Терапевтически активные соединения
Номер патента: 14693
Опубликовано: 30.12.2010
Авторы: Дэк Кевин Нил, Брэдли Пол Энтони, Джонсон Патрик Стефен, Скерратт Сара Элизабет


























Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемое производное,
в которой R1 и R3независимо обозначают Н, С1-6алкил, С3-8циклоалкил или галоген;
R2 обозначает C1-6алкил, CF3 или арил;
а обозначает 1 или 2;
R4, R5, R7 и R8независимо обозначают Н, С1-6алкил, C1-6алкилокси, CN или галоген, или R4и R5 или R7и R8 вместе с кольцом, к которому они присоединены, образуют арил или гетероциклическую конденсированную кольцевую систему;
X обозначает С или N;
Y обозначает СН2 или О и
R6 обозначает Н, CN или галоген, но когда X обозначает N, тогда R6отсутствует.
2. Соединение по п.1, в котором R1 обозначает С1-6алкил или С3-8циклоалкил.
3. Соединение по п.1 или 2, в котором R2 обозначает С1-6алкил.
4. Соединение по любому из пп.1-3, в котором R3 обозначает C1-6алкил или С3-8циклоалкил.
5. Соединение по любому из пп.1-4, в котором R4 обозначает Н.
6. Соединение по любому из пп.1-5, в котором R5 обозначает Н, С1-6алкил или галоген.
7. Соединение по любому из пп.1-4, в котором R4 и R5 вместе обозначают фенильное или пиридинильное кольцо, конденсированное с кольцом, к которому они присоединены.
8. Соединение по любому из пп.1-7, в котором R6 обозначает CN.
9. Соединение по любому из пп.1-8, в котором R7 обозначает Н, C1-6алкил или галоген.
10. Соединение по любому из пп.1-9, в котором R8 обозначает Н.
11. Соединение по любому из пп.1-10, в котором Y обозначает О.
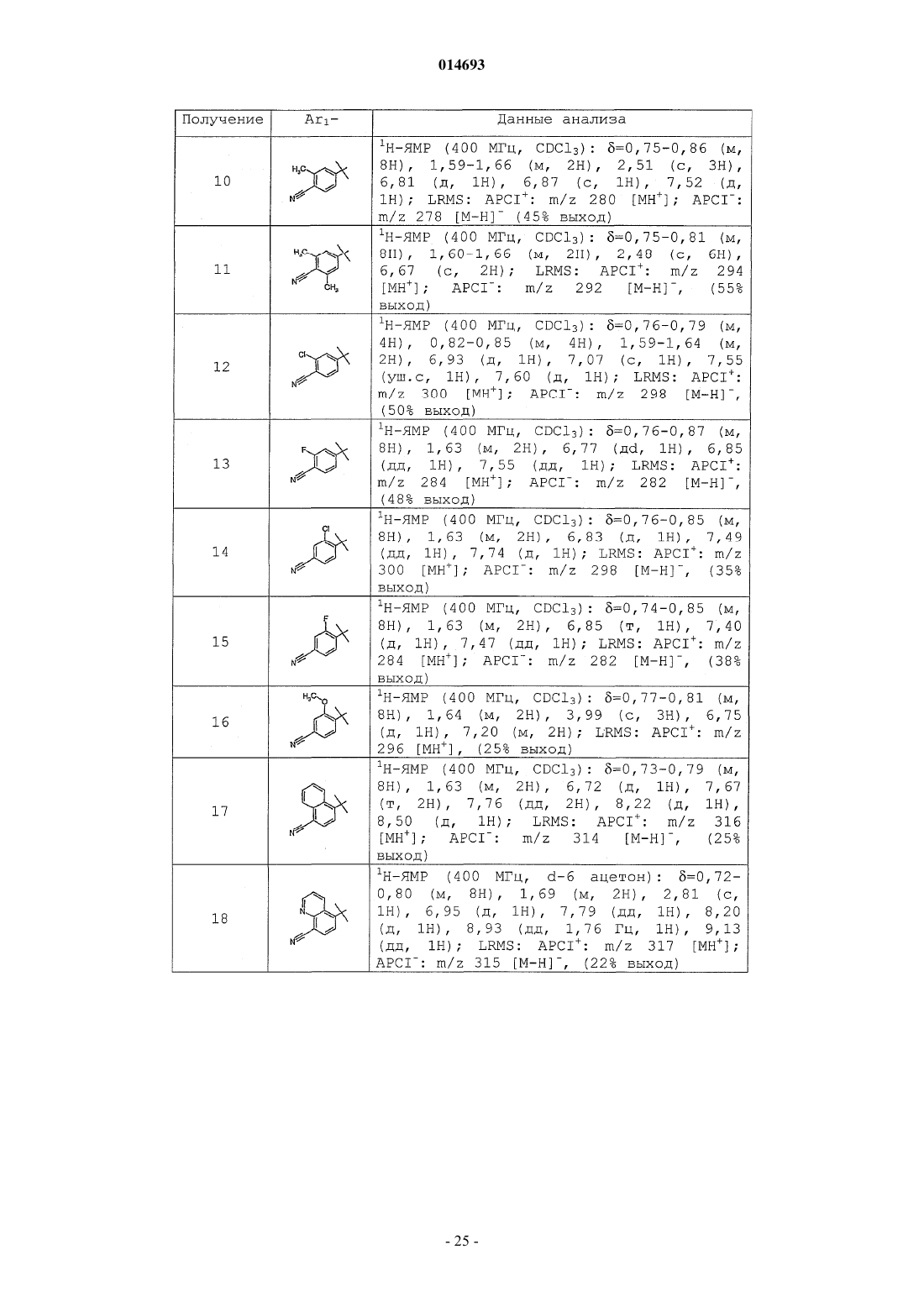
12. Соединение по п.1, выбранное из
4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)бензонитрила;
4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-2-метилбензонитрила;
4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
2-хлор-4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)бензонитрила;
4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-2-фторбензонитрила;
3-хлор-4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)бензонитрила;
4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-3-фторбензонитрила;
4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-3-метоксибензонитрила;
4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)нафталин-1-карбонитрила;
5-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)хинолин-8-карбонитрила;
4-(3,5-дициклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)хинолина;
4-(4-хлор-3-фторфенокси)-3,5-дициклопропил-1-метансульфонилметил-1Н-пиразола;
3,5-дициклопропил-4-(3,4-дифторфенокси)-1-метансульфонилметил-1Н-пиразола;
3,5-дициклопропил-1-метансульфонилметил-4-(3,4,5-трифторфенокси)-1Н-пиразола;
3,5-дициклопропил-4-(3,5-дифторфенокси)-1-метансульфонилметил-1Н-пиразола;
3,5-дициклопропил-1-метансульфонилметил-4-(2,4,5-трифторфенокси)-1Н-пиразола;
4-(3-циклопропил-1-метансульфонилметил-5-метил-1Н-пиразол-4-илокси)-2-метилбензонитрила;
4-(5-циклопропил-1-метансульфонилметил-3-метил-1Н-пиразол-4-илокси)-2-метилбензонитрила;
4-(3-циклопропил-1-метансульфонилметил-5-метил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(5-циклопропил-1-метансульфонилметил-3-метил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(1-метансульфонилметил-3,5-диметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(3,5-диэтил-1-метансульфонилметил-1Н-пиразол-4-илокси)бензонитрила;
4-(3,5-диэтил-1-метансульфонилметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(3,5-ди-трет-бутил-1-метансульфонилметил-1Н-пиразол-4-илокси)бензонитрила;
4-(3-трет-бутил-1-метансульфонилметил-5-метил-1Н-пиразол-4-илокси)бензонитрила;
4-(5-трет-бутил-1-метансульфонилметил-3-метил-1Н-пиразол-4-илокси)бензонитрила;
4-(3-хлор-5-циклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(5-хлор-3-циклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(3-циклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(5-циклопропил-1-метансульфонилметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(3,5-диэтил-1-метансульфонилметил-1Н-пиразол-4-илметил)бензонитрила;
4-(3,5-дициклопропил-1-трифторметансульфонилметил-1Н-пиразол-4-илокси)бензонитрила;
4-(3,5-дициклопропил-1-этансульфонилметил-1Н-пиразол-4-илокси)бензонитрила;
4-[3,5-дициклопропил-1-(пропан-2-сульфонилметил)-1Н-пиразол-4-илокси]бензонитрила;
4-[3,5-дициклопропил-1-(2-метилпропан-2-сульфонилметил)-1Н-пиразол-4-илокси]бензонитрила;
4-(1-бензолсульфонилметил-3,5-дициклопропил-1Н-пиразол-4-илокси)бензонитрила;
4-(3,5-диэтил-1-метансульфинилметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила;
4-(3,5-диэтил-1-метансульфинилметил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрила
и их фармацевтически приемлемых производных.
13. 4-(3-Циклопропил-1-метансульфонилметил-5-метил-1Н-пиразол-4-илокси)-2,6-диметилбензонитрил

или его фармацевтически приемлемое производное.
14. Способ получения соединения по п.1, в котором а обозначает 1, включающий окисление соединения формулы (II)

в которой R1-R8, X и Y имеют значения, определенные в п.1.
15. Способ получения соединения по п.1, в котором а обозначает 2, включающий окисление соединения формулы (II)

в которой R1-R8, X и Y имеют значения, определенные в п.1.
16. Способ получения соединения по п.1, в котором а обозначает 2, включающий окисление соединения формулы (Ia)

в которой R1-R8, X и Y имеют значения, определенные выше.
17. Фармацевтический состав, включающий соединение по любому из пп.1-13 или его фармацевтически приемлемое производное вместе с фармацевтически приемлемым эксципиентом, разбавителем или носителем.
18. Применение соединения по любому из пп.1-13 или его фармацевтически приемлемого производного либо композиции в качестве лекарственного средства.
19. Применение соединения по любому из пп.1-13 или его фармацевтически приемлемого производного либо композиции в получении лекарственного средства для лечения эндометриоза, фибромы матки (лейомиоматозных образований), меноррагии, аденомиоза, первичной и вторичной дисменореи (включая симптомы диспареунии, затрудненной дефекации и хронической тазовой боли) или синдрома хронической тазовой боли.
20. Применение по п.19, где заболеванием или нарушением является эндометриоз и/или фиброма матки (лейомиоматозные образования).
21. Способ лечения эндометриоза, фибромы матки (лейомиоматозных образований), меноррагии, аденомиоза, первичной и вторичной дисменореи (включая симптомы диспареунии, затрудненной дефекации и хронической тазовой боли) или синдрома хронической тазовой боли, включающий введение млекопитающему эффективного количества соединения по любому из пп.1-13 или его фармацевтически приемлемого производного либо композиции.
22. Способ по п.21, где заболеванием или нарушением является эндометриоз и/или фиброма матки (лейомиоматозные образования).
Текст
Брэдли Пол Энтони, Дэк Кевин Нил,Джонсон Патрик Стефен, Скерратт Сара Элизабет (GB) Представитель: или его фармацевтически приемлемое производное, в котором R1 и R3 независимо обозначают Н,C1-6 алкил, С 3-8 циклоалкил или галоген; R2 обозначает C1-6 алкил, CF3 или арил; а обозначает 1 или 2; R4, R5, R7 и R8 независимо обозначают Н, C1-6 алкил, С 1-6 алкилокси, CN или галоген, или R4 иR5, или R7 и R8, вместе с кольцом, к которому они присоединены, образуют арил или гетероциклическую конденсированную кольцевую систему; X обозначает С или N; Y обозначает СН 2 или О; R6 обозначает Н, CN или галоген, но когда X обозначает N, тогда R6 обозначает Н; могут быть использованы для лечения эндометриоза, фибром матки (лейомиоматозных образований), меноррагии, аденомиоза, первичной и вторичной дисменореи (включая симптомы диспареунии,затрудненной дефекации и хронической тазовой боли) или синдрома хронической тазовой боли. 014693 Это изобретение относится к новым соединениям и их производным, которые являются пригодными для использования в терапии, и к способам их получения. Оно также относится к промежуточным соединениям, используемым в получении таких соединений и производных, к содержащим их композициям и к их применению. Эндометриоз представляет собой распространенное гинекологическое заболевание, которое встречается у 10-20% женщин репродуктивного возраста и проявляется в присутствии функциональных эктопических эндометриальных желез и стромы вне полости матки (Prentice A. (2001) Bmj., 323, 93-95). Пациенты с эндометриозом могут проявлять множество различных симптомов разной степени серьезности. Обычно такими симптомами является дисменорея, однако хроническая тазовая боль, диспареуния, затрудненная дефекация, меноррагия, боль внизу живота или в пояснице, бесплодие, вспучивания и боль при мочеиспускании также представляют собой симптомы эндометриоза. Первоначально описанный Von Rokitansky в 1860 (Von Rokitansky С. (1860) Ztsch K.K. Gesellsch derAerzte zu Wien, 37, 577-581) точный патогенез эндометриоза неясен (Witz С.А. (1999) Clinical ObstetricsGynaecology, 42, 566-585; Witz С.A. (2002) GynaecologicObstetric Investigation, 53, 52-62), но наиболее распространенной теорией является теория имплантации, или теория Сэмпсона (Sampson J.A. (1927)American Journal of ObstetricsGynaecology, 14, 422-429). Теория Сэмпсона предполагает, что развитие эндометриоза является следствием ретроградной диссеминации и имплантации ткани эндометрия в брюшную полость в ходе менструального кровотечения. После прикрепления фрагменты эндометрия получают кровоснабжение и переносят циклы пролиферации и секреции под местным и системным гормональным контролем. У женщин с открытыми фаллопиевыми трубами ретроградное менструальное кровотечение, по-видимому, является универсальным явлением (Liu D.T. (Hitchcock A.) British Journal ofObstetricsGynaecology, 93, 859-862). Это заболевание часто проявляется как ректовагинальный эндометриоз или аденомиоз, овариальные кистозные ограниченные разрастания эндометриоидной ткани и,чаще всего, перитонеальный эндометриоз. Основными участками прикрепления и роста поражения в пределах таза являются яичники, широкие и круглые связки, фаллопиевы трубы, шейка, влагалище,брюшина и дивертикул Дугласа. В самом тяжелом случае эндометриоз может вызвать глубокую структурную модификацию брюшной полости, включая полиорганные спайки и фиброз. Симптоматический эндометриоз можно лечить медикаментозно и оперативно, если намерение состоит в том, чтобы удалить ткань эктопического поражения. Хирургическое вмешательство может быть консервативным, когда стремятся сохранить репродуктивный потенциал пациента, или сравнительно радикальным в случае тяжелого заболевания, включая рассечение мочевых путей, кишечника и ректовагинальной перегородки, или полную брюшную гистерэктомию и двустороннюю сальпинго-ооферэктомию. Медицинские фармакологические виды лечения, такие как андрогенотерапия, даназол и гестринон,совокупность агонистов GnRH, бусерелин, госерелин, лейпролид, нафарелин и трипторелин, антагонисты GnRH, цетрореликс и абареликс, а также прогестогены, включая медроксипрогестеронацетат, вызывают атрофию поражения, подавляя продукцию эстрогена. Эти подходы не лишены нежелательных побочных эффектов; даназол и гестринон включают увеличение массы тела, гирсутизм, угри, перемены настроения и метаболические эффекты в отношении сердечно-сосудистой системы. Группа агонистов и антагонистов GnRH, как оказалось, вызывает глубокое подавление эстрогена, что приводит к вазомоторным эффектам (приступообразные ощущения жара) и уменьшению минеральной плотности костей, что ограничивает их использование только шестью месяцами терапии. Группа прогестогенов, включая медроксипрогестеронацетат, подавляет гонадотропины, но не ингибирует овариальную продукцию эстрогена в той же степени, как аналоги GnRH. Побочные эффекты включают нерегулярное кровотечение, вздутия, увеличение массы тела и метаболические эффекты в отношении сердечно-сосудистой системы. Лейомиомы (Flake G.P., et al. (2003) Environmental Health Perspectives, 111, 1037-1054; Walker C.L.(2002) Recent Progress in Hormone Research, 57, 277-294) или фибромы матки являются самыми распространенными доброкачественными опухолями у женщин и встречаются у большинства женщин к моменту менопаузы. Хотя фибромы матки являются наиболее распространенным показанием для гистерэктомии в Соединенных Штатах, наряду с эндометриозом, об основной патофизиологии этого заболевания известно на удивление мало. Как и в случае эндометриоза, наличие увеличенных фибром матки связано с патологическим маточным кровотечением, дисменореей, тазовой болью и бесплодием. Помимо хирургического лечения показано, что такие виды терапии, обычно используемые для лечения эндометриоза, как аналоги GnRH или даназол, подавляют рост фибром, вызывая обратимое гипоэстрогенное состояниеLeo V., et al. (2002) Drug Safety, 25, 759-779; Ishihara H., et al. (2003) FertilitySterility, 79, 735-742). Однако в будущем лечение как фибром матки, так и эндометриоза должно основываться на разработке более эффективных, хорошо переносимых и более безопасных средств, чем те, которые являются доступными в настоящее время. Стероидные прогестины (то есть агонисты рецептора прогестерона) обычно используются в здравоохранении у женщин, например в контрацепции и гормональной терапии и для лечения гинекологических нарушений. Недавние исследования на женщинах и приматах также показывают, что антагонисты-1 014693 рецептора прогестерона могут найти применение в контрацепции и для лечения репродуктивных нарушений, таких как фибромы и эндометриоз. В настоящее время все клинически доступные агонисты и антагонисты рецептора прогестерона представляют собой стероидные соединения. Они часто вызывают различные побочные эффекты вследствие их функциональных взаимодействий с другими рецепторами стероидов или из-за эффектов, связанных с их стероидными метаболитами (Winneker, Richard С. et al.;Endocrinology and Reproductive Disorders Division, Women's Health Research Institute, Collegeville, PA,USA. Seminars in Reproductive Medicine (2005), 23(1), 46-57). Антагонисты рецептора прогестерона [антипрогестины (APs)], включая наиболее значимые члены этого класса мифепристон (RU-486; Roussel UCLAF, Romainville, Франция), онапристон (ZK 98 299;Schering AG), ZK 137 316 и ZK-230 211, являются соединениями, которые связываются с рецепторами прогестерона (PR) и предотвращают вызываемую прогестероном экспрессию генов (Spitz I.M. (2003)Steroids, 68, 981-993). Действуя на активируемый эстрогеном эндометрий, прогестерон играет существенную роль в дифференцировке и протоковом морфогенезе эндометриальной ткани, а также участвует в ингибировании миометриальной сократимости и в ответах поляризации лейкоцита Th1/Th2, которые являются критическими для имплантации эмбриона и поддержании беременности. Было проведено множество исследований потенциальных благоприятных воздействий антипрогестинов на признаки и симптомы эндометриоза (Grow D.R., et al. (1996) Journal of Clinical EndocrinologyMetabolism, 81, 19331939; Kettel L.M., et al. (1996) FertilitySterility, 65, 23-28; Kettel L.M., et al. (1998) American Journal ofObstetricsGynaecology, 178, 1151-1156) и фибром матки (Eisinger S.H., et al. (2003) ObstetricsGynaecology, 101, 243-250; Murphy A.A., and Castellano, P.Z. (1994) Current Opinion in ObstetricsGynaecology,6, 269-278; Murphy A.A., et al. (1995) FertilitySterility, 63, 761-766; Steinauer J., Pritts, et al. (2004) ObstetricsGynaecology, 103, 1331-1336; Yang Y., et al. (1996) Chinese. Chung-Hua Fu Chan Ko Tsa Chih [Chinese Journal of ObstetricsGynaecology], 31, 624-626). В отличие от аналогов GnRH и других обычных фармакологических подходов антипрогестины, особенно мифепристон, по-видимому, способны уменьшать объем поражения или фибромы, поддерживая тонизирующий уровень овариальной секреции эстрогена. Такие антипрогестины вызывают аменорею и уплотнение эндометрия, а также, по-видимому, достаточно защищают против быстрой эстроген-зависимой потери костной массы (Grow D.R., et al. (1996)Journal of Clinical EndocrinologyMetabolism, 81, 1933-1939). По контрасту с ними, аналоги GnRH вызывают быстрое снижение минеральной плотности кости, что является клинической особенностью, которая ограничивает продолжительность лечения с их использованием шестью месяцами. Хотя мифепристон является мощным антипрогестином, он также имеет аналогичную активность антиглюкокортикоида. Вне паллиативного лечения гиперкортизолизма при синдроме Кушинга (Chu J.W., et al. (2001) J. ClinSpitz, I.M. (2003) Steroids, 68, 981-993; Van Look P.F., and von Hertzen, H. (1995) Human Reproduction Update 1, 19-34, антиглюкокортикоидная активность является нежелательной особенностью мифепристона и потенциально многих представителей стероидных классов антипрогестинов. Также был описан следующий класс стероидных и нестероидных соединений, которые называют модуляторами рецептора прогестерона (PRM или мезопрогестины), включая асоприснил (J867, бензальдегид, 4-[(11,17)-17-метокси-17-(метоксиметил)-3-оксоэстра-4,9-диен-11-ил]-, 1 оксим; Jenpharm,ТАР), J912, J956, J1042. В дополнение к их потенциальной пригодности в гормональной заместительной терапии и в качестве противозачаточных средств можно предполагать, что эти классы соединений могут быть использованы в лечении эндометриоза и лейомиомы матки (Chwalisz K., et al. (2004) Semin Reproddiscussion 389-393; DeManno D., et al. (2003) Steroids, 68, 1019-1032). Асоприснил и структурнородственные PRM отличаются от антипрогестинов и прогестинов на моделях животных, демонстрируя частичную прогестогенную активность, например, в эндометрии кролика (тест Макфэйла (McPhail M.K.(1934) Journal of physiology, 83, 145-156 и влагалище гвинейской свинки. Доклинические исследования с асоприснилом на приматах показали, что PRM подавляют рост эндометрия и в отличие от эффектов прогестинов не подавляют эндометриальную экспрессию ER и PR (Chwalisz K., et al. (2000) Steroids 65,741-751; DeManno D., et al. (2003) Steroids, 68, 1019-1032; Elger W., et al. (2000) Steroids, 65, 713-723). Было обнаружено, что соединения согласно настоящему изобретению имеют полезные фармацевтические свойства. Они могут использоваться для лечения эндометриоза, фибром матки (лейомиоматозные образования) и меноррагии, аденомиоза, первичной и вторичной дисменореи (включая симптомы диспареунии, затрудненной дефекации и хронической тазовой боли), синдрома хронической тазовой боли, раннего полового созревания, цервикального созревания, контрацепции (срочные случаи), рака молочной железы, рак яичника, рака эндометрия, рака предстательной железы, рака легкого, рака яичек,рака желудка, менингиомы, тревожных состояний, предменструального синдрома, предменструального дисфорического расстройства, злоупотребления алкоголем и мышечной перонеальной атрофии или болезни Шарко-Маритута. Особенный интерес представляют следующие заболевания или нарушения: эндометриоз, фибромы матки (лейомиоматозные образования), меноррагия, аденомиоз, первичная и вторичная дисменорея(включая симптомы диспареунии, затрудненной дефекации и хронической тазовой боли) и синдром хро-2 014693 нической тазовой боли. В частности, соединения и производные согласно настоящему изобретению демонстрируют активность как антагонисты рецептора прогестерона и могут быть использованы для лечения в случаях, где показан антагонизм рецептора прогестерона. Более конкретно, соединения и производные согласно настоящему изобретению могут быть использованы для лечения эндометриоза и/или фибром матки (лейомиоматозных образований). В международной заявке на патент WO 02/085860 описаны пиразольные производные формулы в которой R1, R2, R3 и R4 имеют определенные там значения, которые являются модуляторами обратной транскриптазы ВИЧ. Согласно настоящему изобретению объектом изобретения является соединение формулы (I) или его фармацевтически приемлемое производное,в которой R1 и R3 независимо обозначают Н, C1-6 алкил, С 3-8 циклоалкил или галоген;R и R8, вместе с кольцом, к которому они присоединены, образуют арил или гетероциклическую конденсированную кольцевую систему;R6 обозначает Н, CN или галоген, но когда X обозначает N, тогда R6 отсутствует. В приведенных выше определениях алкильные группы, содержащие необходимое число атомов углерода, кроме особо обозначенных случаев, могут иметь неразветвленную или разветвленную цепь. Примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил. Примеры алкилокси включают метокси, этокси, н-пропилокси, изопропилокси, н-бутилокси, изобутилокси,втор-бутилокси и трет-бутилокси. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Термин галоген означает фтор, хлор, бром или иод. Арильными кольцами, включенными в определение арила, являются фенил или нафтил. Гетероциклами, включенными в определения гетероциклического ядра, являются пирролил, имидазолил, триазолил, тиенил, фурил, тиазолил, оксазолил, тиадиазолил, оксадиазолил, пиридинил, пиримидинил, пиридазинил, пиразинил, индолил, изоиндолил, хинолинил, изохинолинил, бензимидазолил, хиназолинил, фталазинил, бензоксазолил и хиноксалинил, вместе с их частично или полностью насыщенными версиями, а также азетидинил, пирролидинил, пиперидинил, пиперазинил, гомопиперазинил, оксазепанил и морфолинил. В варианте осуществления изобретения R1 обозначает C1-6 алкил или С 3-8 циклоалкил. В другом варианте осуществления изобретения R2 обозначает С 1-6 алкил. В другом варианте осуществления изобретения R3 обозначает C1-6 алкил или С 3-8 циклоалкил. В другом варианте осуществления изобретения R4 обозначает Н. В другом варианте осуществления изобретения R5 обозначает Н, C1-6 алкил или галоген. В другом варианте осуществления изобретения R4 и R5 вместе обозначают фенильное или пиридинильное кольцо, конденсированное с кольцом, к которому они присоединены. Предпочтительно R4 и R5 вместе обозначают фенильное кольцо, конденсированное с кольцом, к которому они присоединены. В другом варианте осуществления изобретения R6 обозначает CN. В другом варианте осуществления изобретения R7 обозначает Н, C1-6 алкил или галоген. В другом варианте осуществления изобретения R8 обозначает Н. В другом варианте осуществления изобретения R7 и R8 вместе обозначают фенильное или пиридинильное кольцо, конденсированное с кольцом, к которому они присоединены. Предпочтительно R7 и R8 вместе обозначают фенильное кольцо, конденсированное с кольцом, к которому они присоединены. В другом варианте осуществления изобретения Y обозначает О. В другом варианте осуществления изобретения галоген обозначает фтор или хлор. Предпочтительными соединениями согласно настоящему изобретению являются 4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)бензонитрил;-3 014693 4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-2-метилбензонитрил; 4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 2-хлор-4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)бензонитрил; 4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-2-фторбензонитрил; 3-хлор-4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)бензонитрил;. 4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-3-фторбензонитрил; 4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-3-метоксибензонитрил; 4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)нафталин-1-карбонитрил; 5-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)хинолин-8-карбонитрил; 4-(3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)хинолин; 4-(4-хлор-3-фторфенокси)-3,5-дициклопропил-1-метансульфонилметил-1 Н-пиразол; 3,5-дициклопропил-4-(3,4-дифторфенокси)-1-метансульфонилметил-1 Н-пиразол; 3,5-дициклопропил-1-метансульфонилметил-4-(3,4,5-трифторфенокси)-1 Н-пиразол; 3,5-дициклопропил-4-(3,5-дифторфенокси)-1-метансульфонилметил-1 Н-пиразол; 3,5-дициклопропил-1-метансульфонилметил-4-(2,4,5-трифторфенокси)-1 Н-пиразол; 4-(3-циклопропил-1-метансульфонилметил-5-метил-1 Н-пиразол-4-илокси)-2-метилбензонитрил; 4-(5-циклопропил-1-метансульфонилметил-3-метил-1 Н-пиразол-4-илокси)-2-метилбензонитрил; 4-(3-циклопропил-1-метансульфонилметил-5-метил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(5-циклопропил-1-метансульфонилметил-3-метил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(1-метансульфонилметил-3,5-диметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(3,5-диэтил-1-метансульфонилметил-1 Н-пиразол-4-илокси)бензонитрил; 4-(3,5-диэтил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(3,5-ди-трет-бутил-1-метансульфонилметил-1 Н-пиразол-4-илокси)бензонитрил; 4-(3-трет-бутил-1-метансульфонилметил-5-метил-1 Н-пиразол-4-илокси)бензонитрил; 4-(5-трет-бутил-1-метансульфонилметил-3-метил-1 Н-пиразол-4-илокси)бензонитрил; 4-(3-хлор-5-циклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(5-хлор-3-циклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(3-циклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(5-циклопропил-1-метансульфонилметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(3,5-диэтил-1-метансульфонилметил-1 Н-пиразол-4-илметил)бензонитрил; 4-(3,5-дициклопропил-1-трифторметансульфонилметил-1 Н-пиразол-4-илокси)бензонитрил; 4-(3,5-дициклопропил-1-этансульфонилметил-1 Н-пиразол-4-илокси)бензонитрил; 4-[3,5-дициклопропил-1-(пропан-2-сульфонилметил)-1 Н-пиразол-4-илокси]бензонитрил; 4-[3,5-дициклопропил-1-(2-метилпропан-2-сульфонилметил)-1 Н-пиразол-4-илокси]бензонитрил; 4-(1-бензолсульфонилметил-3,5-дициклопропил-1 Н-пиразол-4-илокси)бензонитрил; 4-(3,5-диэтил-1-метансульфинилметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; 4-(3,5-диэтил-1-метансульфинилметил-1 Н-пиразол-4-илокси)-2,6-диметилбензонитрил; и их фармацевтически приемлемые производные. Вышеописанные варианты осуществления изобретения могут быть скомбинированы с одним или более другими вариантами осуществления так, что в других вариантах осуществления две или более переменных определены более конкретно в комбинации. Например, в рамках изобретения находится другой вариант осуществления, в котором переменные R1, R2 и L все имеют более ограниченные определения, которые они имеют в более конкретных вариантах осуществления, описанных выше. Все такие комбинации более конкретных вариантов осуществления, описанных и определенных выше, входят в рамки изобретения. Фармацевтически приемлемые производные соединений формулы (I) согласно изобретению включают их соли, сольваты, комплексы, полиморфы и кристаллические формы, пролекарства, стереоизомеры, геометрические изомеры, таутомерные формы и изотопные варианты соединений формулы (I). Предпочтительно фармацевтически приемлемые производные соединений формулы (I) включают соли,сольваты, сложные эфиры и амиды соединений формулы (I). Более предпочтительно фармацевтически приемлемые производные соединений формулы (I) представляют собой соли и сольваты. Фармацевтически приемлемые соли соединений формулы (I) включают их соли присоединения с кислотой и основанием. Подходящие соли присоединения с кислотой получают из кислот, которые образуют нетоксичные соли. Примеры включают ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдисилат, эзилат, формиат, фумарат, глуцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидроиодид/иодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат,сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинофоат.-4 014693 Подходящие соли с основанием получают из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина,глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Можно также получить полусоли с кислотами и основаниями, например гемисульфат и полусоли кальция. В отношении подходящих солей см. "Handbook of Pharmaceutical Salts: Properties, Selection, and Use"by Stahl and Wermuth (Wiley-VCH, 2002). Фармацевтически приемлемые соли соединений формулы (I) могут быть получены одним или более из трех способов:(i) введением в реакцию соединения формулы (I) с желаемой кислотой или основанием;(ii) удалением лабильной к кислоте или основанию защитной группы от подходящего предшественника соединения формулы (I) или размыканием кольца подходящего циклического предшественника,например лактона или лактама, с использованием желаемой кислоты или основания или(iii) преобразованием одной соли соединения формулы (I) в другую реакцией с подходящей кислотой или основанием или посредством подходящей колонки ионного обмена. Все три реакции обычно выполняют в растворе. Полученная соль может быть осаждена и собрана фильтрацией или может быть восстановлена выпариванием растворителя. Степень ионизации в конечной соли может варьировать от полной ионизации до почти полного отсутствия ионизации. Соединения по изобретению могут существовать в континууме твердых состояний в пределах от полностью аморфного до полностью кристаллического. Термин "аморфный" относится к состоянию, в котором материал неупорядочен на молекулярном уровне и в зависимости от температуры может показывать физические свойства твердого или жидкого вещества. Обычно такие материалы не дают отличительные структуры дифракции в рентгеновских лучах и, показывая свойства твердого вещества, более формально описываются как жидкость. После нагревания происходит изменение свойств от таковых твердого вещества до свойств жидкого вещества, что характеризуется изменением состояния, обычно второго порядка ("стеклование"). Термин "кристаллический" относится к твердой фазе, в которой материал имеет постоянную упорядоченную внутреннюю структуру на молекулярном уровне и дает отличительную структуру преломления в рентгеновских лучах с определенными пиками. Такие материалы при достаточном нагревании также показывают свойства жидкости, но изменение от твердого вещества до жидкости характеризуется фазовым переходом, обычно первого порядка ("точка плавления"). Соединения по изобретению могут также существовать в сольватированных и несольватированных формах. Термин "сольват" обозначает молекулярный комплекс, включающий соединение по изобретению и одну или более молекул фармацевтически приемлемого растворителя, например этанола. Термин"гидрат" используется, когда указанным растворителем является вода. Используемая в настоящее время система классификации для органических гидратов представляет собой систему, которая определяет гидраты изолированного участка, канала или скоординированные ионом металла гидраты - см. "Polymorphism in Pharmaceutical Solids" by K.R. Morris (Ed. H.G. Brittain,Marcel Dekker, 1995). Гидраты изолированного участка представляют собой такие, в которых молекулы воды изолированы от прямого контакта друг с другом органическими молекулами. В гидратах канала молекулы воды лежат в каналах решетки, где они соседствуют с другими молекулами воды. В скоординированных ионом металла гидратах молекулы воды сцеплены с ионом металла. Когда растворитель или вода прочно связаны, комплекс будет иметь четкую стехиометрию независимо от влажности. Когда, однако, растворитель или вода слабо связаны, как в сольватах канала и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и режима сушки. В таких случаях нормой является нестехиометрия. Также в рамки изобретения входят многокомпонентные комплексы (кроме солей и сольватов), в которых лекарственное средство и по меньшей мере один другой компонент присутствуют в стехиометрических или нестехиометрических количествах. Комплексы этого типа включают клатраты (инклюзионные комплексы лекарственное средство-хозяин) и со-кристаллы. Последние обычно определяют как кристаллические комплексы нейтральных молекулярных составляющих, которые связаны через нековалентные взаимодействия, но могут также представлять собой комплексы нейтральной молекулы с солью. Coкристаллы могут быть получены кристаллизацией расплава, перекристаллизацией из растворителей или физически перетиранием компонентов друг о друга - см. Chem. Commun, 17, 1889-1896, О. Almarsson andM.J. Zaworotko (2004). В отношении общего обзора многокомпонентных комплексов см. J. Pharm. Sci, 64(8), 1269-1288, by Haleblian (August, 1975). Соединения по изобретению могут также существовать в мезоморфном состоянии (мезофаза или жидкий кристалл), если они помещены в подходящие условия. Мезоморфное состояние является промежуточным между истинным кристаллическим состоянием и состоянием идеальной жидкости (расплава или раствора). Мезоморфизм, возникающий как результат изменения температуры, описан как "термотропный", а возникающий в результате добавления второго компонента, такого как вода или другой растворитель, описан как "лиотропный". Соединения, которые имеют потенциал для образования лиотропных мезофаз, описаны как "амфифильные" и состоят из молекул, которые имеют ионную (такую как-COO-Na+, -COO-K+ или -SO3-Na+) или неионную (такую как -N-N+(СН 3)3) полярную главную группу. В отношении дополнительной информации см. Crystals and the Polarizing Microscope by N.H. Hartshorne andA. Stuart, 4th Edition (Edward Arnold, 1970). В дальнейшем все ссылки на соединения формулы (I) включают ссылки на их соли, сольваты, многокомпонентные комплексы и жидкие кристаллы и на сольваты, многокомпонентные комплексы и жидкие кристаллы их солей. Как обозначено выше, так называемые "пролекарства" соединений формулы (I) также находятся в рамках изобретения. Так, некоторые производные соединений формулы (I), которые сами по себе могут иметь низкую фармакологическую активность или не иметь такой активности, могут быть преобразованы в соединения формулы (I), имеющие желаемую активность, например, в результате гидролитического расщепления при введении в или нанесении на тело. Такие производные упоминаются "как пролекарства". Дальнейшая информация относительно использования пролекарств может быть найдена в "Pro-drugsas Novel Delivery Systems", vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и "Bioreversible Carriers in Drug Design", Pergamon Press, 1987 (Ed. E.B. Roche, American Pharmaceutical Association). Пролекарства в соответствии с изобретением могут быть получены заменой подходящих функциональных групп, присутствующих в соединениях формулы (I), определенными группами, известными специалистам как "про-группы", как описано, например, в "Design of Prodrugs" by H. Bundgaard (Elsevier,1985). Некоторые примеры пролекарств в соответствии с изобретением включают:(i) если соединение формулы (I) содержит спиртовую функциональную группу (-ОН), его простой эфир, например соединение, в котором атом водорода спиртовой функциональной группы соединения формулы (I) заменен (C1-C6)алканоилоксиметил; и(ii) если соединение формулы (I) содержит первичную или вторичную функциональную аминогруппу (-NH2 или -NHR, где RН), его амид, например соединение, в котором в зависимости от обстоятельств одни или оба атома водорода функциональной аминогруппы соединения формулы (I) заменены(C1-С 10)алканоилом. Другие примеры групп замены в соответствии с предшествующими примерами и примерами других типов пролекарства могут быть найдены в вышеупомянутых ссылках. Кроме того, некоторые соединения формулы (I) могут сами по себе действовать как пролекарства других соединений формулы (I). Также в рамки изобретения включены метаболиты соединений формулы (I), то есть соединения, которые образуются in vivo при введении лекарственного средства. Таким образом, в рамки изобретения входят метаболиты соединений формулы (I), когда они образуются in vivo. Соединения формулы (I), содержащие один или более асимметрических атомов углерода, могут существовать как два или более стереоизомера. Если соединение формулы (I) содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Если структурные изомеры взаимно превращаются друг в друга через низкий энергетический барьер, может иметь место таутомерная изомерия ("таутомерия"). Она может принимать форму протонной таутомерии в соединениях формулы (I), содержащих, например, имино, кето или оксимную группы, или так называемой таутомерии валентности в соединениях, которые содержат ароматическую группу. Из этого следует, что отдельное соединение может показывать более одного типа изомерии. В рамки настоящего изобретения входят все стереоизомеры, геометрические изомеры и таутомерные формы соединений формулы (I), включая соединения, показывающие более одного типа изомерии, и смеси одного или более из них. Также в рамки изобретения входят соли присоединения с кислотой или основанием, в которых противоион оптически активен, например d-лактат или l-лизин, или рацемические формы, например, dl-тартрат или dl-аргинин. Цис/транс-изомеры могут быть разделены обычными методиками, известными специалисту, например хроматографией и фракционной кристаллизацией. Обычные методики получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ). Альтернативно, рацемат (или рацемический предшественник) может быть введен в реакцию с подходящим оптически активным соединением, например спиртом, или в случае, если соединение формулы(I) содержит кислотную или основную группу, с основанием или кислотой, такой как 1-фенилэтиламин или винная кислота. Полученная диастереомерная смесь может быть разделена хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера могут быть превращены в соответствующий чистый энантиомер(ы) средствами, известными специалисту. Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в энантиомерно-обогащенной форме с использованием хроматографии, обычно ВЭЖХ, на асимметричной смоле с мобильной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащей от 0 до 50 об.% изопропанола, обычно от 2 до 20 и от 0 до 5 об.% алкиламина, обычно 0,1% диэтиламина. Кон-6 014693 центрация элюата предоставляет обогащенную смесь. Когда рацемат кристаллизуется, возможны кристаллы двух различных типов. Первый тип представляет собой рацемическое соединение (истинный рацемат), указанное выше, когда образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, когда образуются две формы кристалла в эквимолярных количествах, каждая из которых содержит единственный энантиомер. Хотя обе кристаллические формы в рацемической смеси имеют идентичные физические свойства,они могут иметь разные физические свойства по сравнению с истинным рацематом. Рацемические смеси могут быть разделены обычными методиками, известными специалисту - см., например, "Stereochemistryof Organic Compounds" by E.L. Eliel and S.H. Wilen (Wiley, 1994). Настоящее изобретение включает все фармацевтически приемлемые меченные изотопом соединения формулы (I), в которых один или более атомов заменены атомами, имеющими то же самое атомное число, но атомную массу или массовое число, отличное от атомной массы или массового числа, которое преобладает в природе. Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2 Н и 3 Н, углерода, такие как 11 С, 13 С и 14 С, хлора, такие как 36Cl, фтора, такие как 18F,иода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32 Р, и серы, таких как 35S. Некоторые меченные изотопом соединения формулы (I), например, такие, которые включают радиоактивный изотоп, могут быть использованы в исследованиях распределения лекарственного средства и/или субстрата в ткани. Радиоактивный изотоп тритий, то есть 3 Н, и углерод-14, то есть 14 С, являются особенно пригодными для этой цели ввиду легкости их включения и доступности средств детекции. Замещение более тяжелыми изотопами, такими как дейтерий, то есть 2 Н, может представить определенные терапевтические преимущества вследствие большей метаболической стабильности, например увеличенного период полураспада in vivo или уменьшенной требуемой дозы, и, следовательно, может быть предпочтительным при некоторых обстоятельствах. Замещение изотопами, испускающими позитроны, такими как 11C, 18F, 15O и 13N, может быть использовано в исследованиях Позитронной Эмиссионной Топографии (ПЭТ) для исследования занятия рецептора субстратом. Меченные изотопом соединения формулы (I) могут в целом быть получены обычными способами,известными специалисту, или способами, аналогичными описанным в сопутствующих примерах и примерах получения, с использованием подходящего меченного изотопом реактива вместо немеченого реактива, использованного ранее. Фармацевтически приемлемые сольваты в соответствии с изобретением включают такие, в которых растворитель кристаллизации может быть замещен изотопом, например D2O, d6-ацетон, d6-ДМСО. Соединения формулы (I) должны быть оценены в отношении их биофармацевтических свойств, таких как растворимость и стабильность раствора (в диапазоне рН), проницаемость и т.д., для выбора наиболее подходящей лекарственной формы и пути-введения для лечения предложенного показания. Соединения по изобретению, предназначенные для фармацевтического использования, могут вводиться в форме кристаллических или аморфных продуктов. Они могут быть получены, например, как твердые тампоны, порошки или пленки такими способами, как осаждение, кристаллизация, лиофилизация, сушка распылением или сушка выпариванием. С этой целью может использоваться микроволновая или высокочастотная сушка. Соединения по изобретению могут вводиться индивидуально или в комбинации с одним или более другими соединениями по изобретению или в комбинации с одним или более другими лекарственными средствами (или в форме любой комбинации вышеперечисленного). Соединения согласно настоящему изобретению могут вводиться в комбинации с ингибиторами СОХ. Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и один или более ингибиторов СОХ в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Ингибиторы СОХ, пригодные для комбинации с соединениями согласно настоящему изобретению,включают, но не ограничены ими:(ii) мелоксикам (регистрационный номер CAS 71125-38-7, описанный в патенте США 4233299) или его фармацевтически приемлемую соль или пролекарство;(iii) целекоксиб (патент США 5466823), вальдекоксиб (патент США 5633272), деракоксиб (патент США 5521207), рофекоксиб (патент США 5474995), эторикоксиб (международная публикация заявки на патентWO 98/03484), JTE-522 (Публикация заявки на патент Японии 9052882) или их фармацевтически приемлемую соль или пролекарство;(iv) парекоксиб (описанный в патенте США 5932598), который является терапевтически эффективным пролекарством селективного ингибитора трициклического Сох-2 вальдекоксиба (описанного в патенте США 5633272), в частности парекоксиб натрия;(v) ABT-963 (описанный в международной публикации заявки на патентWO 00/24719),(vi) нимесулид (описанный в патенте США 3840597), флосулид (обсуждаемый в J. Carter, Exp. Opin.Ther. Patents, 8(1), 21-29 (1997, NS-398 (раскрытый в патенте США 4885367), SD 8381 (описанный в патенте США 6034256), BMS-347070 (описанный в патенте США 6180651), S-2474 (описанный в Европейской патентной публикации 595546) и МК-966 (описанный в патенте США 5968974);Wellcome) и S-2474 (Shionogi). Соединения согласно настоящему изобретению могут вводиться в комбинации с ингибиторами ФДЭ 5. Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и один или более ингибиторов ФДЭ 5 в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Ингибиторы PDEV, пригодные для комбинирования с соединениями согласно настоящему изобретению, включают, но не ограничены ими:(i) предпочтительно 5-[2-этокси-5-(4-метил-1-пиперазинилсульфонил)фенил]-1-метил-3-н-пропил 1,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он (силденафил, например, выпускаемый как Viagra),также известный как 1-3-(6,7-дигидро-1-метил-7-оксо-3-пропил-1 Н-пиразоло[4,3-d]пиримидин-5 ил)-4 этоксифенил]сульфонил]-4-метилпиперазин (см. ЕР-А-0463756); 5-(2-этокси-5-морфолиноацетилфенил)1-метил-3-н-пропил-1,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он (см. ЕР-А-0526004); 3-этил-5-[5-(4 этилпиперазин-1-илсульфонил)-2-н-пропоксифенил]-2-(пиридин-2-ил)метил-2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он (см. WO 98/49166); 3-этил-5-[5-(4-этилпиперазин-1-илсульфонил)-2-(2-метоксиэтокси)пиридин-3 ил]-2-(пиридин-2-ил)метил-2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он (см. WO 99/54333); (+)-3-этил-5-[5(4-этилпиперазин-1-илсульфонил)-2-(2-метокси-1(R)-метилэтокси)пиридин-3-ил]-2-метил-2,6-дигидро 7 Н-пиразоло[4,3-d]пиримидин-7-он, также известный как 3-этил-5-5-[4-этилпиперазин-1-илсульфонил]-2([(1R)-2-метокси-1-метилэтил]окси)пиридин-3-ил-2-метил-2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он (см.WO 99/54333); 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он, также известный как 1-6-этокси-5-[3-этил-6,7-дигидро 2-(2-метоксиэтил)-7-оксо-2 Н-пиразоло[4,3-d]пиримидин-5-ил]-3-пиридилсульфонил-4-этилпиперазин(см. WO 01/27113, пример 8); 5-[2-изобутокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил 2-(1-метилпиперидин-4-ил)-2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он (см. WO 01/27113, пример 15); 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-фенил-2,6-дигидро-7 Н-пиразоло[4,3-]пиримидин-7-он (см. WO 01/27113, пример 66); 5-(5-ацетил-2-пропокси-3-пиридинил)-3-этил 2-(1-изопропил-3-азетидинил)-2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он (см. WO 01/27112, пример 124); 5-(5-ацетил-2-бутокси-3-пиридинил)-3-этил-2-(1-этил-3-азетидинил)-2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он (см. WO 01/27112, пример 132); (6R,12aR)-2,3,6,7,12,12 а-гексагидро-2-метил-6(3,4-метилендиоксифенил)пиразино[2',1':6,1]пиридо[3,4-b]индол-1,4-дион (тадалафил, IC-351, Cialis),то есть соединение из примеров 78 и 95 опубликованной международной заявки WO 95/19978, а также соединения из примеров 1, 3, 7 и 8; 2-[2-этокси-5-(4-этилпиперазин-1-ил-1-сульфонил)фенил]-5-метил-7 пропил-3 Н-имидазо[5,1-f][1,2,4]триазин-4-он (варденафил, LEVITRA), также известный как 1-3-(3,4 дигидро-5-метил-4-оксо-7-пропилимидазо[5,1-f]-as-триазин-2-ил)-4-этоксифенил]сульфонил]-4-этилпиперазин,то есть соединение из примеров 20, 19, 337 и 336 опубликованной международной заявки WO 99/24433; соединение из примера 11 опубликованной международной заявки WO 93/07124 (EISAI); соединения 3 и 14 из Rotella D.P., J. Med. Chem., 2000, 43, 1257; 4-(4-хлорбензил)амино-6,7,8-триметоксихиназолин; N3-(4,7-дигидро-1-метил-7-оксо-3-пропил-1 Н-пиразоло[4,3-d]пиримидин-5-ил)-4-пропоксифенил]сульфонил]-1 метил-2-пирролидинпропанамид ["DA-8159" (пример 68 из WO 00/27848)]; и 7,8-дигидро-8-оксо-6-[2-8 014693 пропоксифенил]-1 Н-имидазо[4,5-g]хиназолин и 1-[3-[1-[(4-фторфенил)метил]-7,8-дигидро-8-оксо-1 Нимидазо[4,5-g]хиназолин-6-ил]-4-пропоксифенил]карбоксамид; 4-[(3-хлор-4-метоксибензил)амино]-2[(2S)-2-(гидроксиметил)пирролидин-1-ил]-N-(пиримидин-2-илметил)пиримидин-5-карбоксамид(ТА 1790); 3-(1-метил-7-оксо-3-пропил-6,7-дигидро-1 Н-пиразоло[4,3-d]пиримидин-5-ил)-N-[2-(1-метилпирролидин 2-ил)этил]-4-пропоксибензол сульфамид (DA 8159) и их фармацевтически приемлемые соли;(ii) 4-бром-5-(пиридилметиламино)-6-[3-(4-хлорфенил)пропокси]-3(2 Н)пиридазинон; 1-[4-[(1,3 бензодиоксол-5-илметил)амино]-6-хлор-2-хинозолинил]-4-пиперидинкарбоновую кислоту, однонатриевую соль; (+)-цис-5,6 а,7,9,9,9 а-гексагидро-2-[4-(трифторметил)фенилметил-5-метилциклопент-4,5]имидазо[2,1-b]пурин-4(3 Н)-он; фуразлоциллин; цис-2-гексил-5-метил-3,4,5,6 а,7,8,9,9 а-октагидроциклопент[4,5]имидазо[2,1-b]пурин-4-он; 3-ацетил-1-(2-хлорбензил)-2-пропилиндол-6-карбоксилат; 3-ацетил 1-(2-хлорбензил)-2-пропилиндол-6-карбоксилат; 4-бром-5-(3-пиридилметиламино)-6-(3-(4-хлорфенил)пропокси)-3-(2 Н)пиридазинон; 1-метил-5(5-морфолиноацетил-2-н-пропоксифенил)-3-н-пропил-1,6 дигидро-7 Н-пиразоло(4,3-d)пиримидин-7-он; 1-[4-[(1,3-бензодиоксол-5-илметил)амино]-6-хлор-2-хиназолинил]-4-пиперидинкарбоновую кислоту, однонатриевую соль; Pharmaprojects4516 (Glaxo Wellcome); Pharmaprojects5051 (Bayer); Pharmaprojects5064 (Kyowa Hakko; см. WO 96/26940); Pharmaprojects5069 (Schering Plough); GF-196960 (Glaxo Wellcome); E-8010 и Е-4010 (Eisai); Bay-38-3045 и 38-9456 (Bayer); FR229934 и FR226807 (Fujisawa); и Sch-51866. Предпочтительно ингибитор ФДЭ 5 выбирают из силденафила, тадалафила, варденафила, DA-8159 и 5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2-метоксиэтил]-2,6-дигидро 7 Н-пиразоло[4,3-d]пиримидин-7-она. Наиболее предпочтительно ингибитором ФДЭ 5 является силденафил и его фармацевтически приемлемые соли. Предпочтительной солью является силденафилцитрат. Соединения согласно настоящему изобретению могут вводиться в комбинации с антагонистом V1a. Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и один или более антагонистов V1a в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Подходящим антагонистом рецептора вазопрессина V1a является, например, (4-[4-бензил-5-(4 метокси-пиперидин-1-илметил)-4 Н-[1,2,4]триазол-3-ил]-3,4,5,6-тетрагидро-2 Н-[1,2']бипиридинил), который соответствует примеру 26 в WO 2004/37809. Другим примером подходящего антагониста рецептора вазопрессина V1a является 8-хлор-5-метил-1-(3,4,5,6-тетрагидро-2 Н-[1,2']бипиридинил-4-ил)-5,6 дигидро-4 Н-2,3,5,10b-тетраазобензо[е]азулен, или его фармацевтически приемлемая соль либо сольват,который соответствует примеру 5 в WO 04/074291. Другими примерами антагониста рецептора вазопрессина V1a для использования в изобретении являются SR49049 (релковаптан), атосибан (Tractocile), кониваптан (YM-087), VPA-985, CL-385004, вазотоцин и ОРС 21268. Дополнительно антагонисты рецептора V1a, описанные в WO 01/58880, являются подходящими для использования в изобретении. Соединения согласно настоящему изобретению могут вводиться в комбинации с антагонистом альфа-адренергического рецептора (также известным как блокатор -адренорецептора, блокатор-рецептора или -блокатор). Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и один или более антагонистов альфаадренергического рецептора в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Антагонисты 1-адренергического рецептора, пригодные для использования в настоящем изобретении, включают, но не ограничены ими, теразосин (патент США 4026894), доксазосин (патент США 4188390), празосин (патент США 3511836), буназосин (патент США 3920636), альфузосин (патент США 4315007), нафтопидил (патент США 3997666), тамсулосин (патент США 4703063), силодосин (патент США 5387603), фентоламин и фентоламин мезилат (патент США 2503059), тразодон (патент США 3381009), индорамин (патент США 3527761), феноксибензамин (патент США 2599000), алкалоиды раувольфии (натуральный продукт из куста раувольфии змеиной), Recordati 15/2739 (WO 93/17007), SNAP 1069 (WO 94/08040 например 3, соединение 9, с. 77 и табл. 3, с. 86), SNAP 5089 (WO 94/10989), RS17053(США 5436264), SL 89.0591 (ЕР 435749) и абанохил (ЕР 100200); соединения, раскрытые в международной публикации WO 03/076427, в частности 5-циклопропил-7-метокси-2-(2-морфолин-4-илметил-7,8 дигидро[1,6]-нафтиридин-6(5 Н)-ил)-4(3 Н)хиназолинон (пример 11), и соединения, раскрытые в международной публикации WO 98/30560, в частности 4-амино-6,7-диметокси-2-(5-метансульфонамидо 1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хиназолин (пример 19); и их фармацевтически приемлемые производные. Предпочтительными антагонистами -адренергического рецептора являются доксазосин, 5 циклопропил-7-метокси-2-(2-морфолин-4-илметил-7,8-дигидро[1,6]-нафтиридин-6(5 Н)-ил)-4(3 Н)хиназолинон и 4-амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хиназолин и их фармацевтически приемлемые производные. Особенный интерес представляет 4-амино-6,7 диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хиназолинмезилат(см. WO 01/64672). Антагонисты 2-адренергического рецептора, подходящие для настоящего изобретения, включают-9 014693 дибенамин (DE 824208), толазолин (патент США 2161938), тримазосин (патент США 3669968), эфароксан (ЕР 71368), йохимбин (Goldberg M.R., et al., Pharmacol. Rev., 35, 143-180 (1987, идазоксан (ЕР 33655) и клонидин (патент США 3202660). Неселективные антагонисты -адренергического рецептора, подходящие для настоящего изобретения, включают дапипразол (патент США 4252721). Соединения согласно настоящему изобретению могут вводиться в комбинации с ингибитором 5-альфа-редуктазы. Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и один или более ингибиторов 5-альфаредуктазы в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Ингибиторы 5-альфа-редуктазы включают ингибиторы изофермента 2 5-альфа-редуктазы. Подходящими соединениями для использования в настоящем изобретении являются PROSCAR (также известный как финастерид, патенты США 4377584 и 4760071), соединения, описанные в WO 93/23420,ЕР 0572166, WO 93/23050, 93/23038, 93/23048, 93/23041, 93/23040, 93/23039, 93/23376, 93/23419,ЕР 0572165 и WO 93/23051. Соединения согласно настоящему изобретению могут вводиться в комбинации со средством, которое понижает уровни эстрогена или которое противодействует рецептору эстрогена. Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и одно или более средств, которые понижают уровни эстрогена или противодействуют рецептору эстрогена, в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Средства, которые понижают уровни эстрогена, включают агонисты гонадотропинвысвобождающего гормона (GnRH), антагонисты GnRH и ингибиторы синтеза эстрогена. Средства, которые противодействуют рецептору эстрогена, то есть антагонисты рецептора эстрогена, включают антиэстрогены. Агонисты GnRH, подходящие для настоящего изобретения, включают лейпрорелин (Prostap Wyeth), бусерелин (Suprefact - Shire), госерелин (Zoladex - Astra Zeneca), трипторелин (Decapeptyl Ipsen), нафарелин (Synarel - Searle), деслорелин (Somagard - Shire) и гистрелин/суппрелин (Ortho Pharmaceutical Corp/Shire). Антагонисты GnRH, подходящие для настоящего изобретения, включают тевереликс (также известный как антареликс), абареликс (Plenaxis - Praecis Pharmaceuticals Inc), цетрореликс (Cetrotide - ASTAMedica) и ганиреликс (Orgalutran - Organon). Антиэстрогены, подходящие для настоящего изобретения, включают тамоксифен, Faslodex (Astra(Labrie F., et al., (2001) J. steroid Biochem. Mol. Biol., 79, 213). Ингибиторы синтеза эстрогена, подходящие для настоящего изобретения, включают ингибиторы ароматазы. Примеры ингибиторов ароматазы включают форместан (4-ОН андростендион), экземестан,анастрозол (Arimidex) и летроксол. Соединения согласно настоящему изобретению могут вводиться в комбинации с альфа-2-дельта лигандом. Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и один или более альфа-2-дельта лигандов, в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Примерами альфа-2-дельта лигандов для использования в настоящем изобретении являются соединения или их фармацевтически приемлемые соли, раскрытые, в общем виде или конкретно, в(2S,4S)-4-(3-фторфеноксиметил)пролин,(2S,4S)-4-(2,3 дифторбензил)пролин, (2S,4S)-4-(3-хлорфенокси)пролин и (2S,4S)-4-(3-фторбензил)пролин, ЕР 1178034,1201240, WO-A-99/31074, WO-A-03/000642, WO-A-02/22568, WO-A-02/30871, WO-A-02/30881,WO-A-02/100392, WO-A-02/100347, WO-A-02/42414, WO-A-02/32736 и WO-A-02/28881, которые все включены в настоящее описание путем ссылки. Предпочтительные альфа-2-дельта лиганды для использования в комбинации согласно настоящему изобретению включают габапентин, прегабалин, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6- 10014693 ил]уксусную кислоту, 3-(1-аминометилциклогексилметил)-4 Н-[1,2,4] оксадиазол-5-он, С-[1-(1 Нтетразол-5-илметил)циклогептил]метиламин, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусную кислоту, (1 ,3 ,5 )(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусную кислоту, (3S,5R)-3-аминометил-5 метилоктановую кислоту,(3S,5R)-3-амино-5-метилгептановую кислоту,(3S,5R)-3-амино-5 метилнонановую кислоту, (3S,5R)-3-амино-5-метилоктановую кислоту, (2S,4S)-4-(3-хлорфенокси)пролин и (2S,4S)-4-(3-фторбензил)пролин или их фармацевтически приемлемые соли. Другими предпочтительными альфа-2-дельта лигандами для использования в комбинации согласно настоящему изобретению являются (3S,5R)-3-амино-5-метилоктановая кислота, (3S,5R)-3-амино-5 метилнонановая кислота, (3R,4R,5R)-3-амино-4,5-диметилгептановая кислота и (3R,4R,5R)-3-амино-4,5 диметилоктановая кислота и их фармацевтически приемлемые соли. Особенно предпочтительные альфа 2-дельта лиганды для использования в комбинации согласно настоящему изобретению выбирают из габапентина, прегабалина, (1 ,3 ,5 )(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусной кислоты, (2S,4S)4-(3-хлорфенокси)пролина и (2S,4S)-4-(3-фторбензил)пролина или их фармацевтически приемлемых солей. Соединения согласно настоящему изобретению могут вводиться в комбинации с антагонистом рецептора окситоцина. Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и один или более антагонистов окситоцина, в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Примерами антагонистов рецептора окситоцина, подходящих для настоящего изобретения, являются атосибан (Ferring AB), барусибан (Ferring AB), TT-235 (Northwestern University) и AS-602305 (SeronoSA). Содержание опубликованных заявок на патент, упомянутых выше, и в особенности общие формулы терапевтически активных соединений, заявленных в формулах изобретения, и соединений, показанных в примерах, полностью включены в настоящее описание путем ссылки. Соединения согласно настоящему изобретению могут также вводиться в комбинации с одним или более из следующих веществ:(xvii) модулятор прогестерона. Таким образом, в другом аспекте изобретение относится к фармацевтическому продукту, содержащему антагонист рецептора прогестерона и один или более из следующих веществ:(xvii) модулятор прогестерона в форме комбинированного препарата для одновременного, раздельного или последовательного использования в лечении эндометриоза. Обычно соединения по изобретению вводят в форме состава в комбинации с одним или более фармацевтически приемлемыми эксципиентами. Термин "эксципиент" используется здесь для обозначения любого ингредиента, кроме соединения(й) по изобретению. Выбор эксципиента будет в большой степени зависеть от таких факторов, как конкретный способ введения, влияние эксципиента на растворимость и стабильность и характер лекарственной формы. Фармацевтические композиции, подходящие для доставки соединений согласно настоящему изобретению, и способы их получения очевидны для специалиста. Такие композиции и способы их получения могут быть найдены, например, в "Remington's Pharmaceutical Sciences", 19th Edition (Mack PublishingCompany, 1995). Соединения по изобретению могут вводиться перорально. Пероральное введение может включать проглатывание, такое, чтобы соединение поступило в желудочно-кишечный тракт, и/или щечное, язычное или подъязычное введение, с помощью которого соединение поступает в кровоток непосредственно изо рта. Составы, подходящие для перорального введения, включают твердые, полутвердые и жидкие системы, такие как таблетки; мягкие или твердые капсулы, содержащие мульти- или наночастицы, жидкости или порошки; таблетки для рассасывания (включая заполненные жидкостью); жевательные резинки; гели; быстродиспергирующиеся лекарственные формы; пленки; суппозитории; спреи и буккальные/мукоадгезивные пластыри. Жидкие составы включают суспензии, растворы, сиропы и эликсиры. Такие составы могут использоваться как наполнители в мягких или твердых капсулах (сделанных, например, из желатина или гидроксипропилметилцеллюлозы) и обычно включать носитель, например воду, этанол, полиэтиленгликоль,пропиленгликоль, метилцеллюлозу или подходящее масло, и один или более эмульгаторов и/или суспендирующих агентов. Жидкие составы могут также быть получены восстановлением твердого вещества,например, из пакетика. Соединения по изобретению могут также использоваться в быстрорастворимых, быстрорасщепляющихся лекарственных формах, таких как описанные в Expert Opinion in Therapeutic Patents, 11 (6),981-986, Liang and Chen (2001). Для лекарственных форм в виде таблеток в зависимости от дозы лекарственное средство может составлять от 1 до 80 вес.% лекарственной формы, обычно от 5 до 60 вес.% лекарственной формы. В дополнение к лекарственному средству таблетки обычно содержат дезинтегрирующее средство. Примеры дезинтегрирующих средств включают гликолят крахмала натрия, натриевую соль карбоксиметилцеллюлозы, кальциевую соль карбоксилметилцеллюлозу, кроскармеллозу натрия, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, замещенную низшим алкилом гидроксипропилцеллюлозу, крахмал, предварительно желатинированный крахмал и альгинат натрия. Обычно дезинтегрирующее средство составляет от 1 до 25 вес.%, предпочтительно от 5 до 20 вес.% лекарственной формы. Связующие обычно используются для придания составу таблетки когезивных свойств. Подходящие связующие включают микрокристаллическую целлюлозу, желатин, сахар, полиэтиленгликоль, природные и синтетические камеди, поливинилпирролидон, предварительно желатинированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Таблетки могут также содержать разбавители, такие как лактоза (моногидрат, высушивали распылением моногидрат, безводный и т.п.), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и двухосновный дигидрат фосфата кальция. Таблетки могут также включать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и глиданты, такие как диоксид кремния и тальк. В случае их присутствия поверхностноактивные вещества могут составлять от 0,2 до 5 вес.% таблетки и глиданты могут составлять от 0,2 до 1 вес.% таблетки. Таблетки также обычно содержат лубриканты, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Лубриканты обычно составляют от 0,25 до 10 вес.%, предпочтительно от 0,5 до 3 вес.% таблетки. Другие возможные ингредиенты включают антиоксиданты, красители, ароматизаторы, консерванты и средства для маскировки вкуса. Примеры таблеток содержат до приблизительно 80% лекарственного средства, от приблизительно 10 до приблизительно 90 вес.% связующих, от приблизительно 0 до приблизительно 85 вес.% разбавителей, от приблизительно 2 до приблизительно 10 вес.% дезинтегрирующего средства и от приблизительно 0,25 до приблизительно 10 вес.% лубрикантов. Смеси для таблеток могут прессоваться непосредственно или роллером с образованием таблеток.- 12014693 Смеси или части смесей для таблеток могут альтернативно быть получены влажной, сухой грануляцией или грануляцией в расплаве, заморозкой расплава или экструзией с последующим таблетированием. Конечный состав может включать один или более слоев и может иметь или не иметь покрытие; он может также быть инкапсулирован. Состав таблеток обсуждается в "Pharmaceutical Dosage Forms: Tablets", vol. 1, by H. Lieberman andL. Lachman (Marcel Dekker, New York, 1980). Потребляемые пероральные пленки обычно представляют собой гибкие водорастворимые или набухающие в воде лекарственные формы в виде тонкой пленки, которые могут быстро растворяться или прилипать к слизистой оболочке и обычно включают соединение формулы (I), пленкообразующий полимер, связующее, растворитель, увлажнитель, пластификатор, стабилизатор или эмульгатор, модификатор вязкости и растворитель. Некоторые компоненты состава могут выполнять более одной функции. Пленкообразующий полимер может быть выбран из природных полисахаридов, белков или синтетических гидроколлоидов и обычно присутствует в диапазоне от 0,01 до 99 вес.%, более предпочтительно в диапазоне от 30 до 80 вес.%. Другие возможные ингредиенты включают антиоксиданты, красители, ароматизаторы и усилители аромата, консерванты, средства, стимулирующие слюноотделение, охлаждающие средства, сорастворители (включая масла), смягчающие средства, наполнители, пеногасители, поверхностно-активные вещества и средства для маскировки вкуса. Пленки в соответствии с изобретением обычно получают путем сушки выпариванием тонких водных пленок, нанесенных на отделяемую поддерживающую подложку или бумагу. Это может быть сделано в сушильном шкафу или туннеле, обычно в комбинированном устройстве для нанесения покрытий и сушки или путем сушки сублимацией или вакуумной сушки. Твердые составы для перорального введения могут быть составлены для немедленного и/или модифицированного высвобождения. Составы модифицированного высвобождения включают составы отсроченного, пролонгированного, импульсного, контролируемого, нацеленного и запрограммированного высвобождения. Подходящие составы модифицированного высвобождения в целях изобретения описаны в патенте США 6106864. Детали других подходящих технологий высвобождения, таких как высокоэнергетическая дисперсия и осмотические, и покрытые частицы могут быть найдены в "Pharmaceutical TechnologyOnline", 25(2), 1-14, by Verma et al. (2001). Использование жевательной резинки для обеспечения контролируемого высвобождения описано в WO 00/35298. Соединения по изобретению могут также вводиться непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие средства для парентерального введения включают средства для внутривенного, внутриартериального, интраперитонеального, внутриоболочкового, внутрижелудочкового, интрауретрального, внутригрудинного, внутричерепного, внутримышечного, внутрисиновиального и подкожного введения. Подходящие устройства для парентерального введения включают иглу (включая микроиглу) инжекторы, инжекторы без иглы и методики инфузии. Парентеральные составы представляют собой обычно водные растворы, которые могут содержать эксципиенты, такие как соли, углеводы и средства буферизации (предпочтительно с рН от 3 до 9), но для некоторых применений они могут быть более предпочтительно составлены в форме стерильного неводного раствора или в высушенной форме для использования в сочетании с подходящим носителем, таким как стерильная апирогенная вода. Парентеральные составы можно легко получить в стерильных условиях, например лиофилизацией,используя стандартные фармацевтические методики, известные специалисту. Растворимость соединений формулы (I), используемых в получении парентеральных растворов, может быть увеличена при помощи подходящих методик составления, таких как включение усиливающих растворимость агентов. Составы для парентерального введения могут быть составлены как составы немедленного и/или модифицированного высвобождения. Составы модифицированного высвобождения включают составы отсроченного, пролонгированного, импульсного, контролируемого, нацеленного и запрограммированного высвобождения. Таким образом, соединения по изобретению могут быть составлены в форме суспензии или твердого, полутвердого состава или тиксотропной жидкости для введения с формированием депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких составов включают покрытые лекарственным средством стенты и полутвердые формы и суспензии, включающие нагруженные лекарственным средством микросферы из сополимера dl-молочной и гликолевой кислот (PGLA). Соединения по изобретению могут также вводиться топически, (внутри)кожно или чрескожно через кожу или слизистые оболочки. Типичные составы с этой целью включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, перевязочные материалы, пены, пленки, кожные пластыри, облатки, имплантаты, губки, волокна, бандажи и микроэмульсии. Также могут использоваться липосомы. Типичные носители включают спирт, воду, минеральное масло, вазелиновое масло, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены усилители проникновения - см., например,J. Pharm. Sci, 88 (10), 955-958, by Finnin and Morgan (October, 1999).- 13014693 Другие средства топического введения включают доставку электропорацией, ионтофорезом, фонофорезом, сонофорезом и микроиглой или без иглы (например, Powderject, Bioject и т.д.) инъекцию. Составы для топического введения могут быть составлены как составы немедленного и/или модифицированного высвобождения. Составы модифицированного высвобождения включают составы отсроченного, пролонгированного, импульсного, контролируемого, нацеленного и запрограммированного высвобождения. Соединения по изобретению могут также вводиться интраназально или ингаляцией, обычно в форме сухого порошка (индивидуально, в виде смеси, например в сухой смеси с лактозой, или как компонент смеси в составе частиц, например в смеси с фосфолипидами, такими как фосфатидилхолин) с помощью сухого порошкового ингалятора, в форме аэрозольного спрея из герметичного контейнера, насоса, спрея, пульверизатора (предпочтительно пульверизатора с использованием электрогидродинамических средств с получением тонкого аэрозоля) или небулайзера с или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан, или как капли для носа. Для внутриносового использования порошок может включать биоадгезивное средство, например хитозан или циклодекстрин. Герметичный контейнер, насос, спрей, пульверизатор или небулайзер содержат раствор или суспензию соединения(й) по изобретению, включающие(ую), например, этанол, водный раствор этанола или подходящее альтернативное средство для диспергирования, солюбилизации или пролонгированного высвобождения активного вещества, пропеллент(ы) в качестве растворителя и дополнительное поверхностно-активное вещество, такое как сорбитан триолеат, олеиновая кислота или олигомолочная кислота. До использования в составе в форме сухого порошка или суспензии продукт лекарственного средства измельчают до размера, подходящего для доставки ингаляцией (обычно менее 5 мкм). Это может быть сделано любым подходящим способом измельчения, таким как размалывание спиральной струи,размалыванием струи кипящего слоя, обработка суперкритической жидкости с получением наночастиц,гомогенизация высокого давления или сушка распылением. Капсулы (изготовленные, например, из желатина или гидроксипропилметилцеллюлозы), пузырьки и картриджи для использования в ингаляторе или инсуффляторе могут быть составлены таким образом,чтобы содержать порошковую смесь соединения по изобретению, подходящую порошковую основу, такую как лактоза или крахмал, и модификатор характеристик, такой как l-лейцин, маннит или стеарат магния. Лактоза может быть безводной или в форме моногидрата, предпочтительно в форме моногидрата. Другие подходящие эксципиенты включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу. Подходящий состав раствора для использования в пульверизаторе с использованием электрогидродинамических средств для получения тонкого аэрозоля может содержать от 1 мкг до 20 мг соединения по изобретению на одно срабатывание, и объем на одно срабатывание может варьировать от 1 до 100 мкл. Типичный состав может включать соединение формулы (I), пропиленгликоль, стерилизованную воду,этанол и хлорид натрия. Альтернативные растворители, которые могут использоваться вместо пропиленгликоля, включают глицерин и полиэтиленгликоль. Подходящие ароматизаторы, такие как ментол и левоментол, или подслащивающие вещества, такие как сахарин или сахарин натрия, могут быть добавлены к составам по изобретению, предназначенным для ингаляционного/внутриносового введения. Составы для ингаляционного/внутриносового введения могут быть составлены как составы для немедленного и/или модифицированного высвобождения, например, с использованием PGLA. Составы модифицированного высвобождения включают составы отсроченного, пролонгированного, импульсного,контролируемого, нацеленного и запрограммированного высвобождения. Соединения по изобретению могут вводиться ректально или влагалищно, например, в форме суппозитория, пессария или клизмы. Масло какао является традиционной основой суппозитория, но могут использоваться различные подходящие альтернативы. Составы для ректального/влагалищного введения могут быть составлены как составы для немедленного и/или модифицированного высвобождения. Составы модифицированного высвобождения включают составы отсроченного, пролонгированного, импульсного, контролируемого, нацеленного и запрограммированного высвобождения. Соединения по изобретению могут быть объединены с растворимыми макромолекулярными веществами, такими как циклодекстрин и его подходящие производные, или полиэтилен, гликольсодержащие полимеры, для улучшения их растворимости, скорости растворения, маскировки вкуса, биодоступности и/или стабильности для использования в любом из указанных способов введения. Обнаружено, что комплексы лекарственное средство-циклодекстрин, например, в целом пригодны для большинства лекарственных форм и путей введения. Могут использоваться как комплексы включения, так и комплексы невключения. Как альтернатива прямому комплексообразованию с лекарственным средством, циклодекстрин может использоваться как вспомогательная добавка, то есть как носитель,разбавитель или солюбилизатор. Обычно в этих целях используют альфа-, бета- и гамма-циклодекстрины, примеры которых могут быть найдены в международных заявках на патенты WO 91/11172,- 14014693 94/02518 и 98/55148. Поскольку желательно введение комбинации активных соединений, например, для лечения конкретного заболевания или состояния, в рамках настоящего изобретения две или более фармацевтических композиций, по меньшей мере одна из которых содержит соединение в соответствии с изобретением,могут быть скомбинированы в форме набора, подходящего для совместного введения этих композиций. Таким образом, набор по изобретению включают две или более отдельных фармацевтических композиции, по меньшей мере одна из которых содержит соединение формулы (I) в соответствии с изобретением, и средства для раздельного хранения указанных композиций, такие как контейнер, разделенный баллон или разделенный пакет из фольги. Примером такого набора является обычная блистерная упаковка, используемая для хранения таблеток, капсул и т.п. Набор по изобретению является особенно подходящим для введения различных лекарственных форм, например пероральной и парентеральной, для введения отдельных композиций с разными интервалами или для титрования отдельных композиций друг относительно друга. Для обеспечения комплекса набор обычно включает указания по введению и может поставляться с так называемой инструкцией. Для введения человеку полная суточная доза соединений по изобретению находится обычно в диапазоне от 1 до 1000 мг, но, конечно, зависит от способа введения. Например, пероральное введение может потребовать полной суточной дозы от 1 до 1000 мг, в то время как внутривенное введение может потребовать только от 1 до 500 мг. Полная суточная доза может вводиться в единой дозе или в разделенных дозах и по усмотрению врача может выходить за пределы указанного здесь типичного диапазона. Эти дозы приведены в расчете на среднего человека, имеющего массу тела приблизительно от 60 до 70 кг. Врач легко определит дозы для пациентов, масса тела которых находится вне этого диапазона, таких как дети и люди пожилого возраста. В рамках изобретения термины "лечение" и "для лечения" означают облегчить симптомы, устранить причину, временно или навсегда, или предотвратить либо замедлить проявление симптомов. Термин "лечение" включает облегчение, устранение причины (временно или навсегда) или профилактику симптомов и нарушений, связанных с эндометриозом и/или лейомиомой матки. Лечение может быть профилактическим лечением, а также лечением в начале установления симптомов. Соединения согласно настоящему изобретению могут быть проверены в скринингах, изложенных ниже. 1.0. In vitro функциональный тест на антагонизм к рецептору прогестерона (PR). Тест на антагонизм к PR основан на широко известном эффекте модуляции экспрессии щелочной фосфатазы (АР) в человеческих клетках рака молочной железы T47D (Beck D.P. et al. (1993). The progesterone antagonist RU486 acquires agonist activity upon stimulation of cAMP signalling pathways., Proc. Natl.agonists: synthesis and SAR study of 6-aryl benzoxazines. BioorganicMedicinal Chemistry Letters, 12, 787790; Zhang, Z. et al. (2000) In vitro characterization of trimegestone: a new potent and selective progestin. Steroids 65, 637-643). В присутствии прогестерона эндогенная экспрессия АР индуцируется в клетках T47D и ингибируется соединениями, обладающими антагонистической активностью к PR. В отсутствие прогестерона любая активность агониста также наблюдается как индукция активности АР. При проведении теста в двух форматах (+/-прогестерон (Р 4, могут быть идентифицированы соединения, ведущие себя как антагонисты, агонисты или частичные агонисты PR.- 15014693 Материалы, необходимые для роста клеток T47D и проведения прогестерон-индуцированного теста АР, приведены в табл. 1. Таблица 1 Среды для теста (агонистический формат): DMEM без фенолового красного + 5% CS-FCS + 2 мМGlutamax. Среды для теста (антагонистический формат): DMEM без фенолового красного + 5% CS-FCS + 2 мМ Glutamax + 10 нМ Р 4. Кратко, клетки T47D выращивают размножением в DMEM + 10% FBS + 2 мМ Glutamax при 37 С/5% CO2. При слиянии на 80-90% среды обменивают на DMEM без фенолового красного + 5% CSFCS (среда для теста) и культивируют еще 24 ч при 37 С/5% СО 2. Клетки T47D высеивают в плотности 2,5104 клеток/лунка в 100 мл среды для теста в 96-луночные тестовые планшеты в 3 экземплярах для каждого условия. Например, для 5 точек кривой IC50 на одно соединение это эквивалентно 36 лункам(218 лунок, Р 4). Эти планшеты затем культивируют в течение 24 ч при 37 С/5% СО 2, оставляя наружные лунки для контроля с добавлением 200 мл PBS. 10 мМ сток-раствора соединений получают в ДМСО (хранят при -20 С в аликвотах по 10 мл). 10 мМ сток-раствора RU486 в ДМСО используют как стандартный чистый антагонист PR. К среде для теста добавляют исследуемые соединения или смесь 0,05% плуроновой кислоты в PBS10 нМ Р 4, чтобы получить конечную концентрацию 20 мкМ (то есть 10 мкл 10 мМ сток-раствора на 5 мкл среды для теста 10 нМ Р 4). Образцы тщательно перемешивают и последовательные разведения соединений от 10 мкМ до 0,1 нМ в 96-луночных планшетах получают следующим образом. Внешние лунки оставляют для контроля. К одной половине планшета (-Р 4), ряды 3-8, добавляют среду для теста (225 мкл), а к другой половине планшета добавляют среду для теста + 10 нМ Р 4. К ряду 2 добавляют 250 мкл высшей концентрации соединения (20 мкМ 10 нМ Р 4). 25 мкл 20 мМ сток-раствора удаляют из ряда 2 и добавляют к 225 мкл среды для теста 10 нМ Р 4 в ряду 3 и тщательно перемешивают. Этот процесс повторяют по всему планшету до ряда 7, получая последовательные разведения. Контрольный носитель ректифицируют таким образом, чтобы содержание в нем ДМСО составляло 0,1% (то есть 20 мкл на 10 мл среды для теста 10 нМ Р 4 с получением концентрации диметилсульфоксида 0,2%,250 мкл, в ряду 8). 100 мкл реагента из планшетов для разведения перемещают в соответствующую лунку, содержащую клетки T47D в 100 мкл среды для теста, получая конечную концентрацию от 10 мкМ до 0,1 нМ соединения (5 нМ Р 4 антагонистический формат). Клетки инкубируют в течение 20 ч при 37 С/5% СО 2,затем среды удаляют, клетки промывают PBS (200 мкл) и лизируют, помещая клетки при -80 С на 15 мин и размораживая при комнатной температуре. Лизис замораживанием-оттаиванием повторяют,затем к каждой лунке добавляют PBS (50 мкл). Через 5 мин к каждой лунке добавляют 30 мкл хемилюминесцентного раствора субстрата CSPD (конечная концентрация 0,06125 мМ, 1,25 мМ раствора 20 разведений с усилителем хемилюминесценции, Great EscAPe SEAP Chemiluminescence Detection kit) и перемешивают. Планшеты инкубируют в течение 30 мин при комнатной температуре и люминесценцию измеряют на люминометре (VICTOR, Wallac). Тест проводят трижды, в агонистическом формате (в отсутствие экзогенного Р 4), с построением сигмовидной кривой результатов, выраженных как индукция тестируемыми соединениями щелочной фосфатазы (люминесценция, произвольные единицы или процент с максимальным ответом прогестерона, взятым за 100%). В этом формате значение ЕС 50 определяют как концентрацию лекарственного средства, необходимую для получения 50%-ной индукции активности АР по сравнению с 5 нМ индивидуально. Соединения с агонизмом или частичным агонизмом, т.е. с индукцией активности АР, которая является субмаксимальной по отношению к вызванной Р 4, таким образом исключают. В антагонистическом формате (5 нМ Р 4) выстраивают кривую результатов, выраженных как ингибирование щелочной фосфатазы тестируемыми соединениями. В этом формате значение IC50 определяют как концентрацию лекарственного средства, необходимую для получения 50%-ного ингибирования активности АР по сравнению с 5 нМ индивидуально. В отношении соединений, иллюстрируемых здесь, значения IC50 составляют менее 5 мкМ. В предпочтительном варианте осуществления значение IC50 составляет менее 500 нМ. В более предпочтительном варианте осуществления IC50 составляет менее 50 нМ. 2.0. In vitro функциональный тест глюкокортикоидной активности (GR). В этом тесте линию клеток SW1353, стабильно трансфицированную полноразмерной конструкциейGR, и репортерный ген люциферазы (Luc) мышиного вируса опухоли молочной железы (MMTV) используют для проведения in vitro функционального теста глюкокортикоидной активности. Материалы, необходимые для роста клеток SW1353-MMTV-GR-Luc и проведения теста, указаны ниже или представлены в табл. 1. Клетки SW1353-MMTV-GR-Luc, выращенные в DMEM, содержащий 10% FBS, 2 мМ glutamax иG418 (0,5 мг/мл, Gibco номер по каталогу 10131-027), высеивают в плотности 0,5104 клеток/лунка(384-луночные планшеты для культуры тканей с черными стенками и прозрачным дном (Greiner номер по каталогу 781091 в 30 мл, используя микропипетку Multidrop и инкубируют при 37 С, 5% СО 2 в течение ночи. Среды заменяют средами для теста (30 мкл; DMEM-феноловый красный с содержанием 1 мг/л инсулина, 2 г/л гидролизата лактальбумина и аскорбата (0,5 мг/л), добавленных непосредственно перед использованием) по меньшей мере за 4 ч до введения. Тест проводят в двух форматах, антагонистическом формате, в котором тестируемые соединения оценивают в отношении их способности блокировать эффект 20 нМ дексаметазона на люциферазную активность, и агонистическом формате. Отдельный 384-луночный планшет используют для оценки соединений в обоих форматах. Робот Genesis используют для разбавления и извлечения 1/2 log единицы (11 точек) доза-ответ (начиная с конечного 1 мкМ; 16 соединений/384-луночный планшет) из 96-луночного планшета, содержащего 4 мМ стоковых концентраций тестируемых соединений. Исследуемые соединения разбавляют в- 17014693 среде для теста + 3,75% ДМСО или смеси 0,05% плуроновой кислоты в PBS. Дексаметазон и RU-486(конечная концентрация 1 мкМ) в качестве положительного контроля получают из концентрированных сток-растворов. MATRIX Platemate используют, чтобы перенести 10 мкл разбавленных соединений на планшеты и либо 10 мкл среды, либо стандарты, так, чтобы конечный объем пробы составлял 50 мкл. Клетки и соединения инкубируют при 37 С, 5% СО 2 в течение ночи. Реагент Steady-Glo LuciLite(Promega cat no. E2520) ресуспендируют и добавляют 30 мкл на лунку, оставляют в темноте в течение 5 мин и затем планшет считывают на счетчике люминесценции Wallac. Все пункты данных измеряют дважды. В агонистическом формате выстраивают сигмовидную кривую результатов, выраженных как индукция тестируемыми соединениями люциферазы (% максимального ответа дексаметазона), и определяют значение EC50. В антагонистическом формате результаты выражают как ингибирование люциферазы тестируемыми соединениями и определяют значение IC50. 3.0. In vivo исследование в отношении антагонизма рецептора прогестерона с использованием теста Макфэйла. Классическим количественным исследованием прогестогенной активности является тест Макфэйла,проводимый на незрелом кролике (McPhail, 1934). Все соединения согласно формуле (I) могут быть получены обычными путями, такими как процедуры, описанные в общих способах, представленных ниже, или специфическими способами, описанными в разделе примеров, или подобными им способами. Настоящее изобретение также охватывает один или более этих способов получения соединений формулы (I) в дополнение к любым используемым в них новым промежуточным соединениям. В следующих общих способах R1-R8, X, Y и а имеют значения, определенные для соединения формулы (I), если не указано иное. В схеме 1 ниже соединения формулы (I) могут быть получены окислением соединения формулы(II). Окисление происходит через сульфоксид (Ia) (а=1) с получением сульфона (Ib) (а=2) Множество подходящих оксидантов и условий окисления доступны в научной литературе для преобразования сульфидов в сульфоксиды и сульфоны. В частности, предпочтительным способом является реакция с Oxone в смеси растворителей, такой как смесь метанола и воды, при температурах от температуры окружающей среды до температуры кипения. В схеме 2 ниже сульфиды формулы (II) могут быть получены алкилированием пиразолов формулы(III) с помощью соединений формулы (IV) (где L1 обозначает удаляемую группу, такую как хлор), и подходящего основания, такого как трет-бутоксид калия, в подходящем растворителе, таком как диметоксиэтан, при температурах от температуры окружающей среды до температуры кипения. Альтернативно, сульфиды формулы (II) могут быть получены из соединений формулы (VI), где L2 обозначает удаляемую группу, такую как хлор, реакцией с тиолатной солью формулы R2SM+, где М+ обозначает катион, такой как Na+, K+ или Cu+, в подходящем растворителе, таком как 1,4-диоксан, диметилформамид или тетрагидрофуран. Соединения формулы (IV) могут быть получены активацией гидроксила соединений формулы (V) с образованием удаляемой группы L2. Предпочтительно, когда L2 обозначает хлор, это может быть осуществлено с использованием тионилхлорида в дихлорметане. Соединения формулы (V) могут быть получены реакцией соединений формулы (III) с источником формальдегида, таким как водный формальдегид. Когда в соединениях формулы (III) R1R3, тогда получаемые соединения формулы (II), (V) и (VI) могут существовать как региоизомеры с транспонированными R1 и R3, которые могут быть разделены. В схеме 3 ниже соединения формулы (III), в которых Y обозначает О, могут быть получены конденсацией соединения формулы (VII) с гидразином или его солью или гидратом, в случае необходимости в присутствии кислоты или основания. Основание предпочтительно является основанием третичного амина, такого как триэтиламин. Кислота предпочтительно представляет собой уксусную кислоту. В типичной процедуре раствор соединения формулы (VII) в подходящем растворителе, таком как этанол, обрабатывают гидразином или его солью или гидратом, и, если используется, подходящей кислотой или основанием, при температуре от комнатной температуры до температуры кипения растворителя. В предпочтительной процедуре реакционную смесь нагревают с обратным холодильником в этаноле и уксусной кислоте. Соединения формулы (VII) могут быть получены реакцией соединений формулы (VIII) с соединениями формулы (X) и подходящим основанием, таким как карбонат цезия, в растворителе, таком как ацетон. Соединения формулы (VIII) являются коммерчески доступными или могут быть получены реакцией соединения формулы (IX) с хлорирующим реактивом. В типичной процедуре охлажденный раствор соединения формулы (IV) в подходящем растворителе, таком как ацетонитрил, обрабатывают сначала тетрабутиламмонийбромидом и хлортриметилсиланом и затем сухим диметилсульфоксидом. Функциональные эквиваленты соединений формулы (VII) могут также использоваться в этой реакции. Они включают соединения формулы (XI) или (XII), в которых L3 является подходящей удаляемой группой; предпочтительно -N(C1-C6-алкил)2, более предпочтительно -N(CH3)2. Таким образом, соединение формулы (III) может быть получено конденсацией соединения формулы (XI) или (XII) с гидразином или его солью или гидратом, в случае необходимости в присутствии кислоты или основания (причем основание предпочтительно является основанием третичного амина, таким как триэтиламин, а кислота предпочтительно является уксусной кислотой). В типичной процедуре раствор соединения формулы (XI) или (XII) в подходящем растворителе (таком как этанол) обрабатывают гидразином или его солью или гидратом и, если используется, подходящей кислотой или основанием,при температуре от комнатной температуры до температуры кипения растворителя. В предпочтительной процедуре реакционную смесь нагревают с обратным холодильником. Соединения формулы (XI) или(XII) являются особенно подходящими для синтеза соединений формулы (I), в которой R1 или R3, соответственно, обозначают Н. Соединения формулы (XI), в которой R1 обозначает Н, и соединения формулы (XII), в которой R3 обозначает Н, a L3 обозначает диметиламино, могут быть получены реакцией соединения формулы(XIII), приведенной ниже, с диметилформамиддиметилацеталем при повышенной температуре, предпочтительно при приблизительно 100 С. Соединения формулы (XIII) являются коммерчески доступными или могут быть получены реакцией соединения формулы (XIV) с фенолом формулы (X): В типичной процедуре раствор соединения формулы (XIV) в подходящем растворителе, таком как ацетон, обрабатывают подходящим основанием, таким как карбонат цезия, и соединением формулы (X). В предпочтительной процедуре реакционную смесь нагревают, например, с обратным холодильником. В случае необходимости может быть добавлен нуклеофильный катализатор, такой как иодид натрия или тетрабутиламмонийиодид. В схеме 4 ниже соединения формулы (III), в которой Y обозначает СН 2, могут быть получены способом, подобным описанному в схеме 3, конденсацией соединения формулы (XV) с гидразином. Соединения формулы (XV) могут быть получены алкилированием соединений формулы (IX) с использованием соединений формулы (XVI), где L4 обозначает удаляемую группу, такую как хлор, бром или иод, с подходящим основанием, таким как гидрид натрия или трет-бутоксид калия, в подходящем растворителе,таком как тетрагидрофуран или 2-бутанон, при температурах от комнатной температуры до температуры кипения растворителя. Специалисту понятно, что во многих случаях соединения формулы (I) могут быть преобразованы в другие соединения формулы (I) трансформациями функциональной группы.- 20014693 Таким образом, согласно следующему аспекту изобретение относится к способу получения соединений формулы (I), в которой а=1, который включает окисление соединения формулы (II): в которой R1-R8, X и Y имеют определенные выше значения. Кроме того, изобретение относится к способу получения соединений формулы (I), в которой а=2,который включает окисление соединения формулы (II) в которой R1-R8, X и Y имеют определенные выше значения. Кроме того, изобретение относится к способу получения соединений формулы (I), в которой а=2,который включает окисление соединения формулы (Ia) в которой R1-R8, X и Y имеют определенные выше значения. Также в рамки изобретения входят промежуточные соединения формулы (II), как определено выше,все их соли, сольваты и комплексы и все сольваты и комплексы их солей, как определено выше для соединений формулы (I). Изобретение включает все полиморфы указанных соединений и их кристаллические формы. При получении соединений формулы (I) в соответствии с изобретением специалист может с помощью рутинных методов выбрать форму соединения формулы (II), которая обеспечит лучшую комбинацию соответствующих характеристик. Такие характеристики включают температуру плавления, растворимость, удобство использования и выход промежуточной формы и в результате легкость, с которой продукт может быть очищен после выделения. Соединения по изобретению могут иметь преимущество в том отношении, что они являются более мощными, имеют более долгую продолжительность действия, имеют более широкий диапазон активности, являются более стабильными, имеют меньше побочных эффектов или более селективны, или имеют другие более полезные свойства, чем соединения предшествующего уровня техники. Таким образом, изобретение относится:(i) к соединению формулы (I) или к его фармацевтически приемлемому производному;(ii) к способу получения соединения формулы (I) или его фармацевтически приемлемого производного;(iii) к фармацевтическому составу, включающему соединение формулы (I) или его фармацевтически приемлемое производное вместе с фармацевтически приемлемыми эксципиентами, разбавителем или носителем;(iv) к соединению формулы (I) или к его фармацевтически приемлемому производному или композиции для применения в качестве лекарственного средства;(v) к применению соединения формулы (I) или его фармацевтически приемлемого производного или композиции для получения лекарственного средства для лечения эндометриоза, фибромы матки(лейомиоматозные образования), меноррагии, аденомиоза, первичной и вторичной дисменореи (включая симптомы диспареунии, затрудненной дефекации и хронической тазовой боли), синдрома хронической тазовый боли;(vi) к применению как в (v), где заболевание или нарушение представляет собой эндометриоз и/или фибромы матки (лейомиоматозные образования);(vii) к способу лечения млекопитающего для лечения эндометриоза, фибромы матки (лейомиоматозные образования), меноррагии, аденомиоза, первичной и вторичной дисменореи (включая симптомы диспареуния, затрудненной дефекации и хронической тазовой боли), синдрома хронической тазовой боли, включающему лечение указанного млекопитающего эффективным количеством соединения формулы(I) или его фармацевтически приемлемого производного либо композиции;(viii) к способу как в (vii), где заболевание или нарушение представляет собой эндометриоз и/или фибромы матки (лейомиоматозные образования);(ix) к промежуточным соединениям формул (II). Следующие примеры получения и примеры иллюстрируют получение соединений формулы (I). Спектры 1 Н Ядерного магнитного резонанса (ЯМР) были во всех случаях совместимы с предложенными структурами. Характерные химические сдвигиприведены в частях на миллион в сторону слабого поля от тетраметилсилана с использованием обычных сокращений для обозначения главных пиков: например s, синглет; d, дуплет; t, триплет; q, квартет; m, мультиплет; br, уширенный. Везде использовались следующие сокращения:CD3OD - дейтерометанол Примеры получения и примеры, которые следуют далее, иллюстрируют изобретение, но никоим образом не ограничивают изобретение. Все исходные материалы доступны коммерчески или описаны в литературе. Все температуры приведены в С. Флэш-хроматографию на колонке проводили, используяMerck silica gel 60 (9385). Тонкослойную хроматографию (TLC) проводили на планшетах с Merck silicagel 60 (5729). "R1" обозначает дистанцию, пройденную соединением, разделенную на дистанцию, пройденную растворителем на планшете TLC. Температуру плавления определяли, используя приборGallenkamp MPD350, и не корректировали. ЯМР осуществляли, используя ЯМР-спектрометр VarianUnity Inova 400 МГц или ЯМР-спектрометр Varian Mercury 400 МГц. Масс-спектроскопию проводили,используя отдельный четырехполюсный масс-спектрометр с электрораспылением Finnigan Navigator или масс-спектрометр Finnigan aQa APCI. Если указано, что соединения были получены, как описано в более раннем примере получения или примере, специалисту понятно, что время реакции, количество эквивалентов реагентов и температура реакции могут быть изменены для каждой конкретной реакции, и что может, однако, быть необходимым или желательным использовать другие условия обработки или очистки. Получение 1. 1,3-Дициклопропилпропан-1,3-дион. Метилциклопропанкарбоксилат (20,2 мл, 286,3 ммоль) был добавлен к перемешиваемому раствору 1-циклопропилэтанона (9 мл, 152,4 ммоль) в диметилсульфоксиде (25 мл). Добавляли порошок метилата натрия (10,8 г, 200 ммоль) и реакционную смесь перемешивали при 55 С в течение 8 ч. Реакционную смесь охлаждали, разбавляли толуолом, нейтрализовывали 6 М соляной кислотой и затем экстрагировали толуолом. Объединенные экстракты промывали карбонатом натрия, высушивали над сульфатом магния и упаривали в вакууме, получая целевое соединение (14,9 г, 78%) как смесь 2:1 енольной:кетонной формы. 1 Н ЯМР (CDCl3, 400 МГц): =0,79-0,87 (м, 4 Н), 0,98-1,01 (м, 4 Н), 1,46-1,51 (м, 2 Н-енол), 1,93-1,97 Гидроксид натрия (37,9 г, 947 ммоль) растворяли в воде (770 мл) и при комнатной температуре за 20 мин добавляли к раствору метилового эфира 3-оксобутановой кислоты (100 г, 861 ммоль). Реакционную смесь перемешивали в течение 18 ч, после чего гасили сульфатом аммония (700 г) и медленно подкисляли раствором концентрированной соляной кислоты (21,5 мл) в воде (250 мл) при охлаждении на льду. Реакционную смесь экстрагировали простым диэтиловым эфиром (6200 мл) и объединенные органические экстракты высушивали над сульфатом магния и концентрировали при пониженном давлении,получая целевое соединение (58,2 г, 60%) в форме светло-желтого масла, которое было смесью таутомеров кетон:енол. 1 Н ЯМР (400 МГц, CDCl3): =2,00 (с, 3 Н-енол), 2,30 (с, 3 Н-кетон), 3,51 (с, 2 Н-кетон), 5,02 (с,1 Н-енол). Получение 3. 1-Циклопропил-1,3-бутандион. Магниевую стружку (3,04 г, 125 ммоль), суспендированную в метаноле (145 мл), нагревали с об- 22014693 ратным холодильником в атмосфере азота в течение 1 ч, затем охлаждали до комнатной температуры и добавляли по каплям при охлаждении на льду -кето-кислоту из получения 2 (25,5 г, 250 ммоль), растворенную в метаноле (25 мл). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре и затем растворитель удаляли при пониженном давлении, получая магниевую соль кислоты. Тем временем циклопропанкарбоновую кислоту (9,91 мл, 125 ммоль) растворяли в диметилформамиде (200 мл). Затем в атмосфере азота при 0 С порциями добавляли карбонилдиимидазол (22,4 г, 138 ммоль). Эту реакционную смесь перемешивали в течение 1,5 ч и затем при 0 С добавляли описанную выше магниевую соль в форме раствора в N,N-диметилформамиде (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 92 ч и затем вливали в 2 М водный раствор соляной кислоты (85 мл), после чего разбавляли водой (170 мл). Смесь экстрагировали простым диэтиловым эфиром (6200 мл) и объединенные органические экстракты промывали солевым раствором (3200 мл), высушивали над сульфатом магния и концентрировали при пониженном давлении. Остаточное оранжевое масло очищали флэшхроматографией на силикагеле с элюированием смесью пентан:простой диэтиловый эфир (100:0, затем 90:10, затем 80:20, по объему), получая целевое соединение (7,39 г, 24%) в форме желтого масла. 1 Н ЯМР (400 МГц, CDCl3): =0,83-0,95 (м, 2 Н), 1,06-1,10 (м, 2 Н), 1,54-1,63 (м, 1 Н), 2,00 (с, 3 Н); Хлортриметилсилан (36 мл, 296 ммоль) добавляли по каплям к перемешиваемому раствору тетрабутиламмонийбромида (1,54 г, 5 ммоль) в сухом ацетонитриле (100 мл) при комнатной температуре в атмосфере азота. Полученный раствор охлаждали на льду и добавляли по каплям дикетон из получения 1(15 г, 98,7 ммоль) в форме раствора в ацетонитриле (30 мл), затем сухой диметилсульфоксид (20 мл,296 ммоль). Реакционной смеси давали медленно нагреться до комнатной температуры и перемешивали в течение 18 ч. Смесь разбавляли водой, перемешивали в течение 10 мин и затем экстрагировали простым диэтиловым эфиром (50 мл). Слои разделяли и водный слой снова экстрагировали простым диэтиловым эфиром. Органические слои объединяли, высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонках с силикагелем с элюированием смесью пентан:простой диэтиловый эфир (20:1, по объему), получая целевое соединение как смесь 2:7 таутомеров кетон:енол (12,1 г, 66%). 1 Н ЯМР (400 МГц, CDCl3): =1,01-1,07 (м, 4 Н), 1,16-1,21 (м, 4 Н), 2,23-2,28 (м, 2 Н-кетон), 2,39-2,44 получали способом, подобным описанному в получении 4, используя подходящий дикетон как исходный материал. Стадия 1. Смесь хлордикетона из получения 4 (48,2 г, 258 ммоль), 4-цианофенола (37 г, 310 ммоль), карбоната цезия (101 г, 310 ммоль) и ацетона (1200 мл) нагревали с обратным холодильником в течение 4 ч. Растворитель выпаривали при пониженном давлении и остаток разделяли между простым диэтиловым эфиром (1000 мл) и водой (300 мл). Слои разделяли и органический слой промывали водой (2500 мл). Органический слой высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на колонках с силикагелем с элюированием смесью пентан:дихлорметан в отношении 1:1 до начала элюирования продукта и затем в отношении 1:2, чтобы закончить элюирование. Растворитель выпаривали, получая промежуточный 4-(1-циклопропанкарбонил 2-циклопропил-2-оксоэтокси)бензонитрил в форме твердого вещества (44,1 г) (примечание: это промежуточное соединение может быть очищено хроматографией на силикагеле с элюированием смесью этилацетат:пентан или использовано на стадии 2 в форме сырого дикетона). Стадия 2. 4-(1-Циклопропанкарбонил-2-циклопропил-2-оксоэтокси)бензонитрил (44 г, 162 ммоль) растворяли в уксусной кислоте (500 мл). При комнатной температуре добавляли раствор гидрата гидразина (8,7 мл,179 ммоль) в этаноле (50 мл) и реакционную смесь нагревали при 90 С в течение приблизительно 3 ч. Растворители выпаривали при пониженном давлении, остатки разделяли между простым диэтиловым эфиром (600 мл) и разбавленным водным раствором гидроксида аммония (25 мл концентрированного гидроксида аммония в 500 мл воды) и слои разделяли. Водный слой экстрагировали простым диэтиловым эфиром (3200 мл) и объединенные органические слои промывали водой (250 мл), высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Твердое вещество суспендировали в простом диизопропиловом эфире (50 мл), фильтровали и промывали простым диизопропиловым эфиром (230 мл) и пентаном (2100 мл), получая целевое соединение (39,5 г, 58%) в форме твердого бесцветного вещества. 1 Н ЯМР (400 МГц, CDCl3): =0,76-0,81 (м, 8 Н), 1,59-1,65 (м, 2 Н), 7,01 (д, 2 Н), 7,60 (д, 2 Н); LRMS: Соединения общей формулы, приведенной выше, получали с использованием способа, подобного описанному в получении 9, используя в качестве исходных материалов хлордикетон из получения 4 и подходящий фенол (Ar1OH, доступного коммерчески или из получений 42 или 45). Соединения общей формулы, приведенной выше, получали способом, подобным описанному в получении 9, с использованием в качестве исходных материалов хлордикетона из получения 5 и подходящего фенола (Ar1OH, доступного коммерчески или из приготовлений 42). Соединения общей формулы, приведенной выше, были получены способом, подобным описанному в получении 9, с использованием в качестве исходных материалов коммерческого 3-хлор-2,4 пентандиона и подходящего фенола (Ar1OH). Соединения общей формулы, приведенной выше, были получены способом, подобным описанному в получении 9, с использованием в качестве исходных материалов хлордикетона из получения 6 и подходящего фенола (Ar1OH). Соединения общей формулы, приведенной выше, были получены способом, подобным описанному для получения 9, с использованием в качестве исходных материалов 4-цианофенола и подходящих хлордикетонов, описанных в получениях 7 и 8. Гидрид натрия (60%-ная дисперсия в масле, 3,43 г, 86 ммоль) добавляли в атмосфере азота к раствору 3,5-гептандиона (10 г, 78 ммоль) в 2-бутаноне (200 мл). Небольшой экзотермический эффект наблюдался в течение выделения газа. Затем добавляли иодид натрия (11,7 г, 78 ммоль), после чего 4-цианобензилбромид (15,29 г, 78 ммоль) в 2-бутаноне (20 мл), и образовывался осадок. Реакционную смесь нагревали с обратным холодильником в течение 16 ч. Затем смесь фильтровали, чтобы удалить осадок, и концентрировали при пониженном давлении. Остаток перемешивали в дихлорметане (100 мл) и твердый бромид натрия удаляли фильтрацией. Фильтрат дихлорметана промывали водой (100 мл) и солевым раствором (100 мл). Органический слой собирали, высушивали над сульфатом магния и концентрировали при пониженном давлении, получая твердое вещество оранжевого цвета. Его суспендировали в простом диэтиловом эфире и отфильтровывали, получая целевое соединение(11,01 г, 58%) в форме твердого вещества светло-желтого цвета. 1 Н ЯМР (400 МГц, CDCl3): =0,98 (т, 6 Н), 2,29-2,35 (м, 2 Н), 2,42-2,52 (м, 2 Н), 3,19 (д, 2 Н), 3,97 (т,- 27014693 1 Н), 7,25 (д, 2 Н), 7,56 (д, 2 Н); LRMS: APCI-: m/z 242 [М-Н]. Получение 33. 4-[(3,5-Диэтил-1 Н-пиразол-4-ил)метил]бензонитрил Целевое соединение (4,9 г, 45%) был получено способом, подобным описанному в стадии 2 получения 9, с использованием в качестве исходных материалов дикетона из получения 32 и гидразина. 1 Н ЯМР (400 МГц, CDCl3): =1,16 (т, 6 Н), 2,51 (кв, 4 Н), 3,82 (с, 2 Н), 7,20 (д, 2 Н), 7,55 (д, 2 Н); Бром (12,84 мл, 250 ммоль) добавляли по каплям за 10 мин в атмосфере азота к охлажденному на льду раствору циклопропилметилкетона (21 г, 250 ммоль) в метаноле (150 мл). Реакции давали продолжить протекать при внутренней температуре, поддерживаемой ниже 10 С, до обесцвечивания. Реакционную смесь перемешивали при комнатной температуре в течение еще 30 мин. Добавляли воду (75 мл) и реакционную смесь перемешивали в течение еще 15 мин. Смесь разбавляли водой (225 мл) и экстрагировали 4 раза простым диэтиловым эфиром (4250 мл). Органические слои объединяли, промывали 10%-ным водным раствором бикарбоната натрия (250 мл), затем водой (250 мл), затем солевым раствором (250 мл), затем высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая 2-бром-1-циклопропилэтанон. Карбонат цезия (30,7 г, 111,16 ммоль) добавляли к раствору 4-гидрокси-2,6-диметилбензонитрила(15,27 г, 101,89 ммоль) в ацетоне (377 мл). Затем к суспензии за 5 мин добавляли по каплям 2-бром-1 циклопропилэтанон (15,1 г, 62,6 ммоль) в ацетоне (100 мл) и реакционную смесь нагревали с обратным холодильником в течение 1,5 ч. Реакционную смесь концентрировали при пониженном давлении и остаток разделяли между насыщенным водным раствором карбоната калия (300 мл) и дихлорметаном(300 мл). Органический слой отделяли и промывали солевым раствором (250 мл), высушивали над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан:пентан (от 50:50 до 80:20, по объему), получая целевое соединение (13,5 г, 64%) в форме твердого вещества. 1 Н ЯМР (400 МГц, CDCl3): =0,97-1,01 (м, 2 Н), 1,12-1,15 (м, 2 Н), 2,19 (м, 1 Н), 2,47 (с, 6 Н), 4,71 (с,2 Н), 6,61 (с, 2 Н); LRMS: APCI+: 230 [МН+] Получение 35. 4-[(E/Z)-1-(Циклопропилкарбонил)-2-(диметиламино)винил]окси-2,6-диметилбензонитрил. Бензонитрил из получения 34 (11,8 г, 51,46 ммоль) и N,N-диметилформамиддиметилацеталь(13,7 мл, 102,93 ммоль) нагревали при 105 С в течение 12 ч. Реакционную смесь концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на силикагеле с элюированием смесью дихлорметан:пентан (50:50, затем 80:20, затем 100:0, по объему), получая целевое соединение(11,19 г, 76%) в форме твердого вещества белого цвета. 1 Н ЯМР (400 МГц, CDCl3): =0,63 (ушир.с, 2 Н), 0,91 (ушир.с, 2 Н), 1,93 (м, 1 Н), 2,44 (с, 6 Н), 2,96 (с,6 Н), 6,69 (с, 2 Н); LRMS: APCI+: 285 [МН+]. Получение 36. 4-[(3-Циклопропил-1 Н-пиразол-4-ил)окси]-2,6-диметилбензонитрил. Бензонитрил из получения 35 (11,19 г, 39,3 ммоль) растворяли в уксусной кислоте (62 мл). Добавляли гидрат гидразина (2,11 мл, 43,6 ммоль) и смесь перемешивали при комнатной температуре в течение 12 ч в атмосфере азота. Реакционную смесь концентрировали при пониженном давлении и остаток разделяли между водой (150 мл) и простым диэтиловым эфиром (200 мл). Органический слой высушива- 28014693 ли над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая целевое соединение (9,71 г, 98%) в форме твердого вещества. 1 п-Толуолсульфокислоту (20 мг, 0,12 ммоль) добавляли к раствору бензонитрила из получения 36(1 г, 3,94 ммоль) в тетрагидрофуране (30 мл). По каплям при комнатной температуре добавляли 3,4-дигидро-2 Н-пиран (664 мг, 7,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 15 ч. Затем ее упаривали при пониженном давлении и остаток разделяли между этилацетатом (100 мл) и водным раствором бикарбоната натрия (100 мл). Органический слой промывали солевым раствором (50 мл), высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая целевое соединение (1,33 г, 100%). 1(с, 6 Н), 3,51 (м, 1 Н), 3,68 (м, 1 Н), 3,88 (м, 1 Н), 4,09 (м, 1 Н), 6,70 (с, 2 Н), 7,40 (с, 1 Н), с примесью некоторого количества 3,4-дигидро-2 Н-пирана; LRMS: APCI+: m/z 338 [МН+] и m/z 254 [М-ТНР]. Получения 38 и 39. 4-[(5-Хлор-3-циклопропил-1-тетрагидро-2 Н-пиран-2-ил-1 Н-пиразол-4-ил)окси]2,6-диметаилбензонитрил и 4-[(5-хлор-3-циклопропил-1 Н-пиразол-4-ил)окси]-2,6-диметилбензонитрил.N-Хлорсукцинимид (764 мг, 5,71 ммоль) добавляли к раствору пиразола из получения 37 (1,33 г,3,9 ммоль) в N,N-диметилформамиде (30 мл). Реакционную смесь нагревали при 50 С в течение 15 ч. Затем ее упаривали при пониженном давлении и остаток разделяли между дихлорметаном (50 мл) и водой (50 мл). Органические слои высушивали над сульфатом натрия, фильтровали и затем концентрировали при пониженном давлении. Сырой продукт очищали флэш-хроматографией на силикагеле с элюированием смесью этилацетат:пентан (градиент от 2:98 до 30:70, по объему), получая соединение из получения 38 (388 мг, 18%), элюируемое первым. 1[(М-ТНР)Н+]. В ходе дальнейшего элюирования получали соединение из получения 39 (325 мг, 20%). 1 Н ЯМР (400 МГц, CDCl3): =0,80-0,82 (м, 2 Н), 0,86-0,94 (м, 2 Н), 1,74 (м, 1 Н), 2,49 (с, 6 Н), 6,65 (с,2 Н); LRMS: APCI+: m/z 288 [МН+]; APCI-: m/z 286 [М-Н]. Получение 40. 4-(3,5-Дициклопропил-1-гидроксиметил-1 Н-пиразол-4-илокси)бензонитрил. Смесь пиразола из получения 9 (2,9 г, 10,9 ммоль) и водного раствора формальдегида (37 вес.% раствор в воде, 40 мл) нагревали при 60 С в течение 4 ч. Смесь охлаждали, разделяли между этилацетатом(100 мл) и насыщенным водным раствором хлорида аммония (50 мл) и слои разделяли. Органический слой промывали насыщенным водным раствором хлоридом аммония (330 мл) и затем солевым раствором (30 мл), высушивали над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая сырое целевое соединение в форме полутвердого вещества (4,6 г, 100% вследствие наличия примесей). 1 Н ЯМР (400 МГц, CDCl3): =0,78-0,82 (м, 8 Н), 1,6 (м, 2 Н), 5,55 (с, 2 Н), 7,0 (д, 2 Н), 7,6 (д, 2 Н);
МПК / Метки
МПК: C07D 401/12, A61P 15/00, A61K 31/415, C07D 231/18
Метки: активные, соединения, терапевтически
Код ссылки
<a href="https://eas.patents.su/30-14693-terapevticheski-aktivnye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Терапевтически активные соединения</a>
Соединения, активные в ингибировании пролилолигопептидазы

Номер патента: 10022
Опубликовано: 30.06.2008
Авторы: Кристиаанс Йоханнес, Гюнтер Юкка, Вепсяляйнен Йоуко, Веняляйнен Яркко, Ярхо Элина, Сааринен Тайя, Ярвинен Томи, Мяннисте Пекка, Посо Антти, Форсберг Маркус, Валлен Эрик
МПК: A61K 31/4025, A61K 31/40, A61K 31/401...
Метки: пролилолигопептидазы, соединения, ингибировании, активные
Формула / Реферат:
1. Соединение формулы (I) причем в формуле X представляет собой С; пунктирная линия представляет собой двойную связь; R1 представляет собой прямую или разветвленную алкильную цепь, содержащую 1-5 атомов углерода, незамещенную или содержащую 1-2 заместителя, каждый из которых независимо представляет собой гидрокси, галоген, низший алкокси, арил, арилокси, арил(низший)алкокси, амино, низший алкиламино, ариламино, арил (низший)алкиламино,...
Фармацевтические композиции, содержащие активные соединения витамина d

Номер патента: 10437
Опубликовано: 29.08.2008
Авторы: Сворбрик Джеймс, Лейдлоу Барбара Ф.
МПК: A61K 45/00, A61K 9/107, A61K 31/59...
Метки: композиции, витамина, содержащие, активные, соединения, фармацевтические
Формула / Реферат:
1. Жидкий или полутвердый фармацевтический состав, содержащий соединение активного витамина D, примерно 50% компонента липофильной фазы, примерно 50% поверхностно-активного вещества, примерно от 0,05 до примерно 0,35% бутилированного гидроксианизола (БГА) и примерно от 0,05 до примерно 0,35% бутилированного гидрокситолуола (БГТ). 2. Фармацевтический состав по п.1, отличающийся тем, что указанное соединение активного витамина D представляет собой...
Новый промежуточный продукт для получения терапевтически активных имидазопиридинов

Номер патента: 8746
Опубликовано: 31.08.2007
Авторы: Мюллер Бернд, Циммерманн Петер, Коль Бернхард, Шойфлер Кристиан
МПК: C07D 471/14
Метки: терапевтически, активных, промежуточный, имидазопиридинов, продукт, получения, новый
Формула / Реферат:
1. Молекулярная соль соединения формулы (1) с 1,5-нафталиндисульфоновой кислотой 2. Молекулярная соль соединения формулы (1) с 1,5-нафталиндисульфоновой кислотой по п.1, в которой молярное отношение содержаний 1,5-нафталиндисульфоновой кислоты и соединения формулы (1) составляет от 1:1 до 2:1. 3. Молекулярная соль соединения формулы (1) с 1,5-нафталиндисульфоновой кислотой по п.1, в которой молярное отношение содержаний...
Производные тропенола – исходный материал для получения терапевтически эффективных соединений

Номер патента: 9808
Опубликовано: 28.04.2008
Авторы: Зоботта Райнер, Рапп Армин Вальтер
МПК: C07D 451/10, C07D 491/18
Метки: производные, получения, тропенола, терапевтически, материал, эффективных, соединений, исходный
Формула / Реферат:
1. Применение соединения формулы (II) в которой R представляет собой остаток, выбранный из группы, включающей С1-С4алкил и С1-С4алкиленфенил, каждый из которых может быть замещен гидроксигруппой или С1-С4алкоксигруппой, необязательно в виде его кислотно-аддитивных солей, а также необязательно в виде его гидратов, в качестве исходного материала для получения производных тропенола, обладающих фармакологическими свойствами. 2. Применение по п.1,...
Способ получения твердого терапевтически активного лекарственного средства, содежащего лекарство в малых дозах, и лекарственное средство, полученное этим способом

Номер патента: 740
Опубликовано: 28.02.2000
Авторы: Кумар Раджиндер, Манек Султан Джеймс, Мортимер Нил, Нэппер Джеймс Альберт, Кларк Майкл Сидней Джордж, Лоудон Джулия Мэри, О"браен Карен Триона
МПК: A61K 9/14
Метки: содежащего, лекарственное, твердого, терапевтически, лекарство, способом, малых, средство, способ, этим, полученное, лекарственного, активного, дозах, получения, средства
Формула / Реферат:
1. Способ приготовления твердого терапевтически активного лекарственного средства, содержащего лекарство в малых дозах, который включает смешивание частиц носителя с водным раствором лекарственного вещества, взятым в количестве 1-3% от общего веса смеси. 2. Способ по п.1, при котором носитель является растворимым или нерастворимым, поддающимся прямому прессованию, фармацевтически приемлемым наполнителем. 3. Способ по п.2, при котором носитель...
Предыдущий патент: Способ индуцирования устойчивости к вредным грибам
Следующий патент: Производные карбазола в качестве функциональных 5-ht6 лигандов
Случайный патент: Способ получения обогащенной igg-композиции из плазмы