Производные тропенола – исходный материал для получения терапевтически эффективных соединений







Формула / Реферат
1. Применение соединения формулы (II)

в которой R представляет собой остаток, выбранный из группы, включающей С1-С4алкил и
С1-С4алкиленфенил, каждый из которых может быть замещен гидроксигруппой или
С1-С4алкоксигруппой, необязательно в виде его кислотно-аддитивных солей, а также необязательно в виде его гидратов, в качестве исходного материала для получения производных тропенола, обладающих фармакологическими свойствами.
2. Применение по п.1, где производные тропенола выбирают из группы, включающей тиотропийбромид, ипратропийбромид и ВЕА2108, предпочтительно тиотропийбромид.
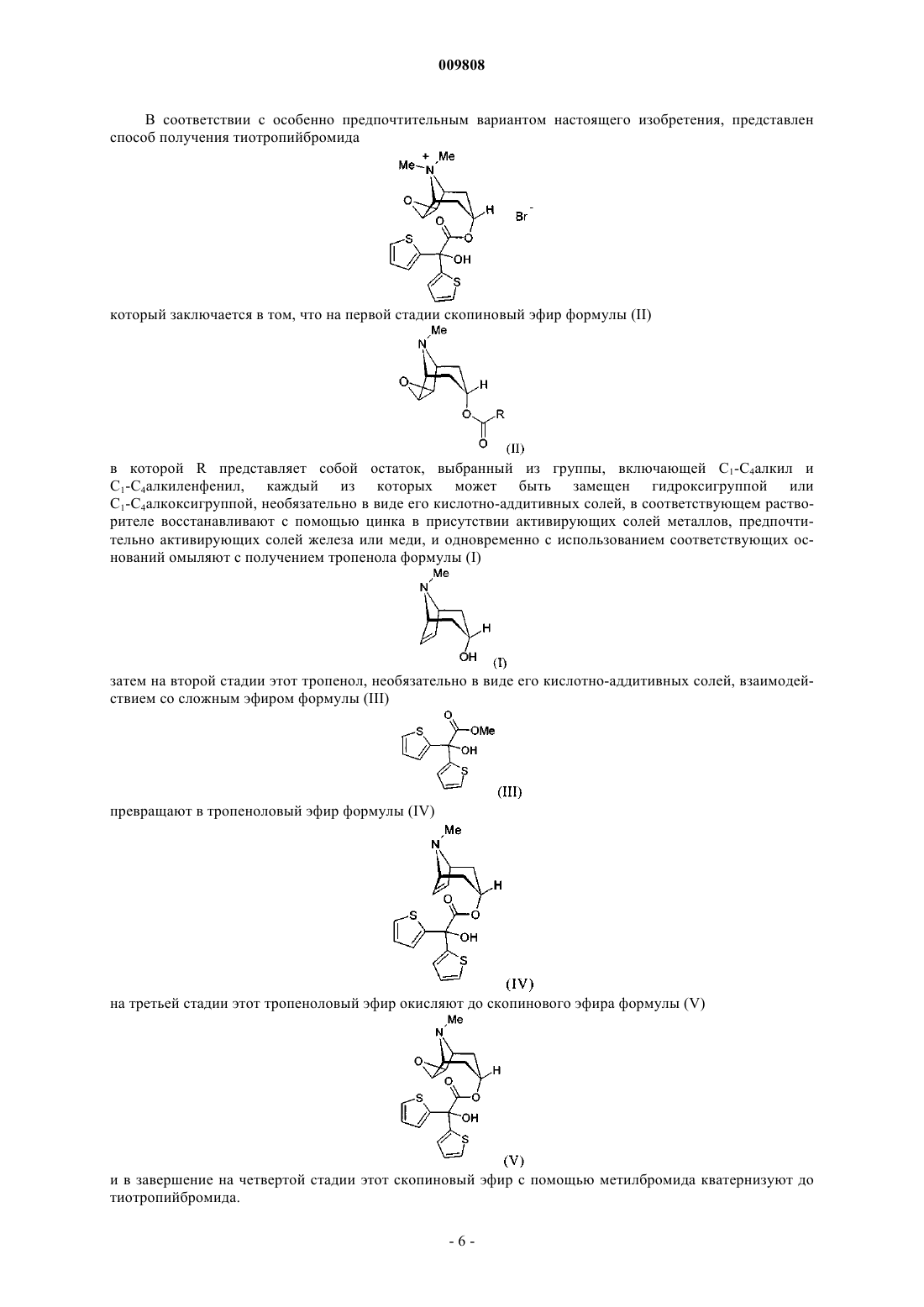
Текст
009808 Настоящее изобретение относится к применению производных тропенола для получения соединений, обладающих ценными фармакологическими свойствами, и промышленному способу получения тропенола и его производных, необязательно в виде их кислотно-аддитивных солей. Предпосылки создания изобретения Соединение тропенол известно из уровня техники и имеет следующую структурную формулу: Это соединение может применяться в качестве исходного для получения соединений, обладающих ценными фармакологическими свойствами. В качестве примера можно назвать такие соединения, как тиотропийбромид, ипратропийбромид или же ВЕА 2108. Названные соединения с ценными фармакологическими свойствами имеют соответственно следующую структурную формулу: Поскольку вышеназванные соединения отличаются высокой степенью активности, для их получения с предельно возможной чистотой необходимы эффективные способы синтеза. Именно требование высокой чистоты у соединений, которое должно, в принципе, всегда соблюдаться при их применении в терапии, допускает наличие лишь предельно минимальных примесей в исходных соединениях. Если же в качестве исходных соединений используют материалы, содержащие относительно большое количество примесей, то очистка конечного продукта часто связана со значительными трудностями, обусловленными, в частности, тем, что присутствующие изначально примеси далеко не во всех случаях удается без проблем отделять и на последующих стадиях синтеза, а если и удается, то лишь при существенном снижении выхода целевого продукта. Это имеет место прежде всего в том случае, когда образующиеся побочные продукты, соответственно присутствующие примеси по своей структуре лишь незначительно отличаются от соответствующих основных продуктов. Исходя из вышеизложенного, в основу настоящего изобретения была положена задача разработать способ синтеза, который позволил бы получать в промышленном масштабе тропенол, предпочтительно в виде его кислотно-аддитивных солей, с высоким выходом и прежде всего с высокой степенью чистоты. Сущность изобретения В соответствии с этим объектом настоящего изобретения является применение соединения формулы (II) в которой R представляет собой остаток, выбранный из группы, включающей С 1-С 4 алкил и С 1-С 4 алкиленфенил, каждый из которых может быть замещен гидроксигруппой или С 1-С 4 алкоксигруппой, необязательно в виде его кислотно-аддитивных солей, а также необязательно в виде его гидратов, в качестве исходного материала для получения производных тропенола, обладающих фармакологическими свойствами. Производные тропенола, обладающие фармакологическими свойствами, представляют собой соединения, выбранные из группы, включающей, например, тиотропийбромид, ипратропийбромид или ВЕА 2108, предпочтительно тиотропийбромид. Таким образом, настоящее изобретение относится к применению производных скопинового эфира формулы (II) для получения терапевтически эффективных соединений, таких, например, как тиотро-1 009808 пийбромид, ипратропийбромид или ВЕА 2108, предпочтительно для получения тиотропийбромида. Подробное описание изобретения Указанную задачу удается решить благодаря предлагаемому изобретению, описанному ниже. В соответствии с этим способ промышленного получения тропенола формулы (I) необязательно в виде его кислотно-аддитивных солей, заключается в том, что скопиновый эфир формулы в которой R представляет собой остаток, выбранный из группы, включающей С 1-С 4 алкил и С 1-С 4 алкиленфенил, каждый из которых может быть замещен гидроксигруппой или С 1-С 4 алкоксигруппой, необязательно в виде его кислотно-аддитивных солей, а также необязательно в виде его гидратов, в соответствующем растворителе восстанавливают с помощью цинка в присутствии активирующих солей металлов, предпочтительно активирующих солей железа или меди, и затем с использованием соответствующих оснований омыляют с получением тропенола формулы (I). Под С 1-С 4 алкилом в контексте настоящего описания имеются в виду разветвленные либо прямоцепочечные алкильные группы, содержащие вплоть до 4 атомов углерода. В качестве примеров можно назвать метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил и трет-бутил. Под С 1-С 4 алкиленфенилом в контексте настоящего описания имеется в виду фенил, который через разветвленный либо прямоцепочечный алкиленовый мостик соединен с атомами углерода, количество которых составляет до 4 включительно. В качестве примеров при этом можно назвать бензил, фенил-2 этил, фенил-1-этил, фенил-3-пропил, фенил-2-пропил и т.д. С 1-С 4 алкильные группы, равно как и С 1 С 4 алкиленфенильные группы могут быть, если не указано иное, замещены одной либо несколькими гидроксигруппами и/или С 1-С 4 алкилоксигруппами. Предпочтительно, способ получения тропенола формулы (I), необязательно в виде его кислотноаддитивных солей, заключается в том, что в качестве производного скопина формулы (II) применяют скополамин формулы (II') необязательно в виде его кислотно-аддитивных солей, а также необязательно в виде гидратов. При осуществлении изобретения способ получения тропенола можно осуществлять следующим образом. В соответствующий реакционный сосуд предварительно загружают растворитель, предпочтительно в атмосфере инертного газа, особенно предпочтительно в атмосфере азота. В качестве пригодных для такой цели растворителей могут использоваться согласно изобретению спирты, выбранные из группы,включающей метанол, этанол и изопропанол, или вода, причем предпочтительной согласно изобретению является вода. На моль применяемого соединения формулы (II) растворитель используют согласно изобретению в количестве от 0,25 до 5, предпочтительно от 0,5 до 3 л, наиболее предпочтительно от 0,75 до 1,5 л. Затем в предварительно загруженный растворитель при интенсивном перемешивании вводят цинк,предпочтительно в виде цинковой пыли, особенно предпочтительно в виде цинковой пыли со средним размером частиц 80 мкм, особенно предпочтительно 70 мкм. На моль применяемого соединения формулы (II) предусматривается использовать по меньшей мере 1 моль цинка. Предпочтительно, однако,использовать цинк согласно изобретению в избытке. Предпочтительно на моль применяемого соедине-2 009808 ния формулы (II) использовать 1,2-3,5 моля, особенно предпочтительно 1,5-3,0 моля, цинка. Согласно одному из особенно предпочтительных вариантов осуществления предлагаемого изобретения, на моль применяемого соединения формулы (II) предусматривается использовать от 1,8 до 2,5 моля цинка. После добавления цинка может оказаться целесообразным провести его активацию. Такую активацию можно обеспечить за счет добавления HI, HBr или, например, HCl. Предпочтителен в качестве активатора HI, предпочтительно в виде водных растворов, особенно предпочтительно в виде концентрированных водных растворов. На моль применяемого соединения формулы (II) может оказаться целесообразным добавлять, например, 0,05-0,25 моля, предпочтительно 0,08-0,2 моля активатора. Кроме того,может оказаться целесообразным перед добавлением активатора повысить температуру образованной смеси. Так, в частности, смесь можно нагреть до температуры выше 50 С, предпочтительно до температуры в интервале от 55 до 90 С, наиболее предпочтительно от 60 до 80 С. Затем к активированной (если такая активация предусмотрена) вышеназванными активаторами суспензии цинка в используемом растворителе добавляют соли металлов. В качестве таких применяемых согласно изобретению солей металлов можно назвать соли железа (предпочтительно соли Fe(III или меди (предпочтительно соли Cu(II, предпочтительно их галогениды. В качестве соли железа предпочтительно применять FeCl3. Особенно предпочтительными при осуществлении предлагаемого изобретения являются соли Cu(II), выбранные из группы, включающей CuCl2, CuI2, CuBr2 и комплекс CuBr2 диметилсульфид, наиболее предпочтителен из них согласно изобретению CuBr2. На моль применяемого соединения формулы (II) во всех случаях согласно изобретению добавляют сверхстехиометрические количества соли металла, предпочтительно от 0,01 до 1 моля. Особенно предпочтительно на моль применяемого исходного соединения формулы (II) использовать от 0,05 до 0,5 моля, наиболее предпочтительно от 0,075 до 0,2 моля, соли металла. Соль металла можно добавлять как таковую либо в растворенном виде к цинковой суспензии. Однако согласно изобретению более целесообразно соль металла сначала растворять в одном из указанных выше растворителей, а затем в растворенном либо суспендированном виде добавлять к цинковой суспензии. Особенно предпочтительно для получения раствора или суспензии соли металла использовать такой же растворитель, который уже использовался для растворения цинка. На моль применяемой соли металла для получения ее раствора или суспензии согласно изобретению рекомендуется использовать от 0,5 до 1,5 л, предпочтительно от 0,6 до 1,0 л растворителя. Затем раствор или суспензию добавляют при перемешивании к предварительно полученной цинковой смеси. Далее к получаемой по описанной технологии цинковой смеси добавляют соединение формулы (II). При этом данное соединение формулы (II) можно добавлять в виде его соответствующей кислотноаддитивной соли. Подобные кислотно-аддитивные соли предпочтительно согласно изобретению выбирать из группы, включающей гидрохлорид, гидробромид, гидрофосфат, гидросульфат, тетрафторборат и гексафторфосфат, особенно предпочтительны из которых гидрохлориды и гидробромиды. При ссылке на кислотно-аддитивные соли соединения формулы (II) имеются в виду также его гидраты, если таковые существуют. Если вышеназванные кислотно-аддитивные соли добавляют к уже образованной цинковой смеси непосредственно, то их можно добавлять как таковые либо в растворенном виде. При добавлении кислотно-аддитивных солей соединений формулы (II) в растворенном виде их рекомендуется растворять в одном из указанных выше растворителей. Предпочтительно для приготовления соответствующего раствора использовать такой же растворитель, который уже использовался для получения цинковой суспензии. Согласно изобретению на моль применяемой кислотно-аддитивной соли соединения формулы (II) предпочтительно использовать от 0,5 до 1,5 л, более предпочтительно от 0,6 до 1,0 л растворителя. Возможен и иной вариант, а именно соединения формулы (II) в виде их свободных оснований с использованием отдельной опытной установки сначала в соответствующем растворителе с помощью соответствующих кислот переводить в растворенные кислотно-аддитивные соли и затем эти соли добавлять к предварительно полученной цинковой смеси. В рассматриваемом случае в качестве растворителя можно использовать один из указанных выше растворителей. Предпочтительно использовать такой же растворитель, который использовали для приготовления цинковой суспензии. Согласно изобретению на моль применяемого свободного основания формулы (II) предпочтительно использовать от 0,5 до 1,5 л, более предпочтительно от 0,6 до 1,0 л растворителя. Полученную таким путем суспензию смешивают затем с соответствующей кислотой, необходимой для образования требующейся кислотно-аддитивной соли, т.е. гидрохлорида, гидробромида, гидрофосфата, гидросульфата, тетрафторбората или гексафторфосфата. На моль применяемого в этих случаях свободного основания формулы (II) предусматривается использовать по меньшей мере 1 моль соответствующей кислоты. Вместе с тем, при осуществлении предлагаемого в изобретении способа допустимо использовать кислоту в избытке (т.е. от 1,1 до порядка 2 молей на моль основания формулы (II. Согласно изобретению предпочтительно использовать гидрохлориды или гидробромиды соединений формулы (II). Соляную кислоту можно добавлять либо в виде водных растворов,либо в газообразном виде, предпочтительно добавление водных растворов. Целесообразно добавлять концентрированную соляную кислоту (36%-ную), растворенную в воде. Если используют бромистоводородную кислоту, что согласно изобретению особенно предпочтительно, то добавлять ее также можно либо в виде водных растворов, либо в газообразном виде, предпочтительным является добавление водных растворов. Целесообразно добавлять концентрированную бромисто-водородную кислоту (62%-3 009808 ную), растворенную в воде. Благодаря добавлению одной из вышеназванных кислот к суспензии свободного основания формулы (II) в соответствующем растворителе значение pH устанавливают на 3,5-5,5,предпочтительно на 4,5-5. Описанный выше и получаемый при определенных условиях различным путем раствор кислотноаддитивных солей соединений формулы (II) добавляют затем к предварительно приготовленной цинковой суспензии. Добавлять раствор можно, например, но необязательно, при повышенной температуре. Следует, однако, учитывать, что такое повышение температуры целесообразно прежде всего в том случае, когда нагрев смеси уже провели перед добавлением активатора. Если процесс добавления проводят при повышенной температуре, то согласно изобретению такая температура должна превышать 50 С,предпочтителен интервал от 55 до 90 С, особенно предпочтителен интервал от 60 до 80 С. По завершении добавления реакционную смесь перемешивают при температурах в интервале от 50 до 100 С, предпочтительно от 60 до 95 С и наиболее предпочтительно от порядка 70 до 85 С. В зависимости от выбора растворителя максимальная температура в указанном интервале может, как очевидно,быть ниже, при условии, что температура кипения используемого растворителя ниже этой максимальной температуры. Процесс перемешивания при постоянной температуре продолжают до полного завершения обменной реакции (продолжительность от 0,5 до 4 ч). Контроль за протеканием реакции можно осуществлять, например, с помощью тонкослойной хроматографии. По завершении обменной реакции реакционную смесь для омыления сложноэфирной функциональной группы смешивают с соответствующим основанием. В качестве такового приемлемы предпочтительно неорганические основания из группы карбонатов щелочных или щелочно-земельных металлов и гидроксидов щелочных и щелочно-земельных металлов. Особенно предпочтительны при этом гидроксиды лития, натрия, калия и кальция, наиболее предпочтительными из них являются гидроксиды натрия или кальция. Согласно изобретению самым предпочтительным для использования в качестве основания является гидроксид натрия. Вышеназванные основания могут применяться в чистом виде или, что особенно предпочтительно, в виде водных концентрированных растворов. Если, например, в качестве особенно предпочтительного, как рекомендуется в изобретении, использовать гидроксид натрия, то его целесообразно добавлять в виде водных растворов с концентрацией по меньшей мере 40 мас.%. На моль первоначально применяемого соединения формулы (II) основание необходимо использовать по меньшей мере в стехиометрических количествах. Вместе с тем основание можно использовать и в избытке. При этом возможны два варианта: либо основание уже добавляют при температуре в интервале от 0 до 50 С,предпочтительно от 10 до 40 С, особенно предпочтительно от порядка 20 до 30 С, либо указанную температуру устанавливают непосредственно после добавления основания. Перемешивание при этой температуре продолжают до полного завершения обменной реакции (в зависимости от объема смеси продолжительность от 12 до 24 ч). При небольших объемах смеси (например в несколько килограммов) омыление можно проводить также при повышенной температуре (при 50-100 С, предпочтительно при 55-90 С,наиболее предпочтительно в интервале от порядка 60 до 80 С). Благодаря этому продолжительность реакции удается сократить до промежутка от примерно 15 мин до 10 ч, предпочтительно до 0,5-3 ч. Контроль за протеканием реакции можно осуществлять, например, с помощью тонкослойной хроматографии. По завершении обменной реакции температуру реакционной смеси понижают при перемешивании до уровня в интервале от 0 до 50 С, предпочтительно от 15 до 45 С, и путем фильтрации удаляют из нее соли цинка. Остаток на фильтре при необходимости можно промывать используемым в реакции растворителем. Для экстракции фильтрата его смешивают с органическим растворителем, вовсе не смешивающимся или смешивающимся лишь в незначительной степени с выбранным для проведения реакции растворителем. Предпочтительно такой органический растворитель выбирают из группы, включающей метил-трет-бутиловый эфир, дихлорметан и хлороформ, предпочтительно дихлорметан. На моль применяемого соединения формулы (II) согласно изобретению для экстракции используют от 0,5 до 5, предпочтительно от 0,75 до 4 л органического растворителя. Согласно изобретению экстракцию целесообразно проводить от 3 до 8, предпочтительно от 4 до 6 раз. После экстракции органические фазы объединяют, а органический растворитель отгоняют под вакуумом. Образующийся сырой продукт растворяют в органическом растворителе, выбранном из группы,включающей метанол, этанол и изопропанол, предпочтительно в изопропаноле. На моль первоначально применяемого соединения формулы (II) указанный органический растворитель используют согласно изобретению в количестве от 0,1 до 2,0 л, предпочтительно от 0,3 до 1,0 л. Из полученного раствора путем фильтрации отделяют выпавшее в осадок твердое вещество (металлические соли кислоты RCOOH,где R может иметь указанные выше значения). Фильтрат содержит тропенол формулы (I) в виде его свободного основания. Если это свободное основание предусматривается использовать в последующей обменной реакции, то используемый растворитель следует предварительно отгонять под вакуумом. Полученное свободное основание можно затем, без дальнейшей очистки, использовать на последующих стадиях синтеза. Согласно изобретению предпочтительно, однако, свободное основание тропенола переводить в одну из его кислотно-аддитивных солей. В качестве кислотно-аддитивных солей тропенола в контексте настоящего описания подразумеваются соли, выбранные из группы, включающей гидрохлорид,-4 009808 гидробромид, гидрофосфат, гидросульфат, тетрафторборат и гексафторфосфат. Особенно предпочтительными из числа названных солей являются гидробромид и гидрохлорид, из которых в свою очередь наиболее предпочтителен согласно изобретению гидрохлорид тропенола. Для получения кислотноаддитивных солей фильтрат охлаждают до температуры в интервале от -10 до +20 С, предпочтительно от-5 до +15 С. Полученную таким путем суспензию смешивают затем с соответствующей кислотой, требуемой для образования предусмотренной кислотно-аддитивной соли, т.е. гидрохлорида, гидробромида,гидрофосфата, гидросульфата, тетрафторбората или гексафторфосфата. На моль первоначально применяемого свободного основания формулы (II) следует использовать по меньшей мере 1 моль соответствующей кислоты. Вместе с тем при осуществлении предлагаемого в изобретении способа предусматривается также возможность использовать кислоту в избытке (т.е. от 1,1 до порядка 2-3 молей на моль первоначально применяемого основания формулы (II. Согласно изобретению предпочтительно получать гидрохлорид тропенола. Необходимую в этих целях соляную кислоту можно добавлять либо в виде растворов, либо в газообразном виде. Предпочтительно использовать хлористый водород, который в газообразном виде вводят в отдельном реакционном сосуде в один из вышеназванных растворителей вплоть до полного насыщения. Наиболее предпочтительно для получения этого HCl-раствора использовать растворитель, который обычно используют для получения тропенолового фильтрата. Для растворения свободного основания тропенола формулы (I) одну из указанных выше кислот добавляют до достижения значения pH, равного 1,5-6,5, предпочтительно 2-6. После добавления кислоты перемешивание при постоянной температуре можно продолжать еще в течение 0,5-2 ч. В завершение выпавшую в осадок кислотноаддитивную соль тропенола отделяют и при необходимости промывают растворителем, выбранным из группы, включающей ацетон, метилизобутилкетон и метилэтилкетон, предпочтительно ацетоном, и сушат под вакуумом. Как уже указывалось в начале описания, тропенол, получаемый по предлагаемому в изобретении способу, представляет собой ценное исходное соединение для получения терапевтически эффективных соединений, таких, например, как тиотропийбромид, ипратропийбромид или ВЕА 2108. Благодаря высокой степени чистоты, с которой тропенол получают согласно настоящему изобретению, создана возможность предложить названные соединения в формах, обеспечивающих их применение в фармацевтике. Предпочтительно настоящее изобретение относится к применению скополамина, необязательно в виде его кислотно-аддитивных солей, а также необязательно в виде его гидратов, в качестве исходного материала для получения терапевтически эффективных соединений, таких, например, как тиотропийбромид, ипратропийбромид или ВЕА 2108, предпочтительно для получения тиотропийбромида. Тиотропийбромид можно получать исходя из тропенола согласно представленной ниже схеме 1. Схема 1 Исходя из получаемого в соответствии с изобретением тропенола формулы (I) сначала взаимодействием с производными ди(2-тиенил)гликолевой кислоты формулы (III) образуют тропеноловый эфир ди(2-тиенил)гликолевой кислоты формулы (IV). Затем этот тропеноловый эфир за счет окисления олефиновой двойной связи переводят в соответствующий скопиновый эфир формулы (V), из которого взаимодействием с метилбромидом получают тиотропийбромид.-5 009808 В соответствии с особенно предпочтительным вариантом настоящего изобретения, представлен способ получения тиотропийбромида который заключается в том, что на первой стадии скопиновый эфир формулы (II) в которой R представляет собой остаток, выбранный из группы, включающей С 1-С 4 алкил и С 1-С 4 алкиленфенил, каждый из которых может быть замещен гидроксигруппой или С 1-С 4 алкоксигруппой, необязательно в виде его кислотно-аддитивных солей, в соответствующем растворителе восстанавливают с помощью цинка в присутствии активирующих солей металлов, предпочтительно активирующих солей железа или меди, и одновременно с использованием соответствующих оснований омыляют с получением тропенола формулы (I) затем на второй стадии этот тропенол, необязательно в виде его кислотно-аддитивных солей, взаимодействием со сложным эфиром формулы (III) превращают в тропеноловый эфир формулы (IV) на третьей стадии этот тропеноловый эфир окисляют до скопинового эфира формулы (V) и в завершение на четвертой стадии этот скопиновый эфир с помощью метилбромида кватернизуют до тиотропийбромида.-6 009808 Ниже предлагаемый в изобретении способ получения тиотропийбромида более подробно поясняется на примерах, которые служат лишь для иллюстрации возможной технологии его осуществления, не ограничивая при этом объем изобретения. Пример 1. Получение тропенола формулы (I) в виде его гидрохлорида (в объеме несколько килограмм). В продутый азотом реактор предварительно загружают 3 л воды и при интенсивном перемешивании добавляют 390 г цинковой пыли (размер частиц 63 мкм), а для активации добавляют 66 мл 57%-ного водного раствора йодистого водорода. Эту смесь перемешивают при комнатной температуре примерно в течение 5 мин. Затем медленно добавляют 67,2 г бромида Cu(II), растворенного в 260 мл воды. Далее к этой смеси медленно добавляют раствор 910,2 г основания скополамина, растворенного в примерно 2,6 л воды, и с помощью 227 мл 62%-ного водного раствора бромистого водорода значениеpH устанавливают на 4,5-5. После добавления смесь нагревают до 75-80 С и при этой температуре перемешивают приблизительно в течение 2 ч. По завершении обменной реакции (контроль с помощью тонкослойной хроматографии (ТСХ охлаждают до температуры порядка 65 С. Затем добавляют 480 мл 45%-ного водного едкого натра и при температуре 65-70 С перемешивают до полного омыления (приблизительно 1 ч). После охлаждения до примерно 40 С Zn-соли отфильтровывают и промывают приблизительно 200 мл воды. Фильтрат несколько раз экстрагируют дихлорметаном (3-5 раз, порциями по 2-4 л дихлорметана), органические фазы объединяют и растворитель отгоняют при пониженном давлении. Образующийся остаток (371 г сырого продукта) растворяют в 1,5 л изопропанола и выпавшее в осадок твердое вещество (металлическая соль троповой кислоты) отфильтровывают. Фильтрат охлаждают до температуры в интервале от -10 до +10 С и медленно при интенсивном перемешивании смешивают со 120 г HCl, растворенными в 780 мл изопропанола. При этом значение pH устанавливается на уровне 2,54. По завершении добавления перемешивание при приблизительно -5 С продолжают еще в течение одного часа. В завершение суспензию фильтруют, остаток на фильтре промывают с помощью примерно 600 мл ацетона и затем сушат под вакуумом при температуре порядка 60 С. Выход: 408,1 г тропенола в виде гидрохлорида (77,4% в пересчете на применяемый скополамин). Пример 2. Получение тропенола формулы (I) в виде его гидрохлорида (в промышленном масштабе). В продутый азотом реактор предварительно загружают 130 л воды и при интенсивном перемешивании добавляют 21,5 кг цинковой пыли (размер частиц 63 мкм). Затем эту смесь нагревают до 65-75 С. Далее к этой смеси добавляют 6,2 кг 57%-ного водного раствора йодистоводородной кислоты. Затем добавляют раствор 3,7 кг бромида Cu(II) в 20-25 л воды. После этого необязательно перемешивают в течение 5 мин и затем добавляют раствор 65,8 мг тригидрата гидробромида скополамина в 140-145 л воды. Полученную смесь нагревают до 75-85 С и перемешивают в течение 2-2,5 ч. По завершении обменной реакции (контроль с помощью ТСХ) добавляют 35,5 кг 45%-ного водного едкого натра. После этого температуру понижают до 20-30 С и смесь продолжают перемешивать еще в течение 20-24 ч. По завершении обменной реакции (контроль с помощью ТСХ) все содержимое реактора фильтруют и образующийся остаток промывают примерно 30 л воды. Фильтрат при температуре, поддерживаемой на постоянном уровне, смешивают с 75 кг хлорида натрия. Для экстракции добавляют 150 л дихлорметана. Органическую фазу отделяют, а водную фазу еще четырежды экстрагируют таким же количеством дихлорметана. Из объединенных органических фаз путем перегонки удаляют растворитель. К образующемуся остатку добавляют приблизительно 100 л изопропанола и температуру устанавливают в интервале от 0 до 10 С. После этого добавляют раствор 5,5 кг хлористого водорода в 38 л изопропанола до момента установления значения pH, равного примерно 2,5-5,5. Выпавший в осадок тропенол в виде гидрохлорида отделяют и промывают с помощью 30 л ацетона. После сушки получают 21,3 кг целевого продукта (выход 81% в пересчете на используемый гидробромид скополамина). Пример 3. Получение тиотропийбромида. а) Получение тропенолового эфира формулы (IV). К 10,9 кг тропенола в виде гидрохлорида (получаемого согласно примеру 1) в толуоле (95 л) при 25 С подают аммиак (1,8 кг). Полученную в результате суспензию перемешивают при постоянной температуре приблизительно в течение 1 ч. Затем образовавшийся гидрохлорид аммония отфильтровывают и промывают толуолом (26 л). При температуре рубашки реактора порядка 50 С часть толуола (примерно 60 л) отгоняют под вакуумом. После охлаждения до приблизительно 25 С добавляют 15,8 кг метилового эфира ди(2-тиенил)гликолевой кислоты и с целью растворения полученную смесь нагревают до 50 С. Одновременно в другой аппарат предварительно загружают толуол (40 л) и в него при температуре порядка 25 С вводят гидрид натрия (2,7 кг). Затем к этому раствору в течение приблизительно 1 ч добавляют при 30 С полученный ранее вышеописанный первый раствор тропенола и метилового эфира гликолевой кислоты. По завершении процесса добавления при пониженном давлении в течение примерно 7 ч нагревают при перемешивании до 75 С. При этом образующийся метанол отгоняют. Оставшуюся смесь охлаждают и добавляют ее к другой смеси, состоящей из воды (958 л) и 36%-ной соляной кислоты(13,2 кг). Водную фазу затем отделяют и промывают метиленхлоридом (56 л). После повторного добав-7 009808 ления метиленхлорида (198 л) значение pH полученной таким путем смеси с помощью приготовленного раствора соды (9,6 кг соды в 45 л воды) устанавливают на 9. Далее метиленхлоридную фазу отделяют, а водную фазу экстрагируют размешиванием с метиленхлоридом (262 л). Затем содержащие метиленхлорид фазы концентрируют при 65 С до образования сухого остатка. Этот остаток растворяют в толуоле(166 л) и нагревают до 95 С. После этого толуольный раствор охлаждают до 0 С. Полученные кристаллы отделяют, промывают толуолом (33 л) и в завершение при температуре порядка 50 С в течение максимум 24 ч сушат в токе азота. Выход: 18,6 кг (83%); tпл примерно 160 С (определение с помощью ДСК (дифференциальной сканирующей калориметрии) при скорости нагрева 10 K/мин). б) Получение скопинового эфира формулы (V). В соответствующий реакционный аппарат предварительно загружают 260 л ДМФ и нагревают до 50 С. Затем добавляют 16,2 кг тропенолового эфира формулы (IV) и перемешивают до момента образования прозрачного раствора. После охлаждения до 40 С последовательно порциями вводят комплекс пероксид водорода-мочевина (10,2 кг), воду (13 л) и оксид ванадия(V) (0,7 кг) и содержимое аппарата нагревают до приблизительно 50 С. После 2-3-часового перемешивания при постоянной температуре охлаждают до примерно 20 С. С помощью соляной кислоты (36%-ной) значение pH полученной реакционной смеси устанавливают на 4,0. Затем к смеси добавляют приготовленный раствор бисульфита натрия(2,4 кг в 24 л воды). При внутренней температуре 35 С растворитель частично отгоняют под вакуумом(приблизительно 210 л). Затем повторно охлаждают до примерно 20 С и смешивают с продуктом Clarcel(3,2 кг). С помощью разбавленной соляной кислоты (36%-ной, 0,8 кг в приблизительно 440 л воды) значение pH устанавливают на порядка 2,0. Далее полученный раствор фильтруют и экстрагируют метиленхлоридом (58 л). Затем метиленхлоридную фазу отбрасывают. К водной фазе повторно добавляют метиленхлорид (130 л) и с помощью приготовленного раствора соды (11,0 кг в 51 л воды) значение pH устанавливают примерно на 10,0. После этого содержащую метиленхлорид фазу отделяют, а водную фазу экстрагируют метиленхлоридом (136 л). Под слабым вакуумом (600-700 мбар) при 40 С из объединенных метиленхлоридных фаз отгоняют метиленхлорид (приблизительно 175 л). Содержимое аппарата охлаждают до 20 С, добавляют ацетилхлорид (приблизительно 0,5 кг) и в течение примерно 40 мин перемешивают при 20 С. Затем реакционный раствор перемещают в другой аппарат. С помощью приготовленного раствора соляной кислоты (4,7 кг 36%-ной соляной кислоты в 460 л воды) при 20 С значениеpH устанавливают на 2,0. Содержащую метиленхлорид фазу отделяют и утилизуют. Водную фазу промывают метиленхлоридом (39 л). Затем добавляют метиленхлорид (130 л) и с помощью приготовленного раствора соды (7,8 кг соды в 38 л воды) при 20 С значение pH устанавливают на 10,0. После 15 минутного перемешивания органическую фазу отделяют, а водную фазу дважды промывают метиленхлоридом (97 л и 65 л). Содержащие метиленхлорид фазы объединяют и под слабым вакуумом часть метиленхлорида (90 л) отгоняют при 30-40 С. Затем добавляют диметилформамид (114 кг) и под вакуумом при 40 С отгоняют остальную часть метиленхлорида. В завершение содержимое аппарата охлаждают до 20 С. в) Получение тиотропийбромида. К получаемому по описанной в разделе б) технологии раствору скопинового эфира при 20 С подают метилбромид (5,1 кг). Содержимое аппарата перемешивают после этого при 30 С примерно в течение 2,5 дней. Затем при 50 С под вакуумом отгоняют 70 мл ДМФ. Раствор перемещают в аппарат меньших размеров. Далее промывают диметилформамидом (10 л). При 50 С под вакуумом продолжают отгонку ДМФ до достижения его отогнанного количества в целом порядка 100 л. Затем охлаждают до 15 С и при этой температуре перемешивают в течение 2 ч. Полученный продукт выделяют с помощью сушилки с нутч-фильтром, промывают охлажденным до 15 С ДМФ (10 л) и охлажденным до 15 С ацетоном (25 л). В завершение продукт при максимум 50 С сушат в течение максимум 36 ч в токе азота. Выход: 13,2 кг (88%); tпл 200-230C (в зависимости от степени чистоты сырого продукта). Полученный таким путем сырой продукт (10,3 кг) вводят в метанол (66 л). С целью растворения эту смесь нагревают с обратным холодильником. Затем образовавшийся раствор охлаждают до 7 С и в течение 1,5 ч перемешивают при этой температуре. Полученный продукт выделяют с помощью сушилки,снабженной нутч-фильтром, промывают охлажденным до 7 С метанолом (11 л) и в завершение в течение максимум 36 ч сушат при температуре порядка 50 С в токе азота. Выход: 9,9 кг (96%); tпл 228C (определение с помощью ДСК при скорости нагрева 10 K/мин). При необходимости полученный описанным выше путем продукт можно переводить в кристаллический моногидрат тиотропийбромида. При этом можно работать следующим образом. В реактор заливают 25,7 кг воды и добавляют 15,0 кг тиотропийбромида. Смесь нагревают до 80-90 С и перемешивают при этой температуре до образования прозрачного раствора. В 4,4 кг воды суспендируют увлажненный водой активированный уголь (0,8 кг), эту смесь добавляют к содержащему тиотропийбромид раствору и дополнительно промывают 4,3 кг воды. Полученную таким путем смесь перемешивают по меньшей мере в течение 15 мин при 80-90 С и затем подают через обогреваемый фильтр в аппарат с рубашкой, предварительно нагретой до температуры 70 С. Фильтр дополнительно промывают 8,6 кг воды. Затем содержимое аппарата охлаждают со скоростью 3-5 С/20 мин до температуры 20-25 С. Далее аппарат охлаж-8 009808 дают до 10-15 С с помощью холодной воды и завершают кристаллизацию путем последующего перемешивания по меньшей мере в течение одного часа. Кристаллизат отделяют пропусканием через сушилку с нутч-фильтром и затем отделенную суспензию кристаллов промывают 9 л холодной воды (10-15 С) и холодным ацетоном (10-15 С). Полученные кристаллы сушат при 25 С в течение 2 ч в токе азота. Выход: 13,4 кг моногидрата тиотропийбромида (86% от теории); tпл 230C (определение с помощью ДСК при скорости нагрева 10 K/мин). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Применение соединения формулы (II) в которой R представляет собой остаток, выбранный из группы, включающей С 1-С 4 алкил и С 1-С 4 алкиленфенил, каждый из которых может быть замещен гидроксигруппой или С 1-С 4 алкоксигруппой, необязательно в виде его кислотно-аддитивных солей, а также необязательно в виде его гидратов, в качестве исходного материала для получения производных тропенола, обладающих фармакологическими свойствами. 2. Применение по п.1, где производные тропенола выбирают из группы, включающей тиотропийбромид, ипратропийбромид и ВЕА 2108, предпочтительно тиотропийбромид.
МПК / Метки
МПК: C07D 491/18, C07D 451/10
Метки: получения, исходный, производные, терапевтически, материал, соединений, эффективных, тропенола
Код ссылки
<a href="https://eas.patents.su/10-9808-proizvodnye-tropenola-ishodnyjj-material-dlya-polucheniya-terapevticheski-effektivnyh-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Производные тропенола – исходный материал для получения терапевтически эффективных соединений</a>
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Крок Вероник, Колладан Колетт, Ларкин Джон Патрик, Руссель Патрик
МПК: C07D 487/04
Метки: кислоты, активных, способ, терапевтически, получения, 1,2, 10-диоксо-6н-пиридазино, применение, 1,2-а, октагидро-6, производные, диазепин-1-карбоновой, соединений
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Абразив на основе церия, исходный материал для него и способы их получения

Номер патента: 3909
Опубликовано: 30.10.2003
Авторы: Ито Терунори, Ютино Йосицугу, Ямасаки Хидехико
МПК: C09K 3/14, C01F 17/00, B24B 37/00...
Метки: него, получения, способы, материал, основе, исходный, церия, абразив
Формула / Реферат:
1. Исходный материал для абразивов на основе церия, включающий карбонат и оксид редкоземельного металла церия и имеющий потери при прокаливании, определенные нагревом при 1000шC в течение 1 ч, в пересчете на сухой материал от 0,5 до 25%. 2. Исходный материал для абразивов на основе церия по п.1, где потери при прокаливании составляют от 5,0 до 20%. 3. Исходный материал для абразивов на основе церия по п.1, где потери при прокаливании составляют...
Способ синтеза тропенола для его получения в промышленном масштабе

Номер патента: 6393
Опубликовано: 29.12.2005
Авторы: Рапп Армин Вальтер, Зоботта Райнер
МПК: C07D 451/10, C07D 471/08
Метки: промышленном, масштабе, тропенола, способ, получения, синтеза
Формула / Реферат:
1. Способ получения тропенола формулы (I) необязательно в виде его кислотно-аддитивных солей, отличающийся тем, что скопиновый эфир формулы (II) в которой R представляет собой остаток, выбранный из группы, включающей C1-C4алкил и C1-C4алкиленфенил, каждый из которых может быть замещен гидроксигруппой или C1-C4алкоксигруппой, необязательно в виде его кислотно-аддитивных солей, а также необязательно в виде его гидратов, в воде восстанавливают с...
Новый промежуточный продукт для получения терапевтически активных имидазопиридинов

Номер патента: 8746
Опубликовано: 31.08.2007
Авторы: Шойфлер Кристиан, Мюллер Бернд, Коль Бернхард, Циммерманн Петер
МПК: C07D 471/14
Метки: терапевтически, имидазопиридинов, активных, получения, продукт, новый, промежуточный
Формула / Реферат:
1. Молекулярная соль соединения формулы (1) с 1,5-нафталиндисульфоновой кислотой 2. Молекулярная соль соединения формулы (1) с 1,5-нафталиндисульфоновой кислотой по п.1, в которой молярное отношение содержаний 1,5-нафталиндисульфоновой кислоты и соединения формулы (1) составляет от 1:1 до 2:1. 3. Молекулярная соль соединения формулы (1) с 1,5-нафталиндисульфоновой кислотой по п.1, в которой молярное отношение содержаний...
Ферментированный высушенный материал, способ его получения и его применение для получения фармацевтической композиции и диетической добавки, а также фармацевтическая композиция и диетическая добавка,содержащие указанный материал

Номер патента: 3090
Опубликовано: 26.12.2002
Авторы: Сенде Бела, Лапиш Карой, Хидвеги Мате, Рашо Эржебет, Темешкезине Фаркаш Рита
МПК: A61K 35/78, A23L 1/172, A61P 37/04...
Метки: добавка,содержащие, способ, диетической, указанный, добавки, ферментированный, композиции, материал, композиция, получения, фармацевтическая, фармацевтической, высушенный, применение, диетическая, также
Формула / Реферат:
1. Ферментированный высушенный материал, являющийся иммуностимулятором и ингибирующий метастазы, который получают из сбраживаемой жидкости посредством ферментации пшеничного зерна с применением Saccharomyces cerevisiae в водной среде, который имеет ВЭЖХ-хроматограмму, по существу соответствующую представленной на фиг. 3, и содержание в нем 2,6-диметоксипарабензохинона составляет 0,12-0,52 мг/г сухого материала, а содержание...
Предыдущий патент: Антитело против igf-i-рецептора
Следующий патент: Способ и устройство нанесения покрытия на имплантаты
Случайный патент: Горизонтально направленное сверление в скважинах