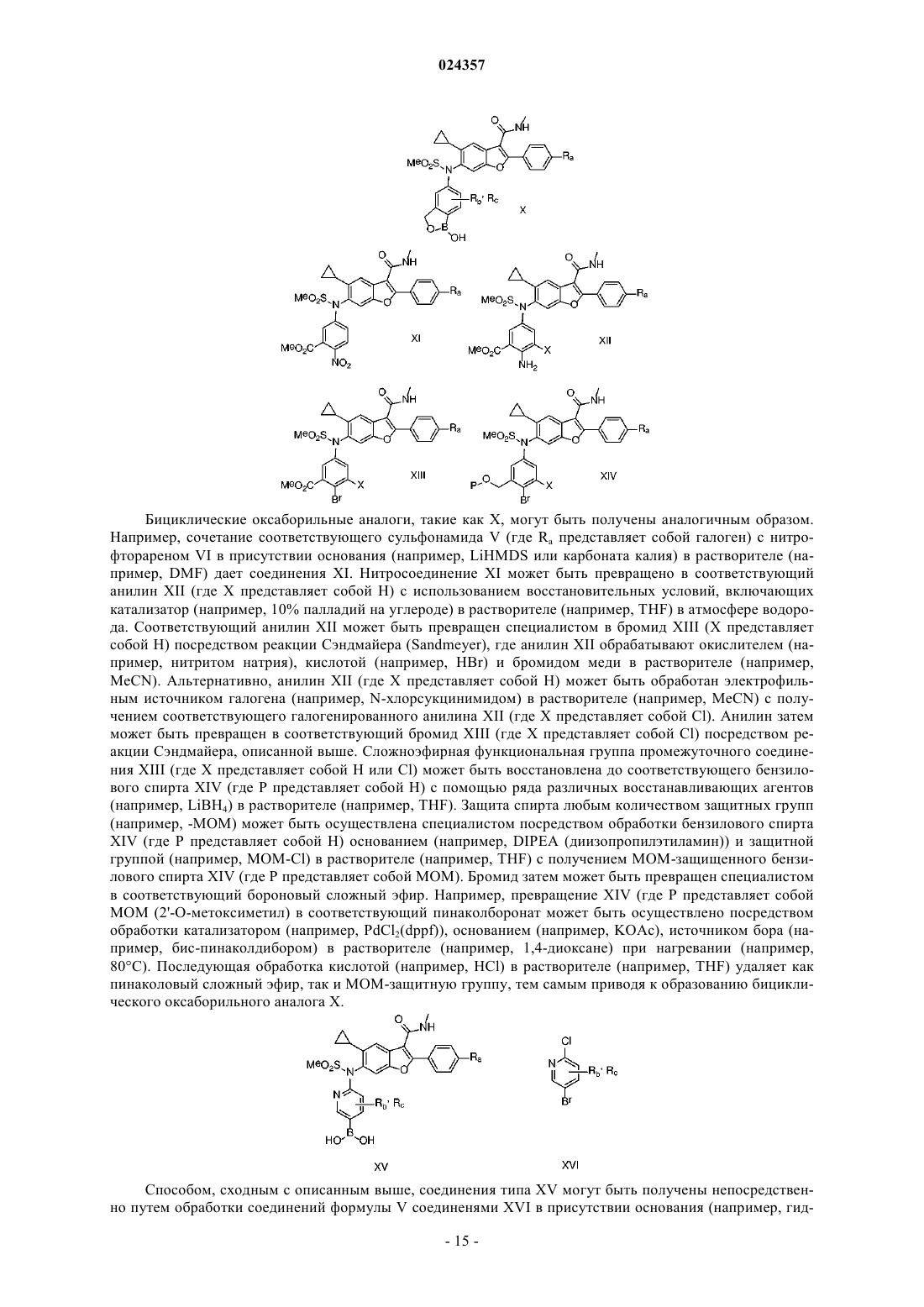
6-(n-(7-хлор-1-гидрокси-1,3-дигидробензо[c][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-n-метилбензофуран-3-карбоксамид





























Формула / Реферат
1. Соединение 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]-оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющее формулу

2. Соединение 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]-оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющее формулу

или его фармацевтически приемлемая соль.
3. Фармацевтически приемлемая соль соединения 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющего формулу

4. Фармацевтическая композиция для лечения или предупреждения вирусных инфекций, содержащая соединение 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющее формулу

совместно по меньшей мере с одним фармацевтически приемлемым эксципиентом.
5. Фармацевтическая композиция для лечения или предупреждения вирусных инфекций, содержащая соединение 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющее формулу

или его фармацевтически приемлемую соль совместно по меньшей мере с одним фармацевтически приемлемым эксципиентом.
6. Фармацевтическая композиция для лечения или предупреждения вирусных инфекций, содержащая фармацевтически приемлемую соль соединения 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющего формулу

совместно по меньшей мере с одним фармацевтически приемлемым эксципиентом.
Текст
Настоящее изобретение относится к 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол 5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамиду и его фармацевтически приемлемым солям, а также содержащим их фармацевтическим композициям, полезным для лечения или предупреждения вирусных инфекций. Перекрестная ссылка на родственные патенты и патентные заявки Данная заявка представляет собой заявку РСТ (Договор о патентной кооперации) и заявляет приоритет предварительной заявки США 61/524429, поданной 17 августа 2011 г., и предварительной заявки США 61/526787, поданной 24 августа 2011 г., каждая из которых включена в данное описание посредством ссылки во всей полноте. Область изобретения Настоящее изобретение относится к 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-5 ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамиду и его фармацевтически приемлемым солям, а также содержащим их фармацевтическим композициям, полезным для лечения или предупреждения вирусных инфекций, таких как инфекции, опосредованные членом семейства вирусов Flaviviridae, таким как вирус гепатита С (HCV). Предпосылки изобретения Хроническая HCV инфекция является серьезной проблемой здоровья, ассоциированной с повышенным риском развития хронического заболевания печени, цирроза, гепатоцеллюлярной карциномы и почечной недостаточности. HCV представляет собой член hepacivirus семейства Flaviviridae РНК вирусов, который поражает животных и людей. Его геном представляет собой одиночную нить РНК приблизительно 9,6 т.н. и состоит из одной открытой рамки считывания, которя кодирует полипротеин из приблизительно 3000 аминокислот, фланкированных нетранслируемыми областями как по 5', так и 3' концам(5'- и 3'-UTR). Этот полипротеин служит предшественником по меньшей мере 10 отдельных вирусных белков, критичных для репликации и сборки потомства вирусных частиц. Организация структурных и неструктурных белков в HCV полипротеине является следующей: C-E1-E2-p7-NS2-NS3-NS4a-NS4bNS5a-NS5b. Поскольку репликативный цикл HCV не включает никакой промежуточной ДНК и вирус не интегрирован в геном хозяина, HCV инфекция теоретически может быть излечена. Хотя как патологияHCV инфекции воздействует главным образом на печень, вирус обнаруживается в других типах клеток в организме, включая лимфоциты периферической крови.HCV является главным этиологическим фактором для пост-трансфузии и для спорадического гепатита. Инфекция HCV медленно развивается у большей части хронически инфицированных (и заразных) носителей, у которых клинические симптомы могут не проявляться в течение многих лет. У установленных 170 миллионов хронических носителей по всему миру существует риск развития заболевания печени, см., например, Szabo, et al., Pathol. Oncol. Res. 2003, 9:215-221, и Hoofnagle J.H., Hepatology 1997,26:15S-20S. Только в Соединенных Штатах Америки имеется 2,7 миллиона хронически инфицированныхHCV, и было установлено, что число случаев смерти, связанной с HCV, в 2000 г. составляло от 8000 до 10000, и ожидается, что это число будет значительно увеличиваться в последующие годы. Исторически стандартным лечением для хронической HCV инфекции являлся интерферон альфа(IFN-), в частности пэгилированный интерферон (PEG-IFN)-, в комбинации с рибавирином, которое требует от шести до двенадцати месяцев лечения. Эта комбинированная схема лечения включала 48 еженедельных инъекций интерферона и ежедневные дозы перорального рибавирина у HCV пациентов, инфицированных вирусом генотипа 1.IFN- принадлежит к семейству природных небольших белков с характерными биологическими эффектами, такими как противовирусная, иммунорегулирующая и противоопухолевые активности. Интерфероны продуцируются и секретируются большинством содержащих ядра клеток животных в ответ на ряд заболеваний, в частности на вирусные инфекции. IFN- представляет собой важный регулятор роста и дифференциации, воздействующий на клеточную коммуникацию и иммунологический контроль. Лечение HCV интерфероном зачастую бывает ассоциировано с тяжелыми побочными эффектами, такими как утомляемость, лихорадка, озноб, головная боль, миалгии, артралгии, умеренная аллопеция, психиатрические эффекты и ассоциированные расстройства, аутоиммунные проявления и ассоциированные расстройства и дисфункция щитовидной железы. Рибавирин, ингибитор инозин-5'-монофосфат-дегидрогеназы (IMPDH), повышает эффективностьIFN- в лечении HCV. Однако несмотря на введение рибавирина, у более чем 50% пациентов вирус не элиминируется при текущей стандартной терапии интерфероном- (IFN) и рибавирином. Кроме того, у большого числа пациентов имеются значительные побочные эффекты, связанные с рибавирином. Рибавирин вызывает существенный гемолиз у 10-20% пациентов, получающих лечение в рекомендуемых в настоящее время дозах, и это лекарственное средство является как тератогенным, так и эмбриотоксичным. Для борьбы с вирусом был предложен ряд дополнительных подходов. Они включают, например,применение антисмысловых олигонуклеотидов или рибозимов для ингибирования репликации HCV. Кроме того, привлекательными стратегиями для борьбы с инфекцией HCV являются низкомолекулярные соединения, которые непосредственно ингибируют белки HCV и препятствуют вирусной репликации. Среди вирусных мишеней наиболее перспективными мишенями HCV вируса для новых лекарственных средств являются протеаза/геликаза NS3/4A, РНК-зависимая РНК полимераза NS5B и неструктурный белок NS5A. Соединения, характеризующиеся как являющиеся полезными для лечения HCV инфекций,-1 024357 раскрыты, например, в WO 2005/051318 (Chunduru, et al.) и WO 2009/023179 (Schmitz, et al.). Эти ссылки раскрывают способы получения соединений, содержащие эти соединения композиции, композиции, содержащие эти соединения и дополнительные соединения, и способы лечения HCV. Недавно в США были предложены два HCV терапевтических лекарственных средства; каждое из них используется как тройные комбинационные терапии в сочетании с пэгилированным интерфероном и рибавирином. Ими являются Vertex's and Johnson and Johnson's ингибитор протеазы NS3/4A, Incivek(телапревир) и Merck's ингибитор протеазы NS3/4A, Victrelis (боцепревир). Более старая двойная схема лечения пэгилированным интерфероном и рибавирином в схеме лечения HCV излечивала только примерно 40% пациентов, инфицированных генотипом 1. Добавление Victrelis к этой схеме сокращает длительность лечения в некоторых случаях и повышает показатель излечивания до более чем 60%. Аналогично, добавление Incivek к этой схеме сокращает лечение и повышает показатели излечивания вплоть до 80%. К несчастью, ни Victrelis, ни Incivek не могут быть использованы сами по себе без включения в дополнение к ним схемы с использованием пэгилированного интерферона и рибавирина, что влечет за собой их сопутствующие неблагоприятные профили побочных эффектов. Эти ингибиторы протеаз также ассоциированы с дополнительными побочными эффектами, такими как сыпь и повышенная нейтропения. Такие лекарственные средства на основе одного активного агента также повышают риск селекции конкретных HCV мутаций в организме пациента, которые являются резистентными к этим ингибиторам протеазы. Даже при таких последних усовершенствованиях существенная часть пациентов не отвечает существенным снижением вирусной нагрузки, и существует явная необходимость в более эффективной противовирусной терапии HCV инфекции. Таким образом, необходимой является стратегия комбинированной терапии для борьбы с вирусом HCV без необходимости включения проблематичного пэгилированного интерферона и рибавирина. Многокомбинационные терапии, которые включают противовирусные средства прямого действия (DAA), нацеленные на более чем один конкретный тип HCV белка, могли бы понизить вероятность развития побочных эффектов. Также важно, что DAA могли бы снизить способность вируса мутировать в организме пациента, что может вести к восстановлению вирусного титраHCV. В виду мирового эпидемического уровня HCV, ограниченных доступных возможностей лечения и необходимости в расширении доступа ко всем пероральным DAA схемам лечения, существует растущая потребность в новых эффективных лекарственных средствах для лечения хронических HCV инфекций. Краткое изложение сущности изобретения Согласно одному из аспектов настоящего изобретения предложено соединение 6-(N-(7-хлор-1 гидрокси-1,3-дигидробензо[с][1,2]-оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющее формулу Согласно другому аспекту настоящего изобретения предложено соединение 6-(N-(7-хлор-1 гидрокси-1,3-дигидробензо[с][1,2]-оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющее формулу или его фармацевтически приемлемая соль. Согласно еще одному аспекту настоящего изобретения предложена фармацевтически приемлемая соль соединения 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-5-ил)метилсульфонамидо)5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющего формулу Кроме того, согласно настоящему изобретению предложена фармацевтическая композиция для лечения или предупреждения вирусных инфекций, содержащая соединение 6-(N-(7-хлор-1-гидрокси-1,3 дигидробензо[с][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющее формулу совместно по меньшей мере с одним фармацевтически приемлемым эксципиентом. Согласно настоящему изобретению также предложена фармацевтическая композиция для лечения или предупреждения вирусных инфекций, содержащая соединение 6-(N-(7-хлор-1-гидрокси-1,3 дигидробензо[с][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющее формулу или его фармацевтически приемлемую соль совместно по меньшей мере с одним фармацевтически приемлемым эксципиентом. Согласно настоящему изобретению также предложена фармацевтическая композиция для лечения или предупреждения вирусных инфекций, содержащая фармацевтически приемлемую соль соединения 6-(N-(7-хлор-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2(4-фторфенил)-N-метилбензофуран-3-карбоксамид, имеющего формулу совместно по меньшей мере с одним фармацевтически приемлемым эксципиентом. Указанные соединения, в которых борсодержащее кольцо непосредственно связано с сульфонамидом, демонстрируют повышенную метаболическую стабильность по сравнению с соединениями изPCT/US 2011/024822 (WO 2011/103063) и PCT/US 2011/024824 (WO 2012/067663), в которых борсодержащее кольцо связано через алкиленовый линкер. Подробное описание репрезентативных воплощений В данной заявке делаются ссылки на разные воплощения, относящиеся к соединениям, композициям и способам. Подразумевается, что различные раскрытые воплощения обеспечивают разнообразие иллюстративных примеров и не должны истолковываться как описания альтернативных разновидностей. Скорее следует отметить, что представленные в данном описании раскрытия различных воплощений могут иметь перекрывающийся объем. Обсуждаемые в данном описании воплощения являются всего лишь иллюстративными и их не следует понимать как ограничивающие объем настоящего изобретения. Следует понимать, что используемая в данном описании терминология предназначена для описания только конкретных воплощений и не предназаначена для ограничения объема настоящего изобретения. В данном описании изобретения будет сделана ссылка на ряд терминов, которые следует определить как имеющие следующие значения. Термин "алкил" относится к прямой или разветвленной углеводородной цепи, содержащей определенное количество атомов углерода. Например, C1-6 алкил означает прямой или разветвленный алкил,содержащий по меньшей мере 1 и максимум 6 атомов углерода. Примеры "алкила", как он используется в данном описании, включают, без ограничения ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 1,1-диметилпропил и н-гексил. Термин "алкокси" относится к алкильному радикалу, содержащему определенное количество атомов углерода, присоединенному посредством связывающего атома кислорода. Например, С 1-6 алкокси относится к углеводородному радикалу с прямой или разветвленной цепью, имеющему по меньшей мере 1 и вплоть до 6 атомов углерода, присоединенному посредством связывающего атома кислорода. Примеры "алкокси", как он используется в данном описании, включают, без ограничения ими, метокси, этокси,проп-1-окси, проп-2-окси, бут-1-окси, бут-2-окси, 2-метилпроп-1-окси, 2-метилпроп-2-окси, пентокси и гексилокси. Термин "галоген" или "галогено" относится к атому фтора (фтор, F), хлора (хлор, Cl), брома (бром,Br) или йода (йод, I). Термины "гидрокси" или "гидроксил" относятся к радикалу или заместителю формулы ОН. Термин "арил" относится к карбоциклической ароматической группировке (такой как фенил или нафтил), содержащей определенное количество атомов углерода, в частности 6-10 атомов углерода. Примеры арильных радикалов включают, без ограничения ими, фенил, нафтил, инденил, азуленил, флуоренил, антраценил, фенантренил, тетрагидронафтил, индалил, фенантридинил и тому подобное. Если не указано иное, термин "арил" также включает любой возможный позиционный изомер ароматического углеводородного радикала, такой как 1-нафтил, 2-нафтил, 5-тетрагидронафтил, 6-тетрагидронафтил, 1 фенантридинил, 2-фенантридинил, 3-фенантридинил, 4-фенантридинил, 7-фенантридинил, 8 фенантридинил, 9-фенантридинил и 10-фенантридинил. В настоящем изобретении раскрыто соединение формулы (I) представляющее собой соединение,имеющее следующую структуру: или его фармацевтически приемлемая соль. Соединение по изобретению также может существовать в стереоизомерных формах (например, они могут содержать один или более асимметричных атомов углерода). Индивидуальные стереоизомеры(энантиомеры и диастереомеры) и их смеси включены в объем настоящего изобретения. Изобретение также охватывает конформационные изомеры соединения по изобретению и любые геометрические (цис и/или транс) изомеры указанных соединений. Понятно, что соединение по изобретению может существовать в таутомерных формах, иных, нежели те, которые показаны в формуле, и они также включены в объем настоящего изобретения. Следует также понимать, что соединения по изобретению, которые существуют как полиморфы, и их смеси входят в объем настоящего изобретения. В настоящем изобретении также раскрыто соединение по изобретению или его фармацевтически приемлемая соль. Как он используется в данном описании, термин "фармацевтически приемлемые соли" относится к солям, которые сохраняют целевую биологическую активность рассматриваемого соединения и проявляют минимальные нежелательные токсикологические эффекты. Обзор подходящих солей см. в Berge et al., J. Pharm. Sci., 1977, 66, 1-19. Термин "фармацевтически приемлемые соли" включает как фармацевтически приемлемые соли присоединения кислот, так и фармацевтически приемлемые соли присоединения оснований. В ряде воплощений соединения по изобретению могут содержать кислотную функциональную группу и могут поэтому быть способны образовывать фармацевтически приемлемые соли присоединения оснований при их обработке подходящим основанием. Фармацевтически приемлемые соли с основаниями включают соли аммония (например, аммония или тетраалкиламмония), соли металлов, например соли щелочных металлов или щелочно-земельных металлов (таких как гидроксиды, натрий, калий, кальций или магний), органических аминов (таких как трис [также известный как трометамин или трис(гидроксиметил)аминометан], этаноламин, диэтиламин, триэтаноламин, холин, изопропиламин, дициклогексиламин или N-метил-D-глюкамин), катионных аминокислот (таких как аргинин, лизин или гистидин) или оснований для нерастворимых солей (таких как прокаин или бензатин). В ряде воплощений соединения по изобретению могут содержать основную функциональную группу и могут поэтому быть способны образовывать фармацевтически приемлемые соли присоединения кислот в результате обработки подходящей кислотой. Фармацевтически приемлемая соль присоединения кислоты может быть образована посредством взаимодействия соединения по изобретению с подходящей сильной неорганической кислотой (такой как бромисто-водородная, соляная, серная, азотная, фосфорная или перхлорная) или подходящей сильной органической кислотой, например сульфоновыми кислотами[такими как п-толуолсульфоновая, бензолсульфоновая, метансульфоновая, этансульфоновая, 2 гидроксиэтансульфоновая, нафталинсульфоновая (например, 2-нафталинсульфоновая)], карбоновыми кислотами (такими как уксусная, пропионовая, фумаровая, малеиновая, бензойная, салициловая или янтарная), анионными аминокислотами (такими как глутаминовая или аспарагиновая), гидроксикислотами(такими как лимонная, молочная, винная или гликолевая), жирными кислотами (такими как капроновая,каприловая, деканоевая, олеиновая или стеариновая) или кислотами для образования нерастворимых солей (такими как памоевая или резиновая [например, полистиролсульфонат]), возможно в подходящем растворителе, таком как органический растворитель, с получением соли, которую обычно выделяют, наример, посредством кристаллизации и фильтрования. В одном воплощении фармацевтически приемлемая соль присоединения кислоты соединения по изобретению представляет собой соль сильной кислоты,например соль гидробромид, гидрохлорид, гидройодид, сульфат, нитрат, перхлорат, фосфат, птолуолсульфонат, бензолсульфонат или метансульфонат. Для специалиста в данной области будет понятно, что органобороновые кислоты и/или их сложные эфиры органоборонаты в присутствии подходящих нуклеофильных комплексообразующих реагентов могут образовывать комплексные аддитивные соли "аты", такие как комплексные аддитивные соли органобораты. Подходящие нуклеофильные комплексообразующие реагенты включают, без ограничения ими, гидроксиды щелочных металлов, например гидроксид лития, гидроксид натрия или гидроксид калия, либо фторид. Примеры органоборатных комплексных аддитивных солей и способы их получения будут очевидны специалисту. Например, одной такой подходящей органоборатной комплексной аддитивной солью является соль тригидроксиорганоборат щелочного металла, такая как соль тригидроксиорганоборат натрия. В качестве иллюстрации комплексные аддитивные соли тригидроксиарилборат натрия и тригидроксиалкилборат натрия и способы их получения описаны в Cammidge, A.N. et al., Org. Lett.,2006, 8, 4071-4074. Фармацевтически приемлемые комплексные аддитивные соли "аты", как они раскрыты в данном описании, также рассматриваются как входящие в объем данного изобретения. В настоящем изобретении раскрыты подходящие фармацевтически приемлемые соли соединений по изобретению, включая их соли в качестве кислот, например соли с натрием, калием, кальцием, магнием, аммонием, тетраалкиламмонием и трисом (трометамин - трис(гидроксиметил)аминометан) и тому подобное, или моно- или диосновные соли с соответствующими кислотами, например органическими карбоновыми кислотами, таким как уксусная, молочная, винная, яблочная, изетионовая, лактобионовая и янтарная кислоты; органическими сульфоновыми кислотами, таким как метансульфоновая, этансульфоновая, бензолсульфоновая и п-толуолсульфоновая кислоты, и неорганическими кислотами, такими как соляная, серная, фосфорная и сульфаминовая кислоты, и тому подобное. В настоящем изобретении раскрыты фармацевтически приемлемые соли присоединения оснований соединения по изобретению, которые представляют собой соли сильного основания, например натрия,лизина, аммония, N-метил-D-глюкамина, калия, холина, аргинина (например, L-аргинина) или магния. В дополнительном аспекте соль представляет собой соль натрия, лизина, аммония, N-метил-D-глюкамина,калия, холина или аргинина (например, L-аргинина). Изобретение включает в свой объем все возможные стехиометрические и нестехиометрические формы солей соединений по изобретению. Для специалистов в области органической химии понятно, что многие органические соединения могут образовывать комплексы с растворителями, в которых они взаимодействуют или из которых они выпадают в осадок или кристаллизуются. Эти комплексы известны как "сольваты". Например, комплекс с водой известен как "гидрат". Сольваты соединений по изобретению и сольваты солей соединений по изобретению включены в объем настоящего изобретения. Для специалистов в данной области очевидно, что некоторые защищенные производные соединений по изобретению, которые могут быть получены перед завершающей стадией снятия защиты, могут сами по себе не обладать фармакологической активностью, но могут в определенных обстоятельствах быть введены перорально или парентерально и после этого метаболизироваться в организме с образованием соединений, как они определены в первом аспекте, которые являются фармакологически активными. Такие производные могут поэтому быть описаны как "пролекарства". Все защищенные производные и пролекарства соединений, определенных в первом аспекте, включены в объем изобретения. Примеры подходящих пролекарств для соединений по настоящему изобретению описаны в Drugs of Today, Volume 19, Number 9, 1983, pp. 499-538, и в Topics in Chemistry, Chapter 31, pp. 306-316, и в "Design of Prodrugs",H. Bundgaard, Elsevier, 1985, Chapter 1 (описания этих документов включены в данное описание посредством ссылки). Для специалистов в данной области также понятно, что некоторые группировки, извест-5 024357 ные специалистам в данной области как "прогруппировки", например, как описано Н. Bundgaard в "Design of Prodrugs" (описание этого документа включено в данное описание посредством ссылки), могут быть размещены на соответствующих функциональных группах, когда такие функциональные группы присутствуют в структуре соединений по изобретению. Подходящие пролекарства для соединений по изобретению включают сложные эфиры, сложные эфиры, представляющие собой карбонаты, гемисложные эфиры, сложные эфиры, представляющие собой фосфаты, нитросложные эфиры, сложные эфиры, представляющие собой сульфаты, сульфоксиды, амиды, карбаматы, азосоединения, фосфамиды, гликозиды, простые эфиры, ацетали, кетали, бороновые сложные эфиры и ангидриды бороновых кислот. Было установлено, что соединения по изобретению обладают противовирусной активностью, в частности HCV ингибиторной активностью, и могут поэтому быть полезными в лечении или предупреждении вирусных инфекций, таких как HCV инфекции, или заболеваний, ассоциированных с такими инфекциями, в частности при введении в комбинации с одним или более альтернативными терапевтическими агентами. Были проведены исследования in vitro, которые продемонстрировали полезность раскрытых в данном описании соединений в качестве противовирусных агентов. Следует понимать, что ссылка в данном описании на терапию или лечение может включать, без ограничения ими, предупреждение, замедление, профилактику и излечивание заболевания. В настоящем изобретении предлагаются соединения и фармацевтические композиции для лечения и предупреждения вирусных инфекций, такой как HCV инфекция, а также заболеваний, ассоциированных с вирусными инфекциями, у живых организмов-хозяев. Следует также понимать, что ссылки в данном описании на лечение или профилактику HCV инфекции включают лечение или профилактику HCV-ассоциированного заболевания, такого как фиброз печени, цирроз и гепатоклеточная карцинома. В контексте настоящего изобретения используемые в данном описании термины, описывающие показания, классифицированы в Merck Manual of Diagnosis и Therapy, 17th Edition и/или International Classification of Diseases 10th Edition (ICD-10). Различные подтипы расстройств, упомянутые в данном описании, предполагаются как часть настоящего изобретения. Соединения по изобретению могут быть получены способами, раскрытыми в данном описании, или с помощью любого способа, известного специалисту в данной области. В изобретении также раскрыты фармацевтические композиции, содержащие соединение по изобретению (далее в данном описании соединение А) и второй терапевтический агент, выбранный из группы,состоящей из ингибитора протеазы NS2 HCV, ингибитора протеазы NS3/4A HCV, ингибитора геликазыNS3 HCV, ингибитора фактора репликации NS4B HCV, ингибитора фактора репликации NS5A HCV,ингибитора полимеразы NS5B HCV, ингибитора проникновения HCV, ингибитора участка внутренней посадки рибосомы HCV, ингибитора микросомального белка-переносчика триглицеридов, ингибитора глюкозидазы, ингибитора каспазы, ингибитора циклофилина, иммуномодулятора, ингибитора метаболического пути, интерферона и нуклеозидного аналога (далее в данном описании соединение В), и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Возможно, такие фармацевтические композиции могут также содержать один или более дополнительных терапевтических агентов, независимо выбранных из группы, состоящей из ингибитора протеазы NS2 HCV, ингибитора протеазы NS3/4A HCV, ингибитора геликазы NS3 HCV, ингибитора фактора репликации NS4B HCV,ингибитора фактора репликации NS5A HCV, ингибитора полимеразы NS5B HCV, ингибитора проникновения HCV, ингибитора участка внутренней посадки рибосомы HCV, ингибитора микросомального белка-переносчика триглицеридов, ингибитора -глюкозидазы, ингибитора каспазы, ингибитора циклофилина, иммуномодулятора, ингибитора метаболического пути, интерферона и нуклеозидного аналога (далее в данном описании соединение С, соединение D и так далее). Следует понимать, что когда они объединены в одной композиции, эти два соединения должны быть стабильными и совместимыми друг с другом и с другими компонентами композиции. Когда они представлены раздельно, они могут быть представлены в любой обычной композиции, как правило, таким образом, как это известно для данного соединения из уровня техники. Примеры подходящих ингибиторов протеазы NS3/4A HCV включают боцепревир (такой как Victrelis), телапревир (такой как Incivek), симепревир (также известный как ТМС-435350), данопревир(также известный как RG7227 или ITMN-191), BI-201335, нарлапревир (также известный как SCH 900518), ванипревир (также известный как MK-7009), асунапревир (также известный как BMS-650032),GS 9256, GS 9451, АСН-0141625, VX-985, ABT-450, PHX1766, IDX320, MK-5172, GNS-227, AVL-192,АСН-2684 и АСН-1095. Примеры подходящих ингибиторов фактора репликации NS4B HCV включают клемизол. Примеры подходящих ингибиторов фактора репликации NS5A HCV включают АВТ-267, BMS790052, BMS-824393, BMS-766, AZD7295, CF102, GS5885, PPI-461, PPI-1301, PPI-437, PPI-668, PPI-833,ACH-2928, EDP-239, IDX380 и IDX719. Примеры подходящих ингибиторов полимеразы NS5B HCV включают силибинин натрия гемисукцинат, тегобувир (также известный как GS-9190), филибувир (также известный как PF-00868554), VX222, VX-759, ANA598, BMS-791325, АВТ-333, АВТ-072, BI 207127, IDX375, мерицитабин (также извест-6 024357 ный как RG7128 ), RG7348 (также известный как МВ-11362), RG7432, PSI-7977, PSI-7851, PSI-352938,PSI-661, ТМС 649128, IDX184, INX-08189, JTK-853, VCH-916, BILB 1941, GS-6620 и GS-9669. Примеры подходящих ингибиторов проникновения HCV включают PRO-206, ITX-5061, ITX4520,REP 9C, SP-30 и JTK-652. Примеры подходящих ингибиторов микросомального белка-переносчика триглицеридов (МТР) включают BMS-201038 и СР-346086. Примеры подходящих ингибиторов -глюкозидазы включают целгосовир (также известный как МХ-3253 или MBI-3253) и кастаноспермин. Примеры подходящих ингибиторов каспазы включают IDN-6556. Примеры подходящих ингибиторов циклофилина включают алиспоривир (также известный какDEBIO-025), NIM811 (также известный как N-метил-4-изолейцин-циклоспорин) и SCY-635 (также известный как [(R)-2-(N,N-диметиламино)этилтио-Sar]3-[4'-гидрокси-MeLeu]4-циклоспорин А). Примеры подходящих иммуномодуляторов включают аллоферон, IMN-6001, NOV-205, МЕ-3738,интерлейкин-7 (такой как CYT 107), ANA-773, IMO-2125 и GS 9620. Примеры подходящих ингибиторов метаболического пути включают ритонавир (такой как Norvir). Примеры подходящих интерферонов включают интерферон -2 а (такой как Roferon-A, Veldona или LBSI5535), пэгинтерферон -2 а (такой как Pegasys), интерферон -2b (такой как Intron А или Locteron), пэгинтерферон -2b (такой как PEG Intron или Р 1101), аналоги интерферона -2b (такие какTRK-560), пэгинтерферон(такой как BMS-914143) и интерферон-5. Примеры подходящих нуклеозидных аналогов включают рибавирин (такой как Copegus, Ravanex,Rebetol, RibaPak, Ribasphere, Vilona и Virazole), тарибавирин (также известный как вирамидин) и изаторибин (также известный как ANA245) и его пролекарства ANA971 и ANA975. Дополнительные примеры включают, без ограничения ими, ALS-2200, ALS-2158, PPI-383, PPI-393,PPI-461, PPI-668 и антисмысловой олиго, называемый mir-122. Носитель(и), разбавитель(и) или эксципиент(ы) должны быть приемлемыми в смысле их совместимости с другими ингредиентами композиции, пригодности для изготовления из них фармацевтических препаратов и безпасности для реципиентов. В соответствии с другим аспектом изобретения также раскрыт способ изготовления фармацевтической композиции, включающий смешивание соединения А и соединения В с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами. Такие элементы используемых фармацевтических композиций могут быть представлены в раздельных фармацевтических комбинациях или могут быть приготовлены совместно в одной фармацевтической композиции. Соответственно, в изобретении также раскрыта комбинация фармацевтических композиций, одна из которых включает соединение А и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов, и фармацевтической композиции, содержащей соединение В и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Возможно,комбинация фармацевтических композиций может дополнительно содержать одну или более дополнительных фармацевтических композиций, одна из которых включает соединение С и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов, и возможно другую, которая включает соединение D и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Фармацевтические композиции могут быть представлены в формах стандартной дозы, содержащих предварительно определенное количество активного ингредиента на стандартную дозу. Как известно специалистам в данной области, количество активного ингредиента на дозу будет зависеть от подлежащего лечению состояния, пути введения и возраста, массы и состояния пациента. Предпочтительными композициями стандартной дозы являются композиции, содержащие суточную дозу или суб-дозу или соответствующую ее часть, активного ингредиента. Кроме того, такие фармацевтические композиции могут быть получены любым из способов, хорошо известных в области фармацевтики. Соединения А, В, С, D и так далее могут быть введены любым подходящим путем. Подходящие пути включают пероральный, ректальный, интраназальный, местный (включая буккальный и сублингвальный), вагинальный и парентеральный (включая подкожный, внутримышечный, внутривенный, внутрикожный, подоболочечный и эпидуральный). Следует понимать, что предпочтительный путь может меняться в зависимости, например, от состояния реципиента комбинации. Следует также иметь в виду, что вводимые агенты могут быть введены одним и тем же или разными путями, и что любая комбинация соединений (например, соединения А и В; соединения А и С; соединения А, В и С) может быть объединена вместе в фармацевтической композиции. Фармацевтические композиции, подходящие для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобные пены или крема; или жидкие эмульсии масло-вводе или жидкие эмульсии вода-в-масле. Например, для перорального введения в форме таблетки или капсулы компонент в виде активного лекарственного средства может быть объединен с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и тому подобное. Порошки готовят посредством измельчения соединения до подходящего мелкого размера и смешивания с таким же образом измельченным фармацевтическим носителем, таким как пищевой углевод, как, например, крахмал или маннит. Также могут присутствовать корригенты, консерванты, диспергирующий агент и краситель. Капсулы могут быть изготовлены посредством приготовления порошоковой смеси, как описано выше, и заполнения ею сформованных желатиновых оболочек капсулы. К порошковой смеси перед операцией заполнения могут быть добавлены скользящие и смазывающие агенты, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль. Для улучшения доступности лекарственного средства при проглатывании капсулы также может быть добавлен разрыхляющий или солюбилизирующий агент, такой как агар-агар, карбонат кальция или карбонат натрия. Таблетки готовят, например, посредством приготовления порошковой смеси, гранулирования или комкования, добавления смазывающего агента и разрыхлителя и прессования в таблетки. Смазывающие агенты, используемые в этих лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Разрыхлители включают, без ограничения ими, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобное. Порошковую смесь готовят посредством смешивания подходящим образом измельченного соединения с разбавителем или основой, как описано выше, и возможно со связующим агентом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, раствором ретарданта, такого как парафин, ускорителем всасывания, таким как четвертичная соль, и/или абсорбционным агентом, таким как бентонит, каолин или дикальций фосфат. Возможные ингредиенты включают другие связующие вещества, такие как крахмал, природные сахара, такие как глюкоза или -лактоза, кукурузные подсластители,природные и синтетические смолы, такие как аравийская камедь, трагакант или альгинат натрия, полиэтиленгликоль, воски и тому подобное. Порошковая смесь может быть подвергнута влажному гранулированию со связующим агентом, таким как сироп, крахмальная паста, акациевый растительный клей или растворы целлюлозных или полимерных материалов, и просеиванию через сито. В качестве альтернативы гранулированию порошковая смесь может быть пропущена через таблеточную машину и результатом в этом случае являются неидеально сформированные фрагменты, разломанные в гранулы. Гранулы могут быть смазаны для предотвращения прилипания к формирующим таблетки пуансонам посредством добавления стеариновой кислоты, стеаратной соли, талька или минерального масла. Смазанную смесь затем прессуют в таблетки. Соединения по настоящему изобретению также могут быть объединены со свободнотекущим инертным носителем и спрессованы в таблетки непосредственно без пропускания через стадии гранулирования или комкования. Могут быть нанесены прозрачные или непрозрачные защитные покрытия, состоящие из уплотняющего покрытия из шеллака, сахарного покрытия или покрытия из полимерного материала, и полирующего покрытия из воска. К этим покрытиям могут быть добавлены красители для того, чтобы различать разные стандартные дозировки. Пероральные жидкости, такие как раствор, сиропы и эликсиры, могут быть получены в стандартной лекарственной форме так, чтобы данная единица содержала предварительно определенное количество соединения. Сиропы могут быть получены посредством растворения соединения в подходящим образом ароматизированном водном растворе, тогда как эликсиры готовят посредством использования нетоксичных спиртовых носителей. Суспензии могут быть получены посредством диспергирования соединения в нетоксичном носителе. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и простые эфиры полиоксиэтиленсорбита, консерванты, корригирующие добавки, такие как мятное масло, или природные подсластители, или сахарин, или другие искусственные подсластители и тому подобное. Где это целесообразно, композиции для перорального введения могут быть подвергнуты микроинкапсуляции. Композиция также может быть приготовлена с возможностью пролонгированного или отсроченного высвобождения, например, посредством нанесения покрытия или погружения материала в виде частиц в полимеры, воск и тому подобное. Агенты для применения согласно настоящему изобретению также могут быть введены в форме липосомальных систем доставки, таких как небольшие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из разнообразных фосфолипидов,таких как холестерин, стеариламин или фосфатидилхолины. Фармацевтические композиции, адаптированные для трансдермального введения, могут быть представлены в виде дискретных пластырей, предназначенных для того, чтобы оставаться в тесном контакте с эпидермисом реципиента в течение пролонгированного периода времени. Например, активный ингредиент может быть доставлен из пластыря посредством ионофореза, как в общем описано в PharmaceuticalResearch, 3(6), 318 (1986). Фармацевтические композиции, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатики и растворенные вещества, которые делают препарат изотоничным с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загутители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например запаянных ампулах и флаконах, и могут храниться в высушенном замораживанием(лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, немедленно перед применением. Стохастические растворы и суспензии для инъекций могут быть получены из стерильных порошков, гранул и таблеток. Следует понимать, что в дополнение к ингредиентам, непосредственно конкретно упомянутым выше, композиции могут включать другие агенты, обычные в области техники, имеющей отношение к приготовлению рассматриваемых препаратов, например композиции, подходящие для перорального введения, могут включать корригенты. Соединения А и В могут быть использованы в комбинации в соответствии с изобретением путем введения одновременно в единой фармацевтической композиции, включающей оба соединения. Альтернативно, комбинация может быть введена по раздельности в отдельных фармацевтических композициях,включающих каждое из соединений А и В, последовательным образом, где, например, соединение А или соединение В вводят первым, а другое соединение вторым. Такое последовательное введение может быть близким по времени (например, одновременным) или разнесенным по времени. Кроме того, не имеет значения, вводят ли соединения в одной и той же лекарственной форме, например одно соединение может быть введено парентерально, а другое соединение может быть введено перорально. Приемлемо,когда оба соединения вводят перорально. Соединение С возможно может быть введено в комбинации с одним из соединений А и В или с ними обоими, либо оно может быть введено отдельно от них в отдельной фармацевтической композиции. Соединение С может быть введено одновременно с любым одним из соединений А и В или с ними обоими, либо оно может быть введено последовательным образом относительно любого из соединений А и В или их обоих. Соединение D возможно может быть введено в комбинации с одним из соединений А и В или с ними обоими, либо оно может быть введено отдельно в отдельной фармацевтической композиции. Соединение D может быть введено одновременно с любым одним из соединений А, В и С или с ними со всеми, либо оно может быть введено последовательным образом относительно любого из соединений А, В и С или с ними со всеми. Таким образом, в одном воплощении одну или более доз соединения А вводят одновременно или раздельно с одной или более доз соединения В. Если не указано иное, во всех схемах введения, раскрытых в данном описании, введение соединений не обязательно должно начинаться с началом лечения и заканчиваться с концом лечения, необходимо только, чтобы количество последовательных суток, в которые вводят оба соединения, и возможное количество последовательных суток, в которые вводят только одно соединение - компонент, или указанная схема введения, включая количество вводимого соединения, имели место в какой-либо временной момент в течение курса лечения. В одном воплощении множественные дозы соединения А вводят одновременно или раздельно с множественными дозами соединения В. В другом воплощении множественные дозы соединения А вводят одновременно или раздельно с одной дозой соединения В. В другом воплощении одну дозу соединения А вводят одновременно или раздельно с множественными дозами соединения В. В другом воплощении одну дозу соединения А вводят одновременно или раздельно с одной дозой соединения В. Во всех вышеуказанных воплощениях соединение А может быть введено первым, либо соединение В может быть введено первым. Комбинации могут быть представлены как комбинационный набор. Термин "комбинационный набор" или "набор", как он используется в данном описании, означает фармацевтическую композицию или композиции, которые используются для введения соединения А и соединения В согласно изобретению. Возможно, набор может дополнительно содержать фармацевтическую композицию или композиции,которые используются для введения соединения С и возможно соединения D. Когда соединение А и соединение В вводят одновременно, комбинационный набор может содержать соединение А и соединение В в единой фармацевтической композиции, такой как таблетка, или в раздельных фармацевтических композициях. Возможно, набор может содержать соединения А, В и С в единой фармацевтической композиции, такой как таблетка, или любые два из соединений А, В и С в единой фармацевтической композиции, или каждое соединений А, В и С в отдельной фармацевтической композиции. Возможно, набор может содержать соединения А, В, С и D в единой фармацевтической композиции, такой как таблетка,или любые три из соединений А, В, С и D в единой фармацевтической композиции, или любые два из соединений А, В, С и D в единой фармацевтической композиции, или каждое из соединений А, В, С и D в отдельной фармацевтической композиции. Когда соединения А и В не вводят одновременно, комбинационный набор содержит соединение А и соединение В в раздельных фармацевтических композициях в единой упаковке, либо соединение А и соединение В в раздельных фармацевтических композициях в раздельных упаковках. Возможно, набор может содержать соединения А, В и С в раздельных фармацевтических композициях в единой упаковке или в раздельных упаковках. Возможно, набор может содержать соединения А, В, С и D в раздельных фармацевтических композициях в единой упаковке или в раздельных упаковках. В одном воплощении изобретения раскрыт набор, содержащий следующие компоненты: соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; и соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем. В другом воплощении изобретения раскрыт набор, содержащий следующие компоненты: соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем и соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем, где компоненты представлены в форме, которая является подходящей для последовательного,раздельного и/или одновременного введения. В другом воплощении изобретения раскрыт набор, содержащий следующие компоненты: первый контейнер, содержащий соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; и второй контейнер, содержащий соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем, и контейнерное приспособление для размещения указанных первого и второго контейнеров. В другом воплощении изобретения раскрыт набор, содержащий следующие компоненты: соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем и соединение С в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем. В другом воплощении изобретения раскрыт набор, содержащий следующие компоненты: соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; соединение С в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем, где компоненты представлены в форме, которая является подходящей для последовательного,раздельного и/или одновременного введения. В другом воплощении изобретения раскрыт набор, содержащий следующие компоненты: первый контейнер, содержащий соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; второй контейнер, содержащий соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; третий контейнер, содержащий соединение С в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем, и контейнерное приспособление для размещения указанных первого, второго и третьего контейнеров. В другом воплощении изобретения раскрыт набор, содержащий следующие компоненты: соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; соединение С в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; и соединение D в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем. В другом воплощении изобретения раскрыт набор, содержащий следующие компоненты: соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; соединение С в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем и соединение D в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем, где компоненты представлены в форме, которая является подходящей для последовательного,раздельного и/или одновременного введения. В другом воплощении изобретения раскрыт набор, содержащий следующие компоненты: первый контейнер, содержащий соединение А в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; второй контейнер, содержащий соединение В в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; третий контейнер, содержащий соединение С в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; и четвертый контейнер, содержащий соединение D в сочетании с фармацевтически приемлемыми эксципиентом, разбавителями или носителем; и контейнерное приспособление для размещения указанных первого, второго, третьего и четвертого контейнеров. Подходящим образом комбинации по данному изобретению вводят в пределах "установленного периода". Под термином "установленный период", как он используется в данном описании, понимают интервал времени между введением, например, одного из соединения А и соединения В и другого из соединения А и соединения В. Если не указано иного, установленный период времени может включать одновременное введение. Когда соединение А и соединение В вводят один раз в сутки, установленный период относится к введению соединения А и соединения В в течение одних суток. Когда одно или оба соединения вводят более чем один раз в сутки, установленный период рассчитывают на основании первого введения каждого соединения в конкретные сутки. Все введения соединения по изобретению, которые являются последующими относительно первого в течение конкретных суток, не учитываются при расчете установленного периода. Соответственно, если соединения вводят в пределах "установленного периода" и не вводят одновременно, то когда их вводят в пределах примерно 24 ч друг от друга - в этом случае установленный период будет составлять примерно 24 ч; соответственно, если они будут введены в пределах примерно 12 ч друг от друга - в этом случае установленный период будет составлять примерно 12 ч; соответственно,если они будут введены в пределах примерно 11 ч друг от друга - в этом случае установленный период будет составлять примерно 11 ч; соответственно, если они будут введены в пределах примерно 10 ч друг от друга - в этом случае установленный период будет составлять примерно 10 ч; соответственно, если они будут введены в пределах примерно 9 ч друг от друга - в этом случае установленный период будет составлять примерно 9 ч; соответственно, если они будут введены в пределах примерно 8 ч друг от друга в этом случае установленный период будет составлять примерно 8 ч; соответственно, если они будут введены в пределах примерно 7 ч друг от друга - в этом случае установленный период будет составлять примерно 7 ч; соответственно, если они будут введены в пределах примерно 6 ч друг от друга - в этом случае установленный период будет составлять примерно 6 ч; соответственно, они будут введены в пределах примерно 5 ч друг от друга - в этом случае установленный период будет составлять примерно 5 ч; соответственно, если они будут введены в пределах примерно 4 ч друг от друга - в этом случае установленный период будет составлять примерно 4 ч; соответственно, если они будут введены в пределах примерно 3 ч друг от друга - в этом случае установленный период будет составлять примерно 3 ч; соответственно, если они будут введены в пределах примерно 2 ч друг от друга - в этом случае установленный период будет составлять примерно 2 ч; соответственно, если они будут введены в пределах примерно 1 ч друг от друга - в этом случае установленный период будет составлять примерно 1 ч. При использовании в данном описании введение соединения А и соединения В с промежутком менее чем примерно 45 мин считают одновременным введением. Соответственно, когда комбинацию по изобретению вводят в течение "установленного периода",эти соединения будут вводиться совместно в течение "промежутка времени". Термин "промежуток времени", как он используется в данном описании, означает, что каждое из соединений по изобретению вводят в течение указанного числа последовательных суток. Относительно введения в течение "установленного периода", когда каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере одних суток - в этом случае промежуток времени будет составлять по меньшей мере одни сутки; когда в течение курса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 3 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 3 суток; когда в течениекурса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 5 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 5 суток; когда в течение курса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 7 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 7 суток; когда в течение курса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 14 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 14 суток; когда в течение курса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 30 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 30 суток; когда в течение курса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 60 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 60 суток; когда в течение курса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 90 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 90 суток; когда в течение курса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 180 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 180 суток; когда в течение курса лечения каждое из соединений будет вводиться в пределах установленного периода в течение по меньшей мере 365 последовательных суток - в этом случае промежуток времени будет составлять по меньшей мере 365 суток. Далее относительно введения в течение "установленного периода". Приемлемо, когда в течение курса лечения соединение А и соединение В будут вводиться в пределах установленного периода в течение от 1 до 4 суток за 7-дневный период, а во время других суток этого 7-дневного периода соединение А будет вводиться одно или возможно с соединением С и возможно соединением D. Приемлемо, когда этот 7-дневный протокол повторяется в течение 2 циклов или в течение 14 суток; приемлемо в течение 4 циклов или 28 суток; приемлемо в течение 12 циклов или 84 суток; приемлемо для постоянного введения. Приемлемо, когда в течение курса лечения соединение А и соединение В будут вводиться в пределах установленного периода в течение 1 суток за 7-дневный период, а в другие сутки этого 7-дневного периода соединение А будет вводиться одно или возможно с соединением С и возможно соединением D. Приемлемо, когда этот 7-дневный протокол повторяют в течение 2 циклов или в течение 14 суток; приемлемо в течение 4 циклов или 28 суток; приемлемо в течение 12 циклов или 84 суток; приемлемо для постоянного введения. Соответственно, если соединения не вводят во время "установленного периода", то их вводят последовательно. Под термином "последовательное введение" и его производными, как они используются в данном описании, понимают, что одно из соединения А и соединения В вводят в течение двух или более последовательных суток, а другое из соединения А и соединения В затем вводят в течение двух или более последовательных суток. Также охватывается данным описанием отдых от лекарства, применяемый между последовательным введением одного из соединения А и соединения В и другого из соединения А и соединения В. Как он используется в данном описании, термин "отдых от лекарства" представляет собой период в сутках после последовательного введения одного из соединения А и соединения В и перед введением другого из соединения А и соединения В, где не вводят ни соединение А, ни соединение В. Соответственно, "отдых от лекарства" будет составлять период в сутках, выбранный из следующего: 1 сутки, 2 суток, 3 суток, 4 суток, 5 суток, 6 суток, 7 суток, 8 суток, 9 суток, 10 суток, 11 суток, 12 суток, 13 суток и 14 суток. В отношении последовательного введения. Приемлемо, когда одно из соединения А и соединения В вводят в течение 2-30 последовательных суток с последующим возможным "отдыхом от лекарства", а затем введением другого из соединения А и соединения В в течение 2-30 последовательных суток. Приемлемо, когда одно из соединения А и соединения В вводят в течение 2-21 последовательных суток с последующим возможным "отдыхом от лекарства", а затем введением другого из соединения А и соединения В в течение 2-21 последовательных суток. Приемлемо, когда одно из соединения А и соединения В вводят в течение 2-14 последовательных суток с последующим "отдыхом от лекарства" в течение 1-14 суток, а затем введением другого из соединения А и соединения В в течение 2-14 последовательных суток. Приемлемо, когда одно из соединения А и соединения В вводят в течение 3-7 последовательных суток с последующим "отдыхом от лекарства" в течение 3-10 суток, а затем введением другого из соединения А и соединения В в течение 3-7 последовательных суток. Приемлемо, когда соединение В будут вводить первым в этой последовательности с последующим возможным "отдыхом от лекарства", а затем введением соединения А. Приемлемо, когда соединение В вводят в течение 2-21 последовательных суток с последующим возможным "отдыхом от лекарства", а затем вводят соединение А в течение 2-21 последовательных суток. Приемлемо, когда соединение В вводят в течение 3-21 последовательных суток с последующим "отдыхом от лекарства" в течение 1-14 суток,а затем вводят соединение А в течение 3-21 последовательных суток. Приемлемо, когда соединение В вводят в течение 3-21 последовательных суток с последующим "отдыхом от лекарства" в течение 3-14 суток, а затем вводят соединение А в течение 3-21 последовательных суток. Приемлемо, когда соединение А будут вводить первым в этой последовательности с последующим возможным "отдыхом от лекарства", а затем введением соединения В. Приемлемо, когда соединение А вводят в течение 2-21 последовательных суток с последующим возможным "отдыхом от лекарства", а затем вводят соединение В в течение 2-21 последовательных суток. Приемлемо, когда соединение А вводят в течение 3-21 последовательных суток с последующим "отдыхом от лекарства" в течение 1-14 суток,а затем вводят соединение В в течение 3-21 последовательных суток. Приемлемо, когда соединение А вводят в течение 3-21 последовательных суток с последующим "отдыхом от лекарства" в течение 3-14 суток, а затем вводят соединение В в течение 3-21 последовательных суток. Следует понимать, что за введением в течение "установленного периода" и "последовательным" введением может следовать повторное дозирование или может следовать альтернативный протокол до- 12024357 зирования, и "отдых от лекарства" может предшествовать повторному дозированию или альтернативному протоколу дозирования. Соответственно, количество соединения А (на основе массы несолевого/несольватированного количества), вводимое как часть комбинации согласно настоящему изобретению, будет находиться в диапазоне от 0,01 до 100 мг на 1 кг массы тела реципиента (например, человека) в сутки; приемлемо, когда количество будет выбираться в диапазоне от 0,1 до 30 мг на 1 кг массы тела в сутки; приемлемо, когда количество будет выбираться в диапазоне от 0,1 до 10 мг на 1 кг массы тела в сутки; приемлемо, когда количество будет выбираться в диапазоне от 0,5 до 10 мг на 1 кг массы тела в сутки. Целевая доза может быть представлена в виде одной, двух, трех, четырех, пяти, шести или более суб-доз, вводимых с соответствующими интервалами на протяжении суток. В некоторых случаях целевая доза может быть введена в альтернативные сутки или по другой подходящей схеме введения, например еженедельно или ежемесячно. Эти суб-дозы могут быть введены в стандартных лекарственных формах, например, содержащих от 0,5 до 100 мг, от 5 до 1000 мг, или от 50 до 500 мг, или от 20 до 500 мг активного ингредиента на стандартную лекарственную форму. Следующие неограничивающие примеры иллюстрируют настоящее изобретение. Примеры Для специалиста в данной области техники будет очевидно, что когда в реакциях используют растворители, желательно использовать безводные растворители. Также желательно проводить взаимодействия в инертной атмосфере, например под азотом или аргоном, как целесообразно. Аббревиатуры мкл - микролитры,мкМ - микромолярный,ЯМР - ядерный магнитный резонанс,br - широкий,d - дублет, - химический сдвиг,С - градусы Цельсия,dd - дублет дублетов,DMEM - модифицированная по Дульбекко среда Игла,DMF - N,N-диметилформамид,DMSO - диметилсульфоксид,г - грамм,ч - часы,HCV - вирус гепатита С,ВЭЖХ - высокоэффективная жидкостная хроматография,Гц - герцы,J - константа спин-спинового взаимодействия (приведена в Гц, если не указано иного),m - мультиплет,М - молярный,М+Н+ - масс-спектрометрический молекулярный пик плюс Н+,мг - миллиграмм,мл - миллилитр,мМ - миллимолярный,ммоль - миллимоль,МС - масс-спектр,нМ -наномолярный,м.д. - миллионная доля,s - синглет,t - триплет. Соединения по настоящему изобретению могут быть получены специалистом в данной области в соответствии с примерами синтеза и указаниями, представленными в следующей заявке: РСТ заявка на патентPCT/US 2012/050268, которая включена в данное описание во всей ее полноте. Соединения по настоящему изобретению также могут быть получены специалистом посредством следующих приведенных ниже примеров синтеза. Для специалиста в данной области техники будет очевидно, что когда в реакциях используют растворители, желательно использовать безводные растворители. Также желательно проводить взаимодействия в инертной атмосфере, например под азотом или аргоном, как целесообразно. Соединения по изобретению могут быть получены следующими способами или посредством любого способа, известного специалисту в данной области. Соединения формулы (II) типа IX (Ra представляет собой галоген, Rb и Rc представляют собой галоген, алкил, алкокси или кольцо) легко получают из соответствующих соединений - бромидов (III) или соответствующих трифлатных соединений (IV, где Р представляет собой OTf), используя условия, известные специалисту в данной области. Например, превращение III в соответствующий пинаколборонат может быть осуществлено посредством обработки катализатором (например PdCl2(dppf) (дихлор[1,1'бис(дифенилфосфино)ферроцен]палладий(II, основанием (например KOAc), источником бора (например, бис-пинаколдибором) в растворителе (например, 1,4-диоксане) при нагревании (например, 80 С). В результате последующей обработки кислотой (например, HCl) в смеси растворителей (например, THF(тетрагидрофуран)/вода) с поглотителем пинакола (например, бензолбороновой кислотой на полимерной подложке) или перйодатом натрия получают IX. Далее для специалиста в данной области понятно, что нитросоединение II может быть превращено в соответствующий анилин, используя восстановительные условия, включающие катализатор (например, 10% палладий на углероде) в растворителе (например,THF) в атмосфере водорода. В результате последующего применения реакции Сандмейера (Sandmeyer),включая окислитель (наприме,р нитрит натрия), кислоту (например HBr) и бромид меди в растворителе(например, MeCN), получают III. Трифлатные соединения (IV, где Р представляет собой OTf) могут быть получены посредством обработки соответствующего фенольного промежуточного соединения IV (где Р представляет собой Н) "трифлирующим" реагентом (например, трифликовым ангидридом). Соединения II, III и IV легко получаются в результате сочетания соответствующего сульфонамида(VII), либо фенолзащищенной бороновой кислотой (VIII, где Р представляет собой бензил. В первом случае прямая обработка V основанием (например, LiHMDS (гексаметилдисилазид лития) или карбонатом калия) в растворителе (например, DMF) с последующим взаимодействием с VI дает соответствующие продукты замещения SNAR II. Кроме того, обработка сульфонамида V арилбороновой кислотой, такой как VII или VIII (где Р представляет собой бензил), используя условия сочетания Чана-Лама (ChanLam), включающие источник меди (например, ацетат меди(II, основание (например, триэтиламин) и поглотитель (например, 3 или 4 молекулярные сита) в растворителе (например, DCM (хлористый метилен, дает соответствующий бромид III или фенолзащищенное промежуточное соединение IV (где Р представляет собой бензил), которое после снятия защиты (например, посредством гидрирования бензильной группы) дает фенольное промежуточное соединение IV (где Р представляет собой Н). Бициклические оксаборильные аналоги, такие как X, могут быть получены аналогичным образом. Например, сочетание соответствующего сульфонамида V (где Ra представляет собой галоген) с нитрофторареном VI в присутствии основания (например, LiHMDS или карбоната калия) в растворителе (например, DMF) дает соединения XI. Нитросоединение XI может быть превращено в соответствующий анилин XII (где X представляет собой Н) с использованием восстановительных условий, включающих катализатор (например, 10% палладий на углероде) в растворителе (например, THF) в атмосфере водорода. Соответствующий анилин XII может быть превращен специалистом в бромид XIII (X представляет собой Н) посредством реакции Сэндмайера (Sandmeyer), где анилин XII обрабатывают окислителем (например, нитритом натрия), кислотой (например, HBr) и бромидом меди в растворителе (например,MeCN). Альтернативно, анилин XII (где X представляет собой Н) может быть обработан электрофильным источником галогена (например, N-хлорсукцинимидом) в растворителе (например, MeCN) с получением соответствующего галогенированного анилина XII (где X представляет собой Cl). Анилин затем может быть превращен в соответствующий бромид XIII (где X представляет собой Cl) посредством реакции Сэндмайера, описанной выше. Сложноэфирная функциональная группа промежуточного соединения XIII (где X представляет собой Н или Cl) может быть восстановлена до соответствующего бензилового спирта XIV (где Р представляет собой Н) с помощью ряда различных восстанавливающих агентов(например, LiBH4) в растворителе (например, THF). Защита спирта любым количеством защитных групп(например, -MOM) может быть осуществлена специалистом посредством обработки бензилового спиртаXIV (где Р представляет собой Н) основанием (например, DIPEA (диизопропилэтиламин и защитной группой (например, МОМ-Cl) в растворителе (например, THF) с получением МОМ-защищенного бензилового спирта XIV (где Р представляет собой MOM). Бромид затем может быть превращен специалистом в соответствующий бороновый сложный эфир. Например, превращение XIV (где Р представляет собойMOM (2'-O-метоксиметил) в соответствующий пинаколборонат может быть осуществлено посредством обработки катализатором (например, PdCl2(dppf, основанием (например, KOAc), источником бора (например, бис-пинаколдибором) в растворителе (например, 1,4-диоксане) при нагревании (например,80 С). Последующая обработка кислотой (например, HCl) в растворителе (например, THF) удаляет как пинаколовый сложный эфир, так и МОМ-защитную группу, тем самым приводя к образованию бициклического оксаборильного аналога X. Способом, сходным с описанным выше, соединения типа XV могут быть получены непосредственно путем обработки соединений формулы V соединенями XVI в присутствии основания (например, гид- 15024357 рида натрия или LHMDS) в растворителе (например, THF). Превращение полученного бромида в соединения формулы XVI может быть осуществлено способом, аналогичным превращению соединения III в соединение IX. Бороновые кислоты типа XVII могут быть получены посредством алкилирования соответствующего фенольного промежуточного соединения IV (где Р представляет собой Н) с помощью 2-(хлорметил)4,4,5,5-тетраметил-1,3,2-диоксаборолана в присутствии основания (например, K2CO3) и в подходящем растворителе (например, DMF). В результате удаления пинакольной группы в раскрытых в данном описании условиях получают соответствующие бороновые кислоты XVII. Для специалиста в данной области понятно, что бензильные бороновые кислоты типа XVIII могут быть получены из соответствующих бороновых сложных эфиров типа IX или из соответствующих бромидов типа III. Например, посредством обработки соответствующим образом защищенного пинаколборонового сложного эфира IX реагентомLiCH2Cl в соответствующем растворителе (например, THF) при низкой температуре (например, -78 С),как описано в литературе (например, J. Med. Chem. 2010, 53, 7852), получают соответствующий бензильный бороновый сложный эфир XVIII (где Rd представляет собой Н). Альтернативно, соответствующие бензильные бороновые сложные эфиры типа XVIII (где Rd представляет собой Н) могут быть получены из соответствующим образом замещенных арилбромидов типа III посредством обмена галоген-металл,используя подходящий алкиллитий (например, трет-BuLi), в подходящем растворителе (например, THF) при низкой температуре (например, -78 С), с последующим добавлением 2-(хлорметил)-4,4,5,5 тетраметил-1,3,2-диоксаборолана. Альтернативно, соответствующие бензильные бороновые сложные эфиры типа XVIII могут быть получены из соответствующим образом замещенных арилбромидов типаIII посредством Pd-катализируемого кросс-сочетания арилбромида с соответствующим образом замещенным бис-бороновым сложным эфиром, таким как XIX (где Rd представляет собой алкил, бензил), как описано в литературе (например, J. Am. Chem. Soc. 2010, 132, 11033). Циклические оксаборилы типа XX (где Re представляет собой Н, алкил) могут быть получены различными способами, как описано в литературе. Например, посредством взаимодействия биспинаколборана (B2pin2) с ,-ненасыщенными сложными эфирами (например, где Rf представляет собой ОМе), амидами (например, где Rf представляет собой NMe2) и кетонами (например, где Rf представляет собой алкил) типа XXII в присутствии металлического катализатора (например, CuCl или Rh(Phebox)XXIII), как описано в литературе (J. Org. Chem. 2011, 76, 3997 или Chem. Commun. 2009, 5987), получают промежуточные соединения типа XXI, которые при восстановлении сложного эфира, амида или кетона и удалении пинакола дают соответствующие циклические оксаборилы XX (где Re представляет собой Н,алкил). Региоизомерные циклические оксаборилы XXIV и XXV легко получаются посредством гидроборирования соответствующих алкенов (XXVI и XXVII соответственно) в стандартных условиях, описанных в литературе. Синтез промежуточных соединений Промежуточное соединение 1. 2-(4-Хлорфенил)-5-циклопропил-N-метил-6-(метилсульфонамидо) бензофуран-3-карбоксамид(25 мл, 0,288 моль), и затем по каплям добавляли DMF (0,5 мл). Реакционную смесь кипятили с обратным холодильником в течение 2 ч. Полученный прозрачный желтый раствор концентрировали в вакууме с получением хлорангидрида в виде желтой жидкости. TEA (триэтиламин, 67 мл) добавляли к раствору малоната этилкалия (41 г, 0,241 моль) в ацетонитриле (537 мл). При охлаждении на бане лед-соль добавляли MgCl2 (27,4 г, 0,288 моль) и полученную смесь перемешивали при этой температуре в течение 3 ч. Добавляли хлорангидрид (полученный, как описано выше) и реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение ночи. Смесь охлаждали в ледяной бане и осторожно добавляли 2 н. HCl (600 мл). Смесь перемешивали в ледяной бане в течение 1,5 ч, затем переносили в делительную воронку и экстрагировали этилацетатом (3200 мл). Объединенные органические слои промывали насыщенным бикарбонатом натрия (450 мл), рассолом (250 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта этил-3-(4-хлорфенил)-3-оксопропаноата (48,6 г), который использовали без очистки. Стадия 2: этил-2-(4-хлорфенил)-5-гидроксибензофуран-3-карбоксилат. Хлорид цинка (28,3 г, 0,207 моль) перемешивали в безводном этаноле (45 мл), затем нагревали до 95 С (возврат флегмы) в атмосфере азота, используя высушенную в сушильном шкафу лабораторную посуду. Одной порцией добавляли этил-4-хлорбензоилацетат (44 г, 0,194 моль) с последующим добавлением по каплям на протяжении 2 ч раствора бензохинона (22,6 г, 0,21 моль) в безводном МТВЕ (метилтрет-бутиловый эфир, 500 мл). Этот процесс осуществляли с одновременной отгонкой МТВЕ из реакционной смеси так, чтобы реакционный объем оставался приблизительно постоянным. Температуру бани 145-155 С и внутреннюю температуру 75-95 С поддерживали в продолжении большей части добавления. В середине процесса добавления добавляли дополнительное количество безводного этанола (45 мл), поскольку реакционная смесь становилась густой и ожидалась потеря некоторой части исходного объема этанола в результате отгонки. После завершения добавления нагревание продолжали в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры и распределяли между водой (100 мл) иEtOAc (250 мл). Нерастворимую твердую фазу удаляли фильтрованием двухфазного раствора и органический слой затем отделяли, промывали дополнительным количеством воды, сушили и выпаривали под вакуумом. Оставшееся коричневое твердое вещество суспендировали в теплом дихлорметане и смесь охлаждали до комнатной температуры и охлаждали дополнительно в холодильнике в течение ночи. Желто-коричневое твердое вещество отфильтровывали из темно-коричневого раствора и промывали неболь- 17024357 шим объемом DCM и сушили под вакуумом с получением этил-2-(4-хлорфенил)-5-гидроксибензофуран 3-карбоксилата (27 г, 44%). Стадия 3: этил-2-(4-хлорфенил)-5-изопропоксибензофуран-3-карбоксилат. К этил-2-(4-хлорфенил)-5-гидроксибензофуран-3-карбоксилату (26 г, 0,051 моль) в NMP (N-метил 2-пирролидон, 160 мл) добавляли изопропилбромид (15 мл), затем добавляли карбонат цезия (33 г, 0,101 моль). Реакционную смесь перемешивали при 60 С в масляной бане в течение 20 ч, затем охлаждали до температуры окружающей среды. Реакционную смесь обрабатывали 5%-ным раствором аммиака и перемешивали в течение 15 мин. Эту смесь затем разбавляли водой и экстрагировали гексаном. Органический слой промывали водой, сушили с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении с получением этил-2-(4-хлорфенил)-5-изопропоксибензофуран-3 карбоксилата (15 г, 82%). Стадия 4: этил-2-(4-хлорфенил)-5-изопропокси-6-нитробензофуран-3-карбоксилат. Этил-2-(4-хлорфенил)-5-изопропоксибензофуран-3-карбоксилат 4 (30 г, 0,084 моль) растворяли в хлороформе (75 мл) и полученный раствор охлаждали в ледяной бане. Азотную кислоту (55 мл) также растворяли в хлороформе (75 мл) и охлаждали в ледяной бане. Раствор кислоты добавляли по каплям в течение 1 ч к раствору этил-2-(4-хлорфенил)-5-изопропоксибензофуран-3-карбоксилата и реакционную смесь затем перемешивали при 0 С в течение 1,5 ч. Реакционную смесь затем разбавляли водой (60 мл) и слои разделяли. Органический слой сушили с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении с получением коричневого масла, которое очищали колоночной хроматографией (5/1 РЕ/ЕА (петролейный эфир/этилацетат с получением этил-2-(4-хлорфенил)5-изопропокси-6-нитробензофуран-3-карбоксилата в виде коричневого твердого вещества (11 г, 32%). Стадия 5: этил-2-(4-хлорфенил)-5-гидрокси-6-нитробензофуран-3-карбоксилат. Этил-2-(4-хлорфенил)-5-изопропокси-6-нитробензофуран-3-карбоксилат (11 г, 27,2 ммоль) растворяли в безводном DCM (150 мл) и охлаждали в ледяной бане в атмосфере азота. В течение приблизительно 20 мин добавляли трихлорид бора (41 мл, 41,0 ммоль). После завершения реакции реакционную смесь гасили выливанием ее в смесь лед/вода. Смесь экстрагировали DCM и объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали под вакуумом с получением этил-2-(4-хлорфенил)-5-гидрокси-6-нитробензофуран-3-карбоксилата (10,2 г, 84%). Стадия 6: этил-2-(4-хлорфенил)-6-нитро-5-(трифторметилсульфонилокси)бензофуран-3 карбоксилат. К раствору этил-2-(4-хлорфенил)-5-гидрокси-6-нитробензофуран-3-карбоксилата (10,2 г, 22,9 ммоль) и DMAP (4-диметиламинопиридин, 0,289 г, 2,3 ммоль) в безводном DCM (300 мл) и безводномTEA (триэтиламин, 4,8 мл) в ледяной бане под азотом добавляли ангидрид трифторметансульфоновой кислоты (5,62 мл, 34 ммоль). Реакционную смесь перемешивали под азотом при 0 С в течение 30 мин,затем гасили при 0 водой (200 мл) и экстрагировали DCM (3200 мл). Объединенные органические слои промывали водой (3600 мл), 2 н. HCl (2300 мл) водой (300 мл), сушили с помощью безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении с получением этил-2-(4 хлорфенил)-6-нитро-5-(трифторметилсульфонилокси)бензофуран-3-карбоксилата (10 г, 80%) в виде желтого твердого вещества, которое использовали без дополнительной очистки. Стадия 7: этил-2-(4-хлорфенил)-5-циклопропил-6-нитробензофуран-3-карбоксилат. В смесь 2-(4-хлорфенил)-6-нитро-5-(трифторметилсульфонилокси)бензофуран-3-карбоксилата (10 г, 18 ммоль), KF (4,64 г, 79,9 ммоль), NaBr (2,48 г, 24 ммоль), циклопропилбороновой кислоты (3,2 г, 37 ммоль) и Pd(Ph3P)4 (1,33 г, 1,15 ммоль) добавляли толуол (130 мл) и воду (2,8 мл). Реакционную колбу вакуумировали в течение приблизительно 3 мин, затем заполняли азотом. Реакционную смесь кипятили с обратным холодильником под азотом в течение 20 ч, затем охлаждали до температуры окружающей среды. Реакционную смесь разбавляли EtOAc (150 мл), промывали водой (3200 мл), рассолом (200 мл),сушили с помощью безводного сульфата натрия, декантировали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (петролейный эфир/этилацетат от 30/1 до приблизительно 10/1) с получением этил-2-(4-хлорфенил)-5-циклопропил-6-нитробензофуран-3 карбоксилата в виде желтого твердого вещества (7,9 г, 99%). Стадия 8: этил-6-амино-2-(4-хлорфенил)-5-циклопропилбензофуран-3-карбоксилат. К раствору этил-2-(4-хлорфенил)-5-циклопропил-6-нитробензофуран-3-карбоксилата (8 г, 18,2 ммоль) в этилацетате (450 мл) добавляли 10% палладий на угле (1,83 г), 1 н. раствор HCl (2,5 мл) и перемешивали под давлением водорода 0,4 МПа при комнатной температуре в течение 8 ч. Реакционную смесь фильтровали через целит и фильтрат выпаривали под вакуумом с получением этил-6-амино-2-(4 хлорфенил)-5-циклопропилбензофуран-3-карбоксилата в виде коричневого твердого вещества (7,4 г,99%). Стадия 9: этил-2-(4-хлорфенил)-5-циклопропил-6-(N-(метилсульфонилметил)метилсульфонамидо) бензофуран-3-карбоксилат. К раствору этил-6-амино-2-(4-хлорфенил)-5-циклопропилбензофуран-3-карбоксилата (7,4 г, 18,06 ммоль) в сухом дихлорметане (170 мл) при -15 С в атмосфере N2 по каплям добавляли сухой TEA (6,73 мл, 45,15 ммоль) и затем метансульфонилхлорид (4,91 мл, 63,2 ммоль). Перемешанный раствор нагревали до комнатной температуры и перемешивали в течение 2 ч. Реакционную смесь разбавляли водой (100 мл) и экстрагировали DCM (3150 мл). Органические слои объединяли, сушили с помощью Na2SO4,фильтровали и выпаривали под вакуумом с получением этил-2-(4-хлорфенил)-5-циклопропил-6-(N(метилсульфонилметил)метилсульфонамидо)бензофуран-3-карбоксилата (9,2 г, 99%). Стадия 10: 5-циклопропил-2-(4-хлорфенил)-6-(метилсульфонамидо)бензофуран-3-карбоновая кислота. Гидроксид калия (15,1 г, 270 ммоль) добавляли к раствору этил-2-(4-хлорфенил)-5-циклопропил-6(N-(метилсульфонилметил)метилсульфонамидо)бензофуран-3-карбоксилата в этаноле (64 мл) и воде (32 мл) в атмосфере азота. Реакционную смесь кипятили с обратным холодильником в течение 1 ч и затем концентрировали в вакууме. Оставшееся твердое вещество растворяли в воде и раствор подкисляли с помощью 1 н. HCl (250 мл) до тех пор, пока не образовывался осадок. Твердое вещество отфильтровывали и затем сушили с получением 5-циклопропил-2-(4-хлорфенил)-6-(метилсульфонамидо)бензофуран-3 карбоновой кислоты (8,7 г, количественный выход). Стадия 11: 2-(4-хлорфенил)-5-циклопропил-N-метил-6-(метилсульфонамидо)бензофуран-3 карбоксамид. К раствору 5-циклопропил-2-(4-хлорфенил)-6-(метилсульфонамидо)бензофуран-3-карбоновой кислоты (5 г, 11,52 ммоль) в сухом DMF (30 мл) при 20 С добавляли DIPEA (3,3 г, 25,34 ммоль) и HATU(гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, 5,15 г, 13,5 ммоль). Спустя 15 мин по каплям добавляли 2 М метиламин в THF (23,04 мл, 46,08 ммоль) и смесь перемешивали в течение еще 2 ч, после чего добавляли воду (60 мл). Смесь экстрагировали ЕА (3200 мл), промывали водой(2200 мл), сушили и концентрировали с получением 2-(4-хлорфенил)-5-циклопропил-N-метил-6(метилсульфонамидо)бензофуран-3-карбоксамида в виде коричневого твердого вещества (4,7 г, 97%). 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 9.32 (br. s., 1 Н), 8.45 (q, 1 Н), 7.90 (d, 2 Н), 7.53 - 7.64 (m, 3 Н),7.16 (s, 1 Н), 3.06 (s, 3 Н), 2.83 (d, 3 Н), 2.21 - 2.37 (m, 1 Н), 0.94 - 1.05 (m, 2 Н), 0.64 - 0.73 (m, 2 Н). ЖХ-МС Стадия 1: метил-2-(4-фторфенил)-5-гидрокси-1-бензофуран-3-карбоксилат. Используя лабораторную посуду, высушенную в сушильном шкафу, в атмосфере азота безводный хлорид цинка (25 г, 183 ммоль) перемешивали в безводном метаноле (60 мл), затем нагревали до внутренней температуры 75 С. Одной порцией добавляли метил-4-фторбензоилацетат (39,6 г, 202 ммоль), а затем в течение 4 ч по каплям добавляли раствор п-бензохинона (19,83 г, 183 ммоль) в безводном диэтиловом эфире (500 мл). Этот процесс осуществляли с отновременной отгонкой эфира из реакционной смеси так, чтобы реакционный объем оставался приблизительно постоянным (температура бани 140 С поддерживала внутреннюю температуру сначала равной 75 С, затем постепенно повышая до максимум 115 С). Через 2,5 ч после начала добавления бензохинона для облегчения перемешивания добавляли дополнительное количество метанола (20 мл). После того как добавление бензохинона было завершено,нагревание реакционной смеси при 100 С (внутренняя температура) продолжали в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и распределяли между водой (500 мл) и этилацетатом (800 мл). Нерастворимую твердую фазу удаляли из двухфазного раствора фильтрованием и органический слой затем отделяли, сушили (Na2SO4), фильтровали и выпаривали под вакуумом. Коричневый остаток суспендировали в теплом дихлорметане (приблизительно 225 мл) и смесь оставляли стоять в холодильнике в течение 18 ч. Полученную твердую фазу отфильтровывали из темно-коричневого раствора,промывали небольшим объемом дихлорметана, затем сушили под вакуумом с получением метил-2-(4 фторфенил)-5-гидрокси-1-бензофуран-3-карбоксилата. ЖХ-МС (m/z, ЭР+) = 285(М-1). Стадия 2: метил-2-(4-фторфенил)-5-[(1-метилэтил)окси]-1-бензофуран-3-карбоксилат. Смесь метил-2-(4-фторфенил)-5-гидрокси-1-бензофуран-3-карбоксилата (18,86 г, 65,9 ммоль), изопропилбромида (24,74 мл, 264 ммоль) и карбоната цезия (42,9 г, 132 ммоль) в сухом N-метил-2 пирролидоне (191 мл) перемешивали при 60 С под азотом в течение 20 ч. Полученную густую суспензию оставляли охлаждаться до комнатной температуры и затем при быстром перемешивании добавляли 7%-ный водный раствор аммиака (200 мл). Эту смесь экстрагировали гептаном (700 мл) и затем водную фазу отделяли. К органической фазе добавляли этилацетат (приблизительно 100 мл) и полученную смесь встряхивали и затем сушили над Na2SO4 и выпаривали с получением коричневого масла, которое кристаллизовалось при стоянии в течение ночи. Этот материал перекристаллизовывали из горячего метанола и твердую фазу собирали фильтрованием, промывали метанолом и в заключение сушили под вакуумом с получением метил-2-(4-фторфенил)-5-[(1-метилэтил)окси]-1-бензофуран-3-карбоксилата. ЖХ-МС (m/z,ЭР+) = 329 (М+1). Маточный раствор от первой перекристаллизации подвергали кристаллизации во второй раз с получением дополнительной партии метил-2-(4-фторфенил)-5-[(1-метилэтил)окси]-1 бензофуран-3-карбоксилата. Стадия 3: метил-2-(4-фторфенил)-5-[(1-метилэтил)окси]-6-нитро-1-бензофуран-3-карбоксилат. К раствору метил-2-(4-фторфенил)-5-[(1-метилэтил)окси]-1-бензофуран-3-карбоксилата (6,16 г,18,76 ммоль) в хлороформе (22 мл) при -15 С добавляли по каплям холодный раствор 70%-ной азотной кислоты (11 мл, 172 ммоль) в хлороформе (22 мл). После перемешивания при 0 С в течение 1 ч реакционную смесь промывали водой (50 мл) и органическую фазу отделяли с помощью гидрофобной фильтрационной трубки, затем выпаривали под вакуумом с получением коричневого твердого вещества. Твердое вещество растирали в метил-трет-бутиловом эфире (25 мл) и полученный бледно-желтый порошок отфильтровывали, промывали гептаном и сушили под вакуумом с получением метил-2-(4-фторфенил)-5[(1-метилэтил)окси]-6-нитро-1-бензофуран-3-карбоксилата. ЖХ-МС (m/z, ЭР+) = 764 (2 М+ NH4)+. Стадия 4: метил-2-(4-фторфенил)-5-гидрокси-6-нитро-1-бензофуран-3-карбоксилат. К перемешиваемому раствору метил-2-(4-фторфенил)-5-[(1-метилэтил)окси]-6-нитро-1-бензофуран 3-карбоксилата (5,237 г, 14,03 ммоль) в сухом дихлорметане (70 мл) при -15 С в атмосфере азота в течение 30 мин с помощью шприцевого насоса добавляли 1 М раствор трихлорида бора в дихлорметане(23,85 мл, 23,85 ммоль). Темную коричнево-красную реакционную смесь выливали на лед (приблизительно 250 мл). Лед оставляли плавиться и смесь экстрагировали дихлорметаном (приблизительно 450 мл). Органическую фазу отделяли с помощью гидрофобной фильтрационной трубки и выпаривали под вакуумом с получением метил-2-(4-фторфенил)-5-гидрокси-6-нитро-1-бензофуран-3-карбоксилата. 1 Н ЯМР (d6-DMSO):10.97 (1 Н, br. s), 8.34 (1 Н, s), 8.07 (2 Н, dd), 7.67 (1 Н, s), 7.43 (2 Н, t), 3.86 (3 Н, s). Стадия 5: метил-2-(4-фторфенил)-6-нитро-5-[(трифторметил)сульфонил]окси-1-бензофуран-3 карбоксилат. К охлаждаемой на льду перемешиваемой смеси метил-2-(4-фторфенил)-5-гидрокси-6-нитро-1 бензофуран-3-карбоксилата (4,915 г, 14,84 ммоль) и 4-(диметиламино)пиридина (0,181 г, 1,484 ммоль) в безводном дихлорметане (130 мл) под азотом добавляли триэтиламин (3,10 мл, 22,26 ммоль), а затем ангидрид трифторметансульфоновой кислоты (3,76 мл, 22,26 ммоль). Через 50 мин при 0 С добавляли воду и органический слой отделяли. Водную фазу экстрагировали дополнительным количеством дихлорметана и объединенную органическую фазу промывали 2 М HCl и водой. Органическую фазу сушили с помощью гидрофобной фильтрационной трубки и выпаривали с получением метил-2-(4-фторфенил)-6 нитро-5-[(трифторметил)сульфонил]окси-1-бензофуран-3-карбоксилата. ЖХ-МС (m/z, ЭР+) = 481 (М+(7,12 г, 15,37 ммоль), циклопропилбороновую кислоту (2,19 г, 25,5 ммоль), фторид калия (3,26 г, 56,1 ммоль), бромид натрия (1,75 г, 17,01 ммоль) и тетракис(трифенилфосфин)палладий(0) (0,85 г, 0,736 ммоль) перемешивали вместе под азотом в смеси толуола (90 мл) и воды (2,25 мл) и нагревали при 100 С в течение 18 ч. Реакционную смесь охлаждали, разбавляли этилацетатом и промывали водой. Органическую фазу отделяли, сушили с помощью гидрофобной фильтрационной трубки и выпаривали под вакуумом. Остаток очищали посредством флэш-хроматографии, элюируя силикагель градиентом 0-5% этилацетата в циклогексане. Содержащие продукт фракции выпаривали под вакуумом с получением метил-5 циклопропил-2-(4-фторфенил)-6-нитро-1-бензофуран-3-карбоксилата. ЖХ-МС (m/z, ЭР+) = 728 (2 М+NH4)+. Стадия 7: метил-6-амино-5-циклопропил-2-(4-фторфенил)-1-бензофуран-3-карбоксилат. Раствор метил-5-циклопропил-2-(4-фторфенил)-6-нитро-1-бензофуран-3-карбоксилата (3,175 г, 8,94 ммоль) в этилацетате (250 мл), содержащий 2 М HCl (17 капель), перемешивали с 10% палладием на углероде (0,951 г, 0,894 ммоль) в атмосфере водорода при 21 С в течение 16 ч. Реакционную смесь фильтровали через целит и фильтрат выпаривали под вакуумом с получением темно-зеленого твердого вещества. Его растворяли в дихлорметане, промывали раствором бикарбоната натрия, отделяли с помощью гидрофобной фритты, затем выпаривали досуха и очищали посредством флэш-хроматографии, элюируя силикагель градиентом 0-30% этилацетата в циклогексане. Соответствующие фракции объединяли и выпаривали под вакуумом с получением метил-6-амино-5-циклопропил-2-(4-фторфенил)-1-бензофуран-3 карбоксилата. ЖХ-МС (m/z, ЭР+) = 326 (М+Н+). Стадия 8: метил-6-[бис(метилсульфонил)амино]-5-циклопропил-2-(4-фторфенил)-1-бензофуран-3 карбоксилат. Раствор метил-6-амино-5-циклопропил-2-(4-фторфенил)-1-бензофуран-3-карбоксилата (1,96 г, 6,02 ммоль) и триэтиламина (2,52 мл, 18,07 ммоль) в сухом дихлорметане (40 мл) охлаждали (ледяная баня) до 0 С, затем обрабатывали метансульфонилхлоридом (1,174 мл, 15,06 ммоль). Реакционную смесь перемешивали при 0 С (ледяная баня) в течение 2 ч. Добавляли воду (100 мл) и органическую фазу экстрагировали 3 раза дихлорметаном, сушили с помощью гидрофобной фритты и выпаривали досуха с получением метил-6-[бис(метилсульфонил)амино]-5-циклопропил-2-(4-фторфенил)-1-бензофуран-3-карбок- 20024357 силата. ЖХ-МС (m/z, ЭР+) = 482 (М+Н+). Стадия 9: 5-циклопропил-2-(4-фторфенил)-6-[(метилсульфонил)амино]-1-бензофуран-3-карбоновая кислота. Суспензию метил-6-[бис(метилсульфонил)амино]-5-циклопропил-2-(4-фторфенил)-1-бензофуран-3 карбоксилата (2,88 г, 5,98 ммоль) в этаноле (50 мл) и воде (25 мл) обрабатывали гидроксидом калия (6,71 г, 120 ммоль) и кипятили с обратным холодильником в течение 1 ч (суспензия переходила в раствор при нагревании). Реакционную смесь концентрировали под вакуумом, добавляли воду (100 мл) и раствор подкисляли с помощью 2 М HCl (50 мл). Полученный осадок отфильтровывали, промывали 0,5 М HCl,затем растворяли в метаноле. Этот раствор упаривали досуха и дважды подвергали азеотропной перегонке с толуолом с получением 5-циклопропил-2-(4-фторфенил)-6-[(метилсульфонил)амино]-1 бензофуран-3-карбоновой кислоты. ЖХ-МС (m/z, ЭР+) = 390 (М+Н+). Стадия 10: 5-циклопропил-2-(4-фторфенил)-N-метил-6-(метилсульфонамидо)бензофуран-3 карбоксамид. Раствор 5-циклопропил-2-(4-фторфенил)-6-[(метилсульфонил)амино]-1-бензофуран-3-карбоновой кислоты (2,52 г, 6,47 ммоль), HATU (2,95 г, 7,77 ммоль) и триэтиламина (1,984 мл, 14,24 ммоль) в сухом дихлорметане (100 мл) перемешивали при комнатной температуре в течение 1 ч, затем обрабатывали метиламином (16,18 мл, 32,4 ммоль). Раствор перемешивали при комнатной температуре под азотом в течение 4 ч, в течение которых образовывался осадок. Реакционную смесь разбавляли дихлорметаном(300 мл) и раствором бикарбоната натрия (200 мл) и перемешивали в течение 10 мин. Слои разделяли и водный слой экстрагировали дополнительным количеством дихлорметана (150 мл). Объединенную органическую фазу промывали рассолом (200 мл), сушили с помощью гидрофобной фритты и выпаривали досуха с получением не совсем белого твердого вещества. Неочищенный продукт очищали посредством флэш-хроматографии (элюируя градиентом 0-100% этилацетат/циклогексан, а затем 10% метанол/ дихлорметан) с получением белого твердого вещества. Твердое вещество растворяли в горячей смеси метанол-хлороформ (10% об./об.), преабсорбировали на силикагеле и очищали посредством флэшхроматографии (0-10% метанол/дихлорметан) с получением 5-циклопропил-2-(4-фторфенил)-N-метил-6[(метилсульфонил)амино]-1-бензофуран-3-карбоксамида. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 9.30 (br s, 1 Н), 8.42 (q, 1 Н), 7.88 - 7.98 (m, 2 Н), 7.60 (s, 1 Н), 7.37(t, 2 Н), 7.16 (s, 1 Н), 3.06 (s, 3 Н), 2.83 (d, 3 Н), 2.23 - 2.36 (m, 1 Н), 0.93 - 1.05 (m, 2 Н), 0.65 - 0.74 (m, 2 Н). ЖХ-МС (m/z, ЭР+) = 403 (М+Н+). Синтез родственных соединений (примеры 1-14) Пример 1. Стадия 1: 6-(N-(3-хлор-4-нитрофенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамид. Смесь 5-циклопропил-2-(4-фторфенил)-N-метил-6-(метилсульфонамидо)-бензофуран-3-карбоксамида (12,5 г, 31,1 ммоль), 2-хлор-4-фторнитробензола (10,9 г, 62,1 ммоль) и карбоната калия (12,9 г, 93,0 ммоль) в 130 мл смеси 4:1 DME/вода в герметично закрытой колбе нагревали до 100 С с перемешиванием. Идентичную реакционную смесь в количестве 12,5 г готовили во втором герметично закрытом сосуде. Реакционные сосуды выдерживали при 100 С в течение 70 ч, охлаждали до к.т. и содержимое перемешивали в течение дополнительных 18 ч. Объединенные реакционные смеси распределяли междуEtOAc (300 мл) и водой (600 мл) и фазы разделяли. Водный раствор экстрагировали двумя дополнительными порциями по 150 мл EtOAc. Объединенные EtOAc растворы промывали полунасыщенным рассолом (1), насыщенным рассолом (1), сушили над сульфатом натрия и концентрировали досуха при пониженном давлении. Полученное желто-коричневое твердое вещество перекристаллизовывали из смесиEtOAc/эфир с получением указанного в заголовке соединения (22,3 г, 64%) в виде не совсем белого твердого вещества. ЖХ-МС (m/z, ЭР+) = 558 (М+Н+). Стадия 2: 6-(N-(4-амино-3-хлорфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамид. Раствор 6-(N-(3-хлор-4-нитрофенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамида (11,0 г, 19,7 ммоль) в смеси 1:1 THF/MeOH (75 мл) подвергали гидрированию при 40 фунт-сила на кв.дюйм (280 кПа) в присутствии 5% сульфидированной платины на углероде (0,560 г). Спустя 4 ч добавляли дополнительное количество катализатора (0,250 г). Спустя еще 16 ч реакционный сосуд продували азотом, катализатор удаляли фильтрованием через целит и фильтрат концентрировали досуха при пониженном давлении. Остаток перекристаллизовывали из смеси гексан/EtOAc с получением указанного в заголовке соединения (10,3 г, 99%) в виде белого твердого вещества. ЖХ-МС(10,0 г, 18,9 ммоль), а затем ацетонитрил (200 мл) и затем 48%-ную водную HBr (200 мл). Полученную густую комковатую суспензию интенсивно перемешивали в течение 30 мин с получением более однородной суспензии. Реакционный сосуд охлаждали в бане лед-вода в течение 30 мин и смесь в течение 5 мин обрабатывали раствором нитрита натрия (1,96 г, 28,4 ммоль) в воде (20 мл) посредством загрузочной воронки. Полученную желтую суспензию перемешивали в ледяной бане в течение 1,5 ч и затем в течение 5 мин обрабатывали небольшими порциями CuBr (4,1 г, 28,4 ммоль). В результате получали темнокоричневый раствор, который нагревали до 60 С (внутренняя температура) при непрерывном перемешивании. После выдерживания смеси в течение 40 мин при повышенной температуре смесь охлаждали до к.т. и вливали в быстро перемешиваемую смесь 5%-ного водного бисульфита натрия (600 мл) и EtOAc(800 мл). Фазы разделяли и водный раствор экстрагировали двумя дополнительными порциями по 150 мл EtOAc. Объединенный EtOAc раствор промывали 5%-ным водным бисульфитом натрия (2150 мл),насыщенным водным бикарбонатом натрия (2300 мл), насыщенным рассолом (1200 мл), сушили над сульфатом натрия и концентрировали досуха при пониженном давлении с получением желтой пены. Этот материал подвергали флэш-хроматографии (силикагель, градиент от 9:1 гексан/EtOAc до EtOAc). В процессе концентрирования чистых фракций кристаллизовалось белое твердое вещество. После концентрирования до густой суспензии смесь разбавляли 150 мл гексана и эту смесь перемешивали при к.т. в течение ночи. Твердое вещество собирали фильтрованием через воронку из средней фритты и сушили в вакууме с получением указанного в заголовке соединения (8,55 г, 76%) в виде белого кристаллического твердого вещества. ЖХ-МС (m/z, ЭР+) = 591,593(М+Н+). Стадия 4: (2-хлор-4-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)фенил)бороновая кислота. В 350 мл колбу, закрытую завинчивающейся крышкой, снабженную магнитной мешалкой, добавляли 6-(N-(4-бром-3-хлорфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид (8,54 г, 14,4 ммоль), бис(пинаколато)дибор (18,3 г, 72,1 ммоль), ацетат калия (7,08 г, 72,1 ммоль), комплекс Pd(II)(dppf)Cl2 и дихлорметана (0,589 г, 0,721 ммоль) и безводный 1,4-диоксан(150 мл). Смесь продували азотом в течение 10 мин. Сосуд герметично закрывали и нагревали на масляной бане при 80 С с перемешиванием. Через 4 ч смесь охлаждали до к.т. и разбавляли EtOAc (400 мл). Полученный черный раствор промывали водой (2), насыщенным рассолом (1) и сушили над сульфатом натрия. В процессе перемешивания с сульфатом натрия добавляли целит для облегчения удаления нерастворимого черного материала, который оставался суспендированным в растворе. Смесь фильтровали через среднюю фритту с получением золотисто-коричневого фильтрата, который концентрировали досуха при пониженном давлении. Остаток растворяли в 300 мл THF и полученный раствор охлаждали в бане лед-вода. Раствор обрабатывали 1 н. водной HCl (120 мл), а затем перйодатом натрия (46,3 г, 216 ммоль). Смесь перемешивали при 0 С в течение 10 мин и затем оставляли нагреваться до к.т. Через 18 ч смесь распределяли между водой и EtOAc и фазы разделяли. Водный раствор экстрагировали EtOAc(2). Объединенный EtOAc раствор промывали 5%-ным водным бисульфитом натрия (4), насыщенным рассолом (2), сушили над сульфатом натрия и концентрировали досуха при пониженном давлении. Остаток подвергали флэш-хроматографии (силикагель, 2-частный градиент: от DCM до EtOAc за 15 мин,затем заменяли растворители на А, представляющий собой смесь 9:1 DCM/MeOH, В, представляющий собой DCM; градиент от 100% В до 65% А за 4 мин, затем 65% А изократическое элюирование в течение 15 мин) с получением светлой желто-коричневой пены (7,28 г). Этот материал растворяли в ацетонитриле (75 мл) и раствор перемешивали с быстрым капельным добавлением 0,25 н. водной HCl (175 мл) на протяжении периода времени 20 мин. Полученную белую суспензию перемешивали при к.т. Спустя 2 ч твердое вещество собирали фильтрованием через воронку из средней фритты. Осадок на фильтре промывали водой (2), сушили отсасыванием на воздухе в течение 30 мин и затем сушили в вакууме в течение ночи с получением указанного в заголовке соединения (5,90 г, 74%) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, метанол-d4)м.д. 7.90 - 7.97 (m, 2 Н), 7.85 (s, 1 Н), 7.29 - 7.41 (m, 4 Н), 7.26 (t, 2 Н),3.34 (s, 3 Н), 2.95 (s, 3 Н), 2.07 - 2.17 (m, 1 Н), 0.69 - 1.08 (m, 3 Н), 0.49 (br s, 1 Н). ЖХ-МС (m/z, ЭР+) = 557 Стадия 1: 6-(N-(3-хлор-4-нитрофенил)метилсульфонамидо)-2-(4-хлорфенил)-5-циклопропил-Nметилбензофуран-3-карбоксамид. Смесь 2-(4-хлорфенил)-5-циклопропил-N-метил-6-(метилсульфонамидо)-бензофуран-3-карбоксамида (2,00 г, 4,77 ммоль), 2-хлор-4-фторнитробензола (1,68 г, 9,55 ммоль) и карбоната калия (1,98 г, 14,3 ммоль) в смеси 4:1 DME/вода (20 мл) в герметично закрытом сосуде при перемешивании нагревали до 100 С. Через 18 ч смесь обрабатывали дополнительной порцией 2,00 г карбоната калия, нагревали при 100 С в течение еще 15 ч и затем перемешивали при к.т. в течение 3 суток. Смесь распределяли междуEtOAc и водой. После разделения фаз водную часть экстрагировали двумя дополнительными порциямиEtOAc. Объединенные EtOAc растворы промывали водой (2), рассолом (1), сушили над сульфатом натрия и концентрировали досуха при пониженном давлении. Неочищенный материал подвергали флэшхроматографии (силикагель, градиент от DCM до 7:3 DCM/EtOAc) с получением липкой желтой пены. Этот материал подвергали кристаллизации из смеси гексан/EtOAc с получением указанного в заголовке соединения (1,78 г, 65%) в виде светло-желтого твердого вещества. ЖХ-МС (m/z, ЭР+) = 574 (М+Н+). Стадия 2: 6-(N-(4-амино-3-хлорфенил)метилсульфонамидо)-2-(4-хлорфенил)-5-циклопропил-Nметилбензофуран-3-карбоксамид. Раствор 6-(N-(3-хлор-4-нитрофенил)метилсульфонамидо)-2-(4-хлорфенил)-5-циклопропил-Nметилбензофуран-3-карбоксамида (0,500 г, 0,870 ммоль) в смеси 3:1 MeOH/THF (30 мл) подвергали гидрированию при 45 фунт-сила на кв.дюйм (310,3 кПа) в присутствии 5% сульфидированной платины на углероде (50 мг). Через 18 ч реакционный сосуд продували азотом, катализатор удаляли фильтрованием через целит и фильтрат концентрировали досуха при пониженном давлении. Остаток перекристаллизовывали из смеси гексан/EtOAc с получением указанного в заголовке соединения (0,45 г, 95%) в виде не совсем белого твердого вещества. ЖХ-МС (m/z, ЭР+) = 544 (М+Н+). Стадия 3: 6-(N-(4-бром-3-хлорфенил)метилсульфонамидо)-2-(4-хлорфенил)-5-циклопропил-Nметилбензофуран-3-карбоксамид. Перемешиваемую суспензию 6-(N-(4-амино-3-хлорфенил)метилсульфонамидо)-2-(4-хлорфенил)-5 циклопропил-N-метилбензофуран-3-карбоксамида (0,421 г, 0,773 ммоль) в 25 мл ацетонитрила обрабатывали 25 мл 48%-ной водной HBr и полученную суспензию охлаждали в бане лед-вода. К смеси добавляли раствор нитрита натрия (0,056 г, 0,812 ммоль) в 2 мл воды. После перемешивания при 0 С в течение 30 мин добавляли дополнительную порцию 14 мг нитрита натрия в 1 мл воды. После перемешивания при 0 С в течение еще 30 мин смесь обрабатывали CuBr (0,130 г, 0,906 ммоль) четырьмя порциями в течение 5 мин. Смесь нагревали до 60 С в течение 30 мин и затем охлаждали до к.т. Смесь распределяли междуEtOAc и 5%-ным водным бисульфитом натрия. EtOAc раствор промывали 5%-ным водным бисульфитом натрия (2), насыщенным водным бикарбонатом натрия (2), рассолом (1), сушили над сульфатом натрия и концентрировали досуха при пониженном давлении. Неочищенный продукт очищали посредством флэш-хроматографии (силикагель, градиент от гексана до 3:7 гексан/EtOAc) с последующей перекристаллизацией из смеси гексан/EtOAc с получением указанного в заголовке соединения (0,212 г, 45%) в виде белого твердого вещества. ЖХ-МС (m/z, ЭР+) = 609 (М+Н+). Стадия 4:(2-хлор-4-(N-(2-(4-хлорфенил)-5-циклопропил-3-(метилкарбамоил)бензофуран-6 ил)метилсульфонамидо)фенил)бороновая кислота. Смесь 6-(N-(4-бром-3-хлорфенил)метилсульфонамидо)-2-(4-хлорфенил)-5-циклопропил-N-метилбензофуран-3-карбоксамида (0,205 г, 0,337 ммоль), бис(пинаколато)дибора (0,428 г, 1,69 ммоль), ацетата калия (0,165 г, 1,69 ммоль) и комплекса Pd(ll)(dppf)Cl2 и дихлорметана (0,0138 г, 0,017 ммоль) в безводном 1,4-диоксане (4 мл) в герметично закрываемой пробирке продували азотом в течение 10 мин. Сосуд герметизировали и при перемешивании нагревали при 80 С в масляной бане. Через 4 ч смесь охлаждали до к.т. и разбавляли EtOAc. Полученный раствор промывали водой (2), насыщенным рассолом (1) и сушили над сульфатом натрия. В процессе перемешивания с сульфатом натрия добавляли целит для облегчения удаления нерастворимого черного материала, который оставался суспендированным в растворе. Смесь фильтровали через среднюю фритту с получением золотисто-коричневого фильтрата, который концентрировали досуха при пониженном давлении. Остаток растворяли в 10 мл THF и полученный раствор охлаждали на бане лед-вода. Раствор обрабатывали 1 н. водной HCl (4 мл), а затем перйодатом натрия (1,08 г, 5,05 ммоль). Смесь перемешивали при 0 С в течение 10 мин и затем оставляли нагреваться до к.т. Спустя 18 ч смесь распределяли между водой и EtOAc и фазы разделяли. Водный раствор экстрагировали EtOAc (2). Объединенные EtOAc растворы промывали 5%-ным водным бисульфитом натрия(4), насыщенным рассолом (2), сушили над сульфатом натрия и концентрировали досуха при пониженном давлении. Остаток подвергали флэш-хроматографии (силикагель, 2-частный градиент: от DCM до EtOAc за 15 мин, затем растворители заменяли на А = 9:1 DCM/MeOH, В = DCM; градиент от 100% В до 65% А за 4 мин, затем 65% А изократическое элюирование в течение 15 мин) с получением светлой желто-коричневой пены (0,142 г). Этот материал растворяли в ацетонитриле (3 мл) и раствор перемешивали при капельном добавлении 0,25 н. водной HCl (10 мл) в течение периода времени 20 мин. Полученную белую суспензию перемешивали при к.т. Через 2 ч твердое вещество собирали фильтрованием через воронку из средней фритты. Осадок на фильтре промывали водой (2), сушили отсасыванием на воздухе в течение 30 мин и затем сушили в вакууме в течение ночи с получением указанного в заголовке соединения (0,119 г, 62%) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 8.50 (q, 1 Н), 8.32 (s, 2 Н), 8.11 (S, 1 Н), 7.92 (d, 2 Н), 7.62 (d, 2 Н),7.30 - 7.45 (m, 3 Н), 7.19 (s, 1 Н), 3.40 (S, 3 Н), 2.83 (d, 3 Н), 2.06 - 2.16 (m, 1 Н), 0.72 - 1.07 (m, 3 Н), 0.52 (br s,1 Н). ЖХ-МС (m/z, ЭР+) = 573 (М+Н+). Пример 3. 4-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)фенилбороновая кислота Стадия 1: 6-(N-(4-бромфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид. Смесь 5-циклопропил-2-(4-фторфенил)-N-метил-6-[(метилсульфонил)амино]-1-бензофуран-3 карбоксамид (500 мг, 1,244 ммоль), 4-бромфенилбороновой кислоты (1,5 г, 7,464 ммоль), моногидрата ацетата меди(II) (372 мг, 1,866 ммоль), триэтиламина (252 мг, 2,488 ммоль) и 4 молекулярных сит (1 g) в дихлорметане (160 мл) перемешивали в течение 2 суток. Раствор фильтровали, концентрировали досуха и очищали колоночной хроматографией с получением 6-(N-(4-бромфенил)метилсульфонамидо)-5 циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (180 мг, 0,323 ммоль, 26%-ный выход) в виде коричневого твердого вещества. ЖХ-МС (m/z, ЭР+) = 557, 559 (М+Н+). Стадия 2: 5-циклопропил-2-(4-фторфенил)-N-метил-6-(N-(4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)фенил)метилсульфонамидо)бензофуран-3-карбоксамид Суспензию 6-(N-(4-бромфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (150 мг, 0,269 ммоль), ацетата калия (80 мг, 0,807 ммоль),бис(пинаколато)дибора (205 мг, 0,807 ммоль) и комплекса Pd(dppf)Cl2 и дихлорметана (22 мг, 0,027 ммоль) в 1,4-диоксане (20 мл) выдерживали при 95 С с перемешиванием в течение ночи. Раствор охлаждали до комнатной температуры, фильтровали, концентрировали досуха и очищали колоночной хроматографией с получением 5-циклопропил-2-(4-фторфенил)-N-метил-6-(N-(4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)фенил)метилсульфонамидо)бензофуран-3-карбоксамида (152 мг, 0,251 ммоль, 94%ный выход) в виде коричневого твердого вещества. ЖХ-МС (m/z, ЭР+) = 605 (М+Н+). Стадия 3: 4-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)фенилбороновая кислота. Суспензию 5-циклопропил-2-(4-фторфенил)-N-метил-6-(N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо)бензофуран-3-карбоксамида (152 мг, 0,251 ммоль), бензолбороновой кислоты на полимерной подложке (480 мг, 1,255 ммоль) и водной 5 н. HCl (0,35 мл, 1,757 ммоль) в тетрагидрофуране (15 мл) перемешивали при комнатной температуре в течение 48 ч. Раствор фильтровали, концентрировали досуха и очищали посредством ВЭЖХ с обращенной фазой с получением 4-(N(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)фенилбороновой кислоты (45 мг, 0,086 ммоль, 34%-ный выход) в виде белого твердого вещества. 1 Н ЯМР (метанол-d4) : 7.98 (d, 1H), 7.96 (d, 1H), 7.87 (S, 1 Н), 7.66 (d, 2H), 7.46 (d, 2H), 7.32 - 7.26 Стадия 1: 6-(N-(3-бромфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид. Смесь 5-циклопропил-2-(4-фторфенил)-N-метил-6-[(метилсульфонил)амино]-1-бензофуран-3 карбоксамида (500 мг, 1,24 ммоль), 3-бромфенилбороновой кислоты (1,5 г, 7,46 ммоль), моногидрата ацетата меди(II) (372 мг, 1,87 ммоль), триэтиламина (252 мг, 2,49 ммоль), 4 А молекулярных сит (1 g) в дихлорметане (160 мл) перемешивали в течение 2 суток. Твердое вещество отфильтровывали, фильтрат концентрировали досуха и очищали колоночной хроматографией с получением 6-(N-(3-бромфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (270 мг, 0,48 ммоль, 39%-ный выход) в виде коричневого твердого вещества. ЖХ-МС (m/z, ЭР+) = 557, 559 (М+Н+). Стадия 2: 5-циклопропил-2-(4-фторфенил)-N-метил-6-(N-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо)бензофуран-3-карбоксамид. Суспензию 6-(N-(3-бромфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (240 мг, 0,43 ммоль), ацетата калия (128 мг, 1,29 ммоль),бис(пинаколато)дибора (328 мг, 1,29 ммоль) и комплекса Pd(dppf)Cl2 и дихлорметана (35мг, 0,043 ммоль) в 1,4-диоксане (20 мл) выдерживали при 95 С с перемешиванием в течение ночи. Реакционную смесь охлаждали до комнатной температуры и твердое вещество отфильтровывали. Фильтрат концентрировали досуха и очищали колоночной хроматографией с получением 5-циклопропил-2-(4 фторфенил)-N-метил-6-(N-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо) бензофуран-3-карбоксамида (269 мг, 0,45 ммоль, 92%-ный выход) в виде коричневого твердого вещества. ЖХ-МС (m/z, ЭР+) = 605 (М+Н+). Стадия 3: 3-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)фенилбороновая кислота. Суспензию 5-циклопропил-2-(4-фторфенил)-N-метил-6-(N-(3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо)бензофуран-3-карбоксамида (269 мг, 0,45 ммоль), бензолбороновой кислоты на полимерной подложке (770 мг, 2,23 ммоль), водной 5 н. HCl (0,62 мл, 3,12 ммоль) в тетрагидрофуране (15 мл) перемешивали при комнатной температуре в течение 48 ч. Смесь фильтровали,фильтрат концентрировали досуха и очищали посредством ВЭЖХ с обращенной фазой с получением 3(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)фенилбороновой кислоты (41 мг, 0,078 ммоль, 17%-ный выход ) в виде белого твердого вещества. 1 Н ЯМР (метанол-d4)7.99 - 7.95 (m, 2 Н), 7.94 (s, 1 Н), 7.77 - 7.38 (m, 4 Н), 7.32 - 7.25 (m, 3 Н), 3.32 (s,3 Н), 2.98 (s, 3 Н), 2.29 - 2.25 (m, 1 Н), 1.10 - 0.37 (m, 4 Н). ЖХ-МС (m/z, ЭР+) = 523 (М+Н+). Пример 5. 4-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)-бензофуран-6-ил)метилсульфонамидо)-2-фторфенилбороновая кислота(0,8 мл) и воде (0,2 мл) обрабатывали карбонатом калия (616,86 мг, 4,47 ммоль) и нагревали до 100 С в течение 24 ч. Реакционную смесь концентрировали и очищали колоночной хроматографией с получением 5-циклопропил-6-(N-(3-фтор-4-нитрофенил)метилсульфонамидо)-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (230 мг, 0,43 ммоль, 29%-ный выход) в виде желтого порошка. ЖХ-МС (m/z, ЭР+) = 542 (М+Н+). Стадия 2: 6-(N-(4-амино-3-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамид. Смесь 5-циклопропил-6-(N-(3-фтор-4-нитрофенил)метилсульфонамидо)-2-(4-фторфенил)-N- 25024357 метилбензофуран-3-карбоксамида (230 мг, 0,43 ммоль) и хлорида олова (291 мг, 1,29 ммоль) в этилацетате (5 мл) и этаноле (5 мл) кипятили с обратным холодильником в течение 3 ч. Смесь охлаждали до комнатной температуры и распределяли между этилацетатом и водой. Органический слой сушили над сульфатом натрия, фильтровали, концентрировали досуха и очищали колоночной хроматографией с получением 6-(N-(4-амино-3-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (200 мг, 0,39 ммоль, 90%-ный выход) в виде желтого порошка. ЖХ-МС (m/z, ЭР+) = 512 (М+Н+). Стадия 3: 6-(N-(4-бром-3-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамид. Суспензию 6-(N-(4-амино-3-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамида (200 мг, 0,39 ммоль) в ацетонитриле (5 мл) и водном растворе бромистого водорода (5 мл) при 0 С с перемешиванием в течение 0,5 ч обрабатывали водным раствором нитрита натрия (29,6 мг, 0,43 ммоль). Затем к раствору порциями при 0 С добавляли бромид меди (64,3 мг,0,45 ммоль) и нагревали до начала флегмообразования в течение 2 ч. Раствор охлаждали до комнатной температуры и распределяли между этилацетатом и водой. Органический слой отделяли, сушили над сульфатом натрия, фильтровали, концентрировали досуха и очищали колоночной хроматографией с получением 6-(N-(4-бром-3-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (140 мг, 0,24 ммоль, 61%-ный выход) в виде белого твердого вещества. ЖХМС (m/z, ЭР+) = 575, 577 (М+Н+). Стадия 4: 5-циклопропил-6-(N-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо)-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид. Суспензию 6-(N-(4-бром-3-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамида (140 мг, 0,24 ммоль) и бис(пинаколато)дибора (914 мг, 3,60 ммоль) в безводном диоксане (5 мл) обрабатывали PdCl2(dppf) (19,6 мг, 0,024 ммоль), ацетатом калия (47,04 мг,0,48 ммоль) и выдерживали под N2 c перемешиванием при 90 С в течение 4 ч. Суспензию охлаждали до комнатной температуры и распределяли между этилацетатом и водой. Органический слой отделяли, сушили над сульфатом натрия, фильтровали, концентрировали досуха и очищали колоночной хроматографией с получением 5-циклопропил-6-(N-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфон-амидо)-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (140 мг, 0,22 ммоль, 56%ный выход ) в виде белого твердого вещества. ЖХ-МС (m/z, ЭР+) = 623 (М+Н+). Стадия 5: 4-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)-2-фторфенилбороновая кислота. Раствор 5-циклопропил-6-(N-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо)-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (140 мг, 0,22 ммоль) и 1 н. водной HCl (2 мл) в безводном THF (10 мл) обрабатывали бензолбороновой кислотой на полимерной подложке (5000 мг) и перемешивали при комнатной температуре в течение 24 ч. Смесь распределяли между этилацетатом и водой. Органический слой отделяли, сушили над сульфатом натрия, фильтровали, концентрировали досуха и очищали посредством ВЭЖХ с обращенной фазой с получением 4-(N-(5 циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)-2-фторфенилбороновой кислоты (50 мг, 0,092 ммоль, 42%-ный выход ) в виде белого твердого вещества. 1 Н ЯМР (300 МГц, метанол-d4)7.96 (dd, 2H), 7.83 (s, 1H), 7.45-7.15 (m, 6H), 3.43 (s, 3 Н), 2.96 (s,3 Н), 2.12 (m, 1 Н), 1.01 -0.52 (m, 4 Н). ЖХ-МС (m/z, ЭР+) = 541 (М+Н+). Пример 6. 4-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)-3-фторфенилбороновая кислота Стадия 1: 5-циклопропил-6-(N-(2-фтор-4-нитрофенил)метилсульфонамидо)-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамид. Смесь 5-циклопропил-2-(4-фторфенил)-N-метил-6-[(метилсульфонил)амино]-1-бензофуран-3 карбоксамида (402 мг, 1 ммоль), 1,2-дифтор-4-нитробензола (318 мг, 2 ммоль) и карбоната калия (414 мг,3 ммоль) в 1,2-диметоксиэтане (20 мл) и воде (5 мл) нагревали при 100 С в течение 48 ч. Раствор охлаждали до комнатной температуры, концентрировали, разбавляли этилацетатом (50 мл) и промывали водой. Органический слой сушили над сульфатом натрия, фильтровали, концентрировали досуха и очищали колоночной хроматографией с получением 5-циклопропил-6-(N-(2-фтор-4-нитрофенил)метилсульфонамидо)-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (420 мг, 0,77 ммоль, 77%-ный выход) в- 26024357 виде желтого твердого вещества. ЖХ-МС (m/z, ЭР+) = 542 (М+Н+). Стадия 2: 6-(N-(4-амино-2-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамид. Суспензию 5-циклопропил-6-(N-(2-фтор-4-нитрофенил)метилсульфонамидо)-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамида (420 мг, 0,77 ммоль) и дигидрата хлорида олова(II) (519 мг, 2,31 ммоль) в этилацетате (15 мл) и этаноле (15 мл) нагревали при 80 С в течение 2 ч. Раствор охлаждали до комнатной температуры, концентрировали, разбавляли этилацетатом (50 мл) и промывали раствором карбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали, концентрировали досуха и очищали колоночной хроматографией с получением 6-(N-(4-амино-2 фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида(360 мг, 0,7 ммоль, 91%-ный выход) в виде желтого твердого вещества. ЖХ-МС (m/z, ЭР+) = 512 (М+Н+). Стадия 3: 6-(N-(4-бром-2-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамид. К раствору 6-(N-(4-амино-2-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамида (360 мг, 0,7 ммоль) в ацетонитриле (20 мл) добавляли бромистоводородную кислоту (5 мл, 40%-ный раствор в воде), а затем нитрит натрия (49 мг, 0,77 ммоль в 2 мл воды) по каплям при 0 С. После перемешивания при 0 С в течение 10 мин добавляли бромид меди (115 мг, 0,8 ммоль). Смесь нагревали до 80 С и перемешивали в течение 1 ч. Смесь охлаждали до комнатной температуры и разбавляли водой (20 мл). Водный слой экстрагировали этилацетатом (320 мл), сушили над сульфатом натрия, фильтровали, концентрировали досуха и очищали колоночной хроматографией с получением 6-(N-(4-бром-2-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамида (240 мг, 0,42 ммоль, 60%-ный выход) в виде не совсем белого твердого вещества. ЖХ-МС (m/z, ЭР+) = 575, 577 (М+Н+). Стадия 4: 5-циклопропил-6-(N-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо)-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамид. Суспензию 6-(N-(4-бром-2-фторфенил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамида (240 мг, 0,42 ммоль), ацетата калия (123 мг, 1,26 ммоль),бис(пинаколато)дибора (533 мг, 2,1 ммоль) и PdCl2(dppf) (68,5 мг, 0,084 ммоль) в 1,4-диоксане (30 мл) нагревали при 100 С под азотом при перемешивании в течение 16 ч. Раствор охлаждали до комнатной температуры, фильтровали, концентрировали и очищали колоночной хроматографией с получением 5 циклопропил-6-(N-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо)2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (200 мг, 0,32 ммоль, 76%-ный выход) в виде светло-желтого твердого вещества. ЖХ-МС (m/z, ЭР+) = 623 (М+Н+). Стадия 5: 4-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)-бензофуран-6-ил)метилсульфонамидо)-3-фторфенилбороновая кислота. Суспензию 5-циклопропил-6-(N-(2-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил) метилсульфонамидо)-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (200 мг, 0,32 ммоль), бензолбороновой кислоты на полимерной подложке (600 мг, 1,60 ммоль) и 5 н. водной HCl (0,45 мл, 2,24 ммоль) в тетрагидрофуране (20 мл) перемешивали при комнатной температуре в течение 24 ч. Раствор фильтровали, концентрировали досуха и очищали посредством ВЭЖХ с обращенной фазой с получением 4-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)-3 фторфенилбороновой кислоты (61 мг, 0,11 ммоль, 35%-ный выход ) в виде белого твердого вещества. 1 Н ЯМР (метанол-d4)8.03 (s, 1H), 7.97 - 7.79 (m, 2H), 7.57 (dd, 3 Н), 7.24 (dd, 3 Н), 3.29 (s, 3 Н), 2.94 Стадия 1: 6-(N-(4-(бензилокси)-3-фторфенил)метилсульфонамидо)-2-(4-хлорфенил)-5-циклопропилN-метилбензофуран-3-карбоксамид. Смесь 2-(4-хлорфенил)-5-циклопропил-N-метил-6-(метилсульфонамидо)бензофуран-3-карбоксамида (2,0 г, 4,77 ммоль), (4-(бензилокси)-3-фторфенил)бороновой кислоты (1,762 г, 7,16 ммоль),Cu(OAc)2 (1,301 г, 7,16 ммоль), триэтиламина (3,33 мл, 23,87 ммоль) и порошкообразных 3 молекулярных сит (2 g) в CH2Cl2 (60 мл) перемешивали в колбе, открытой для доступа воздуха и снабженной осушительной трубкой. Через 12 ч смесь обрабатывали дополнительными количествами (4-(бензилокси)-3- 27024357 фторфенил)бороновой кислоты (1,762 г, 7,16 ммоль) и Cu(OAc)2 (1,301 г, 7,16 ммоль) и перемешивали при комнатной температуре в течение 4 суток. Смесь разбавляли CH2Cl2, фильтровали через целит и темно-коричневый фильтрат концентрировали досуха. Неочищенный материал очищали посредством флэш-хроматографии на силикагеле (0-4% EtOAc/CH2Cl2) с получением целевого соединения, которое далее очищали посредством флэш-хроматографии на элюируемом силикагеле (0-40% EtOAc/гексан) с получением целевого соединения в виде светло-желто-коричневого твердого вещества (406 мг, 14%). ЖХ-МС (m/z, ЭР+) = 619 (М+Н+). Стадия 2: 2-(4-хлорфенил)-5-циклопропил-6-(N-(3-фтор-4-гидроксифенил)метилсульфонамидо)-Nметилбензофуран-3-карбоксамид. 6-(N-(4-(Бензилокси)-3-фторфенил)метилсульфонамидо)-2-(4-хлорфенил)-5-циклопропил-Nметилбензофуран-3-карбоксамид (350 мг, 0,565 ммоль) растворяли в этилацетате (8,0 мл) и этаноле (8,0 мл). Добавляли 10% Pd/C (30,1 мг, 0,283 ммоль), а затем Н 2 (1 атм, баллон). Смесь перемешивали в течение 2 ч при к.т. и фильтровали через целит. Фильтрат концентрировали досуха и дважды ополаскивали гексаном с получением целевого продукта в виде белого твердого вещества, который использовали без дополнительной очистки. ЖХ-МС (m/z, ЭР+) = 529 (М+Н+). Стадия 3: 4-(N-(2-(4-хлорфенил)-5-циклопропил-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)-2-фторфенил-трифторметансульфонат.Tf2O (0,157 мл, 0,930 ммоль) в CH2Cl2 (1 мл) по каплям добавляли к суспензии 2-(4-хлорфенил)-5 циклопропил-6-(N-(3-фтор-4-гидроксифенил)метилсульфонамидо)-N-метилбензофуран-3-карбоксамида(328 мг, 0,620 ммоль) и пиридина (0,251 мл, 3,10 ммоль) в CH2Cl2 (5 мл) под N2 при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 3 ч, затем добавляли воду. Смесь экстрагировали CH2Cl2 и объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали посредством флэш-хроматографии на элюируемом силикагеле (0-40% EtOAc/гексан) с получением целевого соединения в виде белого твердого вещества (410 мг,83%-ный выход). ЖХ-МС (m/z, ЭР+) = 661 (M+H+). Стадия 4: 2-(4-хлорфенил)-5-циклопропил-6-(N-(3-фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан 2-ил)фенил)метилсульфонамидо)-N-метилбензофуран-3-карбоксамид. Смесь 4-(N-(2-(4-хлорфенил)-5-циклопропил-3-(метилкарбамоил)-бензофуран-6-ил)метилсульфонамидо)-2-фторфенил-трифторметансульфоната (170 мг, 0,257 ммоль), ацетата калия (76 мг, 0,772 ммоль), бис(пинаколато)дибора (98 мг, 0,386 ммоль) и аддукта PdCl2(dppf)-CH2Cl2 (10,50 мг, 0,013 ммоль) в 1,4-диоксане (2,0 мл) нагревали при 80 С в герметично закрытой пробирке под N2 в течение ночи. Смесь охлаждали до комнатной температуры, разбавляли EtOAc и фильтровали через набивку из силикагеля и целита. Фильтрат концентрировали досуха с получением неочищенного продукта в виде желто-коричневой пены, который использовали без дополнительной очистки. ЖХ-МС (m/z, ЭР+) = 639(М+Н+). Стадия 5: (4-(N-(2-(4-хлорфенил)-5-циклопропил-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)-2-фторфенил)бороновая кислота. Перйодат натрия (549 мг, 2,57 ммоль) добавляли к смеси 2-(4-хлорфенил)-5-циклопропил-6-(N-(3 фтор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)метилсульфонамидо)-N-метилбензофуран-3 карбоксамида (164 мг, 0,257 ммоль) в THF (6 мл) и 1 н. HCl (3,21 мл, 3,21 ммоль). Смесь перемешивали при комнатной температуре в течение ночи и добавляли EtOAc и воду. Водный слой экстрагировалиEtOAc (2) и объединенные органические фазы промывали 10%-ным водн. Na2S2O3, рассолом, сушили над Na2SO4, фильтровали и концентрировали досуха. Остаток очищали посредством ВЭЖХ с обращенной фазой (MeCN/H2O с 0,1% TFA (трифторуксусная кислота с получением указанного в заголовке соединения в виде белого твердого вещества (80 мг, 56%). 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 8.52 (q, 1 Н), 8.23 (br. s., 1 Н), 8.07 (s, 1 Н), 7.94 (d, 2 Н), 7.64 (d,2 Н), 7.55 (t, 1 Н), 7.22 (s, 1 Н), 7.11 (s, 1 Н), 7.07 - 7.10 (m, 1 Н), 3.45 (s, 4 Н), 2.85 (d, 3 Н), 2.04 - 2.15 (m, 1 Н),1.00 (br. s., 1 Н), 0.82 (d, 2 Н), 0.54 (br. s., 1 Н). ЖХ-МС (m/z, ЭР+) = 557 (М+Н+). Пример 8. 6-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)пиридин-3-илбороновая кислота К раствору 5-циклопропил-2-(4-фторфенил)-N-метил-6-[(метилсульфонил)амино]-1-бензофуран-3 карбоксамида (400 мг, 1,00 ммоль) в диметилформамиде (5 мл) при 0 С добавляли бис(триметилсилил)амид лития (1,5 мл, 1,50 ммоль). Через 1 ч добавляли 5-бром-2-фторпиридин (350 мг,2,00 ммоль) и поддерживали перемешивание при 80 С в течение 16 ч. Раствор охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом (350 мл). Объединенные экстракты промывали рассолом, сушили над сульфатом натрия и концентрировали досуха при пониженном давлении. Неочищенный материал очищали колоночной хроматографией с получением 6-(N-(5-бромпиридин 2-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-N-метилбензофуран-3-карбоксамида (430 мг,0,775 ммоль, 77,5%-ный выход) в виде желтого твердого вещества. ЖХ-МС (m/z, ЭР+) = 559 (М+Н+). Стадия 2: 5-циклопропил-2-(4-фторфенил)-N-метил-6-(N-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метилсульфонамидо)-бензофуран-3-карбоксамид. Суспензию 6-(N-(5-бромпиридин-2-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-Nметилбензофуран-3-карбоксамида (400 мг, 0,72 ммоль), ацетата калия (210 мг, 2,14 ммоль),бис(пинаколато)дибора (2721 мг, 10,71 ммоль) и PdCl2(dppf) (58 мг, 0,071 ммоль) в 1,4-диоксане (10 мл) в толстостенном стеклянном сосуде для работы под давлением выдерживали при 90 С с перемешиванием в течение 16 ч. Раствор охлаждали до комнатной температуры и фильтровали. Фильтраты концентрировали и очищали колоночной хроматографией с получением 5-циклопропил-2-(4-фторфенил)-N-метил-6(N-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метилсульфонамидо)бензофуран-3 карбоксамида (320 мг, 0,53 ммоль, 74%-ный выход) в виде белого твердого вещества. ЖХ-МС (m/z, ЭР+) = 606 (М+Н+). Стадия 3: 6-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо)пиридин-3-ил)бороновая кислота. Суспензию 5-циклопропил-2-(4-фторфенил)-N-метил-6-(N-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)метилсульфонамидо)бензофуран-3-карбоксамида (320 мг, 0,53 ммоль), бензолбороновой кислоты на полимерной подложке (862 мг, 2,65 ммоль) и 5 н. водной HCl (0,74 мл, 3,78 ммоль) в тетрагидрофуране (10 мл) перемешивали при комнатной температуре в течение 48 ч. Раствор фильтровали, концентрировали досуха и очищали посредством ВЭЖХ с обращенной фазой с получением 6-(N-(5-циклопропил-2-(4-фторфенил)-3-(метилкарбамоил)бензофуран-6-ил)метилсульфонамидо) пиридин-3-ил)бороновой кислоты (80 мг, 0,153 ммоль, 29%-ный выход) в виде белого твердого вещества. 1 Н ЯМР (300 МГц, метанол-d4)8.36 (s, 1 Н), 7.72 (m, 3 Н), 7.57 (s, 1 Н), 7.25 - 7.1 (m, 3 Н), 6.90 - 6.87 Стадия 1: 2-(дифторметил)-4-фтор-1-нитробензол. Трифторид диэтиламиносеры (0,78 мл, 5,91 ммоль) медленно добавляли к 0 С раствору 5-фтор-2 нитробензальдегида (1 г, 5,91 ммоль) в дихлорметане (30 мл). Реакционную смесь перемешивали в течение 10 мин при 0C, 2,5 ч при комнатной температуре, охлаждали до 0 С и гасили путем медленного добавления насыщенного водного NaHCO3. Водный слой экстрагировали дихлорметаном. Объединенные органические слои сушили (Na2SO4), фильтровали и упаривали с получением 2-(дифторметил)-4-фтор-1 нитробензола (1,13 г, количеств.) в виде коричневой жидкости. 1 Н ЯМР (400 МГц, хлороформ-d)м.д. 8.26 (dd, 1 Н), 7.60 (dd, 1 Н), 7.28 - 7.56 (m, 2 Н). Стадия 2: 5-циклопропил-6-(N-(3-(дифторметил)-4-нитрофенил)метилсульфонамидо)-2-(4 фторфенил)-N-метилбензофуран-3-карбоксамид. Раствор 5-циклопропил-2-(4-фторфенил)-N-метил-6-(метилсульфонамидо)бензофуран-3-карбоксамида (1,00 г, 2,49 ммоль), 2-(дифторметил)-4-фтор-1-нитробензола (1,13 г, 5,91 ммоль) и K2CO3 (0,85 г,6,15 ммоль) в НМРА (гексаметилфосфорамид) (6,2 мл) перемешивали при 60 С в течение 15 ч. Реакционную смесь разбавляли EtOAc и водой. Органический слой промывали рассолом, сушили (Na2SO4),фильтровали, упаривали и очищали хроматографией на силикагеле (0-80% EtOAc/гексаны) с получением указанного в заголовке соединения (1,45 г, 97%) в виде желтого твердого вещества. ЖХ-МС (m/z, ЭР+) = 574,3 (М+Н+).
МПК / Метки
МПК: A61K 31/69, A01N 55/08
Код ссылки
<a href="https://eas.patents.su/30-24357-6-n-7-hlor-1-gidroksi-13-digidrobenzoc12oksaborol-5-ilmetilsulfonamido-5-ciklopropil-2-4-ftorfenil-n-metilbenzofuran-3-karboksamid.html" rel="bookmark" title="База патентов Евразийского Союза">6-(n-(7-хлор-1-гидрокси-1,3-дигидробензо[c][1,2]оксаборол-5-ил)метилсульфонамидо)-5-циклопропил-2-(4-фторфенил)-n-метилбензофуран-3-карбоксамид</a>
Способ получения 5-[2-циклопропил-1-(2-фторфенил)-2-оксоэтил]-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ила ацетата (прасугреля)

Номер патента: 16207
Опубликовано: 30.03.2012
Авторы: Гайицек Йосеф, Степанкова Гана
МПК: C07D 495/02
Метки: 5-[2-циклопропил-1-(2-фторфенил)-2-оксоэтил]-4,5,6,7-тетрагидротиено[3,2-с]пиридин-2-ила, прасугреля, ацетата, способ, получения
Формула / Реферат:
1. Способ получения 5-[2-циклопропил-1-(2-фторфенил)-2-оксоэтил]-4,5,6,7-тетрагидротиено[3,2-c]пиридин-2-ила ацетата, имеющего MHH прасугрель, формулы Iхарактеризующийся тем, что вещество формулы VIподвергают взаимодействию с циклопропилмагния галогенидом с получением вещества формулы Vкоторое далее подвергают взаимодействию с метансульфонилхлоридом с получением метансульфоната формулы IVкоторый далее подвергают взаимодействию с соединением...
Способ простого получения (3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6-нитрохиназолин-4 -ил] амина или (3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6 -аминохиназолин-4-ил] амина

Номер патента: 5213
Опубликовано: 30.12.2004
Авторы: Шнайдер Зимон, Штайнер Клаус, Барт Хуберт
МПК: C07D 239/94
Метки: аминохиназолин-4-ил, ил, амина, 3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6-нитрохиназолин-4, получения, простого, 3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6, способ
Формула / Реферат:
1. Способ получения (3-хлор-4-фторфенил)-[7-(3-морфолин-4-илпропокси)-6-нитрохиназолин-4-ил]амина (I) или (3-хлор-4-фторфенил)-[7-(3-морфолин-4-ил-пропокси)-6-аминохиназолин-4-ил]амина (VII) отличающийся тем, что в реакции, осуществляемой в одном реакционном сосуде в 3, соответственно 4 стадии, получают вначале 7-фтор-6-нитрохиназолин-4(3H)-он (III) с помощью тионилхлорида превращают в 4-хлор-7-фтор-6-нитрохиназолин (IV) который с помощью...
Кристаллический гидрохлорид n-{(1s)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1н-пиразол-5-ил)-2-тиофенкарбоксамида

Номер патента: 18907
Опубликовано: 29.11.2013
Авторы: Чэнь Пиньюн Й., Голдинг Джеффри
МПК: A61P 35/00, A61K 31/381
Метки: кристаллический, гидрохлорид, n-{(1s)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1н-пиразол-5-ил)-2-тиофенкарбоксамида
Формула / Реферат:
1. Гидрохлорид N-{(1S)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1H-пиразол-5-ил)-2-тиофенкарбоксамида в кристаллической форме.2. Фармацевтическая композиция, обладающая активностью ингибитора АКТ, включающая гидрохлорид N-{(1S)-2-амино-1-[(3-фторфенил)метил]этил}-5-хлор-4-(4-хлор-1-метил-1H-пиразол-5-ил)-2-тиофенкарбоксамида в кристаллической форме и фармацевтически приемлемый носитель или разбавитель.3. Способ получения...
Кристаллическая форма (2s)-(-)-n-(6-хлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамида

Номер патента: 18567
Опубликовано: 30.08.2013
Авторы: Ву Вэньцзю, Мерман Стивен Дж.
МПК: A61P 25/00, A61K 31/357, A61P 25/24...
Метки: кристаллическая, форма, 2s)-(-)-n-(6-хлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамида
Формула / Реферат:
1. Кристаллическая форма (2S)-(-)-N-(6-хлор-2,3-дигидробензо[1,4]диоксин-2-илметил)сульфамида формулы (IS)содержащая следующие пики, полученные дифракцией рентгеновских лучей на порошке:и характеризующаяся температурой начала плавления 100,4°C и/или теплотой плавления 96,2 Дж/г.2. Кристаллическая форма по п.1, содержащая следующие пики, полученные дифракцией рентгеновских лучей на порошке:3. Фармацевтическая композиция для лечения расстройства,...
Кристаллическая модификация ii 2-[2-(1-хлор-циклопропил)- 3 -(2-хлорфенил)-2-гидроксипропил]-2, 4-дигидро-3н-1, 2, 4-триазол-3-тиона

Номер патента: 7384
Опубликовано: 27.10.2006
Авторы: Зайдель Эрика, Хазенак Карин, Фермер Рональд, Оленик Бритта
МПК: C07D 249/12, A01N 43/653
Метки: 4-дигидро-3н-1, 2-хлорфенил)-2-гидроксипропил]-2, 2-[2-(1-хлор-циклопропил, модификация, кристаллическая, 4-триазол-3-тиона
Формула / Реферат:
1. Кристаллическая модификация II 2-[2-(1-хлорциклопропил)-3-(2-хлорфенил)-2-гидроксипропил]-2,4-дигидро-3Н-1,2,4-триазол-3-тиона формулы которая характеризуется а) максимумами пиков в Раман-спектре со следующими волновыми числами, см-1: b) следующими длинами связей, Е, и углами между связями, ш с) элементарной ячейкой со следующими размерами а = 9,8927 (8) Е, a = 90ш, b = 9,5635 (8) Е, b= 92,651 (6)ш, с = 16,4448 (10) Е, g= 90ш, d)...
Предыдущий патент: Электрод для электролитической ячейки
Следующий патент: Способ получения карбида кремния
Случайный патент: Пептидомиметик smac, применимый в качестве ингибитора iap (ингибитор белков апоптоза)