Ингибиторы котранспортера натрий-глюкозы 2 и способы их применения
Номер патента: 16511
Опубликовано: 30.05.2012
Авторы: Харрисон Брайс А., Гудвин Николь К., Мейбон Росс, Роулинс Дэвид Б., Кимболл С.Дэвид







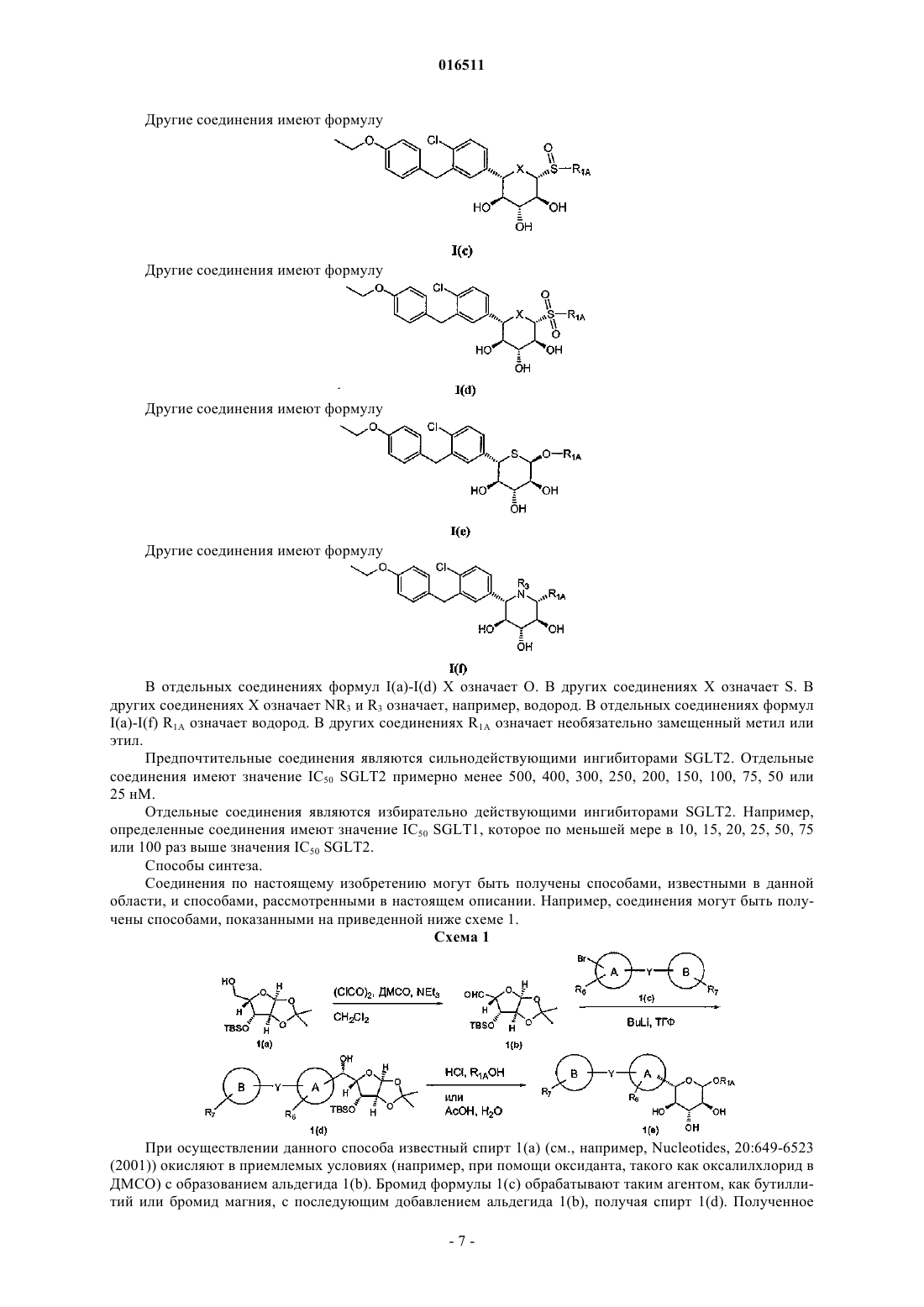


















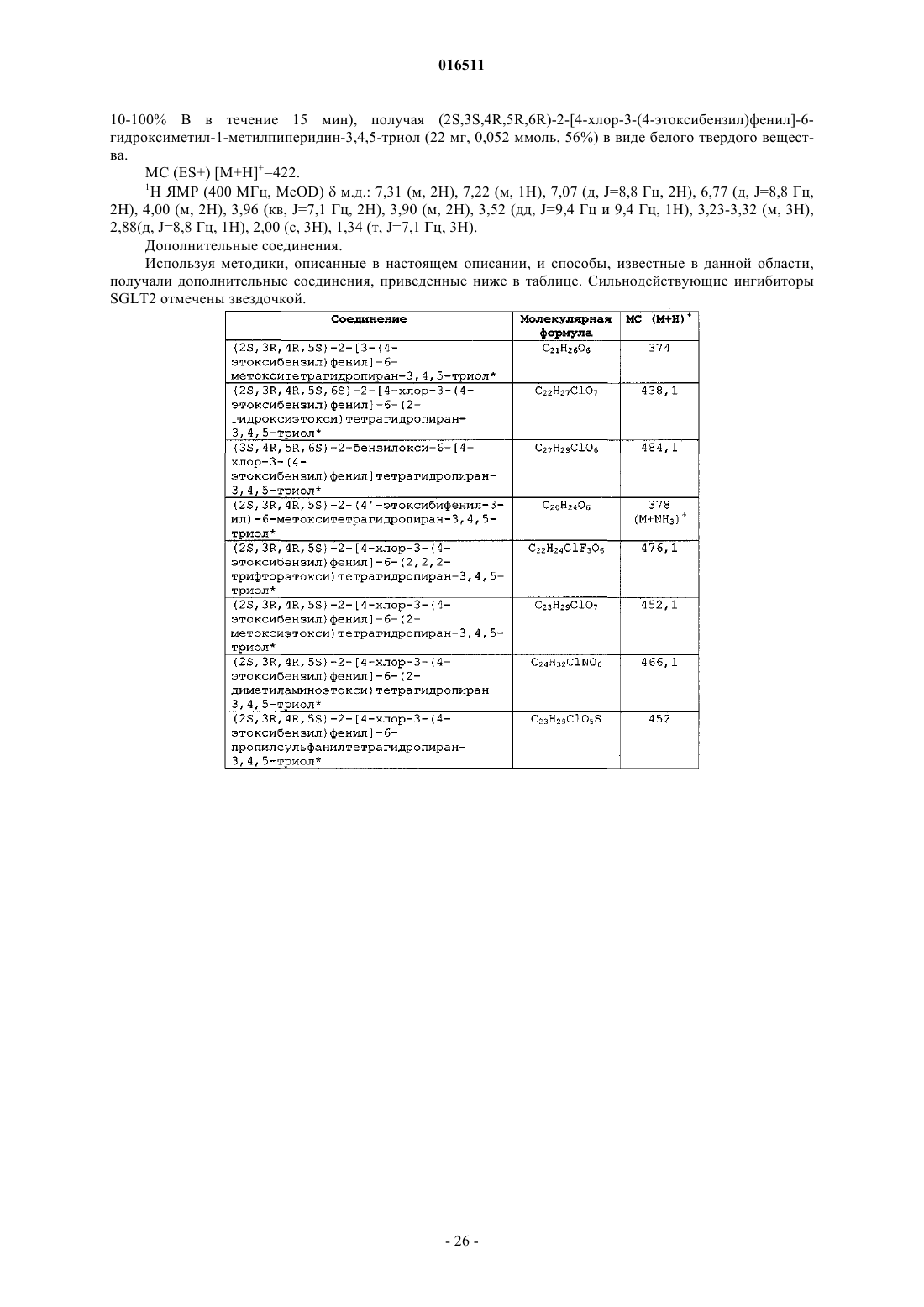



Формула / Реферат
1. Соединение формулы

его фармацевтически приемлемая соль или сольват, где
X означает O, S или NR3;
Y означает O, S, SO, SO2, NR4, (C(R5)2)p, (C(R5)2)q-C(O)-(C(R5)2)q;
когда X означает O, Rx является OR1A, SR1A, SOR1A, SO2R1A или N(R1A)2;
когда X означает S, R1 является водородом, OR1A, SR1A, SOR1A или SO2R1A;
когда X означает NR3, R1 является OR1A, SR1A, SOR1A, SO2R1A или R1A;
каждый R1A независимо означает водород, C1-4алкил или C(O)C1-4алкил;
R2 означает фтор или OR2A;
каждый R2A, R2B и R2C независимо означает водород, C1-4алкил или C(O)C1-4алкил;
R3 означает водород, C(O)R3A, CO2R3A, CON(R3B)2 или C1-4алкил;
R3A независимо означает C1-4алкил;
каждый R3B независимо означает водород, C1-4алкил или C1-4алкенил;
каждый R4 независимо означает водород или C1-4алкил;
каждый R5 независимо означает водород, гидроксил или C1-4алкил;
каждый R6 независимо означает водород, гидроксил, галоген, амино, циано, OR6A, SR6A, SOR6A, SO2R6A, C(O)R6A, CO2R6A, CO2H, CON(R6A)(R6A), CONH(R6A), CONH2, NHC(O)R6A, NHSO2R6A или C1-10алкил, необязательно замещенный альдегидом, амино, галогеном или гидроксилом;
каждый R6A независимо означает C1-4алкил;
каждый R7 независимо означает водород, гидроксил, галоген, амино, циано, OR7A, SR7A, SOR7A, SO2R7A, C(O)R7A, CO2R7A, CO2H, CON(R7A)(R7A), CONH(R7A), CONH2, NHC(O)R7A, NHSO2R7A или C1-10алкил, необязательно замещенный альдегидом, амино, галогеном или гидроксилом;
каждый R7A независимо означает C1-4алкил;
m равен 1-3;
n равен 1-3;
p равен 0-3 и
каждый q независимо равен 0-2.
2. Соединение по п.1, имеющее формулу

3. Соединение по п.2, где X означает O.
4. Соединение по п.2, где X означает S.
5. Соединение по п.2, где X означает NR3.
6. Соединение по п.2, где R1 означает OR1A.
7. Соединение по п.6, где R1A означает водород.
8. Соединение по п.6, где R1A означает C1-4алкил.
9. Соединение по п.2, где R1 означает SR1A.
10. Соединение по п.9, где R1A означает водород.
11. Соединение по п.9, где R1A означает C1-4алкил.
12. Соединение по п.2, где R1 означает SOR1A.
13. Соединение по п.12, где R1A означает водород.
14. Соединение по п.12, где R1A означает C1-4алкил.
15. Соединение по п.2, где R1 означает SO2R1A.
16. Соединение по п.15, где R1A означает водород.
17. Соединение по п.15, где R1A означает C1-4алкил.
18. Соединение по п.2, где R1 означает N(R1A)2.
19. Соединение по п.18, где R1A означает водород.
20. Соединение по п.18, где R1A означает C1-4алкил.
21. Соединение по п.2, где R1 означает водород.
22. Соединение по п.2, где R1 означает R1A и R1A означает C1-4алкил.
23. Соединение по п.2, где R6 означает водород, гидроксил, галоген, OR6A или C1-4алкил.
24. Соединение по п.2, где R7 означает водород, OR7A или C1-4алкил.
25. Соединение по п.2, имеющее формулу

26. Соединение по п.2, имеющее формулу

27. Соединение по п.2, имеющее формулу

28. Соединение по п.2, имеющее формулу

29. Соединение по одному из пп.25-28, где X означает O.
30. Соединение по одному из пп.25-28, где X означает S.
31. Соединение по одному из пп.25-28, где X означает NR3.
32. Соединение по п.2, имеющее формулу

33. Соединение по п.2, имеющее формулу

34. Соединение по одному из пп.25-28, 32 или 33, где R1A означает водород или C1-4алкил.
35. Соединение по п.1, которое представляет собой (2R,3R,4R,5S,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-метилсульфанилтетрагидропиран-3,4,5-триол или его фармацевтически приемлемую соль.
36. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый разбавитель или эксципиент.
37. Фармацевтическая композиция по п.36, в которой соединение представляет собой (2R,3R,4R,5S,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-метилсульфанилтетрагидропиран-3,4,5-триол или его фармацевтически приемлемую соль.
Текст
Настоящее изобретение относится к соединениям и фармацевтическим композициям, содержащим указанные соединения, которые могут быть использованы для лечения таких заболеваний и нарушений, как диабет и ожирение. 016511 По заявке на данный патент испрашивается приоритет по предварительной заявке на патент США 60/848156, поданной 29 сентября 2006 г., предварительной заявке на патент США 60/905714, поданной 8 марта 2007 г., и предварительной заявке на патент США 60/948780, поданной 10 июля 2007 г.,описания которых включены в настоящее описание изобретения во всей своей полноте посредством ссылки. Настоящее изобретение относится к способам лечения метаболических заболеваний и нарушений,таких как диабет, к соединениям и фармацевтическим композициям, полезным при осуществлении вышеуказанных способов. Котранспортер натрий-глюкозы 2 (SGLT2) является транспортером, который реабсорбирует глюкозу из почечного фильтрата и предотвращает выведение глюкозы в моче. Так как конкурентные ингибиторы SGLT2 вызывают экскрецию глюкозы в моче, они могут быть использованы для нормализации высоких уровней глюкозы в крови, обусловленных такими заболеваниями, как диабет. Handlon, A.L., ExpertOpin. Ther. Patents 15 (11):1531-1540 (2005). В научной литературе описан целый ряд ингибиторов SGLT2. См., например, приведенную выше публикацию Handlon; патент США 6515117; публикации заявок на патент СШАUS 2006/0035841, US 2004/0138439. По меньшей мере один ингибитор проходит клинические исследования в качестве лекарственного средства для лечения сахарного диабета типа 2. См., например, Komoroski, В., et al., "Dapagliflozin (BMS-512148), a Selective Inhibitor of the SodiumGlucose Uptake Transporter 2 (SGLT2), Reduces Fasting Serum Glucose and Glucose Excursion in Type 2 Diabetes Mellitus Patients Over 14 Days" American Diabetes Assn. 67th Scientific Sessions. Abstract 0188-OR(2007). Первым известным ингибитором SGLT2 являлся природный продукт флоризин (глюкоза-1-[2-(-Dглюкопиранозилокси)-4,6-дигидроксифенил]-3-(4-гидроксифенил)-1-пропанон), и "все последующие ингибиторы SGLT2 являются гликозидами, выделенными из его структуры", Handlon, см. выше, с. 1533. Флоризин состоит из глюкозной части и двух гидроксилированных ароматических колец, соединенных пропаноновым спейсером. Ehrenkranz, J.R.L., et al., Diabetes Metab. Res. Rev. 21:31-38 (2005). Обзор патентной литературы не позволяет выявить каких-либо синтетических ингибиторов SGLT2, не содержащих глюкозид или его производное; Handlon, см. выше. Действительно, "из-за относительного единообразия гликозидов, представленных в патентной литературе, посвященной SGLT2, изобретателям потенциальных лекарственных средств все труднее найти неисследованную область химического синтеза таких соединений"; там же, с. 1537. Но такие попытки все же предпринимаются, см., например, заявку на патент США 11/168905, поданную на имя Eckhardt et al. и озаглавленную "D-Xylopyranosyl-SubstitutedPhenyl Derivatives, Medicaments Containing Such Compounds, Their Use and Process for Their Manufacture"; заявку на патент США 11/182986, поданную на имя Eckhardt et al. и озаглавленную "Methylidene-DXylopyranosyl- and Oxo-D-Xylopyranosyl-Substituted Derivatives, Medicaments Containing Such Compounds,Their Use and Process for Their Manufacture; и заявку на патент США 11/199962, поданную на имя Eckhardt et al. и озаглавленную "D-Xylopyranosyl-Phenyl-Substituted Cycles, Medicaments Containing SuchCompounds, Their Use and Process for Their Manufacture". Настоящее изобретение относится к новым ингибиторам SGLT2. Один вариант осуществления изобретения относится к соединениям формулы их фармацевтически приемлемым солям или сольватам, гдеR2 означает фтор или OR2A; каждый R2A, R2B и R2C независимо означает водород, C1-4 алкил или C(O)C1-4 алкил;R3A независимо означает C1-10 алкил; каждый R3B независимо означает водород, C1-4 алкил или C1-4 алкенил; каждый R4 независимо означает водород или C1-4 алкил; каждый R5 независимо означает водород, гидроксил или C1-4 алкил; каждый R6 независимо означает водород, гидроксил, галоген, амино, циано, OR6A, SR6A, SOR6A,-1 016511p равен 0-3 и каждый q независимо равен 0-2. Настоящее изобретение относится к фармацевтическим композициям, содержащим соединения,представленные в настоящем описании. Настоящее изобретение относится также к способам ингибирования активности SGLT2, а также к способам лечения, профилактики и устранения рецидива различных заболеваний и нарушений. Определенные объекты настоящего изобретения могут быть поняты со ссылкой на чертеж, где показано воздействие различных соединений по настоящему изобретению на экскрецию глюкозы в моче у мышей. Указанные соединения вводили перорально в дозе 30 мг/кг. В основе настоящего изобретения частично лежит открытие того, что соединения формулы имеющие описанные ниже заместители, могут ингибировать котранспортер натрий-глюкозы 2(SGLT2). Если не указано иное, термин "алкенил" означает прямой, разветвленный и/или циклический углеводород, содержащий 2-20 (например, 2-10 или 2-6) атомов углерода и имеющий по меньшей мере одну углерод-углеродную двойную связь. Конкретные алкенильные группы включают винил, аллил, 1 бутенил, 2-бутенил, изобутиленил, 1-пентенил, 2-пентенил, 3-метил-1-бутенил, 2-метил-2-бутенил, 2,3 диметил-2-бутенил, 1-гексенил, 2-гексенил, 3-гексенил, 1-гептенил, 2-гептенил, 3-гептенил, 1-октенил, 2 октенил, 3-октенил, 1-ноненил, 2-ноненил, 3-ноненил, 1-деценил, 2-деценил и 3-деценил. Если не указано иное, термин "алкокси" означает -O-алкильную группу. Примеры алкоксигрупп включают, но не ограничиваясь ими, -OCH3, -OCH2CH3, -O(CH2)2CH3, -O(CH2)3CH3, -O(CH2)4CH3 и-O(CH2)5CH3. Если не указано иное, термин "алкил" означает прямой и/или разветвленный углеводород, содержащий 1-20 (например, 1-10 или 1-4) атомов углерода. Алкильные группы, содержащие 1-4 атома углерода, определяются как "низший алкил". Примеры алкильных групп включают, но не ограничиваясь ими,метил, этил, пропил, изопропил, н-бутил, трет-бутил, изобутил, пентил, гексил, изогексил, гептил, 4,4 диметилпентил, 2,2,4-триметилпентил, нонил, децил, ундецил и додецил. Циклоалкильные группы могут быть моноциклическими или полициклическими, примеры таких групп включают циклопропил, циклобутил, циклопентил, циклогексил и адамантил. Дополнительные примеры алкильных групп включают группы с линейной, разветвленной и/или циклической структурой (например, 1-этил-4 метилциклогексил). В определение термина "алкил" входят насыщенные углеводороды, а также алкенильные и алкинильные группы. Если не указано иное, термин "алкиларил" означает алкильную группу, связанную с арильной группой. Если не указано иное, термин "алкилгетероарил" означает алкильную группу, связанную с гетероарильной группой. Если не указано иное, термин "алкилгетероцикл" означает алкильную группу, связанную с гетероциклом. Если не указано иное, термин "алкинил" означает прямой, разветвленный или циклический углеводород, содержащий 2-20 (например, 2-20 или 2-6) атомов углерода и имеющий по меньшей мере одну углерод-углеродную тройную связь. Конкретные алкинильные группы включают ацетиленил, пропинил,1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 3-метил-1-бутинил, 4-пентинил, 1-гексинил, 2-гексинил,5-гексинил, 1-гептинил, 2-гептинил, 6-гептинил, 1-октинил, 2-октинил, 7-октинил, 1-нонинил, 2 нонинил, 8-нонинил, 1-децинил, 2-децинил и 9-децинил. Если не указано иное, термин "арил" означает ароматическое кольцо либо ароматическую или частично ароматическую кольцевую систему, состоящую из атомов углерода и водорода. Арильная группа может включать несколько колец, связанных или конденсированных друг с другом. Примеры арильных-2 016511 групп включают, но не ограничиваясь ими, антраценил, азуленил, бифенил, флуоренил, индан, инденил,нафтил, фенантренил, фенил, 1,2,3,4-тетрагидронафталин и толил. Если не указано иное, термин "арилалкил" означает арильную группу, связанную с алкильной группой. Если не указано иное, термин "галоген" означает фтор, хлор, бром и йод. Если не указано иное, термин "гетероалкил" означает алкильную группу (например, линейную, разветвленную или циклическую), в которой по меньшей мере один из атомом углерода заменен гетероатомом (например, N, O или S). Если не указано иное, термин "гетероарил" означает арильную группу, в которой по меньшей мере один из атомов углерода заменен гетероатомом (например, N, O или S). Примеры таких групп включают,но не ограничиваясь ими, акридинил, бензимидазолил, бензофуранил, бензоизотиазолил, бензоизоксазолил, бензохиназолинил, бензотиазолил, бензоксазолил, фурил, имидазолил, индолил, изотиазолил, изоксазолил, оксадиазолил, оксазолил, фталазинил, пиразинил, пиразолил, пиридазинил, пиридил, пиримидинил, пиримидил, пирролил, хиназолинил, хинолинил, тетразолил, тиазолил и триазинил. Если не указано иное, термин "гетероарилалкил" означает гетероарильную группу, связанную с алкильной группой. Если не указано иное, термин "гетероцикл" означает ароматическое, частично ароматическое или неароматическое моноциклическое или полициклическое кольцо или кольцевую систему, состоящую из атомов углерода, водорода и по меньшей мере одного гетероатома (например, N, O или S). Гетероцикл может включать несколько (т.е. два или более) колец, конденсированных или связанных друг с другом. Гетероциклы включают гетероарилы. Примеры таких групп включают, но не ограничиваясь ими, бензо[1,3]диоксолил, 2,3-дигидробензо[1,4]диоксинил, циннолинил, фуранил, гидантоинил, морфолинил,оксетанил, оксиранил, пиперазинил, пиперидинил, пирролидинонил, пирролидинил, тетрагидрофуранил,тетрагидропиранил, тетрагидропиридинил, тетрагидропиримидинил, тетрагидротиофенил, тетрагидротиопиранил и валеролактамил. Если не указано иное, термин "гетероциклоалкил" означает гетероцикл, связанный с алкильной группой. Если не указано иное, термин "гетероциклоалкил" означает неароматический гетероцикл. Если не указано иное, термин "гетероциклоалкилалкил" означает гетероциклоалкильную группу,связанную с алкильной группой. Если не указано иное, термин "ингибирует SGLT2 in vivo" означает ингибирование SGLT2, определяемое при выполнении анализа in vivo, описанного в приведенных ниже примерах. Если не указано иное, термины "устранять рецидив" и "устранение рецидива" означают предотвращение рецидива определенного заболевания или нарушения у субъекта, который ранее страдал указанным заболеванием или нарушением, и/или увеличение продолжительности ремиссии у субъекта, страдающего данным заболеванием или нарушением. Указанные термины означают изменение порога возникновения, развития и/или продолжительности заболевания или нарушения либо изменение реакции субъекта на данное заболевание или нарушение. Если не указано иное, термин "фармацевтически приемлемые соли" означает соли, полученные из фармацевтически приемлемых нетоксичных кислот или оснований, включающих неорганические кислоты и основания и органические кислоты и основания. Фармацевтически приемлемые основноаддитивные соли включают, но не ограничиваясь ими, соли металлов, полученные из алюминия, кальция, лития, магния, калия, натрия и цинка, или органические соли, полученные из лизина, N,N'дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, меглумина (Nметилглюкамина) и прокаина. Приемлемые нетоксичные кислоты включают, но не ограничиваясь ими,неорганические и органические кислоты, такие как уксусная, альгиновая, антраниловая, бензосульфоновая, бензойная, камфорсульфоновая, лимонная, этиленсульфоновая, муравьиная, фумаровая, фуроновая,галактуроновая, глюконовая, глюкуроновая, глутаминовая, гликолевая, бромисто-водородная, хлористоводородная, изэтионовая, молочная, малеиновая, яблочная, миндальная, метансульфоновая, слизевая,азотная, памовая, пантотеновая, фенилуксусная, фосфорная, пропионовая, салициловая, стеариновая,янтарная, сульфаниловая, серная, винная кислота и паратолуолсульфоновая кислоты. Особенно приемлемые нетоксичные кислоты включают хлористо-водородную, бромисто-водородную, фосфорную, серную и метансульфоновую кислоты. Таким образом, примеры особенно приемлемых солей включают гидрохлорид и мезилат. В данной области хорошо известны другие соли. См.,например, публикацииand Practice of Pharmacy, 19th ed. (Mack Publishing, Easton PA: 1995). Если не указано иное, "сильнодействующий ингибитор SGLT2" является соединением, которое имеет значение IC50 SGLT2 примерно менее 500 нМ. Если не указано иное, термины "предотвращать" и "предотвращение" означают меры, предпринимаемые до того, как субъект заболеет определенным заболеванием или нарушением, которые позволяют ингибировать или ослабить тяжесть данного заболевания или нарушения. Другими словами, указанные термины означают профилактику.-3 016511 Если не указано иное, "профилактически эффективное количество" соединения является количеством, достаточным для предотвращения заболевания или состояния, одного или нескольких симптомов,ассоциированных с данным заболеванием или состоянием, или устранения рецидива. "Профилактически эффективное количество" соединения означает количество лекарственного средства, используемого отдельно или в комбинации с другими средствами, которое оказывает профилактическое воздействие по предотвращению заболевания. Термин "профилактически эффективное количество" может означать количество, которое улучшает профилактику или усиливает профилактическую эффективность другого профилактического средства. Если не указано иное, "избирательно действующим ингибитором SGLT2" является соединение, у которого значение IC50 SGLT1 по меньшей мере в 10 раз выше значения IC50 SGLT2. Если не указано иное, термин "IC50 SGLT1" означает значение IC50 соединения, полученное при выполнении анализа ингибирования SGLT1 человека in vitro, описанного в приведенных ниже примерах. Если не указано иное, термин "IC50 SGLT2" означает значение IC50 соединения, полученное при выполнении анализа ингибирования SGLT2 человека in vitro, описанного в приведенных ниже примерах. Если не указано иное, термин "смесь стереоизомеров" означает рацемические смеси, а также смеси с повышенным содержанием стереоизомеров (например, R/S=30/70, 35/65, 40/60, 45/55, 55/45, 60/40,65/35 и 70/30). Если не указано иное, термин "стереомерно чистый" означает композицию, которая содержит один стереоизомер соединения и, по существу, не содержит других стереоизомеров указанного соединения. Например, стереомерно чистая композиция, содержащая соединение с одним стереоцентром, по существу, не должна содержать противоположного стереоизомера данного соединения. Стереомерно чистая композиция, содержащая соединение с двумя стереоцентрами, по существу, не должна содержать других диастереомеров данного соединения. Обычно стереомерно чистое соединение включает примерно более 80 мас.% одного стереоизомера соединения и примерно менее 20 мас.% других стереоизомеров данного соединения, примерно более 90 мас.% одного стереоизомера соединения и примерно менее 10 мас.% других стереоизомеров данного соединения, примерно более 95 мас.% одного стереоизомера соединения и примерно менее 5 мас.% других стереоизомеров данного соединения, примерно более 97 мас.% одного стереоизомера соединения и примерно менее 3 мас.% других стереоизомеров данного соединения или примерно более 99 мас.% одного стереоизомера соединения и примерно менее 1 мас.% других стереоизомеров данного соединения. Если не указано иное, термин "замещенный", используемый при описании химической структуры или группы, означает производное данной структуры или группы, в которой один или несколько атомов водорода замещены химическим фрагментом или функциональной группой, которые включают, но не ограничиваясь ими, гидроксил, альдегид, алкокси, алканоилокси, алкоксикарбонил, алкенил, алкил (например, метил, этил, пропил, трет-бутил), алкинил, алкилкарбонилокси (-OC(O)алкил), амид (-C(O)NHалкил- или алкил-NHC(O)алкил), амидинил (-C(NH)NH-алкил- или -C(NR)NH2), амин (первичный, вторичный или третичный, такой как алкиламино, ариламино, арилалкиламино), ароил, арил, арилокси, азо,карбамоил (-NHC(O)O-алкил- или -OC(O)NH-алкил), карбамил (например, CONH2, а также CONH-алкил,CONH-арил и CONH-арилалкил), карбонил, карбоксил, карбоновую кислоту, ангидрид карбоновой кислоты, хлорангидрид карбоновой кислоты, циано, сложный эфир, эпоксид, простой эфир (например, метокси, этокси), гуанидино, галоген, галогеналкил (например, -CCl3, -CF3, C(CF3)3), гетероалкил, гемиацеталь, имин (первичный и вторичный), изоцианат, изотиоцианат, кетон, нитрил, нитро, оксо, сложный фосфодиэфир, сульфид, сульфонамидо (например, SO2NH2), сульфон, сульфонил (включая алкилсульфонил, арилсульфонил и арилалкилсульфонил), сульфоксид, тиол (например, сульфгидрил, простой тиоэфир) и мочевину (-NHCONH-алкил-). Если не указано иное, "терапевтически эффективное количество" соединения является количеством,достаточным для обеспечения благоприятного терапевтического воздействия при лечении или устранении рецидива заболевания или состояния, для задержки или минимизации одного или нескольких симптомов, ассоциированных с данным заболеванием или состоянием. "Терапевтически эффективное количество" соединения означает количество лекарственного средства, используемого отдельно или в комбинации с другими способами лечения, которое обеспечивает благоприятное терапевтическое воздействие при лечении или устранении рецидива заболевания или состояния. Термин "терапевтически эффективное количество" может означать количество, которое улучшает общее лечение, ослабляет или устраняет симптомы или причины заболевания или состояния, усиливает терапевтическую эффективность другого лекарственного средства. Если не указано иное, термины "лечить" и "лечение" означают меры, предпринимаемые во время болезни субъекта, которые позволяют ослабить тяжесть заболевания или нарушения, задержать или замедлить прогрессирование заболевания или нарушения. Если не указано иное, термин "включают" имеет такое же значение, что и термин "включают, но не ограничиваются ими", и термин "включает" имеет такое же значение, что и термин "включает, но не ограничивается им". Аналогичным образом термин "такой как" имеет такое же значение, что и термин "такой как, но не ограничивается им".-4 016511 Если не указано иное, одно или несколько прилагательных, непосредственно предшествующих нескольким существительным, относятся к каждому из существительных. Например, фраза "необязательно замещенный алкил, арил или гетероарил" имеет такое же значение, что и фраза "необязательно замещенный алкил, необязательно замещенный арил или необязательно замещенный гетероарил". Следует отметить, что химическая группа, образующая часть более крупного соединения, может иметь в настоящем описании название, обычно используемое для определения отдельной молекулы, или название, обычно используемое для определения радикала. Например, термины "пиридин" и "пиридил" имеют одинаковое значение при их использовании для обозначения фрагмента, присоединенного к другим химическим фрагментам. Таким образом, две фразы "XOH, где X означает пиридил" и "XOH, где X означает пиридин" имеют одинаковое значение и означают соединения пиридин-2-ол, пиридин-3-ол и пиридин-4-ол. Следует также отметить, что, если стереохимия структуры или части структуры не отмечена, например, жирными или пунктирными линиями, такая структура или часть структуры предположительно включает все стереоизомеры. Кроме того, считается, что любой атом, показанный на чертеже с ненасыщенными валентностями, присоединен к достаточному числу атомов водорода для насыщения валентностей. Помимо этого, химические связи, изображенные в виде одной сплошной линии, параллельной одной пунктирной линии, означают как простые, так и двойные (например, ароматические) связи, если валентности допускают такую возможность. Соединения. Один вариант осуществления изобретения относится к соединениям формулы их фармацевтически приемлемым солям и сольватам, гдеY означает O, S, SO, SO2, NR4, (C(R5)2)p, (C(R5)2)q-C(O)-(C(R5)2)q; когда X означает O, R1 является OR1A, SR1A, SOR1A, SO2R1A или N(R1A)2; когда X означает S, R1 является водородом, OR1A, SR1A, SOR1A или SO2R1A; когда X означает NR3, R1 является OR1A, SR1A, SOR1A, SO2R1A или R1A; каждый R1A независимо означает водород C1-4 алкил или C(O) C1-4 алкил;R2 означает фтор или OR2A; каждый R2A, R2B и R2C независимо означает водород, C1-4 алкил или C(O)C1-4 алкил;R3A независимо означает C1-10 алкил; каждый R3B независимо означает водород, C1-4 алкил или C1-4 алкенил; каждый R4 независимо означает водород или C1-4 алкил; каждый R5 независимо означает водород, гидроксил или C1-4 алкил; каждый R6 независимо означает водород, гидроксил, галоген, амино, циано, OR6A, SR6A, SOR6A,SO2R6A, C(O)R6A, CO2R6A, CO2H, CON(R6A)(R6A), CONH(R6A), CONH2, NHC(O)R6A, NHSO2R6A илиp равен 0-3 и каждый q независимо равен 0-2. Отдельные соединения имеют формулу-5 016511 Некоторые соединения имеют формулу Некоторые соединения имеют формулуX означает NR3. В некоторых соединениях Y означает (C(R4)2)p и, например, p равно 1. В некоторых соединениях Y означает (C(R5)2)q-C(O)-(C(R5)2)q и, например, каждый q независимо равен 0 или 1. В некоторых соединениях R1 означает OR1A. В других соединениях R1 означает SR1A. В других соединениях R1 означает SOR1A. В других соединениях R1 означает SO2R1A. В других соединениях R1 означает N(R1A)2. В других соединениях R1 означает водород. В других соединениях R1 означает R1A. В некоторых соединениях R1A означает водород. В других соединениях R1A означает необязательно замещенный алкил (например, необязательно замещенный низший алкил). В некоторых соединениях R2 означает фтор. В других соединениях R2 означает OR2A. В некоторых соединения R2A означает водород. В некоторых соединения R2B означает водород. В некоторых соединениях R2C означает водород. В некоторых соединениях R3 означает водород. В других соединениях R3 означает необязательно замещенный низший алкил (например, необязательно замещенный метил). В некоторых соединениях R4 означает водород или необязательно замещенный низший алкил. В некоторых соединениях каждый R5 означает водород или необязательно замещенный низший алкил (например, метил, этил, CF3). В некоторых соединениях R6 означает водород, гидроксил, галоген, OR6A или необязательно замещенный низший алкил (например, необязательно галогензамещенный метил, этил или изопропил). В некоторых соединениях R6 означает водород. В некоторых соединениях R6 означает галоген (например,хлор). В некоторых соединениях R6 означает гидроксил. В некоторых соединениях R6 означает OR6A (например, метокси, этокси). В некоторых соединениях R6 означает необязательно замещенный метил (например, CF3). В некоторых соединениях R7 означает водород, OR7A или необязательно замещенный низший алкил(например, необязательно галогензамещенный метил, этил или изопропил). В некоторых соединениях R7 означает OR7A (например, метокси, этокси). В некоторых соединениях R7 означает необязательно замещенный метил или этил. Отдельные соединения по настоящему изобретению имеют формулу Другие соединения имеют формулу-6 016511 Другие соединения имеют формулу Другие соединения имеют формулу Другие соединения имеют формулу Другие соединения имеют формулу В отдельных соединениях формул I(a)-I(d) X означает O. В других соединениях X означает S. В других соединениях X означает NR3 и R3 означает, например, водород. В отдельных соединениях формулI(a)-I(f) R1A означает водород. В других соединениях R1A означает необязательно замещенный метил или этил. Предпочтительные соединения являются сильнодействующими ингибиторами SGLT2. Отдельные соединения имеют значение IC50 SGLT2 примерно менее 500, 400, 300, 250, 200, 150, 100, 75, 50 или 25 нМ. Отдельные соединения являются избирательно действующими ингибиторами SGLT2. Например,определенные соединения имеют значение IC50 SGLT1, которое по меньшей мере в 10, 15, 20, 25, 50, 75 или 100 раз выше значения IC50 SGLT2. Способы синтеза. Соединения по настоящему изобретению могут быть получены способами, известными в данной области, и способами, рассмотренными в настоящем описании. Например, соединения могут быть получены способами, показанными на приведенной ниже схеме 1. Схема 1 При осуществлении данного способа известный спирт 1(a) (см., например, Nucleotides, 20:649-6523(2001 окисляют в приемлемых условиях (например, при помощи оксиданта, такого как оксалилхлорид в ДМСО) с образованием альдегида 1(b). Бромид формулы 1(c) обрабатывают таким агентом, как бутиллитий или бромид магния, с последующим добавлением альдегида 1(b), получая спирт 1(d). Полученное-7 016511 соединение обрабатывают спиртом или водой в кислотных условиях, с образованием соединения 1(e). При желании для преобразования соединения 1(e) в различные другие соединения, входящие в объем настоящего изобретения (например, соединения формулы I, где один или несколько элементов R2A, R2B иR2C не являются водородом, и/или R1 означает SR1A или NHR1A), могут быть использованы способы, хорошо известные в данной области. Со ссылкой на схему 1 и другие способы синтеза, рассмотренные в настоящем описании, можно отметить, что хорошо известны способы получения фрагментов A и A-Y-B, а также способы их использования для получения ингибиторов SGLT2. Например, синтез связанных диарильных производных при получении ингибиторов SGLT2 описан в патентах США 7045665 и 7053060; в заявках на патент США 10/735179, 10/745075, 11/080150 и 11/182986 и в международных заявках на патент WO 2006/006496 иWO 2006/089872. Синтез ингибиторов SGLT2, содержащих связанные фенилкарбоциклические фрагменты, описан,например, в заявках на патент США 11/190315 и 11/199962. Синтез связанных гетероциклов и их использование при получении ингибиторов SGLT2 описан,например, в заявках на патент США 10/540519, 10/734573, 11/247216, 11/247356 и в международных заявках на патент WO 03/020737, WO 2004/058790, WO 2004/080990, WO 2004/089967, WO 2005/011592,WO 2005/012242, WO 2005/012243, WO 2005/012318, WO 2005/021566 и WO 2005/085265. Соединения на основе пиперидина могут быть получены способами, показанными на приведенной ниже схеме 2. Схема 2 При осуществлении данного способа соединение 2 (a), которое может быть получено способом, показанным на схеме 1, контактируют с азидом (например, дифенилфосфорилазидом) в условиях, обеспечивающих получение азида 2(b). Затем азид обрабатывают в кислотных условиях с образованием незащищенного фурана 2(c), который затем обрабатывают восстановителем (например, водородом в присутствии оксида платины) в кислотных условиях, получая соединение 2(d). При желании для преобразования соединения 2(d) в различные другие соединения, входящие в объем настоящего изобретения (например, соединения формулы I, где один или несколько элементов R2A, R2B и R2C не являются водородом,и/или R1 означает SR1A или NHR1A), могут быть использованы способы, хорошо известные в данной области. Соединения на основе тетрагидротиопирана могут быть получены в соответствии со схемой 3. Схема 3 При осуществлении данного способа соединение 3(a), которое может быть получено в соответствии со схемой 1, контактируют с приемлемым серосодержащим соединением (например, тиоацетатом) в приемлемых условиях (например, в присутствии диэтилазодикарбоксилата) с образованием тиоацетата 3(b). Затем тиоацетат обрабатывают приемлемым основанием (например, гидроксидом цезия) с образованием тиола формулы 3(c), который затем обрабатывают спиртом или водой в кислотных условиях, получая соединение 3(d). При желании для преобразования соединения 3(d) в различные другие соединения, входящие в объем настоящего изобретения (например, соединения формулы I, где один или несколько элементов R2A, R2B и R2C не являются водородом, и/или R1 означает SR1A или NHR1A), могут быть использованы способы, хорошо известные в данной области. Соединения, содержащие фторированный сахар или аналог сахара (соединения формулы I, где R2-8 016511 означает F), могут быть получены из соответствующих замещенных исходных веществ при помощи способов, известных в данной области. См., например, заявку на патент США 10/735179. Соединения с открытой структурой (например, соединения формулы II) могут быть легко получены способами, известными в данной области. Например, указанные соединения могут быть получены способами, показанными на приведенной ниже схеме 4. Схема 4 При осуществлении данного способа соединение 4(a), которое может быть получено в соответствии со схемой 1, контактируют с реакционноспособным соединением (например, метилхлорформиатом) в приемлемых условиях с образованием метилкарбоната 4(b). Затем метилкарбонат обрабатывают спиртом в кислотных условиях с образованием соединения 4(c). При желании для преобразования соединения 4(c) в различные другие соединения, входящие в объем настоящего изобретения (например, соединения формулы II, где один или несколько элементов R2A, R2B и R2C не являются водородом), могут быть использованы способы, хорошо известные в данной области. Используя способы, известные в данной области, можно легко модифицировать представленные выше способы синтеза для получения целого ряда соединений. Стереомерно чистые соединения могут быть получены при помощи хиральной хроматографии и других хорошо известных методов. См., например, Jacques, J., et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen,S.H., et al., Tetrahedron 33:2725 (1977); Eliel, E.L., Stereochemistry of Carbon Compounds (McGraw Hill,N.Y., 1962); и Wilen, S.H., Tables of Resolving Agents and Optical Resolutions, p. 268 (E.L. Eliel, Ed., Univ.of Notre Dame Press, Notre Dame, IN, 1972). Кроме того, при выполнении способов синтеза могут быть использованы хиральные исходные вещества для получения продуктов с повышенным содержанием стереомеров или чистых продуктов. Способы применения. Настоящее изобретение относится к способу ингибирования активности SGLT2, который включает контактирование SGLT2 с эффективным количеством соединения по настоящему изобретению (то есть,нового соединения, раскрытого в настоящем описании). В одном варианте осуществления изобретения белок используют in vivo. В другом варианте осуществления изобретения белок используют ex vivo. Настоящее изобретение относится также к способу снижения уровня глюкозы в крови у субъекта(например, млекопитающего, такого как человек, собака или кошка), который включает введение субъекту эффективного количества соединения по настоящему изобретению. Настоящее изобретение относится также к способу увеличения экскреции глюкозы в моче субъекта,который включает введение субъекту эффективного количества соединения по настоящему изобретению. Настоящее изобретение относится также к способу восстановления или увеличения восприимчивости к инсулину у субъекта, который включает введение субъекту эффективного количества соединения по настоящему изобретению. Настоящее изобретение относится также к способу лечения, устранения рецидива или профилактики заболевания или нарушения у субъекта, который включает введение субъекту терапевтически или профилактически эффективного количества соединения по настоящему изобретению. Примеры заболеваний и нарушений включают атеросклероз, сердечно-сосудистые заболевания, диабет (типа 1 и 2), гипергликемию, гипертензию, нарушение липидного обмена, ожирение и синдром X. Особенно предпочтительным заболеванием является диабет типа 2. Количество вводимого соединения, способ введения и схема дозирования соединения могут зависеть от таких факторов, как конкретное заболевание, подлежащее лечению, предотвращению или устранению рецидива, а также возраст, пол и состояние здоровья субъекта. Значение таких факторов хорошо известно в данной области, условия лечения могут быть приведены в соответствие с указанными факторами путем обычного экспериментирования. Фармацевтические препараты. Настоящее изобретение относится к фармацевтическим композициям, содержащим одно или несколько соединений по настоящему изобретению. Некоторые фармацевтические композиции представляют собой единичные дозированные формы на один прием, предназначенные для перорального введе-9 016511 ния, введения через слизистую оболочку (например, для назального, сублингвального, вагинального,трансбуккального или ректального введения), парентерального введения (например, для подкожного введения, внутривенного введения, инъекции ударной дозы, внутримышечного или внутриартериального введения) или чрескожного введения субъекту. Примеры дозированных форм включают, но не ограничиваясь ими, таблетки; драже; капсулы, такие как мягкие эластичные желатиновые капсулы; крахмальные капсулы; пастилки; лепешки; дисперсии; суппозитории; мази; припарки; пасты; порошки; повязки; кремы; пластыри; растворы; наклейки; аэрозоли (например, назальные аэрозоли или ингаляторы); гели; жидкие дозированные формы для пероральоного введения и введения субъекту через слизистую оболочку, включающие суспензии (например, водные или неводные жидкие суспензии, эмульсии масло-в-воде или жидкие вода-в-масле эмульсии), растворы и эликсиры; жидкие дозированные формы, пригодные для парентерального введения субъекту; и стерильные твердые вещества (например, кристаллические или аморфные твердые вещества), которые могут быть восстановлены с образованием жидких дозированных форм, пригодных для парентерального введения субъекту. Препарат должен соответствовать способу введения. Например, препараты, предназначенные для перорального введения, должны быть защищены энтеросолюбильным покрытием от деградации в желудочно-кишечном тракте. Аналогичным образом препарат может содержать ингредиенты, облегчающие доставку активных ингредиентов к месту воздействия. Например, соединения могут быть введены в липосомных препаратах для их защиты от воздействия расщепляющих ферментов, облегчения переноса к кровеносную систему и проникновения через клеточные мембраны во внутриклеточное пространство. Композиция, форма и тип дозированной формы могут изменяться в зависимости от применения. Например, дозированная форма, используемая для неотложного лечения заболевания, может содержать большие количества одного или нескольких активных ингредиентов по сравнению с дозированной формой, используемой для продолжительного лечения того же заболевания. Аналогично, дозированная форма для парентерального введения может содержать меньшие количества одного или нескольких активных ингредиентов по сравнению с дозированной формой для перорального введения при лечении одного и того же заболевания. Специалистам в данной области известны вышеуказанные и другие факторы, определяющие различие между конкретными дозированными формами по настоящему изобретению. См.,например, публикацию Remington's Pharmaceutical Sciences, 18th ed. (Mack Publishing, Easton PA: 1990). Фармацевтические композиции по настоящему изобретению предпочтительно вводят перорально. Дискретные дозированные формы, предназначенные для перорального введения, включают таблетки(например, жевательные таблетки), драже, капсулы и жидкости (например, ароматизированные сиропы). Такие дозированные формы содержат заранее определенные количества активных ингредиентов и могут быть получены фармацевтическими способами, хорошо известными специалистам в данной области. См., например, публикацию Remington's Pharmaceutical Sciences, 18th ed. (Mack Publishing, Easton PA: 1990). Конкретные дозированные формы для перорального введения получают, объединяя активные ингредиенты в тщательно перемешанной смеси по меньшей мере с одним эксципиентом в соответствии со стандартными фармацевтическими способами смешивания. Эксципиенты могут иметь различные формы в зависимости от формы препарата, требуемой для введения. Благодаря легкости введения таблетки и капсулы являются наиболее предпочтительными дозированными формами для перорального введения. При желании на таблетки может быть нанесено покрытие стандартными водными или неводными методами. Такие дозированные формы могут быть получены стандартными способами, применяемыми в фармации. Как правило, фармацевтические композиции и дозированные формы получают в результате однородного и тщательного смешивания активных ингредиентов с жидкими носителями, тонко измельченными твердыми носителями или теми и другими вместе, с последующим при необходимости приданием продукту соответствующей формы. Для быстрого растворения в твердые дозированные формы могут быть введены дезинтеграторы. В дозированные формы могут быть также введены лубриканты, облегчающие изготовление дозированных форм (например,таблеток). Примеры. Пониманию аспектов настоящего изобретения могут способствовать приведенные ниже примеры,которые не ограничивают объем изобретения. Пример 1. Синтез (2S,3R,4R,5S)-2-[4-Хлор-3-(4-этоксибензил)фенил]-6-метокситетрагидропиран 3,4,5-триола. Указанное в заголовке соединение получали в несколько стадий.A. Получение [(3aS,5S,6R,6aR)-6-(трет-бутилдиметилсиланилокси)-2,2-диметилтетрагидрофуро[2,3d][1,3]диоксол-5-ил]метанола. Указанное соединение синтезировали способами, известными в данной области. См., например,Nucleosides Nucleotides, 20:649-652 (2001) и приведенные там ссылки.B. Получение (3aS,5R,6R,6aS)-6-(трет-бутилдиметилсиланилокси)-2,2-диметилтетрагидрофуро[2,3d][1,3]диоксол-5-карбальдегида. К раствору оксалилхлорида (0,76 мл, 8,7 ммоль) в CH2Cl2 (55 мл) в атмосфере N2 при -78C по каплям добавляли раствор ДМСО (0,84 мл, 11,8 ммоль) в CH2Cl2 (5 мл). Через 15 мин по каплям добавляли спирт со стадии A (2,40 г, 7,9 ммоль) в CH2Cl2 (20 мл). Через 15 мин медленно добавляли NEt3. Реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение 105 мин, затем гасили H2O, разбавляли Et2O и промывали H2O, насыщенным водным раствором NaHCO3 и насыщенным раствором соли. Объединенные органические фазы подвергали обратной экстракции Et2O, который промывали в указанной последовательности. Объединенные органические фазы сушили MgSO4, фильтровали и концентрировали в вакууме, получая (3aS,5R,6R,6aS)-6-(трет-бутилдиметилсиланилокси)-2,2 диметилтетрагидрофуро[2,3-d][1,3]диоксол-5-карбальдегид (2,4 г, чистота около 64% по результатам ЯМР). Продукт использовали на следующей стадии без дальнейшей очистки.C. Получение 4-бром-1-хлор-2-(4-этоксибензил)бензола. Указанное соединение получали, как описано в заявке на патент США 10/745075, поданной на имя Deshpande et al. 23 декабря 2003 г.D. Получение (S)-[(3aS,5S,6R,6aS)-6-(трет-бутилдиметилсиланилокси)-2,2-диметилтетрагидрофуро[2,3-d][1,3]диоксол-5-ил]-[4-хлор-3-(4-этоксибензил)фенил]метанола. К раствору 4-бром-1-хлор-2-(4-этоксибензил)бензола со стадии C (3,6 г, 11,1 ммоль) в ТГФ (60 мл) в атмосфере N2 при -78C по каплям добавляли BuLi (2,5 M раствор в гексанах, 4,4 мл, 11,1 ммоль). Через 30 мин по каплям добавляли альдегид со стадии B (2,4 г, чистота 64%, 5,1 ммоль) в ТГФ (20 мл), реакционную смесь перемешивали в течение 30 мин при -78C, оставляли нагреваться до комнатной температуры и перемешивали в течение 60 мин, гасили насыщенным водным раствором NH4Cl, разбавлялиEt2O и промывали H2O и насыщенным раствором соли. Объединенные промывные воды подвергали обратной экстракции Et2O, который промывали в указанной последовательности. Объединенные органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали флэшхроматографией (120 г SiO2, 0-20% EtOAc: гексаны, 75 мин, 85 мл/мин), получая чистый (S)[(3aS,5S,6R,6aS)-6-(трет-бутилдиметилсиланилокси)-2,2-диметилтетрагидрофуро[2,3-d][1,3]диоксол-5 ил]-[4-хлор-3-(4-этоксибензил)фенил]метанол (0,84 г, 1,5 ммоль, 30%), эпимер C5 (0,83 г) и несколько смешанных фракций (0,51 г). 1E. Получение (2S,3R,4R,5S)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-метокситетрагидропиран-3,4,5 триола. Раствор 0,35 М HCl в MeOH получали добавлением AcCl (0,25 мл, 3,5 ммоль) к MeOH (10 мл) и перемешиванием в течение 15 мин. Спирт со стадии D (0,84 г, 1,5 ммоль) обрабатывали полученным раствором в течение 16 ч при комнатной температуре и в течение 2 ч при 80C в запаянной ампуле. Реакционную смесь охлаждали до комнатной температуры, гасили K2CO3, доводя до основного состояния, разбавляли CH2Cl2, фильтровали и концентрировали в вакууме. Продукт очищали флэш-хроматографией(2S,3R,4R,5S)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-метокситетрагидропиран-3,4,5-триол (0,46 г, 1,1 ммоль,75%) в виде белого твердого вещества. При проведении ЯМР обнаружены - и -аномеры при соотношении 1,2:1. 1(1 мл) при 80C в запаянной ампуле в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc, переносили в колбу и концентрировали в вакууме. Остаток растворяли вCH2Cl2, обрабатывали NaHCO3 и MgSO4 в течение 30 мин, фильтровали и концентрировали в вакууме. Продукт очищали флэш-хроматографией (4 г SiO2, 0-12% MeOH:CH2Cl2, 30 мин, 10 мл/мин), суспендировали в H2O и лиофилизировали, получая (3S,4R,5R,6S)-6-[4-хлор-3-(4-этоксибензил)фенил]тетрагидропиран-2,3,4,5-тетраол (31 мг, 0,079 ммоль, 85%) в виде белого твердого вещества. При проведении ЯМР обнаружены - и -аномеры при соотношении 1:1. 1 Раствор 0,35 М HCl в EtOH получали, добавляя AcCl (0,025 мл, 0,35 ммоль) к EtOH (1 мл) и перемешивая в течение 15 мин. Спирт примера 1, стадия D (61 мг, 0,11 ммоль) обрабатывали полученным раствором в течение 2 ч при 80C в запаянной ампуле. Реакционную смесь охлаждали до комнатной температуры, гасили концентрированным NH4OH, доводя до основного состояния, обрабатывали NaHCO3 в течение 30 мин, разбавляли CH2Cl2, фильтровали и концентрировали в вакууме. Продукт очищали флэшхроматографией (4 г SiO2, 0-10% MeOH:CH2Cl2, 40 мин, 10 мл/мин), суспендировали в H2O и лиофилизировали, получая (2S,3R,4R,5S)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-этокситетрагидропиран-3,4,5 триол (40 мг, 0,095 ммоль, 85%) в виде белого твердого вещества. При проведении ЯМР обнаружены и -аномеры при соотношении 1,75:1. 1 Раствор 0,35 М HCl в i-PrOH получали, добавляя AcCl (0,025 мл, 0,35 ммоль) к i-PrOH (1 мл) и перемешивая в течение 15 мин. Спирт примера 1, стадия D (68 мг, 0,12 ммоль) обрабатывали полученным раствором в течение 2 ч при 80C в запаянной ампуле. Реакционную смесь охлаждали до комнатной температуры, гасили концентрированным NH4OH, доводя до основного состояния, обрабатывали NaHCO3 в течение 30 мин, разбавляли CH2Cl2, фильтровали и концентрировали в вакууме. Остаток очищали флэшхроматографией (4 г SiO2, 0-10% MeOH:CH2Cl2, 40 мин, 10 мл/мин), получая 50 мг вещества, которое затем очищали препаративной ВЭЖХ (колонка C18 1950 мм, 20-70% MeCN:H2O (10 мМ NH4OAc),14 мин, 30 мл/мин), получая (2S,3R,4R,5S,6S)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-изопропокситетра- 12016511 гидропиран-3,4,5-триол (-аномер, 7 мг, 0,016 ммоль) и (2S,3R,4R,5S,6R)-2-[4-хлор-3-(4 этоксибензил)фенил]-6-изопропокситетрагидропиран-3,4,5-триол (-аномер, 25 мг, 0,057 ммоль). Пробу соединения примера 1, стадия E (80 мг) растворяли в 4 мл смеси 30% этанол/гексаны и впрыскивали в виде 400 мкл порций в колонку ChiralPak AD-H (20250 мм, 5,5 мл/мин, смесь 31,55% этанол/гексан в качестве элюента при одинаковом соотношении, комнатная температура, продолжительность процесса 30 мин) для отделения двух изомеров друг от друга. Первый изомер (комнатная температура, 23 мин) идентифицировали как альфа-изомер (6R, 20 мг), и второй изомер (комнатная температура,26 мин, 21 мг) идентифицировали как бета-изомер (6S).(62 мл) при 100C в течение 22 ч. Реакционную смесь концентрировали в вакууме, 3 раза подвергали вихревому перемешиванию с толуолом и помещали в высокий вакуум. Остаток обрабатывали уксусным ангидридом (9,4 мл, 99,2 ммоль) в пиридине (25 мл) в течение 16 ч. Реакционную смесь гасили H2O, перемешивали в течение 1 ч, разбавляли Et2O, промывали 1 М водным раствором NaHSO4, H2O, насыщенным водным раствором NaHCO3 и насыщенным раствором соли (с обратной экстракцией), сушили надMgSO4, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (120 г SiO2,0-50% EtOAc/гексаны), получая (3S,4R,5S,6S)-2,4,5-триацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]тетрагидропиран-3-иловый эфир уксусной кислоты (6,10 г, 10,9 ммоль, 87%). 1B. Получение (2S,3S,4R,5S,6S)-4,5-диацетокси-2-бром-6-[4-хлор-3-(4-этоксибензил)фенил]тетрагидропиран-3-илового эфира уксусной кислоты. Тетраацетат со стадии A (8,08 г, 14,4 ммоль) обрабатывали 33% HBr в AcOH (30 мл) в течение 1 ч. Реакционную смесь разбавляли CH2Cl2 (60 мл), перемешивали в течение 30 мин, разбавляли DCM, трижды промывали охлажденной льдом H2O и насыщенным водным раствором NaHCO3 (с обратной экстракцией), сушили над MgSCU, фильтровали и концентрировали в вакууме, получая (2S,3S,4R,5S,6S)-4,5 диацетокси-2-бром-6-[4-хлор-3-(4-этоксибензил)фенил]тетрагидропиран-3-иловый эфир уксусной кислоты. 1C. Получение (2S,3S,4R,5S,6S)-4,5-диацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]-2-метокситетрагидропиран-3-илового эфира уксусной кислоты. Неочищенный бромид со стадии B (8,4 г, 14,4 ммоль) и ZnO (1,2 г, 14,4 ммоль) растворяли в MeOH(144 мл) и нагревали при 70C в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры,фильтровали через целит с EtOAc и концентрировали в вакууме. Остаток перекристаллизовывали изMeOH в виде двух порций, получая (2S,3S,4R,5S,6S)-4,5-диацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]-2-метокситетрагидропиран-3-иловый эфир уксусной кислоты (5,98 г, 11,2 ммоль, 78%) в виде чистого -аномера. 1D. Получение (2S,3R,4R,5S,6S)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-метокситетрагидропиран 3,4,5-триола. Перекристаллизованный триацетат со стадии C (5,98 г, 11,2 ммоль) обрабатывали K2CO3 (7,7 г,56 ммоль) в MeOH (112 мл) при интенсивном перемешивании в течение 1 ч. Реакционную смесь фильтровали через целит и концентрировали в вакууме. Остаток растворяли в DCM, промывали H2O и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток пропускали через слой силикагеля с 5% MeOH:CH2Cl2, концентрировали в вакууме, суспендировали в H2O и лиофилизировали, получая (2S,3R,4R,5S,6S)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-метокситетрагидропиран-3,4,5-триол (4,37 г, 10,7 ммоль, 96%) в виде белого твердого вещества. 1(0,5 мл) при 40C в течение 1,5 ч. Реакционную смесь продували N2, затем дважды продували CH2Cl2. Остаток обрабатывали уксусным ангидридом (78 мкл, 0,82 ммоль) в пиридине (1 мл) в течение ночи. Реакционную смесь гасили MeOH, перемешивали в течение 30 мин, разбавляли Et2O, промывали 1 М водным раствором NaHSO4, H2O, насыщенным водным раствором NaHCO3 и насыщенным раствором соли(с обратной экстракцией), сушили над MgSO4, фильтровали и концентрировали в вакууме. Промежуточный продукт обрабатывали K2CO3 (14 мг, 0,10 ммоль) в MeOH (1 мл) в течение 1,5 ч. Реакционную смесь фильтровали, концентрировали в вакууме и остаток очищали флэш-хроматографией (12 г SiO2, 0-10%MeOH:CH2Cl2), получая вещество с чистотой 90%. Продукт далее очищали ВЭЖХ (колонка C18 1950 мм, 20-70% MeCN:H2O (10 мМ NH4OAc), 14 мин, 30 мл/мин), суспендировали в H2O и лиофилизировали,получаяN-(2S,3S,4R,5R,6S)-6-[4-хлор-3-(4-этоксибензил)фенил]-3,4,5-тригидрокситетрагидропиран-2-ил-N-пропилацетамид (3 мг, 0,0063 ммоль, 15%) в виде ротамеров при соотношении 2:1. 1 Соединение примера 2 (50 мг, 0,13 ммоль) и гидрохлорид гидроксиамина (26 мг, 0,38 ммоль) растворяли в пиридине (0,65 мл) и перемешивали в течение 3 часов. Реакционную смесь разбавляли EtOAc,промывали 1 М водным раствором NaHSO4, H2O, насыщенным водным раствором NaHCO3 и насыщенным раствором соли (с обратной экстракцией), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток суспендировали в H2O и лиофилизировали, получая оксим (2R,3S,4S,5S)-5-[4-хлор-3(4-этоксибензил)фенил]-2,3,4,5-тетрагидроксипентаналя (46 мг, 0,11 ммоль, 88%) в виде смеси изомеров оксима при соотношении 5:1. Основной изомер. 1A. Получение (3S,4R,5S,6S)-4,5-диацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]-2-гидрокситетрагидропиран-3-илового эфира уксусной кислоты. Тетраацетат примера 5, стадия A (200 мг, 0,36 ммоль) обрабатывали бензиламином (39 мкл, 0,36 ммоль) в ДМФА (1,8 мл) в течение 2 ч. Реакционную смесь разбавляли Et2O, промывали 1 М водным раствором NaHSO4, H2O, насыщенным водным раствором NaHCO3 и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали флэщ-хроматографией (12 гSiO2, 0-50% EtOAc: гексаны), получая (3S,4R,5S,6S)-4,5-диацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]2-гидрокситетрагидропиран-3-иловый эфир уксусной кислоты (142 мг, 0,27 ммоль, 77%) в виде аномеров при соотношении 3:1. 1B. Получение (3S,4R,5S,6S)-4,5-диацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]-2-[(Z)-гидроксиимино]тетрагидропиран-3-илового эфира уксусной кислоты. Соединение со стадии A (142 мг, 0,27 ммоль) и гидрохлорид гидроксиамина (57 мг, 0,82 ммоль) растворяли в пиридине (1,4 мл). Реакционную смесь перемешивали в течение 6 ч, разбавляли EtOAc,промывали 1 М водным раствором NaHSO4, H2O, насыщенным водным раствором NaHCO3 и насыщенным раствором соли (с обратной экстракцией), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток растворяли в CH2Cl2, охлаждали до -78C, обрабатывали DBU (49 мкл, 0,33 ммоль) и затем N-хлорсукцинимидом (44 мг, 0,33 ммоль). Реакционную смесь перемешивали в течение 20 мин при-78C и оставляли нагреваться до комнатной температуры в течение 15 мин. Реакционную смесь разбавляли EtOAc, промывали H2O и насыщенным раствором соли (с обратной экстракцией), сушили надMgSO4, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (12 г SiO2,0-50% EtOAc:гексаны), получая (3S,4R,5S,6S)-4,5-диацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]-2-[(Z)гидроксиимино]тетрагидропиран-3-иловый эфир уксусной кислоты (97 мг, 0,18 ммоль, 67%). 1C. Получение оксима (3S,4R,5R,6S)-6-[4-хлор-3-(4-этоксибензил)фенил]-3,4,5-тригидрокситетрагидропиран-2-она. Соединение со стадии B (97 мг, 0,18 ммоль) обрабатывали 7,0 М раствором NH3 в MeOH (1,8 мл) в течение 1 ч. Реакционную смесь концентрировали в вакууме и остаток очищали флэш-хроматографией(57 мг,0,14 ммоль, 77%) в виде белого твердого вещества. 1(2S,3R,4S)-2-(4-хлор-3-(4-этоксибензил)фенил)-3,4-дигидро-2H-пиран-3,4 диилдиацетата. В колбу, содержащую 282 мг тетраацетата примера 5, стадия A (0,5 ммоль) добавляли 1,25 мл HBr(33% в НОАс). Реакционную смесь перемешивали в течение 1 ч, разбавляли 50 мл дихлорметана и гасили, выливая в смесь воды со льдом. Органический слой отделяли и промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли. Смесь сушили над сульфатом магния и растворители концентрировали в вакууме. Неочищенный остаток поглощали 0,5 мл дихлорметана и добавляли к суспензии сульфата меди(II) (20 мг,0,125 ммоль), порошкообразного Zn (82 мг, 1,25 ммоль) и ацетата натрия (984 мг, 12 ммоль) в 2,5 мл смеси уксусная кислота/вода (3:2, об.:об.). Полученную смесь перемешивали при комнатной температуре в течение 4 ч, затем в реакционную смесь добавляли еще 20 мг сульфата меди(II) и 82 мг порошкообразного Zn и перемешивали в течение 18 ч. Смесь гасили водой, экстрагировали этилацетатом. Органический слой сушили над сульфатом магния и удаляли в вакууме. При проведении флэш-хроматографии получали (2S,3R,4S)-2-(4-хлор-3-(4-этоксибензил)фенил)-3,4-дигидро-2H-пиран-3,4-диилдиацетат (32 мг, выход 16%). 1B. Получение (2S,3R,4R,5R,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-5-фтор-6-метокситетрагидро 2H-пиран-3,4-диола. Селектфлуор (45 мг, 0,128 ммоль) добавляли к раствору соединения со стадии A (38 мг,0,0853 ммоль) в 0,4 мл смеси ацетонитрил:метанол (1:1, об.:об.). Реакционную смесь перемешивали при комнатной температуре и контролировали окончание реакции при помощи ЖХ-МС. Реакционную смесь гасили 2 мл насыщенного водного раствора NH4Cl и экстрагировали диэтиловым эфиром (25 мл). Органический экстракт сушили над сульфатом натрия и концентрировали в вакууме. При проведении флэшхроматографии (5-10% этилацетат/гексаны) получали фторированный продукт. Затем к раствору выделенного продукта в 0,5 мл метанола добавляли карбонат калия (5 мг). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, гасили 2 мл воды и экстрагировали этилацетатом (24 мл). Органический слой фильтровали через слой диоксида кремния и концентрировали, получая 6,3 гH ЯМР (400 МГц, хлороформ-d, : аномеры при соотношении 3:2, осевой:экваториальный фтор при соотношении 2:1, изомеры вследствие незначительного содержания в структуре экваториального фтора обозначены курсивом)м.д.: 7,41 (дд, J=8,34, 2,78 Гц, 1H), 7,20-7,33 (м, 2H), 7,11 (д, J=8,59 Гц,2H), 6,83 (д, J=8,59 Гц, 2H), 4,92-5,02 (м, 1H), 4,30-4,52 (м, 1H), 3,96-4,27 (м, 6H), 3,74 (т, J=9,09 Гц, 0,66HA. Получение [4-(5-бром-2-хлорбензил)фенокси]трет-бутилдиметилсилана. Указанное соединение получали, как описано в публикации заявки на патент США 2006/0251728, поданной на имя Himmelsbach et al. и опубликованной 9 ноября 2006 г.(S)-3-[4-(трет-бутилдиметилсиланилокси)бензил]-4-хлорфенил[(3aS,5S,6R,6aS)-6-(третбутилдиметилсиланилокси)-2,2-диметилтетрагидрофуро[2,3-d][1,3]диоксол-5-ил]метанол. Раствор 0,85 г (2,07 ммоль) соединения со стадии A в 4,14 мл диэтилового эфира охлаждали до-78C в инертной атмосфере. К полученному раствору шприцем добавляли 2,66 мл трет-бутиллития(1,55 М раствор в гексанах, 4,14 ммоль) в течение 5 мин. Реакционную смесь перемешивали при -78C в течение 30 мин. Добавляли раствор 0,5 г(1,65 ммоль) соединения примера 1, стадия B, в 1,65 мл диэтилового эфира. Реакционную смесь перемешивали при -78C в течение 30 мин и затем в течение 1,5 ч при 0C. Неочищенную реакционную смесь фильтровали через слой силикагеля с избытком диэтилового эфира, который затем удаляли в вакууме. Полученный продукт состоял из диастереомеров при соотношении примерно 1,2:1 в свежеприготовленном вторичном спирте. Диастереомеры легко разделялись хроматографией на силикагеле (4-8% градиент смеси этилацетат/гексаны). Выход: 40% (требуемый диастереомер), 58% (нежелательный диастереомер). 1C. Получение (2S,3R,4R,5S)-2-[4-хлор-3-(4-гидроксибензил)фенил]-6-метокситетрагидропиран 3,4,5-триола. Ацетилхлорид (0,17 мл) добавляли к 7 мл метанола и перемешивали в течение 15 мин при комнатной температуре. Полученный раствор переносили в ампулу, содержащую 0,446 г соединения со стадииB, которую затем запаивали и нагревали до 80C в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и гасили 50 мл насыщенного водного раствора бикарбоната натрия. Водный слой экстрагировали этилацетатом (350 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом магния и растворитель удаляли в вакууме. Остаток очищали хроматографией на силикагеле (0-20% градиент смеси метанол/дихлорметан), получая смесь : аномеров при соотношении примерно 1:1. Выход: 65%. 1(S)-(Тетрагидрофуран-3-иловый) эфир толуол-4-сульфоновой кислоты (31 мг, 0,126 ммоль) добавляли к суспензии соединения примера 10, стадия C (16 мг, 0,042 ммоль) и карбоната цезия (46 мг, 0,126 ммоль) в 0,22 мл N,N-диметилформамида. Реакционный сосуд герметизировали и нагревали до 80C в течение 15 ч. Неочищенную реакционную смесь охлаждали до комнатной температуры, гасили 2 мл насыщенного раствора соли и экстрагировали этилацетатом (32 мл). Объединенные органические экстракты сушили над сульфатом натрия и концентрировали в вакууме. При проведении хроматографии на силикагеле (0-10% градиент смеси метанол/дихлорметан) получали (2S,3R,4R,5S)-2-[4-хлор-3-(4 тетрагидрофуран-3-илокси)бензилфенил]-6-метокситетрагидропиран-3,4,5-триола в виде прозрачного вязкого масла, которое концентрировали в дихлорметане с образованием белого твердого веществаA. Получение 3aS,5S,6R,6aS)-5-азидо[(S)-4-хлор-3-(4-этоксибензил)фенил]метил-2,2-диметилтетрагидрофуро[2,3-d][1,3]диоксол-6-илокси)трет-бутилдиметилсилана. К раствору C5-эпимера спирта примера 1, стадия D (682 мг, 1,24 ммоль) и PPh3 (489 мг, 1,87 ммоль) в ТГФ (6,2 мл) добавляли DIAD (366 мкл, 1,87 ммоль) и затем дифенилфосфорилазид (DPPA, 323 мкл,1,49 ммоль). Реакционную смесь перемешивали в течение 1,5 ч, гасили насыщенным водным растворомNH4Cl, разбавляли Et2O, промывали H2O и насыщенным раствором соли (с обратной экстракцией), сушили над MgSO4 и концентрировали в вакууме. Остаток очищали флэш-хроматографией (40 г SiO2, 0-8%B. Получение (2R,3S,4S,5S)-5-азидо[(S)-4-хлор-3-(4-этоксибензил)фенил]метилтетрагидрофуран 2,3,4-триола. Ацетилхлорид (0,175 мл, 2,45 ммоль) добавляли к MeOH (7 мл). Раствор перемешивали в течение 15 мин, затем добавляли к азиду со стадии A (392 мг, 0,68 ммоль). Реакционную смесь перемешивали в течение 16 ч, концентрировали в вакууме, дважды подвергали вихревому перемешиванию с MeOH и помещали в высокий вакуум с образованием белого твердого вещества. Твердое вещество обрабатывалиAcOH:H2O при соотношении 1:1 (7 мл) при 100C в течение 2,5 ч. Реакционную смесь концентрировали в вакууме, дважды подвергали вихревому перемешиванию с толуолом и помещали в высокий вакуум. Остаток очищали флэш-хроматографией (40 г SiO2, 0-6% MeOH:CH2Cl2), получая (2R,3S,4S,5S)-5 азидо[(S)-4-хлор-3-(4-этоксибензил)фенил]метилтетрагидрофуран-2,3,4-триол (223 мг, 0,53 ммоль,78%) в виде смеси аномеров. 1C. Получение (2S,3S,4S,5R)-2-[4-хлор-3-(4-этоксибензил)фенил]пиперидин-3,4,5-триола. Соединение со стадии B (216 мг, 0,52 ммоль) гидрировали при атмосферном давлении H2 над PtO2(примерно 55 мг) очищали препаративной ВЭЖХ (колонка C18 Sunfire 30100 мм, 20-70% MeCN:H2O(10 мМ NH4OAc), 15 мин, 45 мл/мин) и лиофилизировали, получая (2S,3S,4S,5R)-2-[4-хлор-3-(4 этоксибензил)фенил]пиперидин-3,4,5-триол (27 мг, 0,071 ммоль) в виде белого твердого вещества. 1(2S,3R,4R,5S,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-этилсульфанилтетрагидропиран-3,4,5-триола. К раствору бромида примера 5, стадия B (291 мг, 0,50 ммоль) в EtOH (5 мл) при 0C добавлялиNaSEt (84 мг, 1,0 ммоль). Реакционную смесь перемешивали в течение 30 мин, разбавляли EtOAc, промывали разбавленным водным раствором NaOH и насыщенным раствором соли (с обратной экстракцией), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали флэшхроматографией (40 г SiO2, 0-7% MeOH:CH2Cl2), суспендировали в H2O и лиофилизировали, получаяB. Получение (2S,3R,4R,5S,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-этансульфинилтетрагидропиран-3,4,5-триола и (2S,3R,4R,5S,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-этансульфонилтетрагидропиран-3,4,5-триола. К раствору соединения со стадии A (10 мг, 0,023 ммоль) в AcOH (0,5 мл) добавляли H2O2 (35 мас.% раствор в H2O, 3 мг, 0,092 ммоль, 9 мкл). Смесь перемешивали при комнатной температуре в течение 2 ч и концентрировали в вакууме. Смесь очищали хроматографией на силикагеле (5% MeOH/CH2Cl2), получая (2S,3R,4R,5S,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-этансульфинилтетрагидропиран-3,4,5-триол(в виде смеси диастереомеров в положении атома серы) (2 мг, 19%) и (2S,3R,4R,5S,6R)-2-[4-хлор-3-(4 этоксибензил)фенил]-6-этансульфонилтетрагидропиран-3,4,5-триол (5 мг, 46%) в виде белых твердых веществ.A. Получение (2S,3R,4R,5S,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-метилсульфанилтетрагидропиран-3,4,5-триола. К раствору бромида примера 5, стадия B (347 мг, 0, 60 ммоль) в EtOH (6 мл) при 0C добавлялиNaSMe (70 мг, 0,72 ммоль). Реакционную смесь перемешивали в течение 30 мин, затем разбавлялиEtOAc, промывали разбавленным водным раствором NaOH и насыщенным раствором соли (с обратной экстракцией), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали флэш- 19016511 хроматографией (40 г SiO2, 0-7% MeOH:CH2Cl2), суспендировали в H2O и лиофилизировали, получая(2R,3S,4R,5S,6S)-4,5-диацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]-2 метилсульфанилтетрагидропиран-3-илового эфира уксусной кислоты. Триол со стадии A (45 мг, 0,11 ммоль) обрабатывали уксусным ангидридом (60 мкл, 0,64 ммоль) в пиридине (0,5 мл) в течение 16 ч. Реакционную смесь разбавляли Et2O, промывали 1 М водным раствором NaHSO4, H2O, насыщенным водным раствором NaHCO3 и насыщенным раствором соли (с обратной экстракцией), сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали флэшхроматографией (4 г SiO2, 0-25% EtOAc/гексаны), суспендировали в H2O и лиофилизировали, получая(2R,3S,4R,5S,6S)-4,5-диацетокси-6-[4-хлор-3-(4-этоксибензил)фенил]-2-метилсульфанилтетрагидропиран-3-иловый эфир уксусной кислоты (46 мг, 0,087 ммоль, 79%) в виде белого твердого вещества. 1 К раствору соединения примера 14, стадия А (41 мг, 0,097 ммоль) в AcOH (0,5 мл) добавляли H2O2(35 мас.% раствор в H2O, 20 мг, 0,58 ммоль, 57 мкл). Смесь перемешивали при комнатной температуре в течение 18 ч и концентрировали в вакууме. Смесь очищали хроматографией на силикагеле (5%MeOH/CH2Cl2), получая (2S,3R,4R,5S,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-метансульфонилтетрагидропиран-3,4,5-триол (20 мг, 45%) в виде белого твердого вещества. 1 Получение 1-(2S,3S,4S,5R)-2-[4-хлор-3-(4-этоксибензил)фенил]-3,4,5-тригидроксипиперидин-1 илэтанона. К раствору неочищенного соединения примера 12, стадия C (38 мг, 0,1 ммоль) в MeOH (1 мл) добавляли уксусный ангидрид (19 мкл, 0,2 ммоль). Реакционную смесь перемешивали в течение 4 часов,добавляли дополнительное количество уксусного ангидрида (10 мкл, 0,1 ммоль) и продолжали перемешивать смесь в течение ночи. Реакционную смесь разбавляли EtOAc, промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (12 г SiO2, 0-8% MeOH: CH2Cl2), суспендировали в и лиофилизировали,получая 1-(2S,3S,4S,5R)-2-[4-хлор-3-(4-этоксибензил)фенил]-3,4,5H 2O тригидроксипиперидин-1-илэтанон (14 мг, 0,033 ммоль, 33% при выполнении 2-х стадий) в виде белого твердого вещества. 1- 20016511 Пример 17. Синтез метилового эфира (2S,3S,4S,5R)-2-[4-хлор-3-(4-этоксибензил)фенил]-3,4,5 тригидроксипиперидин-1-карбоновой кислоты. К раствору неочищенного соединения примера 12, стадия C (38 мг, 0,1 ммоль) и NaHCO3 (42 мг, 0,5 ммоль) в смеси EtOAc:EtOH:H2O при соотношении 1:1:1 (1,5 мл) при 0C добавляли метилхлорформиат(23 мкл, 0,3 ммоль). Реакционную смесь перемешивали в течение 1 ч, разбавляли EtOAc, промывали H2O и насыщенным раствором соли (с обратной экстракцией), сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (4 г SiO2, 0-10% MeOH:CH2Cl2, суспендировали в H2O и лиофилизировали, получая метиловый эфир (2S,3S,4S,5R)-2-[4-хлор-3-(4 этоксибензил)фенил]-3,4,5-тригидроксипиперидин-1-карбоновой кислоты (12 мг, 0,026 ммоль, 26% для 2-х стадий) в виде белого твердого вещества. 1 К раствору неочищенного соединения примера 12, стадия C (38 мг, 0,1 ммоль) в смеси EtOH:EtOAc при соотношении 1:1 (1 мл) добавляли аллилизоцианат (18 мкл, 0,2 ммоль). Реакционную смесь перемешивали в течение 1 ч и концентрировали в вакууме. Остаток очищали флэш-хроматографией (4 г SiO2, 010% MeOH:CH2Cl2, суспендировали в H2O и лиофилизировали, получая аллиламид (2S,3S,4S,5R)-2-[4(хлор-3-(4-этоксибензил)фенил]-3,4,5-тригидроксипиперидин-1-карбоновой кислоты (14 мг, 0,030 ммоль,30% при выполнении 2-х стадий) в виде белого твердого вещества. 1 К раствору соединения примера 12, стадия C (50 мг, 0,13 ммоль) и K2CO3 (55 мг, 0,40 ммоль) в ДМФА (0,65 мл) добавляли метилиодид (10 мкл, 0,16 ммоль). Реакционную смесь перемешивали в течение 3 ч, разбавляли EtOAc, промывали H2O и насыщенным раствором соли (с обратной экстракцией),сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией(12 г SiO2, 2-12% MeOH:CH2Cl2), суспендировали в H2O и лиофилизировали, получая (2S,3S,4S,5R)-2-[4 хлор-3-(4-этоксибензил)фенил]-1-метилпиперидин-3,4,5-триол (16 мг, 0,040 ммоль, 31%) в виде белого твердого вещества. 1A. Получение (3R,4S,5R,6R)-3,4,5-трис-(бензилокси)-6-(бензилоксиметил)тетрагидро-2H-пирон-2 она. Тетра-O-бензил-D-глюкопиранозу (2,07 г, 3,8 ммоль) растворяли в ДМСО (10,1 мл). К полученной смеси добавляли уксусный ангидрид (7,0 мл) и перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли лед и перемешивали в течение 1 ч. Смесь экстрагировали простым эфиром (320 мл). Экстракт промывали водой (210 мл), водным раствором бикарбоната натрия(210 мл), насыщенным раствором соли, сушили (сульфат натрия) и концентрировали в вакууме. При проведении флэш-хроматографии на колонке с силикагелем со смесью 0-25% этилацетат/гексан получали 1,712 г (3R,4S,5R,6R)-3,4,5-трис-(бензилокси)-6-(бензилоксиметил)тетрагидро-2H-пирон-2-она (83%).(3R,4S,5R,6R)-3,4,5-трис-(бензилокси)-6-(бензилоксиметил)-2-(4-хлор-3-(4 этоксибензил)фенил)тетрагидро-2H-пиран-2-ола. н-Бутиллитий (2,5 н. раствор в гексане) (1,263 мл, 3,16 ммоль) по каплям добавляли к раствору соединения примера 1, стадия C (1,028 г, 3,16 ммоль) в безводном ТГФ (15 мл) при -78C. Реакционную смесь перемешивали в течение 30 мин при -78C, по каплям добавляли раствор соединения со стадии A(1,7 г, 3,16 ммоль) в безводном ТГФ (10 мл) и перемешивали в течение 1 ч, оставляя смесь нагреваться до комнатной температуры. К реакционной смеси добавляли водный раствор хлорида аммония (10 мл), ТГФ удаляли в вакууме и водный слой экстрагировали этилацетатом (220 мл). Объединенные органические фазы промывали насыщенным раствором соли, сушили (сульфат натрия) и концентрировали в вакууме. Неочищенную смесь очищали флэш-хроматографией на колонке с силикагелем, используя смесь 0-20% этилацетат/гексан, с получением 712 мг (3R,4S,5R,6R)-3,4,5-трис-(бензилокси)-6-(бензилоксиметил)-2-(4 хлор-3-(4-этоксибензил)фенил)тетрагидро-2H-пиран-2-ола (29%).C. Получение (2R,3R,4S)-2,3,4,6-тетракис-(бензилокси)-1-(4-хлор-3-(4-этоксибензил)фенил)гексан 1,5-диона. К перемешиваемому раствору реагента Десса-Мартина (500 мг, избыток) в CH2Cl2 (10 мл) добавляли соединение со стадии B (500 мг, 0,6 ммоль) в безводном дихлорметане (10 мл) и перемешивали в течение ночи. Реакционную смесь гасили 1 н. раствором гидроксида натрия (3 мл), экстрагировали дихлорметаном (210 мл), объединенные органические фракции промывали насыщенным раствором соли, сушили над сульфатом натрия, концентрировали при пониженном давлении, получая неочищенный продукт (487 мг). (М+H2O=800,1).(3R,4R,5S)-3,4,5-трис-(бензилокси)-2-(бензилоксиметил)-6-(4-хлор-3-(4-этоксибензил)фенил)пиперидина. Раствор соединения со стадии C (400 мг, 0,5 ммоль), 7 н. раствор аммиака в MeOH (1,0 мл) и только что активированные молекулярные сита 4 (250 мг) в дихлорметане (20 мл) нагревали при кипении с обратным холодильником в течение ночи. Реакционную смесь охлаждали до комнатной температуры,добавляли цианоборогидрид натрия (160 мг, 2,55 ммоль) и нагревали с обратным холодильником еще 2 ч. Реакционную смесь фильтровали, разбавляли дихлорметаном (20 мл), промывали водой, насыщенным раствором соли, сушили (сульфат натрия) и концентрировали при пониженном давлении. При проведении хроматографии на силикагеле (50-100% ацетонитрила, содержащего 0,1% градиент смеси ацетат аммония/вода) получали(3R,4R,5S)-3,4,5-трис-(бензилокси)-2-(бензилоксиметил)-6-(4-хлор-3-(4-этоксибензил)фенил)-1-метилпиперидина. Соединение со стадии D (50 мг, 0,065 ммоль) растворяли в ацетонитриле (1 мл) и обрабатывали карбонатом калия (18 мг, 0,13 ммоль) в течение 30 мин. К полученной смеси добавляли йодметан(20 мкл, 0,32 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли этилацетатом(10 мл), промывали водой, насыщенным раствором соли, сушили (сульфат натрия) и концентрировали в вакууме. При проведении хроматографии на силикагеле (50-100% ацетонитрила, содержащего 0,1% градиент смеси ацетат аммония/вода) получали (3R,4R,5S)-3,4,5-трис-(бензилокси)-2-(бензилоксиметил)-6(4-хлор-3-(4-этоксибензил)фенил)-1-метилпиперидин (29 мг, 56%).F. Получение (2S,3S,4R,5R,6R)-2-[3-(4-этоксибензил)фенил]-6-гидроксиметил-1-метилпиперидин 3,4,5-триола. Соединение со стадии E (50 мг) в метаноле и уксусной кислоте (25 мкл) обрабатывали 5% влажнымPd-C (10 мг) в атмосфере H2 в течение 4 ч. Реакционную смесь фильтровали через слой целита и концентрировали. При проведении хроматографии на силикагеле (10-100% ацетонитрила, содержащего 0,1% градиент смеси ацетат аммония/вода) получали (2S,3S,4R,5R,6R)-2-[3-(4-этоксибензил)фенил]-6 гидроксиметил-1-метилпиперидин-3,4,5-триол (6 мг, 70%). 1A. Получение S-(1S)-3aS,6S,6aS)-6-(трет-бутилдиметилсилилокси)-2,2-диметилтетрагидрофуро[2,3-d][1,3]диоксол-5-ил)(4-хлор-3-(4-этоксибензил)фенил)метилбензотиоата. Диэтилазодикарбоксилат (150 мкл, 0,914 ммоль) добавляли к раствору трифенилфосфина (240 мг,0,914 ммоль) в 1,0 мл ТГФ при комнатной температуре. Через 1 ч шприцем добавляли C5-эпимер примера 1, стадия D (167 мг, 0,305 ммоль) в 0,5 мл ТГФ и затем шприцем добавляли тиобензойную кислоту(110 мкл, 0,914 ммоль). Полученный оранжевый раствор перемешивали в течение 22 ч при комнатной температуре. Растворители удаляли в вакууме, остаток очищали флэш-хроматографией (0-10% градиент смеси этилацетат/гексаны), получая указанное в заголовке соединение в виде светло-желтого масла(2S,3R,4R,5S,6R)-2-(4-хлор-3-(4-этоксибензил)фенил)-6-метокситетрагидро-2Hтиопиран-3,4,5-триола. Метоксид натрия (0,3 мл 4,3 М раствора в метаноле) добавляли к раствору соединения со стадии A(104 мг, 0,152 ммоль) в 6 мл метанола. Через 30 мин реакционную смесь разбавляли 20 мл этилацетата и промывали водой и насыщенным раствором соли (по 20 мл каждого вещества). Органический слой сушили над сульфатом магния, фильтровали и растворители удаляли в вакууме. Остаток быстро очищали флэш-хроматографией (5% этилацетат/гексаны) и продукт использовали немедленно для предотвращения образования дисульфида. Одну каплю ацетилхлорида добавляли к 1 мл метанола и перемешивали в течение 15 мин при комнатной температуре. Полученный кислый раствор добавляли к ранее полученному свободному тиолу и нагревали в течение 42 ч при 80C. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли в вакууме. Неочищенный остаток очищали препаративной ВЭЖХ (колонка C18 30250 мм, смесь 5-75% ацетонитрил:вода (10 мМ ацетата аммония), 15 мин, 45 мл/мин), получая указанное в заголовке соединение (альфа-аномер, t=13,82 мин, 8,7 мг, выход 13% при выполнении 2-х стадий). 1(2R,3R,4S,5R,6S)-3,4,5-трисаллилокси-2-аллилоксиметил-6-метокситетрагидропирана. К раствору -D-метилглюкозида (3 г, 15,45 ммоль) в ДМФА (50 мл) добавляли NaH (60% дисперсия в минеральном масле, 3,34 г, 0,14 моль). К образовавшейся в процессе добавления густой суспензии добавляли дополнительное количество ДМФА (15 мл) для возвращения к раствору. Смесь перемешивали при комнатной температуре в течение 30 мин, охлаждали до 0C и медленно добавляли аллилбромид(17 г, 0,14 моль, 12 мл). Затем смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 ч. К светло-коричневой смеси осторожно добавляли MeOH, чтобы погасить избыток NaH,затем смесь концентрировали. Остаток разбавляли CH2Cl2 и промывали H2O, сушили (MgSO4) и концентрировали с образованием желтого масла. Продукт очищали хроматографией на силикагеле (20%B. Получение (3R,4S,5R,6R)-3,4,5-трисаллилокси-6-аллилоксиметилтетрагидропиран-2-ола. Раствор соединения со стадии A (10 г, 0,028 моль) в AcOH (400 мл) нагревали до 90C. ДобавлялиTfOH (2 н. раствор в H2O, 16,69 г, 0,112 моль, 56 мл) и смесь перемешивали при 90C в течение 75 мин. Раствор охлаждали и разбавляли CH2Cl2, промывали H2O (3 раза), насыщенным раствором NaHCO3, сушили (MgSO4) и концентрировали с образованием желтого твердого вещества. Продукт очищали хроматографией на силикагеле (20%-40% EtOAc/гексаны), получая (3R,4S,5R,6R)-3,4,5-трисаллилокси-6 аллилоксиметилтетрагидропиран-2-ол, представляющий собой смесь аномеров (5,85 г, 17,2 ммоль, 61%),в виде белого твердого вещества. TCX: Rf=0,40, 40% EtOAc/гексаны.-78C. Добавляли ДМСО (3,39 г, 43,4 ммоль, 3,08 мл) в виде раствора в CH2Cl2 (60 мл). Смесь перемешивали при -78C в течение 15 мин и добавляли соединение со стадии B (6,70 г, 19,7 ммоль) в виде раствора в CH2Cl2 (150 мл). Реакционную смесь перемешивали еще 15 мин при -78C и добавляли Et3N (9,97 г,98,5 ммоль, 13,7 мл). Смесь перемешивали при -78C еще 5 мин и оставляли нагреваться до комнатной температуры в течение 30 мин. Реакционную смесь гасили H2O, органический слой отделяли, дважды промывали H2O, сушили и концентрировали с образованием светло-желтого масла. Продукт очищали хроматографией на силикагеле (15% EtOAc/гексаны), получая (3R,4S,5R,6R)-3,4,5-трисаллилокси-6 аллилоксиметилтетрагидропиран-2-он (2,49 г, 7,37 ммоль, 37%) в виде бесцветного масла. TCX: Rf=0,40,20% EtOAc/гексаны.-78C. По каплям добавляли n-BuLi (2,5 н. раствор в гексанах, 0,47 г, 7,31 ммоль, 2,92 мл) и перемешивали раствор в течение 15 мин. Добавляли соединение со стадии C (2,47 г, 7,31 ммоль) в виде раствора в ТГФ (25 мл), реакционную смесь перемешивали при -78C еще 15 мин и оставляли нагреваться до комнатной температуры в течение 30 мин. Реакционную смесь гасили насыщенным раствором NH4Cl и отделяли органический слой. Водный слой подвергали обратной экстракции Et2O, объединенные органические части сушили и концентрировали с образованием желтого масла. Продукт очищали хроматографией на силикагеле (10%-20% EtOAc/гексаны), получая (3R,4S,5R,6R)-3,4,5-трисаллилокси-6-аллилоксиметил 2-[4-хлор-3-(4-этоксибензил)фенил]тетрагидропиран-2-ол (0,95 г, 1,63 ммоль, 22%) в виде бесцветного масла. МС (ES+) [M+NH4]+=602.E. Получение (2R,3R,4S)-2,3,4,6-тетракисаллилокси-1-[4-хлор-3-(4-этоксибензил)фенил]гексан-1,5 диона. К раствору соединения со стадии D (0,93 г, 1,59 ммоль) в CH2Cl2 (25 мл) добавляли периодинан Десса-Мартина (0,68 г, 1,59 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч и добавляли вторую порцию периодинана Десса-Мартина (1 экв.). Реакционную смесь продолжали перемешивать еще один час, гасили 1 н. раствором NaOH (приблизительно 4 мл). Добавляли H2O и отделяли органический слой. Водный слой подвергали обратной экстракции CH2Cl2, сушили и концентрировали с образованием желтого воскообразного твердого вещества. Продукт очищали хроматографией на силикагеле (15-20% EtOAc/гексаны), получая (2R,3R,4S)-2,3,4,6-тетракисаллилокси-1-[4-хлор-3-(4-этокси- 24016511 бензил)фенил]гексан-1,5-дион (0,60 г, 1,03 ммоль, 65%) в виде белого твердого вещества. МС (ES+) [M+NH4]+=600.(2R,3R,4R,5S,6S)-3,4,5-трисаллилокси-2-аллилоксиметил-6-[4-хлор-3-(4 этоксибензил)фенил]пиперидина. К раствору соединения со стадии E (0,60 г, 1,03 ммоль) в MeOH (12 мл) добавляли молекулярные сита 4 и формиат аммония (0,13 г, 2,06 ммоль). Затем одной порцией добавляли NaBH3CN (0,14 г,2,3 ммоль) и смесь перемешивали при комнатной температуре в течение 1 ч 30 мин. Реакционную смесь фильтровали и концентрировали. Продукт очищали хроматографией на силикагеле (10-20%Ir(COD) [PCH3Ph2]PF6 (8 мг, 30 мол.%) в ТГФ (0,3 мл) перемешивали в атмосфере H2 до изменения цвета с красного на светло-желтый (приблизительно 5 мин). Добавляли соединение со стадии F (19 мг,0,033 моль) в ТГФ (0,5 мл), смесь перемешивали при комнатной температуре в течение 45 мин и концентрировали. Продукт очищали хроматографией на силикагеле (20% EtOAc/гексаны), получаяH. Получение (2S,3S,4R,5R,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-гидроксиметилпиперидин 3,4,5-триола. Соединение со стадии G (15 мг, 0,026 ммоль) растворяли в растворе ТГФ/AcOH/1 н. раствор HCl(0,2 мл:0,3 мл:0,15 мл) и нагревали до 70C в течение 30 мин. Смесь концентрировали с образованием светло-желтого масла. Продукт очищали препаративной ВЭЖХ (колонка C18 Sunfire 30100 мм, 5 мкм,10-100% В в течение 15 мин), получая (2S,3S,4R,5R,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6 гидроксиметилпиперидин-3,4,5-триол (5 мг, 0,012 ммоль, 46%) в виде белого твердого вещества. МС (ES+) [M+H]+=408. 1(2R,3R,4R,5S,6S)-3,4,5-трисаллилокси-2-аллилоксиметил-6-[4-хлор-3-(4 этоксибензил)фенил]-1-метилпиперидина. К раствору соединения примера 22, стадия F (135 мг, 0,24 ммоль) в MeCN добавляли K2CO3 (164 мг,1,19 ммоль). Смесь перемешивали в течение 30 мин и добавляли MeI (676 мг, 4,76 ммоль). Смесь продолжали перемешивать при комнатной температуре в течение 8 ч, фильтровали и концентрировали. Продукт очищали хроматографией на силикагеле (10% EtOAc/гексаны), получая (2R,3R,4R,5S,6S)-3,4,5 трисаллилокси-2-аллилоксиметил-6-[4-хлор-3-(4-этоксибензил)фенил]-1-метилпиперидинIr(COD)[PCH3Ph2]PF6 (27 мг, 30 мол.%) в ТГФ (1 мл) перемешивали в атмосфере H2 до изменения цвета с красного на светло-желтый (приблизительно 5 мин). Добавляли соединение со стадии A (62 мг,0,11 моль) в ТГФ (1,5 мл), смесь перемешивали при комнатной температуре в течение 45 мин и концентрировали. Продукт очищали хроматографией на силикагеле (20% EtOAc/гексаны), получая(2S,3S,4R,5R,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6-гидроксиметил-1 метилпиперидин-3,4,5-триола. Соединение со стадии B (54 мг, 0,093 ммоль) растворяли в растворе ТГФ/AcOH/1 н. раствор HCl(0,5 мл:0,6 мл:0,30 мл) и нагревали до 70C в течение 30 мин. Смесь концентрировали с образованием светло-желтого масла. Продукт очищали препаративной ВЭЖХ (колонка C18 Sunfire, 30100 мм, 5 мкм,- 25016511 10-100% В в течение 15 мин), получая (2S,3S,4R,5R,6R)-2-[4-хлор-3-(4-этоксибензил)фенил]-6 гидроксиметил-1-метилпиперидин-3,4,5-триол (22 мг, 0,052 ммоль, 56%) в виде белого твердого вещества. МС (ES+) [M+H]+=422. 1
МПК / Метки
МПК: A61P 13/00, C07D 309/10, A61K 31/435, C07D 335/02, C07D 407/12, A61K 31/35, A61K 31/38, C07D 211/46
Метки: применения, способы, ингибиторы, котранспортера, натрий-глюкозы
Код ссылки
<a href="https://eas.patents.su/30-16511-ingibitory-kotransportera-natrijj-glyukozy-2-i-sposoby-ih-primeneniya.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы котранспортера натрий-глюкозы 2 и способы их применения</a>
Высокоселективные ингибиторы обратного захвата норэпинефрина и способы их применения

Номер патента: 5029
Опубликовано: 28.10.2004
Авторы: Тэйлор Дункан П., Сетера Паскаль, Ахмед Саеедуддин, Вонг Эрик Х.Ф., Маршалл Роберт Клайд, Бирджерсон Ларс, Макартур Роберт
МПК: A61P 25/22, A61K 31/5375
Метки: захвата, ингибиторы, высокоселективные, применения, обратного, норэпинефрина, способы
Формула / Реферат:
1. Применение оптически чистого (S,S)-ребоксетина или его фармацевтически приемлемой соли, указанное соединение, по существу, свободно от (R,R)-ребоксетина для изготовления лекарственного средства для лечения или профилактики заболевания, выбранного из хронической боли, недержания, головных болей при мигрени и фибромиалгии и других соматоформных расстройств и периферической невропатии. 2. Применение по п.1, где (S,S)-ребоксетин вводят в...
Высокоселективные ингибиторы обратного захвата норэпинефрина и способы их применения

Номер патента: 6652
Опубликовано: 24.02.2006
Авторы: Вонг Эрик Х.Ф., Бирджерсон Ларс, Макартур Роберт, Ахмед Саеедуддин, Тэйлор Дункан П., Сетера Паскаль, Маршалл Роберт Клайд
МПК: A61P 25/22, A61P 25/00, A61K 31/5375...
Метки: способы, применения, высокоселективные, захвата, обратного, ингибиторы, норэпинефрина
Формула / Реферат:
1. Применение оптически чистого (S,S)-ребоксетина или его фармацевтически приемлемой соли, указанное соединение, по существу, свободно от (R,R)-ребоксетина для изготовления лекарственного средства для лечения или профилактики заболевания, выбранного из аддиктивного расстройства, нарушения обучения или психического расстройства, связанного с возрастом, дефицита внимания (или другого расстройства познавательной функции, обусловленного общими...
Высокоселективные ингибиторы обратного захвата норэпинефрина и способы их применения

Номер патента: 8381
Опубликовано: 27.04.2007
Авторы: Вонг Эрик Х.Ф., Ахмед Саеедуддин, Тэйлор Дункан П., Маршалл Роберт Клайд, Бирджерсон Ларс, Сетера Паскаль, Макартур Роберт
МПК: A61P 25/00, A61K 31/5375
Метки: способы, захвата, ингибиторы, высокоселективные, обратного, норэпинефрина, применения
Формула / Реферат:
1. Применение оптически чистого (S,S)-ребоксетина или его фармацевтически приемлемой соли, указанное соединение по существу свободно от (R,R)-ребоксетина для изготовления лекарственного средства для лечения или профилактики заболевания, выбранного из регулируемого расстройства, нервной анорексии, апатии, нервной булимии, дистимического расстройства, расстройства дыхания, интоксикации, мании, обсессивно-компульсивных расстройств и связанных с...
Высокоселективные ингибиторы обратного захвата норэпинефрина и способы их применения

Номер патента: 11094
Опубликовано: 30.12.2008
Авторы: Макартур Роберт, Бирджерсон Ларс, Сетера Паскаль, Маршалл Роберт Клайд, Ахмед Саеедуддин, Тэйлор Дункан П., Вонг Эрик Х.Ф.
МПК: A61P 25/00, A61K 31/5375
Метки: применения, ингибиторы, норэпинефрина, способы, высокоселективные, обратного, захвата
Формула / Реферат:
1. Применение рацемического ребоксетина или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или профилактики заболевания, выбранного из периферической невропатии, фибромиалгии или другого соматоформного нарушения, мигреней и синдрома хронической усталости. 2. Применение по п.1, где рацемический ребоксетин вводят в количестве от 2 до 20 мг/день. 3. Применение по п.2, где рацемический ребоксетин вводят в...
Лекарственный препарат для местного применения в виде пластыря, содержащий возбудитель аллергической реакции замедленного типа, и способы его применения

Номер патента: 6208
Опубликовано: 27.10.2005
Авторы: Мори Итиро, Судо Ютаро
МПК: A61P 37/08, A61K 31/04
Метки: реакции, применения, способы, лекарственный, виде, замедленного, пластыря, содержащий, местного, препарат, типа, аллергической, возбудитель
Формула / Реферат:
1. Лекарственный препарат возбудителя аллергической реакции замедленного типа для местного применения в виде пластыря, содержащий сшитую клейкую гелевую композицию, содержащую возбудитель аллергической реакции замедленного типа, и подложку. 2. Лекарственный препарат для местного применения в виде пластыря по п.1, отличающийся тем, что возбудителем аллергической реакции замедленного типа является 1-хлор-2,4-динитробензол (DNCB). 3. Лекарственный...
Предыдущий патент: Аминогетероциклические соединения
Следующий патент: Курительное изделие с концентричной полой сердцевиной в табачном стержне и капсулой, содержащей ароматизатор и аэрозольобразующие вещества в фильтрующей системе
Случайный патент: Косметическое средство для удаления ороговевших пробок из пор и питания кожи и его применение