Комбинация ингибитора протеазы ns3 hcv с интерфероном и рибавирином
Номер патента: 19965
Опубликовано: 30.07.2014
Авторы: Хуан Дейвид, Штайнманн Герхард Густав, Стерн Джерри О.

















Формула / Реферат
1. Комбинация, включающая:
(а) соединение формулы (1) или его фармацевтически приемлемую соль

(б) интерферон альфа;
(в) рибавирин,
предназначенная для применения для лечения инфекции, вызываемой вирусом гепатита С (HCV), или облегчения одного или нескольких ее симптомов.
2. Комбинация по п.1, которая включает:
(а) первую фармацевтическую композицию, содержащую соединение следующей формулы (1) или его фармацевтически приемлемую соль:

(б) вторую фармацевтическую композицию, содержащую интерферон альфа; и
(в) третью фармацевтическую композицию, содержащую рибавирин.
3. Комбинация по п.1 или 2, в которой инфекция HCV относится к генотипу 1.
4. Комбинация по п.1 или 2, которая предназначена для лечения инфекции, вызываемой вирусом гепатита С (HCV), или облегчения одного или нескольких ее симптомов у пациента, не подвергавшегося ранее лечению.
5. Комбинация по п.1 или 2, которая предназначена для лечения инфекции, вызываемой вирусом гепатита С (HCV), или облегчения одного или нескольких ее симптомов у пациента, не чувствительного к комбинированной терапии, основанной на применении рибавирина и интерферона альфа.
6. Комбинация по п.1 или 2, где количество соединения (1) или его фармацевтически приемлемой соли составляет по меньшей мере 40 мг.
7. Комбинация по п.1 или 2, где количество рибавирина составляет 400, 600, 800, 1000 или 1200 мг.
8. Комбинация по п.1 или 2, где интерферон альфа представляет собой пэгилированный интерферон альфа.
9. Комбинация по п.8, где пэгилированный интерферон альфа представляет собой пэгилированный интерферон альфа-2а в дозе от примерно 90 до примерно 200 мкг или пэгилированный интерферон альфа-2b в дозе от примерно 0,5 до примерно 2 мкг.
10. Комбинация по п.1 или 2, предназначенная для лечения инфекции HCV генотипа 1 у пациента, не чувствительного к комбинированной терапии, основанной на применении рибавирина и интерферона альфа, где количество соединения (1) или его фармацевтически приемлемой соли составляет от 48 до 240 мг и где интерферон альфа представляет собой пэгилированный интерферон альфа-2а или пэгилированный интерферон альфа-2b.
Текст
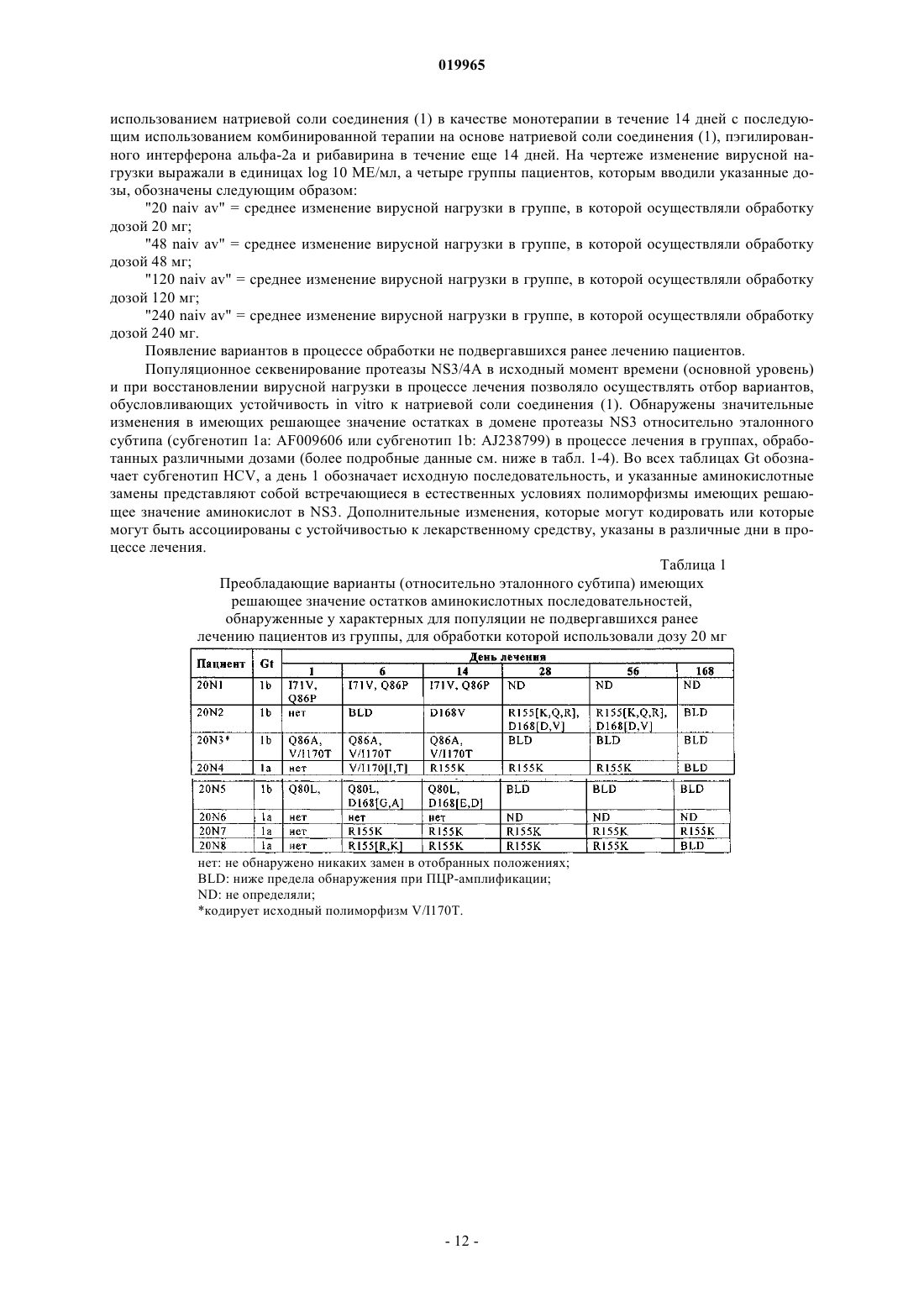
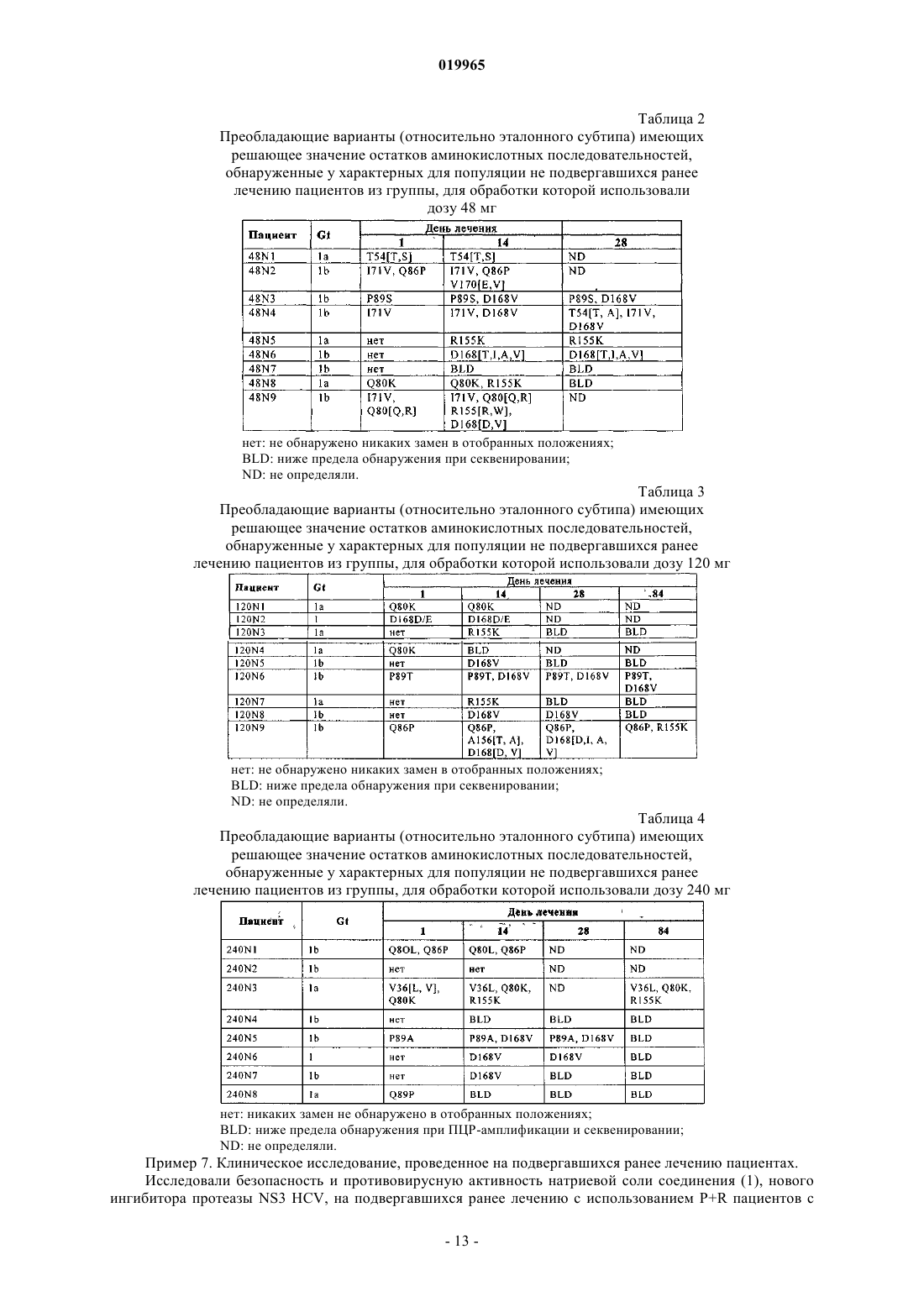
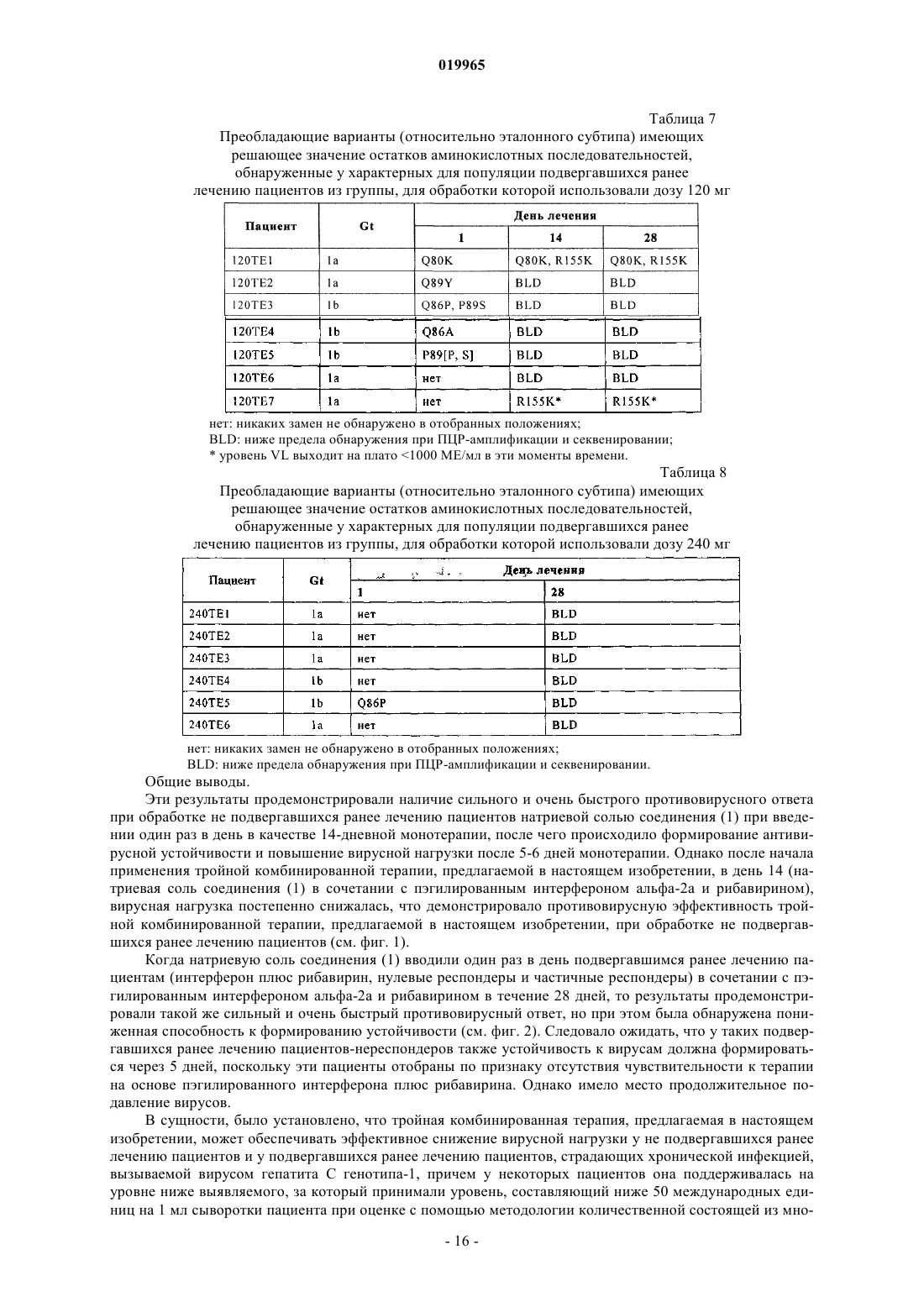
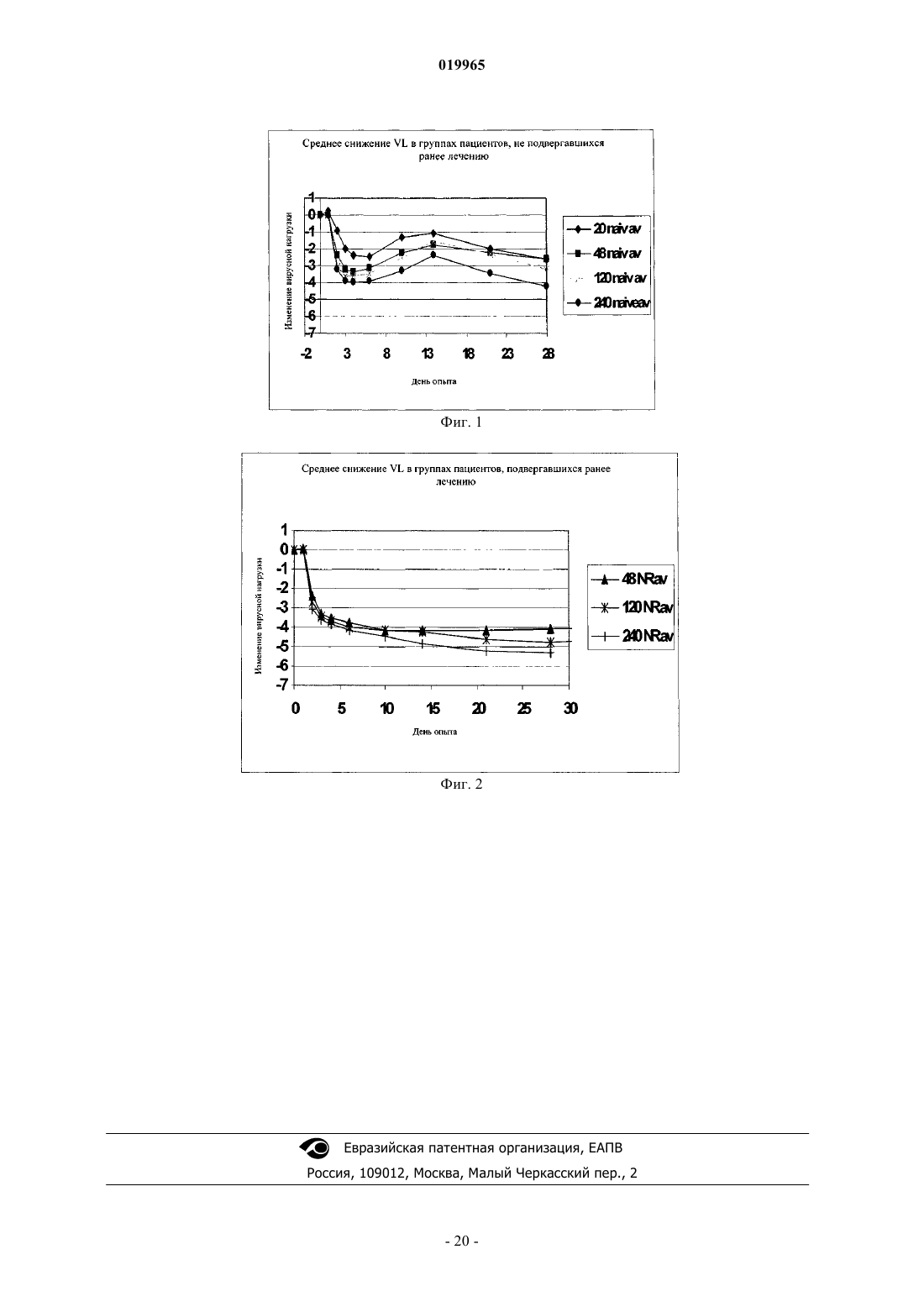
КОМБИНАЦИЯ ИНГИБИТОРА ПРОТЕАЗЫ NS3 HCV С ИНТЕРФЕРОНОМ И РИБАВИРИНОМ Хуан Дейвид (US), Штайнманн Герхард Густав (DE), Стерн Джерри О. В изобретении приведено описание терапевтических комбинаций, содержащих (а) соединение(1) или его фармацевтически приемлемую соль, представленное/представленную в настоящем описании; (б) интерферон-альфа и (в) рибавирин. Соединение (1) представляет собой селективный и эффективный ингибитор сериновой протеазы NS3 HCV. Кроме того, изобретение относится к способам применения указанных терапевтических комбинаций для лечения инфекции, вызываемойHCV, или облегчения одного или нескольких ее симптомов у пациента.(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) Заявка на данное изобретение притязает на приоритет предварительных заявок на патент США 61/097753, зарегистрированной 17 сентября 2008 г., 61/109033, зарегистрированной 28 октября 2008 г., и 61/171935, зарегистрированной 23 апреля 2009 г. Область техники, к которой относится изобретение Настоящее изобретение относится к терапевтическим комбинациям, содержащим соединение (1),представленное в настоящем описании, интерферон альфа и рибавирин. Настоящее изобретение относится также к способам применения указанных терапевтических комбинаций для лечения вызываемойHCV инфекции или облегчения одного или нескольких ее симптомов у пациента. Настоящее изобретение относится также к наборам, содержащим терапевтические комбинации, предлагаемые в настоящем изобретении. Предпосылки создания изобретения Инфекция, вызываемая вирусом гепатита С (HCV), представляет собой глобальную проблему, связанную со здоровьем человека, при этом только в Соединенных Штатах ежегодно фиксируются примерно 150000 новых случаев. HCV представляет собой одноцепочечный РНКовый вирус, который является этиологическим фактором, идентифицированным в большинстве случаев не-А, не-Б гепатита, который передается при переливании крови и при трансплантации, и является обычным причинным фактором острого спорадического гепатита. Установлено, что более 50% пациентов, зараженных HCV, становятся хронически инфицированными и в течение 20 лет у 20% из них развивается цирроз печени. Для лечения хронической инфекции, вызываемой HCV, разрешено применять несколько типов интерферонов, в частности альфа-интерфероны, например интерферон-альфа-2 а (ROFERON-A), интерферон альфа-2b (INTRON-A), консенсусный интерферон (INFERGEN), а также пэгилированные формы этих и других интерферонов типа пэгилированного интерферона альфа-2 а (PEGASYS) и пэгилированного интерферона альфа-2b (PEG-INTRON). Большая часть пациентов не обладает чувствительностью к лечению интерфероном альфа, а среди респондеров обнаружен высокий уровень рецидивов в течение 6 месяцев после прекращения лечения (Liang et al., J. Med. Virol. 40, 1993, p. 69). В клинических опытах было установлено, что рибавирин, аналог гуанозина с широким спектром активности в отношении многих РНКовых и ДНКовых вирусов, обладает эффективностью в отношении хронической вызываемой HCV инфекции при его применении в сочетании с интерферонами альфа (см.,например, Poynard et al., Lancet, 352, 1998, p. 1426-1432; Reichard et al., Lancet, 351, 1998, p. 83-87), и эта комбинированная терапия разрешена при лечении HCV: REBETRON (интерферон альфа-2b плюс рибавирин, фирма Schering-Plough); PEGASYSRBV (комбинированная терапия на основе пэгилированного интерферона альфа-2 а плюс рибавирина, фирма Roche); см. также Manns et al., Lancet, 358, 2001,p. 958-965 и Fried et al., N. Engl. J. Med. 347, 2002, p. 975-982. Однако даже при использовании комбинированной терапии уровень вирусологического ответа все еще находится на уровне 50% или ниже. Кроме того, с такими терапиями, как правило, ассоциированы существенные побочные действия. Недостатками рибавирина являются тератогенная активность, воздействие на развитие спермы, гемолиз,а также на такие факторы, как утомляемость, головная боль, бессонница, тошнота и/или анорексия. С интерфероном альфа при его применении в сочетании с рибавирином или без него связаны многие побочные действия. В течение периода лечения требуется осуществлять тщательный мониторинг пациентов в отношении симптомов типа лихорадки, депрессии, сыпи и аномальной формулы крови. Пациенты,которых можно подвергать лечению интерфероном альфа-2b плюс рибавирином, не должны иметь состояний в виде серьезной почечной дисфункции, и только таких пациентов можно рассматривать как пригодных для лечения гепатита С, при этом требуется осуществлять тщательный мониторинг. Определенные комбинированные терапии, включающие применение интерферона, которые предназначены для лечения вызываемой HCV инфекции, описаны также в следующих опубликованных заявках на патент США: US 2005/0112093; US 2005/0129659 и US 2008/0138316. Приведенное ниже соединение (1) известно в качестве избирательного и эффективного ингибитора сериновой протеазы NS3 HCV и его можно применять для лечения вызываемой HCV инфекции. Соединение (1) относится к группе ациклических пептидных ингибиторов HCV, которые описаны вUS 6323180, 7514557 и 7585845. Соединение (1) описано конкретно как соединение 1055 в US 7585845 и как соединение 1008 в US 7514557. Соединение (1) и включающие его фармацевтические препаративные формы можно получать с помощью общих процедур, которые описаны в процитированных выше ссылках, которые все полностью включены в настоящее описание в качестве ссылки. Предпочтительными формами соединения (1) являются кристаллические формы, в частности кристаллическая форма в виде натриевой соли, которую можно получать согласно методам, представленным в настоящем описании в разделе "Примеры". Соединение (1) можно описывать также с помощью другой химической структуры, которая эквивалентна указанной выше структуре:R2 обозначает Хотя установлено, что соединение (1) в целом обладает эффективностью в отношении снижения вирусной нагрузки и лечения вызываемой HCV инфекции, при этом обнаружен определенный уровень устойчивости вирусов, что приводит к восстановлению вирусной нагрузки. Например, установлено, что при осуществлении 14-дневной монотерапии путем введения соединения (1) один раз в день не подвергавшимся ранее лечению пациентам наблюдается сильное и очень быстро проявляющееся противовирусное действие, после чего через 5-6 дней формируется определенный уровень устойчивости. Таким образом, продолжает сохраняться необходимость в других терапиях для лечения и предупреждения вызываемой HCV инфекции. Краткое изложение сущности изобретения Настоящее изобретение относится к способу лечения вызываемой HCV инфекции или облегчению одного или нескольких ее симптомов у пациента, заключающемуся в том, что вводят пациенту терапевтическую комбинацию, которая содержит соединение (1), представленное в настоящем описании, или его фармацевтически приемлемую соль и интерферон альфа и рибавирин, представленные в настоящем описании. Три действующих вещества, которые входят в комбинацию, можно вводить одновременно или раздельно в качестве части схемы применения. Настоящее изобретение относится также к набору, содержащему первую фармацевтическую композицию, которая включает соединение (1) или его фармацевтически приемлемую соль; вторую фармацев-2 019965 тическую композицию, включающую интерферон альфа; и третью фармацевтическую композицию,включающую рибавирин. Краткое описание чертежей На чертежах показано: на фиг. 1 - среднее изменение вирусной нагрузки HCV в четырех группах пациентов, которые состояли из не подвергавшихся ранее лечению пациентов с хронической инфекцией, связанной с HCV генотипа-1, в процессе лечения путем введения натриевой соли соединения (1) в качестве 14-дневной монотерапии, с последующим применением комбинированной терапии, включающей введение натриевой соли соединения (1), пэгилированного интерферона альфа-2 а и рибавирина в течение еще 14 дней; на фиг. 2 - среднее изменение вирусной нагрузки HCV в трех группах пациентов, которые состояли из подвергавшихся ранее лечению пациентов с хронической инфекцией, связанной с HCV генотипа-1, в процессе лечения путем введения натриевой соли соединения (1), пэгилированного интерферона альфа 2 а и рибавирина в качестве 28-дневной комбинированной терапии. Подробное описание изобретения Определения. Понятие "соединение (1)" относится к описанному выше соединению. Понятие "интерферон" относится к представителю семейства обладающих высокой степенью гомологии видоспецифических белков, которые ингибируют вирусную репликацию и клеточную пролиферацию и модулируют иммунный ответ. Человеческие интерфероны сгруппированы в три класса на основе их клеточного происхождения и антигенности: -интерферон (лейкоциты), -интерферон (фибробласты) и -интерферон (В-клетки). Созданы и поступают в продажу рекомбинантные формы каждой группы. Разделение каждой группы на подтипы основано на антигенных/структурных характеристиках. Идентифицировано по меньшей мере 24 интерферона альфа (сгруппированы в подтипы от А до Н), которые имеют различные аминокислотные последовательности, путем выделения и секвенирования ДНК, кодирующих эти пептиды. Понятия "-интерферон", "альфа-интерферон" и "интерферон альфа" в контексте настоящего описания применяют взаимозаменяемо для описания представителей этой группы. Для воплощения изобретения на практике можно применять как встречающиеся в естественных условиях, так и рекомбинантные альфа-интерфероны, включая консенсусный интерферон. Пригодными для применения согласно настоящему изобретению интерферонами альфа являются(но не ограничиваясь только ими) рекомбинантный интерферон альфа-2b, такой как интерферон INTRON и VIRAFERON; рекомбинантный интерферон альфа-2 а, такой как интерферон ROFERON; рекомбинантный интерферон альфа-2 с, такой как интерферон BEROFOR альфа 2; интерферон альфаn1, очищенная смесь встречающихся в естественных условиях альфа-интерферонов, такая как SUMIFERON, или интерферон альфа-n1 (INS) WELLFERON; или консенсусный альфа-интерферон, например, описанный в US 4897471 и 4695623; или интерферон альфа-n3, смесь встречающихся в естественных условиях альфа-интерферонов, такая как ALFERON. Предпочтительным является применение интерферона альфа-2 а или альфа-2b. Получение интерферона альфа-2b описано в US 4530901. Понятие "интерферон альфа" относится также к интерферону, который включает "пэгилированные" аналоги, т.е. модифицированные полиэтиленгликолем конъюгаты интерферона альфа, предпочтительно интерферона альфа-2 а и -2b. Предпочтительный конъюгат полиэтиленгликоль-интерферон альфа-2b представляет собой ПЭГ 12000-интерферон альфа-2b. Понятие "ПЭГ 12000-IFN альфа" в контексте настоящего описания означает конъюгаты, такие как полученные согласно методам, описанным в международной заявке на патент WO 95/13090, и которые содержат уретановые связи между аминогруппами интерферона альфа-2 а или -2b и полиэтиленгликолем со средней молекулярной массой 12000. Предпочтительный конъюгат ПЭГ 12000-интерферон альфа-2b получают путем присоединения полимера ПЭГ к эпсилон-аминогруппе остатка лизина в молекуле IFN альфа-2b. Одну молекулу ПЭГ 12000 конъюгируют со свободными аминогруппами молекулы IFN альфа-2b через уретановую связь. Этот конъюгат отличается молекулярной массой присоединенного ПЭГ 12000. Конъюгат ПЭГ 12000-IFN альфа-2b приготавливают в виде лиофилизированного порошка для инъекций. Цель конъюгации IFN альфа с ПЭГ заключается в улучшении характеристик вводимого белка путем значительного удлинения времени его полужизни в плазме, обеспечивая, тем самым, пролонгированную активность IFN альфа. Наиболее предпочтительными конъюгатами интерферона альфа, которые можно применять согласно настоящему изобретению, являются пэгилированные альфа-интерфероны, например пэгилированный интерферон альфа-2 а, пэгилированный интерферон альфа-2b, пэгилированный консенсусный интерферон или пэгилированный очищенный продукт интерферона альфа. Пэгилированный интерферон альфа 2 а описан, например, в ЕР 0593868, и он поступает в продажу, например, под товарным знаком PEGASYS (фирма Hoffmann-La Roche). Пэгилированный интерферон альфа-2b описан, например, вUS 5908621 и WO 98/48840, и он поступает в продажу, например, под товарным знаком PEG-INTRON А (фирма Schering Plough). Пэгилированный консенсусный интерферон описан в WO 96/11953. Предпочтительными пэгилированными альфа-интерферонами являются пэгилированный интерферон альфа-2 а и пэгилированный интерферон альфа-2b. Предпочтительным является также пэгилированный консенсус-3 019965 ный интерферон. Понятие "интерферон альфа" относится также к другим конъюгатам интерферона альфа, которые можно получать путем связывания интерферона альфа с водорастворимым полимером. Примерами таких полимеров являются (но не ограничиваясь только ими) другие гомополимеры полиалкиленоксида, такие как полиэтиленгликоль (ПЭГ), полипропиленгликоли, полиоксиэтиленированные полиолы, их сополимеры и их блок-сополимеры. В качестве альтернативы полимеров на основе полиалкиленоксида можно применять эффективные, не обладающие антигенными свойствами материалы, такие как декстран, поливинилпирролидоны, полиакриламиды, поливиниловые спирты, полимеры на основе углеводов и т.п. Такие конъюгаты интерферон альфа-полимер описаны в US 4766106, US 4917888, европейской заявке на патент 0236987, европейских заявках на патент 0510356, 0593868 и 0809996 (пэгилированный интерферон альфа-2 а) и в опубликованной международной заявке на патент WO 95/13090. Понятие "интерферон альфа" относится также к слитым белкам интерферона альфа, например слитым белкам интерферона- -2 а, интерферона- -2b, консенсусного интерферона или очищенного продукта интерферона-, каждый из которых слит с другим белком. Конкретные предпочтительные слитые белки, содержащие интерферон (например, интерферон 2b) и альбумин, описаны в US 6972322 и в опубликованных международных заявках на патент WO 2005/003296 и WO 2005/077042. Предпочтительным интерфероном, конъюгированным с человеческим альбумином, является ALBUFERON, который представляет собой обладающую более длительным действием форму интерферона альфа, созданную с помощью технологии слияния с альбумином. ALBUFERON получают путем генетического слияния человеческого альбумина и интерферона альфа. К ним относятся также консенсусные интерфероны,такие как INFERGEN. Понятие "фармацевтически приемлемая соль" обозначает соль соединения формулы (1), которую с медицинской точки зрения можно использовать в контакте с тканями человека и низших животных, не вызывая при этом признаков повышенной токсичности, раздражения, аллергической реакции и т.п., для которой имеет место приемлемое соотношение польза/риск и которая, как правило, может растворяться или диспергироваться в воде или масле и обладает эффективностью при ее целевом применении. Понятие включает фармацевтически приемлемые кислотно-аддитивные соли и фармацевтически приемлемые соли присоединения оснований. Перечень приемлемых солей приведен, например, у S.M. Birge et al., J.Pharm. Sci., 66, 1977, p. 1-19. Понятие "фармацевтически приемлемая кислотно-аддитивная соль" обозначает соли, которые сохраняют свою биологическую эффективность и свойства свободных оснований и которые не являются нежелательными с биологической точки зрения или по другим причинам, образованные с неорганическими кислотами, такими как соляная, бромисто-водородная, йодисто-водородная, серная, сульфаминовая, азотная, фосфорная кислоты и т.п., и с органическими кислотами, такими как уксусная, трифторуксусная, адипиновая, аскорбиновая, аспарагиновая, бензолсульфоновая, бензойная, масляная, камфорная,камфорсульфоновая, коричная, лимонная, диглюконовая, этансульфоновая, глутаминовая, гликолевая,глицерофосфорная, полусерная, капроновая, муравьиная, фумаровая, 2-гидроксиэтансульфоновая (изэтионовая кислота), молочная, гидроксималеиновая, яблочная, малоновая, миндальная, мезитиленсульфоновая, метансульфоновая, нафталинсульфоновая, никотиновая, 2-нафталинсульфоновая, щавелевая, памовая, пектиновая, фенилуксусная, 3-фенилпропионовая, пивалиновая, пропионовая, пировиноградная,салициловая, стеариновая, янтарная, сульфаниловая, винная, паратолуолсульфоновая, ундекановая кислоты и т.п. Понятие "фармацевтически приемлемая соль присоединения основания" обозначает соли, которые сохраняют свою биологическую эффективность и свойства свободных кислот и которые не являются нежелательными с позиций биологии или по другим причинам, образованные с неорганическими основаниями, такими как аммиак или гидроксид, карбонат или бикарбонат аммония, или катионами металла,такого как натрий, калий, литий, кальций, магний, железо, цинк, медь, марганец, алюминий и т.п. Наиболее предпочтительными являются соли аммония, калия, натрия, кальция и магния. Соли, полученные из фармацевтически приемлемых органических нетоксических оснований, включают соли первичных, вторичных и третичных аминов, четвертичных аминовых соединений, замещенных аминов, включая встречающиеся в естественных условиях замещенные амины, циклические амины и основные ионообменные смолы, такие как метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, изопропиламин, трипропиламин, трибутиламин, этаноламин, диэтаноламин, 2-диметиламиноэтанол,2-диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин,N-этилпиперидин, производные тетраметиламмония, производные тетраэтиламмония, пиридин,N,N-диметиланилин, N-метилпиперидин, N-метилморфолин, дициклогексиламин, дибензиламин,N,N-дибензилфенэтиламин, 1-эфенамин, N,N'-дибензилэтилендиамин, полиаминовые смолы и т.п. Наиболее предпочтительными органическими нетоксическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин. Понятие "рибавирин" относится к 1D-рибофуранозил-1 Н-1,2,4-триазол-3-карбоксамиду, который выпускает фирма ICN Pharmaceuticals Inc., Коста-Меса, шт. Калифорния и который описан в the MerckIndex, 11-е изд. как соединение 8199. Его получают и включают в препаративные формы согласно методу, описанному в US 4211771. Предпочтительными имеющимися на рынке продуктами на основе рибавирина являются REBETOL и COPEGUS. Под понятие подпадают также производные и аналоги рибавирина, например, описанные в US 6063772, 6403564 и 6277830. Например, производные или аналоги включают модифицированные рибавирины, такие как сложные 5'-аминоэфиры, разработанный фирмой ICN Pharmaceutical L-энантиомер рибавирина (ICN 17261), 2'-дезоксипроизводные рибавирина и 3-карбоксамидиновые производные рибавирина, вирамидин (ранее известный как рибамидин) и т.п. Понятие "терапевтическая комбинация" в контексте настоящего описания означает комбинацию,включающую одну или несколько лекарственных субстанций, т.е. соединений, имеющих терапевтическую применимость. Как правило, каждое такое соединение в терапевтических комбинациях, предлагаемых в настоящем изобретении, должно присутствовать в виде фармацевтической композиции, содержащей указанное соединение и фармацевтически приемлемый носитель. Соединения в терапевтической комбинации, предлагаемой в настоящем изобретении, можно вводить одновременно или раздельно в качестве части схемы применения. Варианты осуществления изобретения Основной вариант осуществления настоящего изобретения относится к способу лечения вызываемой HCV инфекции или облегчения одного или нескольких ее симптомов у пациента, заключающемуся в том, что вводят пациенту терапевтическую комбинацию, которая содержит соединение (1), указанное в настоящем описании, или его фармацевтически приемлемую соль в сочетании с интерфероном альфа и рибавирином. Другой вариант осуществления настоящего изобретения относится к применению соединения (1), указанного в настоящем описании, или его фармацевтически приемлемой соли, интерферона альфа и рибавирина для приготовления фармацевтического набора, предназначенного для лечения инфекции, вызываемой вирусом гепатита С (HCV), или облегчения одного или нескольких ее симптомов у пациента. Хотя ожидается, что указанная комбинированная терапия должна обладать эффективностью в отношении всех генотипов HCV, было продемонстрировано, что она обладает эффективностью прежде всего в отношении лечения инфекции, вызываемой генотипом 1 HCV, включая субгенотипы 1 а и 1b. Популяцию пациентов, подлежащих лечению с помощью комбинированной терапии, предлагаемой в настоящем изобретении, можно дополнительно классифицировать как пациентов "не подвергавшихся ранее лечению", т.е. пациентов, которые ранее не подвергались никакому лечению инфекции, вызываемой HCV, и пациентов, "подвергавшихся ранее лечению", т.е. пациентов, которые ранее подвергались лечению инфекции, вызываемой HCV. С помощью комбинированной терапии, предлагаемой в настоящем изобретении, можно лечить пациентов, относящихся к любому из этих классов. Конкретным классом пациентов, которых наиболее предпочтительно лечить таким путем, являются подвергавшиеся ранее лечению пациенты, которых ранее лечили с помощью терапии, включающей применение интерферона плюс рибавирина, но которые не обладали чувствительностью к указанной терапии (обозначены в контексте настоящего описания как "нереспондеры"). Указанных нереспондеров подразделяют на три различные группы пациентов: (1) пациенты, у которых в процессе лечения интерфероном плюс рибавирином максимальное снижение уровней РНК HCV составляло 1log10 ("нулевые респондеры"); (2) пациенты, у которых в процессе лечения интерфероном плюс рибавирином максимальное снижение уровней РНК HCV составляло 1log10, но у которых никогда не достигались уровни РНК HCV ниже предела обнаружения ("частичные респондеры"); и (3) пациенты, у которых удавалось достигать вирусологического ответа с помощью и в период осуществления лечения интерфероном плюс рибавирином, но у которых происходило восстановление вирусной нагрузки либо в процессе лечения (по причине, отличной от несоблюдения пациентом режима и схемы лечения), либо после завершения лечения ("пациент, имеющий рецидив"). Как будет более подробно описано ниже, при создании изобретения при лечении некоторых пациентов-нереспондеров с помощью схемы комбинированной терапии, предлагаемой в настоящем изобретении, были получены особенно неожиданные результаты. Согласно альтернативному варианту осуществления изобретения настоящее изобретение относится к способу снижения уровней РНК HCV у пациента, нуждающегося в этом, заключающемуся в том, что вводят пациенту терапевтическую комбинацию, предлагаемую в настоящем изобретении. Предпочтительно способ, предлагаемый в настоящем изобретении, позволяет снижать уровни РНК HCV у пациентов до уровня, находящегося ниже выявляемого уровня. Понятия "ниже выявляемого уровня" и "ниже уровня обнаружения" в контексте настоящего описания имеют одинаковое значение и означают уровень,более низкий, чем выявляемый уровень РНК HCV. Выявляемый уровень РНК HCV согласно настоящему изобретению соответствует по меньшей мере 50 международным единицам (ME) на 1 мл сыворотки пациента при оценке с помощью методологии количественной, состоящей из многих циклов ПЦР с обратной транскриптазой согласно международному стандарту ВОЗ (Saladanha J., Lelie N. и Heath A.,Establishment of the first international standard for nucleic acid amplification technology (NAT) assays for HCVRNA. WHO Collaborative Study Group. Vox Sang 76, 1999, p. 149-158). Указанные методы хорошо извест-5 019965 ны в данной области. В предпочтительном варианте осуществления изобретения способ, предлагаемый в настоящем изобретении, позволяет снижать уровни РНК HCV у пациентов до величины, составляющей менее чем 25 ME на 1 мл сыворотки, еще более предпочтительно до величины, составляющей менее чем 10 ME на 1 мл сыворотки. Обычная продолжительность лечения с использованием стандартной терапии на основе интерферона плюс рибавирина составляет по меньшей мере 48 недель в случае инфекции, вызываемой HCV генотипа 1, и по меньшей мере 24 недели в случае HCV генотипов 2 и 3. Однако при добавлении соединения(1) или его фармацевтически приемлемой соли при использовании тройной комбинированной терапии,предлагаемой в настоящем изобретении, можно существенно укорачивать продолжительность лечения. При использовании тройной комбинированной терапии, предлагаемой в настоящем изобретении, предполагаемая продолжительность лечения составляет по меньшей мере 4 недели, предпочтительно по меньшей мере 12 недель, например от примерно 12 до примерно 24 недель, хотя также возможно осуществлять лечение в течение вплоть до 48 недель и более. Таким образом, согласно другим вариантам осуществления изобретения продолжительность лечения составляет по меньшей мере 24 недели и по меньшей мере 48 недель. Ожидается, что период лечения инфекции, вызываемой различными генотипамиHCV, например генотипами HCV 2, 3 или 4, является одинаковым. Предполагается также, что за начальной схемой лечения с использованием тройной комбинированной терапии, предлагаемой в настоящем изобретении, следует лечение с использованием двойной комбинированной терапии на основе только интерферона плюс рибавирина. Таким образом, возможными сценариями применения сначала тройной, а затем двойной комбинированной терапии являются: (1) 4 недели применения тройной комбинированной терапии, затем 20-44 недели применения терапии только на основе интерферона плюс рибавирина; (2) 12 недель применения тройной комбинированной терапии, затем 12-36 недель применения терапии только на основе интерферона плюс рибавирина; и (3) 24 недели применения тройной комбинированной терапии, затем 12-24 недели применения терапии только на основе интерферона плюс рибавирина. В состав композиции входит первый компонент терапевтической комбинации, а именно соединение(1) или его фармацевтически приемлемая соль. Указанная композиция содержит соединение (1) или его фармацевтически приемлемую соль и фармацевтически приемлемый адъювант или носитель. Типичные фармацевтические композиции, в составе которых можно применять соединение (1) или его фармацевтически приемлемую соль, описаны в US 7585845. Как правило, соединение (1) или его фармацевтически приемлемую соль можно вводить в дозе, составляющей по меньшей мере 40 мг/день (в виде однократных или разделенных доз). В других вариантах осуществления изобретения уровни и диапазоны доз (при введении в виде однократных или разделенных доз) могут представлять собой:(а) по меньшей мере 48 мг/день;(б) по меньшей мере 100 мг/день;(в) по меньшей мере 120 мг/день;(г) по меньшей мере 200 мг/день;(д) по меньшей мере 240 мг/день;(е) по меньшей мере 360 мг/день;(ж) по меньшей мере 480 мг/день;(т) примерно 480 мг/день. Хотя соединение (1) или его фармацевтически приемлемую соль можно вводить в виде однократных или разделенных суточных доз, предпочтительным является введение суточной дозы один раз в день. Однако, как должно быть очевидно специалисту в данной области, могут потребоваться дозы, более низкие или более высокие по сравнению с вышеуказанными. Конкретные дозы и схемы лечения для любого конкретного пациента должны зависеть от различных факторов, включая возраст, вес тела, общее состояние здоровья, пол, диету, продолжительность введения, скорость экстракции, комбинацию лекарственных средств, серьезность и течение инфекции, предрасположенность пациента к инфекции и рекомендацию лечащего врача. В целом, соединение наиболее предпочтительно применять, используя уровень концентрации, который, как правило, приводит к антивирусному действию, не вызывая при этом вредных или опасных побочных действий. В состав фармацевтической композиции входит второй компонент терапевтической комбинации, а именно интерферон альфа. Как правило, указанные композиции представляют собой предназначенные для инъекций препаративные формы, содержащие интерферон альфа и фармацевтически приемлемый адъювант или носитель, которые известны в данной области, в том числе ряд поступающих в продажу препаративных форм интерферона альфа (см., например, описание различных поступающих в продажу продуктов на основе интерферона альфа и различные патенты и другую литературу, касающуюся интерферона альфа, которые процитированы выше в настоящем описании). Типы интерферонов альфа, которые можно применять в комбинации, указаны выше в настоящем описании в разделе "Определения". В одном из предпочтительных вариантов осуществления изобретения интерферон альфа представляет собой пэгилированный интерферон альфа. В другом варианте осуществления изобретения интерферон альфа представляет собой пэгилированный интерферон альфа-2 а или пэгилированный интерферон альфа-2b. В наиболее предпочтительном варианте осуществления изобретения интерферон альфа представляет собой PEGASYS или PEG-INTRON. При применении известных поступающих в продажу продуктов на основе интерферона альфа указанные продукты можно вводить в указанных на этикетке дозах, которые показаны для применения в комбинированной терапии с использованием комбинации интерферона плюс рибавирина при лечении вызываемой HCV инфекции. Естественно, что при использовании тройной комбинированной терапии,предлагаемой в настоящем изобретении, можно применять более низкие дозы интерферона альфа, например существенно более низкие, чем применяемые при современной стандартной терапии на основе интерферона плюс рибавирина, достигая при этом такой же или более высокой эффективности по сравнению с современной стандартной терапией, что сопровождается меньшими побочными действиями,обычно присущими такой терапии. В одном из вариантов осуществления изобретения интерферон альфа можно вводить парентерально 1-3 раза в неделю, предпочтительно 1 или 2 раза в неделю. Касательно пэгилированных интерферонов альфа следует отметить, что их, как правило, вводят 1 раз в неделю, и диапазоны общих недельных доз составляют, например, от примерно 0,5 до примерно 2 мкг/кг/неделю в случае пэгилированного интерферона альфа-2b, а в случае пэгилированного интерферона альфа-2 а доза зависит от веса тела пациента и, как правило, составляет примерно от 90 до 200 мкг/неделю, более предпочтительно от примерно 160 до примерно 200 мкг/неделю. В комбинации с рибавирином стандартная доза пэгилированного интерферона альфа-2b составляет примерно 1,5 мкг/кг/неделю, а стандартная доза пэгилированного интерферона альфа-2 а составляет примерно 180 мкг/неделю, и ее применяют в сочетании примерно с 600-1200 мг/день, в частности 800-1200 мг/день вводимого орально рибавирина. Согласно другим вариантам осуществления изобретения пэгилированный интерферон альфа-2b можно вводить в следующих дозах:(г) примерно 1,5 мкг/кг/неделю. Согласно другим вариантам осуществления изобретения пэгилированный интерферон альфа-2 а можно вводить в следующих дозах:(в) примерно 180 мкг/кг/неделю. В состав фармацевтической композиции входит третий компонент терапевтической комбинации, а именно рибавирин. Как правило, такие композиции включают рибавирин и фармацевтически приемлемый адъювант или носитель, и они хорошо известны в данной области, включая ряд поступающих в продажу препаративных форм рибавирина. Содержащие рибавирин препаративные формы описаны, например, в US 4211771. Типы рибавирина, которые можно применять в комбинации, указаны выше в настоящем описании в разделе "Определения". В одном из предпочтительных вариантов осуществления изобретения рибавирин представляет собой либо REBETOL, либо COPEGUS, и его можно вводить в указанных на этикетке дозах, которые показаны при применении в комбинированной терапии, включающей комбинацию интерферона плюс рибавирина, для лечения вызываемой HCV инфекции. Естественно, при использовании тройной комбинированной терапии, предлагаемой в настоящем изобретении, можно применять более низкие дозы рибавирина, например более низкие, чем дозы, применяемые при современной стандартной терапии на основе интерферона плюс рибавирина, достигая при этом такой же или более высокой эффективности по сравнению с современной стандартной терапией, что сопровождается меньшими побочными действиями, чем те, которые обычно присущи такой терапией. Согласно различным вариантам осуществления изобретения рибавирин можно вводить в дозах (в виде однократных или разделенных доз), составляющих:(м) примерно 800 мг/день. Согласно одному из вариантов осуществления изобретения включающая рибавирин композиция содержит рибавирин в виде препаративной формы, пригодной для дозирования 1 раз в день, 2 раза в день, 3 раза в день, 4 раза в день, 5 раз в день или 6 раз в день. Например, если терапевтическая комбинация содержит рибавирин в дозе, составляющей примерно 1000 мг/день, и дозирование осуществляют при необходимости 5 раз в день, то терапевтическая комбинация должна содержать рибавирин в препаративной форме, например в таблетке, которая содержит, например, примерно 200 мг рибавирина. Касательно тройной комбинированной терапии, предлагаемой в настоящем изобретении, которая основана на применении соединения (1) или его фармацевтически приемлемой соли в сочетании с интерфероном альфа и в сочетании с рибавирином, следует иметь в виду, что настоящее изобретение относится ко всем комбинациям различных предпочтительных вариантов осуществления и подвариантов осуществления изобретения, которые изложены выше, и все они подпадают под объем изобретения. Например, одним из вариантов осуществления настоящего изобретения является способ лечения вызываемой вирусом гепатита С (HCV) инфекции или облегчения одного или нескольких ее симптомов у пациента, заключающийся в том, что вводят пациенту терапевтическую комбинацию, содержащую:(а) соединение (1) или его фармацевтически приемлемую соль в дозе, составляющей от примерно 48 до примерно 480 мг в день;(б) пэгилированный интерферон альфа-2 а в дозе, составляющей от примерно 160 до примерно 200 мкг/неделю, или пэгилированный интерферон альфа-2b в дозе, составляющей от примерно 0,5 до примерно 2 мкг/кг/неделю; и(в) рибавирин в дозе, составляющей от примерно 400 до примерно 1200 мг/день. Другим вариантом осуществления настоящего изобретения является способ лечения вызываемой вирусом гепатита С (HCV) инфекции или облегчения одного или нескольких ее симптомов у пациента,заключающийся в том, что вводят пациенту терапевтическую комбинацию, содержащую:(а) соединение (1) или его фармацевтически приемлемую соль в дозе, составляющей от примерно 48 до примерно 480 мг в день;(в) рибавирин в дозе, составляющей от примерно 1000 до примерно 1200 мг/день. Другим вариантом осуществления настоящего изобретения является способ лечения вызываемой вирусом гепатита С (HCV) инфекции или облегчения одного или нескольких ее симптомов у пациента,заключающийся в том, что вводят пациенту терапевтическую комбинацию, содержащую:(а) соединение (1) или его фармацевтически приемлемую соль в дозе, составляющей от примерно 48 до примерно 480 мг в день;(в) рибавирин в дозе, составляющей примерно 800 мг/день. Следующие варианты осуществления изобретения включают любой из вышеуказанных вариантов,в которых:(а) вызываемая HCV инфекция ассоциирована с генотипом 1 и пациент представляет собой ранее не подвергавшегося лечению пациента; или(б) вызываемая HCV инфекция ассоциирована с генотипом 1 и пациент представляет собой ранее подвергавшегося лечению пациента, нечувствительного к комбинированной терапии на основе интерферона плюс рибавирина. Следующие варианты осуществления изобретения включают любой из вышеуказанных вариантов и в них соединение (1) или его фармацевтически приемлемую соль вводят один раз в день, интерферон альфа вводят один раз в неделю и рибавирин вводят дважды в день. Согласно другому варианту осуществления терапевтическая схема, предлагаемая в настоящем изобретении, предусматривает обработку пациента в течение по меньшей мере примерно 4 недель, более предпочтительно либо в течение по меньшей мере примерно 12 недель, либо в течение по меньшей мере примерно 24 недель:(I) взятом в терапевтически эффективном количестве соединением (1) или его фармацевтически приемлемой солью один раз в день;(II) взятом в терапевтически эффективном количестве интерфероном альфа один раз в неделю;(III) взятом в терапевтически эффективном количестве рибавирином два раза в день. Другим вариантом осуществления настоящего изобретения являются наборы, предназначенные для лечения вызываемой HCV инфекции у пациента. Наборы, предлагаемые в настоящем изобретении, могут содержать любую из терапевтических комбинаций, предлагаемых в настоящем изобретении. Наборы могут содержать также инструкции по применению терапевтических комбинаций. Наборы могут удовлетворять потребностям различных классов или типов пациентов или другим клинически значимым факторам, таким как возраст, сопутствующие заболевания/состояния, серьезность и стадия вызываемой HCV инфекции, чувствительность или нечувствительность к предшествующему лечению, предрасположенность к побочным действиям и т.д. Согласно другому варианту осуществления изобретения в настоящем изобретении предложен набор, включающий:(а) первую фармацевтическую композицию, содержащую соединение (1) или его фармацевтически приемлемую соль:(г) инструкции по применению вышеуказанных композиций. Кроме того, при создании изобретения неожиданно было обнаружено подавление устойчивости вируса HCV в процессе лечения с использованием комбинированной терапии, предлагаемой в настоящем изобретении. Применение различных доз соединения (1) (натриевая соль) в качестве монотерапии приводило к быстрому восстановлению вирусной нагрузки в первые 14 дней лечения у большинства пациентов из всех обрабатываемых разными дозами групп, не подвергавшихся ранее лечению пациентов, у которых обнаружено снижение вирусной нагрузки. В противоположность этому, из 19 подвергавшихся ранее лечению пациентов, которым вводили соединение (1) (натриевую соль) один раз в день в дозах 48 мг (n=6), 120 мг (n=7) или 240 мг (n=6) в комбинации с пэгилированным интерфероном альфа-2 а и рибавирином (ПЭГ-IFN/RBV) в течение 28 дней, восстановление вирусной нагрузки в течение 28 дней лечения обнаружено только у 2/6 пациентов в группе, обработанной дозой 48 мг, и у 1/7 пациентов в группе, обработанной дозой 120 мг. Важно, что не обнаружено восстановление вирусной нагрузки в течение первых 28 дней лечения в группе подвергавшихся ранее лечению пациентов, которым вводили соединение (1) (натриевую соль) в дозе 240 мг/день в комбинации с ПЭГ-IFN/RBV. Таким образом, согласно другому варианту осуществления изобретения имело место ограничение или отсутствие возникновения устойчивости вируса при использовании комбинированной терапии,предлагаемой в настоящем изобретении. Согласно более конкретному варианту осуществления изобретения имеет место ограничение или отсутствие возникновения вариантов HCV, которые кодируют в протеазе NS3 HCV одну или несколько следующих аминокислотных замен: R155, и/или D168, и/или А 156,при использовании комбинированной терапии, предлагаемой в настоящем изобретении. Следующие варианты осуществления изобретения включают любой из вышеуказанных вариантов,в которых:(а) вызываемая HCV инфекция ассоциирована с генотипом 1 и пациент представляет собой ранее не подвергавшегося лечению пациента; либо(б) вызываемая HCV инфекция ассоциирована с генотипом 1 и пациент представляет собой ранее подвергавшегося лечению пациента, нечувствительного к комбинированной терапии на основе интерферона плюс рибавирина; и в них ограничено или отсутствует возникновение вариантов HCV, которые кодируют замены в протеазе NS3 HCV аминокислоты R155, при использовании комбинированной терапии, предлагаемой в настоящем изобретении. Следующие варианты осуществления изобретения включают любой из вышеуказанных вариантов,в которых:(а) вызываемая HCV инфекция ассоциирована с генотипом 1b и пациент представляет собой ранее не подвергавшегося лечению пациента; либо(б) вызываемая HCV инфекция ассоциирована с генотипом 1b и пациент представляет собой ранее подвергавшегося лечению пациента, нечувствительного к комбинированной терапии на основе интерферона плюс рибавирина; и в них ограничено или отсутствует возникновение вариантов HCV, которые кодируют замены в протеазе NS3 HCV аминокислоты D168, при использовании комбинированной терапии, предлагаемой в настоящем изобретении.I. Методы получения соединения (1). Методы получения аморфного соединения (1) описаны в US 6323180, 7514557 и 7585845, которые включены в настоящее описание в качестве ссылки. В приведенных ниже примерах 1-5 представлены методы получения дополнительных форм соединения (1), которые можно применять согласно настоящему изобретению. Пример 1. Получение кристаллической формы типа А соединения (1). Аморфное соединение (1) (партия 7, 13,80 г) вносили в 1000-миллилитровую трехгорлую колбу. В колбу добавляли абсолютный этанол (248,9 г). При перемешивании содержимое колбы нагревали со скоростью 60 С/ч до 74 С. (Твердые частицы не растворялись при 74 С). Затем к образовавшейся суспензии с линейной скоростью добавляли при перемешивании воду (257,4 г) в течение 4 ч и поддерживали температуру на уровне 74 С. После завершения добавления воды температуру линейно снижали до температуры окружающей среды со скоростью 8 С/ч, а затем смесь выдерживали при температуре окружающей среды в течение 6 ч при перемешивании. Образовавшиеся твердые частицы собирали фильтрацией и промывали 50 мл смеси 1/1 (мас./мас.) EtOH/вода. Влажные твердые частицы сушили в воронке в течение 30 мин путем прокачивания N2 через осадок (XRPD-анализ (рентгеновская порошковая дифракция) этого образца позволил установить, что его структура аналогична EtOH-сольвату). Затем твердые частицы сушили при 65-70 С в вакууме (Р=25 мм рт.ст.) и выпускали азот в течение 1,5 ч. Анализ образовавшихся твердых частиц (12,6 г, скорректированный выход 95,5%) с помощью XRPD подтвердил, что получено соединение (1) типа А. Пример 2. Получение натриевой соли соединения (1). Метод 1. 2,1 г аморфной натриевой соли соединения (1) и 8,90 г ацетона добавляли во флакон и перемешивали при температуре окружающей среды в течение 3 ч. Суспензию отфильтровывали от маточного раствора и образовавшиеся твердые частицы сушили в течение 20 мин в потоке азота. Собирали 1,51 г кристаллической натриевой соли соединения (1). Пример 3. Получение натриевой соли соединения (1). Метод 2. 15,6 г соединения (1) типа А, 175 мл ацетона и 3,6 мл воды вносили в 250-миллилитровый реактор и нагревали до 53 С до растворения твердых частиц. В реактор добавляли 900 мкл 10 н. NaOH и в раствор вносили затравочный кристалл типа А. Содержащий затравку раствор перемешивали при 53 С в течение 10 мин. Добавляли вторую порцию объемом 900 мкл 10 н. NaOH и систему перемешивали при 53 С в течение 30 мин, при этом образовывалась суспензия. Суспензию охлаждали до 19 С со скорость охлаждения 15 С/ч и выдерживали в течение ночи при 19 С. Конечную образовавшуюся суспензию фильтровали и влажные твердые частицы промывали 15 мл ацетона. Частицы сушили в течение 1 ч при 52 С в вакууме в потоке азота и затем твердые частицы выдерживали на воздухе в лаборатории в течение 1 ч. Собирали 12,1 г твердых частиц, представляющих собой кристаллическую натриевую соль соединения(1). Пример 4. Получение натриевой соли соединения (1). Метод 3. 25,4 кг аморфного соединения (1), 228 л ТГФ и 11,1 кг 10 мас.% NaOH (aq) вносили в реактор. Компоненты перемешивали при 25 С до растворения всех твердых частиц. Образовавшийся раствор фильтровали и реактор и фильтр промывали 23 л ТГФ. Удаляли 180 л растворителя с помощью атмосферной перегонки при 65 С. Добавляли 195 л MIBK (метилизобутилкетон) и 166 л растворителя удаляли путем вакуумной перегонки при 44 С. В реактор вновь добавляли 161 л MIBK и 0,41 кг воды и содержимое нагревали до 70 С. 255 г затравочных кристаллов натриевой соли соединения (1) добавляли при 70 С и вносили в течение 1,5 ч 1,42 л воды. После добавления воды суспензию выдерживали при 70 С в течение 45 мин и затем охлаждали до 45 С в течение 1 ч. Образовавшуюся суспензию фильтровали и промывали 64 л MIBK, содержащего 0,8 мас.% воды. Влажный осадок сушили при 55 С, получая 25 кг кристаллической натриевой соли соединения (1). Пример 5. Получение натриевой соли соединения (1). Метод 4. В реактор вносили 2,00 г аморфного соединения (1), 9,96 г ТГФ и 0,11 г воды и перемешивали при температуре окружающей среды до растворения твердых частиц. Добавляли в раствор по каплям при перемешивании 0,820 мл 21 мас.% NaOET в этаноле, получая раствор А. Во второй реактор добавляли 15,9 г н-BuAc и 160 мкл воды и нагревали до 65 С (раствор Б). 2,56 г раствора А добавляли в раствор Б при 65 С и в образовавшуюся смесь вносили 40 мг натриевой соли соединения (1) в качестве затравочных кристаллов. Содержащую затравку смесь выдерживали при 65 С в течение 45 мин. 2,56 г раствора Б добавляли в раствор А и выдерживали в течение 45 мин с использованием четырех отдельных интервалов. После конечного добавления и выдерживания суспензию охлаждали до 50 С в течение 1 ч и фильт- 10019965 ровали. Влажный осадок промывали 6 мл раствора н-BuAc, содержащего 0,5 мас.% воды. Конечные твердые частицы сушили при 50 С в вакууме, применяя продувку азотом. Собирали твердые частицы,представляющие собой кристаллическую натриевую соль соединения (1).II. Результаты клинических исследований. В описанных ниже клинических исследованиях применяемый лекарственный продукт представлял собой раствор натриевой соли соединения (1), предназначенный для орального введения. Натриевую соль соединения (1) поставляли в место(а) проведения клинического(их) исследования(ий) в виде порошка, предназначенного для приготовления орального раствора с помощью поставляемого в комплекте растворителя. Растворитель использовали также в качестве плацебо. Пример 6. Клиническое исследование, проведенное на не подвергавшихся ранее лечению пациентах. Исследовали безопасность и противовирусную активность натриевой соли соединения (1), нового ингибитора протеазы NS3 HCV, на не подвергавшихся ранее лечению пациентах с хроническим гепатитом, вызываемым вирусом гепатита С генотипа-1, при применении в виде монотерапии и в сочетании с ПЭГ-интерфероном альфа-2 а (Р) и рибавирином (R). Краткое описание исследования. Натриевая соль соединения (1) представляет собой ингибитор протеазы NS3 HCV (EC50 3-6 нМ). Проведено исследование нескольких возрастающих доз этого соединения для оценки безопасности и противовирусной активности на не подвергавшихся ранее лечению пациентов (pts) с хроническим гепатитом, вызываемым вирусом гепатита С генотипа-1, при применении в виде монотерапии в течение 14 дней с последующим применением тройной комбинированной терапии с использованием P+R в течение еще 14 дней. Методы. 34 пациента (из Франции, Германии, Испании, США) с индексами фиброза (печени) по шкалеMetavir 0-3 и не подвергавшиеся ранее терапии с применением любого интерферона или R, произвольно разделяли (в группе по 2 пациента, которых обрабатывали плацебо; 6 или 7 пациентов, которых обрабатывали действующими веществами) на 4 группы, которым вводили один раз в день (qd) натриевую соль соединения (1): 20 мг (n=8), 48 мг (n=9), 120 мг (n=9) или 240 мг (n=8). Натриевую соль соединения (1) применяли в виде монотерапии в течение 14 дней. Пациентам, у которых обнаружено снижение вирусной нагрузки (VL), составляющее 1log10, в день 10, прекращали введение натриевой соли соединения(1) после дня 14. Пациентам, у которых обнаружено снижение VL, составляющее 1log10, в день 10,продолжали введение натриевой соли соединения (1) в день 15 и добавляли Р (180 мкг/неделю) + R (в зависимости от веса тела) в качестве тройной комбинированной терапии до дня 28. Первичная конечная точка соответствовала снижению VL, составляющему 2log10, в любой момент времени до дня 14. Уровни в плазме РНК HCV оценивали с помощью анализа Roche COBAS TaqMan (LLOQ (предел определения) 25 МЕ/мл). Через 28 дней по усмотрению исследователя пациенты могли продолжать получать стандартное лечение, т.е. Р+R. Результаты. 33 пациента относились к белой расе, 1 - к азиатской, среди них было 27 мужчин, средний возраст = 48,911,1 лет, средний вес тела 79,117,5 кг, и медианный (с учетом диапазонов) основной уровень VL составлял 6,8 (4,7-7,7) log10. Между группами не существовало никаких значительных демографических различий. Натриевая соль соединения (1) хорошо переносилась. Никто из пациентов не прервал участие в опыте на стадии монотерапии из-за нежелательных явлений (НЯ). Как правило, НЯ обнаруживали при использовании P+R. Одно серьезное НЯ в виде астении обнаружено в группе пациентов, которых обрабатывали дозой 20 мг, через 6 дней после начала применения P+R. Быстрое снижение VL обнаружено у всех пациентов, при этом максимальное снижение обнаружено, как правило, через 2-4 дня после начала применения натриевой соли соединения (1). За исключением 1 пациента из группы пациентов, которых обрабатывали дозой 20 мг, у всех пациентов, которым вводили натриевую соль соединения (1), было обнаружено снижение VL, составляющее 2log10, в период осуществления монотерапии. Медианное (с учетом диапазонов) максимальное снижение VL во время 14-дневной монотерапии в группах, обработанных 20, 48, 120 и 240 мг, составляло 3,0 (1,5-3,9), 3,6 (3,1-3,8), 3,7 (3,3-4,1) и 4,2 (3,6-4,8) log10 МЕ/мл соответственно. Никакого существенного изменения VL не обнаружено в плацебо-группе. Восстановление VL в процессе лечения обнаружено в течение первых 14 дней применения монотерапии у большинства пациентов из групп, обрабатываемых всеми исследуемыми дозами. Заключение. Лечение с применением натриевой соли соединения (1) в качестве 14-дневной монотерапии с последующим применением в сочетании с P+R в течение еще 14 дней хорошо переносилось и вызывало сильный и быстрый противовирусный ответ у не подвергавшихся ранее лечению пациентов. На фиг. 1 представлены данные о среднем изменении вирусной нагрузки HCV, полученные в этом исследовании, у пациентов из групп, которых обрабатывали четырьмя дозами, т.е. у не подвергавшихся ранее лечению пациентов с хронической инфекцией, вызываемой HCV генотипа-1, которых лечили с использованием натриевой соли соединения (1) в качестве монотерапии в течение 14 дней с последующим использованием комбинированной терапии на основе натриевой соли соединения (1), пэгилированного интерферона альфа-2 а и рибавирина в течение еще 14 дней. На чертеже изменение вирусной нагрузки выражали в единицах log 10 МЕ/мл, а четыре группы пациентов, которым вводили указанные дозы, обозначены следующим образом:"20 naiv av" = среднее изменение вирусной нагрузки в группе, в которой осуществляли обработку дозой 20 мг;"48 naiv av" = среднее изменение вирусной нагрузки в группе, в которой осуществляли обработку дозой 48 мг;"120 naiv av" = среднее изменение вирусной нагрузки в группе, в которой осуществляли обработку дозой 120 мг;"240 naiv av" = среднее изменение вирусной нагрузки в группе, в которой осуществляли обработку дозой 240 мг. Появление вариантов в процессе обработки не подвергавшихся ранее лечению пациентов. Популяционное секвенирование протеазы NS3/4A в исходный момент времени (основной уровень) и при восстановлении вирусной нагрузки в процессе лечения позволяло осуществлять отбор вариантов,обусловливающих устойчивость in vitro к натриевой соли соединения (1). Обнаружены значительные изменения в имеющих решающее значение остатках в домене протеазы NS3 относительно эталонного субтипа (субгенотип 1a: AF009606 или субгенотип 1b: AJ238799) в процессе лечения в группах, обработанных различными дозами (более подробные данные см. ниже в табл. 1-4). Во всех таблицах Gt обозначает субгенотип HCV, а день 1 обозначает исходную последовательность, и указанные аминокислотные замены представляют собой встречающиеся в естественных условиях полиморфизмы имеющих решающее значение аминокислот в NS3. Дополнительные изменения, которые могут кодировать или которые могут быть ассоциированы с устойчивостью к лекарственному средству, указаны в различные дни в процессе лечения. Таблица 1 Преобладающие варианты (относительно эталонного субтипа) имеющих решающее значение остатков аминокислотных последовательностей,обнаруженные у характерных для популяции не подвергавшихся ранее лечению пациентов из группы, для обработки которой использовали дозу 20 мг нет: не обнаружено никаких замен в отобранных положениях;BLD: ниже предела обнаружения при ПЦР-амплификации;ND: не определяли; кодирует исходный полиморфизм V/I170T. Таблица 2 Преобладающие варианты (относительно эталонного субтипа) имеющих решающее значение остатков аминокислотных последовательностей,обнаруженные у характерных для популяции не подвергавшихся ранее лечению пациентов из группы, для обработки которой использовали дозу 48 мг нет: не обнаружено никаких замен в отобранных положениях;BLD: ниже предела обнаружения при секвенировании; Таблица 3 Преобладающие варианты (относительно эталонного субтипа) имеющих решающее значение остатков аминокислотных последовательностей,обнаруженные у характерных для популяции не подвергавшихся ранее лечению пациентов из группы, для обработки которой использовали дозу 120 мг нет: не обнаружено никаких замен в отобранных положениях;BLD: ниже предела обнаружения при секвенировании; Таблица 4 Преобладающие варианты (относительно эталонного субтипа) имеющих решающее значение остатков аминокислотных последовательностей,обнаруженные у характерных для популяции не подвергавшихся ранее лечению пациентов из группы, для обработки которой использовали дозу 240 мг нет: никаких замен не обнаружено в отобранных положениях;BLD: ниже предела обнаружения при ПЦР-амплификации и секвенировании; Пример 7. Клиническое исследование, проведенное на подвергавшихся ранее лечению пациентах. Исследовали безопасность и противовирусную активность натриевой соли соединения (1), нового ингибитора протеазы NS3 HCV, на подвергавшихся ранее лечению с использованием P+R пациентов с хроническим гепатитом, вызываемым вирусом гепатита С генотипа-1, при применении в виде комбинированной терапии в сочетании с ПЭГ-интерфероном альфа-2 а (Р) и рибавирином (R) в течение 28 дней. Краткое описание исследования. Натриевая соль соединения (1) представляет собой ингибитор протеазы NS3 HCV (EC50 3-6 нМ). Проведено исследование нескольких возрастающих доз этого соединения для оценки безопасности и противовирусной активности на подвергавшихся ранее лечению с использованием P+R пациентов (pts) с хроническим гепатитом, вызываемым вирусом гепатита С генотипа -1, при применении в виде комбинированной 28-дневной терапии в сочетании с P+R. Методы. Оценивали 19 пациентов (из Франции, Германии, Испании, США) с индексами фиброза (печени) по шкале Metavir 0-3, для которых был выявлен неблагоприятный исход проведенного ранее лечения вирусного заболевания с помощью комбинированной терапии на основе P+R, которым вводили натриевую соль соединения (1) один раз в день (qd) в дозах 48 мг (n=6), 120 мг (n=7) или 240 мг (n=6) в сочетании с Р (180 мкг/неделю)+R (в зависимости от веса тела) в течение 28 дней. Осуществляли мониторинг всех пациентов в отношении безопасности и переносимости исследуемых лекарственных средств. Первичная конечная точка соответствовала снижению вирусной нагрузки (VL) HCV, составляющему 2log10 относительно исходного уровня в любой момент времени до дня 28. Уровни в плазме РНК HCV оценивали с помощью анализа Roche COBAS TaqMan (LLOQ 25 МЕ/мл). Включенные в настоящее исследование подвергавшиеся ранее лечению пациенты включали нулевых P+R-респондеров (т.е. пациентов, которые вообще не давали никакого ответа на комбинированную терапию) и частичных респондеров. Через 28 дней по усмотрению исследователя пациенты могли продолжать получать стандартное лечение, т.е. Р+R. Результаты. 19 пациентов относились к белой расе, среди которых было 11 мужчин, средний возраст = 489 лет,средний вес тела 8115 кг, и медианный (с учетом диапазонов) основной уровень VL составлял 6,9 (5,9-7,4) log10. Между группами не существовало никаких значительных демографических различий. Натриевая соль соединения (1) хорошо переносилась, и у пациентов в этом исследовании не было обнаружено никаких серьезных или тяжелых нежелательных явлений (НЯ). Как правило, НЯ обнаруживали при использовании P+R. Один пациент прервал участие в опыте из-за НЯ (состояние тревоги). У всех пациентов обнаружено быстрое, зависящее от дозы снижение VL. При применении тройной комбинированной терапии у всех пациентов, обработанных натриевой солью соединения (1)+Р+R, обнаружено снижение VL, достигающее 2log 10. Медианное (с учетом диапазонов) максимальное снижение VL во время 28-дневной комбинированной терапии в группах, обработанных дозами 48, 120 и 240 мг, составляло 4,8 (3,4-5,9), 5,2 (3,9-6,0) и 5,3 (4,8-6,1) log 10 МЕ/мл соответственно. Было обнаружено восстановление вирусной нагрузки в процессе лечения в течение первых 28 дней применения натриевой соли соединения (1)+P+Ry 2/6 пациентов при применении дозы 48 и у 1/7 пациентов при применении дозы 120 мг. Популяционное секвенирование у этих пациентов протеазы NS3/4A в исходный момент времени (основной уровень) и при восстановлении вирусной нагрузки в процессе лечения позволяло осуществлять отбор вариантов, обусловливающих устойчивость in vitro к натриевой соли соединения (1). У одного пациента из группы, в которой осуществляли обработку дозой 120 мг, не обнаружено восстановление вирусной нагрузки, но обнаружен выход на плато с уровнем VL 500 копий МЕ/мл ко дню 28 и наличие кодируемого мутанта R155K; этим образом, уровень вирусной нагрузки в указанном образце находился ниже нижнего предела обнаружения в фенотипических анализах устойчивости, проведенных при создании настоящего изобретения. В течение 28-дневного лечения BI 201335 не было обнаружено никакого восстановления в группе,которую обрабатывали дозой 240 мг/день: у 5/6 пациентов уровень VL составлял 25 МЕ/мл в день 28. У шестого пациента уровень VL снижался до 4,7log10 относительно исходного уровня в день 28, и при следующем визите в день 42 уровень VL составлял 25 МЕ/мл. Заключение. Лечение с применением натриевой соли соединения (1) при введении один раз в день в комбинированной терапии с сочетании с Р+R в течение 28 дней хорошо переносилось и вызывало сильный и быстрый противовирусный ответ у подвергавшихся ранее лечению пациентов. На фиг. 2 представлены данные о среднем изменении вирусной нагрузки HCV, полученные в этом исследовании, у пациентов из групп, которых обрабатывали тремя дозами, т.е. у подвергавшихся ранее лечению пациентов с хронической инфекцией, вызываемой HCV генотипа-1, которых лечили с помощью комбинированной терапии, включающей применение натриевой соли соединения (1), пэгилированного интерферона альфа-2 а и рибавирина в течение 28 дней. На чертеже изменение вирусной нагрузки выражали в единицах log 10 МЕ/мл, а три группы пациентов, которым вводили указанные дозы, обозначены следующим образом:"48 NR av" = среднее изменение вирусной нагрузки в группе, в которой осуществляли обработку дозой 48 мг;"120 NR av" = среднее изменение вирусной нагрузки в группе, в которой осуществляли обработку дозой 120 мг;"240 NR av" = среднее изменение вирусной нагрузки в группе, в которой осуществляли обработку дозой 240 мг. В табл. 5 представлены некоторые параметры вирусологического ответа (снижение вирусной нагрузки) у пациентов в этом исследовании (где "D" обозначает день и "QD" обозначает один раз в день;"N" обозначает количество пациентов; "ниже нижнего предела количественного обнаружения" означает менее 25 ME/мл сыворотки). Таблица 5 Появление вариантов в процессе обработки подвергавшихся ранее лечению пациентов. Обнаружены значительные изменения в имеющих решающее значение остатках в домене протеазыNS3 относительно эталонного субтипа (субгенотип 1a: AF009606 или субгенотип 1b: AJ238799) в процессе лечения в группах, обработанных различными дозами (более подробные данные см. в табл. 6-8). Во всех таблицах Gt обозначает субгенотип HCV, а день 1 обозначает исходную последовательность, и указанные аминокислотные замены представляет собой встречающиеся в естественных условиях полиморфизмы имеющих решающее значение аминокислот в NS3. Дополнительные изменения, которые могут кодировать или которые могут быть ассоциированы с устойчивостью к лекарственному средству, указаны в различные дни в процессе лечения. Таблица 6 Преобладающие варианты (относительно эталонного субтипа) имеющих решающее значение остатков аминокислотных последовательностей,обнаруженные у характерных для популяции подвергавшихся ранее лечению пациентов из группы, для обработки которой использовали дозу 48 мг нет: никаких замен не обнаружено в отобранных положениях;BLD: ниже предела обнаружения при ПЦР-амплификации и секвенировании. Таблица 7 Преобладающие варианты (относительно эталонного субтипа) имеющих решающее значение остатков аминокислотных последовательностей,обнаруженные у характерных для популяции подвергавшихся ранее лечению пациентов из группы, для обработки которой использовали дозу 120 мг нет: никаких замен не обнаружено в отобранных положениях;BLD: ниже предела обнаружения при ПЦР-амплификации и секвенировании;уровень VL выходит на плато 1000 МЕ/мл в эти моменты времени. Таблица 8 Преобладающие варианты (относительно эталонного субтипа) имеющих решающее значение остатков аминокислотных последовательностей,обнаруженные у характерных для популяции подвергавшихся ранее лечению пациентов из группы, для обработки которой использовали дозу 240 мг нет: никаких замен не обнаружено в отобранных положениях;BLD: ниже предела обнаружения при ПЦР-амплификации и секвенировании. Общие выводы. Эти результаты продемонстрировали наличие сильного и очень быстрого противовирусного ответа при обработке не подвергавшихся ранее лечению пациентов натриевой солью соединения (1) при введении один раз в день в качестве 14-дневной монотерапии, после чего происходило формирование антивирусной устойчивости и повышение вирусной нагрузки после 5-6 дней монотерапии. Однако после начала применения тройной комбинированной терапии, предлагаемой в настоящем изобретении, в день 14 (натриевая соль соединения (1) в сочетании с пэгилированным интерфероном альфа-2 а и рибавирином),вирусная нагрузка постепенно снижалась, что демонстрировало противовирусную эффективность тройной комбинированной терапии, предлагаемой в настоящем изобретении, при обработке не подвергавшихся ранее лечению пациентов (см. фиг. 1). Когда натриевую соль соединения (1) вводили один раз в день подвергавшимся ранее лечению пациентам (интерферон плюс рибавирин, нулевые респондеры и частичные респондеры) в сочетании с пэгилированным интерфероном альфа-2 а и рибавирином в течение 28 дней, то результаты продемонстрировали такой же сильный и очень быстрый противовирусный ответ, но при этом была обнаружена пониженная способность к формированию устойчивости (см. фиг. 2). Следовало ожидать, что у таких подвергавшихся ранее лечению пациентов-нереспондеров также устойчивость к вирусам должна формироваться через 5 дней, поскольку эти пациенты отобраны по признаку отсутствия чувствительности к терапии на основе пэгилированного интерферона плюс рибавирина. Однако имело место продолжительное подавление вирусов. В сущности, было установлено, что тройная комбинированная терапия, предлагаемая в настоящем изобретении, может обеспечивать эффективное снижение вирусной нагрузки у не подвергавшихся ранее лечению пациентов и у подвергавшихся ранее лечению пациентов, страдающих хронической инфекцией,вызываемой вирусом гепатита С генотипа-1, причем у некоторых пациентов она поддерживалась на уровне ниже выявляемого, за который принимали уровень, составляющий ниже 50 международных единиц на 1 мл сыворотки пациента при оценке с помощью методологии количественной состоящей из мно- 16019965 гих циклов ПЦР с обратной транскриптазой согласно международному стандарту ВОЗ. В предпочтительных вариантах осуществления изобретения тройная комбинированная терапия, предлагаемая в настоящем изобретении, может обеспечивать эффективное снижение вирусной нагрузки у пациентов с хронической инфекцией, вызываемой вирусом гепатита С генотипа-1, до уровня, составлявшего менее 10 международных единиц на 1 мл сыворотки. При создании изобретения получены следующие неожиданные результаты:(1) отсутствие быстро возникающей устойчивости (например, вариантов, кодирующих аминокислотные замены в NS3 в положениях R155 и/или D168) у подвергавшихся ранее лечению пациентов, которым вводили натриевую соль соединения (1) в сочетании с пэгилированным интерфероном альфа-2 а и рибавирином в стандартных дозах (фиг. 2), в отличие от воздействия натриевой соли соединения (1) в такой же дозе индивидуально при обработке не подвергавшихся ранее лечению пациентов, у которых обнаружена устойчивость к вирусу (см. фиг. 1, первые 14 дней). Можно было ожидать, что у таких подвергавшихся ранее лечению пациентов устойчивость к вирусам должна формироваться через 5 дней даже при добавлении пэгилированного интерферона альфа-2 а и рибавирина, поскольку эти пациенты были отобраны по признаку отсутствия чувствительности к комбинированной терапии на основе пэгилированного интерферона плюс рибавирина; и(2) тот факт, что доза, составлявшая всего лишь 120 мг QD (один раз в день), в сочетании с пэгилированным интерфероном и рибавирином индуцировала вирусное истощение до уровня ниже уровня количественного определения (установленного на уровне менее 25 ME на 1 мл сыворотки) более чем у 50% у подвергавшихся ранее лечению пациентов, у которых ранее отсутствовала чувствительность к терапии на основе пэгилированного интерферона плюс рибавирина (см. таблицу). Пример 8. Методы идентификации вариантов NS3 HCV. Экстракция и ПЦР-амплификация вирусной РНК. Вирусную РНК выделяли из плазмы HCV-инфицированных индивидуумов и сначала синтезировали ДНК-фрагмент длиной 2,4 т.п.н., содержащий полную NS3-NS4A-область, с помощью системы для одноступенчатой ОТ-ПЦР SUPERSCRIPT (фирма Invitrogen) и с использованием двух специфических для гена праймеров, охватывающих положениях 3276 в NS2 и 5650 в NS4B. После очистки первого ПЦРпродукта с помощью второго цикла "полугнездовой" РНК с использованием ДНК-полимеразы KOD HotStart (фирма Novagen) создавали два различных ПЦР-продукта длиной либо 2,3, либо 0,7 т.п.н. (которые перекрывали полную NS3/NS4A или только NS3-протеазный домен соответственно). По причинам, связанным с пределом обнаружения метода ОТ-ПЦР-амплификации, анализ образцов из организма пациентов был ограничен образцами с уровнем VL, превышающим 1000 МЕ/мл. Анализ последовательностей. ДНК-продукт длиной 2,3 т.п.н. затем применяли для непосредственного секвенирования на популяционной основе полной NS3-NS4A-области, состоящей из 2055 нуклеотидов, с помощью BIG DYEApplied Biosystems). Последовательности получали с использованием 10 праймеров, достигая по меньшей мере 90%-ного перекрытия двойной цепи NS3-NS4 А-области. Полученные нуклеотидные последовательности анализировали с помощью устройства SEQSCAPE v2.5 (фирма Applied Biosystems). ДНК-фрагмент длиной 0,7 т.п.н. применяли для создания на клональной основе (набор для клонирования ZEROBLUNT ТОРО, фирма Invitrogen) последовательностей состоящей из 543 нуклеотидов области NS3-проеазы, кодирующей первые 181 аминокислот NS3; из каждого образца отбирали по 96 клонов и секвенировали с использованием универсальных праймеров и термоячейки для ПЦРсеквенирования ABI PRISM BIG DYE Terminator Cycle. Осуществляли анализ двух индивидуальных последовательностей каждого клона, обеспечивая 90-100%-ное перекрытие двойной цепи состоящей из 543 нуклеотидов области, с помощью программы MUTATION SURVEYOR, версия 3.0 (фирмаSoftgenetics LLC). Для каждого образца, полученного из организма пациента, клоны с низким качеством последовательности или содержащие делеции, инсерции или стоп-кодоны, не включали в дальнейший анализ, поэтому количество анализируемых клонов варьировало от 74 до 89 (медианное и среднее значение составляло 80 клонов). Полученные последовательности сравнивали с эталонными (референс) последовательностями соответствующих субтипов, которые предварительно определяли на фазе скрининга опыта с использованием генотипирования с помощью TRUEGENE HCV 5'NC. AF009606 служил в качестве эталона для субтипа 1a, a AJ238799 для субтипа 1b. Наибольшее внимание уделяли мутациям, приводящим к аминокислотным заменам в 15 положениях домена NS3-протеазы. Для всех этих положений ранее было установлено,что они потенциально могут обусловливать устойчивость к этому классу соединений. Эти положения представляют собой: 36, 41, 54, 71, 80, 86, 89, 109, 111, 155, 156, 168, 170, 176 и 178.[3] Koev G., Kati W., The emerging field of drug resistance. Expert Opinion Invest Drugs, 17(3), 2008,p. 303-319 (P08-03895). Анализ чувствительности к лекарственным средствам. Бифункциональный вектор, содержащий бицистронный репликон HCV (pIT2), который включал репортерный ген люциферазы и был адаптирован к Con-1 NS3/NS5B-области, модифицировали с целью создания двух уникальных сайтов рестрикции (MluI и SpeI) на кодонах 11 и 225 NS3, которые позволяли осуществлять встраивание совместимых ампликонов NS3, выделенных из образцов плазмы HCVинфицированных пациентов. Полученный в первом цикле ПЦР-продукт, синтезированный на основе очищенной из плазмы пациента РНК (применяемый также для создании фрагментов для секвенирования на популяционной и клональной основе), применяли для амплификации фрагмента длиной 0,65 т.п.н. с помощью пары праймеров, которые содержали соответственно уникальные сайты рестрикции MluI иSpeI для встраивания в бифункциональный вектор. Ампликоны встраивали путем лигирования в бифункциональный вектор pIT2 и реконструированную плазмидную ДНК применяли для создания РНКтранскриптов субгеномного репликона HCV (набор Т 7 RIBOMAX, фирма Promega). Транскрибируемой in vitro РНК кратковременно трансфектировали путем электропорации клетки Huh-7.5, которые затем высевали в 96-луночные планшеты, инкубировали в течение 24 ч и обрабатывали взятой в различных концентрациях натриевой солью соединения (1) (или IFN-) в течение 72 ч. В конце периода инкубации люциферазную активность оценивали с помощью субстрата BRIGHT-GLO для количественной оценки люминесценции (CPS) в каждой лунке культурального планшета, что отражало уровень репликации РНКHCV. Степень ингибирования (% ингибирования) в каждой лунке, содержащей ингибитор, рассчитывали из следующего уравнения: % ингибирования = 100-[100CPS (ингибитор)/CPS (контроль)]. Концентрацию, при которой происходило 50%-ное ингибирование репликации РНК HCV (EC50), определяли с помощью общепринятой процедуры системы статистического анализа (SAS) на основе нелинейной регрессии NLIN. Значение ЕС 50 для мутанта NS3 сравнивали с исходным значением ЕС 50 с получением кратности изменений. Преобладающие мутации генотипа 1 а, обусловливающие устойчивость, которая проявляется в восстановлении вирусной нагрузки в процессе лечения, кодирующие замену R155K и, кроме того, другие минорные варианты, были выявлены путем клонального анализа последовательностей в этом положении. Варианты R155K обусловливали снижение чувствительности к BI 201335, что характеризовалось диапазоном значений ЕС 50 1,8-6,5 мкМ. У вирусов генотипа 1b, как установлено с помощью чувствительного клонального секвенирования, в основном кодировались изменения в положении D168, при этом валин являлся наиболее часто встречающимся заместителем, обнаружены также другие минорные варианты. Значения ЕС 50 для вариантов D168 составляли 3,6-15 мкМ. Такой профиль частично может быть связан с различным барьером к возникновению мутации, обусловливающей устойчивость, в кодоне R155 в генотипе 1 а (традиционные однонуклеотидные замены кодона на лизин) по сравнению с генотипом 1b(в этом случае требуются две нуклеотидные замены для того, чтобы кодировать замену на лизин).(а) соединение формулы (1) или его фармацевтически приемлемую соль(в) рибавирин,предназначенная для применения для лечения инфекции, вызываемой вирусом гепатита С (HCV),или облегчения одного или нескольких ее симптомов. 2. Комбинация по п.1, которая включает:(а) первую фармацевтическую композицию, содержащую соединение следующей формулы (1) или его фармацевтически приемлемую соль:(в) третью фармацевтическую композицию, содержащую рибавирин. 3. Комбинация по п.1 или 2, в которой инфекция HCV относится к генотипу 1. 4. Комбинация по п.1 или 2, которая предназначена для лечения инфекции, вызываемой вирусом гепатита С (HCV), или облегчения одного или нескольких ее симптомов у пациента, не подвергавшегося ранее лечению. 5. Комбинация по п.1 или 2, которая предназначена для лечения инфекции, вызываемой вирусом гепатита С (HCV), или облегчения одного или нескольких ее симптомов у пациента, не чувствительного к комбинированной терапии, основанной на применении рибавирина и интерферона альфа. 6. Комбинация по п.1 или 2, где количество соединения (1) или его фармацевтически приемлемой соли составляет по меньшей мере 40 мг. 7. Комбинация по п.1 или 2, где количество рибавирина составляет 400, 600, 800, 1000 или 1200 мг. 8. Комбинация по п.1 или 2, где интерферон альфа представляет собой пэгилированный интерферон альфа. 9. Комбинация по п.8, где пэгилированный интерферон альфа представляет собой пэгилированный интерферон альфа-2 а в дозе от примерно 90 до примерно 200 мкг или пэгилированный интерферон альфа-2b в дозе от примерно 0,5 до примерно 2 мкг. 10. Комбинация по п.1 или 2, предназначенная для лечения инфекции HCV генотипа 1 у пациента,не чувствительного к комбинированной терапии, основанной на применении рибавирина и интерферона альфа, где количество соединения (1) или его фармацевтически приемлемой соли составляет от 48 до 240 мг и где интерферон альфа представляет собой пэгилированный интерферон альфа-2 а или пэгилированный интерферон альфа-2b.
МПК / Метки
МПК: A61K 45/06
Метки: рибавирином, интерфероном, протеазы, ингибитора, комбинация
Код ссылки
<a href="https://eas.patents.su/21-19965-kombinaciya-ingibitora-proteazy-ns3-hcv-s-interferonom-i-ribavirinom.html" rel="bookmark" title="База патентов Евразийского Союза">Комбинация ингибитора протеазы ns3 hcv с интерфероном и рибавирином</a>
Новая комбинация ингибитора i;f тока синусового узла и ингибитора ангиотензинпревращающего фермента и фармацевтические композиции, её содержащие

Номер патента: 11253
Опубликовано: 27.02.2009
Авторы: Бената Видаль, Леребу-Пижонньере Ги
МПК: A61K 31/55, A61K 31/404, A61P 9/12...
Метки: тока, содержащие, узла, ангиотензинпревращающего, фермента, новая, фармацевтические, композиции, синусового, комбинация, ингибитора
Формула / Реферат:
1. Комбинация избирательного и специфического ингибитора If тока синусового узла, который представляет собой ивабрадин, или 3-{3-[{[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил}(метил)амино]пропил}-7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он, или один(ну) из его гидратов, кристаллических форм или солей присоединения с фармацевтически приемлемой кислотой, и вещества, ингибирующего ангиотензинпревращающий фермент, которое...
Новая комбинация ингибитора i f тока синусового узла и кальциевого ингибитора и фармацевтические композиции, её содержащие

Номер патента: 13536
Опубликовано: 30.06.2010
Авторы: Леребу-Пижонньере Ги, Бената Видаль
МПК: A61K 31/55, A61K 31/4422, A61P 9/10...
Метки: комбинация, содержащие, синусового, новая, тока, композиции, кальциевого, ингибитора, узла, фармацевтические
Формула / Реферат:
1. Фармацевтическая композиция, включающая избирательный и специфический ингибитор If тока синусового узла ивабрадин, или 3-{3-[{[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил}(метил)амино]пропил}-7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он, или один(ну) из его гидратов, кристаллических форм или его солей присоединения с фармацевтически приемлемой кислотой и кальциевый ингибитор, принадлежащий к классу дигидропиридина.2....
Комбинация ингибитора альдозоредуктазы и ингибитора гликогенфосфорилазы

Номер патента: 2365
Опубликовано: 25.04.2002
Авторы: Хувер Деннис Джей, Тредуэй Джудит Ли, Хьюлин Бернард, Майлари Банавара Лэкшмэн
МПК: A61P 3/10, A61K 45/06
Метки: комбинация, ингибитора, гликогенфосфорилазы, альдозоредуктазы
Формула / Реферат:
1. Фармацевтическая композиция, содержащая терапевтически эффективное количество а) ингибитора альдозоредуктазы; б) ингибитора гликогенфосфорилазы; и в) фармацевтический носитель. 2. Фармацевтическая композиция для достижения инсулинсенсибилизирующего эффекта у млекопитающего, содержащая а) некоторое количество первого соединения, причем указанное первое соединение представляет собой ингибитор альдозоредуктазы; и б) некоторое количество второго...
Макроциклическое хиноксалиновое соединение в качестве ингибитора протеазы вгс ns3

Номер патента: 19327
Опубликовано: 28.02.2014
Авторы: Макколи Джон А., Харпер Стивен, Ливертон Найджел Дж., Сумма Винченцо
МПК: A61K 38/07, A61K 38/06, A61K 38/08...
Метки: соединение, макроциклическое, вгс, протеазы, ингибитора, хиноксалиновое, качестве
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль2. Фармацевтическая композиция для лечения пациента, инфицированного ВГС, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.3. Фармацевтическая композиция по п.2, дополнительно содержащая второй терапевтический агент, выбранный из группы, состоящей из ингибитора протеазы ВГС и ингибиторов полимеразы ВГС NS5B.4. Применение соединения по п.1...
Комбинация нспвлс и ингибитора pde-4

Номер патента: 8108
Опубликовано: 27.04.2007
Авторы: Клайн Томас, Эльтце Манфрид, Клей Ханс-Петер, Хатцельманн Армин
МПК: A61K 31/44, A61K 45/06, A61K 31/40...
Метки: pde-4, нспвлс, комбинация, ингибитора
Формула / Реферат:
1. Фармацевтическая композиция, содержащая первое действующее вещество, которым является ингибитор PDE4, выбранный из группы, включающей 3-циклопропилметокси-4-дифторметокси-N-(3,5-дихлорпирид-4-ил)бензамид [МНН рофлумиласт] и его фармацевтически приемлемые производные, в смеси со вторым действующим веществом, которым является традиционное НСПВЛС, выбранное из группы, 2-[(2,6-дихлорфенил)амино]бензолуксусную кислоту [МНН диклофенак] и ее...
Предыдущий патент: Каталитический реактор
Следующий патент: Соединения и композиции в качестве ингибиторов протеинкиназы
Случайный патент: Сборная железобетонная колонна