Ингибиторы вируса гепатита с
Номер патента: 21538
Опубликовано: 30.07.2015
Авторы: Кадов Джон Ф., Ван Гань, Лопез Омар Д., Бендер Джон А., Белема Маконен








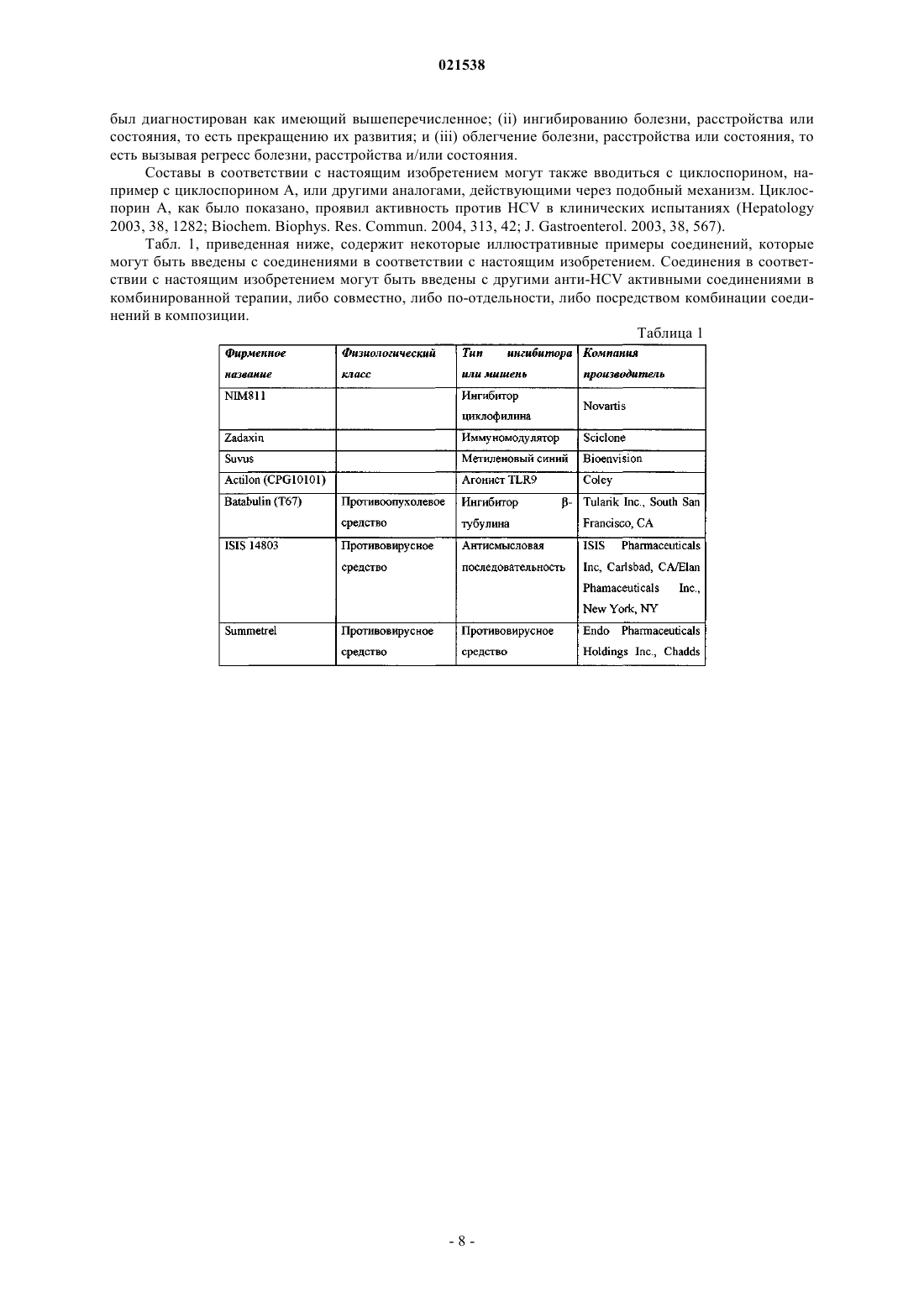
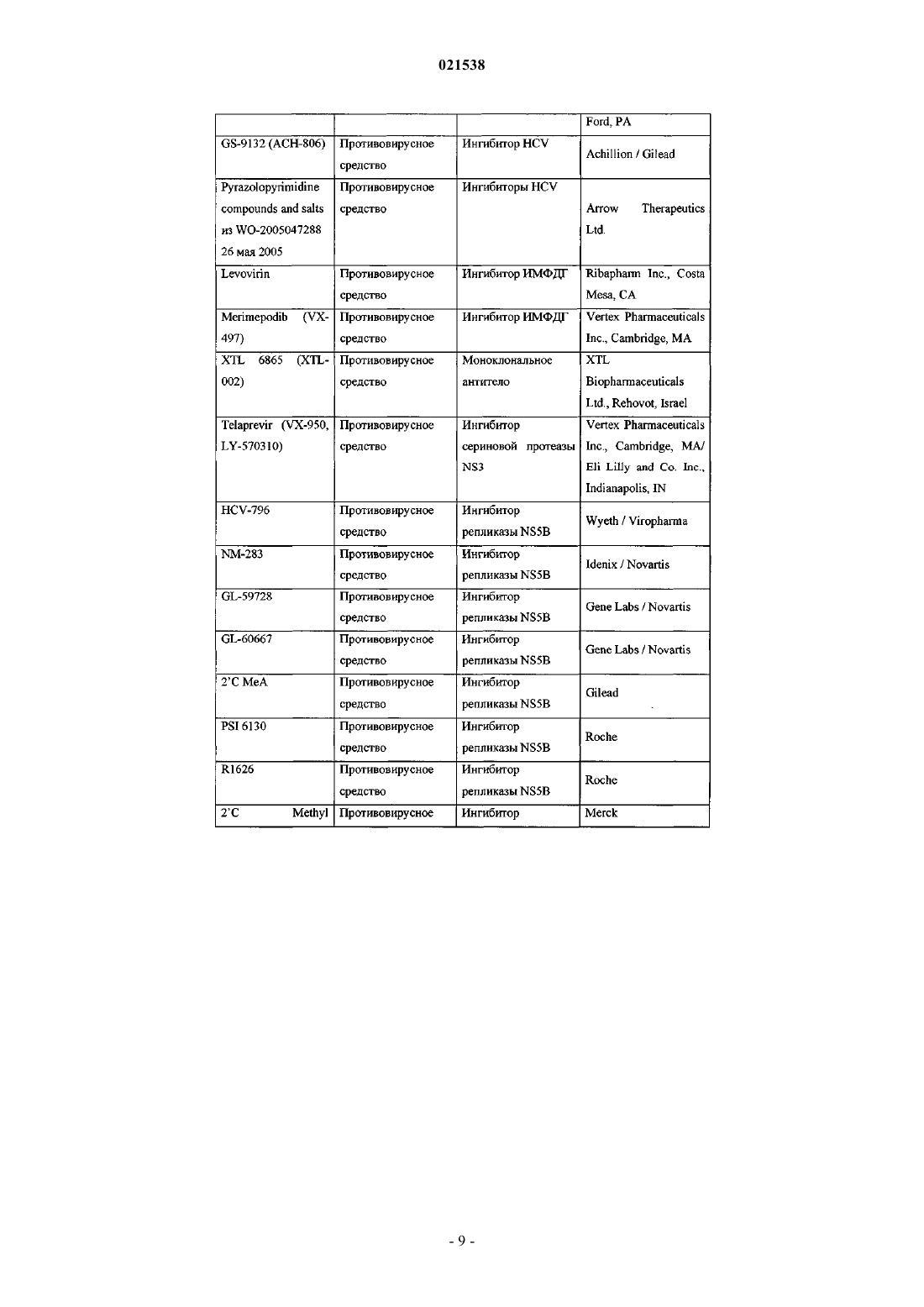
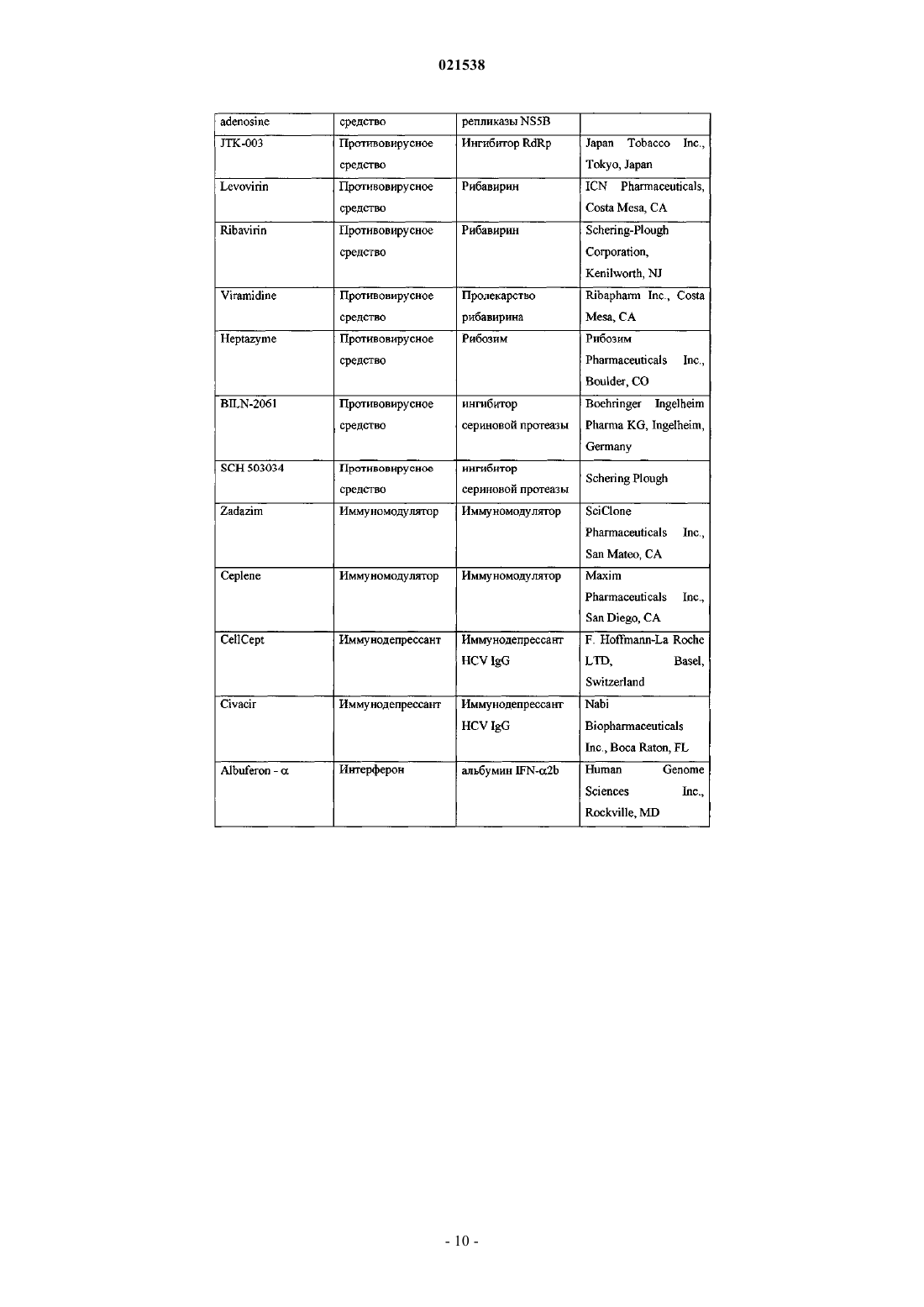
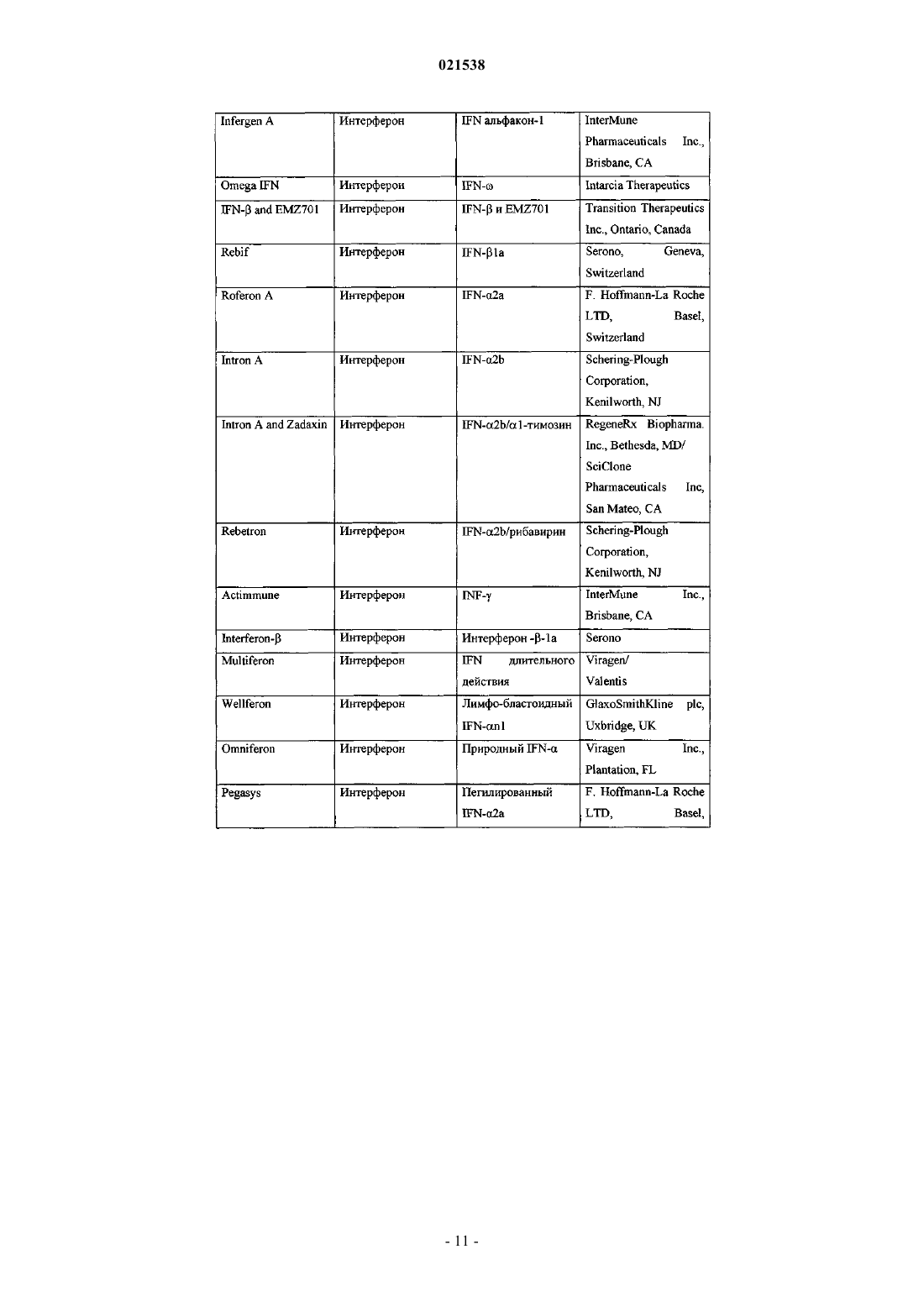
















Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль,
в которой R1 выбирается из водорода, метила и фтора;
R2 выбирается из водорода и метила;
R3 и R4, каждый, представляют собой водород или
R3 и R4 вместе с углеродными атомами, к которым они присоединены, образуют циклопропановое кольцо;
R5 выбирается из водорода и метила.
2. Соединение по п.1 или его фармацевтически приемлемая соль, в котором R1 представляет собой водород.
3. Соединение по п.1 или его фармацевтически приемлемая соль, в котором R1 представляет собой фтор.
4. Соединение по п.3 или его фармацевтически приемлемая соль, в котором R2, R3 и R4, каждый, представляют собой водород и R5 представляет собой метил.
5. Соединение по п.1 или его фармацевтически приемлемая соль, в котором R1 представляет собой метил.
6. Соединение по п.5, в котором R2, R3 и R4, каждый, представляют собой водород и R5 представляет собой метил.
7. Соединение, выбранное из


или их фармацевтически приемлемых солей.
8. Соединение, которое представляет собой

или его фармацевтически приемлемая соль.
9. Соединение, которое представляет собой

или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция для лечения инфекции HCV, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
11. Способ лечения инфекции HCV у пациента, включающий введение пациенту терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
Текст
ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С Изобретение относится к соединениям, композициям и способам лечения инфекции вируса гепатита С (HCV). Также описаны фармацевтические композиции, содержащие такие соединения,и способы применения этих соединений для лечения инфекции HCV. Перекрестная ссылка на родственные заявки По данной заявке испрашивается приоритет в соответствии с предварительной заявкой на патент США 61/467602, поданной 25 марта 2011 г., и предварительной заявкой на патент США 61/440086 поданной 7 февраля 2011 г. Область техники Настоящее изобретение в основном относится к противовирусным соединениям и более определенно относится к соединениям, которые могут ингибировать функцию белка NS5A, кодированного вирусом гепатита С (HCV), к композициям, содержащим такие соединения и способам для того, чтобы ингибировать функцию белка NS5A. Уровень техникиHCV представляет собой главный человеческий патоген, инфицирующий приблизительно 170 млн человек во всем мире - примерно в пять раз больше количества инфицированных вирусом иммунодефицита человека типа 1. У существенной части этих инфицированных HCV людей развиваются серьезные прогрессирующие заболевания печени, включая цирроз печени и гепатоцеллюлярную карциному. Современный стандарт лечения HCV, который использует комбинацию пегилированного интерферона и рибавирина, имеет неоптимальный показатель эффективности в достижении устойчивого вирусологического ответа и вызывает многочисленные побочные эффекты. Таким образом, существует четкая и назревшая необходимость в разработке эффективных методов лечения для решения этой актуальной медицинской потребности.HCV представляет собой вирус с одноцепочечной РНК с положительной полярностью. На основе сравнения расшифрованной аминокислотной последовательности и обширного сходства в 5' нетранслируемой области HCV был классифицирован как отдельный род в семействе Flaviviridae. Все члены семейства Flaviviridae представляют собой вирионы, заключенные в оболочку, которые содержат геном в виде одноцепочечной РНК с положительной полярностью, кодирующий все известные вирусспецифичные белки посредством трансляции единственной непрерывной открытой рамки считывания. Значительная гетерогенность найдена в пределах нуклеотида и кодируемой аминокислотной последовательности всюду по геному HCV вследствие высокой частоты появления ошибок кодирующей РНКзависимой РНК-полимеразы, у которой отсутствует способность коррекции. Были охарактеризованы по меньшей мере шесть главных генотипов, и больше чем 50 подтипов были описаны с распределением во всем мире. Клиническое значение генетической гетерогенности HCV продемонстрировало склонность к мутациям, которые возникают во время лечения монотерапией, таким образом, являются желательными для использования дополнительные варианты лечения. Возможный модулирующий эффект генотипов на патогенез и терапию остается трудным для понимания. Одноцепочечный РНК-геном HCV состоит приблизительно из 9500 нуклеотидов в длину и имеет единственную открытую рамку считывания (ORF), кодирующую единственный большой полипептид приблизительно из 3000 аминокислот. В инфицированных клетках этот полипептид расщепляется в многочисленных сайтах клеточными и вирусными протеазами, что приводит к образованию структурных и неструктурных (NS) белков. В случае HCV образование зрелых неструктурных белков (NS2, NS3, NS4A,NS4B, NS5A и NS5B) осуществляется с помощью двух вирусных протеаз. Первая, как полагают, является металлопротеазой и расщепляет в сочленении NS2-NS3; вторая протеаза представляет собой сериновую протеазу, содержавшуюся в N-концевой области NS3 (также упоминающуюся в настоящем изобретении как протеаза NS3), и опосредует все последующие расщепления ниже по течению NS3, как в цис в сайте расщепления NS3-NS4A, так и в транс, для остающихся сайтов NS4A-NS4B, NS4B-NS5A, NS5ANS5B. Белок NS4A, по всей видимости, выполняет множество функций, действуя и в качестве кофактора для протеазы NS3, так и способствуя в мембранной локализации NS3 и других компонентов вирусной репликазы. Формирование комплекса NS3-NS4A необходимо для надлежащей протеазной активности,приводящей к увеличенной протеолитической эффективности событий расщепления. Белок NS3 также показывает нуклеозид-трифосфатазную и РНК-геликазную активности. NS5B (также упоминающийся в настоящем изобретении как полимераза HCV) представляет собой РНК-зависимую РНК-полимеразу,которая вовлечена в репликацию генома HCV с другими белками HCV, включая NS5A, в репликазном комплексе. Соединения, полезные для лечения HCV-инфицированных пациентов, являются желательно такими, которые селективно ингибируют репликацию вируса HCV. В частности, желательными являются соединения, которые эффективны для ингибирования функции белка NS5A. Белок NS5A HCV описан,например, в следующих ссылках: S.L. Tan, et al., Virology, 284:1-12 (2001); K.-J. Park, et al., J. Biol. Chem.,30711-30718 (2003); T.L. Tellinghuisen, et al., Nature, 435, 374 (2005); R.A. Love, et al., J. Virol., 83, 4395Bachand, et al. в WO2008/021927, опубликованной 21 февраля 2008 г., раскрывает серию бифенильных соединений, которые являются полезными для лечения вируса гепатита С. Новые соединения в соответствии с настоящим изобретением находятся в пределах определения формулы в WO2008/021927 и не раскрываются или описываются Bachand, et al. Неожиданно было отмечено, что эти соединения обла-1 021538 дают уникальными характерными свойствами, которые делают их полезными для лечения от вируса гепатита С. Сущность изобретения В первом аспекте настоящее изобретение обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль,в которой R1 выбирается из водорода, метила и фтора;R2 выбирается из водорода и метила;R3 и R4, каждый, представляют собой водород илиR5 выбирается из водорода и метила. В первом варианте осуществления первого аспекта настоящее изобретение обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль, в которой R1 представляет собой водород. Во втором варианте осуществления первого аспекта настоящее изобретение обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль, в которой R1 представляет собой фтор. В третьем варианте осуществления R2, R3 и R4, каждый, представляют собой водород и R5 представляет собой метил. В четвертом варианте осуществления первого аспекта настоящее изобретение обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль, в которой R1 представляет собой метил. В пятом варианте осуществления R2, R3 и R4, каждый, представляют собой водород и R5 представляет собой метил. Во втором аспекте настоящее изобретение обеспечивает соединение, выбранное из или их фармацевтически приемлемых солей. В третьем аспекте настоящее изобретение обеспечивает композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В первом варианте осуществления третьего аспекта композиция дополнительно содержит одно, два или три дополнительных соединения, имеющих анти-HCV активность. Во втором варианте осуществления третьего аспекта по меньшей мере одно из дополнительных соединений является интерфероном или рибавирином. В третьем варианте осуществления интерферон выбирается из интерферона альфа-2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа-2 А, интерферона лямбда и лимфобластоидного интерферона тау. В четвертом варианте осуществления третьего аспекта настоящее изобретение обеспечивает композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль, фармацевтически приемлемый носитель и одно или два дополнительных соединений, имеющие анти-HCV активность, в которой по меньшей мере одно из дополнительных соединений выбирается из интерлейкина-2,интерлейкина-6, интерлейкина-12, соединения, которое усиливает развитие Т-хелперного ответа 1 типа,интерферирующих РНК, антисмысловых РНК, имиквимода, рибавирина, ингибиторов инозин-5'монофосфат дегидрогеназы, амантадина и римантадина. В пятом варианте осуществления третьего аспекта настоящее изобретение обеспечивает композицию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль, фармацевтически приемлемый носитель и одно или два дополнительных соединений, имеющих анти-HCV активность,в которой по меньшей мере одно из дополнительных соединений является эффективным для ингибирования функции мишени, выбранной из металлопротеазы HCV, сериновой протеазы HCV, полимеразыHCV, геликазы HCV, белка NS4B HCV, входа HCV, сборки HCV, выхода HCV, белка NS5A HCV и ИМФДГ (инозинмонофосфатдегидрогеназа) для лечения инфекции HCV. В четвертом аспекте настоящее изобретение обеспечивает способ лечения инфекции HCV у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В первом варианте осуществления четвертого аспекта способ дополнительно содержит введение одного, двух или трех дополнительных соединений, имеющих анти-HCV активность до, после или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью. Во втором варианте осуществления четвертого аспекта по меньшей мере одно из дополнительных соединений представляет собой интерферон или рибавирин. В третьем варианте осуществления четвертого аспекта интерферон выбирается из интерферона альфа-2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа-2 А, интерферона лямбда и лимфобластоидного интерферона тау. В четвертом варианте осуществления четвертого аспекта настоящее изобретение обеспечивает способ лечения инфекции HCV у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли и одно или два дополнительных соединений, имеющих анти-HCV активность до, после или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью, в котором по меньшей мере одно из дополнительных соединений выбирается из интерлейкина-2, интерлейкина-6, интерлейкина-12, соединения, которое усиливает развитие Т-хелперного ответа 1 типа, интерферирующих РНК, антисмысловых РНК, имиквимода, рибавирина, ингибиторов инозин-5'-монофосфат дегидрогеназы, амантадина и римантадина. В пятом варианте осуществления четвертого аспекта настоящее изобретение обеспечивает способ лечения инфекции HCV у пациента, включающий введение пациенту терапевтически эффективного ко-3 021538 личества соединения формулы (I) или его фармацевтически приемлемой соли и одно или два дополнительных соединений, имеющих анти-HCV активность до, после или одновременно с соединением формулы (I) или его фармацевтически приемлемой солью, в котором по меньшей мере одно из дополнительных соединений является эффективным для ингибирования функции мишени, выбранной из металлопротеазы HCV, сериновой протеазы HCV, полимеразы HCV, геликазы HCV, белка NS4B HCV, входа HCV,сборки HCV, выхода HCV, белка NS5A HCV и ИМФДГ для лечения инфекции HCV. В другом аспекте настоящее изобретение обеспечивает соединение, которое представляет собой или его фармацевтически приемлемую соль. В другом аспекте настоящее изобретение обеспечивает соединение, которое представляет собой или его фармацевтически приемлемую соль. Другие варианты осуществления в соответствии с настоящим изобретением могут содержать подходящие комбинации двух или больше вариантов осуществлений и/или аспектов, раскрытых в настоящем изобретении. Другие варианты осуществления и аспекты изобретения будут очевидны в соответствии с описанием, приведенным ниже. Подробное описание изобретения Соединения в соответствии с настоящим изобретением также существуют как таутомеры; поэтому настоящее изобретение также охватывает все таутомерные формы. Описание в соответствии с настоящим изобретением настоящего изобретения должно быть истолковано в соответствии с законами и принципами химического связывания. Следует понимать, что соединения, охваченные настоящим изобретением, являются теми, которые достаточно устойчивы для использования в качестве фармацевтического агента. Все патенты, заявки на патент и литературные ссылки, процитированные в описании, включены в настоящее изобретение посредством ссылки во всей их полноте. В случае противоречий будет иметь преимущественную силу настоящее изобретение, включая определения. Как использующиеся в настоящем описании, следующие термины имеют указанные значения: В соединениях в соответствии с настоящим изобретением существуют асимметрические центры. Эти центры определяются символами "R" или "S" в зависимости от конфигурации заместителей вокруг хирального атома углерода. Следует понимать, что изобретение охватывает все стереохимические изомерные формы или их смеси, которые обладают способностью ингибировать NS5A. Индивидуальные стереоизомеры соединений могут быть получены синтетически из коммерчески доступных исходных соединений, которые содержат хиральные центры, или посредством получения смесей стереоизомерных веществ, с последующим разделением, такими как превращение в смесь диастереомеров, с последующим разделением либо перекристаллизацией, хроматографическими методами, либо непосредственным разделением на хиральных хроматографических колонках. Исходные соединения с особой стереохимией являются либо коммерчески доступными, либо могут быть изготовлены и разделены методами, известными в данной области техники. Некоторые соединения в соответствии с настоящим изобретением могут также существовать в различных устойчивых конформационных формах, которые могут быть разделяемыми. Торсионная асимметрия вследствие ограниченного вращения вокруг асимметричной одинарной связи, например вследствие стерических затруднений или кольцевого напряжения, может способствовать разделению различных конформеров. Настоящее изобретение включает каждый конформационный изомер этих соединений и их смесей. Термин "соединения в соответствии с настоящим изобретением" и эквивалентные выражения предназначаются, чтобы охватить соединения формулы (I) и фармацевтически приемлемые энантиомеры,диастереомеры и их соли. Точно так же ссылки на промежуточные соединения предназначаются, чтобы охватить их соли, если контекст разрешает это. Настоящее изобретение предназначено, чтобы включить все изотопы атомов, присутствующих в настоящих соединениях. Изотопы включают такие атомы, которые имеют то же самое атомное число, но различные массовые числа. Посредством общего примера и без ограничения изотопы водорода включают дейтерий и тритий. Изотопы углерода включают 13 С и 14 С. Изотопно меченные соединения изобретения могут, как правило, быть получены обычными методами, известными специалистами в данной об-4 021538 ласти техники, или посредством процессов, аналогичных описанным в настоящем изобретении, используя соответствующий изотопно меченный реактив вместо немеченого реактива, обычно используемого. Такие соединения могут иметь множество вариантов потенциального использования, например как стандарты и реактивы для определения биологической активности. В случае стабильных изотопов у таких соединений может быть потенциал для благоприятного изменения биологических, фармакологических или фармакокинетических свойств. Соединения в соответствии с настоящим изобретением могут существовать как фармацевтически приемлемые соли. Термин "фармацевтически приемлемая соль", как используется в настоящем изобретении, представляет соли или цвиттерионные формы соединений в соответствии с настоящим изобретением, которые являются растворимыми в воде или в масле или диспергируемыми, которые являются, в рамках здравого медицинского суждения, подходящими для использования в контакте с тканями пациентов без чрезмерной токсичности, раздражения, аллергической реакции либо другой проблемы или осложнения, соразмерного с разумным отношением выгоды/риска, и эффективны для их намеченного использования. Соли могут быть получены во время заключительного выделения и очистки соединений или отдельно посредством реакции подходящего атома азота с подходящей кислотой. Репрезентативные соли присоединения кислоты включают ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат; диглюконат, дигидробромид, дигидрохлорид,дигидройодид, глицерофосфат, гемисульфат, гептаноат, гексаноат, формиат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, мезитиленсульфонат, метансульфонат,нафтиленсульфонат, никотиноат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3 фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, трихлорацетат, трифторацетат, фосфат,глутамат, бикарбонат, пара-толуолсульфонат и ундеканоат. Примеры кислот, которые могут использоваться, чтобы сформировать фармацевтически приемлемые соли присоединения, включают неорганические кислоты, такие как хлористо-водородную, гидробромистую, серную и фосфорную, и органические кислоты, такие как щавелевую, малеиновую, янтарную и лимонную. Когда это возможно, для использования в терапии терапевтически эффективные количества соединения формулы (I) так же, как их фармацевтически приемлемые соли могут быть введены в виде химического сырья, возможно представить активный ингредиент как фармацевтическую композицию. Соответственно настоящее изобретение дополнительно обеспечивает фармацевтические композиции, которые включают терапевтически эффективные количества соединений формулы (I) или их фармацевтически приемлемых солей и один или большее количество фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ. Термин "терапевтически эффективное количество", как используется в настоящем изобретении, относится к общему количеству каждого активного компонента, которое достаточно, чтобы показать значимую выгоду для пациента, например сокращение вирусной нагрузки. Когда применяемый для индивидуума активный ингредиент вводится один, термин относится только к одному этому компоненту. Когда применяется комбинация, термин относится к объединенному количеству активных ингредиентов, которые приводят к терапевтическому эффекту, вне зависимости, вводятся ли в комбинации, последовательно или одновременно. Соединения формулы (I) и их фармацевтически приемлемые соли являются такими, как описано выше. Носитель(и), разбавитель(и) или вспомогательное вещество(а) должны быть приемлемыми в смысле того, чтобы быть совместимым с другими компонентами состава и не приносить вред их реципиенту. В соответствии с другим аспектом в соответствии с настоящим изобретением также обеспечивается процесс получения фармацевтического состава, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или большим количеством фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ. Термин "фармацевтически приемлемый", как используется в настоящем изобретении, относится к тем соединениям, веществам, композициям и/или лекарственным формам, которые являются в рамках здравого медицинского суждения подходящими для использования в контакте с тканями пациентов без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерного с разумным отношением выгоды/риска, и являются эффективными для их намеченного использования. Фармацевтические составы могут быть представлены в формах одноразовой дозы, содержащей предварительно определенное количество активного ингредиента на одноразовую дозу. Уровни дозировки между около 0,01 и около 250 мг/кг массы тела в день, предпочтительно между около 0,05 и около 100 мг/кг массы тела в день соединений в соответствии с настоящим изобретением являются типичными для монотерапии для профилактики и лечения болезни, опосредованной HCV. Как правило, фармацевтические композиции в соответствии с настоящим изобретением будут вводиться от около 1 около до 5 раз в день или альтернативно в виде непрерывного вливания. Такое введение может использоваться в качестве хронической или острой терапии. Количество активного ингредиента, которое может быть объединено с веществами носителя для получения одноразовой формы дозировки, варьируется в зависимости от состояния, подлежащего лечению, серьезности состояния, времени введения, способа введения, уровня выделения используемого соединения, продолжительности лечения и возраста, пола, массы и состояния пациента. Предпочтительные единичные дозированные лекарственные формы являются такими, которые содержат ежедневную дозу или субдозу, как упомянуто выше в настоящем изобретении, или ее соответствующую часть активного ингредиента. Лечение может быть начато с небольших дозировок, существенно меньших, чем оптимальная доза соединения. После этого дозировка увеличивается небольшими приращениями, пока не будет достигнут оптимальный эффект при этих обстоятельствах. В общем случае соединение наиболее желательно вводить при уровне концентрации, который обычно приводит к антивирусному эффекту, не вызывая опасных или вредных побочных эффектов. Когда соединения в соответствии с настоящим изобретением содержат комбинацию соединения в соответствии с настоящим изобретением и одного или большего количества дополнительных терапевтических или профилактических агентов, как соединение, так и дополнительный агент обычно присутствуют на уровнях дозировки между около 10 и 150% и более предпочтительно между около 10 и 80% дозировки, которую обычно вводят в режиме монотерапии. Фармацевтические составы могут быть приспособлены к введению любым подходящим способом,например пероральным (включая буккальный или сублингвальный), ректальным, назальным, местным(включая буккальный или сублингвальный или трансдермальный), вагинальным, или парентеральным(включая подкожные, внутрикожные, внутримышечные, внутрисуставные, внутрисиновиальные, внутригрудинные, интратекальные, внутриочаговые, внутривенные или интрадермальные инъекции или вливания) способами. Такие составы могут быть получены любым способом, известным в области фармации,например посредством доставки в ассоциации активного ингредиента с носителем(ями) или вспомогательным веществом(ами). Пероральное введение или введение посредством инъекции являются предпочтительными. Фармацевтические составы, приспособленные для перорального введения, могут быть представлены как дискретные единицы, такие как капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобные пены или легкое взбитое кушанье или жидкие эмульсии масло-в-воде или эмульсии вода-в-масле. Например, для перорального введения в форме таблетки или капсулы активный компонент препарата может быть объединен с пероральным, нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Порошки получают путем распыления соединения до подходящего малого размера и смешения со столь же раздробленным фармацевтическим носителем, таким как пищевой углевод, как, например, крахмал или маннит. Вещество, корригирующее вкус и запах лекарственного средства, консервант, диспергирующий агент и окрашивающий агент могут также присутствовать. Капсулы изготавливаются посредством приготовления порошковой смеси, как описано выше, и посредством заполнения сформированных желатиновых оболочек. Скользящие вещества и лубриканты,такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, могут быть добавлены к порошковой смеси перед операцией по заполнению. Дезинтегрирующий или солюбилизирующий агент, такие как агар-агар, карбонат кальция или карбонат натрия, могут также быть добавлены, чтобы улучшить биодоступность лекарства, после проглатывания капсулы. Кроме того, когда желательные или необходимые подходящие связующие, лубриканты, дезинтегрирующие агенты и окрашивающие вещества могут также быть включены в смесь. Подходящие связующие включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подслащивающие вещества, природные и синтетические камеди, такие как гуммиарабик, трагакант или альгинат натрий, карбоксиметилцеллюлоза, полиэтиленгликоль и т.п. Лубриканты, используемые в этих лекарственных формах, включают олеат натрия, хлорид натрия и т.п. Дезинтеграторы включают, без ограничений, крахмал, метилцеллюлозу, агар, бетонит, ксантановую камедь и т.п. Таблетки составляются,например, посредством приготовления порошковой смеси, гранулированием или брикетированием при добавлении лубриканта и дезинтегратора и посредством прессования в таблетки. Порошковую смесь получают посредством смешения соединения, удобным образом раздробленного, с разбавителем или основой, как описано выше, и при необходимости со связующим, таким как карбоксиметилцеллюлоза,альгинат, желирующим веществом или поливинилпирролидоном, замедлителем растворения, таким как парафин, ускорителем всасывания, таким как четвертичная соль и/или и абсорбирующим агентом, таким как бетонит, каолин или дикальция фосфат. Порошковая смесь может гранулироваться посредством увлажнения со связующим, таким как сироп, крахмальная паста, экстракт слизи растительного происхождения или растворами целлюлозных или полимерных веществ и продавливания через сито. В качестве альтернативы гранулированию порошковая смесь может быть пропущена через таблетировочную машину, и результатом является недостаточно хорошо сформированные куски, которые разбиваются на гранулы. Гранулы могут быть смазаны, чтобы предотвратить прилипание к матрице, формирующей таблетку, посредством добавления стеариновой кислоты, соли стеариновой кислоты, талька или минерального масла. Смазанная смесь затем подвергается прессованию в таблетки. Соединения в соответствии с настоящим изобретением могут также комбинироваться с легкосыпучим инертным носителем и спрессованы в таблетки непосредственно, не проходя стадии гранулирования или брикетирования Может быть обеспечено прозрачное или непрозрачное защитное покрытие, состоящее из герметизирующего покрытия из шеллака, покрытия сахаром или полимерным веществом и покрытия полировочным воском. Кра-6 021538 сители могут быть добавлены к этим покрытиям, чтобы отличить различные единицы дозировки. Жидкости для перорального введения, такие как раствор, сиропы и эликсиры, могут быть приготовлены в дозированной лекарственной форме так, чтобы данное количество содержало предварительно определенное количество соединения. Сиропы могут быть приготовлены посредством растворения соединения в водном растворе с соответствующими вкусовыми добавками, в то время как эликсиры получают с помощью нетоксичной основы. Солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленновые эфиры сорбитола, консерванты, вкусовая добавка, такие как мятное масло или естественные подслащивающие вещества либо сахарин или другие искусственные подслащивающие вещества и т.п., могут также быть добавлены. При необходимости составы единицы дозировки для перорального введения могут быть микрокапсулированы. Состав может также быть получен для пролонгированного или постепенного высвобождения, например, посредством покрытия или заключением вещества частицы в полимеры, в воск и т.п. Соединения формулы (I) и их фармацевтически приемлемые соли могут также вводиться в форме липосомальных систем доставки, таких как малые моноламеллярные везикулы, большие моноламеллярные везикулы и мультиламеллярные везикулы. Липосомы могут быть сформированы из множества фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения формулы (I) и их фармацевтически приемлемые соли могут также быть доставлены при помощи моноклональных антител как индивидуальных носителей, с которыми соединены молекулы соединения. Соединения могут также быть соединены с растворимыми полимерами как нацеливаемыми носителями лекарственного препарата. Такие полимеры могут включать поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидофенол или полиэтиленоксидполилизин, замещенный палитоильными остатками. Кроме того, соединения могут быть конъюгированы с классом биоразлагаемых полимеров, полезных для достижения контролируемого высвобождения, лекарственного препарата, например полимолочной кислоты, полиэпсилонкапролактона, полигидроксимасляной кислоты, полиортоэфиров, полиацеталей, полидигидропиранов, полицианоакрилатов и поперечно-сшитых или амфифильных блоксополимеровов гидрогелей. Фармацевтические составы, приспособленные для трансдермального введения, могут быть представлены в виде отдельных пластырей, предназначенных находиться в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Например, активный ингредиент может высвобождаться из пластыря посредством ионтофореза, как в общих чертах описано в Pharmaceutical Research 1986, 3(6), 318. Фармацевтические составы, приспособленные для местного введения, могут составляться как мази,кремы, суспензии, лосьоны, порошки, растворы, пасты, гели, спреи, аэрозоли или масла. Фармацевтические составы, приспособленные для ректального введения, могут быть представлены в виде свечей или в виде клизм. Фармацевтические составы, приспособленные для назального введения, где носитель представляет собой твердое вещество, включают грубый порошок, имеющий размер частиц, например, в диапазоне 20500 мкм, вводится таким образом, при котором осуществляется вдыхание через нос, то есть посредством быстрой ингаляции через носовой канал из контейнера с порошком, поднесенного близко к носу. Подходящие составы, в которых носитель представляет собой жидкость для введения в качестве назального спрея или носовых капель, включает водные или масляные растворы активного ингредиента. Фармацевтические составы, приспособленные для введения посредством ингаляции, включают порошки из мелких частиц или аэрозоли, которые могут быть произведены посредством различных типов дозированных находящихся под давлением аэрозолей, небулайзеров или инсуффляторов. Фармацевтические составы, приспособленные для вагинального введения, могут быть представлены как вагинальные суппозитории, тампоны, кремы, гели, пасты, пены или составы для спрея. Фармацевтические составы, приспособленные для парентерального введения, включают водные и неводные стерильные растворы для инъекции, которые могут содержать антиоксиданты, буферы, бактериостатики и растворенные компоненты, которые доводят состав к изотоническому с кровью намеченного реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Составы могут быть представлены в контейнерах с единственной дозой или мультидозой, например запаянные ампулы и пузырьки, и могут храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Экстемпоральные растворы для инъекции и суспензии могут быть приготовлены из стерильных порошков, гранул и таблеток. Следует понимать, что в дополнение к компонентам, в подробностях упомянутым выше, составы могут включать другие агенты, обычные для данной области техники, учитывающее тип рассматриваемого состава, например составов, предназначенных для перорального введения, могут включать вкусовые агенты. Термин "пациент" включает как человека, так и других млекопитающих. Термин "лечение" относится к (i) предотвращению появления болезни, расстройства или состояния у пациента, который может быть предрасположен к болезни, расстройству и/или состоянию, но еще не был диагностирован как имеющий вышеперечисленное; (ii) ингибированию болезни, расстройства или состояния, то есть прекращению их развития; и (iii) облегчение болезни, расстройства или состояния, то есть вызывая регресс болезни, расстройства и/или состояния. Составы в соответствии с настоящим изобретением могут также вводиться с циклоспорином, например с циклоспорином А, или другими аналогами, действующими через подобный механизм. Циклоспорин А, как было показано, проявил активность против HCV в клинических испытаниях (Hepatology 2003, 38, 1282; Biochem. Biophys. Res. Commun. 2004, 313, 42; J. Gastroenterol. 2003, 38, 567). Табл. 1, приведенная ниже, содержит некоторые иллюстративные примеры соединений, которые могут быть введены с соединениями в соответствии с настоящим изобретением. Соединения в соответствии с настоящим изобретением могут быть введены с другими анти-HCV активными соединениями в комбинированной терапии, либо совместно, либо по-отдельности, либо посредством комбинации соединений в композиции. Таблица 1 Соединения в соответствии с настоящим изобретением могут также использоваться в качестве лабораторных реактивов. Соединения могут служить инструментами для обеспечения исследования для разработки анализа вирусной репликации, валидации систем испытания на животных и структурных биологических исследований для дальнейшего расширения знаний механизмов болезни HCV. Дополнительно соединения в соответствии с настоящим изобретением являются полезными для установления или определения связывающего участка других противовирусных соединений, например, посредством конкурентного ингибирования. Соединения в соответствии с настоящим изобретением могут также использоваться для лечения или предотвращения вирусной контаминации материалов и таким образом снижать риск вирусной инфекции лаборатории или медперсонала или пациентов, которые соприкасаются с такими материалами,например кровью, тканью, хирургическими инструментами и предметами одежды, лабораторными инструментами и предметами одежды, аппаратами и материалами взятия крови или переливания крови. Это изобретение предназначено, чтобы охватить соединения, имеющие формулу (I), если получены посредством синтетических процессов или метаболических процессов, включая такие, которые происходят в теле человека или животного (in vivo), или процессы, происходящие in vitro. Сокращения, используемые в настоящем изобретении, включая в особенности используемые в примерах, которые следуют далее, известны специалистами в данной области техники. Некоторые используемые сокращения являются следующими: ч - час или часы; EtOAc - этилацетат; Hex - гексаны;Rt либо rt или Rt - комнатная температура или время удерживания (определяется контекстом); ON или о/n - на ночь; мин - минуты; DCM - дихлорметан; HATU - О-(7-азабензотриазол-1-ил)-N,N,N',N'тетраметилурониум гексафторфосфат; DMF - N, N-диметилформамид; TFA - трифторуксусная кислота;(1.28 г, 1,21 ммоль). Смесь гидрогенизировали на шейкере Парра под Н 2 (70 фунт на кв.дюйм) при комнатной температуре в течение 72 ч. Реакционную смесь фильтровали через слой диатомовой земли(Celite) и промывали этанолом. Фильтрат концентрировали в вакууме и остаток очищали с помощью флэш-хроматографии (от 10 до 30% EtOAc/Нех). Выделяли две фракции прозрачных маслообразных веществ. Первые элюированные фракции представляли собой смесью (2R,4r,6S)-2,6-диметилтетрагидро 2 Н-пиран-4-ол (Сар 1, стадия а) и (2R,4s,6S)-2,6-диметилтетрагидро-2 Н-пиран-4-ол (1,2 г), в то время как последние элюированные фракции соответствовали исключительно Сар 1, стадия а (10,73 г). 1 Н-ЯМРDEAD (166 мл, 330 ммоль) добавляли по каплям к раствору Сар 1, стадия а (10,73, 82 ммоль), 4 нитробензойную кислоту (48,2 г, 288 ммоль) и Ph3P (86 г, 330 ммоль) в бензоле (750 мл). Отмечали выделение тепла и получающийся янтарный раствор перемешивали при температуре окружающей среды в течение 18 ч. Растворитель удаляли при пониженном давлении и остаток растирали в порошок в Et2O(200 мл), чтобы удалить окись трифенилфосфина (10 г). Остающуюся смесь очищали через Biotage (от 0 до 5% EtOAc/Hex; 300-граммовая колонка X 4). Выделяли белое твердое вещество, соответствующее Сар 1, стадия b (19,36 г). 1H-ЯМР (500 МГц, CDCl3)м.д. 8,27-8,32 (2 Н, м), 8,20-8,24 (2 Н, м), 5,45 (1 Н,квинтет, J=2,82 Гц), 3,92 (2 Н, двойной квартет дублетов, J=11,90, 6,10, 1,53 Гц), 1,91 (2 Н, дд, J=14,80,2,29 Гц), 1,57 (2 Н, дублет триплетов, J=14,65, 3,05 Гц), 1,22 (6 Н, д, J=6,10 Гц). 13 С-ЯМР (126 МГц, CDCl3)м.д. 163,81 (1 С), 150,55 (1 С), 135,94 (1 С), 130,64 (2 С), 123,58 (2 С), 70,20 (1 С), 68,45 (2 С), 36,95 (2 С),21,84 (2 С). LC-MS: анал. вычисл. для [М]+ C14H17NO5: 279,11; найдено 279,12. Сар 1, стадия с Раствор LiOH (8,30 г, 347 ммоль) в воде (300 мл) добавили к раствору Сар 1, стадия b (19,36 г, 69.3 ммоль) в THF (1000 мл) и получающуюся смесь перемешивали при температуре окружающей среды в течение 16 ч. THF удаляли при пониженном давлении, водный слой разбавляли большим количеством воды (200 мл) и экстрагировали EtOAc (3200 мл). Объединенные органические слои сушили (MgSO4),фильтровали и концентрировали под вакуумом. Получали масляный остаток с белым твердым веществом. Смесь растирали в гексанах и твердое вещество удаляли фильтрацией, получая прозрачное масло,соответствующее Сар 1, стадия с (8,03 г). 1 Н-ЯМР (500 МГц, CDCl3)м.д. 4,21 (1 Н, квинтет, J=2,82 Гц),3,87-3,95 (2 Н, м), 1,72 (1 Н, ш.с.), 1,63 (2 Н, дд, J=14,34, 2,14 Гц), 1,39-1,47 (2 Н, м), 1,17 (6 Н, д, J=6,41 Гц). 13 С-ЯМР (126 МГц, CDCl3)м.д. 67,53 (2 С), 64,71 (1 С), 39,99 (2 С), 21,82 (2 С). Сар 1, стадия d ридина (19,96 мл, 247 ммоль) в CH2Cl2 (750 мл) при комнатной температуре и перемешивали в течение 36 ч. Так как реакция не продолжалась до конца, CH2Cl2 удаляли при пониженном давлении и перемешивание продолжали в течение еще 48 ч. Смесь затем добавляли к CH2Cl2 (100 мл) и воде (100 мл) и перемешивали при температуре окружающей среды в течение 2 ч. Смесь отделяли и органический слой промывали водн. 1 н. HCl (250 мл). Органический слой затем высушивали (MgSO4), фильтровали и концентрировали. Выделяли желтое масло, соответствующее Сар 1, стадия d (14,15 г), которое затвердевало под вакуумом в виде грязно-белого твердого вещества. 1H-ЯМР (500 МГц, CDCl3)м.д. 7,80 (2 Н, д, J=8,24 Гц), 7,35 (2 Н, д, J=7,93 Гц), 4,88 (1 Н, квинтет, J=2,82 Гц), 3,79-3,87 (2 Н, м), 2,46 (3 Н, с), 1,76 (2 Н, дд,J=14,50, 2,59 Гц), 1,36 (2 Н, двойной дублет дублетов, J=14,34, 11,60, 2,75 Гц), 1,12 (6 Н, д, J=6,10 Гц). 13 СЯМР (126 МГц, CDCl3)м.д. 144,64 (1 С), 134,24 (1 С), 129,82 (2 С), 127,61 (2 С), 77,34 (1 С), 67,68 (2 С),37,45 (2 С), 21,61 (1 С), 21,57 (2 С). LC-MS: анал. вычисл. для [2 М+Н]+ C28H41O8S2: 569,22; найдено 569,3. Сар 1, стадия еLiHMDS (29,7 мл, 29,7 ммоль, 1 M в THF) добавляли к раствору Сар 1, стадия d (7,05 г, 24,8 ммоль) и бензил 2-(дифенилметиленамино)ацетата (8,57 г, 26,0 ммоль) в толуоле (80 мл) при комнатной температуре в пробирке под давлением и полученную смесь затем перемешивали в течение 5 ч при 100 С. Реакцию останавливали водой (100 мл), экстрагировали EtOAc, промывали водой, высушивали над MgSO4,фильтровали и концентрировали в вакууме. Остаток очищали через Biotage (от 0 до 15% EtOAc/Hex; 240-граммовая колонка) и желтое масло, соответствующее Сар 1, стадия е (8,76 г) выделяли как рацемическую смесь. 1H-ЯМР (400 МГц, CDCl3)м.д. 7,62-7,71 (2 Н, м), 7,30-7,45 (11 Н, м), 7,05 (2 Н, дд, J=7,65,1,63 Гц), 5,13-5,22 (2 Н, м), 3,89 (1 Н, д, J=6,78 Гц), 3,46 (2 Н, двойной квинтет дублетов, J=11,27, 5,90, 2,01 Гц), 2,34-2,45 (1 Н, м), 1,58-1,66 (1 Н, м), 1,34-1,43 (1 Н, м), 1,19 (3 Н, д, J=6,02 Гц), 1,03-1,16 (4 Н, м), 0,830,97 (1 Н, м). 13 С-ЯМР (101 МГц, CDCl3)м.д. 170,84 (1 С), 170,68 (1 С), 139,01 (1 С), 135,96 (1 С), 135,51 Сар 1, стадия е (8,76 г, 19,84 ммоль) растворяли в THF (100 мл) и обрабатывали 2 н. HCl в воде (49,6 мл, 99 ммоль). Получающийся прозрачный раствор перемешивали при температуре окружающей среды в течение 4 ч и затем THF удаляли при пониженном давлении. Остающийся водный слой экстрагировалиEtOAc (330 мл) и концентрировали под вакуумом, получая соответствующий неочищенный амин. Остаток растворяли в CH2Cl2 (100 мл) и загружали DIEA (11,8 мл, 67,6 ммоль) и метилхлорформиатом (1,962 мл, 25,3 ммоль). Получающийся раствор перемешивали при температуре окружающей среды в течение 2 ч. Реакционную смесь разбавляли CH2Cl2 (50 мл) и промывали водой (100 мл) и рассолом (100 мл). Органический слой сушили (MgSO4), фильтровали и концентрировали. Остаток очищали через Biotage (от 15 до 25% EtOAc/Hex; 80-граммовая колонка). Получали прозрачное бесцветное масло, соответствующее рацемическому Сар 1, стадия f (5,27 г). 1 Н-ЯМР (400 МГц, CDCl3)м.д. 7,32-7,41 (5 Н, м), 5,13-5,28 (3 Н,м), 4,36 (1 Н, дд, J=8,16, 4,64 Гц), 3,69 (3 Н, с), 3,30-3,47 (2 Н, м), 2,00-2,16 (1 Н, м), 1,52 (1 Н, д, J=12,55 Гц),1,33 (1 Н, д, J=12,30 Гц), 1,15 (6 Н, дд, J=6,02, 5,02 Гц), 0,88-1,07 (2 Н, м). 13 С-ЯМР (101 МГц, CDCl3)м.д. 171,39 (1 С), 156,72 (1 С), 135,20 (2 С), 128,60 (2 С), 128,57 (1 С), 128,52 (2 С), 72,77 (1 С), 72,74 (1 С), 67,16[М+Н]+ C18H26NO5: 336,18; найдено 336,3. Хиральный способ был разработан для разделения рацемической смеси посредством использования 20%-ного этанола в качестве модификатора на колонке CHIRALPAK (50500 мм, 20 мкм) (длина волны=220 нм, расход=100 мл/мин в течение 22 мин, растворитель А=0,1% диэтиламин в гептанах, растворитель В=EtOH). Два отделенных изомера, которые соответствуют(S)-бензил 2-2R,4r,6S)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-(метоксикарбонил)амино)ацетату (Cap 1, стадия f.1) (Rt=9,8 мин, 2,2 г) и (R)-бензил 2-2R,4r,6S)-2,6-диметилтетрагидро-2 Нпиран-4-ил)-2-(метоксикарбонил)амино)ацетату (Сар 1, стадия f.2) (Rt=16,4 мин, 2,1 г) и каждый из них продемонстрировали одинаковые аналитические данные, как соответствующая смесь (см. выше).Cap 1. (S)-Бензил 2-2R,4r,6S)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-(метоксикарбонил)амино)ацетат (Сар 1, стадия f.1) (2,2 г, 6,6 ммоль) растворяли в МеОН (50 мл) в бомбе Парра и загружали 10% Pd/C (0,349 г, 0,328 ммоль). Суспензию затем помещали в шейкер Парра и смесь продували N2 (3),помещали под 40 фунт на кв.дюйм Н 2 и встряхивали при комнатной температуре в течение 15 ч. Катализатор отфильтровывали через слой диатомовой земли (Celite) и растворитель удаляли при пониженном давлении, получая твердое вещество янтарного цвета, соответствующее Сар 1 (1,6 г). 1H-ЯМР (500 МГц,диметилсульфоксид-d6)м.д. 12,74 (1 Н, ш.с.), 7,35 (1 Н, д, J=6,10 Гц), 3,85 (1 Н, ш.с.), 3,53 (3 Н, с), 3,35(2 Н, двойной дублет дублетов, J=15,95, 9,99, 6,10 Гц), 1,97 (1 Н, ш.с.) 1,48 (2 Н, т, J=13,28 Гц), 1,06 (6 Н, д,J=6,10 Гц), 0,82-1,00 (2 Н, м). 13 С-ЯМР (101 МГц, диметилсульфоксид-d6)м.д. 176,93 (1 С), 156,72 (1 С,),72,10 (1 С), 71,92 (1 С), 58,54 (1 С), 51,35 (1 С), 36,88 (1 С), 35,82 (1 С), 34,71 (1 С), 21,90 (2 С). Примечание: абсолютную стереохимию сар 1 определяли рентгеноструктурным анализом монокристалла сложноэфирного аналога, полученного из сар 1 и (S)-фенетанола. Сар 2 Ссылка: S. Danishefsky; et al. J. Org. Chem, 1982, 47, 1597. Эфират трехфтористого бора (3,81 мл, 30,5 ммоль) добавляли по каплям к перемешиваемому и охлажденному (-78 С) раствору (Е)-(4-метоксибута-1,3-диен-2-илокси)триметилсилана (5,0 г, 29 ммоль) и ацетальдегида (3,28 мл, 58,0 ммоль) в диэтиловом эфире (100 мл) под азотом. Реакцию перемешивали при -78 С в течение 2,5 ч и затем останавливали насыщенным водным NaHCO3 (40 мл), позволяли нагреваться до RT и перемешивали ON. Слои разделяли и водный слой экстрагировали диэтиловым эфиром(250 мл). Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали до желтого/оранжевого масла. Неочищенное масло очищали с помощью Biotage Horizon (110 г SiO2, 25-40% Ссылка: Reddy, D.S.; et al. J. Org. Chem. 2004, 69, 1716-1719. Раствор 1,6 М метиллития в диэтиловом эфире (20,9 мл, 33,4 ммоль) добавили к перемешиваемой суспензии йодида меди(I) (4,25 г, 22,30 ммоль) в диэтиловом эфире (30 мл) при 0 С и под азотом. Реакцию перемешивали при 0 С в течение 20 мин и затем добавляли через 10 мин рацемический 2-метил-2 Нпиран-4-(3 Н)-он (1,25 г, 11,2 ммоль) в диэтиловом эфире (12,0 мл). Реакции позволили нагреться до RT и перемешивали 2 ч. Реакционную смесь выливали в насыщенный NH4Cl (водн.) и перемешивали 20 мин. Раствор экстрагировали диэтиловым эфиром (460 мл) и объединенные органические растворы промывали рассолом (80 мл), сушили (MgSO4), фильтровали и концентрировали, получая рацемический Борогидрид натрия (0,354 г, 9,36 ммоль) добавляли порциями к перемешиваемому раствору рацемического (2R,6R)-2,6-диметилдигидро-2 Н-пиран-4 (3 Н)-она (Сар 2, стадия b) (1,2 г, 9,4 ммоль) в МеОН(30 мл) при 0 С. Раствор перемешивали 10 мин при 0 С, нагревали до RT и перемешивали 1 ч. Реакцию выливали в насыщенный NH4Cl (50 мл), перемешивали 20 мин и затем частично концентрировали (до 1/2 об.). Образовывался осадок, добавляли воду до гомогенного состояния и затем раствор экстрагировали DCM (360 мл). Водный слой подкисляли 1 н. HCl и затем экстрагировали DCM (360 мл). Объединенные органические экстракты сушили Na2SO4, фильтровали и концентрировали, при этом образовалось мутное желтое масло (1,08 г). Неочищенное масло растворяли в DCM (8,0 мл) и затем добавляли птозил-Cl (2,68 г, 14,0 ммоль) и пиридин (1,51 мл, 18,7 ммоль) и реакцию перемешивали при RT в течение 2,5 суток. Реакцию разбавляли насыщенным NH4Cl (60 мл) и экстрагировали DCM (330 мл). Объединенную органическую фазу высушивали (MgSO4), фильтровали и концентрировали до коричневого масла. Масло очищали на Biotage Horizon (80 г SiO2, 10-25% EtOAc/гексаны), получая рацемический(2R,6R)-2,6-демитилтетрагидро-2 Н-пиран-4-ил 4-метилбензолсульфонат (Сар 2, стадия с) (1,63 г) в виде вязкого прозрачного бесцветного масла. Время удерживания в LC-MS 3,321 мин; m/z 284,98 [М+Н]+. Данные LC регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оборудованном колонкойPhenomenex-Luna 3u C18 2,050 мм, с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин,время захвата 1 мин и время анализа 5 мин, где состав растворителя А был 5% МеОН/95% Н 2 О/10 мМ ацетат аммония и состав растворителя В был 5% Н 2 О/95% МеОН/10 мМ ацетат аммония. Данные MS определяли, используя Micromass Platform for LC при ионизации электроспреем. 1H-ЯМР (400 МГц,CDCl3)м.д. 7,81 (д, J=8,3 Гц, 2 Н), 7,36 (д, J=8,0 Гц, 2 Н), 4,81-4,92 (м, 1 Н), 4,17-4,26 (м, 1 Н), 3,78-3,87 (м,1 Н), 2,47 (с, 3 Н), 1,91-1,99 (м, 1 Н), 1,78-1,86 (м, 1 Н), 1,65-1,72 (м, 1 Н), 1,46 (двойной дублет дублетов,J=12,9, 9,4, 9,3 Гц, 1 Н), 1,20 (дд, J=6,5, 4,8 Гц, 6 Н). Рацемическую смесь разделяли на индивидуальные энантиомеры при помощи многократных инъекций, используя хиральную препаративную очистку SFC (Chiralpak препаративная колонка 30250 мм,5 мкм, 10% 1:1 EtOH/гептан в СО 2, 70 мл/мин в течение 10 мин), получали (2R,6R)-2,6 диметилтетрагидро-2 Н-пиран-4-ил 4-метилбензолсульфонат (Сар 2, стадия с.1) (577 мг) в качестве первого элюированного пика и (2S,6S)-2,6-диметилтетрагидро-2 Н-пиран-4-ил 4-метилбензолсульфонат (Сар 2, стадия с.2) (588 мг) в качестве второго элюированного пика. Каждый энантиомер выделяли в виде прозрачного бесцветного масла, которое затвердевало в белое твердое вещество после стояния. Сар 2, стадия d В пробирке под давлением на 48 мл (2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил 4 метилбензолсульфонат (Сар 2, стадия с.1) (575 мг, 2,02 ммоль) и бензил 2-(дифенилметиленамино)ацетат(733 мг, 2,22 ммоль) перемешивали в THF (2 мл) и толуоле (10 мл). Прозрачный бесцветный раствор продували азотом, затем добавляли LiHMDS (1,0 М в THF) (2,22 мл, 2,22 ммоль) и сосуд плотно закрывали и нагревали при 100 С в течение 8 ч. Реакцию охлаждали до RT, выливали в 1/2 от насыщенияNH4Cl (водн.) (50 мл) и экстрагировали EtOAc (330 мл). Объединенные органические слои промывали рассолом, сушили (MgSO4), фильтровали и концентрировали до неочищенного оранжевого масла. Масло очищали на Biotage Horizon (40 г SiO2, 10-25% EtOAc/гексаны), получая содержащее примеси требуемое вещество (501 мг) в виде оранжевого масла. Это вещество повторно очищали на Biotage Horizon(25 г SiO2, 6-12% EtOAc/гексаны), получая 1:1 смесь диастереомеров (Сар 2, стадия d) (306 мг) в виде вязкого оранжевого масла. Смесь разделяли на индивидуальные диастереомеры при помощи многократных инъекций, используя хиральную препаративную очистку SFC (Chiralpak OJ-H препаративная колонка 30250 мм, 5 мкм,10% 1:1 EtOH/гептан в СО 2, 150 бар, 70 мл/мин в течение 10 мин), получая (R)-бензил 2-2R,6R)-2,6 диметилтетрагидро-2 Н-пиран-4-ил)-2-(дифенилметиленамино)ацетат (Сар 2, стадия d.1) (124 мг) в качестве первого элюированного пика и (S)-бензил 2-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2(дифенилметиленамино)ацетат (Сар 2, стадия d.2) (129 мг) в качестве второго элюированного пика. Каждый диастереомер выделяли в виде вязкого желтого масла. Аналитические данные для (R)-бензил 22R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-(дифенилметиленамино)ацетата (Сар 2, стадия d.1): 1H-ЯМР (400 МГц, D4-MeOH)м.д. 7,57-7,61 (м, 2 Н), 7,41-7,48 (м, 4 Н), 7,33-7,40 (м, 7 Н), 7,03-7,08 (м,2 Н), 5,22 (д, J=12,1 Гц, 1 Н), 5,16 (д, J=12,1 Гц, 1 Н), 4,09-4,19 (м, 1 Н), 3,84 (д, J=5,8 Гц, 1 Н), 3,75-3,83 (м,1 Н), 2,53-2,64 (м, 1 Н), 1,58-1,65 (м, 1 Н), 1,33-1,43 (м, 1 Н), 1,26-1,32 (м, 1 Н), 1,24 (д, J=7,0 Гц, 3 Н), 1,10 (д,J=6,0 Гц, 3 Н), 0,98-1,08 (м, 1 Н). Время удерживания LC-MS 4,28 мин; m/z 442,16 [М+Н]+. Данные LC регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оборудованном колонкой PhenomenexLuna 3u C18 2,050 мм, с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин, время захвата 1 мин и время анализа 5 мин, где состав растворителя А был 5% МеОН/95% Н 2 О/10 мМ ацетат аммония и состав растворителя В был 5% Н 2 О/95% МеОН/10 мМ ацетат аммония. Данные MS определяли, используя Micromass Platform for LC при ионизации электроспреем. Аналитические данные для (S)-бензил 2-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2(дифенилметиленамино)ацетат (Сар 2, стадия d.2): 1H-ЯМР (400 МГц, D4-MeOH)м.д. 7,57-7,61 (м, 2 Н),7,41-7,50 (м, 4 Н), 7,33-7,40 (м, 7 Н), 7,04-7,08 (м, 2 Н), 5,22 (д, J=12,1 Гц, 1 Н), 5,16 (д, J=12,1 Гц, 1 Н), 4,20(кв. д., J=6,4, 6,3 Гц, 1 Н), 3,86 (д, J=6,5 Гц, 1 Н), 3,74-3,83 (м, 1 Н), 2,53-2,64 (м, 1 Н), 1,60 (тд, J=12,7, 5,6 Гц, 1 Н), 1,38-1,51 (м, 2 Н), 1,26 (д, J=7,0 Гц, 3 Н), 1,04 (д, J=6,0 Гц, 3 Н), 0,79-0,89 (м, 1 Н). Время удерживания LC-MS 4,27 мин; m/z 442,17 [М+Н]+. Данные LC регистрировали на жидкостном хроматографеShimadzu LC-10AS, оборудованном колонкой Phenomenex-Luna 3u C18 2,050 мм, с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин, время захвата 1 мин и время анализа 5 мин, где состав растворителя А был 5% МеОН/95% Н 2 О/10 мМ ацетат аммония и состав растворителя В был 5% Н 2 О/95% МеОН/10 мМ ацетат аммония. Данные MS определяли, используя Micromass Platform for LC при ионизации электроспреем.(S)-Бензил 2-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-(дифенилметиленамино)ацетат (Сар 2, стадия d.2) (129,6 мг, 0,294 ммоль) растворяли в THF (2 мл) и затем обрабатывали 2 н. HCl (1,0 мл, 2,1 ммоль) в воде. Реакцию перемешивали в течение 2 ч и затем в течение ночи концентрировали под струей азота. Неочищенный остаток растворяли в DCM (2 мл) и DIPEA (0,21 мл, 1,2 ммоль), затем обрабатывали метилхлорформиатом (0,032 мл, 0,41 ммоль) и перемешивали при RT в течение 4 ч. Реакцию разбавляли водой (2,5 мл) и экстрагировали DCM (42 мл). Объединенную органическую фазу в течение ночи концентрировали под струей азота и остаток очищали с помощью Biotage Horizon (4 г SiO2, 10-50%(S)-бензил 2-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2(метоксикарбониламино)ацетат (Сар 2, стадия е) (56 мг) в виде бесцветного стеклообразного вещества. Время удерживания LC-MS 3,338 мин; m/z 335,99 [М+Н]+. Данные LC регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оборудованном колонкой Phenomenex-Luna 3u С 18 2,050 мм, с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин, время захвата 1 мин и время анализа 5 мин, где состав растворителя А был 5% МеОН/95% Н 2 О/10 мМ ацетат аммония и состав растворителя В был 5% Н 2 О/95% МеОН/10 мМ ацетат аммония. Данные MS определяли, используя Micromass PlatformPd/C (12 мг, 0,012 ммоль). Реакционную смесь продували под вакуумом азотом (4) и затем с водородом(4) и перемешивали в атмосфере водорода из баллона в течение ночи. Реакцию отфильтровывали через(м, 1 Н), 1,63 (д, J=13,1 Гц, 1 Н), 1,51-1,60 (м, 1 Н), 1,42-1,49 (м, 1 Н), 1,27 (д, J=7,0 Гц, 3 Н), 1,11 (д, J=6,3 Гц,3 Н), 0,97-1,08 (м, 1 Н). Примечание: абсолютную стереохимию сар 2 определяли рентгеноструктурным анализом монокристалла амидного аналога, полученного из эпимера сар 2 R)-2-2R,6R)-2,6 диметилтетрагидро-2 Н-пиран-4-ил)-2-(метоксикарбонил)амино)уксусной кислоты) и (S)-1-(нафталин-2 ил)этанамина. Альтернативный синтез сар 2 проиллюстрирован на схеме 1. Первые четыре стадии, приводящие к(R)-2-метил-2 Н-пиран-4 (3 Н)-ону, осуществлены, используя адаптацию известной методики (Anderson,K.R.; et al. Org. Proc. Res. Dev. 2010, 14, 58). Превращение (R)-2-метил-2 Н-пиран-4 (3 Н)-она в (2R,6R)2,6-диметилдигидро-2 Н-пиран-4 (3 Н)-он осуществляли адаптацией известной методики (Reddy, D.S.; etal. J. Org. Chem. 2004, 69, 1716). Дополнительные детали приводятся ниже. трет-Бутилхлордиметилсилан (547 г, 3,63 мольы) добавляли к перемешиваемому раствору этил (R)3-гидроксибутаноата (400 г, 3,03 моль) в DCM (800 мл) под азотом при 0 С. Имидазол (412 г, 6,05 моль) добавляли частями в течение 20 мин к реакционной смеси, в это время смесь стала густой белой суспензией. Добавляли дополнительный DCM (175 мл), смеси позволили нагреться до комнатной температуры и перемешивали в течение 16 ч. Белое твердое вещество удаляли фильтрацией, промывали DCM (500 мл), распределяли между водой (1 л) и DCM (500 мл) и водный слой далее экстрагировали дополнительным DCM (500 мл). Фильтрат объединяли с органическими слоями и промывали водой (500 мл) и рассолом, сушили над MgSO4 и фильтровали. Продукт концентрировали и сушили под высоким вакуумом,получая (R)-этил 3-(трет-бутилдиметилсилилокси)бутаноат (777,5 г), содержащий неустановленные примеси. Аналитические данные неочищенного продукта соответствовали данным, известным из литературы. Вещество использовали без дополнительной очистки. 2 М Раствор изопропилмагний хлорида в THF (800 мл, 1,60 моль) добавляли по каплям через канюлю в течение 60 мин к перемешиваемому раствору (R)-этил 3-(трет-бутилдиметилсилилокси)бутаноата(131 г, 532 ммоль) и N,O-диметилгидроксиламина/HCl (80 г, 824 ммоль) в сухом THF (850 мл), поддерживая внутреннюю температуру между -30 и -20 С. Суспензию перемешивали в течение 3 ч между -20 и-10 С и останавливали насыщенным водным раствором хлорида аммония (400 мл), не прекращая охлаждение. Реакционную смесь распределяли между водой (200 мл) и диэтиловым эфиром (500 мл) и водную фазу далее экстрагировали диэтиловым эфиром (1 л). Объединенные органические фазы промывали рассолом, высушивали над MgSO4, фильтровали, концентрировали и высушивали под высоким вакуумом,получая (R)-3-(трет-бутилдиметилсилилокси)-N-метокси-N-метилбутанамид. Реакцию повторяли 6 раз,получая в общей сложности 717 г вещества. Аналитические данные соответствовали данным, известным из литературы. Вещество использовали без дополнительной очистки. Для соответствующих ссылок см. (a) Marques-Lopez, E.; et al. Eur. J. Org. Chem., 2008, 20, 3457; (b)Corey, E.; et al. Chem. Ber., 1978, 111, 1337; (c) Chudek, J.A. et al. J. Chem. Soc. Perkin Trans, 2, 1985, 8,1285. Пиперидин-1-амин (754 мл, 6,99 моль) добавляли по каплям в течение 60 мин к ацетальдегиду, поддерживающемуся при 0 С (304 мл, 5,38 моль), при перемешивании под азотом. [Предостережение: добавление является экзотермическим. В этом масштабе капельная воронка, содержащая пиперидин-1 амин, стала горячей из-за ацетальдегида, который испарился и сконденсировался в капельной воронке.] После 1 ч при 0 С и 1 ч при RT реакционную колбу оборудовали 16-дюймовым обратным холодильником, нагревали до 40 С и перемешивали в течение 20 ч. Реакционную смесь охлаждали до RT и распределяли между диэтиловым эфиром (700 мл) и рассолом (300 мл). Органический слой промывали водой и водный слой экстрагировали диэтиловым эфиром (2500 мл). Объединенные органические слои промывали рассолом, высушивали над MgSO4, фильтровали, концентрировали и высушивали под высоким вакуумом в течение 8 ч, получая (Е)-N-этилилиденпиперидин-1-амин (772 г) (содержавший примеси) в виде желтого масла. Аналитические данные соответствовали данным, известным из литературы. Вещество использовали без дополнительной очистки. н-BuLi в гексанах (2,5 М, 644 мл, 1,61 моль) добавляли в течение 40 мин к перемешиваемому раствору диизопропиламина (245 мл, 1,72 моль) в сухом THF (2,3 л) при 0 С под азотом. Смесь перемешивали при 0 С в течение 30 мин и затем добавили по каплям раствор (Е)-N-этилилиденпиперидин-1-амина(203 г, 1,61 моль) в сухом THF (100 мл). Реакционную смесь перемешивали при 0 С в течение 2 ч, суспензию охлаждали до -78 С и обрабатывали по каплям с раствором (R)-3-(третбутилдиметилсилилокси)-N-метокси-N-метилбутанамида (280,4 г, 1,073 моль) в сухом THF (100 мл). Реакционную смесь перемешивали в течение 3 ч при -78 С, позволяли медленно нагреваться до комнатной температуры и затем перемешивали в течение 16 ч. Реакционную смесь распределяли между водой(600 мл) и диэтиловым эфиром (1,5 л) и водный слой далее экстрагировали диэтиловым эфиром. Объединенные органические слои промывали рассолом, высушивапли над MgSO4, фильтровали и концентрировали, получая масло янтарного цвета. Неочищенное масло очищали через короткую колонку (3 л силикагеля в 4-литровой делительной воронке из спеченного стекла предварительно уравновешивали 5%EtOAc/гексаны, затем неочищенное масло растворяли в 50 мл DCM/гексаны (1:5) и загружали сверху на силикагель, и, наконец, колонку элюировали 5-40% EtOAc/гексаны), получая (Е)-5-R)-третбутилдиметилсилилокси)-1-(пиперидин-1-илимино)гексан-3-он. Загрязненные фракции объединяли,концентрировали и повторно очищали на Biotage Horizon (300 г SiO2, 15-45% EtOAc/гексаны), получая дополнительное вещество (230 г общего количества). Аналитические данные соответствовали данным,известным из литературы. Вещество использовали без дополнительной очистки.Amberlyst-15 (122 г, 372 ммоль) (сухая форма, бледно-коричневые бусинки, Alfa Aesar, Stock89079 or L14146) добавляли одной порцией к перемешиваемому раствору (Е)-5-(R)-третбутилдиметилсилилокси)-1-(пиперидин-1-илимино) гексан-3-она (121 г, 372 ммоль) в сухом THF (1,2 л). Смесь нагревали при кипении в течение 2,5 ч, охлаждали, фильтровали и концентрировали в жидкость янтарного цвета. Неочищенную жидкость очищали через короткую колонку (неочищенный продукт растворяли в 30 мл 5% EtOAc/гексаны и загружали сверху на 1,5 л силикагеля в 4-литровой делительной воронке из спеченного стекла предварительно уравновешивали 5% EtOAc/гексаны, затем неочищенное масло растворяли в 50 мл DCM/гексаны (1:5) и загружали сверху на силикагель, и, наконец, колонку элюировали 5-50% EtOAc/гексаны), приводя к енону (R)-2-метил-2 Н-пиран-4(3 Н)-ону. Примечание: растворитель удаляли тщательным испарением на роторном испарителе (температура бани была меньше или равна температуре окружающей среды), но некоторое количество ожидаемого вещества обнаруживали в улавливающей ловушке. Это вещество концентрировали, чтобы удалить растворитель, используя 20-дюймовую колонку Vigreux с дистилляционной головкой и объединяли с первоначальным веществом. Полностью желаемый продукт дополнительно очищали дистилляцией, получая бесцветное масло (17,8 г) с точкой кипения 68-70 С при 10 мм рт. ст. Оптическое вращение: +212,8 при с=0,46 г на 100 мл CHCl3. Метиллитий в диэтиловом эфире (1,6 М, 218 мл, 349 ммоль) добавляли порциями в 10 мл в течение 20 мин к перемешиваемой суспензии йодида меди(I) (44,3 г, 233 ммоль) в диэтиловом эфире (350 мл), охлажденной до 0 С в атмосфере азота. Реакционную смесь перемешивали при 0 С в течение 30 мин и затем добавляли (R)-2-метил-2 Н-пиран-4 (3 Н)-он (13,5 г, 116 ммоль) в диэтиловом эфире (150 мл) по каплям в течение 30 мин. Холодную баню удаляли и реакционной смеси позволили нагреться до rt и перемешивали в течение 3 ч. Реакционную смесь медленно добавляли (в течение 5 мин) к перемешиваемому раствору насыщенного NH4Cl (водн.) (750 мл) и льда. Дополнительные диэтиловый эфир (-500 мл) и воду (-150 мл) добавляли и раствор, который содержал серый осадок, перемешивали в течение ночи. Слои разделяли и водный слой экстрагировали диэтиловым эфиром (500 мл) и DCM (500 мл). Объединенную органическую фазу высушивали (MgSO4), фильтровали и концентрировали (температуру бани роторного испарителя поддерживали равной температуре окружающей среды или ниже) до оранжевого масла. Неочищенное масло очищали с помощью Biotage Horizon (240 г SiO2, 1-4% диэтиловый эфир вDCM, колонку предварительно уравновешивали DCM), получая (2R,6R)-2,6-диметилдигидро-2 Н-пиран-4(3 Н)-он (13,4 г, 86% чистоты по данным 1 Н-ЯМР) в виде как светло-желтого масла. [Примечание: вещество аккуратно сконцентрировали и оно содержало 10 мас.% DCM по данным Н-ЯМР. Анализ 1H-ЯМР также показал, что вещество загрязнено на -2,5% TBDMSOH, перенесенным из предыдущей реакции, и на 1,6% (2R,6S)-2,6-диметилдигидро-2 Н-пиран-4 (3 Н)-оном]. Аналитические данные соответствовали данным, известным из литературы, и вещество использовали без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3)4,41-4,25 (м, 2 Н), 2,55 (двойной дублет дублетов, J=14,1, 4,8, 1,4 Гц, 2 Н), 2,24 (двойной дублет дублетов, J=14,1, 6,5, 1,5 Гц, 2 Н), 1,28 (д, J=6,5 Гц, 6 Н). Этил 2-2R,6R)-2,6-диметил-2H-пиран-4 (3 Н,5 Н,6 Н)илиден)-2-формамидоацетат Оксид меди (2.21 г, 15.4 ммоль) добавляли к перемешиваемому раствору этил 2-изоцианоацетата(12.7 мл, 116 ммоль) в диэтиловом эфире (300 мл) и суспензию энергично перемешивали в течение 10 мин. (2R,6R)-2,6-Диметилдигидро-2 Н-пиран-4 (3 Н)-он (15,2 г, 103 ммоль, 87%-ной чистоты с 13 мас.%DCM) в диэтиловом эфире (100 мл) добавляли в течение 15 мин и реакционную смесь перемешивали приRT в течение 2 ч. Реакционную смесь охлаждали до 0 С и обрабатывали 1 М KO-tBu (116 мл, 116 ммоль) в THF, добавленном порциями в 10 мл в течение 15 мин, и затем перемешивали при 0 С в течение 1 ч. Раствор уксусной кислоты (6,66 мл, 116 ммоль) в DCM (60 мл) добавляли, реакционной смеси позволили нагреться до RT и перемешивали в течение 2 ч. Неочищенную реакционную смесь распределяли между водой (300 мл) и диэтиловым эфиром (300 мл) и водный слой далее экстрагировали DCM (300 мл). Объединенную органическую фазу высушивали (MgSO4), фильтровали и концентрировали в темнокрасное масло, которое затвердевало под вакуумом. Неочищенное вещество очищали с помощью хроматографии на Biotage Horizon (300 г SiO2; загруженный на колонку с использованием минимального количества DCM; элюировали 40-60% EtOAc/гексаны, поддерживая 40% для четырех объемов колонки). Первые фракции желаемого продукта содержали также цис-стереоизомерную версию целевого продукта в качестве примеси. Эти загрязненные фракции объединяли, концентрировали, получая 8,9 г светложелтого масла, которое повторно очищали с помощью хроматографии на Biotage Horizon (240 г силикагеля; элюировали 40-65% EtOAc/гексаны, поддерживая 40% для пяти объемов колонки). Все фракции с чистым желаемым веществом от обеих очисток, что определяли посредством ТСХ, объединяли и концентрировали, получая этил 2-2R,6R)-2,6-диметил-2 Н-пиран-4 (3 Н, 5 Н, 6 Н)илиден)-2-формамидоацетат(18,8 г) в виде светло-желтого твердого вещества, загрязненного 1% (ВЭЖХ) цис-диметил-изомерным вариантом. 1 Н- и 13 С-ЯМР данные для чистого вещества представляют его как смесь ротамеров амида 2:1: 1 НЯМР (400 МГц, CDCl3)8,24 (д, J=1,3 Гц, 0,66 Н), 7,98 (д, J=11,5 Гц, 0,33 Н), 6,71 (ш. с., 0,66 Н), 6,56 (д,J=12,0 Гц, 0,33 Н), 4,32-4,00 (м, 4 Н), 3,05 (дд, J=14,3, 4,3 Гц, 0,33 Н), 2,92 (д, J=5,3 Гц, 1,33 Н), 2,82 (дд,J=14,3, 6,5 Гц, 0,33 Н), 2,62 (дд, J=13,9, 3,9 Гц, 0,33 Н), 2,45 (дд, J=14,1, 3,8 Гц, 0,66 Н), 2,24 (дд, J=13,8, 7,3 Гц, 0,33 Н), 2,14-2,04 (м, 0,66 Н), 1,37-1,29 (м, 3 Н), 1,28-1,18 (м, 6 Н). 13 С-ЯМР (101 МГц, CDCl3)164,5(0,33 С), 36,2 (0,33 С), 36,0 (0,66 С), 20,5 (0,66 С), 20,1 (0,33 С), 19,5 (0,33 С), 19,2 (0,66 С), 13,8 (с, 1 С). Время удерживания LC-MS 1,85 мин; m/z 242,45 (М+Н)+. Данные LC регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оборудованном колонкой Phenomenex-Luna 3u C18 2,050 мм, с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин, время захвата 1 мин и время анализа 5 мин. Состав растворителя А был 10% ацетонитрила/90% Н 2 О/0,1% трифторуксусной кислоты и состав растворителя В был 10% Н 2 О/90% ацетонитрила/0,1% трифторуксусной кислоты. Данные MS определяли, используя Азот барботировали через раствор этил 2-2R,6R)-2,6-диметил-2 Н-пиран-4 (3 Н,5 Н,6 Н)илиден)-2 формамидоацетата (17,0 г, 70,5 ммоль) в МеОН (480 мл) в течение 10 мин в сосуде для гидрирования Парра на 2,5 л. Затем добавляли (-)-1,2-бис-2S,5S)-2,5-диметилфосфолано)этан(циклооктадиен)родий(I) тетрафторборат (0,706 г, 1,27 ммоль) и сосуд с реагентом плотно закрывали, вакуумировали с продувкой азотом (4) и затем вакуумировали с продувкой водородом (4). Реакционный раствор встряхивали под давлением 60 фунт на кв.дюйм водорода в сечении 3 суток, удаляли из шейкера и концентрировали в темно-красное масло. Неочищенное масло очищали с помощью хроматографии на Biotage Horizon (300 г SiO2, 50-75% EtOAc/гексаны), получая (S)-этил 2-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2 формамидоацетат (17,4 г, содержащий 5,9 мас.% растворителей (EtOAc и DCM) по данным 1H-ЯМР) в виде светло-желтого вязкого масла. 1H-ЯМР данные указывают смесь двух ротамеров амидов 10:1. 1HЯМР (400 МГц, CDCl3)8,26 (с, 0,9 Н), 8,01 (д, J=11,8 Гц, 0,1 Н), 6,15 (д, J=8,5 Гц, 0,9 Н), 5,96-5,84 (м,0,1 Н), 4,68 (дд, J=9,0, 5,0 Гц, 0,9 Н), 4,33-4,17 (м, 3 Н), 3,87 (дд, J=10,2, 6,4 Гц, 0,1H), 3,75 (двойной квартет дублетов, J=11,5, 6,0, 2,0 Гц, 1 Н), 2,41-2,29 (м, 0,9 Н), 2,29-2,19 (м, 0,1H), 1,72-1,28 (м, 3 Н), 1,28-1,23 (м,6 Н), 1,17-1,10 (м, 3 Н), 1,09-0,96 (м, 1H). 13 С-ЯМР (101 МГц, CDCl3)170,6, 160,3, 68,1, 64,0, 61,4, 54,2,36,0, 33,1, 30,6, 21,9, 16,7, 13,9. Время удерживания LC-MS 1,64 мин; m/z 244,25 (М+Н)+. Данные LC регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оборудованном колонкой PhenomenexLuna 3u C18 2,050 мм, с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин, время захвата 1 мин и время анализа 5 мин. Состав растворителя А был 10% ацетонитрила/90% Н 2 О/0,1% трифторуксусной кислоты и состав растворителя В был 10% Н 2 О/90% ацетонитрила/0,1% трифторуксусной кислоты. Данные MS определяли, используя Micromass Platform for LC при ионизации электроспреем. Химическая формула C13H23NO5. Молекулярная масса 273,33. В колбе на 1 л, оборудованной холодильником, который был закрыт мембраной и выпускным отверстием с иглой,раствор(S)-этил 2-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2 формамидоацетата (17, 6 г, 72,3 ммоль) в этаноле (409 мл) и 1,5 н. HCl (водн.) (409 мл) нагревали в предварительно уравновешенной масляной ванне при 52 С в течение 7,5 ч. Летучий компонент удаляли под вакуумом, остаток азеотропно перегоняли с EtOH (250 мл) и сушили под вакуумом в течение ночи, получая белую пену. Белую пену растворяли в DCM (409 мл) и добавляли по каплям N,Nдиизопропилэтиламин (38,0 мл, 218 ммоль) в течение 10 мин с последующим добавлением по каплям метилхлоркарбоната (8,40 мл, 109 ммоль) также в течение 10 мин. Реакционную смесь перемешивали при температуре окружающей среды в течение 4,5 ч и затем останавливали аккуратно посредством 1 н.HCl (100 мл). Слои разделяли, органический слой промывали 1 н. HCl (50 мл) и затем с водным основным раствором (50 мл воды + 2 мл насыщенного NaHCO3). Органический слой сушили (MgSO4), фильтровали и концентрировали. Остаток очищали с помощью Biotage (300 г SiO2, образец загружали на колонку в минимальном количестве хлороформа и элюировали 30% EtOAc/гексаны), получая (S)-этил 2-2R,6R)2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-(метоксикарбониламино)ацетат (13,8 г) в виде вязкого желтого масла. 1H-ЯМР (400 МГц, CDCl3)5,23 (д, J=8,8 Гц, 1H), 4,34-4,15 (м, 4 Н), 3,81-3,72 (м, 1 Н), 3,70 (с, 3 Н),2,35-2,23 (м, 1H), 1,63-1,51 (м, 2 Н), 1,34-1,27 ("м" и "т" перекрывались, J=7,0 Гц, 1H и 3 Н), 1,25 (д, J=6,8 Гц, 3 Н), 1,14 (д, J=6,0 Гц, 3 Н), 1,11-0,99 (м, 1H). Время удерживания LC-MS 2,89 мин; m/z 296,23Phenomenex-серебром 3 колонка С 18 2,050 мм, используя датчик УЛЬТРАФИОЛЕТОВОГО ВИСАSPD-10AV в длине волны датчика 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин, время захвата 1 мин и время анализа 5 мин. Состав растворителя А был 10% МеОН/90% Н 2 О/0,1% трифторуксусной кислоты и состав растворителя В был 10% Н 2 О/90% МеОН/0,1% трифторуксусной кислоты. Данные MS определяли, используя Micromass Platform for LC при ионизации электроспреем. Дополнительное примечание: вышеупомянутый продукт далее очищали с помощью хроматографииSFC (180 мг/ввод пробы, с производительностью 7,2 г/ч) перед стадией гидролиза, чтобы удалить неидентифицированную примесь. Условия: колонка: ChiralPak AD-H 253 см, 5 мкм; температура колонки: 25 С; расход: 150 мл/мин; мобильная фаза: СО 2/МеОН=93/7; вводимый объем: 1,0 мл (180 мг/мл); тип ввода пробы: наложение вводов пробы (время ввода пробы/разделения (мин)=1,47/3,0); длина волны детектора: 220 нм; растворитель для образцов: СН 3 ОН/CH3CN=1:1 (об./об.). Гидроксид лития гидрат (8,51 г, 203 ммоль) в Н 2 О (270 мл) добавляли по каплям к перемешиваемому раствору(S)-этил 2-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-(метоксикарбониламино)ацетату (27,7 г, 101 ммоль) в THF (405 мл) в течение 65 мин, поддерживая внутреннюю температуру 5 С. Реакционную смесь перемешивали в ледяной ванне в течение 40 мин и затем в водяной ванне при RT в течение 5 ч. Реакционную смесь частично концентрировали, чтобы удалить THF, и затем распределяли между водой (250 мл) и DCM (250 мл). Водный слой доводили до рН 2 с помощью 1 н. HCl(205 мл) и затем экстрагировали изопропилацетатом (7250 мл). Объединенные экстракты промывали рассолом (1600 мл), сушили (MgSO4), фильтровали и концентрировали. Получающуюся вязкое масло обрабатывали с толуолом (100 мл) и эфиром (75 мл), упаривали и высушивали в вакууме, получая Сар 2(25,8 г с 0,33 экв. толуола) в виде белой пены. Хиральная чистота по Chiral SFC 99,9% (Chiralpak AD-H(250,46 см, 5 мкм) 5% EtOH в СО 2, 3 мл/мин, 35 С, 220 нм, 100 бар). Чистота по ВЭЖХ 95,6% (колонка:Sunfire C8 754,6 мм ID; 2,5 мкм; 40 С; мобильная фаза: А=вода с 0,1% муравьиная кислота; В=CH3CN с 0,1% муравьиная кислота). LC/MS (ES+) 228,1 (М+Н-Н 2 О)+. По данным 1 Н-ЯМР вещество содержало около 0,33 экв. толуола. 1H-ЯМР (400 МГц, CDCl3)5,29 (д, J=8,6 Гц, 1H), 4,46-4,25 (м, 2 Н), 3,82 (дд,J=9,7, 5,9 Гц, 1 Н), 3,74 (с, 3 Н), 2,46-2,30 (м, 1H), 1,76-1,55 (м, 2 Н), 1,51-1,35 (м, 1 Н), 1,29 (д, J=6,8 Гц, 3 Н),1,18 (д, J=6,2 Гц, 3 Н), 1,18-1,06 (м, 1 Н). 13 С-ЯМР (101 МГц, CDCl3)174,1, 156,7, 68,3, 64,4, 57,5, 52,2,35,8, 32,6, 30,3, 21,6, 16,7. Пример 1 Вышеупомянутое соединение синтезировали в соответствии с литературным протоколом (J. Med.Chem., 2006, 49, 3520) со следующими модификациями очистки: неочищенное вещество перекристаллизовывали из EtOAc/гексаны при температуре окружающей среды, получая соединение примера 1, стадия а в виде белых кристаллов. 1H-ЯМР (400 МГц, CDCl3)м.д. 4,32 (ш. м., 1H), 3,89 (ш. м., 1H), 2,40 (ш. м.,1H), 2,00 (м, 2 Н), 1,65 (м, 1 Н), 1,45 (с, 9 Н), 1,20 (д, J=5,6, 3 Н). LC/MS: анал. вычисл. для [M+Na]+C11H20NO4Na; найдено 252,21. Пример 1, стадия b. К смеси примера 1, стадия а (7,12 г, 31,1 ммоль) и 1,1'-(бифенил-4,4'-диил)-бис-(2-бромэтанона) (6,0 г, 15 ммоль) в ацетонитриле (100 мл) добавляли по каплям i-Pr2EtN (5,56 мл, 31,8 ммоль) и реакцию перемешивали при комнатной температуре в течение 3 ч. Летучий компонент удаляли в вакууме и остаток распределяли между CH2Cl2 (100 мл) и насыщенным водн. NaHCO3 (100 мл). Органический слой отделяли и промывали водой и рассолом, сушили (Na2SO4), фильтровали и концентрировали в вакууме, получая соединение примера 1, стадия b в виде белой пены (9,83 г), которая использовалась на следующей стадии без очистки. 1H-ЯМР (500 МГц, CDCl3)м.д. 8,01 (4 Н, т, J=7,32 Гц), 7,73 (4 Н, д, J=6,71 Гц), 5,18-5,66(160 мл) нагревали в пробирке под давлением при 140 С в течение 4 ч. Летучий компонент удаляли в вакууме и остаток распределяли между CH2Cl2 (140 мл) и водой (100 мл). Органический слой промывали насыщенным NaHCO3 (100 мл), сушили (Na2SO4) и концентрировали в вакууме. Полученное неочищенное вещество очищали с помощью Biotage (от 20 до 50% EtOAc/Нех; 300-граммовая колонка), получая соединение примера 1, стадия с в виде светло-коричневого твердого вещества (4,3 г). 1 Н-ЯМР (500 МГц,диметилсульфоксид-d6)м.д. 11,73 (2 Н, ш.с.), 7,83 (3 Н, д, J=7,63 Гц), 7,62-7,76 (5 H, м), 7,27-7,55 (2 Н, м),4,82 (2 Н, ш.с.), 3,89 (2 Н, ш.с.), 2,10 (6 Н, д, J=15,26 Гц), 1,58-1,89 (2 Н, м), 1,11-1,52 (24 Н, м). LC/MS: анал. вычисл. для [М+Н] + C38H49N6O4: 653,37; найдено 653,60. Альтернативная методика: смесь примера 1, стадия b (157 г, 227 ммоль), ацетат аммония (332 г,4310 ммоль) и имидазол (54,1 г, 795 ммоль) в толуоле (1,2 л) нагревали при 80 С в течение 1 ч, притом что пространство сверху заполняли азотом. Температуру повышали до 85 С и реакционную смесь перемешивали в течение 18 ч. Растворитель удаляли и остаток растворяли в DCM (2 л), промывали водой (1 л) и насыщенным NaHCO3 (3 л) и затем высушивали (MgSO4), фильтровали и концентрировали. Остаток растворяли в МеОН (4 л), концентрировали до 900 мл и оставляли при RT. Через 2 ч выпавшее твердое вещество собирали фильтрацией, промывали МеОН и высушивали, получая белое твердое вещество. Это вещество перекристаллизовывали из метанола (растворяли в 4 л, затем уменьшали до 0,9 л) и высушивали, получая соединение примера 1, стадия с в виде белого твердого вещества (99,5 г). Неочищенное соединение из маточного раствора очищали с помощью хроматографии на силикагеле (300 г SiO2, 2050% EtOAc/гексаны), получая дополнительное вещество в виде белого твердого вещества (13,0 г). Пример 1, стадия d 4 н. HCl в диоксане (8,23 мл, 32,9 ммоль) добавляли по каплям к раствору примера 1, стадия с (4,3 г,6,6 ммоль) в CH2Cl2 (100 мл) и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Удаление летучего компонента в вакууме давало HCl-соль примера 1, стадия d в виде желтого твердого вещества (3,6 г). 1H-ЯМР (400 МГц, диметилсульфоксид-d6)м.д. 10,55 (2 Н, ш.с.), 9,73 (2 Н, ш.с.),8,12-8,26 (2 Н, м), 8,03 (4 Н, д, J=8,03 Гц), 7,92 (4 Н, д, J=6,02 Гц), 5,07 (2 Н, м, J=7,53 Гц), 3,82 (2 Н, ш.с.),2,53-2,65 (4 Н, м), 2,27 (2 Н, дд, J=12,42, 6,40 Гц), 1,85-1,99 (2 Н, м), 1,45 (6 Н, д, J=6,27 Гц). LC/MS: анал. вычисл. для [М+Н]+ C28H33N6: 453,28; найдено 453,21. Степень чистоты вышеупомянутого незащищенного вещества могла быть увеличена посредством использования следующей методики перекристаллизации: 2-пропанол (242 мл) добавляли через дополнительную капельную воронку в течение 15 мин к раствору примера 1, стадия d (60,5 г, чистота по ВЭЖХ 98,9%) в воде (121 мл), перемешиваемому при 60 С, поддерживая внутреннюю температуру между 50 и 60 С. Раствор перемешивали в течение дополнительных 5 мин, колбонагреватель удаляли и смесь в течение ночи перемешивали при температуре окружающей среды. Выпавшее твердое вещество собирали фильтрацией, промывали 2-пропанолом и высушивали при помощи централизованной вакуумной системы в течение ночи, что приводило к желтому твердому веществу (54,4 г) с чистотой по ВЭЖХ 99,7% (колонка: ВЕН С 18 150 мм (L)2,1 мм (ID (внутренний диаметр, 1,7 мкм, мобильная фаза: А: 0,05% TFA в воде; В: 0,05% TFA в ACN). KF (содержание воды по методу Карла Фишера) 15,3 мас.%HATU (1,60 г, 4,21 ммоль) добавляли к раствору HCl-соли соединения примера 1, стадия d (1,2 г,2,0 ммоль), Сар 1 (1,01 г, 4,11 ммоль) и DIEA (2,1 мл, 12 ммоль) в DMF (20 мл) и получающийся желтый раствор перемешивали при RT в течение 3 ч. Смесь разбавляли EtOAc (75 мл) и промывали водой (100 мл). Водный слой еще раз экстрагировали EtOAc (75 мл) и объединенные органические слои промывали 50% от насыщенного водного раствора NaHCO3 (100 мл), водой (100 мл) и рассолом (100 мл). Его затем высушивали (MgSO4), фильтровали и концентрировали. Остающийся остаток разбавляли СН 3 ОН и подвергали очистке посредством ВЭЖХ на обращенной фазе: растворитель А: 5% MeCN/95% воды/10 нМNH4OAc; растворитель В: 95% MeCN/5% воды/10 нМ NH4OAc; колонка: Simfire Prep C18 50300 мм 10 мкм; длина волны: 220 нм; расход: 150 мл/мин; градиент: 0% В до 70% В в течение 25 мин с 5 мин время захвата. Концентрирование под вакуумом давало диметил(4,4'-бифенилдиил-бис-(1H-имидазол-4,2 диил 2S,5S)-5-метил-2,1-пирролидиндиил)1S)-1-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-оксо-2,1-этандиилбискарбамат (пример 1) в виде грязно-белой пены (1,2 г). Кристаллизация изEtOAc/петролейного эфира давала аморфное грязно-белое твердое вещество. 1H-ЯМР (500 МГц, диметилсульфоксид-d6) (для соли с TFA)7,92 (ш. с., 8 Н), 7,59 (д, J=7,9 Гц, 2 Н), 5,03 (т, J=8,5 Гц, 2 Н), 4,694,60 (м, 2 Н), 4,00 (т, J=8,4 Гц, 2 Н), 3,55 (с, 6 Н), 3,36 (ш. с., 1H), 3,33-3,26 (м, 2 Н), 3,21 (ш. с., 2 Н), 2,37 (ш. с., 1H), 2,26 (д, J=11,3 Гц, 1H), 1,95-1,81 (м, 3 Н), 1,65 (д, J=10,7 Гц, 2 Н), 1,48 (д, J=6,4 Гц, 6 Н), 1,28-1,23(м, 4 Н), 1,10-0,99 (м, 16 Н), 0,90-0,75 (м, 4 Н). LC/MS: анал. вычисл. для [М+Н]+ C50H67N8O8: 907,51; найдено 907,8. Rt=3,99 мин 95% индекс гомогенности при следующих условиях LC - колонка: PhenomenexLuna 2,050 мм 3 мкм; начальный % В=0; заключительный % В=100; время градиента=10 мин; расход=4 мл/мин; длина волны=220; растворитель А=Н 2 О:ACN 95%:5% 10 мМ ацетата аммония; растворитель В=Н 2 О:ACN 5%:95% 10 мМ ацетата аммония. Пример 2HATU (63,6 мг, 0,167 ммоль) добавляли к перемешиваемому раствору (S)-2-2R,6R)-2,6 диметилтетрагидро-2 Н-пиран-4-ил)-2-(метоксикарбониламино)уксусной кислоты (Сар 2) (41 мг, 0,17 ммоль) и HCl-соли соединения примера 1, стадия d (45,5 мг, 0,076 ммоль) в DMF (0,9 мл) и DIPEA (0,11 мл, 0,61 ммоль). Реакционную смесь перемешивали при RT в течение 4 ч и затем концентрировали под струей азота ON. Остаток растворяли в МеОН, фильтровали и очищали с помощью препаративной ВЭЖХ Phenomenex Luna С 18(2) 10030 мм, 10 мкм; буфер МеОН/вода с TFA), получая TFA-соль диметил(4,4'-бифенилдиил-бис-(1H-имидазол-4,2-диил 2S,5S)-5-метан-2,1-пирролидиндиил) 1S)-12R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-оксо-2,1-этандиилбискарбамата (пример 2) (62,5 мг) в виде светло-желтого твердого вещества. 1H-ЯМР указывает на присутствие смеси ротамеров; для главного ротамера: 1H-ЯМР (400 МГц, MeOH-d4)8,02-7,93 (м, 3 Н), 7,88 (ш. с., 7 Н), 5,18 (дд, J=10,7, 7,2 Гц,2 Н), 4,77 (квинтет, J=6,6 Гц, 2 Н), 4,27-4,17 (м, 2 Н), 4,15 (д, J=9,3 Гц, 2 Н), 3,72-3,66 (м, 2 Н), 3,67 (с, 6 Н),2,72-2,50 (м, 2 Н), 2,48-2,16 (м, 6 Н), 1,99 (дд, J=12,2, 5,6 Гц, 2 Н), 1,76-1,59 (м, 2 Н), 1,57 (д, J=6,5 Гц, 6 Н),1,52-1,40 (м, 2 Н), 1,30 (дд, J=9,7, 6,7 Гц, 2 Н), 1,22 (д, J=6,8 Гц, 6 Н), 1,06 (д, J=6,0 Гц, 6 Н), 0,97 (кажущийся кв, J=12,0 Гц, 2 Н). Время удерживания LC-MS 3,84 мин; m/z 905,38 [М-Н]-. Данные LC регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оборудованном колонкой Phenomenex-Luna 3u C18 2,050 мм, с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин, время захвата 1 мин и время анализа 5 мин, где состав растворителя А был 5% МеОН/95% Н 2 О/10 мМ ацетат аммония и состав растворителя В был 5% Н 2 О/95% МеОН/10 мМ ацетат аммония. Данные MS определяли, используя Micromass Platform for LC при ионизации электроспреем. Альтернативно, пример 2 мог быть осуществлен следующим образом. Смесь (S)-2-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-(метоксикарбониламино)уксусной кислоты (Сар 2) (25,1 г, 91,0 ммоль), 2-гидроксипиридин-1-оксида (10,12 г, 91 ммоль) и EDC (19,0 г, 99,0 ммоль) в диметилсульфоксиде (400 мл) перемешивали при RT в течение 1 ч. К этому раствору добавляли соединение примера 1, стадия d (HCl-соль; 29,2 г, 41,4 ммоль) и получающийся желтый раствор охлаждали до 10 С в бане вода/лед. Затем добавляли к реакционной смеси i-Pr2EtN (26,7 г, 207 ммоль) и после завершения добавления ледяную баню удаляли. Реакционную смесь перемешивали при температуре окружающей среды в течение 25,5 ч перед тем, как реакцию останавливали, выливая в смесь льда (1600 г) и воды (400 мл). Смесь перемешивали при температуре окружающей среды в течение 3 ч. Белое твердое вещество собирали на воронке Бюхнера посредством фильтрации и затем высушивали при помощи централизованной вакуумной системы в потоке азота, проходящего сверху фильтровальной воронки в течение ночи, получая влажное твердое вещество (124 г). Влажное твердое вещество растворяли в DCM (500 мл), промывали водой (3250 мл), сушили (MgSO4), фильтровали и концентрировали, получая светлокоричневое твердое вещество (39,5 г). Это вещество объединяли с подобным веществом от другой партии (общее количество 44,4 г) и очищали хроматографией, используя устройство ISCO (силикагель 2 х 330 г, 0-30% MeOH/DCM), получая соединение примера 2 (38,1 г) в виде коричневого твердого вещества с чистотой по ВЭЖХ 98,6%. Вещество обесцвечивали следующим образом: 43,1 г вещества растворяли вEtOH (500 мл) и затем обрабатывали активированным углем (8,6 г). Смесь нагревали при 50 С в течение 1 ч, древесный уголь удаляли фильтрованием (3 слоя фильтровальной бумаги) и отфильтрованный осадок промывали дополнительным EtOH (300 мл). Фильтрат упаривали досуха и сушили в вакууме в бане с теплой водой, получая грязно-белое твердое вещество (43,0 г). Вещество могло быть очищено дополнительно, используя протокол SFC. Условия очистки с помощью SFC (сверхкритическая флюидная хроматография): производительность: 1,2 г/ч; количество на инъекцию 30 мг; колонка: Princeton CN 253 см, 5 мкм; температура колонки: 45 С; расход: 200 мл/мин; мобильная фаза: СО 2/[MeOH/DCM=1:1 (об./об.)]=80/20; вводимый объем: 2,5 мл (12 мг/мл); тип ввода пробы: наложение вводов пробы (время ввода пробы/разделения (мин)=1,5/3,2); длина волны детектора: 316 нм; растворитель для образцов: СН 3 ОН/DCM=1:1 (об./об.). Пример 3(24,4 мл, 140 ммоль) и реакционную смесь перемешивали при RT в течение 12 ч. Реакционную смесь затем разбавляли EtOAc (150 мл), промывали водой (3250 мл) и рассолом (150 мл), высушивали надNa2SO4, фильтровали и концентрировали в вакууме, получая неочищенное соединение примера 3, стадия а (9,5 г), который передавали на следующую стадию без очистки. 1 Н-ЯМР (CDCl3, =7,2 м.д., 400 МГц):7,37 (д, J=1,6 Гц, 1H), 7,34 (дд, J=8,0, 1,6 Гц, 1H), 7,14 (д, J=8,0 Гц, 1H), 3,47 (с, 3 Н), 3,30 (с, 3 Н), 2,31 (с,3 Н). LC/MS: анал. вычисл. для [M+H]+ C10H1381BrNO2: 260,01; найдено 260,0. Пример 3, стадия b Соединение примера 3, стадия а (9,5 г, 36,8 ммоль) растворяли в диэтиловом эфире (150 мл) и охлаждали до 0 С. Затем добавляли по каплям в течение 10 мин метилмагнийиодид (3,0 М в диэтиловом эфире, 24,54 мл, 73,6 ммоль). Реакционную смесь перемешивали в течение 6 ч при 40 С и затем доводили до комнатной температуры и перемешивали в течение 12 ч. Реакционную смесь охлаждали до 0 С,реакцию останавливали льдом и затем 1,5 н. HCl (50 мл). Органический слой отделяли и водный слой экстрагировали метил-трет-бутиловым эфиром (2100 мл), сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью флэш-хроматографии (силикагель,60-120, EtOAc: петролейный эфир, 2:98), получая соединение примера 3, стадии b (6,25 г) в виде бледножелтой жидкости. 1 Н ЯМР (CDCl3, =7,26 м.д., 400 МГц):7,55 (д, J=8,0 Гц, 1H), 7,41 (с, 1 Н), 7,40 (д,J=8,0 Гц, 1H), 2,55 (с, 3 Н), 2,50 (с, 3 Н). 4-Ацетилфенилборную кислоту (5,39 г, 32,9 ммоль) добавляли к закупоренной пробирке, содержащей соединение примера 3, стадия b (7,0 г, 32,9 ммоль) в МеОН (75,0 мл), и реакционную смесь продували азотом в течение 10 мин. Затем добавляли K2CO3 (9,08 г, 65,7 ммоль) с последующим добавлениемPd(Ph3P)4 (1,139 г, 0,986 ммоль) и реакционную смесь продували азотом в течение дополнительных 10 мин. Реакционную смесь нагревали до 75 С в течение 12 ч. Затем реакционную смесь концентрировали при пониженном давлении, остаток разбавляли EtOAc (100 мл) и промывали водой (2100 мл), рассолом(50 мл), высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством combiflash (Silicycle, SiO2, 10-15% EtOAc/петролейный эфир), получая соединение примера 3, стадия с (6,5 г) в виде белого твердого вещества. 1 Н-ЯМР (CDCl3, =7,26 м.д., 400 МГц):8,06-8,04 (м, 2 Н), 7,81 (д, J=8,0 Гц, 1H), 7,72-7,70 (м, 2 Н), 7,54-7,49 (м, 2 Н), 2,65 (с, 3 Н), 2,63 (с, 6 Н). Пример 3, стадия d. Бром (1,12 мл, 21,8 ммоль) (разбавленный в 10 мл диоксана) медленно добавляли (в течение 10 мин) к раствору примера 3, стадия с (2,75 г, 10,90 ммоль) в диоксане (50 мл) при 10 С и смесь перемешивали при RT в течение 2 ч. Реакцию останавливали 10% NaHCO3 (25 мл) и экстрагировали DCM (50 мл). Органическую фазу высушивали над Na2SO4 и концентрировали при пониженном давлении, получая неочищенное соединение примера 3, стадия d (5,0 г), которое использовалось без очистки на следующей стадии. LC/MS: анал. вычисл. для [М+Н]+ С 17 Н 1579/81Br2O2: 410,94; найдено 411,0. Пример 3, стадия е К раствору неочищенного соединения примера 3, стадия d (5,1 г, 12 ммоль) в ацетонитриле (75 мл) добавляли (2S,5S)-1-(трет-бутоксикарбонил)-5-метилпирролидин-2-карбоновую кислоту (пример 1, стадия а) (5,70 г, 24,9 ммоль) с последующим добавлением DIPEA (8,69 мл, 49,7 ммоль) при 0 С. Через 10 мин температуру поднимали до RT и перемешивали в течение 2 ч. Затем реакционную смесь разбавлялиEtOAc (100 мл) и промывали 10%-ным NaHCO3 (50 мл), рассолом (50 мл), высушивали над Na2SO4,фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством combiflash(Silicycle, SiO2, 25-30% EtOAc/петролейный эфир), получая соединение примера 3, стадия е (5,8 г) в виде бледно-желтого масла. 1 Н-ЯМР (CDCl3, =7,26 м.д., 400 МГц):8,00 (кажущийся широкий дублет, 2 Н),7,71 (кажущийся дублет, 3 Н), 7,53-7,51 (м, 2 Н), 5,61-5,34 (м, 2 Н), 5,29-5,04 (м, 2 Н), 4,51-4,36 (м, 2 Н),4,09-3,91 (м, 2 Н), 2,59 (с, 3 Н), 2,35-2,21 (м, 4 Н), 2,15-2,04 (м, 2 Н), 1,80-1,63 (м, 2 Н), 1,47-1,44 (с, 18 Н),1,35-1,27 (м, 6 Н). LC/MS: анал. вычисл. для [М-Н]- C39H49N2O10: 705,35; найдено 705,30. Пример 3, стадия f К раствору соединения примера 3, стадия е (5,6 г, 7,92 ммоль) в ксилолах (75 мл) добавлялиNH4OAc (12,21 г, 158 ммоль) и реакционную смесь продували азотом в течение 10 мин. После нагревания в течение 18 ч при 130 С реакционную смесь охлаждали до комнатной температуры и летучие компоненты удаляли при пониженном давлении. Затем реакционную смесь разбавляли EtOAc (100 мл) и промывали 10%-ным NaHCO3 (50 мл), рассолом (50 мл), высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством combiflash (Redi Sep, С-18 column, 30-40% ацетонитрил:10 мМ бикарбонат аммония), получая соединение примера 3, стадия f (2,3 г) в виде бледно-желтого твердого вещества. 1 Н-ЯМР (диметилсульфоксид-d6, =2,50 м.д., 400 МГц):12,27/12,0/11,77/11,71 (с, 2 Н), 7,92-7,63 (м, 5 Н), 7,58-7,47 (м, 3 Н), 7,24 (ш. с., 1H), 4,90-4,75 (м, 2 Н), 3,923,84 (м, 2 Н), 2,54 (с, 3 Н), 2,20-2,01 (м, 6 Н), 1,73-1,65 (м, 2 Н), 1,48-1,12 (м, 24 Н). LC/MS: анал. вычисл. для К раствору соединения примера 3, стадия g (1,55 г, 2,32 ммоль) в МеОН (10 мл) добавляли вHCl/МеОН (4 н., 58,1 мл) и перемешивали при комнатной температуре в течение 2 ч. Летучие компоненты удаляли в вакууме и остаток совместно упаривали с сухим DCM (325 мл). Получающееся твердое вещество выдерживали в высоком вакууме, получая HCl-соль соединения примера 3, стадия g (1,3 г) в виде бледно-желтого твердого вещества. 1H-ЯМР (MeOD, =3,34 м.д., 400 МГц):8,06 (ш. с., 1 Н), 7,98HATU (60,5 мг, 0,159 ммоль) добавляли к перемешиваемому раствору (S)-2-2R,6R)-2,6 диметилтетрагидро-2 Н-пиран-4-ил)-2-(метоксикарбониламино)уксусной кислоте (Сар 2) (39 мг, 0,16 ммоль) и HCl-соли соединения примера 3, стадия g (44,3 мг, 0,072 ммоль) в DMF (0,9 мл) и DIPEA (0,10 мл, 0,58 ммоль). Реакционную смесь перемешивали при RT в течение 2 ч и в течение ночи концентрировали под струей азота. Остаток разбавляли МеОН, фильтровали и очищали посредством препаративной ВЭЖХ (Phenomenex Luna С 18(2) 10030 мм, 10 мкм; буфер МеОН/вода с TFA), получая TFA-соль диметил 3-метил-4,4'-бифенилдиил)-бис-(1 Н-имидазол-4,2-диил 2S,5S)-5-метил-2,1-пирролидиндиил) 1S)-1-2R,6R)-2,6-диметилтетрагидро-2 Н-пиран-4-ил)-2-оксо-2,1-этандиилбискарбамата (пример 3)(кажущийся квартет, J=12,2 Гц, 2 Н). Время удерживания LC-MS 4,050 мин; m/z 461,31 [1/2 М+Н]+. Данные LC регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оборудованном колонкой Phenomenex-Luna 3u C18 2,050 мм, с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: расход 0,8 мл/мин, градиент от 100% растворителя А/0% растворителя В до 0% растворителя А/100% растворителя В, время градиента 4 мин,время захвата 1 мин и время анализа 5 мин, где состав растворителя А был 5% МеОН/95% Н 2 О/10 мМ ацетат аммония и состав растворителя В был 5% Н 2 О/95% МеОН/10 мМ ацетат аммония. Данные MS определяли, используя Micromass Platform for LC при ионизации электроспреем. Пример 4 К раствору 1-(4-бром-2-фторфенил)этанона (5,0 г, 23 ммоль) в диоксане (150 мл) и эфире (150 мл) в бане с водой и льдом при 0 С добавляли по каплям бром (1,18 мл, 23,0 ммоль). Реакцию перемешивали в течение 1 ч, позволяли нагреваться до RT и перемешивали в течение 16 ч. Смесь распределяли междуEtOAc (50 мл) и насыщенным NaHCO3 (50 мл), органический слой промывали водой и высушивали надNa2SO4. Летучий компонент упаривали в вакууме и твердое вещество высушивали под вакуумом в течение ночи, получая соединение примера 4, стадия а (6,94 г) в виде белого твердого вещества. 1H-ЯМР К раствору соединения примера 4, стадия а (2,58 г, 8,72 ммоль) и (2S,5S)-1-(трет-бутоксикарбонил)5-метилпирролидин-2-карбоновой кислоты (2,00 г, 8,72 ммоль) в ацетонитриле (50 мл) добавляли DIEA(2,285 мл, 13,08 ммоль) и смесь перемешивали при комнатной температуре в течение 64 ч. Растворитель удаляли в вакууме и остаток распределяли между EtOAc (40 мл) и водой (30 мл). Органический слой промывали насыщенным NaHCO3 и рассолом, сушили над Na2SO4 и упаривали в вакууме, получая соединение примера 4, стадия b (3,8 г) в виде желтого твердого вещества. 1H-ЯМР (CDCl3, 400 МГц): 7,87 К пробирке под давлением, содержащей раствор примера 4, стадия b (3,8 г, 8,6 ммоль), в ксилолах(40 мл) добавляли ацетат аммония (6,59 г, 86 ммоль) и сосуд с реагентом закрывали и нагревали при 140 С в течение 6 ч. Летучий компонент упаривали в вакууме и остаток распределяли между DCM (80 мл) и водой (50 мл). Органический слой отделяли и промывали насыщенным NaHCO3 и высушивали надNa2SO4. Удаление растворителя в вакууме приводило к красному маслу, которое очищали с помощью флэш-хроматографии (0-40% EtOAc/гексан), что приводило к соединению примера 4, стадия с (2,3 г) в виде коричневого твердого вещества. 1 Н-ЯМР (диметилсульфоксид-d6, =2,5 м.д., 400 МГц): 7,98 (кажущийся триплет, J=8,4 Гц, 1H), 7,65 (дд, J=11, 1,9 Гц, 1H), 7,45 (дд, J=8,3, 2, 1H), 7,36 (м, 1H), 4,85 (м, 1H),3,90 (м, 1H), 2,15-2,07 (м, 3 Н), 1,73 (м, 1H), 1,40-1,17 (м, 12 Н). LC/MS: анал вычисл. для [M+Na]+DIEA (1,524 мл, 8,72 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель удаляли в вакууме и остаток распределяли между EtOAc (40 мл) и водой (30 мл). Органическую фазу промывали насыщенным NaHCO3 и рассолом и высушивали над Na2SO4. Удаление летучего компонента в вакууме приводило к соединению примера 4, стадия d (1,74 г) в виде светло-желтого твердого вещества, которое использовалось без дальнейшей очистки. 1 Н-ЯМР (диметилсульфоксид-d6, =2,5 м.д.,400 МГц): 7,95-7,90 (м, 2 Н), 7,81 (м, 1 Н), 7,79 (м, 1H), 5,63-5,44 (м, 2 Н), 4,36 (м, 1H), 3,99 (м, 1H), 2,27 (м,1 Н), 2,09 (м, 2 Н), 1,63 (м, 1H), 1,41-1,37 (два синглета, 9 Н), 1,19 (м, 3 Н). LC/MS: анал. вычисл. для К пробирке под давлением, содержащей раствор примера 4, стадия d (3,4 г, 8,0 ммоль), в ксилолах(40 мл) добавляли ацетат аммония (6,15 г, 80 ммоль) и смесь нагревали в 140 С в течение 6 ч. Летучий компонент удаляли в вакууме, остаток распределяли осторожно между DCM (60 мл) и насыщенным NaHCO3 (30 мл) и органический слой отделяли и высушивали над Na2SO4. Растворитель удаляли в вакууме,что приводило к красному твердому веществу, которое очищали с помощью флэш-хроматографии (550% EtOAc/гексан), получая соединение примера 4, стадия е (2,65 г) в виде светло-коричневого твердого вещества. 1H-ЯМР (диметилсульфоксид-d6, =2,5 м.д., 400 МГц): 7,73-7,71 (м, 2 Н), 7,59-7,50 (м, 3 Н), 4,80 К раствору примера 4, стадия е (2,64 г, 6,50 ммоль) и 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2 диоксаборолана) (3,30 г, 13,0 ммоль) в диоксане (40 мл) добавляли ацетат калия (1,594 г, 16,24 ммоль). Смесь дегазировали посредством барботирования азота в течение 10 мин, добавляли Pd(Ph3P)4 (0,375 г,0,325 ммоль) и дегазацию продолжали в течение еще 15 мин. Сосуд с реагентом затем плотно закрывали и нагревали при 80 С в течение 16 ч. Летучий компонент упаривали в вакууме и остаток распределяли между DCM (100 мл) и насыщенным наполовину NaHCO3 (50 мл). Органический слой отделяли, высушивали над Na2SO4 и упаривали в вакууме, получая неочищенное красное масло, которое очищали с помощью флэш-хроматографии (10-90% EtOAc/гексаны). Соединение примера 4, стадия f (2,7g) получали в виде желтой пены. 1H-ЯМР (диметилсульфоксид-d6, =2,5 м.д., 400 МГц): 7,77 (д, J=8,3 Гц, 2 Н), 7,64-7,53
МПК / Метки
МПК: A61K 31/4178, A61P 31/14, C07D 405/14
Метки: гепатита, вируса, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-21538-ingibitory-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы вируса гепатита с</a>
Ингибиторы вируса гепатита с

Номер патента: 21194
Опубликовано: 30.04.2015
Авторы: Пак Шон К., Пател Бхарат П., Натали Джр., Тимонко Стивен, Белема Маконен, Кеннет Дж.
МПК: C07D 209/54
Метки: ингибиторы, вируса, гепатита
Формула / Реферат:
1. Способ получения соединения формулы (III)или его фармацевтически приемлемой соли,где R4 и R5 независимо выбраны из атома водорода, (С1-С6)алкила, (C6-C10)арила, (С6-С10)арил(С1-С6)алкила, гетероциклила и гетероциклил(С1-С2)алкила, где гетероциклил представляет собой 5-6-членный гетероциклил, включающий 1-2 гетероатома, выбранных из N, S и О,включающий обработку соединения формулы (IV)ацетатом аммония в присутствии основания.2. Способ по п.1,...
Ингибиторы вируса гепатита с

Номер патента: 18793
Опубликовано: 30.10.2013
Авторы: Белема Маконен, Кэдоу Джон Ф.
МПК: A61K 31/4178, A61K 31/439, A61P 31/14...
Метки: вируса, ингибиторы, гепатита
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, где L выбран из связи,R1 и R2 представляют собойили R1 представляет собойи R2 выбран изгде обозначает точку присоединения к исходной молекуле;R3 и R4 независимо выбраны из водорода и гало;каждый R5 независимо выбран из водорода;каждый R6 независимо выбран из алкила;R6a представляет собой алкил, где алкил необязательно может образовывать конденсированное 3-членное кольцо со смежным...
Ингибиторы протеазы вируса гепатита с

Номер патента: 20235
Опубликовано: 30.09.2014
Авторы: Линь Чу-Чун, Лю Йо-Чин, Кинг Чи-Син Ричард, Лю Чэнь-Фу, Ли Куан-Юань, Чэнь Жун-Цзиунн, Ло Пинь, Чэнь Чих-Мин
МПК: A61P 31/12, C07D 207/16, C07D 207/08...
Метки: вируса, гепатита, ингибиторы, протеазы
Формула / Реферат:
1. Соединение формулы (I)гдеR1 является С3-10циклоалкилом;R2 является C1-6алкилом или С3-10циклоалкилом;U является -NHSO2-;W является -(СН2)m-, -О(СН2)n- или -SO(CH2)n-,m равно 2 иn равно 1;X представляет собой -О-;Y представляет собойгдекаждый из V и Т является -N-;R представляет собой арил или гетероарил икаждый из А1 и А2 независимо являются арилом или гетероарилом, каждый из которых необязательно замещен галогеном, С1-6алкилом или...
Ингибиторы вируса гепатита с

Номер патента: 20815
Опубликовано: 30.01.2015
Авторы: Лопез Омар Д., Смит Майкл Дж., Су Бао-Нин, Сейнт Лорен Денис Р., Ромине Джефри Ли, Минвелл Николас А., Ван Гань, Хевавасам Пиясена, Белема Маконен, Чэнь Ци, Истер Джон А., Ян Чжун, Бендер Джон А., Нгуен Ван Н., Сюй Ниннин
МПК: C07D 405/14, C07D 403/14
Метки: гепатита, вируса, ингибиторы
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеn равно 0,1 или 2;X выбирают из водорода, циано, галогена циклопропила, циклопропилметила, 1-метилциклопропила, аллила, 1-метилпиразол-5-ила, 1-метилимидазол-5-ила и изоксазол-4-ила;R1 представляет собой водород;R2 выбирают из водорода, циклопропила и галогена; илиR1 и R2 вместе образуют -CH=CH- или -CH=CCl-;при условии, что по меньшей мере один из X и R2 не обозначает...
Ингибиторы вируса гепатита с (hcv)

Номер патента: 15415
Опубликовано: 31.08.2011
Авторы: Линк Джон О., Венкатарамани Чандрэсикар, Граупе Михаель
МПК: A61K 31/00, A61K 47/00, A61K 31/74...
Метки: гепатита, вируса, ингибиторы, hcv
Формула / Реферат:
1. Соединение формулы (I)где Е представляет собой -COCONHR6, где R6представляет собой водород, алкил, циклоалкил, аралкил или гетероаралкил, где ароматическое кольцо необязательно замещено одним или двумя галоидами;R1 представляет собой алкил, циклоалкилалкил, где алифатические или алициклические группы в R1необязательно замещены одним или двумя радикалами Rb, которые независимо выбирают из гидрокси, алкокси, арилокси, гетероарилокси, алкилтио,...
Предыдущий патент: Аминозамещенные производные бисфенилпентановой кислоты в качестве ингибиторов nep
Следующий патент: Антитело к склеростину и его применение
Случайный патент: Состав