Производное азола, способ его получения, химический агент, предназначенный для применения в сельском хозяйстве и садоводстве, и агент для защиты промышленных материалов
Номер патента: 24953
Опубликовано: 30.11.2016
Авторы: Араки Нобуюки, Обата Емико, Мияке Таидзи, Имай Еию







Формула / Реферат
1. Производное азола общей формулы (I)

где R1 обозначает незамещенную или замещенную атомом галогена C1-C6-алкильную группу;
R2 обозначает -С(О)OR3 и -C(O)NR3R4;
каждый R3 и R4 обозначает атом водорода, C1-C6-алкильную группу, C2-C6-алкенильную группу или C2-C6-алкинильную группу;
Y обозначает атом галогена;
m равно от 0 до 5 и
А обозначает атом азота.
2. Производное азола по п.1, где в формуле (I) R2 обозначает COOR3 и R3 обозначает C1-C3-алкильную группу, С2-С3-алкенильную группу или С2-С3-алкинильную группу.
3. Производное азола по п.1, где в формуле (I) R2 обозначает CONR3R4 и каждый R3 и R4 независимо обозначает атом водорода, C1-C3-алкильную группу, C2-C3-алкенильную группу или С2-С3-алкинильную группу.
4. Производное азола по любому из пп.1-3, где в формуле (I) R1 обозначает C1-C6-алкильную группу, замещенную атомом галогена.
5. Производное азола по любому из пп.1-3, где в формуле (I) R1 обозначает незамещенную алкильную группу.
6. Производное азола по любому из пп.1-5, где в формуле (I) количество атомов углерода в группе R1 равно от 1 до 4.
7. Производное азола по любому из пп.1-6, где в формуле (I) Y обозначает атом галогена и m равно 1.
8. Способ получения производного азола по п.1, где R2 обозначает COOR3, включающий стадию этерификации карбоксильной группы, содержащейся в соединении карбоновой кислоты общей формулы (Ib)

(в формуле (Ib) значения R1, Y, m и А такие же, как и в формуле (I), и R3 обозначает C1-C6-алкильную группу, C2-C6-алкенильную группу или C2-C6-алкинильную группу).
9. Способ по п.8, который включает стадию окисления, которая представляет собой получение карбоновой кислоты формулы (Ib) путем окисления гидроксиметильной группы в промежуточном соединении общей формулы (III)

(в формуле (III) значения R1, Y, m и А такие же, как и в формуле (Ib)).
10. Способ получения производного азола по п.1, где R2 обозначает COOR3, включающий стадию этерификации карбоксильной группы в соединении общей формулы (XII), и стадию раскрытия цикла сложноэфирного соединения общей формулы (XI), которое получают на вышеуказанной стадии этерификации с помощью галогеноводородной кислоты


(в формулах (XI) и (XII) значения Y, m и А такие же, как и в формуле (I), n равно от 1 до 6, и в формуле (XI) R3 обозначает C1-C6-алкильную группу, C2-C6-алкенильную группу или C2-C6-алкинильную группу).
11. Способ по п.10, включающий стадию окисления, которая представляет собой получение соединения карбоновой кислоты путем окисления гидроксиметильной группы в промежуточном соединении общей формулы (XIII)

(в формуле (XIII) значения Y, m, n и А такие же, как и в формуле (XII)).
12. Способ получения производного азола по п.1, где R2 обозначает COOR3, который включает стадию раскрытия цикла соединения лактона общей формулы (X), с помощью алкоголята металла формулы R3O-Ma+

(в формуле (X) значения R1, Y, m и А такие же, как и в формуле (I); R3 обозначает C1-C6-алкильную группу, C2-C6-алкенильную группу или C2-C6-алкинильную группу и Ма обозначает щелочной металл).
13. Способ получения производного азола по п.1, где R2 обозначает CONR3R4, который включает стадию раскрытия цикла соединения лактона общей формулы (X) с помощью амина формулы NHR3R4

(в формуле (X) значения R1, Y, m и А такие же, как и в формуле (I); каждый R3 и R4 обозначает атом водорода, С1-С6-алкильную группу, С2-С6-алкенильную группу или С2-С6-алкинильную группу).
14. Способ по п.12 или 13, включющий стадию конденсации, которая представляет собой получение соединения общей формулы (X), путем конденсации карбоновой кислоты общей формулы (Ib) с помощью конденсирующего агента

(в формуле (Ib) значения R1, Y, m и А такие же, как и в формуле (X)).
15. Производное азола общей формулы (Ib)

(в формуле (Ib) значения R1, Y, m и А такие же, как и в формуле (I)).
16. Химический агент, обладающий дезинфицирующей активностью в отношении вредных микроорганизмов, предназначенный для применения в сельском хозяйстве и садоводстве или для защиты промышленных материалов, который содержит производное азола по любому из пп.1-7 в качестве активного ингредиента.
17. Химический агент по п.16, предназначенный для применения в сельском хозяйстве и садоводстве для использования при обработке семян.
Текст
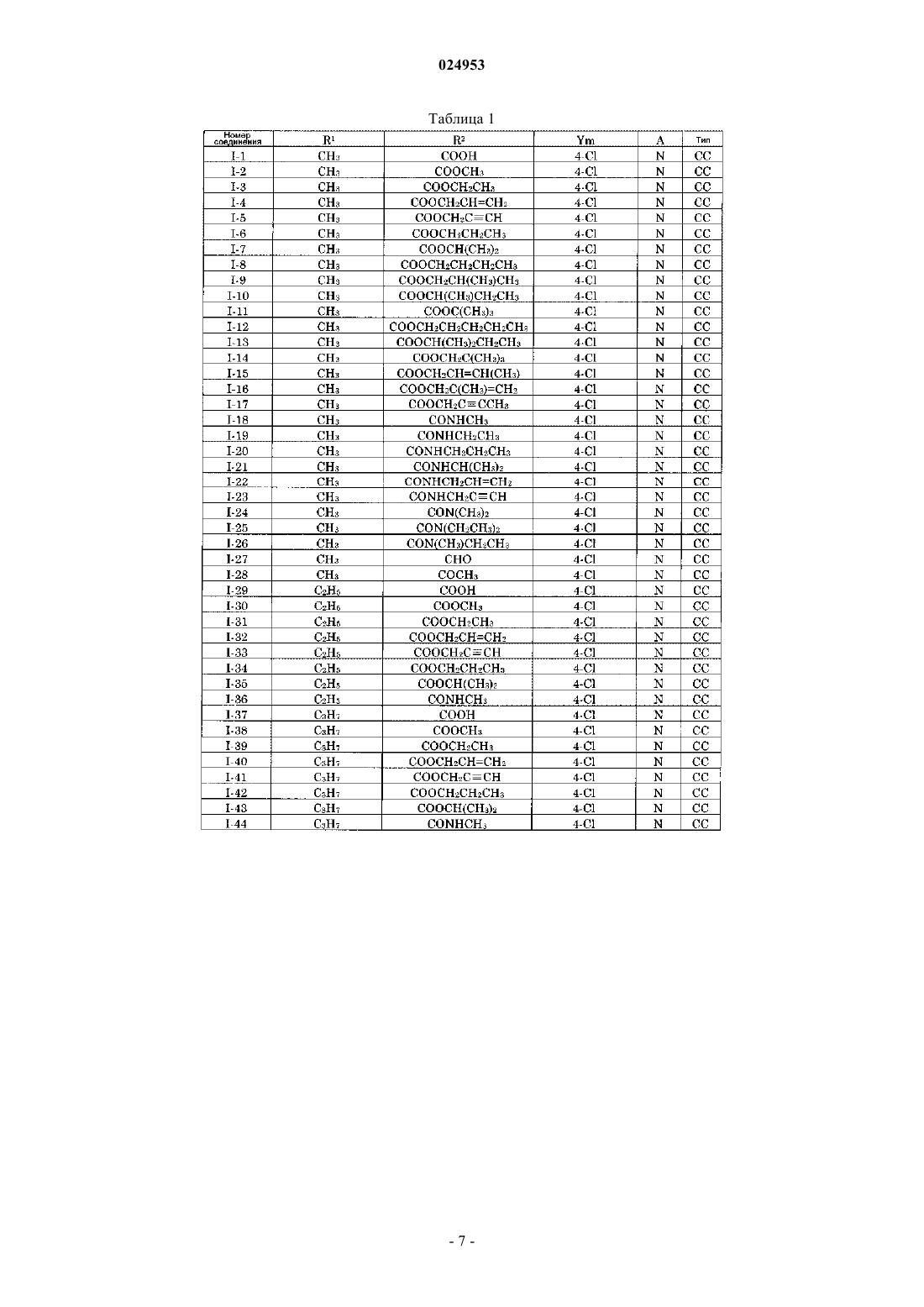
ПРОИЗВОДНОЕ АЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ХИМИЧЕСКИЙ АГЕНТ,ПРЕДНАЗНАЧЕННЫЙ ДЛЯ ПРИМЕНЕНИЯ В СЕЛЬСКОМ ХОЗЯЙСТВЕ И САДОВОДСТВЕ, И АГЕНТ ДЛЯ ЗАЩИТЫ ПРОМЫШЛЕННЫХ МАТЕРИАЛОВ Предлагается производное азола, обладающее лучшей способностью бороться с заболеваниями,которое содержится в качестве активного ингредиента в химических агентах, предназначенных для применения в сельском хозяйстве и садоводстве. Производное азола по настоящему изобретению представлено следующей общей формулой (I): где R1 обозначает незамещенную или замещенную атомом галогена C1-C6-алкильную группу; R2 обозначает -С(О)OR3 и -C(O)NR3R4; каждый R3 и R4 обозначает атом водорода, C1-C6-алкильную группу, C2-C6-алкенильную группу или C2-C6-алкинильную группу; Y обозначает атом галогена; m равно от 0 до 5 и А обозначает атом азота. Область техники, к которой относится изобретение Настоящее изобретение относится к новому производному азола. Оно также относится к химическому агенту, предназначенному для применения в сельском хозяйстве и садоводстве, и к агенту для защиты промышленных материалов, который содержит указанное производное азола в качестве активного ингредиента, и к способу получения указанного производного азола. Уровень техники Известно, что некоторые 2-замещенные производные 5-бензил-1-азолилметилциклопентанола обладают дезинфицирующей способностью (см., например, патентные документы 1 и 2). Кроме того, сообщалось, что некоторые соединения, включенные в 2-(галогенуглеводород)замещенные производные 5-бензил-1-азолилметилциклопентанола, демонстрируют противоэлептическую активность или обладают антифобическим седативным действием (см. Патентный документ 3). В патентном документе 3 конкретно не раскрываются химические агенты, предназначенные для применения в сельском хозяйстве и садоводстве, и агенты и соединения для защиты промышленных материалов, которые включены в объем настоящего изобретения. Перечень ссылок Патентная литература[PTL1] Опубликованная не рассмотренная патентная заявка Японии 1-93574[PTL2] Опубликованная не рассмотренная патентная заявка Японии 1-186871[PTL3] Опубликованная заявка на патент Германии 3902031[PTL4] Опубликованная не рассмотренная патентная заявка Японии 05-271197[PTL5] Опубликованная не рассмотренная патентная заявка Японии 01-301664 Сущность изобретения Техническая проблема Существует потребность в средстве борьбы с заболеваниями сельскохозяйственных или садовых культур, которое менее токсично для человека или животных и обладает улучшенной стабильностью при использовании, а также обладает лучшей способностью бороться с различными заболеваниями растений. Существует также потребность в регуляторе роста растений, который регулирует рост различных сельскохозяйственных культур и садовых растений и тем самым повышает их урожайность и качество, а также в агенте для защиты промышленных материалов, который защищает используемые в промышленности соединения от различных вредных микроорганизмов, разрушающих материалы. Объектами настоящего изобретения являются производное азола, которое удовлетворяет изложенным выше требованиям, способ его получения, его промежуточное соединение и химический агент,предназначенный для применения в сельском хозяйстве и садоводстве, и агент для защиты промышленных материалов, который содержит указанное производное азола. Решение проблемы Для достижения указанных выше целей авторы настоящего изобретения исследовали химическую структуру и физиологическую активность многих производных азола. В итоге, авторы настоящего изобретения обнаружили, что производные азола, представленные следующей общей формулой (I), обладают подходящей активностью, и, таким образом, осуществили настоящее изобретение. Настоящее изобретение, которое основывается на новых полученных данных, содержит следующие объекты изобретения: Производное азола по настоящему изобретению отличается тем, что оно имеет структуру, представленную следующей общей формулой (I): где R1 обозначает незамещенную или замещенную атомом галогена C1-C6-алкильную группу;m равно от 0 до 5. Производное азола по настоящему изобретению в указанной выше конфигурации оказывает благоприятное воздействие и демонстрирует подходящую дезинфицирующую способность по отношению к вызывающим заболевание микробам. В производном азола по настоящему изобретению в приведенной выше общей формуле (I) R2 предпочтительно обозначает COOR3, и R3 обозначает C1-C3-алкильную группу, С 2-С 3-алкенильную группу или С 2-С 3-алкинильную группу. Кроме того, предпочтительно, в соответствии с настоящим изобретением в производном азола согласно приведенной выше общей формуле (I) группа R2 обозначает CONR3R4, и каждый R3 и R4 незави-1 024953 симо обозначает атом водорода, C1-C3-алкильную группу, С 2-С 3-алкенильную группу или C2-C3 алкинильную группу. Кроме того, предпочтительно, в соответствии с настоящим изобретением, в производном азола согласно приведенной выше общей формуле (I) группа R1 обозначает C1-C6-алкильную группу, замещенную атомом галогена. Кроме того, предпочтительно, в соответствии с настоящим изобретением, в производном азола согласно приведенной выше общей формуле (I) группа R1 обозначает незамещенную алкильную группу. Кроме того, в соответствии с настоящим изобретением, в производном азола согласно приведенной выше общей формуле (I) количество атомов углерода в группе R1 предпочтительно равно от 1 до 4. В приведенной выше общей формуле (I) предпочтительно Y обозначает атом галогена и m равно 1. Способ получения производного азола по настоящему изобретению, где R2 обозначает COOR3, отличается тем, что он включает стадию этерификации, которая представляет собой этерификацию карбоксильной группы, содержащейся в соединении карбоновой кислоты следующей общей формулы (Ib). Таким образом, стадия этерификации представляет собой стадию превращения карбоксильной группы в соединении карбоновой кислоты, в группу COOR3. Поскольку R2 обозначает карбоксильную группу, когда R3 обозначает атом водорода, то в данном случае R3 не является атомом водорода.(в формуле (Ib) значения R1, Y, m и А такие же, как и в формуле (I), и R3 обозначает C1-C6 алкильную группу, C2-C6-алкенильную группу или С 2-С 6-алкинильную группу). Вышеуказанный способ получения производного азола предпочтительно включает стадию окисления с целью получения соединения карбоновой кислоты путем окисления гидроксиметильной группы в промежуточном соединении общей формулы (III).(в формуле (III) значения R1, Y, m и А такие же, как и в формуле (Ib).) Способ получения производного азола по настоящему изобретению, где R2 обозначает COOR3, отличается тем, что он включает стадию этерификации карбоксильной группы в соединении карбоновой кислоты следующей общей формулы (XII) и стадию раскрытия цикла сложноэфирного соединения общей формулы (XI), которое получают на стадии этерификации, с помощью галогеноводородной кислоты. Так, стадия этерификации представляет собой стадию превращения карбоксильной группы в соединении карбоновой кислоты, в группу COOR3. Поскольку R2 обозначает карбоксильную группу, когда R3 обозначает атом водорода, то в данном случае R3 не является атомом водорода.(в формулах (XI) и (XII) значения Y, m и А такие же, как и в формуле (I). n равно от 1 до 6. В формуле (XI) R3 обозначает C1-C6-алкильную группу, С 2-С 6-алкенильную группу или С 2-С 6-алкинильную группу). Способ получения вышеуказанного производного азола предпочтительно включает стадию окисления с целью получения соединения карбоновой кислоты следующей общей формулы (XII), путем окисления гидроксиметильной группы, которая содержится в промежуточном соединении следующей общей формулы (XIII).(в формуле (XIII) значения Y, m, n и А такие же, как и Y, m, n и А в формуле (XII. Способ получения производного азола по настоящему изобретению, где R2 обозначает COOR3, отличается тем, что он включает стадию раскрытия цикла соединения лактона, представленного следующей общей формулой (X), с помощью алкоголята металла, представленного формулой R3O-Ma+.(в формуле (X) значения R1, Y, m и А такие же, как и R1, Y, m и А в формуле (I), R3 обозначает C1C6-алкильную группу, С 2-С 6-алкенильную группу или С 2-С 6-алкинильную группу и Ма обозначает щелочной металл). Способ получения производного азола по настоящему изобретению, где R2 обозначает CONR3R4,отличается тем, что он включает стадию раскрытия цикла соединения лактона следующей общей формулы (X), с помощью соединения амина, представленного формулой NHR3R4.(в формуле (X) значения R1, Y, m и А такие же, как и в формуле (I). Каждый R3 и R4 обозначает атом водорода, C1-C6-алкильную группу, С 2-С 6-алкенильную группу или С 2-С 6-алкинильную группу.) Вышеуказанный способ получения производного азола предпочтительно включает стадию конденсации с целью получения соединения, представленного выше общей формулой (X), путем конденсации соединения карбоновой кислоты общей формулы (Ib), с помощью конденсирующего агента.(в формуле (Ib) значения R1, Y, m и А такие же, как и в формуле (X. Кроме того, производное азола представленное следующей общей формулой (Ib), также входит в объем настоящего изобретения.(в формуле (Ib) значения R1, Y, m и А такие же, как и в формуле (I. В объем настоящего изобретения также входят химические агенты, предназначенные для применения в сельском хозяйстве и садоводстве, или агенты для защиты промышленных материалов, которые содержат производное азола по настоящему изобретению в качестве активного ингредиента. Химические агенты, предназначенные для применения в сельском хозяйстве и садоводстве, могут использоваться для обработки семян. Кроме того, обработка семян с помощью химического агента, предназначенного для применения в сельском хозяйстве и садоводстве, также входит в объем настоящего изобретения. Если не указано иное, те же самые обозначения назначаются, соответственно, тем же самым функциональным группам (или атомам) в соответствующих общих формулах, приведенных в данном описании и в др., и подробное их описание здесь опускается. Например, если не указано иное, группа R1 в общей формуле (I) идентична группе R1 в другой общей формуле. То же самое, помимо R1, относится и к другим функциональным группам (или атомам). Преимущества настоящего изобретения Производное азола по настоящему изобретению обладает подходящей дезинфицирующей способностью по отношению ко многим микроорганизмам, вызывающим заболевания растений. Так химические агенты, предназначенные для применения в сельском хозяйстве и садоводстве, которые содержат производное азола по настоящему изобретению в качестве активного ингредиента, оказывают подходящее действие, которое позволяет с высокой эффективностью бороться с широким кругом заболеваний растений. Химические агенты, предназначенные для применения в сельском хозяйстве и садоводстве, которые содержат производное азола по настоящему изобретению в качестве активного ингредиента, оказывают подходящее действие, которое заключается в регулировании роста различных сельскохозяйственных культур и садовых растений, и тем самым повышают урожайность и качество сельскохозяйственных культур и садовых растений. Агенты для защиты промышленных материалов, которые содержат производное азола по настоящему изобретению в качестве активного ингредиента, оказывают дополнительное подходящее действие,которое заключается в эффективной защите используемых в промышленности веществ от вредных микроорганизмов, которые приводят к разрушению используемых в промышленности материалов. Краткое описание чертежей Фиг. 1 представляет собой схему, на которой кратко представлены первый и второй способы получения соединения (I) по настоящему изобретению. Далее будут описаны подходящие варианты осуществления настоящего изобретения. Приведенные ниже варианты осуществления настоящего изобретения являются некоторыми из типичных вариантов осуществления настоящего изобретения, и следует понимать, что они не ограничивают объем настоящего изобретения. 1. Производное азола Описывается производное азола по настоящему изобретению, общей формулы (I) (далее обозначают как соединение формулы (I. Соединение (I) содержит в позиции 2 циклопентанового цикла замещенную или незамещенную алкильную группу и функциональную группу, включающую карбонильную группу.(1) R1 Вначале будет рассмотрена замещенная или незамещенная алкильная группа (R1), присоединенная к позиции 2 циклопентанового цикла. Примеры R1 включают C1-C6-алкильные группы и C1-C6 галогеналкильные группы.C1-C6-алкильную группу специально не ограничивают, при условии, что она является алкильной группой, имеющей от 1 до 6 атомов углерода, однако предпочтительно она представляет собой С 3-С 4 алкильную группу. Типичные ее примеры включают метильную группу, этильную группу, (1 метил)этильную группу, н-пропильную группу, 1-метилпропильную группу, 2-метилпропильную группу,н-бутильную группу, 1-метилбутильную группу, 2-метилбутильную группу, 1-этилпропильную группу,1,1-диметилэтильную группу и т.п. В том случае, когда R1 обозначает С 1-С 6-алкильную группу, соединение (I) представляет собой производное 2-ацил-2-алкил-5-бензил-1-азолилметилциклопентанола.C1-C6-галогеналкильную группу специально не ограничивают, при условии, что она является галогеналкильной группой, имеющей от 1 до 6 атомов углерода, однако предпочтительно она представляет собой С 1-С 4-галогеналкильную группу. Типичными ее примерами являются галогензамещенные C1-C6 алкильные группы, такие как хлорметильная группа, дихлорметильная группа, трихлорметильная группа,2-хлорэтильная группа, 1-хлорэтильная группа, 2,2-дихлорэтильная группа, 1,2-дихлорэтильная группа,2,2,2-трихлорэтильная группа, 3-хлорпропильная группа, 2,3-дихлорпропильная группа, 1-хлор-1 метилэтильная группа, 2-хлор-1-метилэтильная группа, 2-хлорпропильная группа, 4-хлорбутильная группа, 5-хлорпентильная группа, фторметильная группа, дифторметильная группа, трифторметильная группа, 2-фторэтильная группа, 1-фторэтильная группа, 2,2-дифторэтильная группа, 1,2-дифторэтильная группа, 2,2,2-трифторэтильная группа, 3-фторпропильная группа, 2,3-дифторпропильная группа, 1-фтор 1-метилэтильная группа, 2-фтор-1-метилэтильная группа, 2-фторпропильная группа, 3,3,3 трифторпропильная группа, 2,2,3,3-тетрафторпропильная группа, 2,2,3,3,3-пентафторпропильная группа,4-фторбутильная группа, 5-фторпентильная группа, бромметильная группа, дибромметильная группа,трибромметильная группа, 2-бромэтильная группа, 1-бромэтильная группа, 2,2-дибромэтильная группа,1,2-дибромэтильная группа, 2,2,2-трибромэтильная группа, 3-бромпропильная группа, 2,3 дибромпропильная группа, 1-бром-1-метилэтильная группа, 2-бром-1-метилэтильная группа, 2-4 024953 бромпропильная группа, 4-бромбутильная группа, 5-бромпентильная группа, иодметильная группа, дииодметильная группа, 2-иодэтильная группа, 1-иодэтильная группа, 2,2-дииодэтильная группа, 1,2 дииодэтильная группа, 2,2,2-трииодэтильная группа, 3-иодпропильная группа, 2,3-дииодпропильная группа, 1-иод-1-метилэтильная группа, 2-иод-1-метилэтильная группа, 2-иодпропильная группа, 4 иодбутильная группа и т.п. Когда R1 обозначает C1-C6-галогеналкильную группу, соединение (I) представляет собой производное 2-ацил-2-галогеналкил-5-бензил-1-азолилметилциклопентанола.(2) R2 Далее будет рассмотрена функциональная группа, содержащая карбонильную группу (R2), которая присоединена к позиции 2 циклопентанового цикла. Атом углерода карбонильной группы в R2 присоединен к атому углерода в циклопентановом цикле, замещенном группой R1, а также к R3 OR3 или NR3R4. Так, атом углерода карбонильной группы в R2 присоединен к атому углерода, замещенному группой R1. Таким образом, карбонильная группа в R2 находится в позиции, которая наиболее близко расположена к циклопентановому циклу в функциональной группе, обозначенной как R2. Примеры R3 и R4 включают атом водорода, C1-C6-алкильные группы, C2-C6-алкенильные группы,C2-C6-алкинильные группы Когда атом углерода карбонильной группы в R2 присоединен к NR3R4, R3 иR4 могут быть одинаковыми или отличаться друг от друга. Типичные примеры групп, когда атом углерода карбонильной группы в R2 присоединен к R3, OR3 или NR3R4, приведены ниже: Когда атом углерода карбонильной группы в R2 присоединен к атому водорода (когда R3 обозначает атом водорода), R2 обозначает формильную группу (R2=-CHO). Когда атом углерода карбонильной группы в R2 присоединен к OR3 и R3 обозначает атом водорода,2R обозначает карбоксильную группу (R2=-COOH). Когда атом углерода карбонильной группы в R2 присоединен к R3 и R3 отличен от атома водорода,2R представляет собой, например, ацетильную группу, пропионильную группу, бутирильную группу,изобутирильную группу, пентаноильную группу, гексаноильную группу, гептаноильную группу или т.д. Когда атом углерода карбонильной группы в R2 присоединен к OR3 и R3 отличен от атома водорода,2R представляет собой, например, метоксикарбонильную группу, этоксикарбонильную группу, пропоксикарбонильную группу, бутоксикарбонильную группу, пентоксикарбонильную группу, гексаноксикарбонильную группу или т.п. Когда атом углерода карбонильной группы в R2 присоединен к NR3R4, R2 обозначает, например,диметиламидогруппу, этилметиламидогруппу, метилпропиламидогруппу, бутилметиламидогруппу, метилпентиламидогруппу, гексилметиламидогруппу, диэтиламидогруппу, этилпропиламидогруппу, бутилэтиламидогруппу, этилпентиламидогруппу, этилгексиламидогруппу, дипропиламидогруппу, бутилпропиламидогруппу, пентилпропиламидогруппу, гексилпропиламидогруппу, дибутиламидогруппу, бутилпентиламидогруппу, бутилгексиламидо группу, дипропиламидогруппу, гексилпропиламидогруппу, дигексиламидогруппу, метиламидогруппу, этиламидогруппу, пропиламидогруппу, бутиламидогруппу, пентиламидогруппу, гексиламидогруппу или т.п.(3) Ym Примеры Y включают атомы галогенов, такие как атом хлора, атом фтора, атом брома, атом иода и т.п.m обозначает целое число от 0 до 5. Когда m равно 2 или более, каждый Y может быть одинаковым или отличаться друг от друга. В настоящем изобретении m предпочтительно равно от 0 до 3, более предпочтительно от 0 до 2. В частности, наиболее предпочтительно m равно 1.(5) Стереоизомеры Соединение (I) имеет стереоизомеры, представленные следующими общими формулами (СС), (ТТ),(СТ) и (ТС). Соединение (I) может быть либо одним из изомеров, либо смесью изомеров. В следующих общих формулах относительная конфигурация, в которой 1-гидроксильная группа и 2-алкильная группа(R1) имеют цис-ориентацию, и 1-гидроксильная группа и 5-бензильная группа имеют цис-ориентацию,обозначена как (СС). В качестве альтернативы, относительная конфигурация, в которой 1-гидроксильная группа и 2-алкильная группа (R1) имеют трансориентацию, и 1-гидроксильная группа и 5-бензильная группа имеют транс-ориентацию, обозначена как (ТТ). В качестве альтернативы, относительная конфигурация, в которой 1-гидроксильная группа и 2-алкильная группа (R1) имеют цис-ориентацию, и 1 гидроксильная группа и 5-бензильная группа имеют транс-ориентацию, обозначена как (СТ). Наконец, в качестве альтернативы, относительная конфигурация, в которой 1-гидроксильная группа и 2-алкильная группа (R1) имеют трансориентацию, и 1-гидроксильная группа и 5-бензильная группа имеют цисориентацию, обозначена как (ТС). В данном описании или в других атом углерода, связанный с гидроксильной группой, находится в позиции 1 циклопентанового цикла.(6) Типичные примеры Типичные примеры соединения (I), которое имеет различные R1, R2, Ym, А и приведенные выше типы изомеров, включает следующие соединения, приведенные в табл. 1-12. В приведенных ниже таблицах: 1) Столбец R1R1 приведен как замещающая группа. Каждая приведенная в таблице замещающая группа связана с циклопентановым циклом соединения (I) посредством атома углерода левого конца группы R1, где отсутствует атом водорода. 2) Столбец R2R2 приведен как замещающая группа. Каждая приведенная в таблице замещающая группа связана с циклопентановым циклом соединения (I) посредством атома углерода, который соединен с атомом кислорода группы R2. 2) Столбец Ym"-" указывает на то, что соединение не содержит заместителей (m=0). Цифра перед "-" (дефис) указывает на место присоединения заместителя в фенильном цикле, когда фенильный цикл имеет заместитель, по отношению к позиции (позиции 1), связанной с атомом углерода, который присоединен к циклопентановому циклу. 2. Способ получения производного азола Ниже приведен способ получения соединения (I). Соединение (I) можно получить по первому или второму способу получения, которые описаны ниже. В данном описании растворитель, основание, кислота и т.п., которые используют на стадиях указанного способа получения, будут описаны перед конкретным описанием способов получения. Растворитель, основание, кислота и т.п., которые используют на стадиях способов получения по настоящему изобретению, могут быть теми, которые приведены ниже,если не указано иное.(1) Растворитель Используемый растворитель специально не ограничивается, при условии, что он инертен в условиях проведения реакции, и примеры растворителя обычно включают простые эфиры, такие как диэтиловый эфир, тетрагидрофуран (далее в настоящем описании обозначают как ТГФ) и диоксан; спирты, такие как метанол, этанол и изопропанол; ароматические углеводороды, такие как бензол, толуол и ксилол; алифатические углеводороды, такие как петролейный эфир, гексан и метилциклогексан; амиды, такие как N,N-диметилформамид (далее в настоящем описании обозначают ДМФА), N,N-диметилацетамид иN-метил-2-пирролидинон; и т.п. Кроме того, в качестве растворителя могут использоваться, например,вода, ацетонитрил, этилацетат, уксусный ангидрид, уксусная кислота, пиридин или диметилсульфоксид. Указанные растворители могут использоваться в виде смеси двух или большего количества растворителей. Растворитель может представлять собой композицию растворителей, содержащую растворители,которые не образуют гомогенную фазу. В таком случае в реакционную систему может быть добавлен катализатор фазового переноса, такой как обычная четвертичная аммониевая соль или краун-эфир.(2) Основание или кислота В вышеуказанный растворитель может быть добавлено основание или кислота. Используемое основание специально не ограничивается. Примеры оснований включают карбонаты щелочных металлов, такие как карбонат натрия, бикарбонат натрия, карбонат калия и бикарбонат калия; карбонаты щелочно-земельных металлов, такие как карбонат кальция и карбонат бария; гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия; щелочные металлы, такие как литий,натрий и калий; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия и третбутоксид калия; гидриды щелочных металлов, такие как гидрид натрия, гидрид калия и гидрид лития; органические соединения щелочных металлов, такие как н-бутиллитий; щелочные металлы, такие как натрий, калий и литий; амиды щелочных металлов, такие как диизопропиламид лития; органические амины, такие как триэтиламин, пиридин, 4-диметиламинопиридин, N,N-диметиланилин и 1,8 диазабицикло-7-[5.4.0]ундецен и т.п. Используемая кислота специально не ограничивается. Примеры кислот включают неорганические кислоты, такие как хлористо-водородная кислота, бромисто-водородная кислота, иодисто-водородная кислота и серная кислота; органические кислоты, такие как муравьиная кислота, уксусная кислота, масляная кислота, трифторуксусная кислота и п-толуолсульфоновая кислота; кислоты Льюиса, такие как хлорид лития, бромид лития, хлорид родия, хлорид алюминия и трифторид бора; и т.п.(3) Первый способ получения соединения (I) Первый способ получения соединения (I) будет описан ниже со ссылкой на фигуру 1. Фиг. 1 представляет собой схему, на которой представлены первый и второй способы получения соединения (I). В частности, в соответствии с первым способом получения соединения (I), можно селективно получить,среди прочих соединений (I), соединение, представленное следующей общей формулой (Ia) (далее в настоящем описании обозначают как "соединение (Ia)"), соединение, представленное следующей общей формулой (Ib) (далее в настоящем описании обозначают как "соединение (Ib)"), и соединение, представленное следующей общей формулой (Ic) (далее в настоящем описании обозначают как "соединение(Ic)"). Соединение (Ia) представляет собой соединение (I), в котором R2 обозначает COOR3. В качестве альтернативы, соединение (Ib) представляет собой соединение (I), в котором R2 обозначает СООН. В качестве альтернативы, ссоединение (Ic) представляет собой соединение (I), в котором R2 обозначает В указанных формулах R1, R3, R4, Y, m и А такие же, что и приведенные выше. Как показано на фиг. 1, первый способ получения соединения (I) включает стадии 1 А, 1 В, 1 С и 1D. Каждая стадия может дополнительно включать дополнительные стадии. Далее в настоящем описании будет подробно рассмотрена каждая стадия и каждая дополнительная стадия первого способа получения.(3-1) Стадия 1 А Вначале будет кратко рассмотрена стадия 1 А. Стадия 1 А представляет собой стадию получения соединения, представленного следующей общей формулой (VI) (далее в настоящем описании обозначают как "соединение (VI)"). Как показано на фиг. 1, стадия 1 А включает стадии 1 А 1, 1 А 2 и 1 А 3. Стадия 1 А включает стадию гидроксиметилирования с целью гидроксиметилирования кетоэфирного соединения, представленного следующей общей формулой (IX) (далее в настоящем описании обозначают как "соединение (IX)"), стадию введения защитной группы, которая защищает гидроксильную группу полученного соединения, содержащего гидроксиметильную группу (соединения, представленного следующей общей формулой (VIII), и далее в настоящем описании обозначаемого как "соединение(VIII)"), и стадию удаления сложноэфирной группы карбоновой кислоты с получением карбонильного соединения (VI) путем гидролиза и декарбоксилирования соединения, в которое была введена защитная группа (соединения, представленного следующей общей формулой (VII), и далее в настоящем описании обозначаемого как "соединение (VII)") (см. следующую формулу реакции (1.R5 обозначает С 1-С 4-алкильную группу. Типичные примеры алкильной группы R5 включают метильную группу, этильную группу, н-пропильную группу, 1-метилэтильную группу, 2-метилпропильную группу, н-бутильную группу, 1,1-диметилэтильную группу и т.п.G, который обозначает защитную группу, специально не ограничивается, и его примеры включают алкоксиметильные группы, такие как метоксиметильная группа и этоксиметильная группа; низшие алкильные группы, такие как трет-бутильная группа и метильная группа; замещенные и незамещенные бензильные группы и т.п.(3-1-1) Стадия 1 А 1 (стадия гидроксиметилирования) На стадии гидроксиметилирования стадии 1 А преимущественно используют способ взаимодействия соединения (IX) с формальдегидом в присутствии основания. Количество используемого формальдегида по отношению к 1 моль соединения (IX) обычно составляет от 0,5 до 20 моль, предпочтительно от 0,8 до 10 моль. Примеры оснований включают, но этим не ограничиваясь, карбонаты щелочных металлов, такие как карбонат натрия и карбонат калия; гидроксиды щелочных металлов, такие как гидроксид натрия; органические основания, такие как триэтиламин, и т.п. Количество используемого основания по отношению к 1 моль соединения (IX) обычно составляет от 0,1 до 10 моль и предпочтительно от 0,2 до 5 моль. Температура реакции обычно предпочтительно составляет от 0 до 250 С, более предпочтительно от 0 до 100 С. Время реакции обычно предпочтительно составляет от 0,1 ч до нескольких дней, более предпочтительно от 0,5 ч до 2 дней. Используемое соединение (IX) можно получить известным способом (по методу, описанному в патентном документе 1).(3-1-2) Стадия 1 А 2 (стадия введения защитной группы) Далее будет описана стадия (стадия 1 А 2) получения соединения (VII) путем введения защитной группы в гидроксильную группу соединения (VIII), полученного на стадии 1 А. Защитная группа, которая защищает гидроксильную группу, специально не ограничивается, и ее примеры, которые преимущественно используются, включают алкоксиметильные группы, такие как метоксиметильная группа и этоксиметильная группа, и низшую алкильную группу, такую как третбутильная группа. Указанные защитные группы вводят в условиях кислотного катализа. Тем не менее,(а) для введения алкоксиметильной группы преимущественно защищают гидроксильную группу соединения (VIII) путем ацетального обмена, используя диалкилацеталь формальдегида. В качестве альтерна- 20024953 тивы, (b) для введения трет-бутильной группы предпочтительно используют способ присоединения изобутена к гидроксильной группе соединений (VIII). Вначале будет рассмотрен случай (а). Примеры кислот включают неорганические кислоты, такие как хлористо-водородная кислота, фосфорная кислота (включая соединения, образующие кислоту при контакте со спиртом или водой, такие как пентоксид дифосфора) и серная кислота; и органические кислоты, такие как п-толуолсульфоновая кислота. Предпочтительно, используют диалкилацеталь формальдегида в растворителе или без растворителя в присутствии кислоты. В частности, более предпочтительно добавляют соединение, которое способно удалять образующийся спирт, такое как пентоксид дифосфора. Количество используемого диалкилацеталя формальдегида по отношению к 1 моль соединения(VIII) обычно составляет от 0,5 до 50 моль и предпочтительно от 0,8 до 10 моль. Количество используемой кислоты по отношению к 1 моль соединения (VIII) обычно составляет от 0,01 до 10 моль и предпочтительно от 0,05 до 5 моль. Температура реакции обычно предпочтительно составляет от 0 до 250 С и более предпочтительно от 0 до 150 С. Время реакции обычно предпочтительно составляет от 0,1 ч до нескольких дней, более предпочтительно от 0,5 ч до 2 дней. В случае (b) реакцию предпочтительно проводят с изобутеном в растворителе в присутствии неорганической кислоты, такой как хлористо-водородная кислота, фосфорная кислота или серная кислота,или органическая кислота, такая как п-толуолсульфоновая кислота или трифторуксусная кислота. Количество используемого изобутена по отношению к 1 моль соединения (VIII) обычно составляет от 0,5 до 100 моль и предпочтительно от 0,8 до 20 моль. Количество используемой кислоты по отношению к 1 моль соединения (VIII) обычно составляет от 0,01 до 10 моль и предпочтительно от 0,05 до 5 моль. Температура реакции обычно предпочтительно составляет от 0 до 200 С и более предпочтительно от 0 до 100 С. Время реакции обычно предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней.(3-1-3) Стадия 1 А 3 (стадия удаления сложного эфира карбоновой кислоты) Далее будет описана стадия (стадия 1 А 3) получения соединения (VI) из соединения (VII) на стадии 1 А. Реакцию предпочтительно проводят в растворителе в присутствии основания. В качестве основания обычно используют основание щелочного металла, такое как гидроксид натрия или гидроксид калия. Количество используемого основания по отношению к 1 моль соединения (VII) обычно составляет от 0,1 до 50 моль и предпочтительно от 0,2 до 20 моль. В качестве растворителя обычно используют воду, смесь воды и спирта и т.п. или смесь растворителей, которые не образуют гомогенную фазу (таких как вода и толуол; в таком случае в реакционную систему предпочтительно добавляют катализатор фазового переноса, такой как обычная четвертичная аммониевая соль). Температура реакции обычно предпочтительно составляет от 0 С до температуры кипения с обратным холодильником и более предпочтительно от комнатной температуры до температуры кипения с обратным холодильником. Время реакции обычно предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 до 24 ч.(3-2) Стадии 1 В Подробно будет рассмотрена стадия 1 В первого способа получения. Как показано на фиг. 1, стадия 1 В представляет собой стадию получения соединения, представленного общей формулой (III) (далее в настоящем описании обозначают как "соединение (III)"). Стадия 1 В включает стадии 1 В 1, 1 В 2 и 1 В 3. Как дополнительно указано на фиг. 1, стадия 1 В 2 включает два пути: стадию 1 В 2 а и 1 В 2b. Далее в настоящем описании получение соединения (III) по стадиям 1 В 1, 1 В 2 а и дополнительно 1 В 3 будет в основном описано как стадия 1 В, и случай получения по стадии 1 В 2b будет также рассмотрен как часть описания стадии 1 В 2. Соединение (III) можно получить, например, в соответствии с патентным документом 4. Стадия 1 В включает стадию превращения оксирана путем превращения карбонильного соединения,представленного следующей общей формулой (VI) (далее в настоящем описании обозначают как "соединение (VI)"), в оксирановое производное, стадию образования соединения азола путем взаимодействия полученного оксиранового производного (представленного следующей общей формулой (V); далее в настоящем описании обозначают как "соединение (V)") с 1,2,4-триазолом или имидазольным соединением, представленным следующей общей формулой (II) (далее в настоящем описании обозначают как "соединение (II)"), и стадию удаления защитной группы, представляющую собой стадию удаления защитной группы из полученного соединения азола (представленного следующей общей формулой (IV); далее в настоящем описании обозначают как "соединение (IV)") (см. следующую формулу реакции (2. В указанных формулах Y, m, A, R1 и G такие же, что и приведенные выше. М обозначает атом водорода или щелочной металл.(3-2-1) Стадия 1 В 1 (стадия превращения в оксиран) Будет рассмотрена стадия (стадия 1 В 1) получения соединения (V) путем превращения соединения(VI) в оксиран на стадии 1 В. Вначале в качестве первого синтетического способа, пригодного для получения соединения (V),будет рассмотрен способ взаимодействия соединения (VI) с илидом серы (например, метилидом сульфония, таким как метилид диметилсульфония, или метилидом сульфоксония, таким как метилид диметилсульфоксония) в растворителе. Используемый метилид сульфония или метилид сульфоксония может быть получен путем взаимодействия соли сульфония (например, иодида триметилсульфония или бромида триметилсульфония) или соли сульфоксония (например, иодида триметилсульфоксония или бромида триметилсульфоксония) с основанием в растворителе. Количество метилида сульфония или метилида сульфоксония по отношению к 1 моль соединения(VI) предпочтительно составляет от 0,5 до 5 моль и более предпочтительно от 0,8 до 2 моль. Используемый растворитель специально не ограничивается. Примеры растворителей включают амиды, такие как диметилсульфоксид, N-метилпирролидон и N,N-диметилформамид; простые эфиры,такие как тетрагидрофуран и диоксан; их смеси и т.п. Основания, которые используют для получения метилида сульфония или метилида сульфоксония,специально не ограничиваются. Примеры оснований включают гидрид металла, такой как гидрид натрия; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, трет-бутоксид натрия и трет-бутоксид калия; и т.п. Температуру реакции и время реакции можно определить произвольно, например, в соответствии с используемыми типами растворителя, соединения (VI), соли сульфония, соли сульфоксония и основания. Температура реакции предпочтительно составляет от -100 до 200 С и более предпочтительно от -50 до 150 С. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней. Ниже будет описан способ взаимодействия соединения (VI) с дииодидом самария и дииодметаном в растворителе и способ обработки полученной смеси основанием в качестве второго синтетического способа получения соединения (V). Используемое основание специально не ограничивается. Может использоваться, например, гидроксид натрия. Используемый дииодид самария может быть получен по реакции металлического самария с 1,2-дииодэтаном или дииодметаном в безводном растворителе. Используемый растворитель специально не ограничивается, и его примеры включают простые эфиры, такие как тетрагидрофуран и т.д. Количество используемого основания по отношению к 1 моль соединения (VI) специально не ограничивается, но предпочтительно составляет от 0,5 до 10 моль и более предпочтительно от 0,8 до 6 моль. В случае обработки основанием реакционная система может не быть обезвоженной системой, и, таким образом, может быть использован водный раствор гидроксида натрия или т.п. Температуру реакции и время реакции можно определить произвольно, например, в соответствии с используемыми типами растворителя, соединения (VI), основания и т.п. Температура реакции предпочтительно составляет от -100 до 150 С и более предпочтительно от -50 до 100 С. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней.(3-2-2) Стадия 1 В 2 (стадия образования азола) Далее будет описана стадия (стадия 1 В 2) получения соединения (IV) путем превращения соединения (VI) в производное азола с использованием соединения (II) на стадии 1 В. Как указано выше, стадия 1 В 2 включает два пути: стадии 1 В 2 а и 1 В 2b. Ниже вначале будет рассмотрена стадия 1 В 2 а (стадия 1 В 2 а). На стадии 1 В 2 а соединение (IV) получают путем смешивания соединения (V) с соединением (II) в растворителе. В частности, получают соединение (IV), поскольку образуется связь углерод-азот между атомом углерода, составляющим оксирановый цикл соединения (V), и атомом азота в 1,2,4-триазоле или имидазоле. Используемый растворитель специально не ограничивается, и его примеры включают амиды, такие как N-метилпирролидон и N,N-диметилформамид и т.п. Количество используемого соединения (II) по отношению к 1 моль соединения (V) обычно предпочтительно составляет от 0,5 до 10 моль и более предпочтительно от 0,8 до 5 моль. В случае необходимости можно добавить основание. Количество используемого основания по отношению к 1 моль соединения (II) обычно предпочтительно составляет от 0 до 5 моль (0 не включен) и более предпочтительно от 0,5 до 2 моль. Температуру реакции можно определить произвольно в зависимости от используемого растворителя, основания и т.п. Температура реакции предпочтительно составляет от 0 до 250 С и более предпочтительно от 10 до 150 С. Время реакции можно определить произвольно в зависимости от используемого растворителя, основания и т.п. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней.(Стадия 1 В 2b) Далее описывается вариант для соединения (IV), которое получают в соответствии со стадией 1 В 2b. Как указано выше, соединение (IV) можно получить в несколько стадий по реакции полученного соединения (V) с соединением (II). Тем не менее, стадия превращения оксирана первого способа синтеза, если ее проводить отдельно, может привести к образованию оксетанового производного в качестве побочного продукта и к снижению выхода. С целью предотвращения уменьшения выхода, соединение (V) предпочтительно превращают в производное азола, что является обычной процедурой (см. приведенную ниже формулу реакции (3. Формула реакции (3)- 23024953 В указанных формулах Y, m, A, R1, G и М такие же, что и приведенные выше. В данном случае, во-первых, соединение (VI) и соединение (II) растворяют в содержащем амидную связь полярном растворителе, диметилсульфоксиде или смешанном растворителе из полярного растворителя и спирта. Затем попеременно с основанием добавляют соль триметилсульфония или соль триметилсульфоксония, и проводят получение азола, и, таким образом, в системе образуется метилид сульфония, такой как метилид диметилсульфония, или метилид сульфоксония, такой как метилид диметилсульфоксония, и соединение (V). Используемый растворитель специально не ограничивается. Примеры пригодных растворителей включают содержащие амидную связь полярные растворители, такие как N-метилпирролидон и N,Nдиметилформамид, диметилсульфоксид, смешанные растворители из полярного растворителя и спирта и т.п. В качестве спирта можно использовать трет-бутанол. Основание, используемое для генерирования метилида сульфония или метилида сульфоксония,специально не ограничивается. Примеры оснований включают гидриды металлов, такие как гидрид натрия, алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, трет-бутоксид натрия,трет-бутоксид калия и т.п. Могут также использоваться соли щелочных металлов 1,2,4-триазола или имидазола. Температуру реакции можно определить произвольно в зависимости от типа используемого растворителя, соединения (VI), соли сульфония и соли сульфоксония, основания и т.п. Температура реакции предпочтительно составляет от -100 до 250 С и более предпочтительно от -50 до 200 С. Время реакции можно определить произвольно в зависимости от типа используемого растворителя, соединения (VI), соли сульфония и соли сульфоксония, основания и т.п. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней. Частота попеременного добавления галогенида триметилсульфония или галогенида триметилсульфоксония с основанием специально не ограничивается, при условии, что цель достигается. Частота обычно предпочтительно составляет, например, от 2 до 20 мин, и более предпочтительно от 3 до 15 мин. Общее количество используемых солей триметилсульфония или солей триметилсульфоксония по отношению к 1 моль соединения (VI) предпочтительно составляет от 0,5 до 5 моль и более предпочтительно от 0,8 до 2 моль. Количество используемого соединения (II) по отношению к 1 моль соединения (VI) обычно предпочтительно составляет от 0,5 до 10 моль и более предпочтительно от 0,8 до 5 моль. Преимущественно используют соединение (II), в котором М обозначает щелочной металл. Подробные стадии способа получения производного азолилметилциклоалканола, с помощью которого проводят получение азола, по мере того как образуется оксирановое производное, описаны в Патентном документе 5.(3-2-3) Стадия 1 В 3 (удаление защитной группы) Далее будет описана стадия (стадия 1 В 3) получения соединения (III) путем удаления на стадии 1 В защиты у содержащего защитную группу соединения (IV). Условия, пригодные для удаления защитной группы, изменяются в зависимости от типа защитной группы. Например, если используют алкоксиметильную группу, такую как метоксиметильная группа или этоксиметильная группа, или низшую алкильную группу, такую как трет-бутильная группа или метильная группа, то удаление защитной группы преимущественно проводят в кислых условиях, например, в присутствии хлористоводородной кислоты или серной кислоты. Пригодные для использования кислоты включают галогеноводороды, такие как хлористый водород,неорганические кислоты, такие как серная кислота и т.п. Количество используемой кислоты специально не ограничивается, но обычно составляет от 0,5 до 100 моль и более предпочтительно от 0,8 до 2 0 моль по отношению к 1 моль соединения (IV). Температура реакции обычно предпочтительно составляет от 0 до 200 С и более предпочтительно от комнатной температуры до 100 С. Время реакции обычно предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней.(3-3) Стадия 1 С Далее будет кратко описана стадия 1 С. Стадия 1 С представляет собой способ получения соединения (Ib) и соединения (Ia) среди производных азола по настоящему изобретению. Как показано на фиг. 1,стадия 1 С включает четыре промежуточные стадии (1 С 1, 1 С 2, 1 С 3 и 1 С 4). Более конкретно, на стадии 1 С, вначале на стадии 1 С 1 получают соединение (Ib). Возможны два пути: путь (путь 1), при котором соединение (1a) получают по стадиям 1 С 1-1 С 2, и путь (путь 2), при котором соединение (Ia) получают по стадиям 1 С 1-1 С 3 и дополнительно по стадии 1 С 4. Далее более подробно будут описаны пути 1 и 2. Путь 1. Путь 1 включает стадию получения соединения карбоновой кислоты (стадия окисления), которая представляет собой стадию получения соединения карбоновой кислоты путем замещения конкретной функциональной группы соединения (III) карбоксильной группой, и стадию этерификации с получением производного азола, представленного выше общей формулой (Ia), путем этерификации соединения карбоновой кислоты. В данном варианте осуществления настоящего изобретении в качестве примера будет описан случай, когда соединение, представленное следующей общей формулой (III), представляет собой соединение, содержащее гидроксиметильную группу в позиции 2 циклопентанового цикла, и стадия получения соединения карбоновой кислоты представляет собой стадию окисления с образованием карбоксильной группы путем окисления гидроксиметильной группы (см. формулу реакции (4. Формула реакции (4)(3-3-1) Стадия 1 С 1 (стадия окисления) Вначале более детально будет рассмотрена стадия (стадия 1 С 1) получения соединения (Ib) путем окисления соединения (III) на стадии 1 С. Способ окисления специально не ограничивается, и его примеры включают способ с использованием окислительного агента, такого как реагент Джонса (хромовая кислота - серная кислота), дихроматная соль, хлорхромат пиридиния, дихлорхромат пиридиния, или перманганат натрия, и применение реагента Джонса является предпочтительным. Количество используемого окислительного агента по отношению к 1 моль соединения (III) обычно составляет от 0,3 до 2 0 моль и предпочтительно от 0,5 до 10 моль. Растворитель может быть выбран произвольно в зависимости от используемого окислительного агента. В том случае, когда окислительным агентом является реагент Джонса, предпочтительно использовать в качестве растворителя смесь ацетона с водой. Температура реакции обычно предпочтительно составляет от -20 до 250 С и более предпочтительно от -10 до 100 С. Время реакции обычно предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней. (3-3-2) Стадия 1 С 2 (стадия этерификации) Далее будет описана стадия (стадия 1 С 2) получения соединения (Ia) путем этерификации соединения (Ib) на стадии 1 С. Способ этерификации соединения (Ib) специально не ограничивается, и преимущественно можно использовать (а) способ его взаимодействия с диазометаном или производным диазометана или (b) способ его взаимодействия с производным азодикарбоновой кислоты и фосфином с последующим взаимодействием полученного продукта со спиртом, обозначенным как R3OH. Вначале будет описан способ (а). Соединение (Ia) можно получить по реакции с диазометаном или TMS диазометаном в спиртовом растворителе. Предпочтительно в качестве реагента используют TMS диазометан. Количество используемого TMS диазометана по отношению к 1 моль соединения (Ib) обычно составляет от 0,5 до 20 моль и предпочтительно от 0,8 до 10 моль. Температуру реакции и время реакции можно определить произвольно в зависимости от используемых реагентов. Температура реакции предпочтительно составляет от -20 до 200 С и более предпочтительно от -10 до 150 С. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней. Далее будет описан способ (b). Способ (b) представляет собой способ получения соединения (Ia) с использованием этерифицирующего агента. Способ (b) представляет собой способ получения соединения (Ia) путем взаимодействия соединения (Ib) со сложным эфиром азодикарбоновой кислоты, таким как диэтилазодикарбоксилат (DEAD) или диизопропилазодикарбоксилат (DIAD), и соединением фосфора,таким как трифенилфосфин или трибутилфосфин, с последующим взаимодействием полученного продукта со спиртом, обозначенным как R3OH. Этерифицирующим агентом предпочтительно является комбинация DEAD и трифенилфосфина. Используемый растворитель специально не ограничивается, и его примеры включают ТГФ, диэтиловый эфир, толуол, хлороформ и т.п. Спирт, обозначенный как R3OH, который является реагентом,принимающим участие в реакции, может быть, в частности, взят в подходящем количестве вместо использования других растворителей. Количество используемого спирта можно определить произвольно в соответствии с применяемым реагентом и растворителем. Количество спирта, которое используют по отношению к 1 моль соединения(Ib), предпочтительно составляет от 0,5 до 100 моль и более предпочтительно от 0,8 до 5 моль. Температуру реакции и время реакции можно определить произвольно в зависимости от используемых реагентов. Температура реакции предпочтительно составляет от -20 до 200 С и более предпочтительно от -10 до 150 С. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней. Путь 2. Далее подробно будет описан путь 2. Как указано выше, путь 2 представляет собой способ получения соединения (Ia) из соединения (Ib), которое получают на стадии 1 С 1, по стадиям 1 С 3 и 1 С 4. Стадия 1 С 1 идентична со стадией пути 1, и потому ее описание здесь опускается. Путь 2 включает стадию замыкания цикла для получения соединения (X) путем замыкания цикла соединения (Ib), которое получают на стадии образования соединения карбоновой кислоты, при этом соединение карбоновой кислоты получают путем замещения конкретной функциональной группы соединения (III) карбоксильной группой в присутствии конденсирующего агента, и стадию раскрытия цикла для получения производного азола, представленного общей формулой (Ia), по реакции полученного соединения (X) (соединения, представленного следующей общей формулой (X); далее обозначают как В указанных формулах значения Y, m, R1, R3 и А такие же, что и приведенные выше.(3-3-3) Стадия 1 С 3 (стадия конденсации) Будет рассмотрена стадия (стадия 1 С 3) получения соединения (X) путем конденсации соединения(Ib) на стадии 1 А. Способ конденсации специально не ограничивается, и его примеры включают способы с использованием, например, дициклогексилкарбодиимида, 1-этил-3-(3-диметиламинопропил)карбодиимида (далее в настоящем описании обозначают как WSC) или дифенилфосфорилазида в качестве конденсирующего агента. Среди вышеуказанных соединений 1-этил-3-(3-диметиламинопропил)карбодиимид предпочтительно используют в качестве конденсирующего агента. В таком случае может применяться катализатор,такой как гидроксибензотриазол или диметиламинопиридин. Количество используемого конденсирующего агента по отношению к 1 моль соединения (Ib) обычно предпочтительно составляет от 0,5 до 20 моль и более предпочтительно от 0,8 до 10 моль. Растворитель может быть выбран произвольно в соответствии с типом используемого окислительного агента. Например, когда конденсирующим агентом является WSC, то предпочтительно используют ТГФ или метиленхлорид. Температуру реакции и время реакции можно определить и применять произвольно в зависимости от используемых реагентов. Температура реакции предпочтительно составляет от -20 до 200 С и более предпочтительно от -10 до 150 С. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней.(3-3-4) Стадия 1 С 4 (стадия раскрытия цикла) Будет описана стадия (стадия 1 С 4) получения соединения (Ia) по реакции соединения (X) с алкоголятом металла со стадии 1 А. Алкоголят металла представляет собой соединение, обозначенное как R3O-Ma+. R3 такой же, что и указанный выше, за исключением того, что R3 не является атомом водорода. Ма+ обозначает щелочной металл и предпочтительно представляет собой натрий или литий. Алкоголят металла может быть получен по реакции спирта (R3OH) с алкиллитием, металлическим натрием, металлическим литием, гидридом натрия или т.п., в растворителе, таком как ТГФ или диэтиловый эфир. Особенно предпочтительным является способ проведения реакции с металлическим литием, и наиболее предпочтительным является способ проведения реакции с алкиллитием или т.п. Количество используемого алкоголята металла по отношению к 1 моль лактонового соединения (X) обычно составляет от 0,5 до 20 моль и предпочтительно от 0,8 до 10 моль. В качестве растворителя может использоваться ТГФ, диэтиловый эфир, диоксан и т.п., но предпочтительным является использование растворителя, идентичного тому, который применяют при получении алкоголята металла. Температуру реакции и время реакции можно определить и применять произвольно в зависимости от используемых реагентов. Температура реакции предпочтительно составляет от -100 до 200 С и более предпочтительно от -80 до 150 С. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней. Когда R3 обозначает атом водорода, т.е. когда R2 обозначает карбоксильную группу, соединение(Ib), полученное на стадии 1 С 1, является конечным продуктом, и дополнительная стадия после данной стадии не требуется.(304) Стадия 1D (стадия амидирования) Далее будет рассмотрена стадия 1D. Стадия 1D представляет собой стадию получения приведенного ниже соединения (Ic) путем амидирования лактонового соединения (X), полученного на стадии 1 С 3(см. формулу реакции (6. Стадии, предшествующие стадии 1 С 3, получения лактонового соединения (X) в первом способе получения такие же, что и стадии, описанные выше, и потому их описание здесь опускается. Формула реакции (6) В указанных формулах R1, R3, R4, Y, m и А такие же, что и приведенные выше. Способ амидирования лактонового соединения (X) специально не ограничивается, и он представляет собой, например, способ взаимодействия лактонового соединения (X) с соединением амина, представленным формулой R3R4NH. Указанным образом может быть получено соединение (Ic). Количество используемого соединения амина по отношению к 1 моль соединения (X) обычно составляет от 0,5 до 100 моль и предпочтительно от 0,8 до 80 моль. Преимущественно используемые растворители включают такие, как ТГФ, метиленхлорид, хлороформ, толуол и т.п., и ТГФ является более предпочтительным растворителем. Температуру реакции и время реакции можно определить и применять произвольно в зависимости от используемых реагентов. Обычно температура реакции предпочтительно составляет от -20 до 200 С и более предпочтительно от -10 до 150 С. Время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней.(4) Второй способ получения соединения (I) Второй способ получения соединения (I) будет описан ниже со ссылкой на фиг. 1. В частности, в соответствии со вторым способом получения соединения (I) можно получить среди представленных выше соединений (I) соединение, представленное следующей общей формулой (Id) (далее в настоящем описании обозначают как "соединение (Id)"). Вторым способом получения соединения (I) можно также селективно получить соединение (Ia) и соединение (Ib). Соединение (Id) представляет собой соединение(I), в котором R1 обозначает галогеналкильную группу (R6 в соединении (Id и R2 обозначает COOR3. Как показано на фиг. 1, второй способ получения соединения (I) включает стадии 2 А, 2 В, 2 С, 2D и 2 Е. Каждая стадия может дополнительно включать дополнительные стадии. Далее в настоящем описании будет подробно рассмотрена каждая стадия и каждая дополнительная стадия второго способа получения.(4-1) Стадия 2 А Вначале кратко будет рассмотрена стадии 2 А. Стадия 2 А представляет собой стадию получения соединения, представленного следующей общей формулой (XVI) (далее в настоящем описании обозначают как "соединение (XVI)"). Как показано на фиг. 1, стадия 2 А включает стадии 2 А 1, 2 А 2 и 2 А 3. Стадия 2 А включает стадию гидроксиалкилирования, с целью гидроксиалкилирования кетоэфирного соединения, представленного следующей общей формулой (XXI) (далее в настоящем описании обозначают как "соединение (XXI)"), стадию введения защитной группы, которая защищает гидроксильную группу полученного соединения, содержащего гидроксиалкильную группу (соединения, представленного следующей общей формулой (XIX); далее в настоящем описании обозначают как "соединение (XIX)"),и стадию удаления сложноэфирной группы карбоновой кислоты с получением карбонильного соединения (XVI) путем гидролиза и декарбоксилирования соединения, в которое была введена защитная группа(соединения, представленного следующей общей формулой (XVIII); далее в настоящем описании обозначают как "соединение (XVIII)") (см. следующую формулу реакции (7. Соединение (XXI) можно получить путем бензилирования соединения, представленного следующей общей формулой (XXII).(4-1-1) Стадия 2 А 1 (стадия гидроксиалкилирования) Далее вначале будет описана стадия (стадия 2 А 1) получения соединения (XIX) путем гидроксиал- 28024953 килирования соединения (XXI), полученного из соединения, представленного следующей общей формулой (XXII) (далее в настоящем описании обозначают как "соединение (XXII)", на стадии 2 А. Стадия 2 А 1 включает стадию (стадия 2 А 1 а) получения соединения (XX) путем гидроксиалкилирования соединения(XXI) и стадию (стадия 2 А 1b) получения соединения (XIX) путем дополнительного гидроксиметилирования соединения (XX). Далее стадии 2 А 1 а и 2 А 1b будут рассмотрены подробно. Стадия 2 А 1 а: первая стадия гидроксиалкилирования. На стадии 2 А 1 а соединение (XX) может быть получено по реакции соединения (XXI) с гидроксиалкилгалогенидом в растворителе в присутствии основания. Гидроксильную группу используемого гидроксиалкилгалогенида можно предварительно защитить с помощью защитной группы G. Обычно количество используемого гидроксиалкилгалогенида по отношению к 1 моль соединения(XXI) предпочтительно составляет от 0,5 до 20 моль и более предпочтительно от 0,8 до 10 моль. Примеры оснований включают, но этим не ограничиваясь, карбонаты щелочных металлов, такие как карбонат натрия и карбонат калия; гидроксиды щелочных металлов, такие как гидроксид натрия; органические основания, такие как триэтиламин, и т.п. Обычно количество используемого основания по отношению к 1 моль соединения (XXI) предпочтительно составляет от 0,5 до 10 моль и более предпочтительно от 0,2 до 5 моль. Температура реакции обычно предпочтительно составляет от 0 до 250 С и более предпочтительно от 0 до 100 С. Время реакции обычно предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней. Растворитель специально не ограничивается и его примеры включают простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан; ароматические углеводороды, такие как бензол, толуол и ксилол; воду и т.п. В случае необходимости указанные растворители можно применять в виде смесей. В том случае, когда реакционная система образует две фазы, предпочтительно используют катализатор переноса фаз, такой как обычная четвертичная аммониевая соль (в частности, хлорид бензилтриэтиламмония). В том случае, когда вводимой гидроксиалкильной группой является гидроксиметильная группа, соединение (XXI) предпочтительно взаимодействует с формальдегидом или производным формальдегида(далее обозначают как формальдегид или т.п.) в растворителе в присутствии основания. Примеры производного формальдегида включают параформальдегид, 1,3,5-триоксан, диалкилацетали формальдегида и т.п. Используемое соединение (XXI) может представлять собой соединение, которое получают известным способом (в частности, способом, который описан в патентном документе 1). Стадия 2 А 1b: вторая стадия гидроксиалкилирования (стадия гидроксиметилирования). Способ введения гидроксиметильной группы на стадии 2 А 1b может быть способом по стадии 2 А 1 а,где в качестве гидроксиалкильной группы используют гидроксиметильную группу. В том случае, когда гидроксиалкильной группой, которую вводят на стадии 2 А 1 а, является гидроксиметильная группа и когда проводят бисгидроксиметилирование, то стадию 2 А 1b можно пропустить. В таком случае можно проводить гидроксиметилирование за один раз, взяв вдвое большее мольное количество гидроксиметилгалогенида, чем количество соединения (XXI) на стадии 2 А 1 а. Формальдегид или т.п. преимущественно используют в два раза большем мольном количестве, чем соединение (XXI). Хотя выше описан случай, когда соединение (XIX) получают по стадии 2 А 1 а, а затем 2 А 1b, соединение (XIX) можно получить, осуществляя стадию 2 А 1b, а затем 2 А 1 а. Обычно количество используемого формальдегида по отношению к 1 моль соединения (XX) предпочтительно составляет от 1,5 до 20 моль и более предпочтительно от 1,8 до 10 моль. Примеры оснований включают, но этим не ограничиваясь, карбонаты щелочных металлов, такие как карбонат натрия и карбонат калия; гидроксиды щелочных металлов, такие как гидроксид натрия; органические основания, такие как триэтиламин и т.п. Обычно количество используемого основания по отношению к 1 моль соединения (XX) предпочтительно составляет от 0,5 до 10 моль и более предпочтительно от 0,2 до 5 моль. Обычно температура реакции предпочтительно составляет от 0 до 250 С и более предпочтительно от 0 до 100 С. Обычно время реакции предпочтительно составляет от 0,1 ч до нескольких дней и более предпочтительно от 0,5 ч до 2 дней. Используемым соединением (XX) может быть соединение, которое получают известным способом(4-1-2) Стадия 2 А 2 (стадия введения защитной группы) Далее будет описана стадия (стадия 2 А 2) получения соединения (XVIII) путем введения защитной группы для защиты гидроксильной группы соединения (XIX) на стадии 2 А. Защитная группа, которую используют для защиты гидроксильной группы, специально не ограничивается. Защитной группой преимущественно является алкоксиметильная группа, такая как метоксиметильная группа или этоксиметильная группа, или низшая алкильная группа, такая как трет-бутильная группа. Указанные защитные группы вводят аналогично стадии 1 С 2, за исключением того, что две гидроксильные группы вводят одновременно, например, с помощью ацеталя или кеталя, и потому описание
МПК / Метки
МПК: C07D 249/08, C07D 233/60, A01P 3/00, C07D 405/06, A01N 43/653
Метки: азола, способ, сельском, защиты, получения, материалов, применения, производное, предназначенный, промышленных, садоводстве, химический, агент, хозяйстве
Код ссылки
<a href="https://eas.patents.su/30-24953-proizvodnoe-azola-sposob-ego-polucheniya-himicheskijj-agent-prednaznachennyjj-dlya-primeneniya-v-selskom-hozyajjstve-i-sadovodstve-i-agent-dlya-zashhity-promyshlennyh-materialov.html" rel="bookmark" title="База патентов Евразийского Союза">Производное азола, способ его получения, химический агент, предназначенный для применения в сельском хозяйстве и садоводстве, и агент для защиты промышленных материалов</a>
Соединение 4-циклопропил-1,2,3-тиадиазола, агент для борьбы с болезнями растений для применения в сельском и садоводческом хозяйстве и метод его применения

Номер патента: 13120
Опубликовано: 26.02.2010
Авторы: Ода Масацугу, Ямагути Минору, Такемото Цуеси, Симаока Такаси, Киомура Нобуо, Кикутаке Казухико, Уметани Кунихиса
МПК: A01N 43/836, A01N 43/86, A01N 43/828...
Метки: применения, борьбы, соединение, растений, болезнями, агент, метод, хозяйстве, сельском, 4-циклопропил-1,2,3-тиадиазола, садоводческом
Формула / Реферат:
1. Соединение 1,2,3-тиадиазола, представленное формулой (I)где R1, R2, R3, R4 и R5являются одинаковыми или разными и каждый представляет собой атом водорода; атом галогена; циано; (C1-C6)алкил; галоген(C1-C6)алкил; (C1-C6)алкокси(C1-C6)алкил; (C3-C12)циклоалкил; галоген(C3-C12)циклоалкил; (C1-C6)алкилтио(C1-C6)алкил; (C2-C6)алкенил; галоген(C2-C6)алкенил; арил, который может быть замещенным заместителем Z; арил(C1-C6)алкил, который может быть...
Растянутая пластиковая пленка для применения в сельском хозяйстве.

Номер патента: 601
Опубликовано: 29.12.1999
Автор: Джонстоун Питер
МПК: A01G 13/02, B29C 55/02
Метки: сельском, растянутая, хозяйстве, применения, пленка, пластиковая
Формула / Реферат:
1. Пластиковая пленка для покрытия засеянной почвы или почвы, предназначенной для проращивания семян, отличающаяся тем, что пленка вытянута, по меньшей мере, в локализованных областях по длине пленки выше предела текучести пленки для достижения уменьшенной толщины пленки в вытянутой области или областях, в результате чего при применении пленка будет разрушаться для обеспечения проросшему сеянцу прохода через нее. 2. Пленка по п.1, отличающаяся...
Получение полимерных конъюгатов соединений, применяемых в терапии, сельском хозяйстве и в качестве пищевых добавок

Номер патента: 15528
Опубликовано: 31.08.2011
Авторы: Смит Дженифер Л., Конради Андрей В.
МПК: A61K 47/48, A61P 43/00
Метки: терапии, конюгатов, хозяйстве, сельском, качестве, применяемых, добавок, пищевых, получение, соединений, полимерных
Формула / Реферат:
1. Способ получения конъюгатов активных соединений, включающий:(a) обработку первичного или вторичного спиртового заместителя по крайней мере одного активного соединения в условиях реакции Мицунобу или родственных реакций и(b) обработку продукта стадии (а) полимером, содержащим нуклеофильные заместители, активные в условиях реакции Мицунобу или родственных реакций;причем полимер выбирают из группы, состоящей изгде n выбирают для образования...
Соли металлов дигидрожасмоновой кислоты, композиции, включающие их и производные бензойной кислоты, и их применение в сельском хозяйстве

Номер патента: 12413
Опубликовано: 30.10.2009
Автор: Маркс Дэвид
МПК: C07C 59/205, A01N 37/42
Метки: хозяйстве, дигидрожасмоновой, включающие, соли, бензойной, кислоты, производные, металлов, применение, композиции, сельском
Формула / Реферат:
1. Соединение формулы (I) где R1 означает С1-10-алкильную группу и М означает катион щелочно-земельного металла валентности n, при этом М представляет собой магний. 2. Соединение по п.1, где R1 означает н-пентильную группу. 3. Способ получения соединения формулы (I) по п.1 или 2, где способ включает взаимодействие соединения формулы (II) где R1 имеет указанное в случае формулы (I) значение и R2 выбирают из водорода или углеводородного остатка,...
Быстродействующий химический стерилизующий агент

Номер патента: 1112
Опубликовано: 30.10.2000
Авторы: Майнер Норман А., Андерсон Эдвард Л., Хобсон Дэвид В., Воллер Вильям Х.
МПК: A01N 59/00
Метки: агент, стерилизующий, быстродействующий, химический
Формула / Реферат:
1. Водный дезинфицирующий и/или стерилизующий раствор со слабым запахом, быстродействующий при комнатной температуре, имеющий рН примерно от 2,0 до 6,0, включающий примерно 1-30 мас.% пероксида, способного выделять свободные гидроксильные радикалы, и примерно 1-30 мас.% водорастворимой органической кислоты, выбранной из группы, состоящей из малоновой кислоты и янтарной кислоты или их смесей. 2. Раствор по п.1, отличающийся тем, что концентрация...
Предыдущий патент: Бензотиазолы и их применение для лечения вич-инфекции
Следующий патент: Рабочее колесо центробежного насоса и его комбинация с внутренним вкладышем (варианты)
Случайный патент: Плиточный декоративно-отделочный материал и способ его получения