Малатная соль n-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-n’-(4-фторфенил)циклопропан-1,1-дикарбоксамида и ее кристаллические формы для лечения рака
Номер патента: 19959
Опубликовано: 30.07.2014
Авторы: Галлахер Уилльям П., Лэмб Питер, Браун Эйдриан Ст.Клер






Формула / Реферат
1. (D)-Малатная соль N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида.
2. Малатная соль N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.1, в которой упомянутая соль является кристаллической.
3. Малатная соль N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.2, которая находится в кристаллической форме N-1, и упомянутая форма N-1 характеризуется по меньшей мере одним из следующего:
(i) твердофазным 13С ЯМР спектром с четырьмя или более пиками, выбранными из 18,1; 42,9; 44,5; 70,4; 123,2; 156,2; 170,8; 175,7 и 182,1 ppm ±0,2 ppm;
(ii) порошковой рентгенограммой (CuKα l=1,5418 Å), содержащей четыре или более 2θ величин, выбранных из 12,8±0,2°2θ; 13,5±0,2°2θ; 16,9±0,2°2θ; 19,4±0,2°2θ; 21,5±0,2°2θ; 22,8±0,2°2θ; 25,1±0,2°2θ; 27,6±0,2°2θ, в которой измерение кристаллической формы осуществляли при комнатной температуре; и/или
(iii) порошковой рентгенограммой (XRPD) в основном в соответствии с рентгенограммой, показанной на фиг. 1.
4. Малатная соль N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.3, которая содержит минимум 90% по весу формы N-1 в расчете на вес соли.
5. Малатная соль N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.2, которая находится в кристаллической форме N-2, и упомянутая форма N-2 характеризуется по меньшей мере одним из следующего:
(i) твердофазным 13С ЯМР спектром с четырьмя или более пиками, выбранными из 23,0; 25,9; 38,0; 41,7; 69,7; 102,0; 122,5; 177,3; 179,3; 180,0 и 180,3 ppm ±0,2 ppm;
(ii) порошковой рентгенограммой (CuKα l=1,5418 Å), содержащей 2θ величины при 20,9±0,2°2θ и 21,9±0,2°2θ и две или более 2θ величины, выбранные из 6,4±0,2°2θ; 9,1±0,2°2θ; 12,0±0,2°2θ; 12,8±0,2°2θ; 13,7±0,2°2θ; 17,1±0,2°2θ; 22,6±0,2°2θ; 23,72q±0,2°2θ, в которой измерение кристаллической формы осуществляли при комнатной температуре; и/или
(iii) порошковой рентгенограммой (XRPD) в основном в соответствии с рентгенограммой, показанной на фиг. 8.
6. Малатная соль N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.5, которая содержит минимум 90% по весу формы N-2 в расчете на вес соли.
7. Фармацевтическая композиция, содержащая малатную соль N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида согласно одному из пп.1-6 и фармацевтически приемлемое вспомогательное вещество.
8. Применение малатной соли N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида согласно любому одному из пп.1-6 для получения лекарственного средства для лечения рака.
9. Применение малатной соли N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по любому одному из пп.1-6 для лечения рака.
10. Лекарственное средство для лечения рака щитовидной железы у субъекта, содержащее кристаллическую форму (L)-малатной соли N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида согласно любому одному из пп.1-6.
11. Лекарственное средство для лечения глиобластомы у субъекта, содержащее кристаллическую форму (L)-малатной соли N-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида согласно любому одному из пп.1-6.
12. Лекарственное средство по п.10, где рак щитовидной железы представляет собой медуллярный рак щитовидной железы.
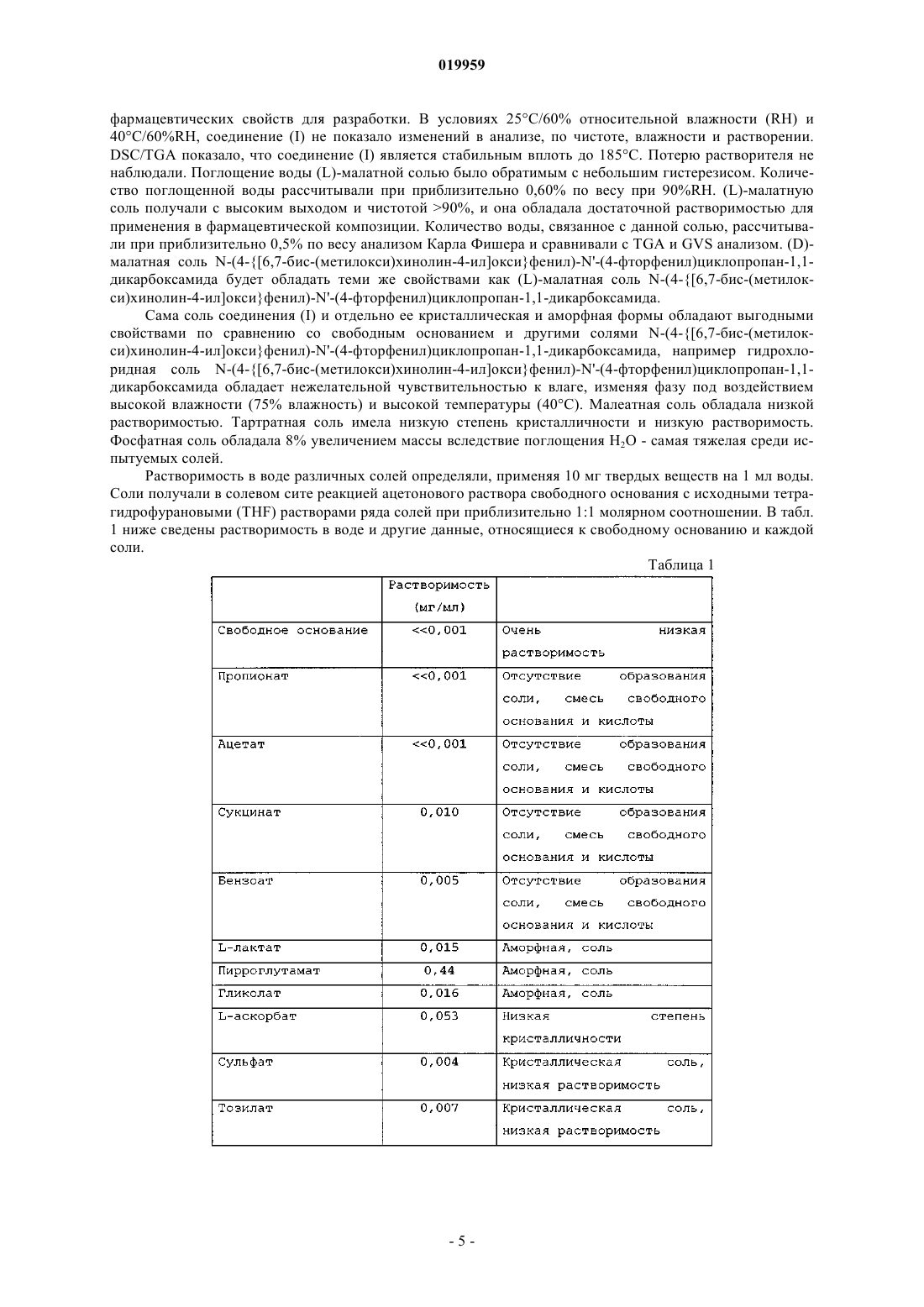
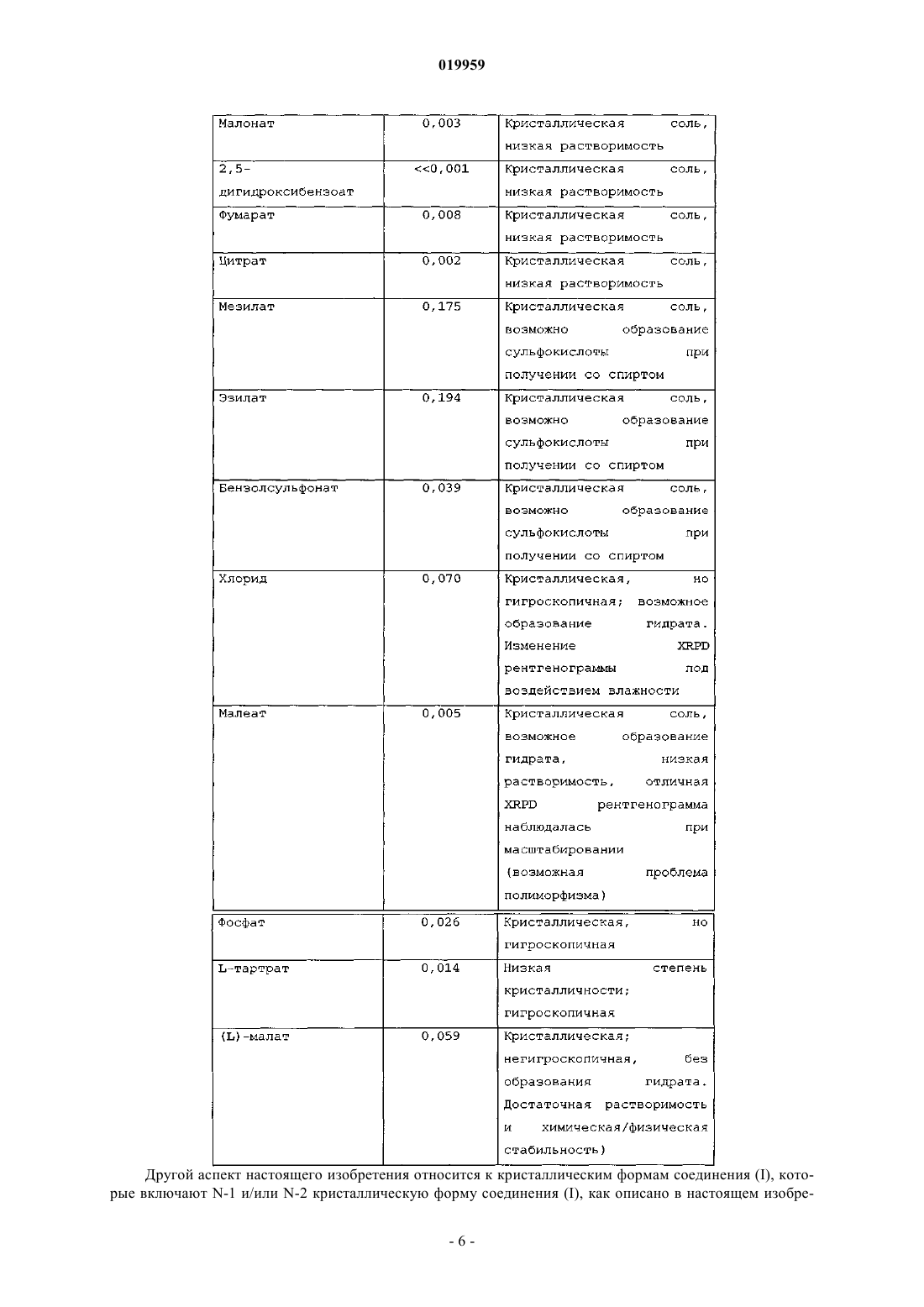
Текст
МАЛАТНАЯ СОЛЬ N-(4-[6,7-БИС-(МЕТИЛОКСИ)ХИНОЛИН-4-ИЛ]ОКСИФЕНИЛ)N'-(4-ФТОРФЕНИЛ)ЦИКЛОПРОПАН-1,1-ДИКАРБОКСАМИДА И ЕЕ КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ДЛЯ ЛЕЧЕНИЯ РАКА Изобретение относится к малатным солям N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, включая (L)-малатную соль, (D)-малатную соль, (DL)-малатную соль и их смеси; и кристаллическим и аморфным формам малатных солей. Изобретение также относится к фармацевтическим композициям, содержащим по меньшей мере одну малатную соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамида; и способам лечения рака, включающим введение по меньшей мере одной малатной соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамида. Перекрестная ссылка на родственные заявки По данной заявке испрашивается приоритет на основании предварительной заявки США 61/145421,внесенной в реестр 16 января 2009, которая включена в настоящее изобретение в качестве ссылки. Область техники, к которой относится изобретение Настоящее изобретение относится к малатным солям N-(4-[6,7-бис-(метилокси)хинолин-4 ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида и к кристаллическим и аморфным формам малатных солей N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. Малатные соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамида включают одну из: (1) (L)-малатной соли, (2) (D)-малатной соли, (3) (D,L)-малатной соли и (4) их смесей. Настоящее изобретение также относится к фармацевтическим композициям, содержащим по меньшей мере одну малатную соль N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. Настоящее изобретение также относится к фармацевтическим композициям, содержащим кристаллическую или аморфную форму по меньшей мере одной малатной соли N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. Настоящее изобретение также относится к способам лечения рака, включающим введение по меньшей мере одной малатной соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамида. Кроме того, настоящее изобретение относится к способам лечения рака, включающим введение кристаллической или аморфной формы по меньшей мере одной малатной соли N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопролан-1,1-дикарбоксамида. Уровень техники Обычно поразительные улучшения в лечении рака связывают с обнаружением терапевтических средств, действующих с помощью новых механизмов. Одним из механизмов, который можно применять для лечения рака, представляет собой модулирование активности протеинкиназы, поскольку передача сигнала посредством активации протеинкиназы является ответственной за многие характеристики опухолевых клеток. Передача сигнала с участием протеинкиназ является особенно важной, например, при раке щитовидной железы, раке желудка, раке головы и шеи, раке легкого, груди, простаты и колоректальном раке, а также при росте и пролиферации опухолевых клеток мозга. Протеинкиназы можно разделить на рецепторный и нерецепторный тип. Тирозинкиназы рецепторного типа состоят из большого количества трансмембранных рецепторов с различной биологической активностью. Что касается подробного обсуждения тирозинкиназ рецепторного типа, см. Plowman et al.,DNP 7(6): 334-339, 1994. Так как протеинкиназы и их лиганды играют значительную роль в различных клеточных активностях, нарушение регуляции ферментативной активности протеинкиназ может приводить к изменениям свойств клетки, таким как неконтролируемый клеточный рост, связанный с раком. Дополнительно к онкологическим показаниям измененная киназная передача сигнала связана с многочисленными другими патологическими заболеваниями, включая, например, иммунологические заболевания, сердечно-сосудистые заболевания, воспалительные заболевания и дегенеративные заболевания. Следовательно, протеинкиназы являются привлекательными мишенями для поиска низкомолекулярных лекарственных средств. Особенно привлекательные мишени для низкомолекулярной модуляции в отношении антиангиогенной и антипролиферативной активности включают тирозинкиназы рецепторного типа Ret, c-Met и VEGFR2. Киназа c-Met представляет собой прототипный член подсемейства гетеродимерных рецепторных тирозинкиназ (RTK), которое включает Met, Ron и Sea. Эндогенным лигандом для c-Met является фактор роста гепатоцитов (HGF), мощный индуктор ангиогенеза.Связывание HGF с с-Met вызывает активацию рецептора посредством аутофосфорилирования, приводя в результате к усилению зависящей от рецептора передачи сигнала, которая способствует росту клетки и инвазии. Показано, что анти-HGF антитела или HGF антагонисты ингибируют метастаз опухоли in vivo (см. Maulik et al. CytokineGrowth FactorReviews 2002 13, 41-59). Сверхэкспрессия c-Met, VEGFR2 и/или Ret была продемонстрирована на большом наборе типов опухолей, включая опухоль груди, толстой кишки, почки, легкого, плоскоклеточную миелоидную лейкемию, гемангиомы, меланомы, астроцитарную опухоль (которая включает глиобластому, гигантоклеточную глиобластому, глиосаркому и глиобластому с олигодендроглиальными компонентами). Ret белок представляет собой трансмембранный рецептор с тирозинкиназной активностью. Ret мутирует в большинстве наследственных форм медуллярного рака щитовидной железы. Данные мутации активируют киназную функцию Ret и превращают его в онкогенный продукт. Ингибирование EGF, VEGF и эфриновой передачи сигнала будет предотвращать клеточную пролиферацию и ангиогенез, два ключевых клеточных процесса, необходимых для роста и выживания опухоли(Matter A. Drug Disc. Techno1. 2001 6, 1005-1024). Киназные KDR (относится к тирозинкиназам рецептора с доменом, содержащим киназную вставку) и flt-4 (fms-подобная тирозинкиназа-4), оба, представляют собой рецепторы фактора роста эндотелия сосудов (VEGF). Ингибирование EGF, VEGF и эфриновой передачи сигнала будет препятствовать клеточной пролиферации и ангиогенезу, двум ключевым клеточным процессам, необходимым для роста и выживания опухоли (Matter A. Drug Disc. Technol. 2001 6,-1 019959 1005-1024). EGF и VEGF рецепторы представляют собой хорошие мишени для ингибирования низкомолекулярными молекулами. Соответственно, низкомолекулярные соединения, которые специфически ингибируют, регулируют и/или модулируют передачу сигнала киназ, в частности включая Ret, c-Met и VEGFR2, описанные выше,являются особенно подходящими в качестве средства для лечения или предотвращения болезненного состояния, связанного с нарушенной клеточной пролиферацией и ангиогенезом. Одной такой малой молекулой является N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан 1,1-дикарбоксамид, который имеет следующую химическую структуру:WO 2005/030140 описывает синтез N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамида (примеры 12, 37, 38 и 48) и также описывает терапевтическую активность данной молекулы по ингибированию, регулированию и/или модулированию передачи сигнала киназ (анализы, табл. 4, строка 289). Пример 48 находится в параграфе [0353] в WO 2005/030140. Помимо терапевтической эффективности, разработчик лекарственных средств приложил усилия по обеспечению подходящей формой терапевтического средства, которая обладает свойствами, относящимися к устойчивости при обработке, получении, хранении и/или пригодности в качестве лекарственного средства. Соответственно обнаружение формы, которая обладает некоторыми или всеми из данных желательных свойств, является крайне необходимым для разработки лекарственного средства. Авторы настоящего изобретения обнаружили солевую форму лекарственного N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, которая обладает подходящими свойствами для применения в фармацевтических композициях для лечения пролиферативных заболеваний, таких как рак. Новая солевая форма настоящего изобретения существует в кристаллической и аморфной формах. Сущность изобретения Настоящее изобретение относится к малатным солям N-(4-[6,7-бис-(метилокси)хинолин-4 ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, как описано в настоящем изобретении, их фармацевтическим композициям, как описано в настоящем изобретении, и их применению, как описано в настоящем изобретении. Другой аспект относится к кристаллическим и аморфным формам малатных солей N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, как описано в настоящем изобретении, их фармацевтическим композициям, как описано в настоящем изобретении, и их применению, как описано в настоящем изобретении. Краткое описание фигур Фиг. 1 показывает экспериментальную XRPD рентгенограмму для кристаллического соединения(I), форма N-1 при 25 С. Фиг. 2 показывает твердофазный 13 С ЯМР-спектр кристаллического соединения (I), форма N-1. Фиг. 3 показывает твердофазный 15N ЯМР-спектр кристаллического соединения (I), форма N-1. Фиг. 4 показывает твердофазный 19F ЯМР-спектр кристаллического соединения (I), форма N-1. Фиг. 5 показывает термический гравиметрический анализ (TGA) кристаллического соединения (I),форма N-1. Фиг. 6 показывает дифференциальную сканирующую калориметрию (DSC) кристаллического соединения (I), форма N-1. Фиг. 7 показывает поглощение влаги кристаллическим соединением (I), форма N-1. Фиг. 8 показывает экспериментальную XRPD рентгенограмму для кристаллического соединения(I), форма N-2 при 25 С. Фиг. 9 показывает твердофазный 13 С ЯМР спектр кристаллического соединения (I), форма N-2. Фиг. 10 показывает твердофазный 15N ЯМР спектр кристаллического соединения (I), форма N-2. Фиг. 11 показывает твердофазный 19F ЯМР спектр кристаллического соединения (I), форма N-2. Фиг. 12 показывает термический гравиметрический анализ (TGA) кристаллического соединения (I),форма N-2. Фиг. 13 показывает дифференциальную сканирующую калориметрию (DSC) кристаллического соединения (I), форма N-2. Фиг. 14 показывает поглощение влаги кристаллическим соединением (I), форма N-2. Фиг. 15 показывает экспериментальную и смоделированную XRPD рентгенограммы для кристаллического соединения (III), форма N-1 при комнатной температуре.-2 019959 Фиг. 16 показывает твердофазный 13 С ЯМР-спектр кристаллического соединения (III), форма N-1. Фиг. 17 показывает твердофазный 15N ЯМР-спектр кристаллического соединения (III), форма N-1. Фиг. 18 показывает твердофазный 19F ЯМР-спектр кристаллического соединения (III), форма N-1. Фиг. 19 показывает термический гравиметрический анализ (TGA) кристаллического соединения(III), форма N-1. Фиг. 20 показывает дифференциальную сканирующую калориметрию (DSC) кристаллического соединения (III), форма N-1. Фиг. 21 показывает поглощение влаги кристаллическим соединением (III), форма N-1. Фиг. 22 показывает XRPD рентгенограмму аморфного соединения (I) при комнатной температуре. Фиг. 23 показывает твердофазный 13 С ЯМР-спектр аморфного соединения (I). Фиг. 24 показывает твердофазный 15N ЯМР-спектр аморфного соединения (I). Фиг. 25 показывает твердофазный 19F ЯМР-спектр аморфного соединения (I). Фиг. 26 показывает дифференциальную сканирующую калориметрию (DSC) аморфного соединения(I). Фиг. 27 показывает поглощение влаги аморфным соединением (I). Подробное описание Настоящее изобретение относится к улучшению физико-химических свойств N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, в результате данное соединение может быть пригодно для разработки лекарственного средства. Описанными в настоящем изобретении являются малатные соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'(4-фторфенил)циклопропан-1,1-дикарбоксамида. Также описывают новые твердые формы данных солей. Малатные соли, а также их кристаллические и аморфные формы, описанные в настоящем изобретении,каждая представляет собой отдельный аспект настоящего изобретения. Хотя малатные соли и их твердые формы описывают в настоящем изобретении, настоящее изобретение также относится к новым композициям, содержащим описанные соли и твердые формы. Терапевтическое применение описанных солей и твердых форм, а также содержащих их терапевтических композиций представляют собой отдельные аспекты настоящего изобретения. Способы, применяемые для характеристики солей и их твердых форм,описывают в примерах ниже. Данные способы, отдельно или в комбинации, можно применять для характеристики солей и их солевых форм, описанных в настоящем изобретении. Соли и их солевые формы можно также характеризовать на основе описанных фигур. Было найдено, что N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамид обладает IC50 величиной для фермента Ret, приблизительно равной 5,2 нМ(наномолярный), и IC50 величиной для фермента c-Met, приблизительно равной 1,3 нМ (наномолярный). Анализ, который применяли для измерения данной c-Met активности, описывают в параграфе [0458] вRET биохимическую активность оценивали, применяя люцифераза-сопряженный хемилюминесцентный киназный анализ (LCCA), как описано в WO2005-030140. Киназную активность измеряли в виде количества АТФ в процентах, оставшегося после киназной реакции. Оставшийся АТФ обнаруживали люцифераза-люциферин-комбинированной хемилюминесценцией. Более определенно, реакцию инициировали смешиванием испытуемых соединений, 2 мкМ АТФ, 1 мкМ поли-EY и 15 нМ RET (человеческийRET киназный домен M700-D1042, экспрессированный бакуловирусом, с (HiS)6 меткой на N-конце) в 20 мкл буфере для анализа (20 мМ Tris-HCL рН 7,5, 10 мМ MgCl2, 0,01% Triton Х-100, 1 мМ DTT, 3 мМMnCl2). Смесь выдерживали при температуре окружающей среды в течение 2 ч, после чего добавляли 20 мкл смеси люцифераза-люциферин и хемилюминисцентный сигнал регистрировали, применяя WallacVictor2 ридер. Смесь люцифераза-люциферин состоит из 50 мМ HEPES, рН 7,8, 8,5 мкг/мл щавелевой кислоты (рН 7,8), 5 мМ DTT, 0,4% Triton Х-100, 0,25 мг/мл коэнзима А, 63 мкМ AMP, 28 мкг/мл люциферина и 40000 единиц света/мл люциферазы. Малатные соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]окси)фенил)-N'-(4-фторфенил)циклопропан-1,1 дикарбоксамида. Настоящее изобретение относится к малатным солям N-(4-[6,7-бис-(метилокси)хинолин-4 ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. Данные малатные соли являются комбинацией N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1 дикарбоксамида с оксиянтарной кислотой, которые образуют 1:1 малатную соль N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. Оксиянтарная кислота имеет следующую структуру: Из-за ее хирального атома углерода существует два энантиомера оксиянтарной кислоты: (L)оксиянтарная кислота и (D)-оксиянтарная кислота.(L)-оксиянтарная кислота имеет следующую структуру: Существуют различные названия или обозначения (L)-оксиянтарной кислоты, которые являются известными в данной области техники. Они включают бутандикислоту, гидрокси-, (2S)-(9CI); бутандикислоту, гидрокси-, (S)-; оксиянтарную кислоту, L-(8CI); оксиянтарную кислоту, 1-(3CI); (-)-(S)оксиянтарную кислоту; (-)-гидроксиянтарную кислоту; (-)-(L)-оксиянтарную кислоту; (-)-оксиянтарную кислоту; (2S)-2-гидроксибутандикислоту; (2S)-2-гидроксиянтарную кислоту; (S)-оксиянтарную кислоту; яблочную кислоту; L-(-)-оксиянтарную кислоту; (L)-оксиянтарную кислоту; NSC 9232; S-(-)оксиянтарную кислоту и S-2-гидроксибутандикислоту.(D)-оксиянтарная кислота имеет следующую структуру: Существует различные названия или обозначения (D)-оксиянтарной кислоты, которые являются известными в данной области техники. Они включают бутандикислоту, 2-гидрокси-, (2R)-, бутандикислоту, гидрокси-, (2R)-(9CI); бутандикислоту, гидрокси-, (R)-; (+)-оксиянтарную кислоту; (2R)-2 гидроксибутандикислоту; (2R)-оксиянтарную кислоту; (R)-(+)-оксиянтарную кислоту; (R)-оксиянтарную кислоту; D-(+)-2-гидроксиянтарную кислоту; D-(+)-оксиянтарную кислоту и D-оксиянтарную кислоту. Как описано выше, химическая структура N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'(4-фторфенил)циклопропан-1,1-дикарбоксамида представляет собой В ее химической структуре нет хиральных атомов углерода. Существуют различные названия дляN-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, которые являются известными для всех, и некоторые из данных различных названий или обозначений включают 1,1-циклопропандикарбоксамид, N'-[4-[(6,7-диметокси-4-хинолинил)окси]фенил]-N-(4-фторфенил)- и 1,1-циклопропандикарбоксамид, N-[4-[(6,7-диметокси-4-хинолинил)окси]фенил]-N'-(4 фторфенил)-(9CI).N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамид можно получить согласно любой из нескольких различных методик или в граммовом масштабе(1 кг), или в килограммовом масштабе (1 кг). Способ в граммовом масштабе изложен в WO 2005030140, который описывает синтез N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида (примеры 25, 37, 38 и 48), который вводится в настоящее изобретение с помощью ссылки. Альтернативно, N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамид, включая активное соединение(я), можно получить в килограммовом масштабе, применяя методику, изложенную в примере 1 ниже. Настоящее изобретение относится к малатным солям N-(4-[6,7-бис-(метилокси)хинолин-4 ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида:(DL)-малатной соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида (Соединение (III. Каждое соединение обладает улучшенными свойствами по сравнению с N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамидом и его другими солями. Названия, применяемые в настоящем изобретении для характеристики конкретной формы,например "N-2" и т.д., не должны быть ограничивающими, чтобы исключать любое другое вещество,обладающее аналогичными или идентичными физическими и химическими характеристиками, но, скорее, данные названия применяют в качестве просто идентификаторов, которые следует толковать в соответствии с информацией о характеристиках, представленной в настоящем изобретении. Малатные соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, и в частности соединение (I), обладают предпочтительными комбинациями фармацевтических свойств для разработки. В условиях 25 С/60% относительной влажности (RH) и 40C/60%RH, соединение (I) не показало изменений в анализе, по чистоте, влажности и растворении.DSC/TGA показало, что соединение (I) является стабильным вплоть до 185 С. Потерю растворителя не наблюдали. Поглощение воды (L)-малатной солью было обратимым с небольшим гистерезисом. Количество поглощенной воды рассчитывали при приблизительно 0,60% по весу при 90%RH. (L)-малатную соль получали с высоким выходом и чистотой 90%, и она обладала достаточной растворимостью для применения в фармацевтической композиции. Количество воды, связанное с данной солью, рассчитывали при приблизительно 0,5% по весу анализом Карла Фишера и сравнивали с TGA и GVS анализом. (D)малатная соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1 дикарбоксамида будет обладать теми же свойствами как (L)-малатная соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. Сама соль соединения (I) и отдельно ее кристаллическая и аморфная формы обладают выгодными свойствами по сравнению со свободным основанием и другими солями N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, например гидрохлоридная соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1 дикарбоксамида обладает нежелательной чувствительностью к влаге, изменяя фазу под воздействием высокой влажности (75% влажность) и высокой температуры (40 С). Малеатная соль обладала низкой растворимостью. Тартратная соль имела низкую степень кристалличности и низкую растворимость. Фосфатная соль обладала 8% увеличением массы вследствие поглощения Н 2 О - самая тяжелая среди испытуемых солей. Растворимость в воде различных солей определяли, применяя 10 мг твердых веществ на 1 мл воды. Соли получали в солевом сите реакцией ацетонового раствора свободного основания с исходными тетрагидрофурановыми (THF) растворами ряда солей при приблизительно 1:1 молярном соотношении. В табл. 1 ниже сведены растворимость в воде и другие данные, относящиеся к свободному основанию и каждой соли. Таблица 1 Другой аспект настоящего изобретения относится к кристаллическим формам соединения (I), которые включают N-1 и/или N-2 кристаллическую форму соединения (I), как описано в настоящем изобре-6 019959 тении. Каждая из форм соединения (I) представляет собой отдельный аспект настоящего изобретения. Аналогично, другой аспект настоящего изобретения относится к кристаллическим формам соединения(II), которые включают N-1 и/или N-2 кристаллическую форму соединения (II), как описано в настоящем изобретении. Каждая из которых также представляет собой отдельный аспект настоящего изобретения. Как известно в данной области техники, кристаллическая (D)-малатная соль будет образовывать ту же кристаллическую форму и иметь те же свойства, что и кристаллическое соединение (I). См. WO 2008/083319, который описывает свойства кристаллических энантиомеров. Смеси кристаллических форм соединений (I) и (II) представляют собой другой аспект настоящего изобретения. Кристаллические N-1 формы соединений (I) и (II), как описано в настоящем изобретении, можно характеризовать по меньшей мере одним из следующего:(iii) порошковой рентгенограммой (CuK =1,5418), содержащей четыре или более пиков, выбранных из 6,4; 9,0; 12,0; 12,8; 13,5; 16,9; 19,4; 21,5; 22,8; 25,1 и 27,62, 0,22, для которой измерение кристаллической формы осуществляли при комнатной температуре;(vi) твердофазным 15N ЯМР-спектром в основном в соответствии со спектром, показанным на фиг. 3. Другие свойства твердого состояния, которые можно применять для характеристики кристаллических N-1 форм соединений (I) и (II), показаны на чертежах и обсуждаются в примерах ниже. Для кристаллического соединения (I) твердая фаза и степень кристалличности остаются без изменения после воздействия 75% RH при 40 С в течение 1 недели. Кристаллические N-2 формы соединений (I) и (II), как описано в настоящем изобретении, можно характеризовать по меньшей мере одним из следующего:(iii) порошковой рентгенограммой (CuK =1,5418), содержащей четыре или более пиков, выбранных из 6,4; 9,1; 12,0; 12,8; 13,7; 17,1; 20,9; 21,9; 22,6 и 23,72, 0,22, для которой измерение кристаллической формы осуществляли при комнатной температуре;(vi) твердофазным 15N ЯМР-спектром в основном в соответствии со спектром, показанным на фиг. 10. Другие свойства твердого состояния, которые можно применять для характеристики кристаллических N-2 форм соединений (I) и (II), показаны на чертежах и обсуждаются в примерах ниже. В другом варианте осуществления настоящее изобретение относится к кристаллической форме соединения (I), как описано в настоящем изобретении, в любом из аспектов и/или вариантов осуществления, которая является практически чистой N-1 формой. В другом варианте осуществления настоящее изобретение относится к кристаллической форме соединения (I), как описано в настоящем изобретении, в любом из аспектов и/или вариантов осуществления, которая является практически чистой N-2 формой. Настоящее изобретение также относится к аморфным формам соединений (I) и (II). Получение и свойства и характеристики твердых состояний аморфной формы соединения (I) описывают в примерах ниже. Аморфные формы соединений (I) и (II) представляют собой другой аспект настоящего изобретения. Еще один аспект настоящего изобретения относится к смеси соединения (I) и соединения (II). Смеси могут содержать от более чем 0 до менее чем 100 вес.% соединения (I) и от менее чем 100 до более чем 100 вес.% соединения (II) на основе суммарного веса соединения (I) и соединения (II). В других вариантах осуществления смесь содержит от приблизительно 1 до приблизительно 99 вес.% соединения (I) и от приблизительно 99 до приблизительно 1 вес.% соединение (II) на основе суммарного веса соединения (I) и соединения (II) в упомянутой смеси. В следующем варианте осуществления смесь содержит от приблизительно 90 до менее чем 100 вес.% соединения (I) и от более чем 0 до приблизительно 10 вес.% соединения (II) на основе суммарного веса соединения (I) и соединения (II). Соответственно смесь может содержать 1-10 вес.% соединения (I); 11-20 вес.% соединения (I); 2130 вес.% соединения (I); 31-40 вес.% соединения (I); 41-50 вес.% соединения (I), 51-60 вес.% соединения(I); 61-70 вес.% соединения (I); 71-80 вес.% соединения (I); 81-90 вес.% соединения (I) или 91-99 вес.% соединения (I), причем оставшиеся массовые проценты малатной соли являются массовыми процентами соединения (II). Другой аспект настоящего изобретения относится к кристаллическим формам (DL)-малатной соли N(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида,соединение (III). (DL)-Малатную соль получают из рацемической оксиянтарной кислоты. Кристаллическую N-1 форму соединения (III), как описано в настоящем изобретении, можно характеризовать по меньшей мере одним из следующего:(i) твердофазным 13 С ЯМР спектром с четырьмя или более пиками, выбранными из 20,8; 26,2; 44,8; 55,7; 70,7; 100,4; 101,0; 114,7; 115,2; 116,0; 119,7; 120,4; 121,6; 124,4; 136,9; 138,9; 141,1; 145,7; 150,3; 156,5; 157,6; 159 6; 165,2; 167,4; 171,2; 176,3 и 182,1 ppm, 0,2 ppm,(ii) твердофазным 13 С ЯМР спектром в основном в соответствии со спектром, показанным на фиг. 16;(iii) порошковой рентгенограммой (CuK =1,5418), содержащей четыре или более 2 величин,выбранных из 12,8; 13,5; 16,9; 19,4; 21,5; 22,8; 25,1 и 27,6, 0,22, для которой измерение кристаллической формы осуществляли при комнатной температуре;(vi) твердофазным 15N ЯМР спектром в основном в соответствии со спектром, показанным на фиг. 17. Другие свойства твердого состояния, которые можно применять для характеристики кристаллической N-1 формы соединения (III), показаны на чертежах и обсуждаются в примерах ниже. В одном варианте осуществления N-1 форма соединения (III) характеризуется параметрами элементарной ячейки,практически равными следующим: Размеры ячейки: а=14,60=90,4=90,0 пространственная группа: P21/n молекулы соединения (I)/единичная ячейка: 4 объем = 2969 3 плотность (рассчитанная) = 1,422 г/см 3 Параметры элементарной ячейки формы N-1 соединения (III) измеряли при температуре приблизительно 25 С, например при комнатной температуре или температуре окружающей среды. Каждая из N-1 и N-2 кристаллических форм соединений (I) и (II) и кристаллическая форма N-1 соединения (III) обладают уникальными характеристиками, которые отличают их друг от друга. Данные характеристики можно понять сравнением физических свойств твердых форм, которые представлены в примерах ниже, например, в табл. 2 перечислены характеристические XRPD положения пиков(20,22) для кристаллического соединения (III), формы N-1 и форм N-1 и N-2 кристаллического соединения (I). Аморфные формы не обладают пиками отражения на своих XRPD рентгенограммах. Таблица 2. Положения характеристических дифракционных пиков (градусы 20,2)RT,на основе рентгенограммы, полученной на дифрактометре (CuK) с вращающимся капилляром Специфические отраженные сигналы между соединением (I), форма N-1, и соединением (I), форма N-2. Специфические отраженные сигналы между формами N-1 и N-2 кристаллического соединения (II) обозначают звездочкой . Как описано выше, соединение (II) представляет собой энантиомер соединения (I) и, таким образом, соединение (II), форма N-1, будет иметь такую же характеристичную дифракционную картину и уникальные пики, как пики, приведенные в табл. 2 для соединения (I), форма N-1. Аналогично соединение (II), форма N-2, будет иметь такую же характеристичную дифракционную картину и уникальные пики, как пики, приведенные в табл. 2 для соединения (I), форма N-2. Соединения (I) и (II) являются отличными друг от друга на основании их абсолютной стереохимии, т.е. (L)-малатной соли относительно (D)-малатной соли соответственно. Кристаллическое соединение (III), форма N-1,является отличной как (D,L)-малатная соль. Характеристичные пики из твердофазной ЯМР могут также служить для различия кристаллической и аморфной форм, описанных в настоящем изобретении. Например, в табл. 3 приведены характеристичные твердофазные 13 С ЯМР пики для кристаллического соединения (III), форма N-1; кристаллического соединения (I), формы N-1 и N-2, и аморфной формы соединения (I). Таблица 3. Твердофазные углерод-13 ЯМР резонансы (ppm, 0,2 ppm) Твердофазные 19F и 15N ЯМР спектры, обсуждаемые ниже, обеспечивают данными для аналогичного сравнения и характеристики. Как описано выше, будучи энантиомером соединения (I), кристаллические формы N-1 и N-2 и аморфная форма соединения (II) будут иметь те же твердофазные ЯМР резонансы и уникальные пики между ними, как те, что перечислены в табл. 3 для форм N-1 и N-2 кристаллического соединения (I). Фармацевтические композиции и способы лечения Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей по меньшей мере одно из соединений (I), (II), (III) или их комбинации, и фармацевтически приемлемое вспомогательное вещество. Количество соединений (I), (II), (III) или их комбинации в фармацевтической композиции может представлять собой терапевтически эффективное количество. Соединение (I), (II) или(III) может отдельно присутствовать в фармацевтической композиции в виде одной из твердых форм,описанных выше, или их комбинаций. Кристаллические формы представляют собой предпочтительные твердые формы. Соответственно другой аспект настоящего изобретения относится к твердой или дис-9 019959 персной фармацевтической композиции, содержащей по меньшей мере терапевтически эффективное количество кристаллической формы одного из соединений (I), (II), (III) или их комбинаций и фармацевтически приемлемое вспомогательное вещество. Другой аспект настоящего изобретения относится к способу лечения рака, включающему введение нуждающемуся в лечении субъекту по меньшей мере одного из соединений (I), (II), (III) или их комбинаций. Вводимое количество соединения (I), (II) или их комбинаций может представлять собой терапевтически эффективное количество. Соединения (I), (II) или (III) можно вводить отдельно в виде одной из твердых форм, описанных выше, или их комбинаций. Кристаллические формы представляют собой предпочтительные твердые формы, причем кристаллическое соединение (I), форма N-1 или N-2, являются предпочтительными. Соответственно другой аспект настоящего изобретения относится к способу лечения рака, включающему введение нуждающемуся в лечении субъекту терапевтически эффективного количества по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, в которых соединение (I), (II) или (III) присутствует в кристаллической форме. В другом аспекте настоящего изобретения,способ лечения можно осуществлять на практике введением фармацевтической композиции по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, так как описано выше. Другой аспект настоящего изобретения относится к способу лечения рака, как описано выше, где рак, подвергаемый лечению, представляет собой рак желудка, рак пищевода, рак почки, рак печени, рак яичника, рак шейки матки, рак толстой кишки, рак тонкой кишки, рак мозга (включая астроцитарную опухоль, которая включает глиобластому, гигантоклеточную глиобластому, глиосаркому и глиобластома с олигодендроглиальными компонентами), рак легкого (включая немелкоклеточный рак легкого), рак костей, рак простаты, рак поджелудочной железы, рак кожи, рак костей, лимфому, солидные опухоли,болезнь Ходжкина, неходжкинскую лимфому или рак щитовидной железы (включая медуллярный рак щитовидной железы). Ингибиторы тирозинкиназ применяют также для лечения немелкоклеточного рака легкого(NSCLC). Гефитиниб и эрлотиниб являются ингибиторами ангиогенеза, которые направлены на рецепторы зпидермального фактора роста, названного тирозинкиназой. Эрлотиниб и гефитиниб в настоящее время применяют для лечения NSCLC. Другой аспект настоящего изобретения относится к способу лечения немелкоклеточного рака легкого (NSCLC) у субъекта, причем способ включает введение нуждающемуся в лечении субъекту терапевтически эффективного количества N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида или его фармацевтически приемлемой соли необязательно в комбинации с эрлотинибом или гефитинибом. В другом варианте осуществления комбинация представляет собой комбинацию с эрлотинибом. Другой аспект настоящего изобретения относится к способу лечения немелкоклеточного рака легкого (NSCLC) у субъекта, причем способ включает введение нуждающемуся в лечении пациенту терапевтически эффективного количества эрлотиниба или гефитиниба в комбинации по меньшей мере с одним из соединений (I), (II), (III) или их комбинаций. Соединение (I), (II) или (III) можно вводить отдельно в виде одной из твердых форм, описанных выше, или их комбинаций. Кристаллические формы представляют собой предпочтительные твердые формы. Соответственно другой аспект настоящего изобретения относится к способу лечения немелкоклеточного рака легкого (NSCLC) у субъекта, причем способ включает введение нуждающемуся в лечении пациенту терапевтически эффективного количества эрлотиниба или гефитиниба в комбинации по меньшей мере с одним из соединений (I), (II), (III) или их комбинаций,в которых соединения (I), (II) или (III) присутствуют в кристаллической форме. В другом аспекте настоящего изобретения данный способ лечения можно осуществлять на практике введением фармацевтической композиции по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, так как описано выше. В другом варианте осуществления вводимая комбинация в данном способе представляет собой эрлотиниб по меньшей мере с одним из соединений (I), (II), (III) или их комбинаций. Другой аспект настоящего изобретения относится к способу лечения астроцитарной опухоли (которая включает глиобластому, гигантоклеточную глиобластому, глиосаркому и глиобластому с олигодендроглиальными компонентами у субъекта) у субъекта, причем способ включает введение нуждающемуся в лечении пациенту терапевтически эффективного количества N-(4-[6,7-бис-(метилокси)хинолин-4 ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. Другой аспект настоящего изобретения относится к способу лечения астроцитарной опухоли (которая включает глиобластому, гигантоклеточную глиобластому, глиосаркому и глиобластому с олигодендроглиальными компонентами у субъекта) у субъекта, причем способ включает введение нуждающемуся в лечении пациенту терапевтически эффективного количества по меньшей мере одного из соединений(I), (II), (III) или их комбинаций. Соединение (I), (II) или (III) можно вводить отдельно в виде одной из твердых форм, описанных выше, или их комбинации. Кристаллические формы представляют собой предпочтительные твердые формы. Соответственно другой аспект настоящего изобретения относится к способу лечения астроцитарной опухоли, включающему введение нуждающемуся в лечении субъекту терапевтически эффективного количества по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, в которых соединение(I), (II) или (III) присутствует в кристаллической форме. В другом аспекте настоящего изобретения дан- 10019959 ный способ лечения можно осуществлять на практике введением фармацевтической композиции по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, так как описано выше. Другой аспект настоящего изобретения относится к способу лечения рака щитовидной железы(включая медуллярный рак щитовидной железы) у субъекта, причем способ включает введение нуждающемуся в лечении пациенту N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида или его фармацевтически приемлемой соли. Вводимое количество может представлять собой терапевтически эффективное количество. Другой аспект настоящего изобретения относится к способу лечения рака щитовидной железы(включая медуллярный рак щитовидной железы) у субъекта, причем способ включает введение нуждающемуся в лечении пациенту по меньшей мере одного из соединений (I), (II), (III) или их комбинаций. Соединение (I), (II) или (III) можно вводить отдельно в виде одной из твердых форм, описанных выше,или их комбинаций. Кристаллические формы представляют собой предпочтительные твердые формы. Соответственно другой аспект настоящего изобретения относится к способу лечения рака щитовидной железы, включающему введение нуждающемуся в лечении субъекту терапевтически эффективного количества по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, в которых соединение (I), (II) или (III) присутствует в кристаллической форме. В другом аспекте настоящего изобретения данный способ лечения можно осуществлять на практике введением фармацевтической композиции по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, так как описано выше. Другой аспект настоящего изобретения относится к способу лечения заболеваний или расстройств,связанных с неконтролируемыми, ненормальными и/или нежелательными клеточными активностями. В данном способе нуждающемуся в лечении субъекту вводят по меньшей мере одно из соединений (I), (II),(III) или их комбинаций. Вводимое количество соединения (I), (II) или их комбинаций может представлять собой терапевтически эффективное количество. Соединение (I), (II) или (III) можно вводить отдельно в виде одной из твердых форм, описанных выше, или их комбинаций. Кристаллические формы представляют собой предпочтительные твердые формы. Соответственно другой аспект настоящего изобретения относится к способу лечения заболеваний или расстройств, связанных с неконтролируемыми, ненормальными и/или нежелательными клеточными активностями, включающему введение нуждающемуся в лечении субъекту терапевтически эффективного количества по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, в которых соединение (I), (II) или (III) присутствует в кристаллической форме. В другом аспекте настоящего изобретения данный способ лечения можно осуществлять на практике введением фармацевтической композиции по меньшей мере одного из соединений (I), (II), (III) или их комбинаций, так как описано выше. Другой аспект настоящего изобретения относится к способу лечения заболеваний или расстройств, связанных с неконтролируемыми, ненормальными и/или нежелательными клеточными активностями. В данном способе нуждающемуся в лечении субъекту вводят кристаллическую форму соединения (I), (II) или любую комбинацию соединения (I) и (II). Вводимое количество соединения (I), (II) или любой комбинации соединения (I) и (II) может представлять собой терапевтически эффективное количество. Другой аспект настоящего изобретения относится к применению малатной соли N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида согласно любому из вышеуказанных вариантов осуществления для получения лекарственного средства для лечения заболевания или расстройства, описанного выше. При растворении кристаллическая или аморфная форма по настоящему изобретению теряет свою структуру твердого состояния, и, следовательно, ее называют раствором, например, соединения (I). По меньшей мере одну кристаллическую форму, описанную в настоящем изобретении, можно применять для получения по меньшей мере одной жидкой рецептуры, в которой по меньшей мере одну кристаллическую форму по настоящему изобретению растворяют и/или суспендируют. Фармацевтическая композиция, такая как описано выше, может представлять собой любую фармацевтическую форму, которая содержит активное соединение (I), (II) и/или (III), включая их твердые формы (далее в настоящем изобретении называемые активное соединение(я). Фармацевтическая композиция может представлять собой, например, таблетку, капсулу, жидкую суспензию, инъецируемую, местную или трансдермальную. Фармацевтические композиции обычно содержат от приблизительно 1 до приблизительно 99 вес.% активного соединения(ий) или кристаллическую форму активного соединения(ий) и 99-1 вес.% подходящего фармацевтического вспомогательного вещества. В одном примере композиция будет содержать от приблизительно 5 до приблизительно 75 вес.% активного соединения, причем остаток представляет собой подходящие фармацевтические вспомогательные вещества или другие вспомогательные вещества, как описано ниже. Терапевтически эффективное количество активного соединения или кристаллической или аморфной формы активного соединения(ий), согласно настоящему изобретению, для ингибирования, регулирования и/или модулирования передачи сигнала киназ (описанные в настоящем изобретении относительно фармацевтических композиций) относится к количеству, достаточному для лечения пациента,страдающего от любого из ряда типов рака, связанных с нарушенной клеточной пролиферацией и ангиогенезом. Терапевтически эффективное количество согласно настоящему изобретению представляет со- 11019959 бой количество, терапевтически пригодное для лечения или предотвращения болезненных состояний и расстройств, описанных в настоящем изобретении. Соединения (I), (II) и/или (III) (включая их твердые формы), обладают терапевтической активностью относительно ингибирования, регулирования и/или модулирования передачи сигнала киназ, так как описано в WO2005-030140. N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамид. Реальное количество, требуемое для лечения любого конкретного пациента, будет зависеть от ряда факторов, включая болезненное состояние, подвергаемое лечению, и его тяжесть; конкретную применяемую фармацевтическую композицию; возраст, вес тела, общее состояние здоровья, пол и рацион пациента; способ введения; время введения; путь введения и скорость выведения активного соединения(ий) или кристаллической формы активного соединения(ий) согласно настоящему изобретению; продолжительность лечения; любые лекарственные средства, применяемые в комбинации или совместно с конкретным применяемым соединением; и другие данные факторы, хорошо известные в медицинской области техники. Данные факторы описывают в Goodman and Gilman's "The Pharmacological Basis of Therapeutics", Tenth Edition, A. Gilman, J. Hardman и L. Limbird, eds., McGraw-Hill Press, 155-173, 2001, которая вводится в настоящее изобретение с помощью ссылки. Активное соединение(я) или кристаллическую форму активного соединения(ий) согласно настоящему изобретению и содержащие их фармацевтические композиции можно применять в комбинации с противораковыми или другими агентами, которые обычно вводят пациенту, который подвергается лечению рака. Их можно также совместно формулировать с одним или более из данных агентов в единичной фармацевтической композиции. В зависимости от типа фармацевтической композиции фармацевтически приемлемый носитель можно выбирать из любого одного или комбинации носителей, известных в данной области техники. Выбор фармацевтически приемлемого носителя частично зависит от требуемого способа введения, который будет применяться. Для фармацевтической композиции настоящего изобретения, т.е. одного активного соединения(ий) или кристаллической формы активного соединения(ий) настоящего изобретения,носитель следует выбирать так, чтобы в основном сохранять конкретную форму активного соединения(ий), будет ли она кристаллической или нет. Другими словами, носитель не должен значительно изменять форму активного соединения(ий). Иначе, носитель не должен быть несовместимым с формой активного соединения(ий), таким как производящим любой нежелательный биологический эффект или,иначе, взаимодействуя неблагоприятным способом с любым другим компонентом(ами) фармацевтической композиции. Фармацевтические композиции настоящего изобретения можно получить способами, известными в области получения фармацевтических средств, например, см. Remington's Pharmaceutical Sciences, 18thEd., (Mack Publishing Company, Easton, Pa., 1990). В твердых лекарственных формах соединение (I) смешивают по меньшей мере с одним фармацевтически приемлемым вспомогательным веществом, таким как цитрат натрия или фосфат дикальция или (а) наполнителем или разбавителем, таким как, например,крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, (b) связующими, такими как, например, производные целлюлозы, крахмал, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь, (с) увлажняющими агентами, такими как, например, глицерин, (d) разрыхлителями, такими как, например, агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, кроскармелоза натрия, комплексные силикаты и карбонат натрия, (е) замедлителями растворения, такими как, например, парафин, (f) ускорителями абсорбции, такими как, например, четвертичные аммониевые соединения, (g) смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, стеарат магния и подобные; (h) адсорбентами, такими как, например, каолин и бентонит, и (i) смазывающими веществами, такими как, например, тальк, стеарат кальция, стеарат магния,твердые полиэтиленгликоли, лаурилсульфат натрия или их смеси. В случае капсулы, таблетки и пилюли лекарственные формы могут также содержать буферные вещества. Фармацевтически приемлемые вспомогательные вещества, известные в области получения фармацевтических средств, можно также применять в фармацевтических композициях настоящего изобретения. Они включают, но не ограничиваются, консерванты, смачивающие агенты, суспендирующие агенты, подсластители, ароматизаторы, ароматизирующие добавки, эмульгаторы и диспергирующие агенты. Предотвращение действия микроорганизмов можно обеспечить различными антибактериальными и антигрибковыми агентами, например парабенами, хлорбутанолом, фенолом, сорбиновой кислотой и подобными. Может также быть желательно включать изотонические агенты, например сахара, хлорид натрия и подобные. По желанию, фармацевтическая композиция настоящего изобретения может также содержать незначительные количества вспомогательных веществ, таких как увлажняющие агенты или эмульгаторы, рН буферные вещества и антиоксиданты, такие как, например, лимонная кислота, монолаурат сорбитана, олеат триэтаноламина и бутилированный гидрокситолуол. Твердые лекарственные формы, как описано выше, можно получить с покрытием и оболочками, такими как энтеросолюбильное покрытие таблетки, и другими, хорошо известными в данной области техники. Они могут содержать замедляющие высвобождение добавки и также могут быть такой композицией, что они высвобождают активное соединение или соединения в определенной части кишечника замедленным способом. Примерами включенных композиций, которые можно применять, являются поли- 12019959 мерные вещества и воски. Активное соединение может быть также в микроинкапсулированной форме,при необходимости, с одним или более из вышеупомянутых вспомогательных веществ. Суспензии в добавление к активным соединениям могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбитол и эфиры сорбитана,микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант или смеси данных веществ и подобные. Композициями для ректального введения являются, например, суппозитории, которые можно получить смешиванием активного соединения(ий) или кристаллической формы активного соединения(ий),например, с подходящими нераздражающими вспомогательными веществами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозитория, которые являются твердыми при обычной температуре, но жидкими при температуре тела и следовательно плавятся в подходящей полости тела и высвобождают в ней активный компонент. Поскольку активное соединение(я) или кристаллическую форму активного соединения(ий) сохраняют в процессе их получения, твердые лекарственные формы являются предпочтительными для фармацевтической композиции настоящего изобретения. Твердые лекарственные формы для перорального введения, которые включают капсулы, таблетки, пилюли, порошки и гранулы, являются особенно предпочтительными. В данных лекарственных формах активное соединение(я), смешивают по меньшей мере с одним инертным фармацевтически приемлемым вспомогательным веществом (также известным как фармацевтически приемлемый носитель). Введение активного соединения(ий) или кристаллической формы активного соединения(ий), в чистой форме или в подходящей фармацевтической композиции,можно осуществлять с помощью любого из приемлемых способов введения или компонентов, служащих для аналогичного применения. Таким образом, введение можно осуществлять, например, перорально,назально, парентерально (внутривенно, внутримышечно или подкожно), местно, трансдермально, интравагинально, интравезикально, интрацистемально или ректально, в форме твердого, полутвердого, лиофилизированного порошка или жидких лекарственных форм, таких как, например, таблетки, суппозитории,пилюли, мягкие эластичные и твердые желатиновые капсулы, порошки, растворы, суспензии или аэрозоли или подобные, предпочтительно в стандартных лекарственных формах, подходящих для простого введения точных доз. Одним предпочтительным путем введения является пероральное введение, применяя общепринятый режим дозирования, который можно приспособить в зависимости от степени тяжести болезненного состояния, которое будут лечить. Способы получения кристаллических форм Кристаллические формы можно получить рядом способов, включая, но не ограничиваясь, например, кристаллизацию или перекристаллизацию из подходящей смеси растворителей; возгонку; рост кристаллов из расплава; твердофазный переход из другой фазы; кристаллизацию из сверхкритической жидкости; и струйное распыление. Способы кристаллизации или перекристаллизации кристаллических форм из смеси растворителей включают, но не ограничиваются, например, упаривание растворителя; понижение температуры смеси растворителей; затравливание кристалла перенасыщенной смеси растворителей соединения и/или его соли; затравливание кристалла из перенасыщенной смеси растворителей соединения и/или соли; лиофилизацию смеси растворителей; и добавление антирастворителей (противорастворители) к смеси растворителей. Способы высокопроизводительной кристаллизации можно применять для получения кристаллических форм, включая полиморфы. Кристаллы лекарственных средств, включая полиморфы, способы получения и характеристику кристаллов лекарственного средства описывают в Solid-State Chemistry of Drugs, S.R. Byrn, R.R. Pfeiffer, иJ.G. Stowel1, 2nd Edition, SSCI, West Lafayette, Indiana (1999). В способе кристаллизации, в котором применяют растворитель, растворитель(и) обычно выбирают исходя из одного или более факторов, включая, но не ограничиваясь, например, растворимость соединения; применяемый способ кристаллизации и давление паров растворителя. Можно применять комбинации растворителей. Например, соединение можно растворять в первом растворителе для получения раствора, к которому затем добавляют антирастворитель для снижения растворимости соединения (I) в растворе, и для того, чтобы вызвать образование кристаллов. Антирастворитель представляет собой растворитель, в котором соединение обладает низкой растворимостью. В одном способе, который можно применять для получения кристаллов, соединения (I), (II) и/или(III) можно суспендировать и/или перемешивать в подходящем растворителе для получения суспензии,которую можно нагревать для того, чтобы способствовать растворению. Термин "суспензия", как применяют в настоящем изобретении, относится к насыщенному раствору соединения, в котором данный раствор может содержать дополнительное количество соединения для получения гетерогенной смеси соединения и растворителя при заданной температуре. Зародыши кристаллов можно добавлять к любой смеси для кристаллизации для того, чтобы способствовать кристаллизации. Затравливание можно применять для контролирования роста конкретного полиморфа и/или контролирования распределения размера частиц кристаллического продукта. Соответственно расчет необходимого количества зародышей кристаллов зависит от размера имеющихся зародышей кристаллов и требуемого размера средних частиц продукта, как описано, например, в ProgrammedCooling Batch Crystallizers, J.W. Mullin and J. Nyvlt, Chemical Engineering Science, 1971, 26, 3690377. В общем, зародыши кристаллов маленького размера требуются для эффективного контроля роста кристаллов в смеси. Зародыши кристаллов маленького размера можно получить грохочением, дроблением или тонким измельчением больших кристаллов или микрокристаллизацией раствора. При дроблении или тонком измельчении кристаллов необходимо проявлять осторожность для того, чтобы избежать изменения кристалличности желательной кристаллической формы (т.е. превращение в аморфную или другую полиморфную форму). Охлажденные кристаллизационные смеси можно фильтровать под вакуумом и отделенный твердый продукт промывать подходящим растворителем, таким как, например, холодный растворитель для перекристаллизации. После промывки продукт сушат продувкой азотом для получения требуемой кристаллической формы. Продукт можно анализировать подходящим спектроскопическим или аналитическим способом, включая, но не ограничиваясь, например, дифференциальную сканирующую калориметрию(DSC); рентгеновскую порошковую дифрактометрию (XRPD); и термогравиметрический анализ (TGA) для того, чтобы убедиться в образовании кристаллической формы соединения. Полученную в результате кристаллическую форму можно получить в количестве, большем чем приблизительно 70 вес.% выход после выделения, на основе веса соединения, первоначально применяемого в способе кристаллизации, и предпочтительно больше, чем приблизительно 90 вес.% выход после выделения. Необязательно, продукт можно размалывать измельчением или пропусканием через грохот с отверстиями. Особенности и преимущества настоящего изобретения будут более понятны специалистам в данной области техники при чтении следующего подробного описания. Должно быть ясно, что определенные особенности настоящего изобретения, которые для ясности описывают выше и ниже в контексте отдельных вариантов осуществления, можно также комбинировать для получения одного варианта осуществления. Кроме того, различные особенности настоящего изобретения, которые для краткости описывают в контексте одного варианта осуществления, можно также комбинировать так, чтобы образовать их субкомбинации. Настоящее изобретение далее иллюстрируется следующими примерами, которые не следует истолковывать как ограничивающие настоящее изобретение по объему или сущности до конкретных способов, описанных в них. Определения, приведенные в настоящем изобретении, имеют преимущество по сравнению с определениями, приведенными в любом патенте, патентной заявке и/или опубликованной патентной заявке,введенной в настоящее изобретение с помощью ссылки. Все измерения имеют экспериментальную ошибку и находятся в пределах сущности настоящего изобретения. Как применяют в настоящем изобретении, "аморфная" относится к твердой форме молекулы и/или иона, которая не является кристаллической. Аморфное твердое вещество не обладает определенной дифракционной рентгенограммой с четкими максимумами. Как применяют в настоящем изобретении, термин "практически чистая" обозначает кристаллическую форму соединения (I), о которой упоминается, что она содержит по меньшей мере приблизительно 90 вес.% на основе веса данной кристаллической формы. Термин "по меньшей мере приблизительно 90 вес.%", не предполагая ограничения применения теории эквивалентов до объема формулы изобретения,включает, но не ограничивается, например, приблизительно 90, приблизительно 91, приблизительно 92,приблизительно 93, приблизительно 94, приблизительно 95, приблизительно 96, приблизительно 97,приблизительно 98, приблизительно 99 и приблизительно 100 вес.%, на основе веса упоминаемой кристаллической формы. Остаток кристаллической формы соединения (I) может содержать другую форму(ы) соединения (I), и/или реакционные примеси, и/или производственные примеси, которые появляются, например, при получении кристаллической формы. Наличие реакционных примесей и/или производственных примесей можно определить аналитическими способами, известными в данной области техники, такими как, например, хроматография, спектроскопия ядерного магнитного резонанса, массспектроскопия и/или ИК-спектроскопия. Примеры получения Пример 1. Получение N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида и его (L)-малатной соли (соединение (I. Синтетическая схема, применяемая для получения N-(4-[6,7-бис-(метилокси)хинолин-4 ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида и его (L)-малатной соли изображена на схеме 1. Способ, показанный на схеме 1, описывают более подробно ниже. 1.1 Получение 4-хлор-6,7-диметоксихинолина. В колбу последовательно помещали 6,7-диметоксихинолин-4-ол (1 л, 10,0 кг) и ацетонитрил (64,0 л). Полученную в результате смесь нагревали до приблизительно 65 С и добавляли оксихлорид фосфора(POCl3, 50,0 кг). После добавления POCl3 температуру реакционной смеси повышали до приблизительно 80 С. Реакцию считали завершенной (приблизительно 9,0 час), когда оставалось 2% исходного вещества (в процессе анализа с помощью высокоэффективной жидкостной хроматографии [ВЭЖХ]). Реакционную смесь охлаждали до приблизительно 10 С и затем быстро охлаждали в охлажденном растворе дихлорметана (DCM, 238,0 кг), 30% NH4OH (135,0 кг) и льде (440,0 кг). Полученную в результате смесь нагревали до приблизительно 14 С и фазы разделяли. Органическую фазу промывали водой (40,0 кг) и концентрировали отгонкой в вакууме с удалением растворителя (приблизительно 190,0 кг). Метил-третбутиловый эфир (МТВЕ, 50,0 кг) добавляли к реакционной смеси и смесь охлаждали до приблизительно 10 С, в процессе чего продукт кристаллизовался. Твердый остаток выделяли центрифугированием, промывали н-гептаном (20,0 кг) и сушили при приблизительно 40 С для получения заявленного в заголовке соединения (8,0 кг). 1.2 Получение 6,7-диметил-4-(4-нитрофенокси)хинолина. В колбу последовательно помещали 4-хлор-6,7-диметоксихинолин (8,0 кг), 4-нитрофенол (7,0 кг), 4 диметиламинопиридин (0,9 кг), и 2,6-лутидин (40,0 кг). Содержимое колбы нагревали до приблизительно 147 С. После завершения реакции (5% оставшегося исходного вещества, как определено в процессе ВЭЖХ анализа, приблизительно 20 ч), содержимое колбы охлаждали до приблизительно 25C. Добавляли метанол (26,0 кг), с последующим добавлением карбоната калия (3,0 кг), растворенного в воде (50,0 кг). Содержимое колбы перемешивали в течение приблизительно 2 ч. Полученный в результате твердый остаток фильтровали, промывали водой (67,0 кг) и сушили при 25 С в течение приблизительно 12 ч для получения заявленного в заголовке соединения(4,0 кг). 1.3 Получение 4-(6,7-диметоксихинолин-4-илокси)фениламина. Раствор, содержащий формиат калия (5,0 кг), муравьиную кислоту (3,0 кг) и воду (16,0 кг), добавляли к смеси 6,7-диметокси-4-(4-нитрофенокси)хинолина (4,0 кг), 10% палладия-на-угле (50% смоченный водой,0,4 кг) в тетрагидрофуране (40,0 кг), которая была нагрета до приблизительно 60 С. Добавление осуществляли так, чтобы температура реакционной смеси сохранялась приблизительно равной 60 С. Когда реакцию считали завершенной, как определено, применяя в процессе ВЭЖХ анализ (2% оставшегося исходного вещества, обычно 15 ч), содержимое колбы фильтровали. Фильтрат концентрировали отгонкой в вакууме при приблизительно 35 С до половины его первоначального объема, что приводило в результате к выпадению продукта. Продукт выделяли фильтрацией, промывали водой (12,0 кг) и сушили в вакууме при приблизительно 50 С для получения заявленного в заголовке соединения (3,0 кг; 97% AUC). 1.4 Получение 1-(4-фторфенилкарбамоил)циклопропанкарбоновой кислоты. Триэтиламин (8,0 кг) добавляли к охлажденному (приблизительно 4 С) раствору имеющейся в продаже циклопропан-1,1-дикарбоновой кислоты (2 1, 10,0 кг) в THF (63,0 кг) с такой скоростью, чтобы температура смеси не превышала 10 С. Раствор перемешивали в течение приблизительно 30 минут и затем добавляли тионилхлорид (9,0 кг), поддерживая температуру смеси ниже 10 С. После завершения добавления добавляли раствор 4-фторанилина (9,0 кг) в THF (25,0 кг) с такой скоростью, чтобы температура смеси не превышала 10 С. Смесь перемешивали в течение приблизительно 4 ч и затем разбавляли изопропилацетатом (87,0 кг). Данный раствор промывали последовательно водным гидроксидом натрия(2,0 кг, растворенные в 50,0 л воды), водой (40,0 л) и водным хлоридом натрия (10,0 кг, растворенные в 40,0 л воды). Органический раствор концентрировали отгонкой в вакууме с последующим добавлением гептана, что приводило в результате к выпадению осадка. Твердый остаток выделяли центрифугированием и затем сушили при приблизительно 35 С в вакууме для получения заявленного в заголовке соединения. (10,0 кг). 1.5 Получение 1-(4-фторфенилкарбамоил)циклопропанкарбонилхлорида. Оксалилхлорид (1,0 кг) добавляли к раствору 1-(4-фторфенилкарбамоил)циклопропанкарбоновой кислоты (2,0 кг) в смеси THF (11 кг) и N,N-диметилформамида (DMF; 0,02 кг) при такой скорости, чтобы температура смеси не превышала 30 С. Данный раствор применяли в следующей стадии без дополнительной обработки. 1.6 Получение N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. Раствор из предыдущей стадии, содержащий 1-(4-фторфенилкарбамоил)циклопропанкарбонилхлорид, добавляли к смеси 4-(6,7-диметоксихинолин-4-илокси)фениламина (3,0 кг) и карбоната калия(4,0 кг) в THF (27,0 кг) и воде (13,0 кг) при такой скорости, чтобы температура смеси не превышала 30 С. После завершения реакции (обычно за 10 мин) добавляли воду (74,0 кг). Смесь перемешивали при 1530 С в течение приблизительно 10 ч, что приводило в результате к выпадению продукта. Продукт выделяли фильтрацией, промывали предварительно полученным раствором THF (11,0 кг) и воды (24,0 кг), и сушили при приблизительно 65 С в вакууме в течение приблизительно 12 ч для получения заявленного в заголовке соединения (свободное основание, 5,0 кг). 1 Н ЯМР (400 МГц, d6-DMSO):10,2 (с, 1 Н), 10,05 (с, 1 Н) , 8,4 (с, 1 Н), 7,8 (м, 2 Н), 7,65 (м, 2 Н), 7,5(с, 1 Н), 7,35 (с, 1 Н), 7,25 (м, 2 Н), 7,15 (м, 2 Н), 6,4 (с, 1 Н), 4,0 (д, 6 Н), 1,5 (с, 4 Н). LC/MS: М+Н=502. 1.7 Получение (b)-малатной соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида (соединение (I. Раствор (L)-оксиянтарной кислоты (2,0 кг) в воде (2,0 кг) добавляли к раствору [4-(6,7 диметоксихинолин-4-илокси)фенил]амид-(4-фторфенил)амида циклопропан-1,1-дикарбоновой кислоты в форме свободного основания (1,5, 5,0 кг) в этаноле, поддерживая температуру смеси приблизительно равной 25 С. Затем добавляли уголь (0,5 кг) и оксид кремния, содержащий тиольные якорные группы(0,1 кг), и полученную в результате смесь нагревали до приблизительно 78 С, при которых добавляли воду (6,0 кг). Затем реакционную смесь фильтровали с последующим добавлением изопропанола (38,0 кг) и охлаждали до приблизительно 25 С. Продукт выделяли фильтрацией, промывали изопропанолом(20,0 кг) и сушили при приблизительно 65 С для получения соединения (I) (5,0 кг). Пример 2: Получение кристаллического соединения (I), форма N-1. Раствор получали добавлением тетрагидрофурана (12 мл/г веса LR (лимитирующий реагент); 1,20 л),N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1 дикарбоксамида (100 г; 1,00 экв; 100,00 г) и (L)-оксиянтарной кислоты (1,2 экв. (молярные); 32,08 г) в 1 л реактор. Добавляли воду (0,5317 мл/г веса LR; 53,17 мл), и раствор нагревали до 60 С и выдерживали при данной температуре в течение 1 ч до полного растворения твердых веществ. Раствор пропускали через фильтр для тонкой очистки. При 60 С добавляли ацетонитрил (12 мл/г веса LR; 1,20 л) в течение 8 ч. Раствор выдерживали при 60 С в течение 10 ч. Затем раствор охлаждали до 20 С и выдерживали в течение 1 ч. Твердые вещества фильтровали и промывали ацетонитрилом (12 мл/г веса LR; 1,20 л). Твердые вещества сушили при 60 С(25 мм рт.ст.) в течение 6 ч для получения соединения (I), форма N-1 (108 г; 0,85 экв.; 108,00 г; 85,22% выход) в виде белого кристаллического твердого остатка. Пример 3. Альтернативное получение кристаллического соединения (I), форма N-1. Раствор получали с 190 мл тетрагидрофурана, 110 мл метилизобутилкетона и 29 мл воды. Затем 20 мл данного раствора переносили в желтый сосуд и затем насыщали добавлением (L)-малата N-(4-[6,7 бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида до образования густой суспензии, и выдерживали в течение по меньшей мере 2 ч с перемешиванием при комнатной температуре. Твердые вещества удаляли фильтрацией через воронку Бюхнера, получая прозрачный насыщенный раствор. Отдельно получали порошковую смесь с известными количествами двух порций соединения (I): (1) 300 мг порции 1, которая содержала приблизительно 41% соединения (I), форма N-1, и 59% соединения(I), форма N-2, на основании анализа с помощью рамановской спектроскопии, и (2) 200 мг порции 2, ко- 16019959 торая имела XPRD рентгенограмму, аналогичную рентгенограмме соединения (I), форма N-2. Порошковую смесь, содержащую соединение (I), форма N-1 и соединение (I), форма N-2, добавляли в насыщенный раствор и суспензию выдерживали с перемешиванием магнитной мешалкой при комнатной температуре в течение 25 дней. Затем суспензию отбирали и фильтровали через воронку Бюхнера для получения 162 мг влажного остатка на фильтре. Влажный остаток сушили в вакуумной печи при 45 С для получения 128 мг кристаллического соединения (I) в N-1 форме. Пример 4. Получение кристаллического соединения (I), форма N-2. 4.1 Получение зародышей кристаллов кристаллического соединения (I), форма N-2. Раствор получали смешиванием 20 мл ацетона и 300 мг свободного основания N-(4-[6,7-бис(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида в 25 мл пробирке с завинчивающейся крышкой. Затем 0,758 мл 0,79 М исходного раствора (L)-оксиянтарной кислоты добавляли в пробирку с перемешиванием магнитной мешалкой. Затем раствор перемешивали в течение 24 ч при температуре окружающей среды. Затем образец фильтровали с подсосом с помощью 0,45 мксPTFE фильтровального картриджа и сушили в вакууме при температуре окружающей среды в течение ночи. 4.2 Получение кристаллического соединения (I), форма N-2. В колбу добавляли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамид (48 г; 1,00 экв.; 48,00 г) и тетрагидрофуран (16,5 мл/г веса LR; 792,00 мл). Содержание воды доводили до 1% по весу воды. Раствор нагревали до 60 С. После растворения раствор пропускали через фильтр для тонкой очистки для получения первого раствора. В отдельной колбе (L)-оксиянтарную кислоту (1,2 экв. (молярные); 15,40 г) растворяли в метилизобутилкетоне (10 мл/г веса LR; 480,00 мл) и тетрагидрофуране (1 мл/г весаLR; 48,00 мл). Затем 50 мл раствора (L)-оксиянтарной кислоты добавляли к первому раствору при 50 С. Добавляли зародыши кристаллов (1%, 480 мг) и раствор оксиянтарной кислоты добавляли при 50 С по каплям через капельную воронку (1,3 мл/мин (3 ч. Суспензию выдерживали при 50 С в течение 18 ч и затем охлаждали до 25 С в течение 30 мин. Твердые вещества фильтровали и промывали 20% тетрагидрофуран/метилизобутилкетон (10V, 480 мл). Твердые вещества сушили в вакууме при 60 С в течение 5 ч для получения соединения (I) (55,7 г; 0,92 экв.; 55,70 г; 91,56% выход) в виде грязно-белого кристаллического остатка. Пример 5. Получение кристаллического соединения (III), форма N-1. 1 мл аликвоту соли (DL)-оксиянтарной кислоты и N-(4-[6,7-бис-(метилокси)хинолин-4 ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, суспендированной в тетрагидрофуране (THF), нагревали до 60 С на горячей плитке в 1/16 унцевой пробирке. Затем добавляли по каплям тетрагидрофуран до получения практически прозрачного раствора. Пробирку закрывали, удаляли с горячей плиты и приводили в равновесие при комнатной температуре без перемешивания. Кристаллизацию наблюдали через несколько часов и раствор оставляли стоять в течение ночи для завершения кристаллизации. Несколько капель полученной в результате суспензии помещали на предметное стекло для микроскопического анализа. Кристаллическое вещество состояло из большого количества вытянутых пластинок, размер которых доходил до 60 мкм в направлении наибольшего размера. Альтернативное получение кристаллического соединения (III), форма N-1. В колбу добавляли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамид (15 г; 1,00 экв.; 15,00 г) и тетрагидрофуран (16,5 мл/г веса LR; 792,00 мл). Содержание воды доводили до 1 вес.% воды. Раствор нагревали до 60 С. После растворения раствор пропускали через фильтр для тонкой очистки для получения первого раствора. В отдельной колбе (DL)-оксиянтарную кислоту (1,2 экв. (молярные); 4,53 г) растворяли в метилизобутилкетоне (8 мл/г веса LR; 120,00 мл) и тетрагидрофуране (1 мл/г веса LR; 15,00 мл). Затем 20 мл раствора добавляли к первому раствору при 50 С. Раствор оксиянтарной кислоты добавляли при 50 С по каплям через капельную воронку (1,3 мл/мин (3 ч. Суспензию выдерживали при 50 С в течение 18 ч и затем охлаждали до 25 С в течение 30 мин. Твердые вещества фильтровали и промывали 20%THF/MIBK (10V, 150 мл). Твердые вещества сушили в вакууме при 60 С в течение 5 ч для получения соединения (III) (15,52 г; 86,68% выход) в виде не совсем белого твердого остатка. Пример 6. Получение аморфного соединения (I). Раствор получали с 5 г (L)-малата N-4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамида и 250 мл 1:1 (об.:об.) смеси метанола и дихлорметана. Мутный раствор фильтровали через 0,45-микронный фильтр для получения прозрачного желтоватого раствора. Раствор откачивали через наконечник для распылительной сушки при скорости 12,9 см 3/мин и распыляли подачей газообразного азота при скорости 10,9 л/мин. Температуру входного отверстия циклона устанавливали на 65 С для сушки влажных капель. Собирали сухой аморфный порошок (1,5 г)H ЯМР (400 МГц, d6-DMSO):1,48 (с, 4 Н), 2,42-2,48 (м, 1 Н), 2,60-2,65 (м, 1 Н), 3,93 (с, 3 Н), 3,96 (с,3 Н), 4,25-4,30 (дд, 1 Н, J=5, 8 Гц), 6,44 (д, J=5 Гц, 1 Н), 7,12-7,19 (м, 2 Н), 7,22-7,26 (м, 2 Н), 7,40 (с, 1 Н),7,51 (с, 1 Н), 7,63-7,68 (м, 2 Н), 7,76-7,80 (м, 2 Н), 8,46-8,49 (м, 1 Н), 10,08 (с, 1 Н), 10,21 (с, 1 Н). 13 С ЯМР (d6-DMSO): 15,36, 31,55, 55,64, 55,67, 66,91, 99,03, 102,95, 107,66, 114,89, 115,07, 115,11,121,17, 122,11, 122,32, 122,39, 135,15, 136,41, 146,25, 148,7, 149,28, 149,38, 152,54, 157,03, 159,42, 160,02,168,07, 171,83, 174,68. Характеристика твердых форм малата N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамидаII. Исследования с помощью порошковой дифракции рентгеновских лучей (XRPD). Порошковые (XRPD) рентгенограммы получали на Bruker AXS C2 GADDS дифрактометре, снабженном автоматическим XYZ координатным лазерным видеомикроскопом для автоматической ориентации образца и HiStar 2-мерным площадным детектором. Применяемым источником радиоактивного излучения была медь (CuK=1,5406 ), в котором напряжение устанавливали на 40 кВ и силу тока устанавливали на 40 мА, рентгеновская оптика состоит из одного Gbel многослойного зеркала в сочетании с 0,3 мм коллиматором типа "пинхол". Расходимость пучка, т.е. эффективный размер рентгеновского луча на образце, была равна приблизительно 4 мм. 9-9 непрерывный сканирующий режим применяли с расстоянием образецдетектор, равном 20 см, которое давало эффективный 2 диапазон 3,2-29,8. Образцы для проведения опытов при температурах окружающей среды (от приблизительно 18 С до приблизительно 25 С) получали в виде плоских образцов, применяя порошок, каким он был получен без измельчения. Приблизительно 1-2 мг образца слегка спрессовывали на предметном стекле для получения плоской поверхности. Обычно образец подвергают действию рентгеновских лучей в течение 120 с. Расходимость пучка, т.е. эффективный размер поверхности, на которую падают рентгеновские лучи, дает величину приблизительно равную 4 мм. Альтернативно, порошковые образцы помещают в герметизированные стеклянные капилляры с диаметром 1 мм или менее; капилляр вращают в процессе сбора данных при расстоянии образец-детектор 15 см. Данные получали для 3235 со временем воздействия на образец по меньшей мере 2000 с. Полученные в результате двухмерные дифракционные линии интегрировали для получения стандартной одномерной XRPD рентгенограммы с размером шага 0,022 в диапазоне 3-3520,22. Программным обеспечением, применяемым для сбора данных, было GADDS дляII.1 Соединение (I), форма N-1. Фиг. 1 показывает экспериментальную XRPD рентгенограмму кристаллического соединения (I),форма N-1, полученную при комнатной температуре (приблизительно 25 С). Перечень пиков показан в табл. 2 выше. 2 величины при 19,4, 21,5, 22,8, 25,1 и 27,6 (0,22) являются подходящими для характеристики кристаллического соединения (I), форма N-1. Полный перечень пиков или их поднабор может быть достаточным для характеристики кристаллического соединения (I), форма N-1.II.2 Соединение (I), форма N-2. Фиг. 8 показывает экспериментальную XRPD рентгенограмму кристаллического соединения (I),форма N-2, полученную при комнатной температуре (приблизительно 25 С). Перечень пиков показан в табл. 2 выше. 2 величины при 20,9 и 21,9 (0,22) являются подходящими для характеристики кристаллического соединения (I), форма N-2. Полный перечень пиков или их поднабор может быть достаточным для характеристики кристаллического соединения (I), форма N-2.II.3 Соединение (III), форма N-1. Фиг. 15 показывает экспериментальную и смоделированную XRPD рентгенограмму кристаллического соединения (III), форма N-1, полученные при 25 С, с применением вращающегося капиллярного образца. Перечень пиков показан в табл. 2 выше. Полный перечень пиков или их поднабор может быть достаточным для характеристики кристаллического соединения (III), форма N-2.II.4 Аморфное соединение (I). Фиг. 22 показывает экспериментальную XRPD рентгенограмму аморфного соединения (I), полученную при комнатной температуре (приблизительно 25 С). Спектр характеризуется широким пиком и отсутствием узких пиков, что согласуется с аморфным веществом.III. Монокристаллическое рентгеноструктурное исследование соединения (III), форма N-1. Данные получали на Bruker-Nonius дифрактометре CAD4 серии. Параметры элементарной ячейки получали с помощью анализа методом наименьших квадратов экспериментальных показаний дифрактометра 25 отраженных сигналов под большим углом. Интенсивности измеряли, применяя CuK излучение (=1,5418 ) при постоянной температуре с -2 переменным способом сканирования и отбирали только для лоренцевых поляризационных факторов. Фоновый счет получали в крайних точках скана для половины времени скана. Альтернативно, данные для монокристалла получали на Bruker-Nonius KappaCCD 2000 системе, применяя CuK излучение (=1,5418 ). Индексирование и обработку данных об измеренных интенсивностях осуществляли с помощью HKL2000 пакета программ (Otwinowski, Z.Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W.C. JrSweet, R.M. (Academic, NY), vol. 276, pp,307-326) в Collect комплекте программ (Collect Data collection and processing user interface: Collect:Data collection software, R. Hooft, Nonius B.V., 1998). Альтернативно, данные для монокристалла получали на Bruker-AXS APEX2 CCD системе, применяя CuK излучение (=1,5418 ). Индексирование и обработку данных об измеренных интенсивностях осуществляли с помощью АРЕХ 2 пакета программ/комплекта программ (АРЕХ 2 Data collection and processing user interface: APEX2 User Manual,v1.27). Когда показано, кристаллы охлаждали в холодном потоке Oxford криосистеме (Oxford Cryosystems Cryostream cooler: J. Cosier и A.M. Glazer, J. Appl. Cryst., 1986, 19, 105) в процессе получения данных. Структуры разрешали прямыми способами и уточняли на основе наблюдаемых отраженных сигналов, применяя или SDP пакет программ (SDP, пакет программ для определения структуры, Enraf-Nonius,Bohemia NY 11716. коэффициенты рассеяния, включая f' и f", в SDP программе брали из "международных кристаллографических таблиц", Kynoch Press, Birmingham, England, 1974; том IV, табл. 2.2 А и 2.3.1) с незначительными локальными модификациями или кристаллографический пакет программ MAXUS(maXus solution and refinement software suite: S. Mackay, C.J. Gilmore, C. Edwards, M. Tremayne, N. Stewart,K. Shankland. maXus: компьютерная программа для разрешения и уточнения кристаллических структур по данным дифракции) или SHELXTL (APEX2 Data collection and processing user interface: APEX2 UserManual, v1.27). Полученные параметры решетки атомного кристалла (координаты и температурные факторы) уточняли с помощью полноматричного способа наименьших квадратов. Минимизированная функция при уточнении представляла собойR определяют как тогда как в которых w представляет собой подходящую весовую функцию на основе ошибок в наблюдаемых интенсивностях. Топограммы сравнения исследовали на всех стадиях уточнения. Атомы водорода вводили в теоретические позиции с изотропными температурными факторами, но параметры атомов водорода не изменяли."Гибрид" смоделированные порошковые рентгенограммы получали, как описано в литературе (Yin.S.; Scaringe, R.P.; DiMarco, J.; Galella, M. and Gougoutas, J.Z., American Pharmaceutical Review, 2003, 6,2,80). Параметры ячейки при комнатной температуре получали проведением уточнения параметров ячейки, применяя CellRefine.xls программу. Вводимые в программу данные включают 2 положения приблизительно 10 отраженных сигналов, полученных из экспериментальной порошковой рентгенограммы при комнатной температуре; соответствующие индексы Миллера, hkl, оценивали на основе данных монокристалла, полученных при низкой температуре. Новую (гибридную) XRPD рассчитывали (с помощью одной из двух компьютерных программ, Alex или LatticeView) вставкой молекулярной структуры, полученной при низкой температуре, в ячейку при комнатной температуре на первой стадии способа. Молекулы вставляли способом, который сохранял размер и форму молекулы и положение молекул относительно исходного положения ячейки, но позволял увеличиваться межмолекулярным расстояниям с ячейкой. Монокристалл с размерами 403010 мкм выбирали из суспензии кристаллов, описанной в примере 5, для монокристаллического дифракционного анализа. Отобранный кристалл прикрепляли к тонкому стекловолокну с небольшим количеством неплотной смазки и помещали при комнатной температуре наBruker ApexII монокристальный дифрактометр, снабженный вращающимся медным анодом. Кристаллическое соединение (III), форма N-1, характеризовали параметрами элементарной ячейки,приблизительно равными параметрам, приведенным в табл. 4. Параметры элементарной ячейки измеряли при температуре, приблизительно равной 25 С. Таблица 4 Разрешение и уточнение структуры раствора были стандартными в моноклинной пространственной группе, P21/n, с четырьмя молекулами в элементарной ячейке. Структура содержит катионы N-(4-[6,7 бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, протонированные по хинолиновому атому азота, и однократно ионизированные анионы оксиянтарной кислоты в отношении 1:1. Кроме того, кристалл содержал 1:1 отношение ионов (L)-оксиянтарной кислоты к ионам (D)-оксиянтарной кислоты. Относительные атомные координаты в табл. 5 для соединения (III), форма N-1, рассчитывали при температуре, приблизительно равной 25 С. На основе монокристаллических данных рентгеновского анализа кристаллическое соединение (III),форма N-1, можно характеризовать смоделированной порошковой рентгенограммой (XRPD) в основном в соответствии со смоделированной рентгенограммой, показанной на фиг. 15, и/или наблюдаемой порошковой рентгенограммой XRPD, в основном в соответствии с экспериментальной рентгенограммой,показанной на фиг. 15. Таблица 5. Относительные атомные координаты для соединения (XII), форма N-1, рассчитанные при температуре, приблизительно равной 25 СIV. Твердофазный ядерный магнитный резонанс (SSNMR). Все твердофазные С-13 ЯМР измерения осуществляли на Bruker DSX-400, 400 МГц ЯМР спектрометре. Спектры высокого разрешения получали, применяя развязку от протонов высокой мощности и ТРРМ импульсный режим и поперечную поляризации с быстро изменяющейся амплитудой (RAMP-CP) с вращением образца под магическим углом (MAS) при приблизительно 12 кГц (А.Е. Bennett et al., J.Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu и S.O. Smith, J. Magn. Reson. A,. 1994, 110, 219-227). Приблизительно 70 мг образца, упакованного в циркониевый ротор с емкостью, применяли в каждом эксперименте. Химические сдвиги (5) приводили относительно внешнего стандарта - адамантана, причем высокочастотный резонанс устанавливали на 38,56 ppm (W.L. Earl and D.L. VanderHart, J. Magn. Reson.,1982, 48, 35-54).IV.1 Соединение (I), форма N-1. Твердофазный 13 С ЯМР спектр кристаллического соединения (I), форма N-1, показан на фиг. 2. Полный перечень пиков или его подгруппа могут быть достаточными для характеристики кристаллического соединения (I), форма N-1.SS 13 С ЯМР пики: 18,1, 20,6, 26,0, 42,9, 44,5, 54,4, 55,4, 56,1, 70,4, 99,4, 100,1, 100,6, 114,4, 114,9,115,8, 119,6, 120,1, 121,6, 123,2, 124,1, 136,4, 138,6, 140,6, 145,4, 150,1, 150,9, 156,2, 157,4, 159,4, 164,9,167,1, 170,8, 175,7 и 182,1 ppm 0,2 ppm. Фиг. 3 показывает твердофазный 15N ЯМР спектр кристаллического соединения (I), форма N-1. Спектр показывает пики при 118,6, 119,6, 120,7, 134,8, 167,1, 176,0 и 180 ppm 0,2 ppm. Полный перечень пиков или его подгруппа могут быть достаточными для характеристики кристаллического соединения(I), форма N-1. Фиг. 4 показывает твердофазный 19F ЯМР спектр кристаллического соединения (I), форма N-1. Спектр показывает пики при -121,6, -120,8 и -118,0 ppm 0,2 ppm.IV.2 Соединение (1), форма N-2. Твердофазный 13 С ЯМР спектр кристаллического соединения (I), форма N-2, показан на фиг. 9. Полный перечень пиков или его подгруппа, могут быть достаточными для характеристики кристаллического соединения (I), форма N-2.SS 13 С ЯМР пики: 20,5, 21,8, 23,0, 25,9, 26,4, 38,0, 41,7, 54,7, 55,8, 56,2, 56,6, 69,7, 99,4, 100,0, 100,4,100,8, 102,3, 114,5, 115,5, 116,7, 119,0, 120,2, 121,1, 121,2, 122,1, 122,9, 124,5, 136,0, 137,3, 138, 1, 138,9,139,5, 140,2, 144,9, 145,7, 146,1, 150,7, 156,7, 157,7, 159,6, 159,7, 165,1, 167,0, 168,0, 171,5, 177,3, 179,3,180,0, и 180,3 ppm, 0,2 ppm Фиг. 10 показывает твердофазный 15N ЯМР спектр кристаллического соединения (I), форма N-2. Спектр показывает пики при 118,5, 120,8, 135,1, 167,3 и 180,1 ррт, 0,2 ppm. Полный перечень пиков или его подгруппа могут быть достаточными для характеристики кристаллического соединения (I), форма N2. Фиг. 11 показывает твердофазный 19F ЯМР спектр кристаллического соединения (I), форма N-2. Спектр показывает пики при -121,0 и -119,1 ppm, 0,2 ppm. Данные пики, отдельно или вместе, могут быть достаточными для характеристики кристаллического соединения (I), форма N-2.IV.3 Соединение (III), форма N-1. Твердофазный 13 С ЯМР спектр кристаллического соединения (III), форма N-1, показан на фиг. 16. Полный перечень пиков или его подгруппа могут быть достаточными для характеристики кристаллического соединения (III), форма N-1.SS 13 С ЯМР пики: 20,8, 26,2, 44,8, 55,7, 70,7, 100,4, 101,0, 114,7, 115,2, 116,0, 119,7, 120,4, 121,6,124,4, 136,9, 138,9, 141,1, 145,7, 150,3, 156,5, 157,6, 159,6, 165,2, 167,4, 171,2, 176,3 и 182,1 ppm 0,2 ppm. Фиг. 17 показывает твердофазный 15N ЯМР спектр кристаллического соединения (III), форма N-1. Спектр показывает пики при 119,6, 134,7 и 175,5 ppm 0,2 ppm. Полный перечень пиков или его подгруппа могут быть достаточными для характеристики кристаллического соединения (III), форма N-1. Фиг. 18 показывает твердофазный 19F ЯМР спектр кристаллического соединения (III), форма N-1. Спектр показывает пики при -120,5 ppm 0,2 ppm.IV.4 Соединение (I), аморфное. Фиг. 23 показывает твердофазный 13 С ЯМР спектр аморфного соединения (I). Полный перечень пиков или его подгруппа могут быть достаточными для характеристики аморфного соединения (I).SS 13 С ЯМР пики (ppm): 12,2, 17,8, 20,3, 21,8, 27,2, 33,8, 41,7, 56,9, 69,9, 99,9, 102,2, 115,6, 122,2,134,4, 137,8, 142,9, 149,1, 150,9, 157,3, 159,7, 167,0, 171,7, 173,1, 177,4 и 179,5 0,2. Фиг. 24 показывает твердофазный 15N ЯМР спектр аморфного соединения (I). Спектр показывает пики при 120,8, 131,8, 174,7 и 178,3 ppm 0,2 ppm. Полный перечень пиков или его подгруппа могут быть достаточными для характеристики аморфного соединения (I). Фиг. 25 показывает твердофазный 19F ЯМР спектр аморфного соединения (I). Спектр показывает пики при -118,9 ppm0,2 ppm.V. Измерение термических характеристик. Термический гравиметрический анализ (T.GA).TGA измерения осуществляли на ТА Instruments модели Q500 или 2950, применяя схему с открытым тигелем. Образец (приблизительно 10-30 мг) помещали в предварительно тарированный платиновый тигель. Вес образца точно измеряли и значения записывали до тысячных миллиграмма с помощью устройства для измерения. Термопечь продували газообразным азотом при 100 мл/мин. Данные регистрировали в диапазоне температур от комнатной температуры до 300 С при скорости нагревания 10 С/мин. Анализ с помощью дифференциальной сканирующей калориметрии (DSC)DSC измерения осуществляли в ТА Instruments модели Q2000, Q1000 или 2920, применяя схему с открытым тиглем. Образец (приблизительно 2-6 мг) взвешивали в алюминиевом тигле и записывали точное значение до сотых миллиграмма и переносили в DSC. Прибор продували газообразным азотом при 50 мл/мин. Данные регистрировали в диапазоне температур от комнатной температуры до 300 С при скорости нагревания 10 С/мин. Построение графика осуществляли с помощью эндотермических пиков,направленных вниз.V.1 Соединение (I), форма N-1 Фиг. 5 показывает TGA термограмму для кристаллического соединения (I), форма N-1, которая показывает потерю веса, приблизительно равную 0,4% по весу при температуре 170 С. Фиг. 6 показывает DSC термограмму кристаллического соединения (I), форма N-1, которая показывает температуру плавления, приблизительно равную 187 С.V.2 Соединение (I), форма N-2. Фиг. 12 показывает TGA термограмму для кристаллического соединения (I), форма N-2, которая показывает потерю веса, приблизительно равную 0,1 вес.% при температуре 170 С. Фиг. 13 показывает DSC термограмму кристаллического соединения (I), форма N-2, которая показывает температуру плавления, приблизительно равную 186 С.V.3 Соединение (III), форма N-1. Фиг. 19 показывает TGA термограмму для кристаллического соединения (III), форма N-1, которая показывает потерю веса, приблизительно равную 0,2 вес.% при температуре 170 С. Фиг. 20 показывает DSC термограмму кристаллического соединения (III), форма N-1, которая показывает температуру плавления, приблизительно равную 186 С.V.2 Соединение (I), аморфное. Фиг. 26 показывает DSC для кристаллического соединения (I).VI. Измерения изотерм водяного пара. Изотермы поглощения влаги получали в VTI SGA-100 Symmetric газоанализаторе, применяя приблизительно 10 мг образца. Образец сушили при 60 С до получения скорости потери веса 0,0005 вес.%/мин в течение 10 мин. Образцы испытывали при 25 С и 3 или 4, 5, 15, 25, 35, 45, 50, 65, 75, 85 и 95% RH. Равновесия при каждой RH достигали при достижении скорости 0,0003 вес.%/мин в течение 35 мин или максимум 600 мин. Фиг. 7 показывает изотерму поглощения водяного пара кристаллическим соединением (I), форма N1.VI.2 Соединение (I), форма N-1. Фиг. 14 показывает изотерму поглощения водяного пара кристаллическим соединением (I), формаVI.3 Соединение (III), форма N-1. Фиг. 21 показывает изотерму поглощения водяного пара кристаллическим соединением (III), формаVI.4 Соединение (I), аморфное. Фиг. 27 показывает изотерму поглощения водяного пара аморфным соединением (I). Вышеизложенная сущность изобретения описана довольно подробно посредством иллюстраций и примеров с целью ясности и понимания. Настоящее изобретение описано со ссылкой на различные конкретные и предпочтительные варианты осуществления и способы. Однако должно быть ясно, что многие изменения и модификации можно осуществлять, оставаясь в пределах объема и сущности настоящего изобретения. Специалистам в данной области техники ясно, что изменения и модификации можно осуществлять на практике в пределах объема прилагаемой формулы изобретения. Следовательно, должно быть ясно, что предполагается, что вышеуказанное описание является иллюстративным и неограничивающим. Следовательно, объем настоящего изобретения должен определяться не ссылкой на вышеуказанное описание, но должен, вместо этого, определяться со ссылкой на следующую прилагаемую формулу изобретения вместе с полным объемом эквивалентов, по отношению к которым данная формула изобретения является правомочной. ФОРМУЛА ИЗОБРЕТЕНИЯ 1.N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида. 2. Малатная соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.1, в которой упомянутая соль является кристаллической. 3. Малатная соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.2, которая находится в кристаллической форме N-1, и упомянутая форма N-1 характеризуется по меньшей мере одним из следующего:(i) твердофазным 13 С ЯМР спектром с четырьмя или более пиками, выбранными из 18,1; 42,9; 44,5; 70,4; 123,2; 156,2; 170,8; 175,7 и 182,1 ppm 0,2 ppm;(ii) порошковой рентгенограммой (CuK =1,5418 ), содержащей четыре или более 2 величин,выбранных из 12,80,22; 13,50,22; 16,90,22; 19,40,22; 21,50,22; 22,80,22; 25,10,22; 27,6 0,22, в которой измерение кристаллической формы осуществляли при комнатной температуре; и/или(iii) порошковой рентгенограммой (XRPD) в основном в соответствии с рентгенограммой, показанной на фиг. 1. 4. Малатная соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.3, которая содержит минимум 90% по весу формы N-1 в расчете на вес соли. 5. Малатная соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.2, которая находится в кристаллической форме N-2, и упомянутая форма N-2 характеризуется по меньшей мере одним из следующего:(i) твердофазным 13 С ЯМР спектром с четырьмя или более пиками, выбранными из 23,0; 25,9; 38,0; 41,7; 69,7; 102,0; 122,5; 177,3; 179,3; 180,0 и 180,3 ppm 0,2 ppm;(ii) порошковой рентгенограммой (CuK =1,5418 ), содержащей 2 величины при 20,90,22 и 21,90,22 и две или более 2 величины, выбранные из 6,40,22; 9,10,22; 12,00,22; 12,80,22; 13,70,22; 17,10,22; 22,60,22; 23,72, 0,22; в которой измерение кристаллической формы осуществляли при комнатной температуре; и/или(iii) порошковой рентгенограммой (XRPD) в основном в соответствии с рентгенограммой, показанной на фиг. 8. 6. Малатная соль N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида по п.5, которая содержит минимум 90% по весу формы N-2 в расчете на вес соли. 7. Фармацевтическая композиция, содержащая малатную соль N-(4-[6,7-бис-(метилокси)хинолин 4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида согласно одному из пп.1-6 и фармацевтически приемлемое вспомогательное вещество. 8. Применение малатной соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамида согласно любому одному из пп.1-6 для получения лекарственного средства для лечения рака. 9. Применение малатной соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4 фторфенил)циклопропан-1,1-дикарбоксамида по любому одному из пп.1-6 для лечения рака. 10. Лекарственное средство для лечения рака щитовидной железы у субъекта, содержащее кристаллическую форму (L)-малатной соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида согласно любому одному из пп.1-6. 11. Лекарственное средство для лечения глиобластомы у субъекта, содержащее кристаллическую форму (L)-малатной соли N-(4-[6,7-бис-(метилокси)хинолин-4-ил]оксифенил)-N'-(4-фторфенил)циклопропан-1,1-дикарбоксамида согласно любому одному из пп.1-6. 12. Лекарственное средство по п.10, где рак щитовидной железы представляет собой медуллярный рак щитовидной железы.
МПК / Метки
МПК: A61K 31/47, C07D 215/227, A61P 35/00
Метки: рака, n-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-n'-(4-фторфенил)циклопропан-1,1-дикарбоксамида, формы, малатная, кристаллические, соль, лечения
Код ссылки
<a href="https://eas.patents.su/30-19959-malatnaya-sol-n-4-67-bis-metiloksihinolin-4-iloksifenil-n-4-ftorfenilciklopropan-11-dikarboksamida-i-ee-kristallicheskie-formy-dlya-lecheniya-raka.html" rel="bookmark" title="База патентов Евразийского Союза">Малатная соль n-(4-{[6,7-бис-(метилокси)хинолин-4-ил]окси}фенил)-n’-(4-фторфенил)циклопропан-1,1-дикарбоксамида и ее кристаллические формы для лечения рака</a>
Соли и кристаллические формы 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила

Номер патента: 15677
Опубликовано: 31.10.2011
Авторы: Штовассер Франк, Бэнцигер Маркус, Гарад Судхакар Девидасрао
МПК: C07D 471/04, A61P 35/00, A61K 31/4745...
Метки: кристаллические, 2-метил-2-[4-(3-метил-2-оксо-8-хинолин-3-ил-2,3-дигидроимидазо[4,5-c]хинолин-1-ил)фенил]пропионитрила, формы, соли
Формула / Реферат:
1. Кристаллическая форма соединения формулы Iили гидрата или сольвата соединения формулы I, или соли соединения формулы I, или гидрата или сольвата соли соединения формулы I.2. Соединение I по п.1 в кристаллической форме A.3. Соединение по п.2, на рентгенограмме которого содержится пик при угле дифракции 2-тэта, равном 8,4±0,3°.4. Соединение I по п.1 в кристаллической форме B.5. Соединение по п.3, на рентгенограмме которого содержится пик при...
N-α-(бензилоксикарбонил)-l-γ-глутамил-3-[[2-[[бис[бис(2-хлорэтил)амино]фосфонил]окси]этил]сульфонил]-l-аланил-2(r)-фенилглицин или его соль, фармацевтическая композиция, содержащая это соединение, применение этого соединения для лечения рака и способ лечения рака с помощью этого соединения

Номер патента: 16052
Опубликовано: 30.01.2012
Авторы: Меклер Харолд, Эрнандес-Абад Педро Э., Колиер Стивен Дж., Поломски Роберт Э., Шоу Стивен Р., Гевель Ронан И., Истам Стивен А., Боуланджер Уилльям А., Иди Деннис Л., Кьэрсгорд Ханс Й., Херр Джейсон Р., Жичкин Павел Э.
МПК: A61P 35/00
Метки: это, соединения, фармацевтическая, рака, способ, содержащая, соль, этого, n-α-(бензилоксикарбонил)-l-γ-глутамил-3-[[2-[[бис[бис(2-хлорэтил)амино]фосфонил]окси]этил]сульфонил]-l-аланил-2(r)-фенилглицин, соединение, помощью, применение, композиция, лечения
Формула / Реферат:
1. Соединение формулыили его соль.2. Фармацевтическая композиция, включающая соединение по п.1 или его соль.3. Применение соединения по п.1 или его соли для приготовления лекарственного средства, предназначенного для лечения рака.4. Способ лечения рака у млекопитающего, включающий введение млекопитающему соединения по п.1 или его...
Кристаллические формы [ r-(r*,r*)]-2-(4-фторфенил)-бета, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино) карбонил]-1h-пиррол-1-гептановой кислоты кальциевой соли (2:1) (аторвастатин)

Номер патента: 5317
Опубликовано: 30.12.2004
Авторы: Крзизэниэк Джозеф Фрэнсис, Ли Женг Джейн, Моррисон Генри Грант Второй, Берн Стивен Роберт, Коутс Дейвид Эндрю, Парк Ири, Влахова Петинка Иванова, Гасхёрст Карен Сью
МПК: A61K 31/40, C07D 207/34
Метки: формы, 2:1, аторвастатин, r-(r*,r*)]-2-(4-фторфенил)-бета, соли, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино, карбонил]-1h-пиррол-1-гептановой, кальциевой, кислоты, кристаллические
Формула / Реферат:
1. Кристаллическая Форма V аторвастатина или его гидрата, дающая дифракцию рентгеновских лучей на порошке, содержащую следующие значения 2Q, измеренные с использованием CuKa-излучения: 4.9 (уширенный), 6.0, 7.0, 8.0 (уширенный), 8.6, 9.9, 16.6, 19.0 и 21.1. 2. Кристаллическая Форма VI аторвастатина или его гидрата, дающая дифракцию рентгеновских лучей на порошке, содержащую следующие значения 2Q, измеренные с использованием CuKa-излучения: 7.2,...
Кристаллические формы 4-[2-(4-метилфенилсульфанил)фенил]пиперидина с объединенным ингибированием повторного поглощения серотонина и норадреналина для лечения невропатической боли

Номер патента: 16054
Опубликовано: 30.01.2012
Авторы: Миллер Зилке, Стенсбёль Тине Брайан, Банг-Андерсен Бенни, Лопес Де Диего Хейди, Фальд Андре
МПК: A61P 25/22, A61P 25/04, A61K 31/451...
Метки: кристаллические, формы, поглощения, норадреналина, ингибированием, невропатической, 4-[2-(4-метилфенилсульфанил)фенил]пиперидина, серотонина, боли, лечения, объединенным, повторного
Формула / Реферат:
1. Аддитивная соль HBr и 4-[2-(4-метилфенилсульфанил)фенил]пиперидинав кристаллической форме.2. Соединение по п.1, где соединение характеризуется пиками на рентгеновской порошковой дифрактограмме (XRPD) в области приблизительно 6,08, 14,81, 19,26 и 25,38°2θ.3. Соединение по п.1, где соединение характеризуется рентгеновской порошковой дифрактограммой (XRPD), представленной на фиг. 1.4. Аддитивная соль DL-молочной кислоты и...
Кристаллическая кальцевая соль [r-(r*,r*)]-2-(4-фторфенил)-β,δ- дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1н-пирролгептановой кислоты

Номер патента: 9795
Опубликовано: 28.04.2008
Авторы: Фаустманн Иржи, Ергов Александр
МПК: C07D 207/327
Метки: соль, r-(r*,r*)]-2-(4-фторфенил)-β,&delta, дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1н-пирролгептановой, кальцевая, кислоты, кристаллическая
Формула / Реферат:
1. Соединение, представляющее кристаллическую форму Fa кальций аторвастатина, имеющее картину дифракции рентгеновских лучей, включающую следующие значения 2q, измеренные с использованием излучения CuK-a: примерно 10,2, примерно 11,3 или примерно 18,9. 2. Соединение по п.1, имеющее спектр 13C ЯМР в твердом состоянии вещества, включающий по меньшей мере один химический сдвиг при примерно 20,7, примерно 23,3, примерно 24,5 или примерно 26,1 м.д. 3....
Предыдущий патент: Скважинное фильтрующее устройство
Следующий патент: Lxr модуляторы
Случайный патент: Способ получения модифицированных полипропиленовых композиций