Ариламидные производные, обладающие антиандрогенными свойствами







Формула / Реферат
1. Ариламидное производное, имеющее формулу (I)

и его стереоизомеры и фармацевтически приемлемые соли;
где R' и R", каждый независимо, выбирают из группы, состоящей из Н и C1-6алкила;
z представляет собой целое число 0 или 1;
R1 выбирают из группы, состоящей из Н, галогена, галоген-С1-6алкила, гидроксигруппы и (CH2)nCHO, где n означает целое число 0-6;
R2 выбирают из группы, состоящей из Н, C1-6алкила, галогена, (галоген)С1-6алкила, гидроксигруппы и (CH2)nCHO, где n означает целое число 0-6;
R3 выбирают из группы, состоящей из NO2, CN, COR, COOH, CONHR, где R представляет собой водород или C1-6алкил; галогена и гидроксигруппы;
R4 и R5, каждый независимо, выбирают из группы, состоящей из Н, С1-6алкила и галогена, или
R6, R9 и R10, каждый, представляют собой Н,
R7 и R8 независимо выбирают из группы, состоящей из Н, Cl, F, циано, метокси и CF3, при условии, что по меньшей мере один из них отличается от Н; или
X выбирают из группы, состоящей из О, S, S(O), SO2, NR12, где R12 выбирают из группы, состоящей из Н, C1-6алкила, СОСН3 и COR, где R является таким же, как он определен выше; или
в том случае, когда z имеет значение 0, тогда X может быть N и образует вместе с R11 гетероциклическое кольцо, выбранное из группы, состоящей из морфолина, 1,2,4-триазола, имидазола и N-замещенного имидазола; и
R11, в том случае, когда не образует кольцо с X, которое определено выше, выбирают из группы, состоящей из C1-6алкила, или фурила, или фенила, необязательно замещенного посредством 1-5 заместителей, выбранных из группы, состоящей из CN, CF3, F и Cl.
2. Ариламидное производное по п.1, где
R1 представляет собой Н, галоген или галоген-C1-6алкил;
R2 представляет собой галоген или галоген-C1-6алкил;
R3 представляет собой нитро или циано;
R4 и R5 представляют собой Н;
R11 представляет собой C1-6алкил, или фурил, или фенил, необязательно замещенный посредством 1-5 заместителей, выбранных из группы, состоящей из CN, CF3, F и Cl.
3. Ариламидное производное по п.1, где
R1, R4 и R5 представляют собой Н;
R2 выбирают из группы, состоящей из галогена и трифторметила;
R3 выбирают из группы, состоящей из NO2 и CN;
R6, R7 и R10 представляют собой Н;
R8 и R9 выбирают из группы, состоящей из Н, Cl, F и трифторметила, при условии, что по меньшей мере один заместитель из R8 и R9 отличается от Н;
X выбирают из группы, состоящей из О и SO2;
R11 выбирают из группы, состоящей из C1-6алкила, фенила, необязательно замещенного посредством 1 или 2 атомов галогена или посредством 1 атома галогена и дополнительного заместителя, выбранного из группы, состоящей из CN, NO2, и фурила.
4. Ариламидное производное по п.3, где
R1, R4 и R5 представляют собой Н;
R2 является трифторметилом;
R3 означает CN;
R6, R7 и R10 представляют собой Н;
R8 представляет собой трифторметил;
R9 означает Н;
X представляет собой SO2;
R11 представляет собой алкил, содержащий вплоть до 4 атомов углерода.
5. Ариламидное производное по п.3, где
R1, R4 и R5 представляют собой Н;
R2 является хлором;
R3 означает CN;
R6, R7 и R10 представляют собой Н;
R8 представляет собой трифторметил;
R9 означает Н;
X представляет собой SO2;
R11 представляет собой 4-фторфенил.
6. Ариламидное производное по п.1, где R8 и R9 представляют собой галогены, или один из R8 и R9 означает галоген, а другой выбирают из группы, состоящей из CN и NO2.
7. Ариламидное производное по п.1, имеющее формулу (I-а)

где R2, R3, R4, R7 и R8 являются такими же, как они определены в п.1;
заместители R12 и R13, каждый независимо, выбирают из группы, состоящей из Н, галогена, цианогруппы и галоген-C1-6алкила,
или его фармацевтически приемлемая соль.
8. Ариламидное производное по п.1, имеющее формулу (I-b)

где R1, R2, R3, R4, R7 и R8 являются такими же, как они определены в п.1;
R11 является таким же, как он определен в п.1,
или его фармацевтически приемлемая соль.
9. Ариламидное производное по п.1, имеющее формулу (I-c)

где R2, R3, R4, R7 и R8 являются такими же, как они определены в п.1;
заместители R12 и R13, каждый независимо, выбирают из группы, состоящей из Н, галогена, цианогруппы и галоген-C1-6алкила,
или его фармацевтически приемлемая соль.
10. Ариламидное производное по п.1, где ариламидное производное выбирают из группы, состоящей из
N-[4-циано-3-(трифторметил)фенил]-3-(этансульфонил)-2-(4-фторфенил)-2-гидроксипропанамида;
N-[4-циано-3-(трифторметил)фенил]-2-(4-фторфенил)-2-гидрокси-3-[(3-метилбутан)сульфонил]пропанамида;
N-[4-циано-3-(трифторметил)фенил]-3-[(фуран-2-илметан)сульфонил]-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-циано-3-фенил)-3-[(4-фторбензол)сульфонил]-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-цианофенил)-2-(3,4-дифторфенил)-3-(этансульфонил)-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-[(3,4-дифторбензол)сульфонил]-2-(4-фторфенил)-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-(этансульфонил)-2-(4-фторфенил)-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-[(3,4-дифторбензол)сульфонил]-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-нитрофенил)-3-[(4-фторбензол)сульфонил]-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-нитрофенил)-3-[(3,4-дифторбензол)сульфонил]-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(4-нитро-3-трифторметилфенил)-3-[(4-циано-3-фторбензол)сульфонил]-2-(4-фторфенил)-2-гидроксипропанамида;
N-(4-нитро-3-трифторметилфенил)-3-[(4-циано-3-фторбензол)сульфонил]-2-(4-хлорфенил)-2-гидроксипропанамида;
N-[4-циано-3-(трифторметил)фенил]-3-(этансульфонил)-2-[4-(трифторметил)фенил]-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-[(4-хлорбензол)сульфонил]-2-(4-хлорфенил)-2-гидроксипропанамида;
N-(3-хлор-4-нитрофенил)-3-[(4-циано-3-фторбензол)сульфонил]-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-цианофенил)-3-[(4-хлорбензол)сульфонил]-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(4-циано-3-(трифторметил)фенил)-3-[(4-фторбензол)сульфонил]-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(4-циано-3-(трифторметил)фенил)-3-[(3,4-дифторбензол)сульфонил]-2-гидрокси-2-[3-фтор-4-(метокси)фенил]пропанамида;
N-(3-хлор-4-цианофенил)-3-(этансульфонил)-2-гидрокси-2-[4-(хлорфенил)пропанамида;
N-(3-хлор-4-цианофенил)-2-((3-фтор-4-метокси)фенил)-3-(этансульфонил)-2-гидроксипропанамида;
N-[3-хлор-4-цианофенил]-3-{[(4-фторфенил)метан]сульфонил}-2-гидрокси-2-[4-(хлор)фенил]пропанамида;
N-[3-хлор-4-цианофенил]-3-{[(4-хлорфенил)метан]сульфонил}-2-гидрокси-2-[4-(хлор)фенил]пропанамида;
N-[4-циано-3-(трифторметил)фенил]-3-(этансульфонил)-2-(4-хлорфенил)-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-{[(4-хлорфенил)метан]сульфонил}-2-гидрокси-2-[4-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-цианофенил)-2-(3-фтор-4-метоксифенил)-3-{[(4-фторфенил)метан]сульфонил}-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-2-(3-фтор-4-метоксифенил)-3-[(3-фторбензол)сульфонил]-2-гидроксипропанамида;
N-[4-циано-3-(трифторметил)фенил]-2-(3,4-дифторфенил)-3-(этансульфонил)-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-(этансульфонил)-2-гидрокси-2-[3-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-цианофенил)-3-{[(4-хлорфенил)метан]сульфонил}-2-(3-фтор-4-метоксифенил)-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-[(3-фторбензол)сульфонил]-2-гидрокси-2-[3-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-цианофенил)-2-(3,4-дифторфенил)-3-[(3-фторбензол)сульфонил]-2-гидроксипропанамида;
N-[4-циано-3-(трифторметил)фенил]-2-(3,4-дифторфенил)-3-[(3-фторбензол)сульфонил]-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-{[(4-хлорфенил)метан]сульфонил}-2-гидрокси-2-[3-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-цианофенил)-3-{[(4-хлорфенил)метан]сульфонил}-2-гидрокси-2-[3-(трифторметил)фенил]пропанамида;
N-(3-хлор-4-цианофенил)-2-(3,4-дифторфенил)-3-[(4-фторбензол)сульфонил]-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-2-(3,4-дифторфенил)-3-{[(4-фторфенил)метан]сульфонил}-2-гидроксипропанамида;
N-(3-хлор-4-цианофенил)-3-{[(4-хлорфенил)метан]сульфонил}-2-(3,4-дифторфенил)-2-гидроксипропанамида;
N-[4-циано-3-(трифторметил)фенил]-2-(4-фторфенил)-2-гидрокси-3-(пропан-1-сульфинил)пропанамида;
N-[4-циано-3-(трифторметил)фенил]-2-(4-фторфенил)-2-гидрокси-3-(пропан-1-сульфонил)пропанамида;
N-(3-хлор-4-циано-2-фторфенил)-2-(3,4-дифторфенил)-3-(этансульфонил)-2-гидроксипропанамида
и их фармацевтически приемлемых солей.
11. Фармацевтическая композиция, содержащая эффективное количество одного или более ариламидных производных или их фармацевтически приемлемых солей по любому из пп.1-10 вместе с подходящим носителем и обычно применяемыми эксципиентами.
12. Применение ариламидного производного или его фармацевтически приемлемой соли по любому из пп.1-10 в качестве лекарственного средства для лечения нарушений, связанных с андрогенным рецептором.
13. Применение ариламидного производного или его фармацевтически приемлемой соли по любому из пп.1-10 в лечении нарушений, связанных с андрогенным рецептором.
14. Применение по п.13, где нарушение представляет собой доброкачественную гиперплазию предстательной железы.
15. Применение по п.13, где нарушение представляет собой раковое заболевание.
16. Применение по п.15, где раковое заболевание выбирают из группы, состоящей из рака предстательной железы и кастрационно-резистентного рака предстательной железы.
17. Применение по любому из пп.13-16, где лечение проводят в комбинации с другим активным средством.
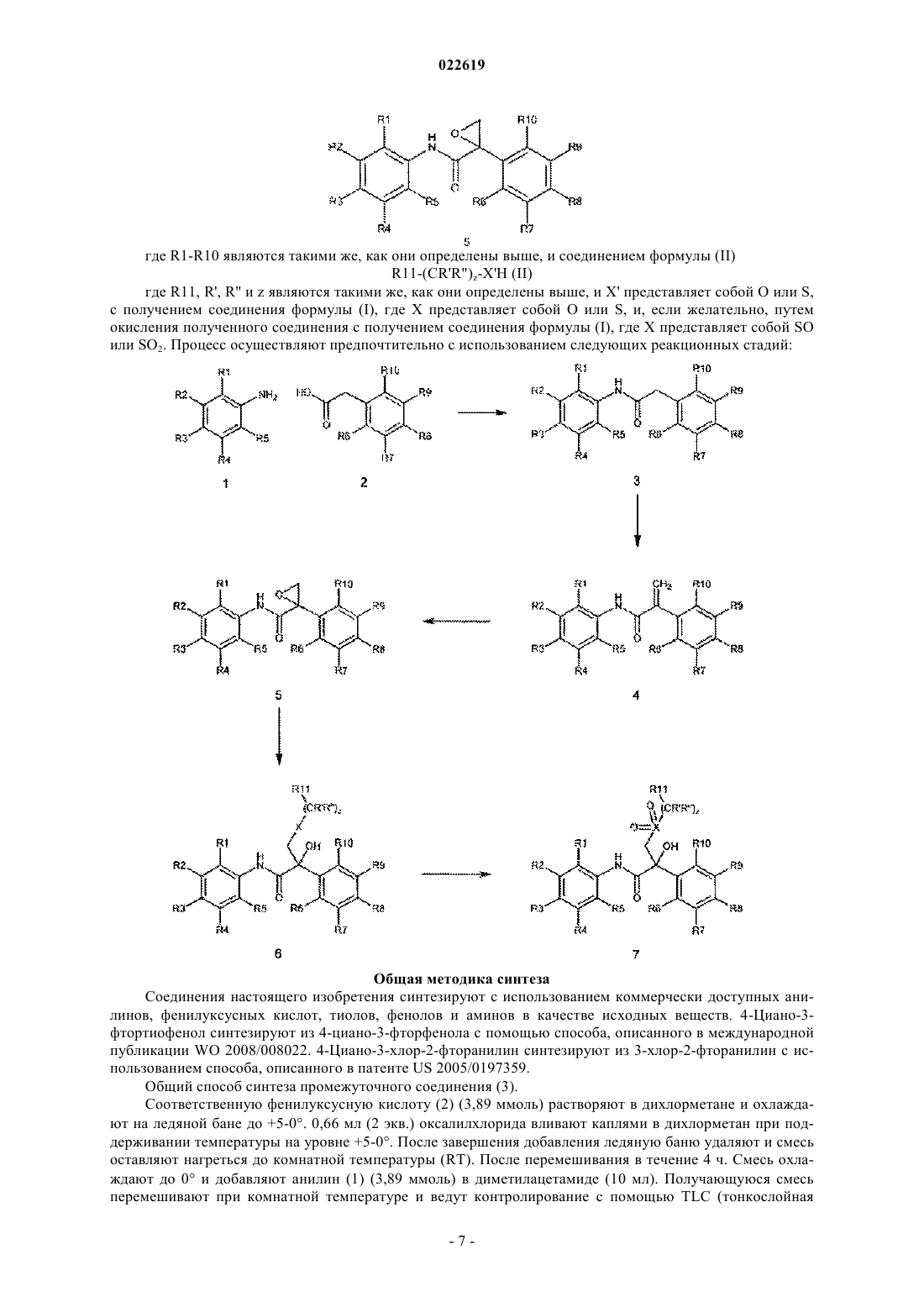
18. Способ получения ариламидного производного формулы (I) по п.1, где X представляет собой О, SO или SO2, включающий проведение реакции между эпоксидным соединением формулы (5)

где R1-R10 являются такими же, как они определены в п.1, и
соединением формулы (II)
где R11, R', R'' и z являются такими же, как они определены в п.1;
X' представляет собой О или S,
с получением соединения формулы (I), где X представляет собой О или S, и, если желательно, окисление полученного соединения с получением соединения формулы (I), где X представляет собой SO или SO2.
19. Способ по п.18, где способ осуществляют, выполняя следующие реакционные стадии:

Текст
Изобретение относится к новым ариламидным производным, имеющим формулу (I), и к их стереоизомерам и фармацевтически приемлемым солям, где R1-R11, R', R, z и X являются такими же, как они определены в формуле изобретения. Ариламидные производные формулы (I) обладают антиандрогенными свойствами. Также изобретение относится к соединениям формулы (I) для применения в качестве лекарственного средства и к фармацевтическим композициям, содержащим их, и к их получению. Область техники, к которой относится изобретение Настоящее изобретение относится к новым ариламидным производным, к их получению, фармацевтическим композициям, содержащим их, и к их применению в лечении нарушений, связанных с андрогенным рецептором, таких как доброкачественная гиперплазия простаты и раковое заболевание, в особенности рак предстательной железы и/или кастрационно-резистентный рак предстательной железы. Предпосылки создания изобретения Андрогены вырабатываются яичками и надпочечными железами, и они играют решающую роль в развитии и физиологии нормальной предстательной железы. Этиология доброкачественной гиперплазии предстательной железы (ВРН) и неоплазии предстательной железы, которые могут прогрессировать в аденокарциному, является андрогензависимой. Предпочтительный метод лечения доброкачественной гиперплазии предстательной железы и рака предстательной железы (РСа) заключается в снижении действия андрогенов в предстательной железе. Фактически, почти 90% мужчин в возрасте от 40 до 90 лет имеют развитие либо ВРН, либо РСа. РСа является второй лидирующей причиной смерти, вызванной раком, и наиболее часто диагностируемым злокачественным новообразованием у мужчин. РСа остается неизлечимым в метастатической стадии рака. Поскольку число случаев РСа увеличивается с возрастом,то число новых диагностируемых случаев возрастает непрерывно вследствие увеличения продолжительности жизни населения. Традиционное первоначальное лечение РСа представляет собой гормональную или андрогендепривационную терапию (ADT). Экспериментальная ADT впервые была описана еще в 1941 г. ADT путем хирургической кастрации или путем химической кастрации с использованием агонистов гормонов,высвобождающих лютеинизирующий гормон, является общепринятой терапией первой линии прогрессирующего РСа. См. Perlmutter M., Lepor H. Androgen deprivation therapy in the treatment of advanced prostate cancer Rev. Urol. 2007; 9 (Suppl. 1): S3-S8, и ссылки, упоминаемые в этой работе. Максимальной блокады андрогенов добиваются путем комбинирования андроген-депривационной терапии с антиандрогенным лечением. Антиандрогены конкурируют с эндогенными андрогенами, тестостероном и дигидротестостероном, за связывание в лигандсвязывающем кармане андрогенного рецептора (AR). AR принадлежит к суперсемейству ядерных гормональных рецепторов и в основном экспрессируется в репродуктивных тканях и мышцах. Связывание лиганда с AR облегчает его диссоциацию от белков теплового шока и других шаперонов, что приводит к димеризации рецептора, фосфорилированию и к последующей транслокации в ядра, где AR связывается с андроген-чувствительными элементами, присутствующими в регуляторных областях с разными генами, участвующими в росте, выживании и дифференциации клеток предстательной железы. Первый нестероидный антиандроген, флутамид, был одобрен для лечения рака предстательной железы (РСа) в 1989 г., и структурно-родственные соединения, бикалутамид и нилутамид, были запущены в производство в 1995 и 1996 гг. соответственно. Нестероидные соединения являются более благоприятно действующими, чем стероидные антиандрогены в клинических применениях вследствие отсутствия перекрестной реактивности с другими стероидными рецепторами и улучшенной пероральной биодоступности. Из этого структурного класса пропанамидных антиандрогенов бикалутамид является наиболее сильнодействующим, наилучшим образом переносимым и основным антиандрогеном на рынке. Бикалутамид описан в патентной литературе, например в Европейском патенте ЕР 0100172. Некоторые ариламидные производные также были описаны в международной публикации WO 2008/011072 А 2 в качестве селективных модуляторов андрогенных рецепторов. К сожалению, хотя андроген-депривационная терапия (ADT) и антиандрогенное лечение обычно приводят к быстрым благотворным реакциям, затем РСа прогрессирует до состояния, в котором андрогенная депривация не дает положительных результатов в контролировании злокачественного новообразования, несмотря на минимальные уровни тестостерона. Такое состояние называют как кастрационнорезистентный рак предстательной железы (CRPC) (или гормонорезистентный рак предстательной железы, HRPC), и является летальной формой заболевания. CRPC, как полагают, проявляет после генетических и/или эпигенетических изменений в предстательной железе раковые клетки и характеризуется реактивизацией роста раковых клеток, которые адаптировались к безгормональной среде в предстательной железе. Рост раковых клеток в CRPC остается зависимым от функции AR, и исследования в последнее десятилетие демонстрируют то, что клетки CRPC используют разные механизмы для реактивизации AR. См. публикацию Chen C.D., Welsbie D.S., Tran C., Baek S.H., Chen R., Vessella R., Rosenfeld M.G., SawyersC.L., Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004 Jan.; 10(1):33-39, и ссылки, упомянутые в ней. Основные механизмы включают амплификацию гена AR или положительную (повышающую) регуляцию мРНК или белка AR, точечные мутации гена AR, которые позволяют активизировать AR посредством неандрогенных лигандов или даже антиандрогенов, изменения уровней экспрессии коактиваторов и корепрессоров транскрипции AR, и экспрессию альтернативно сплайсированных и конститутивно активных вариантов AR. Таким образом, сигнализация AR для направленной доставки лекарственных средств могла бы быть по-прежнему эффективной в предупреждении и лечении CRPC. Ограниченная полезность доступных в настоящий момент антиандрогенов, наиболее вероятно, свя-1 022619 зана с неполным ингибированием AR при некоторых условиях (Taplin M.E. Drug insight: role of the androgen receptor in the development and progression of prostate cancer. Nat. Clin. Pract. Oncol. 2007 Apr.; 4(4): 236-244). Разнообразные молекулярные механизмы могут вносить вклад в неудачу стандартных антиандрогенных методов лечения. Применение антиандрогенов, которые направленно доставляют лигандсвязывающий домен AR, такой как бикалутамид, может приводить к выбору раковых клеток предстательной железы, которые скрывают точечные мутации в лигандсвязывающем домене. В некоторых случаях эти мутации могут побуждать раковые клетки предстательной железы превращать антагонистов в агонисты. Мутации AR обнаружены в 10-40% метастатических опухолей. Было обнаружено более 70 мутаций в AR, которые приводят к повышению базальной активности рецептора или к расширению лигандной специфичности. Например, мутация с заменой треонина на аланин в положении 877 аминокислоты представляет собой наиболее часто обнаруживаемую мутацию у пациентов РСа, и она превращает флутамид, ципротенон (стероидный антиандроген), в прогестерон и эстрогены, агонистичные в отношении AR. Мутация в положении 741 аминокислоты из триптофана либо в лейцин, либо в цистеин предусматривает переключение бикалутамида из антиандрогена в агонист (Hara Т., Miyazaki J., Araki H., Yamaoka M., Kanzaki N.,Kusaka M., Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003 Jan. 1; 63(1):149-153). В дополнение к точечным мутациям в AR, увеличенные уровни рецепторов могут побуждать антиандрогены функционировать в качестве агонистов (Chen C.D., Welsbie D.S., Tran C., Baek S.H., Chen R.,Vessella R., Rosenfeld M.G., Sawyers C.L., Molecular determinants of resistance to antiandrogen therapy. Nat.Med. 2004 Jan.; 10(1): 33-39). Превращение антагонист-агонист имеет существенную клиническую значимость. Приблизительно 30% мужчин с прогрессирующим РСа испытывают парадоксальный перепад уровней простатических специфических антигенов сыворотки крови после прекращения антиандрогенного лечения. До настоящего времени лечение CRPC было разочаровывающим в отношении ожидаемого выживания, которое оценивали через 7-16 месяцев, и в отношении отсутствия значительного улучшения при применении доступных в текущий момент методов лечения. Следовательно, необходимы эффективные, новые средства, которые направленно выбирают в качестве мишени AR. Более конкретно, существует потребность в новых антиандрогенных соединениях, которые являются более сильнодействующими, чем бикалутамид, в антагонизировании активности эндогенных андрогенов на AR. Также существует потребность в новых антиандрогенных соединениях, которые проявляют минимальный агонизм в отношении AR. Важно, существует потребность в новых антиандрогенах, которые не приобретают агонистическую активность в отношении CRPC-связанных мутантных ARs или вCRPC-связанных средах, в которых AR присутствует в больших количествах. Кроме того, существует потребность в нестероидных, нетоксичных молекулах с подобными лекарству свойствами, которые могут быть применены в лечении и в предупреждении ВРН, РСа и CRPC. Теперь неожиданно было обнаружено, что ариламидные производные в соответствии с настоящим изобретением преодолевают недостатки, связанные с бикалутамидом. Краткое изложение сущности изобретения Настоящее изобретение предоставляет новые ариламидные производные, имеющие формулу (I)z представляет собой целое число от 0 до 3;(CH2)nCHO, где n означает целое число 0-6;R2 выбирают из группы, состоящей из Н, алкила, галогена, трифторметила, (галоген)алкила, гидроксигруппы и (CH2)nCHO, где n означает целое число 0-6;R3 выбирают из группы, состоящей из NO2, CN, COR, COOH, CONHR, где R представляет собой водород или алкил; галогена и гидроксигруппы;R4 и R5 образуют вместе с атомами углерода, к которому они присоединены, замещенное или незамещенное алифатическое, гетероалифатическое, ароматическое или гетероароматическое кольцо;R6-R10, каждый независимо, выбирают из группы, состоящей из Н, алкила, галогена,(пер)галогеналкила, CN, NO2, COR, COOH, CONHR, NR2, NHCOCH3, NHCOCF3, NHCOR, NHCONHR,NHCOOR, OCONHR, CONHR, где R является таким же, как он определен выше; NHCSCH3, алкилтиогруппы, алкилсульфинила и алкилсульфонила, при условии, что по меньшей мере одна группа из R6-R10 отличается от Н; или две соседние группы R6-R10 образуют вместе с атомами углерода, к которому они присоединены,замещенное или незамещенное алифатическое, гетероалифатическое, ароматическое или гетероароматическое кольцо;X выбирают из группы, состоящей из О, S, S(О), SO2, NR12, где R12 выбирают из группы, состоящей из Н, алкила, СОСН 3 и COR, где R является таким же, как он определен выше; СН 2 и СО; или в том случае, когда z имеет значение 0, тогда X может быть N и образует вместе с R11 гетероциклическое кольцо, выбранное из группы, состоящей из морфолина, 1,2,4-триазола, имидазола и Nзамещенного имидазола; иR11, в том случае, когда не образует кольцо с X, что определено выше, выбирают из группы, состоящей из алкила, алкенила, (пер)галогеналкила, галогеналкенила, алкил-CN и арила, гетероарила, алифатического или гетероалифатического, 5-7-членного кольца, необязательно замещенного посредством 1-5 заместителей, выбранных из группы, состоящей из алкила, галогена, (пер)галогеналкила, CN, NO2,COR, COOH, CONHR, NR2, NHCOCH3, NHCOCF3, NHCOR, NHCONHR, NHCOOR, OCONHR, NHSO2R,где R является таким же, как он определен выше; NHCSCH3, алкилтиогруппы, алкилсульфинила и алкилсульфонила. Изобретение также относится к фармацевтическим композициям, содержащим эффективное количество одного или более ариламидных производных формулы (I) или их фармацевтически приемлемых солей вместе с подходящим носителем и обычно применяемыми эксципиентами. Дополнительно, изобретение относится к ариламидным производным формулы (I) или к их фармацевтически приемлемым солям для применения в качестве лекарственного средства. Изобретение также относится к ариламидным производным формулы (I) или к их фармацевтически приемлемым солям для применения в лечении заболеваний, связанных с андрогенными рецепторами. В конечном счете, изобретение предоставляет способ получения ариламидных производных формулы (I). Подробное описание изобретения Ариламиды формулы (I) в соответствии с настоящим изобретением обладают по меньшей мере одним асимметричным атомом углерода, т.е. атомом углерода, к которому прикреплен гидроксил. Таким образом, соединения существуют в рацемической форме и в оптически активных формах. Все эти формы охвачены настоящим изобретением. Под термином "алкил", в определении группы соединения формулы (I), понимают линейную или разветвленную, насыщенную углеводородную цепь, содержащую 1-6 атомов углерода. Префикс "галоген" означает, что такая алкильная группа является галогенированной, например, фтором, хлором, бромом или йодом, частично или полностью пер)галоген). Под термином "алкенил" понимают ненасыщенную углеводородную цепь, имеющую одну или более двойных связей и содержащую 2-6 атома углерода. Под термином "алифатическое, гетероалифатическое, ароматическое или гетероароматическое кольцо" понимают насыщенное или ненасыщенное, 4-7 членное кольцо, где 1-3 атома углерода могут быть заменены гетероатомами, выбранными из О, S и N. Такое кольцо может быть замещено одним или более заместителями, выбранными из группы, состоящей из алкила, галогена, (пер)галогеналкила, CN, NO2, COR, COOH, CONHR, NR2, NHCOCH3, NHCOCF3,NHCOR, NHCONHR, NHCOOR, OCONHR, где R представляет собой водород или алкил; NHCSCH3, алкилтиогруппы, алкилсульфинила и алкилсульфонила; где заместитель(и) представляет(ют) собой предпочтительно CN, CF3, F или Cl. Обычными примерами групп, образованных кольцами, подпадающими под термин "алифатическое, гетероалифатическое, ароматическое или гетероароматическое кольцо" и бензольным кольцом, с которым они сконденсированы, являются нафталин, тетрагидронафталин, хинолин и бензофуран. Под термином "арильное, гетероарильное, алифатическое или гетероалифатическое, 5-7-членное кольцо" в определении R11 понимают насыщенное или ненасыщенное кольцо, имеющее 5-7 кольцевых членов, где 0-3 из которых является(ются) гетероатомом, выбранным из О, S и N, где другие члены являются атомами углерода. Обычными примерами R11 в качестве вышеопределенного кольца являются фенил, пиридил, циклопентил, фурил и тетрагидрофурил. Кольцо может быть замещено посредством 1-5 заместителей, выбранных из группы, состоящей из алкила, галогена, (пер)галогеналкила, CN, NO2, COR,COOH, CONHR, NR2, NHCOCH3, NHCOCF3, NHCOR, NHCONHR, NHCOOR, OCONHR, где R представляет собой водород или алкил; NHCSCH3, алкилтиогруппы, алкилсульфинила и алкилсульфонила; где заместитель(и) представляет(ют) собой предпочтительно CN, CF3, F или Cl. Предпочтительные соединения формулы (I) представляют собой соединения, где z имеет значение 0 или 1. Дополнительными предпочтительными соединениями формулы (I) являются соединения, где R2 представляет собой Cl, F или CF3. Также предпочтительными являются соединения, где R3 представляет собой нитрогруппу или цианогруппу. Дополнительные предпочтительные соединения формулы (I) представляют собой соединения, где одну группу или обе группы из R7 и R8 независимо выбирают из группы, состоящей из Н, Cl, F, цианогруппы, метоксигруппы и CF3. Особенно предпочтительными соединениями формулы (I) являются соединения, где R2 представляет собой Cl, F или CF3; R3 представляет собой нитрогруппу или цианогруппу, и одну группу или обе группы из R7 и R8 независимо выбирают из группы, состоящей из Н, Cl, F, цианогруппы, метоксигруппы и CF3. Также предпочтительными являются соединения, где R11 представляет собой этил. Предпочтительные ариламиды настоящего изобретения представляют собой соединения формулы(I), где z имеет значение 0; R1 представляет собой Н, галоген или (пер)галогеналкил; R2 представляет собой галоген или (пер)галогеналкил; R3 представляет собой CN, NO2 или CONH2; R4 и R5 представляют собой Н или алкил, или R4 и R5 образуют вместе с бензольным кольцом нафталиновое кольцо; R6R10 представляют собой Н, (пер)галогеналкил, галоген, NO2, CN или CONH2; X представляет собой SO2 или О; R11 представляет собой алкил, содержащий 2-5 атомов углерода, необязательно замещенный фенил или фурил. Другая предпочтительная группа соединений формулы (I) включает соединения, где z имеет значение 0; R1 и R5 представляют собой Н; R2 выбирают из группы, состоящей из галогена и трифторметила;R3 выбирают из группы, состоящей из NO2, CONH2 и CN; R6, R7 и R10 представляют собой Н; R8 и R9 выбирают из группы, состоящей из Н, галогена и трифторметила, при условии, что по меньшей мере одна группа из R8 и R9 отличается от Н; X выбирают из группы, состоящей из О и SO2; и R11 выбирают из группы, состоящей из алкила, содержащего вплоть до 6 атомов углерода, фенила, необязательно замещенного посредством 1 или 2 атомов галогена или посредством 1 атома галогена и дополнительного заместителя, выбранного из группы, состоящей из CN, NO2, CONHR, NHCOR, NHSO2R, где R является таким же, как он определен в п.1, и алкилсульфонила; и фурила. Более предпочтительными являются соединения формулы (I), где z имеет значение 0; R1, R4 и R5 представляют собой Н; R2 представляет собой трифторметил; R3 представляет собой CN; R6, R7 и R10 представляют собой Н; R8 представляет собой трифторметил; R9 представляет собой Н; X представляет собой SO2; и R11 представляет собой алкил, содержащий вплоть до 4 атомов углерода; и соединения формулы (I), где R1, R4 и R5 представляют собой Н; R2 представляет собой хлор; R3 представляет собойCN; R6, R7 и R10 представляют собой Н; R8 представляет собой трифторметил; R9 представляет собой Н; X представляет собой SO2; и R11 представляет собой 4-фторфенил; и соединения формулы (I), где группы R8 и R9 обе представляют собой галоген, или одна группа из R8 и R9 представляет собой галоген, а другую выбирают из группы, состоящей из CN, NO2, CONHR, NHCOR, NHSO2R и алкилсульфонила. Предпочтительными соединениями являются соединения формулы где R2, R3, R4, R7 и R8 являются такими же, как они определены ранее, и заместители R12 и R13,каждый независимо, выбирают из группы, состоящей из Н, галогена, цианогруппы и (пер)галогеналкила; где R1, R2, R3, R4, R7 и R8 являются такими же, как они определены ранее, и R11 является таким же, как он определен ранее, предпочтительно C1-4 алкилом; и где R2, R3, R4, R7 и R8 являются такими же, как они определены ранее, и заместители R12 и R13,каждый независимо, выбирают из группы, состоящей из Н, галогена, цианогруппы и (пер)галогеналкила,и их фармацевтически приемлемые соли. Предпочтительные I-a, I-b и I-е представляют собой соединения, где R2 является Cl, F или CF3. Предпочтительные I-a, I-b и I-е представляют собой соединения, где R3 является нитрогруппой или цианогруппой. Предпочтительные I-a, I-b и I-е представляют собой соединения, где один или оба заместителя изR7 и R8 независимо выбирают из группы, состоящей из Н, Cl, F, цианогруппы, метоксигруппы и CF3. Особенно предпочтительными I-a, I-b и I-е являются соединения, где R2 представляет собой Cl, F или CF3; R3 представляет собой нитрогруппу или цианогруппу, и один или оба заместителя из R7 и R8 независимо выбирают из группы, состоящей из Н, Cl, F, цианогруппы, метоксигруппы и CF3. Предпочтительные I-а и I-е представляют собой соединения, где R12 и R13, каждый независимо,выбирают из группы, состоящей из Н, Cl, F, цианогруппы и CF3. Предпочтительными соединениями формулы I-b являются соединения, где R11 представляет собой этил. Примеры особенно предпочтительных конкретных соединений включают в себяN-(3-хлор-4-циано-2-фторфенил)-2-(3,4-дифторфенил)-3-(этансульфонил)-2-гидроксипропанамид; и их фармацевтически приемлемые соли. Фармацевтически приемлемые соли и их получение хорошо известны в данной области. Ариламиды изобретения могут быть получены способами, описанными ниже. Например, соединения формулы (I), где X представляет собой О, SO или SO2, могут быть получены путем проведения реакции между эпоксидным соединением формулы (5) где R1-R10 являются такими же, как они определены выше, и соединением формулы (II)R11-(CR'R")z-X'H (II) где R11, R', R и z являются такими же, как они определены выше, и X' представляет собой О или S,с получением соединения формулы (I), где X представляет собой О или S, и, если желательно, путем окисления полученного соединения с получением соединения формулы (I), где X представляет собой SO или SO2. Процесс осуществляют предпочтительно с использованием следующих реакционных стадий: Общая методика синтеза Соединения настоящего изобретения синтезируют с использованием коммерчески доступных анилинов, фенилуксусных кислот, тиолов, фенолов и аминов в качестве исходных веществ. 4-Циано-3 фтортиофенол синтезируют из 4-циано-3-фторфенола с помощью способа, описанного в международной публикации WO 2008/008022. 4-Циано-3-хлор-2-фторанилин синтезируют из 3-хлор-2-фторанилин с использованием способа, описанного в патенте US 2005/0197359. Общий способ синтеза промежуточного соединения (3). Соответственную фенилуксусную кислоту (2) (3,89 ммоль) растворяют в дихлорметане и охлаждают на ледяной бане до +5-0. 0,66 мл (2 экв.) оксалилхлорида вливают каплями в дихлорметан при поддерживании температуры на уровне +5-0. После завершения добавления ледяную баню удаляют и смесь оставляют нагреться до комнатной температуры (RT). После перемешивания в течение 4 ч. Смесь охлаждают до 0 и добавляют анилин (1) (3,89 ммоль) в диметилацетамиде (10 мл). Получающуюся смесь перемешивают при комнатной температуре и ведут контролирование с помощью TLC (тонкослойная хроматография). После завершения реакции смесь выливают в ледяную воду и экстрагируют дихлорметаном. Органическую фазу промывают водой и сушат над Na2SO4 и упаривают, что дает соединение (3). Общий способ синтеза промежуточного соединения (4). 1,7 ммоль соединения (3), 0,075 г (1,8 экв.) параформальдегида и 0,412 г K2CO3 смешивают в NMP(N-метилпирролидон, 2 мл). Смесь нагревают до 90 и перемешивают в течение 3 ч. После охлаждения до комнатной температуры добавляют 10 мл воды и смесь экстрагируют диизопропиловым эфиром (210 мл). Органическую фазу промывают водой (110 мл) и упаривают, что дает соединение (4). Продукт используют в синтезе соединения (5) без дополнительной очистки. Общий способ синтеза промежуточного соединения (5). 1,0 ммоль промежуточного соединения (4) и 10 мг 2,6-ди-трет-бутил-4-метилфенола растворяют вCH2Cl2 (20 мл). Добавляют 0,5 г (2 экв.) МСРВА (мета-хлорпербензойная кислота). Смесь перемешивают при комнатной температуре в течение ночи. Смесь экстрагируют посредством Na2CO2 и воды. Органическую фазу упаривают в вакууме, что дает эпоксидное соединение (5). Продукт используют без дополнительной очистки для синтеза соединения (6). Общий способ синтеза соединения (6). К 3,0 (2 экв.) ммоль K2CO3 в THF (тетрагидрофуран, 5 мл) добавляют 2,2 ммоль (1,5 экв.) соответствующего тиофенола или фенола в THF (7,5 мл) при 0 С. Получающуюся в результате смесь перемешивают при комнатной температуре в течение 2 ч, нагревают вплоть до 50 С и перемешивают в течение 12 ч. После охлаждения реакционную смесь разлагают водой. Получающуюся в результате смесь экстрагируют посредством AcOEt. Органическую фазу концентрируют с получением сырого материала, который используют для синтеза соединения (7) без дополнительной очистки. В случае фенолов, используемых в реакциях, продукты очищают с помощью флэш-хроматографии. Общий способ синтеза соединения (7). 0,45 ммоль соединения (6) растворяют в CH2Cl2 (20 мл). Добавляют МСРВА (метахлорпербензойная кислота, 0,90 ммоль, 2 экв.) и смесь перемешивают при комнатной температуре. После завершения реакции, которую контролируют с помощью TLC (тонкослойная хроматография), реакционную смесь разлагают насыщенным раствором сульфита натрия в воде и экстрагируют дихлорметаном. Органический слой промывают насыщенным раствором сульфита натрия, сушат над Na2SO4 и упаривают. Продукты очищают с помощью флэш-хроматографии. Получение сульфинильных соединений. Сульфинильные соединения настоящего изобретения получают из соответствующего промежуточного соединения (6) в соответствии с методикой, описанной Bhise et al. в публикации журнала Syntheticcommunications, 2009, 39, 1516-1526, с использованием тригидрата пербората натрия в качестве окисляющего средства. Получение ароматических аминов из эпоксида (5). Ароматические амины настоящего изобретения делают из соответствующего промежуточного соединения (5) в соответствии с методикой, описанной Dalton et al. в патенте US 2006/0241180. Получение алифатических аминов из эпоксида (5). Алифатические амины настоящего изобретения делают из соответствующего промежуточного соединения (5) с применением такого же способа, который описан для случая тиолов и фенолов, но в качестве основания в реакциях используют NaH. Примеры Соединения, приведенные в табл. 1 ниже, получают с применением методики синтеза, описанной выше, и иллюстрируют настоящее изобретение. Таблица 1 Названия и 1 Н-ЯМР-характеристики молекул соединений примеров настоящего изобретения Общее описание фармакологических свойств соединений настоящего изобретения Ариламидные производные настоящего изобретения показывают высокую антагонистическую активность в отношении AR. Антагонистическая активность в отношении AR относится к эффективности соединения в конкурировании и/или в ингибировании активности природных лигандов AR, таких как дигидротестостерон (DHT) и тестостерон. Настоящее изобретение обеспечивает соединения, обладающие антагонистической активностью в отношении AR в конкурировании и/или в ингибировании активности неприродных лигандов AR, таких как синтетические андрогены или антиандрогены, используемые в качестве лекарственных препаратов (но которые могут проявлять ухудшающие побочные эффекты). Дополнительно, настоящее изобретение предоставляет соединения, которые демонстрируют силь- 21022619 ную антиандрогенную активность дозозависимым образом. Главным недостатком бикалутамида является неполный антагонизм AR. В случае бикалутамида увеличение концентраций не обеспечивает значительной дополнительной пользы (см. табл. 2). Для лечения поздней стадии РСа, характеризующейся повышением уровней AR, могут потребоваться более сильнодействующие антиандрогены, чем бикалутамид, в связи с чем существует потребность в сильнодействующих антиандрогенах, которые могут компенсировать повышенные уровни AR дозозависимым образом. Настоящее изобретение предоставляет соединения, которые проявляют минимальные агонистические эффекты в отношении AR. Соединения настоящего изобретения могут быть использованы для лечения заболеваний, связанных с AR, таких как ВРН и РСа. Также соединения могут быть применены для лечения CRPC. Дополнительно, соединения могут быть применены в комбинации с другими антиандрогенными методами лечения. Соединения настоящего изобретения не приобретают агонистическую активность в мутациях, связанных с CRPC. Под мутациями, связанными с CRPC, рассматривают все мутации, которые оказывают влияние на развитие, прогрессирование или тяжесть заболевания. Мутация, связанная с CRPC, может получаться в результате индуцированного андрогенной депривацией накопления раковых клеток предстательной железы, скрывающих упомянутую мутацию. Например, рассматривают мутацию в положении 741 из тритофана в лейцин или в цистеин и также мутацию в положении 877 из треонина в аланин. Соединения настоящего изобретения сохраняют свою антагонистическую активность при повышении уровней AR. Следующие испытания и результаты предоставлены для того, чтобы продемонстрировать настоящее изобретение иллюстративным способом, и их не следует рассматривать в качестве ограничения объема изобретения. Дополнительно, концентрации соединений в аналитических образцах являются примерными, и их не следует принимать в качестве ограничения. Специалист в данной области может определить фармацевтически релевантные концентрации с помощью известных в данной области способов. Эксперименты Для выяснения эффективности соединений настоящего изобретения при функционировании в качестве антиандрогенов и для демонстрирования того, что соединения настоящего изобретения сохраняют свою антагонистическую активность в условиях, которые, как известно, придают агонистическую активность антиандрогенным лекарственным препаратам первой линии в клиническом применении (например, флутамиду или бикалутамиду, BIC) был разработан ряд исследований in vitro. Эти исследования основаны на измерении трансактивации андрогенных рецепторов (AR) с использованием анализа с помощью гена-репортера, который является общепризнанным, методом анализа "золотой стандарт" в исследовании AR. В зависимости от присутствия или отсутствия природного лиганда AR, например, тестостерона, этот анализ с помощью гена-репортера может быть использован для определения как антагонистической и агонистической активности соединений. BIC используют в качестве эталонного соединения во всех исследованиях, отражающих доступный в настоящее время стандартный антиандрогенный метод лечения. Анализ трансактивации андрогенных рецепторов (AR).COS-1 (Американская коллекция типовых культур) выращивают в питательной среде DMEM (модифицированная по способу Дульбекко минимальная эссенциальная среда Игла), дополненной 10% FBS(фетальная бычья сыворотка), пенициллином (6,25 Ед./мл) и стрептомицином (6,25 мкг/мл), и высевают на 48-луночные планшеты (50000 клеток/лунку) за один день до трансфекции. Среду для трансфекции,содержащую 2,5% очищенной на активированном угле FBS в DMEM, заменяют на клетки за 4 ч до трансфекции. Клетки подвергают трансфекции с помощью 50 нг плазмиды с встроенным геномрепортером (pPB-28 6/+32-LUC; PB, промотор пробазина), 5 нг плазмиды экспрессии AR (pSG5-hAR), и 5 нг pCMV (внутренний, контроль эффективности трансфекции и роста клеток с помощью бетагалактозидазы) с использованием реагента TranslT-LT1 (Mirus Bio Corporation) в соответствии с инструкциями производителя. Через один день после трансфекции подготовленные в трех экземплярах лунки получают либо (i) среду для лекарства (EtOH-DMSO (этиловый спирт-диметилсульфоксид, (ii) 50 нм тестостерона (эталонный агонист, от Makor или от Steraloids Inc.), (iii) увеличивающиеся концентрацииBIC (эталонный антагонист) или (iv) соединение настоящего изобретения как таковое (для испытания на агонизм) или (v) увеличивающиеся концентрации BIC (эталонный антагонист) или (vi) соединение настоящего изобретения вместе с эталонным агонистом в конкурентной среде (50 нМ; для испытания на антагонизм индуцированной тестостероном транскрипции AR). По истечении 18 ч определяют активность гена-репортера (LUC и бета-галактозидаза) в соответствии со стандартными способами. Данные выражают в виде относительной LUC-активности (световые единицы люциферазы, поделенные на А 420 нм, для контролирования эффективности трансфекции) данного соединения относительно активности эталонного пробного соединения (=100%). Агонизм в отношении андрогенных рецепторов (AR) дикого типа (WT). Агонизм соединений настоящего изобретения в отношении AR WT измеряют в анализе трансактивации AR в клетках COS-1 путем подвергания трансфицированных клеток воздействию испытуемых соединений как таковых, которые описаны выше. Тестостерон используют в качестве эталонного агони- 22022619 ста. Измеряют относительную LUC-активность, отражающую уровень активации AR. Реакцию, получаемую эталонным агонистом, принимают за 100%. Соединения настоящего изобретения не показывают агонизм в отношении андрогенных рецепторов дикого типа. Антагонизм в отношении андрогенных рецепторов (AR) дикого типа (WT). Антагонизм соединений настоящего изобретения в отношении андрогенных рецепторов (AR) дикого типа (WT) измеряют в анализе трансактивации AR в клетках COS-1 в конкурентной среде с использованием тестостерона в качестве агониста, что описано выше. Известный антиандроген BIC используют в качестве эталонного антагониста. Относительную LUC-активность, отражающую AR-зависимую транскрипцию, получаемую в результате воздействия тестостерона как такового, принимают за 100%. Соединения настоящего изобретения являются эффективными антагонистами в отношении андрогенных рецепторов дикого типа (табл. 2). Таблица 2 Антагонизм в отношении андрогенных рецепторов дикого типа (WT AR) Одним из главных ограничений в применении доступных в текущий момент антиандрогенов, таких как флутамид и BIC, является превращение антагонист-агонист, наблюдаемое у мутированных андрогенных рецепторов. Агонизм в отношении мутантного андрогенного рецептора (AR) с мутацией W741L. Агонизм соединений настоящего изобретения в отношении андрогенных рецепторов (AR) с мутацией W741L измеряют в анализе трансактивации AR в клетках COS-1, что описано выше, за исключением того, что вместо андрогенных рецепторов дикого типа (WT AR) используют вектор экспрессии AR,скрывающий мутацию W741L. Трансфицированные клетки подвергают воздействию испытуемых соединений как таковых. BIC используют в качестве эталонного соединения. Как сообщается в литературе,функциональные характеристики BIC в качестве агониста в этом мутантном варианте AR и относительную LUC-активность, отражающую AR-зависимую транскрипцию, индуцированную посредством BIC,принимают за 100%. Соединения настоящего изобретения не показывают агонизм в отношении андрогенных рецепторов с мутацией W741L (табл. 3). Агонизм в отношении мутантных андрогенных рецепторов (AR) с мутацией Т 877 А. Агонизм соединений настоящего изобретения в отношении андрогенных рецепторов (AR) с мутацией Т 877 А измеряют в анализе трансактивации AR в клетках COS-1, что описано выше, за исключением того, что в этом случае используют вектор экспрессии AR, скрывающий мутацию Т 877 А. Трансфицированные клетки подвергают воздействию испытуемых соединений как таковых. Тестостерон используют в качестве эталонного агониста, и его относительную LUC-активность, отражающую AR-зависимую транскрипцию, принимают за 100%. Соединения настоящего изобретения не показывают агонизм в отношении андрогенных рецепторов с мутацией Т 877 А (табл. 3). Таблица 3 Агонизм в отношении мутантных андрогенных рецепторов (AR) с мутацией W741L и Т 877 А Антагонизм в отношении мутантных андрогенных рецепторов (AR) с мутацией Т 877 А. Для подтверждения того, что соединения настоящего изобретения сохраняют антагонизм в отношении мутантных андрогенных рецепторов (AR), соединения настоящего изобретения подвергают анализу трансактивации AR в конкурентной среде вместе с тестостероном, что описано выше, за исключением того, что используют вектор экспрессии AR, скрывающий мутацию Т 877 А. Относительную LUCактивность, отражающую AR-зависимую траскрипцию, получаемую в результате действия тестостерона как такового, принимают за 100%. Соединения настоящего изобретения сохраняют свои антагонистические свойства в отношении мутантных андрогенных рецепторов с мутацией Т 877 А. Экспрессия генов в клетках VCaP. Количественную полимеразную цепную реакцию с обратной транскриптазой используют в изучении способности соединений настоящего изобретения ингибировать экспрессию гена-мишени андрогенных рецепторов (AR). Клетки VCaP высевают на 12-луночные планшеты (105 клеток/лунку), и подготовленные в трех экземплярах лунки обрабатывают либо (i) средой для лекарства (EtOH-DMSO), либо(эталонный антагонист), либо (iv) испытуемым соединением наряду с эталонным агонистом (1 нМ) (все конечные концентрации). По истечении 18 ч экстрагируют тотальную РНК с помощью реагента TRIzol (Invitrogen Life Technologies) и превращают в комплементарную ДНК с помощью набора для синтеза первой цепи кДНК, содержащего траскриптор (Roche Diagnostics GmbH), следуя инструкциям производителя. кДНК используют в качестве матрицы в количественной полимеразной цепной реакции с обратной транскриптазой (RT-qPCR), которую проводят с использованием системы PCR в реальном времени Mx3000P (Stratagene), смеси FastStart SYBR Green Master Mix (Roche) и специфических праймеров для генов-мишеней AR, PSA, TMPRSS2 и FKBP51. Проанализированные уровни мРНК глицеральдегид-3 фосфатдегидрогеназы (GAPDH) используют для нормализации количеств тотальной РНК в образцах. Кратность изменений (индукции лигандов) вычисляют с использованием формулы 2-(Ct), где Ct представляет собой Ct(лиганд)-Ct(EtOH-DMSO), Ct означает Ct(ген x)-Ct(GAPDH), и Ct означает цикл, в котором преодолевается пороговое значение. Данные по экспрессии генов выражают как относительный уровень мРНК (уровень мРНК гена, представляющего интерес, поделенный на уровень мРНК GAPDH) каждого гена для данного соединения. Соединения настоящего изобретения эффективно подавляют экспрессию гена-мишени андрогенных рецепторов в клетках VCaP. Анализ пролиферации LNCaP. Способность соединений настоящего изобретения ингибировать рост раковых клеток предстательной железы изучают на андроген-чувствительной клеточной линии аденокарциномы предстательной железы человека, LNCaP. Клетки высевают на 96-луночные планшеты (5000 клеток/лунку) и выращивают в течение 72 ч. Подготовленные в трех экземплярах лунки обрабатывают либо (i) средой для лекарства(EtOH-DMSO), либо (ii) 0,1 нМ R1881 (эталонный агонист, Perkin-Elmer), либо (iii) увеличивающимися концентрациями BIC (эталонный антагонист), либо (iv) испытуемым соединением наряду с эталонным агонистом (0,1 нМ) (все конечные концентрации) в течение 5 дней. Пролиферацию клеток LNCaP измеряют в день 0, в день 1, в день 3 и в день 5 с помощью набора реагентов для анализа пролиферации клеток в форме одного раствора Cell Titer 96 AQueous от Promega в соответствии с инструкциями производителя. 20 мкл реагента Cell Titer добавляют в 100 мкл питательной среды для клеточных культур в каждой лунке, и клетки оставляют расти в течение одного часа в инкубаторе. Питательную среду для культур переносят в лунки мерного планшета, и регистрируют поглощение на длине волны 492 нм. Соединения настоящего изобретения ингибируют пролиферацию клеток LNCaP. Анализ связывания андрогенных рецепторов (AR). Способность испытуемых соединений связываться с AR измеряют в соответствии с относительным ингибированием связывания (RBI), т.е. в соответствии с их способностью вытеснять 3 Н-меченый синтетический агонист R1881 из AR, экспрессированных в клетках COS-1. Клетки COS-1 подвергают трансфекции так же, как упомянуто выше, для анализа гена-репортера, за исключением тех случаев, когда используют 24-луночные планшеты (100000 клеток/лунку) и 50 нг pSG5-AR (в отсутствии других плазмид). Через 40 ч после трансфекции питательную среду удаляют, лунки промывают однократно посредством PBS (забуференный фосфатом физиологический раствор) и 0,5 мл среды DMEM (без сыворотки), и добавляют 5 мкл [3H]R1881 (Perkin Elmer; 72 Ки/ммоль, 1 микроКи/мкл), которые разбавляют (1+129) средой DMEM (что дает в результате конечную концентрацию 1 нМ [3H]R1881 в лунке). Подготовленные в трех экземплярах лунки получают: (i) не "холодный" лиганд (EtOH+DMEM), (ii) 20 нМ "холодного" синтетического агониста R1881, (iii) 200 нМ "холодного" синтетического агониста R1881, (iv) "холодный" антагонист BIC или (v) "холодное" испытуемое соединение в концентрации 200 нМ, 2000 нМ, и 10000 нМ. После инкубации в течение 2 ч при 37 С измеряют радиоактивность. Относительную 3 Нактивность (число импульсов в минуту (cpm R1881 принимают за 100%, и относительное ингибирование связывания считывают для испытуемых соединений. Соединения настоящего изобретения показывают зависимое от концентрации сродство к связыванию с андрогенным рецептором (AR). Соединения настоящего изобретения проявляют небольшую агонистическую активность или не проявляют агонистическую активность в отношении андрогенного рецептора. Так как эти соединения являются сильнодействующими антагонистами AR, они могут быть использованы не только для лечения рака предстательной железы, но и для лечения других связанных с андрогенным рецептором состояний и заболеваний, таких как доброкачественная гиперплазия предстательной железы, выпадение волос, акне,гирсутизм, мужская гиперсексуальность или синдром поликистозных яичников. В части лечения ракового заболевания, соединения этого изобретения наиболее предпочтительно применяют по отдельности или в комбинации с антиандрогенными средствами лечения раковых заболеваний. Такие соединения также могут быть объединены со средствами, которые подавляют продукцию циркулирующего тестостерона, такими как агонисты или антагонисты LHRH, или с хирургической кастрацией. Настоящее изобретение также предусматривает применение антиэстрогена и/или ингибитора ароматазы в комбинации с соединением настоящего изобретения, например, для содействия в снижении побочных эффектов, сопутствующих антиандрогенной терапии, таких как гинекомастиа.AR принадлежит к суперсемейству ядерных рецепторов, и соединения настоящего изобретения также могут быть применены в качестве остова (центральной части) молекул при разработке лекарства в отношении других ядерных рецепторов гормонов, таких как эстрогенный рецептор или рецептор, активируемый пролифератором пероксисом. Таким образом, соединения настоящего изобретения также могут быть дополнительно оптимизированы с тем, чтобы можно было их применять в лечении других состояний и заболеваний, таких как рак яичников, рак молочной железы, сахарный диабет, кардиологические заболевания, обусловленные нарушением метаболизма заболевания периферической и центральной нервной системы, где ядерные рецепторы играют некоторую роль. Соединения этого изобретения могут быть введены посредством внутривенной инъекции, посредством инъекции в ткань, интраперитонеально, перорально или назально. Композиция может иметь форму, выбранную из группы, состоящей из раствора, дисперсии, суспензии, порошка, капсулы, таблетки,пилюли, капсулы с контролируемым высвобождением, таблетки с контролируемым высвобождением, и пилюли с контролируемым высвобождением.z представляет собой целое число 0 или 1;(CH2)nCHO, где n означает целое число 0-6;R2 выбирают из группы, состоящей из Н, C1-6 алкила, галогена, (галоген)С 1-6 алкила, гидроксигруппы и (CH2)nCHO, где n означает целое число 0-6;R3 выбирают из группы, состоящей из NO2, CN, COR, COOH, CONHR, где R представляет собой водород или C1-6 алкил; галогена и гидроксигруппы;R6, R9 и R10, каждый, представляют собой Н,R7 и R8 независимо выбирают из группы, состоящей из Н, Cl, F, циано, метокси и CF3, при условии,что по меньшей мере один из них отличается от Н; илиX выбирают из группы, состоящей из О, S, S(O), SO2, NR12, где R12 выбирают из группы, состоящей из Н, C1-6 алкила, СОСН 3 и COR, где R является таким же, как он определен выше; или в том случае, когда z имеет значение 0, тогда X может быть N и образует вместе с R11 гетероциклическое кольцо, выбранное из группы, состоящей из морфолина, 1,2,4-триазола, имидазола и Nзамещенного имидазола; иR11, в том случае, когда не образует кольцо с X, которое определено выше, выбирают из группы,состоящей из C1-6 алкила, или фурила, или фенила, необязательно замещенного посредством 1-5 заместителей, выбранных из группы, состоящей из CN, CF3, F и Cl. 2. Ариламидное производное по п.1, гдеR2 представляет собой галоген или галоген-C1-6 алкил;R3 представляет собой нитро или циано;R11 представляет собой C1-6 алкил, или фурил, или фенил, необязательно замещенный посредством 1-5 заместителей, выбранных из группы, состоящей из CN, CF3, F и Cl. 3. Ариламидное производное по п.1, гдеR2 выбирают из группы, состоящей из галогена и трифторметила;R8 и R9 выбирают из группы, состоящей из Н, Cl, F и трифторметила, при условии, что по меньшей мере один заместитель из R8 и R9 отличается от Н;R11 выбирают из группы, состоящей из C1-6 алкила, фенила, необязательно замещенного посредством 1 или 2 атомов галогена или посредством 1 атома галогена и дополнительного заместителя, выбранного из группы, состоящей из CN, NO2, и фурила. 4. Ариламидное производное по п.3, гдеR11 представляет собой алкил, содержащий вплоть до 4 атомов углерода. 5. Ариламидное производное по п.3, гдеR11 представляет собой 4-фторфенил. 6. Ариламидное производное по п.1, где R8 и R9 представляют собой галогены, или один из R8 иR9 означает галоген, а другой выбирают из группы, состоящей из CN и NO2. 7. Ариламидное производное по п.1, имеющее формулу (I-а) где R2, R3, R4, R7 и R8 являются такими же, как они определены в п.1; заместители R12 и R13, каждый независимо, выбирают из группы, состоящей из Н, галогена, цианогруппы и галоген-C1-6 алкила,или его фармацевтически приемлемая соль. 8. Ариламидное производное по п.1, имеющее формулу (I-b) где R1, R2, R3, R4, R7 и R8 являются такими же, как они определены в п.1;R11 является таким же, как он определен в п.1,или его фармацевтически приемлемая соль. 9. Ариламидное производное по п.1, имеющее формулу (I-c) где R2, R3, R4, R7 и R8 являются такими же, как они определены в п.1; заместители R12 и R13, каждый независимо, выбирают из группы, состоящей из Н, галогена, цианогруппы и галоген-C1-6 алкила,или его фармацевтически приемлемая соль. 10. Ариламидное производное по п.1, где ариламидное производное выбирают из группы, состоящей изN-(3-хлор-4-циано-2-фторфенил)-2-(3,4-дифторфенил)-3-(этансульфонил)-2-гидроксипропанамида и их фармацевтически приемлемых солей. 11. Фармацевтическая композиция, содержащая эффективное количество одного или более ариламидных производных или их фармацевтически приемлемых солей по любому из пп.1-10 вместе с подходящим носителем и обычно применяемыми эксципиентами. 12. Применение ариламидного производного или его фармацевтически приемлемой соли по любому из пп.1-10 в качестве лекарственного средства для лечения нарушений, связанных с андрогенным рецептором. 13. Применение ариламидного производного или его фармацевтически приемлемой соли по любому из пп.1-10 в лечении нарушений, связанных с андрогенным рецептором. 14. Применение по п.13, где нарушение представляет собой доброкачественную гиперплазию предстательной железы. 15. Применение по п.13, где нарушение представляет собой раковое заболевание. 16. Применение по п.15, где раковое заболевание выбирают из группы, состоящей из рака предстательной железы и кастрационно-резистентного рака предстательной железы. 17. Применение по любому из пп.13-16, где лечение проводят в комбинации с другим активным средством. 18. Способ получения ариламидного производного формулы (I) по п.1, где X представляет собой О,SO или SO2, включающий проведение реакции между эпоксидным соединением формулы (5) где R1-R10 являются такими же, как они определены в п.1, и соединением формулы (II) где R11, R', R и z являются такими же, как они определены в п.1;X' представляет собой О или S,с получением соединения формулы (I), где X представляет собой О или S, и, если желательно,окисление полученного соединения с получением соединения формулы (I), где X представляет собой SO или SO2. 19. Способ по п.18, где способ осуществляют, выполняя следующие реакционные стадии:
МПК / Метки
МПК: C07D 295/145, C07C 317/46, C07C 255/60, C07C 237/20
Метки: ариламидные, производные, свойствами, обладающие, антиандрогенными
Код ссылки
<a href="https://eas.patents.su/30-22619-arilamidnye-proizvodnye-obladayushhie-antiandrogennymi-svojjstvami.html" rel="bookmark" title="База патентов Евразийского Союза">Ариламидные производные, обладающие антиандрогенными свойствами</a>
Производные дитерпеноидов, обладающие биологическими свойствами

Номер патента: 21536
Опубликовано: 30.07.2015
Авторы: Торри Марко, Черри Альберто, Феррари Патриция, Ферранди Мара, Бьянки Джузеппе, Гоббини Мауро
МПК: A61K 31/15, C07C 251/34, C07C 211/31...
Метки: биологическими, свойствами, производные, дитерпеноидов, обладающие
Формула / Реферат:
1. Соединение, имеющее общую формулу Iгде R1 представляет собой -CH=NOR4 со значением иминокси, -CH2NHOR4, -CH2XR5, -CH=CHR6, -CH=NR7 или амино(С3-С6)алкил;R7 представляет собой гуанидино;R6 представляет собой амино(C1-C6)алкил;R5 представляет собой амино(C1-C6)алкил;R4 представляет собой Н, амино(С2-С6)алкил, (C3-C6)гетероциклоалкил, гидрокси(С2-С6)алкил, или гидрокси(С2-С6)алкилокси(С2-С6)алкил, или карбоксиалкил;X представляет собой О или...
Ингибиторы фарнезилпротеинтрансферазы, обладающие in vivo радиосенсибилизирующими свойствами

Номер патента: 3877
Опубликовано: 30.10.2003
Авторы: Флорен Вим Йоанна, Ван Гинкел Робер Франсискус, Энд Девид Вилльям, Ваутерс Вальтер Баудевейн Леопольд
МПК: A61K 41/00
Метки: радиосенсибилизирующими, ингибиторы, свойствами, фарнезилпротеинтрансферазы, обладающие
Формула / Реферат:
1. Применение по меньшей мере одного ингибитора фарнезилпротеинтрансферазы в качестве радиосенсибилизатора для получения фармацевтической композиции, обладающей радиосенсибилизирующими свойствами, с целью введения ее перед, во время или после облучения опухоли для лечения рака in vivo, причем указанный ингибитор фарнезилпротеинтрансферазы представляет собой соединение формулы (I) или соединение формулы (II) или (III), которые метаболизируются in...
Конденсированные тиазолы, обладающие антибактериальными свойствами

Номер патента: 18104
Опубликовано: 30.05.2013
Авторы: Чаплевски Ллойд Джорж, Хайдон Дэвид Джон
МПК: A61K 31/428, A61P 31/04, C07D 417/04...
Метки: свойствами, обладающие, конденсированные, тиазолы, антибактериальными
Формула / Реферат:
1. Соединение формулы (I), или его соль, или N-оксидгде m равен 1;Q обозначает водород;Alk обозначает двухвалентный (C1-C6)алкилен, алкенилен или алкинилен, который может содержать эфирную группу (-O-), тиоэфирную группу (-S-) или аминогруппу (-NR-), где R обозначает водород, -CN или (C1-C3)алкил;X обозначает -C(=O)NR6- или -С(=О)О-, где R6 обозначает водород, возможно замещенный (C1-C6)алкил, (C2-C6)алкенил или (C2-C6)алкинил;Z1 обозначает –N=...
Реконструированные сурфактанты, обладающие улучшенными свойствами

Номер патента: 15802
Опубликовано: 30.12.2011
Авторы: Йоханссон Ян, Робертсон Бенгт, Курстедт Торе
МПК: A61P 11/00, C07K 14/785, A61K 38/16...
Метки: улучшенными, реконструированные, свойствами, сурфактанты, обладающие
Формула / Реферат:
1. Реконструированный сурфактант, содержащий смесь фосфолипидов и комбинацию полипептидного аналога нативного сурфактантного белка SP-C с полипептидным аналогом нативного сурфактантного белка SP-B, где указанный полипептидный аналог нативного сурфактантного белка SP-C представляет собой полипептид, содержащий по меньшей мере 20 аминокислотных остатков и не более чем 40 аминокислотных остатков, имеющий последовательность, представленную общей...
Композиции, обладающие свойствами регуляторов роста растения

Номер патента: 14908
Опубликовано: 28.02.2011
Авторы: Хаас Ульрих Йоханнес, Миллз Колин Эдвард
МПК: A01P 3/00, A01N 43/653, A01P 21/00...
Метки: свойствами, роста, регуляторов, растения, обладающие, композиции
Формула / Реферат:
1. Композиция, обладающая способностью регуляции роста растения или материала его размножения, включающая в качестве активного ингредиента, регулирующего рост растения, смесь компонента (А) и компонента (В), где компонент (А) представляет собой паклобутразол и компонент (В) представляет собой дифеноконазол, и где компонент (А) и компонент (В) присутствуют в упомянутой композиции в количествах, которые обеспечивают синергический эффект.2....
Предыдущий патент: Наполнители для фильтра в виде блока из оксида алюминия
Следующий патент: Способ удаления карбонилов металлов из потоков газа и сорбент карбонила металла
Случайный патент: Способ получения низших олефинов с использованием катализатора на основе zsm-5