Ингибиторы akt и фармацевтические составы, их содержащие
Номер патента: 20151
Опубликовано: 30.09.2014
Авторы: Беркхолдер Тимоти Пол, Клэйтон Джошуа Райан, Ремпала Марк Эдвард, Генри Кеннет Джеймс Джуниор, Сойер Джейсон Скотт, Эгген Мариджин, Пэй Хуасин, Джонс Дидре Мичелл, Бейт Дуглас Вейд, Партасарати Сараванан





























Формула / Реферат
1. Соединение формулы
 где А представляет собой
где А представляет собой или
или
R1 представляет собой СН3, СН2СН3 или CF3;
R2 представляет собой Н, CF3, CH2CF3, CH2CH2CF3, C1-C4-алкил, C3-C6-циклоалкил, CN, Cl, Br, СН=СН2, СН2СН2ОСН3, С(СН3)2СН2ОСН3 или тетрагидропиран-4-ил, где C3-C6-циклоалкил необязательно замещен метилом в положении 1, а тетрагидропиран-4-ил необязательно замещен метилом в положении 4;
R3 представляет собой Н или
R2 и R3, оба, представляют собой Cl;
R4 представляет собой Н;
R5 представляет собой СН3, С(СН3)3, СН(СН3)2, циклобутил, циклопентил, СН2-циклопропил, С(СН3)2СН2СН3 или тетрагидропиран-4-ил, или
R4 и R5, оба, представляют собой СН3, или
R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин, необязательно замещенный гидроксигруппой в положении 3, или азетидин,
или фармацевтически приемлемая соль указанного соединения.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где А представляет собой

3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль, где R1 представляет собой СН3 или CF3.
4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, где R2 представляет собой CF3, CH2CF3, CH2CH2CF3, СН2СН3, (СН2)2СН3, циклопропил, Br, СН2СН2ОСН3 или тетрагидропиран-4-ил и R3 представляет собой Н.
5. Соединение по п.4 или его фармацевтически приемлемая соль, где R2 представляет собой CH2CF3, CH2CH2CF3 или СН2СН3 и R3 представляет собой Н.
6. Соединение по любому из пп.1-5 или его фармацевтически приемлемая соль, где R4 представляет собой Н; R5 представляет собой C(СН3)3 или R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин или азетидин.
7. Соединение по п.6 или его фармацевтически приемлемая соль, где R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин или азетидин.
8. Соединение по п.1, выбранное из
(R)-5-метил-4-(4-(1-(2-(пирролидин-1-ил)этил)-4-(3,3,3-трифторпропил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5,6-дигидропиридо[2,3-d]пиримидин-7(8Н)-она;
(R)-4-(4-(4-этил-1-(2-(пирролидин-1-ил)этил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-метил-5,6-дигидропиридо[2,3-d]пиримидин-7(8Н)-она;
(R)-4-(4-(1-(2-(азетидин-1-ил)этил)-4-(2,2,2-трифторэтил)-1Н-имидазол-2-ил)пиперидин-1-ил)-5-(трифторметил)-5,6-дигидропиридо[2,3-d]пиримидин-7(8Н)-она
или их фармацевтически приемлемых солей.
9. Фармацевтическая композиция, обладающая способностью ингибировать AKT, содержащая эффективное количество соединения по любому из пп.1-8 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, разбавитель или наполнитель.
10. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли для лечения пролиферативных нарушений.
11. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли для лечения рака легких, рака груди или глиобластомы.
12. Фармацевтическая композиция для применения для лечения пролиферативных нарушений, содержащая соединение по любому из пп.1-8 или его фармацевтически приемлемую соль.
13. Фармацевтическая композиция для лечения рака легких, рака груди или глиобластомы, содержащая соединение по любому из пп.1-8 или его фармацевтически приемлемую соль.
Текст
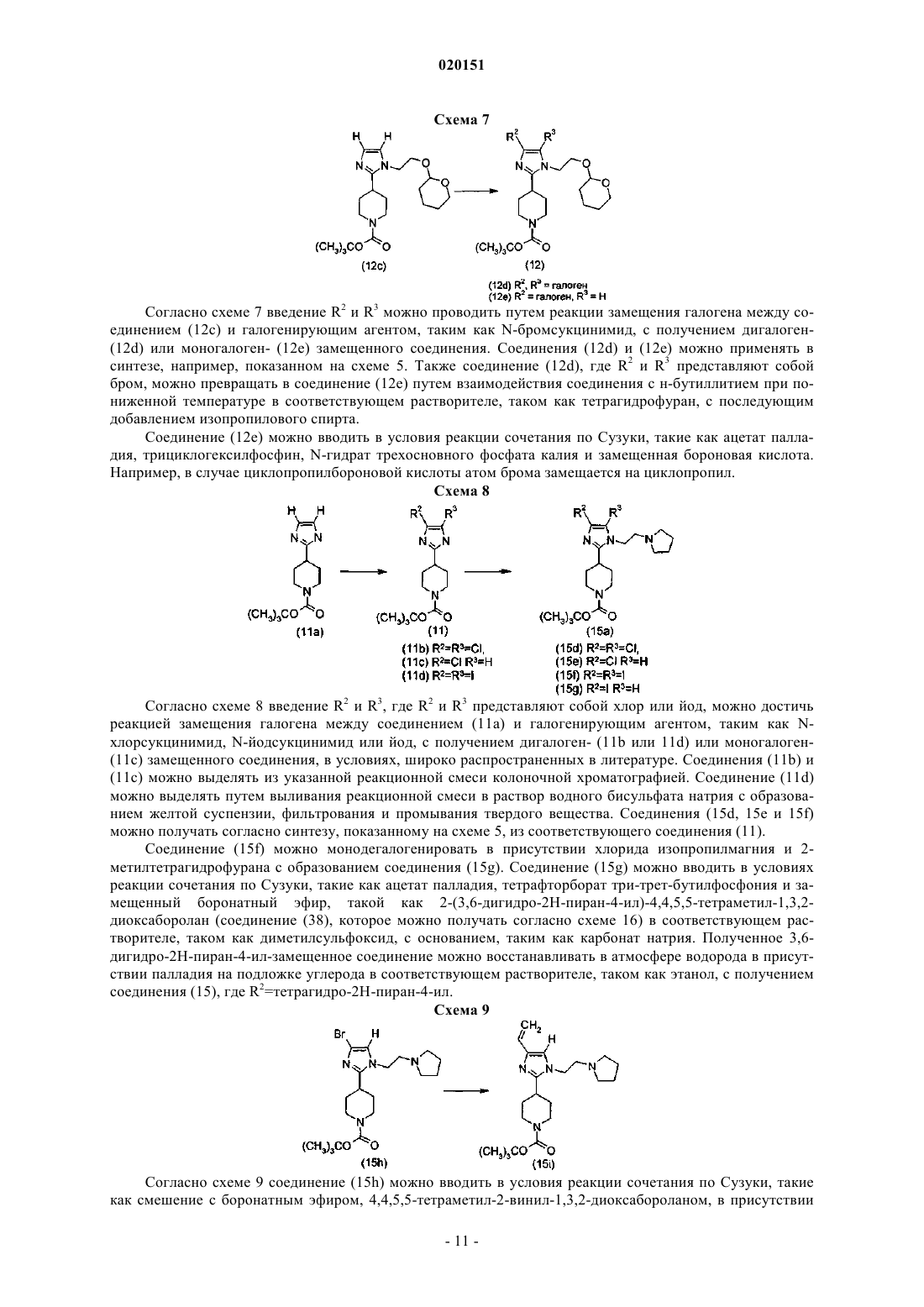
В настоящем изобретении предложены ингибиторы AKT формулы Бэйт Дуглас Вэйд, Беркхолдер Тимоти Пол, Клэйтон Джошуа Райан,Эгген МариДжин, Генри Кеннет Джеймс Джуниор, Джонс Дидре Мичелл, Партасарати Сараванан, Пэй Хуасин, Ремпала Марк Эдвард, Сойер Джейсон Скотт (US) В настоящем изобретении также предложены фармацевтические композиции, содержащие соединения формулы I, применение соединений формулы I и способ применения соединений формулы I.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Путь фосфатидилинозитол-3-киназа (PI3K)/AKT/мишень рапамицина у млекопитающих (mTOR) включает ряд сигнальных точек, имеющих важное значение для контроля роста и выживаемости клеток.AKT, также известная как протеинкиназа В, представляет собой серин-треонин протеинкиназу, которая играет ключевую роль в указанном пути. Активация AKT опосредована PI3K. PI3K вырабатывает фосфолипиды, которые связываются с AKT. После связывания AKT привлекается к плазматической мембране и активируется посредством фосфорилирования. Активация и передача сигнала AKT способствует выживаемости, росту и пролиферации клеток. Повышенная активация AKT вовлечена в широкий круг раковых заболеваний. Ряд замещенных пиперидиновых соединений, имеющих активность в отношении ингибированияAKT, предложен в WO 2008/075109. Эти соединения описаны в качестве подходящих для применения для лечения заболеваний или состояний, включающих или возникающих в результате патологического роста клеток или патологического прекращения гибели клеток, включая рак. По-прежнему существует потребность в создании альтернативных ингибиторов AKT, которые можно применять для лечения пролиферативных нарушений, таких как рак. В настоящем изобретении предложены альтернативные ингибиторы AKT. Некоторые соединения согласно настоящему изобретению являются более высокоактивными ингибиторами AKT по сравнению с соединениями, известными в данной области техники. Некоторые соединения согласно настоящему изобретению имеют низкую активность в отношении киназы 2 (ROCK2) по сравнению с ингибиторами, известными в данной области техники. Некоторые соединения согласно настоящему изобретению имеют улучшенную пероральную эффективность по сравнению с ингибиторами AKT, известными в данной области техники. В настоящем изобретении предложены соединения формулыR1 представляет собой СН 3, СН 2 СН 3 или CF3;R2 представляет собой Н, CF3, CH2CF3, CH2CH2CF3, C1-C4-алкил, C3-C6-циклоалкил, CN, Cl, Br,СН=СН 2, СН 2 СН 2 ОСН 3, С(СН 3)2 СН 2 ОСН 3 или тетрагидропиран-4-ил, где C3-C6-циклоалкил необязательно замещен метилом в положении 1 и тетрагидропиран-4-ил необязательно замещен метилом в положении 4;R5 представляет собой СН 3, C(СН 3)3, СН(СН 3)2, циклобутил, циклопентил, СН 2-циклопропил,С(СН 3)2 СН 2 СН 3 или тетрагидропиран-4-ил, илиR4 и R5, оба, представляют собой СН 3, илиR4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин, необязательно замещенный гидроксигруппой в положении 3, или азетидин,или фармацевтически приемлемые соли указанных соединений. В настоящем изобретении предложен фармацевтический состав, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель. В настоящем изобретении предложено соединение согласно настоящему изобретению или его фар-1 020151 мацевтически приемлемая соль для применения в терапии. В настоящем изобретении предложено соединение согласно настоящему изобретению или его фармацевтически приемлемая соль для применения для лечения рака легких, рака груди или глиобластомы. В настоящем изобретении также предложен способ лечения рака легких, рака груди или глиобластомы у млекопитающего, включающий введение млекопитающему, нуждающемуся в подобном лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Дополнительно в настоящем изобретении предложено применение соединения согласно настоящему изобретению или его фармацевтически приемлемой соли для производства лекарственного средства для лечения рака легких, рака груди или глиобластомы. Кроме того, в настоящем изобретении предложена фармацевтическая композиция для применения в терапии, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль, также предложена фармацевтическая композиция для лечения рака легких, рака груди или глиобластомы, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль. В настоящем изобретении предложена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению совместно с фармацевтически приемлемым носителем и необязательно другими терапевтическими агентами. Общие химические термины, используемые выше в формулах, имеют традиционные значения. Например, термин "C1-C4-алкил" относится к линейной или разветвленной одновалентной насыщенной алифатической цепи, содержащей от 1 до 4 атомов углерода, и включает, но не ограничивается ими, метил, этил, пропил, изопропил, бутил, изобутил и т-бутил. Этил, пропил, изопропил, бутил и изобутил являются предпочтительными алкильными группами. Этил является особенно предпочтительным. Соединения согласно настоящему изобретению представляют собой основания и соответственно взаимодействуют с любыми органическими и неорганическими кислотами с образованием фармацевтически приемлемых солей; настоящее изобретение включает фармацевтически приемлемые соли соединений формулы I. Термин "фармацевтически приемлемая соль", используемый в настоящем описании, относится к солям соединений формулы I, которые, по существу, не являются токсичными для живых организмов. Подобные соли включают фармацевтически приемлемые соли, перечисленные в Journal ofPharmaceutical Science, 66, 2-19 (1977), которые известны специалистам в данной области. В одном из вариантов реализации соединение согласно настоящему изобретению представляет собой свободное основание или гидрохлоридную соль, в частности свободное основание. Некоторые соединения согласно настоящему изобретению имеют один или более хиральный центр и могут существовать в виде ряда стереоизомерных конфигураций. Вследствие наличия хиральных центров соединения согласно настоящему изобретению существуют в виде рацематов, смесей энантиомеров и индивидуальных энантиомеров, а также диастереомеров и смесей диастереомеров. Все указанные рацематы, энантиомеры и диастереомеры включены в объем настоящего изобретения. Конкретные стереоизомеры и энантиомеры соединений формулы I могут быть получены специалистами в данной области хорошо известными способами и процессами, например, описанными Дж. Жаком с соавторами (J.Jacques, et al.), "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981; Э.Л. Эльелем и С.Х. Уиленом (E.L. Eliel and S.H. Wilen), "Stereochemistry of Organic Compounds" (Wiley-Interscience 1994); в европейской заявке на патентЕР-А-838448, опубликованной 29 апреля 1998 г. Примеры разделения включают способы перекристаллизации и хиральной хроматографии. Термины "R" и "S" используют в настоящем описании в традиционном значении, используемом в органической химии, для описания конкретной конфигурации хирального центра. Термин "R" (rectus) относится к конфигурации хирального центра с расположением групп по приоритету (от наивысшего ко второму наименьшему) по часовой стрелке при рассмотрении в направлении связи с группой с наименьшим приоритетом. Термин "S" (sinister) относится к конфигурации хирального центра с расположением групп по приоритету (от наивысшего ко второму по значимости) против часовой стрелки при рассмотрении в направлении связи с группой с наименьшим приоритетом. Приоритетность групп основывается на значении атомного числа (в порядке уменьшения атомного числа). Неполный список приоритетов и обсуждение стереохимии содержится в "Nomenclature of Organic Compounds: Principles and Practice" (под ред. Дж.Х.Флетчера с соавторами (J.H. Fletcher, et al., eds.), 1974) на с. 103-120. Обозначение относится к связи, которая располагается над плоскостью листа. Обозначение относится к связи, которая располагается за плоскостью листа. Термин "энантиомерное обогащение" относится к повышенному содержанию одного из энантиомеров по сравнению с другим. Удобным способом выражения существующего энантиомерного обогащения является понятие энантиомерного избытка, или "эи", которое определяют при помощи следующего уравнения:, где Е 1 представляет собой содержание в процентах первого энантиомера, а Е 2 представляет собой содержание в процентах второго энантиомера. Энантиомерное обогащение легко определяется специалистами в данной области стандартными способами и процедурами, такими как высокоэффективная жидкостная хроматография на хиральной колонке. Предпочтительной является R конфигурация атома углерода, к которой присоединен R1: Термин "R энантиомер", используемый в настоящем описании, означает % эи R энантиомера, который составляет более 90%, предпочтительно более 95% и более предпочтительно более 98%. Специалистам в данной области также очевидно, что соединения формулы I существуют в виде таутомеров, например Несмотря на то что таутомеры имеют различную структуру, специалистам в данной области очевидно, что они существуют в равновесии и легко и быстро превращаются друг в друга в стандартных условиях (см. Марч (March), Advanced Organic Chemistry, Third Edition, Wiley Interscience, New York,New York (1985), p. 66-70; и Эллинджер (Allinger), Organic Chemistry, Second Edition, Worth Publishers,New York, New York (1976), p. 173). Как таковое, представление соединения формулы I в единственной таутомерной форме охватывает индивидуальные таутомерные формы и их смеси. Соединения, представленные в примерах, называли при помощи программы составления названийChem Draw Ultra версии v10 или Chem Bio Viz Ultra версии v11. В одном из вариантов реализации настоящее изобретение включает соединение формулы I, где А В частности, А представляет собой В одном из вариантов реализации R1 представляет собой СН 3 или CF3. В частности, R1 представляет собой СН 3. В альтернативном варианте реализации настоящее изобретение включает соединения формулы I, где А представляет собой В одном из вариантов реализации настоящее изобретение включает соединения формулы I, где R2 представляет собой CF3, CH2CF3, CH2CH2CF3, C1-C4-алкил, C3-C6-циклоалкил, CN, Cl, Br, СН=СН 2,СН 2 СН 2 ОСН 3, С(СН 3)2 СН 2 ОСН 3 или тетрагидропиран-4-ил, где C3-C6-циклоалкил необязательно замещен метилом в положении 1 и тетрагидропиран-4-ил необязательно замещен метилом в положении 4, R3 представляет собой Н или R2 и R3, оба, представляют собой Cl. В частности, R2 представляет собой CF3,CH2CF3, CH2CH2CF3, СН 2 СН 3, (СН 2)2 СН 3, (СН 2)3 СН 3, СН(СН 3)2, СН 2 СН(СН 3)2, С 3-С 6-циклоалкил, Cl, Br,-3 020151 СН=СН 2, СН 2 СН 2 ОСН 3, С(СН 3)2 СН 2 ОСН 3 или тетрагидропиран-4-ил, где C3-C6-циклоалкил необязательно замещен метилом в положении 1 и тетрагидропиран-4-ил необязательно замещен метилом в положении 4, R3 представляет собой Н или R2 и R3, оба, представляют собой Cl. Более конкретно, R2 представляет собой CF3, CH2CF3, CH2CH3 или тетрагидропиран-4-ил и R3 представляет собой Н. Еще более конкретно, R2 представляет собой тетрагидропиран-4-ил и R3 представляет собой Н. В другом варианте реализации настоящее изобретение включает соединения формулы I, где R2 представляет собой CF3, CH2CF3, CH2CH2CF3, СН 2 СН 3, (СН 2)2 СН 3, циклопропил, Br, СН 2 СН 2 ОСН 3 или тетрагидропиран-4-ил и R3 представляет собой Н. В частности, R2 представляет собой CH2CF3,CH2CH2CF3 или СН 2 СН 3 и R3 представляет собой Н. В одном из вариантов реализации настоящее изобретение включает соединения формулы I, где R4 представляет собой Н, R5 представляет собой СН 3, С(СН 3)3, СН(СН 3)2, циклобутил, циклопентил или СН 2-циклопропил, или R4 и R5, оба, представляют собой СН 3, или R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин, необязательно замещенный гидроксигруппой в положении 3, или азетидин. В частности, R4 представляет собой Н, R5 представляет собой СН 3, С(СН 3)3,СН(СН 3)2, циклобутил, циклопентил или СН 2-циклопропил или R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин или азетидин. Более конкретно, R4 и R5 совместно с атомомN, к которому они присоединены, образуют пирролидин. В другом варианте реализации настоящее изобретение включает соединения формулы I, где R4 представляет собой Н, R5 представляет собой С(СН 3)3, или R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин или азетидин. Более конкретно, R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин или азетидин. В другом варианте реализации настоящее изобретение включает соединения формулы I, гдеR1 представляет собой СН 3 или CF3;R2 представляет собой CF3, CH2CF3, CH2CH2CF3, CH2CH3, (СН 2)2 СН 3, (СН 2)3 СН 3, СН(СН 3)2,СН 2 СН(СН 3)2, C3-C6-циклоалкил, Cl, Br, СН=СН 2, CH2CH2OCH3, C(CH3)2CH2OCH3 или тетрагидропиран 4-ил, где C3-C6-циклоалкил необязательно замещен метилом в положении 1, а тетрагидропиран-4-ил необязательно замещен метилом в положении 4;R5 представляет собой СН 3, C(СН 3)3, СН(СН 3)2, циклобутил, циклопентил или СН 2-циклопропил илиR4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин или азетидин,или фармацевтически приемлемые соли указанных соединений. В другом варианте реализации настоящее изобретение включает соединения формулы где R2 представляет собой CF3, CH2CF3, CH2CH3 или тетрагидропиран-4-ил,или их фармацевтически приемлемые соли. В другом варианте реализации настоящее изобретение включает соединения формулы I, гдеR1 представляет собой СН 3, CF3 или СН 2 СН 3;R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин или азетидин,или фармацевтически приемлемые соли указанных соединений. В другом варианте реализации настоящее изобретение включает соединения формулы где R1 представляет собой СН 3 или CF3;R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин или азетидин,или фармацевтически приемлемые соли указанных соединений. В другом варианте реализации предложены следующие соединения или их фармацевтически приемлемые соли:(R)-4-(4-(1-(2-(азетидин-1-ил)этил)-4-(2,2,2-трифторэтил)-1 Н-имидазол-2-ил)пиперидин-1-ил)-5(трифторметил)-5,6-дигидропиридо[2,3-d]пиримидин-7(8 Н)-он. В одном из вариантов реализации соединение согласно настоящему изобретению представляет собой(R)-5-метил-4-(4-(1-(2-(пирролидин-1-ил)этил)-4-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазол-2 ил)пиперидин-1-ил)-5,6-дигидропиридо[2,3-d]пиримидин-7(8 Н)-он или его фармацевтически приемлемую соль. В частности, соединение представляет собой (R)-5-метил-4-(4-(1-(2-(пирролидин-1-ил)этил)-4(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазол-2-ил)пиперидин-1-ил)-5,6-дигидропиридо[2,3-d]пиримидин 7(8 Н)-он. Более конкретно, соединение представляет собой кристаллическую форму III (R)-5-метил-4-(4(1-(2-(пирролидин-1-ил)этил)-4-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазол-2-ил)пиперидин-1-ил)-5,6 дигидропиридо[2,3-d]пиримидин-7(8 Н)-она. Кристаллическая форма III (R)-5-метил-4-(4-(1-(2(пирролидин-1-ил)этил)-4-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазол-2-ил)пиперидин-1-ил)-5,6 дигидропиридо[2,3-d]пиримидин-7(8 Н)-она характеризуется порошковой рентгеновской дифрактограммой (излучение CuK, =1,54056 ), содержащей пик при 8,53 (20,1) и необязательно один или более пик, выбранный из 17,06, 7,97 и 14,17 (20,1), предпочтительно характеризуется порошковой рентгеновской дифрактограммой, содержащей пики при 8,53, 17,06, 7,97 и 14,17 (20,1). Соединения согласно настоящему изобретению являются ингибиторами AKT и, следовательно,подходят для лечения рака, в частности для лечения раковых заболеваний, при которых активируется путь PI3K/AKT/mTOR, включающих рак груди (Carpten et al.), 448: 439-444 (2007), в частности HER2 положительного рака груди (Yakes et al.), Cancer Research, 62: 4132-4141 (2003); колоректального рака(Parsons et al.), Nature, 436: 792 (2005); (Carpten et al.), 448: 439-444 (2007); рака яичников (Carpten et al.),448: 439-444 (2007); рака легких, в частности плоскоклеточной карциномы легких (Malanga et al.), CellCycle, 7:5: 665-669 (2008); карциномы желудка (Byun et al.), Int. J. Cancer, 104: 318-327 (2003); рака поджелудочной железы (Ruggeri et al.), Molecular Carcinogenesis, 21: 81-86 (1998); плоскоклеточной карциномы головы и шеи (Pedrero et al.), Int. J. Cancer, 114: 242-248 (2005); меланомы (Stahl et al.), Cancer Research, 64: 7002-7010 (2004); глиобластомы (The Cancer Genome Atlas Research Network, 455: 1061-1068(2008; рака простаты (Sasaki et al.), Biochem. Biophys. Res. Comm., 399(1): 79-83 (2010); рака мочевого пузыря (Ching et al.), Lab. Invest., Epub. 26 July 2010); мезотелиомы (Mohiuddin et al.), Annals of Sur. Oncol., 9(3): 310-316 (2002); саркомы, в частности саркомы мягких тканей (Zhu et al.), Cancer Res., 68(8): 2895-2903 (2008); и рака почек (Hara et al.), Annals of Oncol., 16: 928-933 (2005). Соединения согласно настоящему изобретению или их фармацевтически приемлемые соли можно применять в способе лечения рака, в частности раковых заболеваний, описанных выше, у млекопитающего, включающем введение млекопитающему, нуждающемуся в подобном лечении, эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли. Дополнительно предложены соединения согласно настоящему изобретению или их фармацевтически приемлемые соли для применения для лечения рака, в частности раковых заболеваний, описанных выше. Кроме того, соединения согласно настоящему изобретению или их фармацевтически приемлемые соли можно применять для производства лекарственного средства для лечения рака, в частности раковых заболеваний, описанных выше. Также предложена фармацевтическая композиция для лечения рака, в частности раковых заболеваний, описанных выше, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль. Соединения согласно настоящему изобретению можно применять в комбинации с другими терапевтическими агентами и, в частности, ингибиторами mTOR (мишени рапамицина у млекопитающих), ингибиторами РЭФР (рецептора эпидермального фактора роста), гемцитабином (Gemzar), цисплатином,тасисуламом(Alimta), доцетакселом (Taxotere), доксорубицином (Doxil), иринотеканом (Campto; Camptosar),паклитакселом (Taxol) или тамоксифеном. Предпочтительные ингибиторы mTOR включают рапамицин (также известный как сиролимус) и его аналоги, такие как эверолимус (42-O-(2-гидрокси)этилрапамицин; описан в ЕР 1413581),темсиролимус(42(диметилфосфинат)рапамицин; описан в WO 03/64383). Предпочтительные ингибиторы РЭФР включают эрлотиниб (Tarceva), цетуксимаб (Erbitux; описан в ЕР 0359282), панитумумаб (Vectibix; описан в ЕР 0359282) и гефитиниб (Iressa; описан в ЕР 0566226). В одном из вариантов реализации в настоящем изобретении предложен продукт, содержащий соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и терапевтический агент, выбранный из перечисленных выше, в виде объединенного препарата для одновременного,раздельного или последовательного применения в терапии. В настоящем изобретении также предложено соединение согласно настоящему изобретению или его фармацевтически приемлемая соль для одновременного, раздельного или последовательного применения в комбинации с терапевтическим агентом, выбранным из перечисленных выше, для лечения рака груди, колоректального рака, рака яичников, рака легких, карциномы желудка, рака поджелудочной железы, плоскоклеточной карциномы головы и шеи,меланомы, глиобластомы, рака простаты, рака мочевого пузыря, мезотелиомы, саркомы и рака почек. В настоящем изобретении также предложен способ лечения рака, выбранного из группы, состоящей из рака груди, колоректального рака, рака яичников, рака легких, карциномы желудка, рака поджелудочной железы, плоскоклеточной карциномы головы и шеи, меланомы, глиобластомы, рака простаты, рака мочевого пузыря, мезотелиомы, саркомы и рака почек, включающий введение пациенту, нуждающемуся в этом, соединения согласно настоящему изобретению или его фармацевтически приемлемой соли и терапевтического агента, выбранного из агентов, перечисленных выше, в количествах, которые в комбинации являются эффективными. В другом варианте реализации в настоящем изобретении предложена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению совместно с фармацевтически приемлемым носителем и, необязательно, другими терапевтическими агентами, в частности терапевтическим агентом, выбранным из перечисленных выше. Пероральное введение соединений согласно настоящему изобретению является предпочтительным. В зависимости от условий другие способы введения, например внутривенный, можно применять или даже являются предпочтительными. Трансдермальное введение может быть желательным для пациентов,которые забывают или предпочитают не принимать пероральные лекарственные средства. Соединения согласно настоящему изобретению также можно вводить при помощи подкожного, внутримышечного,интраназального или ректального способов введения в конкретных случаях. Способ введения можно изменять любым способом, ограничиваясь физическими свойствами лекарственных средств, удобством для пациента и лица, осуществляющего уход, и другими важными условиями (Remington's PharmaceuticalSciences, 18th Edition, Mack Publishing Co. (1990. Соединения формулы I могут быть получены специалистами в данной области известными в данной области способами и процедурами. Более конкретно, соединения формулы I можно получать в соответствии со схемами, примерами получения и примерами, представленными далее. Специалистам в данной области очевидно, что конкретные стадии, представленные на следующих схемах, можно изменять для получения соединений формулы I. Реагенты и исходные вещества легкодоступны для специалистов в данной области. Все заместители, если не указано иное, определены выше. Согласно схеме 1 соединение формулы I можно получать реакцией нуклеофильного замещения между аминогруппой пиперидинового кольца соединения (1) и уходящей хлор-группой соединения (2). Соединение (1) и соединение (2) растворяют в подходящем растворителе, таком как Nметилпирролидинон, метанол или н-пропанол, с подходящим основанием, таким как диизопропилэтиламин, триэтиламин или 1,8-диазабицикло[5.4.0]ундец-7-ен. Реакционную смесь можно нагревать в колбе или пробирке, стойкой к микроволновому излучению. Соединение формулы I можно выделять способами, известными в данной области, такими как водная обработка, которая может включать промывку водной фосфорной кислотой, а затем промывку основанием, водным гидроксидом натрия, и дополнительную очистку, такую как хроматография на силикагеле или высокоэффективная жидкостная хроматография (ВЭЖХ-Chiral AD). В качестве альтернативы после водной обработки соединение формулы I можно выделять перекристаллизацией из растворителя, такого как 75% смесь метил-трет-бутилового эфира и гексана. Соль соединения формулы I можно получать растворением соединения формулы I в соответствующей водной кислоте, такой как 4 М хлористо-водородная кислота, и можно выделять концентрированием при пониженном давлении. В качестве альтернативы для получения дигидрохлоридной или трифторацетатной соли можно применять обращенно-фазовую хроматографию соединения формулы I. Схема 2 На схеме 2 описан альтернативный способ получения соединения формулы I (А определен выше). Этот способ синтеза включает реакцию аминозамещения соединения (16) и соответствующего амина с получением заместителя -NR4R5, определенного для соединения формулы I. Соединение (16) растворяют в соответствующем растворителе, таком как диметилформамид или диметилсульфоксид. Добавляют подходящее основание, такое как триэтиламин. Добавляют соответствующий амин, что приводит к получению заместителя -NR4R5, определенного для соединения формулы I. Реакционную смесь нагревают примерно до 50 С до завершения взаимодействия. Соединение формулы I выделяют при помощи традиционных способов, таких как водная обработка, концентрирование и хроматография органических экстрактов. Соединение (16) получают растворением соединения (17) в соответствующем растворителе, таком как дихлорметан, добавления соответствующего основания, такого как триэтиламин, и охлаждения примерно до 0 С. Метансульфонилхлорид добавляют по каплям. Затем реакцию гасят насыщенным водным бикарбонатом натрия, а затем при помощи традиционных способов, известных в данной области, выделяют соединение (16). Соединение (16) можно применять без очистки для получения соединения формулы I. Соединение (17) можно получать из соединения (18) (n = от 1 до 2) и соединения (2) при помощи способов, описанных на схеме 1. Соединение (17) можно применять без очистки для получения соединения (16). Если полученное соединение формулы I представляет собой рацемат, то его можно разделять на индивидуальные энантиомеры при помощи способов, известных в данной области, таких как хиральная хроматография. Хиральную чистоту индивидуальных энантиомеров соединения формулы I можно определять сравнением двух энантиомеров при помощи ВЭЖХ (Chiralpak AD-H) и сверхкритической флюидной хроматографии (Chiral AD-H). Схема 3 Согласно схеме 3 соединения (2b) и (2 с), энантиомеры рацемической смеси соединения (2 а), 4 хлор-5-R1-6,8-дигидро-5 Н-пиридо[2,3-d]пиримидин-7-она, можно получать при помощи серии взаимодействий, начиная с соединения (5) и соединения (6). Соединение (2 а) получают объединением соединения (3) с водным гидроксидом аммония (20-30%) или раствором газообразного аммиака в изопропаноле при нагревании. Соединение (2 а) можно выделять фильтрованием после охлаждения и последующего промывания холодной водой. Дополнительное разделение соединения (2 а) путем хиральной хроматографии приводит к получению соединения (2b) и соединения (2 с). Соединения (2 а), (2b) или (2 с) можно применять согласно способам синтеза, представленным на схеме 1 или схеме 2, для получения рацемата соединения формулы I или индивидуальных энантиомеров. В качестве альтернативы можно проводить защиту соединения (2 а) при помощи защищающей атом азота группы, такой как трет-бутоксикарбонил (ВОС), с применением трет-бутоксикарбонил третбутилкарбоната и 4-(диметиламино)пиридина в растворителе, таком как дихлорметан. Последующее разделение рацемата хиральной хроматографией приводит к получению соединений (2b) и (2 с) с ВОСзащищенным атомом азота. Снятие защиты способами, известными в данной области, такими как взаимодействие ВОС-защищенного соединения (2b, 2 с) с хлористо-водородной кислотой в диоксане, приводит к получению единственного целевого энантиомера. Соединение (3) можно получать реакцией замещения галогена соединения (4). Соединение (4) растворяют в подходящем растворителе, таком как ацетонитрил или толуол, в присутствии основания, такого как N,N-диэтиланилин, и хлорирующего агента, такого как фосфорилхлорид. После кипячения с обратным холодильником реакционной смеси соединение (3) можно выделять традиционными способами,такими как водная обработка 3 M водным раствором двухосновного фосфата калия, экстракция в соответствующем растворителе, таком как метил-трет-бутиловый эфир, промывание органического слоя водой и концентрирование в вакууме. В качестве альтернативы можно синтезировать соединение (2 а), содержащее защищающую атом азота группу, такую как 2,4-диметоксибензильная группа. Соединение (3) сначала растворяют в подходящем растворителе, таком как диметилформамид. Добавляют основание, такое как диизопропилэтиламин, и реагент, 2,4-диметоксибензиламин. Промежуточное 2,4-диметоксибензильное соединение выделяют способами, известными в данной области, такими как водная обработка. Это промежуточное соединение нагревают в присутствии основания, такого как диизопропилэтиламин, с образованием 2,4 диметоксибензил-защищенного соединения (3). Это соединение можно применять в синтезе согласно схеме 1 с получением 2,4-диметоксибензил-защищенного соединения формулы I. Снятие защиты 2,4 диметоксибензил-защищенного соединения формулы I проводят при помощи традиционных способов с получением рацемата, последующая хиральная хроматография приводит к разделению на индивидуальные энантиомеры. Соединение (4) можно получать реакцией присоединения по Михаэлю и последующей реакцией циклизации in situ. Диметиловый эфир пропандиовой кислоты (соединение (5 и соединение (6) добавляют к смеси метоксида натрия в растворе метанола и формамидинацетата. Соединение (4) можно выделять традиционными способами доведением рН смеси примерно до 3, фильтрованием и промыванием продукта охлажденной смесью растворителей, такой как метанол/вода. Согласно схеме 4 соединение (2d) можно получать при помощи серии взаимодействий, начиная с реакции восстановительного аминирования коммерчески доступных исходных веществ, таких как амин(10), 2,2,2-трифторэтиламин, и коммерчески доступного альдегида, 4-амино-6-хлорпиримидин-5 карбальдегида (7), с получением имина (8), в смеси растворителей, таких как тетрагидрофуран и метанол, в присутствии тетраизопропоксида титана. Имин восстанавливают растворением соединения (8) в растворителе, таком как дихлорметан, охлаждения в азоте, добавления метансульфокислоты и подходящего восстановителя, такого как трет-бутиламин-боран. Амин (9) выделяют основной водной обработкой и сушкой органических веществ в вакууме. Соединение (9) можно применять в следующей реакции непосредственно без дополнительной очистки. Соединение (2d) получают взаимодействием соединения(9) с трифосгеном в присутствии основания, такого как триэтиламин, в соответствующем растворителе,таком как дихлорметан, при охлаждении примерно до 0 С в атмосфере азота и последующего нагревания реакционной смеси в течение ночи примерно при 40 С. Соединение (2d) выделяют способами, известными в данной области, таких как водная обработка, концентрирование и хроматография органических экстрактов. Соединение формулы I можно получать путем применения соединения (2d) согласно схемам 1 и 2. Схема 5 Согласно схеме 5 соединение (1) можно получать при помощи снятия защиты соответствующего трет-бутоксикарбонильного соединения (15) традиционных способов, таких как добавление к соединению (15), которое необязательно растворено в подходящем растворителе, таком как дихлорметан, метанол или изопропанол, хлороводорода в диоксане, изопропаноле, метаноле или этаноле. Реакцию можно проводить при температурах в диапазоне от комнатной температуры до 50 С в течение примерно от 2 до 18 ч. Традиционная обработка может включать выпаривание летучих веществ последующей необязательной стадией подщелачивания при помощи основания, такого как 2 М водный гидроксид натрия, экстракцию в растворителе, таком как этилацетат, и концентрирование в вакууме с получением соединения(1) в виде свободного основания, nHCl или n-ацетатной соли. Соединение (15) можно получать из соединения (14) при помощи реакции, описанной на схеме 2 для получения соединения формулы I из соединения (16). Соединение (14) можно получать из соединения (13) при помощи реакции, описанной на схеме 2 для получения соединения (16) из соединения (17). Соединение (13) можно получать при помощи катализируемой кислотой реакции снятия защиты соединения (12) согласно способу синтеза А. Соединение (12) растворяют в подходящем растворителе,-9 020151 таком как тетрагидрофуран. Добавляют кислотный катализатор, такой как 1 н. водная хлористоводородная кислота. Соединение (13) можно выделять способами, известными в данной области, такими как водная обработка. Соединение (12) изображено в виде одного региоизомера, но также может представлять собой смесь региоизомеров. Синтез и выделение региоизомеров (12 а) и (12b) показаны на схеме 6. Соединение(12) можно получать растворением соединения (11) в подходящем растворителе, таком как диметилсульфоксид, с основанием, таким как гидроксид калия или трет-бутоксид калия. Также можно добавлять йодид натрия. К реакционной смеси добавляют 2-(2-галогенэтокси)тетрагидропиран. Реакционную смесь выдерживают при комнатной температуре в течение примерно 4 ч или нагревают примерно до 45-50 С в течение примерно от 1 до 12 ч. Соединение (12) можно выделять способами, известными в данной области, такими как водная обработка, концентрирование в вакууме и очистка хроматографией. В качестве альтернативы можно проводить способ В для получения соединения (1), где R4 и R5 совместно с атомом N, к которому они присоединены, образуют пирролидин. Соединение (15 а) можно получать при помощи реакции соединения (11) с гидрохлоридом 1-(2-хлорэтил)пирролидина с применением условий реакции, описанных выше для превращения соединения (11) в соединение (12). Соединение(15 а) изображено в виде одного региоизомера, но также может представлять собой смесь региоизомеров. Синтез и выделение региоизомеров (15b) и (15 с) показаны на схеме 6. Защитную ВОС-группу можно удалять традиционными способами, описанными выше. Схема 6 При алкилировании соединения (11), показанном на схеме 5, могут образовываться региоизомеры(12 а) и (12b) соединения (12) и региоизомеры (15b) и (15 с) соединения (15 а) соответственно в различных соотношения, как показано на схеме 6. В некоторых случаях получают только целевой изомер (12 а) или(15b). В других случаях синтез приводит к преимущественному содержанию целевого соединения (12 а) или (15b). В этом случае дополнительная очистка является необязательной и может проводиться на дальнейших стадиях для удаления незначительных примесей. Если, тем не менее, соотношения соединения (12 а) и соединения (12b) или соединения (15b) и соединения (15 с) не соответствуют преимущественному содержанию целевого изомера, то необходима очистка. Очистка для выделения целевого изомера(12 а) или (15b) включает колоночную хроматографию или перекристаллизацию из соответствующего растворителя, такого как изопропиловый спирт, содержащий бутандиовую кислоту, или 1 М, или 3 M хлористо-водородная кислота в метаноле или смеси метанол/этанол, содержащей этилацетат. Согласно схеме 7 введение R2 и R3 можно проводить путем реакции замещения галогена между соединением (12 с) и галогенирующим агентом, таким как N-бромсукцинимид, с получением дигалоген(12d) или моногалоген- (12 е) замещенного соединения. Соединения (12d) и (12 е) можно применять в синтезе, например, показанном на схеме 5. Также соединение (12d), где R2 и R3 представляют собой бром, можно превращать в соединение (12 е) путем взаимодействия соединения с н-бутиллитием при пониженной температуре в соответствующем растворителе, таком как тетрагидрофуран, с последующим добавлением изопропилового спирта. Соединение (12 е) можно вводить в условия реакции сочетания по Сузуки, такие как ацетат палладия, трициклогексилфосфин, N-гидрат трехосновного фосфата калия и замещенная бороновая кислота. Например, в случае циклопропилбороновой кислоты атом брома замещается на циклопропил. Схема 8 Согласно схеме 8 введение R2 и R3, где R2 и R3 представляют собой хлор или йод, можно достичь реакцией замещения галогена между соединением (11 а) и галогенирующим агентом, таким как Nхлорсукцинимид, N-йодсукцинимид или йод, с получением дигалоген- (11b или 11d) или моногалоген(11 с) замещенного соединения, в условиях, широко распространенных в литературе. Соединения (11b) и(11 с) можно выделять из указанной реакционной смеси колоночной хроматографией. Соединение (11d) можно выделять путем выливания реакционной смеси в раствор водного бисульфата натрия с образованием желтой суспензии, фильтрования и промывания твердого вещества. Соединения (15d, 15e и 15f) можно получать согласно синтезу, показанному на схеме 5, из соответствующего соединения (11). Соединение (15f) можно монодегалогенировать в присутствии хлорида изопропилмагния и 2 метилтетрагидрофурана с образованием соединения (15g). Соединение (15g) можно вводить в условиях реакции сочетания по Сузуки, такие как ацетат палладия, тетрафторборат три-трет-бутилфосфония и замещенный боронатный эфир, такой как 2-(3,6-дигидро-2 Н-пиран-4-ил)-4,4,5,5-тетраметил-1,3,2 диоксаборолан (соединение (38), которое можно получать согласно схеме 16) в соответствующем растворителе, таком как диметилсульфоксид, с основанием, таким как карбонат натрия. Полученное 3,6 дигидро-2 Н-пиран-4-ил-замещенное соединение можно восстанавливать в атмосфере водорода в присутствии палладия на подложке углерода в соответствующем растворителе, таком как этанол, с получением соединения (15), где R2=тетрагидро-2H-пиран-4-ил. Схема 9 Согласно схеме 9 соединение (15h) можно вводить в условия реакции сочетания по Сузуки, такие как смешение с боронатным эфиром, 4,4,5,5-тетраметил-2-винил-1,3,2-диоксабороланом, в присутствии основания, как правило, N-гидратом трехосновного фосфата калия, и палладиевого катализатора, как правило, бис-(дибензилиденацетон)палладия(0) или ацетата палладия, и дициклогексил-[2-(2,6 диметоксифенил)фенил]фосфана. Полученное соединение (15i) можно применять согласно схеме 5 для получения соединения (1). В качестве альтернативы эту реакцию можно проводить с гидрокси-этил-замещенным соединением(13) вместо пирролидин-этил-замещенного соединения. Полученный алкен можно подвергать условиям гидрогенирования, которые включают 10% палладий на подложке углерода в соответствующем растворителе, таком как этанол, в атмосфере водорода с образованием алкил-замещенного соединения. Соединение можно выделять при помощи фильтрования через Celite, последующего промывания метанолом и концентрирования в вакууме. Схема 10 Согласно схеме 10 соединение (18) можно получать традиционными способами снятия защиты. Соединение (12) растворяют в подходящем растворителе, таком как метанол, добавляют раствор 4 M хлористо-водородной кислоты в диоксане и перемешивают в течение ночи. Соединение (18) выделяют способами, известными в данной области, такими как концентрирование в вакууме, с получением соединения (18) в виде соли nHCl. Соединение (18) можно применять согласно схеме 2 для получения соединения формулы I. Схема 11 Согласно схеме 11 соединение (11 е) можно получать при помощи двухстадийного способа, который включает реакцию окисления кетона (22) или альдегида (26) с получением соединения (23) и реакции конденсации с аммиаком соединения (23) и альдегидного фрагмента соединения (24). Диоксид селена смешивают в подходящей смеси растворителей, такой как 1,4-диоксан и вода, с кислотой, такой как уксусная кислота. Кетон (22) или альдегид (26) добавляют к окислителю, нагревают примерно до 90 С,перемешивают в течение примерно 2-18 ч и фильтруют с получением соединения (23). Дополнительная обработка может включать фильтрование через Celite, концентрирование в вакууме и растворение остатка в растворителе, таком как метанол. Соединение (24) растворяют в метаноле, содержащем гидроксид аммония или ацетат аммония, и необязательно охлаждают примерно до 0 С. Соединение (23) добавляют по каплям и реакционную смесь перемешивают в течение ночи. Соединение (11e) можно выделять при помощи способов, известных в данной области, таких как фильтрование, водная обработка и очистка хроматографией на силикагеле. Дополнительная обработка может включать разбавление остатка метил-трет-бутиловым эфиром и водой и доведение рН примерно до 2 при помощи добавления водной фосфорной кислоты, отделение водного слоя, промывание водного слоя метил-трет-бутиловым эфиром,доведение рН примерно до 10 при помощи карбоната натрия и конечная экстракция в этилацетате. Органические слои объединяют, промывают насыщенным хлоридом натрия, фильтруют и концентрируют в вакууме с получением соединения (11 е). Соединение (23) можно получать, если оно не является коммерчески доступным, при помощи серии реакций окисления, начиная с соединения (25) или (26). 3,3,3-Триацетокси-3-йодфталид растворяют в подходящем растворителе, таком как дихлорметан. Соединение (25) растворяют в том же растворителе и добавляют по каплям к окислителю. Примерно через 4 ч соединение (26) можно выделять при помощи способов, известных в данной области, таких как фильтрование через Celite, водная обработка с применением промывания водным тиосульфатом натрия и гидроксидом натрия, фильтрование и концентрирование в вакууме. Схема 12 Согласно схеме 12 соединение (22) можно получать при помощи синтеза кетонов по Вайнребу, который включает образование амида Вайнреба и последующую реакцию с металлорганическим нуклеофилом и образование целевого кетона. Сложный эфир (27) растворяют в соответствующем растворителе,таком как метанол, и добавляют основание, такое как гидроксид натрия. Кислоту (28) можно выделять традиционными способами, такими как водная обработка метил-трет-бутиловым эфиром и промывка водной хлористо-водородной кислотой. Твердое вещество можно применять в следующей реакции без очистки. Амид Вайнреба (29) получают перемешиванием кислоты (28) в соответствующем растворителе,таком как дихлорметан, в присутствии 1,1'-карбонилдиимидазола и добавления гидрохлорида N,Oдиметилгидроксиламина. Амид Вайнреба выделяют при помощи водной обработки, включающей промывание водным хлоридом аммония и насыщенным водным хлоридом натрия и концентрирование в вакууме. Соединение (29) можно применять на следующей стадии без дополнительной очистки. Следующая стадия включает растворение амида Вайнреба в растворителе, таком как тетрагидрофуран, охлаждении примерно до 0 С и добавление металлорганического нуклеофила, хлорида метилмагния. Это комплекс подвергают гидролизу путем выливания реакционной смеси в смесь лед/вода или водный хлорид аммония. Водная обработка с применением метил-трет-бутилового эфира и концентрирование в вакууме приводят к получению соединения (22). Соединение (22) можно применять на следующей стадии без очистки или после очистки хроматографией на силикагеле. Альтернативный подход для получения соединения (22) включает взаимодействие соединения (27) с хлоридом изопропилмагния и гидрохлоридом N,O-диметилгидроксиламина в растворителе, таком как тетрагидрофуран, при пониженной температуре с получением амида Вайнреба. Схема 13 Согласно схеме 13 соединение (11 е) также можно получать взаимодействием соединения (30) с ацетатом натрия в воде при повышенной температуре, затем с соединением (24) в соответствующем растворителе, таком как метанол, и водным гидроксидом аммония. Соединение (11 е) можно выделять способами, известными в данной области, такими как водная обработка, и можно применять без дополнительной очистки. Схема 14 Согласно схеме 14 получение соединения (30) начинают с получения аниона 1,1-дибромметана (32). Получение аниона включает получение диизопропиламида лития из диизопропиламина и н-бутиллития способами, распространенными в литературе. После получения диизопропиламида лития соединение(31) и соединение (32) перемешивают в подходящем растворителе, таком как тетрагидрофуран, при пониженной температуре, и диизопропиламид лития добавляют по каплям, поддерживая пониженную температуру. Реакцию гасят водной хлористо-водородной кислотой, затем проводят водную обработку растворителями, такими как метил-трет-бутиловый эфир и гептан. На схеме 15 показан синтез соединения (11f), который начинают с альдегида (33). Синтез проводят,получаяN-метил-N(метиленамино)метанамина с основанием, таким как 2,6-лутидин, и добавления ангидрида трифторуксусной кислоты при пониженной температуре. Промежуточное соединение подвергают взаимодействию с соединением (24) в уксусной кислоте и ацетате аммония с получением соединения (11f). Соединение(11f) можно дополнительно превращать в соединение (11g) превращением трифторметильного заместителя в циано заместитель нагреванием соединения (11f) в гидроксиде аммония, охлаждением и фильтрованием для выделения твердых веществ. Схема 16 Согласно схеме 16 соединение (38) можно получать серией реакций, начиная с синтеза простых эфиров по Вильямсону из соединения (34) и (35) в присутствии основания, такого как гидрид натрия, и растворителя, такого как метил-трет-бутиловый эфир, с получением простого эфира (36). Соединение(36) подвергают стандартным условиям реакции получения боронатного эфира (37). Эти реагенты могут включать хлорид лития, монохлорид меди и бис-(пинаколато)дибор и подходящий растворитель, такой как диметилформамид. Соединение (38) можно получать при помощи катализируемого рутением диспропорционирования олефинов с применением катализатора Граббса второго поколения в соответствующем растворителе, таком как дихлорметан. Соединение (38) можно применять для синтеза соединения формулы I, описанного на схеме 8. Специалистам в данной области очевидно, что не все заместители соединений формулы I могут выдерживать конкретные условия реакций, применяемых для синтеза соединений. Эти фрагменты можно вводить на удобной стадии синтеза или можно защищать, а затем снимать защиту при необходимости или при желании. Специалистам в данной области также очевидно, что защитные группы можно удалять на любой удобной стадии синтеза соединений согласно настоящему изобретению. Способы введения и удаления защищающих атом азота групп хорошо известны в данной области; см., например, Greene andWuts, "Protective Groups in Organic Synthesis", 3rd Ed., John Wiley and Sons, New York, Chapter 7 (1999). Кроме того, специалистам в данной области очевидно, что во многих случаях порядок, в котором вводят фрагменты, не важен. Конкретный порядок стадий, требуемых для получения соединений формулы I,зависит от конкретного синтезируемого соединения, исходного соединения и относительной подверженности воздействиям замещенных промежуточных соединений и продуктов. Пример получения 1. Добавляли раствор 4-амино-6-хлорпиримидин-5-карбальдегида (0,31 г, 1,94 ммоль) в тетрагидрофуране (4 мл) к смеси тетраизопропоксида титана (0,85 мл, 1,5 экв.), 2,2,2-трифторэтиламина (0,76 мл,4,9 экв.) и метанола (3,8 мл). Перемешивали реакционную смесь при комнатной температуре в течение ночи. Добавляли смесь 2:1 гидроксид аммония:вода к реакционной смеси, затем разбавляли этилацетатом. Разделяли слои. Сушили органические вещества над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (0,44 г, 96%). МС (ES) m/z=239 [M]+.(2,30 мл, 2,4 экв.) по каплям через шприц, поддерживая температуру, не превышающую 5 С. Добавляли раствор трет-бутиламиноборанового комплекса (1,86 г, 1,5 экв.) в дихлорметане (10 мл) по каплям через шприц, поддерживая температуру, не превышающую 5 С. Перемешивали реакционную смесь в течение 1 ч при 0 С. Добавляли смесь 2:1 гидроксид аммония:вода, затем разбавляли дихлорметаном. Разделяли слои. Сушили органические вещества над сульфатом натрия, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества (2,39 г, 69%). МС (ES) m/z=241 [M]+. Пример получения 3. 5-Хлор-3-(2,2,2-трифторэтил)-3,4-дигидропиримидо[4,5-d]пиримидин-2(1 Н)-он Смешивали 6-хлор-5-2,2,2-трифторэтиламино)метил)пиримидин-4-амин (9,57 г, 39,78 ммоль),триэтиламин (5,50 мл, 2,0 экв.) и дихлорметан (795 мл). Охлаждали до 0 С в атмосфере азота. Добавляли раствор трифосгена (11,85 г, 1,0 экв.) в дихлорметане (228 мл). Перемешивали при 0 С в течение 30 мин и оставляли нагреваться до комнатной температуры. Нагревали реакционную смесь до 40 С в течение ночи. Добавляли водный бикарбонат натрия и экстрагировали в этилацетате. Сушили органические вещества над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очищали хроматографией на силикагеле, элюируя смесью 9:1 дихлорметан:метанол с получением указанного в заголовке соединения (4,50 г, 42%). 1 Добавляли метоксид натрия (14,69 г, 0,85 экв.) к метанолу (70 мл). Кипятили с обратным холодильником в течение 15 мин, добавляя смесь диметилового эфира пропандиовой кислоты (36,64 мл, 320,00 ммоль) и метилкротонат (34,01 мл, 1,0 экв.). Смесь кипятили с обратным холодильником в течение 40 мин, затем оставляли смесь охлаждаться до комнатной температуры. Добавляли смесь метоксида натрия(19,02 г, 1,1 экв.), метанола (70 мл) и формамидинацетата (39,98 г, 1,2 экв.). Перемешивали при комнатной температуре в течение ночи. Охлаждали смесь на ледяной бане и добавляли 5 М водную хлористоводородную кислоту, доводя рН до 3. Фильтровали с получением указанного в заголовке соединения(41,00 г, 60%). МС (ES) m/z=213 [M]+. Следующие соединения получали по существу согласно описанию получения метил 3-(4,6 дигидроксипиримидин-5-ил)бутаноата.(95 мл). Добавляли фосфорилхлорид (39,50 мл, 2,2 экв.) по каплям в течение 10 мин (наблюдали выделение тепла). Перемешивали смесь в течение 10 мин и добавляли N,N-диэтиланилин (34,00 мл, 1,1 экв.) по каплям в течение 10 мин (наблюдали выделение тепла). Смесь кипятили с обратным холодильником в течение ночи. Охлаждали смесь на ледяной бане. Медленно добавляли предварительно охлажденную смесь водного раствора двухосновного фосфата калия (336,52 г в 500 мл воды, 10 экв.). Экстрагировали водный слой в этилацетате. Промывали органические вещества насыщенным водным хлоридом натрия,сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очищали хроматографией на силикагеле, элюируя 10-40% этилацетата в гексане, с получением указанного в заголовке соединения (44,00 г, 55%). МС (ES) m/z=249 [M]+. Следующие соединения получали по существу согласно описанию получения метил 3-(4,6 дихлорпиримидин-5-ил)бутаноата. Растворяли метил 3-(4,6-дихлорпиримидин-5-ил)пентаноат (10,00 г, 38,01 ммоль) в 28% гидроксида аммония в воде (95 мл) и закрывали в 350-мл пробирке. Нагревали реакционную смесь до 200 С в течение 2 ч. Охлаждали реакционную смесь на ледяной бане, затем фильтровали и промывали холодной водой. Сушили твердые вещества в вакууме с получением рацемата. Хиральное разделение (Chiralpak ASH, 100% этанол (мас.)/0,2% диметилэтиламина) приводило к получению указанного в заголовке соединения в виде энантиомера 2 (3,19 г, 40%) (99% эи). МС (ES) m/z=212 [M]+. Следующие соединения получали по существу согласно описанию получения (R)-4-хлор-5-этил 5,6-дигидропиридо[2,3-d]пиримидин-7(8 Н)-она. роксид аммония (100,00 мл, 7,5 экв.) в герметичной пробирке. Закрывали и перемешивали смесь при 60 С в течение ночи. Охлаждали смесь на ледяной бане. Отфильтровывали твердое вещество и промывали холодной водой с получением 4-хлор-5-метил-5,6-дигидропиридо[2,3-d]пиримидин-7(8 Н)-она (11,35 г, 60%). МС (ES) m/z=198 [M]+. Хиральное разделение (Chiralpak AS, этанол с 0,2% диметилэтиламина) приводило к получению(6 мл). Нагревали при 50 С в течение ночи. Оставляли охлаждаться до комнатной температуры. Разбавляли водой и экстрагировали в метил-трет-бутиловом эфире. Промывали органический слой насыщенным водным хлоридом натрия. Сушили органические вещества над безводным сульфатом магния, фильтровали и концентрировали в вакууме с получением смеси указанного в заголовке соединения и метил 3(4-хлор-6-(2,4-диметоксибензиламино)пиримидин-5-ил)-4,4,4-трифторбутаноата в виде маслянистой жидкости. Смешивали неочищенную смесь (0,78 г), диизопропилэтиламин (0,63 мл) и этанол (7,8 мл). Кипятили с обратным холодильником в течение 4 ч. Оставляли охлаждаться до комнатной температуры. Фильтровали и промывали этанолом с получением указанного в заголовке соединения в виде белого твердого вещества (0,43 г, 55%). Пример получения 15. 5,5,5-Трифторпентаналь Смешивали 3,3,3-триацетокси-3-йодфталид (17,91 г, 1,2 экв.) и дихлорметан (95 мл). Добавляли 5,5,5-трифтор-1-пентанол (5,00 г, 35,18 ммоль) в дихлорметане (238 мл) по каплям в атмосфере азота. Через 4 ч фильтровали реакционную смесь через Celite. Концентрировали фильтрат в вакууме; смешивали с 50 мл дихлорметана и промывали смесью 1:1 10% тиосульфат натрия:водный гидроксид натрия(1 Н). Сушили органические вещества безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветной маслянистой жидкости (2,13 г, 43%). 1 Смешивали 5,5,5-трифторпентаналь (2,01 г, 14,35 ммоль), 1,4-диоксан (10 мл), диоксид селена (1,62 г, 1,0 экв.), воду (0,51 мл) и уксусную кислоту (0,69 мл). Нагревали смесь при 90 С и перемешивали в течение ночи. Оставляли реакционную смесь охлаждаться до комнатной температуры. Фильтровали,промывали твердые вещества диоксаном. Объединяли фильтрат и промывочные растворы с получением указанного в заголовке соединения (2,21 г, 100%). ГХМС m/z=154. Следующие соединения получали по существу согласно описанию получения 5,5,5-трифтор-2 оксопентаналя. Добавляли 2,5 М н-бутиллитий в гексане (281,91 мл, 2,4 экв.) к раствору диизопропиламина (99,70 мл, 2,4 экв.) в тетрагидрофуране (900 мл) при 0 С. Перемешивали в течение 15 мин, затем добавляли раствор циклобутановой кислоты (28,65 мл, 293,66 ммоль) в тетрагидрофуране (100 мл) по каплям, поддерживая температуру, не превышающую 5 С. Перемешивали смесь при 5 С в течение 5 мин. Добавляли метилйодид (18,47 мл, 1,0 экв.) по каплям. Через 2 дня охлаждали смесь до 0 С и подкисляли 10% водной хлористо-водородной кислотой. Экстрагировали водную фазу в диэтиловом эфире. Промывали органические вещества насыщенным водным хлоридом натрия, сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением желтой маслянистой жидкости. Очищали хроматографией на силикагеле, элюируя смесью 5% этилацетата в гексане, с получением указанного в заголовке соединения в виде бесцветной жидкости (15,77 г, 47%). 1 Н ЯМР (400 МГц, CDCl3):11,84 (шир.с, 1 Н), 2,47 (м, 2 Н), 1,86 (м, 4 Н), 1,42 (с, 3 Н). Пример получения 23. 1-(1-Метилциклобутил)этанон Добавляли 1,6 М метиллитий в диэтиловом эфире (176,15 мл, 2,0 экв.) по каплям к раствору 1 метилциклобутанкарбоновой кислоты (15,77 г, 138,16 ммоль) в диэтиловом эфире (500 мл) при 0 С в течение 2 ч. Нагревали реакционную смесь до комнатной температуры и перемешивали в течение 5 ч. Выливали смесь в ледяную 3 М водную хлористо-водородную кислоту. Промывали органические вещества насыщенным водным бикарбонатом натрия, затем насыщенным водным хлоридом натрия. Сушили органические вещества над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветной маслянистой жидкости (11,70 г, выход 76%). 1H ЯМР (400 МГц, CDCl3-d3):2,38 (м, 2 Н), 1,90 (м, 2 Н), 1,70 (м, 2 Н), 1,38 (с, 3 Н), 1,10 (с, 3 Н). Следующие соединения получали по существу согласно описанию получения 1-(1 метилциклобутил)этанона. Добавляли 3-оксо-метиловый эфир бутановой кислоты (18,60 мл, 172,20 ммоль), бис-(2 хлорэтиловый) эфир (20,20 мл, 1,0 экв.), карбонат калия (52,42 г, 2,2 экв.) и йодид натрия (25,88 г, 1,0 экв.) в диметилформамиде (861 мл). Нагревали реакционную смесь при 80 С в течение ночи. Охлаждали до комнатной температуры. Дополнительно добавляли карбонат калия (23,78 г) и йодид натрия (25,88 г). Нагревали реакционную смесь при 80 С в течение 2 ч, затем оставляли смесь охлаждаться до комнатной температуры. Фильтровали через Celite и промывали этилацетатом. Промывали фильтрат водой и солевым раствором. Сушили органические вещества над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением метил 4-ацетилтетрагидро-2 Н-пиран-4-карбоксилата (23,06 г). Смешивали метил 4-ацетилтетрагидро-2 Н-пиран-4-карбоксилат (23,06 г, 123,84 ммоль) с изопропиловым спиртом (124 мл) и водой (124 мл). Добавляли серную кислоту (33,00 мл, 5,0 экв.). Нагревали реакционную смесь до 100 С в течение двух ночей. Охлаждали и медленно добавляли реакционную смесь к смеси бикарбоната натрия (136 г) в воде (1 л). Экстрагировали в дихлорметане. Сушили органические вещества над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очищали хроматографией на силикагеле, элюируя гексаном, смесями 25% этилацетата в гексане и 50% этилацетата в гексане, с получением указанного в заголовке соединения (7,39 г, 34%). 1 Н ЯМР (400 МГц, ДМСО-d6)3,79 (м, 2 Н), 3,27 (м, 2 Н), 2,54 (м, 1 Н), 2,05 (с, 3 Н), 1,66 (м, 2 Н), 1,38 Смешивали диоксид селена (10,45 г, 94,15 ммоль), 1,4-диоксан (60 мл), уксусную кислоту (5 мл) и воду (5 мл). Нагревали до 80 С в атмосфере азота, затем медленно добавляли 4,4,4-трифторбутан-2-он(9,01 мл, 1,0 экв.) по каплям. Нагревали при 90 С в атмосфере азота в течение 12 ч, затем оставляли охлаждаться до комнатной температуры. Фильтровали реакционную смесь с получением оранжевокрасного фильтрата. В отдельную колбу добавляли трет-бутил 4-формилпиперидин-1-карбоксилат (20,08 г, 1,0 экв.) в метаноле (150 мл) и гидроксид аммония (117,84 мл, 10,0 экв.). Охлаждали до 0 С в атмосфере азота. Добавляли фильтрат по каплям через капельную воронку. Оставляли нагреваться до комнатной температуры и перемешивали в течение ночи в атмосфере азота. Концентрировали насухо в вакууме. Добавляли воду и экстрагировали в этилацетате. Сушили органические вещества над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Очищали хроматографией на силикагеле, элюируя гексаном, смесями 4:1 гексан:этилацетат, 2:1 гексан:этилацетат, 1:1 гексан:этилацетат, 1:2 гексан:этилацетат, этилацетатом, с получением указанного в заголовке соединения в виде светлокоричневого твердого вещества (8,06 г, 26%). МС (ES) m/z=334 [M]+. Следующие соединения получали по существу согласно описанию получения трет-бутил 4-(4(2,2,2-трифторэтил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилата. Смешивали диоксид селена (5,72 г, 51,54 ммоль), 1,4-диоксан (52 мл), уксусную кислоту (2,4 мл,0,81 экв.), воду (2,4 мл) и 1-(тетрагидропиран-4-ил)этанон (6,28 г, 1,0 экв.). Перемешивали при 90 С в течение ночи. Охлаждали и фильтровали, затем промывали 1,4-диоксаном. Добавляли полученный фильтрат к раствору трет-бутил 4-формилпиперидин-1-карбоксилата (10,47 г, 1,0 экв.), метанола (78 мл) и 30% водного гидроксида аммония (30,8 мл) при 0 С. Оставляли смесь нагреваться до комнатной температуры и перемешивали в течение ночи. Концентрировали в вакууме и добавляли этилацетат и насыщенный водный хлорид натрия. Разделяли слои. Дополнительно экстрагировали водный слой в смеси 9:1 дихлорметан:изопропиловый спирт. Смешивали диоксид селена (0,91 г, 8,17 ммоль), 1,4-диоксан (8,3 мл), уксусную кислоту (0,4 мл,0,81 экв.), воду (0,41 мл) и 1-(тетрагидропиран-4-ил)этанон (1,00 г, 1,0 экв.). Перемешивали при 90 С в течение ночи. Охлаждали и фильтровали, затем промывали 1,4-диоксаном. Добавляли полученный фильтрат к раствору трет-бутил 4-формилпиперидин-1-карбоксилата (1,66 г, 1,0 экв.), метанола (12,4 мл) и 30% водного гидроксида аммония (4,9 мл) при 0 С. Оставляли смесь нагреваться до комнатной температуры и перемешивали в течение ночи. Концентрировали в вакууме и добавляли этилацетат и насыщенный водный хлорид натрия. Разделяли слои. Дополнительно экстрагировали водный слой в смеси 9:1 дихлорметан:изопропиловый спирт. Сушили объединенные органические слои, полученные в обеих реакциях, над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очищали хроматографией на силикагеле, элюируя гексаном, этилацетатом, смесями 5% метанола в этилацетате, 10% метанола в этилацетате, с получением указанного в заголовке соединения (6,68 г, 35%). МС (ES) m/z=336 [М]+. Пример получения 36. Смешивали диметилгидразин (10,63 мл, 139,77 ммоль) и параформальдегид (4,20 г, 0,33 экв.). Перемешивали реакционную смесь в течение 1 ч. Добавляли гептан (20 мл) и сульфат натрия (20 г). Перемешивали 5 мин, затем отфильтровывали сульфат натрия. Перегоняли фильтрат в устройстве для перегонки, собирая указанное в заголовке соединение в виде 75% (мас./мас.) раствора в гептане (10,3 г, 77%). 1 Добавляли N-метил-N-(метиленамино)метанамин (1,30 г, 18,03 ммоль) в хлороформе (100 мл), затем добавляли 2,6-лутидин (3,2 мл, 1,5 экв.). Охлаждали реакционную смесь до 0 С и добавляли ангидрид трифторуксусной кислоты (3,9 мл, 1,5 экв.) в течение 1 мин. Оставляли реакционную смесь перемешиваться при 0 С в течение 10 мин. Промывали последовательно 0,5 М водной HCl, водой и 0,1 М водным карбонатом натрия. Сушили органические вещества над безводным сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного (Е)-3-(диметилгидразоно)-1,1,1 трифторпропан-2-она (1,80 г). Добавляли часть промежуточного соединения (0,21 г, 1,25 ммоль) и трет-бутил 4 формилпиперидин-1-карбоксилат (0,33 г, 1,24 экв.) в уксусную кислоту (8 мл) и ацетат аммония (3,0 г). Нагревали при 80 С в течение 12 ч, затем оставляли смесь охлаждаться до комнатной температуры. Разбавляли дихлорметаном и насыщенным водным бикарбонатом натрия. Перемешивали в течение 10 мин,затем экстрагировали дважды в дихлорметане. Концентрировали органические вещества в вакууме и очищали хроматографией на силикагеле с получением указанного в заголовке соединения (0,19 г, 28%). 1 Добавляли ацетат натрия (360,8 г, 2,0 экв.) в воду (3,54 л) при 30 С. Добавляли 1,1-дибром-3,3,3 трифторацетон (653,01 г, 1,1 моль) по каплям. Нагревали смесь при 90 С в атмосфере азота в течение 1 ч. Добавляли трет-бутил 4-формилпиперидин-1-карбоксилат (470,00 г, 2,0 экв.) в метанол (10 л) в другой колбе при 30 С. Добавляли 28% водный раствор гидроксида аммония (2,53 л, 8,18 экв.) в раствор в метаноле. Охлаждали первую смесь до 30 С и добавляли по каплям к раствору в метаноле в течение 45 мин. Перемешивали в течение ночи в атмосфере азота. Удаляли растворитель из реакционной смеси. Добавляли воду (2 л) и дихлорметан (6 л) и перемешивали в течение 15 мин при 25 С. Трижды экстрагировали водный слой в дихлорметане (1 л 3). Промывали органические вещества насыщенным водным раствором хлорида натрия. Сушили над безводным сульфатом натрия и концентрировали в вакууме. Добавляли 5 л раствора 2% этилацетата в гексане и перемешивали при 30 С в течение 30 мин. Отфильтровывали твердое вещество, промывали гексаном и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (618,0 г, 88%). 1H ЯМР (400 МГц, CDCl3)10,5 (с, 1 Н), 7,4 (с, 1 Н), 4,18 (с, 2 Н), 2,98 (м, 1 Н), 2,80 (м, 2 Н), 2,01 (м,2 Н), 1,71 (м, 2 Н), 1,45 (с, 9 Н). Следующее соединение получали по существу согласно описанию получения трет-бутил 4-(4(трифторметил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилата. Смешивали трет-бутил 4-(4-(трифторметил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилат (1,50 г,4,70 ммоль) и гидроксид аммония (90 мл). Нагревали реакционную смесь при 60 С в течение двух дней. Оставляли охлаждаться до комнатной температуры, затем фильтровали через воронку Бюхнера. Промывали твердые вещества водой и гексаном с получением указанного в заголовке соединения в виде белого твердого вещества (1,08 г, 83%). МС (ES) m/z=221 [M]+. Пример получения 40. трет-Бутил 4-(1 Н-имидазол-2-ил)пиперидин-1-карбоксилат Смешивали гидроксид аммония (150 мл), трет-бутил 4-формилпиперидин-1-карбоксилат (29,82 г,139,81 ммоль) и метанол (600 мл). Добавляли этандиаль (16,10 мл, 1,0 экв.) (40% в воде) в атмосфере азота. Перемешивали в течение ночи. Затем концентрировали в вакууме для удаления метанола. Разбавляли водой, затем экстрагировали в дихлорметане. Промывали органические вещества насыщенным водным хлоридом натрия. Сушили органические вещества над сульфатом магния, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (33,30 г, 95%). МС (ES) m/z=252 [M]+. Следующие соединения получали по существу согласно описанию получения трет-бутил 4-(1 Нимидазол-2-ил)пиперидин-1-карбоксилата. Добавляли трет-бутил 4-(1 Н-имидазол-2-ил)пиперидин-1-карбоксилат (6,00 г, 23,87 ммоль) в дихлорэтане (200 мл). Добавляли N-бромсукцинимид (3,19 г, 1,0 экв.). Перемешивали при комнатной температуре в атмосфере азота в течение 14 ч. Концентрировали реакционную смесь в вакууме. Очищали хроматографией на силикагеле, элюируя гексаном, 9:1 гексан:этилацетат, 4:1 гексан:этилацетат, 2:1 гексан:этилацетат, 1:1 гексан:этилацетат, с получением двух основных фракций. Концентрировали фракцию, имеющую наибольший Rf, в вакууме с получением трет-бутил 4-(4,5-дихлор-1 Н-имидазол-2 ил)пиперидин-1-карбоксилата (2,25 г, 29%). МС (ES) m/z=321 [M]+. Концентрировали фракцию с меньшим Rf в вакууме. Суспензию, содержащую твердое вещество в смеси диэтиловый эфир/хлороформ, затем фильтровали. Концентрировали фильтрат в вакууме с получением трет-бутил 4-(4-хлор-1 Н-имидазол-2 ил)пиперидин-1-карбоксилата (1,16 г, 17%). МС (ES) m/z=286 [М]+. Пример получения 50. трет-Бутил 4-(4-изобутил-1-(2-(пирролидин-1-ил)этил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилат Смешивали трет-бутил 4-(4-изобутил-1 Н-имидазол-2-ил)пиперидин-1-карбоксилат (1,50 г, 4,88 ммоль) и гидроксид калия (1,65 г, 6,0 экв.) (в виде свежего порошка) в диметилсульфоксиде (30 мл). Нагревали реакционную смесь до 45 С в атмосфере азота. Через 5 мин добавляли гидрохлорид 1-(2 хлорэтил)пирролидина (1,08 г, 1,3 экв.). Прекращали нагревание через 2 ч. Добавляли воду и экстрагировали в этилацетате. Сушили органические вещества над безводным сульфатом магния, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтой маслянистой жидкости (2,11 г, 100%). МС (ES) m/z=405 [M]+. Следующие соединения получали по существу согласно описанию получения трет-бутил 4-(4 изобутил-1-(2-(пирролидин-1-ил)этил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилата (Примечание 1: можно получать смесь 4- и 5-замещенных изомеров в результате алкилирования. В некоторых случаях очистка нормально-фазовой хроматографией может приводить к получению целевого изомера. Примечание 2: применение реагентов 2-(2-бромэтокси)тетрагидро-2 Н-пирана или 2-(2-хлорэтокси)тетрагидро 2 Н-пирана может приводить к получению 2-(тетрагидро-2 Н-пиран-2-илокси)этильных соединений). Смешивали трет-бутил 4-(4-(тетрагидро-2 Н-пиран-4-ил)-1 Н-имидазол-2-ил)пиперидин-1 карбоксилат (5,84 г, 17,40 ммоль) и гидроксид калия (5,91 г, 6,0 экв.) (в виде свежего порошка) в диметилсульфоксиде (30 мл). Нагревали реакционную смесь до 45 С в атмосфере азота. Через 15 мин добавляли 2-(2-бромэтокси)тетрагидро-2 Н-пиран (2,90 мл, 1,1 экв.). Продолжали нагревать реакционную смесь в течение ночи. Добавляли воду и экстрагировали в этилацетате. Промывали органические вещества насыщенным водным хлоридом натрия. Сушили органические вещества над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Очищали хроматографией на силикагеле, элюируя гексаном, смесью 50% этилацетата в гексане, этилацетатом, смесью 10% метанола в этилацетате, с получением указанного в заголовке соединения в виде густой желтой маслянистой жидкости (6,94 г, 86%). МС (ES) m/z=464 [M]+. Пример получения 79. трет-Бутил 4-(4-(трифторметил)-1-(пирролидин-1-илэтил)-1H-имидазол-2-ил)пиперидин-1 карбоксилат сукцинат Добавляли хлорид 1-(2-хлорэтил)пирролидиния (73,50 г, 1,15 экв.) к смеси трет-бутил 4-(4(трифторметил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилата (120,00 г, 375,79 ммоль) и гидроксида калия (54,82 г, 2,60 экв.) в диметилсульфоксиде (1,1 л). Перемешивали полученную суспензию при 50 С в течение ночи. Охлаждали реакционную смесь до комнатной температуры и добавляли смесь лед/вода(1,50 л). Экстрагировали в этилацетате (3500 мл). Промывали органические вещества водой (2300 мл) и насыщенным водным хлоридом натрия (300 мл), сушили над безводным сульфатом натрия. Очищали хроматографией на силикагеле, элюируя 5-15% изопропиловым спиртом в дихлорметане, с получением смеси трет-бутил 4-(4-(трифторметил)-1-(2-пирролидин-1-илэтил)-1 Н-имидазол-2-ил)пиперидин-1 карбоксилата (122,00 г, 78%) и трет-бутил 4-(5-(трифторметил)-1-(2-пирролидин-1-илэтил)-1 Н-имидазол 2-ил)пиперидин-1-карбоксилата (14,00 г, 9%). Добавляли изопропиловый спирт (470 мл) и нагревали до 70 С. Добавляли раствор бутандиовой кислоты (35 г, 1 экв.) в изопропиловом спирте (350 мл), предварительно нагретом до 75 С. Прекращали нагревание и оставляли перемешиваться при комнатной температуре на ночь. Отфильтровывали твердое вещество и промывали изопропиловым спиртом (300 мл). Суспендировали твердое вещество в изопропиловом спирте (500 мл) и перемешивали в течение 15 мин. Отфильтровывали твердое вещество и дополнительно перемешивали в изопропиловом спирте (500 мл) в течение 15 мин, затем отфильтровывали с получением указанного в заголовке соединения (124,00 г, 82%) в виде белого твердого вещества. МС (ES) m/z=417 [M]+. Добавляли трет-бутил 4-(1-(2-(тетрагидро-2 Н-пиран-2-илокси)этил)-1 Н-имидазол-2-ил)пиперидин 1-карбоксилат (121,70 г, 320,69 ммоль) в дихлорметане (1750 мл). Добавляли N-бромсукцинимид (114,15 г, 2,0 экв.). Перемешивали при комнатной температуре в атмосфере азота. Прекращали реакцию через 90 мин. Разбавляли реакционную смесь водой и экстрагировали в дихлорметане. Сушили органические вещества над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Очищали хроматографией на силикагеле, элюируя гексаном, смесью 1:1 гексан:этилацетат, этилацетатом, с получением указанного в заголовке соединения в виде светло-оранжевой вязкой маслянистой жидкости (125,40 г,73%). МС (ES) m/z=538 [M]+. Пример получения 81. трет-Бутил 4-(4-бром-1-(2-(тетрагидро-2 Н-пиран-2-илокси)этил)-1 Н-имидазол-2-ил)пиперидин-1 карбоксилат Добавляли трет-бутил 4-(4,5-дибром-1-(2-(тетрагидро-2 Н-пиран-2-илокси)этил)-1 Н-имидазол-2 ил)пиперидин-1-карбоксилат (46,00 г, 85,61 ммоль) в тетрагидрофуран (1 л). Охлаждали до -78 С в атмосфере азота. Добавляли 1,6 М бутиллитий в гексане (90,97 мл, 1,7 экв.) по каплям в течение 15 мин. Поддерживали внутреннюю температуру, не превышающую -65 С. Через 65 мин добавляли изопропиловый спирт (50 мл). Оставляли нагреваться до комнатной температуры в течение 2 ч. Разбавляли насыщенным водным хлоридом аммония, затем трижды экстрагировали в этилацетате. Промывали органические вещества насыщенным водным хлоридом натрия. Сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (43,00 г, 100%). МС (ES) m/z=460 [M]+. Пример получения 82. трет-Бутил 4-(4-циклопропил-1-(2-(тетрагидро-2 Н-пиран-2-илокси)этил)-1 Н-имидазол-2 ил)пиперидин-1-карбоксилат Добавляли трет-бутил 4-(4-бром-1-(2-(тетрагидро-2 Н-пиран-2-илокси)этил)-1 Н-имидазол-2 ил)пиперидин-1-карбоксилат (1,82 г, 3,97 ммоль), циклопропилбороновую кислоту (0,44 г, 1,3 экв.), трициклогексилфосфин (0,11 г, 0,1 экв.) и фосфат калия (2,95 г, 3,5 экв.) в толуоле (18 мл) и воде (0,9 мл). Дегазировали азотом в течение 5 мин. Добавляли ацетат палладия (0,045 г, 0,05 экв.) и нагревали при 90 С в течение ночи. Прекращали нагревание через 12 ч. Разбавляли водой, затем трижды экстрагировали в этилацетате. Сушили над безводным сульфатом магния и концентрировали в вакууме. Очищали Смешивали трет-бутил 4-(4-этил-1-(2-(тетрагидро-2 Н-пиран-2-илокси)этил)-1 Н-имидазол-2 ил)пиперидин-1-карбоксилат (4,70 г, 11,53 ммоль), дихлорметан (100 мл) и метанол (50 мл). Медленно добавляли хлороводород (20 мл) (4 М в диоксане). Перемешивали в течение ночи в атмосфере азота. Концентрировали в вакууме с получением указанного в заголовке соединения (3,40 г, 100%). МС (ES) m/z=224 [M]+. Следующие соединения получали по существу согласно описанию получения дигидрохлорида 2-(4 этил-2-(пиперидин-4-ил)-1 Н-имидазол-1-ил)этанола. Смешивали трет-бутил 4-(4-бром-1-(2-(тетрагидро-2 Н-пиран-2-илокси)этил)-1 Н-имидазол-2 ил)пиперидин-1-карбоксилат (5,00 г, 10,91 ммоль), тетрагидрофуран (150 мл) и 1 М водную хлористоводородную кислоту (50 мл); оставляли перемешиваться на ночь при комнатной температуре. Разбавляли этилацетатом. Промывали избытком 1 М водного гидроксида натрия, затем насыщенным хлоридом натрия. Сушили органические вещества над безводным сульфатом магния, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтой пены (3,84 г, 94%). МС (ES) m/z=376 [M]+. Следующие соединения получали по существу согласно описанию получения трет-бутил 4-(4-бром 1-(2-гидроксиэтил)-1 Н-имидазол-2-ил)пиперидин-1-карбоксилата. Смешивали трет-бутил 4-(1-(2-(тетрагидро-2 Н-пиран-2-илокси)этил)-4-(тетрагидро-2 Н-пиран-4-ил)1 Н-имидазол-2-ил)пиперидин-1-карбоксилат (6,88 г, 14,84 ммоль), тетрагидрофуран (136 мл) и 1 н. водную хлористо-водородную кислоту (25 мл). Перемешивали реакционную смесь при комнатной температуре в течение ночи. Разбавляли этилацетатом и промывали насыщенным водным хлоридом натрия и насыщенным водным бикарбонатом натрия. Экстрагировали объединенные водные слои в 9:1 смеси дихлорметан:изопропиловый спирт. Сушили объединенные органические слои над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Очищали хроматографией на силикагеле, элюируя гексаном, смесью 50% этилацетата в гексане, этилацетатом, смесями 10% метанола в этилацетате, 10% метанола в дихлорметане, с получением указанного в заголовке соединения в виде белого твердого вещества (4,48 г, 79%). МС (ES) m/z=380 [М]+.
МПК / Метки
МПК: C07D 487/04, A61P 35/00, A61K 31/519, C07D 471/04
Метки: содержащие, ингибиторы, составы, фармацевтические
Код ссылки
<a href="https://eas.patents.su/30-20151-ingibitory-akt-i-farmacevticheskie-sostavy-ih-soderzhashhie.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы akt и фармацевтические составы, их содержащие</a>
Фармацевтические составы, содержащие ингибиторы деацетилазы гистонов

Номер патента: 18982
Опубликовано: 30.12.2013
Авторы: Бастин Ричард Дж., Хагес Николас Дж.
МПК: A61K 31/18, A61K 47/18, A61K 47/26...
Метки: деацетилазы, гистонов, составы, ингибиторы, содержащие, фармацевтические
Формула / Реферат:
1. Фармацевтическая композиция для ингибирования деацетилазы гистонов и лечения состояний, опосредуемых деацетилазой гистонов, у субъекта, представляющего собой животное или человека, включающая:(а) ингибитор деацетилазы гистонов, причем указанный ингибитор деацетилазы гистонов представляет собой соединение следующей формулы или фармацевтически приемлемую соль или сольват указанного соединения:и (b) свободный аргинин или фармацевтически...
Топикальные, содержащие нимесулид фармацевтические составы

Номер патента: 2694
Опубликовано: 29.08.2002
Авторы: Фигероа Рей, Эмбил Корал
МПК: A61K 31/18, A61P 29/00
Метки: фармацевтические, топикальные, составы, нимесулид, содержащие
Формула / Реферат:
1. Состав, включающий нимесулид в фазе растворителя глицерилмоноолеата, содержащей глицерилмоноолеат в количестве 17-59 вес.% состава. 2. Состав, включающий нимесулид в фазе растворителя глицерилмоноолеата, где фаза растворителя глицерилмоноолеата имеет жидкую кристаллическую структуру. 3. Состав, включающий нимесулид в количестве 0,1-5 вес.% состава, глицерилмоноолеат в количестве 17-59 вес.% состава и неводный растворитель в количестве 40-82...
Жидкие составы, содержащие диалкилсульфосукцинат и ингибиторы гидроксифенилпируват-диоксигеназы

Номер патента: 15674
Опубликовано: 31.10.2011
Авторы: Хаазе Детлеф, Боммель Мартин, Шнабель Герхард, Фриш Герхард
МПК: A01N 25/30, A01N 41/10, A01N 43/56...
Метки: ингибиторы, жидкие, гидроксифенилпируват-диоксигеназы, составы, содержащие, диалкилсульфосукцинат
Формула / Реферат:
1. Жидкие составы, содержащие:A) одно или несколько гербицидных биологически активных веществ из группы HPPD-ингибиторов, состоящей из пирасульфатола и топрамезона,B) одно или несколько поверхностно-активных веществ из группы диалкилсульфосукцинатов,C) одно или несколько отличных от В) поверхностно-активных веществ иD) один или несколько растворителей, причем молярное соотношение HPPD-ингибитора к диалкилсульфосукцинату составляет меньше чем 1:6...
Фармацевтические составы, содержащие соединения активного витамина d

Номер патента: 8072
Опубликовано: 27.02.2007
Авторы: Ю Кси-Юн, Фан Жун, Уайтхаус Марта Дж., Чен Эндрю-Ксиан
МПК: A61K 31/59
Метки: активного, составы, соединения, фармацевтические, витамина, содержащие
Формула / Реферат:
1. Жидкий или полутвердый фармацевтический состав, который содержит соединение активного витамина D, 50% компонента липофильной фазы, поверхностно-активное вещество и один или более антиоксидантов. 2. Фармацевтический состав по п.1, в котором указанное соединение активного витамина D представляет собой кальцитриол. 3. Фармацевтический состав по п.1, в котором указанный компонент липофильной фазы представляет собой Miglyol 812. 4....
Производные дигидро- и тетрагидрохинолина, способ их получения и содержащие их фармацевтические составы

Номер патента: 3272
Опубликовано: 27.02.2003
Авторы: Локар Бриан, Казара Патрик, Лестаж Пьер, Дорей Жильбер
МПК: A61P 25/28, A61K 31/4709, C07D 215/06...
Метки: получения, тетрагидрохинолина, способ, производные, дигидро, содержащие, составы, фармацевтические
Формула / Реферат:
1. Соединения общей формулы (I) где R1 представляет собой атом водорода или группу где A представляет собой атом водорода или группу -BNZ1Z2, в которой B представляет собой линейную или разветвленную (C1-C6)алкиленовую группу и Z1 и Z2 независимо представляют собой атом водорода или алкильную, (C3-C8)циклоалкильную или необязательно замещенную арильную группу или вместе с соединяющим их атомом азота образуют необязательно замещенную...