,1'-Бифенил]-4-ил-карбонил)-5,10-дигидро-4 Н-пиразоло[5,1-с] [1,4] бензодиазепин. К перемешиваемому раствору 185 мг 5,10-дигидро-4 Н-пиразоло[5,1-с][1,4]бензодиазепина в 50 мл метиленхлорида, содержащего 2,0 мл триэтиламина, медленно добавляли раствор 300 мг 4-бифенилкарбонилхлорида в 10 мл метиленхлорида при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч и выпаривали под вакуумом с получением остатка. Остаток экстрагировали хлороформом, промывали водой, высушивали над Na2SO4,фильтровали и фильтрат выпаривали под вакуумом с получением остатка, который очищали путем хроматографии на колонке с силикагелем, элюируя 30%-ной смесью этилацетат-гексан, с получением 250 мг желаемого продукта в виде твердого вещества. Массспектр: М+Н:366. Пример 8. 10,11-Дигидро-10-[(2'-(трифторметил)[1,1'-бифенил]-4-ил]карбонил-5 Нпирроло[2,1-с] [1,4] бензодиазепин. К раствору 2,0 г 10,11-дигидро-10-(4 иодбензоил)-5 Н-пирроло[2,1-с][1,4]бензодиазепина и 4,2 г м-трифторметилтрибутил олова в 100 мл толуола и 30 мл N,Nдиметилформамида в атмосфере азота добавляли 0,5 г тетракис(трифенилфосфин)палладия и смесь нагревали до 120 С в течение 12 ч. Толуол выпаривали под вакуумом и маслянистый осадок разбавляли 50 мл хлороформа и фильтровали через диатомовую землю. Фильтрат промывали водой (3 х 50 мл),высушивали (Nа 2SO4) и выпаривали под вакуумом с получением остатка, который хроматографировали на колонке с силикагелем,элюируя смесью этилацетат : гексан от 7:1 до 1:1, с получением 1,75 г желаемого продукта в виде твердого вещества, т. пл. 138-141 С,М+Н=433,4; M+Na=455,4. Пример 9. 10,11-Дигидро-10-[4-(2-пиридинил)бензоил]-5 Н-пирроло[2,1-с][1,4] бензодиазепин. К раствору 2,0 г 10,11-дигидро-10-(4 иодбензоил)-5 Н-пирроло[2,1-с][1,4]безодиазепина и 4,39 r 2-[(три-н-бутил)станнил] пиридина в 30 мл толуола в атмосфере азота добавляли 0,2 тетракис(трифенилфосфин) палладия,и смесь нагревали до 120 С в течение 16 ч. Толуол выпаривали под вакуумом и маслянистый остаток разбавляли 50 мл хлороформа,промывали водой, высушивали (Na2SO4),фильтровали через диатомовую землю и выпаривали под вакуумом с получением остатка,который хроматографировали на колонке с силикагелем, элюируя смесью этилацетат : гексан от 7:1 до 1:1, с получением 1,44 г же 45 лаемого продукта в виде твердого вещества, т. пл. 170-172 С, М+Н=366,1; M+Na=388,l. Пример 10. 10,11-Дигидро-10-[4-(2 тиазолил)бензоил]-5 Н-пирроло[2,1-с][1,4] бензодиазепин. К раствору 1,1 г 10,11-дигидро-10-(4 иодбензоил)-5 Н-пирроло[2,1-с][1,4]бензодиазепина и 1,49 г 2-(трибутил-станнил)тиазола(ссылочный пример 48) в 50 мл толуола в атмосфере азота добавляли 0,2 г тетракис(трифенилфосфин) палладия и смесь нагревали до 120 С в течение 16 ч. Толуол выпаривали под вакуумом до остатка, который подвергали хроматографии на колонке с силикагелем, элюируя смесью этилацетат : гексан от 7:1 до 1:1, с получением 0,62 г желаемого продукта в виде аморфного твердого вещества, М+Н=372,3; M+Na=395,3. Пример 11. 10,11-Дигидро-10-[4-[(4 метилфенил)тио]бензоил]-5 Н-пирроло[2,1-с][1,4] бензодиазепин. К раствору 1,0 г 10,11-дигидро-5 Нпирроло[2,1-с] [1,4]-бензодиазепина в 50 мл сухого метиленхлорида, охлажденного до 0 С,в атмосфере азота добавляли по каплям раствор 2,50 г 4-[(4-метилфенил) тио]бензоилхлорида в 30 мл метиленхлорида. Добавляли часть 5,0 мл триэтиламина и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли 50 мл хлороформа и промывали по 50 мл каждого водой, 2 н НС 1, водой,насыщенным раствором NaHCO3 и водой. Органический слой высушивали (Na2SO4) и хроматографировали на колонке с силикагелем,элюируя смесью этилацетат : гексан от 7:1 до 3:1, с получением 1,96 г желаемого продукта в качестве твердого вещества. М+Н=411,2;M+Na=433,2. Пример 12. 10,11-Дигидро-10-[4-фенилсульфонил]бензоил]-5 Н-пирроло[2,1-с][1,4] бензодиазепин. К раствору 1,0 г 10,11-дигидро-5 Нпирроло[2,1-с] [1,4]-бензодиазепина в 30 мл сухого метиленхлорида, охлажденного до 0 С,в атмосфере азота добавляли по каплям раствор 1,82 г 4-(фенилсульфонил) бензоилхлорида в 30 мл метиленхлорида. Добавляли порцию 7,0 мл триэтиламина и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли 50 мл хлороформа и промывали по 50 мл каждого водой, 2 н НСl, водой, насыщенным раствором NаНСО 3 и водой. Органический слой высушивали (Na2SO4) и хроматографировали на колонке с силикагелем, элюируя смесью этилацетат : гексан от 7:1 до 3:1, с получением 2,2 г желаемого продукта в виде твердого вещества. М+Н=429,2; M+Na=451,2.[2,1-с] [1,4] бензодиазепин. Смесь 2,13 г хлорида 4-[(4 метилфенил)сульфонил]бензоила (ссылочный пример 42) и 1,0 г 10,11-дигидро-5 Нпирроло[2,1-с] [1,4] бензодиазепина в 30 мл сухого метиленхлорида, содержащего 7 мл триэтиламина, перемешивали в атмосфере азота в течение 16 ч. Реакционную смесь разбавляли 7 мл хлороформа и промывали 2 н НСl, водой, насыщенным раствором NaHCO3 и водой. Органический слой высушивали(Na2SO4) и хроматографировали на колонке с силикагелем, элюируя смесью этилацетат : гексан от 7:1 до 3:1, с получением 1,3 г желаемого продукта в виде твердого вещества. М+Н=443,2; M+Na 465,2. Пример 14. 10,11-Дигидро-10-[4'-(2 пропенилокси) [1,1'-бифенил]-4-ил]карбонил]5 Н-пирроло[2,1-с][1,4]бензодиазепин. К раствору 3,3 г 10,11-дигидро-5 Нпирроло[2,1-с] [1,4]-бензодиазепина в 60 мл сухого метиленхлорида, охлажденного до 0 С,в атмосфере азота добавляли 22,0 мл триэтиламина, затем раствор 1,2 эквивалентов 4'-(2 пропенокси)-[1,1'-бифенил]-4-карбонил хлорида в 20 мл метиленхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли 50 мл хлороформа и промывали по 50 мл каждого водой, 2 н НС 1, водой, насыщенным раствором NаНСО 3 и водой. Органический слой высушивали (Na2SO4) и хроматографировали на колонке с силикагелем,элюируя смесью этилацетат : гексан от 7:1 до 3:1, с получением 4,8 г желаемого продукта в виде твердого вещества. М+Н=421,2;M+Na=443,2. Пример 15. 10,11-Дигидро-10-[4-(фенилтио)бензоил]-5 Н-пирроло[2,1-с][1,4]бензодиазепин. К раствору 1,0 г 10,11-дигидро-5 Нпирроло[2,1-с] [1,4]-бензодиазепина в 50 мл сухого метиленхлорида, охлажденного до 0 С,в атмосфере азота добавляли 5,0 мл триэтиламина, затем раствор 2,16 г 4-(фенилтио) бензоил хлорида (ссылочный пример 34) в 30 мл метиленхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли 50 мл хлороформа и промывали по 50 мл каждого водой, 2 н НСl, водой, насыщенным растворомNaHCO3 и водой. Органический слой высушивали (Na2SO4) и хроматографировали на колонке с силикагелем, элюируя смесью этилацетат : гексан от 7:1 до 3:1, с получением 1,92 г желаемого продукта в виде твердого вещества. М+H=397,1; M+Na 419,1. Пример 16. 10-(4-Бензоилбензоил)-10,11 дигидро-5 Н-пирроло[2,1-с][1,4]бензодиазепин.(ссылочный пример 36) и 1,0 г 10,11-дигидро 5 Н-пирроло[2,1-с][1,4]бензодиазепина в 30 мл сухого метиленхлорида, содержащего 7 мл триэтиламина, перемешивали в атмосфере азота в течение 16 ч. Реакционную смесь разбавляли 50 мл хлороформа и промывали 2 н НС 1, водой, насыщенным раствором NаНСО 3 и водой. Органический слой высушивали(Na2SO4) и хроматографировали на колонке с силикагелем, элюируя смесью этилацетат : гексан от 7:1 до 3:1, с получением 1,0 г желаемого продукта в виде твердого вещества. М+Н=393,2; M+Na=415,2. Пример 17. 5-([1,1'-Бифенил]-4-илкарбонил)-4,5-дигидропирроло[1,2-а]хиноксалин. К смеси 0,5 г 4,5-дигидропирроло[1,2-а] хиноксалина и 0,96 г 4-бифенилкарбонил хлорида в 50 мл метиленхлорида добавляли 7,0 мл триэтиламина в атмосфере азота, затем перемешивали в течение 16 ч. Реакционную смесь разбавляли 50 мл хлороформа и промывали водой, 2 н НСl, насыщенным раствором NaHCO3 и водой. Органический слой высушивали (Na2SO4) и хроматографировали на колонке с силикагелем, элюируя смесью этилацетат : гексан от 7:1 до 1:1, с получением 0,87 г желаемого продукта в виде твердого вещества. М+Н=351,2; M+Na 373,2. Пример 18. 10,11-Дигидро-10-[4-(4-метилфенокси)бензоил]-5 Н-пирроло[2,1-с][1,4] бензодиазепин. К перемешиваемому раствору 1,0 г 10,11 дигидро-5 Н-пиразоло[5,1-с][1,4]бензодиазепина в 30 мл метиленхлорида при охлаждении до 0 С в атмосфере азота по каплям добавляли 30 мл триэтиламина, затем раствор 1,2 эквивалентов 4-(4-метилфенокси)бензоил хлорида в 10 мл CH2Cl2 (ссылочный пример 53), полученного по ссылочному примеру 52, и тионилхлорид. Реактивы перемешивали при комнатной температуре в течение 16 ч и концентрировали под вакуумом до остатка. Остаток растворяли в метиленхлориде, промывали водой,2 н HCl, водой, насыщенным растворомNaHCO3, водой, высушивали (MgSO4) и выпаривали под вакуумом с получением 2,2 г желаемого продукта. МС (М+Н=395,1). Пример 19. 10-([1,1'-Бифенил]-4-илкарбонил)-10,11-дигидро-5 Н-пирроло[2,1-с][1,4] бензодиазепин. К раствору 0,9 г 10,11-дигидро-5 Нпиразоло[2,1-с][1,4]бензодиазепина в 30 мл метиленхлорида при охлаждении до 0 С добавляли 1,6 мл триэтиламина. Добавляли раствор с 1,2 эквивалентами 4-бифенилкарбонил хлорида в 10 мл CH2Cl2 и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли 50 мл хлороформа и промывали по 20 мл каждого водой, 2 н НС 1, водой, насыщенным вод 000056 ным раствором NаНСО 3 и водой. Органический слой выпаривали под вакуумом до остатка. Остаток очищали путем хроматографии на колонке с силикагелем, элюируя смесью 3:1 гексан : этилацетат, с получением 1,5 г желаемого продукта. Пример 20. 10-([1,1'-Бифенил]-4-илкарбонил)-10,11-дигидро-N,N-диметил-5 Н-пирроло[2,1-с][1,4]бензодиазепин-3-метанамин. Раствор 0,6 г 10-([1,1'-бифенил]-4-илкарбонил)-10,11-дигидро-5 Н-пиразоло[2,1-с][1,4] бензодиазепина в 50 мл смеси метиловый спирт : тетрагидрофуран 1:1 перемешивали в атмосфере азота при добавлении 10 мл 30%ного раствора формальдегида, 10 мл 40%-ного раствора диметиламина и 2 капель уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч,затем экстрагировали хлороформом (3 х 100 мл) и органический слой промывали насыщенным водным раствором NaHCO3 (2 х 50 мл), водой (2 х 50 мл), высушивали и выпаривали под вакуумом с получением 0,68 г остатка, который перемешивали в петролейном эфире с получением 0,62 г желаемого продукта в виде кристаллического твердого вещества,т. пл. 85-87 С. Пример 21. 10,11-Дигидро-10-[(4'-пропил[2,1-с][1,4]бензодиазепин. К перемешиваемому раствору 2,0 г 10,11 дигидро-5 Н-пиразоло[2,1-с][1,4]бензодиазепина в 30 мл метиленхлорида при охлаждении до 0 С в атмосфере азота по каплям добавляли 30 мл триэтиламина, затем раствор 4'-(пропил)[1,1'-бифенил]-4-карбонил хлорида в 10 мл метиленхлорида. Реактивы перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь вливали в ледяную воду и экстрагировали метиленхлоридом (3 х 50 мл). Органический слой промывали водой, 2 н лимонной кислотой, насыщенным водным раствором NaHCO3, водой, высушивали (МgSO4) и выпаривали под вакуумом с получением остатка, который очищали путем хроматографии на колонке с силикагелем, элюируя смесью этилацетат-гексан, с получением 1,4 г желаемого продукта в виде кристаллического твердого вещества, т. пл. 118-120 С. МС(М+Н=407,3; M+Na=429,3). Пример 22. 10,11-Дигидро-10-[2-(2-тиенил)пиридин-5-ил)карбонил]-5 Н-пирроло[2,1 с] [1,4]бензодиазепин. Раствор 5 ммоль 10,11-дигидро-5 Нпиразоло[2,1-с] [1,4]-бензодиазепина в 30 мл метиленхлорида охлаждали до 0 С и добавляли 10 мл триэтиламина. К реакционной смеси добавляли 6 ммоль соединения по ссылочному примеру 60 и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли 60 мл хло 49 роформа и промывали по 40 мл каждого водой, насыщенным водным раствором NaHСО 3 и водой. Органический слой высушивали и выпаривали под вакуумом до остатка. Остаток очищали путем хроматографии на колонке с силикагелем, элюируя смесью гексан : этилацетат 3:1, с получением желаемого продукта. Исследование полезности. Определение связывания с рецепторами V1 печени крысы. Плазматические мембраны печени крысы, экспрессирующие подтипы рецептора вазопрессина V1, выделяли на градиенте плотности сахарозы, согласно методу, описанномуLesko et al., (1973). Эти мембраны быстро суспендировали в 50,0 мМ буфере Трис-HCl, рН 7,4, содержащем 0,2% бычьего сывороточного альбумина (БСА) и 0,1 мМ фенилметилсульфонилфторида (ФМСФ), и хранили замороженными при -70 С до использования в последующих экспериментах по связыванию. Для экспериментов по связыванию в ячейки микротитровальных плат формата 96 ячеек добавляли: 100 мкл буфера Трис-HCl 100,0 мМ, содержащего 10,0 мМ МgСl2, 0,2%-ного инактивированного нагреванием БСА и смесь ингибиторов протеаз: леупептина, 1,0 мг %; апротинина, 1,0 мг %; 1,10-фенантролина, 2,0 мг %; ингибитора трипсина, 10,0 мг % и 0,1 мМ ФМСФ, 20,0 мкл 0,8 нМ [фенилаланил 3,4,5-3 Н] вазопрессина (Спец. активн. 45,1Ci/ммоль), реакцию начинали добавлением 80 мкл мембран ткани, содержащих 20 мкг белка ткани. Платы содержали, не перемешивая, на верхней подставке при комнатной температуре в течение 120 мин до достижения равновесия. Образцы для неспецифического связывания измеряли в присутствии 0,1 мкМ немеченного антагониста фенилаланил-вазопрессина, добавленного в объеме 20,0 мкл. Исследуемые соединения солюбилизировали в 50%-ном диметилсульфоксиде (ДМСФ) и добавляли в объеме 20,0 мкл до конечного объема инкубационной среды 200 мкл. По завершении связывания содержимое каждой ячейки фильтровали при помощи аппаратаBrandel Cell Harvester (Gaithesburg, MD). Радиоактивность, задержавшуюся на фильтровальном диске вместе с лиганд-рецепторным комплексом, определяли при помощи подсчета в жидкостном сцинтилляторе в счетчике Packard LS с эффективностью счета для трития 65%. Значения IC50 анализировали при помощи программы LUNDON-2 для конкурентного связывания (LUNDON SOFTWARE, ОН). 50 Определение связывания с рецепторами V2 мозгового слоя почки крысы. Ткань мозгового слоя почки крыс вырезали, разрезали на маленькие кусочки и вымачивали в 0,154 мM растворе хлорида натрия,содержащем 1,0 мM ЭДТА, со многими сменами жидкой фазы до тех пор, пока раствор не стал прозрачным от крови. Ткань гомогенизировали в 0,25 М растворе сахарозы, содержащем 1,0 мМ ЭДТА и 0,1 мМ ФМСФ, при помощи гомогенизатора Поттера-Эльвейма с тефлоновым пестиком. Гомогенат фильтровали через несколько слоев(4 слоя) марли. Фильтрат повторно гомогенизировали в даунс гомогенизаторе с плотно подогнанным пестиком. Конечный гомогенат центрифугировали при 1500 х g в течение 15 мин. Осадок ядер отбрасывали, а супернатант повторно центрифугировали при 40 000 х g в течение 30 мин. Образовавшийся конечный осадок содержит темную внутреннюю часть с внешней слегка розовой частью. Розовую внешнюю часть суспендировали в небольшом количестве 50,0 мМ Трис-HCl буфере с рН 7,4. Содержание белка определяли по методу Лоури (Lowry et al., J. Biol. Chem., 1953). Суспензию мембран хранили при -70 С в 50,0 мМ Трис-HCl, содержащем 0,2% инактивированного БСА и 0,1 мМ ФМСФ в аликвотах по 1,0 мл, содержащих 10,0 мг белка в мл суспензии,до использования в последующих экспериментах по связыванию. Для экспериментов по связыванию в ячейки микротитровальных плат формата 96 ячеек добавляли: 100 мкл буфера Трис-HCl 100,0 мМ, содержащего 10,0 мМ MgCl2, 0,2% инактивированного нагреванием БСА и смесь ингибиторов протеаз: леупептина, 1,0 мг%; апротинина, 1,0 мг%; 1,10-фенантролина, 2,0 мг%; ингибитора трипсина, 10,0 мг% и 0,1 мМ ФМСФ, 20,0 мкл 0,8 нМ [3 Н] Аргинин 8 вазопрессина (Спец. активн. 75,0 Ci/ммоль),реакцию начинали добавлением 80,0 мкл мембран ткани (200,0 мкг белка ткани). Платы содержали, не перемешивая, на верхней подставке при комнатной температуре в течение 120 мин до достижения равновесия. Неспецифическое связывание определяли в присутствии 1,0 мкМ немеченного лиганда, добавленного в объеме 20 мкл. Исследуемые соединения солюбилизировали в 50% диметилсульфоксиде (ДМСФ) и добавляли в объеме 20,0 мкл до конечного объема инкубационной среды 200 мкл. По завершении связывания содержимое каждой ячейки фильтровали при помощи аппаратаBrandel Cell Harvester (Gaithesburg, MD). Радиоактивность, задержавшуюся на фильтровальном диске вместе с лиганд-рецепторным комплексом, определяли при помощи подсчета в жидкостном сцинтилляторе в счетчике Pac 51kard LS с эффективностью счета для трития 65%. Значения IC50 анализировали при помощи программы LUNDON-2 для конкурентного связывания (LUNDON SOFTWARE, ОН). Результаты настоящего исследования характерных соединений по настоящему изобретению представлены в табл.1 (см. конец описания). Эксперименты по связыванию радиоактивного лиганда с мембранами тромбоцитов человека.(а) Получение мембран тромбоцитов. Замороженную, обогащенную тромбоцитами плазму (PRP), (Источник плазмы: Служба крови Hudson Valley, Westchester MedicalCenter, Valhalla, NY) оттаивали до комнатной температуры. Пробирки, содержащие PRP,центрифугировали при 16 000 х g в течение 10 мин при 4 С и отбрасывали супернатант. Тромбоциты повторно суспендировали в равном объеме 50,0 мМ Трис-НС 1 буфера, рН 7,5,содержащего 120 мМ NaCl и 20 мМ ЭДТА. Суспензию повторно центрифугировали при 16 000 х g в течение 10 мин. Данный этап отмывки проводили еще один раз. Промывочную жидкость отбрасывали и лизированные тромбоциты гомогенизировали в буфере ТрисHCl, 5 мМ с низкой ионной силой, рН 7,5, содержащего 5,0 мМ ЭДТА. Гомогенат центрифугировали при 39 000 х g в течение 10 мин. Конечный осадок повторно суспендировали в Трис-HCl буфере, 70 мM, рН 7,5 и повторно центрифугировали при 39 000 х g в течение 10 мин. Конечный осадок повторно суспендировали в 50,0 мМ буфере Трис-HCl, рН 7,4, содержащем 120 мМ NaCl и 5,0 мМ КСl с получением суспензии 1,0-2,0 мг белка в мл.(b) Связывание с подтипом V1 рецептора вазопрессина мембран тромбоцитов человека. В ячейки микротитровальной платы формата 96 ячеек добавляли 100 мкл 50,0 мМ Трис-HCl буфера, содержащего 0,2% БСА, и смесь ингибиторов протеаз (апротинина, леупептина и т.п.). Затем добавляли 20 мкл [3 Н] лигандаArg8 Вазопрессин) до получения конечных концентраций от 0,01 до 10,0 нМ. Инициацию связывания проводили путем добавления 80,0 мкл суспензии тромбоцитов (приблизительно, 100 мкг белка). Все реактивы смешивали путем пипетирования смеси вверх и вниз несколько раз. Неспецифическое связывание измеряли в присутствии 1,01 М немеченного лиганда(Manning пептида или Аrg8-Вазопрессина). Смесь оставляли, не перемешивая, при комнатной температуре в течение девяноста (90) мин. По истечении этого времени инкубационную смесь быстро фильтровали при отсасывании под вакуумом через фильтры GF/B, используя аппарат Brandel Cell Harvester. Определение радиоактивности, задержанной на фильтровальном диске, проводили при помощи добавления жидкостного сцинтиллятора и подсчета в жидкостном сцинтилляционном счетчике. Связывание с мембранами клеток фибропластов мыши, трансфецированных кДНК, линииLV-2, экспрессирующих рецептор вазопрессина V2 человека.(а) Получение мембран. Культуральные матрасы, объемом 175 мл, содержащие прикрепленные клетки, выращенные до конфлуентности, освобождали от культуральной среды путем аспирации. Матрасы, содержащие прикрепленные клетки,промывали 2 х 5 мл физраствором на фосфатном буфере (PBS) и каждый раз отсасывали жидкость. В конце добавляли 5 мл раствора Хенкса для диссоциации клеток, без ферментов (Speciality Media, Inc., Lafayette, NJ) и матрасы оставляли на 2 мин. Содержимое всех матрасов вливали в центрифужные пробирки и осаждали клетки при 300 х g в течение 15 мин. Раствор Хенкса аспирировали и клетки гомогенизировали гомогенизатором типа политрон на положении 6 в течение 10 с в буфере Трис-HCl, рН 7,4, содержащем 0,25 М сахарозу и 1,0 мМ ЭДТА. Гомогенат центрифугировали при 1500 х g в течение 10 мин для удаления следов (ghost) мембран. Супернатант центрифугировали при 100 000 х g в течение 60 мин для осаждения белка рецептора. По завершении осадок ресуспендировали в небольшом объеме буфера Трис-HCl, рН 7,4, 50,0 мМ. Содержание белка определяли по методу Лоури, мембранные рецепторы суспендировали в 50,0 мМ буфере Трис-HCl, содержащем 0,1 М фенилметилсульфонилфторид (ФМСФ) и 0,2% бычий сывороточный альбумин (БСА),с получением 2,5 мг белка рецептора на мл суспензии.(b) Связывание с рецептором. Для экспериментов по связыванию в ячейки микротитровальной платы формата 96 ячеек добавляли в мкл: 100,0 мкл буфера Трис-HCl 100 мМ, содержащего 0,2% инактивированного нагреванием БСА, 10,0 мM[3 Н] Аргинин 8-вазопрессина (спец. активн. 75,0 Ci/ммоль), реакцию начинали путем добавления 80,0 мкл мембран ткани (200,0 мкг белка ткани). Платы оставляли в покое на верхней подставке в течение 120 мин для достижения равновесия. Неспецифическое связывание определяли в присутствии 1,0 мкМ немеченного лиганда, добавленного в объеме 20,0 мкл. Исследуемые соединения солюбилизировали в 50% диметилсульфоксиде (ДМСФ) и добавляли в объеме 20,0 мкл до конечного объема 200 мкл. По завершении связывания содержимое каждой ячейки фильтровали при помощи аппарата Brandel Cell Harvester(Gaithesburg). Радиоактивность, задержавшуюся на фильтровальном диске в составе лиганд-рецепторного комплекса, определяли путем подсчета в сцинтилляционной жидкости в счетчике Packard LS Counter с эффективностью для трития 65%. Данные значений IC50 анализировали при помощи программы LUNDON-2 для конкурентного связывания (LUNDON SOFTWARE, ОН). Активность в качестве V2 антагониста вазопрессина на бодрствующих крысах с нагрузкой водой. Бодрствующим гидратированным крысам вводили через рот исследуемые соединения от 0,1 до 100 мг/кг или носитель. Для каждого соединения использовали от двух до четырех крыс. Через один час внутрибрюшинно вводили аргинин-вазопрессин (АВП, антидиуретический гормон, АДГ), растворенный в арахисовом масле в дозе 0,4 мг/кг. Две крысы,служащие в качестве контроля нагрузки водой из каждой исследуемой группы, не получали аргинин-вазопрессин, а только один носитель(арахисовое масло). Двадцать минут спустя каждая крыса получала 30 мл/кг деионизованной воды через рот, через зонд и помещалась отдельно в метаболические клетки, снабженные воронкой и градуированным стеклянным цилиндром для сбора мочи в течение четырех часов. Измеряли объем мочи и анализировали осмоляльность при использовании осмометраFiske One-Ten (Fiske Assoc., Norwood, MA,USA). Натрий, калий и хлорид в моче анализировали при использовании ионспецифических электродов анализатора Beckman ЕЗ (Electrolyte 3). В следующих результатах отмечается пониженный объем мочи и пониженная осмоляльность по отношению к АВП-контролю. Результаты настоящего исследования характерных соединений по настоящему изобретению представлены в табл. 2. Таблица 2. Активность в качестве V2 антагониста на бодрствующих крысах с нагрузкой водойДоза 10 2 7,8 992 10 2 7,5 1005 10 2 8 887 10 2 8,3 857 10 2 11 825 10 2 5,8 1254 10 2 5,5 1380 10 2 4 1247 10 2 4 1278 10 2 6 1022 10 2 10,3 929 10 2 5,3 919 10 2 3,5 1598 10 2 3,5 1650 10 2 4,5 1347 Контроль с нагрузкой водой Контроль с нагрузкой водой + ДМСО(10%)Контроль с Аrg-Вазопрессином Активность в качестве V1 антагониста вазопрессина на бодрствующих крысах Бодрствующих крыс иммобилизовали в лежачем положении при помощи эластичной ленты. Проводили местную анестезию области в основании хвоста путем накожного нанесения 2% прокаина (0,2 мл). Применяя асептический способ, изолировали хвостовую артерию и вводили канюлю, сделанную из полиэтиленовых трубочек 10 и 20 (запаянных при нагревании), в нижнюю брюшную аорту. Канюлю закрепляли для безопасности, гепаринизировали (1000 ед/см 3), запаивали и зашивали рану одним или двумя стежками Dexon 4-0. Для внутривенного введения препаратов в хвостовую вену также вводили канюлю подобным образом. Продолжительность хирургического вмешательства приблизительно 5 мин. Если требовалось, то обеспечивалась дополнительная местная анестезия (2% прокаин или лидокаин). Животного помещали в пластиковую клетку с закрепляющими элементами в нормальном вертикальном положении. Канюлю прикрепляли к датчику измерения давленияStatum P23Db и регистрировали пульсирование давления крови. Увеличение систолического давления крови в ответ на внутрибрюшинное введение аргинин-вазопрессина 0,01 и 0,2 международных единиц (i.u. -ед.) (350 I.U.= 1 мг) регистрировали до введения любого из препаратов (соединения), после чего каждая крыса через рот получала исследуемое соединение в дозе 0,1-100 мг/кг (10 см 3/кг), или при внутривенном введении 0,1-30 мг/кг (1 см 3/кг). Введение вазопрессина повторяли позже на 30, 60, 90, 120, 180, 240 и 300 мин. Антагонистический эффект рассчитывали в процентах,принимая за 100% вазопрессорный ответ на вазопрессин до приема препарата. Результаты этого исследования характерных соединений по настоящему изобретению в конкретной дозе, максимальный процент ингибирования и время в минутах представлены в табл.3. Таблица 3. Вазопрессорный ответ на вазопрессин (ВАЗ)(а) Получение мембран Самок крыс Sprague-Dawley весом, приблизительно, 200-250 г внутримышечно инъецировали (в.м.) диметилстилбэстролом (DES) 0,3 мг/кг веса тела. Крыс забивали через 18 ч после анестезии пентобарбиталом. Матки вырезали, очищали от жира и соединительной ткани и промывали 50 мл обычного физраствора. Ткань, собранную от шести крыс, гомогенизировали в 50 мл 0,01 мМ Трис-HCl, рН 7,4, содержащего 0,5 мМ дитиотреитола и 1,0 мМ ЭДТА, при помощи гомогенизатора типа политрон в положении 6 с тремя повторами по 10 с каждый. Гомогенат процеживали через два (2) слоя марли, а фильтрат центрифугировали при 1000 х g в течение 10 мин. Осветленный супернатант удаляли и повторно центрифугировали при 165 000 х д в течение 30 мин. Конечный осадок, содержащий рецепторы окситоцина, повторно суспендировали в 50,0 мМ Трис-HCl, содержащего 5,0 мМ MgCl2 при рН 7,4, с получением концентрации белка в суспензии ткани 2,5 мг/мл. этот препарат использовали в последующих определениях связывания с [3 Н] Окситоцином.([ Н]ОТ) со своими рецепторами проводили в микротитровальных платах, используя [3H]OT в различных концентрациях, в буфере определения 50,0 мМ Трис-HCI, рН 7,4, содержащего 5,0 мМ MgCl2 и смесь ингибиторов протеаз: БСА, 0,1 мг; апротинина, 1,0 мг; 1,10 фенантролина, 2,0 мг; трипсина, 10,0 мг; и ФМСФ, 0,3 мг на 100 мл раствора буфера. Неспецифическое связывание определяли в присутствии 1,0 мкМ немеченного ОТ. Реакцию связывания останавливали через 60 мин при 22 С путем быстрой фильтрации через стеклофильтры, используя аппарат Brandel, Сell Harvester (Biomedical Research and DevelopmentLaboratories Inc., Gaithersburg, MD). Эксперименты по конкуренции проводили при равновесии, используя 1,0 нМ [3 Н]ОТ и различные концентрации вытесняющих агентов. Концентрацию агента, вытесняющего 50% [3 Н]ОТ из его участков связывания (IC50), рассчитывали при помощи компьютерной программы LUNDON-2 (LUNDON SOFTWARE INC., Ohio,USA). Результаты настоящего исследования характерных примеров представлены в табл.4. Таблица 4. Определение связывания окситоцинаДоза, % ингибиэксп (мкМ) рования при 10 мкМ 2 10 100 3 10 89 4 10 62 6 10 100 7 10 100 8 10 95 9 10 100 10 10 77 11 10 86 12 10 69 13 10 67 14 10 67 15 10 98 16 10 100 17 10 92 18 10 99 19 10 81 20 10 100 21 10 68 22 10 98 Соединения по настоящему изобретению могут быть использованы в форме солей, производных от фармацевтически или физиологически приемлемых солей или оснований. Эти соли включают, но не ограничиваются, следующими: соли неорганических кислот, таких как соляная кислота, серная кислота, азотная 57 кислота, фосфорная кислота, и в виде случая могут быть такими органическими кислотами,как уксусная кислота, щавелевая кислота, янтарная кислота и малеиновая кислота. Другие соли включают соли щелочных металлов или щелочноземельных металлов, таких как натрий, калий, кальций или магний, или органических оснований. Соединения могут быть также использованы в виде эфиров, карбаматов и других общепринятых форм препаратовпредшественников, которые при введении в такой форме превращаются in vivo в активный радикал. Когда соединения используются для вышеуказанных применений, они могут сочетаться с одним или несколькими фармацевтически приемлемыми носителями, например растворителями, разбавителями и тому подобными, и могут быть введены через рот в таком виде как таблетки, капсулы, диспергируемые порошки, гранулы или суспензии, содержащие, например, от около 0,05 до 5% суспендирующего агента, сиропы, содержащие, например, от около 10 до 50% сахара, и эликсиры,содержащие, например, от около 20 до 50% этанола, и подобные, или парентерально в виде стерильного инъекционного раствора или суспензии, содержащей от около 0,05 до 5% суспендирующего агента в изотонической среде. Такие фармацевтические препараты могут содержать, например, от около 0,05 до около 90% активного ингредиента в комбинации с носителем, более обычно, между около 5 и 60% по весу. Эффективная дозировка применяемого активного ингредиента может изменяться в зависимости от конкретного используемого соединения, способа введения и тяжести подлежащего лечению состояния. Однако, в целом, удовлетворительные результаты получаются, когда соединения по изобретению вводятся в дозах от около 0,5 до около 500 мг/кг веса тела животного, предпочтительно даваемых в дозах по частям, от двух до четырех раз в день, или в форме длительного высвобождения. Для наиболее крупных животных общая дневная доза составляет от около 1 до 100 мг,предпочтительно от около 2 до 80 мг. Дозировочная форма, подходящая для внутреннего применения, составляет от около 0,5 до 500 мг активного соединения в хорошо знакомой смеси вместе с твердым или жидким фармацевтически приемлемым носителем. Расписание введения доз может быть приспособлено для получения оптимального терапевтического ответа. Например, могут быть введены несколько раздельных доз в течение дня, или доза может быть пропорционально снижена,как назначено в случае острой необходимости при терапевтической ситуации. Данные активные соединения могут быть введены оральным путем, а также внутривен 000056 но, внутримышечно или подкожным путем. Твердые носители включают крахмал, лактозу,дикальцийфосфат,микрокристаллическую целлюлозу, сахарозу и каолин, тогда как жидкие носители включают стерильную воду, полиэтиленгликоль, неионные поверхностноактивные соединения и съедобные масла, такие как кукурузное, арахисовое и кунжутное масла, что подходит для природы активного ингредиента и конкретного способа желаемого введения. Коммерчески используемые адъюванты для получения фармацевтических композиций могут преимущественно включать такие, как вкусовые агенты, окрашивающие агенты, консервирующие агенты и антиоксиданты, например витамин Е, аскорбиновая кислота, ВНТ и ВНА. Предпочтительными фармацевтическими композициями с точки зрения легкого приготовления и введения являются твердые композиции, предпочтительно таблетки и твердозаполненные и жидкозаполненные капсулы. Предпочтительным является прием через рот. Эти активные композиции могут быть также введены парентерально или внутрибрюшинно. Растворы или суспензии данных активных соединения в виде свободного основания или фармацевтически приемлемой соли могут быть получены в воде, подходящим образом смешанной с поверхностно-активным соединением, таким как гидроксипропилцеллюлоза. Дисперсии могут быть получены в глицерине, жидком полиэтиленгликоле и их смесях в маслах. В обычных условиях хранения и использования данные препараты содержат консервант для предотвращения роста микроорганизмов. Фармацевтические формы, подходящие для инъекционного применения, включают стерильные водные растворы или дисперсии и стерильные порошки для непосредственного приготовления стерильных инъекционных растворов или дисперсий. Во всех случаях форма должна быть стерильной и должна быть жидкой в такой степени, чтобы быть способной для легкого введения через шприц. Она должна быть стабильной в условиях производства и хранения и должна быть предохранена от загрязнения микроорганизмами, такими как бактерии и грибки. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол(например глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла. Новые трициклические непептидные антагонисты вазопрессина применимы для лечения состояний, когда желателен пониженный уровень вазопрессина, таких как застойная сердечная недостаточность, при болезненных состояниях с избыточной реабсорбцией воды в почке и состояниях с увеличенной сосудистой 59 сопротивляемостью и коронарной вазоконстикцией. В частности, антагонисты вазопрессина по настоящему изобретению являются терапевтически применимыми при лечении и/или профилактике гипертонии, сердечной недостаточности, коронарного вазоспазма, ишемии сердца, почечного вазоспазма, цирроза печени,застойной сердечной недостаточности, почечного синдрома, отеке мозга, ишемии мозга,кровоизлиянии в мозг,тромбозахкровотечениях и состоянии нарушения задержки воды. В частности, антагонисты окситоцина по настоящему изобретению являются применимыми при профилактике преждевременных родов и рождении недоношенного, что является существенной причиной проблем со здоровьем у детей и детской смертности. атомом серы; где 5- или 6-членные гетероциклические кольца необязательно замещены (C1 С 3) низшим алкилом, галогеном или (C1-С 3) низшим алкокси; радикал: является пятичленным ароматическим (ненасыщенным), содержащим азот кольцом, где D,E и F являются выбранными из углерода или азота и где атомы углерода могут быть необязательно замещенными галогеном, (C1-С 3) низшим алкилом, гидрокси, -СОСl3, -СОСF3, ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, выбранное среди соединений с общей формулой I:R3 где m является целым числом от 1 до 2; и радикал:-СНО, амино, (C1-С 3) низшим алкокси и (C1 С 3) низшим алкиламином, -CONH-(C1-С 3) низшим алкилом, -СON [низшим алкилом (C1 С 3)]2; q равен 1 или 2; Rb является независимо выбранным из водорода, -СН 3 и -C2H5; R3 является радикалом формулы: представляет: (1) фенил или замещенный фенил, необязательно замещенный одним или двумя заместителями, выбранными из (C1-С 3) низшего алкила, галогена, амина, (C1-С 3) низшего алкокси или (C1-С 3) низшего алкиламина; (2) 5-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один гетеро-атом, выбранный из О, N, или S;(3) 6-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один азот; (4) 5- или 6-членное ароматическое (ненасыщенное) гетероциклическое кольцо,имеющее два атома азота; (5) 5-членное ароматическое (ненасыщенное) гетероциклическое кольцо, имеющее один атом азота, вместе с или одним атомом кислорода, или одним
МПК / Метки
МПК: C07D 487/04, A61K 31/55
Метки: диазепинов, антагонистов, качестве, использованием, вазопрессина, получения, способ, трициклических, трициклические, лечения, диазепины
Код ссылки
<a href="https://eas.patents.su/30-56-triciklicheskie-diazepiny-v-kachestve-antagonistov-vazopressina-sposob-ih-polucheniya-i-sposob-lecheniya-s-ispolzovaniem-triciklicheskih-diazepinov.html" rel="bookmark" title="База патентов Евразийского Союза">Трициклические диазепины в качестве антагонистов вазопрессина, способ их получения и способ лечения с использованием трициклических диазепинов</a>
Производные оксаиндена, способ их получения (варианты), фармацевтический состав на их основе, их применение и способ лечения с их использованием

Номер патента: 25
Опубликовано: 26.02.1998
Авторы: Шимиг Дьюла, Драбант Шандор, Божинг Даниэль, Новак Лайош, Ковач Габор, Ковач Петер, Пирок Дьордь, Такач Габорне, Эдьед Андраш, Сантаи Чаба, Семереди Каталин, Гадо Клара, Гиглер Габор, Блашко Габор, Чёргё Маргит
МПК: A61K 31/34, C07D 307/77
Метки: использованием, получения, фармацевтический, варианты, основе, состав, оксаиндена, лечения, способ, применение, производные
Формула / Реферат:
1. Производные оксаиндена общей формулы (I), где R1 представляет собой низший С1-4 алкил, низшую С1-4 алкоксигруппу или низший С3-6 циклоалкил, R2 представляет собой водород, низший С1-4 алкил или низший С3-6 циклоалкил, R3 представляет собой водород, низший С1-4 алкил, низшую С1-4 алкоксигруппу или бензилоксигруппу, Х представляет собой водород, алифатический С1-4 ацил или бензоиловую или нафтоиловую группу, возможно замещенные одной или...
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Бьенейм Юг, Ансель Жан-Эрик, Мейллян Пьер
МПК: B01J 31/24, A61K 31/355, C07C 39/19...
Метки: использованием, витамина, замещенных, получения, фенолов, cпособ, способ
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Твердое синтетическое топливо в форме поленьев, используемое в качестве заменителя древесины, и способ его получения

Номер патента: 41
Опубликовано: 26.02.1998
Авторы: Гарибальди Пьерпаоло, Пеллегрини Леонардо
МПК: C10L 5/40
Метки: поленьев, способ, древесины, используемое, топливо, форме, качестве, получения, заменителя, синтетическое, твердое
Формула / Реферат:
1. Твердое синтетическое топливо в форме поленьев, используемое в качестве заменителя древесины, отличающееся тем, что оно получено способом, заключающимся в том, что смешивают в расплавленном состоянии: а) термопластичную смолу с высокой молекулярной массой, плотностью от 0,8 до 1,0 г/см3 и точкой плавления, равной или меньшей 230°С, в количестве от 70 до 97% по массе, б) смолу с низкой молекулярной массой в количестве от 3 до 30% по массе, ...
Безводный хлоргидрат цилпатерола в кристаллизованной форме, способ его получения (его варианты), моногидрат и тригидрат хлоргидрата цилпатерола в качестве промежуточных продуктов

Номер патента: 11
Опубликовано: 30.12.1997
Авторы: Годар Жан-Ив, Шевремон Ив
МПК: A23K 1/16, C07D 487/04
Метки: его, продуктов, качестве, промежуточных, кристаллизованной, безводный, способ, моногидрат, форме, хлоргидрата, тригидрат, варианты, получения, цилпатерола, хлоргидрат
Формула / Реферат:
1. Безводный хлоргидрат цилпатерола в кристаллизованной форме с кристаллами размером менее 250 мкм, содержащий менее 5% кристаллов размером менее 15 мкм. 2. Способ получения продукта по п.1, характеризующийся тем, что получают пересыщенный раствор хлоргидрата цилпатерола в воде или 50%-ном водном растворе этанола при температуре 50-70°С, охлаждают полученный раствор до 45°С и выдерживают при этой температуре до окончания образования моногидрата...
Применение олигосахарида и аспирина для лечения тромбоэболических заболеваний, фармацевтическая композиция, способ лечения

Номер патента: 48
Опубликовано: 30.04.1998
Авторы: Кариу Роже, Стикема Якобус
МПК: A61K 31/725
Метки: тромбоэболических, аспирина, фармацевтическая, способ, олигосахарида, применение, лечения, композиция, заболеваний
Формула / Реферат:
1. Применение синтетического олигосахарида, который представляет собой селективный ингибитор фактора Ха, действующий через антитромбин III, в комбинации с аспирином для получения лекарственных средств, предназначенных для предупреждения или лечения тромбоэмболических заболеваний, имеющих место у млекопитающего, подвергающегося чрезкожной внутрипросветной ангиопластике. 2. Применение по п.1, отличающееся тем, что олигосахарид представляет собой...
Предыдущий патент: Способ добычи метана из подземного угольного отложения
Следующий патент: Система скважин для добычи вязкой нефти
Случайный патент: Дубление кожи