Фармацевтическая композиция и фармацевтическая дозированная форма, содержащие соединение пиридазина
Номер патента: 18173
Опубликовано: 28.06.2013
Авторы: Доуди Эрик Д., Кент Кеннет М., Зия Вахид, Том Норма Дж.















Формула / Реферат
1. Фармацевтическая композиция для лечения вирусной инфекции гепатита С, содержащая соединение формулы (1)

и одну или более жирных кислот.
2. Фармацевтическая композиция по п.1, отличающаяся тем, что жирная кислота является насыщенной или ненасыщенной и содержит от 4 до 22 атомов углерода в цепи.
3. Фармацевтическая композиция по п.1, отличающаяся тем, что жирная кислота представляет собой каприновую, линолевую, олеиновую, лауриновую, пальмитиновую или миристиновую кислоты.
4. Фармацевтическая композиция по п.1, отличающаяся тем, что жирная кислота представляет собой олеиновую кислоту.
5. Фармацевтическая композиция по п.4, содержащая 8 мас.% соединения (1) и 92 мас.% олеиновой кислоты.
6. Фармацевтическая композиция по п.4, содержащая 20 мас.% соединения (1) и 80 мас.% олеиновой кислоты.
7. Фармацевтическая композиция по п.1, дополнительно содержащая полиэтиленгликоли или моно-, ди- или триглицериды с короткой, средней или длинной углеводородной цепью или пегилированные жирные кислоты с короткой, средней или длинной углеводородной цепью.
8. Фармацевтическая композиция по п.3, дополнительно содержащая одно или более поверхностно-активных веществ, выбранных из сложных эфиров жирных кислот и полиоксиэтиленсорбита, сложных эфиров сорбита и жирной кислоты, производных полиоксиэтиленкасторового масла, полиоксиэтиленглицерин оксистеарата, полиэтиленгликоля 60, гидрогенизированного касторового масла и блок-сополимеров этиленоксида и пропиленоксида.
9. Фармацевтическая композиция по п.8, отличающаяся тем, что жирная кислота представляет собой олеиновую кислоту, а поверхностно-активное вещество представляет собой полиоксиэтиленсорбит.
10. Фармацевтическая композиция по п.9, содержащая 8 мас.% соединения (1), 87,4 мас.% олеиновой кислоты и 4,6 мас.% полиоксиэтиленсорбита.
11. Фармацевтическая композиция по п.9, содержащая 20 мас.% соединения (1), 76 мас.% олеиновой кислоты и 4 мас.% полиоксиэтиленсорбита.
12. Фармацевтическая композиция по п.9, содержащая 8 мас.% соединения (1), 86,47 мас.% олеиновой кислоты, 4,6 мас.% полиоксиэтиленсорбита, 0,92 мас.% аэросила 200 и 0,01 мас.% бутилгидрокситолуола (БГТ).
13. Фармацевтическая композиция по п.9, содержащая 8 мас.% соединения (1), 85,55 мас.% олеиновой кислоты, 4,6 мас.% полиоксиэтиленсорбита, 1,84 мас.% аэросила 200 и 0,01 мас.% БГТ.
14. Фармацевтическая композиция по п.9, содержащая 10 мас.% соединения (1), 84,55 мас.% олеиновой кислоты, 5 мас.% полиоксиэтиленсорбита, 0,35 мас.% бутилгидроксианизола (БГА) и 0,1 мас.% БГТ.
15. Фармацевтическая композиция по п.9, дополнительно содержащая один или более спиртов, выбранных из этанола, бензилового спирта, глицерина, полиэтиленгликоля 200, полиэтиленгликоля 300 и полиэтиленгликоля 400.
16. Фармацевтическая композиция по п.9, содержащая 8 мас.% соединения (1), 73,6 мас.% олеиновой кислоты, 9,2 мас.% полиоксиэтиленсорбита и 9,2 мас.% этанола.
17. Фармацевтическая дозированная форма для лечения вирусной инфекции гепатита С, содержащая соединение формулы (1)

и одну или более жирных кислот, выбранных из каприновой, олеиновой, лауриновой, пальмитиновой и миристиновой кислот.
18. Фармацевтическая дозированная форма по п.17, содержащая олеиновую кислоту и полиоксиэтиленсорбит.
19. Фармацевтическая дозированная форма по п.17, отличающаяся тем, что указанная дозированная форма представляет собой капсулу.
20. Фармацевтическая дозированная форма по п.19, отличающаяся тем, что указанная капсула представляет собой капсулу с твердой оболочкой.
21. Фармацевтическая дозированная форма по п.19, отличающаяся тем, что указанная капсула представляет собой капсулу с мягкой оболочкой.
22. Фармацевтическая дозированная форма по п.19, отличающаяся тем, что указанная капсула представляет собой капсулу с мягкой желатиновой оболочкой.
23. Фармацевтическая дозированная форма по п.19, отличающаяся тем, что указанная капсула представляет собой гидроксипропилметилцеллюлозную капсулу.
Текст
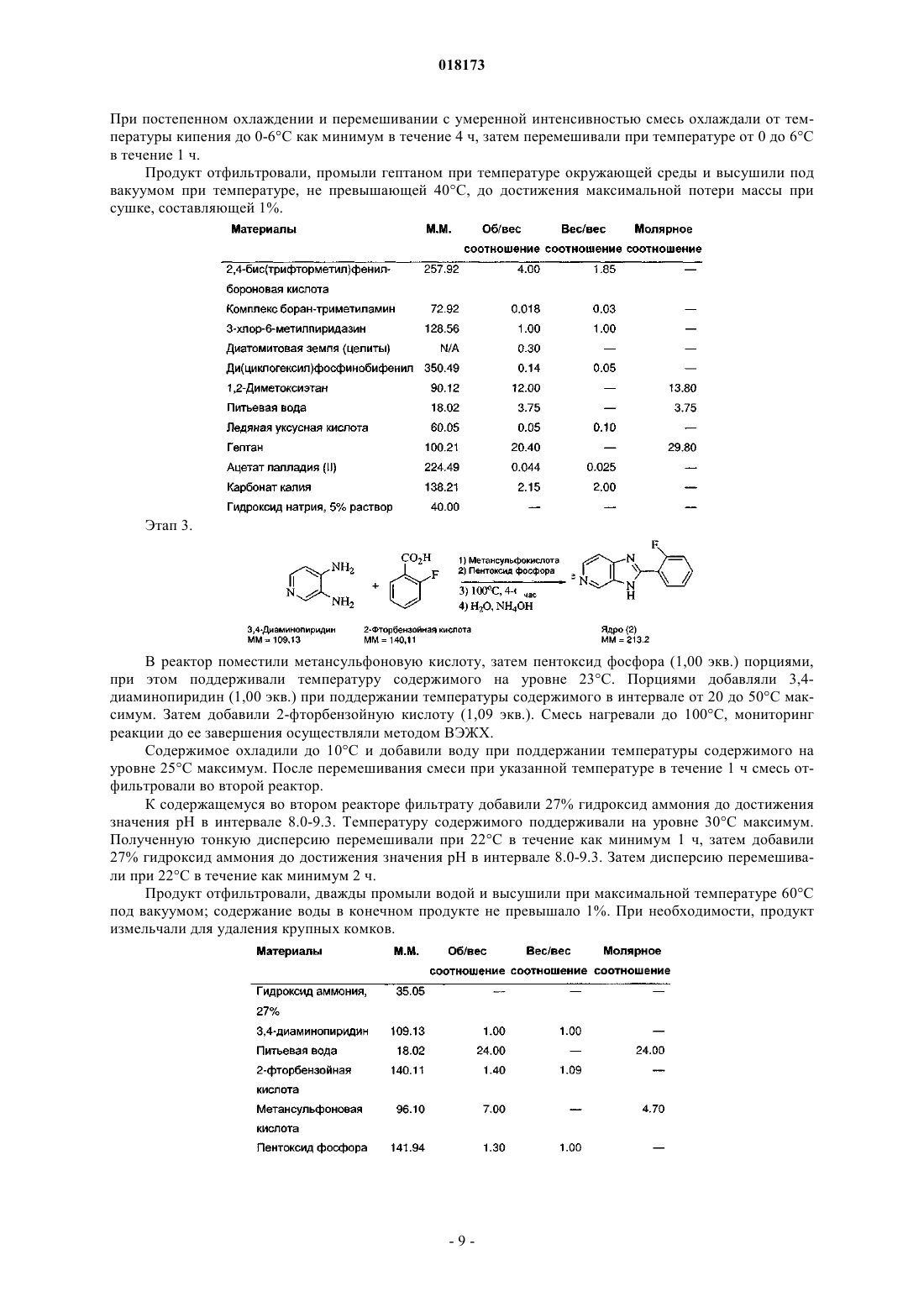
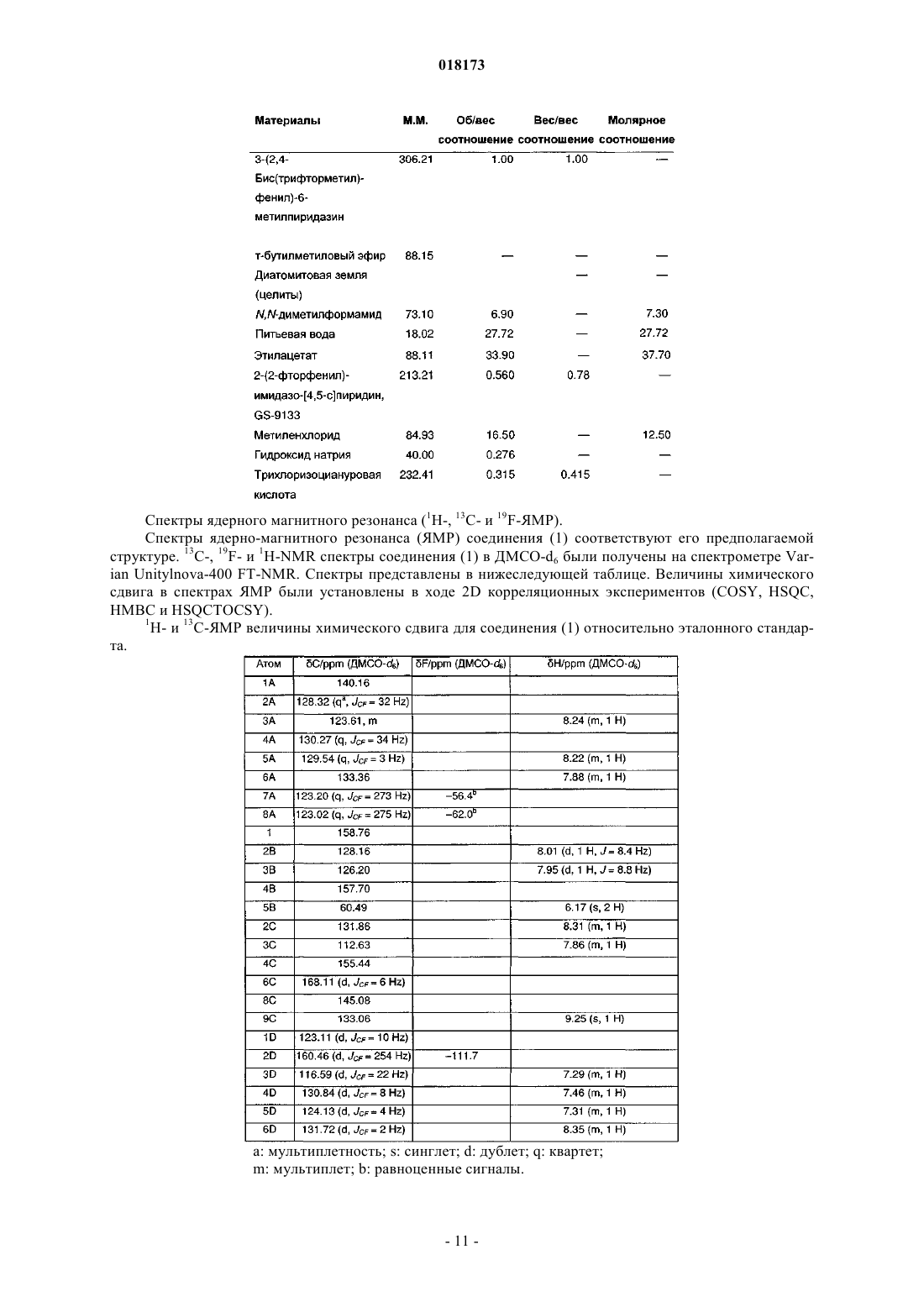
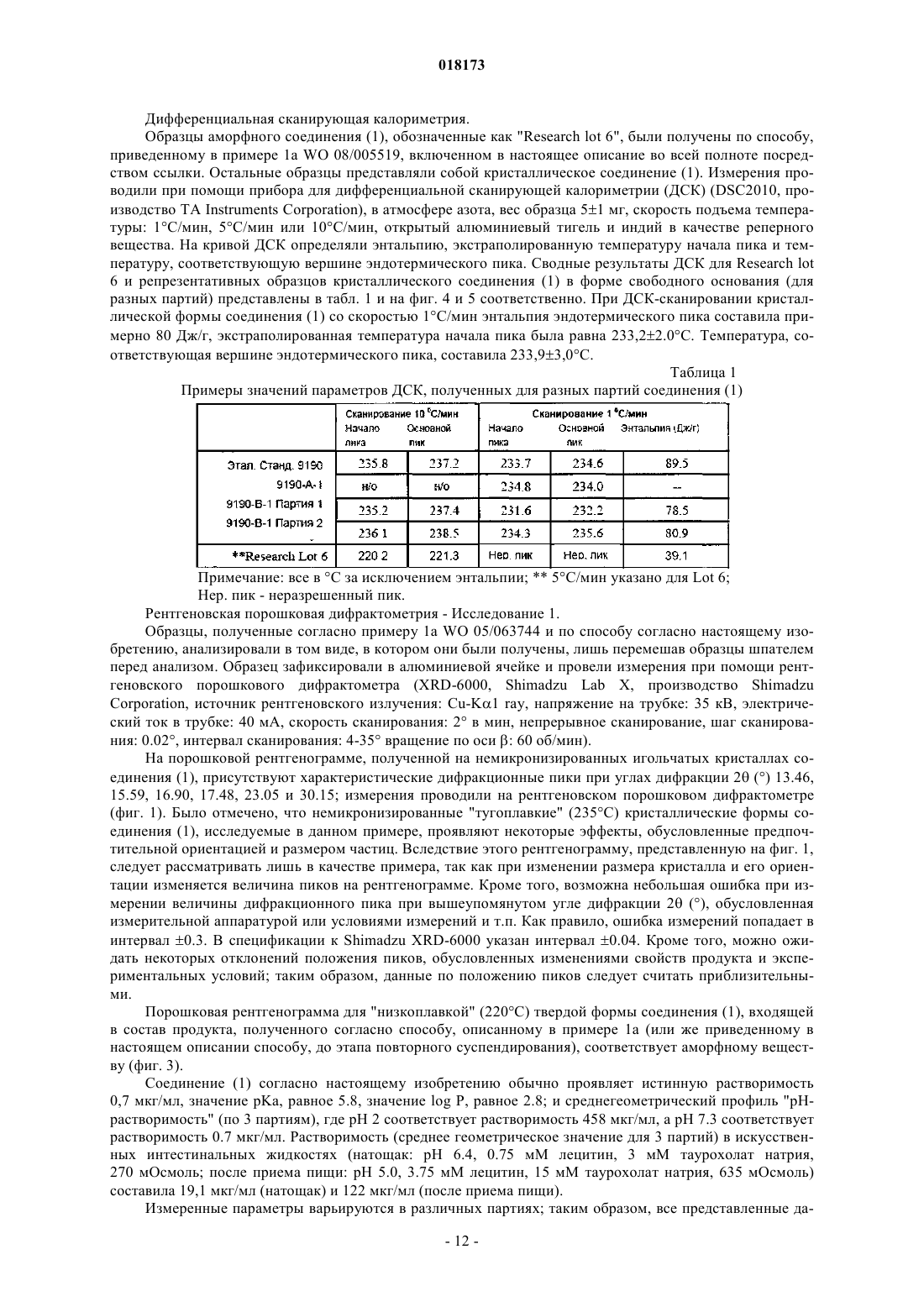
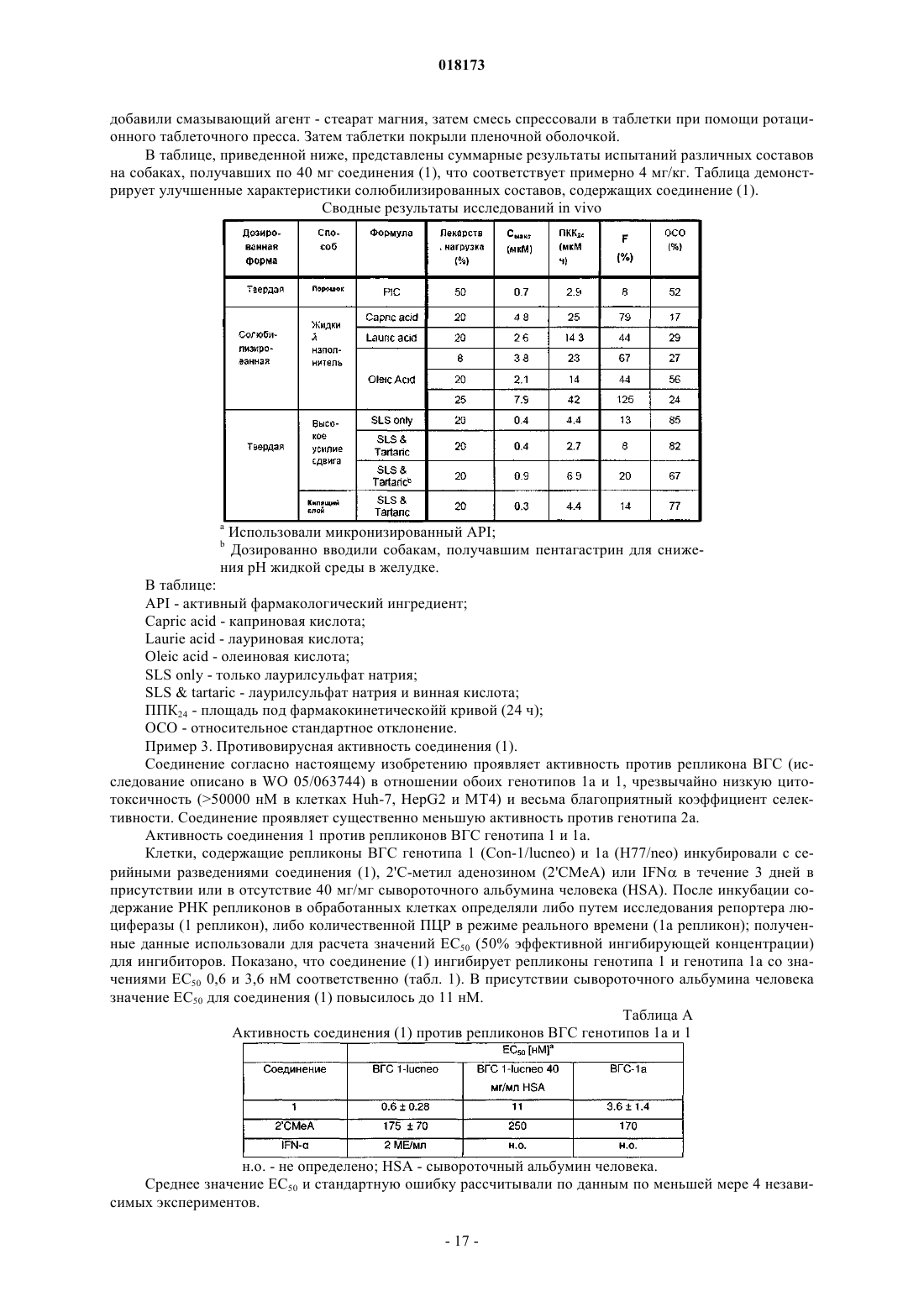
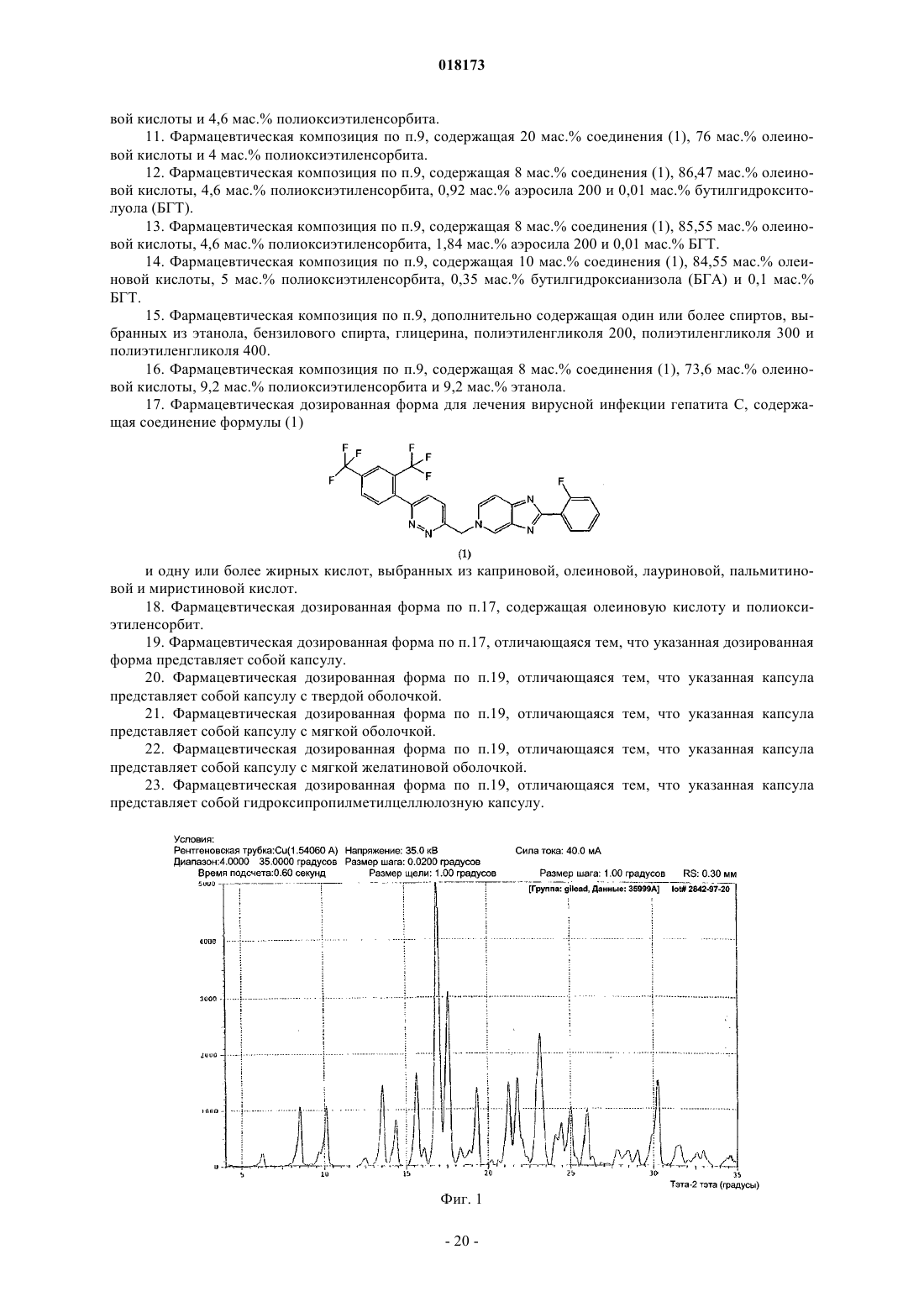
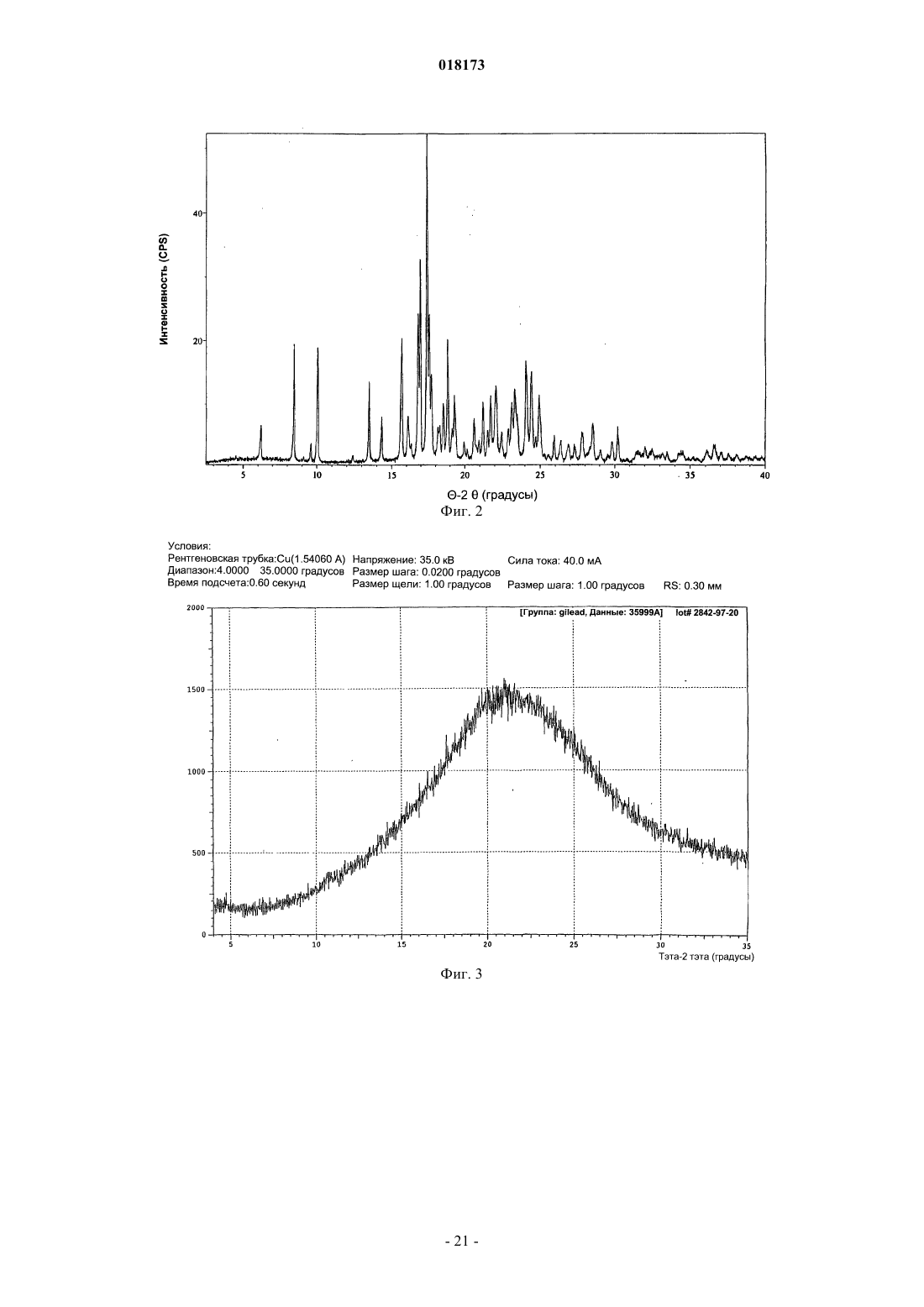
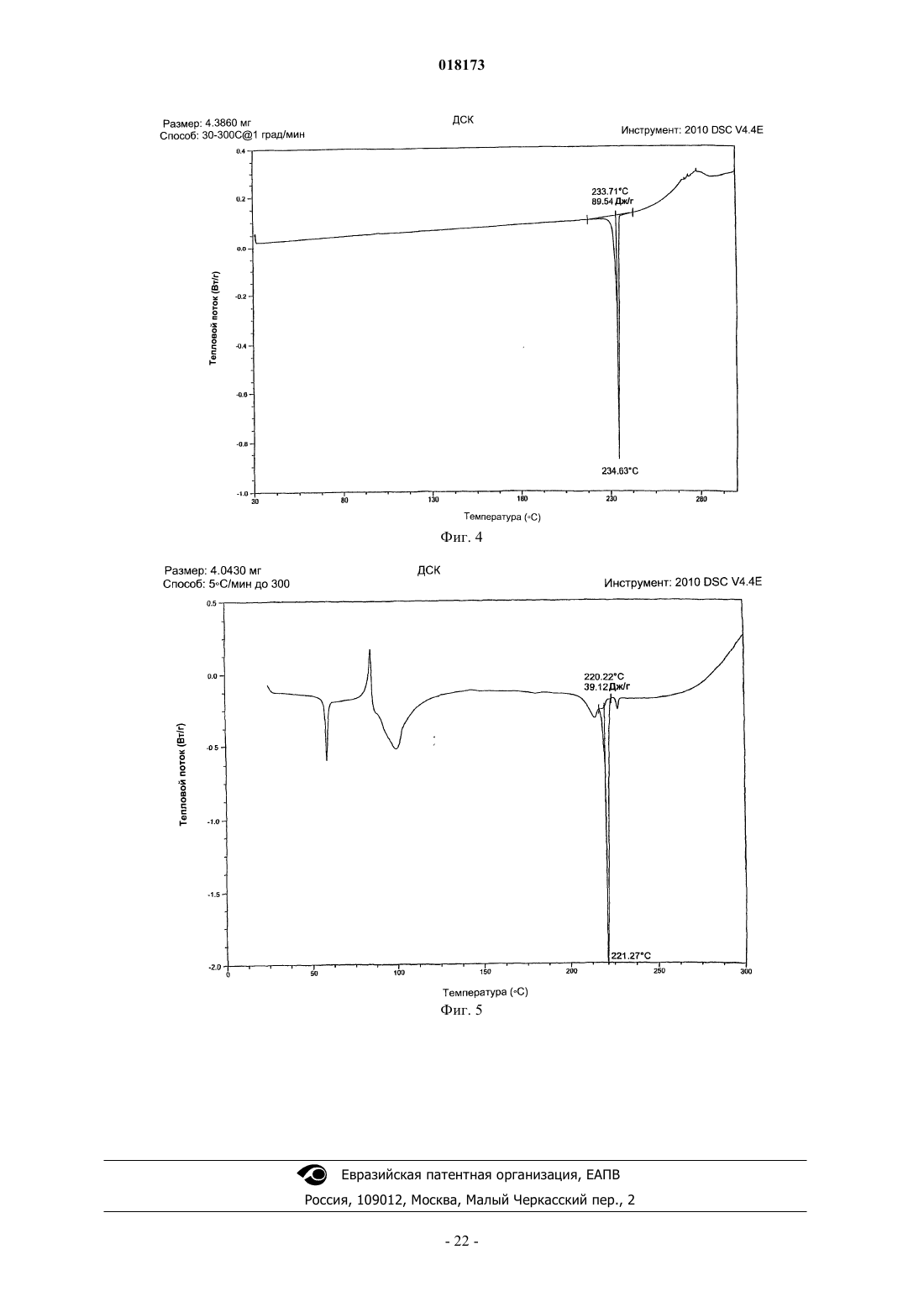
В настоящем изобретении предложена фармацевтическая композиция для лечения вирусной инфекции гепатита С, содержащая соединение формулы (1) Кроме того, предложена фармацевтическая дозированная форма для лечения вирусной инфекции гепатита С, содержащая указанное соединение.(US); К.Ю. ЛЕВЕН РИСЕРЧ ЭНД ДИВЕЛОПМЕНТ (BE); ПЮРСТИНГЕР ГЕРХАРД (AT) Область изобретения Вирус гепатита С представляет собой оболочечный вирус с оболочкой, содержащий однонитевую положительную смысловую (+) РНК, относящийся к семейству Flaviviridae. Репликация вируса гепатита С (ВГС) происходит, главным образом, в гепатоцитах печени. Частицы ВГС в кровотоке связываются с рецепторами на поверхности гепатоцитов и затем проникают в клетки. Проникнув внутрь гепатоцита,ВГС использует внутриклеточные структуры, необходимые для репликации ВГС. Lindenbach, В. Nature 436(7053):932-8 (2005). В результате трансляции генома ВГС образуется единственный белок, включающий примерно 3011 аминокислот. Затем этот "полибелок" подвергается протеолитическому процессингу под действием вирусных и клеточных протеаз с образованием трех структурных (вирионассоциированных) и семи неструктурных (NS) белков. ВГС кодирует две протеазы, цистеиновую аутопротеазу NS2 и сериновую протеазу NS3-4A. Неструктурные (NS) белки далее рекрутируют геном вируса в комплекс репликации РНК, ассоциированный с реаранжированными цитоплазматическими мембранами. Репликация РНК происходит под действием РНК-зависимой РНК-полимеразы NS5B, которая обеспечивает образование промежуточной минус-цепи РНК. Далее указанная минус-цепь РНК выступает в качестве матрицы для образования новых геномов вирусов, состоящих из плюс-цепи РНК. Затем образующиеся геномы могут подвергаться трансляции,дальнейшей репликации или упаковке в новые вирусные частицы. Предположительно, новые вирусные частицы отпочковываются в секреторный путь и высвобождаются на поверхности клетки. ВГС обладает высокой скоростью репликации; в организме инфицированного субъекта продуцируется приблизительно один триллион частиц ежедневно. Вследствие недостаточной коррекции ошибок репликации РНК-полимеразой ВГС ВГС обладает исключительно высокой частотой мутаций, при этом данный фактор может способствовать ускользанию ВГС от иммунного ответа организма хозяина. На основании генетических различий между изолятами ВГС, вид вируса гепатита С подразделяют на шесть генотипов (1-6), при этом каждый генотип включает несколько подтипов. Далее, на основании генетических различий подтипы подразделяют на квазивиды. Распределение и преобладание тех или иных генотипов ВГС различно в разных регионах мира. Например, в Северной Америке преобладает генотип 1 а, за ним следуют 1, 2 а, 2b и 3 а. В Европе преобладает генотип 1, далее следуют 2 а, 2b, 2 с и 3 а. Генотипы 4 и 5 обнаруживают почти исключительно в Африке. Генотип обладает клинической значимостью с точки зрения оценки потенциального ответа на терапию интерфероном и длительности подобного лечения. Генотипы 1 и 4 менее чувствительны к лечению интерфероном, чем другие генотипы(2, 3, 5 и 6). Длительность стандартного курса лечения интерфероном для генотипов 1 и 4 составляет 48 недель, в то время как полный курс лечения для генотипов 2 и 3 составляет 24 недели. По оценке Всемирной организации здравоохранения в мире от 170 до 200 миллионов человек (3% населения Земли) хронически инфицированы вирусом гепатита С. Примерно 75% из них хронически инфицированы и при этом у них в плазме содержатся обнаруживаемые количества РНК ВГС. Эти хронические носители заболевания подвержены риску развития цирроза и/или рака печени. В ходе исследований с врачебным контролем в течение 7-16 лет отмечено развитие цирроза у 7-16% пациентов, развитие гепатоцеллюлярной карциномы у 0,7-13% пациентов, при этом 1,3-3,7% пациентов умерло от заболеваний, связанных с печенью. В настоящее время единственным доступным способом лечения является терапия с применением интерферона -2 (или его пегилированной формы), применяемого в отдельности или в комбинации с рибавирином. Тем не менее, устойчивый ответ наблюдали лишь примерно у 40% пациентов, при этом лечение связано с серьезными неблагоприятными побочными эффектами. В настоящее время существует насущная необходимость в разработке высокоэффективных и селективных ингибиторов ВГС. Релевантные источники включают патенты США 4914108; 4988707; 4990518; 5137896; 5208242; 5227384; 5302601; 5374638; 5405964; 5438063; 5486525; 6479508; и публикацию патента СШАUS 2003/0108862 А 1, патент Канады 2423800 А 1, патенты Германии 4211474 А 1, 4236026, 4309969,4318813, Европейские патенты ЕР 0138552 А 2, ЕР 0706795 А 2, ЕР 1132381 А 1, патент Великобритании 2158440 А, международные публикации WO 00/20416, WO 00/39127, WO 00/40583, WO 03/007945 А 1,WO 03/010140 А 2, WO 03/010141 А 2, WO 93/02080, WO 93/14072, WO 96/11192, WO 96/12703,WO 99/27929, PCT-US 2004/43112, РСТ-ВЕ 2003/000117, PCT-US 2005/26606, Akamatsu et al., "New Efficient Route for Solid-Phase Synthesis of Benzimidazole Derivatives", 4:475-483, J. COMB. CHEM., 2002,Baginski SG et al., Proc. Natl. Acad. Sci. USA 2000 Jul. 5; 97(14):7981-6). Cleve et al., "Derivate des Imidazo[4.5-b]- und Imidazo[4.5-c]pyridins", 747:158-171, JUSTUS LIEBIGS ANNALEN DER CHEMICA, 1971,Kiyama, et al., "Synthesis and Evaluation of Novel Nonpeptide Angiotensin II Receptor Antagonists: Imidazo[4,5-c]pyridine Derivatives with an Aromatic Substituent", 43(3):450-60, CHEM PHARM BULL, 1995,Mederski et al., "Synthesis and Structural Assignment of Some N-substituted Imidazopyridine Derivatives",48(48):10549-58,TETRAHEDRON,1992,Yutiloval.,23(1):56-9,KHIMIKOFARMATSEVTICHESKIIZHURNAL, 1989. Кроме того, см. WO 05/063744. Соединение формулы (1) является объектом изобретения согласно WO 08/005519. Соединение (1), полученное по способу согласно WO 05/063744, является по существу или полностью аморфным. Полагают, что указанное соединение представляет собой гидрат (далее, "аморфное" соединение (1. Краткое описание изобретения В соответствии с указанными выше задачами в одном аспекте настоящего изобретения предложена фармацевтическая композиция для лечения вирусной инфекции гепатита С, содержащая соединение формулы (1) и одну или более жирных кислот. Соединение (1) подходит для применения согласно способу лечения или профилактики инфекции ВГС, включающему введение субъекту терапевтической или профилактической дозы соединения (1). Другой вариант реализации настоящего изобретения относится к фармацевтическим композициям соединения формулы (1), содержащим по меньшей мере один фармацевтически приемлемый наполнитель. Согласно одному варианту реализации готовят состав на основе соединения формулы (1) с использованием органической кислоты, при этом указанный состав может быть получен в виде дозированной фармацевтической формы, такой как капсула. Фармацевтические композиции согласно настоящему изобретению применяют для лечения или профилактики гепатита С. Другие признаки настоящего изобретения, включая новые промежуточные соединения и композиции на основе конечных соединений, станут очевидны при рассмотрении данного патента в целом. Описание чертежей На фиг. 1 представлена порошковая рентгенограмма, полученная для стандартного образца кристаллического соединения (1), полученного по способу согласно примеру 1. На фиг. 2 представлена другая порошковая рентгенограмма, полученная для кристаллического соединения (1). На фиг. 3 представлена порошковая рентгенограмма, полученная для аморфной формы соединения(1) Research Lot 6, полученного по способу, описанному в примере 1 а в WO 08/005519. На фиг. 4 представлена термограмма дифференциальной сканирующей калориметрии для стандартного образца кристаллического соединения (1) (сканирование при скорости нагрева 1 С/мин), полученного по способу, описанному далее в примере 1. На фиг. 5 представлена термограмма дифференциальной сканирующей калориметрии для аморфной формы соединения (1) Research Lot 6 (сканирование при скорости нагрева 5 С/мин), полученного по способу, описанному в примере 1 а в WO 08/005519. Подробное описание изобретения В одном аспекте настоящего изобретения предложена композиция, полученная путем объединения соединения (1) с фармацевтически приемлемым наполнителем с последующим получением фармацевтической дозированной формы, такой как таблетка или капсула. Согласно некоторым вариантам реализации соединение (1) представляет собой полупродукт для растворения в носителе или наполнителе. Соединение (1) согласно настоящему изобретению вводят субъекту, представляющему собой млекопитающее (включая человека), любыми способами, известными в данной области техники, а именно пероральным, интраназальным, подкожным, внутримышечным, интрадермальным, внутривенным, внутриартериальным, парентеральным способом или путем катетеризации в терапевтически эффективном количестве, т.е. в количестве, подавляющем ВГС или репликацию ВГС. Полагают, что указанное количество представляет собой количество, обеспечивающее содержание в плазме, составляющее примерно 100 нМ, в 3 раза превышающий уровень ЕС 90, установленный с применением белка (protein-adjustedEC90). В общем случае полагают, что данный уровень обеспечивается путем ежедневного перорального приема в количестве примерно 5 мг/кг; обычно примерно от 0,7 до 2,2 мг/кг; как правило, примерно 1,2 мг/кг массы тела для человека. Оптимальная доза соединения согласно настоящему изобретению зависит от множества факторов,известных специалисту в данной области техники, в том числе от биодоступности соединения в конкретном составе, метаболизма и распределения соединения в организме субъекта, приема до или после еды,выбора носителей и наполнителей, входящих в состав, и других факторов. Как правило, подходящую дозу определяет специалист в данной области техники в ходе доклинических и клинических исследований. Терапевтически эффективное количество соединения согласно изобретению можно разделить на несколько субъединиц для приема в течение дня или вводить ежедневно или с интервалами, превышающими один день, в зависимости от природы инфекции, общего состояния пациента и конкретного состава на основе соединения согласно настоящему изобретению. Как правило, соединение принимают два раза в день. Соединение согласно настоящему изобретению применяют в сочетании с другими агентами, эффективными против инфекций, вызываемых ВГС. В ходе лечения указанные агенты можно вводить отдельно или в комбинации с соединением (1) в виде единой дозированной формы, такой как таблетка,раствор для внутривенного введения или капсула. Указанные другие агенты включают, например, интерферон-альфа, рибавирин и/или соединения согласно ЕР 1162196, WO 03/010141, WO 03/007945,WO 00/204425 и/или WO 03/010140 (и других заявок, входящих в семейство патентов-аналогов для указанных патентных документов). Другие агенты, подходящие для введения в ходе лечения совместно с соединением согласно настоящему изобретению, включают соединения, проходящие в настоящее время клинические испытания, в частности ингибиторы протеазы ВГС, такие как VX-950 (Vertex Pharmaceuticals), SCH 5030347 (Schering Plough) и BILN-2061 (Boehringer Ingelheim), нуклеозидные ингибиторы ВГС, такие как NM283, NM107 (оба Idenix/Novartis) и R1626 (Hoffmann-LaRoche), а также ненуклеозидные ингибиторы ВГС, включая ВГС-086 и -796 (оба ViroPharma/Wyeth). Дополнительные антивирусные агенты применяют в традиционных количествах. Если эффект от применения соединения согласно изобретению и дополнительного соединения является аддитивным, то возможно соразмерное уменьшение количества каждого активного агента, тем более если агенты действуют синергически. Тем не менее, в общем случае агенты применяют в обычных активных количествах в составе единых комбинированных композиций. Агенты, вводимые совместно, как правило, вводят в состав единых композиций совместно с соединением согласно изобретению, если указанные агенты и соединение химически совместимы и предназначены для введения одинаковым путем. Если это не так, то указанные агенты и соединение можно обеспечить в виде медицинского набора или упаковки, содержащих два агента в разных контейнерах или отделениях. Как правило, соединение согласно изобретению обеспечивают в виде свободного основания, однако возможно получение его в виде соли. Соли обычно получают путем добавления органических и/или неорганических кислот к свободному основанию. Примеры включают (1) неорганические кислоты, такие как галогеноводородные кислоты, например хлористо-водородная или бромисто-водородная кислота,серная кислота, азотная кислота, фосфорная кислота и сульфаминовые кислоты; или (2) органические кислоты, такие как уксусная, пропионовая, гидроксиуксусная (гликолевая), бензойная, 2 гидроксипропионовая, 2-оксопропионовая, молочная, фумаровая, винная, пировиноградная, малеиновая,малоновая, яблочная, салициловая (например, 2-гидроксибензойная), п-аминосалициловая, изэтионовая,лактобионовая, янтарная, щавелевая и лимонная кислоты; органические сульфокислоты, такие как метансульфоновая, этансульфоновая, бензосульфоновая, п-толуолсульфоновая, С 1-С 6 алкилсульфоновые,бензолсульфоновые, п-толуолсульфоновые и цикпогексансульфаминовая кислоты. Типичные соли представляют собой хлориды, сульфаты, бисульфаты, мезилаты, безилаты, эзилаты, фосфаты, оксалаты, малеаты, сукцинаты, цитраты, малонаты и/или фумараты. Кроме того, настоящее изобретение включает соли, образованные соединением согласно настоящему изобретению и одной или несколькими аминокислотами, обычно природными аминокислотами, такими как аминокислоты, содержащиеся в белках. Кислотный противоион желательно является физиологически безопасным и нетоксичным или же фармацевтически приемлемым, если только соль не является полупродуктом, применяемым для получения соединений, когда токсичность не имеет существенного значения. Обычно соединение (1) вводят в форме свободного основания, однако подходящие соли включают мезилаты (соли метансульфокислоты) иHCl. Соединения согласно изобретению включают сольваты, образованные соединением согласно настоящему изобретению или его солями, такие как, например, гидраты, алкоголяты и т.п. Составы согласно изобретению на основе фармацевтического соединения согласно изобретению можно готовить с применением традиционных фармацевтических носителей и наполнителей, выбираемых в соответствии с обычной практикой. Таблетки содержат эксципиенты; агенты, способствующие скольжению; наполнители; связующие агенты и т.п. Водные составы готовят в виде стерильных форм, при этом если указанные составы предназначены для доставки путем, отличным от перорального, то в общем случае они должны быть изотоническими. Составы могут содержать наполнители, например, предложенные в "Handbook of Pharmaceutical Excipi-3 018173ents" (2005), включая аскорбиновую кислоту и другие антиоксиданты; хелатирующие агенты, такие как ЭДТА; углеводы, такие как декстрин, гидроксиалкилцеллюлоза, гидроксиметилцеллюлоза и/или органические кислоты, такие как олеиновая или стеариновая кислота. Термин "фармацевтически приемлемый носитель" в настоящем описании обозначает любой материал или вещество, введенное в состав вместе с активным ингредиентом для облегчения его получения и/или его применения или распространения к месту, подвергаемому лечению. Подходящие фармацевтические носители для применения в составах согласно настоящему изобретению хорошо известны специалистам в данной области техники. Такие носители включают вспомогательные вещества, такие как увлажняющие агенты; диспергирующие агенты; адгезивы; эмульгаторы; растворители; агенты, облегчающие скольжение; материалы покрытия; антибактериальные и противогрибковые агенты (например,фенол, сорбиновая кислота, хлорбутанол) и изотонические агенты (такие как сахара или хлористый натрий), при условии, что эти вещества применимы в фармацевтической практике, т.е. не являются токсичными для млекопитающих. Фармацевтические композиции согласно настоящему изобретению получают любыми подходящими способами, например, путем гомогенного смешивания, нанесения покрытия и/или измельчения активных ингредиентов в один этап или несколько этапов с выбранным веществом-носителем и, там, где это целесообразно, с другими вспомогательными веществами, такими как поверхностно-активные вещества. Композиции, содержащие соединение согласно настоящему изобретению, приготовленное в виде микросфер (обычно диаметр микросферы составляет примерно от 1 до 10 мкм (gm, подходят в качестве составов с замедленным или контролируемым высвобождением. В одном из возможных составов соединение (1) измельчено до тонкоизмельченной формы, при этом обычно средний размер частиц в любой точке варьируется в интервале примерно от 1 до 20 мкм. Продукт согласно примеру 1 представляет собой игольчатые или стержневидные кристаллы, длина которых варьируется в интервале примерно от 25 до 40 мкм. Указанные кристаллы можно микронизировать на вихревой мельнице Jet mill-00 примерно при 60-80 psi (413,7-551,6 кПа) для получения частиц размером примерно 3-4 мкм, площадь поверхности которых составляет 7-8 кв.м/г. Тем не менее, исходные размеры кристаллов различны в разных партиях и степень их измельчения является предметом выбора. Соответственно, микронизированное кристаллическое соединение (1) определяют попросту как кристаллическое или аморфное соединение (1), подвергшееся процедуре микронизации, такое как соединение,представленное в настоящем описании в качестве примера. Ни размер, ни площадь поверхности полученных частиц не имеют существенного значения. Микронизированное соединение (1) суспендируют в водном растворе; возможно, добавляя к нему суспендирующий агент, эмульгаторы и поверхностноактивные вещества, как описано далее. Обычно фармацевтический состав представляет собой солюбилизированную форму соединения (1),где соединение (1) растворено в соответствующем растворителе или солюбилизирующем веществе, или их комбинации. Соединение (1) солюбилизируют в фармацевтически приемлемом наполнителе для введения с терапевтической или профилактической целью. Подходящие растворы соединения (1) для фармацевтических препаратов включают воду вместе с различными органическими кислотами (обычно С 4-С 24), как правило, с жирными кислотами, такими как каприновая, олеиновая, лауриновая, пальмитиновая и/или миристиновая кислота. Жирные кислоты могут быть насыщенными или ненасыщенными или же представлять собой смеси насыщенных и ненасыщенных кислот. Кроме того, в дополнение к органическим кислотам или вместо них можно использовать полиэтиленгликоли (ПЭГи) и/или моно-, ди- или триглицериды с короткой, средней или длинной углеродной цепью. Возможно также использование таким же образом пегилированных жирных кислот с короткой, средней или длинной углеродной цепью. Чаще всего органические кислоты являются карбоновыми кислотами, кислотные свойства которых связаны с карбоксильной группой -СООН. Сульфоновые кислоты, содержащие группу OSO3H, являются сравнительно сильными для применения согласно изобретению. В общем случае, кислота желательно содержит липофильный домен. Подходящими являются моно- и дикарбоновые кислоты. Подходящие поверхностно-активные вещества можно использовать во всех составах согласно настоящему изобретению (любое или несколько из нижеперечисленных веществ, как правило, любое из указанных веществ). Такие вещества известны также как эмульгаторы или эмульсификаторы и подходят для применения в фармацевтических композициях согласно настоящему изобретению. Указанные вещества являются неионными, катионными и/или анионными веществами, обладающими подходящими эмульгирующими, диспергирующими и/или смачивающими свойствами. Подходящие анионные поверхностно-активные вещества включают водорастворимые мыла и водорастворимые синтетические поверхностно-активные вещества. Подходящие мыла представляют собой соли щелочных или щелочноземельных металлов, незамещенные или замещенные соли аммония и высших жирных кислот (С 10-С 22),например, натриевые или калиевые соли олеиновой или стериновой кислоты, или смесь солей природных жирных кислот, полученных из кокосового масла или животного жира (tallow oil). Синтетические поверхностно-активные вещества включают натриевые или кальциевые соли полиакриловых кислот; сульфаты и сульфонаты жиров; сульфонированные производные бензимидазола и алкиларилсульфонаты. Сульфаты или сульфонаты жиров обычно присутствуют в форме солей щелочных или щелочноземельных металлов, незамещенных солей аммония или солей аммония, замещенных алкилом или арилом, содержащим от 8 до 22 атомов углерода, например, в виде натриевой или кальциевой соли лигносульфоновой кислоты или додецилсульфоновой кислоты или в виде смеси сульфатов жирных спиртов,полученных из природных жирных спиртов, солей щелочных или щелочно-земельных металлов сложных эфиров серной или сульфоновой кислот (таких как лаурилсульфат натрия) и сульфоновых кислот с продуктами присоединения жирных спиртов / окиси этилена. Подходящие сульфонированные производные бензимидазола предпочтительно содержат от 8 до 22 атомов углерода. Примеры алкиларилсульфонатов включают соли натрия, кальция и додецилбензосульфоновой кислоты или дибутилнафталинсульфоновой кислоты, или продукт конденсации нафталинсульфоновой кислоты и формальдегида. Кроме того, подходящими являются соответствующие фосфаты, например соли сложных эфиров фосфорной кислоты, и продукты присоединения п-нонилфенола и оксида этилена и/или оксида пропилена, или фосфолипиды. Подходящие для применения с данной целью фосфолипиды представляют собой природные(выработанные клетками организма растения или животного) или синтетические фосфолипиды типа цефалина или лецитина, такие как, например, фосфатидилэтаноламин, фосфатидилсерин, фосфатидилглицерин, лизолецитин, кардиолипин, диоктанилфосфатидил-холин, дипальмитоилфосфатидил-холин и их смеси. Водные эмульсии, содержащие такие вещества, также находятся в рамках настоящего изобретения. Подходящие неионные поверхностно-активные вещества включают полиэтоксилированные и полипропоксилированные производные алкилфенолов, жирные спирты, жирные кислоты, алифатические амины или амиды, содержащие по меньшей мере 12 атомов углерода в молекуле, алкиларенсульфонаты и диалкилсульфосукцинаты, такие как производные эфира полигликоля и алифатических и циклоалифатических спиртов, насыщенных и ненасыщенных жирных кислот и алкилфенолов, при этом указанные производные предпочтительно содержат от 3 до 10 гликолевых эфирных групп и от 8 до 20 атомов углерода в (алифатическом) углеводородном фрагменте алкилфенола. Далее, подходящими неионными поверхностно-активными веществами являются водорастворимые продукты присоединения полиэтиленоксида и полипропиленгликоля, этилендиаминопропиленгликоль, содержащий от 1 до 10 атомов углерода в углеродной цепи, при этом указанные продукты присоединения включают от 20 до 250 эфирных групп этиленгликоля и/или от 10 до 100 эфирных групп пропиленгликоля. Подобные соединения обычно содержат от 1 до 5 единиц этиленгликоля на единицу пропиленгликоля. Репрезентативные примеры неионных поверхностно-активных веществ представляют собой нонилфенол-полиэтоксиэтанол, полигликолевые эфиры касторового масла, продукты присоединения полипропилен/полиэтиленоксида, трибутилфеноксиполиэтоксиэтанол, полиэтиленгликоль и октилфеноксиполиэтоксиэтанол. Кроме того, в качестве неионных поверхностно-активных веществ подходят эфиры жирных кислот полиэтиленсорбитана(такие как полиоксиэтилен сорбитана триолеат), глицерол, сорбитан, сахароза и пентаэритрит. Подходящие катионные поверхностно-активные вещества включают четвертичные аммониевые соли, в частности галоиды, содержащие 4 углеводородных радикала, возможно, содержащие в качестве заместителей галоген, фенил, замещенный фенил или гидрокси; например, четвертичные аммониевые соли, содержащие в качестве заместителя у атома N по меньшей мере один алкильный радикал С 8-С 22(например, цетил, лаурил, пальмитил, мистрил и олеил) и, в качестве других заместителей, незамещенный или галогенированный низший алкил, бензил и/или гидроксинизший алкил. Более подробное описание поверхностно-активных веществ, подходящих для применения с данной целью, см. в "McCutcheon's Detergents and Emulsifiers Annual" (MC Publishing Crop., Ridgewood, New Jersey, 1981), "Tensid-Taschenbucw", 2nd ed. (Hanser Verlag, Vienna, 1981) и "Encyclopedia of Surfactants"(Chemical Publishing Co., New York, 1981). Соединение согласно настоящему изобретению вводят любым путем, подходящим при лечении конкретного состояния, например пероральным, ректальным, назальным, местным (включая окулярный,буккальный и сублингвальный), вагинальным и парентеральным (включая подкожный, внутримышечный, внутривенный, внутрикожный, интратекальный и эпидуральный). Предпочтительные пути введения могут быть различными и зависят, например, от состояния субъекта; однако, как правило, введение осуществляют пероральным путем. Составы на основе соединения согласно настоящему изобретению, предназначенные для перорального приема, обычно представлены в виде дискретных единиц, таких как капсулы, облатки или таблетки,каждая из которых содержит определенное количество активного ингредиента; в порошкообразной или гранулированной форме; в виде раствора или суспензии в водной или неводной жидкости; или в виде жидкой эмульсии типа "масло в воде" или "вода в масле". Соединение согласно изобретению может быть представлено в виде болюса, электуария или пасты. Таблетки изготавливают путем прессования или формовки, возможно, с применением одного или нескольких вспомогательных ингредиентов. Прессованные таблетки изготавливают путем прессования в подходящей машине соединения согласно изобретению в свободнотекучей форме, такой как порошок или гранулы, возможно в смеси с наполнителем, смазывающим веществом, инертным разбавителем,консервантом, поверхностно-активным веществом и/или диспергирующим агентом. Формованные таб-5 018173 летки обычно изготавливают путем формования в подходящей машине смеси порошкообразного соединения, смоченного инертным жидким разбавителем. На таблетки можно нанести покрытие или делительную насечку, или же изготовить их так, чтобы обеспечить замедленное или контролируемое высвобождение содержащихся в них активных ингредиентов. Возможно местное применение составов в виде кремов и мазей, содержащих активный ингредиент(ингредиенты) в количестве, составляющем, например, от 0,075 до 20% вес./вес. (включая активный ингредиент (ингредиенты) в интервале от 0,1 до 20% с шагом 0,1% вес./вес., например 0,6% вес./вес., 0.7% вес./вес. и т.д.), предпочтительно от 0,2 до 15% вес./вес. и наиболее предпочтительно от 0,5 до 10% вес./вес. В составе мази соединение используют вместе с парафиновой или водорастворимой мазевой основой. Альтернативно, соединение вводят в состав крема вместе с кремовой основой типа "вода в масле". При желании водная фаза кремовой основы может содержать, например, по меньшей мере 30% вес./вес. многоатомного спирта, т.е. спирта, содержащего две или более гидроксильных групп, такого как пропиленгликоль, бутан-1,3-диол, маннитол, сорбитол, глицерин и полиэтиленгликоль (включая ПЭГ 400) и их смеси. При желании составы для местного применения могут включать соединение, увеличивающее абсорбцию или проникновение активного ингредиента через кожу и другие поврежденные участки. Примеры подобных агентов, усиливающих кожное проникновение, включают диметилсульфоксид и соответствующие аналоги. Масляная фаза эмульсий согласно настоящему изобретению состоит из традиционных ингредиентов, соединенных известными способами. В то время как данная фаза может содержать лишь эмульгатор(называемый также эмульгентом), желательно включение в состав смеси по меньшей мере одного эмульгатора с жиром или маслом либо и с жиром, и с маслом. Гидрофильный эмульгатор можно ввести в состав вместе с липофильным эмульгатором, который выступает в качестве стабилизатора. Кроме того,предпочтительно совместное введение в состав и масла, и жира. Эмульгатор (эмульгаторы) вместе со стабилизатором (стабилизаторами) образуют так называемый эмульгирующий воск, а воск вместе с маслом и жиром образуют так называемую эмульгирующую мазевую основу, образующую масляную дисперсную фазу состава крема. Подбор подходящих масел и жиров для состава основан на достижении заданных косметических характеристик. Таким образом, указанный крем может представлять собой нежирный, не оставляющий пятен легкосмываемый продукт подходящей консистенции, не вытекающий из туб и других видов контейнеров. Возможно применение одно- или двухосновных алкиловых сложных эфиров с линейной или разветвленной цепью, таких как диизоадипат, изоцетилстеарат, пропиленгликолевый диэфир жирных кислот кокосового ореха, изопропилмиристат, децилолеат, изопропилпальмитат, бутилстеарат, 2 этилгексилпалмитат или смесь сложных эфиров с разветвленной цепью, известная как Кродамол КАП,причем последние три вещества являются предпочтительными сложными эфирами. Их можно применять по отдельности или в комбинации, в зависимости от необходимых свойств. В качестве альтернативы можно использовать липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин, или другие минеральные масла. Составы, подходящие для местного офтальмологического применения, включают также глазные капли, в которых активный ингредиент растворен или суспендирован в соответствующем носителе, в частности в водном растворителе, подходящем для растворения активного ингредиента. Активный ингредиент может содержаться в подобных составах в концентрации от 0,5 до 20%, предпочтительно от 0,5 до 10%, в частности в концентрации примерно 1,5% вес./вес. Составы, подходящие для местного применения в ротовой полости, включают лепешки, содержащие активный ингредиент в ароматизированной основе, в общем случае представляющей собой сахарозу и гуммиарабик или трагакант; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик; жидкость для полоскания рта, содержащую активный ингредиент в подходящем жидком носителе. Составы для ректального применения могут иметь форму суппозиториев с подходящей основой,содержащей, например, кокосовое масло или салицилат. Композиции, подходящие для интраназального введения, в которых носитель является твердым веществом, включают крупнодисперсный порошок с размером частиц в интервале, например, 20-500 мкм (включает размер частиц в интервале от 20 до 500 мкм с шагом в 5 мкм, например 30 мкм, 35 мкм и т.д.), вводимый при помощи аэрозольного или порошкового ингалятора, при этом большое количество примеров таких ингаляторов имеется в продаже. Подходящие составы, в которых носителем является жидкость, для применения, например, в виде назального спрея или капель для носа, включают водные или масляные растворы активного ингредиента. Составы, подходящие для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или аэрозолей, содержащих наряду с активным ингредиентом носители,хорошо известные специалистам в данной области техники. Составы, подходящие для введения парентеральным путем, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферные вещества, бактериостаты и растворенные вещества, которые придают составу изотонические по отношению к крови предполагаемого реципиента свойства; а также водные и неводные стерильные суспензии, возможно содержащие суспендирующие агенты и загустители. Эти составы представлены в контейнерах, содержащих единичную дозу или множество доз, например, герметично закрытые ампулы и пузырьки, и могут храниться в сублимированном (лиофилизированном) виде, когда непосредственно перед использованием необходимо лишь добавить к продукту стерильный жидкий носитель, например воду для инъекций. Растворы и суспензии для инъекций, предназначенные для немедленного применения, можно приготовить из стерильных порошков, гранул и таблеток, подобных вышеописанным. Соединение согласно настоящему изобретению может быть получено в виде композиции с контролируемым высвобождением, в которой высвобождение соединения контролируют и регулируют с целью уменьшения частоты приемов дозированного состава или для улучшения фармакокинетического и токсического профиля соединения согласно изобретению. Композиции с контролируемым высвобождением получают традиционными способами, многие из которых включают введение активного соединения в состав вместе с одним или несколькими полимерными носителями, такими как полиэфир, полиаминокислоты, поливинилпирролидон, сополимер этилена с винилацетатом, метилцеллюлоза, карбоксиметилцеллюлоза и/или протаминсульфат. Скорость высвобождения лекарства и длительность действия можно контролировать путем заключения активного ингредиента внутрь частиц, например в микрокапсулы,полученные из полимерных материалов, таких как гидрогели, полимолочная кислота, гидроксиметилцеллюлоза, полиметилметакрилат и другие вышеописанные полимеры. Кроме того, подходящими являются коллоидные системы доставки лекарственного препарата, такие как липосомы, микросферы, микроэмульсии, наночастицы, нанокапсулы и т.д. В зависимости от пути введения фармацевтическая композиция, например таблетки, может требовать нанесения защитной оболочки. Для обеспечения более полного понимания настоящего изобретения далее приведены примеры, которые являются исключительно иллюстративными и не ограничивают настоящее изобретение. Процентные содержания компонентов композиции являются весовыми процентами, если из контекста явным образом не следует иное. Пример 1. Синтез кристаллического 5-6-(2,4-бис-(трифторметил)фенил)пиридазин-3-ил)метил)-2-(2 фторфенил)-5H-имидазо[4,5-c]пиридина. Схема-10 С внутри реактора. Смесь перемешивали при -10 С до завершения реакции по данным ВЭЖХ. Полученную смесь перенесли во второй реактор, содержащий триметилборат (2,26 экв.) и ТГФ при температуре -10 С внутри реактора. Когда по данным ВЭЖХ-мониторинга содержание 1,3-бис(трифторметил)бензола составило не более 2%, реакцию погасили добавлением водной HCl (водного раствора хлористо-водородной кислоты), полученного из воды и концентрированной 37% хлористоводородной кислоты, при этом температуру содержимого реактора поддерживали на уровне, не превышающем 25 С. После перемешивания содержимого в течение 1-2 ч и отстаивания в течение примерно 30 мин слои разделили. Органический слой промыли солевым раствором, смешанным с водой, затем сконцентрировали под вакуумом. Затем добавили гептан и вновь сконцентрировали содержимое под вакуумом. Все операции повторили еще один раз. Вновь добавили гептан и охладили полученную суспензию до 3 С, затем перемешивали при данной температуре в течение 4-6 ч. Продукт отфильтровали, промыли гептаном дважды и высушили под вакуумом, максимальная температура при сушке составила 40 С. 3-Хлор-6-метилпиридазин (1.00 экв.), 2-(дициклогексилфосфино)бифенил (0.05 экв.), 2,4-бис(трифторметил)фенилбороновую кислоту (1.85 экв.), 1,2-диметоксиэтан и водный раствор карбоната калия поместили в реактор. После трехкратного дегазирования азотом добавили ацетат палладия(0,025 экв.) и нагревали содержимое реактора с обратным холодильником при перемешивании до предполагаемого завершения реакции. Реакционную смесь охладили до 22C. Добавили гептан, затем целит. После примерно 30-минутного перемешивания при 22 С смесь отфильтровали в первый реактор, предварительно ополоснутый смесью 1,2-диметоксиэтана и гептана. Слои фильтрата разделили. К органическому слою добавили комплекс боран-триметиламин (0.03 экв.), воду и уксусную кислоту. Полученную смесь с рН не выше 4 перемешивали в течение 1-2 ч при 22 С, затем нагревали с обратным холодильником при температуре около 80 С в течение 2-3 ч. После повторного охлаждения до 22 С,рН смеси довели до 10-11 добавлением 5% водного гидроксида натрия, поддерживая температуру содержимого на уровне 22 С, затем перемешивали смесь в течение 1-2 ч. Смесь отфильтровали и разделили слои. Водный слой отбросили, а органический слой отфильтровали через картриджи ZetaCarbon в первый реактор, очищаемый в ходе процесса, предварительно ополоснутый 1,2-диметоксиэтаном через угольные картриджи. Фильтрат сконцентрировали под вакуумом при максимальной заданной температуре нагревательной рубашки 60 С. Добавили гептан, затем сконцентрировали содержимое под вакуумом при максимальной заданной температуре нагревательной рубашки 60 С. К концентрату добавили дополнительное количество гептана и при помощи ЯМР проверили содержание 1,2-диметоксиэтана (ДМЭ) в смеси (максимальное содержание составило 0,5%). После нагревания до 85 С и перемешивания в течение примерно 1 ч смесь в горячем виде отфильтровали через фильтр окончательной очистки во второй реактор. Во втором реакторе фильтрат нагрели до температуры кипения и затем перемешивали в течение 1 ч. При постепенном охлаждении и перемешивании с умеренной интенсивностью смесь охлаждали от температуры кипения до 0-6 С как минимум в течение 4 ч, затем перемешивали при температуре от 0 до 6 С в течение 1 ч. Продукт отфильтровали, промыли гептаном при температуре окружающей среды и высушили под вакуумом при температуре, не превышающей 40 С, до достижения максимальной потери массы при сушке, составляющей 1%. В реактор поместили метансульфоновую кислоту, затем пентоксид фосфора (1,00 экв.) порциями,при этом поддерживали температуру содержимого на уровне 23 С. Порциями добавляли 3,4 диаминопиридин (1,00 экв.) при поддержании температуры содержимого в интервале от 20 до 50 С максимум. Затем добавили 2-фторбензойную кислоту (1,09 экв.). Смесь нагревали до 100 С, мониторинг реакции до ее завершения осуществляли методом ВЭЖХ. Содержимое охладили до 10 С и добавили воду при поддержании температуры содержимого на уровне 25 С максимум. После перемешивания смеси при указанной температуре в течение 1 ч смесь отфильтровали во второй реактор. К содержащемуся во втором реакторе фильтрату добавили 27% гидроксид аммония до достижения значения рН в интервале 8.0-9.3. Температуру содержимого поддерживали на уровне 30 С максимум. Полученную тонкую дисперсию перемешивали при 22 С в течение как минимум 1 ч, затем добавили 27% гидроксид аммония до достижения значения рН в интервале 8.0-9.3. Затем дисперсию перемешивали при 22 С в течение как минимум 2 ч. Продукт отфильтровали, дважды промыли водой и высушили при максимальной температуре 60 С под вакуумом; содержание воды в конечном продукте не превышало 1%. При необходимости, продукт измельчали для удаления крупных комков. В реактор добавили соединение 2 а (1.24 экв.), метиленхлорид и трихлоризоциануровую кислоту(0.491 экв.). Смесь нагрели до температуры кипения и перемешивали при нагревании с обратным холодильником до завершения реакции. Реакционную смесь охладили до 22 С и добавили целит. После перемешивания в течение 30 мин(минимум) смесь отфильтровали во второй реактор, предварительно ополоснутый трижды метиленхлоридом. Осадок с фильтра отбросили. К фильтрату во втором реакторе добавили 3% водный раствор гидроксида натрия при поддержании температуры содержимого реактора на уровне 22 С. Смесь перемешивали в течение 1-2 ч и разделили слои. Нижний органический слой перенесли в очищенный в ходе процесса первый реактор и сконцентрировали под вакуумом при температуре нагревательной рубашки 45 С(максимум). Добавили метиленхлорид и отфильтровали смесь через фильтр окончательной очистки во второй реактор, очищенный в ходе процесса. Фильтрат сконцентрировали под вакуумом при температуре нагревательной рубашки 45 С (максимум). Добавили диметилформамид (ДМФ) и продолжили концентрирование содержимого реактора. Температуру смеси довели до 22 С и добавили ДМФ, затем соединение-ядро 2 (1.00 экв.) и 10% водный раствор гидроксида натрия при поддержании температуры содержимого на уровне 22 С. Полученную смесь перемешивали при 22C при ВЭЖХ-мониторинге. В ходе реакции проводили мониторинг рН смеси и добавляли 10% водный раствор гидроксида натрия в количестве, необходимом для достижения рН 11-12, по данным рН-метра. По окончании реакции добавили 10% водный раствор гидроксида натрия при поддержании температуры содержимого на уровне 22 С. Смесь разбавили ДМФ и перемешивали в течение 2 ч. В течение минимум 1 ч смесь отфильтровали в первый реактор, очищенный в ходе процесса,содержащий воду, при поддержании температуры содержимого на уровне 16 С, с последующим промыванием ДМФ. Полученную суспензию перемешивали в течение 1-3 ч при 22 С. Неочищенный продукт отфильтровали, промыли водой и метил-трет-бутиловым эфиром (МТБЭ). Неочищенный влажный продукт с фильтра перенесли в первый реактор, добавили этилацетат (EtOAc). Смесь нагрели до температуры кипения и перемешивали при нагревании с обратным холодильником до полного растворения твердых веществ. Содержание воды не должно быть менее 6.0%. При постепенном охлаждении температуру содержимого довели до 22 С в течение как минимум 4 ч. Кристаллизовавшийся продукт отфильтровали и промыли EtOAc, затем вновь перенесли в первый реактор. Добавили этилацетат (EtOAc). Смесь нагрели до температуры кипения и перемешивали при нагревании с обратным холодильником до полного растворения твердых веществ. Содержание воды не должно быть менее 1.0%. Смесь отфильтровали во второй реактор (предварительно обработанныйEtOAc) в горячем виде через фильтр для окончательной очистки, промыв EtOAc. Продукт сконцентрировали при атмосферном давлении. После доведения температуры до 65 С и добавления EtOAc, содержимое сосуда нагрели до температуры кипения и перемешивали при температуре кипения в течение примерно 30 мин. Проверяли содержание воды и, если содержание воды превышало 0,2%, повторяли этот же цикл. В случае, если содержание воды не превышало 0,2%, содержимое реактора нагревали до температуры кипения, затем перемешивали при температуре кипения в течение 1-3 ч. Содержимое реактора постепенно охлаждали до 22 С в течение как минимум 4 ч, затем перемешивали при данной температуре в течение как минимум 8 ч. Продукт отфильтровали, промыли EtOAc и высушили под вакуумом при температуре 60 С максимум. Затем продукт измельчили. Спектры ядерного магнитного резонанса (1H-, 13 С- и 19F-ЯМР). Спектры ядерно-магнитного резонанса (ЯМР) соединения (1) соответствуют его предполагаемой структуре. 13 С-, 19F- и 1H-NMR спектры соединения (1) в ДМСО-d6 были получены на спектрометре Varian Unitylnova-400 FT-NMR. Спектры представлены в нижеследующей таблице. Величины химического сдвига в спектрах ЯМР были установлены в ходе 2D корреляционных экспериментов (COSY, HSQC,НМВС и HSQCTOCSY). 1H- и 13 С-ЯМР величины химического сдвига для соединения (1) относительно эталонного стандарта. Дифференциальная сканирующая калориметрия. Образцы аморфного соединения (1), обозначенные как "Research lot 6", были получены по способу,приведенному в примере 1 а WO 08/005519, включенном в настоящее описание во всей полноте посредством ссылки. Остальные образцы представляли собой кристаллическое соединение (1). Измерения проводили при помощи прибора для дифференциальной сканирующей калориметрии (ДСК) (DSC2010, производство ТА Instruments Corporation), в атмосфере азота, вес образца 51 мг, скорость подъема температуры: 1 С/мин, 5 С/мин или 10 С/мин, открытый алюминиевый тигель и индий в качестве реперного вещества. На кривой ДСК определяли энтальпию, экстраполированную температуру начала пика и температуру, соответствующую вершине эндотермического пика. Сводные результаты ДСК для Research lot 6 и репрезентативных образцов кристаллического соединения (1) в форме свободного основания (для разных партий) представлены в табл. 1 и на фиг. 4 и 5 соответственно. При ДСК-сканировании кристаллической формы соединения (1) со скоростью 1 С/мин энтальпия эндотермического пика составила примерно 80 Дж/г, экстраполированная температура начала пика была равна 233,22.0 С. Температура, соответствующая вершине эндотермического пика, составила 233,93,0 С. Таблица 1 Примеры значений параметров ДСК, полученных для разных партий соединения (1)Hep. пик - неразрешенный пик. Рентгеновская порошковая дифрактометрия - Исследование 1. Образцы, полученные согласно примеру 1 а WO 05/063744 и по способу согласно настоящему изобретению, анализировали в том виде, в котором они были получены, лишь перемешав образцы шпателем перед анализом. Образец зафиксировали в алюминиевой ячейке и провели измерения при помощи рентгеновского порошкового дифрактометра (XRD-6000, Shimadzu Lab X, производство ShimadzuCorporation, источник рентгеновского излучения: Cu-K1 ray, напряжение на трубке: 35 кВ, электрический ток в трубке: 40 мА, скорость сканирования: 2 в мин, непрерывное сканирование, шаг сканирования: 0.02, интервал сканирования: 4-35 вращение по оси : 60 об/мин). На порошковой рентгенограмме, полученной на немикронизированных игольчатых кристаллах соединения (1), присутствуют характеристические дифракционные пики при углах дифракции 213.46,15.59, 16.90, 17.48, 23.05 и 30.15; измерения проводили на рентгеновском порошковом дифрактометре(фиг. 1). Было отмечено, что немикронизированные "тугоплавкие" (235 С) кристаллические формы соединения (1), исследуемые в данном примере, проявляют некоторые эффекты, обусловленные предпочтительной ориентацией и размером частиц. Вследствие этого рентгенограмму, представленную на фиг. 1,следует рассматривать лишь в качестве примера, так как при изменении размера кристалла и его ориентации изменяется величина пиков на рентгенограмме. Кроме того, возможна небольшая ошибка при измерении величины дифракционного пика при вышеупомянутом угле дифракции 2 , обусловленная измерительной аппаратурой или условиями измерений и т.п. Как правило, ошибка измерений попадает в интервал 0.3. В спецификации к Shimadzu XRD-6000 указан интервал 0.04. Кроме того, можно ожидать некоторых отклонений положения пиков, обусловленных изменениями свойств продукта и экспериментальных условий; таким образом, данные по положению пиков следует считать приблизительными. Порошковая рентгенограмма для "низкоплавкой" (220 С) твердой формы соединения (1), входящей в состав продукта, полученного согласно способу, описанному в примере 1 а (или же приведенному в настоящем описании способу, до этапа повторного суспендирования), соответствует аморфному веществу (фиг. 3). Соединение (1) согласно настоящему изобретению обычно проявляет истинную растворимость 0,7 мкг/мл, значение pKa, равное 5.8, значение log P, равное 2.8; и среднегеометрический профиль "рНрастворимость" (по 3 партиям), где рН 2 соответствует растворимость 458 мкг/мл, а рН 7.3 соответствует растворимость 0.7 мкг/мл. Растворимость (среднее геометрическое значение для 3 партий) в искусственных интестинальных жидкостях (натощак: рН 6.4, 0.75 мМ лецитин, 3 мМ таурохолат натрия,270 мОсмоль; после приема пищи: рН 5.0, 3.75 мМ лецитин, 15 мМ таурохолат натрия, 635 мОсмоль) составила 19,1 мкг/мл (натощак) и 122 мкг/мл (после приема пищи). Измеренные параметры варьируются в различных партиях; таким образом, все представленные да- 12018173 лее параметры, за исключением молекулярной массы, следует считать приблизительными. При титровании с кислотами выявили более высокую растворимость в случае мезилата (20 мг/мл) по сравнению с хлоридом (примерно 0.6 мг/мл) или сульфатом (примерно 0.5 мг/мл) в качестве противоиона. Рентгеновская порошковая дифрактометрия - Исследование 2. Другой образец кристаллического соединения (1), полученный по способу согласно настоящему изобретению, проанализировали по способу, аналогичному приведенному в Исследовании 1, с той разницей, что измерения проводили на рентгеновском порошковом дифрактометре PANalytical X'Pert Pro(1.54059 ), напряжением трубки: 45 кВ, силой тока: 40 мА, интервалом сканирования: 1-55 2, величиной шага: 0.008 2, временем накопления данных измерений: 3373 с, скорость сканирования: 0.9 в мин,щель дифрактометра : DS: 1/2, SS: 1/4, время одного оборота: 0.5 с, режим: пропускание (transmission). Результаты представлены на фиг. 2. Пример 2. Приготовление композиций с применением соединения (1). Кристаллическое соединение (1) применяют в качестве полупродукта для получения фармацевтически приемлемых растворов. В представленных ниже примерах состав композиций является таким, что содержание активного ингредиента составляет 10% вес./вес. Ниже приведены примеры количественных составов на основе соединения (1) в виде капсул по 20 и 40 мг, необходимых для получения 12 кг раствора. Количественный состав композиции капсул, включающих соединение (1), 20 и 40 мг Композиция представляет собой раствор этанол/вода - 1:1 (вес./вес.). Удаляют в процессе герметизации капсул. Емкость/сосуд: 12 кг, нержавеющая сталь. В следующем порядке взвешивают: 0.012 кг бутилгидрокситолуола (0.10%); 0.035 кг бутилгидроксианизола (0.35%); 1.2 кг соединения (1) в форме свободного основания (10%); 0.6 кг полисорбата 80 (5%); 10.153 кг олеиновой кислоты (эквивалентно 84.55 г (84.55%. Капсулы, содержащие солюбилизированное соединение (1) в количестве 20 или 40 мг, получают в ходе последовательностей единичных этапов процесса. Лекарственное вещество на основе соединения(1), олеиновую кислоту, полисорбат 80, бутилгидрокситолуол (БГТ) и бутилгидроксианизол (БГА) смешивали до получения раствора. Раствором заполняли твердые желатиновые капсулы, состоящие из двух частей. Затем закрытые капсулы герметизировали водным раствором спирта, который испаряется в процессе герметизации. Перед упаковкой герметичные капсулы проверяли на протечку в ходе испытания под вакуумом. Альтернативные составы. Возможно применение кристаллического соединения формулы (1) в качестве полупродукта для приготовления солюбилизированной формы вместе со следующими агентами: жирные кислоты (с короткой, средней или длинной углеродной цепью, насыщенные или ненасыщенные), обычно от С 4 до С 22. Типичными жирными кислотами являются линолевая кислота, лауриловая кислота, каприновая кислота или олеиновая кислота; спирты, такие как этанол, бензиловый спирт, глицерин, полиэтиленгликоль 200, полиэтиленгликоль 300, полиэтиленгликоль 400; поверхностно-активные вещества, включая ионные и неионные поверхностно-активные вещества. Примеры неионных поверхностно-активных веществ включают эфиры жирных кислот и полиоксиэтиленсорбита, эфиры сорбита и жирной кислоты, производные полиэтоксиэтиленкасторового масла, полиоксиэтиленглицерина оксистеарат, полиэтиленгликоль 60, гидрогенизированное касторовое масло и/или блок-сополимеры этиленоксида и пропиленоксида; антиоксиданты, например бутилгидроксианизол (БГА), бутилгидрокситолуол (БГТ), аскорбилпальмитат, витамин Е и/или ПЭГ 1000 сукцинат витамина Е для химической стабильности; усилитель вязкости (диоксид кремния, полиэтиленгликоли, оксид титана и т.п.); а также смеси вышеперечисленных компонентов. Возможно инкапсулирование состава в капсулу из мягкого эластичного желатина или твердого желатина или твердой гидроксипропилметилцеллюлозы. Жидкий состав (в виде раствора или инкапсулированного раствора) обеспечивает улучшенную биодоступность при пероральном введении. Заполнение капсул. Композиции и способы получения мягких эластичных желатиновых капсул хорошо известны специалистам в данной области техники. Композиция обычно содержит от 30-50% вес./вес. желатина, 1040% вес./вес. пластификатора или смеси пластификаторов и примерно 25-40% вес./вес. воды. Пластификатор может представлять собой глицерин, сорбит или производные сорбита, пропиленгликоль и т.п. или их комбинации. Для получения и заполнения мягких эластичных желатиновых капсул можно применять различные способы, такие как способ с применением ротационной машины, машины для запечатывания (liner machine), машина для изготовления капсул Accogel (accogel machine) и т.п. Твердые желатиновые капсулы или капсулы из гидроксипропилметилцеллюлозы (ГПМЦ) можно приобрести у Capsugel, Greenwood,S.C. и у других производителей. Капсулы заполняют вручную или при помощи машины для заполнения капсул. Получение составов. В общем случае композиции согласно настоящему изобретению можно получить следующим образом. Ингредиенты смешивают в сосуде подходящего размера при помощи миксера, расположенного сверху (возможно продувание емкости, в которой осуществляют смешивание, азотом). Фармацевтически приемлемые жирные кислоты и фармацевтически приемлемые антиоксиданты смешивают при комнатной температуре (в случае необходимости раствор можно нагреть, например, примерно до 45 С в случае лауриловой кислоты, для перевода жирной кислоты в жидкое состояние). Добавляют соединение формулы (1) и перемешивают до растворения. Фармацевтически приемлемое поверхностно-активное вещество добавляют при перемешивании. Подходящими навесками полученной смеси заполняют твердые желатиновые капсулы. Пример 2 а. Микронизированный состав соединения (1). Микронизированное лекарственное вещество (Jet mill-00 при 60-80 psi; средний размер частиц 3-4 мкм, примерно 7-8 кв.м/г) методом сухого смешивания перемешали с лактозой, микрокристаллической целлюлозой, кросскармеллозой натрия, лаурилсульфатом натрия, винной кислотой и гидроксипропилцеллюлозой. Смесь гранулировали путем распыления раствора смеси. Гранулы высушили в псевдоожиженном слое. Высушенные гранулы измельчили в мельнице, затем смешали с дополнительным количеством микрокристаллической целлюлозы и кросскармеллозой натрия. К порошкообразной смеси добавили смазывающий агент - стеарат магния, затем смесь спрессовали в таблетки при помощи ротационного таблеточного пресса. Затем таблетки покрыли пленочной оболочкой. В таблице, приведенной ниже, представлены суммарные результаты испытаний различных составов на собаках, получавших по 40 мг соединения (1), что соответствует примерно 4 мг/кг. Таблица демонстрирует улучшенные характеристики солюбилизированных составов, содержащих соединение (1). Сводные результаты исследований in vivo Использовали микронизированный API; Дозированно вводили собакам, получавшим пентагастрин для снижения рН жидкой среды в желудке.SLStartaric - лаурилсульфат натрия и винная кислота; ППК 24 - площадь под фармакокинетическойй кривой (24 ч); ОСО - относительное стандартное отклонение. Пример 3. Противовирусная активность соединения (1). Соединение согласно настоящему изобретению проявляет активность против репликона ВГС (исследование описано в WO 05/063744) в отношении обоих генотипов 1 а и 1, чрезвычайно низкую цитотоксичность (50000 нМ в клетках Huh-7, HepG2 и МТ 4) и весьма благоприятный коэффициент селективности. Соединение проявляет существенно меньшую активность против генотипа 2 а. Активность соединения 1 против репликонов ВГС генотипа 1 и 1 а. Клетки, содержащие репликоны ВГС генотипа 1 (Con-1/lucneo) и 1 а (Н 77/neo) инкубировали с серийными разведениями соединения (1), 2'С-метил аденозином (2'СМеА) или IFN в течение 3 дней в присутствии или в отсутствие 40 мг/мг сывороточного альбумина человека (HSA). После инкубации содержание РНК репликонов в обработанных клетках определяли либо путем исследования репортера люциферазы (1 репликон), либо количественной ПЦР в режиме реального времени (1 а репликон); полученные данные использовали для расчета значений ЕС 50 (50% эффективной ингибирующей концентрации) для ингибиторов. Показано, что соединение (1) ингибирует репликоны генотипа 1 и генотипа 1 а со значениями EC50 0,6 и 3,6 нМ соответственно (табл. 1). В присутствии сывороточного альбумина человека значение ЕС 50 для соединения (1) повысилось до 11 нМ. Таблица А Активность соединения (1) против репликонов ВГС генотипов 1 а и 1 н.о. - не определено; HSA - сывороточный альбумин человека. Среднее значение ЕС 50 и стандартную ошибку рассчитывали по данным по меньшей мере 4 независимых экспериментов. Активность соединения (1) против репликона ВГС генотипа 1 а и вируса. Противовирусную активность соединения (1) в отношении ВГС генотипа 2 а протестировали на клетках, хронически инфицированных вирусом генотипа 2 а, а также на клетках, реплицирующих субгеномный репликон 2 а. Клетки линии Huh-7, содержащие хронически реплицирующий вирус ВГС генотипа 2 а (J6/JFH-Rluc) или субгеномные репликоны, культивировали с соединением (1) или 2'СМеА в течение 3 дней в отсутствие сывороточного альбумина человека. По окончании культивирования определили количество люциферазы в клетках, содержащих вирус 2 а, и активность протеазы NS3 ВГС в клетках,содержащих репликон 2 а, путем анализа люциферазы (Promega) и нового исследования флуоресценции с временным разрешением соответственно. Противовирусная активность соединения (1) значительно снизилась в обеих моделях клеточных культур, хронически инфицированных ВГС-2 а (ЕС 50=2.9 мкМ), и в модели субгеномного репликона 2 а(ЕС 50=21.9 мкМ) по сравнению с клетками Huh-7, реплицирующими субгеномный репликон ВГС-1(ЕС 50=0.0006 мкМ) (табл. 2). Анализ данных результатов позволяет предположить, что снижение противовирусного воздействия соединения (1) на ВГС генотипа 2 а возможно объясняется генотипными различиями между генотипом 1 и генотипом 2 ВГС. Таблица В Активность соединения (1) против ВГС генотипов 1 а и 2 а н.о. - не определено; HSA - сывороточный альбумин человека; Среднее значение ЕС 50 и стандартную ошибку рассчитывали по данным по меньшей мере 4 независимых экспериментов. Провели оценку цитотоксичности соединения (1) для ряда типов клеток, включая линии клеток, содержащих репликон ВГС (Huh-7, SL3 и МН 4), и линий клеток, не содержащих репликона (HepG2, MT4),путем люминесцентного анализа жизнеспособности клеток с применением CellTiter-Glo LuminescenceCell Viability assay (Promega). Ни в одной из клеточных линий не было отмечено токсического воздействия при максимальных концентрациях исследуемого вещества (50 мкМ) (табл. С). Эти результаты в сочетании с эффективным противовирусным воздействием (ЕС 50 = 0.62-3.6 нМ) на репликоны ВГС-1 и ВГС-1 а указывают на высокий коэффициент селективности (СС 50/ ЕС 50 13000-80000) для Соединения(1). Таблица С Цитотоксичность соединения (1) в линиях клеток, содержащих репликон ВГС а н.о. - не определено; HSA - сывороточный альбумин человека; Среднее значение ЕС 50 и стандартную ошибку рассчитывали по данным по меньшей мере 4 независимых экспериментов;b Линии клеток, содержащих репликон ВГС. Активность соединения (1) в комбинации с IFN In Vitro против ВГС. Пегилированный интерферон- (ПЭГ-IFN-) в комбинации с рибавирином применяют в современной стандартной терапии пациентов с инфекцией ВГС. Исследования in vitro комбинации соединения (1) и IFN- проводили на клетках, содержащих репликоны. Данные анализировали при помощи моделиMacSynergy template, разработанной Причардом (Prichard) и Шипманом (Shipman). Результаты данного исследования позволяют предположить наличие аддитивного взаимодействия между соединением (1) иIFN-. Пример 4. Противовирусная активность, фармакокинетика и безопасность соединения (1) по данным фазы 1 исследования, проведенного среди субъектов, инфицированных ВГС генотипа 1 (впервые на человеке). Рандомизированное двойное слепое исследование с плацебо-контролем направлено на выявление безопасности/переносимости, фармакокинетики и противовирусной активности разовых (часть А) и множественных (часть В) доз соединения (1) (в растворе олеиновой кислоты, как описано выше) у субъа ектов, хронически инфицированных ВГС генотипа 1 (GT-1) без декомпенсированного цирроза. В исследовании принимали участие субъекты в возрасте 18-60 лет, не получавшие ранее лекарственной терапии,состояние здоровья которых в целом было хорошим. В завершенной части исследования (часть А) пять последовательных групп из 6 субъектов были рандомизированы (5:1) для получения разовых возрастающих доз соединения 1 (40, 120, 240, 240-вместе с пищей или 480 мг) или плацебо. В текущей части исследования (часть В) четыре последовательных группы из 12 субъектов были рандомизированы (10:2) для получения множественных возрастающих доз соединения 1 (40 мг BID (два раза в сутки), 120 мг BID, 240 мг QD, 240 мг BID) или плацебо в течение 8 дней. Средний возраст субъекта (из 31 субъекта, принимавших участие в части А) составлял 43,6 лет,преимущественно в группу входили мужчины (20/31), белой расы (25/31), инфицированные либо ВГС Генотип-1a (24) или 1 (6). Средний (диапазон) базовый уровень вирусной нагрузки ВГС составил 6.6 Log10 РНК ME (международных единиц)/мл (5.2-7.3). Была отмечена хорошая переносимость разовых доз соединения (1) при отсутствии серьезных или препятствующих лечению неблагоприятных явлений(НЯ). Самым частым НЯ была головная боль. Все НЯ по степени тяжести оценили как легкие, за исключением одного эпизода умеренной головной боли. Связанных с лечением отклонений 3 и 4 степени в лабораторных показателях отмечено не было. Среднее значение периода полувыведения соединения (1) из плазмы варьировалось от 10 до 15 ч по группам. Системное воздействие препарата повышалось примерно в два раза в случаях введения соединения (1) вместе пищей с высоким содержанием жира. Средняя концентрация соединения (1) через 24 ч после введения натощак дозы 240 мг примерно в 7 раз превышала значение ЕС 50, рассчитанное по связыванию белка репликона ВГС GT-1 in vitro. После введения разовой дозы максимальный противовирусный эффект наблюдали через 24 ч, с отклонением от медианы в интервале от 0.46 до 1.49 Log10 ВГС РНКME (международных единиц)/мл по группам. Индивидуальные отклонения по РНК ВГС среди всех реципиентов соединения (1) варьируются от 0,19 до 2.54 log10 МЕ/мл после введения разовой дозы. Это исследование представляет собой первую клиническую демонстрацию противовирусной активности соединения (1). При воздействии разовой дозы соединения (1) отмечали хорошую переносимость, благоприятные фармакокинетические свойства и высокую противовирусную активность соединения (1). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Фармацевтическая композиция для лечения вирусной инфекции гепатита С, содержащая соединение формулы (1) и одну или более жирных кислот. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что жирная кислота является насыщенной или ненасыщенной и содержит от 4 до 22 атомов углерода в цепи. 3. Фармацевтическая композиция по п.1, отличающаяся тем, что жирная кислота представляет собой каприновую, линолевую, олеиновую, лауриновую, пальмитиновую или миристиновую кислоты. 4. Фармацевтическая композиция по п.1, отличающаяся тем, что жирная кислота представляет собой олеиновую кислоту. 5. Фармацевтическая композиция по п.4, содержащая 8 мас.% соединения (1) и 92 мас.% олеиновой кислоты. 6. Фармацевтическая композиция по п.4, содержащая 20 мас.% соединения (1) и 80 мас.% олеиновой кислоты. 7. Фармацевтическая композиция по п.1, дополнительно содержащая полиэтиленгликоли или моно-,ди- или триглицериды с короткой, средней или длинной углеводородной цепью или пегилированные жирные кислоты с короткой, средней или длинной углеводородной цепью. 8. Фармацевтическая композиция по п.3, дополнительно содержащая одно или более поверхностноактивных веществ, выбранных из сложных эфиров жирных кислот и полиоксиэтиленсорбита, сложных эфиров сорбита и жирной кислоты, производных полиоксиэтиленкасторового масла, полиоксиэтиленглицерин оксистеарата, полиэтиленгликоля 60, гидрогенизированного касторового масла и блоксополимеров этиленоксида и пропиленоксида. 9. Фармацевтическая композиция по п.8, отличающаяся тем, что жирная кислота представляет собой олеиновую кислоту, а поверхностно-активное вещество представляет собой полиоксиэтиленсорбит. 10. Фармацевтическая композиция по п.9, содержащая 8 мас.% соединения (1), 87,4 мас.% олеино- 19018173 вой кислоты и 4,6 мас.% полиоксиэтиленсорбита. 11. Фармацевтическая композиция по п.9, содержащая 20 мас.% соединения (1), 76 мас.% олеиновой кислоты и 4 мас.% полиоксиэтиленсорбита. 12. Фармацевтическая композиция по п.9, содержащая 8 мас.% соединения (1), 86,47 мас.% олеиновой кислоты, 4,6 мас.% полиоксиэтиленсорбита, 0,92 мас.% аэросила 200 и 0,01 мас.% бутилгидрокситолуола (БГТ). 13. Фармацевтическая композиция по п.9, содержащая 8 мас.% соединения (1), 85,55 мас.% олеиновой кислоты, 4,6 мас.% полиоксиэтиленсорбита, 1,84 мас.% аэросила 200 и 0,01 мас.% БГТ. 14. Фармацевтическая композиция по п.9, содержащая 10 мас.% соединения (1), 84,55 мас.% олеиновой кислоты, 5 мас.% полиоксиэтиленсорбита, 0,35 мас.% бутилгидроксианизола (БГА) и 0,1 мас.% БГТ. 15. Фармацевтическая композиция по п.9, дополнительно содержащая один или более спиртов, выбранных из этанола, бензилового спирта, глицерина, полиэтиленгликоля 200, полиэтиленгликоля 300 и полиэтиленгликоля 400. 16. Фармацевтическая композиция по п.9, содержащая 8 мас.% соединения (1), 73,6 мас.% олеиновой кислоты, 9,2 мас.% полиоксиэтиленсорбита и 9,2 мас.% этанола. 17. Фармацевтическая дозированная форма для лечения вирусной инфекции гепатита С, содержащая соединение формулы (1) и одну или более жирных кислот, выбранных из каприновой, олеиновой, лауриновой, пальмитиновой и миристиновой кислот. 18. Фармацевтическая дозированная форма по п.17, содержащая олеиновую кислоту и полиоксиэтиленсорбит. 19. Фармацевтическая дозированная форма по п.17, отличающаяся тем, что указанная дозированная форма представляет собой капсулу. 20. Фармацевтическая дозированная форма по п.19, отличающаяся тем, что указанная капсула представляет собой капсулу с твердой оболочкой. 21. Фармацевтическая дозированная форма по п.19, отличающаяся тем, что указанная капсула представляет собой капсулу с мягкой оболочкой. 22. Фармацевтическая дозированная форма по п.19, отличающаяся тем, что указанная капсула представляет собой капсулу с мягкой желатиновой оболочкой. 23. Фармацевтическая дозированная форма по п.19, отличающаяся тем, что указанная капсула представляет собой гидроксипропилметилцеллюлозную капсулу.
МПК / Метки
МПК: A61P 31/12, A61K 31/4353, C07D 471/04
Метки: форма, содержащие, соединение, фармацевтическая, дозированная, пиридазина, композиция
Код ссылки
<a href="https://eas.patents.su/23-18173-farmacevticheskaya-kompoziciya-i-farmacevticheskaya-dozirovannaya-forma-soderzhashhie-soedinenie-piridazina.html" rel="bookmark" title="База патентов Евразийского Союза">Фармацевтическая композиция и фармацевтическая дозированная форма, содержащие соединение пиридазина</a>
Фармацевтическая композиция и пероральная фармацевтическая дозированная форма (варианты), проявляющие активность в отношении вич-инфекций, лечебный набор и таблетка и способ лечения или предотвращения симптомов или эффектов вич-инфекции

Номер патента: 15145
Опубликовано: 30.06.2011
Авторы: Меннинг Марк М., Олияй Реза, Дал Теренс К.
МПК: A61K 45/06, A61K 31/675, A61K 31/513...
Метки: эффектов, симптомов, таблетка, активность, пероральная, предотвращения, отношении, лечения, лечебный, способ, вич-инфекции, фармацевтическая, вич-инфекций, дозированная, проявляющие, набор, композиция, варианты, форма
Формула / Реферат:
1. Фармацевтическая композиция, проявляющая активность в отношении ВИЧ-инфекций, содержащая фумарат диизопропоксикарбонилоксиметилового эфира [2-(6-аминопурин-9-ил)-1-метилэтоксиметил]фосфоновой кислоты (тенофовир дизопроксил фумарат) и (2R,5S,цис)-4-амино-5-фтор-1-(2-гидроксиметил-1,3-оксатиолан-5-ил)-(1Н)-пиримидин-2-он (эмтрицитабин).2. Композиция по п.1, дополнительно содержащая один или несколько фармацевтически подходящих носителей или...
Вагинальная мукоадгезивная композиция на основе нитрата сертаконазола, фармацевтический набор на основе композиции, фармацевтическая дозированная форма для лечения вульвовагинального кандидоза и способего лечения

Номер патента: 10155
Опубликовано: 30.06.2008
Авторы: Герреро Марта, Рага Мануэль М., Гугльетта Антонио, Паласин Селия, Ромеро Альфонсо
МПК: A61K 9/06, A61P 15/02, A61K 31/4178...
Метки: кандидоза, форма, сертаконазола, фармацевтическая, композиция, нитрата, мукоадгезивная, вульвовагинального, основе, композиции, дозированная, вагинальная, способего, лечения, набор, фармацевтический
Формула / Реферат:
1. Вагинальная мукоадгезивная композиция для введения в однократной дозе, отличающаяся тем, что она содержит 6-10% микронизированного нитрата сертаконазола с размером частиц меньше 80 мкм. 2. Композиция по п.1, отличающаяся тем, что количество микронизированного нитрата сертаконазола составляет 6-7%. 3. Композиция по п.1 или 2, отличающаяся тем, что представляет собой крем или гель. 4. Композиция по п.3, отличающаяся тем, что крем содержит...
Твердая фармацевтическая дозированная форма

Номер патента: 11924
Опубликовано: 30.06.2009
Авторы: Липольд Бернд, Райнхольд Ульрих, Берндль Гунтер, Розенберг Йорг, Брайтенбах Йорг, Алани Ламан, Гхош Соумоджит
МПК: A61K 31/00, A61K 31/425, A61K 9/14...
Метки: фармацевтическая, твердая, форма, дозированная
Формула / Реферат:
1. Твердая фармацевтическая дозированная форма, которая включает твердую дисперсию одного ингибитора ВИЧ протеазы по крайней мере в одном фармацевтически приемлемом водорастворимом полимере и по крайней мере одно фармацевтически приемлемое поверхностно-активное вещество, причем указанный ингибитор ВИЧ протеазы представляет собой...
Фармацевтическая оральная дозированная форма, включающая в себя нестероидное противовоспалительное лекарственное средство

Номер патента: 9515
Опубликовано: 28.02.2008
Авторы: Маркитто Леонардо, Раньи Лорелла, Марьетти Франческа
МПК: A61K 9/08
Метки: нестероидное, оральная, себя, включающая, дозированная, средство, лекарственное, противовоспалительное, форма, фармацевтическая
Формула / Реферат:
1. Фармацевтическая оральная дозированная форма, содержащая трометамин и НПВС (нестероидное противовоспалительное средство), выбранное из группы, состоящей из ибупрофена, напроксена и флурбипрофена, отличающаяся тем, что включает также соединение, выбранное из группы, включающей глицин, витамин В6 и их смеси. 2. Фармацевтическая оральная дозированная форма по п.1, отличающаяся тем, что она включает от 0,2 до 50 мас.ч. трометамина на 1 мас.ч....
Твердая разовая пероральная фармацевтическая дозированная форма саквинавирмезилата и способ ее изготовления

Номер патента: 15349
Опубликовано: 30.06.2011
Авторы: Шах Навнит-Нарговиндас, Чжан Линь, Альбано Антонио А., Инфелд Мартин Хауард, Пхуапрадит Вантани
Метки: способ, изготовления, саквинавирмезилата, разовая, твердая, дозированная, форма, пероральная, фармацевтическая
Формула / Реферат:
1. Твердая разовая пероральная фармацевтическая дозированная форма саквинавирмезилата, содержащая ядро и включающий ядро элемент, выбранный из пленочного покрытия таблетки, капсулы или покрытия овальной таблетки, включающая от примерно 60 до примерно 80% микронизированного саквинавирмезилата в пересчете на мезилат, от примерно 4 до примерно 8% фармацевтически приемлемого растворимого в воде связующего, фармацевтически приемлемое вещество,...
Предыдущий патент: Соединительный узел и защитное кольцо для него
Следующий патент: Вирус гриппа с нарушенной репликацией для экспрессии гетерологичных последовательностей
Случайный патент: Имплантат для артродеза