Новые соединения 2-меркаптоциклопентанкарбоновой кислоты, способ их получения и содержащие их фармацевтические композиции
Номер патента: 17867
Опубликовано: 29.03.2013
Авторы: Буар Никола, Кирьон Жан-Шарль, Глоанек Филипп, Жийюзи Анне-Франсуа, Рюпэн Алан, Валле Мари-Одиль, Де-Нантой Гийом, Жюбо Филипп, Меннесье Филипп, Парментье Жан-Жилль, Вербёрен Тони







Формула / Реферат
1. Соединение формулы (I)

в которой R1 представляет собой атом водорода, ацетильную группу или группу формулы (А)

R2 представляет собой группу формулы NR5R6, в которой R5 и R6, которые являются одинаковыми или различными, каждый представляет собой атом водорода или метильную группу, или R2 представляет собой пиперидин, азетидин, пиридин, необязательно замещенный аминогруппой или аминометилфенилом,
R3 представляет собой атом водорода,
m представляет собой целое число от 1 до 5 включительно,
n представляет собой 1,
их оптические изомеры, а также их аддитивные соли с фармацевтически приемлемой кислотой.
2. Соединение формулы (I) в соответствии с п.1, в которой R1 представляет собой атом водорода.
3. Соединение формулы (I) в соответствии с любым из пп.1 или 2, в которой R2 представляет собой аминогруппу или группу пиридила.
4. Соединение формулы (I) в соответствии с любым из пп.1 или 2, в которой m представляет собой 3.
5. Соединение формулы (Ia) в соответствии с п.1

в которой n, R1, R3, R5 и R6 являются такими, как определено в п.1.
6. Соединение формулы (I) в соответствии с п.1, выбранное из
(1R,2S)-1-(3-аминопропил)-2-меркаптоциклопентанкарбоновой кислоты, ее оптических изомеров и аддитивных солей с фармацевтически приемлемой кислотой и
(1R,2S)-2-ацетилтио-1-(3-аминопропил)циклопентанкарбоновой кислоты, ее оптических изомеров и аддитивных солей с фармацевтически приемлемой кислотой.
7. Способ синтеза соединений формулы (I) в соответствии с п.1 взаимодействием соединения формулы (II)

в которой n представляет собой 1 и G представляет собой защитную группу для группы карбокси,
с соединением формулы (III)
в которой X представляет собой атом галогена или трифлат, группу тозилата или мезилата,
m представляет собой целое число от 1 до 5 включительно,
a R'2 представляет собой группу формулы NR'5R'6, в которой R'5 и R'6, которые могут быть одинаковыми или различными, каждый представляет собой защитную группу для аминогруппы или метильную группу, или R'2 представляет собой пиперидин, азетидин, пиридин, необязательно замещенный аминогруппой или аминометилфенилом, необязательно замещенную защитной группой для аминогруппы,
с получением соединения формулы (IV)

в которой R'2, G, m и n являются такими, как определено выше,
которое подвергают воздействию восстанавливающего агента для оксогруппы,
с получением соединения формулы (V)

в которой R'2, G, m и n являются такими, как определено выше,
которое подвергают реакции с мезилхлоридом, тозилхлоридом, трифторметансульфоновым ангидридом или галогенирующим реагентом с получением соединения формулы (VI)

в которой R'2, G, m и n являются такими, как определено выше, а X представляет собой мезилат, тозилат, или группу трифлата, или атом галогена,
с возможным выделением соединения формулы (I) в форме единственного диастереоизомера,
и которое потом подвергают реакции с соединением формулы (VII)

в которой М представляет собой калий, натрий или литий, a R'1 представляет собой метильную группу,
с получением соединения формулы (VIII)

в которой R'1, R'2, G, m и n являются такими, как определено выше,
с возможным выделением с помощью хиральной хроматографии соединения формулы (I) в форме единственного энантиомера,
с возможным снятием защиты с групп тиола, амино и карбокси, с получением соединения формулы (I),
с возможным получением аддитивной соли соединения формулы (I) с фармацевтически приемлемой кислотой.
8. Способ получения соединений формулы (Ia) в соответствии с п.5 взаимодействием соединения формулы (II)

в которой n представляет собой 1 и G представляет собой защитную группу для группы карбокси,
с акролеином, в присутствии асимметричного катализатора, с получением соединения формулы (IX) конфигурации (1R) или (1S)

в которой n и G являются такими, как определено выше,
альдегидную группу которого восстанавливают с получением соединения формулы (X)

в которой n и G являются такими, как определено выше,
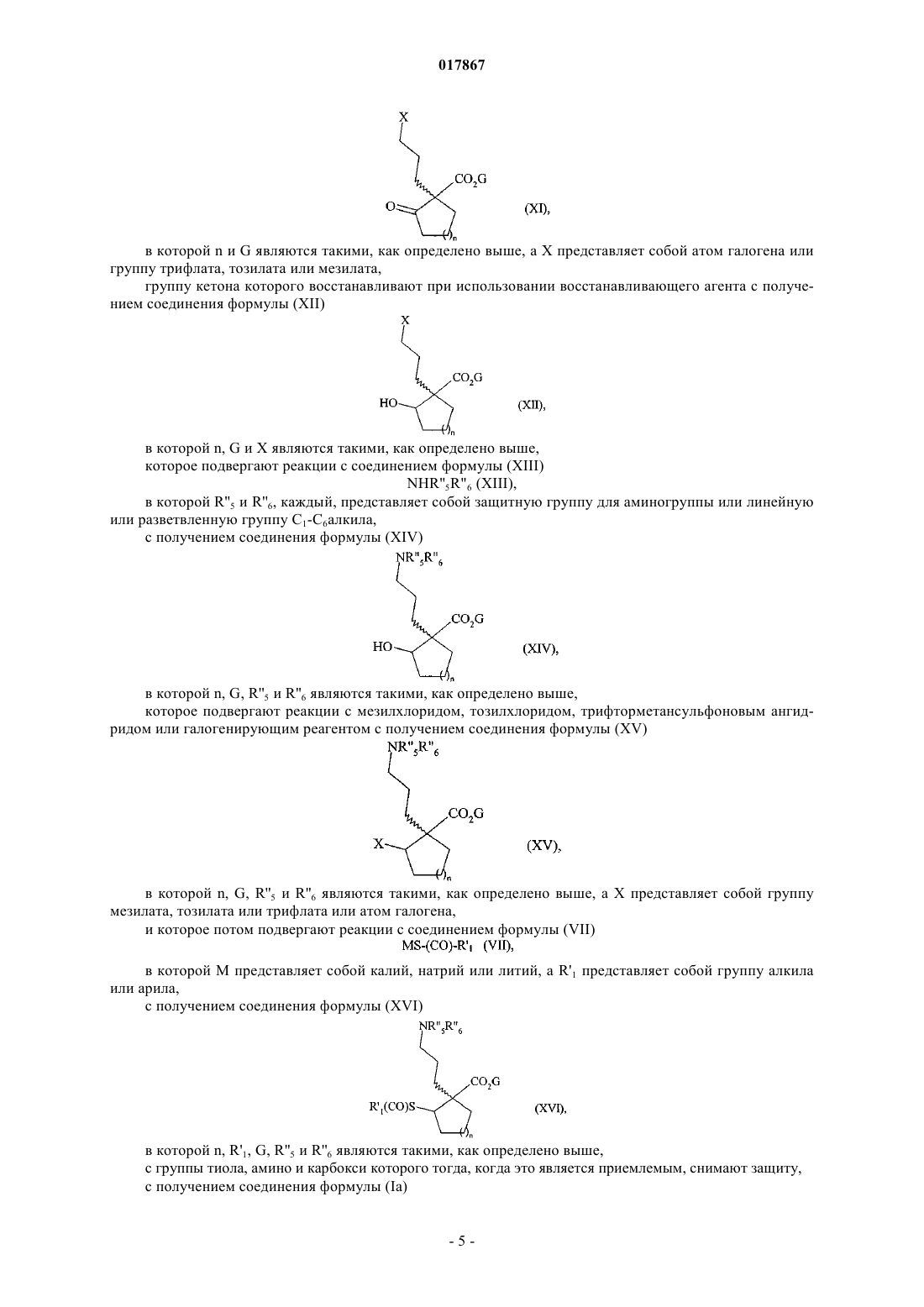
которое подвергают реакции с мезилхлоридом, тозилхлоридом, трифторметансульфоновым ангидридом или галогенирующим реагентом с получением соединения формулы (XI)

в которой n и G являются такими, как определено выше, а X представляет собой атом галогена или трифлат, группу тозилата или мезилата,
группу кетона которого восстанавливают при использовании восстанавливающего агента с получением соединения формулы (XII)

в которой n, G и X являются такими, как определено выше,
которое подвергают реакции с соединением формулы (XIII)
в которой R''5 и R''6, каждый, представляет собой защитную группу для аминогруппы или метильную группу,
с получением соединения формулы (XIV)

в которой n, G, R"5 и R''6 являются такими, как определено выше,
которое подвергают реакции с мезилхлоридом, тозилхлоридом, трифторметансульфоновым ангидридом или галогенирующим реагентом с получением соединения формулы (XV)

в которой n, G, R''5 и R''6 являются такими, как определено выше, а X представляет собой группу мезилата, тозилата, или трифлат, или атом галогена,
и которое потом подвергают реакции с соединением формулы (VII)
в которой М представляет собой калий, натрий или литий, a R'1 представляет собой метильную группу,
с получением соединения формулы (XVI)

в которой n, R'1, G, R''5 и R''6 являются такими, как определено выше,
с возможным снятием защиты с группы тиола, амино и карбокси с получением соединения формулы (Ia)

в которой n, R1, R3, R5 и R6 являются такими, как определено выше,
с возможным получением аддитивной соли соединения формулы (Ia) с фармацевтически приемлемой кислотой.
9. Фармацевтическая композиция, включающая соединение формулы (I) в соответствии с любым из пп.1-6 в комбинации с одним или более инертными, нетоксическими, фармацевтически приемлемыми наполнителями или носителями.
10. Фармацевтическая композиция в соответствии с п.9, отличающаяся тем, что дополнительно включает фибринолитический, антикоагулянтный или антитромбоцитарный агент.
11. Фармацевтическая композиция в соответствии с п.10 в инъецируемой форме, отличающаяся тем, что фибринолитический агент, выбран из рекомбинантного tPA, рекомбинантного uPA и стрептокиназы.
12. Применение соединения в соответствии с любым из пп.1-6 в производстве лекарственных средств для использования в предотвращении, вторичном предотвращении или лечении инфаркта миокарда, стенокардии, артериита нижних конечностей, венозных тромбозов, легочной эмболии, инсультов, васкулярных осложнений диабета, аневризмы аорты или деменции.
13. Применение соединения в соответствии с п.12 в сочетании с фибринолитическим, антикоагулянтным или антитромбоцитарным агентом.
Текст
НОВЫЕ СОЕДИНЕНИЯ 2-МЕРКАПТОЦИКЛОПЕНТАНКАРБОНОВОЙ КИСЛОТЫ,СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ в которой R1 представляет собой атом водорода, ацетильную группу или группу формулы (А)R2 представляет собой группу формулы NR5R6, в которой R5 и R6, которые являются одинаковыми или различными, каждый представляет собой атом водорода или метильную группу, или R2 представляет собой пиперидин, азетидин, пиридин, необязательно замещенный аминогруппой или аминометилфенилом, R3 представляет собой атом водорода, m представляет собой целое число от 1 до 5 включительно, n представляет собой 1, их оптические изомеры, а также их аддитивные соли с фармацевтически приемлемой кислотой. Лекарственные средства.(71)(73) Заявитель и патентовладелец: ЛЕ ЛАБОРАТУАР СЕРВЬЕ; Л'ИНСТИТЮ НАСЬОНАЛЬ ДЕ СЬЯНС АППЛИКЕ ДЕ РУЕН; ЛЕ САНТР НАСЬОНАЛЬ ДЕЛЯ РЕШЕРШ САЙНТИФИК; Л'ЮНИВЕРСИТЕ ДЕ РУЕН (FR) Данное изобретение относится к новым соединениям 2-меркаптоциклопентанкарбоновой кислоты,способу их получения и к содержащим их фармацевтическим композициям. Соединения в соответствии с изобретением представляют собой ингибиторы TAFIa (активированный ингибитор фибринолиза, способный к активации с помощью тромбина).TAFI (также называется прокарбоксипептидаза В, прокарбоксипептидаза R или прокарбоксипептидаза U) представляет собой гликопротеин плазмы крови с молекулярным весом 60 кДа, который вырабатывается печенью и циркулирует в форме зимогена. В процессе коагуляции крови и фибринолиза тромбин и плазмин расщепляют просегмент TAFI в положении Arg92-Ala93 связи с превращением его в активный фермент, TAFIa, который имеет период полураспада от 8 до 15 мин при 37 С. Расщепление просегмента тромбином ускоряется тромбомодулином, кофактором, который присутствует в плазме крови и на поверхности васкулярных эндотелиальных клеток (Bouma B.N. и Meijers J.C., Thrombin-activatablefibrinolysis inhibitor, 2003, Journal of Thrombosis и Haemostasis, 1: 1566-1574). TAFIa негативно регулирует фибринолиз путем отщепления С-терминальных остатков лизина фибриновых волокон, что выявляется в частичной деградации фибрина с помощью первых следовых количеств плазмина. Эти С-терминальные остатки лизина на частично деградированном фибрине действуют как лиганды плазминогена, циркулирующего в плазме крови, и тканевого активатора плазминогена (tPA), вырабатываемого эндотелиальными клетками в случае тромботической ишемии. Они, соответственно, делают возможным локализацию превращения плазминогена в плазмин с помощью tPA при отсутствии взаимодействия либо с циркулирующим в плазме крови ингибитором плазмина 2-антиплазмином, либо с циркулирующим в плазме крове ингибитором активатора тканевого плазминогена (PAI-1). Отщепление С-терминальных сайтов лизина с помощью TAFIa, таким образом, снижает уровень, при котором вырабатывается плазмин. Эндогенный фибринолиз, таким образом, ингибируется и уменьшается лизис фибринозных артериальных и венозных тромбозов, а также терапевтический тромболизис, который осуществляется у пациентов в посттромботической острой ишемической фазе. Ингибиторы TAFIa, таким образом, имеют потенциал для повышения потенциала эндогенного и терапевтического фибринолиза и действуют как антитромботические и профибринолитические агенты при отсутствии основного геморрагического риска, поскольку они не взаимодействуют ни с процессом активации тромбоцитов, ни с процессом коагуляции во время гомеостаза крови. Свойство ингибирования TAFIa, соответственно, делает возможным предполагать применение соединений в соответствии с изобретением в лечении и предотвращении тромботических случаев у пациентов с повышенным риском. Их применение будет ценным при лечении, предотвращении и вторичном предотвращении васкулярных осложнений, в частности сердечно-сосудистых, легочных и и цереброваскулярных осложнений,ассоциированных с атеротромботическими заболеваниями, атеросклерозом, диабетом, гиперлипидемией,гипертензией, хроническими венозными заболеваниями, метаболическим синдромом, ассоциированным с ожирением, и раком. Соединения в соответствии с изобретением являются особенно полезными в лечении, предотвращении и вторичном предотвращении инфаркта миокарда, стенокардии, инсультов, аневризмы аорты,артериита нижних конечностей, венозных тромбозов и легочной эмболии. Факторы сосудистого риска и васкулярных заболеваний, таких как гипертензия, ожирение, диабет,сердечные заболевания, цереброваскулярные заболевания и гиперлипидемия и, таким образом, атеросклероз, играют определенную роль в развитии деменции, такой как болезнь Альцгеймера и васкулярная деменция (Qiu С., De Ronchi D. и Fratiglioni L., The epidemiology of the dementias: an update, 2007, CurrentOpinion in Psychiatry, 20: 380-385). Соединения в соответствии с изобретением, соответственно, будут также полезными в лечении и/или предотвращении деменции, такой как болезнь Альцгеймера и васкулярная деменция.TAFIa снижает эндогенный фибринолитический потенциал. В качестве ингибиторов TAFIa соединения в соответствии с данным изобретением являются, таким образом, полезными как вспомогательные средства для острого лечения с помощью инъецируемых фибринолитических агентов, таких как рекомбинантный tPA (например, альтеплаза, тенектеплаза, ретеплаза), рекомбинантный uPA или стрептокиназа, которые используются при чрезвычайных обстоятельствах (например, инфаркте миокарда, инсульте). Соединения в соответствии с данным изобретением усиливают активность таких инъецируемых фибринолитических агентов и, таким образом, приводят к тому, что они используются с меньшим геморрагическим и нейротоксическим риском (их доза снижается и, таким образом, уменьшаются побочные эффекты). Данное изобретение, в частности, относится к соединениям формулы (I) в которой R1 представляет собой атом водорода или группу формулы COR4, в которой R4 представляет собой линейную или разветвленную группу C1-С 6 алкила или группу арила,или R1 представляет собой группу формулы (А)R2 представляет собой группу формулы NR5R6, в которой R5 и R6, которые являются одинаковыми или различными, каждый представляет собой атом водорода или линейную или разветвленную группуC1-С 6 алкила, или R2 представляет собой азотсодержащую гетероциклическую группу, группу арила или группу гетероарила,R3 представляет собой атом водорода или линейную или разветвленную группу C1-С 6 алкила,m представляет собой целое число от 1 до 6 включительно,n представляет собой 0, 1 или 2,к их оптическим изомерам, а также к их аддитивным солям с фармацевтически приемлемой кислотой. Под группой арила понимают фенил, необязательно замещенный одной или более идентичными или различными группами, выбранными из галогена, линейного или разветвленного (С 1-С 6)алкила, линейного или разветвленного (С 1-С 6)алкокси, амино (необязательно замещенного одним или двумя линейными или разветвленными группами (С 1-С 6)алкила) и аминоалкила, аминогруппы, необязательно замещенной одной или двумя линейными или разветвленными группами (С 1-С 6)алкила. Под группой гетероарила понимают от 5- до 12-членную моно- или бициклическую ароматическую группу, содержащую один, два или три гетероатома, выбранных из кислорода, азота и серы, при этом понятно, что гетероарил может необязательно быть замещенным одной или более идентичными или различными группами, выбранными из галогена, линейного или разветвленного (С 1-С 6)алкила, линейного или разветвленного (С 1-С 6)алкокси, амино (необязательно замещенного одной или двумя линейными или разветвленными группами (С 1-С 6)алкила) и аминоалкила, аминогруппы, необязательно замещенной одной или двумя линейными или разветвленными группами (С 1-С 6)алкила. Среди групп гетероарила могут быть упомянуты без какого-либо ограничения группы тиенила, пиридила, фурила, пирролила, имидазолила, оксазолила, изоксазолила, тиазолила, изотиазолила, пиримидинила, пиразинила, пиридазинила. Под азотсодержащей гетероциклической группой понимают от 4- до 7-членную насыщенную или ненасыщенную моноциклическую группу, содержащую один или более атомов азота и необязательно один или более других гетероатомов, выбранных из кислорода и серы. Среди азотсодержащих гетероциклических могут быть упомянуты без какого-либо ограничения группы азетидинила, пирролидинила, пиперидила и пиперазинила. Под оптическими изомерами понимают как диастереоизомеры, так и энантиомеры. Соединения формулы (I) содержат по крайней мере два асимметричных центра (в положениях 1 и 2 кольца) и могут,таким образом, существовать в форме единственного энантиомера, единственного диастереоизомера или в форме смеси диастереоизомеров. Среди фармацевтически приемлемых кислот могут быть упомянуты без какого-либо ограничения хлористо-водородная кислота, бромисто-водородная кислота, серная кислота, фофорная кислота, уксусная кислота, трифторуксусная кислота, молочная кислота, пировиноградная кислота, малоновая кислота,янтарная кислота, глутаровая кислота, муравьиная кислота, виннокаменная кислота, малеиновая кислота,лимонная кислота, аскорбиновая кислота, щавелевая кислота, метансульфоновая кислота, бензолсульфоновая кислота, камфорная кислота. Один аспект в соответствии с данным изобретением относится к соединениям формулы (I), в которой R1 представляет собой атом водорода. Другой аспект в соответствии с данным изобретением относится к соединениям формулы (I), в которой R2 представляет собой аминогруппу. Другой аспект в соответствии с данным изобретением относится к соединениям формулы (I), в которой R2 представляет собой группу пиридила. Другой аспект в соответствии с данным изобретением относится к соединениям формулы (I), в которой R3 представляет собой атом водорода. Другой аспект в соответствии с данным изобретением относится к соединениям формулы (I), в которой m представляет собой 3. Другой аспект в соответствии с данным изобретением относится к соединениям формулы (I), в которой n представляет собой 1. Другой аспект в соответствии с данным изобретением относится к соединениям формулы (Ia), частному случаю соединений формулы (I) в которой n, R1, R3, R5 и R6 являются такими, как определено в формуле (I). Другой аспект в соответствии с изобретением относится к следующим соединениям формулы (I):(1R,2S)-1-(3-аминопропил)-2-меркаптоциклопентанкарбоновая кислота, а также ее оптические изомеры и аддитивные соли с фармацевтически приемлемой кислотой,(1R,2S)-2-ацетилтио-1-(3-аминопропил)циклопентанкарбоновая кислота, а также ее оптические изомеры и аддитивные соли с фармацевтически приемлемой кислотой. Данное изобретение также относится к способу получения соединений формулы (I), начиная от соединения формулы (II) в которой n представляет собой 0, 1 или 2 и G представляет собой защитную группу для группы карбокси,которое подвергают реакции с соединением формулы (III) в которой X представляет собой атом галогена или трифлат, группу тозилата или мезилата,m представляет собой целое число от 1 до 6 включительно,a R'2 представляет собой группу формулы NR'5R'6, в которой R'5 и R'6, которые могут быть одинаковыми или различными, каждый представляет собой защитную группу для аминогруппы или линейную или разветвленную группу C1-С 6 алкила, или R'2 представляет собой азотсодержащую гетероциклическую группу, необязательно замещенную защитной группой для аминогруппы, или R'2 представляет собой группу арила или гетероарила,с получением соединения формулы (IV) в которой R'2, G, m и n являются такими, как определено выше,которое подвергают воздействию восстанавливающего агента для оксогруппы, с получением соединения формулы (V) в которой R'2, G, m и n являются такими, как определено выше,которое подвергают реакции с мезилхлоридом, тозилхлоридом, трифторметансульфоновым ангидридом или галогенирующим реагентом с получением соединения формулы (VI) в которой R'2, G, m и n являются такими, как определено выше, а X представляет собой группу мезилата, тозилата, трифлата или атом галогена,диастереоизомеры которого отделяют тогда, когда является желательным получить соединения формулы (I) в форме единственного диастереоизомера,и которое потом подвергают реакции с соединением формулы (VII) в которой M представляет собой калий, натрий или литий, a R'1 представляет собой группу алкила или арила,с получением соединения формулы (VIII) в которой R'1, R'2, G, m и n являются такими, как определено выше,энантиомеры которого отделяют с помощью хиральной хроматографии тогда, когда является желательным получить соединения формулы (I) в форме единственного, с групп тиола, амино и карбокси которого, если это является приемлемым, снимают защиту с получением соединения формулы (I),которое тогда, когда является желательным получить аддитивную соль соединения формулы (I) с фармацевтически приемлемой кислотой, подвергают реакции с соответствующей кислотой. Данное изобретение относится также к способу получения соединений формулы (Ia), частного случая соединения формулы (I), в которой m представляет собой 3 и R2 представляет собой NR5R6,начиная от соединения формулы (II) в которой n представляет собой 0, 1 или 2, a G представляет собой защитную группу для группы карбокси,которое подвергают реакции с акролеином,в присутствии ассиметричного катализатора, такого как, например, катализатор Q или QD, в (1R) или (1S) с получением соединения формулы (IX) в конфигурации (1R) или (1S) в которой n и G являются такими, как определено выше,альдегидную группу которого восстанавливают с получением соединения формулы (X) в которой n и G являются такими, как определено выше,которое подвергают реакции с мезилхлоридом, тозилхлоридом, трифторметансульфоновым ангидридом или галогенирующим реагентом с получением соединения формулы (XI) в которой n и G являются такими, как определено выше, а X представляет собой атом галогена или группу трифлата, тозилата или мезилата,группу кетона которого восстанавливают при использовании восстанавливающего агента с получением соединения формулы (XII)NHR5R"6 (XIII),в которой R5 и R6, каждый, представляет собой защитную группу для аминогруппы или линейную или разветвленную группу С 1-С 6 алкила,с получением соединения формулы (XIV) в которой n, G, R5 и R6 являются такими, как определено выше,которое подвергают реакции с мезилхлоридом, тозилхлоридом, трифторметансульфоновым ангидридом или галогенирующим реагентом с получением соединения формулы (XV) в которой n, G, R5 и R6 являются такими, как определено выше, а X представляет собой группу мезилата, тозилата или трифлата или атом галогена,и которое потом подвергают реакции с соединением формулы (VII) в которой М представляет собой калий, натрий или литий, a R'1 представляет собой группу алкила или арила,с получением соединения формулы (XVI) в которой n, R'1, G, R5 и R6 являются такими, как определено выше,с группы тиола, амино и карбокси которого тогда, когда это является приемлемым, снимают защиту,с получением соединения формулы (Ia) в которой n, R1, R3 R5 и R6 являются такими, как определено выше,которое, если является желательным получить аддитивную соль соединения формулы (1 а) с фармацевтически приемлемой кислотой, подвергают реакции с соответствующей кислотой. Оптически чистые соединения формулы (Ia) получают либо путем асимметричного восстановления кетона формулы (XI) при использовании хирального восстанавливающего агента, либо их выделяют из смеси диастереоизомеров на более позднем этапе. Соединения в соответствии с изобретением представляют собой ингибиторы TAFIa. Как таковые они являются полезными в предотвращении или лечении тромботических явлений у пациентов с повышенным риском. Их применение является ценным в лечении и предотвращении васкулярных осложнений, в частности сердечно-сосудистых, легочных и цереброваскулярных, ассоциированных с атеротромботическими заболеваниями, атеросклерозом, диабетом, гиперлипидемией, гипертензией, хроническими венозными заболеваниями, метаболическим синдромом, ассоциированным с ожирением, или раком. Соединения в соответствии с изобретением являются особенно полезными в лечении, предотвращении и вторичном предотвращении инфаркта миокарда, стенокардии, инсультов любого происхождения (в частности, атеротромботического, кардиоэмболического или вызванного атриальной фибрилляцией), аневризма аорты или артериита нижних конечностей, венозных тромбозов (в частности, у катетеризированных раковых пациентов) и легочной эмболии. Факторы васкулярного риска и васкулярные заболевания, такие как гипертензия, ожирение, диабет,сердечное заболевание, цереброваскулярные заболевания и гиперлипидемия, и, таким образом, атеросклероз играют определенную роль в развитии деменции, такой как болезнь Альцгеймера и васкулярная деменция (Qiu С., De Ronchi D. и Fratiglioni L., The epidemiology of the dementias: an update, 2007, CurrentOpinion in Psychiatry, 20: 380-385). Соединения в соответствии с изобретением, таким образом, будут также полезными в лечении и/или предотвращении деменции, такой как болезнь Альцгеймера и васкулярная деменция.TAFIa снижает эндогенный фибринолитический потенциал. В качестве ингибиторов TAFIa соединения в соответствии с данным изобретением являются, таким образом, полезными как вспомогательные средства для острого лечения с помощью инъецируемых фибринолитических агентов, таких как рекомбинантный tPA (например, альтеплаза, тенектеплаза, ретеплаза), рекомбинантный uPA или стрептокиназа, которые используются при чрезвычайных обстоятельствах (например, при инфаркте миокарда, инсульте). Соединения в соответствии с данным изобретением усиливают активность таких инъецируемых фибринолитических агентов и, таким образом, приводят к тому, что они используются с меньшим геморрагическим и нейротоксическим риском (их доза снижается и, таким образом, уменьшаются побочные эффекты). Данное изобретение также относится к фармацевтическим композициям, которые включают соединение формулы (I) в комбинации с одним или более инертными, нетоксическими, фармацевтически приемлемыми наполнителями или носителями. Среди фармацевтических композиций в соответствии с изобретением, в частности, могут быть упомянуты такие, которые являются приемлемыми для перорального, парентерального (внутривенного,внутримышечного или подкожного), чрескожного или транскутанного, назального, ректального, подъязычного, окулярного или респираторного введения, и, в частности, таблетки или драже, подъязычные таблетки, капсулы, твердые желатиновые капсулы, суппозитории, кремы, мази, дермальные гели, инъецируемые или пригодные для питья препараты, аэрозоли, глазные капли и ушные капли. В дополнение к соединению формулы (I) фармацевтические композиции в соответствии с изобретением включают один или более наполнителей или носителей, таких как разбавители, скользящие вещества, связующие вещества, вещества для улучшения распадаемости таблеток, поглощающие вещества,красители, подсластители. В качестве примера наполнителей или носителей могут быть упомянуты в качестве разбавителей лактоза, декстроза, сахароза, маннит, сорбит, целлюлоза, глицерин,в качестве скользящих агентов двуокись кремния, тальк, стеариновая кислота и ее соли кальция и магния, полиэтиленгликоль,в качестве связующих веществ силикат алюминия, силикат магния, крахмал, желатин, трагакантовая камедь, метилцеллюлоза, карбоксиметилцеллюлоза натрия и поливинилпирролидон,-6 017867 в качестве веществ для повышения распадаемости таблеток агар, альгиновая кислота и ее натриевые соли, шипучие смеси. Процентное содержание активного ингредиента формулы (I) в фармацевтической композиции предпочтительно составляет от 5 до 50 вес.%. Полезная дозировка варьирует в зависимости от возраста, веса пациента, способа введения, природы и тяжести расстройства, а также от любого ассоциированного вида лечения и колеблется в интервале от 0,5 до 1000 мг в день в виде одного или более введений. В соответствии с одним аспектом настоящего изобретения фармацевтические композиции не содержат какого-либо активного ингредиента, отличного от соединения формулы (I). В соответствии с другим аспектом настоящего изобретения фармацевтические композиции также включают, кроме соединения формулы (I), фибринолитический агент, в частности инъецируемый фибринолитический агент, такой как рекомбинантный tPA (например, альтеплаза), рекомбинантный uPA или стрептокиназа. В этом случае композиции находятся в инъецируемой форме. Является возможным для дозы tPA варьировать в диапазоне от 0 до 100 мг. В соответствии с другим аспектом настоящего изобретения фармацевтические композиции включают кроме соединения формулы (I) антикоагулянт, такой как, например, варфарин, дабигатран этексилат, ривароксабан. Возможная доза для варфарина будет варьировать в интервале от 1 до 100 мг. В соответствии другим аспектом настоящего изобретения фармацевтические композиции включают кроме соединения формулы (I) антитромбоцитарный агент, такой как, например, аспирин, клопидогрель,прасугрель. Возможная доза для аспирина будет варьировать в интервале от 10 до 1000 мг. Возможная доза для клопидогреля будет варьировать в интервале от 10 до 1000 мг. Примеры, которые приведены ниже, иллюстрируют настоящее изобретение. Структуры соединений, описанных в примерах, были определены в соответствии с обычными спектрофотометрическими методиками (измерение в инфракрасных лучах, ядерно-магнитный резонанс, масс-спектрометрия). Сокращения.(+)-DIPCI: (+)-диизопинокамфеилхлороборан +)-диизопинокамфенил боронхлорид),DMAP: диметиламинопиридин,ДМФ: диметилформамид,ДМСО: диметилсульфоксид,экв.: молярный эквивалент,НМРА: гексаметилфосфорамид,ВЭЖХ: высокоэффективная жидкостная хроматография,ЯМР: ядерный магнитный резонанс,TAFIa: активированный ингибитор фибринолиза, способный к активации с помощью тромбина,ТГФ: тетрагидрофуран,tPA: тканевой активатор плазминогена,uPa: урокиназный активатор плазминогена или таковой урокиназного типа. Пример 1. (1R,2S)-1-(3-Аминопропил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат. Этап А. Бензил 2-оксоциклопентанкарбоксилат. В колбе на 4 л, оснащенной устройством Dean-Stark и холодильником, растворяли метил 2 оксоциклопентанкарбоксилат (380 мл; 3 моль), бензиловый спирт (342 мл; 3,3 моль; 1,1 экв.) и DMAP(18,3 г; 0,15 моль; 0,05 экв.) в 1800 мл циклогексана. Реакционную смесь перемешивали в течение 48 ч при кипении (температура смеси 90 С) и отгоняли образовавшийся метанол. После охлаждения реакционную смесь концентрировали и переносили в 1500 мл дихлорметана. Органическую фазу промывали с помощью 1N хлористо-водородной кислоты, водой, а потом насыщенным раствором хлорида натрия. Потом продукт высушивали, фильтровали и выпаривали. Полученное таким образом масло очищали с помощью фракционной дистилляции под сниженным давлением (128-130 С/410-2 мбар) с получением ожидаемого продукта. Этап В. Бензил 1-(4-трет-бутокси-4-оксобутил)-2-оксоциклопентанкарбоксилат. При комнатной температуре раствор бензил 2-оксоциклопентанкарбоксилата, полученного на этапе, описанном выше (1 г; 5,43 ммоль), в 5 мл безводного ацетона выливали при интенсивном перемешивании в суспензию сухого карбоната калия (3 г; 21,7 ммоль; 4 экв.) в 10 мл безводного ацетона. Интенсивное перемешивание реакционной смеси продолжали в течение 1 ч, а потом быстро вливали в третбутил 4-бромобутаноат (2,04 г; 9,16 ммоль; 2 экв.), растворенный в 2 мл ацетона. Реакционную смесь нагревали при кипении в течение ночи. После охлаждения реакционную смесь фильтровали, а потом переносили в 20 мл дихлорметана. Органическую фазу промывали, высушивали, фильтровали, а потом выпаривали. Сырьевой продукт очищали с помощью флэш-хроматографии на силикагеле при использовании смеси гептана/этилацетата (95/5) в качестве элюента. Этап С. 4-1-[(Бензилокси)карбонил]-2-оксоциклопентилбутановая кислота. 140 мл трифторуксусной кислоты выливали при комнатной температуре в течение 20 мин в раствор трет-бутилового эстера, полученного на этапе, описанном выше (200 г; 0,545 моль), в 700 мл безводного дихлорметана. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Примечание: прибавляли еще 10% трифторуксусной кислоты, если после перемешивания в течение ночи оставалось какое-то количество исходного материла, и продолжали перемешивание в течение 3 ч. Реакционную смесь выпаривали и переносили в толуол (3500 мл) для того, чтобы удалить максимальное количество трифторуксусной кислоты. После высушивания под вакуумным насосом полученное сырьевое масло переносили в раствор гидрокарбоната натрия (84 г/1 л; 2 экв.). Основную водную фазу потом промывали с помощью этера (3300 мл), а потом повторно подкисляли с помощью раствора 4N хлористо-водородной кислоты. Кислую водную фазу экстрагировали с помощью этилацетата (3300 мл). Объединенные органические фазы промывали, высушивали и выпаривали. Продукт использовали без очистки на следующем этапе. Этап D. Бензил 1-(3-[бензилокси)карбонил]аминопропил)-2-оксоциклопентанкарбоксилат. К раствору кислоты, полученной на этапе, описанном выше (123,7 г; 0,406 моль), в 1 л безводного толуола прибавляли по каплям в течение 5 мин при комнатной температуре триэтиламин (84,5 мл; 0,610 моль; 1,5 экв.), бензиловый спирт (75,6 мл; 0,731 моль; 1,8 экв.), а потом дифенилфосфорилазид (96,3 мл; 0,447 моль; 1,1 экв.). Реакционную смесь перемешивали при кипении в течение ночи. После охлаждения реакционную смесь выпаривали и переносили в 1 л этилацетата. Этилацетатную фазу промывали с помощью 1N раствора хлористо-водородной кислоты (3200 мл), воды, насыщенного раствора гидрокарбоната натрия, а потом насыщенного раствора хлорида натрия. Органическую фазу высушивали, фильтровали, а потом выпаривали. Сырьевой продукт очищали путем отгонки загрязняющих веществ (в основном, бензилового спирта) при температуре от 50 до 70 С при давлении 410-2 мбар. Этап Е. Бензил 1-(3-[бензилокси)карбонил]аминопропил)-2-гидроксициклопентанкарбоксилат. К раствору кетоэстера, полученного на этапе, описанном выше (98 г; 0,239 моль), в 600 мл безводного метанола прибавляли при температуре -10 С борогидрид натрия (11,4 г; 0,301 моль; 1,25 экв.) десятью порциями. Реакционную смесь выдерживали при -10 С в течение 1 ч, а потом оставляли для нагревания до комнатнорй температуры и перемешивали при комнатной температуре в течение 2 ч. После выпаривания метанола реакционную смесь переносили в 1 л этилацетата. Органическую фазу промывали с помощью 10% раствора хлорида аммония, воды, а потом насыщенного раствора хлорида натрия. Органическую фазу высушивали, фильтровали, а потом выпаривали. Этап F. Бензил 1-(3-[(бензилокси)карбонил]аминопропил)-2-[(метилсулъфонил)окси]циклопентанкарбоксилат (рацемический транс-диастереоизомер). При комнатной температуре триэтиламин (49,53 мл; 0,357 моль; 1,5 экв.) прибавляли к раствору спирта, полученного на этапе, описанном выше (98 г; 0,238 моль), в 950 мл безводного тетрагидрофурана. Реакционную смесь охлаждали до -30 С, а потом по каплям вливали в мезилхлорид (27,61 мл; 0,357 моль; 1,5 экв.), растворенный в 120 мл безводного тетрагидрофурана. Смесь оставляли для нагревания до комнатной температуры и перемешивали при комнатной температуре в течение 2 ч. После выпаривания тетрагидрофурана реакционную смесь переносили в 1 л этилацетата. Органическую фазу промывали,высушивали, фильтровали, а потом выпаривали. Сырьевой продукт очищали с помощью флэшхроматографии на силикагеле (5 кг) при использовании градиента гептана/этилацетата (от 7/3 до 6/4) в качестве элюента. Транс-диастереоизомер был первым по очередности элюирования. Этап G. Бензил 2-(ацетилтио)-1-(3-[(бензилокси)карбонил]аминопропил)циклопентанкарбоксилат(рацемический цис-диастереоизомер). Получение тиоацетата калия: очень гигроскопичный - не высушивать - получать и использовать немедленно. Суспензию 40 г тиоацетата калия в 350 мл безводного ацетонитрила нагревали при кипении в течение 30 мин и фильтровали и потом операцию проводили второй раз. К раствору (рацемический транс-диастереоизомер) мезилата, полученного на этапе, описанном выше (25 г; 0,051 моль), в 800 мл безводного ацетонитрила прибавляли предварительно промытый тиоацетат калия (45 г не отфильтрованного путем аспирации - теоретически 29,2 г; 0,255 моль; 5 экв.) и 18 С 6 краун этер (13,5 г; 0,051 моль; 1 экв.), а потом реакционную смесь нагревали при кипении в течение 20 ч. После охлаждения реакционную смесь фильтровали и потом выпаривали растворитель. Сырьевой продукт очищали с помощью флэш-хроматографии на силикагеле (2 кг) при использовании градиента гептана/этилацетата (от 8/2 до 5/5) в качестве элюента. Примечание: эта первая колонка позволяет осуществлять предварительную очистку и восстановление непрореагировавшего исходного материала. Порцию, которая содержит ожидаемый продукт (6,5 г), очищали с помощью флэш-хроматографии на силикагеле (200 г) при использовании градиента дихлорметана/этилацетата (от 99/1 до 95/5) в качестве элюента. Этап Н. Бензил (1R,2S)-2-(ацетилтио)-1-(3-[(бензилокси)карбонил]аминопропил)циклопентанкарбоксилат. Энантиомеры рацемического цис-соединения, полученного на этапе, описанном выше, разделяли с помошью препаративной хиральной хроматографии на колонке CHIRALPAK AD-H 5 мкм при использовании смеси CO2/EtOH (80/20) в качестве мобильной фазы. Длина волны для определения 230 нм.(1R,2S)-Энантиомер = энантиомер 2, который является вторым по очередности элюирования. Этап I. Бензил (1R,2S)-1-(3-[(бензилокси)карбонил]аминопропил)-2-меркаптоциклопентанкарбоксилат. К раствору соединения, полученного на этапе, описанном выше (8,16 г; 0,0174 моль), в 150 мл безводного и дегазированного диоксана прибавляли по каплям при 10 С 1N раствор гидроокиси натрия (35 мл; 0,0348 моль; 2 экв.). Смесь нагревали при 60 С в течение 30 мин. После охлаждения реакционную смесь нейтрализовали путем прибавления 35 мл 1N хлористо-водородной кислоты и потом подвергали лиофилизации. Этап J. (1R,2S)-1-(3-Аминопропил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат. 600 мл аммония конденсировали в реакторе на 2 л и при -78 С десятью порциями прибавляли натрий (10 г; 0,435 моль; 25 экв.). Реакционную смесь выдерживали при -78 С в течение 45 мин. Потом прибавляли по каплям в течение 30 мин раствор соединения, полученного на этапе, описанном выше, в предварительно дегазированную смесь тетрагидрофурана/метанола (140 мл/12 мл). Поддерживали перемешивание при -78 С в течение 1 ч, смесь оставляли для нагревания до комнатной температуры и перемешивали при комнатной температуре. После удаления аммония к реакционной смеси прибавляли раствор хлорида аммония (23,3 г в 110 мл воды) в течение 30 мин, выпаривали тетрагидрофуран, а потом осуществляли подкисление при использовании 6 М раствора хлористо-водородной кислоты (15 мл). Проводили фильтрацию, а потом лиофилизацию. Сырьевой продукт обессоливали с помощью препаративной ВЭЖХ на колонке Kromasil при использовании градиента воды/ацетонитрила/трифторуксусной кислоты в качестве мобильной фазы(100/0/0,1 в течение 10 мин, потом от 100/0/0,1 до 70/30/0,1 в течение 20 мин, после чего осуществляли изократическое разделение при 70/30/0,1). Фракции, которые содержали ожидаемый продукт, а также соответствующий димер (дисульфидная связь), подвергали лиофилизации. К раствору указанного выше лиофилизата (4,3 г) в 100 мл уксусной кислоты прибавляли 4,5 г цинка(10 экв.) и потом при интенсивном перемешивании реакционную смесь нагревали при 60 С до исчезновения димера (приблизительно 6 ч). После охлаждения реакционную смесь фильтровали, фильтрат разводили с помощью 400 мл воды и подвергали лиофилизации. Сырьевой продукт очищали с помощью препаративной ВЭЖХ на колонке Kromasil (1 кг - 600 мм 60 мм - 70 мл/мин) при использовании смеси воды/ацетонитрила/трифторуксусной кислоты (85/15/0,1) в качестве мобильной фазы. Фракции, которые содержат ожидаемый продукт, подвергали лиофилизации. Оптическое вращение: растворитель: метанол, C=1, T=21C, L=589 нм, []D = +32,46. CAD/MS/MS спектр [М+Н]+ = 204,1 в соответствии с ожидаемой структурой. Пример 2. (1R,2S)-2-Ацетилтио-1-(3-аминопропил)циклопентанкарбоновая кислота гидрохлорид. К раствору соединения этапа Н примера 1 (2,40 г; 4,72 ммоль) в 20 мл безводного дихлорметана прибавляли по каплям при комнатной температуре 4 М раствор HCl/диоксана (35,5 мл; 141,6 ммоль; 30 экв.). Реакционную смесь оставляли при комнатной температуре при перемешивании до тех пор, пока с двух групп не была снята защита (проверяли с помощью ЖХ/МС - приблизительная длительность: в течение ночи) и потом выпаривали растворители. Полученный продукт очищали на колонке BIOGEL P2 при использовании смеси воды/ацетонитрила/1N хлористо-водородная кислота (1000/1000/2) в качестве мобильной фазы. Фракции, которые содержат ожидаемый продукт, подвергали лиофилизации. Пример 3. (1R,2S)-1-(3-Аминопропил)-2-меркаптоциклопентанкарбоновая кислота гидрохлорид. Раствор соединения примера 2 (4,51 г; 16 ммоль) в 148 мл 4 М раствора HCl нагревали в течение ночи при 45 С. После охлаждения реакционную смесь разводили 150 мл воды и потом подвергали лиофилизации. Сырьевой продукт очищали с помощью препаративной ВЭЖХ на колонке Kromasil (1 кг - 600 мм 60 мм - 70 мл/мин) при использовании смеси воды/ацетонитрила (95/5) в качестве мобильной фазы. Фракции, которые содержат ожидаемый продукт, подвергали лиофилизации.CAD/MS/MS спектр [М+Н]+ = 204,1 в соответствии с ожидаемой структурой. Пример 3 а. (1R,28)-1-(3-Аминопропил)-2-меркаптоциклопентанкарбоновая кислота. Этап А. ди-трет-Бутил адипат. Адипоилхлорид (629,8 г; 3,44 моль) выливали по каплям при комнатной температуре в раствор трет-бутанола (3,27 л, 34,4 моль - 10 экв.) и пиридина (800 мл; 10,32 моль - 3 экв.) в 3 л безводного толуола. Реакционную смесь нагревали при 70 при перемешивании в течение ночи. После охлаждения преципитат хлорида пиридиния отфильтровывали и промывали с помощью толуола. Толуольную фазу промывали с помощью 1N раствора хлористо-водородной кислоты (4500 мл),10% раствора карбоната натрия (1500 мл), воды (1500 мл), а потом насыщенным раствором хлорида натрия (1500 мл). Органическую фазу высушивали над сульфатом натрия, фильтровали, а потом выпаривали. Полученное сырьевое масло очищали с помощью фракционной дистилляции под сниженным давлением (100 С/210-2 мм Hg). Этап В. трет-Бутил 2-оксоциклопентанкарбоксилат. трет-Бутанол (5,66 мл; 0,06 моль - 0,05 экв.), а также 14 г ди-трет-бутил адипата выливали при комнатной температуре в суспензию NaH (108,54 г 60% вещества; 2,71 моль - 2,15 экв.) в 1,1 л безводного толуола. Реакционную смесь нагревали при кипении. При этой температуре по каплям вливали в реакционную смесь раствор ди-трет-бутил адипата (324,6 г; 1,26 моль), растворенного в 500 мл безводного толуола, контролируя при этом выделение газа. Приблизительная длительность прибавления: 3 ч. Примечание: существенное комкование реакционной смеси будет происходить в случае прибавления ди-трет-бутил адипата, однако это будет развиваться в направлении образования суспензии. После окончания прибавления реакционную смесь выдерживали при кипении в течение 5 ч. После охлаждения реакционную смесь подвергали гидролизу при 0 С с использованием 10% раствора уксусной кислоты (1 л). Органическую фазу отделяли и водную фазу экстрагировали с помощью толуола (300 мл). Объединенные толуольные фазы промывали водой (2300 мл), а потом насыщенным раствором хлорида натрия (300 мл). Органическую фазу высушивали над сульфатом натрия, фильтровали, а потом выпаривали. Оранжевое масло очищали с помощью фракционной дистилляции под сниженным давлением (6570 С/110-1 мм Hg). Этап С. трет-Бутил (1S)-2-оксо-1-(3-оксопропил)циклопентанкарбоксилат. Раствор акролеина (4,19 г; 74,83 ммоль - 2,5 экв.) в 10 мл безводного дихлорметана выливали в течение 30 мин при -25 С в атмосфере аргона и со слабым перемешиванием, в раствор трет-бутил 2 оксоциклопентанкарбоксилата (5,51 г; 29,93 ммоль) и катализатора Q (1,46 г; 2,99 ммоль - 0,1 экв.) в 60 мл безводного дихлорметана В конце прибавления реакционную смесь перемешивали в течение 4 ч при этой температуре, а потом в течение ночи при -5 С. Фильтрат выпаривали с получением бесцветного масла. Этап D. трет-Бутил (1S)-1-(3-гидроксипропил)-2-оксоциклопентанкарбоксилат. К раствору альдегида кетоэстера, полученного на этапе, описанном выше (6,5 г; 26,9 ммоль), в 80 мл безводного тетрагидрофурана прибавляли триацетоксиборогидрид натрия (6,84 г; 32,3 ммоль - 1,2 экв.). Реакционную смесь нагревали при кипении, при перемешивании, в течение 5 ч 30 мин. После возвращения к комнатной температуре реакционную смесь выпаривали. Остаток переносили в 100 млCH2Cl2 и промывали с помощью насыщенного раствора бикарбоната натрия (220 мл). Основную водную фазу экстрагировали с помощью CH2Cl2. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, высушивали над сульфатом натрия, а потом выпаривали. Сырьевой продукт очищали с помощью флэш-хроматографии на силикагеле при использовании смесиCH2Cl2/изопропанола (95/5) в качестве элюента. Этап Е. трет-Бутил (1S)-1-(3-бромопропил)-2-оксоциклопентанкарбоксилат. К раствору спирта, полученного на этапе, описанном выше (5,4 г; 22,12 ммоль), и трифенилфосфина (6,7 г; 25,44 ммоль - 1,15 экв.) в 35 мл безводного дихлорметана прибавляли N-бромсукцинимид (4,53 г; 25,44 ммоль - 1,15 экв.) частями, в это время температуру поддерживали на уровне 5 С. После пере- 10017867 мешивания в течение 1 ч при комнатной температуре реакционную смесь выпаривали. Остаток переносили в 100 мл изопропилового этера и растирали в порошок с получением кристаллизации окиси трифенилфосфина. Твердое вещество отфильтровывали и промывали. Фильтрат концентрировали, потом переносили в пентан, а потом вновь фильтровали. Фильтрат, содержащий пентан, выпаривали. Сырьевой продукт фильтровали с помощью флэш-хроматографии на силикагеле при использовании смеси гептана/ этилацетата (8/2) в качестве элюента. Этап F. трет-Бутил (1S,2R)-1-(3-бромопропил)-2-гидроксициклопентанкарбоксилат. При комнатной температуре при перемешивании коммерчески доступный 1,8 М раствор (+)DIPCl в гексане (320 мл; 0,577 моль - 1,5 экв.) выливали на кетоэстер (117,5 г; 0,385 моль), полученный в ходе этапа, описанного выше. Реакционную смесь перемешивали при 55 С в течение ночи при комнатной температуре. К реакционной смеси прибавляли 950 мл этилового этера и потом при перемешивании, при температуре 0 С ацетальдегид (35,9 мл; 0,635 моль - 9,65 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. После охлаждения реакционной смеси до 10 С прибавляли 6N раствор гидроокиси натрия (480 мл; 2,88 моль - 7,5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Органическую фазу отделяли, промывали водой (3300 мл), 10% раствором лимонной кислоты (2300 мл), а потом насыщенным раствором хлорида натрия (3150 мл). Органическую фазу высушивали над сульфатом натрия, а потом выпаривали. Сырьевой продукт очищали с помощью флэш-хроматографии на силикагеле при использовании градиента гептана/этилацетата (от 95/5 до 85/15) в качестве элюента. Этап G. трет-Бутил (1S,2R)-l-3-[бис(трет-бутоксикарбонил)амино]пропилгидроксициклопентанкарбоксилат. К раствору бронированного спирта, полученного на этапе, описанном выше (11,8 г; 36,06 ммоль), в 110 мл безводного ДМФ прибавляли при комнатной температуре ди трет-бутил иминодикарбоксилат(7,83 г; 36,06 ммоль; 1 экв.), а потом карбонат цезия (11,75 г; 36,06 ммоль; 1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь фильтровали. После концентрирования фильтрата маслянистый остаток переносили в 300 мл этилацетата. Этилацетатную фазу промывали водой (3100 мл), а потом насыщенным раствором хлорида натрия (100 мл). Органическую фазу высушивали над сульфатом натрия, фильтровали и выпаривали. Сырьевой продукт очищали с помощью флэш-хроматографии на силикагеле при использовании градиента гептана/этилацетата (от 9/1 до 85/15) в качестве элюента. Этап Н. трет-Бутил (1S,2R)-1-3-[бис(трет-бутоксикарбонил)амино]пропил-2-[(метилсульфонил) окси]циклопентанкарбоксилат. Триэтиламин (4,11 мл; 29,55 ммоль; 1,5 экв.) прибавляли при комнатной температуре к раствору спирта, полученного на этапе, описанном выше (8,70 г; 19,7 ммоль), в 130 мл безводного тетрагидрофурана. Реакционную смесь охлаждали до -30 С и потом по каплям выливали в мезилхлорид (2,29 мл; 29,55 ммоль; 1,5 экв.), растворенный в 10 мл безводного тетрагидрофурана. Смесь оставляли для нагревания до комнатной температуры и перемешивали при комнатной температуре в течение 2 ч. После выпаривания тетрагидрофурана реакционную смесь переносили в 200 мл этилацетата. Этилацетатную фазу промывали водой (220 мл), а потом насыщенным раствором хлорида натрия (120 мл). Органическую фазу высушивали над сульфатом натрия, фильтровали, а потом выпаривали. Сырьевой продукт очищали с помощью флэш-хроматографии на силикагеле при использовании градиента гептана/этилацетата (от 85/15 до 8/2) в качестве элюента. Этап I. трет-Бутил (1R,2S)-2-(ацетилтио)-3-[бис(трет-бутоксикарбонил)амино]пропил)циклопентанкарбоксилат. Ожидаемый продукт получали, начиная с соединения, полученного на этапе, описанном выше, в соответствии с процедурой этапа G примера 1. Этап J. (1R,2S)-1-(3-Аминопропил)-2-меркаптоциклопентанкарбоновая кислота гидрохлорид. Снятие защиты с кислотной и аминной групп. К раствору соединения, полученного на этапе, описанном выше (8,80 г; 17,6 ммоль), в 100 мл безводного CH2Cl2 прибавляли по каплям при комнатной температуре 4 М раствор HCl/диоксана (132 мл; 0,53 моль; 30 экв.). Реакционную смесь оставляли при комнатной температуре при перемешивании до тех пор, пока не снимали защиту с двух групп (проверяли с помощью ЖХ/МС - приблизительная длительность: в течение ночи), и потом выпаривали растворители. Полученный продукт использовали без очистки на следующем этапе. Снятие защиты с тиола. Продукт выпаривания переносили в 200 мл 4 М водного раствора HCl, предварительно подвергнутого дегазации с помощью аргона, и потом нагревали при 45 С в течение ночи. После охлаждения реакционную смесь разводили 100 мл дегазированной воды и потом подвергали лиофилизации. Этап K. (1R,2S)-1-(3-Аминопропил)-2-меркаптоциклопентанкарбоновая кислота. Соединение этапа, описанного выше (10 г, 41,5 ммоль), растворяли в 80 мл воды и перемешивали. Раствор становился прозрачным и имел бледно-желтый цвет (рН приблизительно 1,2). При перемешивании прибавляли приблизительно 11 мл насыщенного раствора бикарбоната натрия. При рН 4 наблюдали образование первого осадка, который отфильтровывали. Потом фильтрат делали более основным, вплоть до значения рН 5,5, отфильтровывали преципитаты, если они образовывались. Фильтрат выпаривали под сниженным давлением (Тбани=55 С). Когда оставалось приблизительно 10 мл раствора, вновь осуществляли фильтрацию. Преципитаты эквивалентного качества собирали и высушивали в вакууме с получением указанного в заглавии соединения. ЯМР спектроскопия.(3 Н, m); 1,32 (1 Н, m). Пример 4. (1S,2R)-1-(3-Аминопропил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах I и J примера 1,начиная с другого энантиомера, полученного на этапе Н примера 1, то есть бензил(1S,2R)-2-(ацетилтио)1-(3-[(бензилокси)карбонил]аминопропил)циклопентанкарбоксилата.CAD/MC/MC спектр [М+Н]+ = 204,1 в соответствии с ожидаемой структурой. Пример 5. (1S,2R)-2-Ацетилтио-1-(3-аминопропил)циклопентанкарбоновая кислота гидрохлорид. Ожидаемый продукт получали в соответствии с процедурой, описанной для примера 2, начиная с другого энантиомера, полученного на этапе H примера 1, то есть бензил(1S,2R)-2-(ацетилтио)-1-(3[(бензилокси)карбонил]аминопропил)циклопентанкарбоксилата. Пример 6. 1-(3-Аминопропил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах I и J примера 1,начиная с соединения, полученного на этапе G примера 1.CAD/MC/MC спектр [М+Н]+ = 204,1 в соответствии с ожидаемой структурой. Пример 7. 1-(4-Аминобутил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический транс-диастереоизомер). Этап А. Бензил 1-(4-бромбутил)-2-оксоциклопентанкарбоксилат. В трехгорлой колбе на 500 мл, оснащенной магнитной мешалкой и холодильником, в атмосфере инертного азота суспендировали 95% гидрид натрия (2,2 г; 87,1 ммоль; 1,25 экв.) в смеси безводного ТГФ (115 мл) и НМРА (14,6 мл). Раствор бензил 2-оксоциклопентанкарбоксилата (15,3 г; 70,3 ммоль) в 45 мл безводного ТГФ прибавляли по каплям так, чтобы поддерживать температуру на уровне 45 С. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Прибавляли 1,4-дибромбутан(12,6 мл; 105,4 ммоль; 1,5 экв.) к прозрачному бледно-желтому раствору. Вновь перемешивали реакционную смесь в течение 14 ч при кипении. После возвращения к комнатной температуре прибавляли 80 мл насыщенного водного раствора хлорида аммония. Смесь выливали в 500 мл этера и органическую фазу пять раз промывали с помощью 100 мл воды, а потом с помощью 100 мл насыщенного водного раствора хлорида натрия. Органическую фазу высушивали над MgSO4, фильтровали и концентрировали. Продукт очищали на силикагеле при использовании смеси циклогексана/этилацетата (градиент от 95/5 до 80/20) в качестве элюента с получением ожидаемого продукта в виде желтого масла. Этап В. Бензил 1-(4-бромобутил)-2-гидроксициклопентанкарбоксилат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе Е примера 1, начиная от соединения, полученного на этапе, описанном выше. Этап С. Бензил 1-(4-азидобутил)-2-гидроксициклопентанкарбоксилат. В колбе в атмосфере инертного азота суспендировали соединение, полученное на этапе, описанном выше (4,41 г; 16,6 ммоль), азид натрия (5,5 г; 84,6 ммоль; 5 экв.) и йодид натрия (0,2 г) в 60 мл этанола. Реакционную смесь перемешивали при кипении в течение 24 ч и потом концентрировали до осушения. Маслянистый остаток растворяли в 150 мл этера и 80 мл воды. Органическую фазу промывали с помощью 80 мл воды и 40 мл насыщенного водного раствора хлорида натрия. Органическую фазу высушивали над MgSO4, фильтровали и концентрировали. Образовавшуюся смесь (Rac)-транс- и (Rac)-цис-азида использовали как таковую в следующей реакции. Этап D. Бензил 1-(4-аминобутил)-2-гидроксициклопентанкарбоксилат. В колбе растворяли смесь (Rac)-Транс- и (Rac)-цис-азида (20,64 г; 62,3 ммоль) и трифенилфосфина(24,4 г; 93 ммоль; 1,5 экв.) в 500 мл ТГФ. Раствор перемешивали в течение 1 ч при 50 С. Прибавляли воду (10 мл; 556 ммоль; 9 экв.). Реакционную смесь перемешивали в течение 4 ч при 50 С и потом концентрировали до осушения. Маслянистый остаток растворяли в 250 мл этера и органическую фазу дважды экстрагировали с помощью 120 мл 1N водного раствора хлористо-водородной кислоты. Водную фазу промывали с помощью 100 мл этера. Значение рН водной фазы доводили до 12-13 путем прибавления твердой гидроокиси калия. Водную фазу экстрагировали трижды с помощью 250 мл этилацетата. Объединенные органические фазы промывали с помощью 100 мл воды, а потом с помощью 100 мл насыщенного водного раствора хлорида натрия. Органическую фазу высушивали над MgSO4, фильтровали и концентрировали. Ожидаемый продукт, полученный в форме цис/транс-смеси, использовали как таковой в следующей реакции. Этап Е. Бензил 1-(4-[бензилокси)карбонил]аминобутил)-2-гидроксициклопентанкарбоксилат. В колбе растворяли продукт, полученный на этапе, описанном выше (62,3 ммоль), в 160 мл диоксана. Прибавляли водный раствор карбоната натрия (13,16 г; 124 ммоль; 2 экв.) в 160 мл воды. При 0 С медленно прибавляли раствор бензилхлорформиата (12 мл; 84,4 ммоль; 1,35 экв.) в 30 мл диоксана. Реакционную смесь перемешивали при комнатной температуре в течение 12 ч и подвергали частичной концентрации. Водную фазу дважды экстрагировали с помощью 250 мл этера. Объединенные органические фазы промывали, высушивали, фильтровали и концентрировали. Продукт очищали на силикагеле (петролейный этер/этилацетат:градиент от 9/1 до 2/8) с получением ожидаемого продукта в форме желтого масла. Этап F. Бензил 1-(4-[бензилокси)карбонил]аминобутил)-2-[(метилсульфонил)оксициклопентанкарбоксилат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе F примера 1, начиная от соединения, полученного на этапе, описанном выше. Цис-диастереоизомер был первым по очередности элюирования. Этап G. Бензил 2-(ацетилтио)-1-(4-[бензилокси)карбонил]аминобутил)циклопентанкарбоксилат(рацемический транс-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе G примера 1, начиная с цис-соединения, полученного на этапе, описанном выше. Этап Н. 1-(4-Аминобутил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический транс-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах I и J примера 1,начиная от соединения, полученного на этапе, описанном выше. Пример 8. 1-(4-Аминобутил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Этап А. Бензил 1-(4-[бензилокси)карбонил]аминобутил)-2-[(метилсулъфонил)оксициклопентанкарбоксилат (рацемический транс-диастереоизомер). Ожидаемый продукт представляет собой другой диастереоизомер, полученный на этапе F примера 7 (транс-диастереоизомер). Этап В. 1-(4-Аминобутил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах G, I и J примера 1,начиная от соединения, полученного на этапе, описанном выше.(цис-диастереоизомер, энантиомер 1). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах G и Н примера 1,начиная от соединения, полученного на этапе А примера 8. Энантиомер 1 был первым по очередности элюирования. Этап В. 1-(4-Аминобутил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (цисдиастереоизомер, энантиомер 1). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах I и J примера 1,начиная от соединения, полученного на этапе, описанном выше.(цис-диастереоизомер, энантиомер 2). Ожидаемый продукт представлял собой другой энантиомер, полученный на этапе А примера 9. Этап В. 1-(4-Аминобутил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (цисдиастереоизомер, энантиомер 2). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах I и J примера 1,начиная от соединения, полученного на этапе, описанном выше.(рацемический цис-диастереоизомер). Этап А. Бензил 1-(4-бромометил)бензил]-2-оксоциклопентанкарбоксилат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе А примера 7, начиная с бензил 2-окс-циклопентанкарбоксилата и 1,4-дибромометилбензола. Этап В. Бензил 1-(4-бромометил)бензил]-2-гидроксициклопентанкарбоксилат (рацемический трансдиастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе Е примера 1, после чего осуществляли диастереоизомерное разделение полученного таким образом соединения с помощью флэш-хроматографии на силикагеле при использовании смеси циклогексана/этилацетата (градиент от 90/10 до 65/35) в качестве элюента. Транс-диастереоизомер был первым по очередности элюирования. Этап С. 1-[(4-Аминометил)бензил]-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах С-Н примера 7,начиная от соединения, полученного на этапе, описанном выше.(рацемический транс-диастереоизомер). Этап А. Бензил 4-(1-[(бензилокси)карбонил]-2-гидроксициклопентилметил)-1-пиперидинкарбоксилат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах А и В примера 7,начиная от бензил 2-оксоциклопентанкарбоксилата и бензил 4-бромометил-1-пиперидинкарбоксилата. Цис-диастереоизомер являлся вторым по очередности элюирования. Этап В. Бензил 4-(1-[(бензилокси)карбонил]-2-(метилсульфонил)окси]циклопентилметил)-1 пиперидинкарбоксилат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе F примера 1, начиная от соединения, полученного на этапе, описанном выше. Этап С. 2-Меркапто-1-(пиперидин-4-илметил)циклопентанкарбоновая кислота трифторацетат (рацемический транс-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах G, I и J примера 1,начиная от соединения, полученного на этапе, описанном выше.(рацемический цис-диастереоизомер). Этап А. Бензил 4-(1-[(бензилокси)карбонил]-2-[(метилсульфонил)окси]циклопентилметил)-1 пиперидинкарбоксилат (рацемический транс-диастереоизомер). Ожидаемый продукт представлял собой другой диастереоизомер, полученный на этапе В примера 12 (транс-диастереоизомер). Этап В. 2-Меркапто-1-(пиперидин-4-илметил)циклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах G, I и J примера 1,начиная от соединения, полученного на этапе, описанном выше. Пример 14. 1-(5-Аминопентил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический транс-диастереоизомер). Этап А. Бензил 1-(5-[бензилокси)карбонил]аминопентил)-2-гидроксициклопентанкарбоксилат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах А-Е примера 7,начиная от бензил 2-оксоциклопентанкарбоксилата и 1,5-дибромпентана. Этап В. Бензил 1-(5-[бензилокси)карбонил]аминопентил)-2-[(метилсульфонил)оксициклопентанкарбоксилат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе F примера 1, начиная от соединения, полученного на этапе, описанном выше. Цис-диастереоизомер являлся вторым по очередности элюирования. Этап С. 1-(5-Аминопентил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический транс-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах G, I и J примера 1,начиная от соединения, полученного на этапе, описанном выше.CAD/MC/MC спектр [М+Н]+ = 232,1 в соответствии с ожидаемой структурой. Пример 15. 1-(5-Аминопентил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Этап А. Бензил 1-(5-[бензилокси)карбонил]аминопентил)-2-[(метилсульфонил)оксициклопентанкарбоксилат (рацемический транс-диастереоизомер). Ожидаемый продукт представлял собой другой диастереоизомер, полученный на этапе В примера 14 (транс-диастереоизомер). Этап В. 1-(5-Аминопентил)-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах G, I и J примера 1,начиная от соединения, полученного на этапе, описанном выше.(рацемический цис-диастереоизомер). Этап А. Бензил 4-(2-1-[(бензилокси)карбонил]-2-гидроксициклопентилэтил)-1-пиперидинкарбоксилат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах А и В примера 7,начиная от бензил 2-оксоциклопентанкарбоксилата и бензил 4-(2-бромоэтил)-1-пиперидинкарбоксилата. Транс-диастереоизомер был первым по очередности элюирования. Этап В. Бензил 4-(2-1-[(бензилокси)карбонил]-2-[(метилсульфонил)окси]циклопентилэтил)-1 пиперидинкарбоксилат (рацемический транс-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе F примера 1, начи- 15017867 ная от соединения, полученного на этапе, описанном выше. Этап С. 2-Меркапто-1-(2-пиперидин-4-илэтил)циклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах G, I и J примера 1,начиная от соединения, полученного на этапе, описанном выше.CAD/MC/MC спектр [М+Н]+ = 258 в соответствии с ожидаемой структурой. Пример 17. 2-Меркапто-1-(пиридин-3-илметил)циклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Этап А. Бензил 2-оксо-1-(3-пиридилметил)циклопентанкарбоксилат. В трехгорлой колбе на 1 л, оснащенной магнитной мешалкой и холодильником, в атмосфере инертного азота, суспендировали гидрид калия 32% (25 г; 200 ммоль; 1,1 экв.) в безводном ТГФ (200 мл). Реакционную смесь охлаждали до -78 С, а потом по каплям прибавляли бензил 2 оксоциклопентанкарбоксилат (39,7 г; 182 ммоль), поддерживая температуру на уровне, ниже -78 С. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Прибавляли раствор основания 3-(хлорметил)пиридина (38,7 г; 226 ммоль; 1,24 экв.) в 100 мл безводного ТГФ к прозрачному желтому раствору. Реакционную смесь перемешивали при кипении в течение 12 ч. После нагревания до комнатной температуры реакционную смесь выпаривали до осушения и переносили в 80 мл воды и 500 мл этилацетата. Водную фазу трижды экстрагировали с помощью 50 мл этилацетата. Объединенные органические фазы промывали насыщенным раствором хлорида натрия, очищали на силикагеле при использовании смеси дихлорметана/этанола (градиент от 98/2 до 95/5) в качестве элюента с получением ожидаемого продукта в форме желтого масла. Этап В. Бензил 2-гидрокси-1-(3-пиридилметил)циклопентанкарбоксилат (рацемический трансдиастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе Е примера 1. Продукт очищали на силикагеле при использовании смеси дихлорметана/этилацетата (градиент от 7/3 до 5/5) в качестве элюента с получением ожидаемого продукта в виде бесцветного масла. Транс-диастереоизомер был вторым по очередности элюирования. Этап С. 2-Меркапто-1-(пиридин-3-илметил)циклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах F, G, I и J примера 1, начиная от соединения, полученного на этапе, описанном выше.CAD/MC/MC спектр [М+Н]+ = 238,1 в соответствии с ожидаемой структурой. Пример 18. 2-Меркапто-1-(2-пиридин-2-илэтил)циклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Этап А. Бензил 2-оксо-1-(2-пиридин-2-илэтил)циклопентанкарбоксилат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе А примера 17, начиная от бензил 2-оксоциклопентанкарбоксилата и 2-пиридин-2-илэтанилметансульфоната. Этап В. Бензил 2-гидрокси-1-(2-пиридин-2-илэтил)циклопентанкарбоксилат (рацемический трансдиастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе В примера 17. Транс-диастереоизомер был вторым по очередности элюирования. Этап С. 2-Меркапто-1-(2-пиридин-2-илэтил)циклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах F, G, I и J примера 1, начиная от соединения, полученного на этапе, описанном выше.CAD/MC/MC спектр [М+Н]+ = 252,1 в соответствии с ожидаемой структурой. Пример 19. 1-[(6-Аминопиридин-3-илметил)]-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Этап А. трет-Бутил 2-оксоциклопентанкарбоксилат. 1) ди-трет-Бутил адипат. Адипоилхлорид (629,8 г; 3,44 моль) вливали по каплям при комнатной температуре в раствор третбутанола (3,27 л, 34,4 моль - 10 экв.) и пиридина (800 мл; 10,32 моль - 3 экв.) в 3 л безводного толуола. Реакционную смесь нагревали до 70 С при перемешивании в течение ночи. После охлаждения преципитат хлорида пиридиния отфильтровывали и промывали с помощью толуола. Толуольную фазу промывали с помощью 1N раствора хлористо-водородной кислоты (4500 мл),10% раствора карбоната натрия (1500 мл), воды (1500 мл), а потом насыщенным раствором хлорида натрия (1500 мл). Органическую фазу высушивали, фильтровали, а потом выпаривали. Полученное сырьевое масло очищали с помощью фракционной дистилляции под сниженным давлением (100 С/210-2 мм Hg). 2) трет-Бутил 2-оксоциклопентанкарбоксилат. трет-Бутанол (5,66 мл; 0,06 моль - 0,05 экв.), а также 14 г ди-трет-бутил адипата выливали при комнатной температуре в суспензию NaH (108,54 г 60% вещества; 2,71 моль - 2,15 экв.) в 1,1 л безводного толуола. Реакционную смесь нагревали при кипении. При этой температуре раствор ди-трет-бутил адипата (324,6 г; 1,26 моль), растворенного в 500 мл безводного толуола, выливали по каплям при контроле выделения газа. Приблизительная длительность прибавления: 3 ч. Примечание: существенное комкование реакционной смеси будет наблюдаться в случае прибавления ди-трет-бутил адипата, однако это будет развиваться в направлении образования суспензии. После окончания прибавления реакционную смесь выдерживали при кипении в течение 5 ч. После охлаждения реакционную смесь подвергали гидролизу при 0 С с использованием 10% раствора уксусной кислоты (1 л). Органическую фазу отделяли и водную фазу экстрагировали с помощью толуола (300 мл). Объединенные толуольные фазы промывали водой (2300 мл), а потом насыщенным раствором хлорида натрия (300 мл). Органическую фазу высушивали над сульфатом натрия, фильтровали, а потом выпаривали. Оранжевое масло очищали с помощью фракционной дистилляции под сниженным давлением (6570 С/110-1 мм Hg). Этап В. трет-Бутил 1-(6-[(трет-бутоксикарбонил)амино]-3-пиридилметил)-2-оксоциклопентанкарбоксилат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе А примера 17, начиная от трет-бутил 2-оксоциклопентанкарбоксилата и трет-бутил [5-(хлорметил)пиридин-2 ил]карбамата. Этап С. трет-Бутил 1-(6-[(трет-бутоксикарбонил)амино]-3-пиридилметил)-2-гидроксициклопентанкарбоксилат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе В примера 17. Продукт очищали на силикагеле при использовании смеси дихлорметана/этанола (градиент от 98/2 до 95/5) с получением ожидаемого продукта в виде бесцветного масла. Транс-диастереоизомер был вторым по очередности элюирования. Этап D. трет-Бутил 1-(6-[(трет-бутоксикарбонил)амино]-3-пиридилметил)-2-[(метилсулъфонил) окси]циклопентанкарбоксилат (рацемический транс-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе F примера 1, начиная от соединения, полученного на этапе, описанном выше. Этап Е. 1-[(6-Аминопиридин-3-илметил)]-2-(ацетилтио)циклопентанкарбоновая кислота (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе G примера 1, начиная от соединения, полученного на этапе, описанном выше. Этап F. 1-[(6-Аминопиридин-3-илметил)]-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Снятие защиты с кислотной и аминной групп. К раствору соединения, полученного на этапе, описанном выше (2,40 г; 4,72 ммоль), в 20 мл безводного CH2Cl2 прибавляли по каплям при комнатной температуре 4 М раствор HCl/диоксана (35,5 мл; 141,6 ммоль; 30 экв.). Реакционную смесь оставляли при комнатной температуре при перемешивании до тех пор, пока не была снята защита с двух групп (кислотной и аминной (проверяли с помощью ЖХ/МС приблизительная длительность: в течение ночи), и потом выпаривали растворитель. Снятие защиты с тиола. Продукт выпаривания переносили в 4 М водный раствор HCl и потом нагревали при 45 С в течение ночи. Реакционную смесь разводили водой и потом подвергали лиофилизации. Продукт очищали на BIOGEL P2 при использовании смеси воды/ацетонитрила (1/1) в качестве элюента. Фракции, которые содержат ожидаемый продукт, подвергали лиофилизации.CAD/MC/MC спектр [М+Н]+ = 253,1 в соответствии с ожидаемой структурой. Пример 20. (1R,2S,1'R,2'S)-2,2'-дисульфандиилбис[1-(3-аминопропил)циклопентанкарбоновая кислота] дигидрохлорид. Раствор соединения, описанного в этапе J примера 1 (0,44 г; 1,39 ммоль), в 3 мл 1N гидроокиси натрия нагревали при 65 С в течение ночи. После охлаждения реакционную смесь нейтрализовали с помощью 3 мл 1N раствора хлористо-водородной кислоты. Продукт очищали на BIOGEL Р 2 при использовании смеси воды/ацетонитрила (1/1) в качестве элюента. Фракции, которые содержат ожидаемый продукт, подвергали лиофилизации. Пример 21. (1R,2S)-1-[3-(Диметиламино)пропил]-2-меркаптоциклопентанкарбоновая кислота трифторацетат. Этап A. (1R,2S,1'R,2'S)-2,2'-Дисульфандиил-бис[1-(3-(диметиламино)пропил)циклопентанкарбоновая кислота. К раствору соединения, полученного в примере 20 (0,57 г; 0,70 ммоль), в 4,5 мл муравьиной кислоты прибавляли по каплям при комнатной температуре 0,2 мл 37% водного раствора формальдегида. Реакционную смесь нагревали при кипении при перемешивании в течение 1 ч. После охлаждения реакционную смесь разводили 10 мл воды, а потом выпаривали растворители. Продукт использовали без очистки на следующем этапе. Этап В. (1R,2S)-1-[3-(Диметиламино)пропил]-2-меркаптоциклопентанкарбоновая кислота трифторацетат. К раствору соединения, полученного на этапе, описанном выше, в 35 мл смеси воды/ТГФ (1/1),прибавляли по каплям при комнатной температуре 2 мл трибутилфосфина. Реакционную смесь нагревали при 50 С при перемешивании в течение ночи. После охлаждения реакционную смесь разводили 100 мл воды и водную фазу потом промывали с помощью этера (325 мл). Водную фазу выпаривали. Сырьевой продукт очищали с помощью препаративной ВЭЖХ на колонке Kromasil (1 кг - 600 мм 60 мм - 70 мл/мин) при использовании смеси воды/ацетонитрила/трифторуксусной кислоты (85/15/0,1) в качестве мобильной фазы. Фракции, которые содержат ожидаемый продукт, подвергали лиофилизации.(рацемический цис-диастереоизомер). Этап А. Бензил 2-[(метилсульфонил)окси]-1-(3-пиридилметил)циклопентанкарбоксилат (рацемический транс-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах А, В и С примера 17, начиная от бензил 2-оксоциклопентанкарбоксилата и 3-(бензилокси)карбонил](метил)амино]пропилметансульфоната. Транс-диастереоизомер был вторым по очередности элюирования. Этап В. 1-[3-(Метиламино)пропил]-2-меркаптоциклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах G, I и J примера 1,начиная от соединения полученного на этапе, описанном выше.CAD/MC/MC спектр [М+Н]+ = 218,1 в соответствии с ожидаемой структурой. Пример 23. 2-Меркапто-1-(азетидин-3-илметил)циклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Этап А. Бензил 3-(1-(трет-бутилоксикарбонил)-2-гидроксициклопентилметил)-1-азетидинкарбоксилат. Ожидаемый продукт получали в соответствии с процедурой, описанной в этапах А и В примера 7,начиная от бензил 2-оксоциклопентанкарбоксилата и трет-бутил 3-[(метилсульфонил)окси]метилазетидин-1-карбамата. Этап В. Бензил 3-(1-(трет-бутилоксикарбонил)-2-[(метилсульфонил)окси]циклопентилметил)-1 азетидинкарбоксилат (рацемический транс-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе D примера 19, начиная от соединения, полученного на этапе, описанном выше. Продукт очищали на силикагеле при использовании смеси дихлорметана/этилацетата (градиент от 9/1 до 8/2) в качестве элюента с получением ожидаемого продукта в виде бесцветного масла. Транс-диастереоизомер был вторым по очередности элюирования. Этап С. 2-Меркапто-1-(азетидин-3-илметил)циклопентанкарбоновая кислота трифторацетат (рацемический цис-диастереоизомер). Ожидаемый продукт получали в соответствии с процедурой, описанной в этапе F примера 19, начиная от соединения, полученного на этапе, описанном выше.CAD/MC/MC спектр [М+Н]+ = 216,1 в соответствии с ожидаемой структурой. Фармакологическое исследование Пример 24. Ингибирование TAFIa. Очищенный человеческий TAFI (25 нг) активировали путем прибавления тромбинтромбомодулинового комплекса в присутствии хлорида кальция. После инкубирования в течение 20 мин при 20 С реакцию останавливали путем прибавления необратимого ингибитора тромбина PPACK (набор для определения активности TAFI, American Diagnostica). Исследуемое соединение прибавляли к раствору (3,2 нМ) TAFIa и инкубировали в течение 5 мин при 20 С. Прибавляли хромогенный TAFIa субстрат, а потом инкубировали в течение 30 мин при 37 С. Останавливали ферментативную реакцию путем прибавления серной кислоты (набор для определения активности TAFI, American Diagnostica). Оптическую плотность (OD) раствора измеряли при 490 нм с помощью спектрофотометра (Spectramax, Molecular Devices). Значение OD ячейки, содержащей реагенты без TAFI, отнимали от каждого из измеренных значений OD. Процентное ингибирование TAFIa при данной концентрации исследуемого соединения определяли при использовании приведенной ниже формулы% ингибирование = 100 - [(OD соединения 100)/OD носителя] Концентрацию соединения в соответствии с изобретением, которая ингибирует 50% ферментативной активности TAFIa (IC50), подсчитывали из процентного ингибирования значений OD, измеренных для повышающихся концентраций исследуемого соединения при использовании нелинейной регрессии в соответствии с сигмоидальным уравнением с четырьмя параметрами (эффект-доза). Значения IC50, полученные с характерными соединениями в соответствии с изобретением, регистрировали в нМ, как представлено в таблице, приведенной ниже Пример 25. Фармацевтическая композиция - таблетка. Формула для получения 1000 таблеток, каждая из которых содержит 10 мг активного ингредиента Пример 26. Фармацевтическая композиция - таблетка, в сочетании с варфарином. Формула для получения 1000 таблеток, каждая из которых содержит 10 мг активного ингредиента Формула для получения 1000 таблеток, каждая из которых содержит 10 мг активного ингредиента Пример 28. Инъецируемый раствор, в сочетании с альтеплазой. Формула для получения 10 мл раствора в которой R1 представляет собой атом водорода, ацетильную группу или группу формулы (А)R2 представляет собой группу формулы NR5R6, в которой R5 и R6, которые являются одинаковыми или различными, каждый представляет собой атом водорода или метильную группу, или R2 представляет собой пиперидин, азетидин, пиридин, необязательно замещенный аминогруппой или аминометилфенилом,R3 представляет собой атом водорода,m представляет собой целое число от 1 до 5 включительно,n представляет собой 1,их оптические изомеры, а также их аддитивные соли с фармацевтически приемлемой кислотой. 2. Соединение формулы (I) в соответствии с п.1, в которой R1 представляет собой атом водорода. 3. Соединение формулы (I) в соответствии с любым из пп.1 или 2, в которой R2 представляет собой аминогруппу или группу пиридила. 4. Соединение формулы (I) в соответствии с любым из пп.1 или 2, в которой m представляет собой 3. 5. Соединение формулы (Ia) в соответствии с п.1(1R,2S)-1-(3-аминопропил)-2-меркаптоциклопентанкарбоновой кислоты, ее оптических изомеров и аддитивных солей с фармацевтически приемлемой кислотой и(1R,2S)-2-ацетилтио-1-(3-аминопропил)циклопентанкарбоновой кислоты, ее оптических изомеров и аддитивных солей с фармацевтически приемлемой кислотой. 7. Способ синтеза соединений формулы (I) в соответствии с п.1 взаимодействием соединения формулы (II) в которой n представляет собой 1 и G представляет собой защитную группу для группы карбокси,с соединением формулы (III) в которой X представляет собой атом галогена или трифлат, группу тозилата или мезилата,m представляет собой целое число от 1 до 5 включительно,R'2 представляет собой группу формулы NR'5R'6, в которой R'5 и R'6, которые могут быть одинаковыми или различными, каждый представляет собой защитную группу для аминогруппы или метильную группу, или R'2 представляет собой пиперидин, азетидин, пиридин, необязательно замещенный аминогруппой или аминометилфенилом, необязательно замещенную защитной группой для аминогруппы,с получением соединения формулы (IV) в которой R'2, G, m и n являются такими, как определено выше,которое подвергают воздействию восстанавливающего агента для оксогруппы,с получением соединения формулы (V) в которой R'2, G, m и n являются такими, как определено выше,которое подвергают реакции с мезилхлоридом, тозилхлоридом, трифторметансульфоновым ангидридом или галогенирующим реагентом с получением соединения формулы (VI) в которой R'2, G, m и n являются такими, как определено выше, а X представляет собой мезилат, тозилат,или группу трифлата, или атом галогена,с возможным выделением соединения формулы (I) в форме единственного диастереоизомера,и которое потом подвергают реакции с соединением формулы (VII) в которой М представляет собой калий, натрий или литий, a R'1 представляет собой метильную группу,с получением соединения формулы (VIII) в которой R'1, R'2, G, m и n являются такими, как определено выше,с возможным выделением с помощью хиральной хроматографии соединения формулы (I) в форме единственного энантиомера,с возможным снятием защиты с групп тиола, амино и карбокси, с получением соединения формулы(I),с возможным получением аддитивной соли соединения формулы (I) с фармацевтически приемлемой кислотой. 8. Способ получения соединений формулы (Ia) в соответствии с п.5 взаимодействием соединения формулы (II) в которой n представляет собой 1 и G представляет собой защитную группу для группы карбокси,с акролеином, в присутствии асимметричного катализатора, с получением соединения формулы в которой n и G являются такими, как определено выше,альдегидную группу которого восстанавливают с получением соединения формулы (X) в которой n и G являются такими, как определено выше,которое подвергают реакции с мезилхлоридом, тозилхлоридом, трифторметансульфоновым ангидридом или галогенирующим реагентом с получением соединения формулы (XI) в которой n и G являются такими, как определено выше, а X представляет собой атом галогена или трифлат, группу тозилата или мезилата,группу кетона которого восстанавливают при использовании восстанавливающего агента с получением соединения формулы (XII) в которой n, G и X являются такими, как определено выше,которое подвергают реакции с соединением формулы (XIII) в которой R5 и R6, каждый, представляет собой защитную группу для аминогруппы или метильную группу,с получением соединения формулы (XIV) ридом или галогенирующим реагентом с получением соединения формулы (XV) в которой n, G, R5 и R6 являются такими, как определено выше, а X представляет собой группу мезилата, тозилата, или трифлат, или атом галогена,и которое потом подвергают реакции с соединением формулы (VII) в которой М представляет собой калий, натрий или литий, a R'1 представляет собой метильную группу,с получением соединения формулы (XVI) в которой n, R'1, G, R5 и R6 являются такими, как определено выше,с возможным снятием защиты с группы тиола, амино и карбокси с получением соединения формулы (Ia) в которой n, R1, R3, R5 и R6 являются такими, как определено выше,с возможным получением аддитивной соли соединения формулы (Ia) с фармацевтически приемлемой кислотой. 9. Фармацевтическая композиция, включающая соединение формулы (I) в соответствии с любым из пп.1-6 в комбинации с одним или более инертными, нетоксическими, фармацевтически приемлемыми наполнителями или носителями. 10. Фармацевтическая композиция в соответствии с п.9, отличающаяся тем, что дополнительно включает фибринолитический, антикоагулянтный или антитромбоцитарный агент. 11. Фармацевтическая композиция в соответствии с п.10 в инъецируемой форме, отличающаяся тем, что фибринолитический агент, выбран из рекомбинантного tPA, рекомбинантного uPA и стрептокиназы. 12. Применение соединения в соответствии с любым из пп.1-6 в производстве лекарственных средств для использования в предотвращении, вторичном предотвращении или лечении инфаркта миокарда, стенокардии, артериита нижних конечностей, венозных тромбозов, легочной эмболии, инсультов,васкулярных осложнений диабета, аневризмы аорты или деменции. 13. Применение соединения в соответствии с п.12 в сочетании с фибринолитическим, антикоагулянтным или антитромбоцитарным агентом.
МПК / Метки
МПК: A61K 31/095, C07C 321/22, A61K 31/265, A61K 31/445, C07C 211/18, C07D 213/24, C07C 327/06, A61K 31/44, A61P 9/10, A61P 7/02
Метки: 2-меркаптоциклопентанкарбоновой, кислоты, соединения, содержащие, получения, новые, способ, фармацевтические, композиции
Код ссылки
<a href="https://eas.patents.su/24-17867-novye-soedineniya-2-merkaptociklopentankarbonovojj-kisloty-sposob-ih-polucheniya-i-soderzhashhie-ih-farmacevticheskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Новые соединения 2-меркаптоциклопентанкарбоновой кислоты, способ их получения и содержащие их фармацевтические композиции</a>
Новые соединения имидазолина, способ их получения и содержащие их фармацевтические композиции

Номер патента: 4536
Опубликовано: 24.06.2004
Авторы: Лякост Жан-Мишель, Миллан Марк, Гобер Алан, Корди Алекс, Ньюмен-Танкреди Адриан
МПК: C07D 233/06, A61K 31/4164, A61P 25/24...
Метки: способ, получения, новые, фармацевтические, композиции, содержащие, имидазолина, соединения
Формула / Реферат:
1. Соединения формулы (I) где A представляет кольцо бензола, незамещенное или замещенное 1-4 одинаковыми или различными группами, выбранными из неразветвленного или разветвленного (C1-C6)алкила, неразветвленного или разветвленного (C1-C6)алкокси, гидрокси, полигалоген (C1-C6)алкила, у которого алкильная часть не разветвлена или разветвлена, циано, нитро, амино, алкиламино, диалкиламино, тиоалкила, сульфонилалкила, сульфинилалкила, карбокси,...
Новые соединения аминокислоты, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 6389
Опубликовано: 29.12.2005
Авторы: Верберен Тони, Глоане Филипп, Де-Нантёй Гийом, Рюпэн Ален
МПК: C07C 257/18, A61K 31/196, A61P 7/04...
Метки: новые, получения, аминокислоты, фармацевтические, соединения, содержащие, способ, композиции
Формула / Реферат:
1. Соединение формулы (I) в которой R1 представляет собой арильную группу, гетероарильную группу или линейную или разветвлённую (C1-C6)алкильную группу, необязательно замещённую одной или более одинаковыми или различными группами, выбранными из арила и гетероарила, или R1 представляет собой группу формулы -(CO)-CR6R7NR8R9, в которой R6 представляет собой атом водорода или группу, выбранную из арила, гетероарила, гетероциклоалкила,...
Новые соединения бензо [b]пирано[3,2-h]акридин-7-она, способ их получения и содержащие их фармацевтические композиции

Номер патента: 5374
Опубликовано: 24.02.2005
Авторы: Кош Мишель, Ренар Пьер, Сеген Элизабет, Эль Омри Абдельхаким, Пьер Ален, Хикмен Джон, Пфейффер Брюно, Мишель Сильви, Тийекен Франсуа
МПК: C07D 221/04, A61P 35/00, A61K 31/4741...
Метки: содержащие, бензо, композиции, способ, фармацевтические, получения, соединения, новые, b]пирано[3,2-h]акридин-7-она
Формула / Реферат:
1. Соединения формулы (I) в которой X и Y, которые могут быть одинаковыми или различными, представляют собой независимо друг от друга группу, выбранную из атомов водорода или галогена, меркапто, циано, нитро, линейной или разветвленной (C1-C6)алкильной, линейной или разветвленной (C1-C6)тригалоалкильной и линейной или разветвленной тригало-(C1-C6)алкилкарбониламиногруппы, групп формул -ORa, -NRaRb, -NRa-C(O)-T1, -O-C(O)-T1, -OT2-NRaRb,...
Новые гидразидные соединения, способ их получения и содержащие их фармацевтические композиции

Номер патента: 2442
Опубликовано: 25.04.2002
Авторы: Альдана Мораса Игнанио, Бутэн Жан, Монхе Вега Антонио, Дюо Жак, Делла Зюана Одиль, Кеньяр Даниель-Энри
МПК: A61K 31/165, A61P 3/04, C07C 271/20...
Метки: гидразидные, фармацевтические, способ, соединения, получения, композиции, содержащие, новые
Формула / Реферат:
1. Соединения формулы (I) R-NH-A-CO-NH-NH-(W)n-Z (I) где n равен 0 или 1, W обозначает группу -СО- или группу S(O)r, где r равен 0, 1 или 2, Z обозначает группу, выбранную из необязательно замещенного арила, необязательно замещенного арилалкила, необязательно замещенного гетероарила и необязательно замещенного гетероарилалкила, R обозначает группу, выбранную из: Z1-T-CO- Z1-O-T-CO- Z1-T-O-CO- Z1-T-S(O)q- где Z1 обозначает необязательно...
Новые соединения 3-имино-1,2-дитиола, способ их получения и фармацевтические композиции, их содержащие

Номер патента: 4291
Опубликовано: 26.02.2004
Авторы: Ле Бо Гийом, Ренар Пьер, Нурриссон Мари-Рене, Кэньяр Даньель-Анри, Бизо-Эспьяр Жан-Ги, Робер Жан-Мишель, Дюфло Мюриель
МПК: A61K 31/444, A61P 29/00, C07D 409/14...
Метки: способ, содержащие, получения, новые, соединения, 3-имино-1,2-дитиола, фармацевтические, композиции
Формула / Реферат:
1. Соединение формулы (I) в которой Het1 и Het2, которые могут быть одинаковы или различны, каждый представляет собой гетероарильную группу, A представляет собой связь или линейную или разветвленную (C1-C6)алкиленовую группу, его оптические изомеры, если они существуют, а также его аддитивные соли с фармацевтически приемлемой кислотой, причем гетероарильную группу следует понимать как обозначающую ароматическую моно- или бициклическую группу,...
Предыдущий патент: Применение производных лактамида в составе препаратов для снижения токсичности
Следующий патент: Модифицирующая добавка
Случайный патент: Устройство для очистки текучей среды в виде пара, поступающего из системы трубопроводов