Соединения карбонилбензоксазина для усиления глутаматергических синаптических ответов
Номер патента: 11034
Опубликовано: 30.12.2008
Авторы: Маррс Кристофер М., Хуанг Джианджи, Аллан Мэттью, Харрис Клейтон, Роджерс Гари А., Мюллер Рудольф, Рахвал Станислав





























Формула / Реферат
1. Соединения, имеющие структуру Ia или Ib

где Q и Q' обозначают независимо водород, -СН2-, -О-, -S-, алкил, гидроксиалкил или алкоксиалкил,
R1 обозначает водород, алкил или вместе с Q может быть кольцом циклоалкила,
R2 может отсутствовать или, если присутствует, может быть -СН2-, -CO-, -СН2СН2-, -СН2СО-,
-СН2О-, -CRR'- и -CONR-,
Y обозначает водород или -OR3 или служит для связывания ароматического кольца с А или А' как единственной связью, =N- или -NR-,
R3 обозначает водород, алкил, замещенный алкил или служит для связывания присоединенного кислорода с А, когда обозначает низший алкилен, такой как метилен или этилен, или замещенный низший алкилен, такой как -CRR'-, связывая ароматическое кольцо с А для образования замещенного или незамещенного 6-, 7- или 8-членного кольца или связи, связывающей кислород с А для образования 5- или 6-членного кольца,
А обозначает -NRR', -OR, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалкилалкил, арил, замещенный арил, гетероцикл или замещенный гетероцикл, содержащий 1 или 2 гетероатома, таких как кислород, азот или сера,
А' обозначает -NRR',
R обозначает водород, арил, арилалкил, замещенный арил, замещенный арилкалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или гетероциклоалкил,
R' отсутствует или обозначает водород, арил, арилалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или может быть соединен с R для образования 4-8-членного кольца, которое может быть замещено X и может быть связано с Y для образования 6-членного кольца и которое может произвольно содержать 1 или 2 гетероатома, например кислород, азот или серу,
X и X' обозначают независимо R, гало, -CO2R, -CN, -NRR', -NRCOR', -NO2, -N3 или -OR.
2. Соединение по п.1 со структурой Ia, как указано выше, где
Q и Q' обозначают независимо водород, -СН2-, -О-, -S-, алкил, гидроксиалкил или алкоксиалкил,
R1 обозначает водород, алкил или вместе с Q может быть кольцом циклоалкила,
R2 может отсутствовать или, если присутствует, может быть -СН2-, -СО-, -СН2СН2-, -СН2СО-,
-СН2О- или -CONR-,
Y обозначает водород или -OR3 или служит для связывания ароматического кольца с А как единственной связью, =N- или -NR-,
R3 обозначает водород, алкил, замещенный алкил или служит для связывания присоединенного кислорода с А, когда является низшим алкиленом, таким как метилен или этилен, или замещенным низшим алкиленом, таким как -CRR'-, связывая ароматическое кольцо с А для образования замещенного или незамещенного 6-, 7- или 8-членного кольца или связи, связывающей кислород с А для образования 5- или 6-членного кольца,
А обозначает -NRR', -OR, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалкилалкил, арил, замещенный арил, гетероцикл или замещенный гетероцикл, содержащий 1 или 2 гетероатома, таких как кислород, азот или сера;
R обозначает водород, арил, арилалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или гетероциклоалкил,
R' отсутствует или обозначает водород, арил, арилкалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или может быть присоединен к R для образования 4-8-членного кольца, которое может замещаться X или связываться с Y и которое может произвольно содержать 1 или 2 гетероатома, таких как кислород, азот или сера,
X и X' обозначают независимо R, гало, -CO2R, CN, -NRR', -NRCOR', -NO2, -N3 или -OR.
3. Соединение по п.1 со структурой Ib, как указано выше, где
Q и Q' обозначают независимо водород, -СН2-, -О-, -S-, алкил, гидроксиалкил или алкоксиалкил,
R1 обозначает водород, алкил или вместе с Q может быть кольцом циклоалкила,
R2 может отсутствовать или, если присутствует, может быть -СН2-, -СО-, -СН2СН2-, -СН2СО-,
-СН2О- или -CONR-,
Y обозначает водород или -OR3 или служит для связывания ароматического кольца с А как единственной связью, =N- или -NR-,
R3 обозначает водород, алкил, замещенный алкил или служит для связывания присоединенного кислорода с А', когда является низшим алкиленом, таким как метилен или этилен, или замещенным низшим алкиленом, таким как -CRR'-, связывая ароматическое кольцо с А' для образования замещенного или незамещенного 6-, 7- или 8-членного кольца или связи, связывающей кислород с А' для образования 5- или 6-членного кольца,
А' обозначает -NRR',
R обозначает водород, арил, арилалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или гетероциклоалкил,
R' обозначает водород, арил, арилкалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или может быть присоединен к R для образования 4-8-членного кольца, которое может замещаться X или связываться с Y для образования 6-членного кольца и которое может произвольно содержать 1 или 2 гетероатома, таких как кислород, азот или сера,
X и X' обозначают независимо R, гало, -CO2R, -CN, -NRR', -NRCOR', -NO2, -N3 или -OR.
4. Соединение по любому из пп.1-3, в котором Q и Q' обозначают -СН2-, a R2 обозначает -СН2-.
5. Соединение по любому из пп.1-4, в котором R1 обозначает водород.
6. Соединение по любому из пп.1-3 или 5, в котором Q и Q' обозначают -СН2-, a R2 обозначает
-СН2СН2-.
7. Соединение по любому из пп.1-3 или 5, в котором Q' обозначает -СН2-, R2 обозначает -СН2- и Q обозначает -О- или -S-.
8. Соединение по любому из пп.1-3, 5 или 7, в котором Q обозначает -О-.
9. Соединение по любому из пп.1-3, в котором Q и Q' обозначают алкил, а R2 отсутствует.
10. Соединение по любому из пп.1-3, в котором Q и Q' обозначают алкил, R2 отсутствует, a R1 обозначает водород.
11. Соединение по любому из пп.1-2 или 10, в котором Y обозначает -OR3 и А обозначает -NRR',
-OR, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалакилалкил, арил, замещенный арил, гетероцикл или замещенный гетероцикл, содержащий 1 или 2 гетероатома, таких как кислород, азот или сера.
12. Соединение по любому из пп.1-2 или 11, в котором А обозначает алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалкилалкил, арил, замещенный арил, гетероцикл или замещенный гетероцикл, содержащий 1 или 2 гетероатома, таких как кислород, азот или сера.
13. Соединение по любому из пп.1-2 или 12, в котором А обозначает алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалкилалкил, гетероцикл или замещенный гетероцикл, содержащий 1 гетероатом, такой как кислород, азот или сера.
14. Соединение по любому из пп.1-13, в котором А или А', если имеется, обозначает -NRR', R обозначает водород, арил, арилалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или гетероциклоалкил, R' обозначает водород, арил, арилалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или может быть присоединен к R для образования 4-8-членного кольца, которое может замещаться X и связываться с Y с помощью R3 и которое может произвольно содержать 1 дополнительный гетероатом, такой как кислород, азот или сера, а X и X' обозначают независимо R, гало, -CO2R, -CN, -NRR',
-NRCOR', -NO2, -N3 или -OR.
15. Соединение по любому из пп.1-13, в котором А или А', если имеется, обозначает -NRR', R обозначает алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или гетероциклоалкил, R' обозначает водород, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или может быть присоединен к R для образования 4-8-членного кольца, которое может замещаться X и связываться с Y с помощью R3 и которое может произвольно содержать 1 дополнительный гетероатом, такой как кислород, азот или сера, а X и X' независимо обозначают R, гало, -CO2R, -CN, -NRR', -NRCOR', -NO2, -N3 или -OR.
16. Соединение по п.15, в котором А или А', если имеется, обозначает -NRR' и R' присоединен к R для образования 4-8-членного кольца, которое может замещаться X и связываться с Y с помощью R3 и которое может произвольно содержать 1 дополнительный гетероатом, такой как кислород, азот или сера, а X и X' обозначают независимо R, гало, -CO2R, -CN, -NRR', -NRCOR', -NO2, -N3 или -OR.
17. Соединение по п.16, в котором А или А', если имеется, обозначает -NRR', и R' присоединен к R для образования 5-членного кольца, которое может замещаться X и связываться с Y с помощью R3 и которое может произвольно содержать 1 дополнительный гетероатом, такой как кислород, азот или сера, а X и X' обозначают независимо R, гало, -CO2R, -CN, -NRR', -NRCOR', -NO2, -N3 или -OR.
18. Соединение по п.17, в котором А или А', если имеется, обозначает -NRR', и R' присоединен к R для образования 5-членного кольца, которое может замещаться X и связываться с Y с помощью R3 и которое может произвольно содержать 1 дополнительный гетероатом, такой как кислород, азот или сера, а X и X' обозначают независимо R, гало, -CO2R, -CN, -NRR', -NRCOR', -NO2, -N3 или -OR.
19. Соединение по п.16, в котором А или А', если имеется, обозначает -NRR', a R' присоединен к R для образования 5-членного кольца, которое связывается с Y с помощью R3.
20. Соединение по п.16, в котором А или А', если имеется, обозначает -NRR', a R' присоединен к R для образования 6-членного кольца, которое может замещаться X и связываться с Y с помощью R3 и которое может произвольно содержать 1 дополнительный гетероатом, такой как кислород, азот или сера, а X и X' обозначают независимо R, гало, -CO2R, -CN, -NRR', -NRCOR', -NO2, -N3 или -OR.
21. Соединение по любому из пп.1-10, в котором Y обозначает -OR3.
22. Соединение по п.21, в котором R3 обозначает водород.
23. Соединение по любому из пп.1-3, в котором Y обозначает водород.
24. Соединение по любому из пп.1-3, в котором Y обозначает =N- или -NR-.
25. Соединение по любому из пп.1-3, в котором Y обозначает =N-.
26. Соединение по п.23, в котором А обозначает -OR, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалкилалкил, гетероцикл или замещенный гетероцикл, содержащий 1 или 2 гетероатома, таких как кислород, азот или сера.
27. Соединение по п.23, в котором А или А', если имеется, обозначает -NRR'.
28. Соединение по п.21, в котором А обозначает -OR, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалкилалкил, гетероцикл или замещенный гетероцикл, содержащий 1 или 2 гетероатома, таких как кислород, азот или сера, и А' отсутствует.
29. Соединение по п.21, в котором А или А', если имеется, обозначает -NRR'.
30. Соединение по п.4, в котором Y обозначает -OR3 или А или А', если имеется, обозначает -NRR'.
31. Соединение по п.30, в котором R' обозначает водород.
32. Способ лечения млекопитающего субъекта, страдающего от гипоглутаматергического состояния или дефицита количества или интенсивности эксцитатных синапсов или количества АМРА-рецепторов, что приводит к нарушению памяти или других когнитивных функций, который включает введение указанному субъекту в фармацевтически приемлемом носителе эффективного количества соединения согласно любому из пп.1-31.
33. Способ лечения млекопитающего, в котором субъекту, страдающему от гипоглутаматергического состояния или дефицита количества или интенсивности эксцитатных синапсов или количества АМРА-рецепторов, в результате чего возникает кортиально/стриарный дисбаланс, приводящий к шизофрении или подобному шизофрении поведению, вводят в фармацевтически приемлемом носителе эффективное количество соединения согласно любому из пп.1-31.
34. Способ по п.33, в котором указанное состояние является шизофренией.
35. Способ по п.33, в котором указанное состояние является болезнью Паркинсона.
36. Фармацевтическая композиция для лечения млекопитающего субъекта, страдающего от гипоглутаматергического состояния или недостатка количества или интенсивности эксцитатных синапсов или количества АМРА-рецепторов, что приводит к нарушению памяти или других познавательных функций, включающая эффективное количество соединения согласно одному из пп.1-31 в комбинации с фармацевтически приемлемым носителем, добавкой или наполнителем.
37. Композиция по п.36, в котором указанное соединение включает приблизительно от 0,5 до
75 вес.% указанного соединения, а указанный носитель, добавка или наполнитель включают приблизительно от 25 до 95,5% указанной композиции.
38. Применение соединения по любому из пп.1-31 для изготовления лекарства для использования при лечении млекопитающего субъекта, страдающего от гипоглутаматергического состояния или недостатка количества или интенсивности эксцитатных синапсов или количества АМРА-рецепторов, что приводит к нарушению памяти или других познавательных функций.
39. Применение соединения по любому из пп.1-31 для изготовления лекарства для применения при лечении шизофрении.
40. Применение соединения по любому из пп.1-31 для изготовления лекарства для применения при лечении болезни Паркинсона.
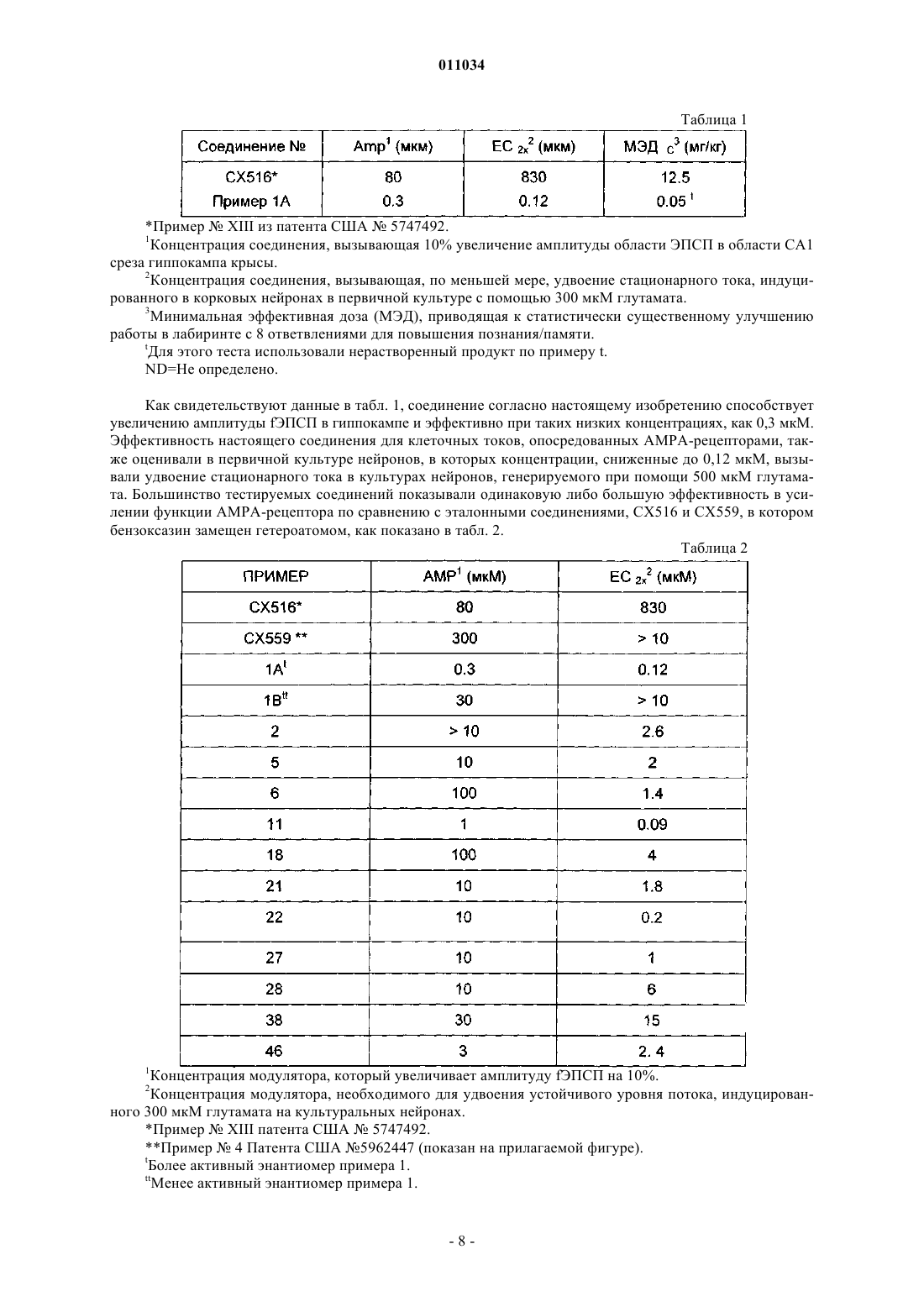
Текст
011034 Область изобретения Настоящее изобретение относится к соединениям, фармацевтическим композициям и способам их использования для профилактики и лечения церебральной недостаточности, включая усиление рецепторов, функционирующих в синапсах в сетях головного мозга, ответственных за поведение более высокого порядка. Эти сети головного мозга, которые участвуют в формировании познавательных способностей,связаны с ухудшением памяти, наблюдаемым при различных видах слабоумия, а также с дисбалансом нейрональной активности между различными областями головного мозга, который, как считается, вызывает такие заболевания, как болезнь Паркинсона, шизофрения и аффективные расстройства или заболевания, влияющие на настроение. В частности, настоящее изобретение касается соединений, используемых для лечения таких состояний, и способов использования этих соединений для такого лечения. Предпосылки создания изобретения Высвобождение глутамата в синапсах на многих участках в переднем мозге млекопитающих стимулирует два класса постсинаптических, ионотропных рецепторов. Эти классы обычно упоминаются как рецепторы АМРА/квисквалат и N-метил-D-аспарагиновой кислоты (NMDA). Рецепторы АМРА/квисквалат опосредуют независимую разность потенциалов быстрого эксцитатного постсинаптического тока(быстрого ЭПСТ), тогда как рецепторы NMDA генерируют зависимую разность потенциалов, медленный эксцитатный ток. Исследования, выполненные в срезах гиппокампа или коры, показывают, что быстрый ЭПСТ, опосредованный АМРА-рецепторами, в основном, является безусловным доминирующим компонентом в большинстве глутаматергических синапсов. АМРА-рецепторы не распределяются равномерно по головному мозгу, а в значительной степени ограничиваются конечным мозгом и мозжечком. Эти рецепторы обнаружены в высоких концентрациях в поверхностных слоях коры головного мозга, в каждой из главных синаптических зон гиппокампа, и в стриарном комплексе, как указывают Monaghan et al., in Brain Research 324: 160-164 (1984). Исследования на животных и человеке показывают, что эти структуры организовывают сложные перцепционныемоторные процессы и обеспечивают основу для поведения более высокого порядка. Таким образом,АМРА-рецепторы опосредуют передачу в тех сетях головного мозга, которые отвечают за познавательную активность хозяина. По вышеприведенным причинам лекарства, которые модулируют и, тем самым, усиливают функционирование АМРА-рецепторов, могут существенно способствовать познавательной и интеллектуальной работе. Такие лекарства должны также облегчать кодирование памяти. Экспериментальные исследования, как указывают Arai and Lynch, Brain Research 598: 173-184 (1992), показывают, что увеличение параметра опосредованного АМРА-рецептором синаптического ответа усиливает индукцию долгосрочного потенцирования (ИДП). ИДП - устойчивое увеличение силы синаптических контактов, которые следуют за повторной физиологической активностью известного типа, происходящей в головном мозге во время обучения. Соединения, которые усиливают функционирование глутаматных рецепторов формы АМРА, облегчают ИДП и усвоение изученных задач, что было измерено с помощью множества образцов. См., например, Granger et al., Synapse 15: 326-329 (1993); Staubli et al., PNAS 91: 777-781 (1994); Arai et al., BrainNeurology 145: 89-92 (1997); Ingvar et al., Exp. Neurology 146: 553-559 (1997); Hampson et al., J. Neurosci. 18: 2748-2763 (1998); and Lynch and Rogers, патент США 5747492. Имется значительное количество свидетельств, показывающих, что ИДП является основой памяти. Например, составы, которые блокируют ИДП, препятствуют формированию памяти у животных, а некоторые лекарства, которые нарушают процесс обучения у человека, противодействуют стабилизации ИДП, как описывает del Cerro and Lynch, Neuroscience 49: 1-6 (1992). Прототип соединения, который усиливает функцию АМРА-рецептора, описан Ito et al., J. Physio. 424: 533-543 (1990). Эти авторы обнаружили, что ноотропный препарат анирацетам (N-анизоил-2-пирролидинон) усиливает токи, опосредованные АМРА-рецепторами мозга, экспрессированными в ооцитах Ксенопуса, не затрагивая ответов -аминомасляной кислоты (GABA), каиновой кислоты (KA) илиNMDA-рецепторов. Инфузия анирацетама в срезы гиппокампа также существенно увеличивала величины быстрых синаптических потенциалов без изменения мембранных свойств в состоянии покоя. С тех пор считалось, что анирацетам усиливает синаптические ответы на нескольких участках в гиппокампе и что он не оказывает никакого эффекта на потенциалы, опосредованные NMDA-рецепторами (Staubli etal., Psychobiology 18: 377-381 (1990) and Xiao et al., Hippocampus 1: 373-380 (1991. Обнаружено, что анирацетам имеет чрезвычайно быстрое начало действия, и быстро выводится, и может применяться неоднократно без очевидных длительных эффектов, которые являются желательными свойствами для лекарств, предназначенных для лечения нарушений поведения. Однако аницетам,действительно, имеет ряд недостатков. Периферийное введение анирацетама не влияет на мозговые рецепторы. Препарат действует только при высоких концентрациях (приблизительно 1000 мкМ), и приблизительно 80% препарата преобразуются в анизоил-GABA после периферийного введения у человека(Guenzi and Zanetti, J. Chromatogr. 530: 397-406 (1990. Обнаружен метаболит, анизоил-GABA, обладающий меньшей синаптической активностью, чем анирацетам. Описан класс соединений, модулирующих АМРА-рецепторы, которые не проявляют низкую эффективность и характерную для анирацетама неустойчивость (Lynch and Rogers, US Patent 5747492). Эти соединения, называемые "Ampakines", можно заменить на бензамиды, которые включают, например, 1(хиноксалин-6-илкарбонил)пиперидин (CX516; Ampalex). Как правило, они химически более устойчивы, чем анирацетам, и проявляют улучшенную биодоступность. CX516 оказывает действие при экспериментах на животных, используемых для обнаружения эффективных лекарств для лечения нарушений памяти, шизофрении и депрессии. В трех отдельных клинических испытаниях СХ 516 проявил эффективность в улучшении различных форм человеческой памяти (Lynch et al., Internat. Clin. Psychopharm. 11: 1319 (1996); Lynch et al., Exp. Neurology 145: 89-92 (1997); Ingvar et al., Exp. Neurology 146: 553-559 (1997. Обнаружено, что бензоксазины - другой класс ампакинов, проявили очень высокую активность в моделях in vitro и in vivo при оценке вероятности их воздействия на усиление процесса познания (Rogersand Lynch; патент США 5736543). Замещенные бензоксазины представляют собой твердые аналоги бензамидов с различными рецепторами, модулирующими свойства гибкого бензамида, СХ 516. Неожиданно было обнаружено, что определенные замещенные бензофуразановые и бензотиадиазоловые соединения действуют значительно сильнее в животной модели шизофрении, чем предыдущие соединения, и что они также эффективны в усилении процесса познания. Структурно эти соединения подобны описанным в Lynch and Rogers, патент США 5736543. Ранее описанные структуры, которые содержат 1,3-бензоксазин-4-он фармакофоры, замещали на части бензола такими гетероатомами, как азот или кислород (патенты США 5736543 и 5962447),замещенными группами алкила (патенты США 5650409 и 5783587) или незамещенными группами алкила (международная заявка WO 99/42456). Еще один класс соединений 1,3-бензоксазина содержит внешний к кольцу оксазина карбонил (патент США 6124278), но не как заместитель на структуре кольца бензола. Недавно был обнаружен новый класс карбонилзамещенных соединений бензоксазина,которые проявляют существенную активность на синаптические ответы гиппокампа и нейрональные токи целых клеток, опосредованные АМРА-рецепторами, а также в животных моделях познания и памяти. Карбонилзамещенные 1,3-бензоксазин-4-он структуры являются первыми молекулами, которые проявили активность как модуляторы АМРА-рецептора и которые имеют 2 тяжелых атома, разветвляющихся от одного и того же альфа-атома до кольца бензола в 6- или 7-положении. Биологическая активность 6- или 7-карбонилзамещенных 1,3-бензоксазинов была неожиданной, а эффективность в АМРА-рецепторе была удивительно высока; причем наиболее сильнодействующие 1,3 бензоксазины являются членами этого класса соединений. Эти соединения описываются далее. Краткое изложение существа изобретения Настоящее изобретение включает, с одной стороны, соединение, представленное структурой I и описанное в разделе II полного описания, которое следует далее. Обнаружено, что введение соединений этого класса усиливает синаптические ответы, опосредованные АМРА-рецепторами. Неожиданно было обнаружено, что соединения согласно настоящему изобретению являются значительно более эффективными, чем ранее описанные соединения, для усиления функции АМРА-рецепторов в первичных нейрональных культурах и в срезах гиппокампа крыс и в усилении когнитивной деятельности, например быстродействии в радиальном лабиринте с 8 ответвлениями. Эта неожиданная активность переносится на фармацевтические соединения и соответствующие способы применения, включая способы лечения, при которых используются значительно более низкие концентрации (из расчета моль-на-моль) настоящих соединений по сравнению с соединениями, известными из предшествующей области техники. Способность соединений согласно изобретению усиливать ответы, опосредованные АМРА-рецептором, позволяет использовать эти соединения для различных целей. Эти цели включают содействие изучению моделей поведения, зависимых от глутаматных рецепторов, рассмотрение состояний, при которых АМРА-рецепторы или синапсы, использующие эти рецепторы, имеют пониженное количество или эффективность, а также увеличение эксцитатной синаптической активности для восстановления дисбаланса между мозговыми подобластями или увеличение уровня нейротропных факторов. С другой стороны, изобретение включает способ лечения относящегося к млекопитающим субъекта, страдающего от гипоглутаматергического состояния, или от дефицита количества или интенсивности эксцитатных синапсов, или от количества АМРА-рецепторов, которые вызывают нарушения памяти или других познавательных функций. Такие состояния могут также вызывать кортикальный/стриарный дисбаланс, приводящий к шизофрении или поведению, подобному форме шизофрении. Согласно данному способу лечение такого субъекта проводят путем назначения эффективного количества соединения,-2 011034 представленного структурой I и описанного в следующем далее разделе II подробного описания, в фармацевтически приемлемом носителе. Эти и другие цели и особенности изобретения становятся более очевидными из следующего ниже подробного описания изобретения с сопровождающими его чертежами. Краткое описание чертежей На прилагаемой фигуре изображены четыре соединения согласно настоящему изобретению, наряду с одним соединением из предшествующей области техники, которые были протестированы на активность и описаны в экспериментальном разделе и в табл. 1 и 2, инфра. Подробное описание изобретенияI. Определения. Нижеприведенные термины имеют следующие определения, если не указано иначе. Другие термины, которые используются для описания настоящего изобретения, имеют те же самые определения, поскольку эти термины обычно используются специалистами в данной области техники. Используемый здесь термин "алкил" относится к полностью насыщенному одновалентному радикалу, содержащему углерод и водород и который может быть прямой цепью, разветвленной или циклической цепью. Примерами алкилированных групп являются метил, этил, н-бутил, н-гептил, изопропил, 2 метилпропил, циклопропил, циклопропилметил, циклобутил, циклопентил, циклопентилэтил и циклогексил. Термин "замещенный алкил" относится к описанному выше алкилу, включающему одну или более функциональных групп, таких как низшие алкилы, содержащие 1-6 атомов водорода, арил, замещенный арил, ацил, галоген (т.е. алкилированные галогены, например, CF3), гидрокси, алкокси, алкоксиалкил,амино, алкил и диалкиламино, ациламино, ацилокси, арилокси, арилоксиалкил, карбоксиалкил, карбоксиамидо, тио, тиоэфиры, как насыщенные, так и ненасыщенные циклические углеводороды, гетероциклы и т.п. Термин "арил" относится к замещенному или незамещенному одновалентному ароматическому радикалу, имеющему одно кольцо (например, фенил) или кратные конденсированные кольца (например,нафтил). Другие примеры включают гетероциклические ароматические кольцевые группы, имеющие 1 или более атомов азота, кислорода или атомы серы в кольце, например имидазолил, фурил, пирролил,пиридил, тиенил и индолил. Термин "замещенный арил" относится к вышеописанному арилу, который содержит одну или более функциональных групп, таких как низший алкил, ацил, арил, галоген, алкилгалогены (например, CF3),гидрокси, алкокси, алкоксиалкил, амино, алкил и диалкиламино, ациламино, ацилокси, арилокси, арилоксиалкил, карбоксиалкил, карбоксамидо, тио, тиоэфиры, например как насыщенные, так и ненасыщенные циклические углеводороды, гетероциклы и т.п. Термины "гетероцикл" или "гетероциклический" относится к карбоциклическому кольцу, где 1 или более атомов углерода замещены 1 или более гетероатомами, такими как азот кислород или сера. Примеры гетероциклов включают, но не ограничены, такими, как пиперидин, пирролидин, морфолин, тиоморфолин, пиперазин, тетрагидрофуран, тетрагидропиран, 2-пирролидинон, -велеролактам, -велеролактон и 2-кетопиперазин. Термин "замещенный гетероцикл" относится к вышеописанному гетероциклу, который содержит одну или более функциональные групп, таких как низший алкил, ацил, арил, циано, галоген, гидрокси,алкокси, алкоксиалкил, амино, алкил и диалкиламино, ациламино, ацилокси, арилокси, арилоксиалкил,карбоксиалкил, карбоксамидо, тио, тиоэфиры, например как насыщенные, так и ненасыщенные циклические углеводороды, гетероцикл и т.п. Используемый здесь термин "соединение" относится к любому конкретному описанному здесь химическому соединению. В пределах его использования в контексте, термин, в целом, относится к отдельному соединению, но в некоторых случаях он может также относиться к стереоизомерам и/или оптическим изомерам (включая рацемические смеси) описанных соединений. Термин "эффективное количество" относится к количеству выбранного соединения формулы I, которое используется для усиления глутаматергического синаптического ответа при повышении активности АМРА-рецептора. Точное используемое количество варьируется в зависимости от специфического выбранного соединения и его планируемого применения, возраста и веса субъекта, способа введения и т.д. и может быть легко определено обычными экспериментами. В случае лечения состояния или стадии заболевания эффективное количество - это то количество, которое используют для эффективного лечения специфического состояния или стадии заболевания. Термин "фармацевтически приемлемый носитель" относится к носителю или наполнителю, который имеет допустимую токсичность по отношению к субъекту, которому его вводят. Фармацевтически приемлемые наполнители описаны подробно Е.W. Martin, in "Remington's Pharmaceutical Sciences"."Фармацевтически приемлемая соль" соединения амина, которая рассматривается в настоящем изобретении, является солью аммония, имеющей в качестве противоиона неорганический анион, например хлорид, бромид, йодид, сульфат, сульфит, нитрат, нитрит, фосфат и т.п., или органический анион, например ацетат, малонат, пируват, пропионат, фумарат, циннамат, тозилат и т.п.-3 011034 Используемый везде в описании термин "пациент" или "субъект" для описания животного, в основном, млекопитающего животного, включает человека, который подвергается лечению с использованием соединения или композиции согласно настоящему изобретению. Для лечения или применения с/или теми состояниями или заболеваниями, которые являются специфическими для определенного животного (в частности, например, человека в качестве субъекта или пациента), термин "пациент" или "субъект" относятся к такому специфическому животному. Термин "сенсорные моторные нарушения" используют для описания нарушения, которое появляется у пациента или объекта из-за неспособности интегрировать внешнюю информацию, получаемую от пяти известных органов чувств таким образом, чтобы посылать соответствующие физические ответы,приводящие к движению и действию. Термин "когнитивная задача" или "когнитивная функция" используется для описания усилий или процесса, совершаемых пациентом или субъектом, при которых требуются мышление или знание. Разнообразные функции совокупности париетальных, темпоральных и фронтальных долей коры головного мозга, которые составляют приблизительно 75% всей ткани мозга человека, отвечают за большую часть обработки информации, которая происходит между сенсорным входом и моторным выходом. Разнообразные функции совокупности коры головного мозга зачастую называют познавательной способностью,которая буквально означает процесс, с помощью которого мы познаем мир. Выборочное проявление внимания к специфическому стимулу, распознавание и идентификация этих релевантных особенностей стимула, а также планирование и испытание ответа на опыте представляют некоторые из процессов или способностей, опосредованных человеческим мозгом, которые связаны с познанием. Термин "сеть головного мозга" используется для описания различных анатомических областей мозга, которые связаны друг с другом через синаптическую активность нейронных клеток. Термин "АМРА-рецептор" относится к совокупности белков, обнаруженных в некоторых мембранах, которые позволяет положительным ионам пересекать мембрану в ответ на связывание глутамата или АМРА (DLамино-3-гидрокси-5-метил-4-изоксазолпропионовая кислота), но не NMDA. Термин "эксцитатный синапс" используется для описания соединения клетка-клетка, в котором высвобождение химического посредника одной клеткой приводит к деполяризации внешней мембраны другой клетки. Эксцитатный синапс описывает постсинаптический нейрон, имеющий обратный потенциал, который является более позитивным, чем пороговый потенциал, и, следовательно, в таком синапсе нейромедиатор усиливает вероятность того, что эксцитатный постсинаптический потенциал приведет к результату (нейрон запустит генерацию потенциала действия). Обратные потенциалы аннулирования и пороговые потенциалы определяют постсинаптическое возбуждение и ингибирование. Если обратный потенциал для постсинаптического потенциала (ПСП) является более позитивным, чем порог потенциала действия, то эффект медиатора является эксцитатным и продуцирует эксцитатный постсинаптический потенциал (ЭПСП) и запуск потенциала действия нейроном. Если обратный потенциал для постсинаптического потенциала является более негативным, чем порог потенциала действия, то медиатор является ингибиторным и может генерировать ингибиторные постсинаптические потенциалы (ИПСП), таким образом сокращая вероятность того, что синапс запустит потенциал действия. Общее правило для постсинаптического действия: если обратный потенциал является более позитивным, чем пороговый, то происходит возбуждение; ингибирование происходит, если обратный потенциал является более отрицательным,чем пороговый. См., например, Chapter 7, NEUROSCIENCE, edited by Dale Purves, Sinauer Associates, Inc.,Sunderland, MA 1997. Термин "моторная задача" используется для описания усилия, предпринимаемого пациентом или субъектом, которое приводит к движению или действию. Термин "перцепцивная задача" используется для описания действия пациента или субъекта, при котором внимание направлено на сенсорные входы. Термин "синаптический ответ" используется для описания биофизических реакций в одной клетке как последствия высвобождения химических посредников другой клеткой, с которой она находится в близком контакте. Термин "гипоглутаматергическое состояние" используется для описания состояния или нарушения,при котором передача, опосредованная глутаматом (или связанная с эксцитатными аминокислотами),ниже нормальных уровней. Передача состоит в высвобождении глутамата, связывании с постсинаптическими рецепторами и открытии каналов, объединеных с этими рецепторами. Конечной точкой гипоглутаматергического состояния является сниженный эксцитатный постсинаптический ток. Это может быть результатом любой из трех вышеуказанных фаз передачи. Состояния или заболевания, которые считаются гипоглутаматергическими состояниями и которые можно лечить, используя соединения, композиции и способы согласно настоящему изобретению, включают, например потерю памяти, слабоумие, депрессию, нарушение внимания, сексуальную дисфункцию, нарушения движения, включая болезнь Паркинсона, шизофрению или поведение, подобное шизофрении, нарушения памяти и способности к обучению,включая те заболевания, которые являются следствием старения, травмы, удара и нейродегенеративных нарушений, а также состояния, обусловленные действием лекарственных средств, нейротоксическими агентами, болезнью Альцгеймера и старением. Эти состояния легко распознаются и диагностируются-4 011034 специалистами в данной области техники. Термин "кортикостриарный дисбаланс" используется для описания состояния, в котором баланс нейронной активности во взаимосвязанных коре головного мозга и основном стриарном комплексе отклоняется от обычного. "Активность" может быть оценена путем электрической регистрации или молекулярными биологическими методами. Дисбаланс может быть установлен путем применения этих методов измерения к двум структурам или с помощью функциональных (поведенческих или физиологических) критериев. Термин "аффективное расстройство" или "расстройство настроения" описывает состояние, когда печаль или восторг чрезмерно интенсивны и продолжаются сверх ожидаемого воздействия стрессового события жизни или возникают внутри организма. Используемый здесь термин "аффективное растройство" охватывает все типы расстройства настроения, как описано, например, в Diagnostic and StatisticalManual of Mental Disorders, Fourth Edition (DSM IV), pages 317-391. Термин "шизофрения" используют для описания состояния, которое является обычным типом психоза, характеризующимся нарушением процессов мышления, таким как бред и галлюцинации, экстенсивным уходом интереса индивидуума от других людей и внешнего мира и сосредоточением на собственном "я". В настоящее время шизофрению рассматривают как группу умственных болезней, а не одну отдельную категорию, и проводят различие между реактивной шизофренией и вялотекущей шизофренией. Используемый здесь термин "шизофрения" или "форма, подобная шизофрении" включает все типы шизофрении, включая амбулаторную шизофрению, кататоническую шизофрению, гебефреническую шизофрению, скрытую шизофрению, вялотекущую шизофрению, псевдоневротическую шизофрению, реактивную шизофрению, простую шизофрению, и связанные с ней психотические расстройства, которые подобны шизофрении, но которые не обязательно диагностируются как шизофрения сами по себе. Шизофрения и другие психотические расстройства могут быть диагностированы путем использования основополагающих принципов, изложенных, например, в руководстве Diagnostic and Statistical Manual ofMental Disorders, Fourth Edition (DSM IV) Sections 293.81, 293.82, 295.10, 295.20, 295.30, 295.40, 295.60,295.70, 295.90, 297.1, 297.3, 298.8. Термин "функция мозга" используют для описания комбинированных задач восприятия, интегрирования, фильтрования и ответа на внешние стимулы и внутренние мотивационные процессы. Термин "нарушенный" используют для описания функции, работающей на уровне, который является ниже нормального уровня. Нарушенные функции могут оказывать значительное влияние, поскольку функция, которая выполняется слабо, фактически не существует либо работает в режиме, который является значительно ниже нормального. Нарушенные функции могут также быть субоптимальными. Серьезность нарушения функции зависит от состояния пациента и заболевания, подлежащего лечению.II. Соединения, которые усиливают функцию АМРА-рецепторов. Настоящее изобретение относится, с одной стороны, к соединениям, имеющим свойство усиливать функции АМРА-рецептора. Эти соединения имеют структуры Ia или Ib, которые приведены ниже:Q и Q' независимо обозначают водород, -СН 2-, -О-, -S-, алкил или замещенный алкил,R1 обозначает водород, алкил или вместе с Q может быть кольцом циклоалкила,R2 может отсутствовать или, если присутствует, может быть -СН 2-, -СО-, -СН 2 СН 2-, -СН 2 СО-,-СН 2 О-, -CRR'- и -CONR-,Y обозначает водород или -OR3 или служит для связывания ароматического кольца с А как с единственной связью, =N- или -NR-,R3 обозначает водород, алкил, замещенный алкил или служит для связывания присоединенного кислорода с А, являющимся низшим алкиленом, таким как метилен или этилен, или замещенным низшим алкиленом, например -CRR'-, связывающий ароматическое кольцо с А для образования замещенного или незамещенного 6-, 7- или 8-членного кольца или связи, связывающей кислород для образования 5- или 6 членного кольца,А обозначает -NRR', -OR, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалкилалкил, арил, замещенный арил, гетероцикл или замещенный гетероцикл, содержащий 1 или 2 гетероатома, например кислород, азот или серу,R обозначает водород, арил, арилалкил, замещенный арил, замещенный арилкалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или гетероциклоалкил,R' отсутствует или обозначает водород, арил, арилалкил, замещенный арил, замещенный арилалкил,алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или может быть соединен с R для обра-5 011034 зования 4-8-членного кольца, которое может быть замещено X и может быть связано с Y для образования 6-членного кольца и которое может произвольно содержать 1 или 2 гетероатома, например кислород,азот или серу,X и X' обозначают независимо R, гало, -CO2R, -CN, -NRR', -NRCOR', -NO2, -N3 или -OR. Синтез 1,3-бензоксазин-4-он структур, замещенных карбонильными половинами в 6- или 7-положении, предпочтительно осуществляют следующими методиками: 1) 6-карбонилзамещенные 1,3-бензоксазин-4-он структуры могут быть синтезированы триалкилалюминий-вспомогательным селективным аминолизисом эфиров 4-гидроксиизофталата, сопровождаемым замыканием кольца, с использованием альдегида. Дальнейшие реакции, известные специалистам в данной области техники, могут выполняться на эфире, остающемся в 6-положении, для преобразования эфира в различные другие карбонилы, содержащие, но не ограниченные половинами, такими как амиды, альдегиды, кетоны, оксимы или другие эфиры. Подобным способом можно синтезировать 7-карбонилзамещенные 1,3-бензоксазин-4-он структуры свободным прямым аминолизисом эфиров 2-гидрокситерефталата с последующей реакцией с альдегидом, как показано. Преобразование непрореагировавшего эфира с другим карбонилом, содержащим половины, как описано выше для 6-положения, входит в пределы объема изобретения. Все соединения,описанные в настоящей заявке, можно синтезировать вышеописанным способом, используя аналогичные синтетические стадии, которые специально представленны в описанных здесь примерах, а также те, которые известны специалистам в данной области техники. Выделение стерео- и/или оптических изомеров в чистом виде можно осуществлять способами, которые известны специалистам в данной области техники.III. Способ лечения. Согласно одному аспекту изобретения предлагается способ лечения млекопитающего субъекта,страдающего от гипоглутаматергического состояния или от недостатка количества или интенсивности эксцитатных синапсов или количества АМРА-рецепторов. У такого субъекта может наблюдаться расстройство памяти или других познавательных функций или может произойти кортикально/стриарный дисбаланс, приводящий к потере памяти, слабоумию, депрессии, расстройству внимания, сексуальной дисфункции, нарушению движения, шизофрении или поведению, подобному шизофрении. Расстройство памяти и способности к обучению, которые поддаются лечению согласно настоящему изобретению,включают те нарушения, которые являются следствием старения, травмы, удара и нейродегенеративных нарушений. Примеры нейродегенеративных нарушений включают, но не ограничены теми, которые вызываются лекарственными препаратами, нейротоксическими агентами, болезнью Альцгеймера и старением. Эти состояния могут сразу распознавать и диагностировать специалитсы в данной области техники и проводить лечение путем введения пациенту эффективного количества одного или более соединений согласно настоящему изобретению. Согласно настоящему изобретению способ лечения нуждающегося в лечении субъекта включает введение в фармацевтически приемлемом носителе эффективного количества соединения формулыQ и Q' независимо обозначают водород, -СН 2-, -О-, -S-, алкил или замещенный алкил,R' обозначает водород, алкил или вместе с Q может быть кольцом циклоалкила,R2 может отсутствовать или, если присутствует, может быть -СН 2-, -СО-, -СН 2 СН 2-, -СН 2 СО-,-СН 2 О-, -CRR'- или -CONR-,Y обозначает водород или -OR3 или служит для связывания ароматического кольца с А как единственной связью, =N- или -NR-,R3 обозначает водород, алкил, замещенный алкил, или низший алкилен, например метилен или этилен, или замещенный низший алкилен, например -CRR'-, соединяющий ароматическое кольцо с А для-6 011034 образования замещенного или незамещенного 5-, 6- или 7-членного кольца, или связь, соединяющую ароматическое кольцо с А для образования 5- или 6-членного кольца,А обозначает -NRR', -OR, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, циклоалкилалкил, арил, замещенный арил, гетероцикл или замещенный гетероцикл, содержащий 1 или 2 гетероатома, например кислород, азот или серу;R обозначает водород, арил, арилалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или гетероциклоалкил,R' обозначает водород, арил, арилкалкил, замещенный арил, замещенный арилалкил, алкил, замещенный алкил, циклоалкил, замещенный циклоалкил или может быть присоединен к R для образования 4-8-членного кольца, которое может замещаться R или X или связываться с Y через R3 и которое может произвольно содержать 1 или 2 гетероатома, например кислород, азот или серу,X и X' независимо обозначают R, гало, -CO2R, -NRR', -NRCOR', -NO2, -N3 или -OR. Как указано выше, лечение субъекта способом согласно изобретению способствует усилению активности АМРА-рецепторов, и поэтому его можно применять для облегчения изучения зависимости различных видов поведения от АМРА-рецепторов и проводить лечение таких заболеваний, как расстройство памяти, при котором снижено количество или эффективность АМРА-рецепторов, или синапсов,использующих эти рецепторы. Способ также используют для усиления эксцитатной синаптической активности для восстановления дисбаланса между мозговыми подобластями, который может проявиться при шизофрении, или подобном шизофрении поведении, или другом вышеописанном поведении. Обнаружено, что соединения, вводимые в соответствии с настоящим способом, являются более эффективными, чем ранее описанные соединения, в усилении эффективности АМРА-рецептора, как показано в inIV. Биологическая активность. А. Усиление функции АМРА-рецепторов. Согласно способу по настоящему изобретению синаптические ответы, опосредованные АМРА-рецепторами, усиливаются при использовании описанных здесь соединений. Эти соединения представлены в следующих ниже примерах и являются значительно более сильнодействующими, чем ранее описанные соединения в усилении опосредованных АМРА токов в здоровых клетках в культурах нейронов и функции АМРА-рецептора в срезах гиппокампа крысы. Тесты in vitro описаны далее, а также ниже в примере 64. Известно, что область ЭПСП (эксцитатного постсинаптического потенциала), зарегистрированная в области СА 1 после возбуждения СА 3 аксонов, обычно опосредуется АМРА-рецепторами, которые присутствуют в синапсах (Kessler et al., Brain Res. 560: 337-341 (1991. Лекарственные препараты, которые избирательно блокируют рецептор, избирательно блокируют область ЭПСП (Muller et al., Science, выше). Анирацетам, который, как обнаружено, повышает среднее время открытия канала АМРА-рецептора, также увеличивает амплитуду синаптического тока и продлевает его продолжительность (Tang et al., Science,выше). Эти эффекты отображены в области ЭПСП (см., например, Staubli et al., Psychobiology, выше;Xiao et al., Hippocampus, выше; Staubli et al., Hippocampus 2: 4958 (1992. Сообщается, что подобные результаты получены для ранее описанных стабильных бензамидных аналогов анирацетама (Lynch andRogers, международная публикация РСТ 94/02475). Для получения данных, показанных в табл. 1, биполярный нихромный стимулирующий электрод помещали в древовидный слой (stratum radiatum) гиппокампальной подобласти СА 1 близко к границе подобласти СА 3, как описано в примере 64. Импульсы тока (0,1 мс) через стимулирующий электрод активизировали популяцию комиссурального волокна Шаффера, которая возникает из нейронов в подотделе СА 3 и заканчивается в синапсах на дендритах СА 1 нейронов. Активация этих синапсов заставляет их высвобождать глутамат медиатора. Глутамат связывается с постсинаптическими АМРА-рецепторами,которые затем временно открывают связанный с ионами канал и позволяют току натрия входить в постсинаптическую клетку. Этот ток создает разность потенциалов во внеклеточном пространстве (область ЭПСП), которую регистрируют с помощью записывающего электрода высокого сопротивления, помещенного в середине stratum radiatum CA1. Интенсивность тока возбуждения регулировали для получения полумаксимальных ЭПСП (как правило, приблизительно 1,5-2,0 мВ). Парные импульсы возбуждения подавали каждые 40 с с межпульсным интервалом в 200 мс, как описано далее в примере 64. Срезы гиппокампа сохраняли в регистрирующей камере, непрерывно перфузируемой искусственной цереброспинальной жидкостью (ИЦСЖ). В течение 15-30-минутных интервалов перфузионную среду переключали на другую среду, содержащую тестируемые соединения различной концентрации. Ответы, собранные сразу до перфузии лекарственного препарата и в конце, накладывали для вычисления процента увеличения в амплитуде ЭПСП. Соединение 1 А согласно изобретению (самый активный изомер продукта по примеру 1, как показано на прилагаемой фигуре) и эталонное соединение СХ 516, описанное в патенте США 5747492, были протестированы в системе физиологического тестирования, описанной выше. Первая колонка данных табл. 1, ниже, показывает оценку концентрации каждого тестируемого соединения, которое должно было бы потребоваться для увеличения амплитуды области ЭПСП до значения 10% выше основного уровня. ПримерXIII из патента США 5747492. 1 Концентрация соединения, вызывающая 10% увеличение амплитуды области ЭПСП в области СА 1 среза гиппокампа крысы. 2 Концентрация соединения, вызывающая, по меньшей мере, удвоение стационарного тока, индуцированного в корковых нейронах в первичной культуре с помощью 300 мкМ глутамата. 3 Минимальная эффективная доза (МЭД), приводящая к статистически существенному улучшению работы в лабиринте с 8 ответвлениями для повышения познания/памяти.t Для этого теста использовали нерастворенный продукт по примеру t.ND=Не определено. Как свидетельствуют данные в табл. 1, соединение согласно настоящему изобретению способствует увеличению амплитуды fЭПСП в гиппокампе и эффективно при таких низких концентрациях, как 0,3 мкМ. Эффективность настоящего соединения для клеточных токов, опосредованных АМРА-рецепторами, также оценивали в первичной культуре нейронов, в которых концентрации, сниженные до 0,12 мкМ, вызывали удвоение стационарного тока в культурах нейронов, генерируемого при помощи 500 мкМ глутамата. Большинство тестируемых соединений показывали одинаковую либо большую эффективность в усилении функции АМРА-рецептора по сравнению с эталонными соединениями, СХ 516 и СХ 559, в котором бензоксазин замещен гетероатомом, как показано в табл. 2. Таблица 2 Концентрация модулятора, который увеличивает амплитуду fЭПСП на 10%. Концентрация модулятора, необходимого для удвоения устойчивого уровня потока, индуцированного 300 мкМ глутамата на культуральных нейронах. ПримерXIII патента США 5747492. Пример 4 Патента США 5962447 (показан на прилагаемой фигуре).t Более активный энантиомер примера 1.Meнee активный энантиомер примера 1. 2-8 011034 Исследования, в которых сравнивали воздействие АМРА модуляторов на моносинаптический (как описано здесь) и полисинаптический ответы, продемонстрировали, что 10% увеличение амплитуды моносинаптического поля ЭПСП давало 300% увеличение на трисинаптических ответах (Servio et al.,Neuroscience 74: 1025-1035 (1996. Кроме того, обнаружено, что концентрация модулятора, который вызывал эти ответы, присутствует в плазме, начиная с поведенчески релевантных доз Granger et al., Synapse,выше). Таким образом, концентрация сединения, достаточного для получения 10% увеличения амплитуды моносинаптического поля ЭПСП, как показано в табл. 1 и 2, вероятно, представляет собой поведенчески релевантную плазменную концентрацию. В. Поведенческое тестирование. Соединения согласно изобретению являются эффективными при поведенческом тестировании релевантных животных, при лечении различных заболеваний, таких как шизофрения, а также в моделях когнитивной работы, например деятельности в радиальном лабиринте с 8 ответвлениями. Третья колонка данных показывает МЭД для получения эффективного улучшения выполнения задачи с радиальным лабиринтом с 8 ветвями, в которой тестируется улучшение памяти и познания(МЭДС). Эта задача была описана ранее (Staubli et al., PNAS 91: 777-781 (1994), а также Lynch and Rogers,PCT публикация международной заявки WO94/02475). В этом тесте соединение по примеру 1 действовало в 250 раз сильнее, чем СХ 516.V. Введение, дозировки и составление композиций. Как отмечено выше, соединения и способ согласно изобретению повышают опосредованные АМРАрецептором ответы и применяются для лечения гипоглутаматергических состояний. Их также применяют для лечения таких нарушений, как ухудшение памяти или других познавательных функций, вызванных дефицитом количества или интенсивности эксцитатных синапсов или количества АМРА-рецепторов. Их также можно использовать для лечения шизофрении или поведения, подобного шизофрении, вызываемого кортикально/стриарным дисбалансом, а также для облегчения изучения поведения, зависимого от АМРА-рецепторов. У субъектов, которых подвергают лечению настоящими соединениями, фармацевтическими составами и способом согласно изобретению, может быть повреждена память или другие познавательные функции либо может возникнуть кортикально/стриарный дисбаланс, приводящий к потере памяти, слабоумию, депрессии, нарушениям внимания, сексуальной дисфункции, нарушениям движения, шизофрении или подобному шизофрении поведению. Нарушения памяти и нарушения поведения, которые поддаются лечению согласно настоящему изобретению, включают нарушения, являющиеся последствием старения, травмы, удара и нейродегенеративных расстройств. Примеры нейродегенеративных рассройств включают, но не ограничиваются, состояниями, вызываемыми лекарственными препаратами, нейротоксическими агентами, болезнью Алцгеймера и старением. Эти состояния сразу распознают и диагностируют специалисты в данной области техники и проводят лечение путем введения пациенту эффективного количества одного или более соединений согласно настоящему изобретению. В целом, дозировки и способы введения настоящего соединения определяются в зависимости от размера субъекта и его состояния согласно стандартным фармацевтическим методам. Используемые уровни доз могут широко варьироваться и могут быть легко определены специалистами в данной области техники. Как правило, используются количества, измеряемые от миллиграммов до граммов. Соединения можно вводить субъекту различными способами, т.е. перорально, трансдермально, периневрально или парентерально, то есть путем внутривенной, подкожной, внутрибрюшинной или внутримышечной инъекции,также включая трансбуккальное, ректальное и трансдермальное введение. Субъекты, подлежащие лечению способом согласно изобретению, включают людей, домашних животных, лабораторных животных и т.п. Композиции, содержащие соединения согласно настоящему изобретению, могут иметь форму твердого вещества, полутвердого вещества, лиофилизированного порошка или жидких форм дозировки, такую как, например, таблетки, капсулы, порошки, композиции с задержанным высвобождением, растворы, суспензии, эмульсии, суппозитории, кремы, мази, лосьоны, аэрозоли, бляшки или т.п., предпочтительно в формах единиц дозировки, приемлемых для простого введения точных дозировок. Фармацевтические соединения согласно настоящему изобретению, как правило, включают обычный фармацевтический носитель или наполнитель и могут дополнительно включать другие лекарственные средства, например носители, адъюванты, добавки и т.п. Предпочтительно соединение должно составлять приблизительно от 0,5 до 75 вес.% соединения или соединений согласно изобретению с остатком, состоящим, в основном, из приемлемых фармацевтических наполнителей. Для перорального введения такие наполнители включают фармацевтически чистые маннит, лактозу, крахмал, стеарат магния,сахарин натрия, тальк, целлюлозу, глюкозу, желатин, сахароз, карбонат магния и т.п. По желанию, композиция может также содержать незначительные количества нетоксичных вспомогательных веществ,таких как смачивающие вещества, эмульгирующие вещества, или буферы. Жидкие композиции можно получать растворением или диспергированием соединений (приблизительно от 0,5 до 20 вес.% или более) и дополнительных фармацевтических адъювантов в носителе, таком как, например, водный соляной раствор, водная декстроза, глицерин или этанол, для получения раствора или суспензии. Для применения в пероральном жидком лекарственном средстве композицию можно получать в виде раствора, суспензии, эмульсии или сиропа, поставляемого как в жидкой, так и в сухой-9 011034 форме, подходящей для гидратации в водном или нормальном соляном растворе. Когда соединение используется в форме твердых препаратов для перорального введения, препараты могут быть таблетками, гранулами, порошками, капсулами или т.п. Для состава таблетки композицию обычно изготавливают с добавками, т.е. такими наполнителями, как препараты сахаридов или целлюлозы, связующим веществом, например пастой крахмала или метилцеллюлозой, наполнителем, дизинтегратором, и другими добавками, обычно используемыми при изготовлении медицинских препаратов. Композиция для инъекции для парентерального введения, как правило, содержит соединение в виде приемлемого внутривенного раствора, например стерильного физиологического раствора соли. Композицию можно также получать в виде суспензии в липидной или фосфолипидной, в липосомальной суспензии либо в водной эмульсии. Способы получения подобных форм дозировки известны или очевидны специалистам в данной области техники; например, см. Remington's Pharmaceutical Sciences (17th Ed., Mack Pub. Co, 1985). Вводимая композиция будет содержать выбранное соединение в фармацевтически эффективном количестве для получения у субъекта повышенных токов АМРА-рецепторов. Следующие далее примеры иллюстрируют, но никоим образом не ограничивают изобретение. Если не указано иначе, все температуры приводятся в градусах Цельсия. Если не указано иначе, все спектры ЯМР представляют спектры 1 Н ЯМР и получены в дейтерохлороформе или дейтеризованном диметилсульфоксиде при использовании в качестве растворителя тетраметилсилана как внутренний стандарт. Инфракрасные (IR) спектры были зарегистрированы как тонкие пленки на кристалле Френеля, на NaCl кристаллах или в KBr грануле в ATI Mattson Gemini в серии FTIR. Все названия соединений соответствуют IUPAC классификации, как предусмотрено программным обеспечением Chemistry 4-D Draw Метод А. 2,5-Дигидрокситерефталевую кислоту (3,75 г, 18,9 ммоль) добавляли к раствору карбонилдиимидазола (6,20 г, 38,2 ммоль) в 50 мл безводного диметилформамида. Раствор перемешивали при комнатной температуре в течение 24 ч, в течение этого времени добавляли 4-аминобутиральдегид диэтилацеталь(6,30 г, 39,0 ммоль). Раствор дополнительно перемешивали в течение 24 ч при комнатной температуре и затем быстро нагревали до 80 С. Растворитель удаляли вакуумной дистилляцией и остаток очищали флэш-хроматографией (гексан:этилацетат 2:1) на силикагеле для получения 4,0 г сырого бис-ацеталь/амида. Промежуточный продукт растворяли в 125 мл метиленхлорида, к которому добавляли 2,0 мл трифторуксусной кислоты кислота, 20 мг камфорсульфоновой кислоты и активизированные 4 молекулярные сита. Как показала тонкослойная хроматография, реакция быстро завершилась. Неочищенный продукт хроматографировали на силикагеле (гексан:этилацетат 95:5) для получения 1,9 г твердого вещества, которое растворяли в метиленхлориде (250 мл) и фильтровали. Концентрированный раствор 50 мл растворяли с диэтиловым эфиром, стимулирующим кристаллизацию. Раствор охлаждали до 0 С и собирали фильтрацией 1,06 г (19%) белого твердого вещества. Т.пл.=270-271 С. IR: 2977, 2880, 1667, 1453,1422, 1335, 1181, 1073 и 780 см-1. 1 Н ЯМР (500 МГц)7,55 (1 Н, s) 7,53 (1 Н, s), 5,47 (2 Н, m) 3,86 (2 Н, m),3,62 (2 Н, m), 2,44 (2 Н, m), 2,27 (2 Н, m), 2,12 (2 Н, m) и 1,96 ppm (2 Н, m). Метод В. Диэтил 2,5-дигидрокситерефталат (500 мг, 1,97 ммоль) и 4-аминобутилальдегид диэтилацеталь(90%) (1,30 г, 7,2 ммоль) комбинировали и нагревали с флегмой в течение 90 с. Реакционную смесь охлаждали до температуры окружающей среды и затем растворяли 10 мл CH2Cl2. Трифторуксусную кислоту (2,0 мл) и 5 молекулярные сита добавляли к реакционной смеси, которые быстро нагревали с флегмой и оставляли в течение 2 ч. Эту смесь концентрировали на силикагеле при пониженном давлении и элюировали с этилацетатом. Фракции концентрировали при пониженном давлении для получения 420 мг(71%) продукта в виде белого твердого вещества с физическими и спектроскопическими характеристиками, в основном, идентичными описанным для продукта, полученного методом А, выше. Пример 2. (R,S),(R,S)-3 аН,6 аН-Пирролидино[2,1-b]пирролидино[2",1"-3',2'](1,3-оксазино)[6',5'-2,1] бензо[4,5-е]-1,3-оксапергидроин-10,12-дион.SOCl2 (8,0 мл, 100 ммоль) и 2 мл сухого CHCl3 добавляли 3 капли DMF. Реакционную смесь нагревали в- 10011034 течение 1,5 ч, в течение этого времени она становилась прозрачной. Раствор концентрировали при пониженном давлении, растворяли с CH2Cl2 и переконцентрировали при пониженном давлении. Остаток растворяли в 10 мл сухого CHCl3, к которому добавляли по каплям 4-аминобутиральдегид диэтилацеталь(1,00 г, 6,2 ммоль). После охлаждения раствора до 0 С к смеси добавляли 2,0 мл триэтиламина, который оставляли нагреваться до температуры окружающей среды. Через 1 ч раствор разбавляли CH2Cl2, промывали последовательно 10% HCl, насыщенным NaHCO3, насыщенным NaCl и затем высушивали надNa2SO4. Полученное масло концентрировали при пониженном давлении и повторно растворяли в 10 мл сухого CHCl3, к которому добавляли 30 мг камфорсульфоновой кислоты. Реакционную смесь перемешивали в течение 12 ч и затем концентрировали на силикагеле при пониженном давлении. После элюирования с EtOAc/MeOH (99:1) получили 270 мг (36%) белого прозрачного порошка со следующими свойствами. Т.пл.=248-252 С. IR: 1673, 1432, 1356 и 1138 см-1. 1 Н ЯМР (500 МГц)8,58 (0,5 Н, s), 8,52 (0,5 Н, s),6,51 (1 Н, m), 5,50 (2 Н, m) 3,80-3,85 (2 Н, m), 3,60-3,65 (2 Н, m), 2,40-2,45 (2 Н, m), 2,20- 2,30 (2 Н, m), 2,052,15 (2 Н, m) и 1,90-2,00 ppm (2H, m). Пример 3. (R,S)-Метил-9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазапергидроин-6-карбоксилат. Триметилалюминий (2,0 мл 2 М толуола, 4,0 ммоль) добавляли к раствору 810 мг диметилгидрокситерефталата (3,85 ммоль) в 15 мл сухого CHCl3, который перемешивали в течение 20 мин. Добавляли 4 аминобутиральдегид диэтилацеталь (750 мг, 4,65 ммоль), и реакционную смесь нагревали с флегмой в течение 6 ч. Трифторуксусную кислоту (2,0 мл) и 5 молекулярные сита добавляли к охлажденному раствору, который быстро нагревали с флегмой и выдерживали в течение 30 мин. Реакционную смесь концентрировали на силикагеле при пониженном давлении и элюировали с гексан/EtOAc (1:1) с получением 387 мг указанного бензоксазина в виде белого твердого вещества. Т.пл.=76-78 С. IR: 1725, 1672,1439, 1291, 1212 и 1088 см-1. 1 Н ЯМР (200 МГц)8,01 (1 Н, d, J=8,0 Гц), 7,77 (1 Н, dd, J=8,0, 1,5 Гц), 7,64 Диметил 4-гидроксиизофталат (1,00 г, 4,8 ммоль) и 4-аминобутиральдегид диэтилацеталь (1,60 г,10 ммоль) комбинировали и нагревали с флегмой в течение 90 с. Смесь охлаждали до температуры окружающей среды и растворяли в 10 мл CHCl3. К раствору добавляли трифторуксусную кислоту (1,0 мл) и 5 молекулярные сита, которые перемешивали в течение 2 ч. После концентрации на силикагеле при пониженном давлении и элюировании с гексан/EtOAc (2:1) получили 500 мг (42%) белого твердого вещества. Т.пл.=164166 С. IR: 1711, 1669, 1615, 1445 и 1285 см-1. 1 Н ЯМР (200 МГц)8,62 (1 Н, d, J=2,2 Гц), 8,12 (1 Н, dd, J=8,7 и 2,2 Гц), 7,02 (1 Н, d, J=8,6 Гц), 5,55 (1 Н, t, J=5,66 Гц), 3,92 (3 Н, s), 3,50-3,90 (3 Н, m) и 1,80-2,60 ppm (3 Н, m). Пример 5. (R,S)-6-(Пиперидилкарбонил)-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксапергидроин-9-он.(2,0 мл 2 М в толуоле, 4,0 ммоль), затем 400 мг пиперидина (4,7 ммоль). Реакционную смесь нагревали с флегмой в течение 3 ч, охлаждали до температуры окружающей среды и гасили уксусной кислотой. После концентрации на силикагеле при пониженном давлении и элюировании с EtOAc/МеОН (99:1) получили 300 мг белого прозрачного твердого вещества. Т.пл.=149-151 С. IR: 1670, 1632 и 1440 см-1. 1 Н ЯМР- 11011034 Гидрокситерефталевую кислоту (4,0 г, 22 ммоль) растворяли в 200 мл тетрагидрофурана, к которому добавляли избыток диазометана в диэтиловом эфире. Реакционную смесь гасили уксусной кислотой,растворяли CH2Cl2, концентрировали на силикагеле при пониженном давлении и элюировали гексаном/EtOAc (4:1) с получением 4,40 г продукта в виде воскового твердого вещества. Т.пл.=69-75 С. IR: 1720, 1678, 1436, 1319, 1212 и 1109 см-1. Диметилгидрокситерефталат (3,00 г, 14,3 ммоль) охлаждали до 5 С и перемешивали с 7 мл кипящей азотной кислоты в течение 40 мин. Реакционную смесь наливали на лед и экстрагировали с этилацетатом. Органический слой промывали рН 7 фосфатным буфером, насыщенным NaCl и высушивали над безводным Na2SO4. Растворитель удаляли при пониженном давлении для получения 3,0 г неочищенного диметил-2-гидрокси-4-нитротерефталата. IR: 1739, 1688, 1549, 1442, 1338 и 1254 см-1. Диметил-2-гидрокси-4-нитротерефталат (3,0 г, 11,8 ммоль) восстанавливали в 25 мл МеОН в бутыли гидрирования Парра при 90 мг 10% Pd-C под водородом (70 фунтов на кв.дюйм) в течение 3 ч. Катализатор удаляли фильтрацией и раствор концентрировали на силикагеле при пониженном давлении. После элюирования с гексаном/EtOAc (2:1) получали 1,20 г диметил 2-амино-4-гидрокситерефталата в виде желтого вещества, которое использовали без дальнейшей очистки.POCl3 (0,95 г, 6,2 ммоль) добавляли к перемешиваемому раствору 2-пирролидинона (1,00 г, 11,8 ммоль) в 10 мл сухого бензола. Через 2 ч добавляли 1,20 г диметил 2-амино-4-гидрокситерефталата (5,3 ммоль) и смесь нагревали с флегмой в течение 5 ч. Реакционную смесь растворяли с CH2Cl2, промывали насыщенным NaHCO3 и концентрировали при пониженном давлении на силикагеле. После элюирования с(2 Н, m). 9-Карбоксиметил-8-гидрокси-2,3-дигидро-1H-пирроло[2,1-b][1,3]хиназолин-6-он (530 мг, 2,0 ммоль) и 4-аминобутиральдегид диэтилацеталь (1,00 г, 6,2 ммоль) комбинировали и нагревали с флегмой в течение 90 с. Охлажденную реакционную смесь растворяли с CH2Cl2, к которому добавляли 2,0 мл трифторуксусной кислоты и 5 молекулярные сита. Смесь перемешивали в течение 1 ч, концентрировали на силикагеле при пониженном давлении и элюировали с EtOAc/MeOH (90:10). В результате обесцвечивания раствора древесным углем в CHCl3/MeOH и удаления растворителей при пониженном давлении получали 600 мг продукта (99%) в виде белого вещества. Т.пл.=245-250 С. IR: 1681, 1660, 1464, 1209 и 1137 см-1. 1 Н ЯМР (200 МГц)8,21 (1 Н, s), 7,80 (1 Н, s), 5,58 (1 Н, t, J=5,8 Гц), 4,20 (2 Н, t, J=7,3 Гц), 3,75-3,95 (1H,m), 3,55-3,70 (1H, m), 3,19 (2 Н, t, J=8,0 Гц) и 1,90-2,60 ppm (6 Н, m). Пример 7. (R,S)-Этил 7-гидрокси-9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-6-карбоксилат. Диэтил 2,5-дигидрокситерефталат (7,00 г, 27,5 ммоль) и 4-аминобутиральдегид диэтилацеталя (7,00 г,43,4 ммоль) помещали в большую пробирку и обезвоживали в течение 5 мин. Реакционную смесь наливали в разделительную воронку и полностью перемешивали с 800 мл EtOAc и 100 мл 6 Н HCl. Через 30 мин органический слой высушивали над Na2SO4 и растворитель выпаривали до получения желтого масла,которое очищали на силикагеле до получения 4,90 г (64%) бесцветных, пушистых кристаллов указанного соединения. Т.пл.=139-141 С. IR: 1676, 1454, 1240 и 1200 см-1. 1 Н ЯМР (200 МГц)10,48 (1 Н, s), 7,54 Триметилалюминий (1,1 мл 2 М в толуоле, 2,2 ммоль) и 400 мг пиперидина (4,7 ммоль) добавляли к раствору 500 мг метил 9-оксо-3 аН-бензо[3,4-е]пирролидино[2,1-b]-1,3-оксазин-7-карбоксилата (2,0 ммоль) в 10 мл сухого CHCl3. Реакционную смесь нагревали с флегмой в течение 4 ч, охлаждали до температуры окружающей среды и гасили уксусной кислотой. Смесь концентрировали на силикагеле при пониженном давлении и элюировали с гексаном/EtOAc (1:1) для получения 537 мг (90%) белого твердого вещества. Т.пл.=134-136 С. IR: 1673, 1626, 1434 и 1255 см-1. 1 Н ЯМР (200 МГц)7,96 (1 Н, d, J=2,0 Гц), 7,53 (1 Н,dd, J=8,4, 2,2 Гц), 7,00 (1 Н, d, J=8,4 Гц), 5,52 (1 Н, t, J=5,7 Гц), 3,20-4,00 (6 Н, m) и 1,40-2,60 ppm (10 Н, m). К 1,6 г (5 ммоль) диметиламингидрохлорида в 60 мл сухого CH2Cl2 добавляли 10 мл 2,0 М AlMe3 в толуоле при перемешивании под Ar. Затем добавляли 5 ммоль (1,4 г) эфира по примеру 7 в 40 мл CH2Cl2 и дефлегмировали в течение 0,5 ч, в течение этого времени CH2Cl2 дистиллировали и замещали 40 мл сухого толуола. После дефлегмирования в течение другого часа реакционную смесь смешивали с 50 мл 3 М HCl. После отделения слоев водный слой экстрагировали с 350 мл CH2Cl2. Объединенные органические слои высушивали над NaSO4 и растворитель удаляли в вакууме для получения 810 мг фенолического амида (58% выхода). IR: 1652 см-1. CI-MS 277,2 amu (M+H). К продукту амида, полученному выше,(810 мг, 2,9 ммоль) в 40 мл безводного диметилформамида под аргоном сразу добавляли 174 мг (4,35 ммоль) 60% NaH при хорошем перемешивании. Через 10 мин при комнатной температуре температуру ванны повышали до 65 С и сразу добавляли 0,25 мл MEI (11,6 ммоль). Через 2 ч реакционную смесь охлаждали до температуры окружающей среды и гасили 5 мл воды. Воду и диметилформамид затем удаляли под вакуумом и остаток хроматографировали на 150 г силикагеля с EtOAc. Объединенные фракции дали 600 мг Этил 7-гидрокси-9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-6-карбоксилат (пример 7 выше) (1,20 г, 4,83 ммоль) и этаноламин (1,00 г, 16,3 ммоль) комбинировали и нагревали с флегмой в течение 90 с. Охлажденную реакционную смесь разбавляли CHCl3, окисляли уксусной кислотой и концентрировали на силикагеле при пониженном давлении. После элюирования с CH2Cl2/MeOH (95:5) получали 1,50 г гидроксиэтиламида в виде воскового твердого вещества. IR: 3000-3500, 1650, 1562, 1470, 1344,1264, 1211 и 1066 см-1. Пример 11. (R,S),(R,S)-2 Н,3 Н,12 аН,6 аН-1,3-Оксазолидино[2,3-b]пирролидино[2",1"-2',3'](1,3-оксазино)[5',6'-2,1]бензо[4,5-е]-1,3-оксапергидроин-4,10-дион.N-(2-Гидроксиэтил)(7-гидрокси-9-оксо(3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазапергидроин-6-ил карбоксамид (продукт из примера 10) (1,50 г, 5,13 ммоль) суспендировали в 20 мл сухого CHCl3 вместе с 4,6 мл триметилортоформата (43 ммоль) и 1,0 мл 96% муравьиной кислоты (21 ммоль) и нагревавали с флегмой в течение 6 ч. Охлажденную реакционную смесь нейтрализовали карбонатом калия и концентрировали на силикагеле при пониженном давлении. После элюирования с гексаном/EtOAc (1:1) и перекристаллизации из этилацетата получали 430 мг белого вещества. Т.пл.=285-287 С. IR: 1675, 1458 и 1421 см-1. 1 Н ЯМР (200 МГц)7,66 (0,5 Н, s), 7,64 (0,5 Н, s), 7,54 (0,5 Н, s), 7,51 (0,5 Н, s), 6,22 (0,5H, s), 6,21 (0,5H, s),5,45-5,53 (1H, m), 4,20-4,40 (3H, m), 3,75-3,95 (1H, m), 3,55-3,70 (2H, m) и 1,90-2,58 ppm (4H, m). Пример 12. (R,S),(R,S)-2H,3H,8H,9H,12aH,6aH-1,3-Оксазолидино[2,3-b]-1,3-оксазолидино[2,3-2',3']- 13011034 Охлажденную смесь растворяли с хлороформом, метанолом и уксусной кислотой и концентрировали на силикагеле при пониженном давлении. Элюирование с CH2Cl2/MeOH (90:10) дало 1,20 г неочищенного бис-амида. Неочищенный амид суспендировали в 20 мл сухого CHCl3 вместе с 4,6 мл триметилортоформиата (43 ммоль) и 1,0 мл 96% муравьиной кислоты (21 ммоль) и дефлегмировали в течение 6 ч. Охлажденную смесь нейтрализовывали с карбонатом калия и концентрировали на силикагеле при пониженном давлении. Продукт элюировали и кристаллизовали из этилацетата для получения 88 мг белого порошка Синтез проводили в основном, как в примере 1, метод А, за исключением замены 5-аминопентанал диэтилацеталя на 4-аминобутиральдегид диэтилацеталь. 1 Н ЯМР (200 МГц)7,502 (s), 7,493 (s), 5,19(соединение примера 3) в 20 мл метанола в колбе Эрленмейера объемом 125 мл добавляли 20 мл 1NNaOH. После энергичного перемешивания в течение 30 мин раствор окисляли до рН 2 с 1N HCl. Полученный белый преципитат отфильтровывали и промывали 25 мл воды. Это твердое вещество повторно растворяли в этилацетате/метаноле (1/1) и высушивали над безводным Na2SO4. Раствор концентрировали с получением 703 мг (81%) белого твердого вещества. IR: 1712 и 1637 см-1. К суспензии 9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-6-карбоновой кислоты (300 мг,1,29 ммоль) в CH2Cl2 добавляли 1,1'-карбонилдиимидазол (209 мг, 1,29 ммоль) в колбу с круглым основанием объемом 25 мл. Через 2 ч добавляли морфолин через шприц. Полученную реакционную смесь перемешивали в течение 30 мин и затем промывали 1N HCl, насыщенным NaHCO3 и соляным раствором. Затем органический слой высушивали над Na2SO4, отфильтровывали и концентрировали для получения 375 мг пены. Продукт очищали хроматографией на силикагеле при элюировании с этилацетатом с получением 330 мг серовато-белых кристаллов (85%). Т.пл.=184-186 С. IR: 1669 и 1636 см-1. 1 Н ЯМР 7,98 К суспензии 9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-6-карбоновой кислоты (160 мг,0,689 ммоль) в 2 мл дихлорметана в грушевидной колбе объемом 25 мл добавляли 1,1'-карбонилдиимидазол (112 мг, 0,689 ммоль). Перемешивание проводили при положительном давлении аргона и в течение 1 ч достигали полного растворения. Раствор перемешивали дополнительные 2 ч и затем медленно добавляли к перемешиваемому раствору через шприц 4-гидроксипиперидин (148 мг, 3,44 ммоль) в 5 млCH2Cl2. Полученную реакционную смесь промывали 1N HCl, насыщенным NaHCO3 и соляным раствором. Органический слой высушивали над безводным Na2SO4, отфильтровывали и концентрировали для получения 174 мг белой пены. Пену кристаллизовали из CH2Cl2/Et2O с получением 158 мг белого твердого вещества (72%). Т.пл.=130-135 С. IR: 3400, 1657 и 1619 см-1. 1 Н ЯМР 7,96 (1 Н, d, J=7,6 Гц), 7,10 (1 Н,d, J=7,6 Гц), 7,00 (1 Н, s), 5,51 (1 Н, t, J=6,0 Гц), 4,16 (1 Н, bs), 3,99 (1 Н, m), 3,86 (1 Н, m), 3,63 (1 Н, m), 3,60 Следуя методике получения соединения по примеру 14, производили замену 1-метилпиперазина на морфолин для получения 378 мг (94%). IR: 1666 и 1633 см-1. 1 Н ЯМР 7,97 (1 Н, d), 7,10 (1 Н, d), 7,00 (1 Н,s), 5,51 (1 Н, t), 3,86 (1 Н, m), 3,79 (2 Н, bs), 3,62 (1 Н, m), 3,39 (1 Н, bs), 2,48 (4 Н, m), 2,32 (3 Н, s), 2,25 (2 Н,m), 2,13 (1 Н, m) и 1,96 ppm (1H, m). Пример 17. Получение (R,S)-6-[(4-аминопиперидил)карбонил]-3 аН-бензо[е]пирролидино[2,1-b]-1,3 оксазин-9-она. Колбу с круглым основанием с 3 горловинами, снабженную термометром, наполняли азидом натрия (3,25 г, 50,0 ммоль) и теплой Н 2 О (3,25 мл). Полученную пасту энергично перемешивали по мере добавления 20 мл бензола. Полученный гетерогенный раствор охлаждали до 5 С и затем по каплям добавляли H2SO4 (1,34 мл, 25,0 ммоль). Температуру раствора поддерживали ниже 10 С и перемешивание продолжали в течение дополнительных 5 мин. Органический слой фильтровали в колбе Эрленмейера,высушивали над безводным NaSO4 и использовали в следующей реакции. Диизопропилазодикарбоксилат добавляли через шприц к раствору 4-гидроксипиперидина (2,0 г, 20 ммоль) и трифенилфосфина (6,75 г, 25,7 ммоль) в CH2Cl2. Сначала добавляли часть раствора гидразойной кислоты из вышеупомянутого полученного кипящего раствора и поэтому раствор охлаждали в ледяной ванне во время добавления остатка. Перемешивание продолжали в ледяной ванне, которую оставляли нагреваться до температуры окружающей среды. После перемешивания при температуре окружающей среды в течение 16 ч раствор промывали Н 2 О, далее 1N HCl. Кислый раствор промывали Et2O/EtOAc (2/1), а с помощью 1 Н NaOH затем получали основной раствор до рН 9. Полученный мутный водный раствор экстрагировали с CH2Cl2. Органический слой высушивали над Na2SO4, фильтровали и концентрировали с получением 1,05 г желтого масла (42%). IR: 2092 см-1. Неочищенный продукт использовали без дальнейшей очистки в следующей стадии, которая следовала за процедурой по примеру 14 выше с замещением 4-азидопиперидина для морфолина. Неочищенный продукт очищали хроматографией на силикагеле элюированием с 3/1 гексаном/этилацетатом, получая 304 мг 6-[(4-азидопиперидил)карбонил]-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-9-он в виде пены (73%). IR: 2095, 1670 и 1636 см-1. В колбу Парра объемом 50 мл, предварительно продутую аргоном, добавляли 10% Pd/C (90 мг), затем раствор 304 мг 6-[(4-азидопиперидил)карбонил]-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-9-она(0,891 ммоль) в 5 мл МеОН. Полученный раствор обрабатывали водородом при 25 фунтов на кв.дюйм в течение 2 ч, фильтровали через целит и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя с 15/1 CH2Cl2/MeOH (1% NH4OH) с получением 220 мг чистого продукта. 1 Н ЯМР (500 МГц)7,96 (1 Н, d, J=7,7 Гц), 7,09 (1 Н, d, J=8,5 Гц), 6,99 (1 Н, s), 5,51 (1 Н, t, J=5,9 Гц), 4,57 (1 Н,bd), 3,85 (1 Н, m), 3,67 (1 Н, bs), 3,63 (1 Н, m), 3,04 (1 Н, bs), 2,98 (1 Н, m), 2,94 (1 Н, bs), 2,45 (1 Н, m), 2,27 В колбу с круглым основанием с 2 горловинами объемом 100 мл, снабженную дефлегматором, добавляли этил 9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксапергидроин-6-карбоксилат (343 мг, 1,41 ммоль) и 20 мл CH2Cl2 под атмосферой аргона. Перемешиваемый раствор наполняли триметилалюминием (1,5 мл,3,0 ммоль) и через 30 мин добавляли через шприц 4-метилпиперидин (0,19 мл, 2,9 ммоль). После того,как раствор нагревали с флегмой в течение 24 ч и выдерживали при температуре окружающей среды в- 15011034 течение дополнительных 72 ч, его гасили Н 2 О, затем медленно добавляли 6N HCl. Органический слой изолировали, а водный слой экстрагировали тремя 50 мл частями CH2Cl2, которые комбинировали с органическим слоем и промывали водой, соляным раствором, высушивали над Na2SO4, отфильтровывали и концентрировали. Полученное масло очищали хроматографией на силикагеле, элюируя с 1:1 гексаном/ этилацетатом. Чистые фракции концентрировали для получения 323 мг пены (54%) IR: 1668 и 1625 см-1. 1 Н ЯМР (500 МГц)7,96 (1 Н, d, J=7,5 Гц), 7,09 (1 Н, d), 6,99 (1 Н, s), 5,51 (1 Н, t, J=6,0 Гц), 4,66 (1 Н, bd),3,86 (1H, m), 3,62 (2 Н, m), 2,96 (1 Н, bt), 2,76 (1 Н, bt), 2,44 (1 Н, m), 2,27 (1 Н, m), 2,12 (1 Н, m), 1,94 (1 Н, m),1,77 (1 Н, bd), 1,65 (2 Н, m), 1,22 (1 Н, m), 1,08 (1H, m) и 0,98 ppm (3 Н, d, J=6,3 Гц). Пример 19. Получение (R,S)-1-[(9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-6-ил)карбонил]пиперидин-4-карбонитрила. Следуя методике для получения соединения из примера 14, за исключением замещения 4-цианопиперидина на морфолин, получали 1-[(9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-6-ил)карбонил]пиперидин-4-карбонитрил в виде белого кристаллического вещества (61%). Т.пл.=154-156 С. IR: 1667 и 1633 см-1. 1 Н ЯМР 7,98 (1 Н, d, J=7,5 Гц), 7,08 (1 Н, d, J=7,4 Гц), 6,98 (1 Н, s), 5,52 (1 Н, t, J=5,96 Гц),3,92 (1 Н, bs), 3,86 (1 Н, m), 2,27 (1 Н, m), 2,14 (1 Н, m), 1,96 (1 Н, m) и 1,85 ррm (4 Н, bs). Пример 20. Получение (R,S),(R,S)-3-(гидроксиметил)-6 аН-хромано[7,6-е]пирролидино[2,1-b]-1,3 оксазин-4,10-диона. Изоксазолин согласно примеру 18 (570 мг, 2,00 ммоль) растворяли в 100 мл метанола. Добавляли раствор борной кислоты (1,00 г) в 20 мл воды и 1,00 г никеля Ренея и полученную смесь обрабатывали водородом при 1 атм в течение 2 ч при комнатной температуре. Метанол выпаривали и остаток экстрагировали с 200 мл EtOAc, который высушивали над Na2SO4. Выпаривание растворителя дало 420 мг масла,которое очищали на силикагеле элюированием 20% гексана/EtOAc. В результате кристаллизации из элюирующего растворителя получили 202 мг кетона в виде желтого твердого вещества. Т.пл.=189-190 С. К 840 мг (3,0 ммоль) диэтил 2,5-дигидрокситерефталата в 100 мл безводного CHCl3 под аргоном при 0 С добавляли при перемешивании 10 мл (20 ммоль) 2,0 М AlMe3 в толуоле. Затем также добавляли 5,0 мл (77 ммоль) безводного этиламина. В течение обоих добавлений происходило выделение газа в большом количестве. Реакционную смесь медленно нагревали с флегмой и затем дефлегмировали под аргоном в течение 3 ч. В этой точке не было обнаружено никакого исходного материала с помощью тонкослойной хроматографии (1:1 EtOAc/гексан). Реакционную смесь охлаждали до температуры окружающей среды и наливали в 200 мл 3 М HCl. Слои отделяли, а водный слой экстрагировали с 3100 млEtOAc. Комбинированные органические слои высушивали над Na2SO4 и растворитель удаляли в вакууме для получения 600 мг диамида (79% выход). 1 Н ЯМР (500 МГц)11,61 (2 Н, s), 6,94 (2 Н, s), 6,28 (2 Н, bs),3,51 (4 Н, q) и 1,28 ppm (6H, t). К 4,9 г (19,4 ммоль) накопившегося диамида 5,0 г (56 ммоль) триоксана и 4,6 г (20 ммоль) камфорсульфоновой кислоты в 340 мл растворителя (17% тетрагидрофуран/CCl4) добавляли 10 капель концентрированной H2SO4. Затем бесцветную реакционную смесь помещали с флегмой под аргоном при перемешивании. Воду удаляли азеотропно из реакционной смеси во время дефлегмирования при включении 250 мл дополнительной трубки с боковым ответвлением с 3 молекулярными ситами (100 г) в паровом канале. Через 18 ч молекулярные сита заменяли, добавляли дополнительные 3,6 г (40 ммоль) триоксана и быстро продолжали дефлегмирование. На следующий день сита вновь заменяли, добавляли 3,8 г (42 ммоль)- 16011034 триоксана и реакционную смесь дефлегмировали с течение следующих 4 ч. Исходное вещество не содержалось в реакционной смеси кофейного цвета, которую охлаждали до 10-15 С в ледяной ванне, и затем смешивали с 200 мл ледяного 0,5 М NaOH. Отделенный органический слой промывали 100 мл Н 2 О и высушивали над Na2SO4. После удаления растворителя в вакууме получили 2,5 г неочищенного материала, который хроматографировали на 150 г силикагеля, используя 2 л EtOAc, с получением 1,5 г (28%) продукта. Т.пл.=236-237 С. После перекристаллизации из 3:1 Et2O/CH2Cl2 получили 1,09 г (20%) из 3,8 диэтил-2H,7H-1,3-оксапергидроино[5',6'-4,5]бензо[e]-1,3-оксазин-4,9-диона. CIMS m/z=220,1 и 277,2 amu. 1 Н ЯМР (500 МГц)7,57 (1 Н, s), 5,16 (2 Н, s), 3,60 (2 Н, q, J=7,23 Гц) и 1,25 (3 Н, t, J=7,23 Гц). Пример 22. Получение (R,S)-3-метил-2 Н,6 аН-пирролидино[2",1"-3',2']-1,3-оксазино[5',6'-4,5]бензо К 1,4 г (5,1 ммоль) этил 7-гидрокси-9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-6-карбоксилата (продукт из примера 7) в 120 мл сухого CHCl3 (0 С водной ванны) добавляли 5,0 мл (10 ммоль) 2,0M AlMe3 в CY2Cl2 и 6,0 мл (12 ммоль) 2,0 М раствора метиламина в тетрагидрофуране. Через 15 мин стабильного выделения газа водную ванну заменяли масляной ванной и реакционную смесь дефлегмировали в течение 1,5 ч, затем быстро перемешивали при комнатной температуре. После того, как добавляли дополнительные 6,0 мл (12 ммоль) AlMe3 раствора и 6,0 мл (12 ммоль) метиламина в тетрагидрофуране, реакционную смесь дефлегмировали в течение 2 ч и наливали в 200 мл 3M HCl с полным перемешиванием. Отделенный водный слой экстрагировали с 3100 мл CHCl3, a комбинированные органические слои высушивали над MgSO4. Растворители удаляли в вакууме для получения 1,2 г фенольного амида в 91% выхода в виде воскового твердого вещества. CI MS: 263,3 amu (M+1). IR: 3319, 1652, 1608 и 1455 см-1. К 1,0 г (3,8 ммоль) вышеупомянутого фенольного амида в 300 мл безводного CHCl3 под аргоном при перемешивании добавляли 900 мг (10,0 ммоль) s-триоксана, затем 2,0 г H2SO4. Реакционную смесь быстро дефлегмировали, охлаждали до комнатной температуры и затем промывали 200 мл насыщенногоNaHCO3. Водный слой возвращали экстракцией с 4100 мл CH2Cl2, а комбинированные органические слои высушивали над MgSO4. Растворитель удаляли в вакууме для получения 900 мг бледно-желтого остатка, который хроматографировали на 400 г силикагеля (EtOAc), с получением 800 мг (77%) бесцветного порошка. Т.пл.=195-196 С. IR 1672 и 1454 см-1. CI-MS amu (М+1). 1 Н-ЯМР (500 МГц)7,57 (1 Н, s),7,54 (1 Н, s), 5,47 (1 Н, t, J=5,7 Гц), 5,16 (2 Н, m), 3,83 (1 Н, m), 3,65 (1 Н, m), 3,12 (3 Н, s), 2,45 (1 Н, m), 2,26 Синтез 3-этил-2 Н,6 аН-пирролидино[2",1"-3',2']-1,3-оксазино[5',6'-4,5]бензо[е]-1,3-оксазапергидроин 4,10-диона проводили, в основном, как описано выше для примера 22, с замещением безводного этиламина на раствор метиламина. Неочищенный материал наносили на 50 г силикагеля и элюировали с EtOAc с получением чистого продукта в 61% выхода. Т.пл.=197-198 С. IR: 1667 и 1451 см-1. CI-MS 289,2 amu Синтез 3-пропил-2 Н,6 аН-пирролидино[2",1"-3',2']-1,3-оксазино[5',6'-4,5]бензо[е]-1,3-оксазапергидроин 4,10-диона проводили, в основном, как описано выше для примера 22 с замещением н-пропиламина на раствор метиламина для получения воскового остатка, который хроматографировали на силикагеле с получением бесцветного твердого вещества. Т.пл.=153-154 С. IR (NaCl) 1667 и 1454 см-1. CI-MS 303,1 amu Синтез 3-(метилэтил)-2 Н,6 аН-пирролидино[2",1"-3',2']-1,3-оксазино[5',6'-4,5]бензо[е]-1,3-оксазапергидроин-4,10-диона проводили, в основном, как описано выше для примера 22, с замещением изопропиламина на раствор метиламина. Заключительную очистку проводили колоночной хроматографией на силикагеле (35% выход). Т.пл.=204-205 С. IR: 1664 и 1452 см-1. CI-MS 303,3 amu (M+1). 1 Н ЯМР (500 МГц)7,58 (1 Н, s), 7,53 (1 Н,s), 5,49 (1 Н, t, J=5,9 Гц), 5,14 (2 Н, m), 4,87 (1 Н, sept, J=4,9 Гц), 3,88 (1 Н, dt, J=11,6 и 7,3 Гц), 3,65 (1 Н, m),2,45 (1 Н, m), 2,27 (1 Н, m), 2,13 (1 Н, m), 1,96 (1 Н, m) и 1,26 ppm (6 Н, m). Пример 26. Получение (R,S)-3-циклопентил-2 Н,6 аН-пирролидино[2",1"-3',2']-1,3-оксазино[5',6'-4,5] бензо[е]-1,3-оксапергидроин-4,10-диона. Синтез (R,S)-3-циклопентил-2 Н,6 аН-пирролидино[2",1"-3',2']-1,3-оксазино[5',6'-4,5]бензо[е]-1,3-оксазапергидроин-4,10-диона проводили, в основном, как описано выше для примера 22, со следующими исключениями: неразбавленный циклопентиламин замещали на раствор метиламина в тетрагидрофуране; толуол использовали как растворитель для реакции; вместо дефлегмации раствор быстро нагревали до 80 С. Окончательную очистку проводили хроматографией на колонке силикагеля (56% выхода). Т.пл.=184-188 С. CI-MS 329,1 amu (M+1). 1 Н-ЯМР (500 МГц)7,58 (1 Н, s), 7,53 (1H, s), 5,47 (1 Н, t, J=6 Гц),5,14 (2 Н, s), 4,94 (1 Н, р, J=8,5 Гц), 3,85 (1 Н, m), 3,63 (1 Н, m), 2,45 (1 Н, m), 2,26 (1 Н, m), 2,13 (1 Н, m), 1,99 Синтез проводили, в основном, как описано для соединения в примере 22, за исключением того, что на заключительной стадии циклизации использовали 5 экв. камфорсульфоновой кислоты. Т.пл.=189190 С. IR: 1667 см-1. CI-MS 386,3 amu (М+1). 1 Н ЯМР (500 МГц)7,5 (2 Н, s), 5.47 (1 Н, t, J=6,0 Гц), 5,22 Синтез 3-(2 Н-бензо[d]-1,3-диоксолен-5-илметил)-2 Н,6 аН-пирролидино[2",1"-3',2']-1,3-оксазино[5',6'-4,5] бензо[е]-1,3-оксазапергидроин-4,10-диона проводили, в основном, тем же способом, как описано выше для примера 22, за исключением того, что пиперониламин замещали метиламином. IR-спектроскопия Методика для синтеза 3-(2,2,2-трифтороэтил)-2 Н,6 аН-пирролидино[2",1"-3',2']-1,3-оксазино[5',6'4,5]бензо[e]-1,3-оксазапергидроин-4,10-диона была, в основном, такой же, как для примера 22, за исключением того, что трифтороэтиламин замещали метиламином. Процедуру для триоксана проводили по шкале 5 ммоль с получением 1,2 г очищенного трифтороэтилбензоксазина, 70% выход с т.пл. 179-180 С. IR-спектроскопия (NaCl): 1672 и 1454 см-1. CI-MS, 343,1 Серийно выпускаемый диэтиловый эфир 2,5-дигидрокситерефталевой кислоты (7,00 г, 27,5 ммоль) и 4-аминобутиральдегид диэтилацеталь (7,00 г, 43,4 ммоль) помещали в пробирку и нагревали с флегмой в течение 5 мин. Реакционную смесь наливали в разделительную трубку с 800 мл EtOAc и 100 мл 6 МHCl. Через 30 мин органический слой отделяли, высушивали над Na2SO4 и растворитель выпаривали для получения желтого масла. Флэш-хроматографией (силикагель, этилацетат/гексан 80/20) получали 4,90 г(64%) бесцветных, пушистых кристаллов со следующими свойствами. Т.пл.=139-141 С. 1 Н ЯМР (300 МГц,CDCl3)10,41 (1 Н, s), 7,49 (1 Н, s), 7,44 (1 Н, s), 5,41 (1 Н, t, J=5,7 Гц), 4,38 (2 Н, q, J=7,2 Гц), 3,86-3,76 (1H,m), 3,64-3,53 (1H, m), 2,46-2,33 (1 Н, m), 2,25-2,02 (2 Н, m), 1,99-1,82 (1H, m), 1,38 ppm (3 Н, t, J=7,2 Гц). К раствору 2,0 г (7,21 ммоль) промежуточного фенола в 50 мл толуола добавляли 15 мл 1,2-дибромэтана и 2,4 г (17,4 ммоль) K2CO3. Смесь дефлегмировали в течение 48 ч до полного использования исходного материала. Растворитель выпаривали и добавляли 200 мл EtOAc и 100 мл воды. Органическую фазу высушивали над Na2SO4 и растворитель выпаривали. Флэш-хроматографией на силикагеле (EtOAc/ гексан 60/40) получали бесцветное масло, которое кристаллизовалось через 1 ч (2,1 г, 76%). Т.пл.=9899 С. 1 Н ЯМР (300 МГц, CDCl3)7,47 (1H, s), 7,36 (1 Н, s), 5,46 (1 Н, t, J=5,9 Гц), 4,45-4,30 (4 Н, m), 3,893,79 (1 Н, m), 3,66 (2 Н, t, J=6,0 Гц), 3,69-3,57 (1 Н, m), 2,50-2,38 (1 Н, m), 2,31-2,06 (2 Н, m), 2,05-1,87 (1H,m), 1,39 ppm (3 Н, t, J=7,1 Гц). 720 мг (1,87 ммоль) промежуточного бромида растворяли в 30 мл этанола. Добавляли раствор 350 мгKOH (6,25 ммоль) в 10 мл воды. Реакция завершалась через 10 мин. Добавляли 100 мл воды и 200 млEtOAc и смесь подкисляли HCl до рН 2. Органическую фазу высушивали над Na2SO4 и растворитель выпаривали, получая бесцветное масло (700 мг). Масло растворяли в 50 мл тетрагидрофурана, и добавляли 1 мл H2NEt, и смесь перемешивали в течение 72 ч при комнатной температуре. Растворитель выпаривали и остаток повторно растворяли в 50 мл тетрагидрофурана. К раствору добавляли 253 мг (1,87 ммоль) НОВТ, 228 мг (1,87 ммоль) DMAP, 189 мг(1,87 моль) NEt3 и 1,08 г (5,61 ммоль) EDCl и смесь перемешивали в течение 24 ч. Растворитель выпаривали и добавляли 100 мл EtOAc и 100 мл водной HCl (рН 2). Органическую фазу высушивали над Na2SO4 и выпаривали. Флэш-хроматографией на силикагеле (EtOAc/гексан 75/25) получали 335 мг бесцветного масла (59%). Кристаллизацией из EtOAc/гексана получали 283 мг белых кристаллов со следующими свойствами. Т.пл.=180-181 С. IR: 1670, 1631, 1475, 1443, 1034 см-1. 1 Н ЯМР (500 МГц, CDCl3)7,58 (1 Н,s), 7,36 (1 Н, s), 5,47 (1H, t, J=6 Гц), 4,36 (1H, ddd, J=10,9, 8,5 и 4,1 Гц), 4,29 (1 Н, ddd, J=10,9, 4,6 и 4,2 Гц),3,88-3,81 (1 Н, m), 3,66 (2 Н, q, J=7,1 Гц), 3,65-3,58 (1 Н, m), 3,52 (1 Н, ddd, J=15,8, 8,5 и 4,2 Гц), 3,40 (1 Н,ddd, J=15,8, 4,6 и 4,1 Гц), 2,48-2,41 (1 Н, m), 2,30-2,22 (1 Н, m), 2,17-2,08 (1 Н, m), 2,00-1,90 (1 Н, m), 1,26CDI под атмосферой аргона при перемешивании. Через 1 ч добавляли 5 мл (избыток) пирролидина. Реакционную смесь перемешивали в течение следующего часа, затем ее промывали 100 мл 1 М водногоHCl, 100 мл насыщенного водного NaHCO3 и высушивали над Na2SO4. Концентрацией в вакууме получали 6,0 г промежуточного амида (одно пятно на тонкослойной хроматографии, EtOAc). Амид растворяли в 50 мл EtOH и сразу гидрогенизировали при 30 фунтов на кв.дюйм с 800 мг 10% Pd/C. После удаления растворителя получали 3,5-граммовый промежуточный амин в виде бледно-желтой восковой жидкости. К 1,3 г (5,22 ммоль) промежуточного продукта салициловой кислоты (см. пример 33 ниже для его синтеза), суспендированного в 40 мл диметилформамида, добавляли 930 мг (5,74 ммоль) CDI. Через 10 мин суспензия стала прозрачным коричневым раствором, и его нагревали при 60-80 С в течение 4 ч. Затем раствор охлаждали до комнатной температуры перед добавлением 1,5 г (10 ммоль) промежуточного амина. Реакционную смесь сразу перемешивали перед удалением растворителя при давлении масляного насоса (40). Остаток растворяли в 200 мл CH2Cl2, промывали 50 мл 1 М HCl, а 50 мл насыщенногоNaHCO3 высушивали над Na2SO4 и концентрировали в вакууме с получением 1,1 г промежуточного амида (одно пятно на тонкослойной хроматографии). К 1,1 г (2,9 ммоль) промежуточного амида в 150 мл 33% CH2Cl2/CHCl3 добавляли 5,4 г (60 ммоль) триоксана, 2,3 г (100 ммоль) камфорсульфоновой кислоты и 10 капель H2SO4 при перемешивании под атмосферой аргона. Полученную двухфазную систему нагревали с флегмой и воду удаляли при 50 г 4 молекулярного сита, находящегося в 125 мл дополнительной воронке с боковым ответвлением. После того, как смесь быстро дефлегмировали, сита заменяли и дефлегмирование продолжали следующие 3 ч. Реакционную смесь охлаждали до комнатной температуры и встряхивали с 100 мл 1 М NaOH. Водный слой экстрагировали с 3100 мл CH2Cl2. Комбинированные органические слои промывали 200 мл воды,высушивали над Na2SO4 и концентрировали в вакууме для получения 1,5 г неочищенного продукта. Флэш-хроматографией на 150 г силикагеля с 20% EtOH в EtOAc получали 750 мг продукта (63%) в виде бледно-зеленого прозрачного вещества, 98% чистого при LC-MS (М+Н)=400 amu со следующими свойствами. IR: 1666, 1638, 1454, 1428 см-1. 1 Н ЯМР (300 МГц, CDCl3)7,54 (1 Н, s), 7,53 (1 Н, s), 5,46 (1 Н, t,J=5,6 Гц), 5,21 (2 Н, s), 3,85 (1 Н, m), 3,62 (1 Н, m), 3,61 (2 Н, t, J=7,0 Гц), 3,40 (4 Н, m), 2,44 (1 Н, m), 2,33 Синтез 3-(3-морфолин-4-илпропил)-2 Н,6 аН-пирролидино[2",1-3',2']-1,3-оксазино[5',6'-4,5]бензо[е]1,3-оксазапергидроин-4,10-диона проводили, в целом, как указано выше для примера 22. В частности, к 1,15 г (4,0 ммоль) этил 7-гидрокси-9-оксо-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазин-6-карбоксилата(продукт из примера 7) в 30 мл CH2Cl2 под аргоном добавляли 3,0 мл (6,0 ммоль) AlMe3 при перемешивании. Добавляли 1-(3-аминопропил)морфолин (0,45 мл; 3,0 ммоль) и полученную смесь сразу перемешивали при комнатной температуре. Дополнительные 0,6 мл из 1-(3-аминопропил)морфолина добавляли в течение периода 3,5 ч, после чего смесь дефлегмировали. Поскольку, как показала тонкослойная хроматография, реакция не завершалась, то реакционную смесь охлаждали до комнатной температуры и добавляли 1,0 мл (2,0 ммоль), 2,0 М AlMe3 в толуоле. Смесь дефлегмировали в течение следующих 2 ч,после чего тонкослойная хроматография показывала завершение. Реакционную смесь охлаждали до 5 С и затем гасили осторожным добавлением 12 М HCl при перемешивании. рН смеси устанавливали до 7 с помощью 10 М NaOH и экстрагировали с 2250 мл тетрагидрофурана. Раствор высушивали над Na2SO4 и после выпаривания растворителя получали 1,8 г неочищенного фенолического амида только со следами первичного амина. К 1,8 г неочищенного промежуточного продукта в 75 мл 88% муравьиной кислоты добавляли 400 мл бензола и полученную двухфазную систему дефлегмировали под аргоном. После удаления 350 мл бензола дистилляцией добавляли дополнительные 400 мл бензола и реакционную смесь сразу дефлегмировали при перемешивании под аргоном. Весь бензол удаляли дистилляцией при атмосферном давлении и затем добавляли 2200 мл бензола и впоследствии удаляли в вакууме для получения 2,2 г масляного остатка. Остаток обрабатывали 50 мл триэтиламина, который затем удаляли в вакууме. Остаток из этой процедуры пропускали через короткую колонку (50 г кремнезема, 50% EtOH/EtAc) для удаления любого первичного амина. После удаления растворителя из хроматографических фракций в вакууме помещали 1,5 г остатка в 1,0 л EtOAc, и затем промывали 250 мл 1,0 М NaOH, 250 мл соляного раствора, и затем высушивали над Na2SO4. Растворитель удаляли в вакууме с получением 700 мг густого коричневого остатка, который хроматографировали на 75 г силикагеля (EtOH/EtOAc, 2:3) для получения бесцветного твердого вещества. Т.пл.=153-155 С. IR-спектроскопия (NaCl): 1665 и 1454 см-1. CI-MS 388 amu (M+1). 1 Н ЯМР (500 МГц)7,56 (1H,s), 7,54 (1H, s), 5,47 (1 Н, m), 5,22 (2 Н, dd), 3,85 (1 Н, s), 3,71 (5 Н, m), 3,66 (3 Н,- 20011034 К 2,5 г (9,0 ммоль) эфира (продукт из примера 7) в 50 мл тетрагидрофурана/МеОН 1:1 добавляли 50 мл 1,0 М водного NaOH. Полученный желтый раствор нагревали до 40 С в течение 1 ч под аргоном при перемешивании. Тонкослойная хроматография показала отсутствие исходного эфира. Реакционную смесь концентрировали до 20 мл в вакууме, затем охлаждали до 4 С в ледяной ванне и устанавливали рН до 4 с помощью концентрированной HCl. Обильный белый осадок собирали в воронке Бухнера, промывали несколькими небольшими порциями холодной воды и сразу высушивали под вакуумом. Выход составлял 1,3 г (54%) промежуточного продукта салициловой кислоты в виде мелкодисперсного бесцветного твердого вещества. Вышеупомянутую салициловую кислоту суспендировали в 20 мл сухого диметилформамида при перемешивании под атмосферой аргона. К сухой суспензии добавляли 0,92 г (5,7 ммоль) CDI и перемешиваемую смесь нагревали до 50 С в течение 1 ч, в этом промежутке времени образовывался желтый раствор. К этому раствору добавляли 1,0 г (8,0 ммоль) глицин метиловый эфир гидрохлорида, затем 5 мл(28,7 ммоль) диизопропилэтиламина при 42 С при перемешивании. Реакционную смесь быстро перемешивали при 40-45 С под аргоном. Концентрацией раствора получали остаток, который помещали под вакуумом в течение 16 ч при 0,1 мм Hg. Сухой остаток (5,8 г) растворяли в 500 мл EtOAc, промывали 100 мл 3 М HCl, 2100 мл воды, высушивали над Na2SO4 и затем фильтровали через 200 г силикагеля. Силикагель промывали 1500 мл EtOAc. Выпариванием EtOAc из очищенного промежуточного продукта фенольного амида получали 1,2 г (72%) белого твердого вещества. CI-MS 321, (М+1); 362 amu, (M+CH3CN+H). К 2,0 г фенольного амида (комбинированный выход двух порций) в 300 мл 33% CH2Cl2/CHCl3 добавляли 4,6 г (20 ммоль) камфорсульфоновой кислоты и 2,0 г (22 ммоль) триоксана. Полученный раствор дефлегмировали в течение 3 ч. К реакционной смеси добавляли 5 капель концентрированной H2SO4, после чего ее дефлегмировали в течение следущих 2 ч. Реакционную смесь охлаждали до 10 С с помощью ледяной ванны, затем наливали в 100 мл 1 М NaOH при перемешивании. Водный слой отделяли и экстрагировали с 350 мл CH2Cl2. Затем комбинированные органические слои промывали 100 мл воды, высушивали над MgSO4 и растворитель удаляли в вакууме, получая 1,1 г сырого бис-бензоксазина в виде воскового твердого вещества. Неочищенный продукт хроматографировали на 150 г силикагеля кварца сEtOAc как элюента для получения 650 мг (31%) прозрачного вещества. Т.пл.=161-162 С. IR (NaCl): 1748,карбониловый эфир; 1665, карбонилы амида и 1453 см-1 С-О. CI-MS 333 amu, (М+1). 1 Н ЯМР (CDCl3)7,60 (1 Н, s), 7,58 (1 Н, s), 5,5 (1 Н, m), 5,30 (2 Н, m), 4,35 (2 Н, m), 4,89 (1 Н, m), 3,78 (3 Н, s), 3,68 (1 Н, s), 2,50 К 450 мг (1,2 ммоль) (R,S)-метил 2-(4,10-диоксо-2 Н,6 аН-пирролидино[2",1-3',2']-1,3-оксазино[5',6'-4,5] бензо[е]-1,3-оксазапергидроин-3-ил)ацетата (пример 33) в 80 мл сухого CHCl3 добавляли 0,15 мл (1,8 ммоль) пирролидина, затем 0,9 мл (1,8 ммоль) 2,0 М AlMe3 в толуоле. Добавление проводили при комнатной температуре под атмосферой аргона при энергичном перемешивании. Реакционную смесь дефлегмировали в течение 1 ч, затем сразу перемешивали при комнатной температуре. Добавляли пирролидин (0,3 мл,3,6 ммоль), затем 1,8 мл (3,6 ммоль) 2,0 М AlMe3 в толуоле. Реакционную смесь дефлегмировали в течение 5 ч, в течение этого периода времени тонкослойная хроматография не обнаруживала исходного вещества. Реакционную смесь охлаждали до 5 С и затем перемешивали с 50 мл 1 М HCl. Водный слой отделяли и экстрагировали с 350 мл CH2Cl2. Комбинированные органические слои промывали 50 мл насыщенного водного NaHCO3, высушивали над Na2SO4 и концентрировали в вакууме. Неочищенный амид(600 мг) хроматографировали на 150 г силикагеля при элюировании с 10% EtOH в EtOAc, затем проводили кристаллизацию из 10 мл EtOAc, для получения 458 мг (99%) прозрачного порошка. Т.пл.=257259 С. IR (NaCl): 1748 карбонил, 1665 амид карбонил и 1453 см-1 C-O. CI-MS 372 (М+Н); 394 (M+Na),435 amu (M+Na+CH3CH). 1H-ЯМР (500 МГц, CDCl3)7,56 (1 Н, s), 7,55 (1 Н, s), 5,50 (1 Н, m), 5,41 (2 Н, m),4,38 (2 Н, m), 3,87 (1 Н, m), 3,68 (1 Н, m), 3,52 (4 Н, m), 2,49 (1 Н, m), 2,28 (1 Н, m), 2,18 (1 Н, m), 2,08 (2 Н, m),2,03 (1 Н, m) и 1,95 ppm (1 Н, m).HC(OEt)3 при комнатной температуре, затем добавляли 3,0 мл 1 М раствора BCl3 в гексане. После того,как смесь перемешивали в течение 30 мин, ее фильтровали через 3 см силикагеля и силикагель промывали смесью 150 мл гексана и 250 мл EtOAc. Полученное оранжевое масло, которое получали после удаления растворителя, растворяли в 40 мл тетрагидрофурана и 3,5 г NaH (87 ммоль, 60% в минеральном масле) добавляли по частям. Смесь нагревали до 170 С в стальном цилиндре в присутствии СО 2 (600 фунтов на кв.дюйм) в течение 2 ч. После охлаждения и удаления избытка СО 2 смесь распределяли между 150 млEtOAc и 200 мл воды. Водную фазу обрабатывали H2SO4 (рН 2), экстрагировали с 250 мл EtOAc и высушивали над Na2SO4. Растворитель выпаривали до получения 9,2 г темно-коричневого масла. Темно-коричневое масло (9,2 г, 38 ммоль) растворяли в 80 мл CH2Cl2 и добавляли 9,2 г (56,7 ммоль)CDI по частям. После активного перемешивания в течение 90 мин при комнатной температуре добавляли 6,8 г (90,5 ммоль) H2N(CH2)2OMe и смесь перемешивали в течение дополнительных 10 ч. Смесь распределяли между 200 мл воды и 200 мл EtOAc. Органическую фазу высушивали над Na2SO4 и после удаления растворителя получили темно-коричневое масло. После флэш-хроматографии на силикагеле(1 Н, s), 7,35 (1H, d, J=8,3 Гц), 7,10 (1 Н, d, J=1,5 Гц), 6,97 (1H, dd, J=8,3 и 1,5 Гц), 6,67 (1 Н, bs), 5,44 (1 Н, s),3,69-3,35 (8 Н, m), 3,40 (3 Н, s) и 1,28-1,20 ppm (6H, m). Защищенный альдегид (5,0 г, 16,8 ммоль) растворяли в 100 мл этанола. Добавляли 40 мл воды и 1 мл 12N HCl. Растворитель удаляли полностью через 30 мин, получив в результате альдегид в виде красноватобежевого твердого вещества (3,6 г). Неочищенный альдегид растворяли в 60 мл CH2Cl2, затем добавляли 10 г (111 ммоль) триоксана и 10 г CuSO4. При энергичном перемешивании добавляли по каплям 1,3 мл концентрированной H2SO4. Через 30 мин смесь продуктов фильтровали через силикагель, далее промывали 300 мл EtOAc. Комбинированный органический раствор промывали 30 мл 1 М NaOH и высушивали над Na2SO4. Удаление растворителя проводили флэш-хроматографией на силикагеле (EtOAc/гексан 40/6060/40), получая 1,7 г (43%) бесцветных кристаллов со следующими свойствами. Т.пл.=71-73 С. 1 Н ЯМР (300 МГц, CDCl3)10,01 (1 Н, s), 8,12 (1 Н, d, J=8,1 Гц), 7,61 (1 Н, dd, J=8,1 и 1,5 Гц), 7,46 (1 Н, d,J=1,5 Гц), 5,31 (2 Н, s), 3,76 (2 Н, t, J=4,8 Гц), 3,60 (2 Н, t, J=4,8 Гц) и 3,36 ppm (3 Н, s). К 500 мг (20,6 ммоль) магния в 10 мл тетрагидрофурана под атмосферой азота добавляли кристаллыI2, 0,2 мл 1,2-дибромэтана и 1 мл раствора 2,45 г (15 ммоль) бромоциклогексана в 10 мл тетрагидрофурана. Остаток раствора добавляли по частям в течение 5 мин после начала реакции Гриньярда. Реакционную смесь сохраняли при комнатной температуре и перемешивали в течение 45 мин, после чего в осадок выпадали несколько солей магния. Раствор надосадочной жидкости добавляли к раствору промежуточного альдегида (1,7 г, 7,22 ммоль) в 20 мл тетрагидрофурана. Получали желтый раствор, который перемешивали в течение дополнительных 5 мин. Добавляли 20 мл воды, затем медленно 200 мл EtOAc и небольшое количество HCl (достаточного для растворения солей магния). Органическую фазу высушивали над Na2SO4 и концентрировали для получения желтого масла. Флэш-хроматографией на силикагеле (ацетон/толуол 20/80) получали 1,3 г (56%) слегка желтоватого масла (который кристаллизовалось через несколько дней) со следующими свойствами, Т.пл.=78-79 С. IR: 3431, 2926, 2852, 1657, 1620, 1438, 1117, 780 см-1. 1 Н ЯМР(7,4 ммоль) РСС. После перемешивания 50 мл смесь фильтровали через 3 см силикагеля, который промывали смесью 175 мл EtOAc/75 мл гексана. Концентрацией и флэш-хроматографией на силикагеле(EtOAc/гексан 40/6050/50) получали 700 мг (70%) бесцветного масла со следующими свойствами. IR: 2930, 2854, 1677, 1449, 1427, 1314, 986 см-1. 1 Н ЯМР (300 МГц, CDCl3)8,03 (1 Н, d, J=7,8 Гц), 7,62 (1 Н,dd, J=7,8 и 1,5 Гц), 7,49 (1 Н, d, J=1,5 Гц), 5,28 (2 Н, s), 3,75 (2 Н, t, J=5,0 Гц), 3,59 (2 Н, t, J=5,0 Гц), 3,35 (3 Н,s), 3,27-3,15 (1H, m) и 1,95-1,25 ppm (10H, m). Пример 36. Получение 7-(циклопентилкарбонил)-3-этил-2 Н-бензо[е]-1,3-оксазапергидроин-4-она.- 22011034 Синтез проводили, в основном, как для примера 35 со следующими исключениями: этиламин замещали 2-метоксиэтиламином, а бромоциклопентан замещали бромоциклопентаном. Флэш-хроматографией на силикагеле (EtOAc/гексан 40/60) получали бесцветное масло, которое кристаллизовали из МТВЕ/гексана для получения белых кристаллов со следующими свойствами. Т.пл.=53-54 С. IR: 1677, 1664, 1491,1427, 1316 см-1. 1H ЯМР (300 МГц, CDCl3)8,04 (1 Н, d, J=8,3 Гц), 7,66 (1 Н, dd, J=8,3 и 1,5 Гц), 7,53 (1 Н, d,J=1,5 Гц), 5,23 (2 Н, s), 3,72-3,58 (1 Н, m), 3,62 (2 Н, q, J=7,2 Гц), 1,98-1,60 (8 Н, m) и 1,26 ppm (3H, t, J=7,2 Гц). Пример 37. Получение 3-этил-7-(пергидро-2 Н-пиран-4-илкарбонил)-2 Н-бензо[е]-1,3-оксапергидроин-4-она. Синтез проводили, в основном, как для примера 36, за исключением замещения 4-хлоротетрагидропирана на бромоциклопентан. Т.пл.=93-94 С. IR: 1682, 1663, 1430, 1311 см-1. 1 Н ЯМР (300 МГц, CDCl3)8,06 (1 Н, d, J=8,1 Гц), 7,63 (1 Н, dd, J=8,1 и 1,5 Гц), 7,50 (1 Н, d, J=1,5 Гц), 5,23 (2 Н, s), 4,10-4,00 (2 Н, m),3,62 (2 Н, q, J=7,2 Гц), 3,60-3,38 (3 Н, m), 1,94-1,73 (4 Н, m) и 1,26 ppm (3 Н, t, J=7,2 Гц). Пример 38. Получение 7-(циклогексилкарбонил)-3-этил-2 Н-бензо[е]-1,3-оксазапергидроин-4-она. Синтез проводили, в основном, как для примера 36, за исключением замещения бромоциклогексана на бромоциклопентан. Кристаллизацией из EtOAc/гексана получали белые кристаллы со следующими свойствами: Т.пл.=69-70 С. IR: 2932,1678, 1667,1427, 1316 см-1. 1 Н ЯМР (300 МГц, CDCl3)8,04 (1 Н, d,J=8,1 Гц), 7,63 (1 Н, dd J=8,1 и 1,8 Гц), 7,49 (1 Н, d, J=1,8 Гц), 5,22 (2 Н, s), 3,62 (2 Н, q, J=7,2 Гц), 3,25-3,14 Синтез выполняли, в основном, как для примера 36, за исключением замещения бромометилциклогексана на бромоциклопентан. Кристаллизацией из МТВЕ/гексана получали белые кристаллы со следующими свойствами. Т.пл.=55-58 С. IR: 2924, 2851, 1670, 1667,1428, 1317, 894 см-1. 1 Н ЯМР (300 МГц,CDCl3)8,04 (1 Н, d, J=8,1 Гц), 7,63 (1 Н, dd, J=8,1 и 1,5 Гц), 7,50 (1 Н, d, J=1,5 Гц), 5,22 (2 Н, s), 3,62 (2 Н, q,J=7,5 Гц), 2,80 (2 Н, d, J=6,6 Гц), 2,03-1,88 (1 Н, m), 1,80-0,92 (10 Н, m) и 1,26 ppm (3H, t, J=7,5 Гц). Пример 40. Получение (R,S),(R,S)-3-этил-7-[(2-гидроксициклогексил)карбонил]-2 Н-бензо[е]-1,3-оксазапергидроин-4-она. Промежуточный альдегид синтезировали, как в примере 36. К 1,2 г (5,85 ммоль) альдегида в 40 мл этанола добавляли раствор 1,04 г (15 ммоль) H2NOHHCl и 1,59 г Na2CO3 (15 ммоль) в 50 мл воды. Раствор перемешивали в течение 30 мин при комнатной температуре, а белый полученный осадок повторно растворяли, нагревая раствор до 50 С. Через дополнительные 30 мин водный этанол удаляли выпариванием при комнатной температуре, и добавляли 50 мл воды. Смесь экстрагировали с 1200 мл и 1100 млEtOAc. Органическую фазу высушивали над Na2SO4 и выпариванием растворителя получали оксим в виде слегка желтого твердого вещества. Оксим (1,2 г, 5,45 ммоль) растворяли в 30 мл диметилформамида, к которому добавляли 800 мг (6,0 ммоль)NCS и 30 мл HCl газа. Смесь перемешивали в течение 60 мин при комнатной температуре до полного использования исходного вещества. Смесь разбавляли 300 мл EtOAc и экстрагировали с 2150 мл воды. Органическую фазу высушивали над Na2SO4, при этом объем уменьшался до 100 мл. К этой смеси добавляли 5 мл диметилформамида, 25 мл циклогексана и раствор 1,5 мл Net3 в 25 мл СН 2 СН 2 (сразу по- 23011034 каплям). Растворитель выпаривали и добавляли 150 мл воды. Смесь экстрагировали с 250 мл EtOAc и органическую фазу концентрировали после высушивания над Na2SO4. Флэш-хроматографией на силикагеле (EtOAc/гексан 50/50) получали бесцветное масло (800 мг, 49%), которое кристаллизовали изEtOAc/гексана. Т.пл.=102-104 С. IR: 2934, 1664, 1621, 1432, 1314, 838 см-1. 1 Н ЯМР (300 МГц, CDCl3)7,98 (1 Н, d, J=8,1 Гц), 7,42 (1 Н, dd, J=8,1 и 1,5 Гц), 7,31 (1 Н, d, J=1,5 Гц), 5,21 (2 Н, s), 4,55-4,50 (1 Н, m),3,61 (2 Н, q, J=6,9 Гц), 3,27-3,20 (1H, m), 2,31-1,20 (8 Н, m) и 1,25 ppm (3 Н, t, J=6,9 Гц). К вышеупомянутому раствору изоксазолина (749 мг; 2,49 ммоль) в метаноле в 100 мл добавляли 1,2 г В(ОН)3 в 70 мл воды и 1 г никеля Ранея. После гидрирования в течение 4 ч смесь отфильтровывали, концентрировали и распределяли между 50 мл соляного раствора и 200 мл EtOAc. Эти две фазы отделяли и водный слой экстрагировали с дополнительными 200 мл EtOAc. Комбинированные органические слои высушивали над Na2SO4 и концентрировали. Флэш-хроматографией на силикагеле с EtOAc/гексаном(50/50) получали 522 мг (69%) бесцветного масла. Кристаллизацией из EtOAc/гексана получали 250 мг белого твердого вещества со следующими свойствами. Т.пл.=106-108 С. IR: 3460, 2933, 1661, 1431, 1319,988 см-1. 1 Н ЯМР (300 МГц, CDCl3)8,06 (1 Н, d, J=8,1 Гц), 7,61 (1 Н, dd, J=8,1 и 1,5 Гц), 7,48 (1 Н, d, J=1,5 Гц),5,23 (2 Н, s), 4,30-4,25 (1 Н, m), 3,62 (2 Н, q, J=7,2 Гц), 4,33-4,27 (1 Н, m), 2,05-1,35 (8 Н, m) и 1,26 ppm (3 Н, t,J=7,2 Гц). Пример 41. Получение 7-(циклогексилкарбонил)-3-метил-2 Н-бензо[е]-1,3-оксапергидроин-4-она. Синтез проводили, в основном, как для примера 38, за исключением замещения метиламина на этиламин. Т.пл.=99-101 С. IR: 2934, 2855, 1676, 1665, 1574, 1497, 1426, 1350, 1263, 984 см-1. 1 Н ЯМР (300 МГц,CDCl3)8,04 (1 Н, d, J=8,1 Гц), 7,63 (1 Н, dd, J=8,1 и 1,5 Гц), 7,49 (1 Н, d, J=1,5 Гц), 5,21 (2 Н, s), 3,23-3,15 Синтез проводили, в основном, как для примера 38, за исключением замещения изопропиламина на этиламин. Кристаллизацией из МТВЕ/гексана получали белые кристаллы со следующими свойствами: Т.пл.=88-90 С. IR: 2931, 2854, 1676, 1654, 1450, 1427, 1335, 1218, 991 см-1. 1 Н ЯМР (300 МГц, CDCl3)8,04 (1 Н, d, J=8,1 Гц), 7,62 (1 Н, dd, J=8,1 и 1,5 Гц), 7,49 (1 Н, d, J=1,5 Гц), 5,19 (2 Н, s), 4,96-4,82 (1 Н, m),3,25-3,14 (1 Н, m), 1,95-1,20 (10 Н, m) и 1,26 ppm (6 Н, d, J=6,9 Гц). Пример 43. Получение (R,S)-6-(циклогексилкарбонил)-3 аН-бензо[е]пирролидино[2,1-b]-1,3-оксазапергидроин-9-она. Синтез проводили, в основном, как для примера 38, со следующими исключениями: 4-аминобутиральдегид замещали на этиламин, получая масло, которое обрабатывали HBr/НОАс в метиленхлориде с получением промежуточного альдегида. Конечный продукт имел следующие свойства. Т.пл.=110111 С. IR: 2927, 2855, 1676, 1665, 1439, 1078 см-1. 1 Н ЯМР (300 МГц, CDCl3)8,00 (1 Н, d, J=7,8 Гц), 7,63 5-Формилсалициловую кислоту (2,8 г, 16,8 ммоль) растворяли в 150 мл метанола и добавляли по каплям 10 мл тионилхлорида. После перемешивания раствора в течение 18 ч при комнатной температуре добавляли по каплям другие 5 мл тионилхлорида и смесь дефлегмировали в течение 5 ч. Растворитель выпаривали и оставшуюся смесь фильтровали через силикагель, который промывали 300 мл EtOAc/гек- 24011034 сана (1:1). Концентрацией соответствующих фракций получали 3,0 г оранжевого твердого вещества, которое растворяли в 30 мл CH2Cl2. Добавляли этиламин (5,0 г) и раствор перемешивали в течение 3 дней при комнатной температуры. Растворитель выпаривали и добавляли 100 мл водной HCl до рН 1. Смесь экстрагировали с EtOAc (250 мл и 100 мл), высушивали над Na2SO4 и концентрировали для получения 1,4 г желтого амида. Неочищенный амид растворяли в 40 мл CH2Cl2, содержащем 6,0 г триоксана (67 ммоль) и 5,0 CuSO4. Полученную смесь энергично перемешивали добавлением по каплям 1,0 мл концентрированной H2SO4. Через 30 мин смесь отфильтровывали через силикагель, который промывали 250 мл EtOAc. Органическую фазу концентрировали, получая бежевое твердое вещество. Флэш-хроматографией на силикагеле(EtOAc/гексан 50/50) получали 550 мг бесцветного масла, которое застывало через 1 ч. Промежуточный альдегид имел следующие свойства: Т.пл.=47-50 С . 1 Н ЯМР (300 МГц, CDCl3)9,97 (1 Н, s), 8,47 (1 Н, d,J=1,8 Гц), 8,0 (1 Н, dd, J=8,4 и 1,8 Гц), 7,9 (1 Н, d, J=1,8 Гц), 5,28 (2 Н, s), 3,63 (2 Н, q, J=7,2 Гц) и 1,27 ppm(3 Н, t, J=7,2 Гц). Преобразование альдегида в циклогексилкетон завершали, в основном, согласно методике по примеру 38. IR: 2933, 2855, 1683, 1614, 1494, 1463, 1372, 1311, 1247, 1209, 978 см-1. 1 Н ЯМР (300 МГц,CDCl3)8,53 (1 Н, d, J=2,1 Гц), 8,09 (1 Н, dd, J=8,7 и 2,1 Гц), 7,03 (1 Н, d, J=8,7 Гц), 5,25 (2 Н, s), 3,63 (2 Н, q,J=7,2 Гц), 3,37-3,25 (1 Н, m), 1,92-1,20 (10 Н, m) и 1,27 ppm (3 Н, t, J=7,2 Гц). Пример 45. Получение (3S)-3-бензил-(R,S)-2 Н,3 Н,4 Н,7 аН-пирроилидино[2",1"-2',3']-оксазапергидроино[5',6'-5,4]бензо[f]-1,4-оксазепин-5,11-диона. Серийно выпускаемый карбонилбензилоксифенилаланин (10,0 г, 33,4 ммоль) растворяли в 150 млCH2Cl2. Добавляли CDI (8,0 г, 49,3 ммоль) и перемешивали смесь в течение 60 мин при комнатной температуре. Добавляли метанол (25 мл) и перемешивали смесь в течение следующих 60 мин при комнатной температуре. Растворитель выпаривали, добавляли 150 мл воды, а смесь экстрагировали с 250 мл EtOAc. Органическую фазу высушивали над Na2SO4 и концентрировали, получая бесцветное масло (10,1 г). Метиловый эфир карбонилбензилоксифенилаланина растворяли в 120 мл тетрагидрофурана и добавляли 1,2 г (55 ммоль) LiBH4, вызывая нагревание смеси до 60 С. Смесь дефлегмировали в течение 5 мин до полного использования исходного вещества. Реакцию гасили медленным добавлением 25 мл 2N HCl,затем 100 мл метанола. После концентрации раствора добавляли 100 мл соляного раствора и смесь экстрагировали с 2200 мл EtOAc/гексана (1:1). Органическую фазу высушивали над Na2SO4 и концентрировали, получая 9,1 г (31,9 ммоль) белого твердого вещества со следующими свойствами. Т.пл.: 85-87 С. 1 Н ЯМР (300 МГц, CDCl3)7,35-7,15 (10 Н, m), 5,07 (2H, s), 5,02-4,93 (1 Н, m), 4,00-3,89 (1 Н, m), 3,74-3,54(2 Н, m) и 2,86 ppm (2 Н, d, J=6,9 Гц). Вышеупомянутый спирт растворяли в 80 мл пиридина, затем при 400 мг DMAP и 4,6 г (40,1 ммоль)CH3SO2Cl при 0 С. После перемешивания смеси в течение 18 ч при комнатной температуре добавляли 700 мл EtOAc и раствор экстрагировали с 3200 мл 2N HCl. Органическую фазу высушивали над Na2SO4 и концентрировали, получая 9,7 г бежевого твердого вещества со следующими свойствами. Т.пл.: 8285 С. 1 Н ЯМР (300 МГц, CDCl3)7,35-7,18 (10 Н, m), 5,08 (2 Н, S), 5,00-4,94 (1 Н, m), 4,29-4,10 (3 Н, m),2,96 (3 Н, s) и 2,95-2,84 ppm (2 Н, m). К раствору 1,5 г (5,4 ммоль) фенола (синтезируемого, как в примере 30) в 100 мл толуола добавляли 2,0 г K2CO3 и 1,0 г метилсульфоната. Смесь дефлегмировали в течение 7 ч, затем добавляли 2,0 г K2CO3 и 2,5 г метилсульфоната. Смесь дефлегмировали в течение 18 ч, затем добавляли 2,0 г K2CO3 и 5,0 г метилсульфоната и затем дефлегмировали смесь в течение 24 ч. Добавляли воду (100 мл) и экстрагировали смесь с EtOAc (250 мл и 150 мл). Органическую фазу высушивали над Na2SO4 и растворитель выпаривали. После флэш-хроматографии на силикагеле (EtOAc/гексан 45/55, затем 66/34) получали 1,6 г слегка желтоватого масла со следующими свойствами. 1 Н ЯМР (300 МГц, CDCl3)7,44-7,18 (12 Н, m), 5,88-5,80(1 Н, m), 5,46 (0,5 Н, t, J=6 Гц), 5,44 (0,5H, t, J=6 Гц), 5,10 (2H, s), 4,39 (2 Н, q, J=6,9 Гц), 4,29-4,00 (2H, m),3,96-3,77 (2H, m), 3,65-3,56 (1H, m), 3,13-2,96 (2H, m), 2,50-2,37 (1 Н, m), 2,30-1,86 (3 Н, m) и 1,38 ppm (3 Н,t, J=6,9 Гц). Это масло растворяли в 100 мл метанола. Добавляли Pd/C (600 мг) и гидрогенизировали смесь в течение 2 ч. Смесь фильтровали и растворитель выпаривали. Полученное масло нагревали до 150 С в течение 5 мин. После флэш-хроматографии полученного масла на силикагеле (EtOAc/гексан 85/15) получали желтоватое масло (250 мг). Кристаллизацией от EtOAc/МТВЕ получали 175 мг белых кристаллов со следующими свойствами. Т.пл.=159-162 С. IR: 1670, 1442 см-1. 1 Н ЯМР (300 МГц, CDCl3) 87,64-7,13 (7 Н,m), 6,28-6,20 (1 Н, m), 5,46 (1 Н, t, J=6 Гц), 4,29-4,14 (2 Н, m), 3,90-3,77 (2 Н, m), 3,66-3,58 (1 Н, m), 2,96-2,76 К раствору N-гидроксифталимида (7,5 г, 44,6 ммоль) в 200 мл диметилформамида, который обрабатывали газом аргоном, добавляли 2,14 г 60% гидрида натрия (53,5 ммоль) по частям в течение 1 ч. Полученный раствор нагревали до 80 С в течение 3 ч и затем охлаждали до комнатной температуры. К этому раствору добавляли хлоропропиональальдегид диэтилацеталь (11,2 мл, 66,9 мммоль) и йодид натрия(10,0 г, 66,9 ммоль). Полученную суспензию нагревали до 80 С в течение 4 ч. Раствор охлаждали до комнатной температуры и затем распределяли между 1,5 л Н 2 О/0,5 л Et2O. Органический слой промывали соляным раствором, высушивали над Na2SO4, отфильтровывали и выпаривали в вакууме. После флэш-хроматографии остатка (EtOAC:гексан 1:4) получали 9,3 г (71%) белого твердого вещества. Промежуточный продукт растворяли в 250 мл безводного этилового спирта, к которому добавляли безводный гидразин (1,99 мл, 63,4 ммоль). Наблюдалось немедленное осаждение пушистого белого твердого вещества. Через 2 ч смесь фильтровали для удаления осажденного вещества, которое промывали этиловым спиртом. Комбинированные растворы этилового спирта выпаривали в вакууме и полученный остаток разделяли между CH2Cl2/Н 2 О. Органический слой промывали соляным раствором, высушивали надNa2SO4, фильтровали и выпаривали в вакууме. Полученное масло дистиллировали под вакуумом (2 мм[2,1-b]-1,3-оксазин-6-карбоксилата (соединение примера 7) в 75 мл 1:1 МеОН:1 М NaOH. Полученный раствор сразу перемешивали и затем удаляли МеОН в вакууме. рН раствора ярко-желтого цвета регулировали до получения 4,0 концентрированной соляной кислоты. Тяжелый белый осадок собирали в воронке Бухнера и промывали небольшими частями воды, получая 6,6 г промежуточной салициловой кислоты (98%) после высушивания в течение нескольких часов при 0,2 мм Hg. К суспензии промежуточной салициловой кислоты (400 мг, 1,59 ммоль) в 10 мл сухого диметилформамида добавляли 1,1'-карбонилдиимидазол (257 мг, 1,59 ммоль). Реакционная смесь становилась гомогенной через 5 мин. После перемешивания при комнатной температуре в течение 18 ч к раствору добавляли амин (0,45 мл, 2,7 ммоль). Далее реакционную смесь перемешивали в течение 1 ч при комнатной температуре и затем промывали EtOAc/H2O. Органический слой промывали пятью 50 мл частямиEtOAc. Комбинированные органические фазы промывали 1N HCl, насыщенным NaHCO3 и соляным раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме. После высушивания при пониженном давлении в течение 1 ч флэш-хроматографией остатка (EtOAc:гексан, 1:1) получали чистое вещество. Этот промежуточный продукт растворяли в 5 мл CH2Cl2, и обрабатывали 3 каплями H2SO4, и перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь промывали насыщенным NaHCO3, высушивали над Na2SO4, фильтровали и выпаривали в вакууме. Твердое вещество кристаллизовали из CH2Cl2/Et2O для получения 240 мг (32%) не совсем белого порошка. Т.пл.: 245250 С. IR: 1666 и 1458 см-1. 1 Н ЯМР (500 МГц)7,58 (0,5 Н, s), 7,56 (0,5 Н, s), 7,54 (0,5 Н, s), 7,52 (0,5 Н, m),5,76 (1H, m), 5,47 (1H, q, J=5,7 Гц), 4,31 (1H, m), 4,24 (1H, m), 3,85 (1H, m), 3,62 (1H, m), 2,80 (1H, m), 2,71 К суспензии промежуточной салициловой кислоты (см. пример 46) (411 мг, 1,65 ммоль) в 10 мл сухого диметилформамида добавляли 1,1'-карбонилдиимидазол (267 мг, 1,65 ммоль). Реакционная смесь сразу становилась гомогенной. После перемешивания при комнатной температуре в течение 16 ч к раствору добавляли O-3,3-диэтоксипропил гидроксиамин (344 мкл, 2,06 ммоль). Через 1 ч реакционную смесь промывали 1N HCl, насыщенным Na2HCO3 и соляным раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме. Промежуточный продукт растворяли в 15 мл сухого CH2Cl2 и обрабатывали камфорсульфоновой кислотой (100 мг, 0,430 ммоль). Через 0,5 ч реакционную смесь промывали насыщенным NaHCO3, высушивали над Na2SO4, фильтровали и концентрировали. После флэшхроматографии масляного остатка (CH2Cl2:МеОН, 50:1) получали 220 мг (38%) желтого твердого вещества. Т.пл.: 138-140 С. IR: 1667 и 1451 см-1. 1 Н ЯМР (500 МГц)10,83 (0,5 Н, s), 10,79 (0,5 Н, s), 7,78 Раствор 3-гидроксипирролидина (1,0 г, 11,5 ммоль) и имидазола (1,72 г, 25,2 ммоль) в 20 мл CH2Cl2 получали в 100 мл колбе с 3 ответвлениями, снабженной термометром, и затем охлаждали до 0 С. Раствор трет-бутилдиметилсилилхлорида (3,80 г, 25,2 ммоль) в 15 мл CH2Cl2 добавляли через шприц с насосом в течение 1 ч. Раствор перемешивали при комнатной температуре, отмечая медленное осаждение белого твердого вещества (гидрохлорид имидазола). Через 18 ч осадок отфильтровывали и промывалиCH2Cl2. Затем раствор концентрировали в вакууме и полученное оранжевое масло повторно растворяли в метаноле. После перемешивания спиртового раствора в течение 2 ч раствор вновь выпаривали в вакууме и затем распределяли между CH2Cl2/H2O. Органический слой промывали соляным раствором, высушивали над Na2SO4, фильтровали и концентрировали. Остаток высушивали под вакуумом в течение нескольких часов, получая 1,9 г (83%) 3-гидроксипирролидин трет-бутилдиметилового эфира в виде вязкого оранжевого масла. IR: 3150, 2953, 2928, 2888, 1413 и 1255 см-1. К суспензии промежуточной салициловой кислоты (см. пример 46 для его синтеза) (430 мг, 1,84 ммоль) в 10 мл сухого CH2Cl2 добавляли 1,1'-карбонилдиимидазола (373 мг, 2,30 ммоль). Реакционная смесь становилась гомогенной очень медленно, и выделение газа СО 2 также происходило медленно. Через 1 ч при комнатной температуре добавляли раствор 3-гидроксипиррилидин трет-бутилдиметилового эфира(556 мг, 2,76 ммоль) в 3 мл CH2Cl2. Через 16 ч реакционную смесь концентрировали в вакууме при комнатной температуре. Остаток распределяли между EtOAc/1N HCl и органический слой промывали насыщенным NaHCO3, соляным раствором, высушивали над Na2SO4, фильтровали и концентрировали в вакууме. Флэш-хроматографией неочищенного твердого вещества (EtOAc:гексан, 1:1-1,5) получали 648 мг(84%) силилового эфира в виде чистого масла. IR: 1671, 1630 и 1434 см-1. К раствору силилового эфира (598 мг, 1,43 ммоль) в 10 мл тетрагидрофурана добавляли тетраэтиламмоний фторид гидрат (321 мг, 2,15 ммоль). Через 1 ч реакционную смесь выпаривали в вакууме и полученный остаток разделяли между CH2Cl2/H2O. Органический слой промывали соляным раствором,высушивали над Na2SO4, фильтровали и концентрировали в вакууме. После флэш-хроматографии остатка (CHCl2:MeOH, 30:1) получали 323 мг (75%) спиртового продукта в виде белого твердого вещества. Т.пл.: 187-188 С. IR: 3400, 1668, 1620 и 1430 см-1. 1 Н ЯМР: 7,98 (1 Н, m), 7,20 (1 Н, m), 7,10 (1 Н, m), 5,50 Концентрированную смесь эфира этилсалициловой кислоты (пример 7) (632 мг, 2,28 ммоль) и N2,2-диметоксиметил-N-метилглицинамида (2,0 г, 11,3 ммоль) нагревали до 130 С. Из смеси получали гомогенный расплав, который нагревали в течение 10 мин. Остаток хроматографировали (EtOAc), а промежуточный продукт растворяли в 20 мл CH2Cl2 и обрабатывали 6 каплями концентрированной H2SO4. Через 6 ч реакционную смесь нейтрализовали насыщенным NaHCO3, экстрагировали с CH2Cl2, высушивали над Na2SO4, фильтровали и выпаривали. После флэш-хроматографии остатка (EtOAc) получали 325 мг[5',6'-2,1]бензо[4,5-е]-1,3-оксазапергидроин-6,12-диона (продукт из примера 1) в 30 мл уксусного ангидрида добавляли по каплям 3,5 мл (43 ммоль) 90% азотной кислоты более чем в течение 10 мин при комнатной температуре. Через 30 мин реакционную смесь распределяли между 100 мл CH2Cl2 и 200 мл насыщенного водного NaHCO3. Водную фазу экстрагировали с 3100 мл CH2Cl2 и объединенные органические слои высушивали с MgSO4. Концентрацией раствора в вакууме получали 408 мг коричневой пены через 4 ч под вакуумом при 0,1 мм Hg. Хроматографией на 200 г кремнезема (элюируемого с EtOAc) получали 220 мг продукта, который повторно растворяли в двух пятнах с подобными значениеми Rf при экспериментах тонкослойной хроматографии с использовании EtOAc. Анализ LC/MS (С-18) показал два пика, содержащих 95,5% вещества: 346 (М+1). IR: 1778, 1548, 1463 и 1426 см-1. Метод В. К 900 мг (3,0 ммоль) продукта из примера 1 в 20 мл 0 С H2SO4 добавляли по каплям свыше 10 мин 2 мл 90% HNO3. Через 30 мин реакционную смесь наливали в 200 мл холодной воды, а органические продукты экстрагировали с 350 мл CH2Cl2. Комбинированные органические слои промывали 100 мл воды и высушивали над MgSO4. Растворитель удаляли в вакууме для получения 1,1 г 5-нитро-3 аН,9 аН-пирролидино[2,1-b]пирролидино[2",1"-3',2'](1,3-оксазапергидроино)[5',6'-2,1]бензо[4,5-е]-1,3-оксазин-6,12-диона. В результате такой нитрации на основе серной кислоты получали продукт нитрации, состоящий,главным образом, из менее мобильного пятна тонкослойной хроматографии, т.е. двух более полярных из четырех возможных диастереомеров. Вышеописанный процесс повторяли с 800 мг (2,7 ммоль) исходного вещества, получая дополнительно 1,0 г продукта. Комбинированное неочищенное нитросоединение (2,1 г) фильтровали через 40 г силикагеля в 50 мл EtOAc, затем 300 мл EtOAc для получения 1,9 г бесцветного вещества, которое давало пену при высушивании. Кристаллизацией из минимального объема EtOAc получали 450 мг очищенного более полярного изомера (86%), по оценке ЯМР. Материнский раствор давал менее полярный изомер (2:1) в виде бесцветного твердого вещества после его выдержки в течение ночи. Пример 51. Получение (R,S),(R,S)-5-амино-3 аН,9 аН-пирролидино[2,1-b]пирролидино[2",1"-3',2']EtOAc добавляли 200 мг 10% Pd/C при окуривании аргоном. Полученную суспензию сразу обрабатывали водородом при 50-60 фунтов на кв.дюйм в гидрогенаторе Парра. Катализатор удаляли вакуумной фильтрацией при помощи целита, а фильтрат концентрировали до твердого вещества в вакууме. После хроматографии силикагелем на остатке с 25% EtOAc в CH2Cl2 получали 253 мг (70% выход) бесцветного твердого вещества со следующими свойствами: IR: 3447, 3333, 1670, 1654, 1595, 1431, 1388, 1341, 1079 и 775 см-1. 1 Н ЯМР (300 МГц, CDCl3)6,73 (1 Н, s), 6,72 (1 Н, s), 6,04 (2 Н, bs s), 5,46 (1H, t, J=6,0 Гц), 5,38 (1H, t,J=6,0 Гц), 3,81 (2 Н, m), 3,58 (2 Н, m), 2,43 (2 Н, m), 2,29 (2 Н, m), 2,15 (2 Н, m) и 1,94 ppm (2H, m). Пример 52. Получение (R,S),(R,S)-5-амино-3 аН,9 аН-пирролидино[2,1-b]пирролидино[2",1-3',2'] К 450 мг (1,30 ммоль) продукта из примера 50, метод В, в 80 мл CH2Cl2 добавляли 180 мг 10% Pd/C под аргоном. Полученную суспензию сразу гидрогенизировали при 50-60 фунтах на кв.дюйм в гидрогенаторе Парра. Катализатор удаляли фильтрованием через прокладку целита и раствор концентрировали для получения 470 мг неочищенного продукта. После хроматографии на силикагеле, используя 25%EtOAc в CH2Cl2, получали 380 мг (93%) в виде бесцветного твердого вещества со следующими свойствами.- 28011034 К 450 мг (1,5 ммоль) продукта из примера 50 в 60 мл EtOAc добавляли 200 мг Pd/C под аргоном и полученную суспензию гидрогенизировали в гидрогенаторе Парра в течение 18 ч при давлении 50-60 фунтов на кв.дюйм. Реакционную смесь фильтровали и растворитель удаляли в вакууме с получением 212 мг(0,67 ммоль, 45%) анилинового промежуточного продукта в виде прозрачного желтого твердого вещества. Тонкослойная хроматография (EtOAc) указывает на полное превращение. ЯМР указывает (1:1:1:1) на наличие смеси диастереомеров. Анализ LC-MS указывает на 98,2% чистоту при полном токе ионов с(М+Н)=315,9 как доминирующим пиком и совместимым со структурой. Анилин растворяли в 25 мл CH2Cl2 (безводный), и 450 мг (2,8 ммоль) Br2 добавляли при 0 С под атмосферой аргона. Первая капля Br2 немедленно обесцвечивалась. Через 15 мин при 0-5 С реакционную смесь растворяли в 50 мл CH2Cl2. Органическую фазу промывали 20 мл 10% водного бисульфита натрия и 30 мл насыщенного водного NaHCO3, высушенного над Na2SO4, и концентрировали в вакууме для получения 240 мг (91%) бледно-зеленого твердого вещества, которое имело следующие свойства. IR: 3473,3333, 2977, 2883, 1652, 1591, 1527, 1451 и 1386 см-1. 1 Н ЯМР (300 МГц, CDCl3)6,1 (2 Н, br s), 3,94 (1H,m), 3,83 (1 Н,m), 3,53 (2 Н, m), 2,42 (2 Н, m), 2,28 (2 Н, m), 2,11 (2 Н, m) и 1,99 ppm (2 Н, m). Пример 54. Получение (R,S),(R,S)-N-(6,12-диоксо-3aH,9aH-пирролидино[2,1-b]пирролидино[2",1"3',2'](1,3-оксазино)[5',6'-4,5]бензо[е]-1,3-оксазапергидроин-5-ил)ацетамида. К 850 мг анилина (промежуточный продукт примера 53) в 20 мл Ac2O добавляли 1 каплю H2SO4. Через 1 ч реакционную смесь перемешивали с 50 мл ледяной воды. Полученные твердые вещества выделяли фильтрацией, промывали несколькими небольшими частями воды и высушивали воздухом для получения 700 мг неочищенного ацетамида. Это неочищенное вещество кристаллизовали из 10% EtOH вEtOAc для получения 400 мг бесцветного твердого вещества (1:1 изомерная композиция LC-MS) со следующими свойствами: IR: 3279, 2983, 1651,1464, 1428, 1391, 1246,1080 и 777 см-1. 1 Н ЯМР (300 МГц,CDCl3)9,76 (0,5 Н, s), 9,67 (0,5 Н, s), 7,40 (0,5 Н, s), 7,38 (0,5H, s), 5,63 (0,5H, t, J=6,0 Гц), 5,46 (1H, m),5,40 (0,5H, t, J=6,0 Гц), 3,84 (2H, m), 3,60 (2H, m), 2,41 (2H, m), 2,22 (2H, m), 2,20 (3H, s), 2,15 (2H, m) и 1,96 ppm (2H, m). Материнский раствор из кристаллизации этилацетата концентрировали в вакууме, а остаток хроматографировали на 100 г силикагеля с 10% EtOH в EtOAc для получения 160 мг более подвижного изомера. Это вещество обладало следующими свойствами: Чистота: 98,8% с изомерным составом 50:1 при С 18 LC/MS; (М+Н)=357,9. IR: 3272, 1656, 1466, 1421, 1367, 1243, 1080 и 779 см-1. 1 Н ЯМР (300 МГц,CDCl3)9,76 (1 Н, s), 7,38 (1 Н, s), 5,63 (1 Н, t, J=5,4 Гц), 5,46 (1 Н, t, J=6,0 Гц), 3,86 (2 Н, m), 3,60 (2 Н, m),2,46 (2 Н, m), 2,27 (2 Н, m), 2,20 (3 Н, s), 2,05 (2 Н, m) и 1,91 ppm (2H, m). Пример 55. Получение (R,S),(R,S)-N-(11-хлоро-6,12-диоксо-3 аН,9 аН-пирролидино[2,1-b]пирролидино[2",1"-3',2'](1,3-оксазино)[5',6'-4,5]бензо[е]-1,3-оксазапергидроин-5-ил)ацетамида. В раствор 250 мг (0,70 ммоль) продукта из примера 54 в 50 мл CH2Cl2 барботировали Cl2 из колбы в течение 2 с. Реакционную смесь промывали 50 мл 20% водного NaHSO3 и 50 мл насыщенного водногоNaHCO3, высушивали над MgSO4 и концентрировали в вакууме. После перекристаллизации из EtOAc выход составлял 250 мг (64%) бесцветного твердого вещества со следующими свойствами: LC-MS (С-18) указывают на чистоту на 96,3% (M(35Cl)+H)=391,8, (M(37Cl)+H)=393,8. IR: 3272, 1673, 1458, 1422, 1246,1064 и 732 см-1. Пример 56. Получение (R,S)-3-(3-оксо-3-пирролидинилпропил)-2 Н,6 аН-пирролидино[2",1-3',2']1,3-оксазино[5',6'-4,5]бензо[e]-1,3-оксазапергидроин-4,10-диона. К 2,5 г (10 ммоль) промежуточного продукта салициловой кислоты (см. пример 46 для гидролиза- 29011034 эфира салициловой кислоты этила из примера 7) в 40 мл диметилформамида добавляли 1,8 г (11 ммоль)CDI под атмосферой аргона при перемешивании. Полученную суспензию нагревали до 85-100 С, при которой она становилась гомогенным коричневым раствором. Через 5 ч при той же температуре добавляли 3,0 г -аланин HCl, далее 5 мл диизопропилэтиламина и полученный раствор сразу перемешивали при комнатной температуре. Реакционную смесь концентрировали при 1 мм Hg/40 С, а остаток разделяли между 200 мл CH2Cl2 и 100 мл 1 М водного HCl. Водный слой промывали 350 мл CH2Cl2. Комбинированные органические слои промывали 100 мл воды, высушивали над Na2SO4 и концентрировали в вакууме для получения 3,8 г коричневого масла. Хроматографией на 75 г силикагеля с 1 л EtOAc получали 1,2 г (34%) промежуточного эфира в виде бесцветного твердого вещества со следующими свойствами:CI-MS, М+1=390 amu. К 1,2 г (3,5 ммоль) эфира, суспендированного в 90 мл EtOH, добавляли 10 мл EtOH, затем 10 мл воды и 1,0 мл (10 ммоль) 10N NaOH. Через 5 мин получали ярко-желтый раствор, а через 30 мин исходного вещества не оставалось (тонкослойная хроматография, EtOAc). Реакционную смесь концентрировали в вакууме и водный остаток экстрагировали с 100 мл EtOAc, окисляли до рН 3,0 с помощью 6N HCl и затем экстрагировали с 3100 мл EtOAc. Органические слои комбинировали, высушивали над MgSO4 и концентрировали в вакууме для получения 1,0 г промежуточного продукта карбоновой кислоты (91%). К 1,0 г (3,1 ммоль) карбоновой кислоты в 20 мл диметилформамида добавляли 0,55 г (3,4 ммоль)CDI, затем 12 мг (0,1 ммоль) DMAP, под атмосферой аргона при перемешивании. Через 4 ч добавляли 2 мл пирролидина и реакцию сразу продолжали. Диметилформамид удаляли в вакууме, а остаток разделяли между 100 мл CH2Cl2 и 50 мл 1 М HCl. После высушивания над MgSO4 растворитель удаляли в вакууме для получения 1,2 г неочищенного промежуточного продукта амида. Колоночной хроматографией на 50 г силикагеля (EtOAc) получали 700 мг чистого промежуточного продукта амида (54%). К 0,60 г (1,9 ммоль) амида в 200 мл 40% CH2Cl2/CHCl3 (безводный) добавляли 11 г триоксана (122 ммоль),затем 4,6 г (20 ммоль) камфорсульфоновой кислоты и 18 капель H2SO4. Реакционную смесь перемешивали с флегмой под атмосферой аргона. Воду, образующуюся в ходе реакции, удаляли при 100 г 3 молекулярного сита, находящегося в боковом ответвлении дополнительной воронки. Через 4 ч реакция завершалась (тонкослойная хроматография, 10% EtOH в EtOAc). Реакционную смесь охлаждали до комнатной температуры и промывали 100 мл ледяного холодного 1 М NaOH. Водный слой промывали 2100 млCHCl3. Комбинированные органические слои промывали 100 мл воды, высушивали над Na2SO4 и концентрировали в вакууме. После быстрого высушивания образца под вакуумом (0,1 мм Hg) получали 600 мг неочищенного бис-бензоксазина. Хроматографией на 75 г силикагеля с EtOAc получали 360 мг бесцветного твердого вещества. Твердое вещество повторно кристаллизовали, растворяя его в 1 мл CHCl3 и медленно добавляя 30 мл эфира. Выход составлял 260 мг (36%) бесцветных кристаллов со следующими свойствами. Т.пл.=170171 С. IR (тонкая пленка): 1665, 1638 и 1453 см-1. 1 Н ЯМР (500 МГц, CDCl3)7,53 (s, 1H), 7,52 (1 Н, s),5,34 (1 Н, t, J=6,0 Гц), 5,33 (2 Н, m), 3,84 (1 Н, m), 3,85 (2 Н, m), 3,63 (1H, m), 3,45 (2 Н, t, J=6,7 Гц), 3,38 (2 Н,t, J=6,6 Гц), 2,71 (2 Н, m), 2,42 (1 Н, m), 2,23 (1 Н, m), 2,14 (1 Н, m), 1,97 (2 Н, m), 1,95 (1 Н, m) и 1,85 ppm Продукт (2,7 г, 10 ммоль) из примера 7 нагревали под атмосферой аргона при перемешивании до 180-200 С в течение 3 мин с 8,0 г (48,4 ммоль) 2,4-диметоксибензиламина. После охлаждения реакционной смеси до 100 С ее растворяли в 100 мл CHCl3 и промывали 100 мл 3 М HCl и 100 мл воды. Органический раствор высушивали над MgSO4 и концентрировали в вакууме для получения 10 г неочищенного промежуточного продукта (одно пятно на тонокслойной хроматографии с EtOAc). Неочищенный промежуточный продукт растворяли в 50 мл TFA под атмосферой аргона. Через 2 ч тонкослойная хроматография не показывала наличие исходного вещества. Раствор TFA наливали медленно в 300 мл насыщенного водного NaHCO, а осадок собирали фильтрацией. После быстрого высушивания на вакуумной линии выход промежуточного продукта салициламида составлял 3,0 г. К 3,0 г салициламида в 20 мл 37% водного формальдегида добавляли 20 мл муравьиной кислоты и полученную суспензию помещали с флегмой под атмосферой аргона. Полученный раствор дефлегмировали в течение 3 ч, после чего большую часть растворителя дистиллировали. После того, как полученный остаток охлаждали до комнатной температуры, его суспендировали в 200 мл холодного 1 М NaOH. Продукт экстрагировали из водного раствора с 5150 мл CHCl3. Комбинированные органические фракции высушивали над NaSO4 и концентрировали в вакууме для получения 1,7 г неочищенного продукта (глав- 30
МПК / Метки
МПК: A61K 31/536, C07D 515/04, A61K 31/5365, C07D 513/04, C07D 491/04, A61P 25/18, C07D 265/24, C07D 498/04
Метки: ответов, карбонилбензоксазина, глутаматергических, усиления, соединения, синаптических
Код ссылки
<a href="https://eas.patents.su/30-11034-soedineniya-karbonilbenzoksazina-dlya-usileniya-glutamatergicheskih-sinapticheskih-otvetov.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения карбонилбензоксазина для усиления глутаматергических синаптических ответов</a>
Комбинированные адъювантные композиции для усиления иммунного ответа у позвоночного животного

Номер патента: 6233
Опубликовано: 27.10.2005
Автор: Хэйджен Майкл
МПК: A61K 39/39
Метки: комбинированные, иммунного, животного, композиции, позвоночного, ответа, усиления, адъювантные
Формула / Реферат:
1. Антигенная композиция, содержащая антиген, выбранный из группы, включающей патогенные вирусы, бактерии, грибы, паразитические организмы, антигены опухолевых клеток, в том числе злокачественных, и эффективное количество адъювантной комбинации из 3-O-дезацилированного монофосфорилированного липида A или монофосфорилированнного липида A и GM-CSF или IL-12, усиливающей иммунный ответ на указанный антиген у позвоночного животного. 2. Антигенная...
Фармацевтическая композиция для усиления трансдермальной доставки андрогена и способы ее применения

Номер патента: 9839
Опубликовано: 28.04.2008
Автор: Гиурик Роберт
МПК: A61K 31/56, A61P 5/26
Метки: способы, усиления, доставки, фармацевтическая, применения, андрогена, трансдермальной, композиция
Формула / Реферат:
1. Фармацевтическая композиция для усиления трансдермальной доставки андрогена, содержащая: (А) андроген; (В) Hsieh энхансер, как он определен в описании; и (С) загущающий агент. 2. Композиция по п.1, где указанный андроген представляет собой тестостерон. 3. Композиция по п.2 в форме разовой дозы, эффективной для трансдермальной доставки тестостерона в кровь пациента мужского пола, где указанная разовая доза содержит приблизительно до 1 мас.%...
Стальной корд для усиления резиновых шин и/или конвейерных лент и способ их изготовления

Номер патента: 4583
Опубликовано: 24.06.2004
Авторы: Ваннесте Стейн, Де Рор Нико, Мерссхаут Дирк, Востин Стивен
МПК: D07B 1/06
Метки: стальной, усиления, способ, корд, изготовления, лент, конвейерных, резиновых, шин
Формула / Реферат:
1. Стальной корд для усиления резиновых шин и/или конвейерных лент, при этом стальной корд содержит прядь металлического сердечника и по меньшей мере один смежный слой стальных элементов вокруг пряди металлического сердечника, отличающийся тем, что прядь металлического сердечника покрыта полимерным материалом, при этом полимерный материал имеет минимальную толщину, причем минимальная толщина больше 0,02 мм. 2. Стальной корд по п.1, в котором...
Система автоматической регулировки усиления для многопользовательских систем цифровой передачи данных по электрической сети с использованием очу-модуляции

Номер патента: 5656
Опубликовано: 28.04.2005
Авторы: Абад Молина Хосе, Бласко Кларет Хорхе Висенте, Фоурен Нильс Хакан, Матас-Бониллья Алехандро, Ирансо Молинеро Сальвадор, Хименес Маркина Франсиско Хавьер, Ривейро Инсуа Хуан Карлос
МПК: H03G 3/20, H04B 3/54, H04J 11/00...
Метки: данных, система, передачи, использованием, усиления, сети, систем, многопользовательских, регулировки, автоматической, электрической, очу-модуляции, цифровой
Формула / Реферат:
1. Система автоматической регулировки усиления для многопользовательской системы цифровой передачи данных по электрической сети с использованием ОЧУ-модуляции (ортогональное частотное уплотнение), имеющая множество пользовательских терминалов (2) и головной узел (1), взаимодействующие между собой через систему двунаправленной связи по электрической сети с независимым управлением восходящим каналом, соединяющим пользовательские терминалы (2) с...
Рекомбинантные тканезащитные цитокины и кодирующие их нуклеиновые кислоты для защиты, восстановления и усиления чувствительных клеток, тканей и органов

Номер патента: 10200
Опубликовано: 30.06.2008
Авторы: Леист Марсель, Геист Мари Ованг, Серами Карла, Нельсен Якоб, Серами Энтони, Педерсен Ларс Остергорд, Сагер Томас, Каллунки Пекка, Брайнс Майкл, Гервен Енс, Баи Катрин, Кристенсен Сорен, Педерсен Ян Торлеиф
МПК: A61K 38/00, C07K 14/505, C12N 15/87...
Метки: защиты, усиления, органов, восстановления, тканезащитные, цитокины, чувствительных, кодирующие, клеток, кислоты, рекомбинантные, нуклеиновые, тканей
Формула / Реферат:
1. Фармацевтическая композиция, содержащая неэритропоэтиновый мутантный рекомбинантный тканезащитный цитокин, содержащий аминокислотную последовательность SEQ ID NO:10, где в указанном цитокине (i) отсутствует или снижена по меньшей мере одна активность, выбранная из группы, состоящей из увеличения гематокрита, вазоактивного действия, гиперактивации тромбоцитов, прокоагулянтных активностей и увеличения продукции тромбоцитов, и указанный цитокин...
Предыдущий патент: 3-замещенные-2(арилалкил)-1-азабициклоалканы и способы их применения
Следующий патент: Производные 4-арилморфолин-3-она, их получение и их применение в терапии
Случайный патент: Искусственная трава с упругим гранулированным верхним поверхностным слоем