Производные бензоизоиндола для лечения боли
Номер патента: 14428
Опубликовано: 30.12.2010
Авторы: Хили Марк Патрик, Прайс Хелен Сьюзанн, Гиблин Джерард Мартин Пол












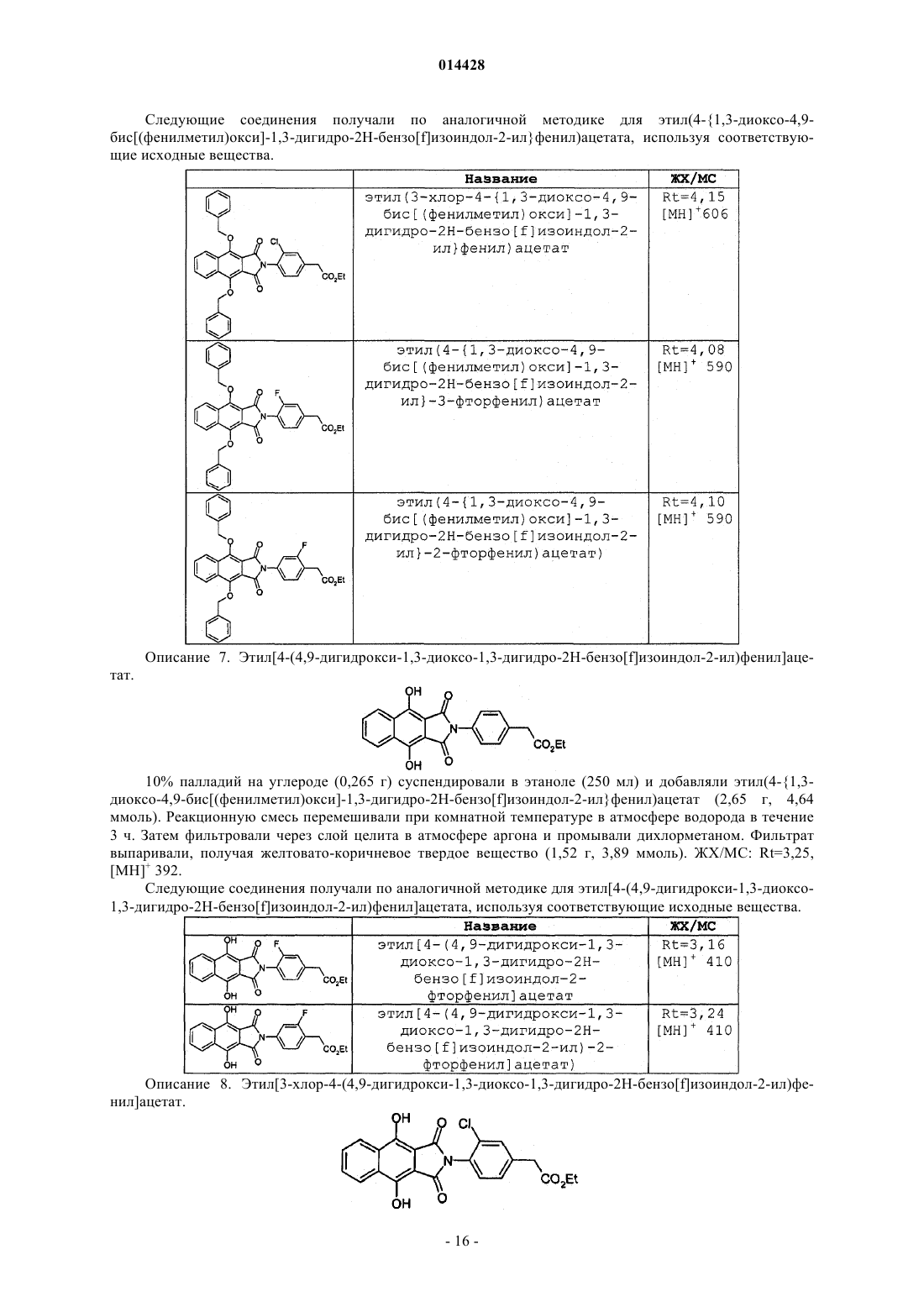

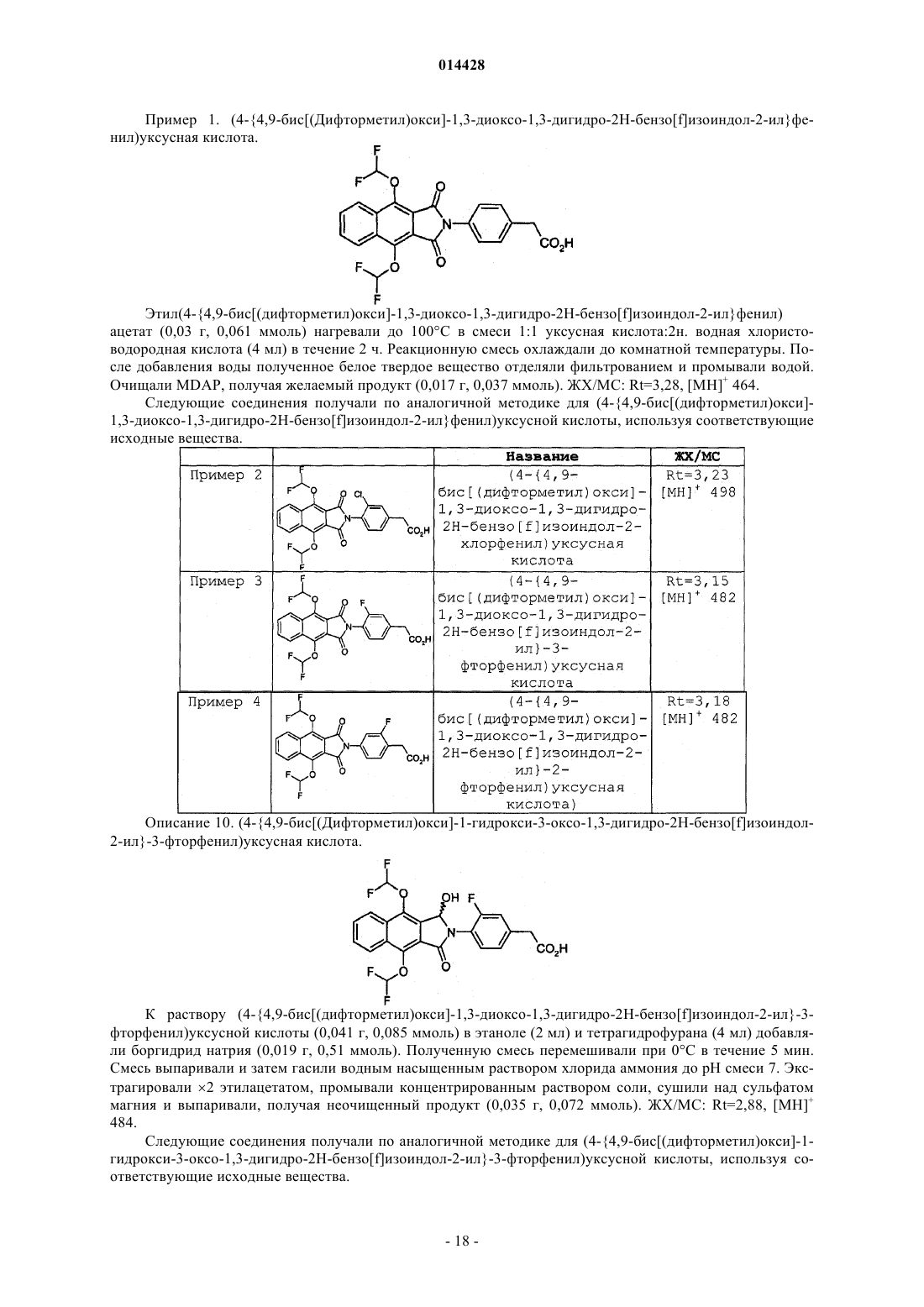
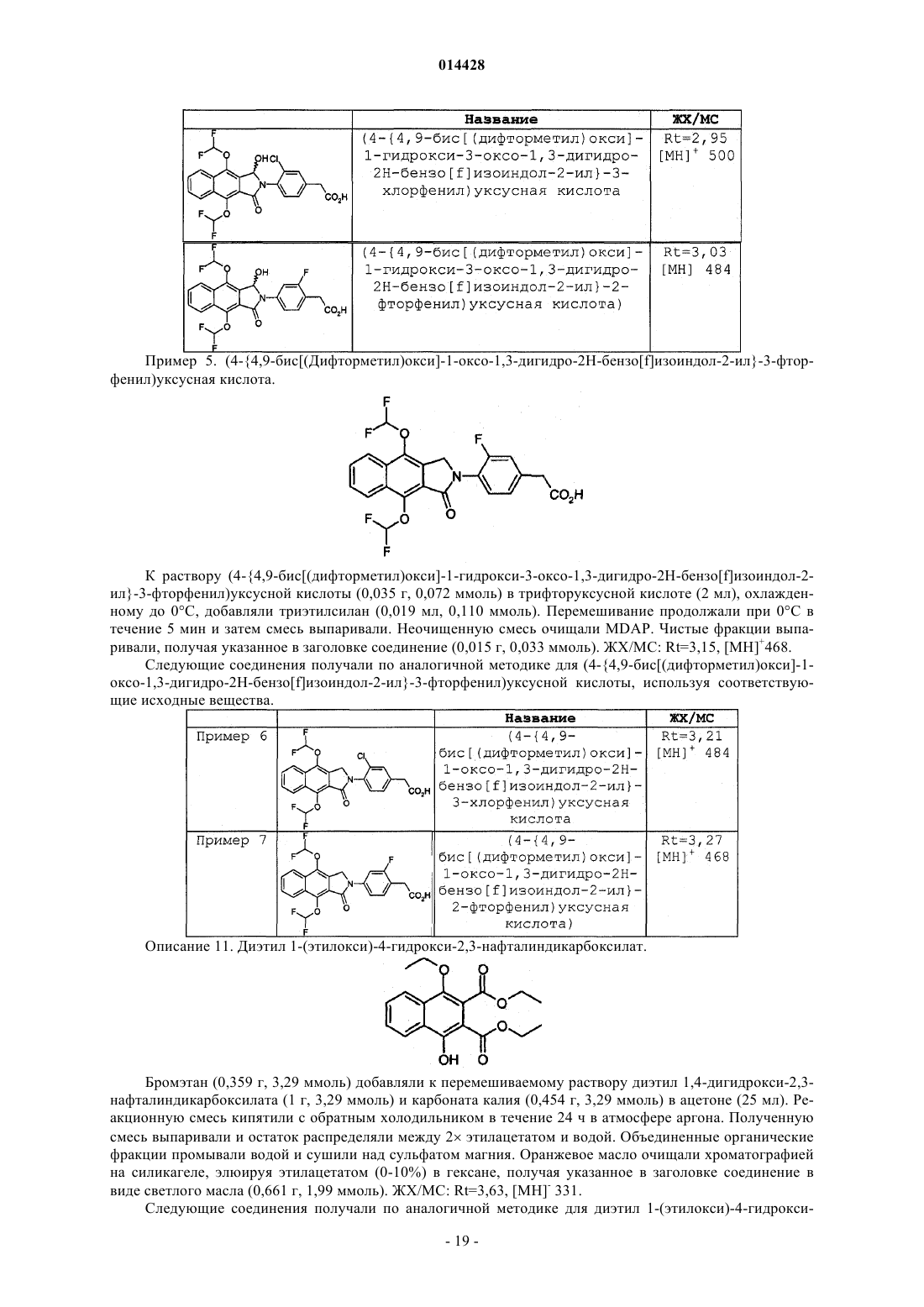
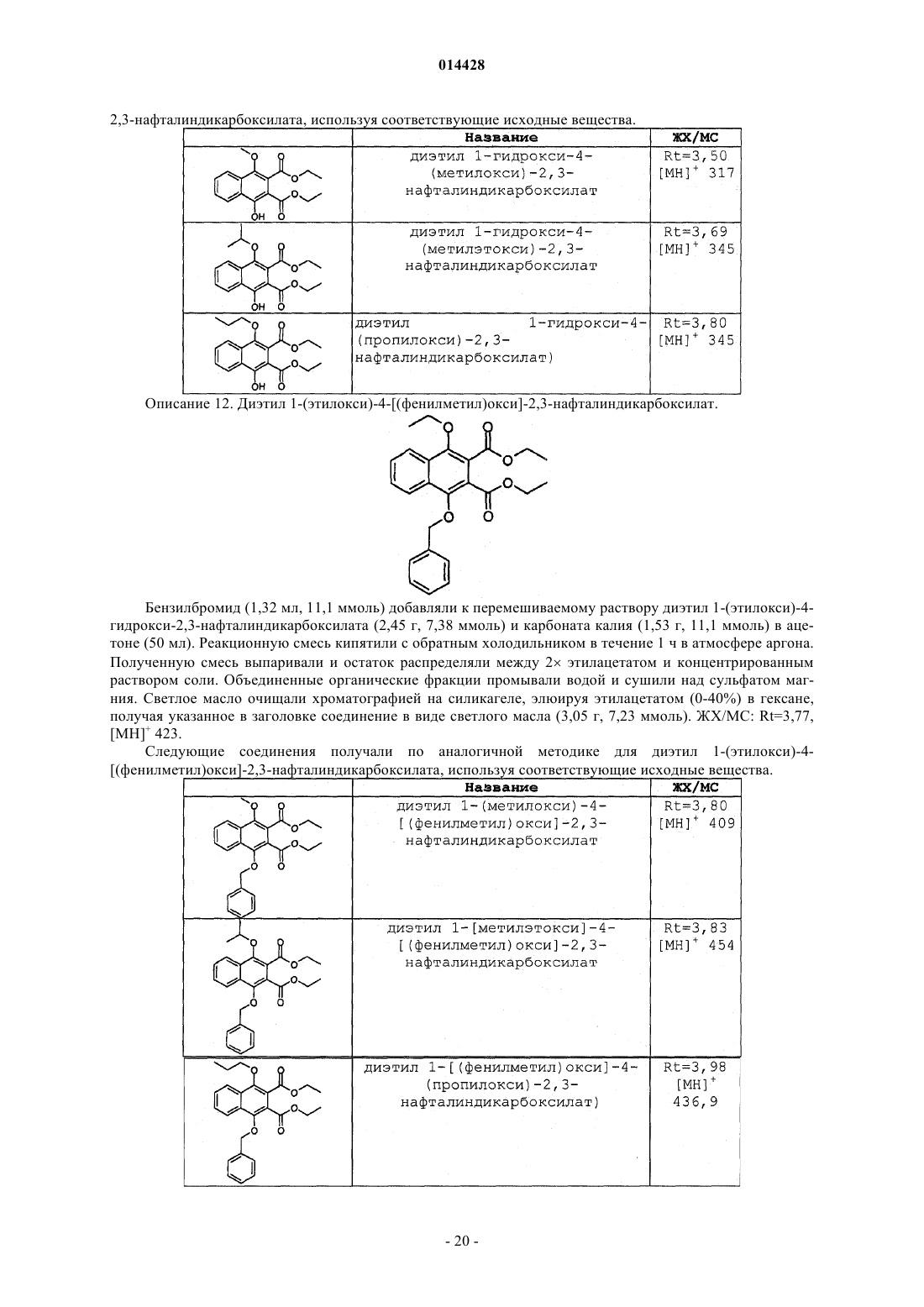
Формула / Реферат
1. Соединение формулы (I) или его фармацевтически приемлемое производное

где R1и R2 независимо представляют С1-4алкил или дифторметил, при условии, что по меньшей мере один из R1и R2 представляет дифторметил;
R3 представляет Н, F, Cl или Br и
X и Y независимо представляют С=O или СН2, при условии, что по меньшей мере один из X и Y представляет С=O.
2. Соединение формулы (I) по п.1, где один из R1 и R2 представляет С1-4алкил и другой представляет дифторметил.
3. Соединение формулы (I) по п.1, где оба R1 и R2 представляют дифторметил.
4. Соединение формулы (I) по любому из пп.1-3, где как X, так и Y представляют С=O.
5. Соединение формулы (I) по любому из пп.1-4, где R3 представляет Н, F или Cl.
6. Соединение формулы (I) по п.1, выбранное из группы, состоящей из следующих
(4-{4,9-бис[(дифторметил)окси]-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}фенил)уксусная кислота;
(4-{4,9-бис[(дифторметил)окси]-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-3-хлорфенил)уксусная кислота;
(4-{4,9-бис[(дифторметил)окси]-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-3-фторфенил)уксусная кислота;
(4-{4,9-бис[(дифторметил)окси]-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-2-фторфенил)уксусная кислота;
(4-{4,9-бис[(дифторметил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-3-фторфенил)уксусная кислота;
(4-{4,9-бис[(дифторметил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-3-хлорфенил)уксусная кислота;
(4-{4,9-бис[(дифторметил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-2-фторфенил)уксусная кислота;
(4-{4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}фенил)уксусная кислота;
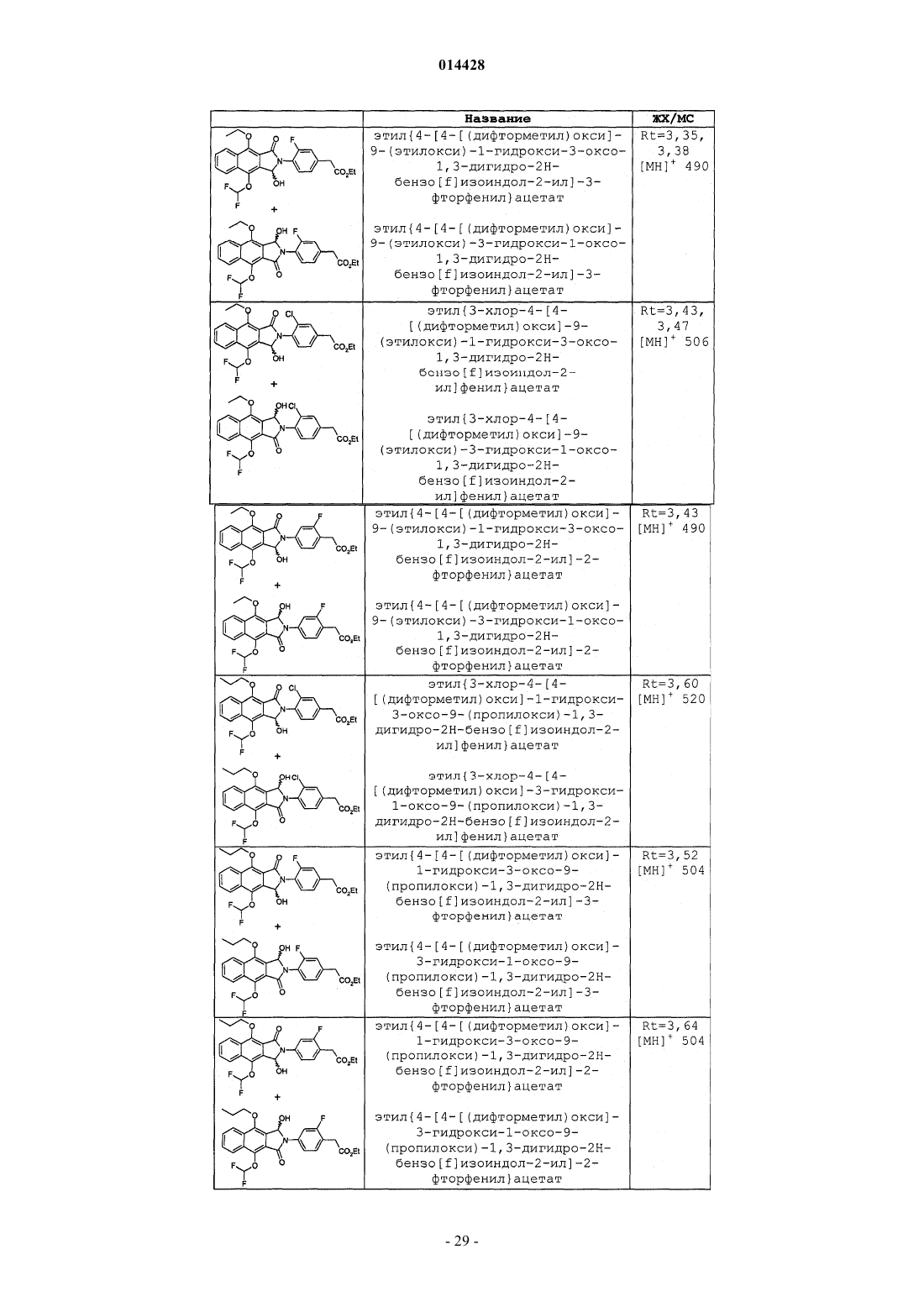
{4-[4-[(дифторметил)окси]-9-(этилокси)-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{4-[4-[(дифторметил)окси]-9-(метилокси)-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{4-[4-[(дифторметил)окси]-9-(этилокси)-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-3-фторфенил}уксусная кислота;
{3-хлор-4-[4-[(дифторметил)окси]-9-(этилокси)-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{4-[4-[(дифторметил)окси]-9-(этилокси)-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-2-фторфенил}уксусная кислота;
{4-[4-[(дифторметил)окси]-1,3-диоксо-9-(пропилокси)-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-3-фторфенил}уксусная кислота;
{3-хлор-4-[4-[(дифторметил)окси]-1,3-диоксо-9-(пропилокси)-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{4-[4-[(дифторметил)окси]-1,3-диоксо-9-(пропилокси)-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-2-фторфенил}уксусная кислота;
(4-{4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-3-фторфенил)уксусная кислота;
(3-хлор-4-{4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}фенил)уксусная кислота;
(4-{4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1,3-диоксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-2-фторфенил)уксусная кислота;
{3-хлор-4-[4-[(дифторметил)окси]-9-(этилокси)-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{3-хлор-4-[9-[(дифторметил)окси]-4-(этилокси)-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{4-[9-[(дифторметил)окси]-4-(метилокси)-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{4-[4-[(дифторметил)окси]-9-(метилокси)-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{4-[9-[(дифторметил)окси]-4-(этилокси)-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-3-фторфенил}уксусная кислота;
{4-[4-[(дифторметил)окси]-9-(этилокси)-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-3-фторфенил}уксусная кислота;
{4-[9-[(дифторметил)окси]-4-(этилокси)-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-2-фторфенил}уксусная кислота;
4-[4-[(дифторметил)окси]-9-(этилокси)-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-2-фторфенил}уксусная кислота;
{3-хлор-4-[4-[(дифторметил)окси]-1-оксо-9-(пропилокси)-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]фенил}уксусная кислота;
{4-[9-[(дифторметил)окси]-1-оксо-4-(пропилокси)-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-3-фторфенил}уксусная кислота;
(4-[4-[(дифторметил)окси]-1-оксо-9-(пропилокси)-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-3-фторфенил}уксусная кислота;
{4-[9-[(дифторметил)окси]-1-оксо-4-(пропилокси)-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-2-фторфенил}уксусная кислота;
{4-[4-[(дифторметил)окси]-1-оксо-9-(пропилокси)-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил]-2-фторфенил}уксусная кислота;
(3-хлор-4-{9-[(дифторметил)окси]-4-[(1-метилэтил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}фенил)уксусная кислота;
(3-хлор-4-{4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}фенил)уксусная кислота;
(4-{9-[(дифторметил)окси]-4-[(1-метилэтил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-3-фторфенил)уксусная кислота;
(4-{4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-3-фторфенил)уксусная кислота;
(4-{9-[(дифторметил)окси]-4-[(1-метилэтил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-2-фторфенил)уксусная кислота;
(4-{4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1-оксо-1,3-дигидро-2Н-бензо[f]изоиндол-2-ил}-2-фторфенил)уксусная кислота;
или их фармацевтически приемлемых производных.
7. Способ получения соединения формулы (I) по п.1, где один из X и Y представляет С=O и другой представляет СН2, R1, R2 и R3имеют значения, указанные в п.1, включающий взаимодействие соединения формулы (II)

где один из X и Y представляет С=O и другой представляет СН2; R1, R2и R3 имеют значения, указанные выше в п.1, и R4 представляет С1-6алкил, с подходящим основанием и необязательно с последующим образованием фармацевтически приемлемого производного полученного таким образом соединения и/или преобразование одного соединения формулы (I) в другое.
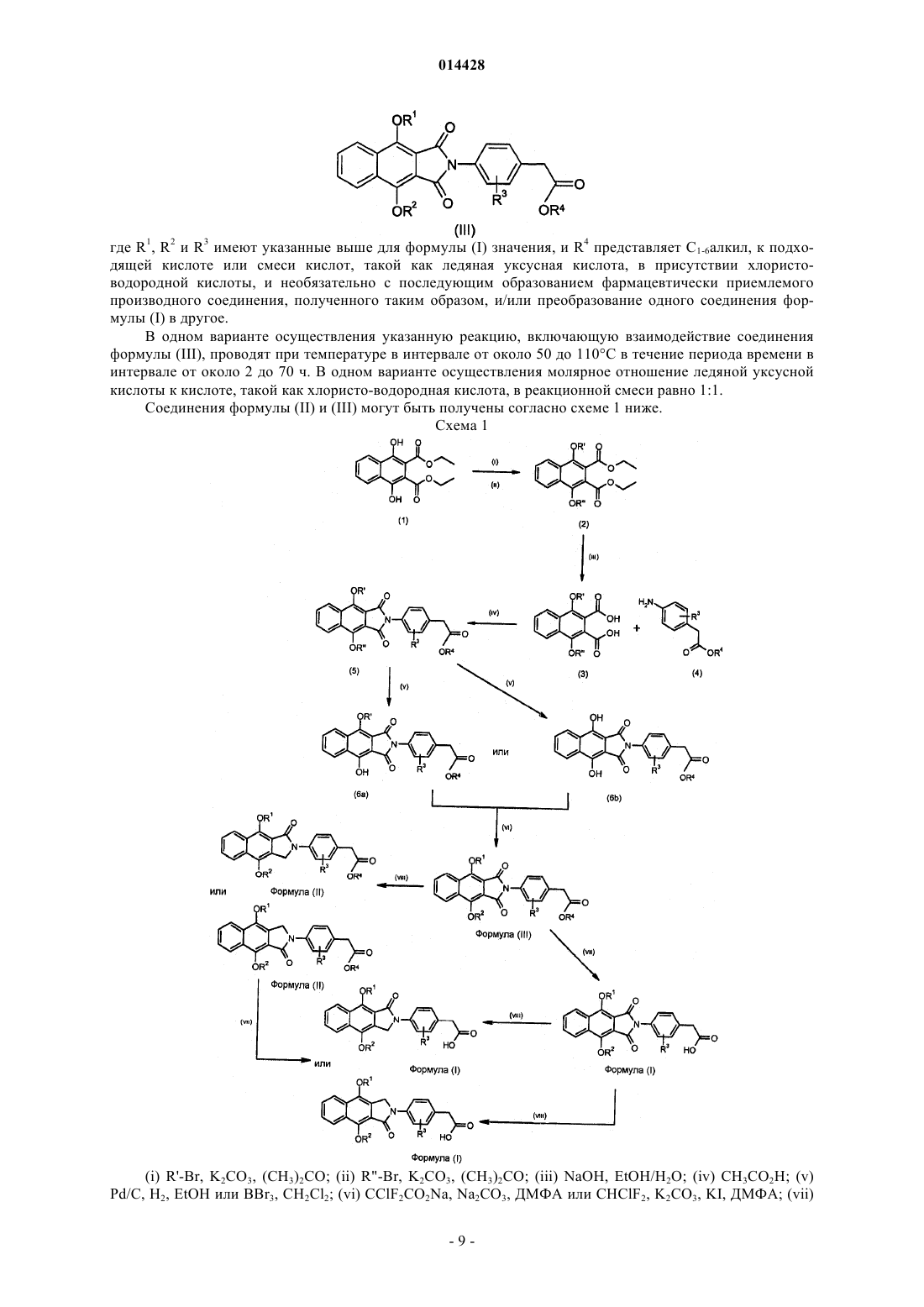
8. Способ получения соединения формулы (I) по п.1, где X и Y представляют С=O и R1, R2и R3 имеют значения, указанные выше в п.1, включающий добавление соединения формулы (III)

где R1, R2 и R3имеют значения, указанные выше в п.1, и R4 представляет C1-6алкил, к подходящей кислоте или смеси кислот и необязательно с последующим образованием фармацевтически приемлемого производного полученного таким образом соединения и/или преобразование одного соединения формулы (I) в другое.
9. Применение соединения формулы (I) по п.1 в медицине и ветеринарии.
10. Применение соединения формулы (I) по п.1 при лечении состояния, которое опосредуется действием или потерей действия PGE2 при рецепторах ЕР4.
11. Способ лечения человека или животного, страдающего от состояния, которое опосредуется действием или потерей действия PGE2 при рецепторах ЕР4, включающий введение указанному субъекту эффективного количества соединения формулы (I) по п.1.
12. Применение соединения формулы (I) по п.1 для производства лекарственного средства для лечения состояния, которое опосредуется действием PGE2при рецепторах ЕР4.
13. Фармацевтическая композиция, содержащая соединение формулы (I) по п.1 и один или несколько приемлемых носителей или разбавителей.
14. Фармацевтическая композиция по п.13, содержащая один или несколько дополнительных терапевтических агентов.
Текст
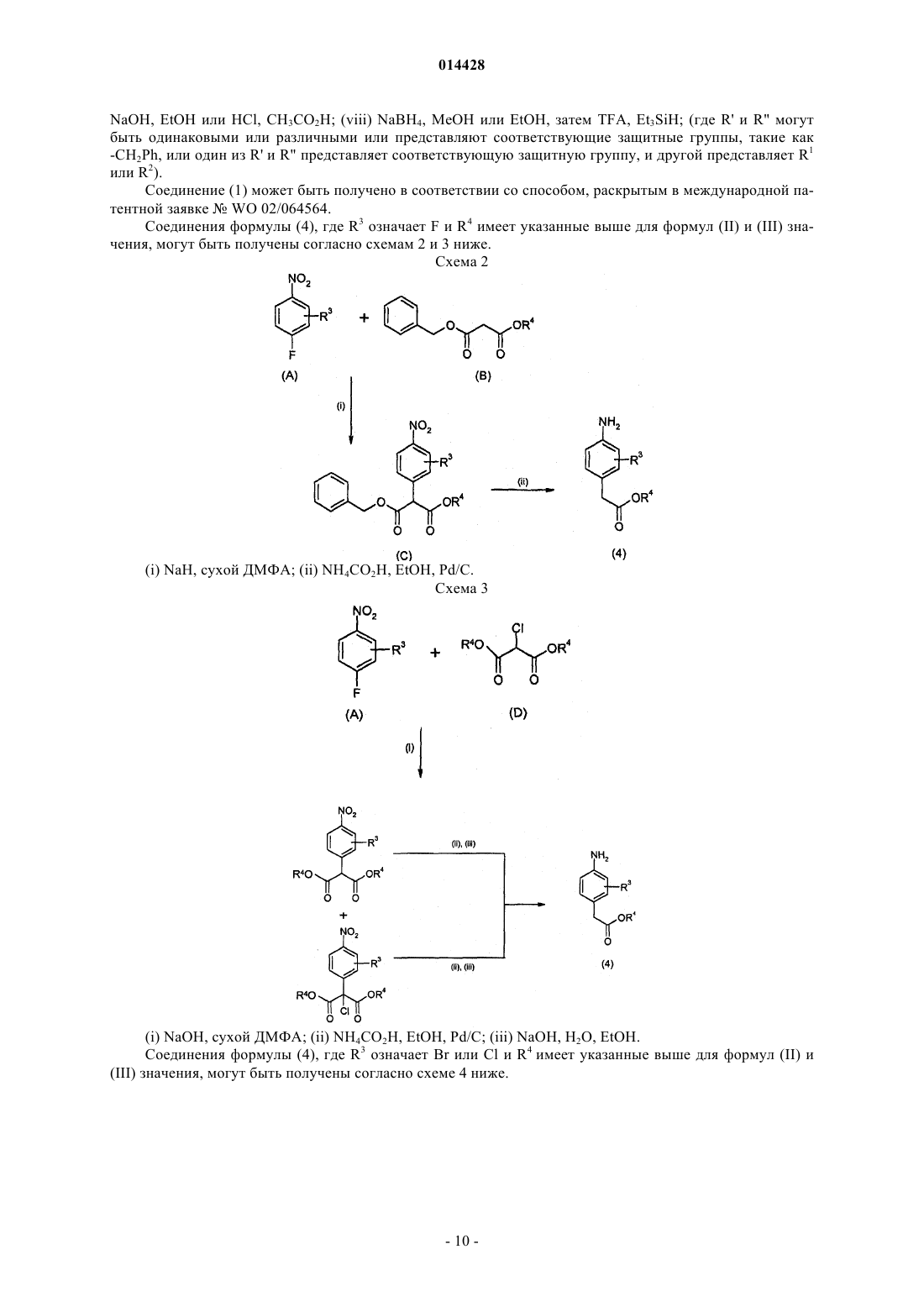
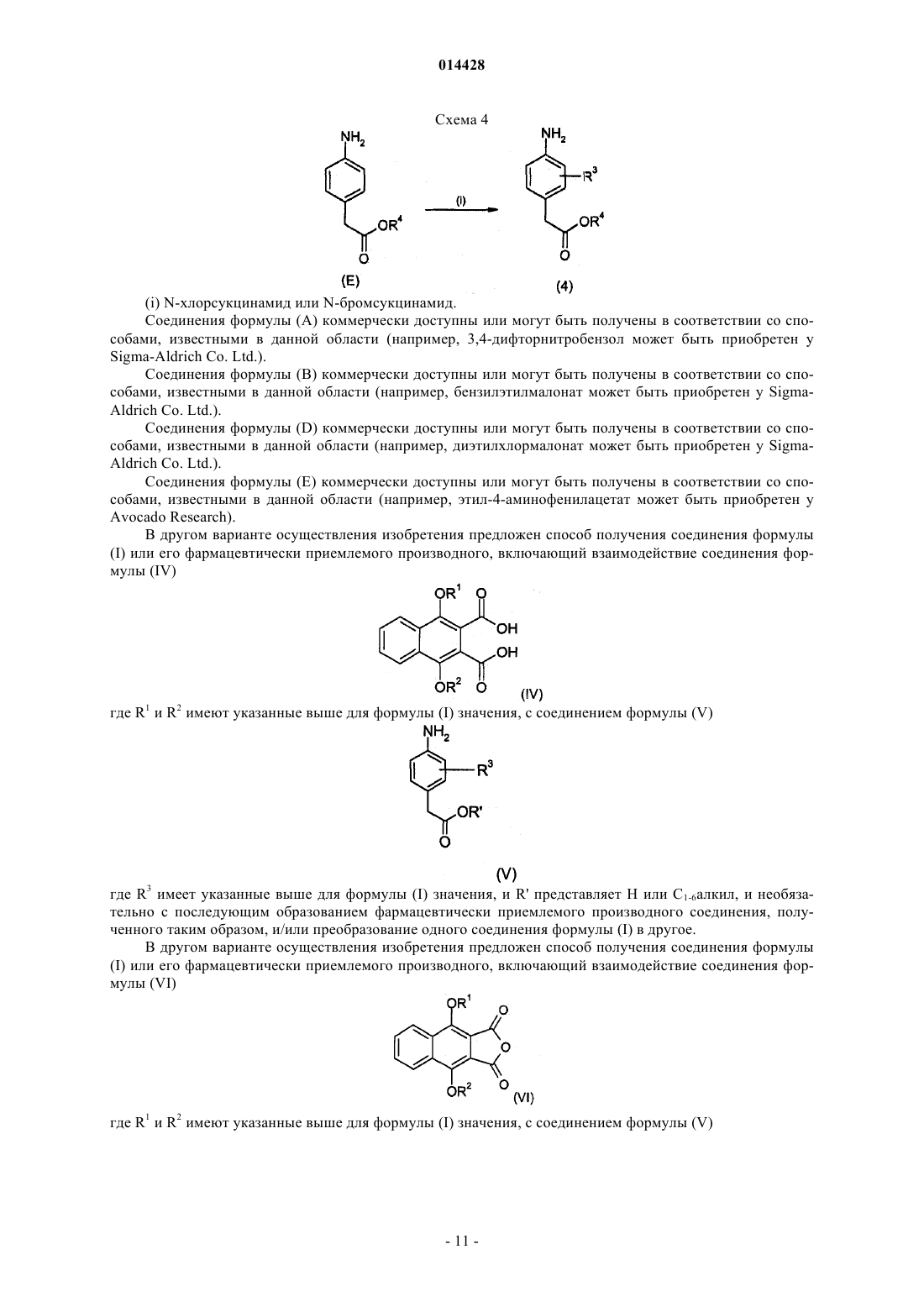
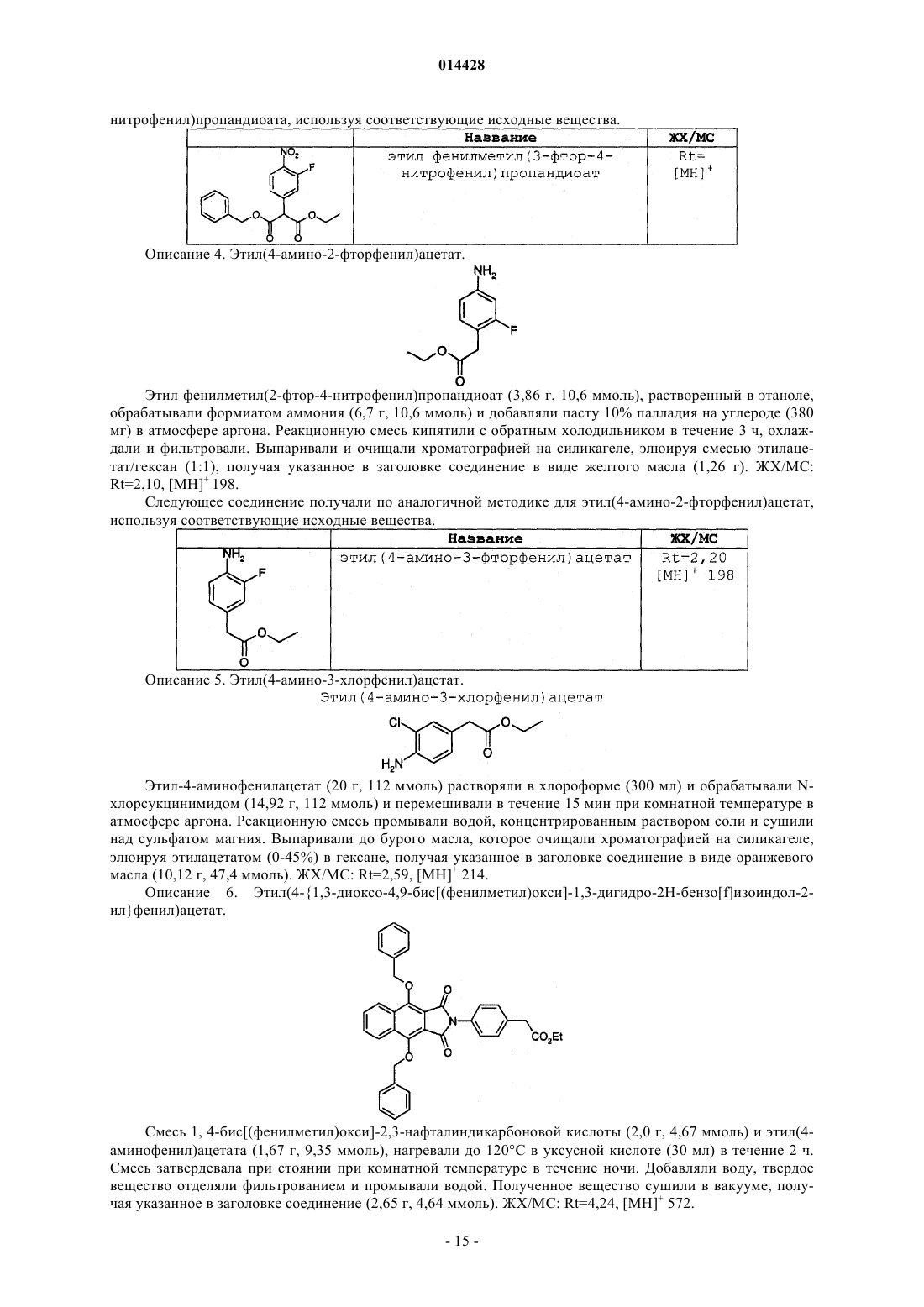
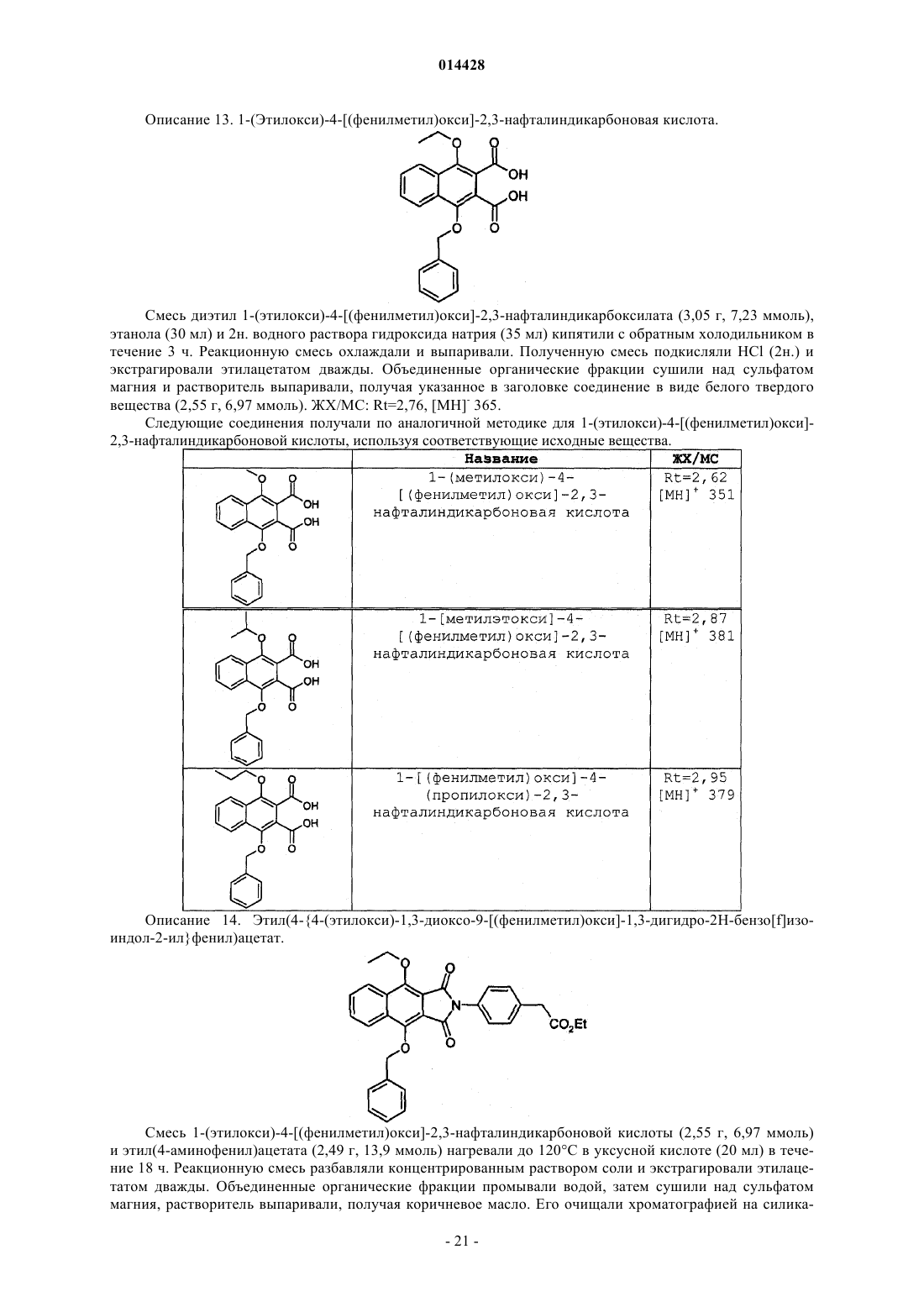
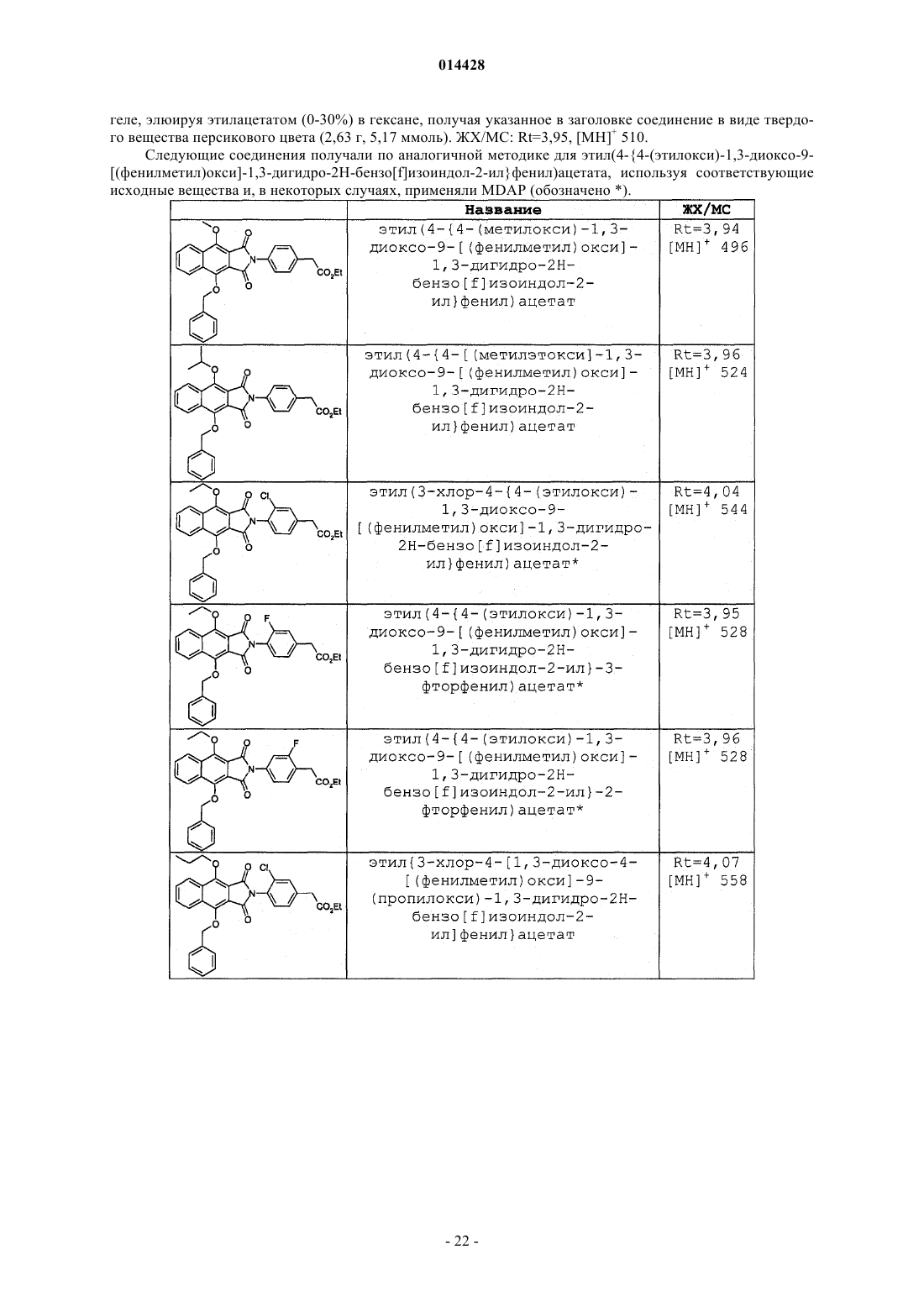
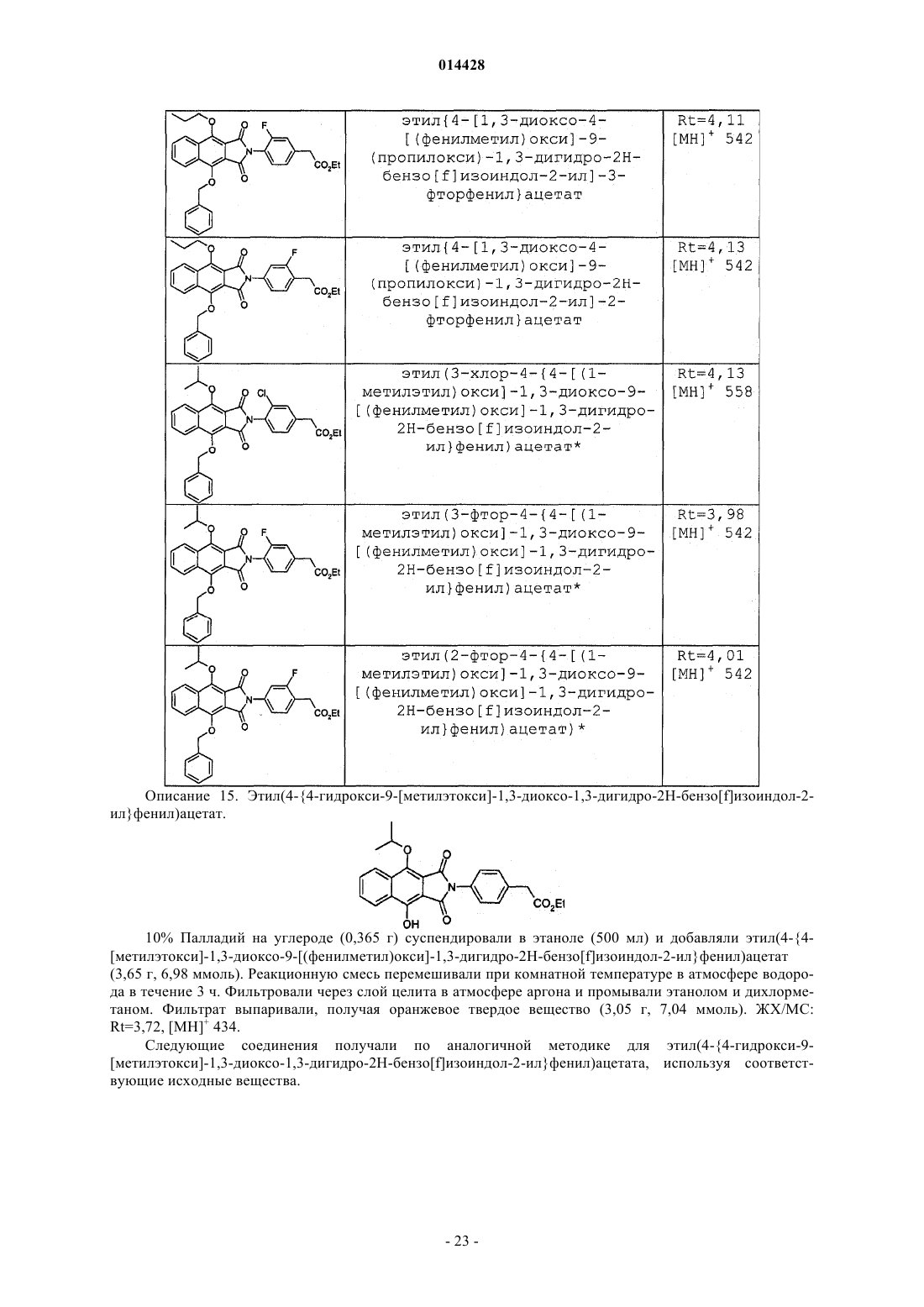
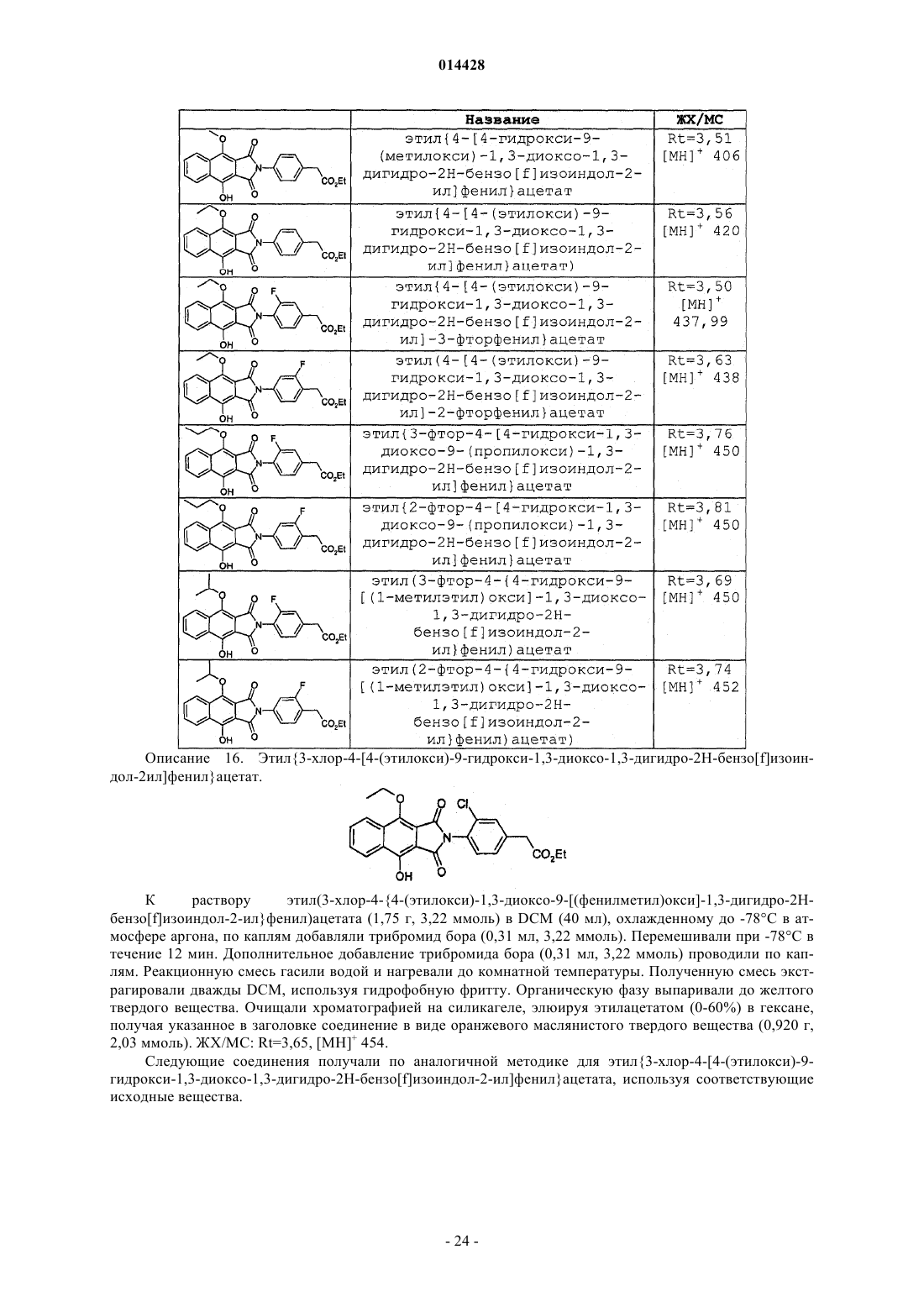
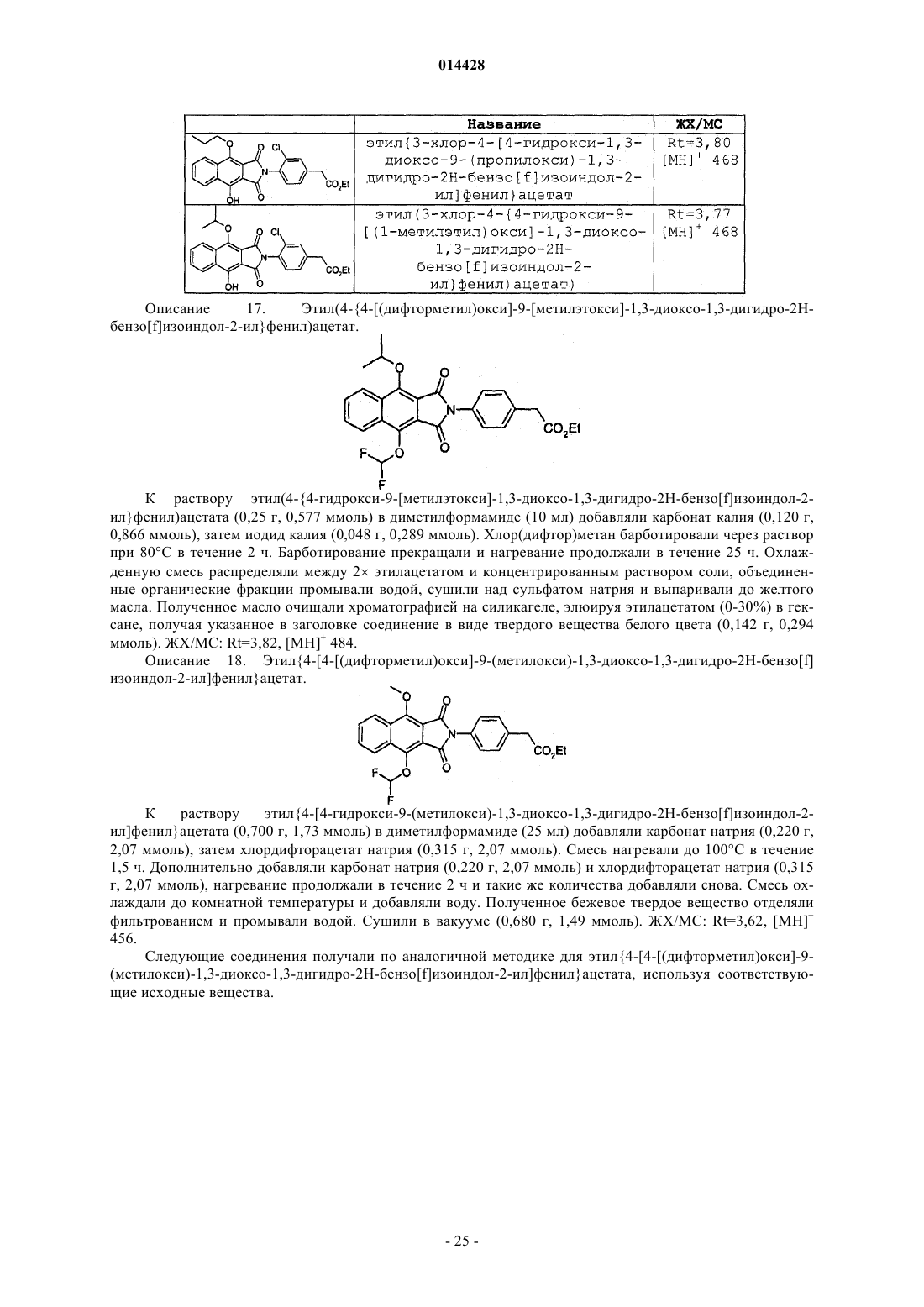
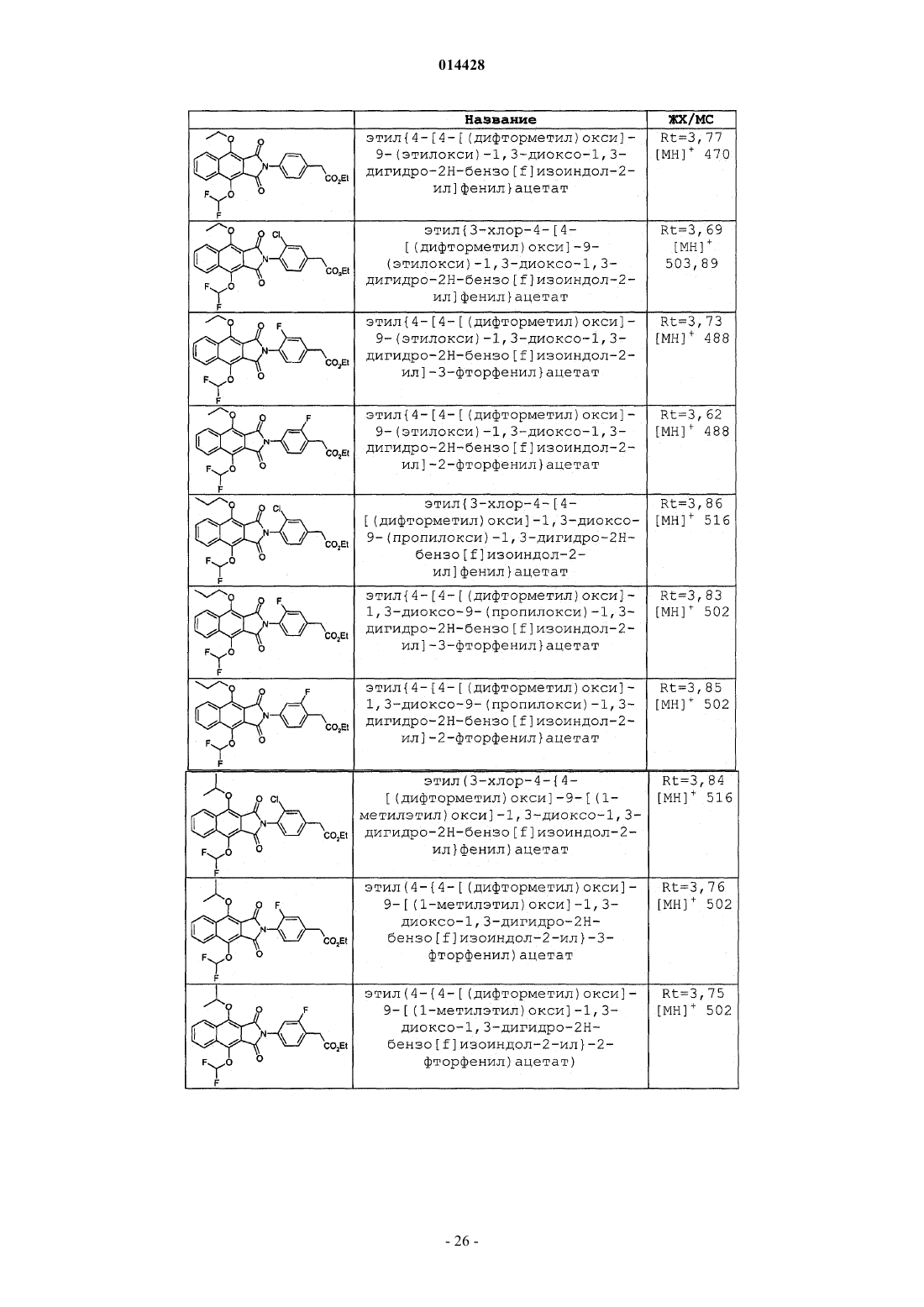
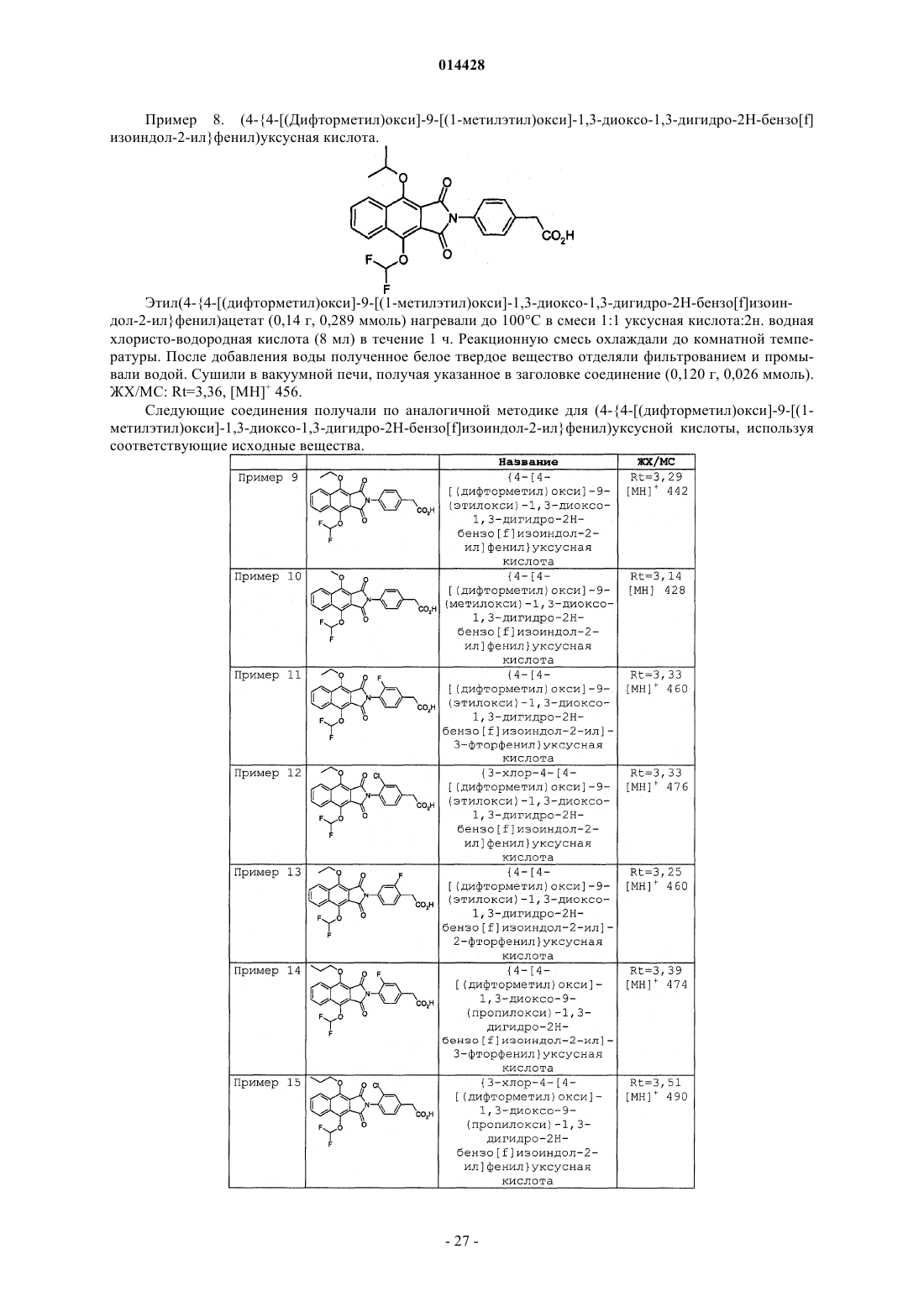
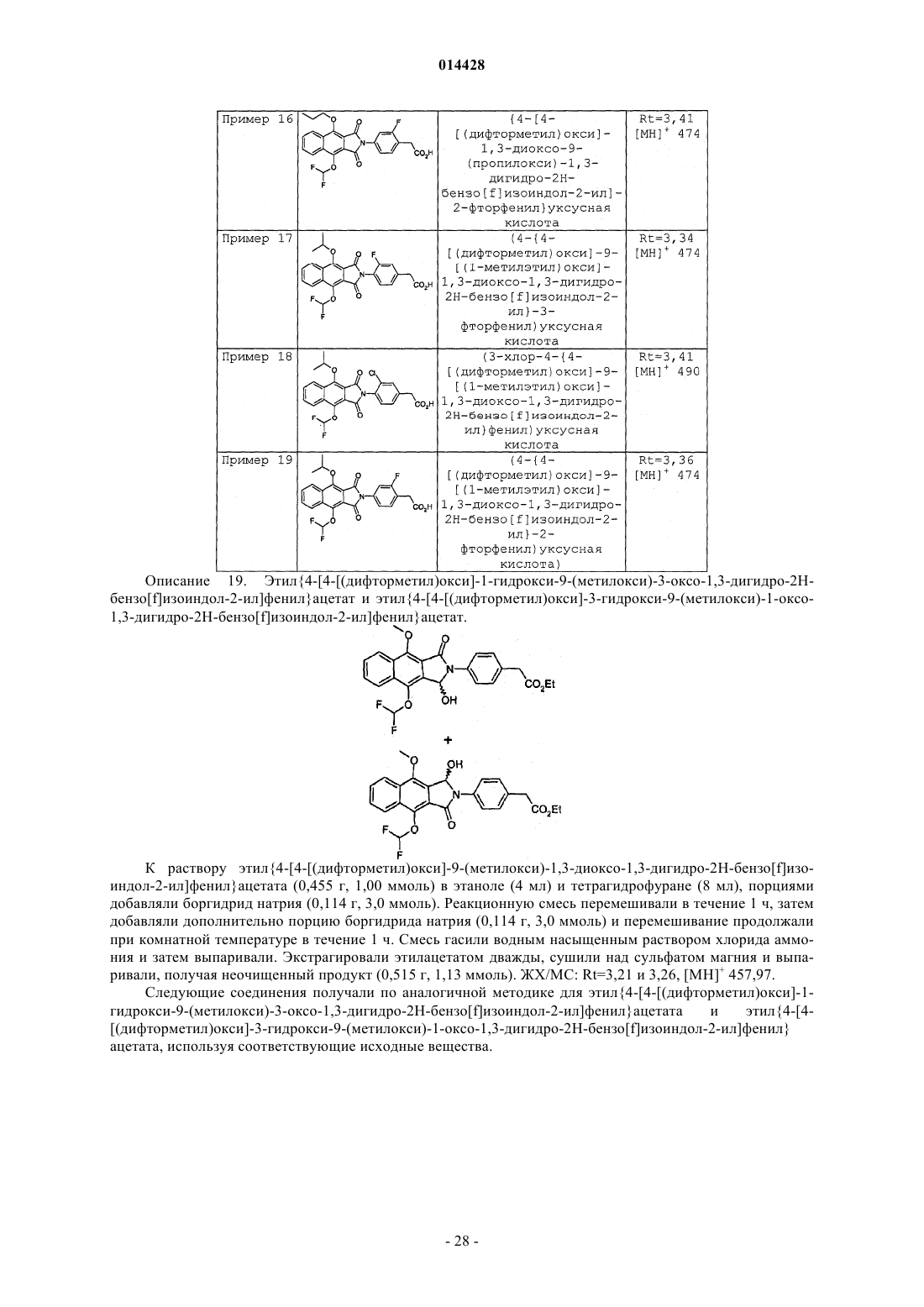
ПРОИЗВОДНЫЕ БЕНЗОИЗОИНДОЛА ДЛЯ ЛЕЧЕНИЯ БОЛИ(71)(73) Заявитель и патентовладелец: ГЛЭКСО ГРУП ЛИМИТЕД (GB) Хили Марк Патрик, Гиблин Джерард Мартин Пол, Прайс Хелен Сьюзанн (GB) Представитель: Соединение формулы (I) или его фармацевтически приемлемое производное, где R1, R2 и R3, X иY имеют указанные в описании значения, способ получения таких соединений,фармацевтическая композиция, содержащая такие соединения, и применение таких соединений в медицине. 014428 Данное изобретение относится к производным нафталина, способам их получения, содержащим их фармацевтическим композициям и к их применению в медицине. Соединения по данному изобретению являются агонистами рецептора ЕР 4. Несколько обзорных статей посвящено характеристике и терапевтической значимости простаноидных рецепторов, а также наиболее широко используемым селективным агонистам и антагонистам Eicosanoids; From Biotechnology to Therapeutic Applications, Folco, Samuelsson, Maclouf, and Velo eds, Plenumand Prostanoid Receptors, Structure, Properties and Function, S. Narumiya et al., Physiological Reviews 1999,79(4), 1193-126. Рецептор EP4 является 7-трансмембранным рецептором, и его природным лигандом является простагландин PGE2. PGE2 также обладает аффинностью в отношении других рецепторов ЕР (типы EP1, ЕР 2 и ЕР 3). Простаноидный рецептор ЕР 4 входит в группу рецепторов, нормально ассоциируемых с повышением уровня внутриклеточного циклического аденозинмонофосфата (сАМР). Рецептор ЕР 4 ассоциируется с релаксацией гладкой мускулатуры, внутриглазным давлением, болью (в частности, болью при воспалении, невропатической и внутренней болью), воспалением, нейрозащитой, дифференциацией лимфоцитов, костными метаболическими процессами, аллергическими активностями, стимулированием сна,почечной регуляцией, желудочной или кишечной слизистой секрецией и дуоденальной бикарбонатной секрецией. Рецептор ЕР 4 играет важную роль в закрывании артериального протока, вазодепрессии, воспалении и трансформировании костной ткани, как рассмотрено Narumiya в ProstaglandinsOther LipidMediators, 2002, 68-69, 557-73. В некоторых публикациях было продемонстрировано, что PGE2, действуя через подтип рецептора ЕР 4, и только агонисты ЕР 4 могут регулировать воспалительные цитокины после воспалительного стимула. Takayama et al. в Journal of Biological Chemistry, 2002, 277(46), 44147-54 показали, что PGE2 модулирует воспаление во время воспалительных заболеваний путем подавления продуцирования выделяемого макрофагами хемокина посредством рецептора ЕР 4. В BioorganicMedicinal Chemistry, 2002, 10(7),2103-2110, Maruyama et al. демонстрируют, что селективный агонист рецептора ЕР 4 (ONO-AE1-437) подавляет TNF-, индуцируемый LPS, в цельной крови человека, повышая в то же время уровни содержания IL-10. В статье в Anesthesiology, 2002, 97, 170-176, подтверждается то, что селективный агонист рецептора ЕР 4 (ONO-AE1-329) эффективно подавляет механическую и термическую гипералгезию и воспалительные реакции при остром и хроническом моноартрите. В двух независимых статьях Sakuma et al. в Journal of Bone and Mineral Research 2000, 15(2), 218-227 и Miyaura et al. в Journal of Biological Chemistry, 2000, 275(26), 19819-23 сообщают о пониженном образовании остеокластов в культивированных клетках от мышей с нокаутированным рецептором ЕР 4. Yoshidaet al. в Proceedings of the National Academy of Sciences of the United States of America 2002, 99(7), 45804585 путем использования мышей, лишенных подтипов ЕР рецептора PGE2, идентифицировали ЕР 4 как рецептор, который опосредует образование кости в ответ на введение PGE2. Они также продемонстрировали, что селективный агонист рецептора ЕР 4 (ONO-4819) равным образом индуцирует образование кости у мышей дикого типа. Дополнительно Terai et al. в Bone 2005, 37(4) 555-562 показали, что присутствие селективного агониста рецептора ЕР 4 (ONO-4819) усиливает способность индуцирования кости терапевтического цитокина rhBMP-2, который может индуцировать образование кости. Дальнейшее исследование, проведенное Larsen et al., показывает, что воздействия PGE2 на секрецию во втором отделе двенадцатиперстной кишки человека опосредуются через рецептор ЕР 4 (Acta.Physiol. Scand. 2005, 185, 133-140). Также было показано на крысах, что селективный агонист рецептора ЕР 4 (ONO-АЕ 1-329) может защищать от колита (Nitta et al. в Scandinavian Journal of Immunology 2002,56(1), 65-75).Dore et al. в European Journal of Neuroscience 2005, 22(9), 2199-206 показали, что PGE2 может защищать нейроны от амилоидной -пептидной токсичности путем воздействия на рецепторы ЕР 2 и ЕР 4. Более того, Dore было продемонстрировано в Brain Research 2005, 1066(1-2), 71-77, что агонист рецептора ЕР 4 (ONO-AE1-329) защищает от нейротоксичности на острой модели экситотоксичности в головном мозге.Woodward et al. в Journal of Lipid Mediators 1993, 6(1-3), 545-53 обнаружили, что внутриглазное давление может быть понижено с помощью селективных простаноидных агонистов. В двух статьях в Investigative OphthalmologyVisual Science показано, что простаноидный рецептор ЕР 4 экспрессируется в эпителиальных клетках хрусталика человека (Mukhopadhyay et al, 1999, 40(1), 105-12), и подтверждена физиологическая роль простаноидного рецептора ЕР 4 в модулировании потока в трабекулярной структуре глаза (Hoyng et al. 1999, 40(11), 2622-6). Соединения, проявляющие активность связывания рецептора ЕР 4, и их применения описаны, например, в WO 98/55468, WO 00/18744, WO 00/03980, WO 00/15608, WO 00/16760, WO 00/21532, WO 01010426, ЕР 0855389, ЕР 0985663, WO 02/047669, WO 02/50031, WO 02/50032, WO 02/50033, WO 02/064564, WO 03/103604, WO 03/077910, WO 03/086371, WO 04/037813, WO 04/067524, WO 04/085430,US 04/142969, WO 05/021508, WO 05/105733, WO 05/105732, WO 05/080367, WO 05/037812, WO-1 014428 05/116010 и WO 06/122403. Производные индопрофена, такие как натриевая соль [4-(1-оксо-1,3-дигидро-2 Н-бензо[f]изоиндол 2-ил)фенил]-2-пропионовой кислоты, описаны Rufer et al. в Eur. J. Med. Chem. - Chimica Therapeutica,1978, 134, 193. Данное изобретение относится к соединениям формулы (I) и/или их фармацевтически приемлемым производным где R1 и R2 независимо представляют С 1-4 алкил или дифторметил, при условии, что по меньшей мере один из R1 и R2 представляет дифторметил;X и Y независимо представляют С=O или СН 2, при условии, что по меньшей мере один из X и Y представляет С=O. В одном варианте осуществления изобретения один из R1 и R2 представляет С 1-4 алкил, и другой представляет дифторметил. В одном варианте осуществления изобретения один из R1 и R2 представляет метил, и другой представляет дифторметил. В одном варианте осуществления изобретения один из R1 иR2 представляет этил, и другой представляет дифторметил. В одном варианте осуществления изобретения один из R1 и R2 представляет н-пропил, и другой представляет дифторметил. В одном варианте осуществления изобретения один из R1 и R2 представляет изопропил, и другой представляет дифторметил. В другом варианте осуществления изобретения оба R1 и R2 представляют дифторметил. В одном варианте осуществления изобретения R3 представляет F. В другом варианте осуществления изобретения R3 представляет Cl. В другом варианте осуществления изобретения R3 представляет Н. В другом варианте осуществления изобретения R3 представляет Н, F или Cl. В другом варианте осуществления изобретения R3 представляет F или Cl. В одном варианте осуществления изобретения X представляет СН 2, и Y представляет С=O. В другом варианте осуществления изобретения X представляет С=O, и Y представляет СН 2. В другом варианте осуществления изобретения оба X и Y представляют С=O. В другом варианте осуществления изобретения предложено соединение формулы (I), выбранное из группы, состоящей из таких соединений, как(4-4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1-оксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил-2 фторфенил)уксусная кислота или их фармацевтически приемлемое производное. Данное изобретение охватывает все комбинации конкретных и предпочтительных вариантов осуществления, которые описаны в данном описании. Используемый в данном описании термин "C1-4 алкил" включает алкильные группы с прямой и разветвленной цепью, содержащие от 1 до 4 атомов углерода, такие как метил, этил, н-пропил, изопропил,бутил и изобутил. Термин "C1-6 алкил" может быть интерпретирован соответственно. Используемый в данном описании термин дифторметил означает -CHF2. Используемый в данном описании F означает фтор, Cl означает хлор и Br означает бром. Под фармацевтически приемлемым производным подразумевается какая-либо фармацевтически приемлемая соль, сольват или сложный эфир, или соль, или сольват такого сложного эфира соединений формулы (I), или какое-либо другое соединение, которое при введении реципиенту способно обеспечивать (непосредственно или опосредованно) соединение формулы (I) или его активный метаболит или остаток. В одном варианте осуществления изобретения фармацевтически приемлемое производное озна-3 014428 чает соль, сольват или сложный эфир или соль или сольват такого сложного эфира. В другом варианте осуществления изобретения фармацевтически приемлемое производное означает соль, или сложный эфир, или соль такого сложного эфира. Следует учитывать, что для фармацевтического применения указанные выше соли должны быть фармацевтически приемлемыми солями, но другие соли могут находить применение, например при получении соединений формулы (I) и их фармацевтически приемлемых солей. Фармацевтически приемлемые соли включают такие, как описано Berge, Bighley and Monkhouse, J.Pharm. Sci., 1977, 66, 1-19. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых оснований, включая неорганические основания и органические основания. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция,меди, железа(III), железа(II), лития, магния, соли марганца(III), марганца(II), калия, натрия, цинка и тому подобное. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов; замещенных аминов, включая встречающиеся в природе замещенные амины, и циклических аминов. Конкретные фармацевтически приемлемые органические основания включают аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2 диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, Nэтилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин,морфолин, пиперазин, пиперидин, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трис(гидроксиметил)аминометан и т.п. Соли также могут быть образованы из ионообменных смол основного характера, например полиаминосмолы. Необходимо учитывать, что соединение формулы (I) может быть образовано in vivo путем метаболизма соответствующего пролекарства. Такими пролекарствами могут быть, например, физиологически приемлемые метаболически лабильные сложные эфиры соединений общей формулы (I). Они могут быть образованы переэтерификацией карбоксильной кислотной группы исходного соединения общей формулы (I), где уместно с предварительной защитой каких-либо других реакционноспособных групп, присутствующих в молекуле, с последующим удалением защитной группы, если требуется. Примеры таких метаболически лабильных сложных эфиров включают С 1-4 алкиловые сложные эфиры, например метилэтиловый или трет-бутиловый сложный эфир, С 3-6 алкениловые сложные эфиры, например замещенные аллилом или незамещенные аминоалкиловые сложные эфиры (например, аминоэтиловый, 2-(N,Nдиэтиламино)этиловый или 2-(4-морфолино)этиловый сложные эфиры или ацилоксиалкиловые сложные эфиры, такие как ацилоксиметиловый или 1-ацилоксиэтиловый, например пивалоилоксиметиловый, 1 пивалоилоксиэтиловый, ацетоксиметиловый, 1-ацетоксиэтиловый, 1-(1-метокси-1-метил)этилкарбонилоксиэтиловый, 1-бензоилоксиэтиловый, изопропоксикарбонилоксиметиловый, 1-изопропоксикарбонилоксиэтиловый, циклогексилкарбонилоксиметиловый, 1-циклогексилкарбонилоксиэтиловый сложный эфир, циклогексилоксикарбонилоксиметиловый, 1-циклогексилоксикарбонилоксиэтиловый, 1-(4 тетрагидропиранилокси)карбонилоксиэтиловый или 1-(4-тетрагидропиранил)карбонилоксиэтиловый. Должно быть понятно, что данное изобретение охватывает все изомеры соединений формулы (I) и их фармацевтически приемлемых производных, включая все геометрические, таутомерные и оптические формы и их смеси (например, рацемические смеси). Так как соединения по данному изобретению, в частности соединения формулы (I), предназначаются для применения в фармацевтических композициях, должно быть понятно, что каждое из них представлено, по существу, в чистой форме, например по меньшей мере 50% чистоты, более подходяще по меньшей мере 75% чистоты и предпочтительно по меньшей мере 95% чистоты (% даны на основе мас./мас.). Загрязненные препараты соединений формулы (I) могут быть использованы для получения более чистых форм, используемых в фармацевтических композициях. Хотя чистота промежуточных соединений по данному изобретению является менее критической, легко можно понять, что, по существу,чистая форма предпочтительна, как для соединений формулы (I). Предпочтительно всякий раз, когда возможно, соединения по данному изобретению получают в кристаллической форме. Когда некоторым соединениям по данному изобретению дают возможность кристаллизоваться или подвергают перекристаллизации из органических растворителей, растворитель кристаллизации может присутствовать в кристаллическом продукте. Данное изобретение включает в свой объем такие сольваты. Подобным образом, некоторые соединения по данному изобретению могут быть кристаллизованы или перекристаллизованы из растворителей, содержащих воду. В таких случаях может образовываться вода гидратации. Данное изобретение включает в свой объем стехиометрические гидраты, а также соединения, содержащие переменное количество воды, которые могут образовываться при таких процессах, как лиофилизация. В дополнение, различные условия кристаллизации могут приводить к образованию различных полиморфных форм кристаллических продуктов. Данное изобретение включает в свой объем все полиморфные формы соединений формулы (I). Настоящее изобретение также включает в свой объем все меченные изотопами соединения формулы (I). Такие соединения идентичны соединениям, обозначенным формулой (I), за исключением того,что один или несколько их атомов заменены атомом, имеющим атомную массу или массовое число, от-4 014428 личное от атомной массы или массового числа, обычно обнаруживаемого в природе. Примеры изотопов,которые могут быть введены в соединения формулы (I), и их фармацевтически приемлемые производные, включают изотопы водорода, углерода, азота, кислорода, фтора и хлора, такие как 2 Н, 3 Н, 11 С, 13 С,14 С, 15N, 17O, 18O, 18F и 36 Сl. Меченные изотопами соединения по данному изобретению, например соединения, в которые введены радиоактивные изотопы, такие как 3 Н, 14 С, полезны в анализах распределения в тканях лекарства и/или субстрата. Изотопы тритий, т.е. 3 Н, и углерод-14, т.е. 14 С, особенно предпочтительны из-за легкости их получения и обнаружения. Изотопы 11 С и 18F особенно полезны в PET (позитронной эмиссионной томографии) и используются для получения изображения головного мозга. Дополнительным замещением более тяжелыми изотопами, такими как дейтерий, т.е. 2 Н, можно достичь некоторых терапевтических преимуществ, являющихся результатом более высокой метаболической устойчивости, например увеличения периода полураспада in vivo или требования уменьшенной дозировки, и, следовательно, могут быть предпочтительны в некоторых обстоятельствах. Меченные изотопами соединения формулы (I) могут быть получены осуществлением способов синтеза, раскрытых на схемах и/или в примерах ниже,путем замещения легко доступным меченым изотопом реагентом не меченного изотопом реагента. Соединения по изобретению являются агонистами рецептора ЕР 4 и поэтому могут быть полезны при лечении заболеваний, опосредуемых рецептором ЕР 4. В частности, соединения формулы (I) могут быть полезны при лечении боли, например, хронической суставной боли (например, ревматоидного артрита, остеоартрита, ревматоидного спондилита, подагрического артрита и юношеского артрита), включая свойства модификации болезни и предохранение структуры сустава; скелетно-мышечной боли; боли нижнего отдела спины и шеи; боли при растяжениях и деформациях; невропатической боли; симпатически поддерживаемой боли; миозита; боли, связанной с раком и фибромиалгией; боли, связанной с мигренью; боли, связанной с гриппом или другими вирусными инфекциями, такими как простуда; ревматической лихорадки; боли, связанной с функциональными расстройствами кишечника, такими как неязвенная диспепсия, несердечная боль грудной клетки и синдром раздраженного кишечника; боли, связанной с ишемией миокарда; постоперационной боли; головной боли; зубной боли и дисменореи. Соединения формулы (I) могут быть особенно полезны при лечении невропатической боли и связанных с этим симптомов. Синдромы невропатической боли включают диабетическую невропатию,ишиас, неспецифическую боль нижнего отдела спины, боль при рассеянном склерозе, фибромиалгию,относящуюся к ВИЧ, невропатию, постгерпетическую невралгию, невралгию тройничного нерва и боль в результате физической травмы, ампутации, рака, токсинов или хронических воспалительных состояний. Симптомы невропатической боли включают спонтанную, стреляющую и колющую боль или длящуюся жгучую боль. В дополнение, включена боль, связанная с обычно неболезненными ощущениями, такими как "шипы и иголки" (параэстезия и дисэстезия), повышенная чувствительность к прикосновению (гиперэстезия), болезненное ощущение, сопровождающее безобидную стимуляцию (динамическая, статическая или термическая аллодиния), повышенная чувствительность к вредным стимулам (тепловая, холодовая, механическая гипералгезия), длящееся болевое ощущение после удаления стимуляции (гиперпатия) или отсутствие или недостаточность селективных сенсорных путей (гипоалгезия). Соединения формулы (I) также могут быть полезны при лечении воспаления, например при лечении кожных состояний (например, солнечного ожога, ожогов, экземы, дерматита, псориаза); глазных болезней, таких как глаукома, ретинопатии, увеит и острое поражение глазной ткани (например, конъюнктивит); легочных расстройств (например, астмы, бронхита, эмфиземы, аллергического ринита, синдрома дыхательной недостаточности, болезни любителей голубей, экзогенного аллергического альвеолита (легкие фермера), COPD; расстройств желудочно-кишечного тракта (например, ящурной язвы, болезни Крона, атопического гастрита, гастриты varialoforme, язвенного колита, заболеваний брюшной полости, регионального илеита, синдрома раздраженного кишечника, воспалительной болезни кишечника,желудочно-кишечного рефлюкса, диарреи, запора); при трансплантации органов; при других состояниях с воспалительной составляющей, таких как болезнь сосудов, мигрень, нодозный периартериит, тироидит,апластическая анемия, болезнь Ходжкина, склеродома, миастения высокой степени, рассеянный склероз,соркоидоз, нефротический синдром, синдром Бехчета, полимиозит, гингивит, ишемия миокарда, пирексия, системная красная волчанка, полимиозит, тендинит, бурсит и синдром Шегрена. Соединения формулы (I) также могут быть полезны при лечении иммунологических заболеваний,таких как аутоиммунные заболевания, заболевания иммунологической недостаточности или при трансплантации органов. Соединения формулы (I) могут быть также эффективны в увеличении латентности инфекции ВИЧ. Соединения формулы (I) также могут быть полезны при лечении заболеваний с избыточной или нежелательной активацией тромбоцитов, таких как перемежающаяся хромота, нестабильная стенокардия, удар и острый коронарный синдром (например, окклюзионные сосудистые заболевания). Соединения формулы (I) также могут быть полезны в качестве лекарственного средства с диуретическим действием или могут быть полезны при лечении синдрома сверхактивного мочевого пузыря. Соединения формулы (I) также могут быть полезны при лечении импотенции или эректильной-5 014428 дисфункции. Соединения формулы (I) также могут быть полезны при лечении заболеваний костей, характеризующихся аномальным костным метаболизмом или резорбцией, таких как остеопороз (особенно постклимактерический остеопороз), гиперкальциемия, гиперпаратироидизм, костные болезни Педжета,остеолиз, гиперкальциемия злокачественного новообразования с костными метастазами или без них,ревматоидный артрит, периодонтит, остеоартрит, остеалгия, калькулез, литиаз (особенно уролитиаз),подагра и анкилозный спондилит, тендинит и бурсит. Соединения формулы (I) также могут быть полезны в моделировании костей, и/или промотировании образования кости, и/или промотировании заживления перелома. Соединения формулы (I) также могут быть полезны для ослабления гемодинамических побочных эффектов NSAID и ингибиторов СОХ-2. Соединения формулы (I) также могут быть полезны при лечении сердечно-сосудистых заболеваний,таких как гипертензия или ишемия миокарда; функциональная или органическая венозная недостаточность; варикозная терапия; геморроидальные узлы и шоковые состояния, связанные с заметным падением артериального давления (например, септический шок). Соединения формулы (I) также могут быть полезны при лечении нейродегенеративных заболеваний, таких как деменция, особенно дегенеративная деменция (включая сенильную деменцию, болезнь Альцгеймера, болезнь Пика, Хорею Хантингтона, болезнь Паркинсона и болезнь Крейтцфельдта-Якоба,амиотрофический боковой склероз (ALS), болезнь двигательных нейронов); сосудистая деменция (включая мультиинфарктную деменцию); а также деменция, связанная с поражениями, занимающими интракраниальное пространство; травма; инфекции и относящиеся к ним состояния (включая инфекцию ВИЧ); метаболизм; токсины; аноксия и недостаточность витаминов и слабое ухудшение познавательной способности, связанное со старением, особенно возрастное ухудшение памяти. Соединения формулы (I) также могут быть полезны при лечении неврологических расстройств и могут быть применимы в качестве нейрозащитных агентов. Соединения по изобретению также могут быть полезны при лечении нейродегенерации после удара, сердечного приступа, легочного шунтирования, травматического повреждения головного мозга, повреждения спинного мозга или т.п. Соединения формулы (I) также могут быть полезны при лечении осложнений диабета типа 1 (например, диабетической микроангиопатии, диабетической ретинопатии, диабетической нефропатии, дегенерации желтого пятна, глаукомы), нефротического синдрома, апластической анемии, увеита, болезни Кавасаки и саркоидоза. Соединения формулы (I) также могут быть полезны при лечении дисфункции почек (нефрита, особенно мезангиального пролиферативного громерулонефрита, нефритного синдрома), дисфункции печени(гепатита, цирроза) и желудочно-кишечной дисфункции (диарреи). Должно быть понятно, что ссылка на лечение включает и лечение установленных синдромов, и профилактические мероприятия. Согласно дополнительному варианту осуществления изобретения предложено соединение формулы(I) или его фармацевтически приемлемое производное для применения в медицине или ветеринарии. Согласно другому варианту осуществления изобретения предложено соединение формулы (I) или его фармацевтически приемлемое производное для применения при лечении состояния, которое опосредуется действием или потерей действия PGE2 при рецепторах ЕР 4. Согласно другому варианту осуществления изобретения предложен способ лечения человека или животного, страдающего от состояния, которое опосредуется действием или потерей действия PGE2 при рецепторах ЕР 4, включающий введение указанному субъекту эффективного количества соединения формулы (I) или его фармацевтически приемлемого производного. Согласно дополнительному варианту осуществления изобретения предложен способ лечения человека или животного, страдающего от боли или от воспалительного, иммунологического или костного заболеваний, нейродегенеративного заболевания или почечной дисфункции, включающий введение указанному субъекту эффективного количества соединения формулы (I) или его фармацевтически приемлемого производного. Согласно другому варианту осуществления изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемого производного для производства лекарственного средства для лечения состояния, которое опосредуется действием PGE2 при рецепторах ЕР 4. Согласно другому варианту осуществления изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемого производного для производства лекарственного средства для лечения или профилактики состояния, такого как боль или воспалительное, иммунологическое, костное, нейродегенеративное или почечное расстройство. Соединения формулы (I) или их фармацевтически приемлемые производные без труда вводят в форме фармацевтических композиций. Такие композиции легко могут быть предоставлены для применения традиционным образом в смеси с одним или несколькими физиологически приемлемыми носителями или эксципиентами. Таким образом в другом аспекте изобретения предложена фармацевтическая композиция, содер-6 014428 жащая соединение формулы (I) или его фармацевтически приемлемое производное, приспособленное для применения в медицине или ветеринарии. Хотя и возможно вводить соединения формулы (I) или их фармацевтически приемлемые производные в виде сырого химиката, предпочтительно предоставлять их в виде фармацевтической композиции. Композиции по данному изобретению содержат соединения формулы (I) или их фармацевтически приемлемые производные вместе с одним или несколькими приемлемыми носителями или разбавителями для них и необязательно другие терапевтические ингредиенты. Носитель (носители) должен быть "приемлемым" в смысле совместимости с другими ингредиентами композиции и безвредности для принимающего его. Композиции включают композиции, подходящие для перорального, парентерального (включая подкожное, например, путем инъекции или посредством депо-таблетки, интрадермальное, интратекальное, внутримышечное, например, посредством депо, и внутривенное), ректальное и местное (включая дермальное, буккальное и сублингвальное) введение, хотя наиболее подходящий путь может зависеть,например, от состояния и расстройства у пациента. Композиции обычно могут быть представлены в единичной дозированной форме и могут быть получены способами, хорошо известными в фармации (см.,например, способы, раскрытые в "Remington - The Science and Practice of Pharmacy", 21th Edition, Lippincott, WilliamsWilkins, USA, 2005 и приведенные там ссылки). Все способы включают стадию объединения соединения формулы (I) или его фармацевтически приемлемого производного ("активный ингредиент") с носителем, который состоит из одного или нескольких вспомогательных ингредиентов. Как правило, композиции готовят путем равномерного и тонкого объединения активного ингредиента с жидкими носителями или тонкоизмельченными твердыми носителями, или и теми, и другими, и затем, если необходимо, формования продукта в виде желаемого препарата. Препараты по данному изобретению, подходящие для перорального введения, могут быть представлены как дискретные единицы, такие как капсулы, саше или таблетки (например, таблетки для жевания, в особенности для введения детям), каждая из которых содержит заданное количество активного ингредиента в виде порошка или гранул, в виде раствора или суспензии в водной или неводной жидкости или в виде жидкой эмульсии типа масло-в-воде или жидкой эмульсии типа вода-в-масле. Активный ингредиент может быть также представлен в виде болюса, электуария или пасты. Таблетка может быть изготовлена прессованием или формованием необязательно с одним или несколькими вспомогательными ингредиентами. Прессованные таблетки могут быть получены прессованием на подходящей машине активного ингредиента в свободно сыпучей форме, такой как порошок или гранулы, необязательно в смеси со связующим, лубрикантом, инертным разбавителем, смазывающим поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки могут быть изготовлены формованием на соответствующей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки необязательно могут быть покрыты или могут иметь насечки, или могут быть составлены таким образом, чтобы обеспечивать замедленное или регулируемое высвобождение из них активного ингредиента. Препараты для парентерального введения включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества,которые поддерживают состав изотоничным с кровью предполагаемого реципиента, и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Препараты могут быть представлены в контейнерах на одну дозу или на несколько доз, например, в герметичных ампулах и пузырьках и могут храниться в высушенном (лиофилизованном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Импровизированные растворы и суспензии для инъекций могут быть приготовлены из стерильных порошков, гранул и таблеток такого вида, как описано ранее. Препараты для ректального введения могут быть представлены в виде суппозитория с обычными носителями, такими как какао-масло, твердый жир или полиэтиленгликоль. Препараты для местного применения через рот, например буккально или сублингвально, включают облатки, содержащие активный ингредиент в ароматизированной основе, такой как сахароза и камедь акации или трагакант, и пастилки, содержащие активный ингредиент в основе, такой как желатин и глицерин или сахароза и камедь акации. Соединения по изобретению также могут быть составлены как препараты депо. Такие композиции пролонгированного действия могут быть введены имплантацией (например, подкожно или внутримышечно) или внутримышечной инъекцией. Так, например, соединения по изобретению могут быть приготовлены с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле), или с ионообменными смолами, или как слабо растворимые производные, например как слабо растворимая соль. В дополнение к ингредиентам, конкретно приведенным выше, композиции могут включать другие агенты, обычные в данной области, имеющие отношение к типу рассматриваемого состава, например,которые подходят для перорального введения, могут включать ароматизирующие агенты. Соединения рецептора ЕР 4 для применения по данному изобретению могут быть использованы в-7 014428 сочетании с другими терапевтическими агентами, например с ингибиторами СОХ-2, такими как целекоксиб, рофекоксиб, валдекоксиб или парекоксиб; с ингибиторами 5-липоксигеназы; аналгетиками, такими как парацетамол; NSAID, такими как диклофенак, индометацин, набуметон, напроксен или ибупрофен; с антагонистами рецептора лейкотриена; DMARD, такими как метотрексат; с блокаторами натриевых каналов, такими как ламотригин; антагонистами кальциевых каналов N-типа; модуляторами рецептораNMDA, такими как антагонисты рецептора глицина, габапентин, прегабалин и родственные соединения; с трициклическими антидепрессантами, такими как амитриптилин; со стабилизирующими нейроны антиэпилептическими лекарственными средствами; ингибиторами моноаминергического усвоения, такими как венлафаксин; опиоидными аналгетиками; местными анестетиками; агонистами 5HT1, такими как триптаны, например суматриптан, наратриптан, золмитриптан, элетриптан, фроватриптан, алмотриптан или ризатриптан; с лигандами рецептора EP1; лигандами рецептора ЕР 2; лигандами рецептора ЕРз; антагонистами EP1; антагонистами ЕР 2 и антагонистами ЕР 3; антагонистами каннабиноидного рецептора; антагонистами VR1. Когда соединения используют в сочетании с другими терапевтическими агентами,соединения могут быть введены или последовательно, или одновременно любым традиционным путем. Изобретение, таким образом, в дополнительном варианте осуществления относится к комбинации,содержащей соединение формулы (I) или его фармацевтически приемлемое производное вместе с дополнительным терапевтическим агентом или агентами. Указанные комбинации легко могут быть представлены для применения в форме фармацевтической композиции, и такие фармацевтические композиции, содержащие комбинацию, которая определена выше, вместе с фармацевтически приемлемым носителем или эксципиентом являются дополнительным аспектом изобретения. Отдельные компоненты таких комбинаций могут быть введены или последовательно, или одновременно в раздельных или комбинированных фармацевтических композициях. Когда соединение формулы (I) или его фармацевтически приемлемое производное используют в комбинации со вторым терапевтическим агентом, действующим против того же заболевания, доза каждого соединения может отличаться от той, когда используют одно соединение. Соответствующие дозы легко могут быть определены специалистами в данной области. Предполагаемая суточная доза соединений формулы (I) или их фармацевтически приемлемых солей для лечения человека составляет от 0,001 до 30 мг/кг массы тела в сутки и более конкретно от 0,1 до 3 мг/кг массы тела в сутки из расчета на свободную кислоту, которая может быть введена как одна или как разделенная доза, например от одного до четырех раз в сутки. Интервал доз для взрослого человека обычно составляет от 0,1 до 1000 мг/сутки, такие как от 10 до 800 мг/сутки, предпочтительно от 10 до 200 мг/сутки из расчета на свободную кислоту. Точное количество соединений формулы (I), вводимых пациенту, в частности человеку, будет относиться к компетенции лечащего врача. Однако используемая доза будет зависеть от множества факторов,включая возраст и пол пациента, точное состояние, требующее лечения, и его тяжесть, путь введения и какую-либо возможную комбинированную терапию, которая может быть предпринята. Данное изобретение относится также к способу получения соединений формулы (I) и их фармацевтически приемлемых производных. Так, в одном варианте осуществления изобретения предложен способ получения соединения формулы (I), где один из X и Y представляет С=O, и другой представляет СН 2, и R1, R2 и R3 имеют указанные выше для формулы (I) значения, включающий взаимодействие соединения формулы (II) где один из X и Y представляет С=O, и другой представляет СН 2, и R1, R2 и R3 имеют указанные выше для формулы (I) значения, и R4 представляет C1-6 алкил, с подходящим основанием, таким как гидроксид натрия, и необязательно с последующим образованием фармацевтически приемлемого производного соединения, полученного таким образом, и/или преобразование одного соединения формулы (I) в другое. В одном варианте осуществления указанную реакцию, включающую взаимодействие соединения формулы (II), проводят в подходящем растворителе, таком как этанол, при кипячении с обратным холодильником. В другом варианте осуществления данного изобретения предложен способ получения соединения формулы (I), где X и Y представляют С=O, и R1, R2 и R3 имеют указанные выше для формулы (I) значения, включающий добавление соединения формулы (III) где R1, R2 и R3 имеют указанные выше для формулы (I) значения, и R4 представляет C1-6 алкил, к подходящей кислоте или смеси кислот, такой как ледяная уксусная кислота, в присутствии хлористоводородной кислоты, и необязательно с последующим образованием фармацевтически приемлемого производного соединения, полученного таким образом, и/или преобразование одного соединения формулы (I) в другое. В одном варианте осуществления указанную реакцию, включающую взаимодействие соединения формулы (III), проводят при температуре в интервале от около 50 до 110 С в течение периода времени в интервале от около 2 до 70 ч. В одном варианте осуществления молярное отношение ледяной уксусной кислоты к кислоте, такой как хлористо-водородная кислота, в реакционной смеси равно 1:1. Соединения формулы (II) и (III) могут быть получены согласно схеме 1 ниже. Схема 1NaOH, EtOH или HCl, CH3CO2H; (viii) NaBH4, МеОН или EtOH, затем TFA, Et3SiH; (где R' и R" могут быть одинаковыми или различными или представляют соответствующие защитные группы, такие как-CH2Ph, или один из R' и R" представляет соответствующую защитную группу, и другой представляет R1 или R2). Соединение (1) может быть получено в соответствии со способом, раскрытым в международной патентной заявкеWO 02/064564. Соединения формулы (4), где R3 означает F и R4 имеет указанные выше для формул (II) и (III) значения, могут быть получены согласно схемам 2 и 3 ниже. Схема 2(i) NaOH, сухой ДМФА; (ii) NH4CO2H, EtOH, Pd/C; (iii) NaOH, H2O, EtOH. Соединения формулы (4), где R3 означает Br или Cl и R4 имеет указанные выше для формул (II) и(III) значения, могут быть получены согласно схеме 4 ниже.(i) N-хлорсукцинамид или N-бромсукцинамид. Соединения формулы (А) коммерчески доступны или могут быть получены в соответствии со способами, известными в данной области (например, 3,4-дифторнитробензол может быть приобретен уSigma-Aldrich Co. Ltd.). Соединения формулы (В) коммерчески доступны или могут быть получены в соответствии со способами, известными в данной области (например, бензилэтилмалонат может быть приобретен у SigmaAldrich Co. Ltd.). Соединения формулы (D) коммерчески доступны или могут быть получены в соответствии со способами, известными в данной области (например, диэтилхлормалонат может быть приобретен у SigmaAldrich Co. Ltd.). Соединения формулы (Е) коммерчески доступны или могут быть получены в соответствии со способами, известными в данной области (например, этил-4-аминофенилацетат может быть приобретен уAvocado Research). В другом варианте осуществления изобретения предложен способ получения соединения формулы(I) или его фармацевтически приемлемого производного, включающий взаимодействие соединения формулы (IV) где R1 и R2 имеют указанные выше для формулы (I) значения, с соединением формулы (V) где R3 имеет указанные выше для формулы (I) значения, и R' представляет Н или С 1-6 алкил, и необязательно с последующим образованием фармацевтически приемлемого производного соединения, полученного таким образом, и/или преобразование одного соединения формулы (I) в другое. В другом варианте осуществления изобретения предложен способ получения соединения формулы(I) или его фармацевтически приемлемого производного, включающий взаимодействие соединения формулы (VI) где R1 и R2 имеют указанные выше для формулы (I) значения, с соединением формулы (V) где R3 имеет указанные выше для формулы (I) значения, и R' представляет Н или C1-6 алкил, и необязательно с последующим образованием фармацевтически приемлемого производного соединения, полученного таким образом, и/или преобразование одного соединения формулы (I) в другое. Соединения формул (IV), (V) и (VI) коммерчески доступны или могут быть получены в соответствии со способами, известными в данной области. Следующие описания и примеры поясняют получение соединений по изобретению. Описания относятся к промежуточным соединениям. Сокращения.Waters Atlantis (4,6 мм 50 мм). Размер частиц стационарной фазы 3 мкм. Растворители. А. Водный растворитель=вода+0,05% муравьиной кислоты. В. Органический растворитель=ацетонитрил+0,05% муравьиной кислоты. Метод. Скорость потока, 3 мл/мин. Объем впрыска, 5 мкл. Температура колонки, 30 С. Диапазон УФ регистрации от 220 до 330 нм. Все периоды времени удерживания измерены в минутах. ЯМР. Спектр 1 Н ЯМР записывали на ЯМР спектрометре Bruker AVANCE 400 или ЯМР спектрометреBruker DPX250. Химические сдвиги выражены в миллионных долях (м.д.,единицы). Постоянные взаимодействия (J) даны в единицах Герц (Гц). Картины расщепления описывают видимые множественности и обозначены как с (синглет), д (дублет), т (триплет), кв (квартет), дд (дублет дублетов), ддд (двойной дублет дублетов), дт (двойной триплет), м (мультиплет), ушир. (широкий). Методы очистки. Очистка соединений примеров может быть проведена традиционными методами, такими как хроматография и/или перекристаллизация с использованием соответствующих растворителей. Хроматографические методы включают колоночную хроматографию, флэш-хроматографию, ВЭЖХ (жидкостную хроматографию высокого разрешения), SFC (надкритическую жидкостную хроматографию) и MDAP(масса-направленное автополучение). Используемый в данном описании термин "Biotage" относится к коммерчески доступным предварительно набитым силикагелем картриджам.Waters Atlantis: 19 мм 100 мм (малый масштаб) и 30 мм 100 мм (большой масштаб). Размер частиц стационарной фазы 5 мкм. Растворители. А. Водный растворитель=вода+0,1% муравьиной кислоты. В. Органический растворитель=ацетонитрил+0,1% муравьиной кислоты. Пополняющий растворитель=метанол:вода 80:20. Растворитель для промывки иглы=метанол. Методы. Использовали пять методов в зависимости от аналитического времени удерживания представляющего интерес соединения:(1) большой/малый масштаб 1,0-1,5=5-30% В,(2) большой/малый масштаб 1,5-2,2=15-55% В,(3) большой/малый масштаб 2,2-2,9=30-85% В,(4) большой/малый масштаб 2,9-3,6=50-99% В. Время опыта 13,5 мин, включая 10-минутный градиент с последующей 3,5-минутной стадией быстрого притока в колонку и повторного установления равновесия.(5) большой/малый масштаб 3,6-5,0=80-99% В. Время опыта 13,5 мин, включая 6-минутный градиент с последующей 7,5-минутной стадией быстрого притока в колонку и повторного установления равновесия. Скорость потока. 20 мл/мин (малый масштаб) или 40 мл/мин (большой масштаб). Подробное описание системы очистки ВЭЖХ. Колонка: Supelco LCABZ, 100 мм 20 мм ID, размер частиц 5 мкм. Скорость потока: 20 мл/мин. Объем впрыска: 0,5 мл. Температура: комнатная. Растворители: А - водный растворитель=вода + 0,1% муравьиной кислоты, В - органический растворитель=MeCN/вода 95:5+0,05% муравьиной кислоты. Время опыта: 9-минутный градиент с последующим 5-минутным быстрым притоком в колонку и повторным установлением равновесия. Градиент: 30-80% В SFC (надкритическая жидкостная хроматография). Колонка: Chiracel OD-H S.F.C. (250 мм 21,2 мм ID, размер частиц 5 мкм), или этилпиридиновая колонка (150 мм длина 21,2 мм ID, размер частиц 6 мкм), или Chiralpak AD (355 и 438) (250 мм длина 21,2 мм ID, размер частиц 10 мкм). Подвижная фаза: А=диоксид углерода (надкритический), В=метанол, А:В (70:30-85:15 мас./мас. изократический). Скорость потока: 20 гмин-1 (=50 млмин-1 приблизительно). Температура: 40 С. Обратное давление: 100 бар. Регистрация: УФ поглощение при 215 нм. Время опыта: 25 или 20 мин. Концентрация образца: 100 мгмл-1 в диметилформамиде. Объем впрыска: переменный. Очистка ВЭЖХ 1. Стационарная фаза: Chiralcel OD (250 мм длина 21,2 мм ID, размер частиц 10 мкм). Подвижная фаза: гептан:этанол (в интервале от 50:50 об./об. изократический до 90:10 об./об. изократический). 320=50:50 об./об. 353=70:30 об./об. 354=90:10 об./об. 356=70:30 об./об. Скорость потока: 17 млмин-1. Температура: окружающая. Регистрация: УФ поглощение при 215 нм. Объем впрыска: 0,9 мл. Очистка ВЭЖХ 2. Колонка: Phenomenex Luna C18 (2) (50 мм 21,2 мм ID, размер частиц 10 мкм). Подвижная фаза: А=ацетонитрил+0,1% об./об. трифторуксусной кислоты, В=вода+0,1% об./об. трифторуксусной кислоты (градиентная и изократическая методологии).- 13014428 Скорость потока: 20 млмин-1 приблизительно. Температура: окружающая. Регистрация: УФ поглощение при 215 нм (ширина полосы 0 нм). Концентрация образца: 100 мгмл-1 в диметилформамиде. Объем впрыска: переменный. Бензилбромид (2,95 мл, 24,7 ммоль) добавляли к перемешиваемому раствору диэтил 1,4 дигидрокси-2,3-нафталиндикарбоксилата (3 г, 9,87 ммоль) и карбоната калия (3,40 г, 24,7 ммоль) в ацетоне (100 мл). Реакционную смесь кипятили с обратным холодильником в течение 18 ч в атмосфере аргона. Полученную смесь выпаривали и реакционную смесь растирали с водой. Полученное желтоватокоричневое твердое вещество отделяли фильтрованием и промывали водой, затем сушили в вакууме,получая указанное в заголовке соединение (5,23 г, 10,8 ммоль). ЖХ/МС: Rt=4,13, [MH]+ 485. Описание 2. 1,4-бис[(Фенилметил)окси]-2,3-нафталиндикарбоновая кислота.(75 мл) и 2 н. водного раствора гидроксида натрия (75 мл) кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали и выпаривали. Подкисляли HCl (2 н.), полученное желтоватое твердое вещество отделяли фильтрованием и промывали водой. Полученное вещество сушили в вакууме, получая указанное в заголовке соединение (3,79 г, 8,86 ммоль). ЖХ/МС: Rt=3,15, [МН]- 427. Описание 3. Этил фенилметил(2-фтор-4-нитрофенил)пропандиоат. Гидрид натрия (504 мг, 12,6 ммоль) добавляли порциями к охлаждаемому на бане со льдом раствору бензилэтилмалоната (2,9 г, 12,6 ммоль) в сухом ДМФА (20 мл) и перемешивали в течение 10 мин. При комнатной температуре 3,4-дифторнитробензол (2 г, 12,6 ммоль) добавляли и перемешивали в атмосфере аргона. Нагревали при 100 С в течение 20 ч. Реакционную смесь охлаждали и распределяли между 2 н. хлористо-водородной кислотой (75 мл) и этилацетатом (75 мл). Водный слой экстрагировали этилацетатом (275 мл) и объединенные органические фракции выпаривали до желтого масла. Очищали хроматографией на силикагеле, элюируя смесью этилацетат/гексан (1:4), получая указанное в заголовке соединение в виде желтого масла (3,86 г, 10,6 ммоль). ЖХ/МС: Rt=3,40, [MH]+ 362. Следующее соединение получали по аналогичной методике для этил фенилметил(2-фтор-4- 14014428 нитрофенил)пропандиоата, используя соответствующие исходные вещества. Этил фенилметил(2-фтор-4-нитрофенил)пропандиоат (3,86 г, 10,6 ммоль), растворенный в этаноле,обрабатывали формиатом аммония (6,7 г, 10,6 ммоль) и добавляли пасту 10% палладия на углероде (380 мг) в атмосфере аргона. Реакционную смесь кипятили с обратным холодильником в течение 3 ч, охлаждали и фильтровали. Выпаривали и очищали хроматографией на силикагеле, элюируя смесью этилацетат/гексан (1:1), получая указанное в заголовке соединение в виде желтого масла (1,26 г). ЖХ/МС:Rt=2,10, [MH]+ 198. Следующее соединение получали по аналогичной методике для этил(4-амино-2-фторфенил)ацетат,используя соответствующие исходные вещества. Этил-4-аминофенилацетат (20 г, 112 ммоль) растворяли в хлороформе (300 мл) и обрабатывали Nхлорсукцинимидом (14,92 г, 112 ммоль) и перемешивали в течение 15 мин при комнатной температуре в атмосфере аргона. Реакционную смесь промывали водой, концентрированным раствором соли и сушили над сульфатом магния. Выпаривали до бурого масла, которое очищали хроматографией на силикагеле,элюируя этилацетатом (0-45%) в гексане, получая указанное в заголовке соединение в виде оранжевого масла (10,12 г, 47,4 ммоль). ЖХ/МС: Rt=2,59, [MH]+ 214. Описание 6. Этил(4-1,3-диоксо-4,9-бис[(фенилметил)окси]-1,3-дигидро-2 Н-бензо[f]изоиндол-2 илфенил)ацетат. Смесь 1, 4-бис[(фенилметил)окси]-2,3-нафталиндикарбоновой кислоты (2,0 г, 4,67 ммоль) и этил(4 аминофенил)ацетата (1,67 г, 9,35 ммоль), нагревали до 120 С в уксусной кислоте (30 мл) в течение 2 ч. Смесь затвердевала при стоянии при комнатной температуре в течение ночи. Добавляли воду, твердое вещество отделяли фильтрованием и промывали водой. Полученное вещество сушили в вакууме, получая указанное в заголовке соединение (2,65 г, 4,64 ммоль). ЖХ/МС: Rt=4,24, [MH]+ 572.- 15014428 Следующие соединения получали по аналогичной методике для этил(4-1,3-диоксо-4,9 бис[(фенилметил)окси]-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)ацетата, используя соответствующие исходные вещества. 10% палладий на углероде (0,265 г) суспендировали в этаноле (250 мл) и добавляли этил(4-1,3 диоксо-4,9-бис[(фенилметил)окси]-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)ацетат (2,65 г, 4,64 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 3 ч. Затем фильтровали через слой целита в атмосфере аргона и промывали дихлорметаном. Фильтрат выпаривали, получая желтовато-коричневое твердое вещество (1,52 г, 3,89 ммоль). ЖХ/МС: Rt=3,25,[МН]+ 392. Следующие соединения получали по аналогичной методике для этил[4-(4,9-дигидрокси-1,3-диоксо 1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил)фенил]ацетата, используя соответствующие исходные вещества.- 16014428 К раствору этил(3-хлор-4-1,3-диоксо-4,9-бис[(фенилметил)окси]-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)ацетата (0,505 г, 0,84 ммоль) в DCM (30 мл), охлажденному до -78 С в атмосфере аргона, по каплям добавляли трибромид бора (0,24 мл, 2,50 ммоль). Перемешивали при -78 С в течение 25 мин. Реакционную смесь гасили водой и нагревали до комнатной температуры. Экстрагировали дваждыDCM, используя гидрофобную фритту. Органическую фазу выпаривали до оранжевого твердого вещества. Горячее растирание в DCM давало в результате желтое твердое вещество, которое отделяли фильтрованием (0,189 г, 0,44 ммоль). Фильтрат очищали хроматографией на силикагеле, элюируя этилацетатом(0-70%) в гексане, получая указанное в заголовке соединение в виде желтого твердого вещества (0,035 г,0,082 ммоль). ЖХ/МС: Rt=3,22, [МН]+ 426. Описание 9. Этил(4-4,9-бис[(дифторметил)окси]-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2 ил]фенил)ацетат. К раствору этил[4-(4,9-дигидрокси-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил)фенил]ацетата (0,200 г, 0,51 ммоль) в диметилформамиде (7 мл) добавляли карбонат натрия (0,163 г, 1,53 ммоль) и затем хлордифторацетат натрия (0,233 г, 1,53 ммоль). Смесь нагревали до 100 С в течение 2,5 ч. Затем добавляли карбонат натрия (0,054 г, 0,51 ммоль) и хлордифторацетат натрия (0,078 г, 0,51 ммоль), нагревание продолжали в течение 1 ч, такие же количества добавляли снова и нагревание продолжали в течение 30 мин. Смесь охлаждали до комнатной температуры и добавляли воду. Полученное коричневое твердое вещество отделяли фильтрованием и промывали водой. Очищали хроматографией на силикагеле, элюируя этилацетатом (0-30%) в гексане, получая указанное в заголовке соединение в виде белого твердого вещества (0,032 г, 0,065 ммоль). ЖХ/МС: Rt=3,61, [MH]+ 492. Следующие соединения получали по аналогичной методике для этил(4-4,9-бис[(дифторметил)окси]-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)ацетата, используя соответствующие исходные вещества. Этил(4-4,9-бис[(дифторметил)окси]-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил) ацетат (0,03 г, 0,061 ммоль) нагревали до 100 С в смеси 1:1 уксусная кислота:2 н. водная хлористоводородная кислота (4 мл) в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры. После добавления воды полученное белое твердое вещество отделяли фильтрованием и промывали водой. Очищали MDAP, получая желаемый продукт (0,017 г, 0,037 ммоль). ЖХ/МС: Rt=3,28, [MH]+ 464. Следующие соединения получали по аналогичной методике для (4-4,9-бис[(дифторметил)окси]1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)уксусной кислоты, используя соответствующие исходные вещества. К раствору (4-4,9-бис[(дифторметил)окси]-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил-3 фторфенил)уксусной кислоты (0,041 г, 0,085 ммоль) в этаноле (2 мл) и тетрагидрофурана (4 мл) добавляли боргидрид натрия (0,019 г, 0,51 ммоль). Полученную смесь перемешивали при 0 С в течение 5 мин. Смесь выпаривали и затем гасили водным насыщенным раствором хлорида аммония до рН смеси 7. Экстрагировали 2 этилацетатом, промывали концентрированным раствором соли, сушили над сульфатом магния и выпаривали, получая неочищенный продукт (0,035 г, 0,072 ммоль). ЖХ/МС: Rt=2,88, [MH]+ 484. Следующие соединения получали по аналогичной методике для (4-4,9-бис[(дифторметил)окси]-1 гидрокси-3-оксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил-3-фторфенил)уксусной кислоты, используя соответствующие исходные вещества. К раствору (4-4,9-бис[(дифторметил)окси]-1-гидрокси-3-оксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2 ил-3-фторфенил)уксусной кислоты (0,035 г, 0,072 ммоль) в трифторуксусной кислоте (2 мл), охлажденному до 0 С, добавляли триэтилсилан (0,019 мл, 0,110 ммоль). Перемешивание продолжали при 0 С в течение 5 мин и затем смесь выпаривали. Неочищенную смесь очищали MDAP. Чистые фракции выпаривали, получая указанное в заголовке соединение (0,015 г, 0,033 ммоль). ЖХ/МС: Rt=3,15, [MH]+468. Следующие соединения получали по аналогичной методике для (4-4,9-бис[(дифторметил)окси]-1 оксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил-3-фторфенил)уксусной кислоты, используя соответствующие исходные вещества. Бромэтан (0,359 г, 3,29 ммоль) добавляли к перемешиваемому раствору диэтил 1,4-дигидрокси-2,3 нафталиндикарбоксилата (1 г, 3,29 ммоль) и карбоната калия (0,454 г, 3,29 ммоль) в ацетоне (25 мл). Реакционную смесь кипятили с обратным холодильником в течение 24 ч в атмосфере аргона. Полученную смесь выпаривали и остаток распределяли между 2 этилацетатом и водой. Объединенные органические фракции промывали водой и сушили над сульфатом магния. Оранжевое масло очищали хроматографией на силикагеле, элюируя этилацетатом (0-10%) в гексане, получая указанное в заголовке соединение в виде светлого масла (0,661 г, 1,99 ммоль). ЖХ/МС: Rt=3,63, [МН]- 331. Следующие соединения получали по аналогичной методике для диэтил 1-(этилокси)-4-гидрокси- 19014428 2,3-нафталиндикарбоксилата, используя соответствующие исходные вещества. Бензилбромид (1,32 мл, 11,1 ммоль) добавляли к перемешиваемому раствору диэтил 1-(этилокси)-4 гидрокси-2,3-нафталиндикарбоксилата (2,45 г, 7,38 ммоль) и карбоната калия (1,53 г, 11,1 ммоль) в ацетоне (50 мл). Реакционную смесь кипятили с обратным холодильником в течение 1 ч в атмосфере аргона. Полученную смесь выпаривали и остаток распределяли между 2 этилацетатом и концентрированным раствором соли. Объединенные органические фракции промывали водой и сушили над сульфатом магния. Светлое масло очищали хроматографией на силикагеле, элюируя этилацетатом (0-40%) в гексане,получая указанное в заголовке соединение в виде светлого масла (3,05 г, 7,23 ммоль). ЖХ/МС: Rt=3,77,[MH]+ 423. Следующие соединения получали по аналогичной методике для диэтил 1-(этилокси)-4[(фенилметил)окси]-2,3-нафталиндикарбоксилата, используя соответствующие исходные вещества. Смесь диэтил 1-(этилокси)-4-[(фенилметил)окси]-2,3-нафталиндикарбоксилата (3,05 г, 7,23 ммоль),этанола (30 мл) и 2 н. водного раствора гидроксида натрия (35 мл) кипятили с обратным холодильником в течение 3 ч. Реакционную смесь охлаждали и выпаривали. Полученную смесь подкисляли HCl (2 н.) и экстрагировали этилацетатом дважды. Объединенные органические фракции сушили над сульфатом магния и растворитель выпаривали, получая указанное в заголовке соединение в виде белого твердого вещества (2,55 г, 6,97 ммоль). ЖХ/МС: Rt=2,76, [МН]- 365. Следующие соединения получали по аналогичной методике для 1-(этилокси)-4-[(фенилметил)окси]2,3-нафталиндикарбоновой кислоты, используя соответствующие исходные вещества. Смесь 1-(этилокси)-4-[(фенилметил)окси]-2,3-нафталиндикарбоновой кислоты (2,55 г, 6,97 ммоль) и этил(4-аминофенил)ацетата (2,49 г, 13,9 ммоль) нагревали до 120 С в уксусной кислоте (20 мл) в течение 18 ч. Реакционную смесь разбавляли концентрированным раствором соли и экстрагировали этилацетатом дважды. Объединенные органические фракции промывали водой, затем сушили над сульфатом магния, растворитель выпаривали, получая коричневое масло. Его очищали хроматографией на силика- 21014428 геле, элюируя этилацетатом (0-30%) в гексане, получая указанное в заголовке соединение в виде твердого вещества персикового цвета (2,63 г, 5,17 ммоль). ЖХ/МС: Rt=3,95, [MH]+ 510. Следующие соединения получали по аналогичной методике для этил(4-4-(этилокси)-1,3-диоксо-9[(фенилметил)окси]-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)ацетата, используя соответствующие исходные вещества и, в некоторых случаях, применяли MDAP (обозначено ). 10% Палладий на углероде (0,365 г) суспендировали в этаноле (500 мл) и добавляли этил(4-4[метилэтокси]-1,3-диоксо-9-[(фенилметил)окси]-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)ацетат(3,65 г, 6,98 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 3 ч. Фильтровали через слой целита в атмосфере аргона и промывали этанолом и дихлорметаном. Фильтрат выпаривали, получая оранжевое твердое вещество (3,05 г, 7,04 ммоль). ЖХ/МС:Rt=3,72, [MH]+ 434. Следующие соединения получали по аналогичной методике для этил(4-4-гидрокси-9[метилэтокси]-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)ацетата, используя соответствующие исходные вещества. К раствору этил(3-хлор-4-4-(этилокси)-1,3-диоксо-9-[(фенилметил)окси]-1,3-дигидро-2 Нбензо[f]изоиндол-2-илфенил)ацетата (1,75 г, 3,22 ммоль) в DCM (40 мл), охлажденному до -78 С в атмосфере аргона, по каплям добавляли трибромид бора (0,31 мл, 3,22 ммоль). Перемешивали при -78 С в течение 12 мин. Дополнительное добавление трибромида бора (0,31 мл, 3,22 ммоль) проводили по каплям. Реакционную смесь гасили водой и нагревали до комнатной температуры. Полученную смесь экстрагировали дважды DCM, используя гидрофобную фритту. Органическую фазу выпаривали до желтого твердого вещества. Очищали хроматографией на силикагеле, элюируя этилацетатом (0-60%) в гексане,получая указанное в заголовке соединение в виде оранжевого маслянистого твердого вещества (0,920 г,2,03 ммоль). ЖХ/МС: Rt=3,65, [МН]+ 454. Следующие соединения получали по аналогичной методике для этил 3-хлор-4-[4-(этилокси)-9 гидрокси-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил]фенилацетата, используя соответствующие исходные вещества. К раствору этил(4-4-гидрокси-9-[метилэтокси]-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2 илфенил)ацетата (0,25 г, 0,577 ммоль) в диметилформамиде (10 мл) добавляли карбонат калия (0,120 г,0,866 ммоль), затем иодид калия (0,048 г, 0,289 ммоль). Хлор(дифтор)метан барботировали через раствор при 80 С в течение 2 ч. Барботирование прекращали и нагревание продолжали в течение 25 ч. Охлажденную смесь распределяли между 2 этилацетатом и концентрированным раствором соли, объединенные органические фракции промывали водой, сушили над сульфатом натрия и выпаривали до желтого масла. Полученное масло очищали хроматографией на силикагеле, элюируя этилацетатом (0-30%) в гексане, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,142 г, 0,294 ммоль). ЖХ/МС: Rt=3,82, [MH]+ 484. Описание 18. Этил 4-[4-[(дифторметил)окси]-9-(метилокси)-1,3-диоксо-1,3-дигидро-2 Н-бензо[f] изоиндол-2-ил]фенилацетат. К раствору этил 4-[4-гидрокси-9-(метилокси)-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2 ил]фенилацетата (0,700 г, 1,73 ммоль) в диметилформамиде (25 мл) добавляли карбонат натрия (0,220 г,2,07 ммоль), затем хлордифторацетат натрия (0,315 г, 2,07 ммоль). Смесь нагревали до 100 С в течение 1,5 ч. Дополнительно добавляли карбонат натрия (0,220 г, 2,07 ммоль) и хлордифторацетат натрия (0,315 г, 2,07 ммоль), нагревание продолжали в течение 2 ч и такие же количества добавляли снова. Смесь охлаждали до комнатной температуры и добавляли воду. Полученное бежевое твердое вещество отделяли фильтрованием и промывали водой. Сушили в вакууме (0,680 г, 1,49 ммоль). ЖХ/МС: Rt=3,62, [MH]+ 456. Следующие соединения получали по аналогичной методике для этил 4-[4-[(дифторметил)окси]-9(метилокси)-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил]фенилацетата, используя соответствующие исходные вещества. Этил(4-4-[(дифторметил)окси]-9-[(1-метилэтил)окси]-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)ацетат (0,14 г, 0,289 ммоль) нагревали до 100 С в смеси 1:1 уксусная кислота:2 н. водная хлористо-водородная кислота (8 мл) в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры. После добавления воды полученное белое твердое вещество отделяли фильтрованием и промывали водой. Сушили в вакуумной печи, получая указанное в заголовке соединение (0,120 г, 0,026 ммоль). ЖХ/МС: Rt=3,36, [MH]+ 456. Следующие соединения получали по аналогичной методике для (4-4-[(дифторметил)окси]-9-[(1 метилэтил)окси]-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-илфенил)уксусной кислоты, используя соответствующие исходные вещества. К раствору этил 4-[4-[(дифторметил)окси]-9-(метилокси)-1,3-диоксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил]фенилацетата (0,455 г, 1,00 ммоль) в этаноле (4 мл) и тетрагидрофуране (8 мл), порциями добавляли боргидрид натрия (0,114 г, 3,0 ммоль). Реакционную смесь перемешивали в течение 1 ч, затем добавляли дополнительно порцию боргидрида натрия (0,114 г, 3,0 ммоль) и перемешивание продолжали при комнатной температуре в течение 1 ч. Смесь гасили водным насыщенным раствором хлорида аммония и затем выпаривали. Экстрагировали этилацетатом дважды, сушили над сульфатом магния и выпаривали, получая неочищенный продукт (0,515 г, 1,13 ммоль). ЖХ/МС: Rt=3,21 и 3,26, [МН]+ 457,97. Следующие соединения получали по аналогичной методике для этил 4-[4-[(дифторметил)окси]-1 гидрокси-9-(метилокси)-3-оксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил]фенилацетата и этил 4-[4[(дифторметил)окси]-3-гидрокси-9-(метилокси)-1-оксо-1,3-дигидро-2 Н-бензо[f]изоиндол-2-ил]фенил ацетата, используя соответствующие исходные вещества.
МПК / Метки
МПК: A61K 31/4035, A61P 25/02, C07D 209/64, C07D 209/66
Метки: лечения, боли, бензоизоиндола, производные
Код ссылки
<a href="https://eas.patents.su/30-14428-proizvodnye-benzoizoindola-dlya-lecheniya-boli.html" rel="bookmark" title="База патентов Евразийского Союза">Производные бензоизоиндола для лечения боли</a>
4-тетразолил-4-фенилпиперидин производные для лечения боли

Номер патента: 8621
Опубликовано: 29.06.2007
Автор: Чен Женгминг
МПК: A61K 31/454, C07D 211/64, A61P 25/00...
Метки: лечения, боли, 4-тетразолил-4-фенилпиперидин, производные
Формула / Реферат:
1. Соединение формулы (Iа) или (Ib) или его фармацевтически пригодная соль, где Ar1 = -С3-C8циклоалкил, фенил, нафтил, антрил, фенантрил или -(5-7-членный) гетероарил, каждый из которых является незамещенным или замещенным одной или несколькими группами R2; Аr2 = фенил, нафтил, антрил, фенантрил или -(5-7-членный) гетероарил, каждый из которых является незамещенным или замещенным одной или несколькими группами R2; G = -Н, -L-(CH2)nCO2R4,...
Производные бензолсульфонамидов, способ их получения и их применение для лечения боли

Номер патента: 8921
Опубликовано: 31.08.2007
Авторы: Массардье Кристин, Луккарини Жан-Мишель, Доде Пьер, Бонду Мишель, Барт Мартин, Тома Дидье
МПК: C07D 417/04, A61K 31/496, A61K 31/44...
Метки: применение, лечения, производные, получения, способ, бензолсульфонамидов, боли
Формула / Реферат:
1. Производное бензолсульфонамида, характеризующееся тем, что оно выбрано из группы, состоящей из: а) соединений формулы где каждый из радикалов R1, R2, R3, R4 независимо представляет собой один или несколько атомов или групп атомов, выбранных из атома водорода, атомов галогенов, C1-C3алкильных групп, С1-С3алкоксильных групп, CF3- или OCF3-групп, Ra представляет собой С1-С4алкильную группу, Y представляет собой насыщенную С2-С5алкиленовую...
Производные изоксазола, их фармацевтические композиции и способ лечения невропатической боли

Номер патента: 13905
Опубликовано: 30.08.2010
Авторы: Моригги Ерманно, Наполетано Мауро
МПК: A61K 31/42, A61P 25/02
Метки: лечения, невропатической, фармацевтические, изоксазола, способ, композиции, боли, производные
Формула / Реферат:
1. Применение производного изоксазола формулы (I)или его фармацевтически приемлемых солей при получении лекарственного средства для лечения невропатической боли.2. Применение фармацевтической композиции, включающей в качестве активного соединения соединение формулы (I) по п.1 либо его фармацевтически приемлемую соль и, по меньшей мере, фармацевтически приемлемый наполнитель при лечении невропатической боли.3. Применение фармацевтической...
Фармацевтическая композиция для лечения острой, хронической боли и/или невропатической боли и мигреней

Номер патента: 4930
Опубликовано: 28.10.2004
Авторы: Харриган Эдмунд Патрик, Уэтски Эрик Джейкоб, Коу Джоутам Уодзуэрт, Сандз Стивен Брэдли, О'нилл Брайан Томас
МПК: A61K 45/06, A61P 25/06
Метки: невропатической, композиция, острой, лечения, хронической, боли, фармацевтическая, мигреней
Формула / Реферат:
1. Фармацевтическая композиция для лечения острой, хронической и/или невропатической боли и мигрени, содержащая (а) частичный агонист никотиновых рецепторов или его фармацевтически приемлемую соль, где частичный агонист никотиновых рецепторов представляет собой арильное конденсированное азаполициклическое соединение; (б) анальгетический агент или его фармацевтически приемлемую соль и (в) фармацевтически приемлемый носитель; где вышеуказанные...
Производные [[ 2-(амино-3,4-диоксо-1-циклобутен-1-ил)амино ]алкил]кислоты, применяемые для лечения боли

Номер патента: 9993
Опубликовано: 30.06.2008
Авторы: Брандт Майкл Ричард, Залеска Маргарет Мария, Мойер Джон Аллен
МПК: A61K 31/197, A61K 31/198, A61K 31/4245...
Метки: 2-(амино-3,4-диоксо-1-циклобутен-1-ил)амино, лечения, алкил]кислоты, боли, производные, применяемые
Формула / Реферат:
1. Способ лечения боли у млекопитающего, включающий введение млекопитающему, нуждающемуся в лечении боли, эффективного для лечения боли количества по меньшей мере одного соединения, имеющего формулу где R1 представляет собой водород, алкил, содержащий 1-6 атомов углерода, или фенилалкил, содержащий 7-12 атомов углерода; R2 представляет собой водород, алкил, содержащий 1-6 атомов углерода, алкенил, содержащий 2-6 атомов углерода, или фенилалкил,...
Предыдущий патент: Соединение бициклического циннамида
Следующий патент: Композиции, содержащие иммуномодулирующие соединения для лечения и управления течением миелодиспластических синдромов, и способы с их использованием
Случайный патент: Способ и устройство для перемещения подводных камней и отложений