Кристаллическая форма дигидрохлорида метил ((1s)-1-(((2s)-2-(5-(4′-(2-((2s)-1-((2s)-2-((метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-1н-имидазол-5-ил)-4-бифенилил)-1н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата














Формула / Реферат
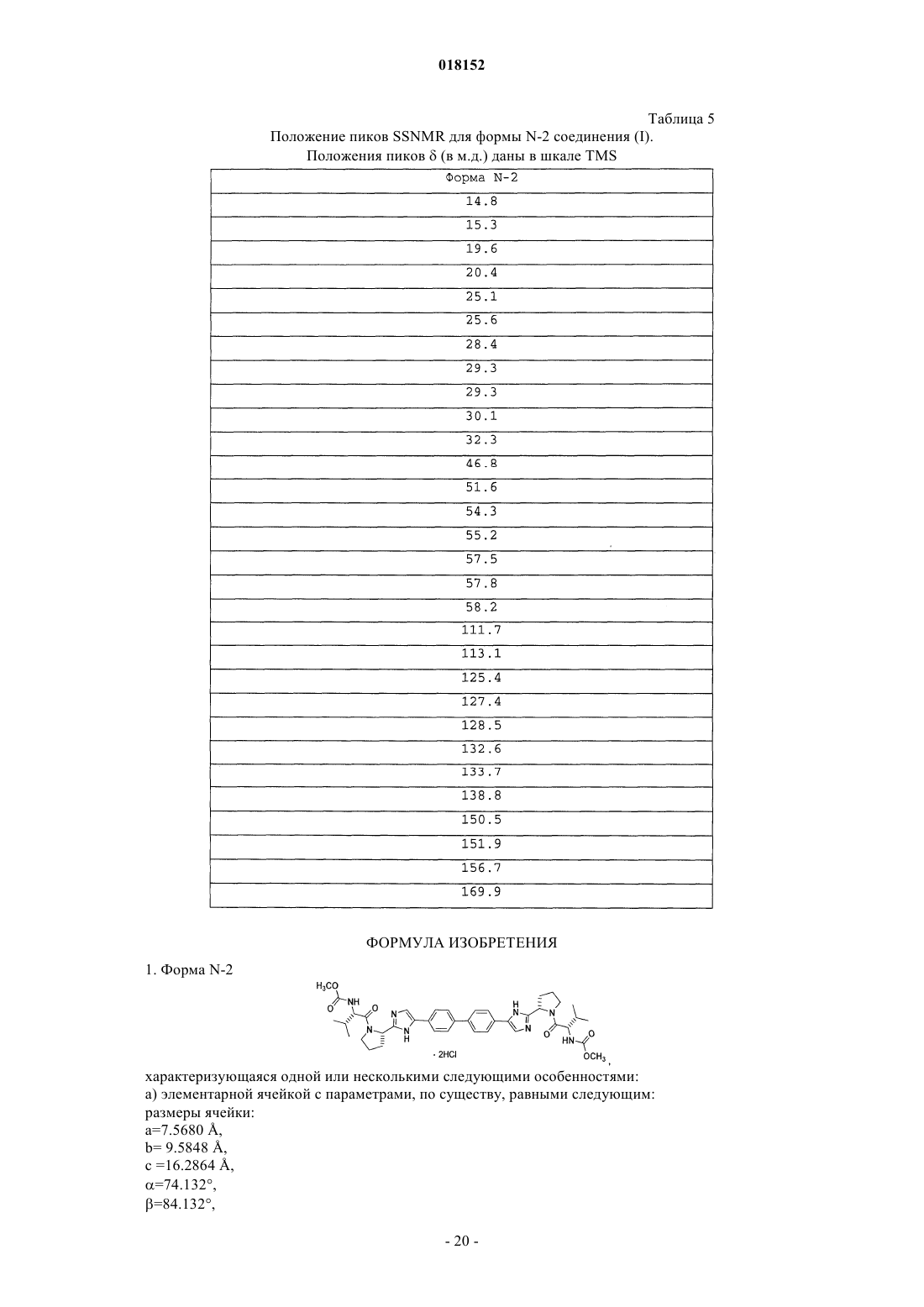
1. Форма N-2

характеризующаяся одной или несколькими следующими особенностями:
a) элементарной ячейкой с параметрами, по существу, равными следующим:
размеры ячейки:
а=7.5680 Å,
b= 9.5848 Å,
с =16.2864 Å,
α=74.132°,
β=84.132°,
γ=70.646°;
пространственная группа Р1:
число молекул в элементарной ячейке 1,
причем определение указанной кристаллической формы проводят при температуре примерно 20-25°С; или
b) координатами атомов в элементарной ячейке, приведенными в табл. 3; или
c) характеристическими пиками на порошковой дифрактограмме при значениях параметра 2θ 10.3±0.1, 12.4±0.1, 12.8±0.1, 13.3±0.1, 13.6±0.1, 15.5±0.1, 20.3±0.1, 21.2±0.1, 22.4±0.1, 22.7±0.1 и 23.7±0.1 при температуре примерно 20-25°С; и/или
d) плавлением с эндотермическим пиком разложения, начинающимся обычно в интервале 225-245°С.
2. Форма по п.1, имеющая чистоту по меньшей мере 95 мас.%.
3. Форма по п.1, имеющая чистоту по меньшей мере 99 мас.%.
4. Фармацевтическая композиция для лечения HCV-инфекции, включающая форму N-2

и фармацевтически приемлемый носитель или разбавитель.
5. Фармацевтическая композиция по п.4, в которой форма N-2 имеет чистоту по меньшей мере 95 мас.%.
6. Фармацевтическая композиция по п.4, в которой форма N-2 имеет чистоту по меньшей мере 99 мас.%.
7. Фармацевтическая композиция, включающая форму N-2

в комбинации с одним или несколькими дополнительными соединениями, обладающими анти-HCV активностью.
8. Фармацевтическая композиция по п.7, в которой форма N-2 имеет чистоту по меньшей мере 90 мас.%.
9. Фармацевтическая композиция по п.7, в которой форма N-2 имеет чистоту по меньшей мере 95 мас.%.
10. Фармацевтическая композиция по п.7, в которой форма N-2 имеет чистоту по меньшей мере 99 мас.%.
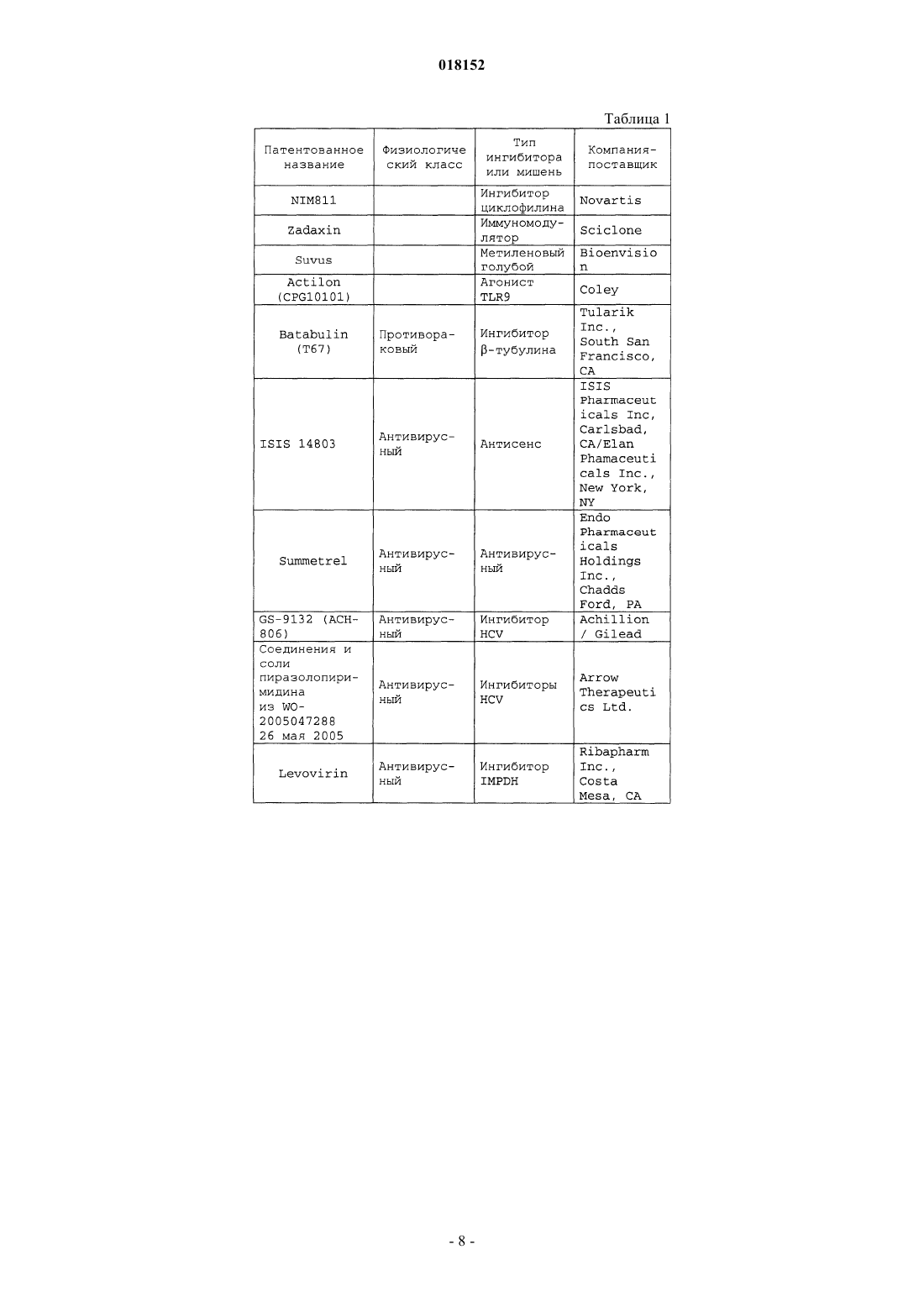
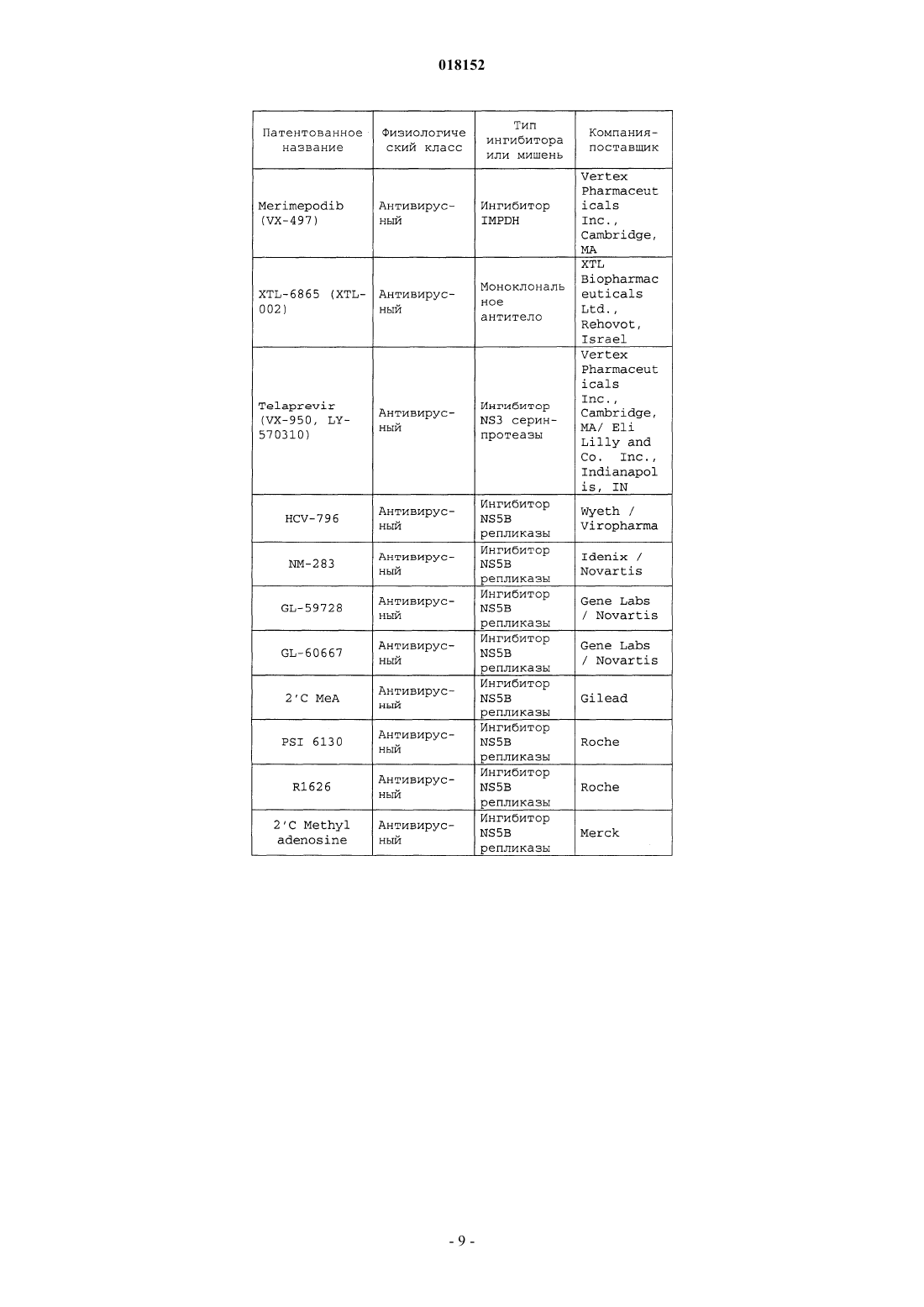
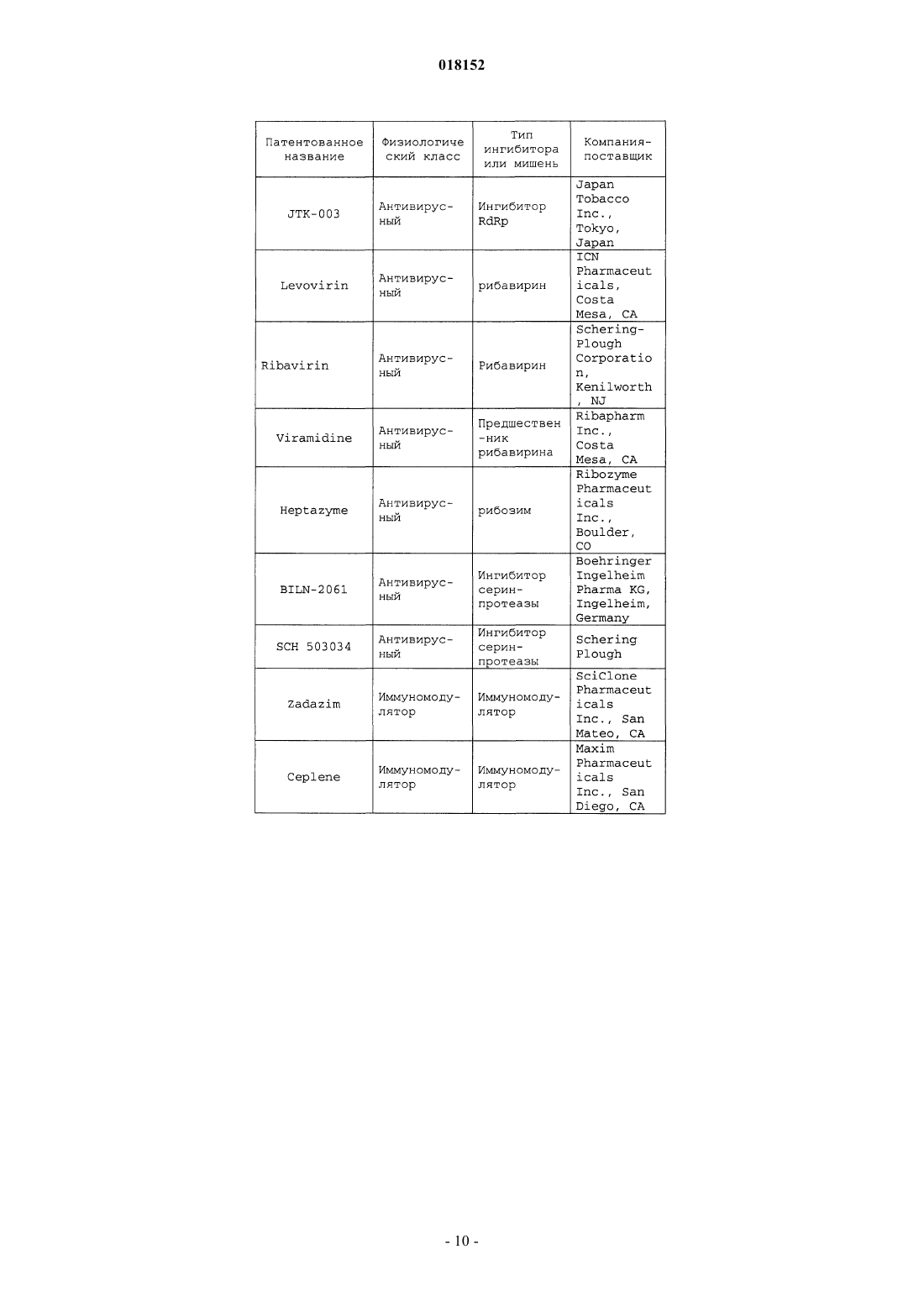
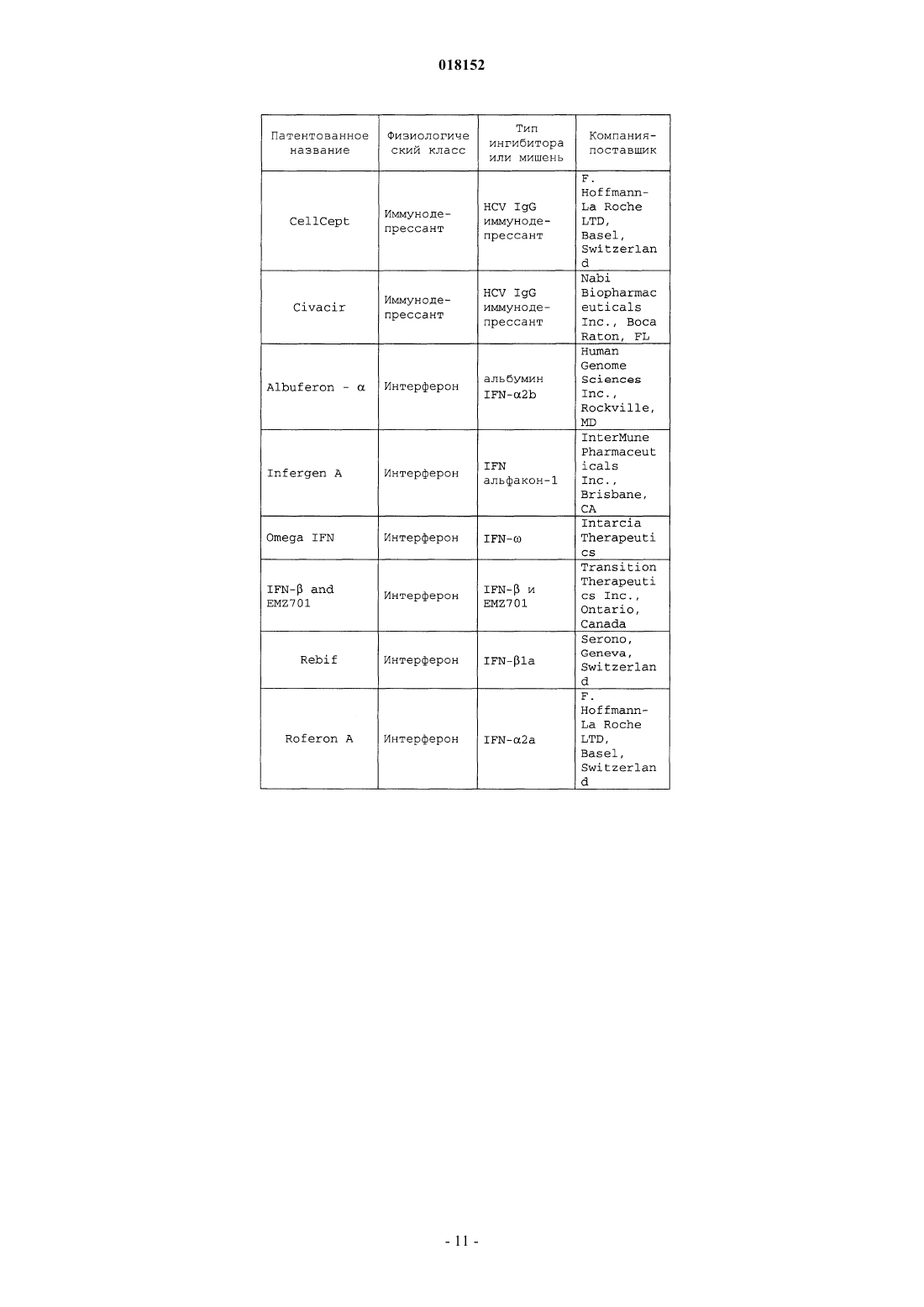
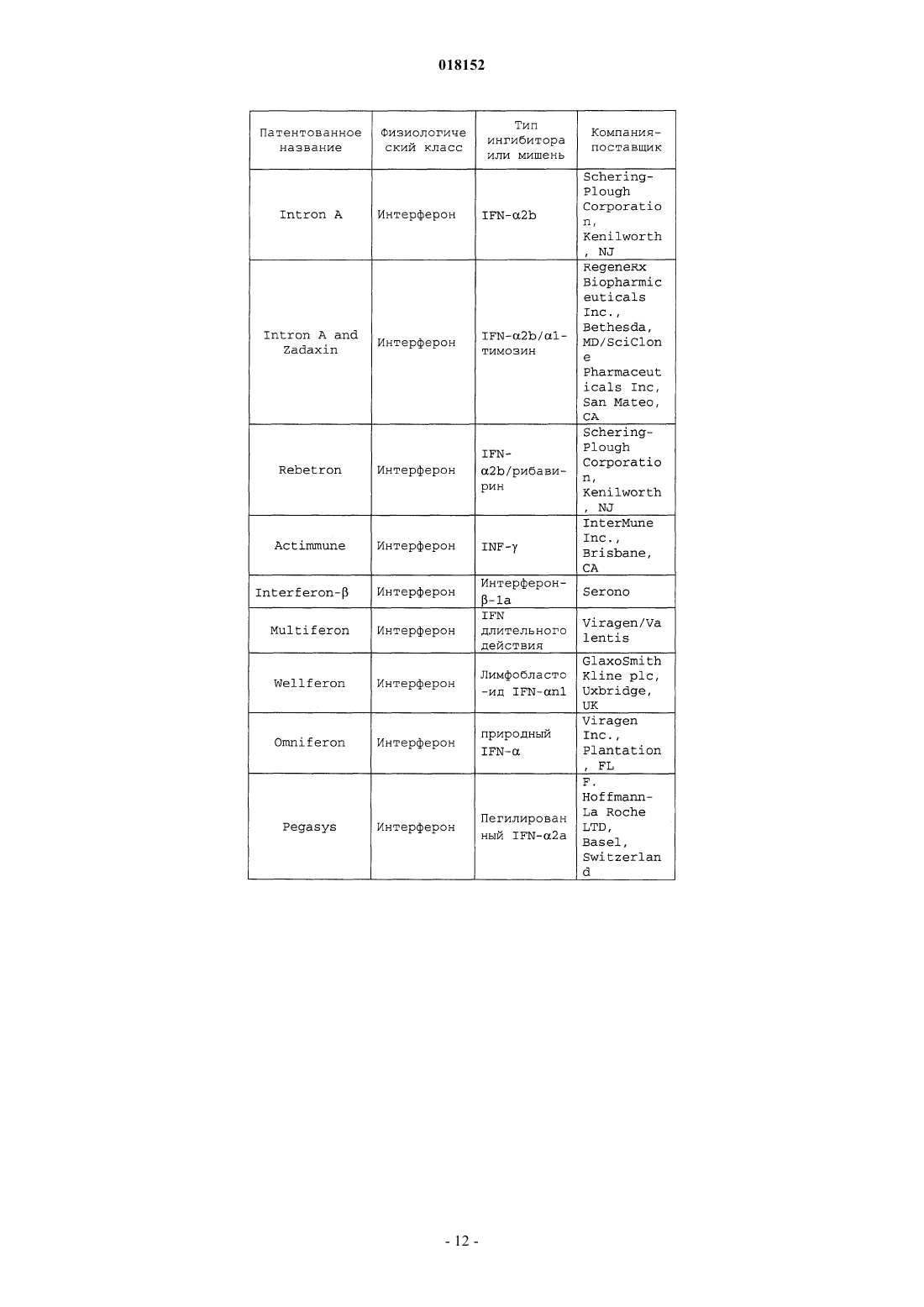
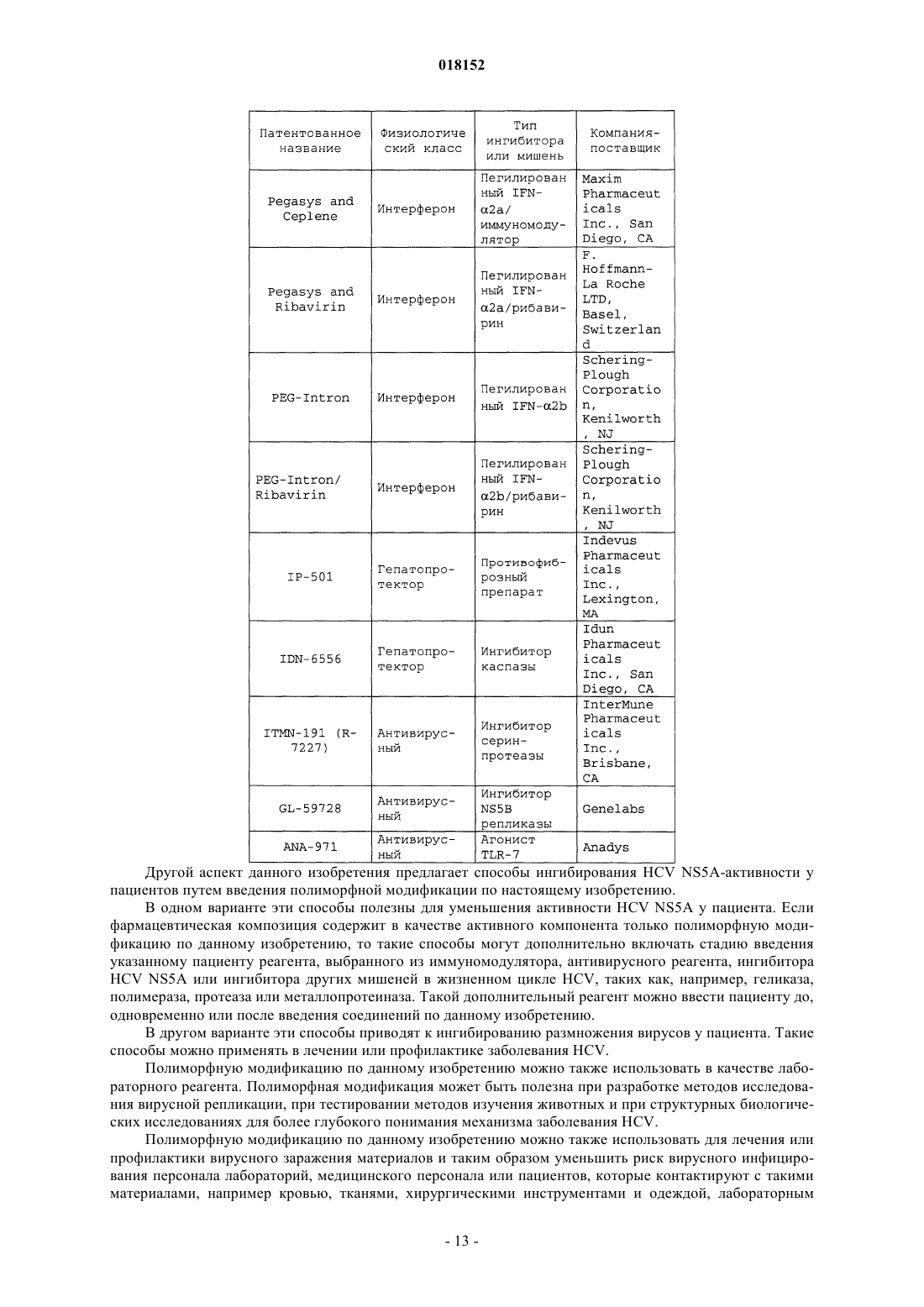
11. Композиция по п.7, в которой по меньшей мере одно из дополнительных соединений с анти-HCV активностью представляет собой интерферон или рибавирин.
12. Композиция по п.11, в которой интерферон выбирают из интерферона альфа 2В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2А и лимфобластоидного интерферона тау.
13. Композиция по п.7, в которой по меньшей мере одно дополнительное соединение выбирают из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения, стимулирующего выработку отклика Т-хелпера клетки типа 1, интерферирующей РНК, антисенс-РНК, имихимода, рибавирина, ингибитора инозин 5'-монофосфатдегидрогеназы, амантадина и римантадина.
14. Способ лечения HCV-инфекции у млекопитающих, включающий введение млекопитающему терапевтически эффективного количества формы N-2

15. Способ по п.14, в котором форма N-2 имеет чистоту по меньшей мере 90 мас.%.
16. Способ по п.14, в котором форма N-2 имеет чистоту по меньшей мере 95 мас.%.
17. Способ по п.14, в котором форма N-2 имеет чистоту по меньшей мере 99 мас.%.
18. Способ по п.14, в котором млекопитающее является человеком.
Текст
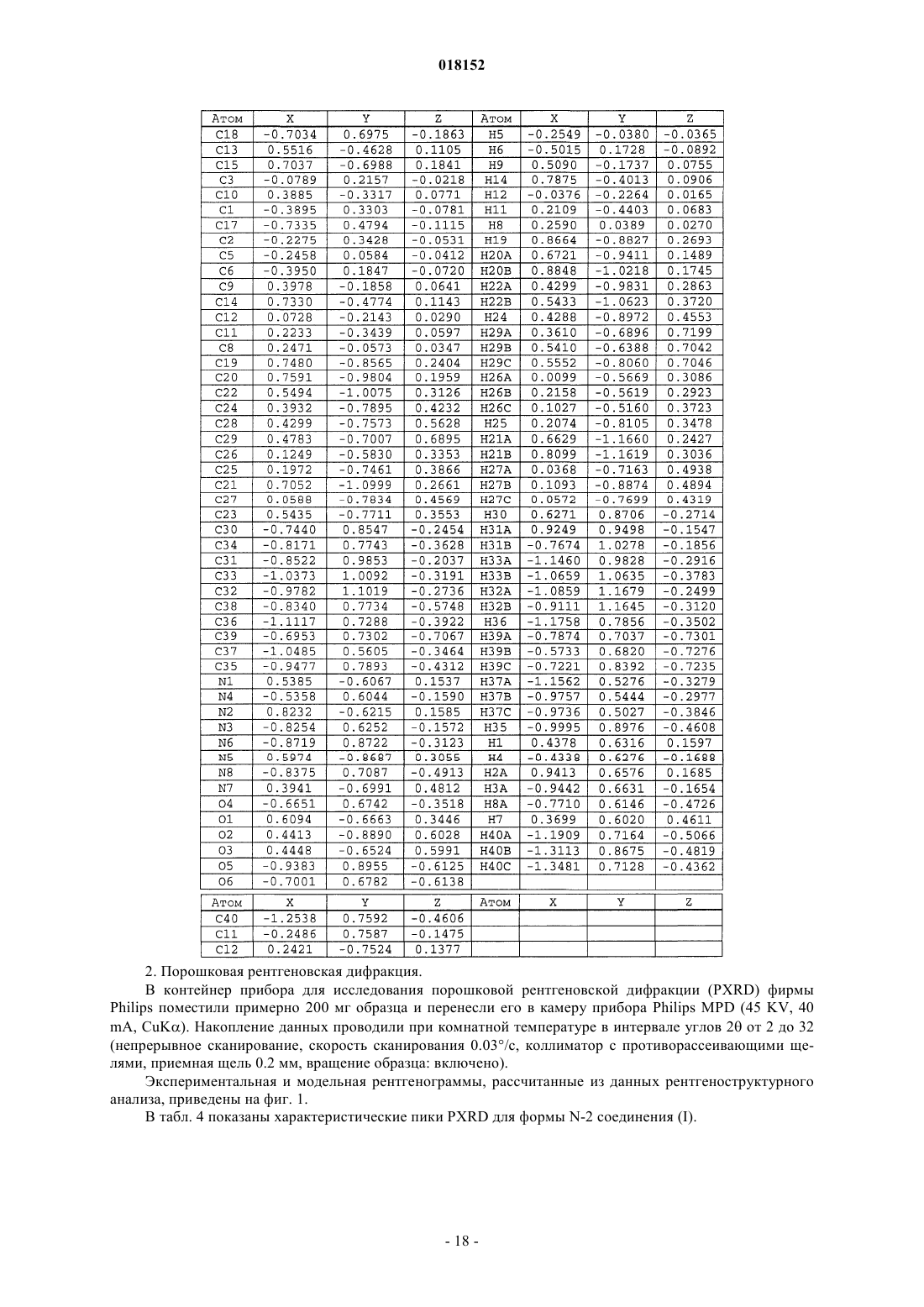
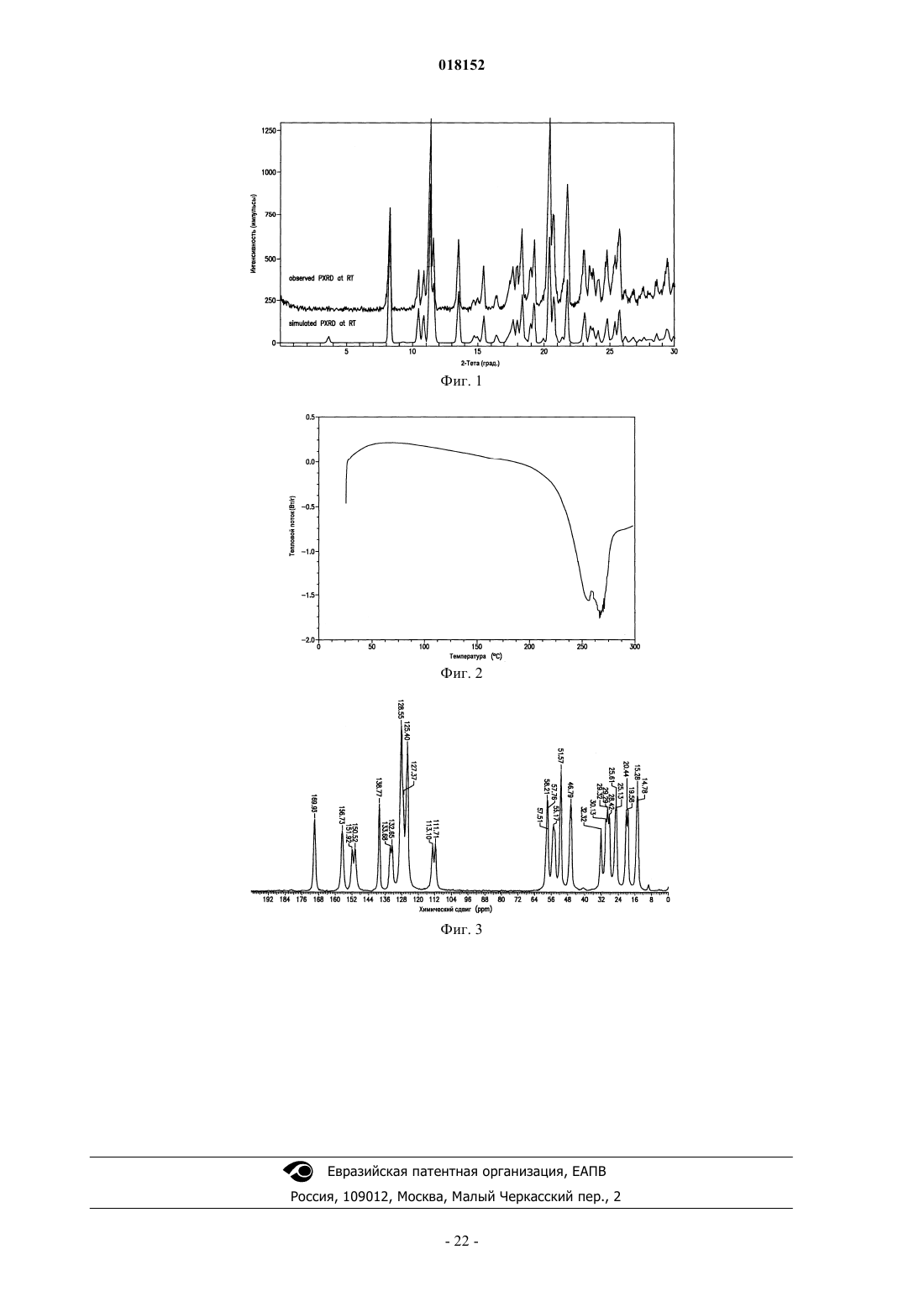
КРИСТАЛЛИЧЕСКАЯ ФОРМА ДИГИДРОХЛОРИДА МЕТИЛ 1S)-1-2S)-2-(5-(4'-(22S)-1-2S)-2-МЕТОКСИКАРБОНИЛ)АМИНО)-3-МЕТИЛБУТАНОИЛ)-2-ПИРРОЛИДИНИЛ)-1 Н-ИМИДАЗОЛ-5-ИЛ)-4-БИФЕНИЛИЛ)-1 Н-ИМИДАЗОЛ-2-ИЛ)-1-ПИРРОЛИДИНИЛ)КАРБОНИЛ)-2-МЕТИЛПРОПИЛ)КАРБАМАТА Описание в целом относится к кристаллической форме дигидрохлорида метил 1S)-1-2S)-2-(5(4'-(2-2S)-1-2S)-2-метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-1 Н-имидазол-5-ил)-4-бифенилил)-1 Н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата. Описание в целом также относится к фармацевтической композиции, включающей данную кристаллическую форму, а также к способам использования кристаллической формы в лечении гепатита С и способам получения такой кристаллической формы. Настоящее изобретение в целом относится к кристаллической форме дигидрохлорида метил 1S)-12S)-2-(5-(4'-(2-2S)-1-2S)-2-метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-1 Н-имидазол-5-ил)-4-бифенилил)-1 Н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата. Настоящее изобретение также в целом относится к фармацевтической композиции, включающей эту кристаллическую форму, а также к способам использования кристаллической формы в лечении вируса гепатита С (HCV) и способам получения кристаллической формы. Вирус гепатита С (HCV) является распространенной патологией человека, поражающей 170 млн человек в мире, примерно в пять раз больше числа инфицированных вирусом иммунодефицита человека типа 1. У значительной части инфицированных HCV людей развиваются серьезные поражения печени,включая цирроз и гепатоцеллюларную карциному. В настоящее время наиболее эффективная терапия HCV включает комбинацию альфа-интерферона и рибавирина, что дает стойкий эффект у 40% пациентов. Недавние клинические данные показывают,что пегилированный альфа-интерферон превосходит немодифицированный альфа-интерферон, если его применяют в качестве единственного средства. Однако даже в экспериментах с терапевтическими режимами, включающими комбинации пегилированного альфа-интерферона и рибавирина, у значительного числа пациентов не отмечается стойкого уменьшения вирусной нагрузки. Таким образом, существует четкая и до сих пор не удовлетворенная потребность в разработке эффективных терапевтических средств для лечения HCV-инфекции. Для лечения HCV-инфекции используют метил 1S)-1-2S)-2-(5-(4'-(2-2S)-1-2S)-2 метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-1 Н-имидазол-5-ил)-4-бифенилил)-1 Нимидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамат. Это соединение плохо кристаллизуется, что ухудшает воспроизводимость при получении чистого продукта. Однако было установлено,что его соль в виде дигидрохлорида, представленная формулой (I) и называемая здесь соединением (I),можно многократно кристаллизовать с образованием единственной полиморфной модификации, называемой здесь формой N-2, которая хорошо растворима в воде и ее легко получить в чистом виде. В первом аспекте настоящее изобретение предлагает форму N-2 Во втором аспекте настоящее изобретение предлагает форму N-2 которая имеет следующие параметры элементарной ячейки: размеры ячейки: а=7.5680 ,b=9.5848 ,с=16.2864 ,=74.132,=84.132,=70.646; пространственная группа Р 1: число молекул в элементарной ячейке 1, причем параметры ячейки указанной кристаллической формы определены при температуре примерно 20-25 С. В третьем аспекте настоящее изобретение предлагает форму N-2 с координатами атомов в элементарной ячейке, приведенными в табл. 3. В четвертом аспекте настоящее изобретение предлагает форму N-2 с характеристическими пиками на порошковой рентгенограмме при значениях 2, равных 10.30.1,12.40.1, 12.80.1, 13.30.1, 13.60.1, 15.50.1, 20.30.1, 21.20.1, 22.40.1, 22.70.1 и 23.70.1, полученной на дифрактометре (CuK) с вращающимся капилляром при температуре примерно 20-25 С, причем значения 2 калибровали по другому подходящему эталону из базы NIST. В пятом аспекте настоящее изобретение предлагает форму N-2a) элементарная ячейка с параметрами, практически равными следующим величинам: размеры ячейки: а=7.5680 ,b= 9.5848 ,с=16.2864 ,=74.132,=84.132,=70.646; пространственная группа Р 1: число молекул в элементарной ячейке 1, причем параметры указанной кристаллической формы определены при температуре примерно 20-25 С:b) характеристические пики на порошковой рентгенограмме при значениях 2, равных 10.30.1,12.40.1, 12.80.1, 13.30.1, 13.60.1, 15.50.1, 20.30.1, 21.20.1, 22.40.1, 22.70.1 и 23.70.1, зарегистрированных при температуре примерно 20-25 С на высококачественной рентгенограмме, полученной на дифрактометре (CuK) с вращающимся капилляром и калибровкой 2 по другому подходящему эталону из базы NIST; и/илиc) расплав с эндотермическим эффектом разложения, начало которого обычно находится в интервале 225-245 С. В шестом аспекте настоящее изобретение предлагает практически чистую форму N-2 В первом варианте шестого аспекта чистота указанной формы N-2 составляет по меньшей мере 95 мас.%. Во втором варианте шестого аспекта чистота указанной формы N-2 составляет по меньшей мере 99 мас.%. В седьмом аспекте настоящее изобретение предлагает практически чистую форму N-2 с характеристическими пиками на порошковой рентгенограмме при значениях 2 10.30.1,12.40.1, 12.80.1, 13.30.1, 13.60.1, 15.50.1, 20.30.1, 21.20.1, 22.40.1, 22.70.1 и 23.70.1, наблюдаемыми на высококачественной рентгенограмме, зарегистрированной при температуре примерно 2025 С на дифрактометре (CuK) с вращающимся капилляром и калибровкой 2 по другому подходящему эталону из базы NIST. В восьмом аспекте настоящее изобретение предлагает фармацевтическую композицию, включающую форму N-2 и фармацевтически приемлемый носитель или разбавитель. В девятом аспекте настоящее изобретение предлагает фармацевтическую композицию, содержащую практически чистую форму N-2 и фармацевтически приемлемый носитель или разбавитель. В первом варианте девятого аспекта чистота указанной формы N-2 составляет по меньшей мере 95 мас.%. Во втором варианте девятого аспекта чистота указанной формы N-2 составляет по меньшей мере 99 мас.%. В десятом аспекте настоящего изобретения предлагается фармацевтическая композиция, содержащая форму N-2 в комбинации с одним или несколькими дополнительными соединениями, обладающими анти-HCV активностью. В первом варианте десятого аспекта чистота указанной формы N-2 составляет по меньшей мере 90 мас.%. Во втором варианте десятого аспекта чистота указанной формы N-2 составляет по меньшей мере 95 мас.%. В третьем варианте десятого аспекта чистота указанной формы N-2 составляет по меньшей мере 99 мас.%. В варианте десятого аспекта настоящего изобретения по меньшей мере одно из дополнительных соединений с анти-HCV активностью представляет собой интерферон или рибавирин. В пятом варианте десятого аспекта интерферон выбирают из интерферона альфа 2 В, пегилированного интерферона альфа,консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. В другом варианте десятого аспекта настоящее изобретение предлагает фармацевтическую композицию, включающую форму N-2 в комбинации с одним или двумя дополнительными соединениями, обладающими анти-HCV активностью, причем по меньшей мере одно из дополнительных соединений выбирают из интерлейкина 2,интерлейкина 6, интерлейкина 12, соединения, стимулирующего отклик Т-хелпера клетки типа 1, интерферирующей РНК,антисенс-РНК,имихимода,рибавирина,ингибитора инозин 5'монофосфатдегидрогеназы, амантадина и римантадина. В одиннадцатом аспекте настоящее изобретение предлагает способ лечения HCV-инфекции у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества формы N-2 В варианте одиннадцатого аспекта указанная форма N-2 имеет чистоту по меньшей мере 90 мас.%. Во втором варианте одиннадцатого аспекта указанная форма N-2 имеет чистоту по меньшей мере 95 мас.%. В третьем варианте одиннадцатого аспекта указанная форма N-2 имеет чистоту по меньшей мере 99 мас.%. В четвертом варианте одиннадцатого аспекта млекопитающее представляет собой человека. Другие варианты настоящего изобретения могут включать подходящие комбинации двух или не-3 018152 скольких приведенных в нем вариантов и/или аспектов. Другие варианты и аспекты изобретения будут очевидны из изобретения, приведенного ниже. Соединения настоящего изобретения также существуют как таутомеры; поэтому настоящее изобретение включает также все таутомерные формы. Фиг. 1 иллюстрирует экспериментальные и модельные порошковые дифрактограммы (CuK=1.54178 при комнатной температуре) кристаллической формы N-2 соединения (I). Фиг. 2 иллюстрирует результаты исследования кристаллической формы N-2 соединения (I) методом дифференциальной сканирующей калориметрии. Фиг. 3 демонстрирует спектры твердофазного ЯМР для кристаллической формы N-2 соединения (I). Настоящее изобретение относится к кристаллической форме соединения (I) Определения Использованный здесь термин "полиморфная модификация" относится к кристаллическим формам одинакового химического состава, но с различным пространственным расположением молекул, атомов и/или ионов, образующих кристалл. Использованный здесь термин "фармацевтически приемлемый" относится к соединениям, материалам, композициям и/или лекарственным формам, которые с точки зрения здравого медицинского смысла пригодны для контакта с тканями человека и животных без избыточной токсичности, раздражения, аллергической реакции или других проблемных осложнений при разумном соотношении успеха и риска. Использованный здесь термин "практически чистый" относится к форме N-2 соединения (I), чистота которой не ниже 90%. Это означает, что полиморфная модификация соединения (I) содержит не более примерно 10% любого другого соединения и, в частности, не более примерно 10% любой другой формы соединения (I). Использованный здесь термин "терапевтически эффективное количество" включает количество кристаллических форм соединения (I), которое эффективно при введении в чистом виде или в комбинации для лечения гепатита С. При использовании комбинации соединения (I) с другим медикаментом описанная здесь комбинация соединений может проявить синергетический эффект. Синергизм, как описано, например, у Chou and Talalay, Adv. Enzyme Regul. 1984, 22, 27-55, проявляется тогда, когда эффект от соединений, вводимых в комбинации, оказывается большим, чем эффект от суммы соединений, вводимых по отдельности. Термин "лечение" относится к (i) профилактике заболевания, расстройства или состояния у пациента, склонного к заболеванию, расстройству или состоянию, которые еще не диагностированы; (ii) ингибированию заболевания, расстройства или состояния, т.е. остановке их развития; и/или (iii) облегчению заболевания, расстройства или состояния, т.е. обратному развитию заболевания, расстройства и/или состояния. В одном варианте данное изобретение предлагает кристаллическую форму соединения (I). Эту кристаллическую форму соединения (I) можно использовать в фармацевтических композициях, которые могут необязательно включать один или несколько компонентов, которые выбирают, например, из группы, состоящей из эксципиентов, носителей, или один из других активных фармацевтических ингредиентов с активными химическими фрагментами другой молекулярной структуры. В одном варианте на фазовую однородность кристаллической формы указывает тот факт, что площадь дополнительных пиков, отсутствующих на модельной рентгенограмме, составляет менее 10%, в другом варианте менее 5% и в еще одном варианте менее 2% от общей площади пиков на полученной экспериментально рентгенограмме кристаллической формы. В другом варианте на фазовую однородность кристаллической формы указывает тот факт, что площадь дополнительных пиков, отсутствующих на модельной рентгенограмме, составляет менее 1% от общей площади пиков на экспериментально определенной рентгенограмме кристаллической формы. В одном варианте предложена композиция, состоящая исключительно из кристаллической формыN-2 соединения (I). Композиция по данному варианту может включать по меньшей мере 90 мас.% кристаллической формы N-2 соединения (I) в расчете на массу соединения (I) в композиции. Остальное вещество представляет собой другие формы соединения и/или примеси от реакции, и/или примеси от обработки, попавшие в ходе приготовления. Присутствие примесей от реакции и/или примесей от обработки можно определить аналитическими методами, известными в данной области, такими как, например, хроматография, масс-спектрометрия или инфракрасная спектроскопия. Получение кристаллических материалов. Кристаллические формы можно получить разными способами, включая, например, кристаллизацию или перекристаллизацию из подходящего растворителя, возгонку, выращивание из расплава, твердофазное превращение из другой фазы, кристаллизацию из сверхкритической жидкости и струйное распыление. Методики кристаллизации или перекристаллизации кристаллических форм из смеси растворителей включают, например, упаривание растворителя, понижение температуры смеси растворителей, внесение в перенасыщенную смесь растворителей затравки кристалла молекулярной формы или ее соли, сушку смеси растворителей с замораживанием и добавление антирастворителей (противорастворителей). Для получения кристаллических форм, включая полиморфные модификации, можно использовать высокопроизводительные способы кристаллизации. Кристаллы лекарственных соединений, включая полиморфные модификации, способы их получения и характеризация кристаллов лекарственных соединений описаны в книге "Твердофазная химия лекарств", S.R. Byrn, R.R. Pfeiffer, and J.G. Stowell, 2nd Edition,SSCI, West Lafayette, Indiana (1999). В методике кристаллизации с применением растворителя выбор растворителя или растворителей обычно зависит от одного или нескольких факторов, таких как растворимость соединения, методика кристаллизации и давление пара растворителя. Можно использовать комбинацию растворителей; например соединение можно растворить в первом растворителе с образованием раствора, затем добавить антирастворитель для уменьшения растворимости соединения в растворе и образования кристаллов. Антирастворитель представляет собой растворитель, в котором данное соединение имеет низкую растворимость. В одном способе получения кристаллов соединение суспендируют и/или перемешивают в подходящем растворителе с образованием суспензии, которую для ускорения растворения можно нагреть. Использованный здесь термин "суспензия" означает насыщенный раствор соединения, который может также содержать дополнительное количество соединения с образованием гетерогенной смеси соединения и растворителя при данной температуре. Затравочные кристаллы можно добавить к любой кристаллизационной смеси для ускорения кристаллизации. Внесение затравки можно использовать для регулирования роста конкретной полиморфной модификации или для регулирования распределения частиц кристаллического продукта по размерам. Соответственно расчет необходимого количества затравки зависит от размера доступной затравки и нужного среднего размера частиц, как описано, например, в работе "Программированное охлаждение периодических кристаллизаторов", J.W. Mullin and J. Nyvlt, Chemical Engineering Science, 1971, 26, 369377. В целом затравки из частиц малого размера нужны для регулирования эффективности роста кристаллов в данной порции. Частицы затравки малого размера можно получить путем отсеивания, измельчения или дробления крупных кристаллов до микронных размеров или микрокристаллизацией из растворов. Следует обращать внимание на то, чтобы измельчение или дробление до микронных размеров не приводили к изменениям кристалличности нужной кристаллической формы (т.е. изменению до аморфной фазы или другой полиморфной модификации). Охлажденную кристаллизационную смесь можно отфильтровать в вакууме и промыть выделенное твердое вещество подходящим растворителем, таким как холодный растворитель перекристаллизации, и высушить в атмосфере азота с образованием нужной кристаллической формы. Выделенные твердые вещества можно проанализировать подходящими спектроскопическими или аналитическими методами,такими как твердофазный ядерный магнитный резонанс, дифференциальная сканирующая калориметрия,рентгеновская порошковая дифракция и т.п., для подтверждения образования предпочтительной кристаллической формы продукта. Конечную кристаллическую форму обычно получают с выходом более примерно 70 мас.%, предпочтительно более 90 мас.% в расчете на массу исходного соединения, введенного в процедуру кристаллизации. Если нужно, продукт можно измельчить или пропустить через сито. Кристаллические формы можно приготовить непосредственно в реакционной смеси на конечной стадии процесса приготовления соединения (I). Это можно осуществить, например, применяя на конечной стадии растворитель или смесь растворителей, из которых можно кристаллизовать соединение (I). Альтернативно кристаллические формы можно получить способами дистилляции или введения растворителя. Подходящие растворители для этой цели включают, например, приведенные выше неполярные растворители и полярные растворители, в том числе протонные полярные растворители типа спиртов и апротонные полярные растворители типа кетонов. Присутствие в образце более одной полиморфной модификации можно установить такими методами, как порошковая рентгеновская дифракция (PXRD) или спектроскопия твердофазного ядерного магнитного резонанса (SSNMR). Например, присутствие дополнительных пиков на экспериментальной рентгенограмме PXRD образца по сравнению с модельной рентгенограммой PXRD может указать на наличие более одной полиморфной модификации. Модельную рентгенограмму PXRD можно рассчитать из рентгеновских данных для монокристалла; см. Smith, D.K., "Программа FORTRAN для расчета рентгеновских порошковых дифрактограмм", Lawrence Radiation Laboratory, Livermore, California, UCRL7196 (April 1963). Характеризация. Форму N-2 соединения (I) можно охарактеризовать разными методами, хорошо известными специалистам в данной области. Примеры способов характеризации включают, но не ограничиваются ими,рентгеноструктурный анализ, порошковую рентгеновскую дифракцию (PXRD), модельные порошковые дифрактограммы (Yin, S.; Scaringe, R.P.; DiMarco, J.; Galella, M. and Gougoutas, J. Z., American Pharmaceutical Review, 2003, 6, 2, 80), дифференциальную сканирующую калориметрию (DSC), твердофазный 13C ЯМР (Earl, W.L. and Van der Hart, D.L., J. Magn. Reson., 1982, 48, 35-54), спектроскопию комбинационного рассеяния, инфракрасную спектроскопию, изотермы сорбции влаги, термогравиметрический анализ(TGA) и методы с использованием нагревательного столика. Кристаллические формы можно охарактеризовать и различить методом рентгеновской дифракции,основанным на определении параметров элементарной ячейки в монокристалле формы N-2. Подробное описание элементарных ячеек содержится в работе StoutJensen, X-Ray Structure Determination: A Practical Guide, Macmillan Co., New York (1968), Chapter 3, которая включена здесь ссылкой. Помимо этого можно определить пространственное расположение атомов в кристаллической решетке по наблюдаемым координатам атомов. Другим способом характеризации кристаллической структуры является метод порошковой рентгеновской дифракции, в котором экспериментальную рентгенограмму сравнивают с модельной дифрактограммой для чистого порошкового материала при условии, что обе дифрактограммы получены при одной и той же температуре в виде набора значений углов 2. Специалисту в данной области понятно, что рентгенограмма может быть получена с определенной погрешностью, которая зависит от условий измерений. В частности, общеизвестно, что интенсивность пиков на рентгеновской дифрактограмме может быть различной в зависимости от условий измерения. Кроме того, понятно, что относительные интенсивности могут также изменяться в зависимости от экспериментальных условий и соответственно точный порядок интенсивности не следует принимать во внимание. Кроме того, ошибка в измерении угла дифракции для традиционной рентгеновской дифрактограммы обычно составляет примерно 5% или менее и такую величину ошибки измерения надо учитывать при определении указанных выше углов дифракции. Следовательно, следует понимать, что кристаллические формы в данном изобретении не ограничиваются только теми кристаллическими формами,которые дают рентгенограммы, полностью идентичные с рентгенограммами, приведенными на сопровождающих чертежах. Любая кристаллическая форма, которая дает рентгенограмму, DSC-термограмму или ЯМР-спектр, практически идентичные с теми, которые приведены на сопровождающих чертежах,попадают в объем настоящего изобретения. Способность оценить практическую идентичность рентгенограмм входит в компетенцию специалиста в данной области. Применение. Форму N-2 соединения (I), одну или в комбинации с другими соединениями, можно использовать для лечения инфекции HCV. Настоящее изобретение также предлагает композиции, включающие терапевтически эффективное количество формы N-2 соединения (I) и по меньшей мере один фармацевтически приемлемый носитель. Количество активного ингредиента, т.е. формы N-2 соединения (I), в таких композициях обычно составляет 0.1-99.9 мас.% от всей композиции и часто составляет примерно 5-95 мас.%. В некоторых случаях для повышения стабильности соединения или его выделенной формы рН препарата можно установить с помощью фармацевтически приемлемых модификаторов (таких как карбонат кальция и оксид магния). Препараты полиморфной модификации настоящего изобретения могут также содержать добавки для повышения абсорбции и биологической доступности. Фармацевтические композиции по данному изобретению можно вводить перорально, парентерально или через имплантированный резервуар. Использованный здесь термин "парентерально" включает методики подкожного, внутрикожного, внутривенного, внутримышечного, внутрисуставного, внутрисиновиального, интрастенального, интратекального и внутрирубцового введения или инфузии. Фармацевтические композиции могут быть в форме стерильного препарата для инъекций, например в виде стерильной водной или масляной суспензии для инъекций. Эту суспензию можно приготовить по методикам, известным в данной области, с использованием подходящих диспергирующих или смачивающих реагентов и суспендирующих реагентов. Подробности, касающиеся приготовления таких соединений, известны специалистам в данной области. При пероральном введении фармацевтические композиции по данному изобретению можно вводить в любой приемлемой лекарственной форме, включая, но не ограничиваясь этим, капсулы, таблетки и водные суспензии и растворы. В случае таблеток для перорального применения обычно используемые носители включают лактозу и кукурузный крахмал. Можно также добавить смазывающие компоненты,такие как стеарат магния. Для перорального введения в виде капсул пригодные носители/разбавители включают лактозу, высоко- и низкомолекулярные полиэтиленгликоли и высушенный кукурузный крахмал. При пероральном введении водных суспензий активный ингредиент комбинируют с эмульгирующими и суспендирующими реагентами. При желании можно добавить подсластители, и/или отдушки,и/или красители. Другие подходящие носители для указанных выше композиций можно найти в стандартных фарма-6 018152 цевтических текстах, например в "Remington's Pharmaceutical Sciences", 19th ed., Mack Publishing Company, Easton, Penn., 1995. Другие подробности относительно разработки и приготовления подходящих лекарственных форм фармацевтических композиций настоящего изобретения известны специалистам в данной области. В монотерапии для профилактики и/или лечения заболеваний с участием HCV типичными являются дозы соединений по настоящему изобретению примерно 0.05-100 мг/кг массы тела в сутки, более конкретно примерно 0.1-50 мг/кг массы тела в сутки. Обычно фармацевтические композиции по данному изобретению вводят примерно 1-3 раза в сутки или альтернативно путем непрерывного вливания. Такое введение можно использовать в терапии хронического или острого течения болезни. Количество активного ингредиента, которое можно комбинировать с носителями для получения одной лекарственной формы, будет варьироваться в зависимости от объекта лечения и конкретного способа введения. Специалисту очевидно, что могут потребоваться меньшие или большие дозы. Конкретная доза и режим лечения пациента будут зависеть от многих факторов, в том числе активности конкретного используемого соединения, возраста, массы тела, общего состояния здоровья, пола, диеты, времени введения, продолжительности лечения, скорости выведения, сочетания лекарств, остроты и течения болезни,реакции пациента на инфекцию и заключения лечащего врача. В одном варианте препараты с единичной дозой содержат суточную дозу или ее часть, как указано выше, или соответствующую часть активного ингредиента. Обычно лечение начинают с малых доз, значительно меньших, чем оптимальная доза белка. Затем дозу понемногу увеличивают до достижения оптимального эффекта в данных обстоятельствах. В целом наиболее желательно вводить соединение в такой концентрации, которая приводит к эффективному антивирусному результату без вредных или опасных побочных эффектов. Поскольку композиции по данному изобретению включают комбинацию полиморфной модификации по данному изобретению и одного или нескольких дополнительных терапевтических или профилактических реагентов, то как соединение, так и дополнительный реагент обычно составляют примерно 10100% и более предпочтительно примерно 10-80% от дозы, обычно вводимой в режиме монотерапии. Один или несколько дополнительных реагентов можно вводить до, после или одновременно с полиморфной модификацией по настоящему изобретению. Поскольку полиморфная модификация находится вместе с фармацевтически приемлемым носителем, полученную композицию можно вводить in vivo млекопитающим, например человеку, для ингибирования NS5A или лечения либо профилактики вирусной инфекции HCV. Такое лечение можно также провести с использованием полиморфной модификации по данному изобретению в комбинации с реагентами, которые включают, но не ограничиваются ими: иммуномодуляторы, такие как интерфероны; другие антивирусные реагенты типа рибавирина, амантадина; другие ингибиторы NS5A; ингибиторы других мишеней жизненного цикла HCV, такие как геликаза, протеаза, полимераза, металлопротеиназа или внутренний сайт связывания рибосомы; или их комбинации. Для создания единой лекарственной формы к полиморфной модификации по данному изобретению можно добавить дополнительные реагенты. Альтернативно, эти дополнительные реагенты можно вводить млекопитающему отдельно в виде части сложной лекарственной формы. Ниже в табл. 1 приведены иллюстрирующие примеры соединений, которые можно вводить вместе с соединениями по данному изобретению. Соединения по данному изобретению можно вводить с другими соединениями, обладающими анти-HCV активностью, вместе или отдельно или комбинируя соединения в композицию. Другой аспект данного изобретения предлагает способы ингибирования HCV NS5A-активности у пациентов путем введения полиморфной модификации по настоящему изобретению. В одном варианте эти способы полезны для уменьшения активности HCV NS5A у пациента. Если фармацевтическая композиция содержит в качестве активного компонента только полиморфную модификацию по данному изобретению, то такие способы могут дополнительно включать стадию введения указанному пациенту реагента, выбранного из иммуномодулятора, антивирусного реагента, ингибитораHCV NS5A или ингибитора других мишеней в жизненном цикле HCV, таких как, например, геликаза,полимераза, протеаза или металлопротеиназа. Такой дополнительный реагент можно ввести пациенту до,одновременно или после введения соединений по данному изобретению. В другом варианте эти способы приводят к ингибированию размножения вирусов у пациента. Такие способы можно применять в лечении или профилактике заболевания HCV. Полиморфную модификацию по данному изобретению можно также использовать в качестве лабораторного реагента. Полиморфная модификация может быть полезна при разработке методов исследования вирусной репликации, при тестировании методов изучения животных и при структурных биологических исследованиях для более глубокого понимания механизма заболевания HCV. Полиморфную модификацию по данному изобретению можно также использовать для лечения или профилактики вирусного заражения материалов и таким образом уменьшить риск вирусного инфицирования персонала лабораторий, медицинского персонала или пациентов, которые контактируют с такими материалами, например кровью, тканями, хирургическими инструментами и одеждой, лабораторным инструментарием и одеждой, а также аппаратурой и материалами для забора и переливания крови. Следующие неограничивающие примеры иллюстрируют данное изобретение. Примеры В трехгорлую круглодонную колбу на 1 л с механической мешалкой, термопарой и трубкой для подачи азота загрузили 20 г (83.9 ммоль, 1 экв.) 1,1'-(бифенил-4,4'-диил)диэтанона, 200 мл CH2Cl2 и 8.7 мл(27.1 г, 169.3 ммоль, 2.02 экв.) брома. Смесь перемешивали в атмосфере азота в течение примерно 20 ч при обычных условиях. К полученной суспензии добавили 200 мл CH2Cl2 и сконцентрировали до объема примерно 150 мл вакуумной дистилляцией. Затем растворитель отогнали в вакууме и добавили 200 мл ТГФ. Суспензию охладили до 20-25 С в течение 1 ч и перемешивали при 20-25 С еще в течение 1 ч. Отфильтровали желтоватые кристаллы и промыли 150 мл CH2Cl2. Продукт высушили в вакууме при 60 С и получили 27.4 г (69.2 ммоль, 82%) целевого соединения: 1 Н ЯМР (400 МГц, CDCl3)7.95-7.85 (м, 4 Н),7.60-7.50 (м, 4 Н), 4.26 (с, 4 Н); 13 С ЯМР (100 МГц, CDCl3)191.0, 145.1, 133.8, 129.9, 127.9, 30.8; ИК В 500-мл колбу с рубашкой, термопарой, механической мешалкой и трубкой для подачи азота загрузили 20 г (50.5 ммоль, 1 экв.) соединения 2, 22.8 г (105.9 моль, 2.10 экв.) 1-(трет-бутоксикарбонил)-Lпролина и 200 мл ацетонитрила. Суспензию охладили до 20 С и добавили 18.2 мл (13.5 г, 104.4 ммоль,2.07 экв.) DIPEA. Суспензию нагрели до 25 С и перемешивали в течение 3 ч. Полученный прозрачный органический раствор промыли 3100 мл 13 мас.% водным NaCl. Затем ацетонитрил отогнали в вакууме и добавили толуол до объема 215 мл, так что концентрация ацетонитрила была ниже 0.5 об.%. К раствору соединения 3 в толуоле добавили 78 г (1.011 моль, 20 экв.) ацетата аммония и нагрели до 95-100 С. Смесь перемешивали при 95-100 С в течение 15 ч. После завершения реакции смесь охладили до 70-80 С и добавили 7 мл уксусной кислоты, 40 мл н-бутанола и 80 мл 5 об.% водной уксусной кислоты. Полученный двухфазный раствор расслоился при температуре 50 С. К органическому слою добавили 80 мл 5 об.% водного раствора уксусной кислоты, 30 мл уксусной кислоты и 20 мл н-бутанола при температуре 50 С. Полученный двухфазный раствор расслоился при температуре 50 С и органический слой промыли дополнительно 80 мл 5 об.% водной уксусной кислоты. Растворитель отогнали в вакууме и добавили 215 мл толуола. При температуре 60 С добавили 64 мл метанола. Полученную суспензию нагрели до 70-75 С и выдержали в течение 1 ч. Суспензию охладили до 20-25 С в течение 1 ч и выдержали при этой температуре еще 1 ч. Суспензию профильтровали и осадок на фильтре промыли 200 мл смеси толуол:метанол 10:3. Продукт высушили в вакууме при 70 С и получили 19.8 г (31.7 ммоль,63%) целевого продукта: 1 Н ЯМР (400 МГц, ДМСО-d6)13.00-11.00 (с, 2 Н), 7.90-7.75 (м, 4 Н), 7.75-7.60 В 250-мл реактор, снабженный механической мешалкой и трубкой для подачи азота, загрузили 25.0 г соединения 4 (40.01 ммоль, 1 экв.) и затем 250 мл метанола и 32.85 мл (400.1 ммоль, 10 экв.) 6 М водного раствора HCl. Температуру повысили до 50 С и перемешивали при 50 С в течение 5 ч. Полученную суспензию охладили до 20-25 С и перемешивали примерно 18 ч. Отфильтровали и получили твердое вещество, которое промыли последовательно 100 мл 90% смеси метанол/вода (V/V) и 2100 мл метанола. Влажный осадок высушили в вакуумном сушильном шкафу при 50 С в течение ночи и получили 18.12 г (31.8 ммоль, 79.4%) целевого соединения. Перекристаллизация соединения 5. В 250-мл реактор с механической мешалкой и трубкой для подачи азота загрузили 17.8 г указанного выше соединения 5 и затем 72 мл метанола. Полученную суспензию перемешивали при 50 С в течение 4 ч, охладили до 20-25 С и перемешивали при 20-25 С в течение 1 ч. Кристаллическое твердое вещество отфильтровали и промыли 60 мл метанола. Полученный влажный осадок высушили в вакуумном сушильном шкафу при 50 С в течение 4 дней и получили 14.7 г (25.7 ммоль, 82.6%) очищенного продукта: 1 В 1-л колбу с рубашкой, механической мешалкой и трубкой для подачи азота загрузили последовательно 100 мл ацетонитрила, 13.69 г (89.4 ммоль, 2.5 экв.) гидрата гидроксибензотриазола, 15.07 г (86 ммоль, 2.4 экв.) N-(метоксикарбонил)-L-валина, 16.46 г (85.9 ммоль, 2.4 экв.) 1-(3-диметиламинопропил)3-этилкарбодиимид гидрохлорида и еще 100 мл ацетонитрила. Полученный раствор перемешивали при 20 С в течение 1 ч и затем добавили 20.4 г (35.8 ммоль, 1 экв.) очищенного соединения 5. Суспензию охладили до примерно 0 С и добавили 18.47 г (142.9 ммоль, 4 экв.) диизопропилэтиламина в течение 30 мин при температуре ниже 10 С. Раствор медленно нагрели до 15 С в течение 3 ч и выдержали при 15 С в течение 12 ч. К полученному раствору добавили 120 мл 13 мас.% водного раствора NaCl и нагрели до 50 С в течение 1 ч. После охлаждения до 20 С добавили 100 мл изопропилацетата. Двухфазный раствор профильтровали через 0.45-мкм фильтр, чтобы смесь расслоилась. Органический слой промыли двумя порциями по 240 мл 0.5 N раствора NaOH, содержащего 13 мас.% NaCl, и затем 120 мл 13 мас.% водного раствора NaCl. Растворитель отогнали в вакууме и добавили изопропилацетат до объема 400 мл. Полученный мутный раствор охладили до 20 С и профильтровали через 0.45-мкм фильтр. Из прозрачного раствора отогнали в вакууме растворитель и добавили этанол дообъема 140 мл. При температуре 50 С добавили 66.4 мл (82.3 ммоль, 2.3 экв.) 1.24 М раствора HCl в этаноле. Затем к смеси добавили 33 мг(0.04 ммоль, 0.001 экв.) затравочных кристаллов соединения (I) (см. получение ниже) и полученную суспензию перемешивали при 50 С в течение 3 ч. Смесь охладили до 20 С в течение 1 ч и выдержали при этой температуре еще 22 ч. Суспензию отфильтровали и влажный осадок промыли 100 мл смеси ацетон:этанол 2:1. Твердое вещество высушили в вакуумном сушильном шкафу при 70 С и получили 22.15 г Обработка углем и перекристаллизация соединения (I). Приготовили раствор соединения (I) растворением 3.17 г описанного выше соединения (I) в 22 мл метанола. Раствор профильтровали через 47-мм фильтр Cuno Zeta Carbon 53SP при 5 фунт/кв. дюйм и скорости потока 58 мл/мин. Угольный фильтр смочили 32 мл метанола. Раствор сконцентрировали вакуумной дистилляцией до объема 16 мл. Поддерживая температуру 40-50 С, добавили 15.9 мл ацетона и 5 мг затравочных кристаллов соединения (I) (см. методику ниже). Затем к полученной суспензии добавили 32 мл ацетона в течение 30 мин. Суспензию выдержали при 50 С в течение 2 ч, охладили до 20 С в течение примерно 1 ч и выдержали при 20 С в течение примерно 20 ч. Твердое вещество отфильтровали,промыли 16 мл смеси ацетон:метанол 2:1, высушили в вакуумном сушильном шкафу при 60 С и получили 2.14 г (67.5%) очищенного соединения (I): 1 Н ЯМР (400 МГц, ДМСО-d6, 80 С): 8.02 (д, J=8.34 Гц, 4 Н), 7.97 (с, 2 Н), 7.86 (д, J=8.34 Гц, 4 Н), 6.75 (с, 2 Н), 5.27 (т, J=6.44 Гц, 2 Н), 4.17 (т, J=6.95 Гц, 2 Н), 3.974.11 (м, 2 Н), 3.74-3.90 (м, 2 Н), 3.57 (с, 6 Н), 2.32-2.46 (м, 2 Н), 2.09-2.31 (м, 6 Н), 1.91-2.07 (м, 2 Н), 0.88 (д,J=6.57 Гц, 6 Н), 0.79 (д, J=6.32 Гц, 6 Н); 13 С ЯМР (75 МГц, ДМСО-d6):170.9, 156.9, 149.3, 139.1, 131.7,127.1, 126.5, 125.9, 115.0, 57.9, 52.8, 51.5, 47.2, 31.1, 28.9, 24.9, 19.6, 17.7; ИК (неразб., см-1): 3385, 2971,2873, 2669, 1731, 1650. Анализ: вычисл. для C40H52N8O6Cl2: С, 59.18; Н, 6.45; N, 13.80; Cl, 8.73. Найдено С, 59.98; Н, 6.80; N, 13.68; Cl, 8.77. Т.пл. 267 С (разл.). Получение затравочных кристаллов соединения (I). В 250-мл круглодонную колбу загрузили 6.0 г (10.5 ммоль, 1 экв.) соединения 5, 3.87 г (22.1 ммоль,2.1 экв.) N-(метоксикарбонил)-L-валина, 4.45 г (23.2 ммоль, 2.2 экв.) 1-(3-диметиламинопропил)-3 этилкарбодиимид гидрохлорида, 0.289 г (2.14 ммоль, 0.2 экв.) 1-гидроксибензотриазола и 30 мл ацетонитрила. К полученной суспензии добавили 7.33 мл (42.03 ммоль, 4 экв.) диизопропилэтиламина и перемешивали при 24-30 С в течение примерно 18 ч. К смеси добавили 6 мл воды и нагрели до 50 С в течение 5 ч. Смесь охладили и добавили 32 мл этилацетата и 30 мл воды. Слои разделили и органический слой промыли 30 мл 10 мас.% водного NaHCO3, 30 мл воды и 20 мл 10 мас.% водного раствора NaCl. Органический слой высушили над MgSO4, отфильтровали и упарили досуха. Сырой материал очистили флэш-хроматографией (силикагель, 0-10% метанола в дихлорметане) и получили свободное основание соединения (I). Свободное основание соединения (I) (0.03 г) растворили в 1 мл изопропанола при 20 С. Добавили безводный этанольный раствор HCl (70 мкл, концентрация примерно 1.25 М) и реакционную смесь перемешали. К раствору добавили метил-трет-бутиловый эфир (1 мл) и полученную суспензию энергично перемешивали при 40-50 С в течение 12 ч. Суспензию кристаллов охладили до 20 С и отфильтровали. Влажный осадок высушили на воздухе при 20 С. Получили белое кристаллическое вещество (форма N-2 соединения (I. Форму N-2 анализировали одним или несколькими описанными ниже методами тестирования. 1. Рентгеноструктурный анализ кристаллов. Для получения данных по дифракции при комнатной температуре использовали дифрактометрBruker APEX2 Kappa CCD с вращающимся анодным генератором CuK-излучения (=1.54178 ). Индексацию и обработку полученных данных по интенсивности пиков провели с помощью программного пакета АРЕХ 2 (АРЕХ 2 Data collection and processing user interface: APEX2 User Manual, vl. 27; BRUKERAXS, INc, 5465 East Cheryl Parkway, Madison, WI 53711 USA). Конечные параметры элементарной ячейки определили с использованием полного массива данных. Структуру расшифровали прямыми методами и уточнили полноматричным методом наименьших квадратов с использованием программного пакета SHELXTL (Sheldrick, G.M., 1997, SHELXTL. StructureDetermination Programs. Version 5.10, Bruker AXS, Madison, Wisconsin, USA.). Минимизированная функция при уточненииR определили как и где w представляет собой соответствующую взвешенную функцию отклонений наблюдаемых интенсивностей. На всех этапах уточнения проводили разностный синтез Фурье. Для всех неводородных атомов уточнили анизотропные тепловые параметры. Водородно-связанные атомы водорода локализовали из конечного разностного синтеза Фурье, а положения остальных атомов водорода определили на основе идеализированной геометрии со стандартными значениями длин связей и углов. Им были приписаны изотропные тепловые параметры, которые были использованы в качестве фиксированных значений при расчете структурных параметров. Кристаллографические данные для кристалла формы N-2 приведены в табл. 2. Координаты атомов даны в табл. 3. Специалисту в данной области следует понимать, что возможны небольшие отклонения в координатах, которые, тем не менее, позволяют включить их в объем настоящего изобретения. Таблица 2 Кристаллографические данные для кристалла формы N-2 2. Порошковая рентгеновская дифракция. В контейнер прибора для исследования порошковой рентгеновской дифракции (PXRD) фирмыmA, CuK). Накопление данных проводили при комнатной температуре в интервале углов 2 от 2 до 32(непрерывное сканирование, скорость сканирования 0.03/с, коллиматор с противорассеивающими щелями, приемная щель 0.2 мм, вращение образца: включено). Экспериментальная и модельная рентгенограммы, рассчитанные из данных рентгеноструктурного анализа, приведены на фиг. 1. В табл. 4 показаны характеристические пики PXRD для формы N-2 соединения (I). Таблица 4 Положения характеристических дифракционных пиков (градусы 20.1) при комнатной температуре на высококачественной дифрактограмме, полученной на дифрактометре (CuK) с вращающимся капилляром и значениях угла 2,откалиброванных с помощью эталонов из базы NIST 3. Дифференциальная сканирующая калориметрия. Опыты по дифференциальной сканирующей калориметрии (DSC) проводили на приборе ТА модели Q2000, Q1000 или 2920. Массу образца (примерно 2-6 мг) взвесили в алюминиевой чашке с точностью до сотых долей миллиграмма и затем перенесли в DSC калориметр. Прибор продули азотом со скоростью 50 мл/мин. Данные записывали в интервале температур от комнатной до 300 С при скорости нагрева 10 С/мин. Эндотермические пики на кривой DSC показаны обращенными вниз. Результаты показаны на фиг. 2. 4. Твердофазный ЯМР (SSNMR). Анализ методом С-13 ЯМР проведен на спектрометре Bruker DSX-400, 400 МГц. Спектры высокого разрешения регистрировали с развязкой от протонов, ТРРМ-последовательностью импульсов и наклонной амплитудной кросс-поляризацией (RAMP-CP) с вращением под магическим углом (MAS) при примерно 12 кГц (А.Е. Bennett et al. J. Chem. Phys. 1995, 103, 6951; G. Metz, X. Wu, and S.O. Smith, J. Magn.Reson. A., 1994, 110, 219-227). В каждом опыте использовали примерно 70 мг образца, упакованного в пустотелом роторе из оксида циркония. Химические сдвигирегистрировали с использованием внешнего образца - адамантана, для которого резонансная частота равна 38.56 м.д. (W.L. Earl and D.L. VanderHart, J. Magn. Reson., 1982, 48, 35-54). Спектр SSNMR приведен на фиг. 3. В табл. 5 показаны характеристические пики SSNMR для формы N-2 соединения (I). характеризующаяся одной или несколькими следующими особенностями:=70.646; пространственная группа Р 1: число молекул в элементарной ячейке 1,причем определение указанной кристаллической формы проводят при температуре примерно 2025 С; илиc) характеристическими пиками на порошковой дифрактограмме при значениях параметра 2 10.30.1, 12.40.1, 12.80.1, 13.30.1, 13.60.1, 15.50.1, 20.30.1, 21.20.1, 22.40.1, 22.70.1 и 23.70.1 при температуре примерно 20-25 С; и/илиd) плавлением с эндотермическим пиком разложения, начинающимся обычно в интервале 225245 С. 2. Форма по п.1, имеющая чистоту по меньшей мере 95 мас.%. 3. Форма по п.1, имеющая чистоту по меньшей мере 99 мас.%. 4. Фармацевтическая композиция для лечения HCV-инфекции, включающая форму N-2 и фармацевтически приемлемый носитель или разбавитель. 5. Фармацевтическая композиция по п.4, в которой форма N-2 имеет чистоту по меньшей мере 95 мас.%. 6. Фармацевтическая композиция по п.4, в которой форма N-2 имеет чистоту по меньшей мере 99 мас.%. 7. Фармацевтическая композиция, включающая форму N-2 в комбинации с одним или несколькими дополнительными соединениями, обладающими анти-HCV активностью. 8. Фармацевтическая композиция по п.7, в которой форма N-2 имеет чистоту по меньшей мере 90 мас.%. 9. Фармацевтическая композиция по п.7, в которой форма N-2 имеет чистоту по меньшей мере 95 мас.%. 10. Фармацевтическая композиция по п.7, в которой форма N-2 имеет чистоту по меньшей мере 99 мас.%. 11. Композиция по п.7, в которой по меньшей мере одно из дополнительных соединений с антиHCV активностью представляет собой интерферон или рибавирин. 12. Композиция по п.11, в которой интерферон выбирают из интерферона альфа 2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. 13. Композиция по п.7, в которой по меньшей мере одно дополнительное соединение выбирают из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения, стимулирующего выработку отклика Тхелпера клетки типа 1, интерферирующей РНК, антисенс-РНК, имихимода, рибавирина, ингибитора инозин 5'-монофосфатдегидрогеназы, амантадина и римантадина. 14. Способ лечения HCV-инфекции у млекопитающих, включающий введение млекопитающему терапевтически эффективного количества формы N-2 15. Способ по п.14, в котором форма N-2 имеет чистоту по меньшей мере 90 мас.%. 16. Способ по п.14, в котором форма N-2 имеет чистоту по меньшей мере 95 мас.%. 17. Способ по п.14, в котором форма N-2 имеет чистоту по меньшей мере 99 мас.%. 18. Способ по п.14, в котором млекопитающее является человеком.
МПК / Метки
МПК: C07D 403/12, A61K 31/4178, A61P 31/12
Метки: форма, метил, кристаллическая, дигидрохлорида, 1s)-1-(((2s)-2-(5-(4'-(2-((2s)-1-((2s)-2-((метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-1н-имидазол-5-ил)-4-бифенилил)-1н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата
Код ссылки
<a href="https://eas.patents.su/23-18152-kristallicheskaya-forma-digidrohlorida-metil-1s-1-2s-2-5-4-2-2s-1-2s-2-metoksikarbonilamino-3-metilbutanoil-2-pirrolidinil-1n-imidazol-5-il-4-bifenilil-1n-imidazol-2-il-1-pirrolidi.html" rel="bookmark" title="База патентов Евразийского Союза">Кристаллическая форма дигидрохлорида метил ((1s)-1-(((2s)-2-(5-(4′-(2-((2s)-1-((2s)-2-((метоксикарбонил)амино)-3-метилбутаноил)-2-пирролидинил)-1н-имидазол-5-ил)-4-бифенилил)-1н-имидазол-2-ил)-1-пирролидинил)карбонил)-2-метилпропил)карбамата</a>
Кристаллическая форма 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2н)-пиримидинил]-4-фтор-n-[[метил-(1-метилэтил) амино]сульфонил]бензамида

Номер патента: 15518
Опубликовано: 31.08.2011
Авторы: Кокс Герхард, Сакселл Хейди Эмилиа, Эрк Петер, Цагар Цирилл, Веверс Ян-Хендрик, Шмидт Томас, Хампрехт Герхард, Зайтц Вернер, Кайл Михаэль, Зиверних Бернд, Райнхард Роберт, Вольф Бернд, Майер Гуидо, Михель Альфред, Гебхардт Йоахим, Лёр Зандра
МПК: A01N 43/48, C07D 239/54
Метки: 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2н)-пиримидинил]-4-фтор-n-[[метил-(1-метилэтил, амино]сульфонил]бензамида, форма, кристаллическая
Формула / Реферат:
1. Кристаллическая, по существу, не содержащая растворитель форма II 2-хлор-5-[3,6-дигидро-3-метил-2,6-диоксо-4-(трифторметил)-1-(2Н)-пиримидинил]-4-фтор-N-[[метил-(1-метилэтил)амино]сульфонил]бензамида, которая в рентгеновской порошковой дифрактограмме при 25°С и Cu-Ka-излучении показывает по меньшей мере два из следующих приведенных как 2q-значения отражений: 6,3±0,3°, 9,4±0,3°, 10,9±0,3°, 11,9±0,3°, 12,6±0,3°, 15,0±0,3°, 15,8±0,3°,...
Кристаллическая форма [3-(4,5-дигидро-3-изоксазолил)-2-метил-4-(метилсульфонил)фенил]-(5-гидрокси-1-метил-1h-пиразол-4-ил)метанона

Номер патента: 16821
Опубликовано: 30.07.2012
Авторы: Братц Маттиас, Крёль Томас, Сакселль Хейди Эмилия, Гебхардт Йоахим, Эрк Петер
МПК: C07D 413/10, A01N 43/80
Метки: 3-(4,5-дигидро-3-изоксазолил)-2-метил-4-(метилсульфонил)фенил]-(5-гидрокси-1-метил-1h-пиразол-4-ил)метанона, форма, кристаллическая
Формула / Реферат:
1. Кристаллическая форма I [3-(4,5-дигидро-3-изоксазолил)-2-метил-4-(метилсульфонил)фенил]-(5-гидрокси-1-метил-1H-пиразол-4-ил)метанона, которая на рентгеновской порошковой дифрактограмме при 30°С с использованием CuKα облучения демонстрирует по крайней мере 5 из следующих изображений, указанных как 2θ значения: 7,7±0,2°, 10,3±0,2°, 12,7±0,2°, 13,8±0,2°, 16,9±0,2°, 18,8±0,2°, 20,7±0,2°, 22,2±0,2°, 28,0±0,2° и 31,4±0,2°.2....
Предназначенная для перорального применения лекарственная форма для этилового эфира 3-[(2-{[4-(гексилоксикарбониламиноиминометил) фениламино]-метил}-1-метил-1h-бензимидазол-5-карбонил)пиридин-2- иламино]пропионовой кислоты и его солей

Номер патента: 9664
Опубликовано: 28.02.2008
Авторы: Хойль Норберт, Зигер Петер, Браунс Ульрих
МПК: A61K 31/4439, A61K 47/12, A61K 47/38...
Метки: кислоты, лекарственная, 3-[(2-{[4-(гексилоксикарбониламиноиминометил, применения, солей, эфира, перорального, иламино]пропионовой, предназначенная, форма, фениламино]-метил}-1-метил-1h-бензимидазол-5-карбонил)пиридин-2, этилового
Формула / Реферат:
1. Фармацевтическая композиция для перорального применения, содержащая по меньшей мере: а) этиловый эфир 3-[(2-{[4-(гексилоксикарбониламиноиминометил)фениламино]метил}-1-метил-1H-бензимидазол-5-карбонил)пиридин-2-иламино]пропионовой кислоты или одну из его фармацевтически приемлемых солей и б) одну или несколько фармацевтически приемлемых органических кислот с растворимостью в воде более 1 г на 250 мл при 20шС. 2. Фармацевтическая композиция по...
Кристаллическая форма гемигидрата 1-(b-d-глюкопиранозил)-4-метил-3-[5-(4-фторфенил)-2-тиенилметил]бензола

Номер патента: 17103
Опубликовано: 28.09.2012
Авторы: Номура Сумихиро, Каваниси Еидзи
МПК: C07H 7/04, A61P 5/00, A61K 31/70...
Метки: форма, кристаллическая, 1-(b-d-глюкопиранозил)-4-метил-3-[5-(4-фторфенил)-2-тиенилметил]бензола, гемигидрата
Формула / Реферат:
1. Кристаллическая форма гемигидрата 1-(β-D-глюкопиранозил)-4-метил-3-[5-(4-фторфенил)-2-тиенилметил]бензола, имеющая, по существу, такой тип порошковой дифракции рентгеновских лучей, как показано на фиг. 1.2. Кристаллическая форма гемигидрата 1-(β-D-глюкопиранозил)-4-метил-3-[5-(4-фторфенил)-2-тиенилметил]бензола, имеющая, по существу, такой инфракрасный спектр, как показано на фиг. 2.3. Способ получения кристаллической формы...
Кристаллическая форма 4-[5-метил-3-фенилизоксазол-4-ил]бензолсульфонамида

Номер патента: 1472
Опубликовано: 23.04.2001
Авторы: Год Генри Т., Маклофлин Кэтлин Т., Йонан Эдвард Э., Тэлли Джон Дж., Медич Джон Р.
МПК: C07D 261/08, A61K 31/421
Метки: форма, кристаллическая, 4-[5-метил-3-фенилизоксазол-4-ил]бензолсульфонамида
Формула / Реферат:
1. Кристаллическая форма В 4-[5-метил-3-фенилизоксазол-4-ил]бензолсульфонамида, имеющая температуру плавления около 170-174шС. 2. Форма по п.1, отличающаяся тем, что на ИК спектре присутствуют следующие пики: 1170, 925, 844 и 729 см-1. 3. Форма по п.1, отличающаяся тем, что ИК спектр не содержит значительного пика при 723 см-1. 4. Форма по п.1, отличающаяся тем, что ее дифракционная картина рентгеновских лучей соответствует картине,...
Предыдущий патент: Способ и система для улучшения производительности или эффективности скважинного насоса
Следующий патент: Липидацилтрансфераза и содержащая ее пищевая или кормовая композиция
Случайный патент: Способ получения углеводородов