Способ асимметрического алкилирования карбонильной группы
Номер патента: 16650
Опубликовано: 30.06.2012
Авторы: Штурм Хуберт, Альберт Мартин, Бергер Андреас, Креммингер Петер




















Формула / Реферат
1. Способ асимметрического алкилирования карбонильной группы в соединении (соединении K), содержащем карбонильную группу и связывающую группу, выбранную из группы, включающей гидроксигруппу, аминогруппу и сульфгидрильную группу, включающий стадии:
a) смешивания соединения K, хирального вспомогательного соединения (соединения А) и производного борной или бороновой кислоты с образованием замещенного производного борной или бороновой кислоты, в котором атом бора связывает хиральное вспомогательное соединение с соединением K; и
b) прибавления металлорганического соединения;
где соединение K выбрано из группы, включающей альфа-, бета-, гамма- и дельта-гидроксикетоны или альдегиды, альфа-, бета-, гамма- и дельта-аминокетоны или альдегиды и альфа-, бета-, гамма- и дельта-сульфгидрилкетоны или альдегиды; а производное борной или бороновой кислоты представляет собой соединение формулы (VI)

в которой R1 обозначает водород, C1-C10-алкил, C2-C10-алкенил, C2-C10-алкинил, C6-C10-арил, C7-C16-алкиларил, 4-10-членный гетероциклический остаток, C1-C10-алкоксигруппу, C1-C10-алкиламиногруппу, C1-C10-алкилтиогруппу, гидроксигруппу или цианогруппу;
R2 обозначает галоген, гидроксигруппу, C1-C10-алкоксигруппу, C6-C10-арилоксигруппу, C1-C10-диалкиламиногруппу или 4-10-членный гетероциклический остаток, связанный с атомом бора с помощью атома S, N или О;
R3 обозначает галоген, аминогруппу, гидроксигруппу, C1-C10-алкоксигруппу, C6-C10-арилоксигруппу, C1-C10-диалкиламиногруппу или 4-10-членный гетероциклический остаток, связанный с атомом бора с помощью атома S, N или О; или
R2 и R3 связаны друг с другом с образованием 5-10-членной циклической структуры, включающей атом бора, к которой присоединены R2 и R3, и циклическая структура может содержать 1 или 2 дополнительных атома бора, и/или кислорода, и/или азота.
2. Способ по п.1, который осуществляют в однореакторном режиме.
3. Способ по любому из пп.1 или 2, в котором из реакционной смеси в основном удаляют побочные продукты стадии а) перед проведением стадии b).
4. Способ по любому из пп.1-3, в котором R1 обозначает C1-C10-алкил или C1-C10-алкоксигруппу.
5. Способ по п.4, в котором R1 обозначает C1-C6-алкил.
6. Способ по п.5, в котором R1 обозначает метил или этил.
7. Способ по любому из пп.1-6, в котором R2 и R3 являются одинаковыми и обозначают гидроксигруппу или C1-C10-алкоксигруппу.
8. Способ по п.7, в котором реакционную смесь подвергают перегонке до проведения стадии b) для удаления воды или C1-C10-алканола.
9. Способ по любому из пп.1-8, в котором производное борной или бороновой кислоты выбрано из группы, включающей фенилбороновую кислоту, триметилборат, триизопропилборат, диизопропилбутилборонат, диизопропилметилборонат, метилбороновую кислоту и триметилбороксин.
10. Способ по п.9, в котором производное борной или бороновой кислоты выбрано из группы, включающей диизопропилметилборонат, метилбороновую кислоту и триметилбороксин.
11. Способ по п.9, в котором связывающая группа представляет собой гидроксигруппу.
12. Способ по любому из пп.1-11, в котором атом углерода карбонильной группы отделен от атома, несущего связывающую группу, с помощью 2-3 атомов углерода.
13. Способ по любому из пп.1-12, в котором хиральное вспомогательное соединение представляет собой хиральный спирт.
14. Способ по п.13, в котором хиральный спирт включает структурный элемент формулы (VII)

где С* обозначает хиральный атом углерода;
n является целым числом, равным от 0 до 3; и
X обозначает гетероатом, обладающий неподеленной электронной парой.
15. Способ по п.14, в котором n равно 1 или X обозначает азот.
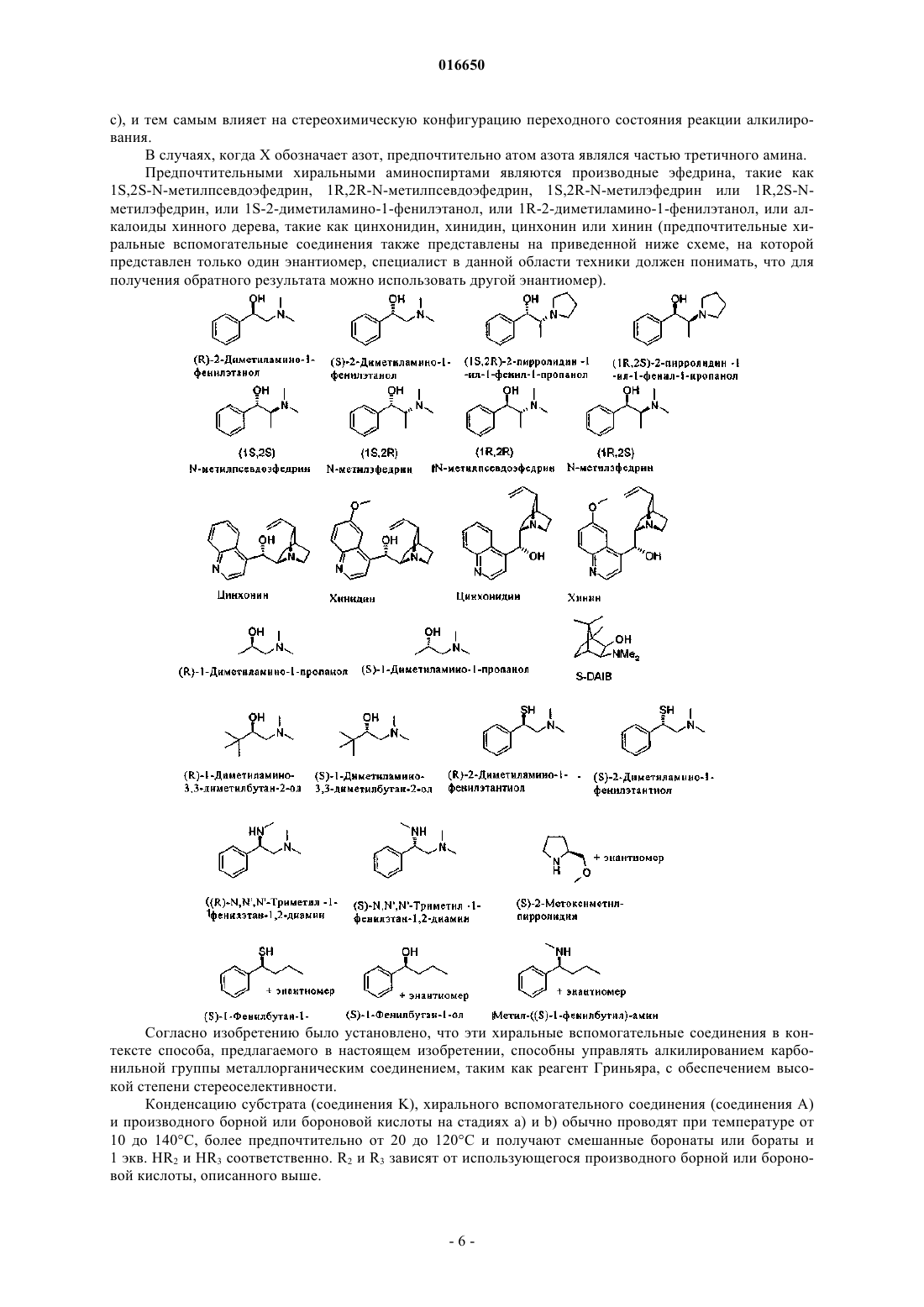
16. Способ по п.15, в котором хиральный спирт представляет собой хиральный аминоспирт.
17. Способ по п.16, в котором хиральный аминоспирт выбран из группы, включающей N-метилэфедрин, N-метилпсевдоэфедрин, 2-диметиламино-1-фенилэтанол, хинин, хинидин, цинхонидин и цинхонин.
18. Способ по любому из пп.1-17, в котором металлорганическое соединение представляет собой магнийорганическое соединение, цинкорганическое соединение, кадмийорганическое соединение, церийорганическое соединение, литийорганическое соединение, титанорганическое соединение, марганецорганическое соединение, железоорганическое соединение, алюминийорганическое соединение или оловоорганическое соединение.
19. Способ по пп.1-18, в котором металлорганическое соединение представляет собой магнийорганическое соединение.
20. Способ по п.18, в котором магнийорганическое соединение представляет собой алкилмагний, алкенилмагний или алкинилмагний.
21. Способ по любому из пп.1-20, в котором соединение K выбрано из группы, включающей гамма-гидроксикетоны, гамма-аминокетоны и гамма-сульфгидрилкетоны.
22. Способ по любому из пп.1-21, в котором соединение K является соединением формулы (III)

где Y обозначает цианогруппу или группу, которую можно превратить в цианогруппу.
23. Способ по п.22, в котором металлорганическое соединение представляет собой металлорганическое соединение формулы (VIII)

где пунктирная линия обозначает одинарную, двойную или тройную связь;
М обозначает металл или производное металла;
Z обозначает -CH2-N(CH3)2 или группу, которую можно превратить в -CH2-N(CH3)2.
24. Способ по п.23, где М обозначает Mg и Z обозначает -CH2-N(CH3)2 или группу, которую можно превратить в -CH2-N(CH3)2, и где диол формулы (II) получают в энантиомерно обогащенной или энантиомерно чистой форме

где Y обозначает цианогруппу или группу, которую можно превратить в цианогруппу.
25. Способ по п.24, дополнительно включающий стадию замыкания цикла диола формулы (II) с образованием соединения формулы (IX)

в которой Y и Z являются такими, как определено в п.24
26. Способ по п.25, в котором соединение формулы (IX) представляет собой эсциталопрам.
Текст
СПОСОБ АСИММЕТРИЧЕСКОГО АЛКИЛИРОВАНИЯ КАРБОНИЛЬНОЙ ГРУППЫ В изобретении приведены описания способов и промежуточных продуктов для стереоселективного алкилирования карбонильных групп. Изобретение, в частности, позволяет стереоселективно синтезировать антидепрессивное лекарственное средство эсциталопрам. Согласно изобретению было установлено, что производные борной или бороновой кислоты являются подходящими мостиковыми элементами для присоединения хиральной группы к соединению, содержащему алкилируемую карбонильную группу. Таким образом, указанные бораты и боронаты применимы в способе асимметрического алкилирования карбонильной группы в соединении, содержащем карбонильную группу и связывающую группу, способную взаимодействовать с производным борной или бороновой кислоты. Асимметрическое алкилирование можно провести путем смешивания соединения, содержащего алкилируемую карбонильную группу и связывающую группу, способную взаимодействовать с производным борной или бороновой кислоты,прибавления хирального спирта и прибавления металлорганического соединения. После реакции алкилирования борат и боронат можно легко удалить с помощью гидролиза. 016650 Область техники, к которой относится изобретение Настоящее изобретение относится к способам и промежуточным продуктам стереоселективного алкилирования карбонильных групп. Настоящее изобретение, в частности, позволяет стереоселективно синтезировать антидепрессивное лекарственное средство эсциталопрам. Уровень техники Существует немного методик получения четвертичных атомов углерода. В частности, это относится к синтезу третичных спиртов, который все еще весьма затруднителен для органиков-синтетиков. Самой прямой методикой получения асимметрических третичных спиртов является стереоселективное присоединение металлорганического реагента к кетону. Однако существует лишь несколько методик, в которых используют регулирование реагентом и катализ (см.: Ramon, D.J.; Yus, M. Angew. Chem. Int. Ed. 2004, 43, 284-287). Таким образом, необходимы новые методики получения хиральных третичных спиртов. Третичным спиртом, представляющим особый интерес, является соединение формулы (II) - основной промежуточный продукт синтеза лекарственного средства эсциталопрама (I), представляющего собой общеизвестное антидепрессивное средство. Он является селективным ингибитором повторного поглощения обладающего центральным действием серотонина (5-гидрокситриптамин; 5-ГТ) и поэтому обладает антидепрессивной активностью Эсциталопрам впервые раскрыт в ЕР 347066, выданном Н. Lundbeck A/S. В этой публикации патента заявлено соединение и описаны два способа его получения, основанные на разделении R- и Sэнантиомеров промежуточного продукта с последующим превращением энантиомерно чистого диола (II) или его нестабильных сложных эфиров в эсциталопрам (I). Первый способ включает превращение рацемического 4-[4-диметиламино-1-(4-фторфенил)-1 гидроксибутил]-3-гидроксиметилбензонитрила (формула (II в соответствующие два диастереоизомерных сложных эфира (путем использования хирального хлорангидрида кислоты), которые можно разделить с помощью хроматографии на ахиральной стационарной фазе или фракционированной кристаллизации. Сложный эфир, обладающий необходимой стереохимической конфигурацией, превращают в эсциталопрам путем замыкания цикла с помощью основания. Рацемический диол формулы (II) и его использование для синтеза циталопрама описаны в US 4650884. Второй способ, описанный в ЕР 347066, основан на разделении рацемического диола формулы (II) по классической методике разделения с использованием (+)-ди-O',О'-толуолилвинной кислоты в качестве разделяющего реагента. По данным ЕР 347066 выход такого разделения составляет 55% (27,5% в пересчете на рацемический диол (II. Энантиомерно чистый диол вводят в последующую реакцию дегидративного замыкания цикла (MsCl, Et3N) и получают эсциталопрам. В WO 03/006449 описано разделение энантиомеров диола (II) с помощью препаративной хроматографии на хиральной стационарной фазе. С помощью такой методики разделения можно обеспечить ЭИ(энантиомерный избыток), составляющий более 99%, и выход - более 95% (47,5% в пересчете на рацемический диол (II. Крупномасштабную хроматографию проводят с помощью технологии ПДС(ПДС=псевдодвижущийся слой) на стационарной фазе на основе углевода. Превращение энантиомерно чистого диола (II) в эсциталопрам проводят в соответствии с ЕР 347066. В WO 04/014821 описан четвертый способ, который основан на использовании ферментов (эстераз и липаз) для разделения рацемического диола формулы (II). Кинетическое ферментативное ацилирование или дезацетилирование рацемического диола (II) или сложных эфиров рацемического диола (II) соответственно приводит к смеси, предпочтительно содержащей один энантиомер в виде диола формулы(II) и второй энантиомер в виде сложного эфира диола (II). После разделения можно провести замыкание цикла, как это описано выше. Во всех четырех описанных подходах получение энантиомерно чистого эсциталопрама начинают с рацемического диола формулы (II). Полный теоретический выход эсциталопрама, полученного по любой из этих методик, не превышает 50% в пересчете на рацемический диол (II). До настоящего времени не описана настоятельно необходимая методика асимметрического синтеза энантиомерно обогащенного или чистого диола формулы (II), которая не основана на разделении рацемического диола (II). Такая методика привела бы к значительному повышению полного выхода эсциталопрама.-1 016650 Согласно изобретению было установлено, что производные борной или бороновой кислоты являются подходящими мостиковыми элементами для присоединения хиральной группы к соединению, содержащему алкилируемую карбонильную группу. Таким образом, бораты и боронаты применимы в способе асимметрического алкилирования карбонильной группы в соединении, содержащем карбонильную группу и функциональную группу (далее называемую "связывающей" группой), способную взаимодействовать с производным борной или бороновой кислоты. Асимметрическое алкилирование можно провести путем смешивания соединения, содержащего алкилируемую карбонильную группу и связывающую группу, способную взаимодействовать с производным борной или бороновой кислоты с производным борной или бороновой кислоты, прибавления хирального спирта и прибавления металлорганического соединения. После реакции алкилирования борат и боронат можно легко удалить с помощью гидролиза. С помощью способа, предлагаемого в настоящем изобретении, искомый S-энантиомер диола (II)(или соответствующий R-энантиомер) можно получить с высоким выходом. Таким образом, эсциталопрам можно синтезировать без необходимости разделения рацемического диола (II). Описание изобретения Объектом настоящего изобретения является способ асимметрического алкилирования карбонильной группы в соединении (соединении K), содержащем карбонильную группу и связывающую группу,выбранную из группы, включающей гидроксигруппу, аминогруппу и сульфгидрильную группу, где способ включает стадииa) смешивания соединения K, хирального вспомогательного соединения (соединения А) и производного борной или бороновой кислоты с образованием замещенного производного борной или бороновой кислоты, в котором атом бора связывает хиральное вспомогательное соединение с соединением K; иb) прибавления металлорганического соединения (R-M), где соединение K выбрано из группы,включающей альфа-, бета-, гамма- и дельта-гидроксикетоны или альдегиды, альфа-, бета-, гамма- и дельта-аминокетоны или альдегиды и альфа-, бета-, гамма- и дельта-сульфгидрилкетоны или альдегиды. На схеме 1 приведен пример способа, предлагаемого в настоящем изобретении, в котором используется предпочтительное вспомогательное соединение, хиральный спирт. Способ, предлагаемый в настоящем изобретении, обладает теми преимуществами, что он является быстрым, экономичным, простым и позволяет получить хиральные третичные спирты с высоким выходом и высоким энантиомерным избытком. Другим преимуществом является то, что способ, предлагаемый в настоящем изобретении, можно осуществить в однореакторном режиме. Схема 1 Связь, отмеченная волнистой линией, является связью с одним из остатков, дополнительно описанных ниже. Полуокружность в соединении K показывает, что связывающая группа и карбонильная группа находятся в одной молекуле. Предпочтительные схемы присоединения этих двух групп приведены ниже. Предпочтительными производными борной или бороновой кислоты являются фенилбороновая кислота, триметилборат, триизопропилборат, диизопропилбутилборонат, диизопропилметилборонат, метилбороновая кислота или триметилбороксин, предпочтительно диизопропилметилборонат, метилбороновая кислота или триметилбороксин.-2 016650 Предпочтительными связывающими группами (А-Н), способными взаимодействовать с производным борной или бороновой кислоты, являются гидроксигруппа, аминогруппа, карбоксигруппа и сульфгидрильная группа, и гидроксигруппа является особенно предпочтительной. Предпочтительными хиральными вспомогательными соединениями (соединением А) являются хиральные спирты, предпочтительно такие хиральные спирты, которые включают структурный элемент формулы (VII) в которой С обозначает хиральный атом углерода;X обозначает азот,особенно предпочтительными являются хиральные аминоспирты - N-метилэфедрин, N-метилпсевдоэфедрин, 2-диметиламино-1-фенилэтанол, хинин, хинидин, цинхонидин или цинхонин. Предпочтительными металлорганическими соединениями (R-M) для стереоселективного алкилирования являются магнийорганические соединения. В частности, для синтеза диола формулы (II) предпочтительным магнийорганическим соединением является N,N-диметиламинопропилмагнийхлорид. Диол формулы (II) можно дополнительно обработать, так чтобы произошло замыкание цикла с образованием эсциталопрама. Настоящее изобретение также относится к различным промежуточным продуктам, образующимся во время осуществления нового способа, к применению бората и бороната в качестве мостикового элемента между исходным соединением, содержащем функциональную группу, способную к нуклеофильному замещению карбанионом, и связывающую группу, способную взаимодействовать с производным борной или бороновой кислоты, и хиральным вспомогательным веществом, способным управлять стереоселективной реакцией карбонильной группы в исходном соединении. Настоящее изобретение также относится к гидроксикетону формулы (III) в кристаллической форме. Подробное описание изобретения Настоящее изобретение относится к способу асимметрического алкилирования карбонильной группы, предпочтительно кетогруппы, в соединении K, которое содержит карбонильную группу и связывающую группу, способную взаимодействовать с производным борной или бороновой кислоты, включающему стадии: а) смешивания соединения K, хирального вспомогательного соединения, такого как хиральный спирт, и производного борной или бороновой кислоты;b) прибавления металлорганического соединения, где соединение K выбрано из группы, включающей альфа-, бета-, гамма- и дельта-гидроксикетоны или альдегиды, альфа-, бета-, гамма- и дельтааминокетоны или альдегиды и альфа-, бета-, гамма- и дельта-сульфгидрилкетоны или альдегиды. На схеме 1 в качестве примера приведен предпочтительный вариант осуществления способа, предлагаемого в настоящем изобретении. Предпочтительно, если после алкилирования карбонильной группы на стадии b) дополнительно проводят заключительную стадию гидролиза и получают третичный спирт,содержащий продукт (соединение Р). Асимметрическое алкилирование означает, что предпочтительно получают один из двух возможных энантиомеров образующегося диола. Прибавление металлорганического соединения обеспечивает стереоспецифическое регулирование с предпочтительным получением одного энантиомера образующегося диола. Это означает, что прибавление металлорганического соединения (стадия b) к хиральному смешанному борату и боронату (соединению MB), полученному после стадии а), является диастереоселективным. Состав смеси, полученной после прибавления металлорганического соединения к смешанному борату и боронату (соединению MB), зависит от конкретного использующегося хирального вспомогательного соединения (соединения А) и условий, при которых проводят реакцию. Особенностью асимметрического присоединения, предлагаемого в настоящем изобретении, является то, что образуется значительно большее количество одного энантиомера формулы (II), т.е. продукта (соединения Р), чем другого. Отношение количества S-энантиомера к количеству R-энантиомера (или R к S) отличается от 1:1, обычно составляет не менее 10:1, предпочтительно более 15:1. Стадии а) и b) и, если ее проводят, стадию гидролиза предпочтительно проводят в одной и той же инертной среде, предпочтительно, если средой является апротонный растворитель. Подходящими органическими растворителями являются толуол, тетрагидрофуран, ацетонитрил, ДМФ (диметилформамид),ДМСО (диметилсульфоксид), диоксан, ДМЭ (диметоксиэтан), диглим, нитрометан, метил-третбутиловый эфир, CH2Cl2 или NMP (N-метилпирролидон) и их смеси, и толуол и смеси диметоксиэтан/тетрагидрофуран являются особенно предпочтительными.-3 016650 Обычно субстрат K, производное борной или бороновой кислоты и хиральное вспомогательное соединение (соединение А), например хиральный спирт, перемешивают в апротонном растворителе в мягких условиях в течение времени, достаточного для присоединения и связывающей группы субстрата, и хирального вспомогательного соединения, например хирального спирта, к производному борной или бороновой кислоты и тем самым образования замещенной борной или бороновой кислоты, в которой атом бора связывает хиральное вспомогательное соединение с субстратом. Порядок проведения стадий а) и b) не является критически важным. Прибавление субстрата K, хирального вспомогательного соединения и производного борной или бороновой кислоты в реакционную систему можно провести в произвольном порядке. Соединение K можно сначала смешать с производным борной или бороновой кислоты и затем можно прибавить хиральное вспомогательное вещество, такое как хиральный спирт, или соединение K можно сначала смешать с хиральным вспомогательным веществом, таким как хиральный спирт, и затем можно прибавить производное борной или бороновой кислоты, или хиральное вспомогательное соединение, такое как хиральный спирт, и производное борной или бороновой кислоты можно прибавить одновременно с субстратом в инертной среде. Во всех случаях будет образовываться замещенное производное борной или бороновой кислоты, в которой атом бора связывает хиральное вспомогательное соединение с субстратом. Конденсацию субстрата K, хирального вспомогательного соединения (соединения А) и производного борной или бороновой кислоты проводят с использованием от 0,8 до 1,8 экв. производного борной или бороновой кислоты в пересчете на субстрат K, более предпочтительно от 1,0 до 1,2 экв. Конденсацию субстрата K, хирального вспомогательного соединения (соединения А) и производного борной или бороновой кислоты проводят с использованием от 0,8 до 2,0 экв. хирального вспомогательного соединения в пересчете на субстрат K, более предпочтительно от 1,0 до 1,4 экв. В зависимости от конкретного производного борной или бороновой кислоты, использующегося на стадии а), на стадиях а) и b) образуются вода, спирт, амин или НХ, где X=галоген. Эти побочные продукты предпочтительно удаляют, например, путем азеотропной перегонки или образования соли (в случае,если побочным продуктом является НХ) с последующим фильтрованием до прибавления металлорганического соединения на стадии с), чтобы сместить равновесие в сторону смешанного бороната (соединения MB). Специалист в данной области техники должен понимать, что в некоторых случаях принудительное удаление не всегда обязательно, например, если образующийся побочный продукт является газом, нерастворимым в растворителе, использующемся на стадиях способа. Однако в основном полное удаление побочного продукта из реакционной смеси обычно является предпочтительным. В предпочтительном варианте осуществления побочным продуктом является вода или спирт, и азеотропную перегонку проводят до стадии с) и побочные продукты стадий а) и b) предпочтительно в основном удалять. Если побочным продуктом является вода, то ее предпочтительно удаляют так, чтобы содержание составляло менее 0,5% мас./об. и более предпочтительно менее 0,1% мас./об. по данным титрования по Карлу Фишеру. Если побочным продуктом является спирт, то его предпочтительно удаляют так, чтобы содержание составляло менее 0,5% мас./об. и более предпочтительно менее 0,1% мас./об. по данным газовой хроматографии. Удаление воды или спирта также можно провести по альтернативным методикам, известным специалисту в данной области техники, например путем прибавления молекулярных сит или реагентов, способным удалять воду, таких как, например, осушающие реагенты. Если побочным продуктом является НХ, предпочтительным является удаление путем образования соли. Это можно выполнить путем прибавления подходящего основания, такого как третичный амин. Азеотропную перегонку предпочтительно проводить при пониженном давлении. Такая стадия перегонки обычно продолжается до 3 ч. Предпочтительными производными борной или бороновой кислоты для способа, предлагаемого в настоящем изобретении, являются производные формулы VIR2 и R3 связаны друг с другом с образованием 5-10-членной циклической структуры, включающей атом бора, к которой присоединены R2 и R3, и циклическая структура может содержать 1 или 2 дополнительных атома бора, и/или кислорода, и/или азота. Более предпочтительно, если R1 обозначает C1-C10-алкил или C1-C10-алкоксигруппу, еще более предпочтительно, если R1 обозначает C1-C6-алкил, где алкилом является метил или этил, и наиболее предпочтительным является метил. В предпочтительных боратах и боронатах R2 и R3 являются одинаковыми и обозначают гидроксигруппу или C1-C10-алкоксигруппу, например метокси-, этокси-, пропокси- или изопропоксигруппу. В другом предпочтительном варианте осуществления настоящего изобретения R1 обозначает метил и R2 иR3 обозначают гидроксигруппу или C1-C10-алкилоксигруппу. Альтернативно, R1 обозначаетC1-C10-алкоксигруппу и R2 и R3 обозначают гидроксигруппу или C1-C10-алкоксигруппу. Таким образом,особенно предпочтительными боратами и боронатами являются фенилбороновая кислота, диметоксиметилборан, триметилборат, триизопропилборат, диизопропилбутилборонат, диизопропилметилборонат,метилбороновая кислота и триметилбороксин, и наиболее предпочтительными являются диизопропилметилборонат, метилбороновая кислота и триметилбороксин. Применение этих предпочтительных боратов и боронатов характеризуется тем преимуществом, что воду или спирты, которые образуются на стадиях а) и b), можно легко удалить из реакционной смеси до проведения стадии с). Это можно, например, выполнить путем азеотропной перегонки, необязательно при пониженном давлении при температуре окружающей среды или немного повышенной (примерно до 35-90 С) температуре, или путем прибавления молекулярных сит. Что касается функции связывающей группы, то эта группа выступает в качестве центра присоединения производного борной или бороновой кислоты. Предпочтительными связывающими группами для способа, предлагаемого в настоящем изобретении, являются гидроксигруппа, моно- или дизамещенная или незамещенная аминогруппа или сульфгидрильная группа, предпочтительно гидроксигруппа. Если не ограничиваться какими-либо теоретическими соображениями, то можно полагать, что в предпочтительных вариантах осуществления настоящего изобретения временно связанная с атомом бора вспомогательная группа влияет на стереохимическую конфигурацию переходного состояния реакции алкилирования. Для облегчения образования системы, пригодной для регулирования стереохимии реакции алкилирования на стадии с), предпочтительными являются такие субстраты, в которых атом углерода алкилируемой карбонильной группы находится на расстоянии от 1 до 6 от атома углерода, содержащего связывающую группу, предпочтительно - на расстоянии от 1,3 до 3 . Для выполнения этого требования карбонильную группу и связывающую группу в молекуле субстрата можно разделить несколькими атомами, если существует доступная конфигурация молекулы субстрата, в которой выполняется такое требование. Однако предпочтительно, если атом углерода карбонильной группы отделен от атома, содержащего связывающую группу, с помощью от 0 до 4 атомов, предпочтительно с помощью от 1 до 4 атомов углерода, более предпочтительно с помощью от 2 до 3 атомов углерода. Например, в словеBUT буква В отделена от буквы Т одной буквой, буквой U. Соединение K выбирают из группы, включающей альфа-, бета-, гамма- и дельта-гидроксикетоны или -альдегиды, альфа-, бета-, гамма- и дельта-аминокетоны или -альдегиды и альфа-, бета-, гамма- и дельта-сульфгидрилкетоны или -альдегиды, предпочтительно гамма-гидроксикетоны или -альдегиды,гамма-аминокетоны или -альдегиды и гамма-сульфгидрилкетоны или -альдегиды. Кетоны предпочтительнее альдегидов. Предпочтительно, если фенильный заместитель находится рядом с кетогруппой. Специалист в данной области техники должен понимать, что другие функциональные группы, содержащиеся в субстрате (соединении K), которые несовместимы с металлорганическими реагентами или с борной или бороновой кислотой, необходимо защищать. Хиральное вспомогательное соединение, применяющееся на стадии b) способа, предлагаемого в настоящем изобретении, может представлять собой хиральный амин или хиральный тиол, но предпочтительно хиральный спирт, предпочтительно включающий структурный элемент формулы (VII) в которой С обозначает хиральный атом углерода;X обозначает гетероатом, обладающий неподеленной электронной парой. Такими гетероатомами являются, например, кислород, сера и азот, причем азот является особенно предпочтительным. Предпочтительно, если n равно 1 или если X обозначает азот, и наиболее предпочтительно, если n равно 1 и X обозначает азот. Если не ограничиваться какими-либо теоретическими соображениями, то можно полагать, что в предпочтительных вариантах осуществления настоящего изобретения гетероатом присоединенной к атому бора вспомогательной группы является частью системы, образующей хелат с металлом металлорганического соединения, использующегося на стадии алкилирования-5 016650 с), и тем самым влияет на стереохимическую конфигурацию переходного состояния реакции алкилирования. В случаях, когда X обозначает азот, предпочтительно атом азота являлся частью третичного амина. Предпочтительными хиральными аминоспиртами являются производные эфедрина, такие как 1S,2S-N-метилпсевдоэфедрин, 1R,2R-N-метилпсевдоэфедрин, 1S,2R-N-метилэфедрин или 1R,2S-Nметилэфедрин, или 1S-2-диметиламино-1-фенилэтанол, или 1R-2-диметиламино-1-фенилэтанол, или алкалоиды хинного дерева, такие как цинхонидин, хинидин, цинхонин или хинин (предпочтительные хиральные вспомогательные соединения также представлены на приведенной ниже схеме, на которой представлен только один энантиомер, специалист в данной области техники должен понимать, что для получения обратного результата можно использовать другой энантиомер). Согласно изобретению было установлено, что эти хиральные вспомогательные соединения в контексте способа, предлагаемого в настоящем изобретении, способны управлять алкилированием карбонильной группы металлорганическим соединением, таким как реагент Гриньяра, с обеспечением высокой степени стереоселективности. Конденсацию субстрата (соединения K), хирального вспомогательного соединения (соединения А) и производного борной или бороновой кислоты на стадиях а) и b) обычно проводят при температуре от 10 до 140 С, более предпочтительно от 20 до 120 С и получают смешанные боронаты или бораты и 1 экв. HR2 и HR3 соответственно. R2 и R3 зависят от использующегося производного борной или бороновой кислоты, описанного выше.HR2 и HR3 предпочтительно удаляют с помощью перегонки, азеотропной перегонки, химической реакции или адсорбции/абсорбции. Удаление HR2 и HR3 обычно проводят при комнатной температуре или немного повышенной температуре, такой как предпочтительно равная от 30 до 70 С. При необходимости удаление HR2 и HR3 можно провести при пониженном давлении. В зависимости от объема партии перегонка обычно продолжается до 3 ч. В предпочтительном варианте осуществления настоящего изобретения R2 и R3 обозначают гидроксигруппу и Н 2 О, которая затем образуется во время конденсации субстрата (соединения K), хирального вспомогательного соединения (соединения А) и производного борной или бороновой кислоты, удаляют с помощью азеотропной перегонки или путем прибавления молекулярных сит. Удаление воды также можно провести по любой другой методике, которая известна специалисту в данной области техники. В другом предпочтительном варианте осуществления R2 и R3 обозначают C1-C10-алкоксигруппу. Соответствующий спирт, который образуется во время конденсации субстрата (соединения K), хирального вспомогательного соединения (соединения А) и производного борной или бороновой кислоты удаляют с помощью перегонки или азеотропной перегонки. Удаление R2H и R3H также можно провести по любой другой методике, которая известна специалисту в данной области техники. Основным преимуществом настоящего изобретения является то, что для осуществления способа можно использовать обычное реакторное оборудование. По сравнению с не-асимметрическим присоединением металлорганического реагента к карбонильной группе технология требует лишь не намного больше времени, что обусловлено необходимостью получения соединения MB до прибавления металлорганического реагента. Металлорганическое соединение, использующееся на стадии с), предпочтительно представляет собой магнийорганическое соединение, цинкорганическое соединение, кадмийорганическое соединение,церийорганическое соединение, литийорганическое соединение, титанорганическое соединение, марганецорганическое соединение, алюминийорганическое соединение, железоорганическое соединение или оловоорганическое соединение. Металлорганические соединения, для которых известно, что их реакции зависят от образования хелата [и содержат такие металлы, как магний, титан, церий, железо, марганец,цинк, олово, алюминий], предпочтительнее реагента, не образующего хелаты, который содержит такие металлы, как литий или алюминий. Предпочтительно, чтобы металлорганическое соединение обладало относительно высокой реакционной способностью по отношению к карбонильной группе. По этой причине в качестве металлорганического соединения наиболее предпочтительным является магнийорганическое соединение, такое как алкилмагний, алкенилмагний или алкинилмагний. Если металлорганический реагент переносит алкильный или алкенильный остаток к карбонильной группе, то стадию алкилирования с) обычно проводят при температуре от -100 до 20 С, более предпочтительно от -60 до -30 С. При более низкой температуре наблюдается более высокая селективность присоединения металлорганического соединения к карбонильной группе субстрата K. Однако по практическим соображениям предпочтительно проводить реакцию при температуре от -80 до -30 С. Для обеспечения полного превращения используют от 1 до 3 экв. металлорганического соединения. Предпочтительно прибавлять 2 экв. металлорганического соединения. Металлорганическое соединение можно прибавлять в неразбавленном виде или в растворе. В предпочтительном варианте осуществления металлорганическое соединение прибавляют в растворе. Растворителем может быть любой органический апротонный растворитель. Подходящими органическими растворителями являются толуол, тетрагидрофуран, диоксан, диметоксиэтан, диглим,метил-трет-бутиловый эфир или диметоксиметан. Наиболее предпочтительным растворителем является тетрагидрофуран. В предпочтительном варианте осуществления используют реагент Гриньяра. При использовании таких реагентов присоединение к карбонильной группе является быстрым и в зависимости от объема партии на нее уходит примерно 30 мин. Последующее прибавление воды, водных растворов солей, водного раствора кислоты или водного раствора основания к реакционной смеси после прибавления металлорганического соединения дает энантиомерно обогащенное соединение Р, хиральное вспомогательное соединение А и борную или бороновую кислоту. Прибавление воды, водного раствора кислоты или водного раствора основания сразу же приводит к гидролизу смешанного бората или бороната. Вместо воды можно использовать избыток C1-C10-спиртов. Таким образом, в дополнение к соединению Р и соединению А получают соответствующийC1-C10-алкиловый эфир борной или бороновой кислоты. Выделение продукта - соединения Р из реакционной смеси можно провести по методикам, известным специалисту в данной области техники, и методика выделения зависит от использующегося хирального вспомогательного соединения. Такие методики включают экстракцию, перегонку, кристаллизацию и хроматографию.-7 016650 Точный состав смеси, полученной после прибавления металлорганического соединения к смешанному борату и боронату после стадий а) и b), зависит от конкретного использующегося хирального вспомогательного соединения и условий, при которых проводят реакцию. Особенностью асимметрического присоединения, предлагаемого в настоящем изобретении, является то, что образуется значительно большее количество одного энантиомера, т.е. продукта, соединения Р, чем другого. Продукт (соединение Р) обычно получают с энантиомерным избытком (ЭИ), составляющим 50%. В предпочтительном варианте осуществления ЭИ превышает 90%. Степень превращения субстрата (соединения K) в продукт (соединение Р) составляет более 50%,обычно более 95%, более предпочтительно более 98%. Прибавление более 1 экв. металлорганического реагента (стадия с) приводит к большей степени превращения. Предпочтительно прибавляют 2 экв. металлорганического соединения. Металлорганическое соединение можно прибавить в неразбавленном виде или в растворе. Оптическую чистоту диола, полученного после выделения, можно дополнительно повысить перед последующей обработкой. Повышение оптической чистоты можно обеспечить, например, путем кристаллизации диастереоизомерных сложных эфиров или солей с оптически активными кислотами, как это описано в US 4943590, или с помощью хроматографии, как это описано в WO 03/011278, или по другим методикам. В предпочтительном варианте осуществления настоящее изобретение относится к способу получения основного промежуточного продукта для синтеза эсциталопрама, диола формулы (II). Таким образом, в предпочтительном варианте осуществления соединение K, использующееся на стадии а), представляет собой соединение формулы (III) в которой Y обозначает цианогруппу или группу, которую можно превратить в цианогруппу,металлорганическое соединение, использующееся на стадии с), представляет собой металлорганическое соединение формулы (VIII) в которой М обозначает металл;Z обозначает -CH2-N(CH3)2 или группу, которую можно превратить в -CH2-N(CH3)2, такую как-CONHR18, где Pg обозначает защитную группу гидроксигруппы, Pg1 и Pg2 обозначают защитные группы аминогруппы, R11 и R12 независимо выбраны из группы, включающей C1-C6-алкил, C2-C6-алкенил,C2-C6-алкинил и необязательно C1-C6-алкилзамещенный арил или арил-C1-C6-алкил или R11 и R12 вместе образуют цепь, содержащую 2-4 атома углерода, каждый из R13-R17 независимо выбран из группы, включающей C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил и необязательно C1-C6-алкилзамещенный арил или арил-C1-C6-алкил, R18 обозначает водород или метил и Q1, Q2 и Q3 выбраны из группы, включающей О иS; L обозначает отщепляющуюся группу, такую как галоген или -O-SO2-R11, и R11 определен выше. Такие превращения (Z в -CH2-N(CH3)2) описаны в WO 01/43525, WO 01/51478, WO 01/68631 иWO CH04/014821. Пунктирная линия в металлорганическом соединении формулы VIII может обозначать ординарную,двойную или тройную связь, и М обозначает любой металл или производное металла и предпочтительноMg и Z обозначает -CH2-N(CH3)2 или группу, которую можно превратить в -CH2-N(CH3)2 Предпочтительно, если пунктирная линия обозначает ординарную связь, М обозначает магний или хлорид магния и Z обозначает -CH2-М(CH3)2-. В другом предпочтительном варианте осуществления пунктирная линия в металлорганическом реагенте формулы (VIII) обозначает тройную связь и М обозначает магний или хлорид или бромид магния. Если пунктирная линия обозначает двойную или тройную связь, то превращение смешанного бороната V в диол (II) проводят при температуре от -40 до 40 С, более предпочтительно от 0 до 30 С. Полученное соединение можно превратить в диол формулы (II) путем восстановления. Диол формулы (II) получают в энантиомерно обогащенной или энантиомерно чистой форме в которой Y обозначает цианогруппу или группу, которую можно превратить в цианогруппу с помощью этого особенно предпочтительного способа, предлагаемого в настоящем изобретении.-8 016650 Диол формулы (II) затем можно использовать для синтеза эсциталопрама. Таким образом, настоящее изобретение также относится к способу, включающему дополнительную стадию замыкания цикла диола формулы (II) с образованием соединения формулы (IX), в которой Y и Z являются такими, как определено в настоящем изобретении. Предпочтительным соединением формулы (IX) является эсциталопрам. Превращение диола формулы (II) в эсциталопрам можно провести так, как это описано вUS 4943590. Более предпочтительно замыкание цикла соединения формулы (II) можно провести путем обработки сложного эфира угольной, карбоновой, сульфиновой или сульфоновой кислоты с помощью основания, такого как KOC(CH3)3 и другие алкоксиды, NaH или другие гидриды, третичные амины, такие как триэтиламин, этилдиизопропиламин или пиридин, при более низкой температуре в инертном органическом растворителе, таком как тетрагидрофуран, толуол, ДМСО, ДМФ, метил-трет-бутиловый эфир, диметоксиэтан, диметоксиметан, диоксан, ацетонитрил или CH2Cl2. Если Z не обозначает -CH2-N(CH3)2, то превращение группы Z в -CH2-N(CH3)2 можно провести до или после замыкания цикла, и его проводят по методикам, известным специалисту в данной области техники. Если Y не обозначает цианогруппу, то превращение Y в цианогруппу можно провести до или после замыкания цикла, и его проводят по методикам, известным специалисту в данной области техники. Если пунктирная линия обозначает двойную или тройную связь, то гидрирование можно провести до или после замыкания цикла по методикам, известным специалисту в данной области техники. Настоящее изобретение также относится к промежуточным продуктам способа, предлагаемого в настоящем изобретении, например к соединению формулы (V), которое является полезным промежуточным продуктом синтеза эсциталопрамаY обозначает цианогруппу или группу, которую можно превратить в цианогруппу;O-R обозначает остаток хирального спирта. Группой, которую можно превратить в цианогруппу, может быть хлор, бром, йод илиCF3-(CF2)n-SO2-O-, где n равно 0-8, CH2OH или защищенная CH2OH, CH2NH2 или защищенная CH2NH2,-CH2Cl, -CH2Br, -CH3, -NHR2, -OR2, в которой R2 обозначает водород или C1-C6-алкилкарбонил;C1-C6-алкил, арил-C1-C6-алкил или арил, или R3 и R4 связаны друг с другом с образованием 5- или 6-членного кольца, необязательно включающего S, О или дополнительный атом N; или CHOR5OR6, в которой R5 и R6 независимо выбраны из группы, включающей алкил, арил, гетероарил, или R5 и R6 связаны друг с другом с образованием 5- или 6-членного кольца; или защищенные группы -CHO. Y может обозначать необязательно замещенную оксазольную, 4,5-дигидрооксазольную, тиазольную или 4,5-дигидротиазольную группу. Смешанный борат или боронат формулы (V) можно выделить или превратить в диол формулы (II) в однореакторном режиме без выделения. Выделение соединения формулы (V) можно провести по методикам, известным специалисту в данной области техники. В предпочтительном варианте осуществления борат и боронат формулы V выделяют путем удаления растворителя при пониженном давлении и кристаллизации соединения путем прибавления другого растворителя. Растворителем для кристаллизации может быть, например, диэтиловый эфир или трет-бутилметиловый эфир. Однако настоящее изобретение не ограничивается этими двумя растворителями. В зависимости от аминоспирта и борной или бороновой кислоты, использующихся для получения соединения V, методика выделения может меняться.-9 016650 Смешанный боронат (V) можно выделить или превратить in situ в диол формулы (II). В предпочтительном варианте осуществления настоящего изобретения смешанный боронат/борат формулы (V) превращают непосредственно в диол (II). В другом варианте осуществления настоящее изобретение относится к гидроксикетону формулы(III) в кристаллической форме. Гидроксикетон (III) можно получить из 5-фталидных производных, в которых Y обозначает цианогруппу или группу, которую можно превратить в цианогруппу. Группы, которые можно превратить в цианогруппу, включают галоген, такой как хлор, бром или йод, предпочтительно хлор или бром. Другие группы, которые можно превратить в цианогруппу, включают CF3-(CF2)n-SO2-O-, где n равно 0-8, -OH, -CHO, -CH2OH, -CH2NH2, -CH2NO2, -CH2Cl, -CH2Br, -CH3,-NHR8, -CHNOH, -COOR9, -CONR9R10, в которой R8 обозначает водород или C1-C6-алкилкарбонил и R9 иR10 выбраны из группы, включающей водород, необязательно замещенный C1-C6-алкил,арил-C1-C6-алкил и арил. Группы, которые можно превратить в цианогруппу, также включают необязательно замещенные оксазольные, 4,5-дигидрооксазольные, тиазольные или 4,5-дигидротиазольные группы. Гидроксикетон (III) можно, например, получить из 5-цианофталида путем прибавления 4-фторфенилмагнийгалогенида, как это описано в ЕР 0171943. Галогенидом может быть хлорид, бромид или йодид. Реакцию можно провести в простом эфире в качестве растворителя, в смеси простых эфиров,в алифатических или ароматических растворителях или в их смесях. В одном варианте осуществления настоящего изобретения гидроксикетон (III) выделяют путем кристаллизации после водной обработки. Растворителем, применяющимся для кристаллизации, может быть простой эфир, алифатический или ароматический растворитель, спирт, вода или их смеси. В предпочтительном варианте осуществления Y обозначает цианогруппу и гидроксикетон (III) кристаллизуют из диизопропилового эфира, толуола или этилбензола. Наиболее предпочтительно, если соединение (III) кристаллизуют из толуола. Примеры Приведенные ниже примеры подробно описывают настоящее изобретение, однако их не следует рассматривать в качестве каким-либо образом ограничивающих настоящее изобретение. Пример 1. 1S,2S-N-Метилпсевдоэфедрин в качестве аминоспирта, -80 С в толуоле, диизопропоксиметилборан в качестве мостикового элемента, выделение однозамещенной соли [(S)-(4-диметиламино)-1(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила и (+)-ди-о-толуоилвинной кислоты. 1,44 г 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (5,6 ммоль, 1,0 экв.) и 1,01 г 1S,2S-N-метилпсевдоэфедрина (5,6 ммоль, 1,0 экв.) растворяют в двухгорлой круглодонной колбе в 20 мл толуола в инертной атмосфере (N2). При комнатной температуре прибавляют 1,17 мл диизопропоксиметилборана 97% (6,3 ммоль, 1,13 экв.). Через 2 мин получают прозрачный раствор. Реакционную смесь нагревают при 70 С в течение 30 мин. Затем реакционную смесь охлаждают до 45 С и 18 мл смеси толуол/2-пропанол осторожно удаляют при пониженном давлении (60 мбар) в течение 30 мин. Прибавляют 20 мл толуола и реакционную смесь охлаждают до -80 С. Медленно прибавляют 4,16 мл (2 экв.) 2,7 М раствора диметиламинопропилмагнийхлорида в ТГФ (тетрагидрофуран) (длительность: 5 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) указывает на степень превращения, равную 98%. Отношение количества S-диола, (4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3 гидроксиметилбензонитрила), к количеству R-диола, (4-[(R)-4-диметиламино-1-(4-фторфенил)-1 гидроксибутил]-3-гидроксиметилбензонитрила), составляет 95,0:5,0 (энантиомерный избыток=90,0%). Реакционную смесь медленно прибавляют к 12 мл холодного 2 М водного раствора H2SO4. Слои разделяют и толуольный слой один раз промывают с помощью 3 мл холодного 2 М водного раствора H2SO4. Толуольный слой отбрасывают. Водные слои объединяют и прибавляют 15 мл МТБЭ (метил-третбутиловый эфир). Значение рН доводят до 9 с помощью 5 М водного раствора NaOH. После разделения фаз водный слой еще раз экстрагируют с помощью 10 мл МТБЭ при рН 9. МТБЭ удаляют при пониженном давлении. Неочищенный продукт очищают с помощью колоночной хроматографии (элюент этилацетат/циклогексан/Et3N 1/1/0,1) на силикагеле. Фракции, содержащие продукт, объединяют и растворитель удаляют при пониженном давлении. Кристаллизация в 12 мл 2-пропанола с 1,05 г- 10016650 Пример 2. 1S,2S-N-Метилпсевдоэфедрин в качестве аминоспирта, -60 С в толуоле, диизопропоксиметилборан в качестве мостикового элемента. 143 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (0,56 ммоль, 1,0 экв.) и 101 мг 1S,2S-N-метилпсевдоэфедрина (0,56 ммоль, 1,0 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 5 мл толуола. При комнатной температуре прибавляют 118 мкл диизопропоксиметилборана 97% (0,63 ммоль, 1,14 экв.). Прозрачный раствор нагревают при 70 С в течение 30 мин. Затем реакционную смесь охлаждают до 45 С и 4,5 мл смеси толуол/2-пропанол осторожно удаляют при пониженном давлении (60 мбар) в течение 30 мин. Прибавляют 20 мл толуола и реакционную смесь охлаждают до -60 С. Медленно прибавляют 420 мкл (2 экв.) 2,7 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 5 мин). Перемешивание продолжают в течение 10 мин при -60 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 98%. Отношение количества S-диола,(4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), к количествуR-диола,(4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила),составляет 91,0:9,0 (энантиомерный избыток=82,0%). Пример 3. 1S,2S-N-Метилпсевдоэфедрин в качестве аминоспирта, -60 С в ТГФ, диизопропоксиметилборан в качестве мостикового элемента. 278 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (1,09 ммоль, 1,0 экв.) и 250 мг 1S,2S-N-метилпсевдоэфедрина (1,39 ммоль, 1,3 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 5 мл толуола. При комнатной температуре прибавляют 286 мкл диизопропоксиметилборана 97% (1,55 ммоль, 1,4 экв.). Прозрачный раствор нагревают при 70 С в течение 30 мин. Затем реакционную смесь охлаждают до 45 С и 4,5 мл смеси толуол/2-пропанол осторожно удаляют при пониженном давлении (60 мбар) в течение 30 мин. Прибавляют 5 мл тетрагидрофурана и реакционную смесь охлаждают до -60 С. Медленно прибавляют 840 мкл (2 экв.) 2,7 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 5 мин). Перемешивание продолжают в течение 10 мин при-60 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 98%. Отношение количества S-диола, (4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3 гидроксиметилбензонитрила), к количеству R-диола, (4-[(R)-4-диметиламино-1-(4-фторфенил)-1 гидроксибутил]-3-гидроксиметилбензонитрила), составляет 91,9:8,1 (энантиомерный избыток=83,8%). Пример 4. 1S,2S-N-Метилпсевдоэфедрин в качестве аминоспирта, выделение смешанного бороната,метилбороновая кислота в качестве мостикового элемента. 1,44 г 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (5,64 ммоль, 1,0 экв.) и 1,01 г 1S,2S-N-метилпсевдоэфедрина (5,64 ммоль, 1,1 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 20 мл толуола. При комнатной температуре прибавляют 348 мг метилбороновой кислоты (5,81 ммоль, 1,03 экв.). Гетерогенную смесь нагревают при 70 С. В течение 30 мин раствор становится гомогенным. Затем реакционную смесь охлаждают до 45 С и 12 мл смеси толуол/Н 2 О осторожно удаляют при пониженном давлении (60 мбар) в течение 30 мин. Прибавляют 12 мл толуола и 12 мл смеси толуол/Н 2 О осторожно удаляют при пониженном давлении (60 мбар) в течение 30 мин. Прибавляют 20 мл диэтилового эфира и реакционную смесь охлаждают до 0 С. Через 30 мин начинают осаждаться белые кристаллы. Кристаллизация завершается через 15 ч. Кристаллы отделяют фильтрованием в инертной атмосфере и получают 2,3 г (91%) смешанного бороната. 1 Н-ЯМР (CDCl3):-0,2 (s, 3H), 0,99 (d, 3H, J=7,0 Гц), 2,31 (s, 6H), 3,1-3,3 (m, 1H), 4,44 (d, 1 Н,J=9,6 Гц), 4,64 (d, 1H, J=14,9 Гц), 4,71 (d, 1H, J=14,9 Гц), 7,0-8,1 (m, 12H). 2,3 г смешанного бороната растворяют в 20 мл толуола. При -60 С медленно прибавляют 2,8 мл(2,0 экв.) 3,6 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 5 мин). Перемешивание продолжают в течение 10 мин при -60 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 95%. Отношение количества S-диола,(4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), к количествуR-диола,(4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила),составляет 89,1:10,9 (энантиомерный избыток=78,2%). Пример 5. 1S,2S-N-Метилпсевдоэфедрин в качестве аминоспирта, -65 С в толуоле, диизопропоксиметилборан в качестве мостикового элемента, выделение однозамещенной соли [(S)-(4-диметиламино)-1(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила и (+)-ди-о-толуоилвинной кислоты. 1,42 г 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (5,6 ммоль, 1,0 экв.) и 1,00 г 1S,2S-N-метилпсевдоэфедрина (5,6 ммоль, 1,0 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 20 мл толуола. При комнатной температуре прибавляют 1,17 мл диизопропоксиметилборана 97% (6,3 ммоль, 1,13 экв.). Через 2 мин получают прозрачный раствор. Реакционную смесь нагревают при 50 С в течение 30 мин. Затем реакционную смесь охлаждают до 45 С и 20 мл смеси толуол/2-пропанол осторожно удаляют при пониженном давлении (70 мбар) в течение 20 мин. Прибавляют 20 мл толуола и реакционную смесь охлаждают до -65 С. Медленно прибавляют 3,36 мл(2 экв.) 3,25 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 10 мин). Перемешивание продолжают в течение 10 мин при -65 С. Исследование протекания реакции с помощью- 11016650 ВЭЖХ указывает на степень превращения, равную 99%. Отношение количества S-диола,(4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), к количествуR-диола,(4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила),составляет 95,2:4,8 (энантиомерный избыток=90,4%). Реакционную смесь медленно прибавляют к 12 мл холодного 2 М водного раствора H2SO4. Слои разделяют и толуольный слой один раз промывают с помощью 3 мл холодного 2 М водного раствораH2SO4. Толуольный слой отбрасывают. Водные слои объединяют и прибавляют 15 мл метил-третбутилового эфира. Значение рН доводят до 9 с помощью 5 М водного раствора NaOH. После разделения фаз водный слой экстрагируют с помощью 10 мл метил-трет-бутилового эфира при рН 9. Объединенные органические слои дважды промывают с помощью 0,2 М водного раствора триметилуксусной кислоты. Объединенные слои, содержащие триметилуксусную кислоту дважды экстрагируют с помощью 10 мл метил-трет-бутилового эфира. Объединенные слои, содержащие метил-трет-бутиловый эфир (40 мл),промывают с помощью 5 мл 5 М водного раствора NaOH. После разделения фаз основную часть метилтрет-бутилового эфира удаляют при пониженном давлении. Прибавляют 12 мл 2-пропанола. При 35 С прибавляют 1,04 г (+)-ди-о-толуоилвинной кислоты. В течение 5 мин начинают образовываться кристаллы. Через 4 ч белый осадок удаляют фильтрованием и получают 2,2 г искомого соединения (выход 69%,ЭИ=99%, температура плавления: 134 С). Пример 6. 1S,2R-N-Метилэфедрин в качестве аминоспирта, -80 С, метилбороновая кислота в качестве мостикового элемента. 142 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (0,56 ммоль, 1,0 экв.) и 100 мг 1S,2R-N-метилэфедрина (0,56 ммоль, 1,0 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 2 мл толуола. При комнатной температуре прибавляют 36,1 мг метилбороновой кислоты (0,60 ммоль, 1,08 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 5 мин. 2 мл толуола прибавляют и повторно удаляют при пониженном давлении. Прибавляют 2 мл толуола и реакционную смесь охлаждают до -80 С. Медленно прибавляют 410 мкл (2 экв.) 2,7 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 10 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 90%. Отношение количества S-диола, (4-[(S)-4-диметиламино-1-(4-фторфенил)-1 гидроксибутил]-3-гидроксиметилбензонитрила), к количеству R-диола, (4-[(R)-4-диметиламино-1-(4 фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), составляет 88,0:12,0 (энантиомерный избыток=76,0%). Пример 7. 1S,2R-N-Метилэфедрин в качестве аминоспирта, -80 С, триметилбороксин в качестве мостикового элемента. 142 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (0,56 ммоль, 1,0 экв.) и 100 мг 1S,2R-N-метилэфедрина (0,56 ммоль, 1,0 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 2 мл толуола. При комнатной температуре прибавляют 26 мкл триметилбороксина(0,60 ммоль, 0,3 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 5 мин. Прибавляют 2 мл толуола и его повторно удаляют при пониженном давлении. Прибавляют 2 мл толуола и реакционную смесь охлаждают до -80 С. Медленно прибавляют 410 мкл (2 экв.) 2,7 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 10 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 98%. Отношение количества S-диола, (4-[(S)-4-диметиламино-1-(4-фторфенил)-1 гидроксибутил]-3-гидроксиметилбензонитрила), к количеству R-диола, (4-[(R)-4-диметиламино-1-(4 фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), составляет 84,2:15,8 (энантиомерный избыток=68,4%). Пример 8. (-)-Цинхонидин в качестве аминоспирта, -80 С, диизопропоксиметилборан в качестве мостикового элемента. 83 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрил (0,28 ммоль, 1,0 экв.) и 100 мг(-)-цинхонидина 96% (0,32 ммоль, 1,16 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 2 мл толуола. При комнатной температуре прибавляют 81 мкл диизопропоксиметилборана 97% (0,36 ммоль, 1,3 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 15 мин. Прибавляют 2 мл толуола и реакционную смесь охлаждают до -80 С. Медленно прибавляют 240 мкл (2 экв.) 2,7 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 10 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 96%. Отношение количества S-диола, (4-[(S)-4 диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), к количеству Rдиола, (4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), составляет 15,9:84,1 (энантиомерный избыток=68,2%).- 12016650 Пример 9. Хинидин в качестве аминоспирта, -80 С в смеси толуол/метиленхлорид, диизопропоксиметилборан в качестве мостикового элемента. 79 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (0,31 ммоль, 1,0 экв.) и 100 мг (-)-хинидина 98% (0,31 ммоль, 1,0 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 2 мл толуола. При комнатной температуре прибавляют 62 мкл диизопропоксиметилборана 97%(0,32 ммоль, 1,03 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 15 мин. Прибавляют 2 мл толуола и 2 мл метиленхлорида и реакционную смесь охлаждают до -80 С. Медленно прибавляют 230 мкл (2 экв.) 2,7 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 10 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 97%. Отношение количестваR-2-диметиламино-1-фенилэтанола 90% (0,46 ммоль, 0,9 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 5 мл толуола. При комнатной температуре прибавляют 108 мкл диизопропоксиметилборана 97% (0,57 ммоль, 1,1 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 15 мин. Прибавляют 5 мл толуола и реакционную смесь охлаждают до -80 С. Медленно прибавляют 312 мкл (2 экв.) 3,3 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 1 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 97%. Отношение количестваS-1-диметиламино-2-пропанола 98% (0,80 ммоль, 1,02 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 3 мл толуола. При комнатной температуре прибавляют 160 мкл диизопропоксиметилборана 97% (0,86 ммоль, 1,1 экв.). Температуру реакционной смеси доводят до 45 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 15 мин. Прибавляют 3 мл толуола и реакционную смесь охлаждают до -80 С. Медленно прибавляют 2,5 мл (3,1 экв.) 0,8 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 1 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 95%. Отношение количестваS-диола, (4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), к количеству R-диола, (4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), составляет 75,5:24,5 (энантиомерный избыток=51,0%). Пример 12. 1S,2R-N-Метилэфедрин в качестве аминоспирта, -80 С, триметилборат в качестве мостикового элемента. 145 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (0,56 ммоль, 1,0 экв.) и 100 мг 1S,2R-N-метилэфедрина 99% (0,56 ммоль, 1,0 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 3 мл толуола. При комнатной температуре прибавляют 160 мкл диизопропоксиметилборана (0,59 ммоль, 1,05 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 15 мин. Прибавляют 3 мл толуола и реакционную смесь охлаждают до -80 С. Медленно прибавляют 800 мкл (2 экв.) 1,3 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 2 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 90%. Отношение количества S-диола,(4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), к количествуR-диола,(4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила),составляет 71,6:28,4 (энантиомерный избыток=43,2%). В качестве побочного продукта образуется примерно 10% 4-[(4-фторфенил)гидроксиметил]-3-гидроксиметилбензонитрила.- 13016650 Пример 13. 1S,2R-N-Метилэфедрин в качестве аминоспирта, -80 С, триизопропилборат в качестве мостикового элемента. 142 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (0,56 ммоль, 1,0 экв.) и 100 мг 1S,2R-N-метилэфедрина 99% (0,56 ммоль, 1,0 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 2 мл толуола. При комнатной температуре прибавляют 135 мкл диизопропоксиметилборана (0,59 ммоль, 1,05 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 15 мин. Прибавляют 2 мл толуола и реакционную смесь охлаждают до -80 С. Медленно прибавляют 412 мкл (2 экв.) 2,7 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 2 мин). Перемешивание продолжают в течение 10 мин при -80 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 50%. Отношение количества S-диола,(4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила), к количествуR-диола,(4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила),составляет 72,2:27,8 (энантиомерный избыток=44,4%). Пример 14. 1S,2S-N-Метилэфедрин в качестве аминоспирта, -80 С в толуоле, 3,3-диметиламино-1 пропин в качестве нуклеофильного реагента, диизопропоксиметилборан в качестве мостикового элемента. 120 мкл 3,3-диметиламино-1-пропина (1,13 ммоль, 2,0 экв.) растворяют в 1 мл ТГФ. При 0 С прибавляют 372 экв. 3 М раствора диметиламинопропилмагнийхлорида в ТГФ. Полученный раствор перемешивают в течение 20 мин. Во второй двухгорлой круглодонной колбе 142 мг 4-(4-фторбензоил)-3 гидроксиметилбензонитрила (0,56 ммоль, 1,0 экв.) и 100 мг 1S,2R-N-метилэфедрина 99% (0,56 ммоль,1,0 экв.) растворяют в инертной атмосфере (N2) в 2 мл толуола. При комнатной температуре прибавляют 135 мкл диизопропоксиметилборана (0,59 ммоль, 1,05 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 15 мин. Прибавляют 2 мл толуола и реакционную смесь охлаждают до-20 С. Затем в течение 5 мин прибавляют раствор, содержащий магниевую соль 3,3-диметиламино-1 пропина. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 24 ч. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 70%. Отношение количества 4-[(S)-4-диметиламино-1-(4-фторфенил)-1-гидроксибут-2-инил]-3 гидроксиметилбензонитрила к количеству 4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибут-2 инил]-3-гидроксиметилбензонитрила составляет 90:10 (энантиомерный избыток=80%). Пример 15. Синтез 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (пример синтеза). 1048 г 10% раствора 4-фторфенилмагнийбромида в тетрагидрофуране при -10 С в течение 3 ч прибавляют к суспензии 60,0 г 5-цианофталида в 390 мл 1,2-диметоксиэтана. После перемешивания в течение 30 мин при -10 С холодную реакционную смесь в течение примерно 5 мин выливают в 1 л водного раствора NH4Cl (180 г в 1000 мл воды, 20 С). Слои разделяют и водный слой экстрагируют с помощью 300 мл тетрагидрофурана. Органические слои объединяют и летучие вещества удаляют при пониженном давлении при 45 С. Остаток растворяют в смеси 1000 мл CH2Cl2 и 200 мл воды, содержащей 2,5 г карбоната натрия (рН 9). Слои разделяют и органическую фазу сушат над 40 г карбоната натрия. Сухой раствор в CH2Cl2 обрабатывают с помощью 6 г древесного угля, перемешивают в течение 10 мин и древесный уголь удаляют фильтрованием. Осадок на фильтре промывают с помощью 50 мл CH2Cl2. Фильтрат и промывочную жидкость объединяют и растворитель удаляют при пониженном давлении. К остатку прибавляют 300 мл диизопропилового эфира. После перемешивания в течение 1 ч при 22 С суспензию кристаллов охлаждают до 0 С и перемешивают в течение еще 2 ч, затем охлаждают до -10 С и перемешивают в течение 14 ч. Продукт выделяют фильтрованием и промывают с помощью 40 мл охлажденного диизопропилового эфира, 80 мл 1:1 смеси диизопропиловый эфир/циклогексан и 80 мл циклогексана. После сушки в течение 3 ч при 50 С в вакууме получают 83,0 г (86,2 % от теоретического, чистота(ВЭЖХ по площади пика): 99,8%) белого кристаллического порошкообразного искомого соединения(т.пл. 85 С). 1 Н-ЯМР (CDCl3, 300 МГц):3,01 (t, J=6,30 Гц, 0,8 Н, ОН), 3,66 (s, 0,2 Н, ОН), 4,66 (d, J=6,11 Гц,1,6 Н, CH2-О), 5,33 (m, CH2-O, 0,4 Н, лактольный изомер), 7,03-7,93 (m, 7 Н, ArH). Пример 16. 1S,2S-N-Метилпсевдоэфедрин в качестве аминоспирта, -60 С, метилбороновая кислота в качестве мостикового элемента. 250 мг 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (0,98 ммоль, 1,0 экв.) и 263 мг 1S,2R-N-метилэфедрина 99% (1,45 ммоль, 1,48 экв.) растворяют в двухгорлой круглодонной колбе в инертной атмосфере (N2) в 15 мл толуола. При комнатной температуре прибавляют 67 мг метилбороновой кислоты (1,12 ммоль, 1,14 экв.). Температуру реакционной смеси доводят до 70 С и перемешивают при этой температуре в течение 30 мин. Растворитель осторожно удаляют при пониженном давлении в течение 15 мин. Прибавляют 10 мл толуола и его повторно осторожно удаляют при пониженном давлении. Прибавляют 5 мл толуола и реакционную смесь охлаждают до -60 С. Медленно прибавляют 2,3 мл(2 экв.) 0,84 М раствора диметиламинопропилмагнийхлорида в ТГФ (длительность: 2 мин). Перемешивание продолжают в течение 10 мин при -60 С. Исследование протекания реакции с помощью ВЭЖХ указывает на степень превращения, равную 98%. Отношение количества S-диола, (4-[(S)-4- 14016650 диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила),к количествуR-диола,(4-[(R)-4-диметиламино-1-(4-фторфенил)-1-гидроксибутил]-3-гидроксиметилбензонитрила),составляет 96,3:3,7 (энантиомерный избыток=92,6%). Пример 17. Синтез и выделение однозамещенной соли (S)-4-(4-диметиламино)-1-(4-фторфенил)-1 гидроксибутил-3-гидроксиметилбензонитрила и(S)-2-N,N-диметиламинофенилэтанола (ММ: 165,24, 47,0 ммоль, 1,24 экв.) и 2,51 г метилбороновой кислоты (ММ: 59,86, результат анализа: 98%, 41,1 ммоль, 1,08 экв.). Раствор становится немного мутным и быстро образуются капельки воды. Смесь нагревают при 50 С. При пониженном давлении (60-70 мбар) осторожно удаляют 100 мл смеси толуол/вода. Прибавляют 100 мл толуола и удаляют 100 мл смеси толуол/вода. Прибавляют 120 мл толуола и удаляют примерно 20 мл смеси толуол/вода и получают раствор смешанного бороната (80 ммоль) примерно в 250 мл растворителя. По данным титрования по Карлу Фишеру содержание воды составляет менее 0,1%. Реакционную смесь охлаждают до -65 С. В течение примерно 10-20 мин прибавляют 38,0 мл 2 М раствора диметиламинопропилмагнийхлорида в ТГФ(2 экв.). При этом температура не превышает -50 С. Раствор перемешивают в течение еще 30 мин. За протеканием реакции следят с помощью ВЭЖХ (ЭИ=90%). После полного превращения (по площади пика в % содержание 4-(4-фторбензоил)-3-гидроксиметилбензонитрила составляет 2%) реакционную смесь обрабатывают далее. Прибавляют 20 мл воды и 35 мл 2 водного раствора H2SO4 (70 ммоль) и значение рН водного слоя становится равным 1,5. После разделения фаз органический слой один раз промывают с помощью 20 мл воды, значение рН доводят до 1 с помощью 2 М водного раствора H2SO4. К объединенным водным слоям прибавляют 150 мл CH2Cl2. Затем прибавляют 7 М водный раствор NH3 (29 мл), пока значение рН не станет равным 9,0. После разделения фаз водный слой дважды промывают с помощью 25 мл MED (при рН 9,0). Объединенные слои, содержащие CH2Cl2, промывают с помощью 15 мл воды. 70 мл Н 2 О прибавляют. 10,7 мл 2 М водного раствора H2SO4 прибавляют для доведения значения рН до 6,4. После перемешивания в течение 10 мин слои разделяют (рН 6,4). К слою, содержащему CH2Cl2, прибавляют 35 мл воды. Прибавление 1,5 мл 2 М водного раствора H2SO4 приводит к рН 6,4. После перемешивания в течение 10 мин слои разделяют. К объединенным водным слоям прибавляют 80 мл CH2Cl2. Прибавление 2 мл 7 М водного раствора NH3 приводит к рН 6,4 (после установления равновесия). Слои разделяют. Объединенные органические слои промывают с помощью 20 мл воды. Слои разделяют. Объединенный слой,содержащий CH2Cl2, содержит энантиомерно обогащенный диол. Объединенный водный слой содержит 90% хирального вспомогательного соединения. Удаляют 200 мл CH2Cl2. Прибавляют 60 мл 2-пропанола. 30 мл смеси 2-пропанол/CH2Cl2 удаляют при пониженном давлении. Прибавляют 30 мл 2-пропанола и получают 13 г CIT-ДИОЛА в 60 мл ISO. К этому раствору прибавляют 6,59 г(+)-дитолуолилвинной кислоты (ММ: 386,36; результат анализа: 99%; 17,1 ммоль, 0,45 экв.), растворенной в 42 мл 2-пропанола и 8 мл CH2Cl2. Продукт начинает кристаллизоваться через 5 мин (или сразу же после внесения загравки). Смесь перемешивают в течение 90 мин при 35 С, в течение 10 мин при 60 С и затем медленно охлаждают до комнатной температуры (в течение 5 ч) и кристаллизуют без перемешивания в течение 10 ч. Продукт выделяют фильтрованием и после сушки в течение 10 ч при 40 С и 20 мбар получают 17,2 г S-CIT-ДИОЛ. 1/2(+)-ДТВК.1/2ISO.1/2 Н 2 О (выход: 81,0%; ЭИ: 99,0%, результат анализа: 61,0%). Пример 18. Синтез и выделение однозамещенной соли (S)-4-(4-диметиламино)-1-(4-фторфенил)-1 гидроксибутил-3-гидроксиметилбензонитрила и(+)-ди-п-толуоилвинной кислоты; 1S,2S-Nметилпсевдоэфедрин в качестве вспомогательного соединения. 20,68 г 4-(4-фторбензоил)-3-гидроксиметилбензонитрила (255,25, результат анализа: 96,7%; 81,0 ммоль) растворяют в 280 мл толуола. Прибавляют 16,05 г 1S,2S-N-метилпсевдоэфедрина (ММ: 179,26, результат анализа: 99,1%, 89,5 ммоль, 1,1 экв.) и 5,02 г метилбороновой кислоты (ММ: 59,86, результат анализа: 98%, 82,2 ммоль, 1,02 экв.). Раствор становится немного мутным и быстро образуются капельки воды. Смесь нагревают при 50 С. При пониженном давлении (60-70 мбар) осторожно удаляют 200-220 мл смеси толуол/вода. Прибавляют 200 мл толуола и удаляют 190-210 мл смеси толуол/вода. Прибавляют 200 мл толуола и удаляют примерно 30 мл толуола и получают раствор смешанного бороната (80 ммоль) примерно в 250 мл растворителя (содержание воды 0,1%). Реакционную смесь охлаждают до -40 С. В течение примерно 10-20 мин прибавляют 78,5 мл 2 М раствора диметиламинопропилмагнийхлорида в ТГФ (2 экв.). При этом температура не превышает -35 С. Раствор перемешивают в течение еще 30 мин. За протеканием реакции следят с помощью ВЭЖХ (ЭИ=92%). После завершения превращения реакционную смесь обрабатывают далее. В течение 5 мин прибавляют 85 мл 2 М водного раствора H2SO4 (170 ммоль). Конечное значение рН водного слоя равно 2. Органический слой дважды промывают с помощью 5 мл 2 М водного раствора H2SO4. К объединенным водным слоям прибавляют 300 мл метил-трет-бутилового эфира. Затем прибавляют 7 М водный раствор NH3, пока значение рН не станет равным 9,2. После разделения фаз водный слой дважды промывают с помощью 100 мл- 15016650 метил-трет-бутилового эфира (при рН 9,2). Объединенные слои, содержащие метил-трет-бутиловый эфир, дважды промывают с помощью 20 мл 7 М водного раствора NH3. Примерно 400 мл метил-третбутилового эфира удаляют при пониженном давлении. Прибавляют метил-трет-бутиловый эфир до полного объема, равного примерно 250 мл. 24,8 г триметилуксусной кислоты (ММ: 102,14; 242 ммоль,3,0 экв.) растворяют в 80 мл метил-трет-бутилового эфира. Этот раствор прибавляют к раствору диола в метил-трет-бутиловом эфире. Быстро кристаллизуется соль 1S,2S-N-метилпсевдоэфедрина с триметилуксусной кислотой. Смесь осторожно перемешивают в течение 30 мин при комнатной температуре и 30 мин при 0 С. NMPE.PIVOH удаляют фильтрованием. Осадок на фильтре промывают с помощью 75 мл метил-трет-бутилового эфира. После сушки (20 мбар, 40 С, 1 ч) получают 19,8 г соли 1S,2S-N-метилпсевдоэфедрина с триметилуксусной кислотой (выход: 79%; результат анализа: 98,8%). Объединенные слои, содержащие метил-трет-бутиловый эфир, промывают с помощью 60 мл 7 М водного раствора NH3 и 20 мл воды. Слои разделяют. Примерно 2/3 метил-трет-бутилового эфира удаляют при пониженном давлении и получают концентрированный раствор энантиомерно обогащенного диола в метил-трет-бутиловом эфире (60 мл). 100 мл 2-пропанол прибавляют и смесь метил-третбутиловый эфир/2-пропанол удаляют до конечного объема, равного примерно 60 мл. Прибавляют 90 мл 2-пропанола и получают 26 г диола примерно в 120 мл 2-пропанола. К этому раствору прибавляют 15,5 г (+)-дитолуолилвинной кислоты (ММ: 386,36; результат анализа: 99%; 40,1 ммоль, 0,49 экв.), растворенной в 80 мл 2-пропанола. Продукт начинает кристаллизоваться через 5 мин (или сразу же после внесения затравки). Смесь перемешивают в течение 1 ч при 30 С, в течение 10 мин при 60 С и затем медленно охлаждают до комнатной температуры (в течение 5 ч) и кристаллизуют без перемешивания в течение 10 ч. Продукт выделяют фильтрованием и после сушки в течение 6 ч при 40 С и 20 мбар получают 33,86 г однозамещенной соли (S)-4-(4-диметиламино)-1-(4-фторфенил)-1-гидроксибутил-3 гидроксиметилбензонитрила и (+)-ди-п-толуолилвинной кислоты (выход: 76,7%; ЭИ: 99,3%, результат анализа: 60,7%). Пример 19. Синтез и выделение эсциталопрамоксалата. Однозамещенную соль (S)-4-(4-диметиламино)-1-(4'-фторфенил)-1-гидроксибутил-3-гидроксиметилбензонитрила и (+)-ди-п-толуоилвинной кислоты (16,64 г; 29,7 ммоль) суспендируют в смеси 180 мл воды и 180 мл дихлорметана. После проведенного с помощью водного раствора аммиака доведения значения рН до 9 фазы разделяют. К охлажденной и высушенной органической фазе (100 мл) прибавляют триэтиламин (5,7 мл; 41 ммоль), а затем п-толуолсульфонилхлорид (6,1 г; 32 ммоль) и полученный раствор перемешивают в течение 1 ч при температуре ниже 5 С. Затем реакционную смесь промывают водой при рН 6 и рН 12, а затем проводят стадию концентрирования при пониженном давлении и разбавления ацетоном. К конечному раствору прибавляют щавелевую кислоту (2,52 г; 28 ммоль) и кристаллизуется эсциталопрамоксалат. Кристаллы собирают фильтрованием и промывают холодным ацетоном. Осадок на фильтре сушат в вакууме и получают 11,4 г эсциталопрамоксалата (чистота ВЭЖХ: 99,7%; ЭИ=98,8%). 1 Н-ЯМР (ДМСО-d6, 300 МГц):1,39-1,60 (m, 2H, CH2), 2,21-2,27 (t, 2H, CH2), 2,50 (s, 3H, CH3), 2,51(s, 3H, CH3), 2,94-2,99 (t, 2H, CH2), 5,13-5,26 (q, 2H, CH2), 7,11-7,19 (m, 2 Н, арил), 7,54-7,61 (m, 2 Н, арил),7,61-7,68 (m, 3H, арил). Пример 20. Асимметрический синтез энантиомерно обогащенного 5-(диметиламино)-2 фенилпентан-1,2-диола 500 мг 2-гидроксиацетофенона (3,60 ммоль, 1,00 экв.), 798 мг (1S,2S)-2-диметиламино-1 фенилпропан-1-ола (1S,2S-NMPE, 4,45 ммоль, 1,20 экв.) и 231 мг метилбороновой кислоты (3,89 ммоль,1,05 экв.) растворяют в 10 мл толуола. При температуре бани, равной 25 С, и 15 мбар 8 мл смеси толуол/вода удаляют при пониженном давлении. Прибавляют 8 мл толуола и 800 мг молекулярных сит типа 5. Суспензию перемешивают в течение 15 ч при -20 С. Молекулярные сита удаляют фильтрованием и в течение 130 мин при температуре бани, равной-70 С, прибавляют 5,8 мл 1,5 М раствора диметиламинопропилмагнийхлорида в ТГФ (8,7 ммоль,2,42 экв.). Реакцию останавливают путем прибавления 17 мл 1 М водного раствора KHSO4. Слои разделяют. Значение рН водного слоя доводят до 10 путем прибавления 5 М водного раствора NaOH и дважды экстрагируют с помощью 5 мл метиленхлорида. Объединенный органический слой сушат над Na2SO4,фильтруют и растворитель удаляют при пониженном давлении. Неочищенный продукт растворяют в 10 мл Et2O. Прибавляют 1,6 г целита и растворитель удаляют при пониженном давлении. Неочищенный продукт на целите дополнительно очищают с помощью колоночной хроматографии на силикагеле (20 г SiO2, элюент: этилацетат/триэтиламин 200+5). Выход: 760 мг, 95%; белое кристаллическое твердое вещество; температура плавления: 50 С.- 16016650 ЭИ=80% (определено после получения производного по описанию в публикации: Kelly, A.M.;Prez-Fuertes, Y.; Arimori, S.; Bull, S.D. Org. Lett. 2006, 8, 1971). 1 Н-ЯМР (CDCl3):1,23-1,40 (m, 2H), 1,90-2,15 (m, 2H), 2,08 (s, 6H), 2,30 (m, 2H), 3,53 (d, 1H,J=10,8 Гц), 3,55 (d, 1H, J=10,8 Гц), 7,21 (m, 1H), 7,31 (m, 2H), 7,41 (m, 2H). 13 С-ЯМР (CDCl3): 21,8, 37,6, 44,9 (2C), 60,0, 72,3, 75,6, 126,0 (2C), 126,5, 128,2 (2C), 145,3. Пример 21. Асимметрический синтез энантиомерно обогащенного 3-(4-фторфенил)бутан-1,3-диола(5,66 ммоль, 1,05 экв.) растворяют в 15 мл толуола. При 25 С и при пониженном давлении (10 мбар) 8 мл смеси толуол/вода удаляют с помощью азеотропной перегонки. Прибавляют 8 мл толуола и его повторно удаляют при пониженном давлении. Прибавляют 5 мл толуола и 300 мг молекулярных сит типа 5. Реакционную смесь перемешивают в течение 10-15 ч при -20 С. Молекулярные сита удаляют фильтрованием. При температуре, равной -60 С, 13 мл прибавляют 0,83 М 4-фторфенилмагнийбромида в ТГФ (10,8 ммоль, 2,00 экв.). Степень превращения определяют с помощью ТСХ (тонкослойная хроматография) (элюент: этилацетат/циклогексан 1+1). Реакцию останавливают через 45 мин путем прибавления 10 мл 2,5 М водного раствора NaOH. Слои разделяют и органический слой дважды промывают с помощью 10 мл 1 М водного раствора KHSO4 и один раз с помощью 10 мл насыщенного водного раствора NaHCO3. Объединенный органический слой сушат над Na2SO4,фильтруют и растворитель удаляют при пониженном давлении. Неочищенный продукт растворяют в 10 мл Et2O. Прибавляют 2 г целита и растворитель удаляют при пониженном давлении. Неочищенный продукт на целите дополнительно очищают с помощью колоночной хроматографии на силикагеле (14 гSiO2, элюент: этилацетат/циклогексан 1+10). Выход: 665 мг, 83%; белое кристаллическое твердое вещество; температура плавления: 73 С. ЭИ=55% (ВЭЖХ). 1 Н-ЯМР (ДМСО-d6):1,42 (s, 3 Н), 1,90 (t, 2 Н, J=7,3 Гц), 2,50 (s, ОН, 1H), 3,27 (m, 1H), 3,42 (m, 1H),7,09 (m, 2 Н), 7,44 (m, 2 Н). 13 С-ЯМР (ДМСО-d6):30,8, 46,2, 57,8, 72,7, 114,3, 114,6, 126,9, 127,0, 142,0, 161,7 (d, JCF=242,6 Гц). Пример 22. Асимметрический синтез энантиомерно обогащенного 4-бутил-2-метил-4-фенил-1,3,2 диоксаборолана (ссылочный пример) 500 мг 2-гидроксиацетофенона (3,60 ммоль, 1,00 экв.), 798 мг (1S,2S)-2-диметиламино-1 фенилпропан-1-ола (1S,2S-NMPE, 4,45 ммоль, 1,20 экв.) и 231 мг метилбороновой кислоты (3,89 ммоль,1,05 экв.) растворяют в 10 мл толуола. При 25 С и при пониженном давлении (10-15 мбар) 8 мл смеси толуол/вода удаляют с помощью азеотропной перегонки. Прибавляют 8 мл толуола и его повторно удаляют при пониженном давлении. Прибавляют 5 мл толуола и 400 мг молекулярных сит типа 5. Реакционную смесь перемешивают в течение 5 ч при 20 С. При температуре, равной -70 С, прибавляют 3,6 мл 2 М раствора бутилмагнийхлорида в ТГФ(7,20 ммоль, 2,00 экв.). Степень превращения определяют с помощью ТСХ (элюент: этилацетат/циклогексан 1+10). Реакцию останавливают через 45 мин путем прибавления 15 мл 1 М водного раствора KHSO4. Слои разделяют и органический слой промывают с помощью 10 мл 1 М водного раствораKHSO4, затем дважды с помощью 10 мл насыщенного водного раствора NaHCO3. Объединенный органический слой сушат над Na2SO4, фильтруют и растворитель удаляют при пониженном давлении. Неочищенный продукт растворяют в 10 мл Et2O. Прибавляют 1,1 г целита и растворитель удаляют при пониженном давлении. Неочищенный продукт на целите дополнительно очищают с помощью колоночной хроматографии на силикагеле (20 г SiO2, элюент: этилацетат/циклогексан 1+10). Выход: 110 мг; масло. 1 Н-ЯМР (CDCl3):0,44 (s, 3 Н), 0,89 (t, 3H, J=7,0 Гц), 1,15 (m, 1H), 1,36 (m, 3 Н), 1,91 (m, 2 Н), 4,24 (d,1H, J=8,8 Гц), 4,33 (d, 1 Н, J=8,8 Гц), 7,34 (m, 5 Н). 13 С-ЯМР (CDCl3):14,1, 22,9, 25,8, 42,8, 77,2, 85,4, 124,6, 125,6, 127,1, 127,4, 128,4, 145,7. Для определения ЭИ боронат гидролизуют водным раствором Н 2 О 2. Значение ЭИ определяют с помощью ВЭЖХ (ЭИ=40%).- 17016650 Пример 23. Асимметрический синтез энантиомерно обогащенного 4-(1-(4-фторфенил)-1-гидрокси 3-метилбутил)-3-(гидроксиметил)бензонитрила(1S,2S)-2-диметиламино-1-фенилпропан-1-ола (1S,2S-NMPE, 2,37 ммоль, 1,20 экв.) и 126 мг метилбороновой кислоты (2,10 ммоль, 1,05 экв.) растворяют в 10 мл толуола. При 35 С и при пониженном давлении (10-15 мбар) 7 мл смеси толуол/вода удаляют с помощью азеотропной перегонки. Прибавляют 7 мл толуола и его повторно удаляют при пониженном давлении. Прибавляют 5 мл толуола и 200 мг молекулярных сит типа 5. Реакционную смесь перемешивают в течение 3 ч при 25 С. При температуре,равной -70 С, прибавляют 2,5 мл 2 М раствора изобутилмагнийхлорида в ТГФ (5,0 ммоль, 2,6 экв.). Степень превращения определяют с помощью ТСХ (элюент: этилацетат/циклогексан 1+1). Реакцию останавливают через 120 мин путем прибавления 15 мл 1 М водного раствора KHSO4. Слои разделяют и органический слой промывают с помощью 10 мл 1 М водного раствора KHSO4, затем дважды с помощью 10 мл насыщенного водного раствора NaHCO3. Объединенный органический слой сушат над Na2SO4,фильтруют и растворитель удаляют при пониженном давлении. Неочищенный продукт растворяют в 10 мл Et2O. Прибавляют 1,1 г целита прибавляют и растворитель удаляют при пониженном давлении. Неочищенный продукт на целите дополнительно очищают с помощью колоночной хроматографии на силикагеле (20 г SiO2, элюент: этилацетат/циклогексан 1+10). Выход: 180 мг 4-(1-(4-фторфенил)-1-гидрокси-3-метилбутил)-3-(гидроксиметил)бензонитрила; ЭИ=60%; белое кристаллическое твердое вещество; температура плавления: 82 С. 1 Н-ЯМР (CDCl3):0,73 (d, 3H, J=6,7 Гц), 0,96 (d, 3H, J=6,7 Гц), 1,51 (m, 1H), 2,17 (m, 2 Н), 3,60 (s,ОН), 4,21 (d, 1H, J=12,9 Гц), 4,29 (d, 1H, J=12,9 Гц), 6,99 (m, 2 Н), 7,26 (m, 2 Н), 7,58-7,74 (m, 3 Н). 13 С-ЯМР (CDCl3):23,8, 24,9, 27,0, 51,5, 63,6, 79,2, 111,6, 115,0, 115,3, 118,5, 127,5, 127,5, 127,6,131,4, 135,0, 140,7, 141,4, 151,0, 161,7 (d, JCF=247,2 Гц). 295 мг 4-4-фторфенил)(гидрокси)метил)-3-(гидроксиметил)бензонитрила, бесцветное масло; ЭИ=60% (определено после получения производного по описанию в публикации: Kelly, A.M.;Prez-Fuertes, Y.; Arimori, S.; Bull, S.D. Org. Lett. 2006, 8, 1971). 1 Н-ЯМР (CDCl3):0,73 (d, 3H, J=6,7 Гц), 0,96 (d, 3H, J=6,7 Гц), 1,51 (m, 1H), 2,17 (m, 2H), 3,60 (s,OH), 4,21 (d, 1H, J=12,9 Гц), 4,29 (d, 1H, J=12,9 Гц), 6,99 (m, 2H), 7,26 (m, 2H), 7,58-7,74 (m, 3H). 13 С-ЯМР (CDCl3):23,78, 24,87, 27,00, 51,53, 63,57, 79,18, 111,58, 115,04, 115,33, 118,50, 127,48,127,54, 127,58, 131,42, 134,96, 140,66, 141,37, 150,95, 161,74 (d, JCF=247,2 Гц). Пример 24. Асимметрический синтез энантиомерно обогащенного 4-(1-(4-фторфенил)-1 гидроксипентил)-3-(гидроксиметил)бензонитрила(1S,2S)-2-диметиламино-1-фенилпропан-1-ола (1S,2S-NMPE, 4,68 ммоль, 1,20 экв.) и 250 мг метилбороновой кислоты (4,20 ммоль, 1,05 экв.) растворяют в 30 мл толуола. При 65 С и при пониженном давлении (85-110 мбар) 20 мл смеси толуол/вода удаляют с помощью азеотропной перегонки. Прибавляют 20 мл толуола и 500 мг молекулярных сит типа 5. Реакционную смесь перемешивают в течение 60 мин при 25 С. При температуре, равной -50 С, прибавляют 3,9 мл 2 М раствора бутилмагнийхлорида в ТГФ. Степень превращения определяют с помощью ТСХ (элюент: этилацетат/циклогексан 1+1). Через 50 мин смесь фильтруют и затем реакцию останавливают путем прибавления 10 мл 1 М водного раствораKHSO4. Слои разделяют и органический слой дважды промывают с помощью 10 мл насыщенного водного раствора NaHCO3. Объединенный органический слой сушат над Na2SO4, фильтруют и растворитель удаляют при пониженном давлении. Неочищенный продукт растворяют в 15 мл метиленхлорида. Прибавляют 4 г целита и растворитель удаляют при пониженном давлении. Неочищенный продукт на целите дополнительно очищают с помощью колоночной хроматографии на силикагеле (40 г SiO2, элюент: этилацетат/циклогексан 1+5). Выход: 675 мг (57%); ЭИ=91%; желтоватое масло. 1 Н-ЯМР (CDCl3):0,90 (t, 3H, J=7,0 Гц), 1,11 (m, 1H), 1,32 (m, 3 Н), 2,2 (m, 2 Н), 4,14 (d, 1H,J=12,9 Гц), 4,26 (d, 1H, J=12,9 Гц), 6,97 (t, 2 Н, J=8,6 Гц), 7,20 (m, 2 Н), 7,52 (s, 1H), 7,64 (m, 2 Н). 13 С-ЯМР (CDCl3):14,0, 23,0, 25,5, 42,9, 63,4, 78,6, 111,4, 114,8, 115,1, 118,5, 127,3, 127,4, 127,6,131,4, 134,8, 140,8, 141,3, 150,5, 161,7 (d, JCF=247,2 Гц).- 18016650 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ асимметрического алкилирования карбонильной группы в соединении (соединении K),содержащем карбонильную группу и связывающую группу, выбранную из группы, включающей гидроксигруппу, аминогруппу и сульфгидрильную группу, включающий стадии:a) смешивания соединения K, хирального вспомогательного соединения (соединения А) и производного борной или бороновой кислоты с образованием замещенного производного борной или бороновой кислоты, в котором атом бора связывает хиральное вспомогательное соединение с соединением K; иb) прибавления металлорганического соединения; где соединение K выбрано из группы, включающей альфа-, бета-, гамма- и дельта-гидроксикетоны или альдегиды, альфа-, бета-, гамма- и дельта-аминокетоны или альдегиды и альфа-, бета-, гамма- и дельта-сульфгидрилкетоны или альдегиды; а производное борной или бороновой кислоты представляет собой соединение формулы (VI)R2 и R3 связаны друг с другом с образованием 5-10-членной циклической структуры, включающей атом бора, к которой присоединены R2 и R3, и циклическая структура может содержать 1 или 2 дополнительных атома бора, и/или кислорода, и/или азота. 2. Способ по п.1, который осуществляют в однореакторном режиме. 3. Способ по любому из пп.1 или 2, в котором из реакционной смеси в основном удаляют побочные продукты стадии а) перед проведением стадии b). 4. Способ по любому из пп.1-3, в котором R1 обозначает C1-C10-алкил или C1-C10-алкоксигруппу. 5. Способ по п.4, в котором R1 обозначает C1-C6-алкил. 6. Способ по п.5, в котором R1 обозначает метил или этил. 7. Способ по любому из пп.1-6, в котором R2 и R3 являются одинаковыми и обозначают гидроксигруппу или C1-C10-алкоксигруппу. 8. Способ по п.7, в котором реакционную смесь подвергают перегонке до проведения стадии b) для удаления воды или C1-C10-алканола. 9. Способ по любому из пп.1-8, в котором производное борной или бороновой кислоты выбрано из группы, включающей фенилбороновую кислоту, триметилборат, триизопропилборат, диизопропилбутилборонат, диизопропилметилборонат, метилбороновую кислоту и триметилбороксин. 10. Способ по п.9, в котором производное борной или бороновой кислоты выбрано из группы,включающей диизопропилметилборонат, метилбороновую кислоту и триметилбороксин. 11. Способ по п.9, в котором связывающая группа представляет собой гидроксигруппу. 12. Способ по любому из пп.1-11, в котором атом углерода карбонильной группы отделен от атома,несущего связывающую группу, с помощью 2-3 атомов углерода. 13. Способ по любому из пп.1-12, в котором хиральное вспомогательное соединение представляет собой хиральный спирт. 14. Способ по п.13, в котором хиральный спирт включает структурный элемент формулы (VII) где С обозначает хиральный атом углерода;X обозначает гетероатом, обладающий неподеленной электронной парой. 15. Способ по п.14, в котором n равно 1 или X обозначает азот. 16. Способ по п.15, в котором хиральный спирт представляет собой хиральный аминоспирт. 17. Способ по п.16, в котором хиральный аминоспирт выбран из группы, включающей- 19016650 18. Способ по любому из пп.1-17, в котором металлорганическое соединение представляет собой магнийорганическое соединение, цинкорганическое соединение, кадмийорганическое соединение, церийорганическое соединение, литийорганическое соединение, титанорганическое соединение, марганецорганическое соединение, железоорганическое соединение, алюминийорганическое соединение или оловоорганическое соединение. 19. Способ по пп.1-18, в котором металлорганическое соединение представляет собой магнийорганическое соединение. 20. Способ по п.18, в котором магнийорганическое соединение представляет собой алкилмагний,алкенилмагний или алкинилмагний. 21. Способ по любому из пп.1-20, в котором соединение K выбрано из группы, включающей гаммагидроксикетоны, гамма-аминокетоны и гамма-сульфгидрилкетоны. 22. Способ по любому из пп.1-21, в котором соединение K является соединением формулы (III) где Y обозначает цианогруппу или группу, которую можно превратить в цианогруппу. 23. Способ по п.22, в котором металлорганическое соединение представляет собой металлорганическое соединение формулы (VIII) где пунктирная линия обозначает одинарную, двойную или тройную связь; М обозначает металл или производное металла;Z обозначает -CH2-N(CH3)2 или группу, которую можно превратить в -CH2-N(CH3)2. 24. Способ по п.23, где М обозначает Mg и Z обозначает -CH2-N(CH3)2 или группу, которую можно превратить в -CH2-N(CH3)2, и где диол формулы (II) получают в энантиомерно обогащенной или энантиомерно чистой форме где Y обозначает цианогруппу или группу, которую можно превратить в цианогруппу. 25. Способ по п.24, дополнительно включающий стадию замыкания цикла диола формулы (II) с образованием соединения формулы (IX) в которой Y и Z являются такими, как определено в п.24 26. Способ по п.25, в котором соединение формулы (IX) представляет собой эсциталопрам.
МПК / Метки
МПК: C07C 215/32, C07D 307/87, C07C 215/30
Метки: алкилирования, способ, асимметрического, карбонильной, группы
Код ссылки
<a href="https://eas.patents.su/21-16650-sposob-asimmetricheskogo-alkilirovaniya-karbonilnojj-gruppy.html" rel="bookmark" title="База патентов Евразийского Союза">Способ асимметрического алкилирования карбонильной группы</a>
Антиокислители, содержащие группы фенола и группы ароматического амина

Номер патента: 646
Опубликовано: 29.12.1999
Авторы: Кноблох Геррит, Майер Ганс Рудольф
МПК: C07C 323/60, C09K 15/28, C08K 5/20...
Метки: антиокислители, группы, фенола, ароматического, содержащие, амина
Формула / Реферат:
1. Соединение формулы I где R обозначает C1-C18-алкил, С5-С8-циклоалкил, С7-С9-фенилалкил, -CH2-A-R2 или группу формулы II, R1 обозначает C1-C18-алкил, С5-С8-циклоалкил, С7-С9-фенилалкил или группу формулы (II), R2 обозначает С4-С18-алкил, -(CH2)m-COOR5 или группу формулы III, R3 обозначает водород, С5-С8-циклоалкил или С1-С12-алкил, R4 обозначает C1-C18-алкил, С5-С8-циклоалкил, С7-С9-фенилалкил или группу формулы II или IV ...
Способ алкилирования затрудненных сульфонамидов

Номер патента: 2277
Опубликовано: 28.02.2002
Автор: Хокинс Джоел Майкл
МПК: A61P 19/00, C07D 309/14, C07C 311/19...
Метки: затрудненных, сульфонамидов, способ, алкилирования
Формула / Реферат:
1. Соединение формулы где R1 представляет собой возможно замещенный бензил; R2 и R3 независимо представляют собой (С1-С6)алкил, либо R2 и R3, взятые вместе, образуют 3-7-членное циклоалкильное, пиран-4-ильное кольцо или бициклическое кольцо формулы где звездочка указывает на углеродный атом, общий для R2 и R3; Q представляет собой (С1-С6)алкил, (С6-С10)арил, (С2-С9)гетероарил, (С6-С10)арил(С1-С6)алкил, (С2-С9)гетероарил(С1-С6)алкил,...
Способ осуществления алкилирования

Номер патента: 10795
Опубликовано: 30.12.2008
Авторы: Грэй Роберт М., Ховис Кит В.
МПК: C07C 2/60
Метки: способ, осуществления, алкилирования
Формула / Реферат:
1. Способ осуществления алкилирования, включающий: a) смешение i) первого, содержащего изобутан, потока поступающего материала, выбранного из группы, включающей часть потока нового изобутана, часть потока рециклированного изобутана и их комбинации; и ii) первого потока олефина с потоком катализатора алкилирования в первой реакционной зоне с образованием вследствие этого выходного потока первой реакционной зоны; b) подачу упомянутого выходного...
Способ алкилирования ароматических углеводородов с реакционной дистилляцией

Номер патента: 6785
Опубликовано: 28.04.2006
Авторы: Шура Дэниел П., Мьюррей Джой Л., Виндер Дж.Бэрри, Уэрри Доналд Л., Соренсен Вэйн Л., Браун Мэри Дж., Хоув Ричард С., Скелл Джон Р.
МПК: C07C 2/64
Метки: способ, углеводородов, алкилирования, ароматических, реакционной, дистилляцией
Формула / Реферат:
1. Способ непрерывного получения моноалкилированных ароматических соединений в реакционно-дистилляционной колонне, содержащей реакционную зону, первую зону ректификации в верхней части дистилляционной колонны и вторую зону ректификации ниже указанной реакционной зоны и дополнительно содержащей твердый кислотный катализатор алкилирования на носителе в реакционной зоне, причем указанный способ включает A) введение в дистилляционную колонну в точке...
Способ алкилирования с реакционной дистилляцией, включающий в себя регенерацию катализатора in situ

Номер патента: 6786
Опубликовано: 28.04.2006
Авторы: Гейтс Фрэнк, Уэрри Доналд Л., Шура Дэниел П., Мьюррей Джой Л., Браун Мэри Дж., Скелл Джон Р., Хоув Ричард С., Соренсен Вэйн Л., Виндер Дж.Бэрри
МПК: B01J 38/38, B01J 38/56, B01J 38/50...
Метки: реакционной, дистилляцией, себя, алкилирования, включающий, катализатора, регенерацию, способ
Формула / Реферат:
1. Способ регенерации твердого кислотного катализатора алкилирования в процессе алкилирования с реакционной дистилляцией, имеющей место в дистилляционной колонне высокого давления, содержащей промежуточную зону каталитической реакции, в которой имеется твердый кислотный катализатор алкилирования на носителе, первую ректификационную зону в верхней части дистилляционной колонны и вторую ректификационную зону, содержащую ребойлер, ниже указанной...