Способ получения ривароксабана и промежуточные соединения, получаемые в указанном способе
Номер патента: 24685
Опубликовано: 31.10.2016
Авторы: Шипош Эва, Кованьине Лакс Дьёрдь, Лукач Дьюла, Вольк Балаж, Печи Эва, Таборине Тот Мариа Юлиа, Баркоци Йожеф, Хаваши Балаж, Хегедюш Ласло Йожеф, Ружич Дьёрдь, Краснаи Дьёрдь, Боза Андраш, Тотне Лауриц Мариа







Формула / Реферат
1. Способ получения фармацевтически активного ингредиента 5-хлор-N-({(5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-ил}метил)тиофен-2-карбоксамида (ривароксабана) формулы

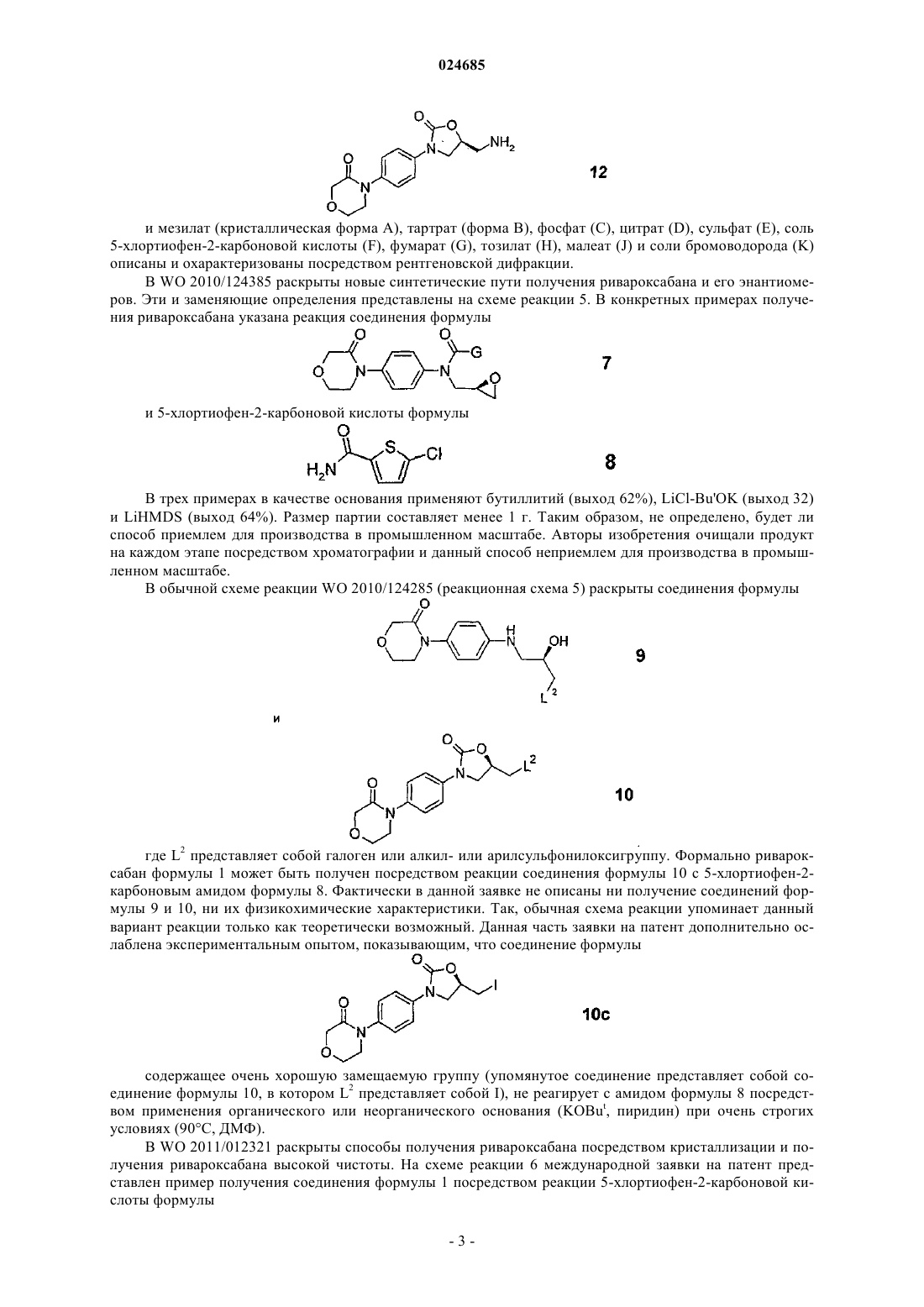
включающий взаимодействие 4-(4-((5S-(5-аминометил-2-оксо-1,3-оксазолидин-3-ил)фенил)морфолин-3-она формулы 12

или его соли общей формулы 3

(где HnX представляет собой органическую или неорганическую кислоту, n представляет собой 1, 2 или 3 и X представляет собой ион кислотного остатка) с 5-хлортиофен-2-карбоновой кислотой формулы 15:

в присутствии агента реакции сочетания, выбранного из хлорэтилформиата, N,N'-диизопропилкарбодиимида (ДИК), N,N'-дициклогексилкарбодиимида (ДЦК), ангидрида трипропилфосфоновой кислоты (ТФА) или N,N'-карбонилдиимидазола (КДИ), при условии, что если в соединении общей формулы 3 n представляет собой 1 и X представляет собой хлор, то агент реакции сочетания не является N,N'-карбонилдиимидазолом.
2. Способ по п.1, где соединение формулы 12 или его соль формулы 3 получают удалением защитной групп(ы) с S-энантиомерного соединения общей формулы 14

(где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом) с последующим выделением полученного таким образом S-энантиомерного основания формулы 12 или его соли формулы 3.
3. Способ по п.2, где соединение формулы 14 получают взаимодействием R-энантиомерного соединения общей формулы 20

(где Z1 и Z2 имеют значения, указанные в п.2) с агентом, способным ввести карбонильную группу, представляющим собой N,N'-карбонилдиимидазол, фосген, дифосген или трифосген.
4. Способ по п.3, где соединение формулы 20 получают взаимодействием R-энантиомерного соединения общей формулы 19

(где Z1 и Z2 имеют значения, указанные в п.2) с 4-(4-аминофенил)морфолин-3-оном формулы 5

5. Способ по п.4, где соединение формулы 19 получают взаимодействием S-энантиомерного соединения формулы 11

с соединением общей формулы Z1Z2NH (где Z1 и Z2 имеют значения, указанные в п.2).
6. Способ по любому из пп.1 или 2, где n представляет собой 1 и X представляет собой ацетатный ион.
7. Способ по любому из пп.2-5, где Z1 и Z2 представляют собой бензил.
8. Способ по любому из пп.1-7, где при взаимодействии S-энантиомерного основания формулы 12

или его соли формулы 3 с 5-хлортиофен-2-карбоновой кислотой формулы 15 в качестве агента реакции сочетания применяют хлорэтилформиат или N,N'-карбонилдиимидазол (КДИ), при условии, что если в соединении общей формулы 3 n представляет собой 1 и X представляет собой хлор, то агент реакции сочетания не является N,N'-карбонилдиимидазолом, взаимодействие необязательно проводят в присутствии органического или неорганического основания в органическом растворителе или смеси указанного растворителя с водой при 0-110°С.
9. Способ по п.8, где органическое или неорганическое основание выбрано из триэтиламина, диизопропилэтиламина, карбоната натрия или гидрокарбоната натрия, органический растворитель выбран из ацетонитрила, дихлорметана, ацетона, толуола, тетрагидрофурана или их смеси и реакцию осуществляют при температуре 40-70°С.
10. Способ по любому из пп.2 или 3, где соединение формулы 14 представляет собой соединение формулы 13

которое получают путем удаления защитных бензильных групп посредством восстановления, проводимого в C1-4 алифатическом спирте, ледяной уксусной кислоте, воде или смеси упомянутых растворителей друг с другом или с дополнительными органическими растворителями, посредством каталитической гидрогенизации или химического восстановления.
11. Способ по п.3, где в качестве агента, способного ввести карбонильную группу, применяют N,N'-карбонилдиимидазол и взаимодействие осуществляют в приемлемом растворителе.
12. Способ по п.11, где растворитель представляет собой толуол.
13. Способ по п.4, где взаимодействие R-энантиомерного соединения общей формулы 19 и 4-(4-аминофенил)морфолин-3-она формулы 5 проводят в протонном растворителе или смеси растворителей или в смеси протонного растворителя и воды при 0-150°С в течение 0,5-60 ч.
14. Способ по п.13, где взаимодействие осуществляют при 60-90°С в течение 20-40 ч.
15. Способ по п.5, где взаимодействие S-энантиомерного соединения формулы 11 и соединения общей формулы Z1Z2NH проводят в отсутствие или в присутствии органического растворителя, или в воде, или в смеси указанных растворителей.
16. Способ по п.15, где соединение общей формулы Z1Z2NH представляет собой N-бензил-1-фенилметанамин формулы 16

и реакцию проводят в присутствии вещества, связывающего неорганические кислоты.
17. Уксуснокислая соль 4-{4-[(5S)-5-(аминометил)-2-оксо-1,3-оксазолидин-3-ил]фенил}морфолин-3-она формулы

18. Способ получения S-энантиомерного соединения формулы 12

или его соли общей формулы 3

включающий удаление защитной групп(ы) с S-энантиомерного соединения общей формулы 14

(где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом) с последующим выделением полученного таким образом S-энантиомерного основания формулы 12 или его соли общей формулы 3.
19. Способ по п.18, где энантиомерная соль общей формулы 3 представляет собой уксуснокислую соль формулы 3b, a S-энантиомерное соединение общей формулы 14 представляет собой S-энантиомерное соединение общей формулы 13.
20. 4-(4-{(5S)-5-Дибензиламинометил-2-оксо-1,3-оксазолидин-3-ил}фенил)морфолин-3-он формулы 13

21. Способ получения S-энантиомерных соединений общей формулы 14

(где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом и Z1 и Z2), включающий взаимодействие R-энантиомерного соединения общей формулы 20

(где Z1 и Z2 представляют собой такие, как указано выше), с агентом, способным ввести карбонильную группу, представляющим собой N,N'-карбонилдиимидазол, фосген, дифосген или трифосген.
22. 4-(4-{[(2R)-3-Дибензиламино-2-гидроксипропил]амино}фенил)морфолин-3-он формулы

23. Способ получения R-энантиомерных соединений общей формулы 20

(где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом), включающий взаимодействие S-энантиомерного соединения общей формулы 19

(где Z1 и Z2 представляют собой такие, как указано выше) с 4-(4-аминофенил)морфолин-3-оном формулы 5

24. Способ по п.21 или 23, где как Z1, так и Z2 в каждом случае представляют собой бензил.
Текст
СПОСОБ ПОЛУЧЕНИЯ РИВАРОКСАБАНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ,ПОЛУЧАЕМЫЕ В УКАЗАННОМ СПОСОБЕ Изобретение относится к способу получения 5-хлор-N-5S)-2-оксо-3-[4-(3-оксоморфолин-4 ил)фенил]-1,3-оксазолидин-5-илметил)тиофен-2-карбоксамида,имеющего международное непатентованное название ривароксабан. Способ характеризуется реакциями по п.1. Изобретение также относится к промежуточным соединениям, получаемым в данном способе. Шипош Эва, Кованьине Лакс Дьрдь,Хаваши Балаж, Вольк Балаж,Краснаи Дьрдь, Ружич Дьрдь,Баркоци Йожеф, Тотне Лауриц Мариа, Лукач Дьюла, Боза Андраш,Хегедюш Ласло Йожеф, Таборине Тот Мариа Юлиа, Печи Эва (HU) Харин А.В. (RU)(71)(73) Заявитель и патентовладелец: ЭГИШ ДЬДЬСЕРДЬЯР НЬИЛЬВАНОШАН МЮКД РЕСВЕНЬТАРШАШАГ (HU) Область техники Настоящее изобретение относится к способу получения фармацевтически активного ингредиента и промежуточным соединениям, получаемым в данном способе. В частности, изобретение относится к новому способу получения 5-хлор-N-5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5 илметил)тиофен-2-карбоксамида формулы имеющего МНН (международное непатентованное название) ривароксабан. Изобретение также относится к промежуточным соединениям, получаемым в упомянутом способе. Уровень техники Известно, что 5-хлор-N-5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-илметил)тиофен-2-карбоксамид, имеющий МНН ривароксабан, представляет собой фармацевтически активный ингредиент ингибиторного механизма фактора коагуляции Ха, приемлемый для лечения инфаркта при тромбозе глубоких вен, стенокардии, окклюзии артерии, артериального рестеноза и легочной эмболии. Ривароксабан был впервые описан Straub et al. в ЕР 1261606. В упомянутом европейском патенте раскрыта широкая группа замещенных оксазолидинов и способ их получения. В частности, не раскрыты ни получение ривароксабана, ни его физико-химические свойства. Согласно общему замыслу патента получение ривароксабана формулы 1 осуществляют согласно схеме реакции 1. Таким образом, конфигурацию, соответствующую ривароксабану, получают из (S)-глицидилфталимида формулы имеющего систематическое химическое название 2-[(2S)-оксиран-2-ил-метил]-1 Н-изоиндол 1,3(2 Н)-дион, который может быть получен из (S)-эпихлоргидрина. На последнем этапе синтеза согласно обычным способам гидрохлорид 4-4-[(5S)-5-аминометил)-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы с образованием ривароксабана формулы 1. Реакцию конденсации проводят в пиридине. Недостатком способа является то, что конечную реакцию конденсации проводят посредством применения очень дорогого хлорангидрида 5-хлортиофен-2-карбоновой кислоты и применения высокотоксичного пиридина в качестве растворителя. Дополнительным недостатком является то, что хлорангидрид 5-хлортиофен 2-карбоновой кислоты является вязким маслом, сложным для работы. Оно легко разлагается и при хранении при 0-5 С быстро гидролизуется до 5-хлортиофен-2-карбоновой кислоты. Таким образом, упомянутый исходный материал не может долго храниться, а должен быть немедленно использован, что делает технологию сложной, в частности, при производстве в промышленном масштабе. Хлорангидрид 5 хлортиофен-2-карбоновой кислоты получают из 5-хлортиофен-2-карбоновой кислоты при помощи тионилхлорида последний реагент также является коррозийным веществом, обладает неприятным запахом и вреден для здоровья и окружающей среды. В конце реакции остаточный тионилхлорид должен быть удален, что требует дополнительного этапа дистилляции. Побочные продукты хлороводород и диоксид серы, образующиеся в процессе получения хлорангидрида, должны быть адсорбированы и удалены в ручную, что представляет дополнительную технологическую трудность и наносит вред здоровью. В ЕР 1583761 описан совершенно иной путь синтеза, приведенный на схеме реакции 2. Так, ривароксабан формулы 1 получают посредством применения в качестве исходного материала (2S)-3-аминопропан-1,2-диолхлористо-водородной соли. В упомянутом ЕР раскрыты определенные этапы способа и некоторые промежуточные соединения. Общий выход способа составляет только 37%. Структурное звено, 5-хлортиофен-2-карбонил, вводят при помощи хлорангидрида 5-хлортиофен-2-карбоновой кислоты. Его недостатки обсуждены выше. В WO 2005/0261335 описано получение промежуточного соединения 4-(4-аминофенил)морфолин 3-она формулы применяемого в ранее упомянутых двух заявках на патент, как показано на схеме реакции 3. В упомянутой международной заявке на патент 4-(4-нитрофенил)-3-морфолинон формулы в алифатический спирт защищен от каталитической гидрогенизации. Упомянутый способ отличается от способа, изложенного в ЕР 1261606, при котором каталитическую гидрогенизацию проводят в тетрагидрофуране. Согласно WO 2005/0068456 (эквивалентен DE 102004002044) ривароксабан получают согласно способу синтеза, приведенному на схеме реакции 4. Упомянутый способ практически идентичен способу, раскрытому в основном патенте и приведенному на схеме реакции 1, за исключением того, что выходы некоторых этапов улучшены за счет оптимизации условий реакции. В упомянутой международной заявке на патент описано получение ривароксабана за счет конденсации гидрохлорида 4-4-[(5S)-5 аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 3 а с хлорангидридом 5-хлортиофен-2-карбоновой кислоты формулы 4 либо в спиртовом или в кетонном растворителе, либо в их смеси в присутствии неорганического основания. В качестве растворителя для реакции конденсации предпочтительно может быть применена смесь ацетона и воды и в качестве неорганического основания предпочтительно гидроксид натрия, гидрокарбонат натрия или карбонат натрия, в частности может быть применен карбонат натрия. В примере, показывающем этап конденсации соли формулы 3 а с карбонатом натрия, основание выделяют в свободном виде, после чего добавляют воду и ацетон и при 8-12 С добавляют по капле приблизительно 30 мас.% раствора хлорангидрида 5-хлортиофен-2-карбоновой кислоты формулы 4 в толуоле. Затем при 50 С добавляют ацетон, реакцию продолжают, реакционную смесь охлаждают до 25 С и ривароксабан отфильтровывают. Недостатки реагента хлорангидрида 5-хлор-тиофен 2-карбоновой кислоты, применяемого в указанном способе, обсуждены выше. Дополнительный недостаток способа заключается в том, что удаление фталильной защитной группы при помощи большого излишка (4,4 экв) метиламина выполняют только на последней стадии способа. Поскольку применяют большой излишек метиламина и строгие условия реакции, образование примесей неизбежно. Выходы показывают, что упомянутый способ является неэкономичным.WO 2005/068456 не упоминает о ВЭЖХ-чистоте конкретных промежуточных соединений, не говоря уже о конечном продукте ривароксабане формулы 1. Хлористо-водородную соль формулы 3 а получают согласно международной заявке на патент с выходом 82,7%, в которой не раскрыта ее чистота. При воспроизводстве примера вышеупомянутой заявки на патент мы обнаружили, что полное удаление защитной группы требует по меньшей мере 20 ч вместо 2 ч, как указано в международной заявке на патент. После удаления фталильной защитной группы качество промежуточного соединения формулы 3 а является неприемлемым для получения конечного продукта фармацевтической степени чистоты. Поэтому промежуточное соединение формулы За должно быть подвергнуто перекристаллизации, которая снизит общий выход способа. Статья, опубликованная в IP. com. Journal в 2010 (IPCOM0000195906D), получение 4-(4-аминофенил)морфолин-3-она формулы 5 начинается с 4-(4-нитрофенил)-3-морфолинона формулы 6. Для того,чтобы избежать каталитической гидрогенизации авторы разработали новый способ восстановления, в соответствии с которым восстановление осуществляют при помощи металла или металлической соли в присутствии кислоты. В качестве металла предпочтительно применяли цинк или железо, и в качестве кислоты предпочтительно применяли соляную кислоту: восстановительная среда представляла собой спирт или смесь спирта и воды. Наилучший выход составил 78%. В статье IP.com. Journal, опубликованной в IP.com. Journal (IPCOM000199058D) в 2009, I, II, III и IV кристаллические формы 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы и мезилат (кристаллическая форма А), тартрат (форма В), фосфат (С), цитрат (D), сульфат (Е), соль 5-хлортиофен-2-карбоновой кислоты (F), фумарат (G), тозилат (Н), малеат (J) и соли бромоводорода (K) описаны и охарактеризованы посредством рентгеновской дифракции. В WO 2010/124385 раскрыты новые синтетические пути получения ривароксабана и его энантиомеров. Эти и заменяющие определения представлены на схеме реакции 5. В конкретных примерах получения ривароксабана указана реакция соединения формулы В трех примерах в качестве основания применяют бутиллитий (выход 62%), LiCl-Bu'OK (выход 32) и LiHMDS (выход 64%). Размер партии составляет менее 1 г. Таким образом, не определено, будет ли способ приемлем для производства в промышленном масштабе. Авторы изобретения очищали продукт на каждом этапе посредством хроматографии и данный способ неприемлем для производства в промышленном масштабе. В обычной схеме реакции WO 2010/124285 (реакционная схема 5) раскрыты соединения формулы где L2 представляет собой галоген или алкил- или арилсульфонилоксигруппу. Формально ривароксабан формулы 1 может быть получен посредством реакции соединения формулы 10 с 5-хлортиофен-2 карбоновым амидом формулы 8. Фактически в данной заявке не описаны ни получение соединений формулы 9 и 10, ни их физикохимические характеристики. Так, обычная схема реакции упоминает данный вариант реакции только как теоретически возможный. Данная часть заявки на патент дополнительно ослаблена экспериментальным опытом, показывающим, что соединение формулы содержащее очень хорошую замещаемую группу (упомянутое соединение представляет собой соединение формулы 10, в котором L2 представляет собой I), не реагирует с амидом формулы 8 посредством применения органического или неорганического основания (KOBut, пиридин) при очень строгих условиях (90 С, ДМФ). В WO 2011/012321 раскрыты способы получения ривароксабана посредством кристаллизации и получения ривароксабана высокой чистоты. На схеме реакции 6 международной заявки на патент представлен пример получения соединения формулы 1 посредством реакции 5-хлортиофен-2-карбоновой кислоты формулы и N,N'-карбонилдиимидазола (1,2 экв.) и триэтиламина (1,2 экв.) и хлористо-водородной соли формулы 3 а (1,03 экв.) в диметилформамиде. Однако согласно данному способу неочищенный ривароксабан может быть получен только с высокими потерями с выходом 72% (относительно соединения формулы 3 а) и полученный неочищенный продукт требует дополнительной очистки. Выход очищенного продукта,составляющий 61% (относительно соединения формулы 3 а) является только средним и много ниже, чем выход реакции алкилирования, выполненной при помощи хлорангидрида формулы 4. Заявка на патент не упоминает в раскрытии данных, связанных с чистотой продукта. Краткое описание изобретения Целью настоящего изобретения является разработка синтетического способа получения ривароксабана формулы 1 в промышленном масштабе, где указанный способ обеспечивает лучшие выходы и где в способе используют кристаллические и легко очищаемые промежуточные соединения. Вышеупомянутой цели достигают посредством следующего способа. На первом этапе способа соединение общей формулы(где Z1 и Z2 представляют собой водород или обычную аминозащитную группу, например, бензил,замещенный бензил, п-метоксибензил, бензилоксикарбонил или третбутоксикарбонил при условии, что,по меньшей мере, Z1 не является водородом и Z1 и Z2 предпочтительно представляют собой бензил), получают посредством реакции соединения формулы с соединением общей формулы Z1Z2NH. На втором этапе способа соединение общей формулы 19 подвергают взаимодействию с соединением формулы 5 с выходом соединения общей формулы(где Z1 и Z2 представляют собой указанные выше). На третьем этапе способа соединение общей формулы(где Z1 и Z2 представляют собой указанные выше) получают посредством реакции соединения общей формулы 20 с агентом, способным ввести карбонильную группу. На четвертом этапе способа из соединения общей формулы 14 получают основание формулы 12 или его соль общей формулы(где HnX представляет собой моно- или бивалентную органическую или неорганическую кислоту; n представляет собой 1, 2 или 3 и X представляет собой ион кислотного остатка) посредством удаления защитной группы(п). В качестве органической кислоты может быть применена, например, сульфоновая кислота, карбоновая кислота, включая моновалентные карбоновые кислоты, например, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, масляную кислоту и т.д., и в качестве неорганических кислот может быть применена, например, серная кислота, сернистая кислота, азотная кислота, фосфор-4 024685 ная кислота, бромоводород, йодоводород. На последнем этапе способа основание формулы 12 или его соль общей формулы 3 (где HnX является таковым, указанным выше) реагирует с 5-хлортиофен-2-карбоновой кислотой с выходом ривароксабана формулы 1. На последнем этапе ацилирования при получении ривароксабана мы применяем кислоту формулы 15, которая существенно менее реактивна, чем хлорангидрид 5-хлортиофен-2-карбоновой кислоты формулы 4, ранее применяемый на последнем этапе ацилирования при получении ривароксабана формулы 1. Упомянутую реакцию проводят в присутствии агента реакции сочетания и органического или неорганического основания в инертном растворителе: при желании применением кислого агента реакции сочетания можно пренебречь. Способ может быть осуществлен посредством применения рацемического исходного материала,при желании. В данном случае вначале посредством разделения получают соединения, имеющие хиральность, показанные на схемах реакции 7 а, 7b, 8 а и 8b. Неожиданно соединения общей формулы 14 также могут быть получены таким образом, что на первом этапе из соединения общей формулы 9 посредством реакции с агентом, способным ввести карбонильную группу, получают соединение общей формулы 10 (где L2 представляет собой хлор, бром, йод,алкилсульфонилокси или арилсульфонилокси, например, метансульфонилокси, бензолсульфонилокси или п-толуолсульфонилокси, предпочтительно хлор, бром или йод). На втором этапе способа соединение общей формулы 10 (где L2 имеет такое же значение, как указано выше) реагирует с соединением общей формулы Z1Z2NH (где Z1 и Z2 представляют собой водород или обычную аминозащитную группу, например, бензил, замещенный бензил, п-метоксибензил, бензилоксикарбонил или третбутоксикарбонил, при условии, что по меньшей мере один из символов Z1 и Z2 не является водородом, и Z1 и Z2 предпочтительно представляют собой бензил). Изобретение также относится к промежуточным соединениям, применяемым в вышеупомянутом способе, соответствующим указанным общим формулам, и к промежуточным соединениям, получаемым в предпочтительных воплощениях вышеупомянутого способа. Подробное описание изобретения Изобретение относится к способу получения фармацевтически активного ингредиента 5-хлор-N5S)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-илметил)тиофен-2-карбоксамида формулы 1, содержащему подвергание разделению 4-4-[(5S)-5-(аминометил)-2-оксо-1,3-оксазолидин-3 ил]фенилморфолин-3-она формулы 12 или его рацемата формулы rac12 или его S-энантиомерной соли формулы 3 или его рацемата общей формулы rac3 (где HnX представляет собой моно- или поливалентную органическую или неорганическую кислоту: n представляет собой 1, 2 или 3: X представляет собой ион кислотного остатка), в случае применения рацемического исходного материала, с последующей реакцией с 5-хлортиофен-2-карбоновой кислотой формулы 15 в присутствии агента реакции сочетания; или на первом этапе подвергание S-энантиомерного соединения общей формулы 14 или его рацемата (где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом) разделению, в случае применения рацемического исходного материала, с последующим удалением защитной группы(п), и отделением полученного таким образом S-энантиомерного основания формулы 12 или его рацемата или, возможно, его соли; на втором этапе подвергание полученного продукта разделению, в случае применения рацемического исходного материала, и последующему взаимодействию с 5-хлортиофен-2-карбоновой кислотой формулы 15 в присутствии агента реакции сочетания; или на первом этапе подвергание R-энантиомерного соединения общей формулы 20 или его рацемата(где Z1 и Z2 представляют собой указанные выше) разделению, в случае применения рацемического исходного материала, с последующей реакцией с агентом, способным ввести карбонильную группу; на втором этапе подвергание полученного S-энантиомерного соединения общей формулы 14 или его рацемата разделению, в случае применения рацемического исходного материала, с последующим восстановлением продукта, отделением полученного таким образом S-энантиомерного основания формулы 12 или его рацемата; на третьем этапе подвергание полученного продукта разделению, в случае применения рацемического исходного материала, с последующей реакцией с 5-хлортиофен-2-карбоновой кислотой формулы 15 в присутствии агента реакции сочетания; или на первом этапе подвергание R-энантиомерного соединения общей формулы 19 или его рацемата (где Z1 и Z2 представляют собой указанные выше) разделению, в случае применения рацемических исходных материалов, с последующей реакцией с 4-(4-аминофенил)морфолин-3-оном, соединением формулы 5, на втором этапе подвергание R-энантиомерного соединения общей формулы 20 или его рацемата (где Z1 и Z2 представляют собой указанные выше) разделению, в случае применения рацемического исходного материала, с последующей реакцией с агентом, способным ввести карбонильную группу, на третьем этапе подвергание полученного Sэнантиомерного соединения общей формулы 14 или его рацемата (где Z1 и Z2 представляют собой указанные выше) разделению, в случае применения рацемического исходного материала, удалению защитной группы(п), и отделению полученного таким образом S-энантиомерного основания формулы 12 или его рацемата или, возможно, его соли; на четвертом этапе подвергание полученного таким образом продукта разделению, в случае применения рацемического исходного материала, и последующей реакции с 5-хлортиофен-2-карбоновой кислотой формулы 15 в присутствии агента реакции сочетания; или на первом этапе реакция S-энантиомерного соединения формулы 11 или его рацемата с соединением общей формулы Z1Z2NH (где Z1 и Z2 представляют собой указанные выше), на втором этапе подвергание полученного таким образом соединения общей формулы 19 или его рацемата (где Z1 и Z2 представляют собой указанные выше) разделению, в случае применения рацемического исходного материала, с последующей реакцией с 4-(4-аминофенил)морфолин-3-оном формулы 5, на третьем этапе подвергание полученного Rэнантиомерного соединения общей формулы 20 или его рацемата (где Z1 и Z2 представляют собой указанные выше) разделению, в случае применения рацемического исходного материала, с последующей реакцией с агентом, способным ввести карбонильную группу; на четвертом этапе подвергание Sэнантиомерного соединения общей формулы 14 или его рацемата (где Z1 и Z2 представляют собой указанные выше) разделению, в случае применения рацемического исходного материала с последующим удалением защитной группы(п) и отделением полученного таким образом S-энантиомерного основания формулы 12 или его рацемата или, возможно, его соли, на пятом этапе подвергание полученного продукта разделению, в случае применения рацемического исходного материала, с последующей реакцией с 5 хлортиофен-2-карбоновой кислотой формулы 15 в присутствии агента реакции сочетания; или на первом этапе подвергание R-энантиомерного соединения общей формулы 9 или его рацемата (где L2 представляет собой указанный выше), разделению в случае применения рацемического исходного материала с последующей реакцией с агентом, способным ввести карбонильную группу, превращением полученного Rэнантиомерного соединения общей формулы 10 или его рацемата (где L2 представляет собой указанный выше), при желании R-энантиомерного соединения формулы(где L2 представляет собой хлор или бром) или его рацемата в R-энантиомерное соединение формулы или его рацемат, или в R-энантиомерное соединение формулы 10 с или его рацемат,на втором этапе подвергание R-энантиомерного соединения общей формулы 10 или его рацемата(где L2 имеет то же значение, как указано ранее) разделению, в случае применения рацемического исходного материала, с последующей реакцией с соединением общей формулы Z1Z2NH, на третьем этапе подвергание S-энантиомерного соединения общей формулы 14 или его рацемата (где Z1 и Z2 представляют собой указанные выше) разделению, в случае применения рацемического исходного материала с последующим удалением защитной группы(п) и отделением полученного таким образом Sэнантиомерного основания общей формулы 12 или, возможно, его соли; на четвертом этапе подвергание полученного таким образом продукта разделению, в случае применения рацемического исходного материала, и реакции продукта с 5-хлор-тиофен-2-карбоновой кислотой формулы 15 в присутствии агента реакции сочетания, при условии, что в общей формуле 3 n представляет собой 1, и X представляет собой хлор, кроме того, агент реакции сочетания не является N,N'-карбонилдиимидазолом. Согласно предпочтительному воплощению изобретения n представляет собой 1, и X представляет собой ион ацетата. Согласно предпочтительному воплощению изобретения в общей формуле 14 Z1 и Z2 представляют собой бензил. Согласно предпочтительному воплощению изобретения в общей формуле 20 Z1 и Z2 представляют собой бензил. Согласно предпочтительному признаку изобретения в общей формуле 19 Z1 и Z2 представляют собой бензил. Согласно предпочтительному признаку изобретения в общей формуле Z1Z2NH Z1 и Z2 представляют собой бензил. Согласно предпочтительному воплощению изобретения S-энантиомерное основание формулы 12 или его рацемат или возможно его соль реагирует с 5-хлортиофен-2-карбоновой кислотой формулы 15 в присутствии хлорформиата, N,N'-диизопропилкарбодиимида (ДИК), N,N'-дициклогексилкарбодиимида(ДЦК), ангидрида трипропилфосфоновой кислоты (ТФА) или N,N'-карбонилдиимидазола (КДИ), предпочтительно хлорэтилформиата или КДИ в качестве агента реакции сочетания. Реакцию проводят предпочтительно в присутствии органического или неорганического основания, предпочтительно триэтила-6 024685 мина, диизопропилэтиламина, карбоната натрия или гидрокарбоната натрия, в органическом растворителе, предпочтительно ацетонитриле, дихлорметане, ацетоне, толуоле, тетрагидрофуране и его смесях или смесях упомянутых растворителей с водой. Реакцию проводят при температуре 0-110 С, предпочтительно 40-70 С. Согласно дополнительному предпочтительному воплощению изобретения защитную группу(пы) Sэнантиомерного соединения формулы удаляют посредством восстановления и упомянутую реакцию восстановления проводят C1-4 алифатическом спирте, ледяной уксусной кислоте, воде или смеси упомянутых растворителей, полученной друг с другом или с другими органическими растворителями. Могут быть применены каталитическая гидрогенизация или химическое восстановление. Согласно еще одному предпочтительному воплощению настоящего изобретения R-энантиомерное соединение общей формулы 20 или его рацемат превращают в S-энантиомерное соединение общей формулы 14 или его рацемат (где Z1 и Z2 представляют собой водород или защитную группу, при условии,что, по меньшей мере, Z1 не является водородом) посредством применения в качестве агента, способного ввести карбонильную группу, N,N'-карбонилдиимидазола, фосгена, дифосгена или трифосгена, предпочтительно N,N'-карбонилдиимидазола. Реакцию проводят в приемлемом растворителе, предпочтительно толуоле. Согласно следующему предпочтительному воплощению способа реакцию R-энантиомерного соединения формулы 19 или его рацемата (где Z1 и Z2 представляют собой водород или защитную группу,при условии, что, по меньшей мере, Z1 не является водородом) и 4-(4-аминофенил)морфолин-3-она формулы 5 проводят предпочтительно в смеси протонного растворителя и воды при температуре 0-150 С,предпочтительно 60-90 С, предпочтительно в течение периода 20-40 ч. Согласно дополнительному предпочтительному воплощению изобретения S-энантиомерное соединение формулы 11 реагирует с соединением общей формулы Z1Z2NH (где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом), предпочтительно реакцию с N-бензил-1-фенилметанамином формулы проводят в отсутствие растворителя или в органическом растворителе или воде или их смеси, предпочтительно в присутствии вещества, связывающего органические или неорганические кислоты. Согласно предпочтительному воплощению изобретения превращение R-энантиомерного соединения формулы 10 а или его рацемата в R-энантиомерное соединение формулы 10b или его рацемат проводят посредством реакции с бромидом щелочного металла, предпочтительно с бромидом натрия. Конверсию в соединение формулы 10 с или его рацемат выполняют посредством реакции с йодидом щелочного металла, предпочтительно йодидом натрия и упомянутую реакцию проводят в приемлемом органическом растворителе или его смеси с водой при 0-150 С, предпочтительно 80-130 С. Согласно еще одному предпочтительному воплощению изобретения реакцию с соединением общей формулы Z1Z2NH (где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом), предпочтительно с N-бензил-1-фенилметанамином формулы 16) проводят в присутствии или отсутствие растворителя, в присутствии вещества, связывающего органические или неорганические кислоты, предпочтительно карбоната цезия при 0-150 С, предпочтительно 60-100 С. Согласно дополнительному предпочтительному воплощению изобретения R-энантиомерное соединение общей формулы 9 или его рацемат превращают в R-энантиомерное соединение общей формулы 10 или его рацемат посредством применения в качестве агента, приемлемого для введения карбонильной группы N,N'-карбонилдиимидазола, фосгена, дифосгена или трифосгена, предпочтительно N,N'-карбонилдиимидазола. Реакцию проводят в растворителе или смеси растворителей, предпочтительно толуоле,1-метил-2-пирролидоне или их смеси, при 0-150 С, предпочтительно при температуре флегмы растворителя или смеси растворителя. Изобретение также относится к 4-4-[(5S)-5-(аминометил)-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-ону или его рацемату формулы 12 и его S-энантиомерным солям общей формулы 3 или его ра-7 024685 цемическим солям (где HnX представляет собой моно- или поливалентную органическую или неорганическую кислоту, n представляет собой 1, 2 или 3, и X представляет собой ион кислотного остатка, при условии, что n представляет собой 1, а X не является хлором). Изобретение также относится к уксуснокислой соли 4-4-[(5S)-5-(аминометил)-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы или его рацемату. Изобретение также относится к способу получения S-энантиомерного соединения формулы 12 и его рацемата или S-энантиомерной соли общей формулы 3 или ее рацемической соли, предпочтительно уксуснокислой соли формулы 3d или ее рацемической уксуснокислой соли, содержащему подвергание Sэнантиомерного соединения общей формулы 14 или его рацемата (где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом) разделению, в случае применения рацемического исходного материала, предпочтительно S-энантиомерного соединения формулы 13 или его рацемата (в случае, когда Z1 и Z2 представляют собой бензил) разделению, в случае применения рацемического исходного материала, с последующим удалением защитной группы(п) и отделением S-энантиомерного основания формулы 12 или его рацемата, или, возможно, S-энантиомерной соли общей формулы 3 или ее рацемической соли, предпочтительно соли уксусной кислоты формулы 3b или рацемической соли уксусной кислоты. Согласно изобретению также предложены S-энантиомерные соединения общей формулы 14 и их рацематы (где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом и Z1 и Z2 вместе представляют собой группу иную чем фталимидо). Изобретение также относится к 4-4-[(5S)-5-[(дибензиламино)метил]-2-оксо-1,3-оксазолидин-3 ил]фенилморфолин-3-ону формулы 13 и его рацемату. Согласно изобретению также предложен способ получения S-энантиомерных соединений общей формулы 14 и их рацематов (где Z1 и Z2 представляют собой водород или защитную группу, при условии,что, по меньшей мере, Z1 не является водородом и Z1 и Z2 представляют собой бензил), содержащий подвергание соединения общей формулы 20 (где Z1 и Z2 представляют собой упомянутые выше) разделению,в случае применения рацемического исходного материала, и последующей реакции с агентом, способным ввести карбонильную группу. Изобретение также относится к R-энантиомерным соединениям общей формулы 20 и их рацематам(где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом и Z1 и Z2 вместе не могут представлять фталимидо). Изобретение также относится к 4-4-[(2R)-33-(дибензиламино)-2-гидропропил]аминофенил)морфолин-3-ону формулы Изобретение также относится к способу получения R-энантиомерных соединений общей формулы 20 и их рацематов (где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом, и предпочтительно Z1 и Z2 представляют собой бензил), содержащему подвергание S-энантиомерного соединения общей формулы 19 или его рацемата (где Z1 и Z2 представляют собой, упомянутые выше) разделению, в случае применения рацемического исходного материала, и последующей реакции с 4-(4-аминофенил)морфолин-3-оном формулы 5. Изобретение также относится к S-энантиомерным соединениям общей формулы 19 и их рацематам(где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом и в случае когда Z1 представляет собой бензил, Z2 не является бензилом). Изобретение также относится к получению S-энантиомерных соединений общей формулы 14 и их рацематам (где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом и Z1 и Z2 предпочтительно представляют собой бензил), содержащему подвергание R-энантиомерного соединения общей формулы 10 или его рацемата (где L2 представляет собой хлор, бром, йод, алкилсульфонилокси или арилсульфонилокси) разделению, в случае применения рацемического исходного материала, и последующей реакции продукта с соединением общей формулыZ1Z2NH (где Z1 и Z2 представляют собой упомянутые выше). Изобретение также относится к R-энантиомерным соединениям общей формулы 10 и их рацематам(где L2 представляет собой хлор, бром, йод, алкилсульфонилокси или арилсульфонилокси, предпочтительно хлор, бром или йод). Изобретение также относится к способу получения R-энантиомерных соединений формулы 10 и их рацематов, где L2 представляет собой хлор, бром, йод, алкилсульфонилокси или арилсульфонилокси,предпочтительно хлор, бром или йод, содержащему подвергание R-энантиомерного соединения формулы 9 (где L2 представляют собой, упомянутые выше) или его рацемата разделению, в случае применения рацемического исходного материала, и последующей реакции с агентом, способным ввести карбонильную группу (где L2 представляет собой упомянутый выше). Изобретение также относится к способу получения R-энантиомерного соединения формулы 10b или его рацемата формулы rac10b или R-энантиомерного соединения формулы 10 с или его рацемата формулы rac10 с, содержащему реакцию R-энантиомерного соединения формулы 10 а или его рацемата формулы rac10 а с бромидом щелочного металла, предпочтительно с бромидом натрия или йодидом щелочного металла, предпочтительно йодидом натрия. Изобретение также относится к R-энантиомерным соединениям общей формулы 9 и их рацематам,где L2 представляет собой хлор, бром, йод, алкилсульфонилокси или арилсульфонилокси, предпочтительно хлор, бром или йод. Наиболее распространенные формы реализации синтеза ривароксабана формулы 1 приведены на схемах реакции 7 а и 7b. Определения в формуле на схемах реакции 7 а и 7b представляют собой следующее: в общих формулах Z1Z2NH, 19, 20 и 14, Z1 и Z2 представляют собой водород или защитную группу,а именно обычную аминозащитную группу, например, бензил, замещенный бензил, п-метоксибензил,бензилоксикарбонил или третбутоксикарбонил, при условии, что, по меньшей мере, Z1 не является водородом, и Z1 и Z2 предпочтительно представляют собой бензил; в общей формуле 3 HnX представляет собой моно- или бивалентную органическую или неорганическую кислоту (где n представляет собой 1, 2 или 3 и X представляет собой ион кислотного остатка). В качестве органической кислоты могут быть применены, например, сульфоновые кислоты и карбоновые кислоты, предпочтительно моновалентные карбоновые кислоты, например, муравьиная кислота, уксусная кислота, пропионовая кислота, масляная кислота, и в качестве неорганических кислот могут быть применены, например, серная кислота, сернистая кислота, азотная кислота, фосфорная кислота, соляная кислота, бромоводород или йодоводород; в общих формулах 9 и 10, где L2 представляет собой хлор, бром, йод, алкилсульфонилокси или арилсульфонилокси, например, метансульфонилокси, бензолсульфонилокси или п-толуолсульфонилокси, предпочтительно хлор, бром или йод. На первом этапе реакционного пути, показанного на схеме 7 а, соединение формулы 11 реагирует с соединением общей формулы Z1Z2NH (где Z1 и Z2 представляют собой упомянутые выше) в отсутствие растворителя или в органическом растворителе или воде или их смеси, предпочтительно в присутствии вещества, связывающего органические или неорганические кислоты. На втором этапе реакционного пути, показанного на схеме 7 а, соединение общей формулы 19 (гдеZ1 и Z2 представляют собой упомянутые выше) реагирует с соединением формулы 5. Реакцию проводят предпочтительно в протонном растворителе или смеси растворителей или в смеси протонного растворителя и воды при 0-150 С, предпочтительно при 60-90 С, в течение 0,5-60 ч, предпочтительно 20-40 ч. На третьем этапе реакционного пути, показанного на схеме 7 а, из полученного соединения общей формулы 20 получают соединение общей формулы 14 (где Z1 и Z2 представляют собой упомянутые выше). Реакцию проводят посредством применения агента, способного ввести карбонильную группу, предпочтительно N,N'-карбонилдиимидазола, фосгена, дифосгена или трифосгена, предпочтительно N,N'карбонилдиимидазола, в приемлемом растворителе или смеси растворителей, предпочтительно толуоле. На четвертом этапе реакционного пути, приведенного на схеме реакции 7 а, из соединения общей формулы 14 (где Z1 и Z2 представляют собой упомянутые выше) получают основание формулы 12 или его соль общей формулы 3 посредством удаления защитной группы, предпочтительно посредством восстановления (где HnX представляет собой упомянутый выше). Восстановление проводят в С 1-С 4 алифатическом спирте, ледяной уксусной кислоте, воде или смеси упомянутых растворителей друг с другом или дополнительным органическим растворителем. Могут быть применены каталитическая гидрогенизация или химическое восстановление. Соединение формулы 3b выделяют напрямую или из уксуснокислой соли формулы 3b, основание формулы 12 выделяют известным способом или, возможно, полученную уксуснокислую соль превращают в соль общей формулы 3 известным способом (где HnX представляют собой упомянутый выше). На последнем этапе реакционного пути, приведенного на схеме реакции 7 а, основание формулы 12 или его соль общей формулы 3 (где HnX представляет собой, упомянутый выше) реагирует с 5-хлортиофен-2-карбоновой кислотой формулы 15 в присутствии агента реакции сочетания и органического или неорганического основания в органическом растворителе. В качестве агента реакции сочетания могут быть применены N,N'-диизопропилкарбодиимид (ДИК), N,N'-дициклогексилкарбодиимид (ДЦК),-9 024685 ангидрид трипропилфосфоновой кислоты (ТФА) или N,N'-карбонилдиимидазол (КДИ), предпочтительно хлорэтилформиат или КДИ. В качестве растворителя могут быть применены ацетонитрил, дихлорметан,ацетон, толуол, тетрагидрофуран или их смесь друг с другом или водой. В качестве органического или неорганического основания могут быть применены триэтиламин, диизопропилэтиламин, карбонат натрия или гидрокарбонат натрия. Реакцию проводят при 0-100 С, предпочтительно при 40-70 С. Применение вещества, связывающего кислоту может быть исключено. На первом этапе реакционного пути, приведенного на схеме реакции 7b из соединения общей формулы 9 получают соединение общей формулы 10 (где L2 означает упомянутое выше). В реакции в качестве агента, способного ввести карбонильную группу, могут быть применены предпочтительно N,N'карбонилдиимидазол, фосген, дифосген или трифосген, предпочтительно N,N'-карбонилдиимидазол. Реакцию проводят в приемлемом растворителе или смеси растворителей, предпочтительно в толуоле, 1 метил-2-пирролидоне или их смеси, при 0-150 С, предпочтительно при температуре флегмы растворителя или смеси растворителей. В случае соединений общей формулы 9, приведенной на схеме реакции 7b L2 представляет собой хлор, т.е. соединение представляет собой формулу и бром- или йод-аналоги соединений общей формулы 9, т.е. соединений формулы могут быть приготовлены из соединения формулы 9 а известным способом предшествующего уровня техники. На втором этапе реакционного пути схемы реакции 7b получают соединения общей формулы 14 посредством реакции соединения общей формулы 10 (где L2 представляет собой упомянутое выше) с соединением общей формулы Z1Z2NH (где Z1 и Z2 представляют собой упомянутые выше). Реакцию проводят в отсутствие или присутствии растворителя, в присутствии вещества, связывающего органические или неорганические кислоты, предпочтительно карбоната цезия при температуре 0-150 С, предпочтительно 60-100 С. Дополнительные реакционные этапы, два последних этапа, приведенные на схеме реакции 7b,идентичны двум последним этапам, приведенным на схеме реакции 7 а. Предпочтительное воплощение способа получения ривароксабана формулы 1 по изобретению приведено на схеме реакции 8 а. На схемах реакции 7 а и 8 а мы указали хиральность промежуточных соединений согласно номенклатуре Кана-Ингольда-Прелога. Новый путь синтеза также осуществляют посредством применения рацемических форм соединений, приведенных на схеме реакции 8 а, начиная с эпихлоргидрина. Рацемические соединения, соответствующие промежуточным соединениям формулы 18, 13 и 3b приемлемы для образования соли и таким образом их соответствующие энантиомеры могут быть получены посредством разделения. Разделение проводят при помощи обычных способов уровня техники при помощи фермента или кинетического разделения или образования диастериомерной соли и последующего отделения диасте- 10024685 риомерных производных посредством хроматографии или фракционной кристаллизации или физических способов. Из соединений, приведенных на схеме реакции 8 а 4-(4-аминофенил)морфолин-3-он формулы 5,(2S)-хлорметилоксиран, широкоизвестное название эпихлоргидрин, формулы 11,N-бензил-1-фенилметинамин, широкоизвестное название дибензиламин, формулы 16,(2S)-N,N-дибензил-1-оксиран-2-ил-метанамин формулы 17,5-хлор-тиофен-2-карбоновая кислота формулы 15 и ривароксабан формулы 1,широкоизвестные в области техники соединения 1, 5, 11, 16 и 15 коммерчески доступны. 4-(4-Аминофенил)морфолин-3-он формулы 5 раскрыт в ЕР 11261606, WO 2004/101556, WO 2005/026135, WO 2006/0632193 и статье IPCOM0000195906D, опубликованной в 2010 в IP.com.Journal.(S)-эпихлоргидрин формулы 11 и его рацемическая форма, дибензиламин формулы 16 и 5-хлортиофен-2 карбоновая кислота формулы 15 коммерчески доступны. (2S)-N,N-дибензилоксиран-2-ил-метанамин формулы 17 описан в СР 2003200582. Получение рацемической формы соединения формулы 17 раскрыто среди прочего в US 4656180. Из промежуточных соединений, приведенных на реакционной схеме 8 а 4-(4-[(2R)-3-(дибензиламино)-2-гидрокси-пропил]аминофенил)морфолин-3 формулы 18 4-(4-(5S)-5-[(дибензиламино)метил]-2-оксо-1,3-оксазолидин-3-илфенил)морфолин-3-он формулы 13 и соль 4-4-[(5S)-5-(аминометил)-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-уксусной кислоты формулы 3d и их рацемические формы не известны из уровня техники. Соединения формул 18 и 13 приемлемы для образования соли. Кроме того, соединения формул 18 и 13 и их рацемические эквиваленты могут существовать в виде сольватов и могут образовывать сокристаллы, соответственно. Изобретение также относится к солям, сольватам и гидратам соединений формул 18 и 13 и их рацематам и также к со-кристаллам упомянутых соединений. Изобретение также относится к 4-(4-[(2R)-3-дибензиламино-2-гидроксипропил]аминофенилморфолин-3-ону формулы 18 и его рацемической форме. Изобретение также относится к получению соединения формулы 18, исходя из соединения формулы 17. Изобретение также относится к получению рацемической формы соединения формулы 18, исходя из рацемической формы соединения формулы 17. Изобретение также относится к соединениям общей формулы 20 и их рацемическим формам (где Z1 2 и Z представляют собой водород или защитную группу, при условии, что,по меньшей мере, Z1 не является водородом). Изобретение также относится к способу получения соединений общей формулы 20 и их рацемическим формам, исходя из соединения общей формулы 19 или его рацемической формы. Изобретение также относится к 4-(4-(5S)-5-[(дибензиламино)метил]-2-оксо-1,3-оксазолидин-3 илфенилморфолин-3-ону формулы 13 и его рацемической форме. Также изобретение относится к способу получения соединения формулы 13, исходя из соединения формулы 18. Изобретение также относится к способу получения рацемической формы соединения формулы 13,исходя из рацемической формы соединения формулы 18. Также изобретение относится к соединениям общей формулы 14 и их рацемическим формам (где Z1 2 и Z представляют собой водород или защитную группу, при условии, что, по меньшей мере, Z1 не является водородом). Также изобретение относится к способу получения соединений общей формулы 14 и их рацемическим формам, исходя из соединений общей формулы 20 и его рацемической формы. Также изобретение относится к способу получения соединения формулы 12 и его рацемической формы, начиная с соединения формулы 14 или его рацемической формы. Также изобретение относится к уксуснокислой соли 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 3b и ее рацемической форме. Изобретение также относится к способу получения уксуснокислой соли 4-4-[(5S)-5-аминометил-2 оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 3b или ее рацемической форме, начиная с соединения формул 13 или 14 или их рацемических форм. Изобретение также относится к способу получения ривароксабана формулы 1 посредством реакции соединения общей формулы 3 (где HnX представляет собой моно- или поливалентную органическую или неорганическую кислоту; n представляет собой 1, 2 или 3, и X представляет собой ион кислотного остатка) или соединения формулы 12 и 5-хлортиофен-2-карбоновой кислоты формулы 15. Согласно предпочтительному воплощению способа (см. реакционную схему 8 а) соединение формулы 17 получают посредством реакции соединения формулы 11 с дибензиламином формулы 16 (в дан- 11024685 ном случае Z1 и Z2 представляют собой бензил). Реакцию проводят в отсутствие растворителя или в органическом растворителе или воде или их смеси и предпочтительно в присутствии вещества, связывающего органическую или неорганическую кислоту. Согласно предпочтительному воплощению способа по изобретению соединение формулы 18 получают посредством реакции 4-аминофенилморфолинона формулы 5 с (2S)-N,N-дибензил-1-оксиран-2-илметанамином формулы 17. Реакцию предпочтительно проводят в протонном растворителе или смеси растворителей или в смеси протонного растворителя и воды при 0-150 С, предпочтительно 60-90 С в течение периода 0,5-60 ч, предпочтительно 20-40 ч. Соединение формулы 18 обладает высокой температурой плавления, быстро кристаллизуется и в частности приемлемо для применения в качестве промежуточного соединения при фармацевтическом производстве промышленного масштаба. Согласно способу по настоящему изобретению указанное соединение может быть получено без перекристаллизации с химической чистотой выше 98% и с очень высокой энантиомерной чистотой 99%. Из предшествующего уровня техники известно, что раскрытие кольца соединений эпоксидного типа при помощи нуклеофильных агентов приводит к образованию смеси региоизомеров в зависимости от того какой из нуклеофильных центров будет атакован (в случае присутствия аминогрупп соединения формулы 5), что приводит к положительной поляризации углеродного атома эпоксигруппы соединения формулы 17. Кроме того, в реакции данного типа, в зависимости от условий реакции существует опасность частичной или полной рацемизации хирального центра. Неожиданно оказалось, что при применении в способе по настоящему изобретению оптимизированных условий реакции нежелательный региоизомер образуется только в очень небольшой степени, рацемизация практически не происходит и полученное соединение формулы 18 может быть охарактеризовано химической чистотой 98,4% и значением энантиомерной чистоты 99%. Согласно предпочтительному конкретному воплощению согласно настоящего изобретения рацемическую форму соединения формулы 18 получают посредством реакции 4-амино-фенил-морфолинона формулы 5 с рацемической формой N,N-дибензил-1-оксиран-2-ил-метанамина формулы 17. Реакцию проводят предпочтительно в протонном растворителе или смеси растворителей или в смеси протонного растворителя и воды при 0-150 С, предпочтительно при 40-80 С в течение периода времени 0,5-60 ч,предпочтительно 40-50 ч. Согласно способу по настоящему изобретению рацемическая форма соединения формулы 18 может быть приготовлена с очень хорошим выходом. Соединение обладает высокой температурой плавления,может быть быстро кристаллизовано и приемлемо в качестве промежуточного соединения при способах производства в фармацевтической промышленности. Упомянутый продукт, в случае получения согласно способу по настоящему изобретению, может быть применен на дополнительных этапах синтеза без перекристаллизации. При оптимизированных реакционных условиях настоящего изобретения нежелательный региоизомер образуется только в очень небольшом количестве и полученное промежуточное соединение (рацемическая форма соединения формулы 18) может быть применено на дополнительных этапах синтеза без перекристаллизации. Согласно предпочтительному конкретному воплощению способа по настоящему изобретению соединение формулы 13 получают посредством взаимодействия 4-(4-[(2R)-3-дибензиламино-2-гидроксипропил]аминофенил)морфолин-3-она формулы 18 и агента, способного ввести карбонильную группу,предпочтительно N,N'-карбонилдиимидазола, фосгена, дифосгена или трифосгена, предпочтительноN,N'-карбонилдиимидазола в приемлемом растворителе, предпочтительно толуоле. Соединение формулы 13 обладает высокой температурой плавления, может быть легко кристаллизовано и высоко приемлемо в качестве промежуточного соединения при способах производства в фармацевтической промышленности. Согласно способу по настоящему изобретению соединение формулы 13 может быть получено с очень высоким выходом, химической чистотой 99,9% и практически энантиомерной чистотой 99,9%. Из предшествующего уровня техники известно, что в реакциях данного типа вместо желаемой внутримолекулярной циклизации в ходе межмолекулярной реакции могут образовываться побочные продукты, поскольку применяемый реагент Cl связывает две молекулы друг с другом через их NH- и ОН- функциональные группы. Кроме того, поскольку в исходном материале формулы 18 гидроксильная группа прикреплена к хиральному центру, потеря воды может привести к образованию побочных продуктов, содержащих двойную связь, а также частичной или полной рацемизации. Неожиданно обнаружено, что при применении оптимизированных условий реакции в способе по настоящему изобретению образование побочных продуктов является очень низким, и полученное соединение формулы 13 может быть охарактеризовано химической чистотой 99,9% и значением энантиомерной чистоты 99,9%. Предпочтительное конкретное воплощение способа получения ривароксабана формулы 1 по изобретению приведено на схеме реакции 8b. На схемах реакции 7b и 8b указана хиральность промежуточных соединений согласно КанИнгольд-Прелогу. Как видно из схемы реакции 8b исходный материал способа представляет собой (2R)хлорметилоксиран (хорошо известное название (R-эпихлоргидрин) формулы 11, и из упомянутого соединения в шесть этапов получают ривароксабан формулы 1, имеющий (S)-конфигурацию. На этапе реакции 10b(10 с)13 конфигурацию очевидно изменяют, однако в действительности это не является инверсионным этапом, а конверсия (R)-хиральности в (S)-хиральность является только изменением номенклатуры в результате новой иерархии групп при введении нового структурного звена. Из соединений, приведенных на схеме реакции 8b,4-(4-аминофенил)морфолин-3-он формулы 5,(2S)-хлорметилоксиран (широкоизвестное название (S)-эпихлоргидрин) формулы 11,N-бензил-1-фенилметанамин (широко известное название дибензиламин) формулы 16,5-хлортиофен-2-карбоновая кислота формулы 15 и ривароксабан формулы 1 известны из области техники и коммерчески доступны. Из промежуточных соединений и их непосредственных аналогов, приведенных на схеме реакции 8b,4-4-[2R)-3-хлор-2-гидроксипропил)амино]фенилморфолин-3-он формулы 9 а,4-4-[2R)-3-бром-2-гидроксипропил)амино]фенилморфолин-3-он формулы 9b (представляет собой соединение формулы 9, где L2 представляет собой бром),4-4-[2R)-3-йод-2-гидроксипропил)амино]фенил]морфолин-3-он (соединение общей формулы 9 с,где L2 представляет собой йод),4-4-[(5R)-5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы 10 а,4-4-[(5R)-5-бромметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы 10b и 4-4-[(5R)-5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы 10 с подпадают под общую формулу WO 2010/124385, однако отсутствует ссылка на их получение и не раскрыты физические константы, приемлемые для идентификации этих соединений. Из промежуточных соединений, приведенных на схеме реакции 8b,4-(4-(5S)-5-(дибензиламинометил]-2-оксо-1,3-оксазолидин-3-илфенилморфолин-3-он формулы 13 и уксуснокислая соль 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 3 не известны из области техники. Для справки, мы проводили последовательность реакции, начиная с коммерчески доступного (S)эпихлоргидрина формулы 11S, и получали энантиомер ривароксабана формулы 1R, а именно (5-хлор-N5R)-2-оксо-3-[4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-илметил(метил)тиофен-2-карбоксамид (схема реакции 9). Из промежуточных соединений, приведенных на схеме реакции 9 4-4-[2S)-3-хлор-2-гидроксипропил)амино]фенилморфолин-3-он формулы 9aS,4-4-[(5S)-5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы 10aS,4-4-[(5S)-5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы 10cS,4-(4-5R-5-[(дибензиламинометил]-2-оксо-1,3-оксазолидин-3-илфенил)морфолин-3-он формулы 13R и уксуснокислая соль 4-4-[5R-5-аминометил-2-оксо-1,3-оксазолидин-3-илфенилморфолин-3-она формулы 3bR не описаны в предшествующем уровне техники. Также мы проводили последовательность синтеза, начиная с коммерчески доступного эпихлоргидрина формулы rac11, и получали рацемический эквивалент ривароксабана формулы rac1 (см. реакционную последовательность 10). Из промежуточных соединений, приведенных на схеме реакции 10,4-4-[(3-хлор-2-гидроксипропил)амино]фенилморфолин-3-он формулы rac9 а,4-4-[5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы rac10 а,4-4-[5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы rac10 с,4-4-[5-(дибензиламинометил)-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы rac13 и уксуснокислая соль 4-4-[5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы rac3b не описаны в предшествующем уровне техники. Соединения формул rac9a, rac13 и rac3b содержат азотистое основание и таким образом посредством разделения могут быть получены потенциально приемлемые энантиомеры, которые могут быть применены для получения ривароксабана формулы 1, как приведено на реакционной схеме 8b посредством применения способов, известных в области техники. Разделение проводят посредством обычных способов, например, посредством применения фермента, кинетического разделения, образования диастериомерной соли и последующего отделения диасте- 13024685 риомерных производных посредством хроматографии или фракционированной кристаллизации или посредством физических способов. Соединения формул 9 а и 13 и их энантиомеры и рацемические формы могут формировать соли. Кроме того, соединения формул 9 а, 10 а, 10b, 10 с и 13 и их энантиомеры и рацемические формы также могут существовать в виде гидратов и других сольватов и также могут образовывать ко-кристаллы. Изобретение также относится к соединениям формул 9 а, 10 а, 10b, 10 с и 13 и их энантиомерам и их рацемическим формам и их солям, гидратам, сольватам и ко-кристаллам. Изобретение также относится к 4-4-[2R)-3-хлор-2-гидроксипропил)амино]фенилморфолин-3 ону формулы 9 а (соединение формулы 9, где L2 представляет собой хлор) и его рацемической форме. Изобретение также относится к 4-4-[2R)-3-бром-2-гидроксипропил)амино]фенилморфолин-3 ону формулы 9b (соединение формулы 9, где L2 представляет собой бром) и его рацемической форме. Изобретение также относится к 4-4-[2R)-3-йод-2-гидроксипропил)амино]фенилморфолин-3-ону формулы 9 с (соединение формулы 9, где L2 представляет собой йод) и его рацемическим формам. Согласно настоящему изобретению предложен способ получения соединения формулы 9 а и его рацемической формы, начиная с соединения формулы 11 и его рацемической формы. Согласно настоящему изобретению также предложено соединение 4-4-[(5R)-5-хлорметил-2-оксо 1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы 10 а и его рацемической формы. Согласно настоящему изобретению также предложено соединение 4-4-[(5R)-5-бромметил-2-оксо 1,3-оксазолидин-3-ип]фенилморфолин-3-он формулы 10b и его рацемической формы. Согласно настоящему изобретению также предложено соединение 4-4-[(5R)-5-йодметил-2-оксо 1,3-оксазолидин-3-ил]фенилморфолин-3-он формулы 10 с и его рацемической формы. Согласно настоящему изобретению также предложен способ получения соединение 10 а и его рацемической формы, начиная с соединения формулы 9 а или его рацемической формы. Согласно настоящему изобретению также предложен способ получения соединения 10b и его рацемической формы, начиная с соединения формулы 9b или его рацемической формы. Согласно настоящему изобретению также предложен способ получения соединения 10 с и его рацемической формы, начиная с соединения формулы 9 с или его рацемической формы. Согласно настоящему изобретению также предложен способ получения соединения формулы 10b и его рацемической формы, начиная с соединения формулы 10 а или его рацемической формы. Согласно настоящему изобретению также предложен способ получения соединения формулы 10 с и его рацемической формы, начиная с соединения формулы 10 а или его рацемической формы. Согласно настоящему изобретению также предложен способ получения соединения формулы 13 и его рацемической формы, начиная с соединения формулы 10 а, 10b или 10 с или его рацемической формы. Согласно настоящему изобретению также предложен способ получения соединения формулы 14 и его рацемической формы, начиная с соединения формулы 10 а, 10b или 10 с или его рацемической формы. Согласно предпочтительному конкретному воплощению способа по изобретению (см. схему реакции 8b) соединение формулы 9 а получают посредством реакции 4-аминофенилморфолина формулы 5 с эпихлоргидрином формулы 11 в органическом растворителе или смеси водорастворимого органического растворителя и воды при температуре 0-150 С, предпочтительно при температуре флегмы растворителя или смеси растворителей в течение 0,5-60 ч. Соединения общей формулы 9 имеют высокую температуру плавления, могут быть быстро кристаллизованы и в частности приемлемы для применения в качестве промежуточного соединения способов производства в фармацевтической промышленности. Согласно способу по настоящему изобретению соединение формулы 9 а может быть приготовлено без перекристаллизации с химической чистотой выше 95% и выдающейся энантиомерной чистотой свыше 99%. Из предшествующего уровня техники известно, что раскрытие кольца соединений эпоксидного типа при помощи нуклеофильных агентов приводит к образованию смеси региоизомеров в зависимости от того какой из нуклеофильных центров (в случае присутствия аминогрупп соединения формулы 5) атакован, что приводит к положительной поляризации атома углерода эпоксигруппы соответствующего соединения формулы 11. Кроме того, в реакциях данного типа, в случае оптически активных исходных материалов, в зависимости от условий реакции существует риск частичной или полной рацемизации хирального центра. Неожиданно было обнаружено, что при оптимизированных условиях реакции согласно настоящему изобретению нежелательный региоизомер образуется только в очень небольшом количестве. В случае применения оптически активных исходных материалов рацемизация практически не происходит. Согласно другому предпочтительному конкретному воплощению изобретения соединение формулы 10 а получают посредством реакции соединения формулы 9 а с агентом, способным ввести карбонильную группу, предпочтительно N,N'-карбонилдиимидазолом, фосгеном, дифосгеном или трифосгеном, предпочтительно N,N'-карбонилдиимидазолом в приемлемом растворителе или смеси растворителей. Предпочтительно в толуоле, 1-метил-2-пирролидоне или их смеси при 0-150 С, предпочтительно при температуре флегмы растворителя или смеси растворителей. Соединения общей формулы 10 имеют высокую температуру плавления, могут быть быстро кри- 14024685 сталлизованы и, в частности, приемлемы для применения в качестве промежуточных соединений способов производства в фармацевтической промышленности. Согласно способу по настоящему изобретению эти соединения могут быть получены с химической чистотой 95% и энантиомерной чистотой свыше 99% без перекристаллизации. Соединение формулы 10 с может быть получено из соединения формулы 10 а с высоким выходом 95,2%. Из предшествующего уровня техники известно, что в процессе реакций данного типа вместо желаемого внутримолекулярного закрытия кольца имеет место межмолекулярная реакция и могут образовываться побочные продукты, поскольку применяемый агент Cl соединяет две молекулы через их NH- и ОН-функциональные группы. Кроме того, поскольку в исходных материалах формулы 9 гидроксигруппа прикреплена к хиральному центру, потеря и дополнительное поглощение воды могут привести к образованию побочных продуктов, содержащих двойную связь, а также может иметь место частичная или полная рацемизация. Неожиданным образом мы обнаружили, что при оптимизированных условиях реакции настоящего изобретения побочные продукты образуются только в очень небольшой степени и рацемизация даже не происходит. Согласно предпочтительному конкретному воплощению настоящего изобретения соединение формулы 10 с получают из соединения формулы 10 а посредством реакции упомянутого соединения формулы 10 а с йодидом щелочного металла, предпочтительно йодидом натрия, в приемлемом растворителе или смеси растворителей, предпочтительно в смеси органического растворителя и воды, при 0-150 С, предпочтительно 80-130 С. Согласно другому предпочтительному конкретному воплощению настоящего изобретения соединение формулы 10b получают из соединения формулы 10 а посредством реакции упомянутого соединения формулы 10 а с бромидом щелочного металла, предпочтительно бромидом натрия, в приемлемом органическом растворителе или смеси растворителей, при 0-150 С, предпочтительно 80-130 С. Согласно предпочтительному конкретному воплощению настоящего изобретения соединение формулы 13 получают посредством реакции соединения формулы 10 а, 10b или 10 с приемлемой хиральности, предпочтительно соединение формулы 10 с приемлемой хиральности, с дибензиламином формулы 16 (в данном случае Z1 и Z2 представляют собой бензил) в присутствии или отсутствие растворителя, в присутствии карбоната цезия, при 0-150 С, предпочтительно 60-100 С, после чего реакционную смесь обрабатывают и полученный продукт подвергают при желании дополнительной очистке. Соединение формулы 13 обладает высокой температурой плавления, может быть быстро кристаллизовано и в частности приемлемо для применения в качестве промежуточного соединения способов производства фармацевтической промышленности. Согласно другому способу настоящего изобретения(схема реакции 8b) данное соединение может быть получено после перекристаллизации с выдающейся химической чистотой свыше 99,5% и энантиомерной чистотой свыше 99,9%. Из предшествующего уровня техники известно, что при реакции нуклеофильного замещения (SN) данного типа, в случае оптически активных исходных материалов, в зависимости от условий реакции,существует опасность частичной или полной рацемизации хирального центра или инверсии, соответственно. Учитывая характерную побочную реакцию галид водорода отделяют из-за основности нуклеофильного агента (в случае присутствия дибензиламина). Неожиданным образом мы обнаружили, что при применении оптимизированных условий реакции по настоящему изобретению имеет место недетектируемая элиминация побочной реакции, и в случае оптически активного исходного материала не происходит ни инверсия, ни рацемизация. Последние два этапа способа, приведенные на схемах реакции 8 а и 8b, идентичны и по этой причине два последних этапа двух предпочтительных конкретных воплощений настоящего изобретения обсуждаются вместе. Согласно предпочтительному конкретному воплощению способа по настоящему изобретению соединение формулы 3b получают посредством подвергания 4-(4-(5S)-[дибензиламинометил]-2-оксо-1,3 оксазолидин-3-илфенил)морфолин-3-она формулы 13 каталитической гидрогенизации или химическому восстановлению в C1-4-алифатическом спирте, ледяной уксусной кислоте, воде или смеси упомянутых растворителей, образованной ими друг с другом или с другими органическими растворителями, обработке реакционной смеси и отделению продукта посредством добавления уксусной кислоты в виде соли формулы 3b. Согласно другому предпочтительному конкретному воплощению соединение формулы 3b получают посредством превращения полученного 4-(4-(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 12 посредством какого-либо пути синтеза, известного в области техники или описанного в настоящей заявке на патент, в соль формулы 3b посредством добавления уксусной кислоты. Соединение формулы 3b обладает высокой температурой плавления, может быть легко кристаллизовано и в частности приемлемо в качестве промежуточного соединения способов фармацевтического производства. В случае получения согласно способу по настоящему изобретению упомянутое соединение получают с очень высоким выходом (98%), выдающейся химической чистотой (99,5%) и высокой энантиомерной чистотой (99,9%) без перекристаллизации. Согласно предпочтительному конкретному воплощению способа по настоящему изобретению ривароксабан формулы 1 получают посредством реакции уксуснокислой соли 4-(4-5S(-5-аминометил-2 оксо 1,3-оксазолидин-3-ил)фенил)морфолин-3-она формулы 3b с 5-хлортиофен-2-карбоновой кислотой формулы 15 в присутствии агента реакции сочетания и органического или неорганического основания, в органическом растворителе. В качестве агента реакции сочетания могут быть применены хлорэтилформиат, N,N'-диизопропилкарбодимид (ДИК), N,N'-дициклогексилкарбодиимид (ДЦК), трипропилфосфоновый ангидрид (ТФА) или N,N'-карбонилдиимидазол (КДИ), предпочтительно хлорэтилформиат или КДИ. В качестве растворителя могут быть применены ацетонитрил, дихлорметан, ацетон, толуол, тетрагидрофуран или их смесь или смесь упомянутого органического растворителя, образованная с водой. В качестве органического или неорганического основания могут быть применены триэтиламин, диизопропилэтиламин, карбонат натрия или гидрокарбонат натрия. Реакцию проводят при температуре 0-110 С,предпочтительно 40-70 С. Возможно, применение вещества, связывающего кислоту, может быть опущено. Преимуществом способа по настоящему изобретению является то, что промежуточные соединения обладают высокой температурой плавления, могут быть быстро кристаллизованы и высоко приемлемы в качестве промежуточных соединений способов фармацевтического производства. Упомянутые соединения могут быть получены согласно способу по настоящему изобретению с наивысшей химической и энантиомерной чистотой без перекристаллизации. Таким образом, например, соединения формул 13 и 3b могут быть получены согласно способу, приведенному на реакционной схеме 8b, без перекристаллизации с химической чистотой свыше 95% для соединения формулы 13, предпочтительно 99,4% и с энантиомерной чистотой свыше 99% для соединений формул 13 и 3b, предпочтительно 99,9%. Дополнительным преимуществом способа по изобретению являются высокие выходы этапов реакции. Дополнительным преимуществом способа по изобретению является то, что из промежуточного соединения 4-4-[(5S)-5-(дибензиламинометил)-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 13 защитная группа может быть удалена при намного более мягких условиях, чем фталильная защитная группа, применяемая согласно WO 2005/068456 (см. схему реакции 4) и поэтому последнее промежуточное соединение формулы 3b пути синтеза может быть получено с более высокой чистотой, чем описанная в уровне техники. Дополнительное преимущество способа по настоящему изобретению заключается в том, что реагент формулы 15, применяемый на последнем этапе (ацилирование) получения ривароксабана формулы 1 является более выигрышным по некоторым свойствам, чем хлорангидрид формулы 4. Эти преимущества представляют собой следующее: соединение формулы 15 является твердым и кристаллическим, в то время как соединение формулы 4 представляет собой вязкую субстанцию, сложную в обращении,соединение формулы 15 является стабильным, в то время как соединение формулы 4 легко гидролизуется, содержание его эффективного количества постепенно снижается при хранении, даже в случае хранения при 0-5 С,при получении соединения формулы 15 применение корродирующего вещества, тионилхлорида,которое обладает неприятным запахом и может быть сложно удалимым, может быть элиминировано, и можно избежать обработки токсичными газами, неблагоприятными для окружающей среды, образующимися при реакции тионилхлорида,соединение формулы 15 является значительно более дешевым реагентом, чем соединение формулы 4. Дополнительные преимущества способа по настоящему изобретению по сравнению со способом,описанным в WO 2011/012321, который также применяет 5-хлортиофен-2-карбоновую кислоту формулы 15, представляют собой следующее: посредством выбора более приемлемого растворителя и применения более выигрышных условий реакции и уксуснокислой соли формулы 3b вместо солянокислой соли согласно способу по изобретению может быть достигнут выход 87%, в отличии от выхода неочищенного продукта 72%, раскрытого в WO 2011/012321. После перекристаллизации общий выход согласно WO 2011/012321 составляет 61%, в то время как общий выход согласно настоящему изобретению составляет 70%. В WO 2011/012321 не приведены данные по чистоте, в то время как ВЭЖХ чистота продукта, полученного согласно способу по изобретению составляет 99,32%. Дополнительным преимуществом способа по изобретению является то, что из уксуснокислой среды, применяемой при каталитической реакции дебензилирования (133b), уксуснокислая соль формулы 3b может быть непосредственно выделена с исключительно высоким выходом (98%), химической чистотой 99,91% и энантиомерной чистотой 99,9%. Таким образом способ по настоящему изобретению напрямую приемлем для получения конечного продукта формулы 1, являющегося чистым и удовлетворяющим требованиям чистоты фармацевтической промышленности. Дополнительным преимуществом способа, приведенного на реакционной схеме 8 а является то, что при оптимизированных условиях реакции, применяемых при получении соединения формулы 18, неже- 16024685 лательные региоизомеры образуются, но в очень небольшой степени и рацемизация практически не происходит. Таким образом, соединение формулы 18 получают с химической чистотой 98,4% и оно может быть охарактеризовано значением энантиомерной чистоты 99%. Дополнительным преимуществом способа, приведенного на схеме реакции 8 а является то, что при оптимизированных условиях реакции, применяемых при получении соединения формулы 13, побочные продукты образуются только в очень небольшой степени и рацемизация вовсе не происходит. Таким образом соединение формулы 18 получают с химической чистотой 99,9%, и оно может быть охарактеризовано значением энантиомерной чистоты 99%. Дополнительным преимуществом способа, приведенного на реакционных схемах 7b и 8b является то, что при оптимизированных условиях реакции, применяемых при реакции соединений формулы 11 и 5, образование нежелательного региоизомера является очень незначительным, и в случае применения оптически активного исходного материала, рацемизация практически не происходит. Дополнительным преимуществом способа, приведенного на схемах реакции 7b и 8b является то,что при оптимизированных условиях реакции, применяемых при получении соединения формулы 10,побочные продукты образуются только в очень небольшой степени и рацемизация вовсе не происходит. Дополнительным преимуществом способа, приведенного на схемах реакции 7b и 8b является то,что при оптимизированных условиях реакции, применяемых при получении соединения формулы 13 детектируемая побочная реакция элиминации не происходила и, в случае применения оптически активных исходных материалов, рацемизация не происходит вовсе. Дополнительным преимуществом способа по настоящему изобретению, приведенному на реакционных схемах 7 а, 7b, 8 а и 8b, является то, что он предлагает путь синтеза для получения ривароксабана формулы 1, который является более эффективным и более приемлем для производства в промышленном масштабе, чем способы получения ривароксабана формулы 1, известные в области техники. Дополнительные детали настоящего изобретения будут приведены в примерах, без ограничения объема правовой охраны упомянутыми примерами. Пример 1 Получение (2S)-N,N-дибензил-1-оксиран-2-ил-метанамина формулы 17 К раствору 49,25 г (48 мл, 0,25 моль) дибензиламина (соединение формулы 16) в 40 мл 2-пропанола при 0 С добавляют по капле 25,44 г (21,56 мл, 0,275 моль) (S)-эпихлоргидрина (соединение формулы 11) при перемешивании. После окончания добавления реакционной смеси позволяют нагреваться до комнатной температуры и перемешивают в течение 24 ч. К реакционной смеси добавляют 90 мл 2-пропанола и смесь охлаждают до 0 С. К реакционной смеси по частям добавляют 112,0 г (2,0 моль) гидроксида калия, температуру поддерживают при 0 С, после окончания добавления реакционную смесь перемешивают в течение дополнительного периода 30 мин, после чего добавляют 250 мл дистиллированной воды и 150 мл гексана. Фазы разделяют, водный слой экстрагируют три раза при помощи 100 мл гексана каждый, объединенные органические фазы промывают дважды при помощи 150 мл воды каждый, высушивают над сульфатом магния и растворитель удаляют под вакуумом. Таким образом, получают 62,0 г(dd, J1=6,2 Гц, J2=13,7 Гц, 1 Н), 2,39 (dd, J1=2,7 Гц, J2=4,9 Гц, 1 Н) ppm. 13 С ЯМР: (CDCl3, 125 МГц): 139,3, 128,8, 128,2, 126,9, 58,9, 55,8, 51,0, 45,0 ppm. Элементарный анализ для формулы C17H19NO (M:253,35): С 80,60: Н 7,56 :N 5,53%. Обнаружено: С 78,90: Н 8,04: N 5,45%. Вращение: []20D=+5,12 (588 нм/20 С; с=0,05 г/10 см 3 ДМСО). Пример 2 Получение 4-(4-[(2R)-3-дибензиламино-2-гидроксипропил]аминофенил)морфолин-3-она формулы 18 19,2 г 4-Аминофенилморфолинона (соединение формулы 5) суспендируют в смеси 150 мл 2 пропанола и 5 мл дистиллированной воды при 25 С, после чего добавляют 25,3 г (0,1 моль) (2S)-N,Nдибензил-1-оксиран-2-ил-метанамина (соединение формулы 17). Реакционную смесь нагревают до 8082 С и перемешивают при данной температуре в течение 44 ч. На 18-м часу к реакционной смеси добавляют 12,6 г (0,05 моль) (2S)-N,N-дибензил-1-оксиран-2-ил-метанамина. После 44 ч реакционной смеси позволяют остыть до комнатной температуры, перемешивают в ледяной бане в течение часа и осажденное твердое вещество отфильтровывают. Продукт промывают на фильтре при помощи 50 мл 2 пропанола и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 23,4 г (53%) белого твердого продукта, ВЭЖХ чистота 98,4%: энантиомерная чистота 99,40%. Тпл.: 179180 С. Тпл.:179-180 С. ИК (KBr): 3372, 1623, 1528, 1103, 749, 699. Н ЯМР (ДМСО, а 400): 7,37 (м, 4 Н), 7,32 (м, 4 Н), 7,24 (м, 2 Н), 7,01 (d, J=8,7 Гц, 2 Н), 6,53 (d, J=8,5 Гц, 2 Н), 5,37 (bt, J=5,8 Гц, 1 Н), 4,68 (d, J=4,8 Гц, 1 Н), 4,13 (s, 2 Н), 3,93 (м, 2 Н), 3,85 (м, 1 Н), 3,67 (d, J=13,7 Гц, 2 Н), 3,60 (м, 2 Н), 3,53 (d, J=13,7 Гц, 2 Н), 3,15 (м, 1 Н), 2,75 (м, 1 Н), 2,50 (м, 2 Н). 13 С ЯМР: 165,89, 147,66, 139,42, 130,34, 128,82, 128,31, 126,98, 126,54, 111,98, 67,91, 67,10, 63,72,58,57, 57,87, 49,76, 48,25, 39,70. Элементарный анализ для формулы C27H31N3O3 (М:445,57): С 72,78; Н 7,01; N 9,43%. Обнаружено: С 72,39; Н 6,97; N 9,49%. Вращение: []20D=-13,83 (588 нм/20 С; с=0,1 г/10 см 3 ДМСО). Пример 3 Получение 4-(4-(5S)-5-(дибензиламинометил]-2-оксо-1,3-оксазолидин-3-илфенил)морфолин-3 она формулы 13(соединение формулы 18) 10,19 г (63 милимоль) N,N'-карбонилдиимидазола суспендируют в 70 мл толуола при 25 С. Смесь нагревают до 80-83 С и перемешивают при данной температуре в течение 20 мин. Реакционную смесь охлаждают до 60 С и добавляют по капле 230 мл этанола. Реакционной смеси позволяют остыть до комнатной температуры, перемешивают в ледяной бане в течение часа, после чего осажденное твердое вещество отфильтровывают и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 22,3 г (93%) белого твердого вещества, ВЭЖХ чистота 99,92%, энантиомерная чистота 100%, Тпл.: 154-155 С. ИК (KBr): 1732, 1653, 1521, 1415 см-1. 1 Н ЯМР (ДМСО-d6, 400 МГц): 7,48 (d, J=9,1 Гц, 2 Н), 7,39 (d, J=9,1 Гц, 2 Н), 7,36 (м, 4 Н), 7,32 (м, 4 Н),7,24 (м, 2 Н), 4,83 (м, 1H), 4,20 (s, 2 Н), 4,03 (м, 1H), 3,97 (м, 2 Н), 3,72 (м, 2 Н), 3,68 (d, J=4,39 Гц, 2 Н), 3,65(d, J=13,8 Гц, 2 Н), 3,57 (м, 1 Н), 2,80 (м, 1 Н), 2,74 (м, 1 Н) ppm. 13 С ЯМР (ДМСО-d6, 100 МГц): 166,1, 154,3, 139,0, 137,2, 136,6, 128,9, 128,4, 127,2, 126,0, 118,5, 71,3,67,9, 63,6, 58,3, 55,9, 49,2, 48,2 ppm. Элементарный анализ для формулы C28H29N3O4 (М: 471,56): С 71,32; Н 6,20; N 8,91%. Обнаружено: С 70,95; Н 6,30; N 9,09%. Вращение: []20D=-19,7 (588 нм/20 С; с=0,1 г/10 см 3 ДМСО).II Тот же протокол, что и согласно способу, описанному в I, исключая то, что после суспендирования 23 г (51 милимоль) 4-(4-[(2R)-3-дибензиламино-2-гидроксипропи]аминофенил)морфолин-3-она (соединение формулы 18) и 10,19 г (63 милимоль) N,N'-карбонилдиимидазола суспендируют в 70 см 3 толуола при 25 С, смесь немедленно нагревают до температуры кипения. Пример 4 Получение соли 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она, образованной при помощи уксусной кислоты (соединение формулы 3b) 18,28 г (0,039 моль) 4-(4-(5S)-5-(дибензиламинометил]-2-оксо-1,3-оксазолидин-3-илфенил)морфолинон-3-она формулы 13 растворяют в 200 мл ледяной уксусной кислоты при комнатной температуре. К раствору добавляют 1,8 г 10% катализатора палладированного угля и проводят гидрогенизирование в автоклаве при давлении водорода 10 бар в течение 24 ч. После завершения гидрогенизирования катализатор отфильтровывают, фильтрат выпаривают до сухости и из осадка четыре раза отгоняют по 100 мл этанола каждый раз при давлении 75 мбар. К таким образом полученному осадку добавляют 50 мл этанола, смесь перемешивают в охлаждающей бане в течение 30 мин. Осажденное твердое вещество отфильтровывают, промывают на фильтре при помощи 30 мл этанола и высушивают под инфракрасной лампой до постоянной массы. Таким образом получают 13,28 г (98%) белого продукта. Тпл.: 142-143 С.IR (KBr): 2557, 1747, 1725, 1650, 1524, 1413 см-1. 1 Н ЯМР (ДМСО-d6, 500 МГц): 7,58 (d, J=8,8 Гц, 2 Н), 7,40 (d, J=8,6 Гц, 2 Н), 5,27 (br s, 3 Н), 4,65 (м,1 Н), 4,19 (s, 2 Н), 4,08 (м, 1 Н), 3,96 (м, 2 Н), 3,87 (м, 1 Н), 3,71 (м, 2 Н), 2,88 (м, 2 Н), 1,88 (s, 3 Н) ppm. 13 С ЯМР (ДМСО-d6, 125 МГц): 172,5, 166,1, 154,5, 137,1, 136,9, 126,1, 118,4, 73,7, 67,9, 63,7, 49,2,47,3, 44,1, 21,6 ppm. Элементарный анализ для формулы C16H21N3O6 (М: 351,36): С 54,70; Н 6,02; N 11,96%. Обнаружено: С 54,42; Н 6,05; N 12,00%. Вращение: []20D=-29,65 (588 нм/20 С; с=0,1 г/10 см 3 ДМСО). Пример 5 Получение ривароксабана формулы 1 Раствор 0,13 г (11,25 милимоль) карбоната натрия в 1,6 мл воды охлаждают до температуры 10 С при перемешивании, после чего добавляют 0,35 г (1,0 милимоль) уксуснокислой соли 4-4-[(5S)-5 аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она (соединение формулы 3b) и 0,7 мл ацетона. Реакционную смесь хранят при температуре 8-12 С и добавляют 0,62 г 32,5 объем.% толуольного раствора хлорангидрида 5-хлортиофен-2-карбоновой кислоты (4) и затем добавляют 0,7 мл толуола. Ре- 18024685 акционную смесь нагревают до 50 С, добавляют 0,7 мл ацетона и смесь перемешивают в течение периода 30 мин при 50-55 С. Реакционную смесь охлаждают до 26 С, осажденный продукт отфильтровывают,промывают при помощи 5 мл воды и 2,5 мл ацетона и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 0,38 г (87%) неочищенного продукта, ВЭЖХ чистота 98,8%, Тпл.: 227 С. При перекристаллизации продукта из 5,7-кратного количества ледяной уксусной кислоты получают 0,32 г белого конечного продукта, ВЭЖХ чистота 99,5%, Тпл.: 229 С. Пример 6 Получение ривароксабана формулы 1I 11,9 г (12 милимоль) 5-хлортиофен-2-карбоновой кислоты (15) и 1,9 г (12 милимоль) N,N'-карбонилдиимидазола растворяют в 50 мл сухого ацетонитрила. Реакционную смесь перемешивают при 5055 С в течение часа, после чего при данной температуре добавляют 0,78 г (7,5 милимоль) карбоната натрия, 3,4 г (9,67 милимоль) уксуснокислой соли 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3 ил]фенилморфолин-3-она (3b) и 1 мл дистиллированной воды. Реакционную смесь перемешивают при 50 С в течение часа и позволяют охлаждаться до комнатной температуры. Твердое вещество отфильтровывают и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 3,65 г(86,5%) неочищенного продукта. Неочищенный продукт перекристаллизуют из 22 мл ледяной уксусной кислоты с выходом 3,24 г (89%) белого конечного продукта, ВЭЖХ чистота 99,3%, Тпл.: 230-231 С.II Те же протоколы, которые описаны в части I, за исключением того, что в качестве основания применяют 0,8 г (8 милимоль) триэтиламина. Качество полученного неочищенного продукта (33,42 г, 81%) идентично таковому неочищенного продукта, полученного согласно способу способа I.III Те же протоколы, как описанные в части I, за исключением того, что не применяют агент, связывающий кислоту (в способе I применяют карбонат натрия). Качество полученного неочищенного продукта (3,25 г, 77%) идентично таковому неочищенного продукта, полученного по способу I. Пример 7 Получение рацемического 4-(4-[(3-дибензиламино-2-гидроксипропил]аминофенил)морфолин-3 она формулы 18 (рацемическая форма соединения формулы 18) 10,0 г (50 милимоль) 4-аминофенилморфолинона (соединение формулы 5) и 2,0 г бромида лития суспендируют в смеси 65 мл 2-пропанола и 26 мл дистиллированной воды при 25 С, после чего добавляют 13,0 г (50 милимоль) N,N-дибензил-1-оксиран-2-ил-метанамина (рацемическую форму соединения формулы 17). Реакционную смесь нагревают до 58-62 С и перемешивают при данной температуре в течение 48 ч. На 18-м часу добавляют дополнительные 4 г (15,7 милимоль) и на 42-м часу добавляют дополнительные 2,0 г (7,9 милимоль) N,N-дибензил-1-оксиран-2-ил-метанамина. После 48 ч реакционной смеси позволяют остыть до комнатной температуры, осажденный продукт отфильтровывают, промывают 15 мл 2:5 смеси дистиллированной воды и 2-пропанола и высушивают под инфракрасной лампой до постоянной массы Таким образом получают 18,8 г (84%) белого твердого вещества. Мр.: 154-155 С. Пример 8 Рацемический 4-(4-(5S)-5-(дибензиламинометил]-2-оксо-1,3-оксазолидин-3-илфенил)морфолин 3-он формулы 13 (рацемическая форма соединения формулы 13)I 0,90 г (2 милимоль) 4-(4-[2R/-3-дибензиламино-2-гидроксипропил]аминофенил)морфолин-3-она,рацемической формы соединения формулы 18 и 0,338 г (3 милимоль) N,N'-карбонилдиимидазола суспендируют в 10 мл толуола при 25 С. Реакционную смесь нагревают до кипения, затем нагревают до кипения в течение 2 ч, охлаждают до комнатной температуры и добавляют 2,5 мл этанола. Реакционную смесь перемешивают в ледяной водной бане в течение часа. Осажденное твердое вещество отфильтровывают и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 0,3 г (68%) белого твердого вещества. Тпл.: 133137 С.II 3,3 г (0,083 моль) 4-4-[5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она (rac10 с) перемешивают в 87 см 3 дибензиламина (16), после чего добавляют 1,73 г (0,0053 моль) карбоната цезия. Реакционной смеси дают прореагировать при 80 С в течение 35 ч, карбонат цезия отфильтровывают и дибензиламин отгоняют. Маслянистый осадок (7,9 г) перемешивают с 40 см 3 диэтилового эфира и позволяют кристаллизоваться при перемешивании при 25 С в течение ночи. Продукт отфильтровывают, промывают при помощи эфира и высушивают. Таким образом, получают 3,6 г (93,5%) неочищенного продукта, плавящегося при 126-128 С. Неочищенный продукт перекристаллизуют из 105 мл метанола. Получают 1,7 г (75%) белого желаемого продукта. Тпл.: 146-148 С. Данные ИК, 1 Н ЯМР и 13 С идентичны таковым соединения 13R. Пример 9 Получение рацемического формиата 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]-фенилморфолин-3-она формулы 0,30 г (0,64 милимоль) 4-(4-5-[(дибензиламино)метил]-2-оксо-1,3-оксазолидин-3-илфенил)морфолин-3-она формулы 13 (рацемическая форма) растворяют в смеси 12 см 3 метанола и 0,12 см 3 муравьиной кислоты при комнатной температуре. К раствору добавляют 0,03 г 10% катализатора палладированного угля и смесь гидрогенизируют в автоклаве при давлении водорода 10 бар. Гидрогенизацию проводят в течение 44 ч, после чего катализатор отфильтровывают и фильтрат выпаривают до сухости. К осадку добавляют 10 см 3 этанола, осажденный продукт отфильтровывают и высушивают под инфракрасной лампой до постоянной массы. Таким образом получают 0,12 г (67%) белого твердого соединения, указанного в заголовке. Тпл.: 113-115 С. Пример 10 Получение рацемической уксуснокислой соли 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3 ил]фенилморфолин-3-она (рацемическая форма соединения формулы 3b) 5,15 г (10,90 милимоль) 4-4-[(5S)-5-[дибензиламинометил]-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 13 (рацемическая форма) растворяют в 100 мл ледяной уксусной кислоты при комнатной температуре. К раствору добавляют 0,5 г 10% катализатора палладированного угля. Гидрогенизацию проводят в автоклаве при комнатной температуре под давлением водорода 10 бар в течение 24 ч. Затем реакционную смесь нагревают до 40 С и катализатор отфильтровывают. Фильтрат выпаривают до сухости и из осадка четыре раза отгоняют 50 см 3 этанола каждый раз при давлении 75 милибар. К таким образом полученному осадку добавляют 30 см 3 этанола, смесь перемешивают в охлаждающей ванне в течение 30 мин. Осажденное твердое вещество отфильтровывают, промывают 25 см 3 этанола на фильтре и высушивают под инфракрасной лампой до постоянной массы. Таким образом получают 3,80 г(99,2%) белого твердого соединения, указанного в заголовке. ВЭЖХ чистота 99,7%, Тпл.: 145-146 С. Пример 11 Получение ривароксабана (соединение формулы 1) из соединения формулы 3bI 1,9 г (11,7 милимоль) 5-хлортиофен-2-карбоновой кислоты (15) и 1,9 г (11,7 милимоль) N,N'карбонилдиимидазола растворяют в 50 мл сухого ацетонитрила. Реакционную смесь перемешивают при 50-55 С в течение часа, затем оставляют остывать при комнатной температуре. К раствору при данной температуре добавляют по капле раствор 0,78 г (9,3 милимоль) карбоната натрия и 3,2 г (69,1 милимоль) ацетата 4-4-[(5S)-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она (3b) в 17 мл воды и 10 мл ацетонитрила комнатной температуры. Реакционную смесь перемешивают при 50-55 С в течение часа, затем охлаждают в ванне с ледяной водой и перемешивают в течение получаса. Осажденное твердое вещество отфильтровывают, промывают три раза 10 мл дистиллированной воды каждый раз и 10 мл ацетона и высушивают под инфракрасной лампой до постоянной массы. Таким образом получают 2,33 г(59%) соединения, указанного в заголовке, ВЭЖХ чистота 99,47%. Неочищенный продукт кристаллизуют из 14 мл ледяной уксусной кислоты с выходом 2,04 г (70%) ривароксабана, ВЭЖХ чистота 99,85%. Тпл.: 230-231 С.II Такие же протоколы, как описано в части I, за исключением того, что в качестве основания применяют 1,23 г (9,5 милимоль) N,N'-диизопропилэтиламина. Качество полученного таким образом неочищенного продукта (2,17 г, 55%) идентично таковому неочищенного продукта, полученного посредством способа I. Пример 12 Получение ривароксабана (соединение формулы 1) из соединения формулы 3 а К смеси 1,51 г (15 милимоль) триэтиламина и 15 мл дихлорметана добавляют по капле 0,81 г (7,5 милимоль) хлорэтилформиата при комнатной температуре. К реакционной смеси добавляют по капле смесь 0,8 г (5 милимоль) 5-хлортиофен-2-карбоновой кислоты (15) и 5 мл дихлорметана. Реакционную смесь перемешивают при комнатной температуре в течение часа, после чего добавляют 1,64 г (5 милимоль) гидрохлорида 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она (3 а). Реакционную смесь перемешивают в течение получаса при комнатной температуре. Образованную соль гидрохлорида триэтиламина отфильтровывают и фильтрат выпаривают до сухости. Осадок суспендируют в 10 мл ледяной уксусной кислоты, суспензию нагревают до кипячения и перемешивают в течение 10 мин. Раствору позволяют остыть до комнатной температуры, затем инокулируют при помощи ложечки количеством кристаллов ривароксабана (1) и перемешивают на льду в течение 20 мин. Осажденный белый твердый продукт отфильтровывают и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получат 0,80 г (37%) соединения, указанного в заголовке. Тпл.: 230-233 С. Пример 13 Получение ривароксабана (соединения формулы 1) из соединения формулы 3 а 0,16 г (1 милимоль) 5-хлортиофен-2-карбоновой кислоты (15) добавляют к 4 мл сухого ацетонитрила. К суспензии под аргоном добавляют 0,13 г (1 милимоль) N,N'-диизопропилкарбодиимида. Реакционную смесь перемешивают при комнатной температуре в течение 30 мин, после чего раствор добавляют по капле к смеси 0,33 г (1 милимоль) гидрохлорида 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3 ил]фенилморфолин-3-она (3 а) и 0,10 г (1,25 милимоль) гидрокарбоната натрия в 4 см 3 сухого ацетонитрила при комнатной температуре. Реакционную смесь перемешивают при 50 С в течение 4,5 ч, затем нагревают до кипения и перемешивают при данной температуре в течение 6 ч. Реакционной смеси позволяют остыть до комнатной температуры, твердый продукт промывают дважды 25 см 3 дистиллированной воды каждый раз и высушивают под инфракрасной лампой до постоянной массы. Таким образом,получают 0,24 г (56%) соединения, указанного в заголовке. Тпл.: 230-233 С. Пример 14 Получение ривароксабана (соединение формулы 1) из соединения формулы 3 а 0,16 г (1 милимоль) 5-хлортиофен-2-карбоновой кислоты (15) и 0,25 г (2,5 милимоль) триэтиламина взвешивают в 8 мл сухого дихлорметана. К таким образом полученной смеси по капле добавляют 0,16 г(1 милимоль) хлорэтилформиата при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 30 мин, после чего полученный раствор добавляют к смеси 0,33 г (1 милимоль) 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он-гидрохлорида(3 а) в 4 см 3 высушенного дихлорметана. Реакционной смеси позволяют остыть до комнатной температуры, образовавшийся гидрохлорид триэтиламина отфильтровывают, фильтрат выпаривают до сухости. Таким образом полученный осадок перемешивают в смеси 2 см 3 ацетона и 3 см 3 дистиллированной воды в течение 20 мин. Полученное бежевое твердое вещество отфильтровывают, дважды промывают 0,5 см 3 дистиллированной воды каждый и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 0,10 г (23%) соединения, указанного в заголовке. Тпл.: 230-235 С. Пример 15 Получение ривароксабана (соединение формулы 1) из соединения формулы 3 а 1,9 г (11,7 милимоль) 5-хлортиофен-2-карбоновой кислоты (15) и 1,9 г (11,7 милимоль) N,N'-карбонилдиимидазола растворяют в 50 мл сухого ацетонитрила. Реакционную смесь перемешивают при 5055 С в течение часа, после чего добавляют 0,78 г (9,3 милимоль) карбоната натрия, 3,27 г (10 милимоль) гидрохлорида 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она и 1 мл дистиллированной воды. Реакционную смесь перемешивают при 50 С в течение часа, охлаждают в ледяной ванне и перемешивают в течение дополнительных 0,5 ч. Осажденное твердое вещество отфильтровывают, промывают трижды 10 мл воды каждый раз и 10 мл ацетона и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 3,9 г (77%) соединения, указанного в заголовке. Данный неочищенный продукт перекристаллизуют из 24 мл ледяной уксусной кислоты. Таким образом получают 3,34 г (86%) ривароксабана с ВЭЖХ чистотой 99,0%. Тпл.: 229-230 С. Пример 16 Получение ривароксабана (соединение формулы 1) из соединения формулы 3 а В 8 мл сухого тетрагидрофурана взвешивают 0,16 г (1 милимоль) 5-хлортиофен-2-карбоновой кислоты (15), 0,16 г (1 милимоль) N,N'-карбонилдиимидазола, 0,10 г (11,25 милимоль) гидрокарбоната натрия и 0,33 г (1 милимоль) 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-онгидрохлорида (3 а). Реакционную смесь нагревают до 50 С и перемешивают при данной температуре в течение 13 ч. Реакционную смесь выпаривают до сухости. Осадок перемешивают в смеси 5 мл ацетона, 3 см 3 толуола и 1 см 3 дистиллированной воды в течение 20 мин. Осажденное белое твердое вещество отфильтровывают, промывают при помощи 1:1 смеси ацетона и дистиллированной воды и высушивают под инфракрасной лампой до постоянной массы. Таким образом получают 0,32 г (74%) соединения, указанного в заголовке. ВЭЖХ чистота - 98,7%. Тпл.: 230-233 С. Пример 17 Получение ривароксабана (соединение формулы 1) из соединения формулы 3bI 0,16 г (1 милимоль) 5-хлортиофен-2-карбоновой кислоты (15) и 0,35 г (1 милимоль) 4-4-[(5S)-5 аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он-ацетата (3b) растворяют в 6 мл N,N'диметилформамида при комнатной температуре. К раствору добавляют 0,078 г (0,93 милимоль) карбоната натрия, после чего по капле добавляют 50% раствор 1,35 мл пропилфосфонового ангидрида (ПФА) вN,N'-диметилформамиде. Реакционную смесь нагревают до 50-55 С и перемешивают при данной температуре в течение 19 ч. К реакционной смеси добавляют 20 см 3 дистиллированной воды температуры 2 С и смесь перемешивают в ледяной водной ванне в течение часа. Осажденное твердое вещество отфильтровывают, промывают три раза 1 мл дистиллированной воды каждый раз и 1 мл ацетона и высушивают под инфракрасной лампой до постоянной массы Таким образом получают 0,01 г (23%) соединения, указанного в заголовке.II Такой же протокол, как и согласно части I, за исключением того, что в качестве основания применяют 0,106 г (1,05 милимоль) триэтиламина. Качество полученного продукта (0,09 г, 21%) идентично таковому, полученному согласно способу I. Пример 18 Получение 4-4-[2S)-3-хлор-2-гидроксипропил)амино]фенилморфолин-3-она формулы 9aS 25 г (0,13 моль) 4-(4-аминофенил)морфолин-3-она (5) суспендируют в смеси 200 мл ацетонитрила и 40 мл дистиллированной воды при перемешивании, после чего добавляют 0,5 мл (11,93 г, 0,13 моль) (S)эпихлоргидрина. Смесь нагревают до 50-55 С, после чего светло-коричневый раствор перемешивают в течение 6 ч и добавляют дополнительное количество 10,5 мл (S)-эпихлоргидрина. После 6 ч к смеси добавляют четыре раза каждый 6-й час 2,62 мл (2,3 г) (S)-эпихлоргидрина (11S). После добавления последней части смесь перемешивают в течение 3 ч. Ацетонитрил отгоняют в вакууме и воду удаляют из двуфазной системы посредством азеотропной дистилляции при добавлении этилацетата. При успешной дистилляции продукт осаждается из смеси в виде светло-бежевой суспензии. Продукт кристаллизуют ночь при температуре (-15)-(-20)С, затем отфильтровывают, промывают 0-5 С этилацетатом и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 25,81 г (70%) светлобежевого продукта, имеющего температуру плавления 135-137 С, химическую чистоту 94,9% и хиральную чистоту (ВЭЖХ) 99,5%. Неочищенный продукт суспендируют в 150 мл гексана, суспензию нагревают и к горячей суспензии при перемешивании добавляют 500 мл ацетона до растворения. Смеси позволяют остыть до 25 С и перемешивают при 0-2 С в течение часа до кристаллизации. Смесь отфильтровывают и продукт высушивают под инфракрасной лампой. Таким образом, получают 17,8 г (69%) соединения, указанного в заголовке. Тпл.: 138-139 С, химическая чистота 98,2%, хиральная чистота (ВЭЖХ) 99,5%. ИК (KBr): 3377, 1627, 1604, 1531, 1345, 1126. 1 Н-ЯМР (ДМСО-d6, 500 МГц): 7,03 (d, J=8,4 Гц, 2 Н), 6,61 (d, J=8,6 Гц, 2 Н), 5,68 (t, J=5,8 Гц, 1 Н),5,32 (d, J=5,1 Гц, 1H), 4,13 (s, 2 Н), 3,92 (м, 2 Н), 3,85 (м, 1 Н), 3,68 (м, 1 Н), 3,60 (м, 3 Н), 3,18 (м, 1H), 3,06(м, 1 Н). 13 С-ЯМР (ДМСО-d6, 125 МГц): 165,91, 147,44, 130,56, 126,63, 112,09, 68,94, 67,89, 63,71, 49,74,48,10, 46,67. Элементарный анализ формулы C13H17ClN2O3 (М: 284,75): С 54,84%; Н 6,02%; Cl 12,45%; N 9,84%. Обнаружено: С 54,87%; Н 6,15%; Cl 12,35; N 9,88%. Вращение: []20D=+2,0 (588 нм/20 С; с=0,1 г/10 мл ДМСО). Пример 19 Получение 4-4-[(5S)-5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 10aS 21,2 г (0,07 моль) 4-4-[2S)-3-хлор-2-гидроксипропил)амино]фенилморфолин-3-она формулы(9aS) суспендируют в смеси 190 см 3 толуола и 22,3 см 3 1-метил-2-пирролидона при перемешивании, после чего добавляют 15,09 г (0,099 моль) CDI. Смесь перемешивают при температуре 80-82 С в течение 20 мин, затем позволяют остыть до 60 С, добавляют по капле 43 мл этанола, смесь постепенно охлаждают до 25 С, при этом продукт начинает осаждаться. Смесь перемешивают при данной температуре в течение 50 ч, после чего смесь выпаривают до половины ее объема и кристаллизуют при температуре между (-15)-(-20)С в течение ночи. Продукт отфильтровывают, промывают 0-5 С ацетоном и высушивают под инфракрасной лампой. Таким образом, получают 17,1 г (74%) соединения, указанного в заголовке. Тпл.: 148-150 С, химическая чистота 99,5%, хиральная чистота (ВЭЖХ) 99,76%. ИК (KBr): 1743, 1660, 1521, 1313, 1229, 1122. 1 Н-ЯМР (ДМСО-d6, 500 МГц): 7,59 (d, J=8,8 Гц, 2 Н), 7,41 (d, J=8,8 Гц, 2 Н), 5,02 (м, 1 Н), 4,22 (м, 1 Н),4,19 (s, 2 Н), 4,01 (м, 1 Н), 3,97 (м, 2 Н), 3,95 (м, 1 Н), 3,85 (м, 1 Н), 3,72 (м, 2 Н). 13 С-ЯМР (ДМСО-d6, 125 МГц): 166,10, 153,94, 137,31, 136,37, 126,12, 118,46, 71,27, 67,87, 63,61,49,14, 47,54, 46,31. Элементарный анализ формулы C14H15ClN2O4 (М: 310,74): С 54,11%; Н 4,87%; Cl 11,41%; N 9,02%. Обнаружено: С 54,01%; Н 4,89%; Cl 11,37%; N 9,05%. Вращение: []20D=+52,26(588 нм/20 С; с=0,1 г/10 мл ДМСО). Пример 20 Получение 4-4-[(5S)-5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 10cS 117 г (0,05 моль) 4-4-[(5S)-5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она(10aS) перемешивают в 310 мл ацетонитрила, после чего добавляют 122,65 г (0,082 моль) йодида натрия. Реакционную смесь нагревают до кипения в течение 15 ч, добавляют дополнительное количество 41,3 г(0,28 моль) йодида натрия. Реакционную смесь нагревают до кипячения в течение 15 ч, добавляют до- 22024685 полнительное количество 20,65 г (0,14 моль) йодида натрия. Реакционную смесь нагревают до кипячения в течение 7 ч, отфильтровывают, ацетонитрил отгоняют и к желтому кристаллическому осадку добавляют 200 мл дихлорметана и 100 мл воды при перемешивании. Фазы разделяют и водный слой промывают дважды 100 мл дихлорметана каждый раз. Объединенные органические фазы промывают три раза 100 мл воды каждый раз, высушивают над сульфатом магния и выпаривают. Полученное желтое вещество перемешивают в 130 мл воды в течение ночи. Практически белую суспензию отфильтровывают, промывают водой и высушивают под инфракрасной лампой. Таким образом получают 21,2 г (96%) соединения указанного в заголовке. Тпл.: 158-161 С, химическая чистота (ВЭЖХ) 95,4%, хиральная чистота 99,4%. ИК (KBr): 1738, 1659, 1518, 1312, 1231, 1121. 1 Н-ЯМР (ДМСО-d6, 500 МГц): 7,58 (d, J=9,0 Гц, 2 Н), 7,41 (d, J=8,8 Гц, 2 Н), 4,73 (м, 1 Н), 4,21 (м, 1 Н),4,19 (s, 2 Н), 3,97 (м, 2 Н), 3,72 (м, 2 Н), 3,68 (м, 1 Н), 3,62 (dd, J1=5,1 Гц, J2=10,8 Гц, 1 Н), 3,57 (dd, J1=4,6 Гц,J2=10,8 Гц, 1 Н). 13 С-ЯМР (ДМСО-d6, 125 МГц): 166,09, 153,81, 137,31, 136,38, 126,11, 118,52, 70,94, 67,87, 63,61,50,56, 49,15, 9,92. Элементарный анализ формулы C14H15N2O4 (М: 402,19): С 41,81%; Н 3,76%; N 6,97%. Обнаружено: С 42,2%; Н 3,75%; N 7,09%. Вращение: []20D=+52,74(588 нм/20 С; с=0,1 г/10 мл ДМСО). Пример 21 Получение 4-(4-(5R)-5-(дибензиламинометил]-2-оксо-1,3-оксазолидин-3-илфенил)морфолин-3 она формулы 13R 20,0 г (0,05 моль) 4-4-[(5S)-5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он (10cS) перемешивают в 533 мл дибензиламина (12), после чего добавляют 10,33 г (0,03 моль) карбоната цезия. Реакцию проводят при 80 С в течение 35 ч. Карбонат цезия отфильтровывают и дибензиламин отгоняют при 140 С и давлении 0,2 бар. Коричневый маслянистый осадок (55,65 г) помещают в 15 мл диэтилового эфира и позволяют кристаллизоваться при перемешивании при 25 С в течение ночи. Светло-бежевое вещество отфильтровывают, промывают диэтиловым эфиром и высушивают. Таким образом, получают 20,15 г (86%) соединения, указанного в заголовке. Тпл.: 128-133 С, химическая чистота 85,5%, хиральная чистота (ВЭЖХ) 97,5%. Неочищенный продукт перекристаллизуют из 260 мл этанола, отфильтровывают и высушивают под инфракрасной лампой. Таким образом, получают 17,97 г (87%) очищенного продукта, темп. плавления 153-155 С, химическая чистота 99,9% и хиральная чистота (ВЭЖХ) 99,9%. ИК (KBr): 1733, 1653, 1521, 1415, 1128. 1 Н-ЯМР (ДМСО-d6, 400 МГц): 7,48 (d, J=9,1 Гц, 2 Н), 7,39 (d, J=9,1 Гц, 2 Н), 7,36 (м, 4 Н), 7,32 (м, 4 Н),7,24 (м, 2 Н), 4,83 (м, 1H), 4,20 (s, 2 Н), 4,03 (м, 1 Н), 3,97 (м, 2 Н), 3,72 (м, 2 Н), 3,68 (d, 2 Н), 3,65 (d, J=13,8 Гц, 2 Н), 3,57 (м, 1 Н), 2,80 (м, 1 Н), 2,74 (м, 1 Н). 13 С-ЯМР (ДМСО-d6, 100 МГц): 166,11, 154,32, 139,00, 137,18, 136,64, 128,88, 128,41, 127,20, 126,03,118,46, 71,27, 67,89, 63,63, 58,32, 55,92, 49,18, 48,19. Элементарный анализ формулы C28H29N3O4 (М: 471,56): С 71,32%; Н 6,20%; N 8,91%. Обнаружено: С 70,86%; Н 6,28%; N 8,92%. Вращение: []20D=+19,32(588 нм/20 С; с=0,1 г/10 мл ДМСО). Пример 22 Получение ацетатной соли 4-4-[(5R)-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин 3-она формулы 3bR 17,48 г (0,037 моль) 4-(4-(5R)-5-(дибензиламинометил]-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 13R растворяют в 190 см 3 ледяной уксусной кислоты при комнатной температуре. К раствору добавляют 11,72 г 10% катализатора палладированного угля. Гидрогенизацию проводят в автоклаве при давлении водорода 10 бар при комнатной температуре в течение 24 ч. При завершении гидрогенизации катализатор отфильтровывают, фильтрат выпаривают до сухости и из осадка три раза отгоняют 100 см 3 этанола каждый раз при давлении 75 мбар. Полученную белую суспензию отфильтровывают, влажный фильтрат перемешивают в 100 см 3 этанола посредством применения 0-5 С охлаждающей ванны в течение 30 мин. Снежно-белый продукт отфильтровывают, промывают 30 мл 0-5 С этанола на фильтре и высушивают под инфракрасной лампой до постоянной массы. Таким образом получают 11,97 г (92%) белого продукта, ВЭЖХ чистота 99,91%, энантиомерная чистота 99,9%, Тпл.: 142-152 С. ИК, 1 Н ЯМР и 13 С ЯМР спектральные характеристики продукта идентичны таковым соединения 3b. Элементарный анализ формулы C16H21N3O6 (М: 351,36): С 54,7%; Н 6,02%; N 11,96%. Обнаружено С 54,27%; Н 6,11%; N 118%. Вращение: []20D=+28,77(588 нм/20 С; с=0,1 г/10 мл ДМСО). Пример 23 Получение 5-хлор-N-5R)-2-оксо-3-(4-(3-оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-илметилтиофен-2-карбоксамида формулы 1R (энантиомер ривароксабана) Раствор 3,7 г (0,035 моль) карбоната натрия в 51 мл воды охлаждают до 10 С при перемешивании,- 23024685 после чего добавляют 10,0 г (0,0028 моль) 4-4-[(5R)-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он-ацетата (3bR), 3 см 3 воды и 23 см 3 ацетона. Раствор отфильтровывают, после чего при температуре 8-12 С добавляют 16,6 см 3 толуольного раствора хлорангидрида 5-хлортиофен-2 карбоновой кислоты концентрации 36,1 г/100 мл (соответствует 5,97 г, 0,33 моль хлорангидрида 5-хлортиофен-2-карбоновой кислоты формулы 4). Реакционную смесь нагревают до температуры 50 С, после чего добавляют 25 см 3 ацетона и смесь перемешивают в течение дополнительного периода 30 мин при 50-53 С. Реакционную смесь охлаждают до 25 С, осажденный продукт отфильтровывают, промывают три раза 20 мл ацетона каждый раз, три раза 20 см 3 воды каждый раз и снова дважды 20 мл ацетона каждый. Таким образом, получают 11,98 г (97%) неочищенного продукта (мр.: 231-234 С), который перекристаллизуют из 6,2-кратного количества ледяной уксусной кислоты. Таким образом получают 20,7 г (93%) конечного продукта, ВЭЖХ чистота 99,93%, энантиомерная чистота 99,9%, Тпл.: 231-234 С. Элементарный анализ формулы C19H18ClN3O5S (М 435,89): С 52,36%; Н 4,16%; Cl 8,13%; N 9,64%, S 7,36%. Обнаружено С 52,08%; Н 4,29%; Cl 8,17; N 9,37%; S 7,43%. Вращение: []20D=+41,02(588 нм/20 С, с=0,1 г/10 мл ДМСО). Пример 24 Получение 4-4-[2R)-3-хлор-2-гидроксипропил)амино]фенилморфолин-3-она формулы 9 а 10,4 г (0,0544 моль) 4-(4-аминофенил)-морфолин-3-она (5) суспендируют в смеси 84 мл ацетонитрила и 17 мл дистиллированной воды при перемешивании, после чего добавляют 4,4 мл (5,0 г, 0,054 моль) (R)-эпихлоргидрина (11). Реакционную смесь нагревают до 50-52 С, светло-коричневый раствор перемешивают в течение 6 ч, после чего добавляют дополнительные 4,4 мл (R)-эпихлоргидрина. После 6 ч в интервале 6 ч четыре раза добавляют 1,1 мл (1,25 г, 00,0135 моль) (R)-эпихлоргидрина. После введения последней части реакцию продолжали в течение дополнительного периода 3 ч. Смесь нагревают до 60 С и перемешивают при данной температуре в течение дополнительных 3 ч. Ацетонитрил отгоняют в вакууме, к двуфазному остатку добавляют этилацетат и проводят азеотропную дистилляцию. В случае успешной дистилляции продукт осаждается в виде светло-желтой суспензии. Кристаллизацию проводят при температуре между (-15) и (-20)С в течение ночи. Продукт отфильтровывают, промывают 0-5 С этилацетатом и высушивают под инфракрасной лампой до постоянной массы. Таким образом получают 10,3 г (67%) желаемого неочищенного продукта, темп. плавления.: 132-134 С, химическая чистота 95,1%, хиральная чистота (ВЭЖХ) 98,2%. Неочищенный продукт суспендируют в 35 мл гексана, суспензию нагревают и к горячей суспензии добавляют по капле 169 мл ацетона при перемешивании до растворения продукта. Смеси позволяют остыть до 25 С и кристаллизацию проводят при 0-2 С при перемешивании в течение часа. Продукт отфильтровывают и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 8,16 г (82%) соединения, указанного в заголовке. Тпл:137-139 С, химическая чистота 98%, хиральная чистота (ВЭЖХ) 98,2%. Данные ИК, 1 Н ЯМР и 13 С ЯМР спектральных характеристик продукта идентичны таковым соединения 9aS. Элементарный анализ формулы C13H17ClN2O3 (М: 284,75%): С 54,84%; Н 6,02%; Cl 12,45%, N 9,84%. Обнаружено: С 54,77%, Н 6,09%; Cl 12,50; N 9,87%. Вращение []20D=-2,5(588 нм/20 С, с=0,1 г/10 мл ДМСО). Пример 25 Получение 4-4-[(5R)-5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 10 а 7,9 г (0,028 моль) 4-4-[2R)-3-хлор-2-гидроксипропил)амино]фенилморфолин-3-она формулы(9 а) суспендируют в смеси 71 мл толуола и 12 мл 1-метил-2-пирролидона при перемешивании, после чего добавляют 5,6 г (0,035 моль) CDI. Смесь оставляют реагировать при 80-82 С в течение 20 мин и затем нагревают до кипячения в течение часа. Смеси позволяют остыть до 60 С и добавляют по капле 15 мл этанола. Смесь постепенно охлаждают до 25 С, после чего продукт начинает осаждаться из смеси. Смесь перемешивают при данной температуре в течение 50 ч, после чего выпаривают до половины объема. Позволяют произойти кристаллизации при температуре между -15 С и -20 С в течение ночи. Продукт отфильтровывают, промывают 0-5 С ацетоном и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 6,9 г (80%) соединения, указанного в заголовке. Тпл.: 143-146 С,химическая чистота 94,6%, хиральная чистота (ВЭЖХ) 99,59%. Данные ИК, 1 Н ЯМР и 13 С ЯМР продукта идентичны таковым соединения 10aS. Элементарный анализ формулы C14H15ClN2O4 (М: 310,74): С 54,11%; Н 4,87%; Cl 11,41%, N 9,02%. Обнаружено: С 54,17%; Н 5,05%; Cl 11,13%; N 9,39%. Вращение: []20D=-53,49(588 нм/20 С; с=0,1 г/10 мл ДМСО). Пример 26 Получение 4-4-[(5R)-5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 10 с 6,5 г (0,02 моль) 4-4-[(5R)-5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он (10) перемешивают в 130 мл ацетонитрила, после чего добавляют 46,89 г (0,31 моль) йодида натрия. Реакционную смесь нагревают до кипячения в течение 15 ч, после чего добавляют 15,79 г (0,11 моль) йодида натрия. Реакционную смесь перемешивают в течение дополнительного периода 15 ч, добавляют дополнительные 7,89 г (0,053 моль) йодида натрия и смесь нагревают до кипячения в течение 7 ч. Смесь фильтруют, ацетонитрил отгоняют и к желтому кристаллическому осадку добавляют 100 мл дихлорметана и 200 мл воды при перемешивании. Фазы разделяют, водный слой экстрагируют три раза 30 мл дихлорметана каждый. Объединенные органические фазы промывают три раза 70 мл воды каждый, высушивают над сульфатом магния и выпаривают. Полученное желтое кристаллическое вещество перемешивают в 60 мл воды в течение ночи. Практически белую суспензию фильтруют, промывают три раза 20 мл воды каждый и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 8,0 г(95,2%) соединения, указанного в заголовке. Тпл.: 154-157 С. Химическая чистота 95,87%, хиральная чистота 99,7% (ВЭЖХ). 1 Н ЯМР и 13 С ЯМР спектральные характеристики продукта идентичны таковым соединения 10cS Элементарный анализ формулы C14H15IN2O4 (M: 402,19): С 41,81%; Н 3,76%; N 6,97%. Обнаружено С 42,04%; Н 3,84%; N 7,17%. Вращение []20D=-53,55(588 нм/20 С; с =0,1 г/10 мл ДМСО). Пример 27 Получение 4-(4-(5S)-5-(дибензиламинометил]-2-оксо-1,3-оксазолидин-3-илфенил)морфолин-3 она формулы 13 7,0 г (0,017 моль) 4-4-[(5R)-5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она (10 с) перемешивают в 187 мл дибензиламина (12), после чего добавляют 3,62 г (0,011 моль) карбоната цезия и реакцию продолжают при 80 С в течение 35 ч. Карбонат цезия отфильтровывают и дибензиламин отгоняют при 140 С под давлением 0,2 мбар. Коричневый маслянистый осадок кристаллизуют при перемешивании с 70 мл диэтилового эфира в течение ночи. Светло-бежевое вещество фильтруют, промывают дважды 30 мл эфира каждый и высушивают Таким образом, получают 7,78 г (94,8%) соединения, указанного в заголовке. Тпл.: 140-145 С, химическая чистота 90,3%, хиральная чистота (ВЭЖХ) 98,5%. Неочищенный продукт (7,5 г) перекристаллизуют из 115 мл этанола, фильтруют и высушивают под инфракрасной лампой до постоянной массы. Выход 6,2 г (82,7%), Тпл.: 154-156 С, химическая чистота 99,4%, хиральная чистота (ВЭЖХ) 99,9%. ИС, 1 Н ЯМР и 13 С ЯМР спектральные характеристики продукта идентичны таковым соединения 13R. Элементарный анализ формулы C28H29N3O4 (М: 471,66): С 71,32%; Н 6,20%; N 8,91%. Обнаружено: С 71,59%; Н 6,28%; N 8,91%. Вращение: []20D=-20,02(588 нм/20 С; с=0,1 г/10 мл ДМСО). Пример 28 Получение ривароксабана формулы 1 Раствор 1,11 г (0,01 моль) карбоната натрия в 16,5 мл воды охлаждают до 10 С при перемешивании,после чего добавляют 3,0 г (0,0085 моль) 4-4-[(5S)-5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-он-ацетата (3b), 0,9 мл дистиллированной воды и 6,9 мл ацетона. К реакционной смеси при 8-12 С добавляют 5 мл 36,1% толуольного раствора хлорангидрида 5-хлортиофен-2-карбоновой кислоты (4). Реакционную смесь нагревают до 50 С, добавляют 7,5 мл ацетона и смесь перемешивают при 50-55 С в течение дополнительного периода 30 мин. Реакционную смесь охлаждают до 25 С, осажденный продукт фильтруют, промывают три раза 15 мл ацетона каждый, три раза 15 мл воды каждый и снова три раза 15 мл ацетона каждый и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 3,4 г (91,4%) неочищенного продукта, ВЭЖХ чистота 99,61%, Тпл.: 230-232 С. Неочищенный продукт перекристаллизуют из 21 мл ледяной уксусной кислоты. Получают 2,99 гrac9a 2,4 г 4-(4-аминофенил)морфолин-3-она (5) суспендируют в смеси 424 мл этанола и 12 мл дистиллированной воды при перемешивании, после чего добавляют 0,98 мл (1,16 г, 0,013 моль) рацемического эпихлоргидрина. Смесь хранят при 25 С в течение 72 ч, после чего добавляют 0,42 мл рацемического эпихлоргидрина. Реакцию продолжают в течение 24 ч, реакционную смесь вливают в 145 мл воды и смесь экстрагируют 145 мл этилацетата при перемешивании. Слои разделяют, водную фазу экстрагируют три раза 75 мл этилацетата каждый, общие органические слои высушивают над сульфатом магния,очищают активированным углем и фильтруют. Фильтрат выпаривают до образования густой суспензии. Смесь охлаждают до 25 С, промывают 0-5 С ацетоном и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 2,0 г (62%) соединения, указанного в заголовке. Тпл.: 152153 С. Данные ИС, 1 Н ЯМР и 13 С ЯМР продукта идентичны таковым соединения 9aS. Пример 30 Получение рацемического 4-4-[5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы rac10a 1,66 г (0,0058 моль) рацемического 4-4-[(3-хлор-2-гидроксипропил)амино]фенилморфолин-3-она формулы (rac9a) суспендируют в смеси 16,5 мл толуола и 2,5 мл 1-метил-2-пирролидона при перемешивании, после чего добавляют 11,15 г (0,007 моль) CDI. Смеси дают прореагировать при 80-82 С в течение 20 мин, после чего нагревают до кипячения в течение часа. Смеси позволяют остыть до 60 С и добавляют по капле 3 мл этанола. Смесь постепенно охлаждают до 25 С. Продукт начинает осаждаться. Смесь перемешивают при данной температуре в течение 48 ч, фильтруют, промывают 0-5 С ацетоном и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 1,69 г (94%) соединения, указанного в заголовке. Тпл.: 188-191 С. Данные ИС, 1 Н ЯМР и 13 С ЯМР продукта идентичны таковым соединения 10aS. Пример 31 Получение рацемического 4-4-[5-йодметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы (rac10c) 4,0 г (0,013 моль) 4-4-[(5-хлорметил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она (rac10 а) перемешивают в 100 мл ацетонитрила, после чего добавляют 28,92 г (0,087 моль) йодида натрия. Реакционную смесь нагревают до кипячения в течение 15 ч, добавляют дополнительные 13,02 г (0,087 моль) йодида натрия и реакционную смесь нагревают до кипячения в течение дополнительного периода 15 ч. После фильтрования ацетонитрил отфильтровывают и к желтому кристаллическому осадку добавляют при перемешивании 50 мл дихлорметана и 25 мл воды. Слои разделяют и водную фазу экстрагируют дважды 25 мл дихлорметана каждый. Общие органические соли промывают три раза 25 мл воды каждый,высушивают над сульфатом магния и высушивают. Полученное таким образом желтое кристаллическое вещество перемешивают в 30 мл воды в течение ночи. Почти белую суспензию фильтруют, промывают водой и высушивают под инфракрасной лампой до постоянной массы. Таким образом, получают 4,18 г(80%) соединения, указанного в заголовке Тпл.: 167-168 С. Данные ИС, 1 Н ЯМР и 13 С ЯМР продукта идентичны таковым соединения 10cS. Пример 32 Получение основного рацемата 4-4-[5-(аминометил)-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она формулы 12 0,5 г (0,0015 моль) рацемической ацетатной соли 4-4-[5-аминометил-2-оксо-1,3-оксазолидин-3 ил]фенилморфолин-3-она (rac3b) суспендируют в 10 мл дистиллированной воды при перемешивании. Смесь охлаждают до 0-5 С и рН подводят до 10-11 посредством добавления 1 М гидроксида натрия. Реакционную смесь перемешивают при 25 С в течение 20 ч, добавляют 20 мл дихлорметана. Слои разделяют после перемешивания в течение 30 мин. Водную фазу экстрагируют три раза 10 мл дихлорметана каждый. Объединенные органические соли выпаривают. Таким образом, получают 0,33 г (80%) соединения, указанного в заголовке. ВЭЖХ чистота 99,12% Тпл.: 147-149 С. Элементарный анализ формулы C14H17N3O4 (М 291,31) С 57,72%, Н 5,88%, N 14,42%. Обнаружено С 57,35%, Н 6,04%, N 14,32%. Пример 33 Получение рацемического ривароксабана формулы rac1 Раствор 0,64 г (0,006 моль) карбоната натрия в 9 мл воды охлаждают до 10 С при перемешивании,после чего добавляют 1,8 г (0,005 моль) ацетата 4-4-[5-аминометил-2-оксо-1,3-оксазолидин-3-ил]фенилморфолин-3-она (rac3b), 0,6 мл воды и 1,8 мл ацетона. Раствор фильтруют и при температуре 8-12 С добавляют 3 мл толуольного раствора хлорангидрида 5-хлортиофен-2-карбоновой кислоты (соединение формулы 4), имеющего концентрацию 36,1 г/100 мл (это соответствует 1,07 г, 0,059 моль хлорангидрида 5-хлортиофен-2-карбоновой кислоты). Реакционную смесь нагревают до 50 С, после чего добавляют 5 мл ацетона и смесь перемешивают при 50-53 С в течение дополнительного периода 30 мин. Реакционную смесь охлаждают до 25 С. Осажденный продукт фильтруют, промывают трижды 5 мл ацетона каждый, три раза 5 мл воды каждый и снова дважды 55 мл ацетона каждый. Таким образом получают 1,75 г(83%) соединения, указанного в заголовке. Тпл.: 229-231 С. Продукт перекристаллизуют из 6,2-кратного количества ледяной уксусной кислоты. Таким образом, получают 1,50 г (86%) соединения, указанного в заголовке. Тпл.: 230-233 С. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения фармацевтически активного ингредиента 5-хлор-N-5S)-2-оксо-3-[4-(3 оксоморфолин-4-ил)фенил]-1,3-оксазолидин-5-илметил)тиофен-2-карбоксамида (ривароксабана) формулы 4-(4-5S-(5-аминометил-2-оксо-1,3-оксазолидин-3-ил)фенил)мор или его соли общей формулы 3(где HnX представляет собой органическую или неорганическую кислоту, n представляет собой 1, 2 или 3 и X представляет собой ион кислотного остатка) с 5-хлортиофен-2-карбоновой кислотой формулы 15: в присутствии агента реакции сочетания, выбранного из хлорэтилформиата, N,N'-диизопропилкарбодиимида (ДИК), N,N'-дициклогексилкарбодиимида (ДЦК), ангидрида трипропилфосфоновой кислоты (ТФА) или N,N'-карбонилдиимидазола (КДИ), при условии, что если в соединении общей формулы 3 n представляет собой 1 и X представляет собой хлор, то агент реакции сочетания не являетсяN,N'-карбонилдиимидазолом. 2. Способ по п.1, где соединение формулы 12 или его соль формулы 3 получают удалением защитной групп(ы) с S-энантиомерного соединения общей формулы 14(где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере,Z не является водородом) с последующим выделением полученного таким образом S-энантиомерного основания формулы 12 или его соли формулы 3. 3. Способ по п.2, где соединение формулы 14 получают взаимодействием R-энантиомерного соединения общей формулы 20 1(где Z1 и Z2 имеют значения, указанные в п.2) с агентом, способным ввести карбонильную группу,- 27024685 представляющим собой N,N'-карбонилдиимидазол, фосген, дифосген или трифосген. 4. Способ по п.3, где соединение формулы 20 получают взаимодействием R-энантиомерного соединения общей формулы 19 5. Способ по п.4, где соединение формулы 19 получают взаимодействием S-энантиомерного соединения формулы 11 с соединением общей формулы Z1Z2NH (где Z1 и Z2 имеют значения, указанные в п.2). 6. Способ по любому из пп.1 или 2, где n представляет собой 1 и X представляет собой ацетатный ион. 7. Способ по любому из пп.2-5, где Z1 и Z2 представляют собой бензил. 8. Способ по любому из пп.1-7, где при взаимодействии S-энантиомерного основания формулы 12 или его соли формулы 3 с 5-хлортиофен-2-карбоновой кислотой формулы 15 в качестве агента реакции сочетания применяют хлорэтилформиат или N,N'-карбонилдиимидазол (КДИ), при условии, что если в соединении общей формулы 3 n представляет собой 1 и X представляет собой хлор, то агент реакции сочетания не является N,N'-карбонилдиимидазолом, взаимодействие необязательно проводят в присутствии органического или неорганического основания в органическом растворителе или смеси указанного растворителя с водой при 0-110 С. 9. Способ по п.8, где органическое или неорганическое основание выбрано из триэтиламина, диизопропилэтиламина, карбоната натрия или гидрокарбоната натрия, органический растворитель выбран из ацетонитрила, дихлорметана, ацетона, толуола, тетрагидрофурана или их смеси и реакцию осуществляют при температуре 40-70 С. 10. Способ по любому из пп.2 или 3, где соединение формулы 14 представляет собой соединение формулы 13 которое получают путем удаления защитных бензильных групп посредством восстановления, проводимого в C1-4 алифатическом спирте, ледяной уксусной кислоте, воде или смеси упомянутых растворителей друг с другом или с дополнительными органическими растворителями, посредством каталитической гидрогенизации или химического восстановления. 11. Способ по п.3, где в качестве агента, способного ввести карбонильную группу, применяют N,N'карбонилдиимидазол и взаимодействие осуществляют в приемлемом растворителе. 12. Способ по п.11, где растворитель представляет собой толуол. 13. Способ по п.4, где взаимодействие R-энантиомерного соединения общей формулы 19 и 4-(4 аминофенил)морфолин-3-она формулы 5 проводят в протонном растворителе или смеси растворителей или в смеси протонного растворителя и воды при 0-150 С в течение 0,5-60 ч. 14. Способ по п.13, где взаимодействие осуществляют при 60-90 С в течение 20-40 ч. 15. Способ по п.5, где взаимодействие S-энантиомерного соединения формулы 11 и соединения общей формулы Z1Z2NH, проводят в отсутствие или в присутствии органического растворителя, или в воде, или в смеси указанных растворителей. 16. Способ по п.15, где соединение общей формулы Z1Z2NH представляет собой N-бензил-1-фенилметанамин формулы 16 или его соли общей формулы 3 включающий удаление защитной групп(ы) с S-энантиомерного соединения общей формулы 14(где Z1 и Z2 представляют собой водород или защитную группу, при условии, что, по меньшей мере,Z не является водородом) с последующим выделением полученного таким образом S-энантиомерного основания формулы 12 или его соли общей формулы 3. 19. Способ по п.18, где энантиомерная соль общей формулы 3 представляет собой уксуснокислую соль формулы 3b, a S-энантиомерное соединение общей формулы 14 представляет собой S-энантиомерное соединение общей формулы 13. 20. 4-(4-(5S)-5-Дибензиламинометил-2-оксо-1,3-оксазолидин-3-илфенил)морфолин-3-он формулы 13 1
МПК / Метки
МПК: C07D 413/10, C07D 413/14, C07D 303/36
Метки: получения, способе, ривароксабана, соединения, указанном, получаемые, промежуточные, способ
Код ссылки
<a href="https://eas.patents.su/30-24685-sposob-polucheniya-rivaroksabana-i-promezhutochnye-soedineniya-poluchaemye-v-ukazannom-sposobe.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения ривароксабана и промежуточные соединения, получаемые в указанном способе</a>
Способ получения иопамидола и новые промежуточные соединения, получаемые в этом способе

Номер патента: 5922
Опубликовано: 25.08.2005
Авторы: Броккетта Марино, Лукс Джованна, Каппеллетти Энрико, Анелли Пьер Лучио
МПК: C07C 237/46
Метки: промежуточные, способе, новые, иопамидола, соединения, способ, этом, получения, получаемые
Формула / Реферат:
1. Способ получения (S)-N,N'-бис[2-гидрокси-1-(гидроксиметил)этил]-5-[(2-гидрокси-1-оксопропил)амино]-2,4,6-трииодо-1,3-бензолдикарбоксамида формулы (I) исходя из соединения формулы (II) причем указанный способ включает а) введение соединения формулы (II) в реакцию с подходящим защитным агентом с образованием соединения формулы (III) где -R - группа формулы A или B где R1 - атом водорода, C1-C4 линейная или разветвленная алкильная группа или...
Способ получения ингибитора цитохром p450 монооксигеназы и промежуточные соединения, применяемые в этом способе

Номер патента: 22739
Опубликовано: 29.02.2016
Авторы: Корде Даг, Истон Лия, Дауди Эрик, Каллен Аарон, Трэн Дуонг, Чжоу Чжунсинь, Польняшек Ричард, Пфайфер Стивен, Юй Ричард, Кент Кеннет
МПК: C07C 307/06, C07D 417/12
Метки: получения, способ, этом, монооксигеназы, применяемые, способе, ингибитора, промежуточные, цитохром, соединения
Формула / Реферат:
1. Способ получения соединения формулы Iили его соли, включающий:а) стадию димеризации соответствующего соединения формулы IIпри этом R1 представляет собой -S(=O)2NRaRb, где каждый из Ra и Rb независимо представляет собой (С1-С8)алкил; или Ra и Rb вместе с азотом, к которому они присоединены, образуют насыщенное кольцо, выбранное из азиридина, азетидина, пиперидина, морфолина, тиоморфолина, пирролидина, гомопиперазина, гомопиперидина или...
Трициклические соединения, обладающие активностью в отношении интегринов, в частности, в отношении интегрина альфаvбета 3, способ их получения и промежуточные соединения, используемые в этом способе,их применение в качестве медикаментов и содержащие их фармацевтические композиции.

Номер патента: 2271
Опубликовано: 28.02.2002
Авторы: Питти Роберт М., Штильц Ханс-Ульрих, Бодари Сара С., Гадек Томас Р., Карниато Дени, Кнолле Йохен, Бернар Серж, Гурвест Жан-Франсуа, Тетш Жан-Жорж, Венер Фолькмар, Макдауэлл Роберт С.
МПК: A61P 9/10, A61K 31/19, C07C 281/12...
Метки: медикаментов, соединения, применение, фармацевтические, обладающие, композиции, содержащие, интегринов, интегрина, частности, активностью, способ, промежуточные, трициклические, качестве, способе,их, альфаvбета, этом, отношении, используемые, получения
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает группу -О-[А]-[В]-COR6, в которой R6 обозначает -ОН, C1-С6алкокси, -О-СН2-СН(ОН)-СН2OН, [A] обозначает группу C1-С6алкилен, возможно замещенный оксогруппой, [B] обозначает радикал -CH(Z)- или простую связь, Z обозначает группу -NHCO2Rc, или -NHSO2Rc, где Rc обозначает радикал фенил(C1-С4)алкил-, хинолинил или пиридинилимидазолил(C1-С4)алкил-, R2 и R3, одинаковые или разные, обозначают атом...
Способ получения имидазо[1,2-c][2,3]бензодиазепинов и получаемые по этому способу промежуточные продукты

Номер патента: 5570
Опубликовано: 28.04.2005
Авторы: Шнайдер Маттиас, Винтер Эрик, Тильстам Ульф
МПК: C07C 69/614, C07D 491/14, C07D 487/04...
Метки: продукты, промежуточные, способ, способу, получения, имидазо[1,2-c][2,3]бензодиазепинов, этому, получаемые
Формула / Реферат:
1. Способ получения имидазо[1,2-c][2,3]бензодиазепинов общей формулы 1 в которой R1 обозначает водород, C1-C6алкил, нитрогруппу, галоген, цианогруппу, C1-C4алкоксигруппу, -CF3, гидроксигруппу или C1-C6алканоилоксигруппу, R2 и R3 имеют идентичные или разные значения и обозначают водород, галоген, C1-C6алкоксигруппу, гидроксигруппу, цианогруппу, C1-C6алканоил, C2-C6алкинил, C2-C6алкенил, необязательно замещенный галогеном, гидроксигруппой...
Промышленный способ синтеза 17a-ацетокси-11b-[4-(n,n-диметиламино)фенил]-19-норпрегна-4,9-диен-3,20-диона и новые промежуточные соединения, полученные в данном способе

Номер патента: 15302
Опубликовано: 30.06.2011
Авторы: Данчи Лайошне, Мадьяри Эндрене, Туба Золтан, Виски Дьёрдь, Чёргей Янош, Молнар Чаба
МПК: C07J 41/00, C07J 21/00, C07J 31/00...
Метки: соединения, промышленный, полученные, синтеза, данном, промежуточные, 17a-ацетокси-11b-[4-(n,n-диметиламино)фенил]-19-норпрегна-4,9-диен-3,20-диона, новые, способ, способе
Формула / Реферат:
1. Способ синтеза несольватированного 17a-ацетокси-11b-[4-(N,N-диметиламино)фенил]-19-норпрегна-4,9-диен-3,20-диона формулы (I), включающий следующие стадии:i) взаимодействие 3-(этилендиокси)эстра-5(10),9(11)диен-17-она формулы (X) с ацетилидом калия, образующимся in situ в сухом тетрагидрофуране,ii) взаимодействие полученного 3-(этилендиокси)-17a-этинил-17b-гидроксиэстра-5(10),9(11)диена формулы (IX) с фенилсульфенилхлоридом в дихлорметане в...
Предыдущий патент: Способ производства олефинов
Следующий патент: Горелочное устройство и горелочный блок
Случайный патент: Бис-(2-галоидэтил)аминофенилзамещенные производные дистамицина и их применение в качестве противоопухолевого и противовирусного средства