Замещенные 7-азаиндазолы, содержащие их композиции, способ получения и применение
Номер патента: 19302
Опубликовано: 28.02.2014
Авторы: Суай Катрин, Бьергард Кирстен, Наир Анил, Патек Марсель, Ронан Батист, Леруа Венсан, Смрчина Мартин, Бак Эрик, Додсон Марк, Табар Мишель, Акерман-Беррьер Марта, Вивьяни Фабрис






















Формула / Реферат
1. Соединение следующей общей формулы (I):
в которой:
1) А представляет собой фенил, необязательно замещенный одним или двумя заместителями, выбранными из галогена, (С1-С4)-алкила, (C1-C3)-алкилгалогена, О(C1-C4)-алкила, S-(C1-C4)-алкила, О(С1-С4)-алкилгалогена, S-(C1-C4)-алкилгалогена;
2) Ar выбирают из группы, состоящей из фенила, тиазолила, тиенила, фурила, пирролила, оксазолила, изоксазолила, изотиазолила, тиадиазолила, пиразолила, имидазолила, индолила, индазолила, бензимидазолила, бензоксазолила и бензотиазолила;
3) L выбирают из группы, состоящей из NH-SO2 и NH-CO-NH;
5) R3 выбирают из группы, состоящей из Н, NH2 и NHC(O)R"3, где R"3 является тиенилом;
6) R4 выбирают из группы, состоящей из Н, CON(R"5)(R"6), где R"5 и R"6 независимо выбирают из группы, состоящей из Н, (C1-С6)-алкила, замещенного (C1-С6)-алкила, (C1-С6)-алкилгетероциклила, замещенного (C1-С6)-алкилгетероциклила, (C1-С6)-алкилгетероарила, гетероарила, или же R"5 и R"6 связаны друг с другом с образованием 4-8-членного насыщенного цикла, содержащего 1-3 гетероатома, выбираемых среди О, S и N, необязательно замещенного;
при этом гетероциклил выбран из морфолинила, пиперазинила и пиперидинила и гетероарил выбран из имидазолила, пиразолила и пиридинила;
7) R5 представляет собой Н;
где "замещенный" относится к заместителю, отличающемуся от Н, который представляет собой галоген, (С1-С4)-алкил, (С1-С3)-алкилгалоген, О(С1-С3)-алкил, S-(C1-C4)-алкил, O(С1-С4)-алкилгалоген, S-(C1-C4)-алкилгалоген.
2. Соединение по п.1, отличающееся тем, что Ar выбирают из группы, состоящей из тиазолила, тиенила, фурила, пирролила, оксазолила, изоксазолила, изотиазолила, тиадиазолила, пиразолила, имидазолила, индолила, индазолила, бензимидазолила, бензоксазолила и бензотиазолила.
3. Соединение по п.1, отличающееся тем, что R4 означает Н или CON(R"5)(R"6), где R"5 и R"6 имеют вышеуказанное значение.
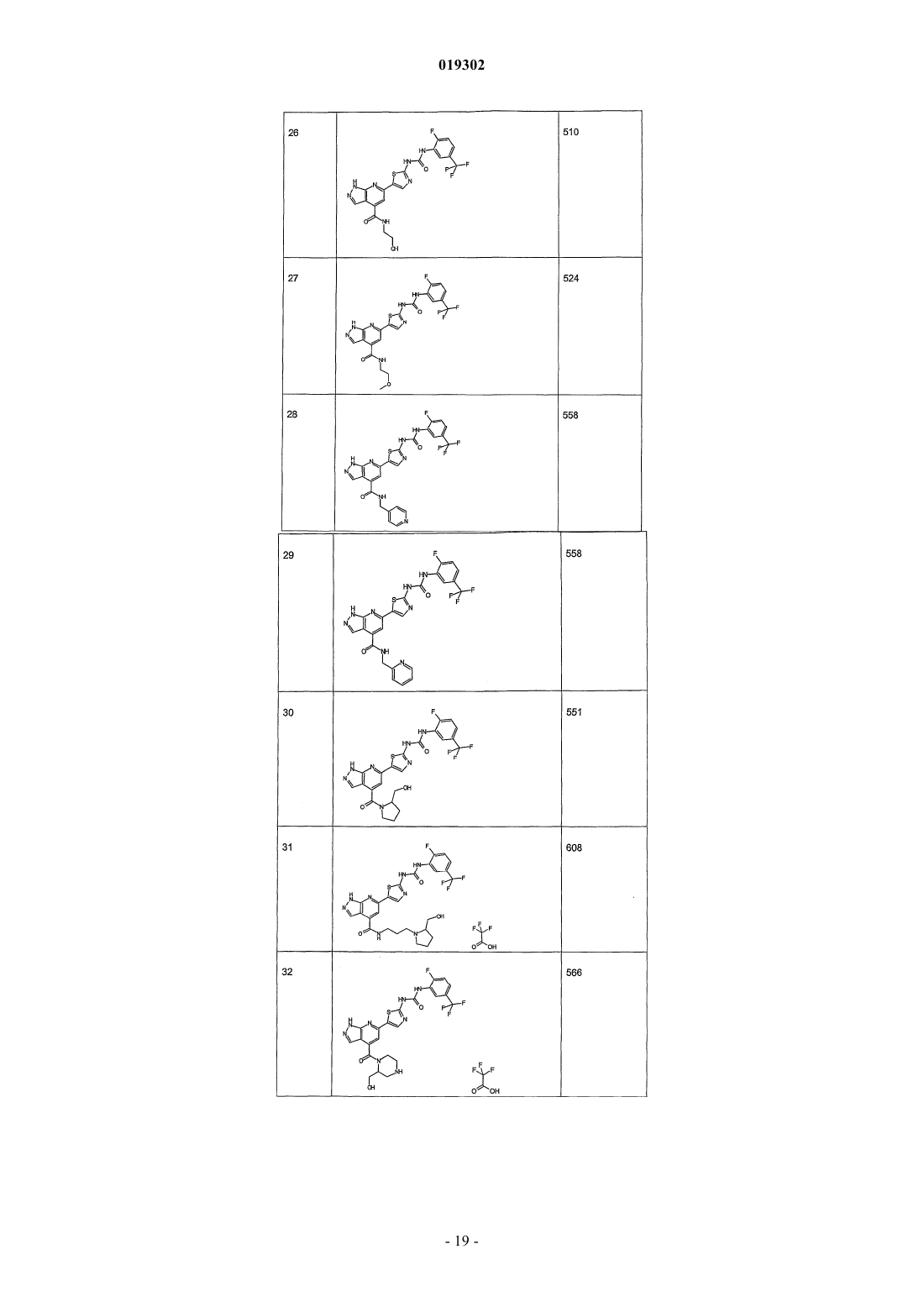
4. Соединение по любому из пп.1-3, отличающееся тем, что его выбирают среди
1-[4-(3-амино-1Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-3-(2-фтор-5-трифторметилфенил)мочевины;
(6-{4-[3-(2-фтор-5-трифторметилфенил)уреидо]фенил}-1Н-пиразоло[3,4-b]пиридин-3-ил)амида тиофен-3-карбоновой кислоты;
трифторацетата (2-морфолин-4-илэтил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
амида 6-{4-[3-(2-фтор-5-трифторметилфенил)уреидо]фенил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
метиламида 6-{4-[3-(2-фтор-5-трифторметилфенил)уреидо]фенил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{4-[3-(2-фтор-5-трифторметилфенил)уреидо]фенил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-[2-(3-фенилуреидо)тиазол-5-ил]-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
метиламида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-диметиламиноэтил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(3-диметиламино-2,2-диметилпропил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
[2-(3Н-имидазол-4-ил)этил]амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
1-(2-фтор-5-трифторметилфенил)-3-{5-[4-(морфолин-4-карбонил)-1Н-пиразоло[3,4-b]пиридин-6-ил]тиазол-2-ил}мочевины;
1-(2-фтор-5-трифторметилфенил)-3-{5-[4-(пиперазин-1-карбонил)-1Н-пиразоло[3,4-b]пиридин-6-ил]тиазол-2-ил}мочевины;
1-(2-фтор-5-трифторметилфенил)-3-{5-[4-(4-метилпиперазин-1-карбонил)-1Н-пиразоло[3,4-b]пиридин-6-ил]тиазол-2-ил}мочевины;
(2,3-дигидроксипропил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2Н-пиразол-3-ил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
метиламида 6-{6-[3-(2-фтор-5-трифторметилфенил)уреидо]пиридин-3-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{6-[3-(2-фтор-5-трифторметилфенил)уреидо]пиридин-3-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(3-морфолин-4-илпропил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-пиперазин-1-илэтил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-пиперидин-4-илэтил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
[3-(4-метилпиперазин-1-ил)пропил]амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-гидроксиэтил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-метоксиэтил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(пиридин-4-илметил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(пиридин-2-илметил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
1-(2-фтор-5-трифторметилфенил)-3-{5-[4-(2-гидроксиметилпирролидин-1-карбонил)-1Н-пиразоло[3,4-b]пиридин-6-ил]тиазол-2-ил}мочевины;
[3-(2-гидроксиметилпирролидин-1-ил)пропил]амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
1-(2-фтор-5-трифторметилфенил)-3-{5-[4-(2-гидроксиметилпиперазин-1-карбонил)-1Н-пиразоло[3,4-b]пиридин-6-ил]тиазол-2-ил}мочевины;
метиламида 6-{3-[3-(2-фтор-5-трифторметилфенил)уреидо]фенил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{3-[3-(2-фтор-5-трифторметилфенил)уреидо]фенил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{5-[3-(2-фтор-5-трифторметилфенил)уреидо]изоксазол-3-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(пиридин-3-илметил)амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
[2-(4-метилпиперазин-1-ил)этил]амида 6-{2-[3-(2-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{5-[3-(2-фтор-5-трифторметилфенил)уреидо]-[1,3,4]тиадиазол-2-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{5-[3-(2-фтор-5-трифторметилфенил)уреидо]тиофен-2-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(3-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(3-трифторметилсульфанилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло [3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(3-фтор-5-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(4-фтор-3-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(2-хлорфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(3-хлорфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(2-фтор-3-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(3-хлор-4-фторфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(2-метоксифенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(2,5-дифторфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(2,4-дифторфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(4-трифторметилфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(2-фторфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(3,4-дихлорфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(4-трифторметоксифенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(3-цианофенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(3-метоксифенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(4-хлорфенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты;
(2-морфолин-4-илэтил)амида 6-{2-[3-(2,4-диметоксифенил)уреидо]тиазол-5-ил}-1Н-пиразоло[3,4-b]пиридин-4-карбоновой кислоты.
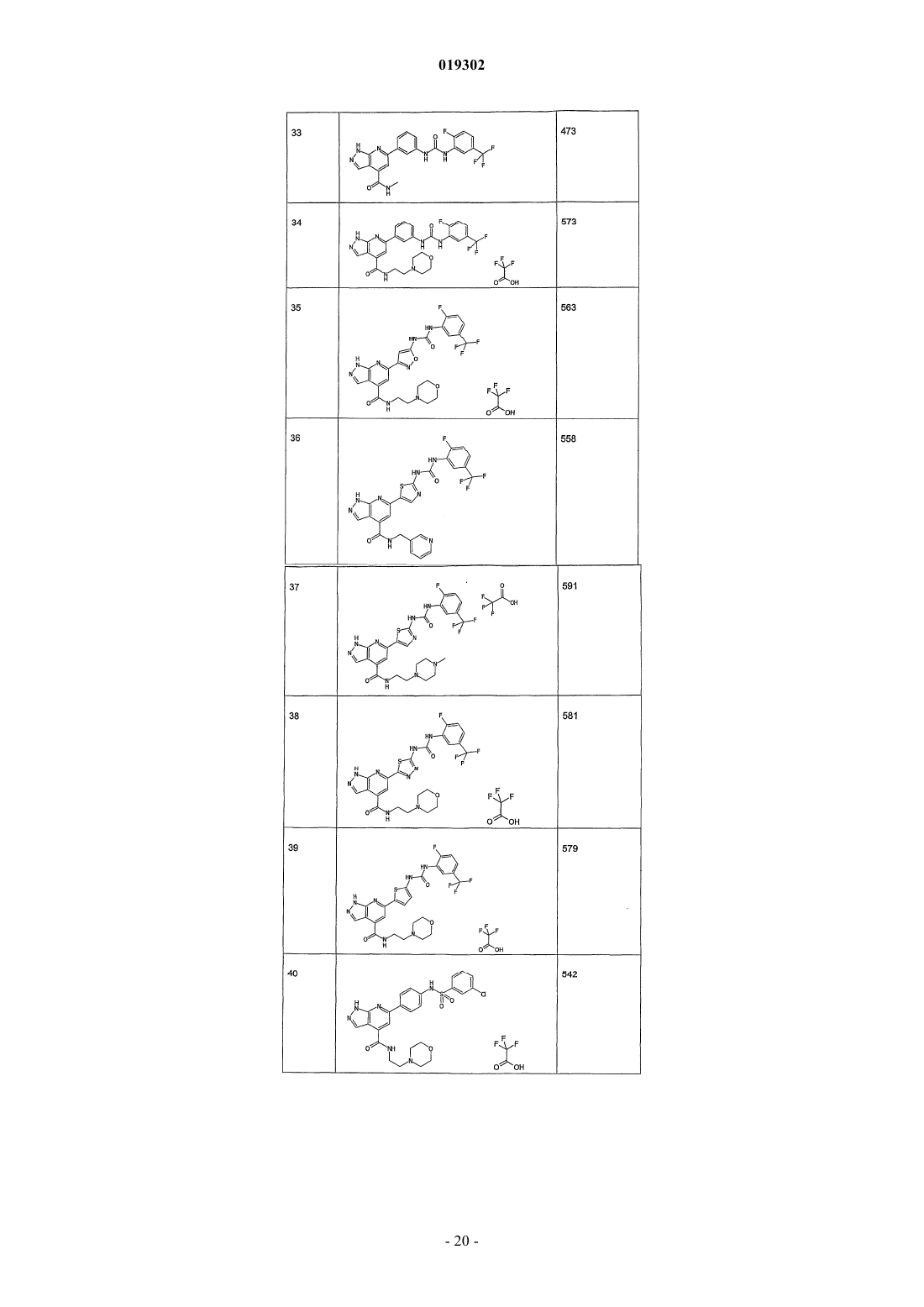
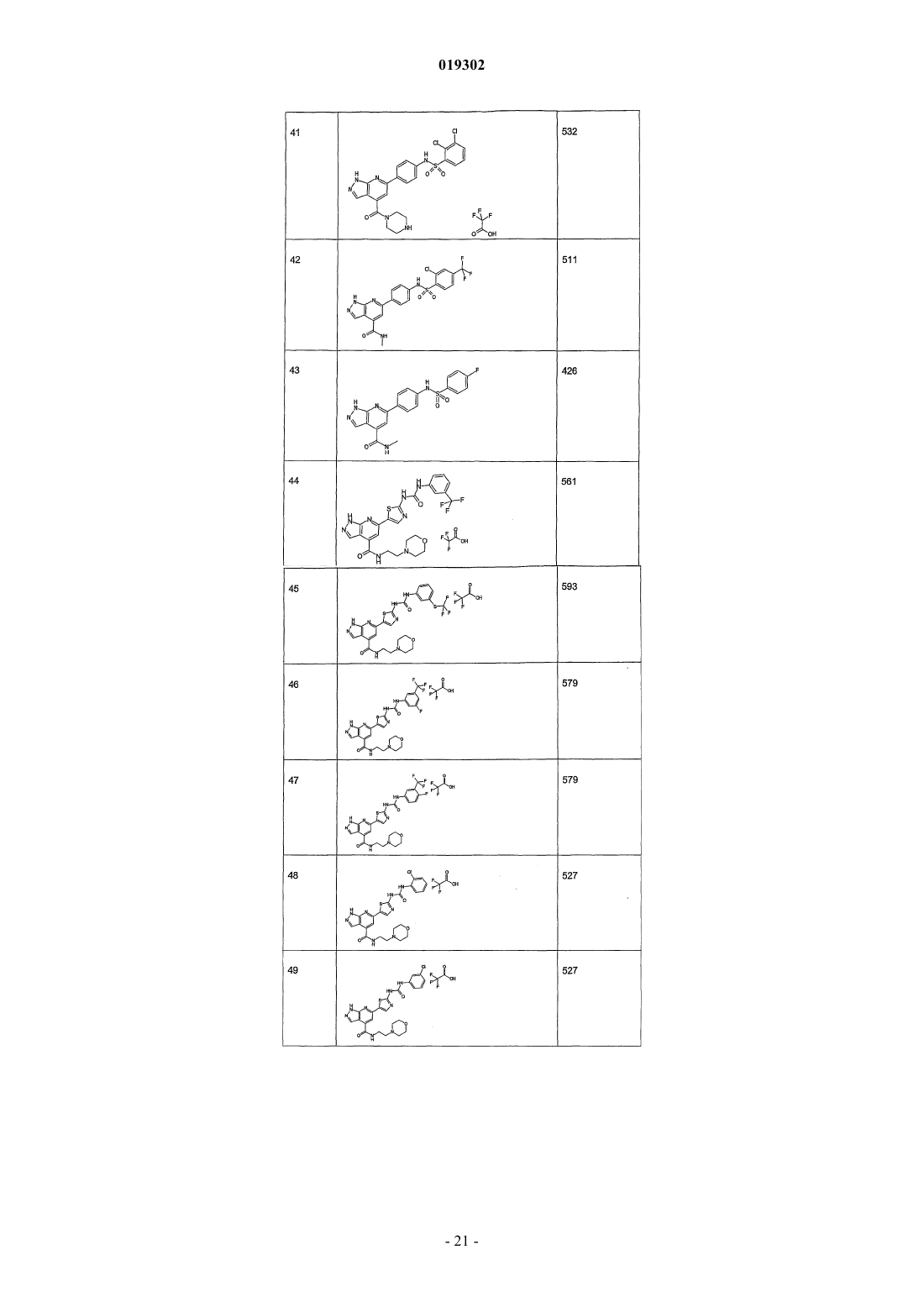
5. Соединение по любому из пп.1-4, отличающееся тем, что его выбирают среди
N-[4-(4-(2-морфолин-4-илэтил)аминокарбонил-1Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-3-хлорбензолсульфонамида;
N-[4-(4-(пиперазин-1-карбонил)-1Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-2,3-дихлорбензолсульфонамида;
N-[4-(4-метиламинокарбонил-1Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-2-хлор-4-трифторметилбензолсульфонамида;
N-[4-(4-метиламинокарбонил-1Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-4-фторбензолсульфонамида;
N-[4-(3-амино-1Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-2,3-дихлорбензолсульфонамида;
N-[4-(3-амино-4-метиламинокарбонил-1Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-2,3-дихлорбензолсульфонамида.
6. Соединение по любому из пп.1-5, отличающееся тем, что оно находится
1) в нехиральной форме, или
2) в рацемической форме, или
3) в обогащенной одним стереоизомером форме, или
4) в обогащенной одним энантиомером форме,
в виде основания или соли.
7. Лекарственное средство, отличающееся тем, что оно включает соединение формулы (I) по любому из пп.1-6 или аддитивную соль этого соединения с фармацевтически приемлемой кислотой.
8. Фармацевтическая композиция, включающая соединение по любому из пп.1-6 в комбинации с фармацевтически приемлемым эксципиентом.
9. Применение соединения по любому из пп.1-6 в качестве ингибитора реакции, катализируемой одной или несколькими киназами, выбранными из FAK, KDR и Tie2.
10. Применение соединения по любому из пп.1-6 для получения лекарственного средства, пригодного для лечения патологического состояния, где патологическое состояние выбирают среди рака, ревматоидного артрита, остеоартрита и/или ассоциированных с ним болей, воспалительных заболеваний кишечника, патологий глаза, диабетических ретинопатий, хронического воспаления, псориаза.
11. Применение по п.10 соединения по любому из пп.1-6 для лечения или профилактики патологического состояния, отличающееся тем, что соединение вводят индивидуально или в комбинации с другими противораковыми средствами.
12. Соединения следующей общей формулы (II):
в которой R'4 означает R4, -СООН или -COO-(C1-С6)-алкил; R'3 означает Н, -NH2 или -NHCO-тиенил и R'6 означает группу -фенил-NH2 или группу Ar-L-A, где Ar, L и А имеют указанное в п.1 значение.
13. Соединения следующей общей формулы (III):
в которой R'4 означает R4 или Н, -СООН или -COO-(C1-С6)-алкил и R'6 означает группу -Ar-NH2, где Ar имеет указанное в п.1 значение, или группу Ar-L-A, где Ar, L и А имеют указанное в п.1 значение.
Текст
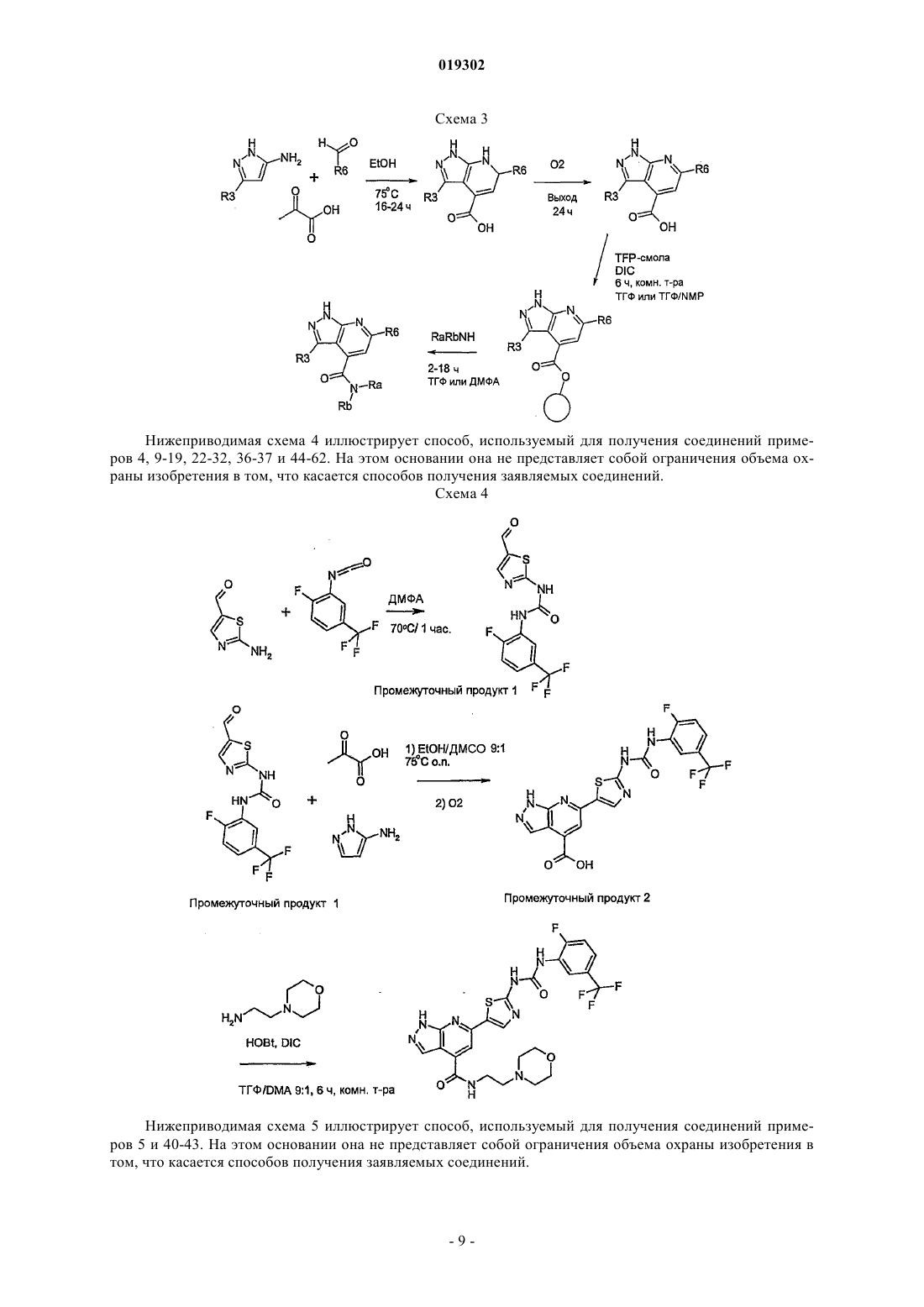
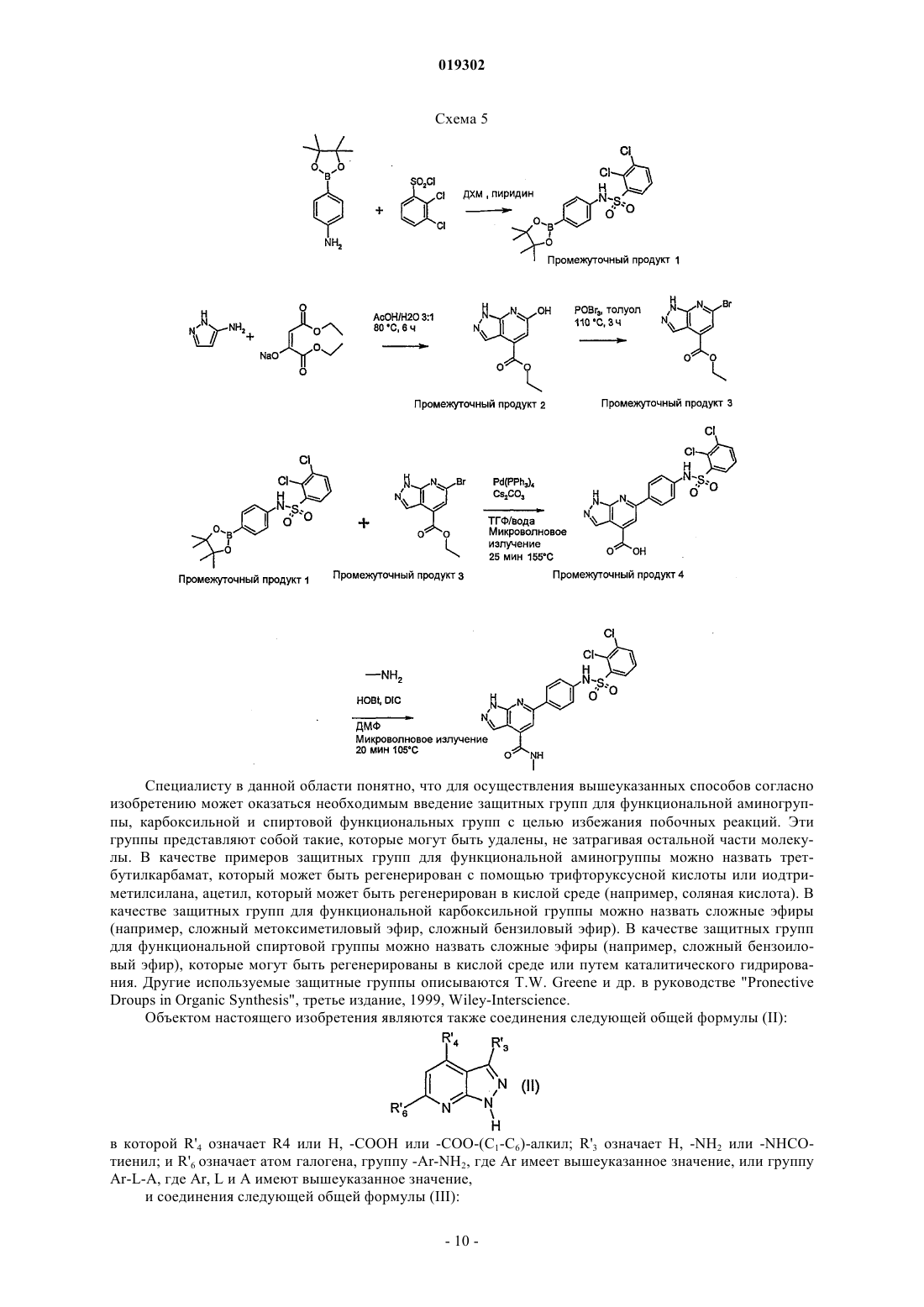
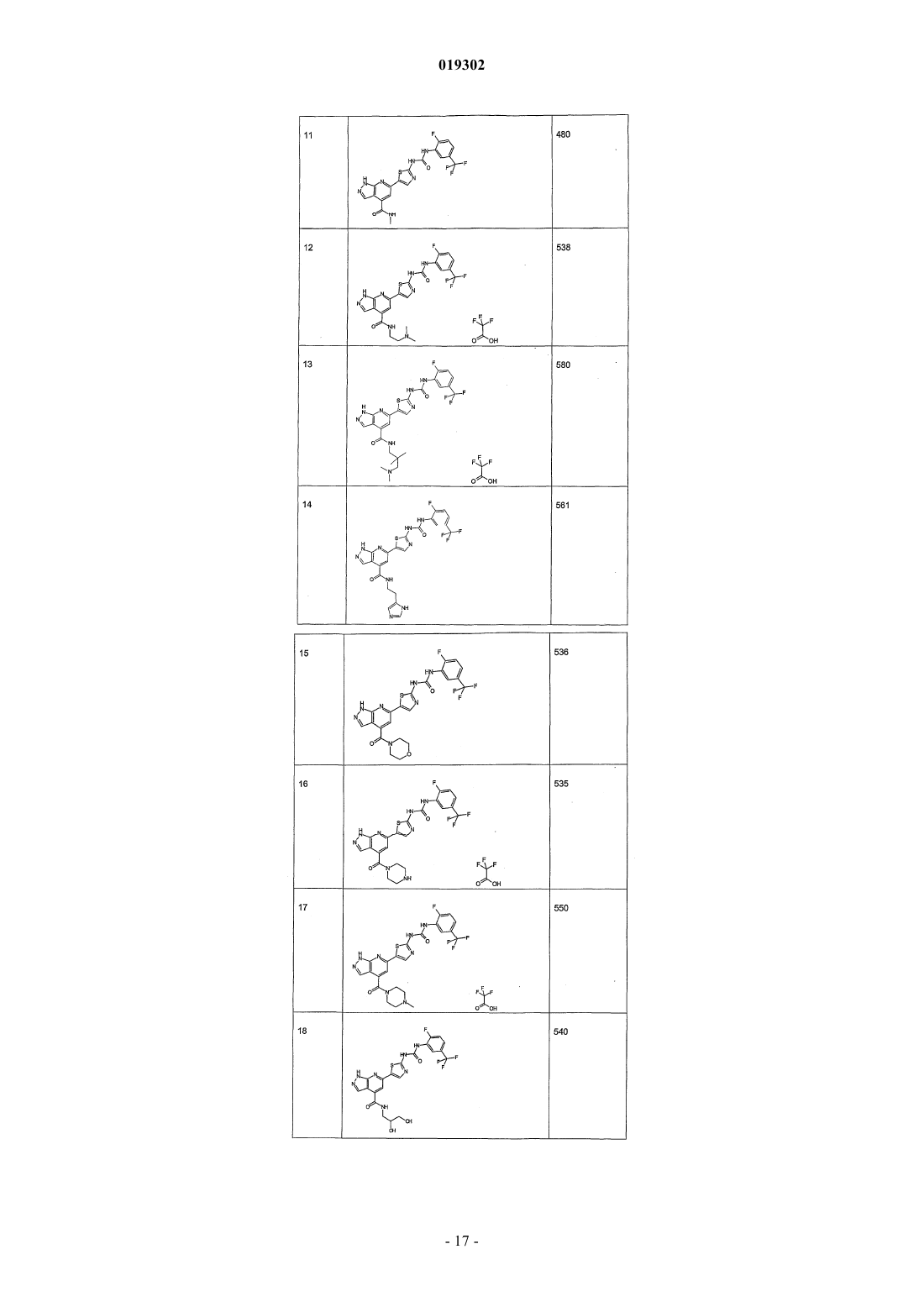
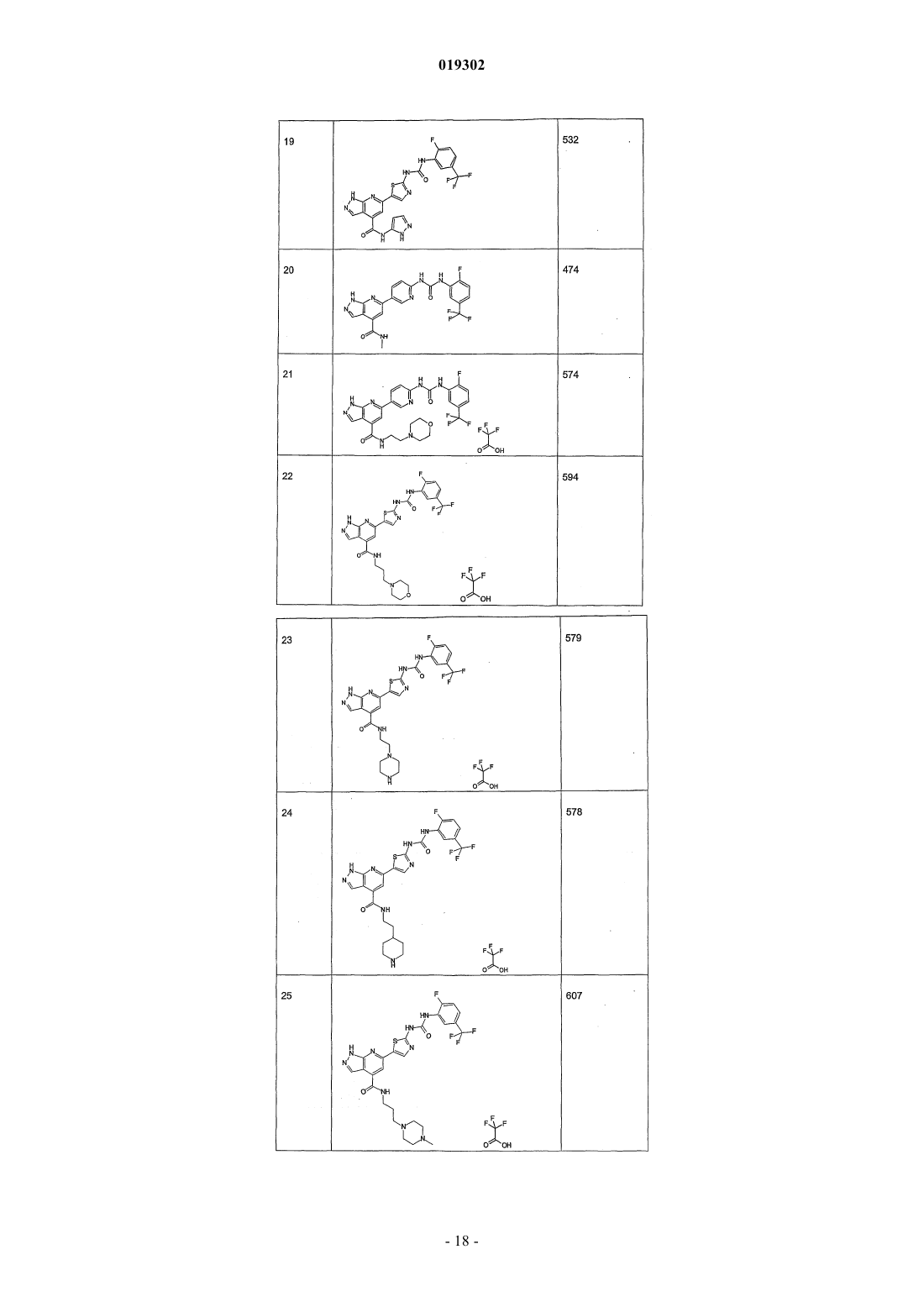
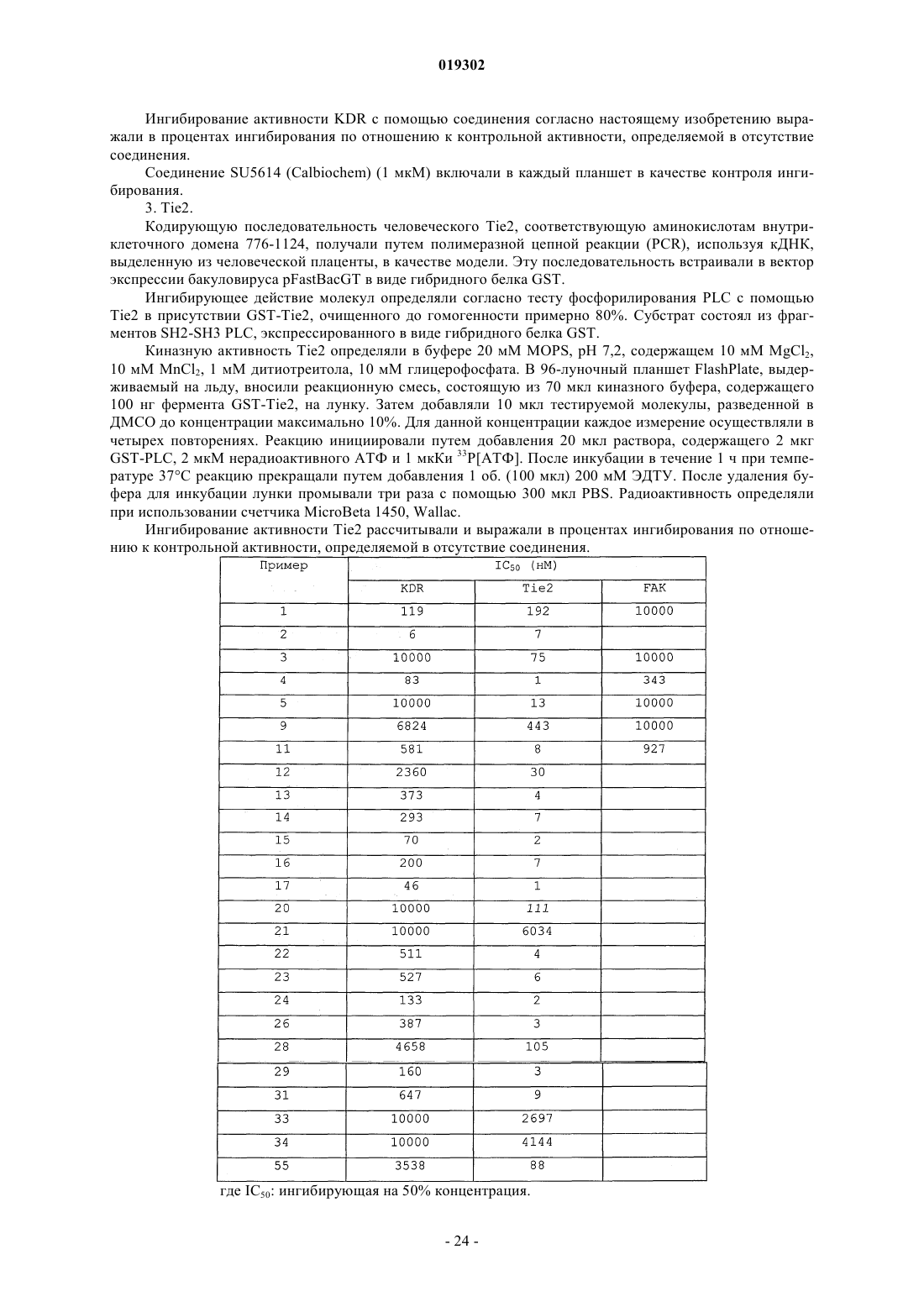
JMCMAR; ISSN: 0022-2623, 1967, XP002186552, 3cyano-2,6-dichloro-4-trif1uoromethylpyridine, page 153, Настоящее изобретение относится к новым специфическим 7-азаиндолам формулы (I), обладающим активностью, модулирующей активность белков, в частности киназ, к содержащим их композициям и их применению в качестве лекарственного средства, в особенности в качестве противораковых средств. Настоящее изобретение относится, в частности, к новым химическим соединениям, в частности к новым замещенным 7-азаиндазолам, содержащим их композициям и их применению в качестве лекарственных средств. Более конкретно, изобретение относится к новым специфическим 7-азаиндазолам, обладающим противораковой активностью за счет модуляции активности белков, в особенности киназ. На сегодняшний день большинство коммерчески доступных соединений, используемых в химиотерапии, вызывает значительные проблемы в отношении побочных эффектов и толерантности у пациентов. Эти эффекты могут быть ограничены, если используемые лекарственные средства селективно воздействуют на раковые клетки, за исключением здоровых клеток. Одно из решений для ограничения нежелательных воздействий химиотерапии, следовательно, может состоять в использовании лекарственных средств, воздействующих на метаболические пути или конститутивные элементы этих путей, в большинстве своем экспрессирующихся в раковых клетках, и которые не экспрессируются или мало экспрессируются в здоровых клетках. Протеинкиназы представляют собой семейство ферментов, которые катализируют фосфорилирование гидроксильных групп специфических остатков белков, таких как тирозиновые, сериновые или треониновые остатки. Такие фосфорилирования могут в высокой степени модифицировать функцию белков; таким образом, протеинкиназы играют важную роль в регуляции большого разнообразия клеточных процессов, включая, в частности, метаболизм, клеточную пролиферацию, клеточную дифференцировку,клеточную миграцию или сохранение клеток. Среди различных клеточных функций, в которых принимает участие активность протеинкиназы, некоторые процессы представляют собой аттрактивные мишени для лечения раковых заболеваний, а также других заболеваний. Таким образом, одним из объектов настоящего изобретения является получение композиций, обладающих противораковой активностью за счет воздействия, в частности, на киназы. Из киназ, в случае которых стремятся достичь модуляции активности, предпочтительны FAK, KDR и Tie2. Эти соединения отвечают следующей формуле (I): в которой: 1) А представляет собой фенил, необязательно замещенный одним или двумя заместителями выбранными из галогена, (C1-C4)-алкила, (C1-C3)-алкилгалогена, О(C1-C4)-алкила, S-(C1-C4)-алкила, О(C1C4)-алкилгалоген, S (C1-C4)-алкилгалоген; 2) Ar выбирают из группы, состоящей из фенила, тиазолила, тиенила, фурила, пирролила, оксазолила, изоксазолила, изотиазолила, тиадиазолила, пиразолила, имидазолила, индолила, индазолила, бензимидазолила, бензоксазолила и бензотиазолила; 3) L выбирают из группы, состоящей из NH-SO2 и NH-CO-NH; 5) R3 выбирают из группы, состоящей из Н, NH2, и NHC(O)R"3, где R"3 является тиенилом; 6) R4 выбирают из группы, состоящей из Н, CON(R"5)(R"6), где R"5 и R"6 независимо выбирают из группы, состоящей из Н, (C1-С 6)-алкила, замещенного (C1-С 6)-алкила, (C1-С 6)-алкилгетероциклила, замещенного (C1-С 6)-алкилгетероциклила, (C1-С 6)-алкилгетероарила, гетероарила, или же R"5 и R"6 связаны друг с другом с образованием 4-8-членного насыщенного цикла, содержащего 1-3 гетероатома, выбираемых среди О, S и N, необязательно замещенного; при этом гетероциклил выбран из морфолинила, пиперазинила и пиперидинила и гетероарил выбран из имидазолила, пиразолила и пиридинила,7) R5 представляет собой Н; где "замещенный" относится к заместителю, отличающемуся от Н, который представляет собой галоген, (С 1-С 4)-алкил, (С 1-С 3)-алкилгалоген, О(С 1-С 3)-алкил, S-(C1-C4)-алкил, O(С 1-С 4)-алкилгалоген, S(C1-C4)-алкилгалоген. В некоторых вариантах осуществления Ar выбирают из группы, состоящей из тиазолила, тиенила,фурила, пирролила, оксазолила, изоксазолила, изотиазолила, тиадиазолила, пиразолила, имидазолила,индолила, индазолила, бензимидазолила, бензоксазолила и бензотиазолила. В некоторых вариантах осуществления R4 означает Н или CON(R"5)(R"6), где R"5 и R"6 имеют вышеуказанное значение. Предпочтительно соединения по изобретению выбирают из 1-[4-(3-амино-1 Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-3-(2-фтор-5-трифторметилфенил)мочевины;(2-морфолин-4-илэтил)амида 6-2-[3-(2,4-диметоксифенил)уреидо]тиазол-5-ил-1 Н-пиразоло[3,4b]пиридин-4-карбоновой кислоты. Предпочтительно соединения по изобретению выбирают средиN-[4-(3-амино-4-метиламинокарбонил-1 Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-2,3-дихлорбензолсульфонамида. Соединения по изобретению могут находиться 1) в нехиральной форме, или 2) в рацемической форме, или 3) в обогащенной одним стереоизомером форме, или 4) в обогащенной одним энантиомером форме в виде основания или соли. Соединение согласно изобретению может быть использовано для получения лекарственного средства, пригодного для лечения патологического состояния, в особенности рака. Объектом настоящего изобретения является лекарственное средство, отличающееся тем, что оно включает соединение формулы (I), или аддитивную соль этого соединения с фармацевтически приемлемой кислотой, или гидрат, или сольват продукта формулы (I). Настоящее изобретение относится также к терапевтическим композициям, включающим соединение согласно изобретению в комбинации с фармацевтически приемлемым эксципиентом согласно выбранному способу введения. Фармацевтическая композиция может находиться в твердой форме, жидкой форме или в форме липосом. Из твердых композиций можно назвать порошки, желатиновые капсулы, таблетки. В число пероральных форм также можно включать твердые формы, защищенные по отношению к кислой среде желудка. Используемые для твердых форм носители представляют собой, в частности, минеральные носители, как фосфаты, карбонаты, или органические носители, как лактоза, целлюлозы, крахмал или полимеры. Жидкие формы образованы растворами, суспензиями или дисперсиями. В качестве диспергирующего носителя они содержат либо воду, либо органический растворитель (этанол, N-метилпирролидон или другие), либо смеси поверхностно-активных веществ и растворителей или комплексообразователей и растворителей. Жидкие формы предпочтительно являются инъецируемыми, и вследствие этого готовая лекарственная форма является приемлемой для такого использования. Приемлемые пути введения путем инъекции включают внутривенный, интраперитонеальный, внутримышечный и подкожный пути введения, причем обычно предпочтительным является внутривенный путь введения. Вводимая доза соединений согласно изобретению подбирается практикующим врачом в зависимости от пути введения пациенту и состояния этого последнего. Благодаря своей слабой токсичности и своим фармакологическим и биологическим свойствам соединения согласно настоящему изобретению находят свое применение при лечении любой карциномы,имеющей значительную степень васкуляризации, или индуцирующую метастазу, или, наконец, в случае патологий типа лимфом и лейкозов. Эти соединения используют для выбранной терапии либо индивидуально, либо в сочетании с адаптируемой химиотерапией или лучевой терапией и/или в сочетании с другими соединениями, обладающими антиангиогенными активностями, как ингибиторы VEGFs или FGFs. Таким образом, соединения общей формулы (I), в частности, пригодны для лечения или профилактики патологического состояния,отличающегося тем, что соединение вводят индивидуально или в сочетании с другими действующими началами, в особенности с противораковыми средствами, такими как цитотоксические, цитостатические,антиангиогенные или антиметастатические соединения. Соединения согласно настоящему изобретению, следовательно, могут быть введены индивидуально или в смеси с другими противораковыми средствами. Из возможных сочетаний можно назвать алкилирующие агенты и, в частности, циклофосфамид, мелфалан, ифосфамид, хлорамбуцил, бусульфан, тиотепа, преднимустин, кармустин, ломустин, семустин, стептозотоцин, декарбазин, темозоломид, прокарбазин и гексаметилмеламин; производные платины, такие как, в частности, цисплатин, карбоплатин или оксалиплатин; антибиотики, такие как, в частности, блеомицин, митомицин, дактиномицин; антимикроканальциевые агенты, такие как, в частности, винбластин, винкристин, виндезин, винорелбин, таксоиды (паклитаксел и доцетаксел); антрациклины, такие как, в частности, доксорубицин, даунорубицин, идарубицин, эпирубицин, митоксантрон, лозоксантрон; ингибиторы топоизомераз I и II групп, такие как этопозид, тенипозид, амсакрин, иринотекан, топотекан и томудекс; фторпиримидины, такие как 5-фторурацил, UFT, флоксуридин; аналоги цитидина, такие как 5-азацитидин, цитарабин, гемцитабин, 6-меркаптомурин, 6-тиогуанин; аналоги аденозина, такие как пентостатин, цитарабин или флударабинфосфат; метотрексат и фолиновую кислоту; различные ферменты и соединения, такие как L-аспарагиназа, гидроксимочевина, транс-ретиноевая кислота, сурамин, дексразоксан, амифостин, герцептин, а также эстрогенные гормоны, андрогенные гормоны; антиваскулярные агенты, такие как производные комбретастатина, например СА 4 Р, халконов или колхицина, например ZD6126, и их пролекарственные формы; антиангиогенные агенты, такие как бевацизумаб, сорафениб или сунитинибмалат; терапевтические агенты, ингибиторы других тирозинкиназ, такие как иматиниб, гефитиниб и эрлотиниб. Когда соединения согласно настоящему изобретению сочетают с другой терапией или с лучевой терапией, эти терапии тогда могут быть осуществлены одновременно, раздельно, последовательно. Терапия адаптируется практикующим врачом в зависимости от заболевания, подлежащего лечению. Соединения согласно изобретению пригодны в качестве ингибиторов реакции, катализируемой од-4 019302 ной или несколькими киназами. FAK, KDR и Tie2 представляют собой киназы, в случае которых соединения согласно изобретению являются особенно пригодными в качестве ингибиторов. Причины, по которым выбраны эти киназы, приводятся ниже.FAK (фокальная адгезивная киназа) представляет собой цитоплазматическую тирозинкиназу, играющую важную роль в трансдукции сигнала, транслируемого интегринами, семейства гетеродимерных рецепторов клеточной адгезии. FAK и интегрины солокализованы в перимембранных структурах, называемых адгезивными бляшками. В случае многочисленных типов клеток показано, что активация FAK, а также ее фосфорилирование по тирозиновым остаткам и, в особенности, ее автофосфорилирование по тирозину 397 зависят от связывания интегринов с их внеклеточными лигандами и, следовательно, индуцируются во время клеточной адгезии [L. Kornberg и др., J. Biol. Chem., 267 (33), 23439-442 (1992)]. Автофосфорилирование по тирозину 397 FAK выполняет сайт связывания другой тирозинкиназы, Src, через ее домен SH2 [Schaller и др., Mol. Cell. Biol., 14, 1680-1688 (1994); Xing и др., Mol. Cell. Biol., 5, 413-421(1994)]. Src тогда может фосфорилировать FAK по тирозину 925, рекрутируя таким образом адаптивный белок Grb2 и индуцируя в некоторых клетках активацию пути белка ras и МАР-киназы, участвующих в контролировании клеточной пролиферации [Schlaepfer и др., Nature, 372, 786-791 (1994); Schlaepfer и др.,Prog. Biophy. Mol. Biol., 71, 435-478 (1999); Schlaepfer и Hunter, J. Biol. Chem., 272, 13189-13195 (1997)]. Активация FAK также может индуцировать сигнальный путь jun NH2-концевой киназы (JNK) и приводить к прогрессированию клеток к фазе G1 клеточного цикла [Oktay и др., J. Cell. Biol., 145, 1461-1469(1999)]. Фосфатидилинозит-3-ОН-киназа (Р 13-киназа) также связывается с FAK по тирозину 397, и это взаимодействие может быть необходимым для активации Р 13-киназы Chen и Guan, Proc. Nat. Acad. Sci.USA, 91, 10148-10152 (1994); Ling и др., J. Cell. Biochem., 73, 533-544 (1999)]. Комплекс FAK/Src фосфорилирует различные субстраты, как паксиллин и p130CAS в фибробластах [Vuori и др., Mol. Cell. Biol.,16, 2606-2613 (1996)]. Результаты многочисленных исследований подтверждают гипотезу, что ингибиторы FAK могут быть пригодны при лечении рака. Исследования наводят на мысль, что FAK может играть важную роль в пролиферации и/или сохранении клеток in vitro. Например, в случае клеток СНО, некоторыми авторами показано, что суперэкспрессия p125FAK приводит к ускорению транзиции от G1 к S, что наводит на мысль о том, что p125FAK благоприятствует клеточной пролиферации J.-H. Zhao и др., J. Cell. Biol.,143, 1997-2008 (1998)]. Другими авторами показано, что опухолевые клетки, обработанные антисмысловыми олигонуклеотидами FAK, утрачивают свою адгезию и подвергаются апоптозу [Xu и др., CellGrowth Differ., 4, 413-418 (1996)]. Также показано, что FAK промотирует миграцию клеток in vitro. Так,дефицитные для экспрессии FAK фибробласты ("нокаутированная" по FAK мышь) обладают округлой морфологией, дефицитами клеточной миграции в ответ на хемотаксические сигналы, и эти дефициты ликвидируются повторной экспрессией FAK [D.J. Sieg и др., J. Cell Science, 112, 2677-2691 (1999)]. Суперэкспрессия С-терминального домена FAK (FRNK) блокирует "растягивание" адгезивных клеток и уменьшает клеточную миграцию in vitro [A. Richardson и J.Т. Parsons, Nature, 380, 538-540 (1996)]. Суперэкспрессия FAK в клетках СНО, COS и в клетках человеческой астроцитомы благоприятствует миграции клеток. Участие FAK в промотировании пролиферации и миграции клеток в случае многочисленных типов клеток in vitro наводит на мысль о потенциальной роли FAK в неопластических процессах. Недавно проведенное исследование действительно показало увеличение пролиферации опухолевых клеток in vivo после индукции экспрессии FAK в клетках человеческой астроцитомы [L.A. Cary и др., J. Cell.Sci., 109, 1787-1794 (1996); D. Wang и др., J. Cell. Sci., 113, 4221-4230 (2000)]. Кроме того, иммуногистохимические исследования человеческих биопсий показали, что FAK суперэкспрессируется в злокачественных опухолях предстательной железы, молочной железы, щитовидной железы, ободочной кишки,меланомы, головного мозга и легких, причем уровень экспрессии FAK прямо коррелирует с опухолями,обладающими наиболее агрессивным фенотипом [Т.М. Weiner и др., Lancet, 342 (8878), 1024-1025 (1993);KDR (рецептор с встроенным киназным доменом), также называемый VEGF-R2 (рецептор-2 фактора васкулярного эндотелиального роста) экспрессируется только в эндотелиальных клетках. Этот рецептор фиксируется на факторе ангиогенного роста VEGF и служит, таким образом, медиатором трансдукционного сигнала за счет активации его внутриклеточного киназного домена. Прямое ингибирование киназной активности VEGF-R2 позволяет уменьшать феномен ангиогенеза в присутствии экзогенногоVEGF (Vascular Endothelial Growth Factor: фактор васкулярного эндотелиального роста) (Strawn и др.,Cancer Research., 56, 3540-3545 (1996. Этот процесс наглядно объяснен, в частности, с помощью мутантов VEGF-R2 (Millauer и др., Cancer Research., 56, 1615-1620 (1996. Рецептор VEGF-R2, по-видимому,не выполняет никакой другой функции у взрослого, чем таковая, связанная с ангиогенной активностьюVEGF. Следовательно, селективный ингибитор киназной активности VEGF должен проявлять токсичность только в незначительной степени. Кроме этой центральной роли в динамическом ангиогенном процессе недавно полученные резуль-5 019302 таты наводят на мысль, что экспрессия VEGF способствует сохранению опухолевых клеток после химиотерапии и лучевой терапии, что подчеркивает потенциальный синергизм ингибиторов KDR с другими агентами (Lee и др., Cancer Research., 60, 5565-5570 (2000.Tie2 (TEK) является членом семейства тирозинкиназных рецепторов, специфичным для эндотелиальных клеток. Tie2 является первым рецептором с тирозинкиназной активностью, известным одновременно как агонист (ангиопоэтин 1 или Ang1), который стимулирует автофосфорилирование рецептора и клеточную сигнализацию [S. Davis и др., Cell., 87, 1161-1169 (1996)], и антагонист (ангиопоэтин 2 илиAng2) [P.C. Maisonpierre и др., Science, 277, 55-60 (1997)]. Ангиопоэтин 1 может давать синергический эффект с VEGF на последних стадиях неоангиогенеза [Т. Asahara, Circ. Res., 233-240 (1998)]. Эксперименты по "нокаутированию" и трансгенные манипуляции в отношении экспрессии Tie2 или Ang1 приводят к животным, которые обладают дефицитами васкуляризации [D.J. Dumont и др., Genes Dev., 8, 18971909 (1994); и С. Suri, Cell., 87, 1171-1180 (1996)]. Связывание Ang1 с его рецептором приводит к автофосфорилированию киназного домена Tie2, который является существенным для неоваскуляризации, а также для рекрутмента и взаимодействия сосудов с перицитами и гладкомышечными клетками; эти явления способствуют созреванию и стабильности вновь образующихся сосудов [Р.С. Maisonpierre и др.,Science, 277, 55-60 (1997)]. Авторами Lin и др., J. Clin. Invest., 100 (8), 2072-2078 (1997) и Lin P., PNAS,95, 8829-8834 (1998), показано ингибирование опухолевого роста и опухолевой васкуляризации, а также уменьшение метастаз легкого во время аденовирусных инфекций или инъекций внеклеточного доменаTie2 (TEK) в случае моделей ксенотрансплантатов опухоли молочной железы и меланомы. По нижеследующим причинам, ингибиторы Tie2 могут быть использованы в случае состояний, где неоваскуляризация или ангиогенез протекает несоответствующим образом, т.е. вообще в случае раковых заболеваний, однако, также в случае конкретных раковых заболеваний, таких как саркома Капоши или инфантильная гемоангиома, ревматоидный артрит, остеоартрит и/или ассоциированные с ним боли, воспалительные заболевания кишечника, такие как язвенный неспецифический колит или болезнь Крона,патологии глаза, такие как макулярная дегенерация, связанная с возрастом, диабетические ретинопатии,хроническое воспаление, псориаз. Ангиогенез представляет собой процесс генерации новых капилляров из ранее существовавших сосудов. Опухолевый ангиогенез (образование новых кровеносных сосудов), необходимый для опухолевого роста, также является одним из существенных факторов метастатической диссеминации (Oncogene,22(20), 3172-9 (2003, 19 мая); Nat. Med., 1(1), 27-31 (июнь 1995. Эта неоваскуляризация возникает вследствие миграции, затем пролиферации и дифференцировки эндотелиальных клеток под влиянием ангиогенных факторов, секретируемых карциноматозными клетками и клетками стромы (Recent Prog Horm Res., 55, 15-35; 35-6 (2000. Система ангиопоэтин 1/рецептор Tie2 играет решающую роль в созревании сосудов, позволяя осуществляться рекрутменту периэндотелиальных клеток для стабилизации сосуда (Cell., 87(7), 1161-9(1996, 27 декабря); Recent Prog Horm Res., 59, 51-71 (2004. Так, было показано, что введение растворимой рекомбинантной формы внеклеточного домена рецептора Tie2 (exTEK) ингибирует опухолевый ангиогенез в случае моделей мышиных опухолей, а также развитие метастаз (Proc. Natl. Acad. Sci. USA,25(15), 8829-34 (1998, 21 июля); Cancer Immunol. Immunother., 53(7), 600-8 (2004, июль. В эндотелиальных клетках, в виде культуры, стимуляция Tie-2 активирует путь Р 13-киназы, пути р 42/р 44, принимающие участие в клеточной пролиферации и миграции; путь синтеза PAF (Cell. Signal, 2006, 14 апреля; в печати), принимающий участие в провоспалительной активности. Стимуляция Tie2 стимулирует путьAkt и ингибирует апоптоз (Ехр. Cell. Res., 298(1), 167-77 (2004, 1 августа, путь трансдукции, известный своей значимостью в сохранении клеток. Добавление ExTEK (растворимый рецептор Tie2) ингибирует образование псевдоканальцев эндотелиальных клеток в матригеле (Cancer Immunol. Immunother., 53(7), 600-8 (2004, июль. Эти исследования указывают на то, что система Tie-2/ангиопоэтин необходима во время первых стадий образования васкулярных зачатков в зрелых тканях и что функцией рецептора Tie-2 является повышение сохранения эндотелиальных клеток в процессе образования кровеносных сосудов. Кроме того, ангиопоэтин-1 стимулирует пролиферацию лимфатических эндотелиальных клеток, а также лимфангиогенез (развитие новых лимфатических сосудов), особенно подходящий путь доступа к развитию метастаз (Blood., 105(12), 464956 (2005, 15 июня. Процесс ангиогенеза также играет решающую роль в прогрессировании многочисленных солидных опухолей. Кроме того, показано, что вероятность появления метастаз очинь сильно увеличивается с повышением васкуляризации первичной опухоли (Br. J. Cancer, 86(10), 1566-77 (2002, 20 мая. Также и сравнительно недавно была подтверждена потеницальная роль проангиогенных агентов в случае лейкозов и лимфом. В самом деле, как правило, сообщалось, что клеточные клоны в случае этих патологий могут быть либо естественным путем уничтожены иммунной системой, либо изменены в ангиогенном фенотипе, который благоприятствует их сохранению, затем их пролиферации. Это изменение фенотипа индуцируется суперзкспрессией ангиогенных факторов, в частности макрофагами, и/или мобилизацией этих факторов из внеклеточного матрикса (Thomas D.A., Glies F.J., Cortes J., Albitar M., Kantarjian H.M.,-6 019302Acta Haematol., 207, 106-190 (2001. Существует корреляция между процессом ангиогенеза костного мозга и "экстрамедуллярными заболеваниями" в случае CML (хронический моеломоноцитарный лейкоз). Различные исследования показывают, что ингибирование ангионеза может представлять собой выбранную терапию в случае этой патологии (Leuk. Res., 30(1), 54-9 (2006, январь); Histol. Histopathol., 19(4), 1245-60 (2004, октябрь. Кроме того, это в высокой степени наводит на мысль, что активация системы Tie2/ангиопоэтин участвует в развитии ангиогенеза костного мозга у пациентов, страдающих множественной миеломой (Blood, 102(2),638-45 (2003, 15 июля. Ревматоидный артрит (RA) представляет собой хроническое заболевание с неизвестной этиологией. Тогда как оно поражает многочисленные органы, наиболее тяжелой формой RA является прогрессирующее синовиальное воспаление суставов, приводящее к разрушению. Ангиогенез, по-видимому, значительным образом влияет на прогрессирование этой патологии. Так, было показано, что активация Tie2 регулирует ангиогенез в синовиальных тканях, благоприятствуя развитию ревматоидного артрита (Arthritis Rheum., 48(9), 2461-71 (2003, сентябрь. Также показана суперзкспрессия ангиопоэтина 1 и Tie2 в синовиальных тканях пациентов, страдающих остеоартритом, коррелируемое с активной неоваскуляцией (Shahrara S. и др., Arthritis Res., 4(3)exTEK (растворимый рецептор Tie2) аденовируса, достигают ингибирования ангиогенеза, развития артроза и защиты от разрушения костей в случае модели мыши, где артроз индуцирован коллагеном (Arthritis Rheum., 52(5), 1346-8 (2005, май. Воспалительные заболевания пищеварительного тракта (IBD) включают две формы хронических болезней кишечника: UC (язвенный колит) и болезнь Крона (CD). IBD характеризуются относящейся к иммунитету дисфункцией, выражающейся в несоответствующем продуцировании воспалительных цитокинов, вызывающей появление локальной микроваскулярной системы. Следствием этого ангиогенеза воспалительного происхождения является интестинальная ишемия, индуцируемая вазоконстрикцией(Inflamm. Bowel Dis., 12(6), 515-23 (2006, июнь. Патологии глаза в связи с явлениями неоваскуляризации, как макулярная дегенерация, связанная с возрастом, являются ответственными в подавляющем большинстве случаев слепоты в развитых странах. Молекулярные факторы, которые контролируют явления неоваскуляризации в глазу, такие как VEGFs или ангиопоэтины, являются выбранными мишенями в случае этих патологий (Campochiaro P.A., Expert.Opin. Biol. Ther., 4(9) (2004, сентябрь. Так, было показано, что блокируя активацию Tie2 путем использования продуцирующего exTEK (растворимый рецептор Tie2) аденовируса, ингибируют относящуюся к сетчатой оболочке и собственно сосудистой оболочке неоваскуляризацию, которая является наиболее частой причиной потери зрения (Hum. Gene Ther., 12(10), 1311-21 (2001, июль. Определения. Термин "галоген" относится к элементу, выбираемому среди F, Cl, Br и I. Термин "алкил" относится к насыщенному, линейному или разветвленному, углеводородному заместителю с 1-12 атомами углерода. Примерами алкильного заместителя являются заместители метил,этил, пропил, 1-метилэтил, бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил, пентил, 1 метилбутил, 2-метилбутил, 3-метилбутил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил,1-этилпропил, гексил, 1-метилпентил, 2-метилпентил, 1-этилбутил, 2-этилбутил, 3,3-диметилбутил, гептил, 1-этилпентил, октил, нонил, децил, ундецил и додецил. Термин "алкилен" относится к линейному или разветвленному углеводородному заместителю с одной или несколькими ненасыщенными связями и с 2-12 атомами углерода. Примерами алкиленового заместителя являются этиленил, 1-метилэтиленил, проп-1-енил, проп-2-енил, Z-1-метилпроп-1-енил, Е-1 метилпроп-1-енил,Z-1,2-диметилпроп-1-енил,Е-1,2-диметилпроп-1-енил,бут-1,3-диенил,1 метилиденилпроп-2-енил, Z-2-метилбут-1,3-диенил, Е-2-метилбут-1,3-диенил, 2-метил-1-метилиденилпроп-2-енил, ундец-1-енил и ундец-10-енил. Термин "алкинил" относится к линейному или разветвленному углеводородному заместителю по меньшей мере с двумя ненасыщенными связями, которые несет пара вицинальных атомов углерода, и с 2-12 атомами углерода. Примерами алкинильного заместителя являются этинил, проп-1-инил, проп-2 инил и бут-1-инил. Термин "арил" относится к моно- или полициклическому ароматическому заместителю с 6-14 атомами углерода. Примерами арильного заместителя являются фенил, нафт-1-ил, нафт-2-ил, антрацен-9-ил,1,2,3,4-тетрагидронафт-5-ил и 1,2,3,4-тетрагидронафт-6-ил. Термин "гетероарил" относится к моно- или полициклическому гетероароматическому заместителю с 1-13 атомами углерода и 1-4 гетероатомами. Примерами гетероарильного заместителя являются пиррол-1-ил, пиррол-2-ил, пиррол-3-ил, фурил, тиенил, имидазолил, оксазолил, тиазолил, изоксазолил, изотиазолил, 1,2,4-триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридил, пиримидил, пиразинил,1,3,5-триазинил, индолил, бензо[b]фурил, бензо[b]тиенил, индазолил, бензимидазолил, азаиндолил, хинолеил, изохинолеил, карбазолил и акридил. Термин "гетероатом" в данном контексте относится по меньшей мере к двухвалентному атому, от-7 019302 личающемуся от атома углерода. Примерами гетероатома являются N, О, S и Se. Термин "циклоалкил" относится к насыщенному или частично ненасыщенному циклическому углеводородному заместителю с 3-12 атомами углерода. Примерами циклоалкильного заместителя являются циклопропил, циклобутил, циклопентил, циклопентенил, циклопентадиенил, циклогексил, циклогексенил, циклогептил, бицикло[2,2,1]гептил, циклооктил, бицикло[2,2,2]октил, адамантил и пергидронафтил. Термин "гетероциклил" относится к насыщенному или частично ненасыщенному циклическому углеводородному заместителю с 1-13 атомами углерода и 1-4 гетероатомами. Предпочтительно насыщенный или частично ненасыщенный циклический углеводородный заместитель является моноциклическим и включает 4 или 5 атомов углерода и 1-3 гетероатома. Термин "замещенный" относится к одному или нескольким заместителям, отличающимся от Н, таким как, например, галоген, алкил, арил, гетероарил, циклоалкил, гетероциклил, алкилен, алкинил, ОН,О-алкил, алкил-ОН, О-алкилен, О-арил, О-гетероарил, NH2, NH-алкил, NH-арил, NH-гетероарил, Nалкил-алкил, SH, S-алкил, S-арил, S(O2)H, S(O2)-алкил, S(O2)-арил, SO3H, SO3-алкил, SO3-арил, СНО,С(О)-алкил, С(О)-арил, С(О)ОН, С(О)О-алкил, С(О)О-арил, ОС(О)-алкил, ОСО(O)-арил, C(O)NH2,C(O)NH-алкил, С(О)NH-арил, NHCHO, NHC(О)-алкил, NHC(O)-арил, NH-циклоалкил, NHгетероциклил. Объектом настоящего изобретения являются также способы получения соединений формулы (I). Соединения согласно изобретению могут быть получены обычными способами органической химии. Нижеприводимая схема 1 иллюстрирует способ, используемый для получения соединений примеров 1 и 3. На этом основании она не представляет собой ограничения объема охраны изобретения в том,что касается способов получения заявляемых соединений. Схема 1 Нижеприводимая схема 2 иллюстрирует альтернативный способ, используемый для получения соединения примера 1. На этом основании она не представляет собой ограничения объема охраны изобретения в том, что касается способов получения заявляемых соединений. Схема 2 Нижеприводимая схема 3 иллюстрирует способ, используемый для получения соединений примеров 6-8, 20-21, 33-35 и 38-39. На этом основании она не представляет собой ограничения объема охраны изобретения в том, что касается способов получения заявляемых соединений. Нижеприводимая схема 4 иллюстрирует способ, используемый для получения соединений примеров 4, 9-19, 22-32, 36-37 и 44-62. На этом основании она не представляет собой ограничения объема охраны изобретения в том, что касается способов получения заявляемых соединений. Схема 4 Нижеприводимая схема 5 иллюстрирует способ, используемый для получения соединений примеров 5 и 40-43. На этом основании она не представляет собой ограничения объема охраны изобретения в том, что касается способов получения заявляемых соединений. Специалисту в данной области понятно, что для осуществления вышеуказанных способов согласно изобретению может оказаться необходимым введение защитных групп для функциональной аминогруппы, карбоксильной и спиртовой функциональных групп с целью избежания побочных реакций. Эти группы представляют собой такие, которые могут быть удалены, не затрагивая остальной части молекулы. В качестве примеров защитных групп для функциональной аминогруппы можно назвать третбутилкарбамат, который может быть регенерирован с помощью трифторуксусной кислоты или иодтриметилсилана, ацетил, который может быть регенерирован в кислой среде (например, соляная кислота). В качестве защитных групп для функциональной карбоксильной группы можно назвать сложные эфиры(например, сложный метоксиметиловый эфир, сложный бензиловый эфир). В качестве защитных групп для функциональной спиртовой группы можно назвать сложные эфиры (например, сложный бензоиловый эфир), которые могут быть регенерированы в кислой среде или путем каталитического гидрирования. Другие используемые защитные группы описываются T.W. Greene и др. в руководстве "PronectiveDroups in Organic Synthesis", третье издание, 1999, Wiley-Interscience. Объектом настоящего изобретения являются также соединения следующей общей формулы (II):Ar-L-A, где Ar, L и А имеют вышеуказанное значение,и соединения следующей общей формулы (III):-Ar-NH2, где Ar имеет вышеуказанное значение, или группу Ar-L-A, где Ar, L и А имеют вышеуказанное значение. Эти соединения пригодны, в частности, в качестве промежуточных соединений синтеза в способах получения соединений общей формулы (I). Соединения формулы (I) выделяют и они могут быть очищены обычными известными методами,такими как, например, кристаллизация, хроматография или экстракция. Энантиомеры, диастереоизомеры соединений формулы (I) также составляют часть изобретения. Соединения формулы (I), включающие основный остаток, могут быть, в случае необходимости,превращены в аддитивные соли с неорганической или органической кислотой путем воздействия такой кислоты в растворителе, например органическом, таком как спирт, кетон, простой эфир или хлорированный растворитель. Соединения формулы (I), включающие кислотный остаток, в случае необходимости, могут быть превращены в соли металлов или в аддитивные соли с азотсодержащими основаниями согласно само по себе известным способам. Эти соли могут быть получены путем воздействия металлсодержащего основания (как, например, щелочной или щелочно-земельный металл), аммиака, амина или соли амина на соединение формулы (I) в растворителе. Образовавшуюся соль выделяют обычными способами. Эти соли также составляют часть настоящего изобретения. Когда продукт согласно настоящему изобретению имеет по меньшей мере одну свободную основную функциональную группу, могут быть получены фармацевтически приемлемые соли путем реакции между вышеуказанным продуктом и неорганической или органической кислотой. Фармацевтически приемлемые соли включают хлориды, нитраты, сульфаты, гидросульфаты, пиросульфаты, бисульфаты,сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты,ацетаты, пропионаты, акрилаты, 4-гидроксибутираты, каприлаты, капроаты, деканоаты, оксалаты, малонаты, сукцинаты, глутараты, адипаты, пимелаты, малеаты, фумараты, цитраты, тартраты, лактаты, фенилацетаты, манделаты, себацинаты, субераты, бензоаты, фталаты, метансульфонаты, птолуолсульфонаты, пропансульфонаты, ксилолсульфонаты, салицилаты, циннаматы, глутаматы, аспартаты, глюкуронаты, галактуронаты. Когда продукт согласно настоящему изобретению имеет по меньшей мере одну свободную кислотную функциональную группу, могут быть получены фармацевтически приемлемые соли путем реакции между вышеуказанным продуктом и неорганическим или органическим основанием. Фармацевтически приемлемые основания включают гидроксиды катионов щелочных или щелочно-земельных металлов,таких как Li, Na, K, Mg, Ca; основные аминосоединения, такие как аммиак, аргинин, гистидин, пиперидин, морфолин, пиперазин, триэтиламин. Изобретение также описывается нижеследующими примерами, приводимыми для пояснения изобретения. Анализы путем LC/MS (жидкостная хроматография/масс-спектрометрия) осуществляли на прибореMicromass, модель LCT, который связан с устройством HP 1100. Относительное содержание соединений определяли с помощью диодного детектора HP G1315A в диапазоне длин волн 200-600 нм и детектора по светорассеянию Sedex 65. Регистрацию масс-спектров (Mass spectra) осуществляли в диапазоне 180-800. Данные анализировали, используя программное обеспечение Micromass MassLynx. Разделение осуществляли на колонке Hypersil BDS C18, 3 мкм (504,6 мм), элюируя линейным градиентом 5-90% ацетонитрила, содержащего 0,05% (об./об.) трифторуксусной кислоты (ТФУК), в воде, содержащей 0,05%(об./об.) ТФУК, в течение 3,5 мин с расходом 1 мл/мин. Общее время анализа, включая период повторного уравновешивания колонки, составляет 7 мин. Масс-спектры получали при использовании ионизации электронным распылением (ES+) на прибореPlatform II (Micromass). Описаны обнаруженные основные ионы. Температуры плавления измеряли в капилляре на приборе Mettler FP62 в диапазоне от 30 до 300 С,при подъеме на 2 С/мин. Очистка путем LC/MS. Соединения могут быть очищены путем LC/MS, используя систему Waters FractionLynx, состоящую из градиентного насоса Waters, модель 600, регенерационного насоса Waters, модель 515, насоса для разбавления Waters Reagent Manager, автоинжектора Waters, модель 2700, двух вентилей Rheodyne, модельLabPro, диодного детектора Waters, модель 996, масс-спектрометра Waters, модель ZMD и сборника фракций Gilson, модель 204. Система контролировалась программным обеспечением WatersFractionLynx. Разделение осуществляли альтернативно при использовании двух колонок Waters Symme- 11019302try (C18, 5 мкм, 1950 мм, регистрационный номер по каталогу 186000210), причем одна колонка находится в процессе выполнения регенерации с помощью смеси вода/ацетонитрил в соотношении 95:5(об./об.), содержащей 0,07% (об./об.) трифторуксусной кислоты, в то время как другая колонка находится в процессе выполнения разделения. Элюирование колонок осуществляли, используя линейный градиент 5-95% ацетонитрила, содержащего 0,07% (об./об.) трифторуксусной кислоты, в воде, содержащей 0,07%(об./об.) трифторуксусной кислоты, с расходом 10 мл/мин. На выходе из разделительной колонки одну тысячную часть эфлюента отделяли с помощью LC Packing Accurate, разбавляли метиловым спиртом с расходом 0,5 мл/мин и направляли к детекторам из расчета 75% к диодному детектору, а остальные 25% к масс-спектрометру. Остальную часть эфлюента (999/1000) направляли к сборнику фракций, куда поток выводили, пока массу ожидаемого продукта не детектировали с помощью программного обеспеченияFractionLynx. Молекулярные формулы ожидаемых соединений получали при использовании программного обеспечения FractionLynx, согласно которому сбор продукта начинается тогда, когда сигнал детектируемой массы соответствует иону [М+Н]+ и/или [M+Na]+. В некоторых случаях, в зависимости от результатов аналитического метода LC/MS, когда был определен интенсивный ион, соответствующий[М+2 Н], также при использовании программного обеспечения FractionLynx определяли значение, соответствующее половине рассчитанной молекулярной массы (MW/2). В этих условиях сбор также начинали, когда были определены сигнал массы иона [М+2 Н] и/или сигнал массы иона [M+Na+H]. Соединения собирали в тарированные стеклянные пробирки. После сбора растворители выпаривали в центрифужном выпарном аппарате Savant AES 2000 или Genevac HT8 и массы продуктов определяли путем взвешивания пробирок после выпаривания растворителей. Пример 1. 1-[4-(3-Амино-1 Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-3-(2-фтор-5-трифторметилфенил)мочевина 100 мг (0,44 ммоль) 6-(4-аминофенил)-1 Н-пиразоло[3,4-b]пиридин-3-иламина частично растворяют в 2,5 мл безводного ТГФ. Смесь после этого дегазируют в течение 15 мин с помощью аргона. Затем добавляют 61,55 мкл (44,93 мг, 0,44 ммоль, 1 экв.) триэтиламина. Раствор еще раз дегазируют в течение 15 мин. Наконец, добавляют по каплям 64,22 мкл (91 мг, 0,44 ммоль, 1 экв.) 2-фтор-5(трифторметил)фенилизоцианата. Смесь перемешивают в течение 2 ч при комнатной температуре в инертной атмосфере. По окончании реакции смесь фильтруют. Фильтрат концентрируют. Выделяют твердое вещество бежевого цвета. Этот сырой продукт затем очищают при использовании колонки с диоксидом кремния (13 г), с С 18 обращенной фазой, при использовании градиента элюирования 5-95% ацетонитрила в воде. Содержащие ожидаемое соединение фракции лиофилизируют. Выделяют твердое вещество белого цвета (10 мг). В микроволновом реакторе соответствующего размера 0,97 ммоль (222 мг) 6-(4-аминофенил)-2 хлорникотиннитрила суспендируют в 3,3 мл 1-пропанола. Добавляют 0,28 мл (0,290 г, 5,80 ммоль, 6 экв.) гидразингидрата. Суспензию нагревают в течение 45 мин при температуре 130 С в микроволновой печи. Смесь фильтруют и твердое вещество высушивают, получая твердое вещество зеленого цвета (215 мг). МС (ионизция электронным ударом (IE M+ m/z=225. 1 2,89 ммоль (500 мг) 2,6-дихлорникотиннитрила и 3,18 ммоль (551 мг, 1,1 экв.) 4 аминофенилбороновой кислоты растворяют в инертной атмосфере в 33,3 мл диоксана. Затем добавляют 680 мг (8,09 ммоль, 2,8 экв.) бикарбоната натрия, потом 8,3 мл воды. Смесь перемешивают в течение 5 мин в инертной атмосфере, затем добавляют 334 мг (0,29 ммоль, 0,1 экв.) тетракис(трифенилфосфин)палладия. Реакционную смесь кипятят с обратным холодильником (100 С) в течение 2 ч, после чего охлаждают до комнатной температуры. Реакционную среду фильтруют, потом экстрагируют четыре раза этилацетатом. Органические фазы объединяют, промывают два раза водным насыщенным раствором хлорида натрия, сушат над сульфатом магния и концентрируют. После этого получают твердое вещество желтого цвета. Это сырое вещество очищают при использовании колонки с 90 г диоксида кремния с помощью градиента элюирования от 20 до 50% этилацетата в гептане. Получают ожидаемое соединение в виде твердого вещества желтого цвета (531 мг).H-HMP (ДМСО-d6)(м.д.): 5,94 (уш.с, 2 Н); 6,65 (д, J=8 Гц, 2 Н); 7,89 (д, J=8 Гц, 2 Н); 7,93 (д, J=8 Гц, 1 Н); 8,27 (д, J=8 Гц, 1 Н). Альтернативный путь получения 1-[4-(3-амино-1 Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-3-(2-фтор 5-трифторметилфенил)мочевины (схема 2) 0,46 ммоль (200 мг) 1-[4-(6-хлор-5-цианопиридин-2-ил)фенил]-3-(2-фтор-5-трифторметилфенил) мочевины суспендируют в 3,2 мл 1-пропанола. Добавляют 0,13 мл (0,14 г, 2,76 ммоль, 6 экв.) гидразингидрата. Суспензию нагревают при температуре 80 С в течение 10 ч. Смесь фильтруют и твердое вещество высушивают, получая 140 мг ожидаемого продукта. 1-[4-(6-Хлор-5-цианопиридин-2-ил)фенил]-3-(2-фтор-5-трифторметилфенил)мочевина 0,578 ммоль (100 мг) 2,6-Дихлорникотиннитрила и 0,64 ммоль (270 мг, 1,1 экв.) 1-(2-фтор-5 трифторметилфенил)-3-[4-(4,4,5,5-тетраметил-[1,3,2]диоксоборолан-2-ил)фенил]мочевины растворяют в инертной атмосфере в 6,6 мл диоксана. Затем добавляют 136 мг (1,62 ммоль, 2 экв.) бикарбоната натрия,потом 1,6 мл воды. Смесь перемешивают в течение 5 мин в инертной атмосфере, после чего добавляют 33,4 мг (0,029 ммоль, 0,05 экв.) тетракис(трифенилфосфин)палладия. Смесь кипятят с обратным холодильником (100 С) в течение 2 ч, затем охлаждают до комнатной температуры. Реакционную среду экстрагируют два раза этилацетатом. Органические фазы объединяют, промывают два раза водным насыщенным раствором хлорида натрия, затем сушат над сульфатом магния и концентрируют. Тогда получают твердое вещество желтого цвета. Сырое вещество очищают при использовании колонки с 30 г диоксида кремния с помощью градиента элюирования от 20 до 40% этилацетата в гептане. Получают твердое вещество желтого цвета (175 мг).H-ЯМР (ДМСО-d6)(м.д.): 7,40 (м, 2 Н); 7,49 (д, J=8 Гц, 2 Н); 8,13 (д, J=8 Гц, 2 Н); 8,16 (д, J=8 Гц,1 Н); 8,45 (д, J=8 Гц, 1 Н); 8,56 (м, 1 Н). 1-(2-Фтор-5-трифторметилфенил)-3-[4-(4,4,5,5-тетраметил-[1,3,2]диоксоборолан-2-ил)фенил]мочевину получают согласно следующей методике. К раствору 1 г 4-(4,4,5,5-тетраметил-[1,3,2]диоксоборолан-2-ил)анилина в 15 мл тетрагидрофурана при комнатной температуре добавляют 936 мг 2-фтор-5-трифторметилфенилизоцианата, затем 0,64 мл триэтиламина. Реакционную смесь перемешивают при комнатной температуре в течение 18 ч, после чего обрабатывают метанолом и, наконец, выпаривают досуха при пониженном давлении. Таким образом полученный остаток очищают хроматографией на диоксиде кремния, используя в качестве элюента смесь дихлорметан/метанол (99,5:0,5, затем 90:10). Фракции, содержащие ожидаемый продукт, концентрируют досуха,получая 1,45 г 1-(2-фтор-5-трифторметилфенил)-3-[4-(4,4,5,5-тетраметил[1,3,2]диоксоборолан-2-ил)фенил]мочевины в виде твердого вещества белого цвета. Раствор 1-[4-(3-амино-1 Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-3-(2-фтор-5-трифторметилфенил) мочевины (80,0 мг, 0,186 ммоль) в пиридине (2 мл) охлаждают до температуры 5 С в атмосфере аргона и добавляют хлорангидрид тиофен-3-карбоновой кислоты (27 мг, 0,186 ммоль, 1,0 экв.). Смесь перемешивают при комнатной температуре в течение 5,5 ч. Добавляют другой эквивалент хлорангидрида тиофен 3-карбоновой кислоты (27 мг, 0,186 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь обрабатывают водой, затем экстрагируют этилацетатом. Органическую фазу промывают два раза солевым раствором, сушат над сульфатом магния и концентрируют. Получают тогда твердое вещество желто-оранжевого цвета. Сырое соединение очищают при использовании колонки с диоксидом кремния с помощью градиента элюирования от 0 до 5% метанола в дихлорметане. Получают твердое вещество бежевого цвета: 13,5 мг. 100 мг (0,44 ммоль) 6-(4-Аминофенил)-1 Н-пиразоло[3,4-b]пиридин-3-иламина растворяют в 3 мл безводного диметилформамида. После этого раствор дегазируют в течение 15 мин с помощью аргона. Затем добавляют 123,8 мкл (89,9 мг, 0,89 ммоль, 2 экв.) триэтиламина. Раствор еще раз дегазируют в течение 15 мин. Наконец, добавляют 0,109 г (0,444 ммоль, 1 экв.) 2,3-дихлорфенилсульфонилхлорида. Раствор перемешивают при комнатной температуре в атмосфере аргона в течение ночи. По окончании реакции добавляют 10 мл воды. Выпадает осадок желто-коричневого цвета. Смесь фильтруют. Твердое вещество высушивают и очищают при использовании колонки с 4 г диоксида кремния с помощью градиента элюирования от 0 до 10% метанола в дихлорметане. Выделяют твердое вещество бежевого цвета (11 мг). Соединение примера 4 может быть получено способом, описанным выше на схеме 4. Синтез промежуточного продукта 1. К раствору 2-аминотиазол-5-карбоксальдегитда (2,5 г, 19,5 ммоль) в диметилформамиде (120 мл) добавляют 2-фтор-(5-трифторметил)фенилизоцианат (4 г, 19,5 ммоль). Раствор нагревают при температуре 70 С в течение 1 ч, выпаривают и очищают хроматографией на силикагеле, элюируя смесью гексан/этилацетат в соотношении 1:1. Содержащие промежуточный продукт 1 фракции объединяют и выпаривают, получая промежуточный продукт 1 в виде твердого вещества желтого цвета (4,28 г). Синтез промежуточного продукта 2. К раствору промежуточного продукта 1 (88 мг, 0,26 ммоль) в смеси этанол/диметилсульфоксид в соотношении 9:1 (2 мл) добавляют раствор 1 Н-пиразол-3-иламина (22 мг, 0,26 ммоль) в 2 мл той же самой смеси растворителей, содержащей пирувиновую кислоту (23 мг, 0,26 ммоль). Смесь нагревают при температуре 75 С в течение 24 ч в присутствии кислорода, затем реакционную смесь выпаривают досуха, получая промежуточный продукт 2 в виде твердого вещества оранжевого цвета, который используют таким, какой есть, на следующей стадии. Получение продукта примера 4. Промежуточное соединение 2 растворяют в безводном тетрагидрофуране (2 мл), добавляют HOBt(39 мкл, 0,3 ммоль). Перемешивание продолжают в течение 5 ч при комнатной температуре, затем реакционную среду выпаривают при пониженном давлении. Остаток очищают хроматографией на силикагеле, элюируя смесью этилацетат/метанол/триэтиламин в соотношении 94:5:1 при использовании градиента этилацетат/метанол/триэтиламин=90:9:1. Содержащие соединение фракции объединяют и выпаривают, затем очищают хроматографией LC/MS (обращенная фаза), элюируя водой+0,1% трифторуксусной кислоты при использовании градиента 25-85% ацетонитрила в течение промежутка времени 8 мин. Содержащие ожидаемое соединение фракции объединяют и выпаривают при пониженном давлении, получая соединение в виде твердого вещества желтого цвета (10,6 мг). Пример 5. N-[4-(3-Амино-4-метиламинокарбонил-1 Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-2,3 дихлорбензолсульфонамид Продукт примера 5 может быть получен способом, описанным выше на схеме 5. Синтез промежуточного продукта 1. 219 мг 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина вместе с 245 мг (1 ммоль) 2,3 дихлорбензолсульфонилхлорида растворяют в 2 мл дихлорметана и 120 мкл (1,5 мкмоль) пиридина. Смесь перемешивают в течение 6 ч при комнатной температуре, затем высушивают в вакууме, получая желательный продукт в виде твердого вещества светло-красного цвета.MC (ES) MH+ m/z=428. Синтез промежуточного продукта 2. 84 г натриевой соли диэтилового эфира щавелевоуксусной кислоты вместе с 33,2 г 2-аминопиразола растворяют в 600 мл смеси уксусной кислоты и воды (1:3). Смесь нагревают в течение 6 ч при температуре 80 С. Продукт осаждается после охлаждения, его отфильтровывают и высушивают в вакууме (17,9 г). 1 Н-ЯМР (600 МГц, ДМСО-d6)(м.д.): 13,74 (с, 1 Н); 12,39 (с, 1 Н); 8,22 (с, 1 Н); 6,70 (с, 1 Н); 4,40 (кв,J=7,1 Гц, 2 Н); 1,40 (т, J=7,1 Гц, 3 Н). Синтез промежуточного продукта 3. 2,07 г (10 ммоль) промежуточного продукта 2, 20 мл толуола и 2,87 г (10 ммоль) POBr3 нагревают на масляной бане при температуре 110 С и при перемешивании. Смесь выпаривают и очищают при использовании колонки с диоксидом кремния, элюируя смесью 20% этилацетата и 80% гексана, получаяH-ЯМР (600 МГц, ДМСО-d6)(м.д.): 14,45 (с, 1 Н); 8,41 (с, 1 Н); 7,79 (с, 1 Н); 4,49 (кв, J=7,1 Гц, 2 Н); 1,46 (т, J=7,1 Гц, 3 Н). Синтез промежуточного продукта 4. 74 мкмоль промежуточного продукта 3 (20 мг), 74 мкмоль промежуточного продукта 1, 185 мкмоль карбоната цезия и 7,4 мкмоль Pd(PPh3)4 растворяют в тетрагидроуране и 20% H2O (общий объем 750 мкл). Смесь нагревают при микроволновом облучении в течение 25 мин при температуре 155 С (мощность 55 Вт). Продукт получают выпариванием и очищают хроматографией LC/MS при использованииMC (ES) MH+ m/z=463. Получение продукта примера 5. Промежуточный продукт 4 (20 мг, 43,3 мкмоль) вместе с HOBt (14,6 мг, 108,25 мкмоль) и DIC (16,9 мкл, 108,25 мкмоль) растворяют в диметилформамиде (0,5 мл). Добавляют 0,5 ммоль метиламина (1 М раствор в тетрагидрофуране, 0,5 мл). Смесь нагревают при микроволновом облучении в течение 20 мин при температуре 105 С (мощность 25 Вт). Продукт получают выпариванием и очищают хроматографиейLC/MS при использовании воды с 0,1% трифторуксусной кислоты и градиента от 25 до 95% ацетонитрила в течение 9 мин. Продукт высушивают в вакууме, получая твердое вещество светло-желтого цвета.H-ЯМР (600 МГц, ДМСО-d6)(м.д.): 13,79 (с, 1 Н), 11,13 (с, 1 Н), 8,35 (с, 1 Н), 8,02 (с, 1 Н), 8,13 (д,J=7,8 Гц, 2 Н), 8,09 (д, J=8,27 Гц, 1 Н), 7,94 (д, J=8,05 Гц, 1 Н), 7,28 (д, J=4,31 Гц, 2 Н), 2,88 (д, J=4,31 Гц,3 Н), 7,58 (кв, J=8,08 Гц, 1 Н), 8,86 (т, J=4,43 Гц, 1 Н). Примеры 6-62. Следуя вышеописанным методикам получают соединения, указанные в нижеследующей таблице. Определение активности соединений - эксперментальные протоколы 1. FAK. Ингибирующее действие соединений в отношении FAK устанавливали путем определения ингибирования автофосфорилирования фермента, используя тест разрешенной во времени флуоресценции(HTRF). Полную кДНК человеческой FAK, N-конец которой маркирован по гистидину, клонировали в вектор экспрессии бакуловируса pFastBac НТс. Белок экспрессировали и очищали до гомогенности примерно 70%. Киназную активность определяли путем инкубирования фермента (6,6 мкг/мл) с различными концентрациями тестируемого соединения в буфере 50 мМ Гепес, рН 7,2, 10 мМ MgCl2, 100 мкМ Na3VO4, 15 мкМ АТФ, в течение 1 ч при температуре 37 С. Ферментативную реакцию прекращали путем добавления буфера Гепес, рН 7,0, содержащего 0,4 мМ KF, 133 мМ этилендиаминтетрауксусной кислоты (ЭДТУ), 0,1% бычьего сывороточного альбумина и маркировку осуществляли в течение 1-2 ч при комнатной температуре путем добавления в этот буфер антитела против гистидина, маркированного с помощью XL665, и моноклонального фосфоспецифического к тирозину антитела, конъюгированного с криптатом европия (EuK). Характеристики двух флуорофоров приводятся G. Mathis и др. в Anticancer Research, 17, 3011-3014(1997). Перенос энергии от возбужденного криптата европия на XL665-акцептор пропорционален степени автофосфорилирования FAK. Сигнал специфической стабильной длительности XL665 измеряли при использовании счетчика бляшек Packard Discovery. Все тесты осуществляли в двух повторениях и рассчитывали среднее из двух тестов. Ингибирование активности автофосфорилирования FAK с помощью соединений согласно изобретению выражали в процентах ингибирования по отношению к контролю,активность которого определяли в отсутствие тестируемого соединения. Для расчета процента ингибирования рассматривали соотношение [сигнал при 665 нм/сигнал при 620 нм]. 2. KDR. Ингибирующее действие соединений определяли согласно тесту фосфорилирования субстрата с помощью фермента KDR in vitro методом сцинтилляции (96-луночные планшеты, NEN). Цитоплазматический домен человеческого фермента KDR клонировали в виде гибрида GST в вектор экспрессии бакуловируса pFastBac. Белок экспрессировали в клетках SF21 и очищали до гомогенности примерно 60%. Киназную активность KDR определяли в буфере 20 мМ MOPS, 10 мМ MgCl2, 10 мМ MnCl2, 1 мМ дитиотреитола, 2,5 мМ EGTA, 10 мМ -глицерофосфата, рН 7,2, в присутствии 10 мМ MgCl2, 100 мкМNa3VO4, 1 мМ NaF. 10 мкл Соединения добавляли к 70 мкл киназного буфера, содержащего 100 нг фермента KDR, при температуре 4 С. Реакцию инициировали путем добавления 20 мкл раствора, содержащего 2 мкг субстрата (фрагмент SH2-SH3 PLC, экспрессированного в виде гибридного белка GST), 2 мкКи 33 Р[АТФ] и 2 мкМ нерадиоактивного АТФ. После инкубации в течение 1 ч при температуре 37 С реакцию прекращали путем добавления 1 об. (100 мкл) 200 мМ ЭДТУ. Буфер для инкубации удаляли и лунки промывали три раза с помощью 300 мкл забуференного фосфатом физиологического раствора(PBS). Радиоактивность измеряли в каждой лунке, используя счетчик радиоактивности Top Count NXT(Packard). Фон определяли путем измерения радиоактивности в четырех разных лунках, содержащих только радиоактивный АТФ и субстрат. Контроль полной активности оценивали в четырех разных лунках, содержащих все реагенты (33 Р[АТФ], KDR и субстрат PLC), но в отсутствие соединения. Ингибирование активности KDR с помощью соединения согласно настоящему изобретению выражали в процентах ингибирования по отношению к контрольной активности, определяемой в отсутствие соединения. Соединение SU5614 (Calbiochem) (1 мкМ) включали в каждый планшет в качестве контроля ингибирования. 3. Tie2. Кодирующую последовательность человеческого Tie2, соответствующую аминокислотам внутриклеточного домена 776-1124, получали путем полимеразной цепной реакции (PCR), используя кДНК,выделенную из человеческой плаценты, в качестве модели. Эту последовательность встраивали в вектор экспрессии бакуловируса pFastBacGT в виде гибридного белка GST. Ингибирующее действие молекул определяли согласно тесту фосфорилирования PLC с помощьюTie2 в присутствии GST-Tie2, очищенного до гомогенности примерно 80%. Субстрат состоял из фрагментов SH2-SH3 PLC, экспрессированного в виде гибридного белка GST. Киназную активность Tie2 определяли в буфере 20 мМ MOPS, рН 7,2, содержащем 10 мМ MgCl2,10 мМ MnCl2, 1 мМ дитиотреитола, 10 мМ глицерофосфата. В 96-луночный планшет FlashPlate, выдерживаемый на льду, вносили реакционную смесь, состоящую из 70 мкл киназного буфера, содержащего 100 нг фермента GST-Tie2, на лунку. Затем добавляли 10 мкл тестируемой молекулы, разведенной в ДМСО до концентрации максимально 10%. Для данной концентрации каждое измерение осуществляли в четырех повторениях. Реакцию инициировали путем добавления 20 мкл раствора, содержащего 2 мкгGST-PLC, 2 мкМ нерадиоактивного АТФ и 1 мкКи 33 Р[АТФ]. После инкубации в течение 1 ч при температуре 37 С реакцию прекращали путем добавления 1 об. (100 мкл) 200 мМ ЭДТУ. После удаления буфера для инкубации лунки промывали три раза с помощью 300 мкл PBS. Радиоактивность определяли при использовании счетчика MicroBeta 1450, Wallac. Ингибирование активности Tie2 рассчитывали и выражали в процентах ингибирования по отношению к контрольной активности, определяемой в отсутствие соединения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение следующей общей формулы (I): в которой: 1) А представляет собой фенил, необязательно замещенный одним или двумя заместителями, выбранными из галогена, (С 1-С 4)-алкила, (C1-C3)-алкилгалогена, О(C1-C4)-алкила, S-(C1-C4)-алкила, О(С 1 С 4)-алкилгалогена, S-(C1-C4)-алкилгалогена; 2) Ar выбирают из группы, состоящей из фенила, тиазолила, тиенила, фурила, пирролила, оксазолила, изоксазолила, изотиазолила, тиадиазолила, пиразолила, имидазолила, индолила, индазолила, бензимидазолила, бензоксазолила и бензотиазолила; 3) L выбирают из группы, состоящей из NH-SO2 и NH-CO-NH; 5) R3 выбирают из группы, состоящей из Н, NH2 и NHC(O)R"3, где R"3 является тиенилом; 6) R4 выбирают из группы, состоящей из Н, CON(R"5)(R"6), где R"5 и R"6 независимо выбирают из группы, состоящей из Н, (C1-С 6)-алкила, замещенного (C1-С 6)-алкила, (C1-С 6)-алкилгетероциклила, замещенного (C1-С 6)-алкилгетероциклила, (C1-С 6)-алкилгетероарила, гетероарила, или же R"5 и R"6 связаны друг с другом с образованием 4-8-членного насыщенного цикла, содержащего 1-3 гетероатома, выбираемых среди О, S и N, необязательно замещенного; при этом гетероциклил выбран из морфолинила, пиперазинила и пиперидинила и гетероарил выбран из имидазолила, пиразолила и пиридинила; 7) R5 представляет собой Н; где "замещенный" относится к заместителю, отличающемуся от Н, который представляет собой галоген, (С 1-С 4)-алкил, (С 1-С 3)-алкилгалоген, О(С 1-С 3)-алкил, S-(C1-C4)-алкил, O(С 1-С 4)-алкилгалоген, S(C1-C4)-алкилгалоген. 2. Соединение по п.1, отличающееся тем, что Ar выбирают из группы, состоящей из тиазолила, тиенила, фурила, пирролила, оксазолила, изоксазолила, изотиазолила, тиадиазолила, пиразолила, имидазолила, индолила, индазолила, бензимидазолила, бензоксазолила и бензотиазолила. 3. Соединение по п.1, отличающееся тем, что R4 означает Н или CON(R"5)(R"6), где R"5 и R"6 имеют вышеуказанное значение. 4. Соединение по любому из пп.1-3, отличающееся тем, что его выбирают среди 1-[4-(3-амино-1 Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-3-(2-фтор-5-трифторметилфенил)мочевины;(2-морфолин-4-илэтил)амида 6-2-[3-(2,4-диметоксифенил)уреидо]тиазол-5-ил-1 Н-пиразоло[3,4b]пиридин-4-карбоновой кислоты. 5. Соединение по любому из пп.1-4, отличающееся тем, что его выбирают средиN-[4-(3-амино-4-метиламинокарбонил-1 Н-пиразоло[3,4-b]пиридин-6-ил)фенил]-2,3-дихлорбензолсульфонамида. 6. Соединение по любому из пп.1-5, отличающееся тем, что оно находится 1) в нехиральной форме, или 2) в рацемической форме, или 3) в обогащенной одним стереоизомером форме, или 4) в обогащенной одним энантиомером форме,в виде основания или соли. 7. Лекарственное средство, отличающееся тем, что оно включает соединение формулы (I) по любому из пп.1-6 или аддитивную соль этого соединения с фармацевтически приемлемой кислотой. 8. Фармацевтическая композиция, включающая соединение по любому из пп.1-6 в комбинации с фармацевтически приемлемым эксципиентом. 9. Применение соединения по любому из пп.1-6 в качестве ингибитора реакции, катализируемой одной или несколькими киназами, выбранными из FAK, KDR и Tie2. 10. Применение соединения по любому из пп.1-6 для получения лекарственного средства, пригодного для лечения патологического состояния, где патологическое состояние выбирают среди рака, ревматоидного артрита, остеоартрита и/или ассоциированных с ним болей, воспалительных заболеваний кишечника, патологий глаза, диабетических ретинопатий, хронического воспаления, псориаза. 11. Применение по п.10 соединения по любому из пп.1-6 для лечения или профилактики патологического состояния, отличающееся тем, что соединение вводят индивидуально или в комбинации с другими противораковыми средствами. 12. Соединения следующей общей формулы (II): в которой R'4 означает R4, -СООН или -COO-(C1-С 6)-алкил; R'3 означает Н, -NH2 или -NHCO-тиенил и R'6 означает группу -фенил-NH2 или группу Ar-L-A, где Ar, L и А имеют указанное в п.1 значение. 13. Соединения следующей общей формулы (III):
МПК / Метки
МПК: C07D 213/85, A61P 35/00, C07D 471/04, A61K 31/437
Метки: применение, содержащие, композиции, способ, замещенные, получения, 7-азаиндазолы
Код ссылки
<a href="https://eas.patents.su/29-19302-zameshhennye-7-azaindazoly-soderzhashhie-ih-kompozicii-sposob-polucheniya-i-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные 7-азаиндазолы, содержащие их композиции, способ получения и применение</a>
Замещённые индолы, содержащие их композиции, способ получения и применение

Номер патента: 12613
Опубликовано: 30.10.2009
Авторы: Ронан Батист, Алле Франк, Бак Эрик, Филош-Ромм Брюно, Леталлек Жан-Филипп, Суай Катрин, Табар Мишель, Вивьяни Фабрис
МПК: A61K 31/404, A61K 31/407, C07D 209/12...
Метки: содержащие, получения, композиции, применение, замещённые, способ, индолы
Формула / Реферат:
1. Продукт, отвечающий следующей формуле (I) где a) Ar обозначает фенил; b) А обозначает фенил, пиразол или изоксазол, в случае необходимости замещенный; c) R1 означает Н; d) X означает N или N-оксид или СН; e) L обозначает NH-CO-NH или NH-SO2; f) R5, R6, R7 и R12, каждый независимо, выбирают из группы, состоящей из Н, NO2, O(R2), N(R2)(R3), NHC(О)R2N(R3)(R4); где каждый R2, R3, R4 независимо выбирают из группы, состоящей из Н, (C1-C4)алкила, в...
Замещённые пироллы, содержащие их композиции, способ получения и применение

Номер патента: 13060
Опубликовано: 26.02.2010
Авторы: Табар Мишель, Демазо Паскаль, Ронан Батист, Летталек Жан-Филипп, Вивьяни Фабрис, Суай Катрин, Бак Эрик
МПК: C07D 207/34, A61K 31/40, A61P 35/00...
Метки: замещённые, пироллы, способ, применение, содержащие, получения, композиции
Формула / Реферат:
1. Соединение, отвечающее следующей формуле (I)в которой1) Ar является фенилом;2) A является необязательно замещенным фенилом;3) L является NH-CO-NH;4) Ra является Н;5) R1 является Н;6) R2 независимо выбраны из группы, состоящей из Н, галогена, R'2, O(R'2), NH(R'2), NHCO(R'2), NHCONH(R'2), CONH2; причем каждый из R'2 независимо выбран из группы, состоящей из Н, (С1-С3)алкила, (С1-С3)алкилена, (С3-С5)циклоалкила, замещенного (С1-С3)алкила,...
Замещённые пирролы и имидазолы, содержащие их композиции, способ получения и применение

Номер патента: 12267
Опубликовано: 28.08.2009
Авторы: Суай Катрин, Бак Эрик, Табар Мишель, Алле Франк, Ронан Батист, Эль Амад Юссеф, Вивьяни Фабрис
МПК: A61K 31/4164, A61K 35/00, A61K 31/40...
Метки: пирролы, способ, содержащие, композиции, замещённые, применение, получения, имидазолы
Формула / Реферат:
1. Соединение, отвечающее следующей формуле (I): в которой: 1) А обозначает фенил, необязательно замещенный одним или несколькими одинаковыми или разными заместителями, независимо выбранными из группы, включающей F, Cl, CH3, OCH3, OCF3; 2) Ar обозначает фенил; 3) L обозначает NH-CO-NH; 4) Ra обозначает Н; 5) R1 обозначает Н; 6) X выбирают из CR3 и N; 7) R2 выбирают из Н и галогена, 8) R3 выбирают из группы, состоящей из Н, галогена, NH2,...
Стероиды, замещенные в положении 11, способ их получения, их применение в качестве лекарственного средства и содержащие их фармацевтические композиции

Номер патента: 1868
Опубликовано: 22.10.2001
Авторы: Ник Франсуа, Буали Иамина, Ван Де Вельд Патрик, Тетш Жан-Жорж
МПК: A61P 19/10, C07J 41/00, A61K 31/565...
Метки: средства, лекарственного, содержащие, получения, способ, стероиды, качестве, замещенные, фармацевтические, положении, применение, композиции
Формула / Реферат:
1. Соединения общей формулы в которой n - целое число, равно 2 или 3, либо R1 и R2, идентичные или разные, означают атом водорода или алкил, содержащий от 1 до 4 атомов углерода, либо R1 и R2 образуют вместе с атомом азота, с которым они связаны, моно- или полициклический гетероцикл, имеющий от 5 до 15 звеньев, ароматический или не ароматический, содержащий, при необходимости, от 1 до 3 дополнительных гетероатомов, выбираемых из кислорода,...
Новые 19-норстероиды, замещенные в положении 11&beta, способ и промежуточные продукты для их получения, применение в качестве лекарственных средств и содержащие их фармацевтические композиции

Номер патента: 3133
Опубликовано: 27.02.2003
Автор: Ник Франсуа
МПК: A61K 31/566, C07J 41/00, A61P 19/10...
Метки: композиции, положении, новые, средств, способ, качестве, продукты, промежуточные, применение, содержащие, 11&beta, лекарственных, получения, замещенные, 19-норстероиды, фармацевтические
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает атом водорода иди радикал ацил, R2 обозначает радикал (C1-C4)алкил, X обозначает атом галогена или атом водорода, n равно 3, 4 или 5, R3 и R4 обозначает (C1-C4)алкил или R3 и R4 образуют вместе с атомом азота, с которым они связаны, группу пироолидинил или пиперидинил, R5 обозначает OH и R6 обозначает H, (C1-C4)алкил возможно замещенный одним или тремя атомами галогена или R5 и R6...
Предыдущий патент: Фунгицидная композиция для протравливания семян
Следующий патент: Способ получения мемантина и его гидрохлорида
Случайный патент: Блок для хранения отходов